Fabricante de medicamentos: Genentech, Inc. (Updated: 2024-11-20)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
XOLAIR® (omalizumab) inyección, para uso subcutáneo
XOLAIR® (omalizumab) para inyección, para uso subcutáneo
Aprobación inicial en EE. UU.: 2003
ADVERTENCIA: ANAFILAXIA
Consulte la información completa de prescripción para ver la advertencia completa en recuadro.
Se ha notificado la aparición de anafilaxia, que se presenta como broncoespasmo, hipotensión, síncope, urticaria y/o angioedema de la garganta o la lengua, después de la administración de XOLAIR. La anafilaxia se ha producido después de la primera dosis de XOLAIR, pero también después de más de 1 año de iniciado el tratamiento. Inicie el tratamiento con XOLAIR en un centro sanitario, observe de cerca a los pacientes durante un período de tiempo adecuado después de la administración de XOLAIR y esté preparado para controlar la anafilaxia, que puede ser mortal. Informe a los pacientes sobre los signos y síntomas de la anafilaxia y pídales que busquen atención médica inmediata si aparecen síntomas. La selección de pacientes para la autoadministración de XOLAIR debe basarse en criterios para mitigar el riesgo de anafilaxia. (2.6, 5.1, 6.1, 6.2)
CAMBIOS IMPORTANTES RECIENTES
INDICACIONES Y USO
XOLAIR es un anticuerpo anti-IgE indicado para:
- Asma persistente de moderada a grave en adultos y pacientes pediátricos de 6 años de edad o mayores con una prueba cutánea positiva o reactividad in vitro a un aeroalérgeno perenne y síntomas que no están adecuadamente controlados con corticosteroides inhalados (1.1)
- Rinosinusitis crónica con pólipos nasales (CRSwNP) en pacientes adultos de 18 años de edad o mayores con respuesta inadecuada a los corticosteroides nasales, como tratamiento de mantenimiento complementario (1.2)
- Alergia alimentaria mediada por IgE en pacientes adultos y pediátricos de 1 año de edad o mayores para la reducción de reacciones alérgicas (Tipo I), incluida la anafilaxia, que pueden ocurrir con la exposición accidental a uno o más alimentos. Para ser usado junto con la evitación de alérgenos alimentarios (1.3)
- Urticaria espontánea crónica (UEC) en adultos y adolescentes de 12 años de edad o mayores que permanecen sintomáticos a pesar del tratamiento con antihistamínicos H1 (1.4)
Limitaciones de Uso:
POSOLOGÍA Y ADMINISTRACIÓN
Sólo para administración subcutánea (SC). (2.2, 2.3, 2.4, 2.5)
Consulte la información completa de prescripción para obtener instrucciones de administración (2.6, 2.7, 2.8).
- Asma: XOLAIR 75 a 375 mg SC cada 2 o 4 semanas. Determine la dosis (mg) y la frecuencia de dosificación según el nivel de IgE sérica total (UI/mL), medido antes del inicio del tratamiento, y el peso corporal (kg). Consulte las tablas de determinación de dosis. (2.2)
- Rinosinusitis crónica con pólipos nasales: XOLAIR 75 a 600 mg SC cada 2 o 4 semanas. Determine la dosis (mg) y la frecuencia de dosificación según el nivel de IgE sérica total (UI/mL), medido antes del inicio del tratamiento, y el peso corporal (kg). Consulte las tablas de determinación de dosis. (2.3)
- Alergia alimentaria mediada por IgE: XOLAIR 75 mg a 600 mg SC cada 2 o 4 semanas. Determine la dosis (mg) y la frecuencia de dosificación según el nivel de IgE sérica total (UI/mL), medido antes del inicio del tratamiento, y el peso corporal (kg). Consulte la tabla de determinación de dosis. (2.4)
- Urticaria espontánea crónica: XOLAIR 150 o 300 mg SC cada 4 semanas. La dosificación en la UEC no depende del nivel de IgE sérica ni del peso corporal. (2.5)
PRESENTACIONES Y CONCENTRACIONES
- Inyección: solución de 75 mg/0,5 mL, 150 mg/mL y 300 mg/2 mL en una jeringa precargada de dosis única. (3)
- Inyección: solución de 75 mg/0,5 mL, 150 mg/mL y 300 mg/2 mL en un autoinyector precargado de dosis única. (3)
- Para inyección: polvo liofilizado de 150 mg en un vial de dosis única para reconstitución. (3)
CONTRAINDICACIONES
ADVERTENCIAS Y PRECAUCIONES
- Anafilaxia: Inicie el tratamiento con XOLAIR en un entorno sanitario preparado para controlar la anafilaxia, que puede ser potencialmente mortal, y observe a los pacientes durante un período de tiempo adecuado después de la administración. (5.1)
- Neoplasias malignas: Se han observado neoplasias malignas en estudios clínicos. (5.2)
- Síntomas agudos de asma: No usar para el tratamiento del broncoespasmo agudo o el asma aguda grave. (5.3)
- Reducción de corticosteroides: No suspenda bruscamente los corticosteroides al iniciar el tratamiento con XOLAIR. (5.4)
- Afecciones eosinofílicas: Esté atento a la eosinofilia, la erupción vasculítica, el empeoramiento de los síntomas pulmonares, las complicaciones cardíacas y/o la neuropatía, especialmente al reducir los corticosteroides orales. (5.5)
- Fiebre, artralgia y erupción cutánea: Suspenda XOLAIR si los pacientes desarrollan signos y síntomas similares a los del suero. (5.6)
- Posible error de medicación relacionado con el tratamiento de urgencia de la anafilaxia: XOLAIR no debe utilizarse para el tratamiento de urgencia de reacciones alérgicas, incluida la anafilaxia. (5.9)
REACCIONES ADVERSAS
- Asma: Las reacciones adversas más frecuentes (≥ 1% de los pacientes) en los estudios clínicos con pacientes adultos y adolescentes ≥12 años fueron artralgia, dolor (general), dolor en las piernas, fatiga, mareos, fracturas, dolor en los brazos, prurito, dermatitis y dolor de oído. En los estudios clínicos con pacientes pediátricos de 6 a <12 años, las reacciones adversas más frecuentes (≥3% de los pacientes) fueron nasofaringitis, cefalea, pirexia, dolor abdominal superior, faringitis estreptocócica, otitis media, gastroenteritis vírica, picaduras de artrópodos y epistaxis. (6.1)
- Rinosinusitis crónica con pólipos nasales: Las reacciones adversas más frecuentes (≥ 3% de los pacientes) en los estudios clínicos con pacientes adultos incluyeron las siguientes: cefalea, reacción en el lugar de la inyección, artralgia, dolor abdominal superior y mareos. (6.1)
- Alergia alimentaria mediada por IgE: Las reacciones adversas más frecuentes (≥3% de los pacientes) fueron reacciones en el lugar de la inyección y pirexia. (6.1)
- Urticaria crónica espontánea: Las reacciones adversas más frecuentes (≥2% de los pacientes) incluyeron las siguientes: náuseas, nasofaringitis, sinusitis, infección de las vías respiratorias superiores, infección vírica de las vías respiratorias superiores, artralgia, cefalea y tos. (6.1)
Para notificar SOSPECHAS DE REACCIONES ADVERSAS, póngase en contacto con Genentech en el 1-888-835-2555 o con la FDA en el 1-800-FDA-1088 o en www.fda.gov/medwatch.
INTERACCIONES MEDICAMENTOSAS
No se han realizado estudios formales de interacción medicamentosa. (7)
Ver 17 para obtener INFORMACIÓN PARA EL PACIENTE y Guía de medicamentos.
Revisado: 2/2024
Tabla de contenido
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDO*
ADVERTENCIA: ANAFILAXIS
1 INDICACIONES Y USO
1.1 Asma
1.2 Rinosinusitis crónica con pólipos nasales
1.3 Alergia alimentaria mediada por IgE
1.4 Urticaria espontánea crónica
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Descripción general de la determinación de la dosis
2.2 Dosis recomendada para el asma
2.3 Dosis recomendada para la rinosinusitis crónica con pólipos nasales
2.4 Dosis recomendada para la alergia alimentaria mediada por IgE
2.5 Dosis recomendada para la urticaria espontánea crónica
2.6 Descripción general de la administración
2.7 Jeringa precargada y autoinyector XOLAIR
2.8 Preparación para el uso e inyección de polvo liofilizado XOLAIR
3 FORMAS Y CONCENTRACIONES FARMACÉUTICAS
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Anafilaxis
5.2 Neoplasias malignas
5.3 Síntomas de asma aguda y empeoramiento de la enfermedad
5.4 Reducción de corticosteroides
5.5 Afecciones eosinofílicas
5.6 Fiebre, artralgia y erupción cutánea
5.7 Infección parasitaria (helmíntica)
5.8 Pruebas de laboratorio
5.9 Posible error de medicación relacionado con el tratamiento de emergencia de la anafilaxis
6 REACCIONES ADVERSAS
6.1 Experiencia en ensayos clínicos
6.2 Experiencia postcomercialización
7 INTERACCIONES MEDICAMENTOSAS
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.4 Uso pediátrico
8.5 Uso geriátrico
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
12.2 Farmacodinamia
12.3 Farmacocinética
12.6 Inmunogenicidad
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, mutagénesis, deterioro de la fertilidad
14 ESTUDIOS CLÍNICOS
14.1 Asma
14.2 Rinosinusitis crónica con pólipos nasales
14.3 Alergia alimentaria mediada por IgE
14.4 Urticaria espontánea crónica
16 PRESENTACIÓN/ALMACENAMIENTO Y MANEJO
17 INFORMACIÓN PARA EL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información completa de prescripción no se enumeran.
ADVERTENCIA EN EL RECUADRO
ADVERTENCIA: ANAFILAXIS
Se ha reportado anafilaxis presentándose como broncoespasmo, hipotensión, síncope, urticaria y/o angioedema de la garganta o lengua, después de la administración de XOLAIR. La anafilaxis ha ocurrido tan pronto como después de la primera dosis de XOLAIR, pero también ha ocurrido después de 1 año de haber comenzado el tratamiento administrado regularmente. Debido al riesgo de anafilaxis, inicie la terapia con XOLAIR en un entorno de atención médica y observe de cerca a los pacientes durante un período de tiempo apropiado después de la administración de XOLAIR. Los proveedores de atención médica que administran XOLAIR deben estar preparados para manejar la anafilaxis, que puede ser potencialmente mortal. Informe a los pacientes sobre los signos y síntomas de la anafilaxis e instrúyalos para que busquen atención médica inmediata en caso de que se presenten síntomas. La selección de pacientes para la autoadministración de XOLAIR debe basarse en criterios para mitigar el riesgo de anafilaxis [ver Dosificación y Administración (2.6), Advertencias y Precauciones (5.1) y Reacciones Adversas (6.1, 6.2)].
1 INDICACIONES Y USO
1.1 Asma
XOLAIR está indicado para adultos y pacientes pediátricos de 6 años de edad o mayores con asma persistente de moderada a grave que tienen una prueba cutánea positiva o reactividad in vitro a un aeroalérgeno perenne y cuyos síntomas no están adecuadamente controlados con corticosteroides inhalados.
1.2 Rinosinusitis crónica con pólipos nasales
XOLAIR está indicado como tratamiento de mantenimiento adicional de la rinosinusitis crónica con pólipos nasales (CRSwNP) en pacientes adultos de 18 años de edad o mayores con respuesta inadecuada a los corticosteroides nasales.
1.3 Alergia alimentaria mediada por IgE
XOLAIR está indicado para la reducción de las reacciones alérgicas (Tipo I), incluida la anafilaxia, que pueden ocurrir con la exposición accidental a uno o más alimentos en pacientes adultos y pediátricos de 1 año de edad o mayores con alergia alimentaria mediada por IgE.
XOLAIR debe utilizarse junto con la evitación de alérgenos alimentarios.
1.4 Urticaria espontánea crónica
XOLAIR está indicado para el tratamiento de adultos y adolescentes de 12 años de edad o mayores con urticaria espontánea crónica (CSU) que permanecen sintomáticos a pesar del tratamiento con antihistamínicos H1.
2 DOSIS Y ADMINISTRACIÓN
2.1 Resumen de la determinación de la dosis
Asma, y rinosinusitis crónica con pólipos nasales, y alergia alimentaria mediada por IgE
- Determine la dosis de XOLAIR según el nivel de IgE sérica total (UI/mL) medido antes del inicio del tratamiento y según el peso corporal (kg).
- Para pacientes con asma, rinosinusitis crónica con pólipos nasales (CRSwNP) y alergia alimentaria mediada por IgE, la determinación de la dosis debe basarse en el diagnóstico principal para el cual se prescribe XOLAIR.
- Ajuste las dosis para cambios significativos en el peso corporal durante el tratamiento.
- Consulte las Tablas 1 y 2 para la dosis recomendada para el tratamiento del asma, la Tabla 3 para el tratamiento de la CRSwNP y la Tabla 4 para el tratamiento de la alergia alimentaria mediada por IgE.
-
Los niveles de IgE total están elevados durante el tratamiento y permanecen elevados hasta un año después de la interrupción del tratamiento. Por lo tanto, la repetición de las pruebas de los niveles de IgE durante el tratamiento con XOLAIR no se puede utilizar como guía para la determinación de la dosis.
- Interrupciones de menos de un año: Dosis basada en los niveles de IgE sérica obtenidos en la determinación de la dosis inicial.
- Interrupciones de un año o más: Vuelva a analizar los niveles de IgE sérica total para la determinación de la dosis (Tabla 1 o 2 para el tratamiento del asma, según la edad del paciente, Tabla 3 para el tratamiento de la CRSwNP y Tabla 4 para el tratamiento de la alergia alimentaria mediada por IgE).
Urticaria espontánea crónica
La dosis de XOLAIR en pacientes con urticaria espontánea crónica (UEC) no depende del nivel de IgE sérica (libre o total) o del peso corporal [ver Dosis y administración (2.5)].
2.2 Dosis recomendada para el asma
La dosis recomendada para el asma es XOLAIR 75 mg a 375 mg por inyección subcutánea cada 2 o 4 semanas según el nivel de IgE sérica total (UI/mL) medido antes del inicio del tratamiento y según el peso corporal (kg) [ver Dosis y administración (2.1)].
- Pacientes adultos y adolescentes de 12 años o más: Inicie la dosificación según la Tabla 1.
- Pacientes pediátricos de 6 a <12 años de edad: Inicie la dosificación según la Tabla 2.
 |
 |
2.3 Dosis recomendada para la rinosinusitis crónica con pólipos nasales
La dosis recomendada para la rinosinusitis crónica con pólipos nasales (CRSwNP) es XOLAIR 75 mg a 600 mg por inyección subcutánea cada 2 o 4 semanas según el nivel de IgE sérica total (UI/mL) medido antes del inicio del tratamiento y según el peso corporal (kg) [ver Dosis y administración (2.1)]. Consulte la Tabla 3 para obtener la dosis recomendada según el nivel de IgE sérica total y el peso corporal para pacientes con CRSwNP.
 |
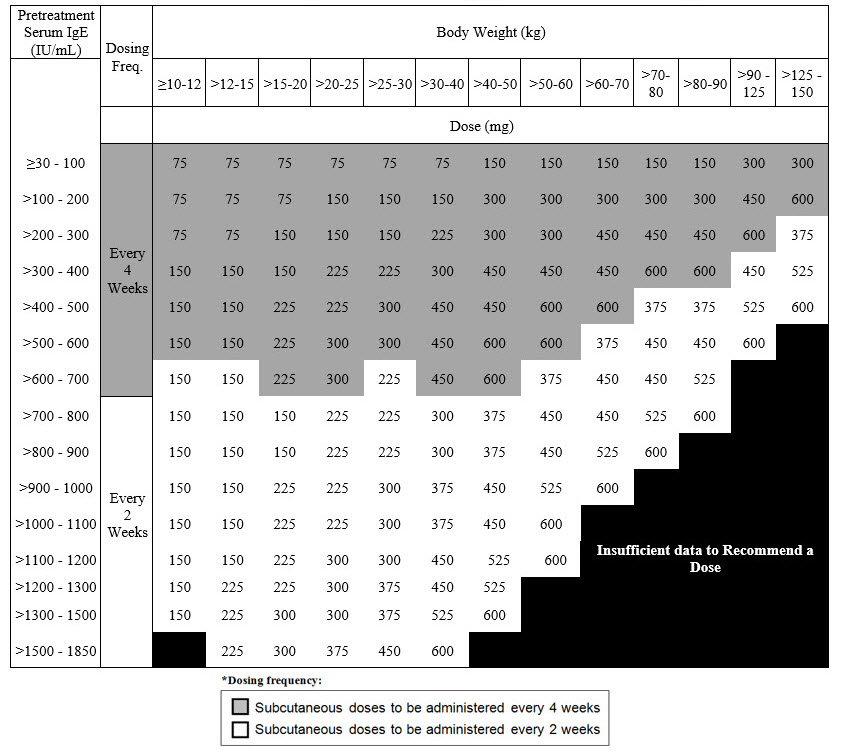
2.4 Dosificación recomendada para la alergia alimentaria mediada por IgE
La dosificación recomendada para la alergia alimentaria mediada por IgE es XOLAIR 75 mg a 600 mg por inyección subcutánea cada 2 o 4 semanas, en función del nivel de IgE total sérica (UI/mL), medido antes del inicio del tratamiento, y del peso corporal [véase Dosificación y administración (2.1)]. Consulte la Tabla 4 para obtener la dosificación recomendada en función del nivel de IgE sérica y del peso corporal para pacientes con alergia alimentaria mediada por IgE.
 |
2.5 Dosis recomendada para la urticaria espontánea crónica
La dosis recomendada para la urticaria espontánea crónica (UEC) es XOLAIR 150 mg o 300 mg por inyección subcutánea cada 4 semanas.
- La dosis de 300 mg se puede administrar como una inyección subcutánea de 300 mg/2 ml o como dos inyecciones subcutáneas de 150 mg/ml.
- La dosificación de XOLAIR en pacientes con UEC no depende del nivel de IgE sérica (libre o total) ni del peso corporal.
2.6 Resumen de la administración
- Administre XOLAIR mediante inyección subcutánea.
- XOLAIR está destinado a usarse bajo la supervisión de un profesional sanitario.
- Inicie el tratamiento en un centro sanitario y, una vez que se haya establecido el tratamiento de forma segura, el profesional sanitario podrá determinar si la autoadministración de la jeringa precargada o el autoinyector de XOLAIR por parte del paciente o cuidador es adecuada, en función de una evaluación cuidadosa del riesgo de anafilaxia y las estrategias de mitigación.
Selección de pacientes para la autoadministración de la jeringa precargada o el autoinyector de XOLAIR
Los profesionales sanitarios deben tener en cuenta los factores de riesgo conocidos de anafilaxia a XOLAIR [véase Advertencias y precauciones (5.1)] y las estrategias de mitigación al seleccionar pacientes para la autoadministración. Deben considerarse los factores específicos del paciente, incluidos los siguientes criterios:
- 1a)
- Asma, RSEcNP y UEC: El paciente no debe tener antecedentes de anafilaxia a XOLAIR u otros agentes, como alimentos, medicamentos, productos biológicos, etc.
- 1b)
- Alergia alimentaria mediada por IgE: El paciente no debe tener antecedentes de anafilaxia a XOLAIR u otros agentes (excepto alimentos), como medicamentos, productos biológicos, etc.
- 2)
- El paciente debe recibir al menos 3 dosis de XOLAIR bajo la supervisión de un profesional sanitario sin reacciones de hipersensibilidad
- 3)
- El paciente o cuidador puede reconocer los síntomas de anafilaxia
- 4)
- El paciente o cuidador puede tratar la anafilaxia adecuadamente
- 5)
- El paciente o cuidador puede realizar inyecciones subcutáneas con la jeringa precargada o el autoinyector de XOLAIR con la técnica adecuada según el régimen de dosificación prescrito y las Instrucciones de uso
2.7 Jeringa precargada y autoinyector de XOLAIR
Las dosis de inyección de XOLAIR están disponibles como jeringa precargada o como autoinyector. Indique a los pacientes o cuidadores que sigan las instrucciones que se proporcionan en las “Instrucciones de uso” para la preparación y administración de la jeringa precargada o el autoinyector de XOLAIR [véase Instrucciones de uso].
Jeringa precargada de XOLAIR
- Adolescentes de 12 años o más: La jeringa precargada de XOLAIR puede autoadministrarse bajo la supervisión de un adulto.
- Pacientes pediátricos de 1 a 11 años: La jeringa precargada de XOLAIR debe ser administrada por un cuidador.
Autoinyector de XOLAIR
- Adolescentes de 12 años o más: El autoinyector de XOLAIR puede autoadministrarse bajo la supervisión de un adulto. Los autoinyectores de XOLAIR (todas las dosis) están destinados únicamente para su uso en adultos y adolescentes de 12 años o más.
- Pacientes pediátricos de 1 a 11 años: Los autoinyectores de XOLAIR (todas las dosis) no están destinados para su uso en pacientes pediátricos menores de 12 años.
Instrucciones de administración para la jeringa precargada y el autoinyector
- Las personas con alergia al látex no deben manipular la jeringa precargada XOLAIR porque la tapa de la aguja de las jeringas precargadas XOLAIR de 75 mg/0,5 mL y 150 mg/mL contiene un derivado del látex de caucho natural que puede causar reacciones alérgicas en individuos sensibles al látex [ver Cómo se suministra/Almacenamiento y manipulación (16)].
- Inspeccione visualmente el contenido de la jeringa precargada o autoinyector en busca de partículas y decoloración antes de la administración. La solución de la jeringa precargada o autoinyector XOLAIR debe ser transparente e incolora a amarillo parduzco pálido. No utilice la jeringa precargada o el autoinyector si el medicamento está turbio, decolorado o contiene partículas.
- Determine el número de jeringas precargadas o autoinyectores necesarios para la dosis del paciente (ver Tabla 5). Para pacientes pediátricos de 1 a 11 años de edad, se debe tener en cuenta el número de inyecciones con jeringas precargadas necesarias y el volumen que se va a inyectar en relación con el peso corporal del paciente.
- Para los pacientes que necesitan más de 1 inyección para completar una dosis completa, administre cada inyección a por lo menos 1 pulgada de distancia de otros sitios de inyección.
- Administre la inyección subcutánea en el muslo o el abdomen, evitando el área de 2 pulgadas (5 cm) directamente alrededor del ombligo. El área externa de la parte superior de los brazos solo se puede utilizar si la inyección la administra un cuidador o un proveedor de atención médica [ver Instrucciones de uso]. La administración de la inyección puede tardar hasta 15 segundos.
| Dosis de XOLAIR‡ | 75 mg | 150 mg | 300 mg‡ | Volumen total inyectado |
|---|---|---|---|---|
|
||||
| 75 mg | 1 | 0 | 0 | 0.5 mL |
| 150 mg | 0 | 1 | 0 | 1 mL |
| 225 mg | 1 | 1 | 0 | 1.5 mL |
| 300 mg | 0 | 0 | 1 | 2 mL |
| 375 mg | 1 | 0 | 1 | 2.5 mL |
| 450 mg | 0 | 1 | 1 | 3 mL |
| 525 mg | 1 | 1 | 1 | 3.5 mL |
| 600 mg | 0 | 0 | 2 | 4 mL |
2.8 Preparación para el uso e inyección de XOLAIR en polvo liofilizado
El polvo liofilizado de XOLAIR solo debe ser preparado e inyectado por un profesional sanitario. El polvo liofilizado de XOLAIR suministrado debe reconstituirse con Agua Estéril para Inyección (AEI) USP, siguiendo las siguientes instrucciones:
- 1)
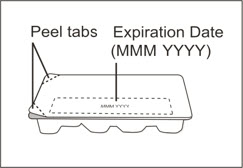
- Antes de la reconstitución, determine el número de viales que necesitarán ser reconstituidos (cada vial contiene 150 mg de XOLAIR en 1,2 mL) (véase Tabla 6).
| Dosis de XOLAIR* | Número de viales | Número de inyecciones | Volumen total inyectado |
|---|---|---|---|
|
|||
| 75 mg | 1 | 1 | 0,6 mL |
| 150 mg | 1 | 1 | 1,2 mL |
| 225 mg | 2 | 2 | 1,8 mL |
| 300 mg | 2 | 2 | 2,4 mL |
| 375 mg | 3 | 3 | 3,0 mL |
| 450 mg | 3 | 3 | 3,6 mL |
| 525 mg | 4 | 4 | 4,2 mL |
| 600 mg | 4 | 4 | 4,8 mL |
- 2)
- Aspire 1,4 mL de AEI, USP, en una jeringa de 3 mL equipada con una aguja de 18 G de 1 pulgada.
- 3)
- Coloque el vial en posición vertical sobre una superficie plana y, utilizando una técnica aséptica estándar, inserte la aguja e inyecte la AEI, USP, directamente sobre el producto.
- 4)
- Manteniendo el vial en posición vertical, gire suavemente el vial durante aproximadamente 1 minuto para humedecer uniformemente el polvo. No agitar.
- 5)
- Gire suavemente el vial durante 5 a 10 segundos aproximadamente cada 5 minutos para disolver cualquier sólido restante. El producto liofilizado tarda de 15 a 20 minutos en disolverse. Si tarda más de 20 minutos en disolverse completamente, gire suavemente el vial durante 5 a 10 segundos aproximadamente cada 5 minutos hasta que no queden partículas gelatinosas visibles en la solución. No lo utilice si el contenido del vial no se disuelve completamente en 40 minutos.
- 6)
- Después de la reconstitución, la solución de XOLAIR es algo viscosa y tendrá un aspecto transparente o ligeramente opalescente. Es aceptable si hay algunas burbujas pequeñas o espuma alrededor del borde del vial; no debe haber partículas gelatinosas visibles en la solución reconstituida. No lo utilice si hay partículas extrañas presentes.
- 7)
- Invierta el vial durante 15 segundos para permitir que la solución drene hacia el tapón.
- 8)
- Utilice la solución de XOLAIR dentro de las 8 horas posteriores a la reconstitución cuando se almacene en el vial a 2 ºC a 8 ºC (36 ºF a 46 ºF), o dentro de las 4 horas posteriores a la reconstitución cuando se almacene a temperatura ambiente. Los viales de XOLAIR reconstituidos deben protegerse de la luz solar.
- 9)
- Utilizando una nueva jeringa de 3 mL equipada con una aguja de 18 G de 1 pulgada, inserte la aguja en el vial invertido. Coloque la punta de la aguja en la parte inferior de la solución en el tapón del vial al aspirar la solución en la jeringa. El producto reconstituido es algo viscoso. Extraiga todo el producto del vial antes de expulsar el aire o el exceso de solución de la jeringa. Antes de retirar la aguja del vial, tire del émbolo hasta el final del cilindro de la jeringa para extraer toda la solución del vial invertido.
- 10)
- Reemplace la aguja de 18 G por una aguja de 25 G para la inyección subcutánea.
- 11)
- Expulse el aire, las burbujas grandes y cualquier exceso de solución para obtener un volumen de 1,2 mL correspondiente a una dosis de 150 mg de XOLAIR. Para obtener un volumen de 0,6 mL correspondiente a una dosis de 75 mg de XOLAIR, expulse el aire, las burbujas grandes y deseche 0,6 mL de la jeringa. Puede quedar una fina capa de pequeñas burbujas en la parte superior de la solución de la jeringa.
- 12)
- Administre XOLAIR mediante inyección subcutánea. La administración de la inyección puede tardar de 5 a 10 segundos debido a que la solución es ligeramente viscosa. No administre más de 150 mg (contenido de un vial) por sitio de inyección. Divida las dosis superiores a 150 mg entre dos o más sitios de inyección. Elija un sitio de inyección diferente para cada nueva inyección, a una distancia mínima de 1 pulgada del área utilizada para otras inyecciones.
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
Inyección:
- 75 mg/0.5 mL es una solución transparente a ligeramente opalescente e incolora a amarillo parduzco pálido en una jeringa precargada de dosis única con protector de aguja o autoinyector precargado de dosis única
- 150 mg/mL es una solución transparente a ligeramente opalescente e incolora a amarillo parduzco pálido en una jeringa precargada de dosis única con protector de aguja o autoinyector precargado de dosis única
- 300 mg/2 mL es una solución transparente a ligeramente opalescente e incolora a amarillo parduzco pálido en una jeringa precargada de dosis única con protector de aguja o autoinyector precargado de dosis única
- Para inyección: 150 mg de polvo liofilizado blanco en un vial de dosis única para reconstitución
4 CONTRAINDICACIONES
XOLAIR está contraindicado en pacientes con reacción de hipersensibilidad grave a XOLAIR o a cualquier componente de XOLAIR [ver Advertencias y precauciones (5.1)].
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Anafilaxia
Se ha notificado la aparición de anafilaxia después de la administración de XOLAIR en ensayos clínicos previos a la comercialización y en notificaciones espontáneas posteriores a la comercialización [véase la Advertencia en recuadro y las Reacciones adversas (6.2)]. Los signos y síntomas en estos casos notificados han incluido broncoespasmo, hipotensión, síncope, urticaria y/o angioedema de la garganta o la lengua. Algunos de estos eventos han sido potencialmente mortales. En los ensayos clínicos previos a la comercialización en pacientes con asma, se notificó anafilaxia en 3 de 3507 (0,1%) pacientes. La anafilaxia se produjo con la primera dosis de XOLAIR en dos pacientes y con la cuarta dosis en un paciente. El tiempo de inicio de la anafilaxia fue de 90 minutos después de la administración en dos pacientes y de 2 horas después de la administración en un paciente.
Un estudio de casos y controles en pacientes con asma mostró que, entre los usuarios de XOLAIR, los pacientes con antecedentes de anafilaxia a alimentos, medicamentos u otras causas tenían un mayor riesgo de anafilaxia asociada con XOLAIR, en comparación con aquellos sin antecedentes previos de anafilaxia [véase Reacciones adversas (6.1)].
En las notificaciones espontáneas posteriores a la comercialización, la frecuencia de anafilaxia atribuida al uso de XOLAIR se estimó en al menos el 0,2% de los pacientes, basándose en una exposición estimada de aproximadamente 57 300 pacientes desde junio de 2003 hasta diciembre de 2006. La anafilaxia se ha producido tan pronto como después de la primera dosis de XOLAIR, pero también se ha producido después de un año de haber comenzado el tratamiento programado regularmente. Aproximadamente del 60% al 70% de los casos de anafilaxia se han notificado que ocurren dentro de las tres primeras dosis de XOLAIR, con casos adicionales que ocurren esporádicamente más allá de la tercera dosis.
Inicie XOLAIR solo en un entorno de atención médica equipado para manejar la anafilaxia, que puede ser potencialmente mortal. Observe a los pacientes de cerca durante un período de tiempo apropiado después de la administración de XOLAIR, teniendo en cuenta el tiempo de inicio de la anafilaxia observado en los ensayos clínicos previos a la comercialización y en las notificaciones espontáneas posteriores a la comercialización [véase Reacciones adversas (6.1), 6.2)]. Informe a los pacientes sobre los signos y síntomas de la anafilaxia e indíqueles que busquen atención médica inmediata si aparecen signos o síntomas.
Una vez que se haya establecido la terapia con XOLAIR, la administración de la jeringa precargada o el autoinyector de XOLAIR fuera de un centro de atención médica por parte de un paciente o un cuidador puede ser adecuada para pacientes seleccionados. La selección del paciente, determinada por el proveedor de atención médica en consulta con el paciente, debe tener en cuenta el patrón de eventos de anafilaxia observados en los ensayos clínicos previos a la comercialización y en las notificaciones espontáneas posteriores a la comercialización, así como los factores de riesgo individuales del paciente (por ejemplo, antecedentes de anafilaxia), la capacidad para reconocer los signos y síntomas de la anafilaxia y la capacidad para realizar inyecciones subcutáneas con la jeringa precargada o el autoinyector de XOLAIR con la técnica adecuada de acuerdo con el régimen de dosificación prescrito y las instrucciones de uso [véase Dosificación y administración (2.6), Reacciones adversas (6.1), 6.2)].
Suspenda XOLAIR en pacientes que experimenten una reacción de hipersensibilidad grave [véase Contraindicaciones (4)].
5.2 Neoplasia maligna
Se observaron neoplasias malignas en 20 de 4127 (0,5%) pacientes tratados con XOLAIR en comparación con 5 de 2236 (0,2%) pacientes control en estudios clínicos de adultos y adolescentes ≥12 años con asma y otros trastornos alérgicos. Las neoplasias malignas observadas en pacientes tratados con XOLAIR fueron de diversos tipos, con mama, piel no melanoma, próstata, melanoma y parótida ocurriendo más de una vez, y otros cinco tipos ocurriendo una vez cada uno. La mayoría de los pacientes fueron observados durante menos de 1 año. Se desconoce el impacto de una exposición más prolongada a XOLAIR o su uso en pacientes con mayor riesgo de neoplasias malignas (por ejemplo, ancianos, fumadores actuales).
En un estudio observacional posterior de 5007 pacientes adolescentes y adultos tratados con XOLAIR y 2829 pacientes no tratados con XOLAIR con asma persistente de moderada a grave y una reacción positiva de la prueba cutánea o reactividad in vitro a un aeroalergeno perenne, los pacientes fueron seguidos durante un máximo de 5 años. En este estudio, las tasas de incidencia de neoplasias malignas primarias (por 1000 años-paciente) fueron similares entre los pacientes tratados con XOLAIR (12,3) y los pacientes no tratados con XOLAIR (13,0) [véase Reacciones adversas (6.1)]. Sin embargo, las limitaciones del estudio impiden descartar definitivamente un riesgo de neoplasia maligna con XOLAIR. Las limitaciones del estudio incluyen: el diseño del estudio observacional, el sesgo introducido al permitir la inscripción de pacientes previamente expuestos a XOLAIR (88%), la inscripción de pacientes (56%) mientras que los antecedentes de cáncer o una afección premaligna fueron criterios de exclusión del estudio, y la alta tasa de abandono del estudio (44%).
5.3 Síntomas agudos de asma y empeoramiento de la enfermedad
No se ha demostrado que XOLAIR alivie las exacerbaciones del asma de forma aguda. No utilice XOLAIR para tratar el broncoespasmo agudo o el estado asmático. Los pacientes deben buscar asesoramiento médico si su asma permanece incontrolada o empeora después de iniciar el tratamiento con XOLAIR.
5.4 Reducción de corticosteroides
No suspenda abruptamente los corticosteroides sistémicos o inhalados al iniciar el tratamiento con XOLAIR para el asma o la RCEcNP. Disminuya los corticosteroides gradualmente bajo la supervisión directa de un médico. En pacientes con CSU, no se ha evaluado el uso de XOLAIR en combinación con corticosteroides.
5.5 Afecciones eosinofílicas
En casos raros, los pacientes con asma en terapia con XOLAIR pueden presentar eosinofilia sistémica grave, a veces con características clínicas de vasculitis consistentes con el síndrome de Churg-Strauss, una afección que a menudo se trata con terapia con corticosteroides sistémicos. Estos eventos generalmente, pero no siempre, se han asociado con la reducción de la terapia con corticosteroides orales. Los médicos deben estar atentos a la eosinofilia, la erupción vasculítica, el empeoramiento de los síntomas pulmonares, las complicaciones cardíacas y/o la neuropatía que se presenten en sus pacientes. No se ha establecido una asociación causal entre XOLAIR y estas afecciones subyacentes.
5.6 Fiebre, artralgia y erupción
En el uso posterior a la aprobación, algunos pacientes han experimentado un conjunto de signos y síntomas que incluyen artritis/artralgia, erupción, fiebre y linfadenopatía con un inicio de 1 a 5 días después de la primera o subsiguientes inyecciones de XOLAIR. Estos signos y síntomas han recurrido después de dosis adicionales en algunos pacientes. Aunque no se observaron complejos inmunitarios circulantes o una biopsia de piel consistente con una reacción de Tipo III en estos casos, estos signos y síntomas son similares a los observados en pacientes con enfermedad del suero. Los médicos deben suspender XOLAIR si un paciente desarrolla este conjunto de signos y síntomas [ver Reacciones adversas (6.2)].
5.7 Infección parasitaria (helmíntica)
Controle a los pacientes con alto riesgo de infección por geohelmintos mientras estén en terapia con XOLAIR. No hay datos suficientes disponibles para determinar la duración del control requerido para las infecciones por geohelmintos después de suspender el tratamiento con XOLAIR.
En un ensayo clínico de un año realizado en Brasil en pacientes adultos y adolescentes con alto riesgo de infecciones geohelmínticas (lombriz intestinal, anquilostoma, tricocéfalo, oxiuro), el 53% (36/68) de los pacientes tratados con XOLAIR experimentaron una infección, según el diagnóstico mediante examen de heces estándar, en comparación con el 42% (29/69) de los controles con placebo. La estimación puntual de la razón de posibilidades de infección fue de 1,96, con un intervalo de confianza del 95% (0,88, 4,36), lo que indica que en este estudio, un paciente que tuvo una infección tenía entre 0,88 y 4,36 veces más probabilidades de haber recibido XOLAIR que un paciente que no tuvo una infección. La respuesta al tratamiento antigeohelmíntico apropiado de la infección, medida por el recuento de huevos en las heces, no fue diferente entre los grupos de tratamiento.
5.8 Pruebas de laboratorio
Los niveles séricos totales de IgE aumentan después de la administración de XOLAIR debido a la formación de complejos XOLAIR:IgE [ver Farmacología clínica (12.2)]. Los niveles séricos totales de IgE elevados pueden persistir hasta por 1 año después de la interrupción de XOLAIR. No utilice los niveles séricos totales de IgE obtenidos menos de 1 año después de la interrupción para reevaluar el régimen de dosificación para pacientes con asma, CRSwNP o alergia alimentaria mediada por IgE, ya que estos niveles pueden no reflejar los niveles de IgE libre en estado estacionario [ver Dosificación y administración (2.2, 2.3, 2.4)].
5.9 Posible error de medicación relacionado con el tratamiento de emergencia de la anafilaxia
XOLAIR no debe utilizarse para el tratamiento de emergencia de reacciones alérgicas, incluida la anafilaxia. En estudios para simular el uso, algunos pacientes y cuidadores no entendieron que XOLAIR no está destinado al tratamiento de emergencia de reacciones alérgicas, incluida la anafilaxia. No se ha establecido la seguridad y eficacia de XOLAIR para el tratamiento de emergencia de reacciones alérgicas, incluida la anafilaxia. Indique a los pacientes que XOLAIR es para uso de mantenimiento para reducir las reacciones alérgicas, incluida la anafilaxia, mientras se evitan los alérgenos alimentarios.
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas clínicamente significativas se describen en otras partes del etiquetado:
- Anafilaxia [ver Advertencia en recuadro y Advertencias y precauciones (5.1)]
- Neoplasias malignas [ver Advertencias y precauciones (5.2)]
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se realizan en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no se pueden comparar directamente con las tasas en los ensayos clínicos de otro fármaco y pueden no reflejar las tasas observadas en la práctica clínica.
Reacciones adversas de estudios clínicos en pacientes adultos y adolescentes de 12 años de edad o mayores con asma
Los datos que se describen a continuación reflejan la exposición a XOLAIR para 2076 pacientes adultos y adolescentes de 12 años o mayores, incluidos 1687 pacientes expuestos durante seis meses y 555 expuestos durante un año o más, en estudios de asma controlados con placebo u otros controlados. La edad media de los pacientes que recibieron XOLAIR fue de 42 años, con 134 pacientes de 65 años de edad o mayores; el 60% eran mujeres y el 85% caucásicos. Los pacientes recibieron XOLAIR de 150 mg a 375 mg cada 2 o 4 semanas o, para los pacientes asignados a grupos de control, terapia estándar con o sin placebo.
Las reacciones adversas que con mayor frecuencia dieron lugar a una intervención clínica (por ejemplo, la interrupción de XOLAIR o la necesidad de medicación concomitante para tratar una reacción adversa) fueron la reacción en el lugar de la inyección (45%), las infecciones víricas (23%), la infección de las vías respiratorias superiores (20%), la sinusitis (16%), el dolor de cabeza (15%) y la faringitis (11%). Estas reacciones se observaron a tasas similares en los pacientes tratados con XOLAIR y en los pacientes de control.
Tabla 7 muestra las reacciones adversas de cuatro ensayos de asma controlados con placebo que ocurrieron ≥1% y con mayor frecuencia en pacientes adultos y adolescentes de 12 años de edad o mayores que recibieron XOLAIR que en aquellos que recibieron placebo. Las reacciones adversas se clasificaron utilizando términos preferidos del diccionario de la nomenclatura médica internacional (IMN). Las reacciones en el lugar de la inyección se registraron por separado de la notificación de otras reacciones adversas.
| Reacción adversa | XOLAIR n=738 |
Placebo n=717 |
|---|---|---|
| Organismo en su conjunto | ||
| Dolor | 7% | 5% |
| Fatiga | 3% | 2% |
| Sistema musculoesquelético | ||
| Artralgia | 8% | 6% |
| Fractura | 2% | 1% |
| Dolor en las piernas | 4% | 2% |
| Dolor en los brazos | 2% | 1% |
| Sistema nervioso | ||
| Mareo | 3% | 2% |
| Piel y anexos | ||
| Prurito | 2% | 1% |
| Dermatitis | 2% | 1% |
| Sentidos especiales | ||
| Otalgia | 2% | 1% |
No hubo diferencias en la incidencia de reacciones adversas según la edad (entre pacientes menores de 65 años), el sexo o la raza.
Estudio de control de casos de anafilaxia
Un estudio retrospectivo de control de casos investigó los factores de riesgo de anafilaxia a XOLAIR entre pacientes tratados con XOLAIR para el asma. Los casos con un historial adjudicado de anafilaxia a XOLAIR se compararon con los controles sin dicho historial. El estudio encontró que un historial autodeclarado de anafilaxia a alimentos, medicamentos u otras causas fue más común entre los pacientes con anafilaxia a XOLAIR (57% de 30 casos) en comparación con los controles (23% de 88 controles) [OR 8.1, IC del 95% 2.7 a 24.3]. Debido a que este es un estudio de control de casos, el estudio no puede proporcionar la incidencia de anafilaxia entre los usuarios de XOLAIR. De otras fuentes, se observó anafilaxia a XOLAIR en el 0,1% de los pacientes en los ensayos clínicos y al menos en el 0,2% de los pacientes según los informes posteriores a la comercialización. Aproximadamente del 60% al 70% de los casos se informaron que ocurrieron dentro de las tres primeras dosis de XOLAIR, con casos adicionales que ocurrieron esporádicamente más allá de la tercera dosis. Se informó que el tiempo de inicio de la anafilaxia ocurrió dentro de las 2 horas para la mayoría de los casos (aproximadamente el 75%) [ver Advertencias y precauciones (5.1), Reacciones adversas (6.2)].
Reacciones en el lugar de la inyección
En adultos y adolescentes, las reacciones en el lugar de la inyección de cualquier gravedad ocurrieron a una tasa del 45% en pacientes tratados con XOLAIR en comparación con el 43% en pacientes tratados con placebo. Los tipos de reacciones en el lugar de la inyección incluyeron: hematomas, enrojecimiento, calor, ardor, escozor, picazón, formación de urticaria, dolor, induraciones, masa e inflamación.
Las reacciones graves en el lugar de la inyección ocurrieron con más frecuencia en pacientes tratados con XOLAIR en comparación con los pacientes del grupo placebo (12% versus 9%).
La mayoría de las reacciones en el lugar de la inyección ocurrieron dentro de 1 hora después de la inyección, duraron menos de 8 días y generalmente disminuyeron en frecuencia en las visitas de dosificación posteriores.
Reacciones adversas de los estudios clínicos en pacientes pediátricos de 6 a <12 años de edad con asma
Los datos que se describen a continuación reflejan la exposición a XOLAIR para 926 pacientes de 6 a <12 años de edad, incluidos 583 pacientes expuestos durante seis meses y 292 expuestos durante un año o más, en estudios de asma controlados con placebo u otros controlados. La edad promedio de los pacientes pediátricos que recibieron XOLAIR fue de 8,8 años; el 69% eran hombres y el 64% eran caucásicos. Los pacientes pediátricos recibieron XOLAIR de 75 mg a 375 mg cada 2 o 4 semanas o, para los pacientes asignados a grupos de control, terapia estándar con o sin placebo. No se informaron casos de malignidad en pacientes tratados con XOLAIR en estos ensayos.
Las reacciones adversas más comunes que ocurrieron en ≥3% en los pacientes pediátricos que recibieron XOLAIR y con más frecuencia que en los pacientes tratados con placebo fueron nasofaringitis, dolor de cabeza, pirexia, dolor abdominal superior, faringitis estreptocócica, otitis media, gastroenteritis viral, picadura de artrópodos y epistaxis.
Las reacciones adversas que con mayor frecuencia provocaron intervención clínica (por ejemplo, interrupción de XOLAIR o la necesidad de medicación concomitante para tratar un evento adverso) fueron bronquitis (0,2%), dolor de cabeza (0,2%) y urticaria (0,2%). Estas reacciones se observaron a tasas similares en pacientes tratados con XOLAIR y pacientes control.
Reacciones adversas de los estudios clínicos en pacientes adultos con rinosinusitis crónica con pólipos nasales
Los datos que se describen a continuación reflejan la exposición a XOLAIR para 135 pacientes ≥ 18 años de edad, expuestos durante seis meses en dos estudios controlados con placebo. La edad promedio de los pacientes que recibieron XOLAIR fue de 49,7 años; el 64% eran hombres y el 94% eran caucásicos. Los pacientes recibieron XOLAIR o placebo SC cada 2 o 4 semanas, con dosis y frecuencia según la Tabla 3. Todos los pacientes recibieron terapia de fondo con mometasona nasal durante todo el estudio. La Tabla 8 enumera las reacciones adversas que ocurrieron en ≥3% de los pacientes tratados con XOLAIR y con más frecuencia que en los pacientes tratados con placebo en los ensayos 1 y 2 de rinosinusitis crónica con pólipos nasales (CRSwNP); los resultados fueron agrupados.
| Reacción adversa | XOLAIR n=135 |
Placebo n=130 |
|---|---|---|
| CRSwNP = Rinosinusitis crónica con pólipos nasales. | ||
|
||
| Trastorno gastrointestinal | ||
| Dolor abdominal superior | 4 (3,0%) | 1 (0,8%) |
| Trastornos generales y afecciones en el lugar de administración | ||
| Reacciones en el lugar de la inyección* | 7 (5,2%) | 2 (1,5%) |
| Trastornos del sistema musculoesquelético y del tejido conjuntivo | ||
| Artralgia | 4 (3,0%) | 2 (1,5%) |
| Trastornos del sistema nervioso | ||
| Cefalea | 11 (8.1%) | 7 (5.4%) |
| Mareo | 4 (3.0%) | 1 (0.8%) |
Reacciones adversas de un estudio clínico en pacientes con alergia alimentaria mediada por IgE
La seguridad de XOLAIR en pacientes con reacciones alérgicas mediadas por IgE (Tipo I), incluida la anafilaxia, que pueden ocurrir con la exposición accidental a uno o más alimentos, se basó en datos del ensayo de alergia alimentaria (FA), un ensayo aleatorizado, doble ciego, controlado con placebo en 168 pacientes (165 pacientes pediátricos y 3 adultos) que eran alérgicos al cacahuete y al menos a otros dos alimentos [ver Estudios clínicos (14.3)]. Los pacientes recibieron una dosis de XOLAIR o placebo por vía subcutánea cada 2 o 4 semanas durante 16 a 20 semanas según la dosis recomendada basada en el nivel de IgE (UI/mL), medido antes del inicio del tratamiento, y por el peso corporal (kg) proporcionado en la Tabla 4 [ver Posología y administración (2.4)]. Los datos de seguridad proporcionados en la Tabla 9 provienen de la población de análisis principal de pacientes pediátricos de 1 a 17 años de edad. Los datos de seguridad obtenidos de adultos (n=3) en este ensayo fueron limitados. La Tabla 9 enumera las reacciones adversas que ocurrieron en ≥3% de los pacientes pediátricos tratados con XOLAIR y con mayor frecuencia que en los pacientes tratados con placebo en el ensayo FA. No hubo interrupciones debido a reacciones adversas.
| Reacción adversa | XOLAIR n=110 |
Placebo n=55 |
|---|---|---|
|
||
| Trastornos generales y alteraciones en el sitio de administración | ||
| Reacciones en el sitio de inyección* | 17 (15.5%) | 6 (10.9%) |
| Pirexia | 7 (6.4%) | 2 (3.6%) |
Reacciones adversas de los estudios clínicos en pacientes con urticaria espontánea crónica
La seguridad de XOLAIR para el tratamiento de la urticaria espontánea crónica (UEC) se evaluó en tres ensayos clínicos controlados con placebo, de dosis múltiples, con una duración de 12 semanas (Ensayo UEC 2) y 24 semanas (Ensayos UEC 1 y 3). En los Ensayos UEC 1 y 2, los pacientes recibieron XOLAIR 75 mg, 150 mg o 300 mg o placebo cada 4 semanas además de su nivel basal de terapia con antihistamínicos H1 durante todo el período de tratamiento. En el Ensayo UEC 3, los pacientes fueron aleatorizados a XOLAIR 300 mg o placebo cada 4 semanas además de su nivel basal de terapia con antihistamínicos H1. Los datos que se describen a continuación reflejan la exposición a XOLAIR de 733 pacientes inscritos y que recibieron al menos una dosis de XOLAIR en los tres ensayos clínicos, incluidos 684 pacientes expuestos durante 12 semanas y 427 expuestos durante 24 semanas. La edad media de los pacientes que recibieron XOLAIR 300 mg fue de 43 años, el 75% eran mujeres y el 89% eran blancos. Los perfiles demográficos de los pacientes que recibieron XOLAIR 150 mg y 75 mg fueron similares.
Tabla 10 muestra las reacciones adversas que ocurrieron en ≥2% de los pacientes que recibieron XOLAIR (150 o 300 mg) y con más frecuencia que los que recibieron placebo. Las reacciones adversas se han agrupado de los Ensayos UEC 2 y las primeras 12 semanas de los Ensayos UEC 1 y 3.
| Reacciones adversas* | Ensayos UEC 1, 2 y 3 Agrupados | ||
|---|---|---|---|
| 150mg (n=175) |
300mg (n=412) |
Placebo (n=242) |
|
|
|||
| Trastornos gastrointestinales | |||
| Náuseas | 2 (1.1%) | 11 (2.7%) | 6 (2.5%) |
| Infecciones e infestaciones | |||
| Nasofaringitis | 16 (9.1%) | 27 (6.6%) | 17 (7.0%) |
| Sinusitis | 2 (1.1%) | 20 (4.9%) | 5 (2.1%) |
| Infección de las vías respiratorias superiores | 2 (1.1%) | 14 (3.4%) | 5 (2.1%) |
| Infección viral de las vías respiratorias superiores | 4 (2.3%) | 2 (0.5%) | (0.0%) |
| Trastornos musculoesqueléticos y del tejido conjuntivo | |||
| Artralgia | 5 (2.9%) | 12 (2.9%) | 1 (0.4%) |
| Trastornos del sistema nervioso | |||
| Cefalea | 21 (12.0%) | 25 (6.1%) | 7 (2.9%) |
| Trastornos respiratorios, torácicos y mediastínicos | |||
| Tos | 2 (1.1%) | 9 (2.2%) | 3 (1.2%) |
Otras reacciones comunicadas durante el período de tratamiento de 24 semanas en los ensayos CSU 1 y 3 [≥2% de los pacientes que recibieron XOLAIR (150 mg o 300 mg) y con mayor frecuencia que los que recibieron placebo] incluyeron: dolor de muelas, infección fúngica, infección del tracto urinario, mialgia, dolor en las extremidades, dolor musculoesquelético, edema periférico, pirexia, migraña, cefalea sinusal, ansiedad, dolor orofaríngeo, asma, urticaria y alopecia.
Reacciones en el lugar de inyección en pacientes con CSU
Las reacciones en el lugar de inyección de cualquier gravedad ocurrieron durante los estudios en más pacientes tratados con XOLAIR [11 pacientes (2,7%) a 300 mg, 1 paciente (0,6%) a 150 mg] en comparación con 2 pacientes tratados con placebo (0,8%). Los tipos de reacciones en el lugar de inyección incluyeron: hinchazón, eritema, dolor, hematomas, picazón, sangrado y urticaria. Ninguno de los eventos provocó la interrupción del estudio o la interrupción del tratamiento.
Eventos cardiovasculares y cerebrovasculares de estudios clínicos en pacientes con asma
Se realizó un estudio de cohorte observacional de 5 años en pacientes ≥12 años de edad con asma persistente de moderada a grave y una reacción positiva de la prueba cutánea a un aeroalergeno perenne para evaluar la seguridad a largo plazo de XOLAIR, incluido el riesgo de malignidad [ver Advertencias y precauciones (5.2)]. Un total de 5007 pacientes tratados con XOLAIR y 2829 pacientes no tratados con XOLAIR se inscribieron en el estudio. Porcentajes similares de pacientes en ambas cohortes eran fumadores actuales (5%) o ex fumadores (29%). Los pacientes tenían una edad promedio de 45 años y se les realizó un seguimiento durante un promedio de 3,7 años. Se diagnosticó asma grave a más pacientes tratados con XOLAIR (50%) en comparación con los pacientes no tratados con XOLAIR (23%) y el 44% de los pacientes interrumpieron prematuramente el estudio. Además, el 88% de los pacientes en la cohorte tratada con XOLAIR habían estado expuestos previamente a XOLAIR durante un promedio de 8 meses.
Se observó una tasa de incidencia más alta (por 1000 pacientes-año) de eventos adversos graves (EAG) cardiovasculares y cerebrovasculares generales en pacientes tratados con XOLAIR (13,4) en comparación con pacientes no tratados con XOLAIR (8,1). Se observaron aumentos en las tasas de ataque isquémico transitorio (0,7 versus 0,1), infarto de miocardio (2,1 versus 0,8), hipertensión pulmonar (0,5 versus 0), embolia pulmonar/trombosis venosa (3,2 versus 1,5) y angina inestable (2,2 versus 1,4), mientras que las tasas observadas para accidente cerebrovascular isquémico y muerte cardiovascular fueron similares entre ambas cohortes de estudio. Los resultados sugieren un posible aumento del riesgo de eventos cardiovasculares y cerebrovasculares graves en pacientes tratados con XOLAIR. Sin embargo, el diseño del estudio observacional, la inclusión de pacientes previamente expuestos a XOLAIR (88%), los desequilibrios iniciales en los factores de riesgo cardiovascular entre los grupos de tratamiento, la incapacidad para ajustar los factores de riesgo no medidos y la alta tasa de interrupción del estudio limitan la capacidad para cuantificar la magnitud del riesgo.
Se realizó un análisis agrupado de 25 ensayos clínicos aleatorizados, doble ciego, controlados con placebo, con una duración de 8 a 52 semanas para evaluar más a fondo el desequilibrio en los EAG cardiovasculares y cerebrovasculares observados en el estudio de cohorte observacional anterior. Un total de 3342 pacientes tratados con XOLAIR y 2895 pacientes tratados con placebo se incluyeron en el análisis agrupado. Los pacientes tenían una edad promedio de 38 años y se les realizó un seguimiento durante una duración promedio de 6,8 meses. No se observaron desequilibrios notables en las tasas de EAG cardiovasculares y cerebrovasculares enumeradas anteriormente. Sin embargo, los resultados del análisis agrupado se basaron en un número bajo de eventos, pacientes ligeramente más jóvenes y una duración de seguimiento más corta que el estudio de cohorte observacional; por lo tanto, los resultados no son suficientes para confirmar o rechazar los hallazgos observados en el estudio de cohorte observacional.
Reacciones adversas del estudio clínico en adultos sanos
Reacciones en el lugar de inyección en adultos sanos
En un ensayo abierto en adultos sanos, en el que el autoinyector de 300 mg/2 ml se comparó con la jeringa precargada de 300 mg/2 ml, se observaron reacciones en el lugar de inyección (p. ej., induración, dolor, eritema, hemorragia, hinchazón, malestar, hematomas, hipoestesia, edema, prurito) en el 24% (16/66) de los sujetos tratados con el autoinyector en comparación con el 14% (9/64) de los sujetos tratados con la jeringa precargada.
6.2 Experiencia postcomercialización
Las siguientes reacciones adversas se han identificado durante el uso posterior a la aprobación de XOLAIR. Debido a que estas reacciones se notifican voluntariamente a partir de una población de tamaño incierto, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal con la exposición al fármaco.
Anafilaxia: Según los informes espontáneos y una exposición estimada de aproximadamente 57 300 pacientes desde junio de 2003 hasta diciembre de 2006, la frecuencia de anafilaxia atribuida al uso de XOLAIR se estimó en al menos el 0,2% de los pacientes.
Los criterios de diagnóstico de anafilaxia fueron la afectación de los tejidos cutáneos o de las mucosas y, ya sea compromiso de las vías respiratorias, y/o reducción de la presión arterial con o sin síntomas asociados, y una relación temporal con la administración de XOLAIR sin otra causa identificable. Los signos y síntomas en estos casos notificados incluyeron broncoespasmo, hipotensión, síncope, urticaria, angioedema de la garganta o la lengua, disnea, tos, opresión en el pecho y/o angioedema cutáneo. Se notificó afectación pulmonar en el 89% de los casos. Se notificó hipotensión o síncope en el 14% de los casos. El quince por ciento de los casos notificados requirieron hospitalización. Se notificó un antecedente de anafilaxia no relacionada con XOLAIR en el 24% de los casos.
De los casos notificados de anafilaxia atribuidos a XOLAIR, el 39% se produjo con la primera dosis, el 19% con la segunda dosis, el 10% con la tercera dosis y el resto después de dosis posteriores. Un caso se produjo después de 39 dosis (después de 19 meses de terapia continua, se produjo anafilaxia cuando se reinició el tratamiento después de un intervalo de 3 meses). El tiempo de aparición de la anafilaxia en estos casos fue de hasta 30 minutos en el 35%, superior a 30 y hasta 60 minutos en el 16%, superior a 60 y hasta 90 minutos en el 2%, superior a 90 y hasta 120 minutos en el 6%, superior a 2 horas y hasta 6 horas en el 5%, superior a 6 horas y hasta 12 horas en el 14%, superior a 12 horas y hasta 24 horas en el 8%, y superior a 24 horas y hasta 4 días en el 5%. En el 9% de los casos, se desconocía el tiempo de aparición.
Veintitrés pacientes que experimentaron anafilaxia fueron reexpuestos a XOLAIR y 18 pacientes presentaron una recurrencia de síntomas similares de anafilaxia. Además, se produjo anafilaxia tras la reexposición a XOLAIR en 4 pacientes que previamente solo habían experimentado urticaria.
Afecciones eosinofílicas: Se han notificado afecciones eosinofílicas [véase Advertencias y precauciones (5.5)].
Fiebre, artralgia y erupción cutánea: Se ha notificado en el uso posterior a la aprobación de XOLAIR un conjunto de signos y síntomas que incluyen artritis/artralgia, erupción cutánea (urticaria u otras formas), fiebre y linfadenopatía similares al suero [véase Advertencias y precauciones (5.6)].
7 INTERACCIONES MEDICAMENTOSAS
No se han realizado estudios formales de interacción medicamentosa con XOLAIR.
En pacientes con asma, SCP con poliposis nasal y alergia alimentaria mediada por IgE, no se ha evaluado el uso concomitante de XOLAIR e inmunoterapia con alérgenos.
En pacientes con CSU, no se ha estudiado el uso de XOLAIR en combinación con terapias inmunosupresoras.
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de riesgos
Un estudio de registro de la exposición a XOLAIR durante el embarazo no mostró un aumento en la tasa de defectos congénitos importantes o abortos espontáneos. Hubo una mayor tasa de bajo peso al nacer entre los bebés del registro en comparación con los bebés de las otras cohortes, a pesar de la edad gestacional promedio al nacer; sin embargo, las mujeres que tomaron XOLAIR durante el embarazo también tenían asma más grave, lo que dificulta determinar si el bajo peso al nacer se debe al medicamento o a la gravedad de la enfermedad [ver Datos]. Existen riesgos asociados con el asma mal o moderadamente controlada en el embarazo [ver Consideraciones clínicas].
Se sabe que los anticuerpos IgG humanos atraviesan la barrera placentaria; por lo tanto, XOLAIR puede transmitirse de la madre al feto en desarrollo.
En estudios de reproducción animal, no se observó evidencia de daño fetal en monos Cynomolgus con dosis subcutáneas de omalizumab de hasta aproximadamente 5 veces la dosis máxima recomendada en humanos (DMRH) [ver Datos].
Se desconoce el riesgo de fondo estimado de defectos congénitos importantes y aborto espontáneo para la(s) población(es) indicada(s). Todos los embarazos tienen un riesgo de fondo de defectos congénitos, pérdida u otros resultados adversos. En la población general de EE. UU., el riesgo de fondo estimado de defectos congénitos importantes y aborto espontáneo en embarazos clínicamente reconocidos es del 2% al 4% y del 15% al 20%, respectivamente.
Consideraciones clínicas
Riesgo materno y/o embrionario/fetal asociado a la enfermedad
En mujeres con asma mal o moderadamente controlada, la evidencia demuestra que existe un mayor riesgo de preeclampsia en la madre y de prematuridad, bajo peso al nacer y pequeño para la edad gestacional en el neonato. El nivel de control del asma debe ser monitoreado de cerca en mujeres embarazadas y el tratamiento debe ajustarse según sea necesario para mantener un control óptimo.
Datos
Datos humanos
Un estudio prospectivo de registro de exposición al embarazo de cohorte, realizado en los EE. UU. de 2006 a 2018, incluyó a 250 mujeres embarazadas con asma tratadas con XOLAIR. De estas, 246 pacientes estuvieron expuestas a XOLAIR en el primer trimestre del embarazo, y la duración media de la exposición fue de 8,7 meses.
Los hallazgos del registro para los subgrupos de madres y bebés aplicables se compararon con las frecuencias ajustadas por edad en una cohorte externa emparejada por enfermedad de 1153 mujeres embarazadas con asma (sin exposición a XOLAIR) identificadas a partir de bases de datos de atención médica de residentes en la provincia canadiense de Quebec, y denominada Cohorte Comparadora Externa de Quebec (“cohorte comparadora”).
Entre los bebés del registro aplicables, la prevalencia de anomalías congénitas importantes (8,1 %) fue similar a la de los bebés de la cohorte comparadora (8,9 %). Entre los embarazos del registro aplicables, el 99,1 % dio lugar a nacidos vivos, similar al 99,3 % de la cohorte comparadora. Hubo una mayor tasa de bajo peso al nacer entre los bebés del registro (13,7 %) en comparación con la cohorte comparadora (9,8 %); sin embargo, las mujeres que tomaron XOLAIR durante el embarazo también tenían asma más grave, lo que dificulta determinar si el bajo peso al nacer se debe al medicamento o a la gravedad de la enfermedad.
El estudio de registro no puede establecer definitivamente la ausencia de ningún riesgo debido a limitaciones metodológicas, incluida la naturaleza observacional del registro, el pequeño tamaño de la muestra y las posibles diferencias entre la población del registro y la cohorte comparadora.
Datos animales
Se han realizado estudios de reproducción en monos Cynomolgus. No hubo evidencia de toxicidad materna, embriotoxicidad o teratogenicidad cuando se administró omalizumab durante el período de organogénesis a dosis que produjeron exposiciones aproximadamente 5 veces la DMRH (en mg/kg con dosis subcutáneas maternas de hasta 75 mg/kg/semana). El omalizumab no provocó efectos adversos sobre el crecimiento fetal o neonatal cuando se administró durante la última etapa de la gestación, el parto y la lactancia.
8.2 Lactancia
Resumen de riesgos
No hay información sobre la presencia de omalizumab en la leche humana ni sobre los efectos en la producción de leche. Sin embargo, el omalizumab es un anticuerpo monoclonal humano (IgG1 kappa) y la inmunoglobulina (IgG) está presente en la leche humana en pequeñas cantidades.
La mayoría de los bebés (80,9 %, 186/230) en el registro de exposición al embarazo fueron amamantados. Los eventos clasificados como “infecciones e infestaciones” no aumentaron significativamente en los bebés que estuvieron expuestos a XOLAIR a través de la lactancia materna en comparación con los bebés que no fueron amamantados o los bebés que fueron amamantados sin exposición a XOLAIR.
Se deben considerar los beneficios para el desarrollo y la salud de la lactancia materna junto con la necesidad clínica de la madre de XOLAIR y cualquier posible efecto adverso sobre el niño amamantado por el omalizumab o por la afección materna subyacente.
8.4 Uso pediátrico
Asma
Se ha establecido la seguridad y eficacia de XOLAIR para el asma persistente de moderada a grave que tuvo una prueba cutánea positiva o reactividad in vitro a un aeroalergeno perenne y cuyos síntomas no se controlan adecuadamente con corticosteroides inhalados, en pacientes pediátricos de 6 años o mayores. El uso de XOLAIR para esta indicación está respaldado por evidencia de estudios adecuados y bien controlados. XOLAIR se evaluó en 2 ensayos en 926 (XOLAIR 624; placebo 302) pacientes pediátricos de 6 a <12 años de edad con asma persistente de moderada a grave que tuvieron una prueba cutánea positiva o reactividad in vitro a un aeroalergeno perenne. Un ensayo fue un ensayo pivotal de diseño y conducta similar al de los Ensayos de Asma 1 y 2 en adultos y adolescentes. El otro ensayo fue principalmente un estudio de seguridad e incluyó la evaluación de la eficacia como resultado secundario. En el ensayo pivotal, los pacientes tratados con XOLAIR tuvieron una reducción estadísticamente significativa en la tasa de exacerbaciones (la exacerbación se definió como el empeoramiento del asma que requirió tratamiento con corticosteroides sistémicos o una duplicación de la dosis inicial de ICS) [see Clinical Studies (14.1)].
No se ha establecido la seguridad y eficacia en pacientes pediátricos con asma menores de 6 años.
Chronic Rhinosinusitis with Nasal Polyps
No se ha establecido la seguridad y la eficacia en pacientes pediátricos con rinosinusitis crónica con pólipos nasales (CRSwNP) menores de 18 años.
IgE-Mediated Food Allergy
Se ha establecido la seguridad y la eficacia de XOLAIR para la reducción de las reacciones alérgicas (Tipo I), incluida la anafilaxia, que pueden ocurrir con la exposición accidental a uno o más alimentos en pacientes pediátricos de 1 año o mayores con alergia alimentaria mediada por IgE. El uso de XOLAIR para esta indicación está respaldado por la evidencia de un estudio adecuado y bien controlado que incluyó un total de 165 pacientes pediátricos; 61 pacientes de 1 año a menos de 6 años de edad y 104 pacientes de 6 a menos de 18 años de edad. Un porcentaje significativamente mayor de pacientes tratados con XOLAIR en comparación con los pacientes tratados con placebo pudo consumir una dosis única de alimento (maní, anacardo, leche, huevo) sin síntomas limitantes de la dosis [see Clinical Studies (14.3)].
No se ha establecido la seguridad y la eficacia en pacientes pediátricos con alergia alimentaria mediada por IgE menores de 1 año.
Chronic Spontaneous Urticaria
La seguridad y la eficacia de XOLAIR para la urticaria espontánea crónica (CSU) que permanece sintomática a pesar del tratamiento con antihistamínicos H1 se ha establecido en pacientes pediátricos de 12 años o mayores. El uso de XOLAIR en esta población está respaldado por evidencia de estudios adecuados y bien controlados. Los pacientes adolescentes con CSU se evaluaron en 39 pacientes de 12 a 17 años de edad (XOLAIR 29, placebo 10) incluidos en tres ensayos de CSU aleatorizados, controlados con placebo. Se observó una disminución numérica en la puntuación semanal de picazón y las reacciones adversas fueron similares a las informadas en pacientes de 18 años o mayores.
No se ha establecido la seguridad y la eficacia en pacientes pediátricos con CSU menores de 12 años.
8.5 Geriatric Use
En estudios clínicos, 134 pacientes con asma, 20 pacientes con CRSwNP, 37 pacientes con CSU y ningún paciente con alergia alimentaria mediada por IgE de 65 años o mayores fueron tratados con XOLAIR. Aunque no se observaron diferencias aparentes relacionadas con la edad en estos estudios, el número de pacientes de 65 años o más no es suficiente para determinar si responden de manera diferente a los pacientes más jóvenes.
11 DESCRIPCIÓN
Omalizumab es un anticuerpo monoclonal humanizado IgG1κ derivado de ADN recombinante que se une selectivamente a la inmunoglobulina E humana (IgE). El anticuerpo tiene un peso molecular de aproximadamente 149 kiloDaltons. XOLAIR se produce mediante un cultivo celular en suspensión de ovario de hámster chino.
XOLAIR (omalizumab) se administra como inyección subcutánea (SC) y está disponible en jeringa precargada, autoinyector y en viales.
Inyección XOLAIR (Jeringa precargada o autoinyector)
La inyección de XOLAIR (omalizumab) se suministra como una solución estéril, sin conservantes, transparente a ligeramente opalescente e incolora a amarillo parduzco pálido para inyección subcutánea. La inyección de XOLAIR (omalizumab) está disponible como una jeringa precargada de dosis única o un autoinyector de dosis única.
Cada jeringa precargada o autoinyector de 75 mg proporciona 75 mg de omalizumab en 0.5 mL y contiene clorhidrato de arginina (21.05 mg), histidina (0.68 mg), monohidrato de clorhidrato de L-histidina (1.17 mg) y polisorbato 20 (0.2 mg) en Agua estéril para inyección (SWFI), USP.
Cada jeringa precargada o autoinyector de 150 mg proporciona 150 mg de omalizumab en 1 mL y contiene clorhidrato de arginina (42.1 mg), histidina (1.37 mg), monohidrato de clorhidrato de L-histidina (2.34 mg) y polisorbato 20 (0.4 mg) en SWFI, USP.
Cada jeringa precargada o autoinyector de 300 mg proporciona 300 mg de omalizumab en 2 mL y contiene clorhidrato de arginina (84.2 mg), histidina (2.74 mg), monohidrato de clorhidrato de L-histidina (4.68 mg) y polisorbato 20 (0.8 mg) en SWFI, USP.
La tapa de la aguja de la jeringa precargada XOLAIR de 75 mg/0.5 mL y 150 mg/mL con aguja de calibre 26 contiene un derivado del látex de caucho natural que puede causar reacciones alérgicas en individuos sensibles al látex [ver Cómo se suministra/Almacenamiento y manipulación (16)].
El autoinyector XOLAIR no está hecho con látex de caucho natural.
XOLAIR para inyección (vial)
XOLAIR (omalizumab) para inyección es un polvo liofilizado estéril, blanco, sin conservantes, en un vial de dosis única. Después de la reconstitución con 1.4 mL de Agua estéril para inyección, USP, el vial contiene 150 mg de omalizumab por 1.2 mL de solución reconstituida para inyección subcutánea. Cada 1.2 mL de solución reconstituida también contiene histidina (1.3 mg), monohidrato de clorhidrato de L-histidina (2.1 mg), polisorbato 20 (0.4 mg) y sacarosa (108 mg).
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
Asma, rinosinusitis crónica con pólipos nasales y alergia alimentaria mediada por IgE
El omalizumab inhibe la unión de la IgE al receptor de alta afinidad para IgE (FcεRI) en la superficie de los mastocitos, basófilos y células dendríticas, lo que resulta en una disminución de la regulación de FcεRI en estas células.
En pacientes asmáticos alérgicos, el tratamiento con omalizumab inhibe la inflamación mediada por IgE, como lo demuestra la reducción de eosinófilos en sangre y tejido y la reducción de mediadores inflamatorios, incluidos IL-4, IL-5 e IL-13.
Urticaria espontánea crónica
El omalizumab se une a la IgE y disminuye los niveles de IgE libre. Posteriormente, los receptores de IgE (FcεRI) en las células disminuyen su regulación. Se desconoce el mecanismo por el cual estos efectos del omalizumab producen una mejoría de los síntomas de la urticaria espontánea crónica (UEC).
12.2 Farmacodinamia
Asma
En estudios clínicos, los niveles séricos de IgE libre se redujeron de manera dosis-dependiente dentro de 1 hora después de la primera dosis y se mantuvieron entre dosis. La disminución media de la IgE sérica libre fue superior al 96% utilizando las dosis recomendadas. Los niveles séricos totales de IgE (es decir, unidos y no unidos) aumentaron después de la primera dosis debido a la formación de complejos omalizumab:IgE, que tienen una tasa de eliminación más lenta en comparación con la IgE libre. A las 16 semanas después de la primera dosis, los niveles séricos totales de IgE promedio fueron cinco veces más altos en comparación con el tratamiento previo cuando se utilizan ensayos estándar. Después de la interrupción de la administración de XOLAIR, el aumento inducido por XOLAIR en la IgE total y la disminución de la IgE libre fueron reversibles, sin observar un rebote en los niveles de IgE después de la eliminación del fármaco. Los niveles totales de IgE no volvieron a los niveles previos al tratamiento hasta un año después de la interrupción de XOLAIR.
Rinosinusitis crónica con pólipos nasales
En estudios clínicos en pacientes con rinosinusitis crónica con pólipos nasales (CRSwNP), el tratamiento con omalizumab provocó una reducción en la IgE sérica libre y un aumento en los niveles séricos totales de IgE, similar a las observaciones en pacientes con asma. Las concentraciones medias totales de IgE en la línea de base fueron 168 UI/ml y 218 UI/ml en los ensayos CRSwNP 1 y 2, respectivamente. Después de la administración repetida cada 2 o 4 semanas, con la dosis y la frecuencia según la Tabla 3, las concentraciones medias de IgE libre antes de la dosis en la semana 16 fueron 10,0 UI/ml en el ensayo CRSwNP 1 y 11,7 UI/ml en el ensayo CRSwNP 2 y se mantuvieron estables en las 24 semanas de tratamiento. Los niveles totales de IgE en suero aumentaron debido a la formación de complejos omalizumab-IgE, que tienen una tasa de eliminación más lenta en comparación con la IgE libre. Después de la administración repetida cada 2 o 4 semanas, con la dosis y la frecuencia según la Tabla 3, los niveles séricos totales de IgE medios y medianos antes de la dosis en la semana 16 fueron de 3 a 4 veces más altos en comparación con los niveles previos al tratamiento, y se mantuvieron estables entre las 16 y las 24 semanas de tratamiento.
Alergia alimentaria mediada por IgE
En un estudio clínico en pacientes con alergia alimentaria mediada por IgE, el tratamiento con omalizumab provocó una reducción en la IgE sérica libre y un aumento en los niveles séricos totales de IgE, similar a las observaciones en pacientes con asma. La concentración media total de IgE en la línea de base fue de 810 UI/ml. Después de la administración repetida cada 2 o 4 semanas, con la dosis y la frecuencia según la Tabla 4, la concentración media de IgE libre antes de la dosis en la semana 16 fue de 10,0 UI/ml. Los niveles medios totales de IgE en suero aumentaron aproximadamente 2,4 veces debido a la formación de complejos omalizumab-IgE, que tienen una vida media más larga en comparación con la IgE libre.
Urticaria espontánea crónica
En estudios clínicos en pacientes con urticaria espontánea crónica (UEC), el tratamiento con XOLAIR provocó una reducción dosis-dependiente de la IgE sérica libre y un aumento de los niveles séricos totales de IgE, similar a las observaciones en pacientes con asma. La supresión máxima de la IgE libre se observó 3 días después de la primera dosis subcutánea. Después de la administración repetida una vez cada 4 semanas, los niveles séricos de IgE libre antes de la dosis se mantuvieron estables entre las 12 y las 24 semanas de tratamiento. Los niveles totales de IgE en suero aumentaron después de la primera dosis debido a la formación de complejos omalizumab-IgE, que tienen una tasa de eliminación más lenta en comparación con la IgE libre. Después de la administración repetida una vez cada 4 semanas a 75 mg hasta 300 mg, los niveles séricos totales de IgE promedio antes de la dosis en la semana 12 fueron de dos a tres veces más altos en comparación con los niveles previos al tratamiento, y se mantuvieron estables entre las 12 y las 24 semanas de tratamiento. Después de la interrupción de la administración de XOLAIR, los niveles de IgE libre aumentaron y los niveles totales de IgE disminuyeron hacia los niveles previos al tratamiento durante un período de seguimiento de 16 semanas.
12.3 Farmacocinética
Después de la administración SC, el omalizumab se absorbió con una biodisponibilidad absoluta promedio del 62%. Después de una dosis SC única en pacientes adultos y adolescentes con asma, el omalizumab se absorbió lentamente, alcanzando concentraciones séricas máximas después de un promedio de 7 a 8 días. En pacientes con UEC, la concentración sérica máxima se alcanzó en un tiempo similar después de una dosis SC única. La farmacocinética del omalizumab fue lineal a dosis superiores a 0,5 mg/kg. En pacientes con asma, después de dosis múltiples de XOLAIR, las áreas bajo la curva de concentración-tiempo sérica del día 0 al día 14 en estado estacionario fueron hasta 6 veces mayores que las después de la primera dosis. En pacientes con UEC, el omalizumab exhibió una farmacocinética lineal en el rango de dosis de 75 mg a 600 mg administrados como dosis subcutánea única. Después de la administración repetida de 75 a 300 mg cada 4 semanas, las concentraciones séricas mínimas de omalizumab aumentaron proporcionalmente con los niveles de dosis.
In vitro, el omalizumab formó complejos de tamaño limitado con IgE. No se observaron complejos precipitantes ni complejos mayores de 1 millón de daltons de peso molecular in vitro o in vivo. Los estudios de distribución tisular en monos Cynomolgus no mostraron captación específica de 125I-omalizumab por ningún órgano o tejido. El volumen aparente de distribución de omalizumab en pacientes con asma después de la administración SC fue de 78 ± 32 mL/kg. En pacientes con CSU, según la farmacocinética poblacional, la distribución de omalizumab fue similar a la de los pacientes con asma.
El aclaramiento de omalizumab implicó procesos de aclaramiento de IgG, así como el aclaramiento a través de la unión específica y la formación de complejos con su ligando diana, IgE. La eliminación hepática de IgG incluyó la degradación en el sistema reticuloendotelial (SRE) hepático y las células endoteliales. La IgG intacta también se excretó en la bilis. En estudios con ratones y monos, los complejos omalizumab:IgE se eliminaron mediante interacciones con los receptores Fcγ dentro del SRE a velocidades que generalmente fueron más rápidas que el aclaramiento de IgG. En pacientes con asma, la semivida de eliminación sérica de omalizumab promedió 26 días, con un aclaramiento aparente promedio de 2,4 ± 1,1 mL/kg/día. Duplicar el peso corporal aproximadamente duplicó el aclaramiento aparente. En pacientes con CSU, en estado estacionario, según la farmacocinética poblacional, la semivida de eliminación sérica de omalizumab promedió 24 días y el aclaramiento aparente promedió 240 mL/día (correspondiente a 3,0 mL/kg/día para un paciente de 80 kg).
Poblaciones específicas
Asma
Se analizó la farmacocinética poblacional de omalizumab para evaluar los efectos de las características demográficas en pacientes con asma. Los análisis de estos datos sugirieron que no son necesarios ajustes de dosis para la edad (6 a 76 años), raza, etnia o sexo.
Rinosinusitis crónica con pólipos nasales
Los análisis de farmacocinética poblacional de omalizumab sugirieron que la farmacocinética de omalizumab en la rinosinusitis crónica con pólipos nasales (CRSwNP) fue consistente con la del asma. Se realizaron análisis gráficos de covariables para evaluar los efectos de las características demográficas y otros factores en la exposición a omalizumab y las respuestas clínicas. Estos análisis demuestran que no son necesarios ajustes de dosis para la edad (18 a 75 años) o el sexo. Los datos de raza y etnia son demasiado limitados en los estudios de CRSwNP para informar sobre el ajuste de la dosis.
Alergia alimentaria mediada por IgE
Los análisis farmacocinéticos (PK) poblacionales de omalizumab sugirieron que la PK de omalizumab en pacientes con alergia alimentaria mediada por IgE fue generalmente consistente con la de los pacientes con asma. Se realizaron análisis de covariables para evaluar los efectos de las características demográficas y otros factores en la exposición a omalizumab y las respuestas clínicas. Estos análisis demuestran que no son necesarios ajustes de dosis para la edad (1 año o más), raza, etnia o sexo.
Urticaria espontánea crónica
Se analizó la farmacocinética poblacional de omalizumab para evaluar los efectos de las características demográficas y otros factores en la exposición a omalizumab en pacientes con urticaria espontánea crónica (CSU). Los efectos de las covariables se evaluaron analizando la relación entre las concentraciones de omalizumab y las respuestas clínicas. Estos análisis demuestran que no son necesarios ajustes de dosis para la edad (12 a 75 años), raza/etnia, sexo, peso corporal, índice de masa corporal o nivel basal de IgE.
12.6 Inmunogenicidad
La incidencia observada de anticuerpos anti-fármaco depende en gran medida de la sensibilidad y especificidad del ensayo. Las diferencias en los métodos de ensayo impiden comparaciones significativas de la incidencia de anticuerpos anti-fármaco en los estudios descritos a continuación con la incidencia de anticuerpos anti-fármaco en otros estudios, incluidos los de XOLAIR o de otros productos de omalizumab.
Se detectaron anticuerpos contra XOLAIR en aproximadamente 1/1723 (<0,1%) de los pacientes tratados con XOLAIR en los estudios clínicos evaluados para el asma en pacientes de 12 años de edad o más. En tres estudios pediátricos, se detectaron anticuerpos contra XOLAIR en un paciente de 581 pacientes de 6 a <12 años de edad tratados con XOLAIR y evaluados para detectar anticuerpos. No hubo anticuerpos detectables en los pacientes tratados en los ensayos clínicos de CSU, pero debido a los niveles de XOLAIR en el momento del muestreo de anticuerpos antiterapéuticos y a las muestras faltantes para algunos pacientes, los anticuerpos contra XOLAIR solo pudieron determinarse en el 88% de los 733 pacientes tratados en estos estudios clínicos. Los datos reflejan el porcentaje de pacientes cuyos resultados de las pruebas se consideraron positivos para anticuerpos contra XOLAIR en ensayos ELISA y dependen en gran medida de la sensibilidad y especificidad de los ensayos.
Los anticuerpos anti-fármaco no se midieron en los ensayos de CRSwNP o alergia alimentaria mediada por IgE.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
No se han realizado estudios a largo plazo en animales para evaluar el potencial carcinogénico de XOLAIR.
No hubo efectos sobre la fertilidad y el rendimiento reproductivo en monos Cynomolgus machos y hembras que recibieron XOLAIR en dosis subcutáneas de hasta 75 mg/kg/semana (aproximadamente 5 veces la dosis máxima recomendada en humanos sobre una base mg/kg).
14 ESTUDIOS CLÍNICOS
14.1 Asma
Pacientes adultos y adolescentes de 12 años de edad o mayores
La seguridad y eficacia de XOLAIR se evaluaron en tres ensayos aleatorizados, doble ciego, controlados con placebo y multicéntricos.
Los ensayos incluyeron pacientes de 12 a 76 años de edad, con asma persistente de moderada a grave (criterios NHLBI) durante al menos un año, y una reacción positiva de la prueba cutánea a un aeroalérgeno perenne. En todos los ensayos, la dosificación de XOLAIR se basó en el peso corporal y la concentración basal de IgE sérica total. Todos los pacientes debían tener una IgE basal entre 30 y 700 UI/ml y un peso corporal no superior a 150 kg. Los pacientes fueron tratados de acuerdo con una tabla de dosificación para administrar al menos 0,016 mg/kg/UI (IgE/ml) de XOLAIR o un volumen equivalente de placebo durante cada período de 4 semanas. La dosis máxima de XOLAIR por 4 semanas fue de 750 mg.
En los tres ensayos, una exacerbación se definió como un empeoramiento del asma que requirió tratamiento con corticosteroides sistémicos o una duplicación de la dosis de ICS basal. La mayoría de las exacerbaciones se manejaron en el ámbito ambulatorio y la mayoría se trataron con esteroides sistémicos. Las tasas de hospitalización no fueron significativamente diferentes entre los pacientes tratados con XOLAIR y con placebo; sin embargo, la tasa general de hospitalización fue pequeña. Entre los pacientes que experimentaron una exacerbación, la distribución de la gravedad de la exacerbación fue similar entre los grupos de tratamiento.
Ensayos de asma 1 y 2
En la selección, los pacientes de los ensayos de asma 1 y 2 tenían un volumen espiratorio forzado en un segundo (FEV1) entre el 40% y el 80% del valor predicho. Todos los pacientes tuvieron una mejora del FEV1 de al menos el 12% después de la administración de beta2-agonista. Todos los pacientes presentaban síntomas y estaban siendo tratados con corticosteroides inhalados (ICS) y beta2-agonistas de acción corta. Se excluyó a los pacientes que recibían otros medicamentos controladores concomitantes, y se prohibió el inicio de medicamentos controladores adicionales durante el estudio. Se excluyó a los pacientes que fumaban actualmente.
Cada ensayo constó de un período de rodaje para lograr una conversión estable a un ICS común (dipropionato de beclometasona), seguido de la aleatorización a XOLAIR o placebo. Los pacientes recibieron XOLAIR durante 16 semanas con una dosis de corticosteroides sin cambios a menos que una exacerbación aguda necesitara un aumento. Los pacientes luego ingresaron a una fase de reducción de ICS de 12 semanas durante la cual se intentó la reducción de la dosis de ICS de manera escalonada.
La distribución del número de exacerbaciones de asma por paciente en cada grupo durante un estudio se analizó por separado para los períodos de esteroides estables y de reducción de esteroides.
Tanto en los ensayos de asma 1 como en el 2, el número de exacerbaciones por paciente se redujo en los pacientes tratados con XOLAIR en comparación con el placebo (Tabla 11).
También se evaluaron las medidas del flujo de aire (FEV1) y los síntomas del asma en estos ensayos. Se desconoce la relevancia clínica de las diferencias asociadas al tratamiento. Los resultados de la fase de esteroides estables del ensayo de asma 1 se muestran en la Tabla 12. Los resultados de la fase de esteroides estables del ensayo de asma 2 y las fases de reducción de esteroides de los ensayos de asma 1 y 2 fueron similares a los presentados en la Tabla 12.
| Fase de esteroides estables (16 sem) | ||||
| Ensayo de asma 1 | Ensayo de asma 2 | |||
| Exacerbaciones por paciente | XOLAIR N=268 |
Placebo N=257 |
XOLAIR N=274 |
Placebo N=272 |
| 0 | 85.8% | 76.7% | 87.6% | 69.9% |
| 1 | 11.9% | 16.7% | 11.3% | 25.0% |
| ≥2 | 2.2% | 6.6% | 1.1% | 5.1% |
| p-Valor | 0.005 | <0.001 | ||
| Número medio de exacerbaciones/paciente | 0.2 | 0.3 | 0.1 | 0.4 |
| Fase de reducción de esteroides (12 sem) | ||||
| Exacerbaciones por paciente | XOLAIR N=268 |
Placebo N=257 |
XOLAIR N=274 |
Placebo N=272 |
| 0 | 78.7% | 67.7% | 83.9% | 70.2% |
| 1 | 19.0% | 28.4% | 14.2% | 26.1% |
| ≥2 | 2.2% | 3.9% | 1.8% | 3.7% |
| p-Valor | 0.004 | <0.001 | ||
| Número medio de exacerbaciones/paciente | 0.2 | 0.4 | 0.2 | 0.3 |
| XOLAIR N=268* |
Placebo N=257* |
|||
|---|---|---|---|---|
| Variable principal | Media basal | Cambio mediano (basal a la semana 16) | Media basal | Cambio mediano (basal a la semana 16) |
| Escala de síntomas de asma: puntuación total de 0 (menor) a 9 (mayor); puntuaciones nocturnas y diurnas de 0 (menor) a 4 (mayor número de síntomas). | ||||
| Puntuación total de los síntomas del asma | 4.3 | –1.5† | 4.2 | –1.1† |
| Puntuación nocturna del asma | 1.2 | –0.4† | 1.1 | –0.2† |
| Puntuación diurna del asma | 2.3 | –0.9† | 2.3 | –0.6† |
| FEV1 % predicho | 68 | 3† | 68 | 0† |
Ensayo de asma 3
En el Ensayo de asma 3, no hubo restricción en la selección de FEV1, y a diferencia de los Ensayos de asma 1 y 2, se permitieron los beta2-agonistas de acción prolongada. Los pacientes estaban recibiendo al menos 1000 µg/día de propionato de fluticasona y un subgrupo también estaba recibiendo corticosteroides orales. Se excluyó a los pacientes que recibían otros medicamentos controladores concomitantes, y se prohibió la iniciación de medicamentos controladores adicionales durante el estudio. Se excluyó a los pacientes que fumaban actualmente.
El ensayo consistió en un período de rodaje para lograr una conversión estable a un ICS común (propionato de fluticasona), seguido de la aleatorización a XOLAIR o placebo. Los pacientes fueron estratificados por el uso de solo ICS o ICS con el uso concomitante de esteroides orales. Los pacientes recibieron XOLAIR durante 16 semanas con una dosis de corticosteroides sin cambios a menos que una exacerbación aguda necesitara un aumento. Luego, los pacientes ingresaron a una fase de reducción de ICS de 16 semanas durante la cual se intentó la reducción de la dosis de ICS o esteroides orales de manera gradual.
El número de exacerbaciones en pacientes tratados con XOLAIR fue similar al de los pacientes tratados con placebo (Tabla 13). La ausencia de un efecto de tratamiento observado puede estar relacionada con las diferencias en la población de pacientes en comparación con los Ensayos de asma 1 y 2, el tamaño de la muestra del estudio u otros factores.
| Fase de esteroides estable (16 sem) | ||||
| Inhalado solamente | Oral + Inhalado | |||
| XOLAIR N=126 |
Placebo N=120 |
XOLAIR N=50 |
Placebo N=45 |
|
| % Pacientes con ≥1 exacerbaciones | 15.9% | 15.0% | 32.0% | 22.2% |
| Diferencia (IC del 95%) |
0.9 (–9.7, 13.7) |
9.8 (–10.5, 31.4) |
||
| Fase de reducción de esteroides (16 sem) | ||||
| XOLAIR N=126 |
Placebo N=120 |
XOLAIR N=50 |
Placebo N=45 |
|
| % Pacientes con ≥1 exacerbaciones | 22.2% | 26.7% | 42.0% | 42.2% |
| Diferencia (IC del 95%) |
–4.4 (–17.6, 7.4) |
–0.2 (–22.4, 20.1) |
||
En los tres ensayos, no se observó una reducción de las exacerbaciones del asma en los pacientes tratados con XOLAIR que tenían un FEV1 > 80% en el momento de la randomización. Tampoco se vieron reducciones en las exacerbaciones en los pacientes que requerían esteroides orales como terapia de mantenimiento.
Pacientes pediátricos de 6 a < 12 años de edad
La seguridad y eficacia de XOLAIR en pacientes pediátricos de 6 a < 12 años de edad con asma moderada a grave se basa en un ensayo clínico aleatorizado, doble ciego, controlado con placebo, multicéntrico (Ensayo de Asma 4 [NCT00079937]) y un estudio de apoyo adicional (Ensayo de Asma 5).
El Ensayo de Asma 4 fue un estudio de 52 semanas que evaluó la seguridad y eficacia de XOLAIR como terapia adicional en 628 pacientes pediátricos de 6 a < 12 años de edad con asma moderada a grave que no estaba controlada adecuadamente a pesar del uso de corticosteroides inhalados (propionato de fluticasona DPI ≥ 200 mcg/día o equivalente) con o sin otros medicamentos controladores del asma. Los pacientes elegibles eran aquellos con un diagnóstico de asma > 1 año, una prueba cutánea positiva a al menos un alérgeno aeroalérgeno perenne y un historial de características clínicas como síntomas diurnos y/o nocturnos y exacerbaciones dentro del año anterior al inicio del estudio. Durante las primeras 24 semanas de tratamiento, las dosis de esteroides se mantuvieron constantes desde la línea de base. Esto fue seguido por un período de 28 semanas durante el cual se permitió el ajuste de los corticosteroides inhalados.
La variable de eficacia primaria fue la tasa de exacerbaciones del asma durante la fase de tratamiento con esteroides fijos de 24 semanas. Una exacerbación del asma se definió como un empeoramiento de los síntomas del asma según la evaluación clínica del investigador, que requería el doble de la dosis de corticosteroides inhalados de la línea de base durante al menos 3 días y/o el tratamiento con corticosteroides sistémicos de rescate (orales o IV) durante al menos 3 días. A las 24 semanas, el grupo de XOLAIR tuvo una tasa de exacerbaciones del asma estadísticamente significativamente menor (0,45 frente a 0,64) con una razón de tasas estimada de 0,69 (95% IC: 0,53, 0,90).
El grupo de XOLAIR también tuvo una tasa de exacerbaciones del asma menor en comparación con el placebo durante todo el período de tratamiento doble ciego de 52 semanas (0,78 frente a 1,36; razón de tasas: 0,57; 95% IC: 0,45, 0,72). Otras variables de eficacia como las puntuaciones de síntomas nocturnos, el uso de beta-agonistas y las medidas de flujo aéreo (FEV1) no fueron significativamente diferentes en los pacientes tratados con XOLAIR en comparación con el placebo.
El Ensayo de Asma 5 fue un estudio aleatorizado, doble ciego, controlado con placebo de 28 semanas que evaluó principalmente la seguridad en 334 pacientes pediátricos, 298 de los cuales tenían de 6 a < 12 años de edad, con asma moderada a grave que estaba bien controlada con corticosteroides inhalados (dipropionato de beclometasona 168 – 420 mcg/día). Un período de tratamiento con esteroides de 16 semanas fue seguido por un período de reducción de la dosis de esteroides de 12 semanas. Los pacientes tratados con XOLAIR tuvieron menos exacerbaciones del asma en comparación con el placebo tanto durante el período de tratamiento con esteroides fijos de 16 semanas (0,18 frente a 0,32; razón de tasas: 0,58; 95% IC: 0,35, 0,96) como durante el período de tratamiento de 28 semanas (0,38 frente a 0,76; razón de tasas: 0,50; 95% IC: 0,36, 0,71).
14.2 Rinossinusitis crónica con pólipos nasales
Pacientes adultos de 18 años de edad y mayores
La seguridad y eficacia de XOLAIR se evaluó en dos ensayos clínicos aleatorizados, multicéntricos, doble ciego, controlados con placebo (Ensayo 1 de CRSwNP [NCT03280550] y Ensayo 2 de CRSwNP [NCT03280537]) que incluyeron a pacientes con rinossinusitis crónica con pólipos nasales (CRSwNP) que no respondieron adecuadamente a los corticosteroides nasales (Ensayo 1 de CRSwNP, n = 138; Ensayo 2 de CRSwNP, n = 127). Los pacientes recibieron XOLAIR o placebo SC cada 2 o 4 semanas, con la dosis y la frecuencia de XOLAIR de acuerdo con la Tabla 3, durante 24 semanas seguidas de un período de seguimiento de 4 semanas. Todos los pacientes recibieron terapia nasal de mometasona de fondo durante el período de tratamiento y durante un período de 5 semanas de periodo de entrada. Antes de la randomización, se requería que los pacientes tuvieran evidencia de pólipos bilaterales según lo determinado por una puntuación de pólipos nasales (NPS) ≥ 5 con NPS ≥ 2 en cada fosa nasal, a pesar del uso de mometasona nasal durante el periodo de entrada. La NPS se midió mediante endoscopia y se puntuó (rango 0 – 4 por fosa nasal: 0 = sin pólipos; 1 = pólipos pequeños en el meato medio que no alcanzan por debajo del borde inferior del cornete medio; 2 = pólipos que alcanzan por debajo del borde inferior del cornete medio; 3 = pólipos grandes que alcanzan el borde inferior del cornete inferior o pólipos mediales al cornete medio; 4 = pólipos grandes que causan obstrucción completa de la cavidad nasal inferior) para una NPS total (rango 0 – 8). Además, se requería que los pacientes tuvieran un promedio semanal de puntuación de congestión nasal (NCS) > 1 antes de la randomización, a pesar del uso de mometasona nasal. La congestión nasal se midió mediante una evaluación diaria en una escala de severidad de 0 a 3 puntos (0 = ninguno, 1 = leve, 2 = moderado, 3 = severo). No se requirió cirugía sinusal previa o uso previo de corticosteroides sistémicos para la inclusión en los ensayos y no se realizaron tomografías computarizadas de los senos paranasales para evaluar la opacificación sinusal. Las características demográficas y basales, incluidas las comorbilidades alérgicas, se describen en la Tabla 14.
| Parámetro | Ensayo de CRSwNP 1 (n = 138) |
Ensayo de CRSwNP 2 (n = 127) |
|---|---|---|
| CRSwNP = rinossinusitis crónica con pólipos nasales; SD = desviación estándar; NPS = puntuación de pólipos nasales; IgE = Inmunoglobulina E; IU = unidades internacionales. Para NPS, NCS, sentido del olfato, goteo postnasal y moqueo, puntuaciones más altas indican mayor gravedad de la enfermedad. | ||
| Edad media (años) (SD) | 51 (13) | 50 (12) |
| % Masculino | 64 | 65 |