
Fabricante de medicamentos: Astellas Pharma US, Inc. (Updated: 2025-01-22)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
XTANDI® (enzalutamida) cápsulas, para administración oral
XTANDI® (enzalutamida) comprimidos, para administración oral
Aprobación inicial en EE. UU.: 2012
CAMBIOS IMPORTANTES RECIENTES
INDICACIONES Y USO
XTANDI es un inhibidor del receptor de andrógenos indicado para el tratamiento de pacientes con:
POSOLOGÍA Y ADMINISTRACIÓN
- •
- Tome XTANDI 160 mg administrado por vía oral una vez al día con o sin alimentos. (2.1)
- •
- Aconseje a los pacientes que tomen cada cápsula o comprimido entero con una cantidad suficiente de agua para asegurar que todo el medicamento se trague correctamente. (2.1, 5.7)
- •
- Los pacientes que reciben XTANDI para el cáncer de próstata resistente a la castración o el cáncer de próstata sensible a la castración metastásico también deben recibir un análogo de la hormona liberadora de gonadotropina (GnRH) de forma concomitante o deben haberse sometido a una orquiectomía bilateral. (2.1)
- •
- Los pacientes con cáncer de próstata sensible a la castración no metastásico con recurrencia bioquímica con alto riesgo de metástasis pueden tratarse con o sin un análogo de GnRH. (2.1)
CONTRAINDICACIONES
Ninguna. (4)
ADVERTENCIAS Y PRECAUCIONES
- •
- Se produjeron convulsiones en el 0,6 % de los pacientes que recibieron XTANDI. En pacientes con factores predisponentes, se notificaron convulsiones en el 2,2 % de los pacientes. Suspenda permanentemente XTANDI en pacientes que presenten una convulsión durante el tratamiento. (5.1)
- •
- Síndrome de encefalopatía posterior reversible (PRES): Suspenda XTANDI. (5.2)
- •
- Hipersensibilidad: Suspenda XTANDI. (5.3)
- •
- Cardiopatía isquémica: Optimice el control de los factores de riesgo cardiovascular. Suspenda XTANDI para reacciones adversas de grado 3-4 (5.4)
- •
- Caídas y fracturas: Evalúe a los pacientes para detectar el riesgo de fracturas y caídas, y trate a los pacientes con agentes dirigidos al hueso de acuerdo con las pautas establecidas. (5.5)
- •
- Toxicidad embriofetal: XTANDI puede causar daño fetal y pérdida del embarazo. Aconseje a los hombres con parejas femeninas en edad fértil que utilicen métodos anticonceptivos eficaces. (5.6, 8.1, 8.3)
- •
- Disfagia grave o atragantamiento relacionado con el tamaño del producto: Considere el uso de comprimidos de XTANDI más pequeños en pacientes que tienen dificultad para tragar. Suspenda XTANDI para los pacientes que no puedan tragar cápsulas o comprimidos. (2.1, 5.7)
REACCIONES ADVERSAS
Las reacciones adversas más comunes (≥ 10 %) que ocurrieron con más frecuencia (≥ 2 % sobre placebo) en los pacientes tratados con XTANDI son dolor musculoesquelético, fatiga, sofocos, estreñimiento, disminución del apetito, diarrea, hipertensión, hemorragia, caídas, fracturas y cefalea. (6.1)
Para notificar SOSPECHAS DE REACCIONES ADVERSAS, póngase en contacto con Astellas Pharma US, Inc. al 1-800-727-7003 o con la FDA al 1-800-FDA-1088 o www.fda.gov/medwatch.
INTERACCIONES MEDICAMENTOSAS
- •
- Inhibidores potentes del CYP2C8: Evite los inhibidores potentes del CYP2C8. Si no se puede evitar la administración conjunta, reduzca la dosis de XTANDI. (2.3, 7.1)
- •
- Inductores potentes del CYP3A4: Evite los inductores potentes del CYP3A4. Si no se puede evitar la administración conjunta, aumente la dosis de XTANDI. (2.3, 7.1)
- •
- Evite la administración conjunta con ciertos sustratos del CYP3A4, CYP2C9 o CYP2C19 para los que una disminución mínima de la concentración puede provocar un fracaso terapéutico del sustrato. En los casos en que se formen metabolitos activos, puede haber una mayor exposición a los metabolitos activos. (7.2)
Consulte el apartado 17 para obtener información sobre el asesoramiento al paciente y el etiquetado del paciente aprobado por la FDA.
Revisado: 1/2025
Tabla de contenido
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDO*
1 INDICACIONES Y USO
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Dosis Recomendada
2.2 Modificaciones de la Dosis para Reacciones Adversas
2.3 Modificaciones de la Dosis para Interacciones Medicamentosas
3 FORMAS Y CONCENTRACIONES FARMACÉUTICAS
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Convulsiones
5.2 Síndrome de Encefalopatía Posterior Reversible (PRES)
5.3 Hipersensibilidad
5.4 Cardiopatía Isquémica
5.5 Caídas y Fracturas
5.6 Toxicidad Embrio-Fetal
5.7 Disfagia o Atragantamiento
6 REACCIONES ADVERSAS
6.1 Experiencia en Ensayos Clínicos
6.2 Experiencia Post-Comercialización
7 INTERACCIONES MEDICAMENTOSAS
7.1 Efecto de Otros Medicamentos sobre XTANDI
7.2 Efecto de XTANDI sobre Otros Medicamentos
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.3 Mujeres y Hombres en Edad Reproductiva
8.4 Uso Pediátrico
8.5 Uso Geriátrico
8.6 Insuficiencia Renal
8.7 Insuficiencia Hepática
10 SOBREDOSIFICACIÓN
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de Acción
12.2 Farmacodinámica
12.3 Farmacocinética
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
14 ESTUDIOS CLÍNICOS
16 PRESENTACIÓN/ALMACENAMIENTO Y MANEJO
17 INFORMACIÓN PARA EL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información completa de prescripción no se enumeran.
1 INDICACIONES Y USO
XTANDI® está indicado para el tratamiento de pacientes con:
- •
- cáncer de próstata resistente a la castración (CRPC)
- •
- cáncer de próstata sensible a la castración metastásico (mCSPC)
- •
- cáncer de próstata sensible a la castración no metastásico (nmCSPC) con recurrencia bioquímica con alto riesgo de metástasis (BCR de alto riesgo)
2 DOSIS Y ADMINISTRACIÓN
2.1 Dosis recomendada
La dosis recomendada de XTANDI es de 160 mg administrada por vía oral una vez al día con o sin alimentos [ver Farmacología clínica (12.3)] hasta la progresión de la enfermedad o toxicidad inaceptable.
Indique a los pacientes que tomen cada cápsula o comprimido entero con una cantidad suficiente de agua para asegurar que todo el medicamento se trague correctamente. NO mastique, disuelva ni abra las cápsulas. NO corte, triture ni mastique los comprimidos.
Los pacientes con CPRC o mCSPC que reciben XTANDI también deben recibir un análogo de la hormona liberadora de gonadotropina (GnRH) de forma concomitante o deben haberse sometido a una orquiectomía bilateral.
Los pacientes con nmCSPC con BCR de alto riesgo pueden ser tratados con XTANDI con o sin un análogo de GnRH. Para los pacientes que reciben XTANDI con o sin un análogo de GnRH, el tratamiento puede suspenderse si el PSA es indetectable (< 0,2 ng/mL) después de 36 semanas de terapia. Reinicie el tratamiento cuando el PSA haya aumentado a ≥ 2,0 ng/mL para los pacientes que se sometieron a una prostatectomía radical previa o ≥ 5,0 ng/mL para los pacientes que se sometieron a radioterapia primaria previa [ver Estudios clínicos (14)].
2.2 Modificaciones de la dosis para reacciones adversas
Si un paciente experimenta una reacción adversa ≥ Grado 3 o intolerable, suspenda XTANDI durante una semana o hasta que los síntomas mejoren a ≤ Grado 2, luego reanude con la misma dosis o una dosis reducida (120 mg u 80 mg) si es necesario [ver Advertencias y precauciones (5.1, 5.2)].
2.3 Modificaciones de la dosis para interacciones medicamentosas
Inhibidores potentes del CYP2C8
Evite la coadministración de inhibidores potentes del CYP2C8. Si no se puede evitar la coadministración de un inhibidor potente del CYP2C8, reduzca la dosis de XTANDI a 80 mg una vez al día. Si se suspende la coadministración del inhibidor potente, aumente la dosis de XTANDI a la dosis utilizada antes del inicio del inhibidor potente del CYP2C8 [ver Farmacología clínica (12.3)].
Inductores potentes del CYP3A4
Evite la coadministración de inductores potentes del CYP3A4. Si no se puede evitar la coadministración de un inductor potente del CYP3A4, aumente la dosis de XTANDI de 160 mg a 240 mg por vía oral una vez al día. Si se suspende la coadministración del inductor potente del CYP3A4, disminuya la dosis de XTANDI a la dosis utilizada antes del inicio del inductor potente del CYP3A4 [ver Farmacología clínica (12.3)].
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
Las cápsulas de XTANDI 40 mg son cápsulas blandas de gelatina oblongas, de color blanco a blanquecino, impresas con tinta negra con las siglas ENZ.
Los comprimidos de XTANDI 40 mg son de color amarillo, redondos, recubiertos con película y con la inscripción E 40.
Los comprimidos de XTANDI 80 mg son de color amarillo, ovalados, recubiertos con película y con la inscripción E 80.
4 CONTRAINDICACIONES
Ninguna.
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Convulsiones
Las convulsiones se produjeron en el 0,6% de los pacientes que recibieron XTANDI en ocho ensayos clínicos aleatorizados. En estos ensayos, generalmente se excluyó a los pacientes con factores predisponentes para las convulsiones. Las convulsiones se produjeron de 13 a 2250 días después del inicio del tratamiento con XTANDI. Los pacientes que experimentaron convulsiones se suspendieron permanentemente del tratamiento, y todos los episodios convulsivos se resolvieron.
En un ensayo de un solo brazo diseñado para evaluar el riesgo de convulsiones en pacientes con factores predisponentes para las convulsiones, 8 de 366 (2,2%) pacientes tratados con XTANDI experimentaron una convulsión. Tres de los 8 pacientes experimentaron una segunda convulsión durante el tratamiento continuado con XTANDI después de que se resolviera su primera convulsión. Se desconoce si los medicamentos antiepilépticos prevendrán las convulsiones con XTANDI. Los pacientes del estudio tenían uno o más de los siguientes factores predisponentes: el uso de medicamentos que pueden disminuir el umbral convulsivo (~ 54%), antecedentes de traumatismo craneoencefálico (~ 28%), antecedentes de accidente cerebrovascular o ataque isquémico transitorio (~ 24%), y enfermedad de Alzheimer, meningioma o enfermedad leptomeníngea por cáncer de próstata, pérdida de conocimiento inexplicable en los últimos 12 meses, antecedentes de convulsiones, presencia de una lesión ocupante de espacio en el cerebro, antecedentes de malformación arteriovenosa o antecedentes de infección cerebral (todos < 5%). Aproximadamente el 17% de los pacientes tenían más de un factor de riesgo.
Avise a los pacientes del riesgo de desarrollar una convulsión mientras reciben XTANDI y de participar en cualquier actividad en la que una pérdida repentina del conocimiento pueda causar daño grave a sí mismos o a otros.
Suspenda permanentemente XTANDI en pacientes que desarrollen una convulsión durante el tratamiento.
5.2 Síndrome de encefalopatía posterior reversible (PRES)
Se han notificado casos de síndrome de encefalopatía posterior reversible (PRES) en pacientes que reciben XTANDI [véase Reacciones adversas (6.2)]. El PRES es un trastorno neurológico que puede presentarse con síntomas de evolución rápida, como convulsiones, dolor de cabeza, letargo, confusión, ceguera y otros trastornos visuales y neurológicos, con o sin hipertensión asociada. El diagnóstico de PRES requiere confirmación mediante imágenes cerebrales, preferiblemente resonancia magnética (RM). Suspenda XTANDI en pacientes que desarrollen PRES.
5.3 Hipersensibilidad
Se han observado reacciones de hipersensibilidad, incluido edema de la cara (0,5%), la lengua (0,1%) o el labio (0,1%) con enzalutamida en ocho ensayos clínicos aleatorizados. Se ha notificado edema faríngeo en casos posteriores a la comercialización. Avise a los pacientes que experimenten cualquier síntoma de hipersensibilidad para que suspendan temporalmente XTANDI y busquen atención médica inmediata. Suspenda permanentemente XTANDI en caso de reacciones de hipersensibilidad graves.
5.4 Cardiopatía isquémica
En los datos combinados de cinco estudios clínicos aleatorizados y controlados con placebo, la cardiopatía isquémica se produjo con más frecuencia en los pacientes del grupo de XTANDI que en los pacientes del grupo de placebo (3,5% frente a 2%). Los eventos isquémicos de grado 3-4 se produjeron en el 1,8% de los pacientes del grupo de XTANDI en comparación con el 1,1% del grupo de placebo. Los eventos isquémicos provocaron la muerte en el 0,4% de los pacientes del grupo de XTANDI en comparación con el 0,1% del grupo de placebo.
Controle los signos y síntomas de cardiopatía isquémica. Optimice el manejo de los factores de riesgo cardiovascular, como la hipertensión, la diabetes o la dislipidemia. Suspenda XTANDI en caso de cardiopatía isquémica de grado 3-4.
5.5 Caídas y fracturas
Se produjeron caídas y fracturas en pacientes que recibieron XTANDI. Evalúe a los pacientes para detectar el riesgo de fracturas y caídas. Controle y maneje a los pacientes con riesgo de fracturas de acuerdo con las pautas de tratamiento establecidas y considere el uso de agentes dirigidos al hueso.
En los datos combinados de cinco estudios clínicos aleatorizados y controlados con placebo, las caídas se produjeron en el 12% de los pacientes tratados con XTANDI en comparación con el 6% de los pacientes tratados con placebo. Las caídas no se asociaron con pérdida del conocimiento o convulsiones. Las fracturas se produjeron en el 13% de los pacientes tratados con XTANDI y en el 6% de los pacientes tratados con placebo. Las fracturas de grado 3-4 se produjeron en el 3,4% de los pacientes tratados con XTANDI y en el 1,9% de los pacientes tratados con placebo. El tiempo medio hasta el inicio de la fractura fue de 420 días (rango: 1 a 2348 días) para los pacientes tratados con XTANDI. En los estudios no se realizó una evaluación rutinaria de la densidad ósea ni el tratamiento de la osteoporosis con agentes dirigidos al hueso.
5.6 Toxicidad embriofetal
No se ha establecido la seguridad y eficacia de XTANDI en mujeres. Según los estudios de reproducción en animales y el mecanismo de acción, XTANDI puede causar daño fetal y pérdida del embarazo cuando se administra a una mujer embarazada. Avise a los hombres con parejas femeninas en edad fértil que utilicen métodos anticonceptivos eficaces durante el tratamiento con XTANDI y durante 3 meses después de la última dosis de XTANDI [véase Uso en poblaciones específicas (8.1, 8.3)].
5.7 Disfagia o atragantamiento
La disfagia o el atragantamiento graves, incluidos los eventos que podrían ser potencialmente mortales y requerir intervención médica o ser fatales, pueden producirse debido al tamaño del producto XTANDI. Avise a los pacientes que tomen cada cápsula o comprimido entero con una cantidad suficiente de agua para asegurar que todo el medicamento se trague correctamente. Considere el uso de un comprimido de menor tamaño de XTANDI en pacientes que tengan dificultad para tragar. Suspenda XTANDI en pacientes que no puedan tragar cápsulas o comprimidos.
6 REACCIONES ADVERSAS
Los siguientes puntos se discuten con más detalle en otras secciones del etiquetado:
- •
- Convulsiones [ver Advertencias y precauciones (5.1)]
- •
- Síndrome de encefalopatía posterior reversible (PRES) [ver Advertencias y precauciones (5.2)]
- •
- Hipersensibilidad [ver Advertencias y precauciones (5.3)]
- •
- Cardiopatía isquémica [ver Advertencias y precauciones (5.4)]
- •
- Caídas y fracturas [ver Advertencias y precauciones (5.5)]
- •
- Disfagia o asfixia [ver Advertencias y precauciones (5.7)]
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se realizan bajo condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un medicamento no pueden compararse directamente con las tasas en los ensayos clínicos de otro medicamento y es posible que no reflejen las tasas observadas en la práctica.
Los datos de ADVERTENCIAS Y PRECAUCIONES reflejan ocho ensayos aleatorizados y controlados [AFFIRM, PREVAIL, TERRAIN, PROSPER, ARCHES, EMBARK, Asian PREVAIL (NCT02294461) y STRIVE (NCT01664923)] que se agruparon para realizar análisis de seguridad en pacientes con CRPC (N = 3651), mCSPC (N = 752) o nmCSPC con BCR de alto riesgo (N = 707) tratados con XTANDI. Los pacientes recibieron XTANDI 160 mg (N = 5110) o placebo por vía oral una vez al día (N = 2829) o bicalutamida 50 mg por vía oral una vez al día (N = 387). En estos ocho ensayos, la mediana de la duración del tratamiento fue de 22,1 meses (rango: < 0,1 a 95,0) en pacientes que recibieron XTANDI.
En cinco ensayos controlados con placebo (AFFIRM, PROSPER, PREVAIL, ARCHES y EMBARK), la mediana de la duración del tratamiento fue de 19,4 meses (rango: < 0,1 a 90,4) en el grupo XTANDI [ver Estudios clínicos (14)]. En estos cinco ensayos, las reacciones adversas más comunes (≥ 10 %) que ocurrieron con mayor frecuencia (≥ 2 % sobre placebo) en los pacientes tratados con XTANDI fueron dolor musculoesquelético, fatiga, sofocos, estreñimiento, disminución del apetito, diarrea, hipertensión, hemorragia, caídas, fracturas y dolor de cabeza.
AFFIRM: XTANDI versus placebo en CRPC metastásico después de la quimioterapia
AFFIRM incluyó a 1199 pacientes con CRPC metastásico que habían recibido docetaxel previamente. La mediana de la duración del tratamiento fue de 8,3 meses con XTANDI y 3,0 meses con placebo. Durante el ensayo, el 48 % de los pacientes en el brazo de XTANDI y el 46 % de los pacientes en el brazo de placebo recibieron glucocorticoides.
Se informaron reacciones adversas de grado 3 y superior entre el 47 % de los pacientes tratados con XTANDI. Se informaron interrupciones debido a reacciones adversas para el 16 % de los pacientes tratados con XTANDI. La reacción adversa más común que condujo a la interrupción del tratamiento fueron las convulsiones, que ocurrieron en el 0,9 % de los pacientes tratados con XTANDI en comparación con ninguno (0 %) de los pacientes tratados con placebo. La Tabla 1 muestra las reacciones adversas informadas en AFFIRM que ocurrieron con una frecuencia ≥ 2 % mayor en el brazo de XTANDI en comparación con el brazo de placebo.
| XTANDI (N = 800) |
Placebo (N = 399) |
|||
|---|---|---|---|---|
| Grado 1-4* (%) |
Grado 3-4 (%) |
Grado 1-4 (%) |
Grado 3-4 (%) |
|
|
||||
|
Trastornos Generales |
||||
|
Asthenic Conditions† |
51 |
9 |
44 |
9 |
|
Edema Periférico |
15 |
1 |
13 |
0.8 |
|
Trastornos Musculoesqueléticos y del Tejido Conjuntivo |
||||
|
Dolor de Espalda |
26 |
5 |
24 |
4 |
|
Artralgia |
21 |
2.5 |
17 |
1.8 |
|
Dolor Musculoesquelético |
15 |
1.3 |
12 |
0.3 |
|
Debilidad Muscular |
10 |
1.5 |
7 |
1.8 |
|
Rigidez Musculoesquelética |
2.6 |
0.3 |
0.3 |
0 |
|
Trastornos Gastrointestinales |
||||
|
Diarrea |
22 |
1.1 |
18 |
0.3 |
|
Trastornos Vasculares |
||||
|
Sofocos |
20 |
0 |
10 |
0 |
|
Hipertensión |
6 |
2.1 |
2.8 |
1.3 |
|
Trastornos del Sistema Nervioso |
||||
|
Dolor de Cabeza |
12 |
0.9 |
5 |
0 |
|
Dizziness‡ |
9 |
0.5 |
7 |
0.5 |
|
Spinal Cord Compression and Cauda Equina Syndrome |
7 |
7 |
4.5 |
3.8 |
|
Parestesia |
7 |
0 |
4.5 |
0 |
|
Trastornos del Deterioro Mental§ |
4.3 |
0.3 |
1.8 |
0 |
|
Hipoestesia |
4 |
0.3 |
1.8 |
0 |
|
Infecciones e Infestaciones |
||||
|
Infección de las Vías Respiratorias Superiores¶ |
11 |
0 |
6 |
0.3 |
|
Infección de las Vías Respiratorias Inferiores y Pulmones# |
8 |
2.4 |
4.8 |
1.3 |
|
Trastornos Psiquiátricos |
||||
|
Insomnio |
9 |
0 |
6 |
0.5 |
|
Ansiedad |
6 |
0.3 |
4 |
0 |
|
Trastornos Renales y Urinarios |
||||
|
Hematuria |
7 |
1.8 |
4.5 |
1 |
|
Polaquiuria |
4.8 |
0 |
2.5 |
0 |
|
Lesiones, Intoxicaciones y Complicaciones de Procedimientos |
||||
|
Caída |
4.6 |
0.3 |
1.3 |
0 |
|
Fracturas No Patológicas |
4 |
1.4 |
0.8 |
0.3 |
|
Trastornos de la Piel y del Tejido Subcutáneo |
||||
|
Prurito |
3.8 |
0 |
1.3 |
0 |
|
Piel Seca |
3.5 |
0 |
1.3 |
0 |
|
Trastornos Respiratorios |
||||
|
Epistaxis (Hemorragia nasal) |
3.3 |
0.1 |
1.3 |
0.3 |
PREVAIL: XTANDI versus Placebo en CRPC Metastásico sin Quimioterapia Previa
PREVAIL incluyó a 1717 pacientes con CRPC metastásico que no habían recibido quimioterapia citotóxica previa, de los cuales 1715 recibieron al menos una dosis del fármaco del estudio. La duración media del tratamiento fue de 17,5 meses con XTANDI y 4,6 meses con placebo. Se informaron reacciones adversas de grado 3-4 en el 44% de los pacientes tratados con XTANDI y en el 37% de los pacientes tratados con placebo. Se informaron interrupciones debido a reacciones adversas para el 6% de los pacientes tratados con XTANDI. La reacción adversa más común que condujo a la interrupción del tratamiento fue fatiga/astenia, que ocurrió en el 1% de los pacientes en cada brazo de tratamiento. Tabla 2 incluye las reacciones adversas informadas en PREVAIL que ocurrieron con una frecuencia ≥ 2% mayor en el brazo de XTANDI en comparación con el brazo de placebo.
| XTANDI (N = 871) |
Placebo (N = 844) |
|||
|---|---|---|---|---|
| Grado 1-4* (%) |
Grado 3-4 (%) |
Grado 1-4 (%) |
Grado 3-4 (%) |
|
|
||||
|
Trastornos Generales |
||||
|
Condiciones Asténicas† |
47 |
3.4 |
33 |
2.8 |
|
Edema Periférico |
12 |
0.2 |
8 |
0.4 |
|
Trastornos Musculoesqueléticos y del Tejido Conjuntivo |
||||
|
Dolor de Espalda |
29 |
2 |
22 |
3 |
|
Artralgia |
21 |
1.6 |
16 |
1.1 |
|
Trastornos Gastrointestinales |
||||
|
Estreñimiento |
23 |
0.7 |
17 |
0.4 |
|
Diarrea |
17 |
0.3 |
14 |
0.4 |
|
Trastornos Vasculares |
||||
|
Sofocos |
18 |
0.1 |
8 |
0 |
|
Hipertensión |
14 |
7 |
4.1 |
2.3 |
|
Trastornos del sistema nervioso |
||||
|
Mareos‡ |
11 |
0.3 |
7 |
0 |
|
Dolor de cabeza |
11 |
0.2 |
7 |
0.4 |
|
Disgeusia |
8 |
0.1 |
3.7 |
0 |
|
Trastornos del deterioro mental§ |
6 |
0 |
1.3 |
0.1 |
|
Síndrome de piernas inquietas |
2.1 |
0.1 |
0.4 |
0 |
|
Trastornos respiratorios |
||||
|
Disnea¶ |
11 |
0.6 |
8 |
0.6 |
|
Infecciones e infestaciones |
||||
|
Infección de las vías respiratorias superiores# |
16 |
0 |
11 |
0 |
|
Infección de las vías respiratorias inferiores y pulmonesÞ |
8 |
1.5 |
4.7 |
1.1 |
|
Trastornos psiquiátricos |
||||
|
Insomnio |
8 |
0.1 |
6 |
0 |
|
Trastornos renales y urinarios |
||||
|
Hematuria |
9 |
1.3 |
6 |
1.3 |
|
Lesiones, intoxicaciones y complicaciones de procedimientos |
||||
|
Caída |
13 |
1.6 |
5 |
0.7 |
|
Fractura No Patológica |
9 |
2.1 |
3 |
1.1 |
|
Metabolism and Nutrition Disorders |
||||
|
Disminución del Apetito |
19 |
0.3 |
16 |
0.7 |
|
Investigations |
||||
|
Pérdida de Peso |
12 |
0.8 |
8 |
0.2 |
|
Reproductive System and Breast Disorders |
||||
|
Ginecomastia |
3.4 |
0 |
1.4 |
0 |
TERRAIN: XTANDI versus Bicalutamide en CRPC Metastásico sin Quimioterapia Previa
TERRAIN incluyó a 375 pacientes con CRPC metastásico que no habían recibido quimioterapia citotóxica previa, de los cuales 372 recibieron al menos una dosis del fármaco del estudio. La duración media del tratamiento fue de 11,6 meses con XTANDI y de 5,8 meses con bicalutamida. Se notificaron interrupciones con una reacción adversa como razón principal en el 8% de los pacientes tratados con XTANDI y en el 6% de los pacientes tratados con bicalutamida. Las reacciones adversas más comunes que llevaron a la interrupción del tratamiento fueron dolor de espalda y fractura patológica, que ocurrieron en el 3,8% de los pacientes tratados con XTANDI para cada evento y en el 2,1% y el 1,6% de los pacientes tratados con bicalutamida, respectivamente. Tabla 3 muestra las reacciones adversas generales y comunes (≥ 10%) en pacientes tratados con XTANDI.
| XTANDI (N = 183) |
Bicalutamide (N = 189) |
|||
|---|---|---|---|---|
| Grado 1-4* (%) |
Grado 3-4 (%) |
Grado 1-4 (%) |
Grado 3-4 (%) |
|
|
General |
94 |
39 |
94 |
38 |
|
Trastornos Generales |
||||
|
Afecciones Asténicas† |
32 |
1.6 |
23 |
1.1 |
|
Trastornos Musculoesqueléticos y del Tejido Conjuntivo |
||||
|
Dolor de Espalda |
19 |
2.7 |
18 |
1.6 |
|
Dolor Musculoesquelético‡ |
16 |
1.1 |
14 |
0.5 |
|
Trastornos Vasculares |
||||
|
Sofocos |
15 |
0 |
11 |
0 |
|
Hipertensión |
14 |
7 |
7 |
4.2 |
|
Trastornos Gastrointestinales |
||||
|
Náuseas |
14 |
0 |
18 |
0 |
|
Estreñimiento |
13 |
1.1 |
13 |
0.5 |
|
Diarrea |
12 |
0 |
9 |
1.1 |
|
Infecciones e Infestaciones |
||||
|
Infección de las Vías Respiratorias Superiores§ |
12 |
0 |
6 |
0.5 |
|
Investigational |
||||
|
Pérdida de Peso |
11 |
0.5 |
8 |
0.5 |
PROSPER: XTANDI versus Placebo en Pacientes con CRPC No Metastásico
PROSPER incluyó a 1401 pacientes con CRPC no metastásico, de los cuales 1395 recibieron al menos una dosis del fármaco del estudio. Los pacientes fueron aleatorizados 2:1 y recibieron XTANDI a una dosis de 160 mg una vez al día (N = 930) o placebo (N = 465). La mediana de la duración del tratamiento en el momento del análisis fue de 18.4 meses (rango: 0.0 a 42 meses) con XTANDI y 11.1 meses (rango: 0.0 a 43 meses) con placebo.
En general, 32 pacientes (3.4%) que recibieron XTANDI murieron por reacciones adversas. Las razones de muerte con ≥ 2 pacientes incluyeron trastornos de las arterias coronarias (n = 7), muerte súbita (n = 2), arritmias cardíacas (n = 2), deterioro general de la salud física (n = 2), accidente cerebrovascular (n = 2) y neoplasia maligna secundaria (n = 5; una de leucemia mieloide aguda, neoplasia cerebral, mesotelioma, cáncer de pulmón de células pequeñas y neoplasia maligna de sitio primario desconocido). Tres pacientes (0.6%) que recibieron placebo murieron por reacciones adversas de paro cardíaco (n = 1), insuficiencia ventricular izquierda (n = 1) y carcinoma pancreático (n = 1). Se informaron reacciones adversas de Grado 3 o superior entre el 31% de los pacientes tratados con XTANDI y el 23% de los pacientes tratados con placebo. Se informaron interrupciones con una reacción adversa como razón principal para el 9% de los pacientes tratados con XTANDI y el 6% de los pacientes tratados con placebo. De estas, la reacción adversa más común que condujo a la interrupción del tratamiento fue la fatiga, que ocurrió en el 1.6% de los pacientes tratados con XTANDI en comparación con ninguno de los pacientes tratados con placebo. Tabla 4 muestra las reacciones adversas informadas en PROSPER que ocurrieron con una frecuencia ≥ 2% mayor en el brazo de XTANDI que en el brazo de placebo.
| XTANDI (N = 930) |
Placebo (N = 465) |
|||
|---|---|---|---|---|
| Grado 1-4* (%) |
Grado 3-4 (%) |
Grado 1-4 (%) |
Grado 3-4 (%) |
|
|
Trastornos del Metabolismo y la Nutrición |
||||
|
Disminución del Apetito |
10 |
0.2 |
3.9 |
0.2 |
|
Trastornos del Sistema Nervioso |
||||
|
Mareo† |
12 |
0.5 |
5 |
0 |
|
Dolor de Cabeza |
9 |
0.2 |
4.5 |
0 |
|
Trastornos Cognitivos y de Atención‡ |
4.6 |
0.1 |
1.5 |
0 |
|
Trastornos Vasculares |
||||
|
Sofocos |
13 |
0.1 |
8 |
0 |
|
Hipertensión |
12 |
4.6 |
5 |
2.2 |
|
Trastornos gastrointestinales |
||||
|
Náuseas |
11 |
0.3 |
9 |
0 |
|
Estreñimiento |
9 |
0.2 |
7 |
0.4 |
|
Trastornos generales y alteraciones en el lugar de administración |
||||
|
Asthenic Conditions§ |
40 |
4 |
20 |
0.9 |
|
Investigaciones |
||||
|
Pérdida de peso |
6 |
0.2 |
1.5 |
0 |
|
Lesiones, intoxicaciones y complicaciones de procedimientos |
||||
|
Caída |
11 |
1.3 |
4.1 |
0.6 |
|
Fracturas¶ |
10 |
2 |
4.9 |
1.7 |
|
Trastornos psiquiátricos |
||||
|
Ansiedad |
2.8 |
0.2 |
0.4 |
0 |
ARCHES: XTANDI versus Placebo en Pacientes con CSPC Metastásico
ARCHES aleatorizó a 1150 pacientes con mCSPC, de los cuales 1146 recibieron al menos una dosis del fármaco del estudio. Todos los pacientes recibieron un análogo de la hormona liberadora de gonadotropina (GnRH) simultáneamente o se sometieron a una orquiectomía bilateral. Los pacientes recibieron XTANDI en una dosis de 160 mg una vez al día (N = 572) o placebo (N = 574). La mediana de la duración del tratamiento fue de 12,8 meses (rango: 0,2 a 26,6 meses) con XTANDI y de 11,6 meses (rango: 0,2 a 24,6 meses) con placebo.
En general, 10 pacientes (1,7%) que recibieron XTANDI murieron por reacciones adversas. Las razones de muerte en ≥ 2 pacientes incluyeron enfermedad cardíaca (n = 3), sepsis (n = 2) y embolia pulmonar (n = 2). Ocho pacientes (1,4%) que recibieron placebo murieron por reacciones adversas. Las razones de muerte en ≥ 2 pacientes incluyeron enfermedad cardíaca (n = 2) y muerte súbita (n = 2). Se informaron reacciones adversas de Grado 3 o superior en el 24% de los pacientes tratados con XTANDI. La interrupción permanente debido a reacciones adversas como la razón principal se informó en el 4,9% de los pacientes tratados con XTANDI y en el 3,7% de los pacientes tratados con placebo. Las reacciones adversas más comunes que resultaron en la interrupción permanente en los pacientes tratados con XTANDI fueron aumento de la alanina aminotransferasa, elevación de la aspartato aminotransferasa y convulsiones, cada una en un 0,3%. Las reacciones adversas más comunes que llevaron a la interrupción permanente en los pacientes tratados con placebo fueron artralgia y fatiga, cada una en un 0,3%.
Las reducciones de dosis debido a una reacción adversa ocurrieron en el 4,4% de los pacientes que recibieron XTANDI. La fatiga/astenia fue la reacción adversa más frecuente que requirió una reducción de la dosis en el 2,1% de los pacientes tratados con XTANDI y en el 0,7% de los pacientes tratados con placebo.
Tabla 5 muestra las reacciones adversas informadas en ARCHES que ocurrieron con una frecuencia ≥ 2% mayor en el grupo de XTANDI que en el grupo de placebo.
| XTANDI (N = 572) |
Placebo (N = 574) |
|||
|---|---|---|---|---|
| Grado 1-4* (%) |
Grado 3-4 (%) |
Grado 1-4 (%) |
Grado 3-4 (%) |
|
|
||||
|
Trastornos del Metabolismo y la Nutrición |
||||
|
Disminución del Apetito |
4.9 |
0.2 |
2.6 |
0 |
|
Trastornos del Sistema Nervioso |
||||
|
Deterioro Cognitivo y de la Memoria† |
4.5 |
0.7 |
2.1 |
0 |
|
Síndrome de Piernas Inquietas |
2.4 |
0 |
0.3 |
0 |
|
Trastornos Vasculares |
||||
|
Sofocos |
27 |
0.3 |
22 |
0 |
|
Hipertensión |
8 |
3.3 |
6 |
1.7 |
|
Trastornos generales y alteraciones en el lugar de administración |
||||
|
Condiciones asténicas‡ |
24 |
1.7 |
20 |
1.6 |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
||||
|
Dolor musculoesquelético |
6 |
0.2 |
4 |
0.2 |
|
Lesiones, intoxicaciones y complicaciones de procedimientos terapéuticos |
||||
|
Fracturas§ |
6 |
1.0 |
4.2 |
1 |
EMBARK: XTANDI versus Placebo en Pacientes con CSPC No Metastásico con BCR de Alto Riesgo
EMBARK inscribió a 1068 pacientes con BCR de alto riesgo, de los cuales 1061 pacientes recibieron al menos una dosis del fármaco del estudio. Los pacientes recibieron XTANDI a una dosis de 160 mg una vez al día simultáneamente con leuprolida (N = 353), XTANDI a una dosis de 160 mg una vez al día como monoterapia abierta (N = 354) o placebo simultáneamente con leuprolida (N = 354). En la semana 37, se suspendió el tratamiento para los pacientes cuyos valores de PSA no eran detectables (<0.2 ng/mL) en la semana 36. El tratamiento se reinició cuando los valores de PSA aumentaron a ≥2.0 ng/mL para pacientes con prostatectomía previa o ≥5.0 ng/mL para pacientes sin prostatectomía previa. Para los pacientes cuyos valores de PSA eran detectables (≥0.2 ng/mL) en la semana 36, el tratamiento continuó sin suspensión hasta que se cumplieron los criterios de interrupción permanente del tratamiento.
Tabla 6 muestra la duración total del tratamiento para los tres grupos de tratamiento.
| XTANDI + Leuprolide (N = 353) |
Placebo + Leuprolide (N = 354) |
XTANDI
(N = 354) |
|
|---|---|---|---|
|
|||
|
Duración Total del Tratamiento* |
|||
|
Mediana, meses |
60.6 |
55.6 |
60.4 |
|
Rango, meses |
0.1 – 90.4 |
0.7 – 94.1 |
0.4 – 95.0 |
|
Duración del Tratamiento con el Fármaco |
|||
|
Mediana, meses |
32.4 |
35.4 |
45.9 |
|
Rango, meses |
0.1 – 83.4 |
0.7 – 85.7 |
0.4 – 88.9 |
|
Duración de la Suspensión del Tratamiento con el Fármaco |
|||
|
Mediana, meses |
20.2 |
16.8 |
11.1 |
|
Rango, meses |
5.7 – 87.9 |
3.4 – 83.0 |
2.3 – 84.9 |
|
Pacientes a los que se les Suspendió el Tratamiento con el Fármaco en la Semana 37 |
|||
|
Número de Pacientes (%) |
321 (90.9) |
240 (67.8) |
304 (85.9) |
En general, las muertes por reacciones adversas durante la duración total del tratamiento ocurrieron en 6 pacientes (1.7%) que recibieron XTANDI más leuprolida, 8 pacientes (2.3%) que recibieron XTANDI como agente único y 3 pacientes (0.8%) que recibieron placebo más leuprolida. La razón de la muerte en ≥ 2 pacientes que recibieron XTANDI más leuprolida fue infección (n = 2), y la razón de la muerte en ≥ 2 pacientes que recibieron XTANDI como agente único fue tromboembolismo arterial (n = 2).
Se informaron reacciones adversas de Grado 3 o superior durante la duración total del tratamiento en el 46% de los pacientes tratados con XTANDI más leuprolida, el 50% de los pacientes que recibieron XTANDI como agente único y el 43% de los pacientes que recibieron placebo más leuprolida. La interrupción permanente del tratamiento debido a reacciones adversas durante la duración total del tratamiento como razón principal se informó en el 21% de los pacientes tratados con XTANDI más leuprolida, el 18% de los pacientes que recibieron XTANDI como agente único y el 10% de los pacientes que recibieron placebo más leuprolida. Las reacciones adversas más comunes que resultaron en la interrupción permanente incluyeron fatiga (3.4% de los pacientes tratados con XTANDI más leuprolida, 3.7% de los pacientes que recibieron XTANDI como agente único y 1.4% de los pacientes que recibieron placebo más leuprolida), sofocos (2% de los pacientes tratados con XTANDI más leuprolida, 0% de los pacientes que recibieron XTANDI como agente único y 1.1% de los pacientes que recibieron placebo más leuprolida), náuseas (1.1% de los pacientes tratados con XTANDI más leuprolida, 0.6% de los pacientes que recibieron XTANDI como agente único y 0.3% de los pacientes que recibieron placebo más leuprolida) y trastorno cognitivo (1.1% de los pacientes tratados con XTANDI más leuprolida, 1.4% de los pacientes que recibieron XTANDI como agente único y 0.8% de los pacientes que recibieron placebo más leuprolida).
Las reducciones de dosis debido a una reacción adversa ocurrieron en el 7% de los pacientes que recibieron XTANDI más leuprolida, el 16% de los pacientes que recibieron XTANDI como agente único y el 4.5% de los pacientes que recibieron placebo más leuprolida. La fatiga fue la reacción adversa más frecuente que requirió una reducción de la dosis en el 3.1% de los pacientes tratados con XTANDI más leuprolida, el 10% de los pacientes que recibieron XTANDI como agente único y el 1.7% de los pacientes que recibieron placebo más leuprolida.
La Tabla 7 muestra las reacciones adversas informadas en EMBARK que ocurrieron con una frecuencia ≥ 5% (Grado 1-4) o ≥ 2% (Grado 3-4) mayor en cualquiera de los brazos de XTANDI que en el brazo de placebo.
| XTANDI + Leuprolide (N = 353) |
Placebo + Leuprolide (N = 354) |
XTANDI
(N = 354) |
||||
|---|---|---|---|---|---|---|
| Grade 1-4* (%) |
Grade 3-4 (%) |
Grade 1-4 (%) |
Grade 3‑4 (%) |
Grade 1-4 (%) |
Grade 3-4 (%) |
|
|
Trastornos del Sistema Nervioso |
||||||
|
Cognitive Disorder† |
10 |
0.3 |
4.8 |
0.6 |
10 |
0.3 |
|
Síncope |
4.8 |
4.2 |
2.3 |
1.7 |
2.5 |
2 |
|
Trastornos Vasculares |
||||||
|
Sofocos |
69 |
0.6 |
57 |
0.8 |
22 |
0.3 |
|
Hemorragia† |
20 |
3.4 |
15 |
1.7 |
21 |
3.7 |
|
Trastornos Gastrointestinales |
||||||
|
Diarrea† |
15 |
0.6 |
9 |
0.8 |
14 |
0.3 |
|
Náuseas |
12 |
0.3 |
8 |
0.3 |
15 |
0.6 |
|
Investigaciones |
||||||
|
Pérdida de Peso |
7 |
0.3 |
3.4 |
0 |
11 |
0.3 |
|
Trastornos Generales y Afecciones en el Sitio de Administración |
||||||
|
Fatiga† |
50 |
4 |
38 |
1.7 |
54 |
4.8 |
|
Trastornos Musculoesqueléticos y del Tejido Conjuntivo |
||||||
|
Dolor Musculoesquelético† |
50 |
4.8 |
43 |
2.3 |
48 |
3.1 |
|
Osteoartritis |
6 |
2.8 |
4.2 |
0.6 |
5 |
0.6 |
|
Lesiones, Intoxicaciones y Complicaciones de Procedimientos |
||||||
|
Caída |
21 |
1.1 |
14 |
1.1 |
16 |
2 |
|
Fractura† |
18 |
4 |
13 |
2.5 |
11 |
2 |
|
Trastornos del Sistema Reproductivo y de las Mamas |
||||||
|
Ginecomastia† |
9 |
0 |
10 |
0 |
49 |
0.8 |
|
Sensibilidad en las mamas† |
5 |
0 |
2.8 |
0 |
35 |
0 |
|
Trastornos Cardíacos |
||||||
|
Cardiopatía Isquémica† |
5 |
4 |
6 |
3.1 |
9 |
6 |
Las reacciones adversas clínicamente relevantes que no cumplieron con los criterios para su inclusión en la Tabla 7 incluyen hipertensión (XTANDI más leuprolida, 25%; XTANDI como agente único, 21%), angioedema (XTANDI más leuprolida, 2.5%; XTANDI como agente único, 2%) y convulsiones (XTANDI más leuprolida, 1.1%; XTANDI como agente único, 0.8%).
Anomalías de Laboratorio
La Tabla 8 muestra las anomalías de laboratorio que ocurrieron en ≥ 5% de los pacientes y con mayor frecuencia (> 2%) en el grupo de XTANDI en comparación con el placebo en los estudios combinados, aleatorizados y controlados con placebo.
| XTANDI (N = 3526) |
Placebo (N = 2636) |
|||
|---|---|---|---|---|
| Grado 1-4 (%) |
Grado 3-4 (%) |
Grado 1-4 (%) |
Grado 3-4 (%) |
|
|
Hematología |
||||
|
Disminución de la hemoglobina |
50 |
1.8 |
47 |
1.5 |
|
Disminución del recuento de neutrófilos |
20 |
1 |
17 |
0.5 |
|
Disminución de los glóbulos blancos |
18 |
0.5 |
11 |
0.2 |
|
Química |
||||
|
Hiperglucemia |
86 |
3.7 |
78 |
4.3 |
|
Hipermagnesemia |
17 |
0.1 |
14 |
0.3 |
|
Hiponatremia |
14 |
1.6 |
9 |
1.4 |
|
Hipofosfatemia |
10 |
1.4 |
7 |
0.8 |
|
Hipercalcemia |
8 |
0.1 |
5 |
0.1 |
Hipertensión
En los datos combinados de cinco ensayos clínicos aleatorizados controlados con placebo, se reportó hipertensión en el 14% de los pacientes que recibieron XTANDI y en el 7% de los pacientes que recibieron placebo. El historial médico de hipertensión fue equilibrado entre los brazos. La hipertensión condujo a la interrupción del estudio en < 1% de los pacientes en cada brazo.
6.2 Experiencia Posterior a la Comercialización
Las siguientes reacciones adversas adicionales se han identificado durante el uso posterior a la aprobación de XTANDI. Debido a que estas reacciones se reportan voluntariamente de una población de tamaño incierto, no siempre es posible estimar de forma fiable su frecuencia o establecer una relación causal con la exposición al fármaco.
Trastornos Gastrointestinales: vómitos, disfagia (incluyendo asfixia relacionada con el tamaño del producto XTANDI)
Trastornos del Sistema Inmunitario: hipersensibilidad (edema de la cara, lengua, labio o faringe)
Trastornos Neurológicos: síndrome de encefalopatía posterior reversible (PRES), disgeusia
Trastornos de la Piel y del Tejido Subcutáneo: erupción cutánea, reacciones adversas cutáneas graves (incluyendo síndrome de Stevens-Johnson (SJS), eritema multiforme, necrólisis epidérmica tóxica (TEN), reacción farmacológica con eosinofilia y síntomas sistémicos (DRESS) y pustulosis exantemática aguda generalizada (AGEP))
7 INTERACCIONES MEDICAMENTOSAS
7.1 Efecto de otros medicamentos sobre XTANDI
Inhibidores potentes del CYP2C8
La administración conjunta de XTANDI con gemfibrozil (un inhibidor potente del CYP2C8) aumenta las concentraciones plasmáticas de enzalutamida más N-desmetil enzalutamida, lo que puede aumentar la incidencia y la gravedad de las reacciones adversas de XTANDI. Evite la administración conjunta de XTANDI con inhibidores potentes del CYP2C8. Si no se puede evitar la administración conjunta de XTANDI con un inhibidor potente del CYP2C8, reduzca la dosis de XTANDI [ver Posología y administración (2.3), Farmacología clínica (12.3)].
Inductores potentes del CYP3A4
La administración conjunta de XTANDI con rifampicina (un inductor potente del CYP3A4 y un inductor moderado del CYP2C8) disminuye las concentraciones plasmáticas de enzalutamida más N-desmetil enzalutamida, lo que puede disminuir la eficacia de XTANDI. Evite la administración conjunta de XTANDI con inductores potentes del CYP3A4. Si no se puede evitar la administración conjunta de XTANDI con un inductor potente del CYP3A4, aumente la dosis de XTANDI [ver Posología y administración (2.3), Farmacología clínica (12.3)].
7.2 Efecto de XTANDI sobre otros medicamentos
Ciertos sustratos del CYP3A4, CYP2C9 o CYP2C19
XTANDI es un inductor potente del CYP3A4 y un inductor moderado del CYP2C9 y del CYP2C19. La administración conjunta de XTANDI disminuye las concentraciones de ciertos sustratos del CYP3A4, CYP2C9 o CYP2C19 [ver Farmacología clínica (12.3)], lo que puede reducir la eficacia de estos sustratos. Evite la administración conjunta de XTANDI con ciertos sustratos del CYP3A4, CYP2C9 o CYP2C19 para los que una disminución mínima de la concentración puede provocar un fracaso terapéutico del sustrato. Si no se puede evitar la administración conjunta, aumente la dosis de estos sustratos de acuerdo con su información de prescripción. En los casos en que se formen metabolitos activos, puede haber una mayor exposición a los metabolitos activos.
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de riesgos
No se ha establecido la seguridad y eficacia de XTANDI en mujeres. De acuerdo con los estudios de reproducción animal y el mecanismo de acción, XTANDI puede causar daño fetal y pérdida del embarazo. No existen datos en humanos sobre el uso de XTANDI en mujeres embarazadas. En estudios de reproducción animal, la administración oral de enzalutamida en ratones preñadas durante la organogénesis causó efectos adversos en el desarrollo a dosis menores que la dosis máxima recomendada en humanos (ver Datos).
Datos
Datos en animales
En un estudio de toxicidad en el desarrollo embriofetal en ratones, la enzalutamida causó toxicidad en el desarrollo cuando se administró en dosis orales de 10 o 30 mg/kg/día durante todo el período de organogénesis (días de gestación 6-15). Los hallazgos incluyeron letalidad embriofetal (mayor pérdida y resorciones posteriores a la implantación) y disminución de la distancia anogenital a ≥ 10 mg/kg/día, y paladar hendido y ausencia de hueso palatino a 30 mg/kg/día. Las dosis de 30 mg/kg/día causaron toxicidad materna. Las dosis probadas en ratones (1, 10 y 30 mg/kg/día) resultaron en exposiciones sistémicas (AUC) de aproximadamente 0.04, 0.4 y 1.1 veces, respectivamente, las exposiciones en pacientes. La enzalutamida no causó toxicidad en el desarrollo en conejos cuando se administró durante todo el período de organogénesis (días de gestación 6-18) a niveles de dosis de hasta 10 mg/kg/día (aproximadamente 0.4 veces las exposiciones en pacientes según el AUC).
En un estudio farmacocinético en ratas preñadas con una sola administración oral de 30 mg/kg de enzalutamida en el día 14 de gestación, la enzalutamida y/o sus metabolitos estuvieron presentes en el feto a una Cmáx que fue aproximadamente 0.3 veces la concentración encontrada en el plasma materno y ocurrió 4 horas después de la administración.
8.2 Lactancia
Resumen de riesgos
No se ha establecido la seguridad y la eficacia de XTANDI en mujeres. No hay información disponible sobre la presencia de XTANDI en la leche humana, los efectos del medicamento en el lactante o los efectos del medicamento en la producción de leche. La enzalutamida y/o sus metabolitos estuvieron presentes en la leche de ratas lactantes (ver Datos).
Datos
Después de una sola administración oral en ratas lactantes en el día 14 posnatal, la enzalutamida y/o sus metabolitos estuvieron presentes en la leche a una Cmáx que fue 4 veces mayor que las concentraciones en el plasma y ocurrió 4 horas después de la administración.
8.3 Mujeres y hombres con potencial reproductivo
Anticoncepción
Hombres
Según los hallazgos en estudios de reproducción animal, se debe aconsejar a los pacientes masculinos con parejas femeninas con potencial reproductivo que utilicen anticonceptivos efectivos durante el tratamiento y durante 3 meses después de la última dosis de XTANDI [ver Uso en poblaciones específicas (8.1)].
Infertilidad
Hombres
Según estudios en animales, XTANDI puede afectar la fertilidad en hombres con potencial reproductivo [ver Toxicología no clínica (13.1)].
8.4 Uso pediátrico
No se ha establecido la seguridad y la eficacia de XTANDI en pacientes pediátricos.
8.5 Uso geriátrico
De los 5110 pacientes que recibieron XTANDI en ocho ensayos clínicos aleatorizados y controlados, el 78% tenía 65 años o más, mientras que el 33% tenía 75 años o más. No se observaron diferencias generales en la seguridad o la eficacia entre estos pacientes y los pacientes más jóvenes. Otra experiencia clínica informada no ha identificado diferencias en las respuestas entre los pacientes ancianos y los más jóvenes, pero no se puede descartar una mayor sensibilidad de algunas personas mayores.
8.6 Insuficiencia renal
No se recomienda modificar la dosis en pacientes con insuficiencia renal leve a moderada (aclaramiento de creatinina [CLcr] ≥ 30 ml/min). XTANDI no se ha estudiado en pacientes con insuficiencia renal grave (CLcr < 30 ml/min) o enfermedad renal en etapa terminal [ver Farmacología clínica (12.3)].
8.7 Insuficiencia hepática
No se recomienda modificar la dosis en pacientes con insuficiencia hepática leve, moderada o grave [ver Farmacología clínica (12.3)].
10 SOBREDOSIS
En caso de sobredosis, suspenda el tratamiento con XTANDI e inicie medidas de soporte general teniendo en cuenta la vida media de 5.8 días. En un estudio de aumento de dosis, no se reportaron convulsiones a < 240 mg diarios, mientras que se reportaron 3 convulsiones, 1 en cada dosis de 360 mg, 480 mg y 600 mg diarios. Los pacientes pueden tener un mayor riesgo de convulsiones después de una sobredosis.
11 DESCRIPCIÓN
Enzalutamide es un inhibidor del receptor de andrógenos. El nombre químico es 4-{3-[4-ciano-3-(trifluorometil)fenil]-5,5-dimetil-4-oxo-2-sulfanylideneimidazolidin-1-il}-2-fluoro-N-metilbenzamida.
El peso molecular es 464.44 y la fórmula molecular es C21H16F4N4O2S. La fórmula estructural es:

Enzalutamide es un sólido cristalino blanco no higroscópico. Es prácticamente insoluble en agua.
XTANDI está disponible como cápsulas de gelatina blanda llenas de líquido para administración oral. Cada cápsula contiene 40 mg de enzalutamida en solución en polioxilglicéridos de caprilocaproílo. Los ingredientes inactivos son polioxilglicéridos de caprilocaproílo, butilhidroxianisol, butilhidroxitolueno, gelatina, solución de sorbitol sorbitán, glicerina, agua purificada, dióxido de titanio y óxido de hierro negro.
XTANDI también está disponible como comprimidos recubiertos con película para administración oral. Cada comprimido contiene 40 mg u 80 mg de enzalutamida. Los ingredientes inactivos son succinato de acetato de hipromelosa, celulosa microcristalina, dióxido de silicio coloidal, croscarmelosa sódica y estearato de magnesio. La película de recubrimiento del comprimido contiene hipromelosa, talco, polietilenglicol, dióxido de titanio y óxido férrico.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
Enzalutamida es un inhibidor del receptor de andrógenos que actúa en diferentes etapas de la vía de señalización del receptor de andrógenos. Se ha demostrado que la enzalutamida inhibe competitivamente la unión de andrógenos a los receptores de andrógenos; y, en consecuencia, inhibe la translocación nuclear de los receptores de andrógenos y su interacción con el ADN. Un metabolito principal, la N-desmetil enzalutamida, exhibió una actividad in vitro similar a la enzalutamida. La enzalutamida disminuyó la proliferación e indujo la muerte celular de las células de cáncer de próstata in vitro, y disminuyó el volumen tumoral en un modelo de xenotrasplante de cáncer de próstata en ratón.
12.2 Farmacodinamia
La dosis diaria de 160 mg de enzalutamida además de ADT redujo los niveles de PSA a niveles indetectables (< 0,2 ng/mL) en el 68% de los pacientes con mCSPC (ARCHES).
Basándose en los resultados de eficacia de AFFIRM después de una dosis diaria de 160 mg de enzalutamida, no se pudo identificar ninguna relación exposición-respuesta para el criterio de valoración de eficacia de la supervivencia global. Además, no hubo una relación exposición-respuesta clínicamente significativa para los efectos adversos (p. ej., fatiga, rubor, dolor de cabeza o hipertensión) dentro del rango de exposición limitado para 160 mg/día.
Electrofisiología cardíaca
A la dosis recomendada, XTANDI no causa aumentos medios grandes (es decir, > 20 mseg) en el intervalo QT.
12.3 Farmacocinética
Enzalutamida alcanza el estado estacionario en el día 28 y su AUC se acumula aproximadamente 8,3 veces en relación con una dosis única. En estado estacionario, la concentración máxima media (%CV) (Cmax) para enzalutamida y N-desmetil enzalutamida son 16,6 µg/mL (23%) y 12,7 µg/mL (30%), respectivamente, y las concentraciones mínimas medias (%CV) (Cmin) son 11,4 µg/mL (26%) y 13,0 µg/mL (30%), respectivamente.
Enzalutamida mostró una farmacocinética aproximadamente proporcional a la dosis en el rango de dosis diarias de 30 (0,2 veces la dosis recomendada aprobada) a 360 mg (2,25 veces la dosis recomendada aprobada).
Absorción
La Tmax mediana es de 1 hora (0,5 a 3 horas) después de una dosis única de 160 mg de cápsulas y de 2 horas (0,5 a 6 horas) después de una dosis única de 160 mg de comprimidos.
Efecto de los alimentos
No hubo un efecto clínicamente significativo en la farmacocinética de enzalutamida o N-desmetil enzalutamida después de la administración de XTANDI con una comida alta en grasas (aproximadamente 150 calorías de proteínas, 250 calorías de carbohidratos y 500 a 600 calorías de grasas).
Distribución
El volumen de distribución medio (%CV) después de una dosis oral única es de 110 L (29%).
Enzalutamida está unida en un 97% a 98% a las proteínas plasmáticas, principalmente albúmina. La N-desmetil enzalutamida está unida en un 95% a las proteínas plasmáticas.
Eliminación
Enzalutamida se elimina principalmente por metabolismo hepático.
El aclaramiento aparente medio (CL/F) de enzalutamida después de una dosis única es de 0,56 L/h (0,33 a 1,02 L/h). La semivida terminal media (t1/2) para enzalutamida después de una dosis oral única es de 5,8 días (2,8 a 10,2 días). La t1/2 terminal media para N-desmetil enzalutamida es de aproximadamente 7,8 a 8,6 días.
Metabolismo
Enzalutamida se metaboliza por CYP2C8 y CYP3A4. CYP2C8 es el principal responsable de la formación del metabolito activo (N-desmetil enzalutamida). La carboxilesterasa 1 metaboliza la N-desmetil enzalutamida y la enzalutamida al metabolito inactivo del ácido carboxílico.
Poblaciones específicas
No se observaron diferencias clínicamente significativas en la farmacocinética de enzalutamida en función de la edad (41 a 92 años), la raza (blanca, china y japonesa), el peso corporal (46 kg a 163 kg), la insuficiencia renal leve a moderada (CLcr ≥ 30 mL/min) y la insuficiencia hepática (Child-Pugh A, B y C). No se ha estudiado la insuficiencia renal grave y la enfermedad renal en etapa terminal (CLcr < 30 mL/min).
Estudios de interacción medicamentosa
Estudios clínicos
Efecto de los inhibidores de CYP2C8 en XTANDI: La coadministración de 160 mg de XTANDI con gemfibrozilo (inhibidor potente de CYP2C8) aumentó el AUC de enzalutamida más N-desmetil enzalutamida en 2,2 veces con un efecto mínimo en Cmax.
Efecto de los inductores de CYP3A4 y CYP2C8 en XTANDI: La coadministración de 160 mg de XTANDI después de múltiples dosis orales de rifampicina (inductor potente de CYP3A4 e inductor moderado de CYP2C8) disminuyó el AUC de enzalutamida más N-desmetil enzalutamida en un 37% sin afectar a Cmax.
Efecto de los inhibidores de CYP3A4 en XTANDI: La coadministración de 160 mg de XTANDI después de múltiples dosis orales de itraconazol (inhibidor potente de CYP3A4) aumentó el AUC de enzalutamida más N-desmetil enzalutamida en 1,3 veces sin afectar a Cmax.
Efecto de XTANDI en otros medicamentos:
La coadministración de 160 mg de XTANDI por vía oral una vez al día con midazolam (un sustrato sensible de CYP3A4) disminuyó el AUC de midazolam en un 86% y la Cmax en un 77%.
La coadministración de 160 mg de XTANDI por vía oral una vez al día con warfarina (un sustrato sensible de CYP2C9) disminuyó el AUC de S-warfarina en un 56% y la Cmax en un 17%.
La coadministración de 160 mg de XTANDI por vía oral una vez al día con omeprazol (un sustrato sensible de CYP2C19) disminuyó el AUC de omeprazol en un 72% y la Cmax en un 62%.
La coadministración de 160 mg de XTANDI por vía oral una vez al día con digoxina (un sustrato de P-glicoproteína) aumentó el AUC de digoxina en un 33% y la Cmax en un 17%.
No se observaron cambios clínicamente significativos en la exposición a pioglitazona (un sustrato sensible de CYP2C8), cafeína (un sustrato sensible de CYP1A2), dextrometorfano (un sustrato sensible de CYP2D6) o rosuvastatina (un sustrato de BCRP) después de la coadministración con XTANDI.
Estudios in vitro
Enzimas del citocromo P450 (CYP): Enzalutamida induce CYP2B6 a concentraciones clínicamente alcanzables.
Sistemas de transporte: Enzalutamida, N-desmetil enzalutamida y el principal metabolito inactivo del ácido carboxílico no son sustratos de P-glicoproteína o BCRP.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
Se realizó un estudio de carcinogenicidad de dos años en ratas macho y hembra con dosis orales de enzalutamida de 10, 30 y 100 mg/kg/día. La enzalutamida aumentó la incidencia de tumores benignos de células de Leydig en los testículos en todos los niveles de dosis probados (≥ 0,3 veces la exposición humana basada en el AUC) e incidencia combinada de papiloma y carcinoma urotelial en la vejiga urinaria en ratas macho a 100 mg/kg/día (1,4 veces la exposición humana basada en el AUC). Los hallazgos en los testículos se consideran relacionados con la actividad farmacológica de la enzalutamida. Las ratas se consideran más sensibles que los humanos al desarrollo de tumores de células intersticiales en los testículos. La administración de enzalutamida a ratones transgénicos rasH2 machos y hembras por sonda gástrica oral diariamente durante 26 semanas no produjo un aumento en la incidencia de neoplasias en dosis de hasta 20 mg/kg/día.
La enzalutamida no indujo mutaciones en el ensayo de reversión de mutación bacteriana (Ames) y no fue genotóxica ni en el ensayo de mutación del gen de la timidina quinasa (Tk) del linfoma de ratón in vitro ni en el ensayo de micronúcleos de ratón in vivo.
Según los hallazgos no clínicos en estudios de toxicología de dosis repetidas, que fueron consistentes con la actividad farmacológica de la enzalutamida, la fertilidad masculina puede verse afectada por el tratamiento con XTANDI. En un estudio de 26 semanas en ratas, se observó atrofia de la próstata y las vesículas seminales a ≥ 30 mg/kg/día (igual a la exposición humana basada en el AUC). En estudios de 4, 13 y 39 semanas en perros, se observó hipospermatogénesis y atrofia de la próstata y los epidídimos a ≥ 4 mg/kg/día (0,3 veces la exposición humana basada en el AUC).
14 ESTUDIOS CLÍNICOS
La eficacia de XTANDI en pacientes con CRPC (N = 4692), mCSPC (N = 1150) o nmCSPC con BCR de alto riesgo (N = 1068) se demostró en seis ensayos clínicos aleatorizados y multicéntricos. Los pacientes recibieron terapia concomitante con GnRH o se les había realizado una orquiectomía bilateral previa, a menos que se indique lo contrario.
AFFIRM (NCT00974311): XTANDI versus Placebo en CRPC metastásico después de la quimioterapia
En AFFIRM, un total de 1199 pacientes que habían recibido quimioterapia previa basada en docetaxel fueron aleatorizados 2:1 para recibir XTANDI por vía oral a una dosis de 160 mg una vez al día (N = 800) o placebo por vía oral una vez al día (N = 399). El tratamiento del estudio continuó hasta la progresión de la enfermedad (evidencia de progresión radiográfica, un evento relacionado con el esqueleto o progresión clínica), el inicio de un nuevo tratamiento antineoplásico sistémico, toxicidad inaceptable o retiro. Los pacientes con antecedentes de convulsiones, que tomaban medicamentos que se sabe que disminuyen el umbral convulsivo o con otros factores de riesgo de convulsiones no eran elegibles [ver Advertencias y precauciones (5.1)].
Las siguientes características demográficas y basales de la enfermedad de los pacientes estaban equilibradas entre los grupos de tratamiento. La mediana de edad fue de 69 años (rango 41-92) y la distribución racial fue 92.7% blanca, 3.9% negra, 1.1% asiática y 2.1% otras. El noventa y dos por ciento de los pacientes tenían una puntuación de estado funcional ECOG de 0-1 y el 28% tenía una puntuación media del Inventario Breve del Dolor de ≥ 4. El noventa y uno por ciento de los pacientes tenían metástasis en los huesos y el 23% tenía afectación visceral en el pulmón y/o el hígado. El cincuenta y nueve por ciento de los pacientes tenían evidencia radiográfica de progresión de la enfermedad y el 41% tenía progresión solo de PSA al ingresar al estudio. Todos los pacientes habían recibido terapia previa basada en docetaxel y el 24% había recibido dos regímenes de quimioterapia citotóxica. Durante el ensayo, el 48% de los pacientes en el grupo de XTANDI y el 46% de los pacientes en el grupo de placebo recibieron glucocorticoides.
Se demostró una mejora estadísticamente significativa en la supervivencia general en el análisis intermedio preespecificado en el momento de 520 muertes en pacientes en el grupo de XTANDI en comparación con los pacientes en el grupo de placebo (Tabla 9 y Figura 1).
| NR = No alcanzado. | ||
|
||
|
XTANDI (N = 800) |
Placebo (N = 399) |
|
|
Número de muertes (%) |
308 (38.5) |
212 (53.1) |
|
Supervivencia media, meses (IC del 95%) |
18.4 (17.3, NR) |
13.6 (11.3, 15.8) |
|
Valor P* |
p < 0.0001 |
|
|
Hazard Ratio (IC del 95%)† |
0.63 (0.53, 0.75) |
|

PREVAIL (NCT01212991): XTANDI versus Placebo en CRPC Metastásico sin Quimioterapia Previa
En PREVAIL, 1717 pacientes sin quimioterapia previa fueron aleatorizados 1:1 para recibir XTANDI por vía oral a una dosis de 160 mg una vez al día (N = 872) o placebo por vía oral una vez al día (N = 845). Se permitieron pacientes con metástasis viscerales, pacientes con antecedentes de insuficiencia cardíaca leve a moderada (clase I o II de la NYHA) y pacientes que tomaban medicamentos asociados con la reducción del umbral convulsivo. Se excluyeron los pacientes con antecedentes de convulsiones o una afección que pudiera predisponer a las convulsiones y los pacientes con dolor moderado o intenso por cáncer de próstata. El tratamiento del estudio continuó hasta la progresión de la enfermedad (evidencia de progresión radiográfica, un evento relacionado con el esqueleto o progresión clínica) y el inicio de una quimioterapia citotóxica o un agente en investigación, toxicidad inaceptable o retiro. Se evaluaron la supervivencia global y la supervivencia libre de progresión radiográfica (SLPr). La progresión radiográfica se evaluó mediante imágenes secuenciales y se definió mediante la identificación mediante gammagrafía ósea de 2 o más nuevas lesiones óseas con confirmación (criterios del Grupo de Trabajo 2 de Ensayos Clínicos de Cáncer de Próstata) y/o criterios de Evaluación de la Respuesta en Tumores Sólidos (RECIST v 1.1) para la progresión de lesiones de tejidos blandos. El análisis primario de la SLPr utilizó la evaluación radiográfica centralizada de la progresión.
Las características demográficas de los pacientes y las características basales de la enfermedad estaban equilibradas entre los grupos de tratamiento al ingreso. La mediana de edad fue de 71 años (rango 42-93) y la distribución racial fue 77% blanca, 10% asiática, 2% negra y 11% otra. La puntuación del estado funcional ECOG fue 0 para el 68% de los pacientes y 1 para el 32% de los pacientes. La evaluación basal del dolor fue 0-1 (asintomático) en el 67% de los pacientes y 2-3 (levemente sintomático) en el 32% de los pacientes, según la definición del Breve Formulario de Inventario del Dolor (peor dolor en las últimas 24 horas al ingreso al estudio). El cincuenta y cuatro por ciento de los pacientes tenía evidencia radiográfica de progresión de la enfermedad y el 43% tenía progresión solo del PSA. El doce por ciento de los pacientes tenía afectación visceral (pulmón y/o hígado). Durante el estudio, el 27% de los pacientes en el grupo de XTANDI y el 30% de los pacientes en el grupo de placebo recibieron glucocorticoides por diversas razones.
Se demostró una mejora estadísticamente significativa en la supervivencia global en el análisis intermedio preespecificado, realizado después de 540 muertes, en pacientes tratados con XTANDI en comparación con los tratados con placebo (Tabla 10). El cuarenta por ciento de los pacientes tratados con XTANDI y el 70% de los pacientes tratados con placebo recibieron terapias posteriores para el CRPC metastásico que pueden prolongar la supervivencia global. Se realizó un análisis de supervivencia actualizado cuando se observaron 784 muertes. El tiempo medio de seguimiento fue de 31 meses. Los resultados de este análisis fueron consistentes con los del análisis intermedio preespecificado (Tabla 10, Figura 2). En el análisis actualizado, el 52% de los pacientes tratados con XTANDI y el 81% de los pacientes tratados con placebo habían recibido terapias posteriores que pueden prolongar la supervivencia global en el CRPC metastásico. XTANDI se usó como terapia posterior en el 2% de los pacientes tratados con XTANDI y en el 29% de los pacientes tratados con placebo.
| NR = No alcanzado. | ||
|
||
|
XTANDI (N = 872) |
Placebo (N = 845) |
|
|
Análisis Interino Preespecificado* |
||
|
Número de Muertes (%) |
241 (28) |
299 (35) |
|
Supervivencia Media, meses (IC del 95%) |
32.4 (30.1, NR) |
30.2 (28.0, NR) |
|
Valor P† |
p < 0.0001 |
|
|
Hazard Ratio (IC del 95%)‡ |
0.71 (0.60, 0.84) |
|
|
Análisis de Supervivencia Actualizado§ |
||
|
Número de muertes (%) |
368 (42) |
416 (49) |
|
Mediana de supervivencia, meses (IC del 95%) |
35.3 (32.2, NR) |
31.3 (28.8, 34.2) |
|
Hazard Ratio (IC del 95%)‡ |
0.77 (0.67, 0.88) |
|

Se demostró una mejora estadísticamente significativa en la rPFS en pacientes tratados con XTANDI en comparación con pacientes tratados con placebo (Tabla 11, Figura 3).
| NR = No alcanzado. Nota: A la fecha de corte para el análisis de rPFS, 1633 pacientes habían sido aleatorizados. |
||
|
XTANDI (N = 832) |
Placebo (N = 801) |
|
|
Número de progresiones o muertes (%) |
118 (14) |
320 (40) |
|
rPFS mediana (meses) (IC del 95%) |
NR (13.8, NR) |
3.7 (3.6, 4.6) |
|
Valor P* |
p < 0.0001 |
|
|
Hazard Ratio (IC del 95%)† |
0.17 (0.14, 0.21) |
|

El tiempo hasta el inicio de la quimioterapia citotóxica se prolongó después del tratamiento con XTANDI, con una mediana de 28.0 meses para los pacientes en el brazo de XTANDI versus una mediana de 10.8 meses para los pacientes en el brazo de placebo [HR = 0.35 (IC del 95%: 0.30, 0.40), p < 0.0001].
El tiempo medio hasta el primer evento relacionado con el esqueleto fue de 31.1 meses para los pacientes en el brazo de XTANDI versus 31.3 meses para los pacientes en el brazo de placebo [HR = 0.72 (IC del 95%: 0.61, 0.84), p < 0.0001]. Un evento relacionado con el esqueleto se definió como radioterapia o cirugía ósea para el cáncer de próstata, fractura ósea patológica, compresión de la médula espinal o cambio de la terapia antineoplásica para tratar el dolor óseo.
TERRAIN (NCT01288911): XTANDI versus Bicalutamida en CRPC Metastásico sin Quimioterapia Previa
TERRAIN se realizó en 375 pacientes sin quimioterapia previa que fueron aleatorizados 1:1 para recibir XTANDI por vía oral a una dosis de 160 mg una vez al día (N = 184) o bicalutamida por vía oral a una dosis de 50 mg una vez al día (N = 191). Se excluyeron los pacientes con antecedentes de convulsiones o una afección que pudiera predisponer a las convulsiones y los pacientes con dolor moderado a intenso por cáncer de próstata. Los pacientes podrían haber recibido bicalutamida previamente, pero se excluyeron aquellos cuya enfermedad había progresado con la terapia antiandrogénica previa (p. ej., bicalutamida). El tratamiento del estudio continuó hasta la progresión de la enfermedad (evidencia de progresión radiográfica, un evento relacionado con el esqueleto), el inicio del agente antineoplásico posterior, la toxicidad inaceptable o la retirada. La progresión radiográfica de la enfermedad se evaluó mediante una Revisión Central Independiente (ICR) utilizando los criterios del Grupo de Trabajo 2 de Ensayos Clínicos de Cáncer de Próstata y/o los criterios de los Criterios de Evaluación de la Respuesta en Tumores Sólidos (RECIST v 1.1) para la progresión de las lesiones de tejidos blandos. La supervivencia libre de progresión radiográfica (rPFS) se definió como el tiempo desde la aleatorización hasta la primera evidencia objetiva de progresión radiográfica según la evaluación de ICR o la muerte, lo que ocurriera primero.
Las características demográficas y basales de la enfermedad de los pacientes estaban equilibradas entre los brazos de tratamiento al inicio. La mediana de edad fue de 71 años (rango 48-96) y la distribución racial fue 93% blanca, 5% negra, 1% asiática y 1% otra. La puntuación del estado funcional ECOG fue 0 para el 74% de los pacientes y 1 para el 26% de los pacientes. La evaluación del dolor basal fue de 0‑1 (asintomático) en el 58% de los pacientes y de 2‑3 (levemente sintomático) en el 36% de los pacientes, según la definición de la Pregunta 3 del Formulario Corto del Inventario Breve del Dolor (peor dolor en las últimas 24 horas al inicio del estudio). El noventa y ocho por ciento de los pacientes tenía evidencia objetiva de progresión de la enfermedad al inicio del estudio. El cuarenta y seis por ciento de los pacientes había recibido tratamiento previo con bicalutamida, mientras que ningún paciente recibió tratamiento previo con XTANDI.
Se demostró una mejora en la rPFS en pacientes tratados con XTANDI en comparación con pacientes tratados con bicalutamida (Tabla 12, Figura 4).
| XTANDI (N = 184) |
Bicalutamida (N = 191) |
|
|---|---|---|
| NR = No alcanzado. | ||
|
||
|
Número de Progresiones o Muertes (%) |
72 (39) |
74 (39) |
|
rPFS Mediana (meses) (IC del 95%) |
19.5 (11.8, NR) |
13.4 (8.2, 16.4) |
|
Hazard Ratio (IC del 95%)* |
0.60 (0.43, 0.83) |
|

PROSPER (NCT02003924): XTANDI versus Placebo en CRPC No Metastásico
PROSPER reclutó a 1401 pacientes con CRPC no metastásico que fueron aleatorizados 2:1 para recibir XTANDI por vía oral a una dosis de 160 mg una vez al día (N = 933) o placebo por vía oral una vez al día (N = 468). Todos los pacientes en el ensayo PROSPER recibieron un análogo de la hormona liberadora de gonadotropina (GnRH) o se les había realizado una orquiectomía bilateral previa. Los pacientes fueron estratificados por el Tiempo de Duplicación del Antígeno Prostático Específico (PSADT) y el uso de agentes dirigidos a los huesos. Se requirió que los pacientes tuvieran un tiempo de duplicación del PSA ≤ 10 meses, PSA ≥ 2 ng/mL y confirmación de enfermedad no metastásica mediante revisión central independiente ciega (BICR). Los resultados del PSA fueron cegados y no se utilizaron para la interrupción del tratamiento. Los pacientes aleatorizados a cualquiera de los brazos interrumpieron el tratamiento por progresión radiográfica de la enfermedad confirmada por BICR, inicio de un nuevo tratamiento, toxicidad inaceptable o retirada.
Las siguientes características demográficas y basales de los pacientes estaban equilibradas entre los dos brazos de tratamiento. La mediana de edad al momento de la aleatorización fue de 74 años (rango 50-95) y el 23% tenía 80 años o más. La distribución racial fue 71% blancos, 16% asiáticos y 2% negros. La mayoría de los pacientes tenían una puntuación de Gleason de 7 o superior (77%). La mediana del PSADT fue de 3.7 meses. El cincuenta y cuatro por ciento (54%) de los pacientes recibieron tratamiento previo para el cáncer de próstata con cirugía o radiación. El sesenta y tres por ciento (63%) de los pacientes recibió tratamiento previo con un antiandrógeno; el 56% de los pacientes recibió bicalutamida y el 11% de los pacientes recibió flutamida. Todos los pacientes tenían una puntuación del Eastern Cooperative Oncology Group Performance Status (ECOG PS) de 0 o 1 al ingresar al estudio.
El principal resultado de eficacia del estudio fue la supervivencia libre de metástasis (MFS), definida como el tiempo desde la aleatorización hasta que ocurriera primero cualquiera de los siguientes eventos: 1) progresión radiográfica loco-regional y/o distante según BICR o 2) muerte hasta 112 días después de la interrupción del tratamiento sin evidencia de progresión radiográfica. Se demostró una mejora estadísticamente significativa en la MFS y la OS en pacientes aleatorizados para recibir XTANDI en comparación con los pacientes aleatorizados para recibir placebo. Se observaron resultados de MFS consistentes al considerar solo eventos de progresión radiográfica distante o muertes independientemente de la fecha de corte. También se observaron resultados de MFS consistentes en subgrupos de pacientes preespecificados y estratificados de PSADT (< 6 meses o ≥ 6 meses) y el uso previo de un agente dirigido a los huesos (sí o no). Los resultados de eficacia de PROSPER se resumen en la Tabla 13, Figura 5 y Figura 6.
| NR = No alcanzado. | ||
|
||
|
XTANDI |
Placebo |
|
|
Supervivencia libre de metástasis |
||
|
Número de Eventos (%) |
219 (23.5) |
228 (48.7) |
|
Mediana, meses (IC del 95%)* |
36.6 (33.1, NR) |
14.7 (14.2, 15.0) |
|
Hazard Ratio (IC del 95%)† |
0.29 (0.24, 0.35) |
|
|
Valor p† |
p < 0.0001 |
|
|
Supervivencia global‡ |
||
|
Número de Eventos (%) |
288 (30.9) |
178 (38.0) |
|
Mediana, meses (IC del 95%)* |
67.0 (64.0, NR) |
56.3 (54.4, 63.0) |
|
Hazard Ratio (95% CI)† |
0.73 (0.61, 0.88) |
|
|
Valor p† |
p = 0.0011 |
|


El resultado primario de eficacia también fue respaldado por un retraso estadísticamente significativo en el tiempo hasta el primer uso de una nueva terapia antineoplásica (TTA) para los pacientes en el brazo de XTANDI en comparación con aquellos en el brazo de placebo. La mediana de TTA fue de 39,6 meses para los pacientes con XTANDI y de 17,7 meses para los pacientes con placebo (HR = 0,21; IC del 95%: [0,17, 0,26], p < 0,0001).
ARCHES (NCT02677896): XTANDI versus Placebo en CSPC Metastásico
ARCHES inscribió a 1150 pacientes con mCSPC que fueron aleatorizados 1:1 para recibir XTANDI por vía oral a una dosis de 160 mg una vez al día (N = 574) o placebo por vía oral una vez al día (N = 576). Todos los pacientes en el ensayo recibieron un análogo de GnRH o se les había realizado una orquiectomía bilateral previa. Los pacientes fueron estratificados por volumen de enfermedad (bajo vs alto) y terapia previa con docetaxel para el cáncer de próstata (sin docetaxel previo, 1-5 ciclos o 6 ciclos previos). El alto volumen de enfermedad se define como metástasis que involucran las vísceras o, en ausencia de lesiones viscerales, debe haber 4 o más lesiones óseas, al menos 1 de las cuales debe estar en una estructura ósea más allá de la columna vertebral y el hueso pélvico. No se permitió el tratamiento con docetaxel concurrente. Los pacientes continuaron el tratamiento hasta la progresión radiográfica de la enfermedad, el inicio de un nuevo tratamiento, la toxicidad inaceptable o el retiro.
Las siguientes características demográficas y basales de los pacientes se equilibraron entre los dos brazos de tratamiento. La mediana de edad al momento de la aleatorización fue de 70 años (rango: 42-92) y el 30% tenía 75 años o más. La distribución racial fue 81% blanca, 14% asiática y 1% negra. El sesenta y seis por ciento (66%) de los pacientes tenía una puntuación de Gleason de ≥ 8. El treinta y siete por ciento (37%) de los pacientes tenía un volumen bajo de enfermedad y el 63% de los pacientes tenía un volumen alto de enfermedad. El ochenta y dos por ciento (82%) de los pacientes no había recibido tratamiento previo con docetaxel; el 2% de los pacientes había recibido de 1 a 5 ciclos de docetaxel y el 16% de los pacientes había recibido 6 ciclos previos de tratamiento con docetaxel. El doce por ciento (12%) de los pacientes recibió agentes dirigidos a los huesos concomitantes (bisfosfonatos o inhibidores de RANKL) que incluían indicaciones de cáncer de próstata y no de próstata. La puntuación del Eastern Cooperative Oncology Group Performance Status (ECOG PS) fue 0 para el 78% de los pacientes y 1 para el 22% de los pacientes al ingresar al estudio.
La principal medida de resultado de eficacia fue la supervivencia libre de progresión radiográfica (rPFS) basada en la revisión central independiente ciega (BICR). La supervivencia libre de progresión radiográfica se definió como el tiempo desde la aleatorización hasta la progresión radiográfica de la enfermedad en cualquier momento o la muerte dentro de las 24 semanas posteriores a la interrupción del fármaco del estudio. La progresión radiográfica de la enfermedad se definió mediante la identificación de 2 o más nuevas lesiones óseas en una gammagrafía ósea con confirmación (criterios del Prostate Cancer Working Group 2) y/o progresión de la enfermedad en tejidos blandos. El tiempo hasta una nueva terapia antineoplásica y la SG fueron criterios de valoración de eficacia adicionales.
XTANDI demostró una mejora estadísticamente significativa en la rPFS y la SG en comparación con el placebo. Se observaron resultados consistentes de rPFS en pacientes con volumen alto o bajo de enfermedad y pacientes con y sin terapia previa con docetaxel. Los resultados de eficacia para rPFS y SG de ARCHES se resumen en la Tabla 14, Figura 7 y Figura 8.
| NR = No alcanzado. | ||
|
||
|
XTANDI |
Placebo |
|
|
Supervivencia libre de progresión radiográfica* |
||
|
Número de eventos (%) |
89 (15.5) |
198 (34.4) |
|
Progresión radiográfica de la enfermedad |
77 (13.4) |
185 (32.1) |
|
Muerte dentro de las 24 semanas posteriores a la interrupción del tratamiento |
12 (2.1) |
13 (2.3) |
|
Mediana, meses (IC del 95%)† |
NR |
19.4 (16.6, NR) |
|
Hazard ratio (IC del 95%)‡ |
0.39 (0.30, 0.50) |
|
|
Valor p§ |
p < 0.0001 |
|
|
Supervivencia global |
||
|
Número de eventos (%) |
154 (26.8) |
202 (35.1) |
|
Mediana, meses (IC del 95%)† |
NR (NR, NR) |
NR (49.7, NR) |
|
Hazard ratio (IC del 95%)‡ |
0.66 (0.53, 0.81) |
|
|
Valor p§ |
p < 0.0001 |
|


También se reportó una mejora estadísticamente significativa en el brazo de XTANDI en comparación con el placebo en el tiempo hasta el inicio de una nueva terapia antineoplásica (HR = 0.28 [IC del 95%: 0.20, 0.40]; p < 0.0001).
EMBARK (NCT02319837): XTANDI versus Placebo en CSPC No Metastásico con BCR de Alto Riesgo
EMBARK inscribió a 1068 pacientes con nmCSPC con BCR de alto riesgo que fueron aleatorizados 1:1:1 para recibir XTANDI por vía oral a una dosis de 160 mg una vez al día simultáneamente con leuprolida (N = 355), XTANDI por vía oral a una dosis de 160 mg una vez al día como etiqueta abierta como agente único (N = 355), o placebo por vía oral una vez al día simultáneamente con leuprolida (N = 358). Todos los pacientes tenían terapia definitiva previa con prostatectomía radical o radioterapia (incluida braquiterapia) con intención curativa, o ambas. Los pacientes no eran candidatos para radioterapia de rescate en el momento de la inscripción. Se requirió que los pacientes tuvieran confirmación de enfermedad no metastásica por BICR, BCR de alto riesgo (definido por un tiempo de duplicación del PSA ≤ 9 meses) y valores de PSA ≥1 ng/mL si habían tenido prostatectomía radical previa (con o sin radioterapia) como tratamiento primario para el cáncer de próstata o valores de PSA al menos 2 ng/mL por encima del nadir si habían tenido radioterapia previa únicamente.
Los pacientes fueron estratificados por PSA de detección (≤10 ng/mL vs. >10 ng/mL), tiempo de duplicación del PSA (≤3 meses versus >3 meses a ≤ 9 meses) y terapia hormonal previa. Para los pacientes cuyos valores de PSA fueron indetectables (<0.2 ng/mL) en la semana 36, el tratamiento se suspendió en la semana 37 y luego se reinició cuando los valores de PSA aumentaron a ≥2.0 ng/mL para pacientes con prostatectomía previa o ≥5.0 ng/mL para pacientes sin prostatectomía previa. Para los pacientes cuyos valores de PSA fueron detectables (≥0.2 ng/mL) en la semana 36, el tratamiento continuó sin suspensión hasta que se cumplieron los criterios de interrupción permanente del tratamiento. Para todos los pacientes, el tratamiento se interrumpió permanentemente al progresar la enfermedad radiográfica confirmada por BICR, al iniciar un nuevo tratamiento, por toxicidad inaceptable o por retiro.
La mediana de edad en la aleatorización fue de 69 años (rango: 49-93) y el 23% tenía 75 años o más. La distribución racial fue 83% blancos, 7% asiáticos, 4% negros, 2.3% otros y 2.7% no reportados; el 5.5% de los pacientes eran hispanos o latinos. La mediana del PSADT fue de 4.9 meses. El setenta y cuatro por ciento (74%) de los pacientes tenían terapia definitiva previa con prostatectomía radical, el 34% de los pacientes tenían radioterapia primaria previa (incluida braquiterapia) y el 49% de los pacientes tenían terapia previa con cirugía y radioterapia (incluida radioterapia adyuvante y de rescate). El treinta y dos por ciento (32%) de los pacientes tenía una puntuación de Gleason de ≥ 8. La puntuación ECOG PS fue 0 para el 92% de los pacientes y 1 para el 8% de los pacientes al ingresar al estudio.
La principal medida de resultado de eficacia fue la supervivencia libre de metástasis (MFS) en pacientes aleatorizados para recibir XTANDI más leuprolida en comparación con pacientes aleatorizados para recibir placebo más leuprolida. La MFS se definió como el tiempo desde la aleatorización hasta que ocurriera primero lo siguiente 1) progresión radiográfica según BICR o 2) muerte. La MFS en pacientes aleatorizados para recibir XTANDI como agente único en comparación con pacientes aleatorizados para recibir placebo más leuprolida y la supervivencia global (OS) fueron medidas de resultado de eficacia adicionales.
Se demostró una mejora estadísticamente significativa en la MFS en pacientes aleatorizados para recibir XTANDI más leuprolida en comparación con pacientes aleatorizados para recibir placebo más leuprolida. También se demostró una mejora estadísticamente significativa en la MFS en pacientes aleatorizados para recibir XTANDI como agente único en comparación con pacientes aleatorizados para recibir placebo más leuprolida. Los resultados se resumen en la Tabla 15 y la Figura 9.
| NR = No alcanzado. | |||
|
|||
|
XTANDI +
Leuprolida |
Placebo +
Leuprolida |
XTANDI (N = 355) |
|
|
Supervivencia libre de metástasis |
|||
|
Número de eventos (%)* |
45 (12.7) |
92 (25.7) |
63 (17.7) |
|
Mediana, meses (IC del 95%)† |
NR (NR, NR) |
NR (85.1, NR) |
NR (NR, NR) |
|
Hazard Ratio relativo a Placebo más Leuprolide (IC del 95%)‡ |
0.42 (0.30, 0.61) |
— |
0.63 (0.46, 0.87) |
|
Valor p para la comparación con Placebo + Leuprolide§ |
p < 0.0001 |
— |
p = 0.0049 |
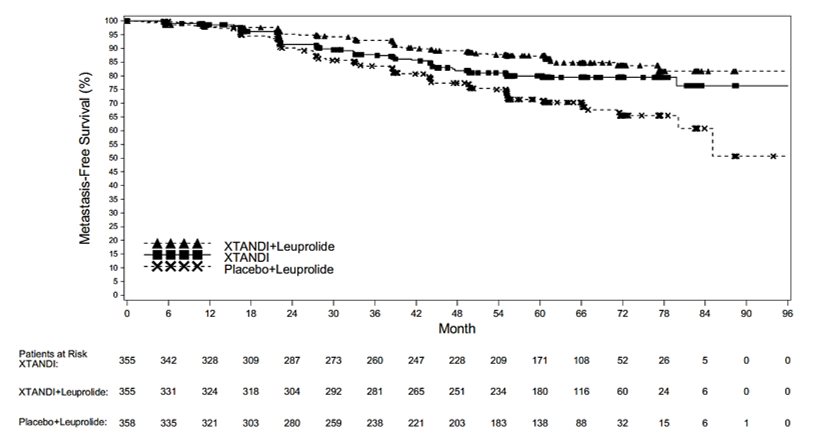
Figura 9. Curvas de Kaplan-Meier de supervivencia libre de metástasis en los brazos de tratamiento con XTANDI más Leuprolide vs. Placebo más Leuprolide vs. XTANDI en EMBARK
Los datos de SG no estaban maduros en el momento del análisis de SLM (12.2% de muertes en la población general de 1068 pacientes).
16 SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
Las cápsulas de XTANDI (enzalutamida) de 40 mg se suministran como cápsulas de gelatina blanda oblongas de color blanco a blanquecino, impresas con tinta negra con ENZ y están disponibles en el siguiente tamaño de envase:
- •
- Frascos de 120 cápsulas con cierre a prueba de niños (NDC 0469-0125-99)
Los comprimidos de XTANDI (enzalutamida) de 40 mg se suministran como comprimidos recubiertos con película, redondos y amarillos, con la inscripción E 40, y están disponibles en el siguiente tamaño de envase:
- •
- Frascos de 120 comprimidos con cierre a prueba de niños (NDC 0469-0625-99)
Los comprimidos de XTANDI (enzalutamida) de 80 mg se suministran como comprimidos recubiertos con película, ovalados y amarillos, con la inscripción E 80, y están disponibles en el siguiente tamaño de envase:
- •
- Frascos de 60 comprimidos con cierre a prueba de niños (NDC 0469-0725-60)
Conserve las cápsulas y los comprimidos de XTANDI a 20°C a 25°C (68°F a 77°F) en un lugar seco y mantenga el envase bien cerrado. Se permiten excursiones de 15°C a 30°C (59°F a 86°F).
17 INFORMACIÓN DE ASESORAMIENTO AL PACIENTE
Aconseje al paciente que lea el etiquetado del paciente aprobado por la FDA (Información para el paciente).
Convulsiones
- •
- Informe a los pacientes que XTANDI se ha asociado con un mayor riesgo de convulsiones. Analice las afecciones que pueden predisponer a las convulsiones y los medicamentos que pueden disminuir el umbral convulsivo. Advierta a los pacientes sobre el riesgo de participar en cualquier actividad en la que una pérdida repentina del conocimiento pueda causar daño grave a sí mismos o a otros. Informe a los pacientes que deben comunicarse con su proveedor de atención médica de inmediato si experimentan pérdida del conocimiento o convulsiones [ver Advertencias y precauciones (5.1)].
Síndrome de encefalopatía posterior reversible (PRES)
- •
- Informe a los pacientes que deben comunicarse con su proveedor de atención médica de inmediato si experimentan síntomas que empeoran rápidamente, posiblemente indicativos de PRES, como convulsiones, dolor de cabeza, disminución del estado de alerta, confusión, disminución de la vista o visión borrosa [ver Advertencias y precauciones (5.2)].
Hipersensibilidad
- •
- Informe a los pacientes que XTANDI puede estar asociado con reacciones de hipersensibilidad que incluyen hinchazón de la cara, labios, lengua o garganta [ver Advertencias y precauciones (5.3)]. Aconseje a los pacientes que experimenten estos tipos de síntomas de hipersensibilidad que interrumpan el tratamiento con XTANDI y se comuniquen rápidamente con su proveedor de atención médica.
Cardiopatía isquémica
- •
- Informe a los pacientes que XTANDI se ha asociado con un mayor riesgo de cardiopatía isquémica. Aconseje a los pacientes que busquen atención médica inmediata si se produce algún síntoma que sugiera un evento cardiovascular [ver Advertencias y precauciones (5.4)].
Caídas y fracturas
- •
- Informe a los pacientes que XTANDI se asocia con una mayor incidencia de mareos/vértigo, caídas y fracturas. Aconseje a los pacientes que informen estas reacciones adversas a su proveedor de atención médica [ver Advertencias y precauciones (5.5)].
Disfagia o atragantamiento
- •
- Informe a los pacientes que el tamaño de las cápsulas y comprimidos de XTANDI se ha asociado con disfagia o atragantamiento graves.
- •
- Aconseje a los pacientes que tomen cada cápsula o comprimido según las instrucciones de Posología y administración.
- •
- Aconseje a los pacientes que informen a su proveedor de atención médica si tienen dificultad para tragar XTANDI [ver Advertencias y precauciones (5.7)].
Hipertensión
- •
- Informe a los pacientes que XTANDI se asocia con una mayor incidencia de hipertensión [ver Reacciones adversas (6.1)].
Posología y administración
- •
- Informe a los pacientes que no se han sometido a orquiectomía bilateral y que están recibiendo terapia con GnRH que deben mantener este tratamiento durante el curso del tratamiento con XTANDI.
- •
- Indique a los pacientes que tomen su dosis a la misma hora todos los días (una vez al día). XTANDI se puede tomar con o sin alimentos. Aconseje a los pacientes que tomen cada cápsula o comprimido entero con una cantidad suficiente de agua para asegurar que todo el medicamento se trague correctamente. No mastique, disuelva ni abra las cápsulas. No corte, triture ni mastique los comprimidos [ver Advertencias y precauciones (5.7)].
- •
- Informe a los pacientes que no deben interrumpir, modificar la dosis o suspender XTANDI sin consultar primero a su proveedor de atención médica.
- •
- Informe a los pacientes que si olvidan una dosis, deben tomarla tan pronto como lo recuerden. Si olvidan tomar la dosis durante todo el día, deben tomar su dosis normal al día siguiente. No deben tomar más de la dosis prescrita por día [ver Posología y administración (2.1)].
Toxicidad embriofetal
- •
- Informe a los pacientes que XTANDI puede ser perjudicial para un feto en desarrollo y puede causar pérdida del embarazo.
- •
- Aconseje a los pacientes varones con parejas femeninas en edad fértil que utilicen métodos anticonceptivos eficaces durante el tratamiento y durante 3 meses después de la última dosis de XTANDI. Aconseje a los pacientes varones que usen un condón si tienen relaciones sexuales con una mujer embarazada [ver Advertencias y precauciones (5.6)].
Infertilidad
- •
- Informe a los pacientes varones que XTANDI puede afectar la fertilidad [ver Uso en poblaciones específicas (8.3)].
Fabricado para y distribuido por: Astellas Pharma US, Inc., Northbrook, IL 60062
Comercializado por:
Astellas Pharma US, Inc., Northbrook, IL 60062 Pfizer Inc., New York, NY 10017
415196-XTA-USA
Rx solamente
© 2025 Astellas Pharma Inc. o sus afiliados
PROSPECTO PARA EL PACIENTE
|
INFORMACIÓN PARA EL PACIENTE |
|
|
XTANDI® (ex TAN dee) (enzalutamida) cápsulas |
XTANDI® (ex TAN dee) (enzalutamida) comprimidos |
|
¿Qué es XTANDI®? XTANDI es un medicamento recetado que se usa para tratar a hombres con cáncer de próstata que:
No se sabe si XTANDI es seguro y eficaz en mujeres. No se sabe si XTANDI es seguro y eficaz en niños. |
|
|
Antes de tomar XTANDI, informe a su proveedor de atención médica sobre todas sus afecciones médicas, incluso si:
Informe a su proveedor de atención médica sobre todos los medicamentos que toma, incluidos los medicamentos recetados y de venta libre, las vitaminas y los suplementos herbales. XTANDI puede afectar la forma en que funcionan otros medicamentos, y otros medicamentos pueden afectar la forma en que funciona XTANDI. No debe comenzar ni suspender ningún medicamento antes de hablar con el proveedor de atención médica que le recetó XTANDI. Conozca los medicamentos que toma. Lleve una lista de ellos para mostrársela a su proveedor de atención médica y farmacéutico cuando reciba un medicamento nuevo. |
|
|
¿Cómo debo tomar XTANDI?
|
|
|
¿Cuáles son los posibles efectos secundarios de XTANDI? XTANDI puede causar efectos secundarios graves, incluyendo:
Su proveedor de atención médica interrumpirá el tratamiento con XTANDI si tiene efectos secundarios graves. |
|
|
Los efectos secundarios más comunes de XTANDI incluyen: |
|
|
|
|
XTANDI puede causar problemas de fertilidad en los hombres, lo que puede afectar la capacidad de engendrar hijos. Hable con su proveedor de atención médica si tiene inquietudes sobre la fertilidad. Estos no son todos los posibles efectos secundarios de XTANDI. Llame a su médico para obtener asesoramiento médico sobre los efectos secundarios. Puede informar los efectos secundarios a la FDA al 1-800-FDA-1088. |
|
|
¿Cómo debo guardar XTANDI?
Mantenga XTANDI y todos los medicamentos fuera del alcance de los niños. |
|
|
Información general sobre el uso seguro y eficaz de XTANDI. A veces, los medicamentos se recetan para fines distintos de los que figuran en un prospecto de información para el paciente. No use XTANDI para una afección para la que no se lo recetaron. No le dé XTANDI a otras personas, incluso si tienen los mismos síntomas que usted. Puede causarles daño. Puede pedirle a su proveedor de atención médica o farmacéutico información sobre XTANDI que esté escrita para profesionales de la salud. |
|
|
¿Cuáles son los ingredientes de XTANDI? Cápsulas XTANDI Ingrediente activo: enzalutamida Ingredientes inactivos: caprylocaproyl polyoxylglycerides, butilhidroxi anisol, butilhidroxitolueno, gelatina, solución de sorbitol sorbitán, glicerina, agua purificada, dióxido de titanio, óxido de hierro negro. Tabletas XTANDI Ingrediente activo: enzalutamida Ingredientes inactivos: acetato de hipromelosa succinato, celulosa microcristalina, dióxido de silicio coloidal, croscarmelosa sódica y estearato de magnesio. La cubierta pelicular de la tableta contiene hipromelosa, talco, polietilenglicol, dióxido de titanio y óxido férrico. Fabricado para y distribuido por: Astellas Pharma US, Inc., Northbrook, IL 60062 Para obtener más información, visite www.Xtandi.com o llame al 1-800-727-7003. |
|
|
Esta información para el paciente ha sido aprobada por la Administración de Alimentos y Medicamentos de los EE. UU. Revisado: enero de 2025 |
|
PANEL DE VISUALIZACIÓN PRINCIPAL DEL PAQUETE/ETIQUETA

NDC 0469-0125-99
Rx Only
Xtandi®
(enzalutamide)
cápsulas
40 mg
Tragar las cápsulas enteras con una cantidad suficiente
de agua. No mastique,
disuelva ni abra las cápsulas.
120 Cápsulas
Fabricado por
Catalent Pharma Solutions, LLC para
Astellas Pharma US, Inc.,
Northbrook, IL 60062
111607-04
415196-XTA-USA
4006396
PANEL DE VISUALIZACIÓN PRINCIPAL DEL PAQUETE/ETIQUETA

NDC 0469-0625-99
Rx Only
Xtandi®
(enzalutamide)
tablets
40 mg
Tragar los comprimidos enteros con
una cantidad suficiente de agua. No
corte, triture ni mastique los comprimidos.
120 Comprimidos
Distribuido por Astellas Pharma US, Inc., Northbrook, IL 60062
75104449
415196-XTA-USA
4006397
PANEL DE VISUALIZACIÓN PRINCIPAL DEL PAQUETE/ETIQUETA

NDC 0469-0725-60
Rx Only
Xtandi®
(enzalutamida)
comprimidos
80 mg
Tragar los comprimidos enteros con
una cantidad suficiente de agua. No
partir, triturar ni masticar los comprimidos.
60 Comprimidos
Distribuido por Astellas Pharma US, Inc., Northbrook, IL 60062
75104452
415196-XTA-USA
4006399
PANEL DE VISUALIZACIÓN PRINCIPAL DEL PAQUETE/ETIQUETA

NDC 0469-1125-56
Rx solamente
Xtandi®
40 mg
56 Cápsulas blandas
MUESTRA
MUESTRA – NO APTO PARA LA VENTA
PANEL DE VISUALIZACIÓN PRINCIPAL DEL PAQUETE/ETIQUETA

NDC 0469-1725-56
Rx Only
Xtandi®
(enzalutamide)
tablets
40 mg
Tragar los comprimidos enteros con
una cantidad suficiente de agua. No
corte, triture ni mastique los comprimidos.
56 Comprimidos
Muestra – No para la venta
Distribuido por Astellas Pharma US, Inc., Northbrook, IL 60062
75104450
415196-XTA-USA
4006398