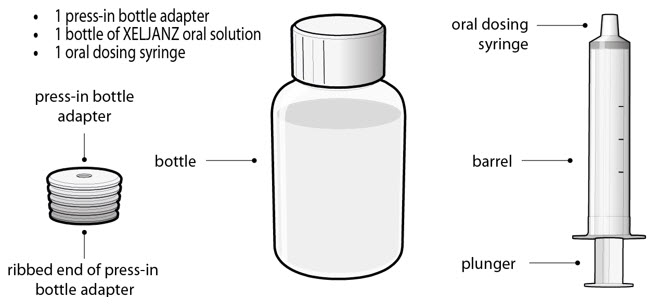
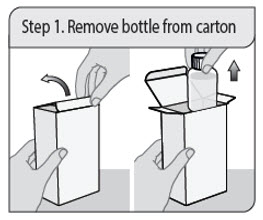
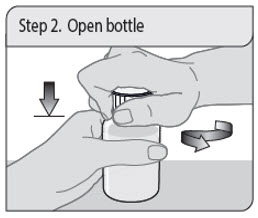
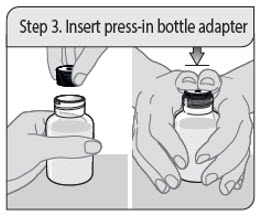
Fabricante de medicamentos: U.S. Pharmaceuticals (Updated: 2024-12-31)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
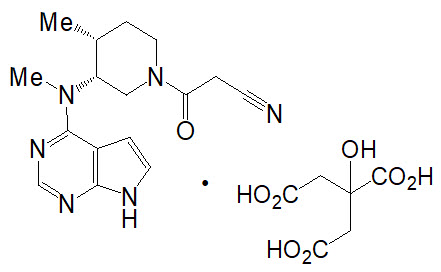
XELJANZ® (tofacitinib) comprimidos, para administración oral
XELJANZ® XR (tofacitinib) comprimidos de liberación prolongada, para administración oral
XELJANZ® (tofacitinib) Solución oral
Aprobación inicial en EE. UU.: 2012
ADVERTENCIA: INFECCIONES GRAVES, MORTALIDAD, NEOPLASIAS, EVENTOS CARDIOVASCULARES ADVERSOS MAYORES (ECAM) Y TROMBOSIS
Consulte la información completa de la advertencia en recuadro.
- •
- Mayor riesgo de infecciones bacterianas, micóticas, víricas y oportunistas graves que pueden provocar hospitalización o muerte, incluida la tuberculosis (TB). Interrumpa el tratamiento con XELJANZ/XELJANZ XR/Solución oral de XELJANZ si se produce una infección grave hasta que la infección esté controlada. Realice pruebas de tuberculosis latente antes y durante el tratamiento; trate la tuberculosis latente antes del uso. Controle a todos los pacientes en busca de tuberculosis activa durante el tratamiento, incluso a los pacientes con una prueba inicial de tuberculosis latente negativa. (5.1)
- •
- Mayor tasa de mortalidad por todas las causas, incluida la muerte cardiovascular súbita con XELJANZ frente a los bloqueadores del TNF en pacientes con artritis reumatoide (AR). (5.2)
- •
- Se han producido neoplasias en pacientes tratados con XELJANZ. Mayor tasa de linfomas y cánceres de pulmón con XELJANZ frente a los bloqueadores del TNF en pacientes con AR. (5.3)
- •
- Mayor tasa de ECAM (definidos como muerte cardiovascular, infarto de miocardio y accidente cerebrovascular) con XELJANZ frente a los bloqueadores del TNF en pacientes con AR. (5.4)
- •
- Se ha producido trombosis en pacientes tratados con XELJANZ. Mayor incidencia de embolia pulmonar, trombosis venosa y arterial con XELJANZ frente a los bloqueadores del TNF en pacientes con AR. (5.5)
INDICACIONES Y USO
XELJANZ/XELJANZ XR/Solución oral de XELJANZ es un inhibidor de la Janus quinasa (JAK) indicado para:
- •
-
Artritis reumatoide: XELJANZ/XELJANZ XR está indicado para el tratamiento de pacientes adultos con artritis reumatoide moderada a gravemente activa que han tenido una respuesta inadecuada o intolerancia a uno o más bloqueadores del TNF.
- o
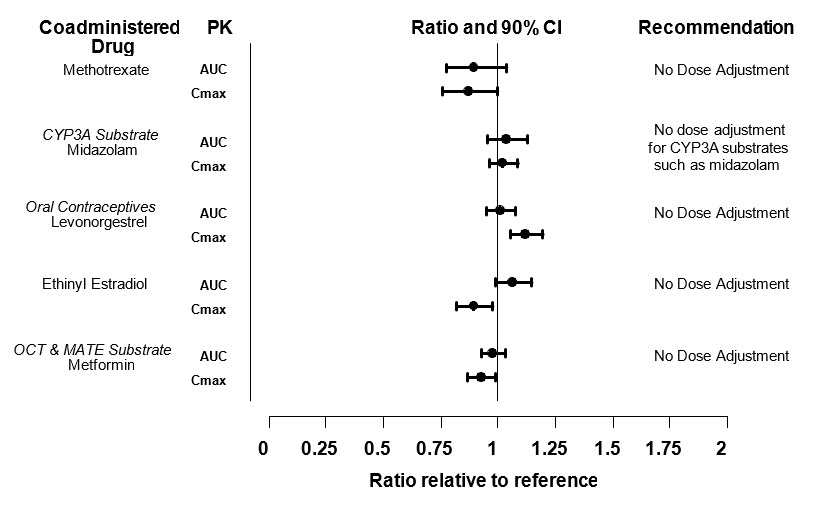
- Limitaciones de uso: No se recomienda el uso de XELJANZ/XELJANZ XR en combinación con fármacos antirreumáticos modificadores de la enfermedad (DMARD) biológicos o inmunosupresores potentes como la azatioprina y la ciclosporina. (1.1)
- •
-
Artritis psoriásica: XELJANZ/XELJANZ XR está indicado para el tratamiento de pacientes adultos con artritis psoriásica activa que han tenido una respuesta inadecuada o intolerancia a uno o más bloqueadores del TNF.
- o
- Limitaciones de uso: No se recomienda el uso de XELJANZ/XELJANZ XR en combinación con fármacos antirreumáticos modificadores de la enfermedad (DMARD) biológicos o inmunosupresores potentes como la azatioprina y la ciclosporina. (1.2)
- •
-
Espondilitis anquilosante: XELJANZ/XELJANZ XR está indicado para el tratamiento de pacientes adultos con espondilitis anquilosante activa que han tenido una respuesta inadecuada o intolerancia a uno o más bloqueadores del TNF.
- o
- Limitaciones de uso: No se recomienda el uso de XELJANZ/XELJANZ XR en combinación con fármacos antirreumáticos modificadores de la enfermedad (DMARD) biológicos o inmunosupresores potentes como la azatioprina y la ciclosporina. (1.3)
- •
-
Colitis ulcerosa: XELJANZ/XELJANZ XR está indicado para el tratamiento de pacientes adultos con colitis ulcerosa (CU) moderada a gravemente activa, que han tenido una respuesta inadecuada o intolerancia a uno o más bloqueadores del TNF.
- o
- Limitaciones de uso: No se recomienda el uso de XELJANZ/XELJANZ XR en combinación con terapias biológicas para la CU o con inmunosupresores potentes como la azatioprina y la ciclosporina. (1.4)
- •
-
Artritis idiopática juvenil de curso poliarticular: La solución oral de XELJANZ/XELJANZ está indicada para el tratamiento de la artritis idiopática juvenil de curso poliarticular (AIJcp) activa en pacientes de 2 años de edad o mayores que han tenido una respuesta inadecuada o intolerancia a uno o más bloqueadores del TNF.
- o
- Limitaciones de uso: No se recomienda el uso de la solución oral de XELJANZ/XELJANZ en combinación con fármacos antirreumáticos modificadores de la enfermedad (DMARD) biológicos o inmunosupresores potentes como la azatioprina y la ciclosporina. (1.5)
POSOLOGÍA Y ADMINISTRACIÓN
Instrucciones de administración
- •
- XELJANZ XR (comprimidos de liberación prolongada de tofacitinib) no es intercambiable ni sustituible con la solución oral de XELJANZ. (2.1)
- •
- Los cambios entre XELJANZ y XELJANZ XR deben ser realizados por el profesional sanitario. (2.1)
- •
- No inicie XELJANZ/XELJANZ XR/Solución oral de XELJANZ si el recuento absoluto de linfocitos es <500 células/mm3, el recuento absoluto de neutrófilos (RAN) es <1000 células/mm3 o la hemoglobina es <9 g/dL. (2.1)
Posología recomendada
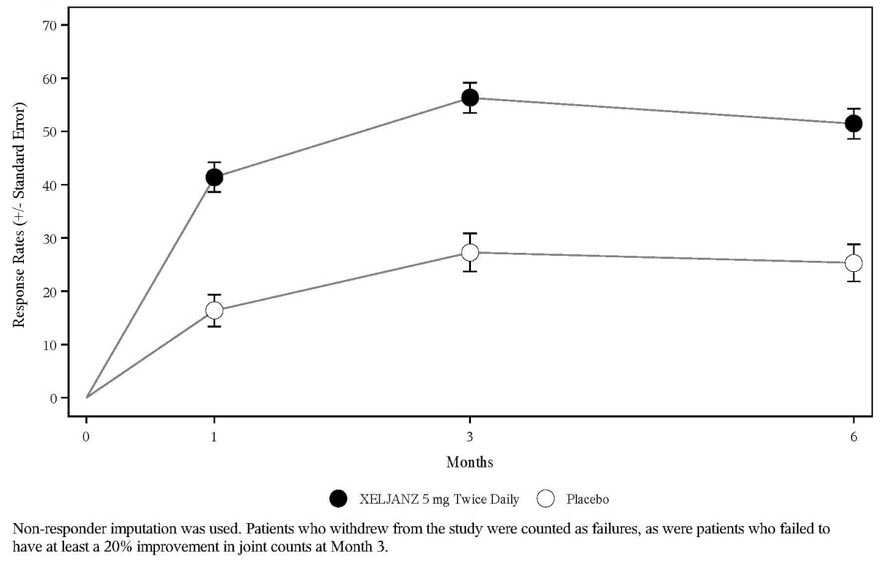
Artritis reumatoide
- •
- XELJANZ 5 mg dos veces al día o XELJANZ XR 11 mg una vez al día. (2.2)
- •
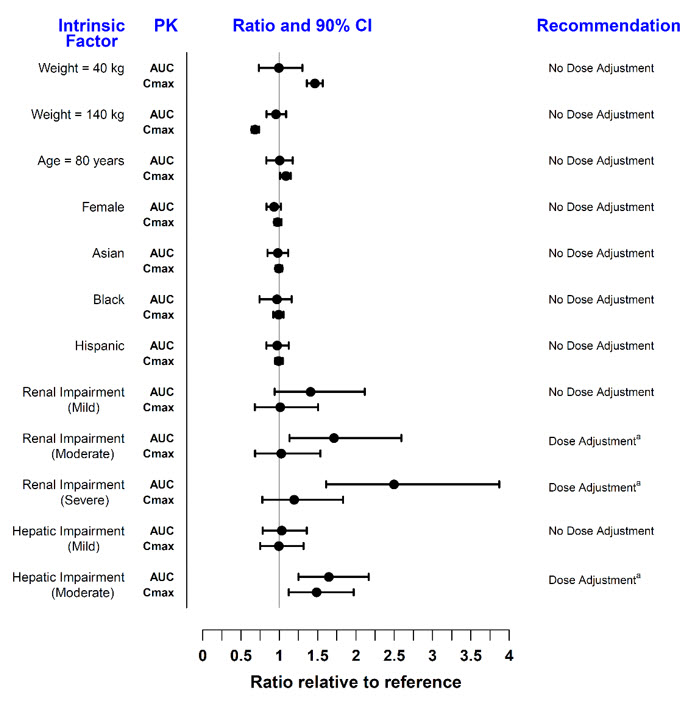
- La dosis recomendada en pacientes con insuficiencia renal moderada y grave o insuficiencia hepática moderada es XELJANZ 5 mg una vez al día. (2, 8.7, 8.8)
Artritis psoriásica (en combinación con DMARD no biológicos)
- •
- XELJANZ 5 mg dos veces al día o XELJANZ XR 11 mg una vez al día. (2.2)
- •
- La dosis recomendada en pacientes con insuficiencia renal moderada y grave o insuficiencia hepática moderada es XELJANZ 5 mg una vez al día. (2, 8.7, 8.8)
Espondilitis anquilosante
- •
- XELJANZ 5 mg dos veces al día o XELJANZ XR 11 mg una vez al día. (2.2)
- •
- La dosis recomendada en pacientes con insuficiencia renal moderada y grave o insuficiencia hepática moderada es XELJANZ 5 mg una vez al día. (2, 8.7, 8.8)
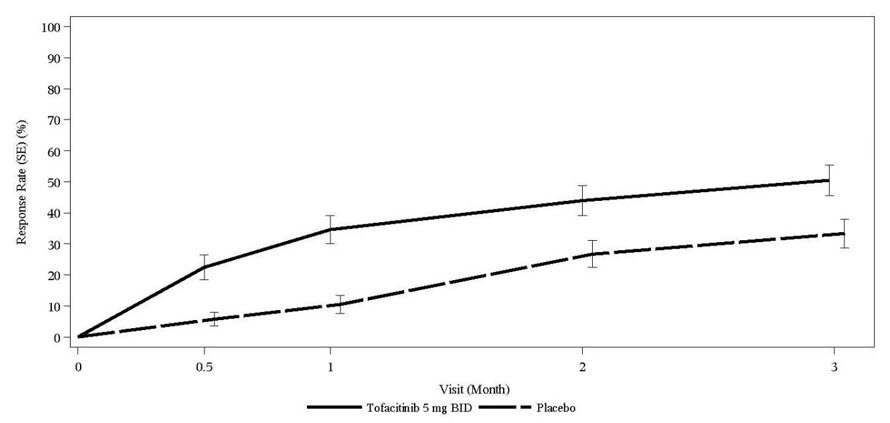
Colitis ulcerosa
- •
- Inducción: XELJANZ 10 mg dos veces al día o XELJANZ XR 22 mg una vez al día durante 8 semanas; evaluar a los pacientes y pasar a la terapia de mantenimiento en función de la respuesta terapéutica. Si es necesario, continuar con XELJANZ 10 mg dos veces al día o XELJANZ XR 22 mg una vez al día durante un máximo de 16 semanas. Suspender XELJANZ 10 mg dos veces al día o XELJANZ XR 22 mg una vez al día después de 16 semanas si no se logra una respuesta terapéutica adecuada. (2.3)
- •
- Mantenimiento: XELJANZ 5 mg dos veces al día o XELJANZ XR 11 mg una vez al día. Para pacientes con pérdida de respuesta durante el tratamiento de mantenimiento, se puede considerar XELJANZ 10 mg dos veces al día o XELJANZ XR 22 mg una vez al día y limitarlo a la duración más corta, teniendo en cuenta cuidadosamente los beneficios y riesgos para el paciente individual. Utilizar la dosis efectiva más baja necesaria para mantener la respuesta. (2.3)
- •
- Se necesita un ajuste de la dosis en pacientes con insuficiencia renal moderada y grave o insuficiencia hepática moderada: ver la información de prescripción completa. (2.3)
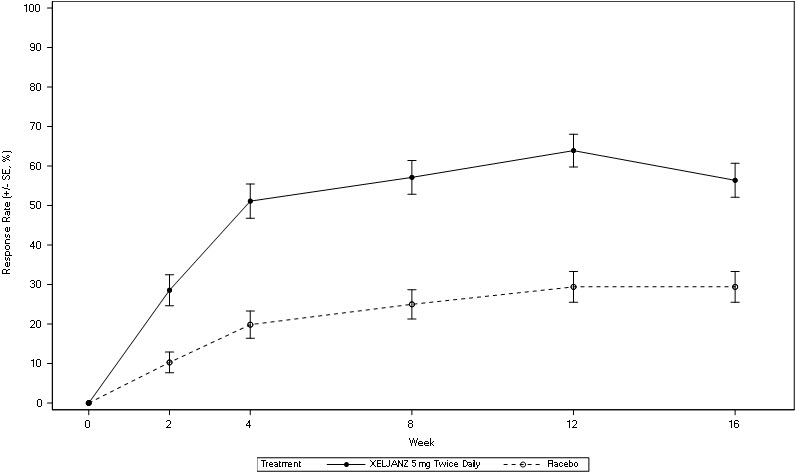
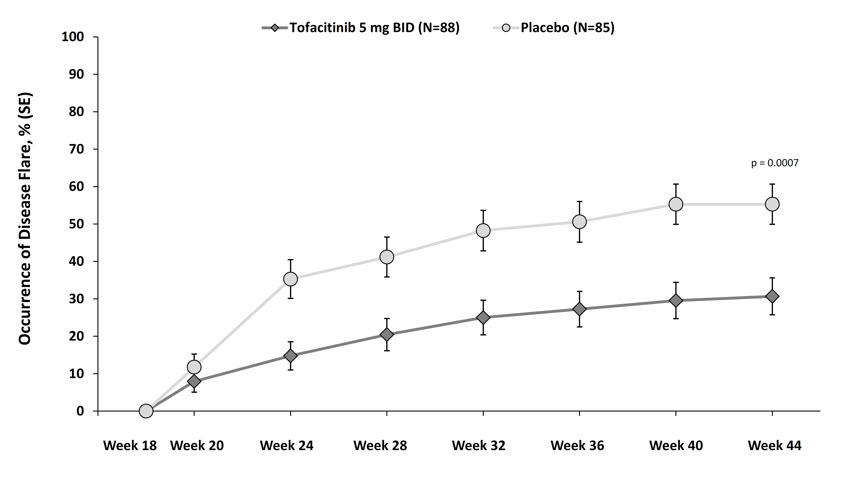
Artritis idiopática juvenil poliarticular
- •
- XELJANZ / XELJANZ Solución Oral 5 mg dos veces al día o el equivalente basado en el peso dos veces al día. (2.4)
- •
- Se necesita un ajuste de la dosis en pacientes con insuficiencia renal moderada y grave o insuficiencia hepática moderada: ver la información de prescripción completa. (2.4)
Ajuste de dosis
- •
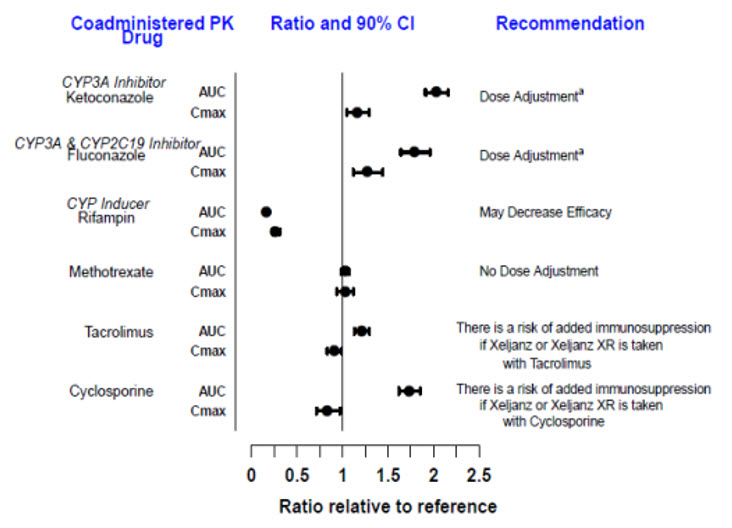
- Consulte la información de prescripción completa para los ajustes de dosis por indicación en pacientes que reciben inhibidores de CYP2C19 y / o CYP3A4; en pacientes con insuficiencia renal moderada o grave o insuficiencia hepática moderada; y pacientes con linfopenia, neutropenia o anemia. (2.2, 2.3, 2.4, 8.7, 8.8)
- •
- No se recomienda el uso de XELJANZ / XELJANZ XR / XELJANZ Solución Oral en pacientes con insuficiencia hepática grave en ninguna población de pacientes. (2.2, 2.3, 2.4, 8.8)
FORMAS Y POTENCIAS DE DOSIFICACIÓN
CONTRAINDICACIONES
Ninguna (4)
ADVERTENCIAS Y PRECAUCIONES
- •
- Infecciones graves: Evitar el uso de XELJANZ / XELJANZ XR / XELJANZ Solución Oral durante una infección grave activa, incluidas las infecciones localizadas. (5.1)
- •
- Perforaciones gastrointestinales: Usar con precaución en pacientes que pueden estar en mayor riesgo. (5.6)
- •
- Monitorización de laboratorio: Recomendada debido a posibles cambios en linfocitos, neutrófilos, hemoglobina, enzimas hepáticas y lípidos. (5.8)
- •
- Inmunizaciones: Vacunas vivas: Evitar el uso con XELJANZ / XELJANZ XR / XELJANZ Solución Oral. (5.9)
REACCIONES ADVERSAS
Las reacciones adversas más comunes son:
- •
- Artritis reumatoide, artritis psoriásica y espondilitis anquilosante: Reportadas durante los primeros 3 meses en ensayos clínicos controlados con placebo en artritis reumatoide y que ocurren en ≥2% de los pacientes tratados con monoterapia de XELJANZ o en combinación con DMARDs: infección del tracto respiratorio superior, nasofaringitis, diarrea y dolor de cabeza. (6.1)
- •
- Colitis ulcerosa: Reportadas en ≥5% de los pacientes tratados con 5 mg o 10 mg dos veces al día de XELJANZ y ≥1% mayor que lo reportado en pacientes que recibieron placebo en los ensayos clínicos de inducción o mantenimiento: nasofaringitis, niveles elevados de colesterol, dolor de cabeza, infección del tracto respiratorio superior, aumento de la creatina fosfocinasa en sangre, erupción cutánea, diarrea y herpes zoster. (6.1)
- •
- Artritis idiopática juvenil poliarticular: Consistente con las reacciones adversas comunes reportadas en pacientes adultos con artritis reumatoide. (6.1)
Para reportar REACCIONES ADVERSAS SOSPECHOSAS, comuníquese con Pfizer, Inc. al 1-800-438-1985 o con la FDA al 1-800-FDA-1088 o www.fda.gov/medwatch.