Fabricante de medicamentos: Takeda Pharmaceuticals America, Inc. (Updated: 2023-04-07)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
Estos puntos destacados no incluyen toda la información necesaria para usar VONVENDI de manera segura y eficaz. Consulte la información completa de prescripción de VONVENDI.
VONVENDI [factor de von Willebrand (recombinante)] polvo liofilizado para solución, para inyección intravenosa
Aprobación inicial en EE. UU.: 2015
INDICACIONES Y USOS
VONVENDI [factor de von Willebrand (recombinante)] es un factor de von Willebrand recombinante (rVWF) indicado para su uso en adultos (de 18 años o más) diagnosticados con la enfermedad de von Willebrand (VWD) para:
- Tratamiento a demanda y control de episodios de sangrado. (1)
- Gestión perioperatoria del sangrado. (1)
- Profilaxis rutinaria para reducir la frecuencia de episodios de sangrado en pacientes con VWD tipo 3 grave que reciban terapia a demanda. (1)
DOSIS Y ADMINISTRACIÓN
Solo para uso intravenoso después de la reconstitución.
Tratamiento y control a demanda de episodios de sangrado
- Para cada episodio de sangrado, administre la primera dosis de VONVENDI con un factor VIII recombinante aprobado (que no contenga factor de von Willebrand), si los niveles basales de factor VIII están por debajo del 40% o son desconocidos. (2.1)
- La dosis inicial es de 40 a 80 Unidades Internacionales (UI) por kg de peso corporal (PC). Ajuste la dosis según la extensión y la localización del sangrado. (2.1)
| Episodio de sangrado |
Dosis inicial |
Dosis subsiguiente |
| Menor |
40 a 50 UI/kg |
40 a 50 UI/kg cada 8 a 24 horas |
| Mayor |
50 a 80 UI/kg |
40 a 60 UI/kg cada 8 a 24 horas durante aproximadamente 2 a 3 días |
Gestión perioperatoria del sangrado
Para procedimiento quirúrgico electivo
- Se puede administrar una dosis de VONVENDI de 12 a 24 horas antes de la cirugía para permitir que los niveles endógenos de factor VIII aumenten a al menos 30 UI/dL (cirugía menor) o 60 UI/dL (cirugía mayor). (2.1)
- Evalúe los niveles de FVIII:C dentro de las 3 horas antes de la cirugía. Si los niveles de FVIII:C están en o por encima de los niveles mínimos objetivo recomendados, administre una dosis de VONVENDI sola dentro de 1 hora antes del procedimiento. Si los niveles de FVIII:C están por debajo de los niveles mínimos objetivo recomendados, administre factor VIII recombinante además de VONVENDI para elevar VWF:RCo y FVIII:C. (2.1)
Para cirugía de emergencia
- Evalue los niveles basales de VWF:RCo y FVIII:C dentro de las 3 horas antes de la cirugía. Si no están disponibles, use el cálculo de dosificación basado en el peso. (2.1)
- Administre VONVENDI 1 hora antes de la cirugía con o sin factor VIII recombinante y ajuste la dosis para elevar VWF:RCo y FVIII:C al nivel adecuado. (2.1)
| Tipo de cirugía |
Nivel plasmático máximo objetivo |
Cálculo de la dosis de rVWF
(UI VWF:RCo requeridas) |
| VWF:RCo |
FVIII:C |
| Menor |
50 a 60 UI/dL |
40 a 50 UI/dL |
∆ VWF:RCo × PC (kg)/IR |
| Mayor |
100 UI/dL |
80 a 100 UI/dL |
∆ VWF:RCo × PC (kg)/IR |
- Continuar monitorizando los niveles plasmáticos de VWF:RCo y FVIII:C después del procedimiento quirúrgico. (2.1)
Profilaxis rutinaria para reducir la frecuencia de episodios hemorrágicos en pacientes con VWD tipo 3 grave que reciben tratamiento según la demanda
La dosis inicial de VONVENDI es de 40 a 60 UI/kg de peso corporal que se administrará dos veces por semana. La dosis puede ajustarse hasta 60 UI/kg dos veces por semana según la frecuencia de episodios hemorrágicos. (2.1)
FORMAS DE DOSIFICACIÓN Y FUERZAS
VONVENDI está disponible como polvo liofilizado en frascos de dosis única que contienen nominalmente 650 o 1300 unidades internacionales de VWF:RCo. (3)
CONTRAINDICACIONES
No utilizar en pacientes que hayan tenido reacciones de hipersensibilidad potencialmente mortales a VONVENDI o sus componentes (citrato trisódico-dihidrato, glicina, manitol, trehalosa-dihidrato, polisorbato 80 y proteínas de hámster o ratón). (4)
ADVERTENCIAS Y PRECAUCIONES
- Las reacciones tromboembólicas pueden ocurrir, particularmente en pacientes con factores de riesgo de trombosis. Supervisar las primeras señales de trombosis y tener medidas de profilaxis contra la tromboembolismo instituídas de acuerdo con las recomendaciones actuales. Una de cada 100 personas con VWD tratadas con VONVENDI en ensayos clínicos desarrolló trombosis venosa profunda proximal en el periodo perioperatorio después de someterse a una cirugía de reemplazo total de cadera. En pacientes que requieran dosis frecuentes de VONVENDI en combinación con factor VIII recombinante, monitorizar los niveles plasmáticos de FVIII:C porque los niveles plasmáticos de factor VIII excesivos sostenidos pueden aumentar el riesgo de eventos tromboembólicos. (5.1)
- Las reacciones de hipersensibilidad, incluidas la anafilaxia, pueden ocurrir. Interrumpir VONVENDI si se presentan síntomas de hipersensibilidad y administrar el tratamiento de emergencia apropiado. (5.2)
- Los inhibidores del factor von Willebrand (VWF) y/o del factor VIII pueden ocurrir. Si no se alcanzan los niveles plasmáticos esperados de actividad de VWF (VWF:RCo), o si el sangrado no se controla con una dosis apropiada, realizar un ensayo apropiado para determinar si están presentes inhibidores anti-VWF o anti-factor VIII. (5.3)
REACCIONES ADVERSAS
- Las reacciones adversas más comunes observadas (≥ 2% de los sujetos) fueron cefalea, vómito, náuseas, mareos, artralgia, lesión articular, vértigo, aumento de ALT y prurito generalizado. (6.1)
- Un sujeto tratado con VONVENDI en el entorno perioperatorio desarrolló trombosis venosa profunda después de someterse a una cirugía de reemplazo total de cadera. (6.1)
Para reportar las REACCIONES ADVERSAS SOSPECHADAS, comuníquese con Takeda Pharmaceuticals U.S.A., Inc. al 1-877-TAKEDA-7 (1-877-825-3327) o con la FDA al 1-800-FDA-1088 o en www.fda.gov/medwatch.
Ver 17 para INFORMACIÓN DE CONSEJO AL PACIENTE y la etiqueta del paciente aprobada por la FDA.
Revisión: 3/2023
Tabla de Contenido
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDO*
1 INDICACIONES Y USO
VONVENDI [factor de von Willebrand (recombinante)] es un factor de von Willebrand recombinante (rVWF) indicado para su uso en adultos (mayores de 18 años) diagnosticados con enfermedad de von Willebrand (VWD) para:
- Tratamiento a demanda y control de episodios de sangrado.
- Manejo perioperatorio de sangrado.
- Profilaxis rutinaria para reducir la frecuencia de episodios de sangrado en pacientes con VWD de tipo 3 grave que reciben terapia a demanda.
2 DOSIS Y ADMINISTRACIÓN
2.1 Dosis recomendada
Sólo para uso intravenoso después de la reconstitución.
- Cada frasco de VONVENDI está etiquetado con la cantidad real de actividad de rVWF en Unidades Internacionales (IU), medida con el ensayo del cofactor de ristocetina (VWF:RCo).
- Individualizar la dosis y la frecuencia según el juicio clínico y basándose en el peso del sujeto, el tipo y la gravedad de los episodios de hemorragia/intervención quirúrgica y en base al seguimiento de las medidas clínicas y de laboratorio apropiadas.
- Para pacientes con hemorragia, la hemostasia no puede asegurarse hasta que la actividad de la coagulación del factor VIII (FVIII:C) haya alcanzado 40 UI/décilitro (dL) (es decir, el 40% de la actividad normal). Si el nivel plasmático basal de FVIII:C del paciente está por debajo de 40%, o es desconocido, administrar un factor VIII recombinante aprobado (que no contenga factor de von Willebrand) (rFVIII) con la primera infusión de VONVENDI para lograr un nivel plasmático hemostático de FVIII:C. Sin embargo, si no es necesario un aumento inmediato de FVIII:C o si el nivel basal de FVIII:C es suficiente para asegurar la hemostasia, administrar VONVENDI sin rFVIII.
- En función del nivel basal de FVIII:C del paciente, se espera que una sola infusión de VONVENDI, en la mayoría de los pacientes, provoque un aumento de la actividad endógena de FVIII:C por encima del 40% en un plazo de 6 horas.
- Cuando se requieran infusiones repetidas, monitorizar los niveles de factor VIII para determinar si se necesita rFVIII con las siguientes infusiones.
Tratamiento a demanda y control de los episodios de hemorragia
Administrar una dosis inicial de 40 a 80 UI/kg de peso corporal (PC). Las pautas de dosificación para el tratamiento de hemorragias leves y graves se proporcionan en Tabla 1.
Administrar VONVENDI dentro de los rangos designados en base al juicio clínico, teniendo en cuenta la gravedad, el lugar de la hemorragia y los antecedentes médicos del paciente. Ajustar la dosis en función de la extensión y la ubicación del episodio de hemorragia. Administrar dosis posteriores siempre que sea clínicamente necesario. Monitorizar las medidas clínicas y de laboratorio apropiadas [ver Advertencias y Precauciones (5.2, 5.3)].
La dosis inicial de VONVENDI debería lograr más del 60 % de los niveles del factor de von Willebrand (VWF) (según VWF:RCo mayor a 60 UI/dL) y la infusión de rFVIII debería lograr niveles del factor VIII mayores al 40 % (FVIII:C mayor a 40 UI/dL). En episodios de hemorragia mayor, mantener niveles mínimos de VWF:RCo mayores al 50 % durante el tiempo que se considere necesario.
Administrar VONVENDI con rFVIII si el nivel de FVIII:C es menor al 40 %, o es desconocido, para controlar la hemorragia. La dosis de rFVIII debe calcularse de acuerdo a la diferencia entre el nivel basal de FVIII:C en plasma del paciente y el nivel máximo deseado de FVIII:C, para lograr un nivel de FVIII:C en plasma adecuado basándose en la recuperación media aproximada de 2 (UI/dL)/(UI/kg). Administrar la dosis completa de VONVENDI seguida de rFVIII dentro de los 10 minutos.
Cálculo de la dosis
Dosis de VONVENDI [IU] = dosis en [IU/kg] × peso corporal [kg]
En un episodio de hemorragia mayor cuando el nivel basal del factor VIII es desconocido, se debe administrar rFVIII para alcanzar un nivel máximo objetivo de FVIII:C de 80 a 100 UI/dL, basándose en la recuperación media aproximada de 2 (UI/dL)/(UI/kg). Consultar la etiqueta para el cálculo de la dosis basada en el peso de rFVIII que se utiliza.
Si los niveles plasmáticos esperados de actividad de VWF no se alcanzan, o si el episodio de hemorragia no se controla con una dosis adecuada, realizar un análisis que mida la presencia de inhibidores de VWF o factor VIII [ver Advertencias y Precauciones (5.3)].
Gestión perioperatoria de la hemorragia
Procedimientos quirúrgicos electivos
Se puede administrar una dosis preoperatoria de VONVENDI de 12 a 24 horas antes de la cirugía para permitir que los niveles endógenos del factor VIII aumenten a al menos 30 UI/dL (cirugía menor) o 60 UI/dL (cirugía mayor) antes de que se administre la dosis inicial (dosis preoperatoria de 1 hora) de rVWF, con o sin rFVIII.
Asegurar que el nivel basal de FVIII:C esté disponible antes de determinar la necesidad de la dosis preoperatoria de 12 a 24 horas. El nivel de FVIII:C también debe evaluarse dentro de las 3 horas anteriores al inicio del procedimiento quirúrgico. Si el nivel está en los niveles objetivo mínimos recomendados (30 UI/dL para cirugía menor y 60 UI/dL para cirugía mayor), administrar una dosis de VONVENDI solo (sin tratamiento con factor VIII) dentro de la hora anterior al procedimiento. Si el nivel de FVIII:C está por debajo del nivel objetivo mínimo recomendado, administrar la dosis completa de VONVENDI seguida de rFVIII dentro de los 10 minutos para elevar VWF:RCo y FVIII:C.
Consultar Tabla 2 para niveles máximos objetivo recomendados del plasma de VWF:RCo y FVIII:C y pautas de dosificación para la gestión perioperatoria de la hemorragia.
Evaluar los niveles basales de VWF:RCo dentro de las 3 horas de administración de la dosis preoperatoria de 12 a 24 horas. Si la dosis preoperatoria de 12 a 24 horas no se administra, entonces evaluar el nivel basal de VWF:RCo antes de la cirugía.
Cuando sea posible, medir la recuperación incremental (IR) de VONVENDI antes de la cirugía. Para el cálculo de IR, medir el VWF:RCo en plasma basal. Luego infundir una dosis de 50 UI/kg de VONVENDI. Medir VWF:RCo a los 30 minutos después de la infusión de VONVENDI.
Usar la siguiente fórmula para calcular IR:
IR = [VWF:RCo en plasma a los 30 minutos (UI/dL) – VWF:RCo en plasma basal (UI/dL)]/Dosis (IU/kg)
Cirugía de emergencia
La dosis preoperatoria de 12 a 24 horas puede no ser factible en sujetos que requieren cirugía de emergencia. Los niveles basales de VWF:RCo y FVIII:C deben evaluarse dentro de las 3 horas anteriores al inicio del procedimiento quirúrgico si es factible. La dosis inicial (dosis preoperatoria de 1 hora) puede calcularse como la diferencia en los niveles máximos objetivos y los niveles basales de VWF:RCo en plasma dividido por la IR. Si la IR no está disponible, suponer una IR de 2.0 UI/dL por UI/kg.
Si los niveles basales de VWF:RCo y FVIII:C no están disponibles, como guía general se debe administrar una dosis inicial (dosis preoperatoria de 1 hora) de VONVENDI, de 40 a 60 UI/kg de VWF:RCo. Además, rFVIII en una dosis de 30 a 45 UI/kg puede infundirse secuencialmente, preferentemente dentro de los 10 minutos después de la infusión de VONVENDI en pacientes cuyo nivel de factor VIII en plasma ya es (o es muy probable que sea) menor a 40 a 50 UI/dL para cirugía menor o 80 a 100 UI/dL para cirugía mayor.
Consultar Tabla 2 para niveles máximos objetivos recomendados del plasma de VWF:RCo y FVIII:C y pautas de dosificación para la gestión perioperatoria de la hemorragia.
En ausencia de valores de referencia disponibles de FVIII:C, VWF:RCo y Recuperación Incremental, se recomienda utilizar la dosificación basada en el peso corporal tal como se describe a continuación en Tabla 3.
Monitorizar los niveles plasmáticos de VWF:RCo y FVIII:C empezando 12 a 24 horas después de la cirugía y al menos cada 24 horas durante el período perioperatorio para ajustar la dosis de VONVENDI o los niveles de rFVIII. Los niveles plasmáticos de VWF:RCo y FVIII:C deben monitorizarse y el régimen de mantenimiento intra y postoperatorio debe individualizarse según los resultados farmacocinéticos (PK) y la intensidad y duración del desafío hemostático. La frecuencia de la dosificación de VONVENDI debe oscilar entre dos veces al día y cada 48 horas.
Consulte Tabla 4 para los niveles plasmáticos objetivo de VWF:RCo y FVIII:C y la duración mínima de tratamiento para las dosis de mantenimiento posteriores a la cirugía.
Profilaxis rutinaria para reducir la frecuencia de episodios hemorrágicos en pacientes con VWD grave de tipo 3 que reciben terapia bajo demanda
Para iniciar el tratamiento profiláctico, administrar 40 a 60 UI de VONVENDI por kilogramo de peso corporal dos veces por semana. Ajustar la dosis de profilaxis hasta 60 UI/kg dos veces por semana si ocurre hemorragia repentina en las articulaciones o si se produce hemorragia grave. Tratar la hemorragia repentina de acuerdo con las pautas de dosificación en Tabla 1.
2.2 Preparación y reconstitución
- Permitir que VONVENDI y Agua estéril para inyección (diluyente) alcancen la temperatura ambiente.
- Si el paciente necesita más de un frasco de VONVENDI por inyección, reconstituir cada frasco de acuerdo con las siguientes instrucciones.
- Algunos copos o partículas pueden permanecer en el frasco reconstituido. El filtro incluido en el dispositivo Mix2Vial eliminará los copos o partículas extraños, y la solución resultante en la jeringa debe ser clara e incolora. No use la solución en la jeringa si está turbia o contiene copos o partículas después de la filtración desde el frasco a la jeringa.
Reconstitución
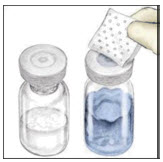
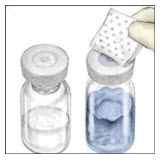
- Retire las tapas de plástico de los frascos de VONVENDI y agua para inyección.
- Limpie los tapones de goma con un hisopo de alcohol estéril.
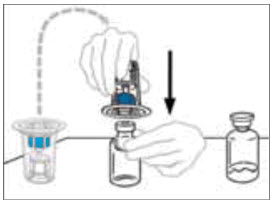
- Despegue la cubierta del dispositivo de transferencia Mix2Vial. Para mantener la esterilidad, deje el dispositivo Mix2Vial en el empaque de plástico transparente.
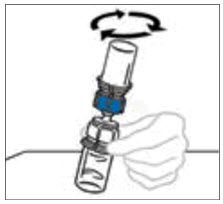
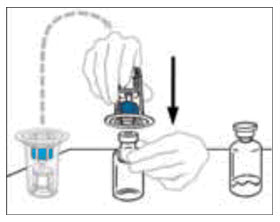
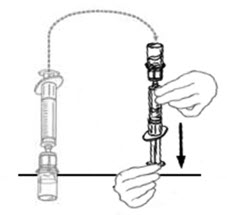
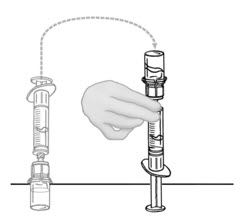
- Mientras sostiene firmemente el frasco del diluyente en una superficie nivelada, tome el Mix2Vial en su empaque de plástico e invierta sobre el frasco del diluyente. Empuje firmemente la cánula de plástico azul del Mix2Vial hacia abajo a través del tapón de goma. Retire cuidadosamente el empaque de plástico dejando el Mix2Vial fijado firmemente al frasco del diluyente.
- Sostenga firmemente el frasco de VONVENDI en una superficie nivelada, invierta rápidamente el frasco del diluyente con el Mix2Vial adjunto y empuje el extremo de la cánula de plástico transparente del Mix2Vial firmemente hacia abajo a través del tapón del frasco de VONVENDI. El diluyente se absorberá en el frasco de VONVENDI por el vacío.
- Verifique que la transferencia del diluyente esté completa. No use si se ha perdido el vacío.
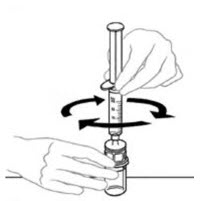
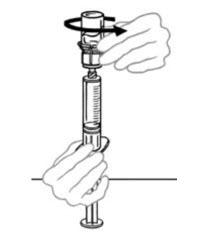
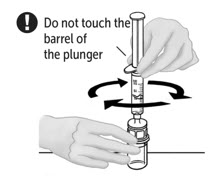
- Con ambos frascos aún unidos, gire suavemente los frascos o deje que el producto reconstituido repose durante 5 minutos y luego gire suavemente para asegurar que el polvo esté completamente disuelto.
No agite. Agitar afectará negativamente la integridad del producto.
Nota: Algunos copos o partículas pueden permanecer en el frasco reconstituido. El filtro incluido en el dispositivo Mix2Vial eliminará los copos o partículas extraños, y la solución resultante en la jeringa debe ser clara e incolora. No use la solución en la jeringa si está turbia o contiene copos o partículas después de la filtración desde el frasco a la jeringa.
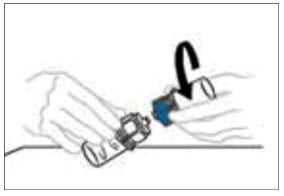
- Una vez que el contenido esté completamente disuelto, sostenga firmemente las partes transparentes y azules del Mix2Vial. Desenrosque el Mix2Vial en dos piezas separadas y deseche el frasco de diluyente vacío y la parte azul del Mix2Vial. Nota: El Mix2Vial se destina a un solo uso con un frasco individual de VONVENDI y diluyente únicamente. Si la dosis requiere más de un frasco de VONVENDI, reconstituir cada frasco por separado.
No refrigerar después de la reconstitución.
2.3 Administración
Solo para administración intravenosa.
- Administre VONVENDI inmediatamente después de la reconstitución. Si no, almacene a temperatura ambiente no superior a 25°C (77°F) durante un máximo de 3 horas. Deséchelo después de 3 horas.
- No se pueden agrupar más de dos frascos de VONVENDI en una sola jeringa. La agrupación de más de dos frascos en una jeringa puede provocar la formación de filamentos, lo que requiere el desecho de la solución en la jeringa. Si un paciente tiene que recibir más de un frasco de VONVENDI, deje la jeringa conectada al frasco o cubra la punta de la jeringa con una tapa estéril adecuada hasta que esté listo para infundirse para reducir el riesgo de contaminación.
- Use jeringas de plástico con este producto porque las proteínas en el producto tienden a pegarse a la superficie de las jeringas de vidrio.
- No mezcle VONVENDI con otros productos medicinales.
Administración
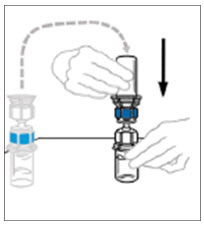
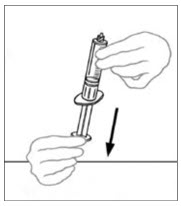
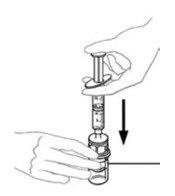
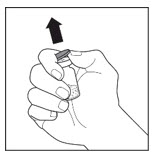
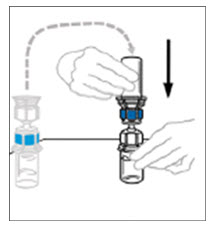
- Inyecte aire en una jeringa desechable plástica estéril vacía. La cantidad de aire debe ser igual a la cantidad de VONVENDI reconstituido que se va a extraer del frasco.
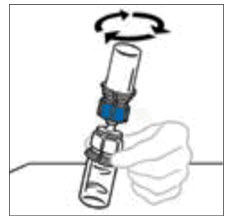
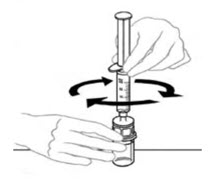
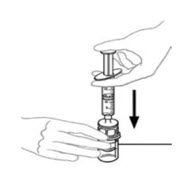
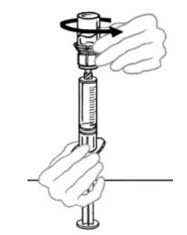
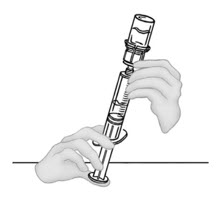
- Dejando el frasco de VONVENDI (que contiene el producto disuelto) sobre su superficie de trabajo plana, conecte la jeringa al conector de plástico transparente fijándolo y girándolo en sentido horario (Figura A). Sostenga el frasco con una mano y use la otra mano para empujar la totalidad del aire de la jeringa hacia el frasco (Figura B). La cantidad requerida de producto no se extraerá en la jeringa si no se empuja todo el aire hacia el frasco.
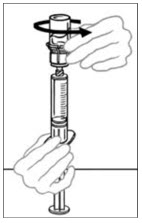
- Voltee la jeringa conectada y el frasco de VONVENDI de modo que el frasco esté en la parte superior. Asegúrese de mantener presionada la émbolo de la jeringa. Extraiga el VONVENDI a la jeringa tirando lentamente hacia atrás de la émbolo. No empuje y tire la solución de un lado a otro entre la jeringa y el frasco. Hacer esto puede dañar la integridad del producto.
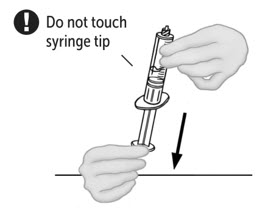
- Inspeccione el VONVENDI después de la filtración/extracción a la jeringa en busca de decoloración y partículas antes de la administración. La solución debe ser clara o ligeramente opalescente en apariencia. No administre si se observan partículas, decoloración o turbidez y notifique a la Información Médica de Takeda al 1-877-TAKEDA-7 (1-877-825-3327).
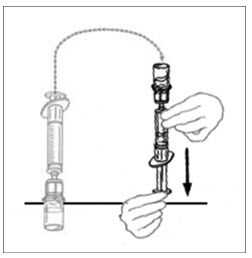
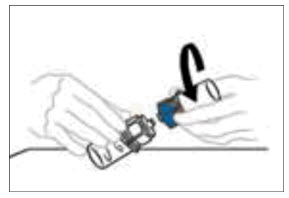
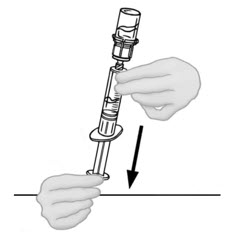
- Cuando esté listo para infundir, sostenga firmemente el cilindro de la jeringa (manteniendo la émbolo de la jeringa hacia abajo) y separe el Mix2Vial de la jeringa. Descarte el Mix2Vial (parte plástica transparente) y el frasco vacío de VONVENDI. Si un paciente va a recibir más de un frasco de VONVENDI, el contenido de hasta dos frascos puede extraerse a una sola jeringa. Al introducir aire en un segundo frasco de VONVENDI para agrupado en una jeringa, coloque el frasco y la jeringa conectada de modo que el frasco esté en la parte superior.
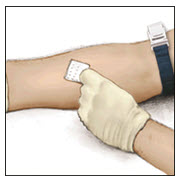
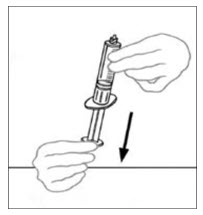
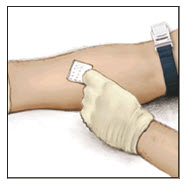
- Limpie el sitio de inyección previsto con una torunda de alcohol estéril.
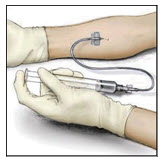
- Coloque una aguja de infusión adecuada en la jeringa. Infunda por vía intravenosa a una velocidad lo suficientemente lenta como para garantizar la comodidad del paciente, hasta un máximo de 4 mL por minuto.
- Si se produce taquicardia, la velocidad de inyección debe reducirse o la administración debe interrumpirse.
- Deseche cualquier producto no utilizado o material de desecho de acuerdo con los requisitos locales.
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
VONVENDI está disponible como un polvo liofilizado no pirogénico, de color blanco a blanquecino, para reconstitución en frascos de una dosis que contiene nominalmente 650 o 1300 UI de VWF:RCo/frasco.
Cada frasco de VONVENDI está etiquetado con el número de unidades de VWF:RCo expresadas en UI, que se basan en el estándar actual de la Organización Mundial de la Salud (OMS) para el concentrado de VWF.
4 CONTRAINDICACIONES
VONVENDI está contraindicado en pacientes que hayan tenido reacciones de hipersensibilidad que pongan en peligro la vida debido a VONVENDI o a los componentes del producto (citrato trisódico-dihidrato, glicina, manitol, trehalosa-dihidrato, polisorbato 80 y proteínas de hámster o ratón) [véase Descripción (11)].
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Embolismo y trombosis
Pueden ocurrir reacciones tromboembólicas, incluyendo coagulación intravascular diseminada (DIC), trombosis venosa, embolia pulmonar, infarto de miocardio y accidente cerebrovascular, especialmente en pacientes con factores de riesgo conocidos de trombosis, incluyendo niveles bajos de ADAMTS13 [ver Descripción (11)]. Vigile los signos y síntomas tempranos de trombosis como dolor, hinchazón, decoloración, disnea, tos, hemoptisis y síncope, y establezca medidas profilácticas contra el tromboembolismo de acuerdo con las recomendaciones actuales y las normas de atención.
En pacientes que requieran dosis frecuentes de VONVENDI en combinación con rFVIII, controle los niveles plasmáticos de la actividad de FVIII:C, ya que los niveles plasmáticos excesivos sostenidos del factor VIII pueden aumentar el riesgo de complicaciones tromboembólicas.
De los 100 sujetos tratados con VONVENDI en ensayos clínicos, un sujeto fue diagnosticado con trombosis venosa profunda, que se reveló por imágenes realizadas como parte de la norma de atención del hospital para pacientes de alto riesgo, 3 días después de la cirugía de reemplazo total de cadera mientras recibía VONVENDI.
5.2 Reacciones de hipersensibilidad
Se han producido reacciones de hipersensibilidad con VONVENDI. Estas reacciones pueden incluir shock anafiláctico, urticaria generalizada, angioedema, opresión torácica, hipotensión, shock, letargo, náuseas, vómitos, parestesia, prurito, inquietud, visión borrosa, sibilancias y/o dificultad respiratoria aguda. Si se presentan signos y síntomas de reacciones alérgicas graves, interrumpa de inmediato la administración de VONVENDI y brinde atención de apoyo adecuada.
VONVENDI contiene cantidades traza de inmunoglobulina G de ratón (MuIgG) y proteínas de hámster inferiores o iguales a 2 ng/IU VONVENDI. Los pacientes tratados con este producto pueden desarrollar reacciones de hipersensibilidad a proteínas de mamíferos no humanos.
5.3 Anticuerpos neutralizantes
Pueden ocurrir anticuerpos neutralizantes (inhibidores) a VWF y/o factor VIII. Si no se alcanzan los niveles plasmáticos esperados de la actividad de VWF (VWF:RCo), realice un análisis adecuado para determinar si están presentes inhibidores anti-VWF o anti-factor VIII. Considere otras opciones terapéuticas y dirija al paciente a un médico con experiencia en la atención de la enfermedad de von Willebrand (VWD) o la hemofilia A.
En pacientes con niveles altos de inhibidores a VWF o factor VIII, la terapia con VONVENDI puede no ser eficaz y la infusión de esta proteína puede provocar reacciones de hipersensibilidad graves. Dado que los anticuerpos inhibidores pueden ocurrir concomitantemente con reacciones anafilácticas, valore a los pacientes que experimentan una reacción anafiláctica para la presencia de inhibidores.
5.4 Monitorización de pruebas de laboratorio
Vigile los niveles plasmáticos de VWF:RCo y de la actividad del factor VIII en pacientes que reciben VONVENDI para evitar niveles de actividad de VWF y/o factor VIII excesivos sostenidos, lo que puede aumentar el riesgo de eventos trombóticos, especialmente en pacientes con factores de riesgo clínicos o de laboratorio conocidos.
Vigile el desarrollo de inhibidores de VWF y/o factor VIII cuando se sospeche. Realice análisis de inhibidores apropiados para determinar si están presentes inhibidores de VWF y/o factor VIII si el sangrado no se controla con la dosis esperada de VONVENDI.
6 REACCIONES ADVERSAS
Las reacciones adversas más comunes observadas en más del 2% de los sujetos en ensayos clínicos con VONVENDI (n = 100) fueron cefalea, vómitos, náuseas, mareos, artrogia, lesión articular, vértigo, aumento de ALT y prurito generalizado.
Un sujeto tratado con VONVENDI en el estudio de cirugía para el manejo perioperatorio del sangrado desarrolló trombosis venosa profunda proximal postoperatoriamente [véase Advertencias y Precauciones (5.1)].
6.1 Experiencia con Ensayos Clínicos
Debido a que los ensayos clínicos se realizan en condiciones muy variadas, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no se pueden comparar directamente con las tasas en ensayos clínicos de otro fármaco y puede que no reflejen las tasas observadas en la práctica clínica.
El perfil de seguridad de VONVENDI se evaluó en cinco ensayos prospectivos, multicéntricos; cuatro se llevaron a cabo en sujetos con VWD (n = 100) y uno en sujetos con hemofilia A (n = 12). Los términos de reacción adversa a medicamentos (RAM) que se indican a continuación (Tabla 5) fueron evaluados por el patrocinador/empresa como que tenían una relación causal plausible con el medicamento en estudio en tres ensayos clínicos (n = 80) durante los cuales VONVENDI se utilizó para la evaluación PK, el tratamiento bajo demanda y el manejo perioperatorio de episodios de sangrado en pacientes sometidos a cirugía.
Un paciente individual tratado con Vonvendi en un ensayo clínico desarrolló una reacción relacionada con la infusión. Este paciente había estado expuesto anteriormente a Vonvendi sin presentar ningún síntoma. Tres minutos después de iniciar la infusión, desarrolló molestias en el pecho y aumento del ritmo cardíaco. El paciente fue tratado con cuidados de apoyo y los síntomas se resolvieron en 3 horas.
En el estudio clínico de profilaxis completado, veintidós sujetos adultos de 18 años y mayores que recibieron terapia bajo demanda o profiláctica con VWF derivado del plasma antes de la entrada del estudio recibieron tratamiento profiláctico con VONVENDI durante el estudio. Los términos de reacción adversa a la droga (RAD) se enumeran a continuación (Tabla 6).
6.2 Experiencia poscomercialización
Las siguientes reacciones adversas a la droga (RAD) han sido identificadas durante el uso posterior a la aprobación de VONVENDI. Debido a que estas reacciones son reportadas voluntariamente desde una población de tamaño incierto, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal con la exposición a la droga.
Las RAD reportadas poscomercialización en asociación con el tratamientoo con VONVENDI incluyen reacciones relacionadas con la infusión (IRI) que pueden manifestarse clínicamente por síntomas como taquicardia, rubor, erupciones, disnea y visión borrosa. En los casos poscomercialización reportados, los síntomas se resolvieron y los pacientes se recuperaron completamente dentro de 4 horas después de detener la infusión.
Se ha informado de una reacción anafiláctica con el uso de Vonvendi en el entorno poscomercialización.
6.3 Inmunogenicidad
La inmunogenicidad de VONVENDI se evaluó en ensayos clínicos evaluando el desarrollo de anticuerpos neutralizantes contra VWF y FVIII, así como anticuerpos de unión contra VWF, Furin, proteína de ovario de hámster chino (CHO) e IgG de ratón. De los 100 sujetos que recibieron VONVENDI en los ensayos clínicos, un sujeto que fue tratado con VONVENDI perioperatoriamente desarrolló anticuerpos de unión emergentes de tratamientoo frente a VWF después de una cirugía, para el que no se informaron eventos adversos o falta de eficacia hemostática. No se desarrollaron anticuerpos de unión contra impurezas potenciales como rFurin, proteína CHO o IgG de ratón después del tratamientoo con VONVENDI.
Dos sujetos incluidos en el estudio tenían anticuerpos de unión específicos de alto título preexistentes contra VWF. Los anticuerpos de unión anti-VWF de alto título estaban asociados con una actividad significativamente disminuida de VWF:Ag post-infusion de VWF derivado del plasma (pdVWF) o rVWF y, en consecuencia, la disminución de la actividad de VWF:RCo, VWF:CB y FVIII:C. Este hallazgo indica la importancia clínica potencial de los anticuerpos de unión (no neutralizantes) preexistentes: Los pacientes con VWD que previamente hayan sido tratados con concentrados de pdVWF pueden estar en riesgo de expresar un anticuerpo de unión preexistente contra VWF antes de la primera exposición a rVWF, lo que podría resultar potencialmente en una respuesta hemostática disminuida a rVWF. Dichos pacientes podrían ser manejados clínicamente mediante la administración de dosis más altas de rVWF basadas en los datos PK de cada paciente individual.
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen del riesgo
No se han realizado estudios adecuados y bien controlados con VONVENDI en mujeres embarazadas. No se han realizado estudios de toxicidad para el desarrollo y la reproducción animal con VONVENDI. No se sabe si VONVENDI puede causar daño fetal cuando se administra a una mujer embarazada o si puede afectar la capacidad de reproducción.
El riesgo basal de defectos congénitos graves y aborto espontáneo en la población indicada es desconocido; sin embargo, en la población de los Estados Unidos, el riesgo basal estimado de defectos congénitos graves y aborto espontáneo es del 2 al 4% y del 15 al 20%, respectivamente.
8.2 Lactancia
Resumen del riesgo
No se dispone de información sobre la presencia de VONVENDI en la leche materna, los efectos en el lactante amamantado o los efectos en la producción de leche.
Los beneficios del desarrollo y la salud de la lactancia materna deben considerarse junto con la necesidad clínica de la madre de VONVENDI y cualquier efecto adverso potencial en el lactante amamantado debido a VONVENDI o a la afección materna subyacente.
8.4 Uso pediátrico
No se ha establecido la seguridad y eficacia en pacientes pediátricos menores de 18 años.
8.5 Uso geriátrico
Los ensayos clínicos de VONVENDI no incluyeron un número suficiente de sujetos de 65 años o más para determinar si responden de manera diferente en comparación con los sujetos más jóvenes.
11 DESCRIPCIÓN
VONVENDI es un rVWF purificado expresado en células de ovario de hámster chino (CHO). VONVENDI se produce y formula sin la adición de ningún material crudo exógeno de origen humano o animal en el cultivo celular, la purificación o la formulación del producto final. Las proteínas presentes en el producto final del contenedor que no sean rVWF son cantidades traza de inmunoglobulina de ratón (IgG, de la purificación por inmunoadsorción), proteína de células huésped (es decir, CHO), rFurin (utilizado para procesar aún más rVWF) y factor VIII recombinante (rFVIII).
El factor de Von Willebrand es una glicoproteína multimérica grande que se encuentra normalmente en el plasma y se almacena como multímeros ultra grandes en los gránulos alfa de las plaquetas y en orgánulos intracelulares conocidos como cuerpos de Weibel-Palade, antes de su secreción en la sangre.1 Una vez que el VWF se libera al torrente sanguíneo y entra en contacto con ADAMTS13 (una enzima proteolítica en la sangre), se divide en tamaños más pequeños que se pueden detectar con geles de agarosa SDS como bandas de multímeros, que representan las diversas especies de VWF en la circulación. VONVENDI es rVWF que contiene multímeros ultra grandes además de todos los multímeros que se encuentran en el plasma, ya que no está expuesto a la proteólisis por ADAMTS13 durante el proceso de fabricación.
VONVENDI se formula como un polvo estéril, no pirogénico, blanco a blanco amarillento para inyección intravenosa después de la reconstitución. VONVENDI en un vial monodosis contiene nominalmente 650 o 1300 UI de VWF:RCo.
El producto no contiene conservantes. Cuando se reconstituye con el agua estéril para inyección proporcionada, la solución final contiene los siguientes estabilizadores y excipientes (Tabla 7) en cantidades objetivo:
Cada vial de VONVENDI está etiquetado con el número específico de unidades de VWF:RCo expresadas en UI, que se basan en la norma actual de la Organización Mundial de la Salud (OMS) para el concentrado de VWF. Después de la reconstitución del polvo liofilizado y la filtración/aspiración en una jeringa, todas las concentraciones de dosis producen una solución clara, incolora y libre de partículas.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
En pacientes con VWD, VONVENDI actúa 1) para promover la hemostasia mediando la adhesión de las plaquetas a la matriz sub-endotelial vascular dañada (por ejemplo, el colágeno) y la agregación plaquetaria, y 2) como una proteína portadora del factor VIII, protegiéndolo de la proteólisis rápida. La actividad adhesiva de VWF depende del tamaño de sus multímeros, siendo los multímeros más grandes los más efectivos para apoyar las interacciones con colágeno y receptores plaquetarios.1 La capacidad de unión y la afinidad de VONVENDI al factor VIII en plasma es comparable a la de VWF endógeno, permitiendo que VONVENDI reduzca la depuración del factor VIII.2
12.2 Farmacodinámica
La farmacodinámica (PD) de VWF después del tratamiento profiláctico con VONVENDI fue investigada en un ensayo clínico. En individuos adultos previos OD con Tipo 3 de VWD, una sola infusión de VONVENDI condujo a un aumento de FVIII:C con niveles pico observados aproximadamente 24 horas después de la infusión (media [DE] de la dosis: 50.2 [3.33] UI/kg). Después de 1 año de infusiones repetidas de 50 ± 10 UI/kg de VONVENDI dos veces por semana, la mediana (rango) de FVIII:C previa a la dosis aumentó de 2.0 (2.0 a 4.0) UI/dL (basal después del lavado, N=10) a 10.5 (6.0 a 31.0) UI/dL (Mes 12, N=8). Los niveles de FVIII:C previos a la dosis (mediana [rango] ≥10.5 [1.0 a 148] UI/dL) fueron observados durante las visitas profilácticas en los meses 1, 2, 3, 6, 9 y 12. En el Mes 12, la media (DE) Cmax y AUC(0-96 horas) de FVIII:C fueron 106 (37.2) UI/dL y 5962 (2671) UI*h/dL (N=8), respectivamente.
12.3 Farmacocinética
El perfil farmacocinético (PK) de VONVENDI se determinó basado en los análisis de datos de dos ensayos clínicos mediante la evaluación de VWF:RCo, VWF:Ag y VWF:CB después de una dosis única. Los sujetos fueron evaluados en el estado de no hemorragia.
Tabla 8 a continuación resume los parámetros PK de VONVENDI después de las infusiones de 50 UI/kg (PK50) o 80 UI/kg VWF:RCo (PK80) VONVENDI.
Se investigó el PK de VWF después del tratamiento profiláctico con VONVENDI en un ensayo clínico. Los parámetros PK en estado estacionario de VWF:RCo se presentan en Tabla 9 para los sujetos con VWD tipo 3 que anteriormente habían sido tratados bajo demanda (OD) con cualquier producto de VWF antes de la entrada al estudio (grupo Anterior OD) y los sujetos que anteriormente habían sido tratados profilácticamente con un producto de VWF derivado del plasma (grupo de Cambio) antes del inclusión en el estudio. La farmacocinética de VWF después de la dosificación única y múltiple (en el Mes 12) fue similar en el grupo Anterior OD.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Alteración de la Fertilidad
In vitro y estudios de genotoxicidad in vivo indicaron que VONVENDI no tiene potencial mutagénico. No se realizaron estudios a largo plazo en animales para evaluar el potencial carcinogénico de VONVENDI. No se llevaron a cabo estudios en animales para evaluar la toxicidad para el desarrollo y la reproducción de VONVENDI.
14 ESTUDIOS CLÍNICOS
Tratamiento bajo demanda y control de los episodios hemorrágicos
La eficacia hemostática de VONVENDI se evaluó en un ensayo multidisciplinario y de etiqueta abierta que investigaba diferentes estrategias de dosificación con y sin rFVIII para el tratamiento bajo demanda y el control de los episodios hemorrágicos en adultos (de 18 años o más) diagnosticados con enfermedad de von Willebrand. En este ensayo, todos los sujetos que requerían rFVIII recibieron ADVATE [Factor antihemofílico (recombinante)].
Los episodios hemorrágicos se trataron inicialmente con una infusión de VONVENDI y ADVATE en una proporción de 1,3:1 respectivamente (es decir, un 30% más de VONVENDI que de ADVATE), y posteriormente con VONVENDI con o sin ADVATE, basada en los niveles de FVIII:C. El objetivo de la dosis inicial de VONVENDI con ADVATE era alcanzar niveles plasmáticos objetivo de más de 60 UI/dL (60%) VWF:RCo y más de 40 UI/dL (40%) de FVIII:C.
Se reportaron un total de 193 episodios hemorrágicos en 22/37 sujetos expuestos a VONVENDI para el tratamiento bajo demanda. Las características demográficas y basales se enumeran en Tabla 10.
El punto final de eficacia primaria fue el número de sujetos con éxito terapéutico en el control de los episodios hemorrágicos. El éxito terapéutico se definió como un puntaje promedio de eficacia de menos de 2.5 para todos los episodios hemorrágicos en un sujeto tratado con VONVENDI (con o sin ADVATE) durante el período del ensayo. La evaluación de la eficacia se realizó utilizando una escala de calificación de 4 puntos pre-especificada que comparaba el número prospectivo estimado de infusiones necesarias para tratar los episodios hemorrágicos según la evaluación del investigador con el número real de infusiones administradas. Las definiciones de cada una de las escalas de calificación de 4 puntos se proporcionan en Tabla 11.
Las medidas de eficacia secundarias fueron el número de episodios de sangrado tratados con una evaluación de eficacia de “excelente” o “buena”, el número de infusiones y el número de unidades de VONVENDI, administradas con o sin ADVATE, por episodio de sangrado.
La evaluación de eficacia primaria excluyó a los sujetos con hemorragias gastrointestinales (n = 2) y a los sujetos en los que el número de infusiones para controlar un episodio de sangrado se estimó retrospectivamente (n = 2). La tasa de sujetos (n = 18) con éxito del tratamientoo fue del 100% (95% CI, 81,5 a 100). Los análisis de sensibilidad del éxito del tratamientoo para los episodios de sangrado que incluyeron hemorragias gastrointestinales y aquellos episodios de sangrado en los que el investigador tuvo que hacer una evaluación retrospectiva del número de infusiones requeridas (n = 22: 17 con VWD tipo 3, 4 con VWD tipo 2A y 1 con VWD tipo 2N) confirmaron el análisis primario, con una tasa de éxito del tratamientoo del 100% en cada escenario.
Todos los episodios de sangrado tratados con VONVENDI y ADVATE o VONVENDI solo se controlaron con una evaluación de eficacia de excelente (96,9%) o buena (3,1%). El control de los episodios de sangrado fue consistente en todos los grados de severidad.
Para una visión general de la eficacia hemostática por gravedad del sangrado y el número de infusiones requeridas para tratar un episodio de sangrado, consulte Tabla 12.
La dosis acumulada media de VONVENDI administrada por episodio de sangrado (con o sin ADVATE) fue de 48,2 UI/kg (90% CI, 43,9 a 50,2) IU/kg. En relación con la gravedad del sangrado, la dosis acumulada media para tratar un episodio de sangrado fue de 43,3 (rango, 25,2 a 158,2) UI/kg para episodios de sangrado leve (n = 122), de 52,7 (rango, 23,8 a 184,9) UI/kg para episodios de sangrado moderado (n = 61), de 100 (rango, 57,5 a 135) UI/kg para episodios de sangrado mayor (n = 7).
Tabla 13 resume los datos obtenidos para el número de infusiones y la evaluación de eficacia por episodio de sangrado por ubicación.
Gestión perioperatoria de las hemorragias
La eficacia hemostática de VONVENDI se evaluó en un ensayo prospectivo, abierto y multicéntrico para evaluar la eficacia y la seguridad de VONVENDI con o sin ADVATE en procedimientos quirúrgicos electivos en adultos (de 18 años o más) diagnosticados con VWD grave y los sujetos se siguieron durante 14 días después de la cirugía.
Un total de 15 sujetos con VWD completaron el ensayo y el 93% de los sujetos tenían menos de 65 años (rango, de 20 a 70 años), de los cuales el 53,3% eran mujeres y el 53% (8/15) eran pacientes con VWD tipo 3. De los 15 sujetos, 10 sujetos se sometieron a cirugías mayores y cinco sujetos se sometieron a cirugías menores.
Las cirugías mayores incluyeron cirugías ortopédicas: reemplazo total de cadera, reemplazo total de rodilla, endoprótesis de rodilla, prótesis de tobillo, cirugía del ligamento cruzado anterior y meniscectomía. Otras cirugías mayores incluyeron colecistectomía laparoscópica, cistectomía laparoscópica y extracciones dentales complejas. Las cirugías menores/procedimientos incluyeron nasofaringoscopia, extracciones dentales, colonoscopia y sinovectomía con radioisótopos.
A todos los sujetos se les administró una dosis preoperatoria de 12 a 24 horas de 40 a 60 UI/kg de VONVENDI para aumentar los niveles de factor VIII a los niveles objetivo. Dentro de las 3 horas anteriores a la cirugía, se evaluaron los niveles de FVIII:C de los sujetos para asegurar que se alcanzara el objetivo de 30 UI/dL para las cirugías menores y 60 UI/dL para las cirugías mayores. Dentro de la 1 hora anterior a la cirugía, los sujetos recibieron una dosis de VONVENDI. ADVATE (factor VIII recombinante) se administró en función de los niveles de FVIII:C realizados 3 horas antes de la cirugía. La recuperación incremental de VWF y factor VIII se utilizó para guiar las dosis iniciales y posteriores.
Seis de los 10 sujetos que se sometían a cirugía mayor recibieron la dosis de carga especificada en el protocolo. Cabe señalar que la dosis de carga especificada en el protocolo se basó en los niveles de VWF:RCo evaluados antes de la dosis preoperatoria de 12 a 24 horas. Cuatro de los 10 sujetos que se sometían a cirugía mayor y cuatro de los cinco sujetos que se sometían a cirugía menor recibieron una dosis de carga de VONVENDI en función de VWF:RCo evaluada antes de la dosis de carga y después de la administración de la dosis preoperatoria de 12 a 24 horas. A diferencia de la dosis de carga especificada en el protocolo basada en los niveles evaluados antes de la dosis preoperatoria entre 12 y 24 horas de la cirugía, las dosis de carga en estos ocho sujetos se calcularon en función de los niveles de VWF:RCo después de la dosis preoperatoria y, por lo tanto, fueron dosis más bajas que la dosis de carga especificada en el protocolo. No se observaron diferencias en la seguridad o la eficacia entre los dos grupos.
La medida de resultado primaria fue la eficacia hemostática global evaluada 24 horas después de la última infusión perioperatoria de VONVENDI o al finalizar la visita del estudio, lo que ocurriera primero, utilizando una escala ordinal de eficacia de 4 puntos descrita en Tabla 14 (“excelente”, “buena”, “moderada” y “ninguna”) basada en la pérdida de sangre esperada estimada frente a la real, los requisitos de transfusión y la hemorragia y el goteo postoperatorios. Se requería una calificación de excelente o buena para declarar el resultado un éxito.
La eficacia hemostática global para cirugías mayores y menores fue del 100% (15/15) con un intervalo de confianza del 90% de 81,9% a 100%. Fue excelente en el 60% de las cirugías y buena en el 40% de las cirugías.
La eficacia hemostática intraoperatoria fue un punto final secundario. Para cirugías mayores y menores, fue del 100% con un intervalo de confianza del 90% de 81,9% a 100%. Fue excelente en el 73,3% de las cirugías y buena en el 26,7% de las cirugías.
Para más detalles sobre la eficacia hemostática para cirugías menores y mayores, consulte Tabla 15.
La dosificación se individualizó según los resultados de recuperación incremental realizados antes de la cirugía.
La dosis media total preoperatoria de 12 a 24 fue de 50,9 UI/kg (mediana 55,0 UI/kg; rango, 36,1 a 59,9 UI/kg).
La dosis media total de carga (dosis preoperatoria de 1 hora) por infusión fue de 38,6 UI/kg (mediana 35,8 UI/kg; rango, 8,0 a 82,7 UI/kg). Las cirugías mayores requirieron una dosis media de carga de 42,8 UI/kg (mediana 37,6 UI/kg; rango, 15,7 a 82,7 UI/kg) en comparación con una dosis media de carga de 30,2 UI/kg (mediana 34,2 UI/kg; rango, 8,0 a 46,4 UI/kg) para las cirugías menores.
Para los sujetos tratados con VONVENDI (con o sin ADVATE), la dosis media total postoperatoria dentro de los primeros 7 días después de la cirugía fue de 114,2 UI/kg con un rango de 23,8 a 318,9 UI/kg (n = 13) y 76,2 UI/kg con un rango de 23,8 a 214,4 UI/kg para los siguientes 7 días postoperatorios (n = 8).
Profilaxis rutinaria para reducir la frecuencia de episodios de sangrado en pacientes con VWD Tipo 3 grave que reciben terapia bajo demanda
VONVENDI se estudió en un estudio prospectivo, de un solo brazo, abierto, multicéntrico internacional para evaluar la eficacia, seguridad, PK y farmacodinámica (PD) del tratamiento profiláctico en la reducción de la frecuencia de episodios de sangrado en sujetos adultos (de 18 años o más) diagnosticados con VWD.
Los sujetos se inscribieron en dos grupos de tratamiento en función del tratamiento que recibieron para el manejo del sangrado antes de la entrada al estudio, consistiendo en un grupo Prior OD en el que los sujetos recibieron solo tratamiento bajo demanda (OD) y el grupo Switch en el que los sujetos habían recibido tratamiento profiláctico con VWF derivado del plasma (pdVWF). La eficacia se evaluó en función de la tasa media anualizada de sangrado para todos los sangrados, sangrados espontáneos y sangrados articulares y se basó en estadísticas descriptivas.
Veintidós sujetos evaluables de eficacia recibieron una mediana de 92,5 dosis, 12 de estos sujetos habían recibido tratamiento bajo demanda anteriormente (grupo Prior OD) y 10 sujetos habían recibido tratamiento profiláctico con pdVWF (grupo Switch) antes de inscribirse en este estudio. Nueve sujetos en el grupo OD y 8 sujetos en el grupo Switch completaron el estudio, que requirió 12 meses de tratamiento.
La edad media de los sujetos en el estudio fue de 34 años (18 – 77 años), el 54,5% eran varones y el 96% de los sujetos eran blancos y el 86,4% no eran hispanos o latinos. De los 12 sujetos en el grupo Prior OD, uno tenía Tipo 1 severo, uno tenía Tipo 2 severo de VWD y diez tenían Tipo 3 severo de VWD. De los 10 sujetos en el grupo Switch, uno tenía Tipo 1 severo, uno tenía Tipo 2 severo de VWD y ocho tenían Tipo 3 severo de VWD.
Los sujetos en el grupo Prior OD comenzaron el tratamiento a 50 ± 10 UI/kg por infusión dos veces por semana. Un sujeto en el grupo Prior OD requirió ajustes de dosis para el manejo del sangrado repentino. La mayoría de los sujetos (9/10) con Tipo 3 de VWD en el grupo Prior OD recibieron dosis dos veces por semana con Vonvendi y tuvieron una dosis máxima mediana de 55,9 (UI/kg).
El estudio no demostró adecuadamente la eficacia de Vonvendi como tratamiento profiláctico para reducir los eventos de sangrado en sujetos con Tipo 3 de VWD que recibieron tratamiento profiláctico antes de la entrada al estudio (grupo Switch).
Los parámetros de eficacia de Vonvendi como tratamiento profiláctico en sujetos con Tipo 3 de VWD que recibieron tratamiento bajo demanda antes de la entrada al estudio se proporcionan en Tabla 16. El porcentaje medio de cambio de ABRs desde históricos a los del estudio fueron: para todos los sangrados -54,7%, para sangrados espontáneos -75,9% y para sangrados articulares -100,0%.