ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
Estos puntos destacados no incluyen toda la información necesaria para usar TRODELVY de forma segura y eficaz. Consulte la información completa de prescripción de TRODELVY
TRODELVY® (sacituzumab govitecan-hziy) para inyección, para uso intravenoso Aprobación inicial en EE. UU.: 2020
ADVERTENCIA: NEUTROPENIA Y DIARREA
Consulte la información completa de prescripción para ver la advertencia completa en recuadro.
Puede producirse neutropenia grave o potencialmente mortal. Suspenda TRODELVY si el recuento absoluto de neutrófilos es inferior a 1500/mm3 o si hay fiebre neutropénica. Controle periódicamente los recuentos sanguíneos durante el tratamiento. Considere el uso de G-CSF para profilaxis secundaria. Inicie el tratamiento antiinfeccioso en pacientes con neutropenia febril sin demora. (2.3, 5.1)
Puede producirse diarrea grave. Controle a los pacientes con diarrea y administre líquidos y electrolitos según sea necesario. Al inicio de la diarrea, evalúe las causas infecciosas y, si son negativas, inicie rápidamente loperamida. Si se produce diarrea grave, suspenda TRODELVY hasta que se resuelva a ≤ Grado 1 y reduzca las dosis posteriores. (2.3, 5.2)
CAMBIOS IMPORTANTES RECIENTES
Indicaciones y uso, cáncer urotelial localmente avanzado o metastásico – Aprobación acelerada (texto eliminado) (1.2)
11/2024
INDICACIONES Y USO
TRODELVY es un conjugado de anticuerpo dirigido a Trop-2 e inhibidor de la topoisomerasa indicado para el tratamiento de pacientes adultos con:
Cáncer de mama localmente avanzado o metastásico
Cáncer de mama triple negativo (mTNBC) localmente avanzado o metastásico irresecable que han recibido dos o más tratamientos sistémicos previos, al menos uno de ellos para la enfermedad metastásica. (1.1, 14.1)
Cáncer de mama localmente avanzado o metastásico irresecable con receptor hormonal (RH)-positivo y receptor 2 del factor de crecimiento epidérmico humano (HER2)-negativo (IHC 0, IHC 1+ o IHC 2+/ISH–) que han recibido terapia basada en hormonas y al menos dos tratamientos sistémicos adicionales en el contexto metastásico. (1.1, 14.2)
POSOLOGÍA Y ADMINISTRACIÓN
NO sustituya TRODELVY ni lo use con otros medicamentos que contengan irinotecán o su metabolito activo SN-38. (2.1)
Solo para infusión intravenosa. No administre como inyección intravenosa rápida o bolo.
La dosis recomendada es de 10 mg/kg una vez por semana los días 1 y 8 de ciclos de tratamiento continuos de 21 días hasta la progresión de la enfermedad o la toxicidad inaceptable. (2.2)
Se recomienda la premedicación para la prevención de reacciones a la infusión y la prevención de náuseas y vómitos inducidos por quimioterapia. (2.2)
Controle a los pacientes durante la infusión y durante al menos 30 minutos después de la finalización de la infusión. Puede ser necesaria la interrupción del tratamiento y/o la reducción de la dosis para controlar las reacciones adversas. (2.2)
Consulte la información completa de prescripción para obtener instrucciones de preparación y administración. (2.4)
FORMAS Y CONCENTRACIONES FARMACÉUTICAS
Para inyección: 180 mg de polvo liofilizado en viales unidosis para reconstitución. (3)
CONTRAINDICACIONES
Reacción de hipersensibilidad grave a TRODELVY. (4, 5.3)
ADVERTENCIAS Y PRECAUCIONES
Hipersensibilidad y reacciones relacionadas con la infusión: Se han observado reacciones de hipersensibilidad, incluidas reacciones anafilácticas graves. Controle a los pacientes para detectar reacciones relacionadas con la infusión. Suspenda permanentemente TRODELVY si se producen reacciones graves o potencialmente mortales. (5.3)
Náuseas/Vómitos: Utilice un tratamiento preventivo antiemético y suspenda TRODELVY en pacientes con náuseas de Grado 3 o vómitos de Grado 3-4 en el momento del tratamiento programado. (5.4)
Pacientes con actividad reducida de UGT1A1: Las personas homocigóticas para el alelo uridina difosfato-glucuronosil transferasa 1A1 (UGT1A1)*28 tienen un mayor riesgo de neutropenia, neutropenia febril y anemia después del inicio del tratamiento con TRODELVY. (5.5)
Toxicidad embriofetal: TRODELVY puede causar daño fetal. Avise a las pacientes del riesgo potencial para un feto y del uso de métodos anticonceptivos eficaces. (5.6, 8.1, 8.3)
REACCIONES ADVERSAS
Las reacciones adversas más comunes (incidencia ≥25%) son (incluidas las alteraciones de laboratorio): disminución del recuento de leucocitos, disminución del recuento de neutrófilos, disminución de la hemoglobina, diarrea, náuseas, disminución del recuento de linfocitos, fatiga, alopecia, estreñimiento, aumento de la glucosa, disminución de la albúmina, vómitos, disminución del apetito, disminución del aclaramiento de creatinina, aumento de la fosfatasa alcalina, disminución del magnesio, disminución del potasio y disminución del sodio. (6.1)
Para notificar SOSPECHAS DE REACCIONES ADVERSAS, póngase en contacto con Gilead Sciences, Inc. al 1-888-983-4668 o con la FDA al 1-800-FDA-1088 o www.fda.gov/medwatch.
INTERACCIONES FARMACOLÓGICAS
Inhibidores o inductores de UGT1A1: Evite el uso concomitante. (7)
Las secciones o subsecciones omitidas de la información completa de prescripción no se enumeran.
ADVERTENCIA EN EL RECUADRO
ADVERTENCIA: NEUTROPENIA Y DIARREA
Puede producirse neutropenia grave o potencialmente mortal. Suspenda TRODELVY si el recuento absoluto de neutrófilos es inferior a 1500/mm3 o si hay fiebre neutropénica. Controle periódicamente los recuentos sanguíneos durante el tratamiento. Considere el uso de G-CSF para profilaxis secundaria [véase Posología y administración (2.3)]. Inicie el tratamiento antiinfeccioso en pacientes con neutropenia febril sin demora [véase Advertencias y precauciones (5.1)].
Puede producirse diarrea grave. Controle a los pacientes con diarrea y administre líquidos y electrolitos según sea necesario. Al inicio de la diarrea, evalúe las causas infecciosas y, si son negativas, inicie rápidamente loperamida [véase Advertencias y precauciones (5.2)]. Si se produce diarrea grave, suspenda TRODELVY hasta que se resuelva a ≤ Grado 1 y reduzca las dosis posteriores [véase Posología y administración (2.3)].
1 INDICACIONES Y USO
1.1 Cáncer de mama localmente avanzado o metastásico
TRODELVY está indicado para el tratamiento de pacientes adultas con cáncer de mama triple negativo (mTNBC) localmente avanzado irresecable o metastásico que han recibido dos o más terapias sistémicas previas, al menos una de ellas para la enfermedad metastásica.
TRODELVY está indicado para el tratamiento de pacientes adultas con cáncer de mama localmente avanzado irresecable o metastásico con receptor hormonal (HR) positivo y receptor 2 del factor de crecimiento epidérmico humano (HER2) negativo (IHC 0, IHC 1+ o IHC 2+/ISH–) que han recibido terapia endocrina y al menos dos terapias sistémicas adicionales en el contexto metastásico.
2 DOSIS Y ADMINISTRACIÓN
2.1 Información Importante sobre el Uso
NO sustituya TRODELVY ni lo use con otros medicamentos que contengan irinotecán o su metabolito activo SN-38.
2.2 Dosis Recomendada
La dosis recomendada de TRODELVY es de 10 mg/kg administrada como infusión intravenosa una vez por semana los días 1 y 8 de ciclos de tratamiento de 21 días. Continúe el tratamiento hasta la progresión de la enfermedad o la toxicidad inaceptable. No administre TRODELVY a dosis superiores a 10 mg/kg.
Administre TRODELVY únicamente como infusión intravenosa. No administre como inyección intravenosa rápida o bolo.
Primera infusión: Administre la infusión durante 3 horas. Observe a los pacientes durante la infusión y durante al menos 30 minutos después de la dosis inicial, para detectar signos o síntomas de reacciones relacionadas con la infusión [véase Advertencias y precauciones (5.3)].
Infusiones posteriores: Administre la infusión durante 1 a 2 horas si las infusiones anteriores se toleraron. Observe a los pacientes durante la infusión y durante al menos 30 minutos después de la infusión.
Premedicación
Antes de cada dosis de TRODELVY, se recomienda la premedicación para la prevención de reacciones a la infusión y la prevención de náuseas y vómitos inducidos por quimioterapia (CINV).
Premedique con antipiréticos, bloqueadores H1 y H2 antes de la infusión, y se pueden usar corticosteroides para pacientes que tuvieron reacciones previas a la infusión.
Premedique con un régimen de combinación de dos o tres fármacos (por ejemplo, dexametasona con un antagonista del receptor 5-HT3 o un antagonista del receptor NK1, así como otros fármacos según esté indicado).
2.3 Modificaciones de la Dosis para las Reacciones Adversas
Reacciones relacionadas con la infusión
Ralentice o interrumpa la velocidad de infusión de TRODELVY si el paciente desarrolla una reacción relacionada con la infusión. Suspenda permanentemente TRODELVY para reacciones relacionadas con la infusión que pongan en peligro la vida [véase Advertencias y precauciones (5.3)]
Modificaciones de la dosis para reacciones adversas
Suspenda o interrumpa TRODELVY para controlar las reacciones adversas como se describe en la Tabla 1. No vuelva a aumentar la dosis de TRODELVY después de que se haya realizado una reducción de la dosis por reacciones adversas.
Tabla 1: Modificaciones de la dosis para reacciones adversas
Neutropenia de grado 4 ≥ 7 días O Neutropenia febril de grado 3–4, O En el momento del tratamiento programado, neutropenia de grado 3–4 que retrasa la dosificación durante 2 o 3 semanas para la recuperación a ≤ Grado 1
Primera
Reducción de la dosis del 25% y administrar factor estimulante de colonias de granulocitos (G-CSF).
Segunda
Reducción de la dosis del 50% y administrar G-CSF.
Tercera
Interrumpir el tratamiento y administrar G-CSF.
En el momento del tratamiento programado, neutropenia de grado 3–4 que retrasa la dosificación más de 3 semanas para la recuperación a ≤ Grado 1
Primera
Interrumpir el tratamiento y administrar G-CSF.
Toxicidad grave no neutropénica
Toxicidad no hematológica de grado 4 de cualquier duración, O Cualquier náusea, vómito o diarrea de grado 3–4 debido al tratamiento que no se controla con antieméticos y agentes antidiarreicos [véase Advertencias y precauciones (5.2), 5.4)], O Otra toxicidad no hematológica de grado 3–4 que persiste > 48 horas a pesar del manejo médico óptimo, O En el momento del tratamiento programado, toxicidad hematológica o no hematológica de grado 3–4 no neutropénica, que retrasa la dosis durante 2 o 3 semanas para la recuperación a ≤ Grado 1
Primera
Reducción de la dosis del 25%
Segunda
Reducción de la dosis del 50%
Tercera
Interrumpir el tratamiento
En caso de toxicidad hematológica o no hematológica de grado 3–4 no neutropénica, que no se recupera a ≤ Grado 1 dentro de las 3 semanas
Primera
Interrumpir el tratamiento
2.4 Preparación y administración
Reconstitución
TRODELVY es un fármaco peligroso.
Siga los procedimientos de manipulación y eliminación especiales aplicables1.
Calcule la dosis requerida (mg) de TRODELVY en función del peso corporal del paciente al comienzo de cada ciclo de tratamiento (o con más frecuencia si el peso corporal del paciente ha cambiado en más del 10 % desde la administración anterior) [véase Posología y administración (2.2)].
Deje que el número requerido de viales se caliente a temperatura ambiente.
Utilizando una jeringa estéril, inyecte lentamente 20 mL de inyección de cloruro de sodio al 0,9 %, USP, en cada vial de 180 mg de TRODELVY. Cada vial contiene un exceso para compensar la pérdida de líquido durante la preparación y después de la reconstitución; el volumen total resultante proporciona una concentración de 10 mg/mL.
Gire suavemente los viales y deje que se disuelvan hasta durante 15 minutos. No agite. Los productos farmacéuticos parenterales deben inspeccionarse visualmente para detectar partículas y decoloración antes de la administración, siempre que la solución y el recipiente lo permitan. La solución debe estar libre de partículas visibles, transparente y amarilla. No utilice la solución reconstituida si está turbia o decolorada.
Úselo inmediatamente para preparar una solución de infusión de TRODELVY diluida.
Dilución
Calcule la cantidad requerida de la solución reconstituida de TRODELVY necesaria para obtener la dosis adecuada según el peso corporal del paciente.
Determine el volumen final de la solución de infusión para administrar la dosis adecuada en un rango de concentración de TRODELVY de 1,1 mg/mL a 3,4 mg/mL.
Utilice únicamente inyección de cloruro de sodio al 0,9 %, USP, ya que la estabilidad de la solución reconstituida de TRODELVY no se ha determinado con otras soluciones basadas en infusión. Utilice una bolsa de infusión de cloruro de polivinilo, polipropileno/polietileno, poliolefina o etilvinilacetato.
Extraiga y deseche el volumen de inyección de cloruro de sodio al 0,9 %, USP, de la bolsa de infusión final que sea necesario para lograr la concentración de TRODELVY indicada después de añadir la cantidad calculada de solución reconstituida de TRODELVY.
Extraiga la cantidad calculada de la solución reconstituida de TRODELVY de los viales utilizando una jeringa. Deseche cualquier porción no utilizada que quede en los viales.
Para minimizar la formación de espuma, inyecte lentamente la cantidad calculada de solución reconstituida de TRODELVY en la bolsa de infusión. No agite el contenido.
Si no se utiliza inmediatamente, la bolsa de infusión que contiene la solución de TRODELVY puede almacenarse refrigerada a 2 °C a 8 °C (36 °F a 46 °F) hasta 24 horas, protegida de la luz. Después de la refrigeración, administre la solución diluida a temperatura ambiente hasta 25 °C (77 °F) en un plazo de 8 horas (incluido el tiempo de infusión).
No congelar ni agitar.
Administración
Administre TRODELVY como infusión intravenosa. Proteja la bolsa de infusión de la luz. La bolsa de infusión debe cubrirse durante la administración al paciente hasta que se complete la dosificación. No es necesario cubrir el tubo de infusión ni utilizar un tubo de protección contra la luz durante la infusión.
Se puede utilizar una bomba de infusión.
No mezcle TRODELVY ni lo administre como infusión con otros productos medicinales.
Una vez finalizada la infusión, enjuague la vía intravenosa con 20 mL de inyección de cloruro de sodio al 0,9 %, USP.
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
Para inyección: 180 mg de polvo liofilizado de color blanquecino a amarillento en un vial de dosis única.
4 CONTRAINDICACIONES
TRODELVY está contraindicado en pacientes que han experimentado una reacción de hipersensibilidad grave a TRODELVY [ver Advertencias y precauciones (5.3)].
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Neutropenia
Puede producirse neutropenia grave, potencialmente mortal o mortal en pacientes tratados con TRODELVY. La neutropenia se produjo en el 64 % de los pacientes tratados con TRODELVY. La neutropenia de grado 3-4 se produjo en el 49 % de los pacientes. La neutropenia febril se produjo en el 6 % de los pacientes. El tiempo medio hasta la aparición de la primera neutropenia (incluida la neutropenia febril) fue de 16 días y se ha producido antes en algunas poblaciones de pacientes [véase Advertencias y precauciones (5.5)]. La colitis neutropénica se produjo en el 1,4 % de los pacientes.
Suspenda la administración de TRODELVY si el recuento absoluto de neutrófilos es inferior a 1500/mm3 el día 1 de cualquier ciclo o si el recuento de neutrófilos es inferior a 1000/mm3 el día 8 de cualquier ciclo. Suspenda la administración de TRODELVY en caso de fiebre neutropénica. Es posible que se requieran modificaciones de la dosis debido a la neutropenia. Administre G-CSF según esté clínicamente indicado o según se indica en la Tabla 1 [véase Posología y administración (2.3)].
5.2 Diarrea
TRODELVY puede causar diarrea grave. La diarrea se produjo en el 64 % de todos los pacientes tratados con TRODELVY. La diarrea de grado 3-4 se produjo en el 11 % de todos los pacientes tratados con TRODELVY. Un paciente presentó perforación intestinal tras la diarrea. La diarrea que provocó deshidratación y posterior lesión renal aguda se produjo en el 0,7 % de todos los pacientes.
Suspenda la administración de TRODELVY en caso de diarrea de grado 3-4 en el momento de la administración del tratamiento programado y reanude la administración cuando se haya resuelto hasta ≤grado 1 [véase Posología y administración (2.3)].
Al inicio de la diarrea, evalúe las causas infecciosas y, si son negativas, inicie inmediatamente la administración de loperamida, 4 mg inicialmente seguidos de 2 mg con cada episodio de diarrea hasta un máximo de 16 mg diarios. Suspenda la administración de loperamida 12 horas después de que desaparezca la diarrea. También se pueden emplear otras medidas de apoyo (p. ej., sustitución de líquidos y electrolitos) según esté clínicamente indicado.
Los pacientes que presenten una respuesta colinérgica excesiva al tratamiento con TRODELVY (p. ej., calambres abdominales, diarrea, salivación, etc.) pueden recibir premedicación adecuada (p. ej., atropina) para tratamientos posteriores.
5.3 Hipersensibilidad y reacciones relacionadas con la infusión
Se han producido reacciones de hipersensibilidad graves, incluidas reacciones anafilácticas potencialmente mortales, con el tratamiento con TRODELVY. Los signos y síntomas graves incluyeron paro cardíaco, hipotensión, sibilancias, angioedema, hinchazón, neumonitis y reacciones cutáneas [véase Contraindicaciones (4)].
Las reacciones de hipersensibilidad en las 24 horas posteriores a la administración se produjeron en el 35 % de los pacientes tratados con TRODELVY. La hipersensibilidad de grado 3-4 se produjo en el 2 % de los pacientes tratados con TRODELVY. La incidencia de reacciones de hipersensibilidad que llevaron a la interrupción permanente de TRODELVY fue del 0,2 %. La incidencia de reacciones anafilácticas fue del 0,2 %.
Se recomienda la premedicación para las reacciones a la infusión en pacientes que reciben TRODELVY. Tenga a mano los medicamentos y el equipo de emergencia para tratar las reacciones relacionadas con la infusión, incluida la anafilaxia, para su uso inmediato al administrar TRODELVY [véase Posología y administración (2.2)].
Vigile estrechamente a los pacientes para detectar reacciones de hipersensibilidad y reacciones relacionadas con la infusión durante cada infusión de TRODELVY y durante al menos 30 minutos después de la finalización de cada infusión [véase Posología y administración (2.3)].
Interrompa permanentemente la administración de TRODELVY en caso de reacciones relacionadas con la infusión de grado 4 [véase Posología y administración (2.3)].
5.4 Náuseas y vómitos
TRODELVY es emético. Las náuseas se produjeron en el 64 % de todos los pacientes tratados con TRODELVY. Las náuseas de grado 3-4 se produjeron en el 3 % de los pacientes.
Los vómitos se produjeron en el 35 % de todos los pacientes tratados con TRODELVY. Los vómitos de grado 3-4 se produjeron en el 2 % de estos pacientes.
Administre premedicación con un régimen de combinación de dos o tres fármacos (p. ej., dexametasona con un antagonista del receptor 5-HT3 o un antagonista del receptor NK1, así como otros fármacos según esté indicado) para la prevención de las náuseas y los vómitos inducidos por quimioterapia (CINV) [véase Posología y administración (2.2)].
Suspenda las dosis de TRODELVY en caso de náuseas de grado 3 o vómitos de grado 3-4 en el momento de la administración del tratamiento programado y reanude la administración con medidas de apoyo adicionales cuando se haya resuelto hasta ≤grado 1 [véase Posología y administración (2.3)].
También se pueden emplear antieméticos adicionales y otras medidas de apoyo según esté clínicamente indicado. A todos los pacientes se les deben administrar medicamentos para llevar a casa con instrucciones claras para la prevención y el tratamiento de las náuseas y los vómitos.
5.5 Mayor riesgo de reacciones adversas en pacientes con actividad reducida de UGT1A1
Los pacientes homocigotos para el alelo uridina difosfato-glucuronosil transferasa 1A1 (UGT1A1)*28 presentan un mayor riesgo de neutropenia, neutropenia febril y anemia; y pueden presentar un mayor riesgo de otras reacciones adversas cuando se les trata con TRODELVY.
Se analizó la incidencia de neutropenia y anemia en 948 pacientes que recibieron TRODELVY y tenían resultados de genotipo UGT1A1. En los pacientes homocigotos para el alelo UGT1A1 *28 (n=112), la incidencia de neutropenia de grado 3-4 fue del 58 %. En los pacientes heterocigotos para el alelo UGT1A1*28 (n=420), la incidencia de neutropenia de grado 3-4 fue del 49 %. En los pacientes homocigotos para el alelo de tipo salvaje (n=416), la incidencia de neutropenia de grado 3-4 fue del 43 % [véase Farmacología clínica (12.5)]. En los pacientes homocigotos para el alelo UGT1A1 *28, la incidencia de anemia de grado 3-4 fue del 21 %. En los pacientes heterocigotos para el alelo UGT1A1*28, la incidencia de anemia de grado 3-4 fue del 10 %. En los pacientes homocigotos para el alelo de tipo salvaje, la incidencia de anemia de grado 3-4 fue del 9 %.
El tiempo medio hasta la primera neutropenia, incluida la neutropenia febril, fue de 9 días en pacientes homocigotos para el alelo UGT1A1*28, 15 días en pacientes heterocigotos para el alelo UGT1A1*28 y 20 días en pacientes homocigotos para el alelo de tipo salvaje. El tiempo medio hasta la primera anemia fue de 21 días en pacientes homocigotos para el alelo UGT1A1*28, 25 días en pacientes heterocigotos para el alelo UGT1A1*28 y 28 días en pacientes homocigotos para el alelo de tipo salvaje.
Monitorice estrechamente a los pacientes con actividad reducida conocida de UGT1A1 para detectar reacciones adversas. Suspenda o interrumpa permanentemente TRODELVY en función de la evaluación clínica del inicio, la duración y la gravedad de las reacciones adversas observadas en pacientes con evidencia de reacciones adversas agudas de inicio temprano o inusualmente graves, que pueden indicar una actividad enzimática reducida de UGT1A1 [ver Posología y administración (2.3)].
5.6 Toxicidad embriofetal
Basándose en su mecanismo de acción, TRODELVY puede causar teratogenicidad y/o letalidad embriofetal cuando se administra a una mujer embarazada. TRODELVY contiene un componente genotóxico, SN-38, y se dirige a células que se dividen rápidamente [ver Farmacología clínica (12.1) y Toxicología no clínica (13.1)]. Avise a las mujeres embarazadas y a las mujeres en edad fértil del riesgo potencial para el feto. Avise a las mujeres en edad fértil que utilicen un método anticonceptivo eficaz durante el tratamiento con TRODELVY y durante 6 meses después de la última dosis. Avise a los pacientes varones con parejas femeninas en edad fértil que utilicen un método anticonceptivo eficaz durante el tratamiento con TRODELVY y durante 3 meses después de la última dosis [ver Uso en poblaciones específicas (8.1, 8.3)].
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas se describen con mayor detalle en otras secciones de la etiqueta:
Debido a que los ensayos clínicos se realizan en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no se pueden comparar directamente con las tasas en los ensayos clínicos de otro fármaco y pueden no reflejar las tasas observadas en la práctica clínica.
La población de seguridad agrupada que se describe en la sección de Advertencias y precauciones refleja la exposición a TRODELVY en 1063 pacientes, que incluyó 366 pacientes con mTNBC y 322 pacientes con cáncer de mama con receptor hormonal positivo (HR+)/receptor 2 del factor de crecimiento epidérmico humano negativo (HER2-) de IMMU-132-01, ASCENT y TROPiCS-02; y 375 pacientes con otros tipos de tumores. TRODELVY se administró como una infusión intravenosa una vez por semana los días 1 y 8 de ciclos de tratamiento de 21 días a dosis de 10 mg/kg hasta la progresión de la enfermedad o la toxicidad inaceptable. Entre los 1063 pacientes tratados con TRODELVY, la duración media del tratamiento fue de 4,1 meses (rango: 0 a 63 meses). En esta población de seguridad agrupada, las reacciones adversas más comunes (≥ 25%), incluidas las anormalidades de laboratorio, fueron disminución del recuento de leucocitos (84%), disminución del recuento de neutrófilos (75%), disminución de la hemoglobina (69%), diarrea (64%), náuseas (64%), disminución del recuento de linfocitos (63%), fatiga (51%), alopecia (45%), estreñimiento (37%), aumento de glucosa (37%), disminución de albúmina (35%), vómitos (35%), disminución del apetito (30%), disminución del aclaramiento de creatinina (28%), aumento de fosfatasa alcalina (28%), disminución de magnesio (27%), disminución de potasio (26%) y disminución de sodio (26%).
Cáncer de mama triple negativo localmente avanzado o metastásico
Estudio ASCENT
La seguridad de TRODELVY se evaluó en un estudio aleatorizado, controlado activo, abierto (ASCENT) en pacientes con mTNBC que habían recibido previamente un taxano y al menos dos quimioterapias previas. Los pacientes se asignaron al azar (1:1) para recibir TRODELVY (n=258) o quimioterapia en monoterapia (n=224) y se trataron hasta la progresión de la enfermedad o la toxicidad inaceptable [ver Estudios clínicos (14.1)]. Para los pacientes tratados con TRODELVY, la duración media del tratamiento fue de 4,4 meses (rango: 0 a 23 meses).
Se produjeron reacciones adversas graves en el 27% de los pacientes que recibieron TRODELVY. Las reacciones adversas graves en > 1% de los pacientes que recibieron TRODELVY incluyeron neutropenia (7%), diarrea (4%) y neumonía (3%). Se produjeron reacciones adversas mortales en el 1,2% de los pacientes que recibieron TRODELVY, incluida la insuficiencia respiratoria (0,8%) y la neumonía (0,4%). TRODELVY se suspendió permanentemente por reacciones adversas en el 5% de los pacientes. Las reacciones adversas que llevaron a la interrupción permanente en ≥ 1% de los pacientes que recibieron TRODELVY fueron neumonía (1%) y fatiga (1%).
Las reacciones adversas que llevaron a una interrupción del tratamiento con TRODELVY se produjeron en el 63% de los pacientes. Las reacciones adversas más frecuentes (≥5%) que llevaron a una interrupción del tratamiento fueron neutropenia (47%), diarrea (5%), infección respiratoria (5%) y leucopenia (5%).
Las reacciones adversas que llevaron a una reducción de la dosis de TRODELVY se produjeron en el 22% de los pacientes. Las reacciones adversas más frecuentes (>4%) que llevaron a una reducción de la dosis fueron neutropenia (11%) y diarrea (5%).
Se utilizó factor estimulante de colonias de granulocitos (G-CSF) en el 44% de los pacientes que recibieron TRODELVY.
Los cuadros 2 y 3 resumen las reacciones adversas y las anormalidades de laboratorio, respectivamente, en el estudio ASCENT.
Tabla 2: Reacciones adversas en ≥ 10% de pacientes con mTNBC en ASCENT
TRODELVY (n=258)
Quimioterapia en monoterapia (n=224)
Reacción adversa
Todos los grados %
Grado 3 – 4 %
Todos los grados %
Grado 3 – 4 %
*La quimioterapia en monoterapia incluyó uno de los siguientes agentes en monoterapia: eribulina (n=139), capecitabina (n=33), gemcitabina (n=38) o vinorelbina (excepto si el paciente tenía ≥ neuropatía de grado 2, n=52).
Trastornos musculoesqueléticos y del tejido conjuntivo
Dolor de espalda
16
1
14
2
Artralgia
12
0.4
7
0
Trastornos del sistema nervioso
Cefalea
18
0.8
13
0.4
Mareo
10
0
7
0
Trastornos psiquiátricos
Insomnio
11
0
5
0
Trastornos respiratorios, torácicos y mediastínicos
Tos
24
0
18
0.4
Trastornos de la piel y del tejido subcutáneo
Alopecia
47
0
16
0
Erupción
12
0.4
5
0.4
Prurito
10
0
3
0
Tabla 3: Anormalidades de laboratorio en > 10% de pacientes con mTNBC en ASCENT
Anormalidad de laboratorio
TRODELVY (n=258)
Quimioterapia en monoterapia (n=224)
Todos los grados (%)
Grado 3 – 4 (%)
Todos los grados (%)
Grado 3 – 4 (%)
Hematología
Disminución de hemoglobina
94
9
57
6
Disminución del recuento de linfocitos
88
31
40
24
Disminución del recuento de leucocitos
86
41
53
25
Disminución del recuento de neutrófilos
78
49
48
36
Disminución del recuento de plaquetas
23
1.2
25
2.7
Bioquímica
Aumento de glucosa
49
2.3
43
2.8
Disminución de calcio
36
1.6
21
1.4
Disminución de magnesio
33
0.4
20
0
Disminución de potasio
33
4.3
28
0.9
Aumento de albúmina
32
0.8
25
1.4
Aumento de aspartato aminotransferasa
27
1.2
32
1.4
Aumento de alanina aminotransferasa
26
1.2
26
1.8
Aumento de fosfatasa alcalina
26
0
17
0.5
Disminución de fosfato
26
7.8
20
3.3
Disminución de sodio
22
0.4
17
0.5
Aumento de lactato deshidrogenasa
18
0
22
0
Disminución de glucosa
10
0
3.2
0
Estudio IMMU-132-01
La seguridad de TRODELVY se evaluó en un estudio de un solo brazo, abierto (IMMU-132-01) en pacientes con mTNBC y otras neoplasias malignas, que incluyó a 108 pacientes con mTNBC que habían recibido al menos dos terapias anticancerosas previas para la enfermedad metastásica [ver Estudios clínicos (14.1)]. TRODELVY se administró como una infusión intravenosa una vez por semana los días 1 y 8 de ciclos de tratamiento de 21 días a dosis de hasta 10 mg/kg hasta la progresión de la enfermedad o toxicidad inaceptable. La duración media del tratamiento en estos 108 pacientes fue de 5,1 meses (rango: 0 a 51 meses).
Se produjeron reacciones adversas graves en el 31% de los pacientes. Las reacciones adversas graves en > 1% de los pacientes que recibieron TRODELVY incluyeron neutropenia febril (6%), vómitos (5%), náuseas (3%), disnea (3%), diarrea (4%), anemia (2%), derrame pleural, neutropenia, neumonía, deshidratación (cada una 2%).
TRODELVY se suspendió permanentemente por reacciones adversas en el 2% de los pacientes. Las reacciones adversas que llevaron a la suspensión permanente fueron anafilaxia, anorexia/fatiga, cefalea (cada una 0,9%). El cuarenta y cinco por ciento (45%) de los pacientes experimentaron una reacción adversa que provocó la interrupción del tratamiento. La reacción adversa más común que provocó la interrupción del tratamiento fue la neutropenia (33%). Las reacciones adversas que llevaron a la reducción de la dosis se produjeron en el 33% de los pacientes tratados con TRODELVY, con un 24% que tuvo una reducción de la dosis y un 9% con dos reducciones de la dosis. La reacción adversa más común que provocó reducciones de la dosis fue la neutropenia/neutropenia febril.
Los cuadros 4 y 5 resumen las reacciones adversas y las anormalidades de laboratorio que se producen en ≥10% de los pacientes con mTNBC en el estudio IMMU-132-01.
Tabla 4: Reacciones adversas en ≥ 10% de los pacientes con mTNBC en IMMU-132-01
Incluyendo infección de las vías respiratorias superiores e inferiores, neumonía, gripe, infección viral de las vías respiratorias superiores, bronquitis e infección por virus respiratorio sincitial
Tabla 5: Anormalidades de laboratorio observadas en ≥ 10% de los pacientes mientras recibían TRODELVY en IMMU-132-01
Anormalidad de laboratorio
TRODELVY (n=108)
Todos los grados (%)
Grado 3–4 (%)
Hematología
Disminución de hemoglobina
93
6
Disminución del recuento de leucocitos
91
26
Disminución del recuento de neutrófilos
82
32
Aumento del tiempo de tromboplastina parcial activado
60
12
Disminución del recuento de plaquetas
30
3
Química
Aumento de fosfatasa alcalina
57
2
Disminución de magnesio
51
3
Disminución de calcio
49
3
Aumento de aspartato aminotransferasa
45
3
Disminución de albúmina
39
1
Aumento de alanina aminotransferasa
35
2
Aumento de glucosa
31
2.8
Disminución de fosfato
29
5
Disminución de magnesio
27
1.9
Disminución de fosfato
27
6.5
Disminución de sodio
25
4.7
Disminución de potasio
24
3.7
Disminución de glucosa
19
2
Cáncer de mama metastásico o localmente avanzado, HR-positivo, HER2-negativo
Estudio TROPiCS-02
La seguridad de TRODELVY se evaluó en un estudio aleatorizado, controlado activo, abierto (TROPiCS-02) en pacientes con cáncer de mama metastásico o localmente avanzado, HR-positivo, HER2-negativo cuya enfermedad había progresado después de lo siguiente en cualquier contexto: un inhibidor de CDK 4/6, terapia endocrina y un taxano; las pacientes recibieron al menos dos quimioterapias previas en el contexto metastásico (una de las cuales podría ser en el contexto neoadyuvante o adyuvante si la progresión se producía en un plazo de 12 meses). Las pacientes se asignaron aleatoriamente (1:1) para recibir TRODELVY (n=268) o quimioterapia en monoterapia (n=249) y se las trató hasta la progresión de la enfermedad o una toxicidad inaceptable [véase Estudios clínicos (14.2)]. En las pacientes tratadas con TRODELVY, la duración media del tratamiento fue de 4,1 meses (intervalo: 0 a 63 meses).
Se produjeron reacciones adversas graves en el 28 % de las pacientes que recibieron TRODELVY. Las reacciones adversas graves en >1 % de las pacientes que recibieron TRODELVY incluyeron diarrea (5 %), neutropenia febril (4 %), neutropenia (3 %), dolor abdominal, colitis, colitis neutropénica, neumonía y vómitos (cada una, 2 %). Se produjeron reacciones adversas mortales en el 2 % de las pacientes que recibieron TRODELVY, incluidas arritmia, COVID-19, trastorno del sistema nervioso, embolia pulmonar y shock séptico (cada una, 0,4 %). TRODELVY se interrumpió permanentemente debido a reacciones adversas en el 6 % de las pacientes. Las reacciones adversas más frecuentes (≥0,5 %) que llevaron a la interrupción permanente en las pacientes que recibieron TRODELVY fueron astenia, deterioro general del estado de salud físico y neutropenia (cada una, 0,7 %).
Las reacciones adversas que llevaron a una interrupción del tratamiento con TRODELVY se produjeron en el 66 % de las pacientes. La reacción adversa más frecuente (≥5 %) que llevó a la interrupción del tratamiento fue la neutropenia (50 %).
Las reacciones adversas que llevaron a una reducción de la dosis de TRODELVY se produjeron en el 33 % de las pacientes. Las reacciones adversas más frecuentes (>5 %) que llevaron a una reducción de la dosis fueron la neutropenia (16 %) y la diarrea (8 %). Se utilizó G-CSF en el 54 % de las pacientes que recibieron TRODELVY.
Los cuadros 6 y 7 resumen las reacciones adversas y las anomalías de laboratorio en el estudio TROPiCS-02.
Cuadro 6: Reacciones adversas en ≥ 10 % de las pacientes con mBC HR+/HER2- en TROPiCS-02
TRODELVY (n=268)
Quimioterapia en monoterapia (n=249)
Reacción adversa
Todos los grados %
Grado 3 – 4 %
Todos los grados %
Grado 3 – 4 %
*La quimioterapia en monoterapia incluyó uno de los siguientes agentes en monoterapia: eribulina (n=130), vinorelbina (n=63), gemcitabina (n=56) o capecitabina (n=22).
Otras reacciones adversas clínicamente significativas en TROPiCS-02 (≤ 10%) incluyen: hipotensión (5%), dolor (5%), rinorrea (5%), hipocalcemia (3%), congestión nasal (3%), hiperpigmentación de la piel (3%), colitis o colitis neutropénica (2%), hiponatremia (2%), neumonía (2%), proteinuria (1%), enteritis (0,4%).
Tabla 7: Anormalidades de laboratorio en > 10% de pacientes con mBC HR+/HER2- en TROPiCS-02
Anormalidad de laboratorio
TRODELVY (n=268)
Quimioterapia en monoterapia (n=249)
Todos los grados (%)
Grado 3 – 4 (%)
Todos los grados (%)
Grado 3 – 4 (%)
Hematología
Disminución del recuento de leucocitos
88
38
73
26
Disminución del recuento de neutrófilos
83
53
67
40
Disminución de la hemoglobina
73
8
59
5
Disminución del recuento de linfocitos
65
21
47
14
Disminución del recuento de plaquetas
21
1
30
4
Eosinofilia
13
0
4
0
Química
Aumento de glucosa
37
0
31
0
Disminución de albúmina
32
0
27
0.4
Disminución del aclaramiento de creatinina
24
2
19
1
Aumento de fosfatasa alcalina
23
0
23
1
Disminución de potasio
22
3
12
0.4
Aumento de alanina aminotransferasa
21
1
31
2
Disminución de sodio
19
1
17
0.4
Disminución de magnesio
18
0
15
0
Disminución de fosfato
17
0
10
0
Aumento de fosfato
16
0
16
0
Aumento de lactato deshidrogenasa
16
0
28
0
Aumento de aspartato aminotransferasa
15
2
25
1
Aumento de potasio
14
2
9
0
7 INTERACCIONES MEDICAMENTOSAS
7.1 Efecto de otros medicamentos en TRODELVY
Inhibidores de UGT1A1
La administración concomitante de TRODELVY con inhibidores de UGT1A1 puede aumentar la incidencia de reacciones adversas debido a un posible aumento en la exposición sistémica a SN-38 [ver Advertencias y precauciones (5.5) y Farmacología clínica (12.3, 12.5)]. Evite administrar inhibidores de UGT1A1 con TRODELVY.
Basándose en su mecanismo de acción, TRODELVY puede causar teratogenicidad y/o letalidad embriofetal cuando se administra a una mujer embarazada. No hay datos disponibles en mujeres embarazadas para informar sobre el riesgo asociado al fármaco. TRODELVY contiene un componente genotóxico, SN-38, y es tóxico para las células que se dividen rápidamente [ver Farmacología Clínica (12.1) y Toxicología No Clínica (13.1)]. Se debe advertir a las mujeres embarazadas y a las mujeres en edad fértil sobre el riesgo potencial para el feto.
Se desconoce el riesgo de base estimado de defectos congénitos importantes y aborto espontáneo para la población indicada. En la población general de EE. UU., el riesgo de base estimado de defectos congénitos importantes y aborto espontáneo en embarazos clínicamente reconocidos es del 2 al 4 % y del 15 al 20 %, respectivamente.
Datos
Datos en animales
No se realizaron estudios de toxicología reproductiva y del desarrollo con sacituzumab govitecan-hziy.
8.2 Lactancia
Resumen de Riesgo
No hay información sobre la presencia de sacituzumab govitecan-hziy o SN-38 en la leche materna, los efectos en el niño amamantado o los efectos en la producción de leche. Debido a la posibilidad de reacciones adversas graves en un niño amamantado, se debe aconsejar a las mujeres que no amamanten durante el tratamiento y durante 1 mes después de la última dosis de TRODELVY.
8.3 Mujeres y Hombres en Edad Fértil
Prueba de Embarazo
Verificar el estado de embarazo de las mujeres en edad fértil antes de iniciar el tratamiento con TRODELVY.
Anticoncepción
Mujeres
TRODELVY puede causar daño fetal cuando se administra a una mujer embarazada [ver Uso en Poblaciones Específicas (8.1)]. Se debe aconsejar a las mujeres en edad fértil que utilicen métodos anticonceptivos eficaces durante el tratamiento con TRODELVY y durante 6 meses después de la última dosis.
Hombres
Debido a la posibilidad de genotoxicidad, se debe aconsejar a los pacientes varones con parejas femeninas en edad fértil que utilicen métodos anticonceptivos eficaces durante el tratamiento con TRODELVY y durante 3 meses después de la última dosis.
Infertilidad
Mujeres
Basándose en los hallazgos en animales, TRODELVY puede afectar la fertilidad en mujeres en edad fértil [ver Toxicología No Clínica (13.1)].
8.4 Uso Pediátrico
No se ha establecido la seguridad y eficacia de TRODELVY en pacientes pediátricos.
8.5 Uso en Geriatría
De los 366 pacientes con cáncer de mama triple negativo que fueron tratados con TRODELVY, el 19 % de los pacientes tenían 65 años o más y el 3 % tenían 75 años o más. No se observaron diferencias generales en la seguridad y la eficacia entre los pacientes ≥ 65 años de edad y los pacientes más jóvenes.
De los 322 pacientes con cáncer de mama HR+/HER2- que fueron tratados con TRODELVY, el 26 % de los pacientes tenían 65 años o más y el 6 % tenían 75 años o más. No se observaron diferencias generales en la eficacia entre los pacientes ≥ 65 años de edad y los pacientes más jóvenes. Hubo una mayor tasa de interrupción debido a reacciones adversas en pacientes de 65 años o más (14 %) en comparación con pacientes más jóvenes (3 %).
8.6 Insuficiencia Hepática
No se requiere ajuste de la dosis inicial cuando se administra TRODELVY a pacientes con insuficiencia hepática leve [ver Farmacología Clínica (12.3)].
No se ha establecido la seguridad de TRODELVY en pacientes con insuficiencia hepática moderada (bilirrubina total > 1,5 a 3,0 × LSN) o grave (bilirrubina total > 3,0 × límite superior de la normalidad [LSN]). TRODELVY no se ha probado en pacientes con AST o ALT > 3 LSN sin metástasis hepáticas, o AST o ALT > 5 LSN con metástasis hepáticas. No se pueden hacer recomendaciones para la dosis inicial en estos pacientes.
10 SOBREDOSIS
En un ensayo clínico, se administraron dosis planificadas de hasta 18 mg/kg (aproximadamente 1,8 veces la dosis máxima recomendada de 10 mg/kg) de TRODELVY. En estos pacientes, se observó una mayor incidencia de neutropenia grave.
11 DESCRIPCIÓN
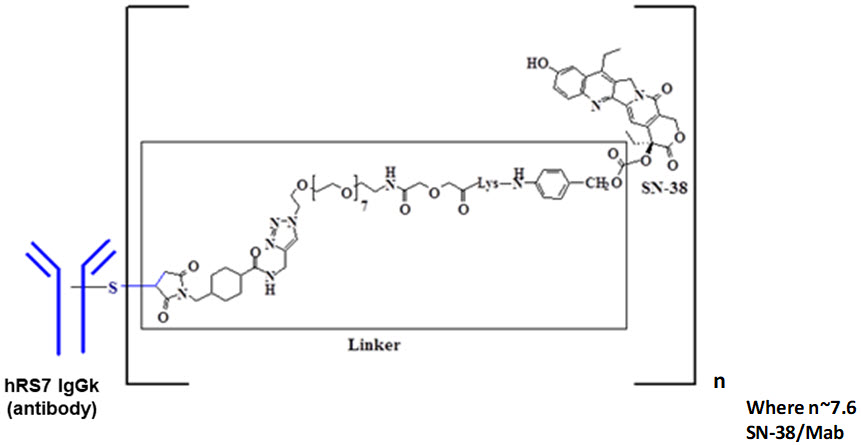
Sacituzumab govitecan-hziy es un conjugado de anticuerpo dirigido a Trop-2 e inhibidor de la topoisomerasa, compuesto de los siguientes tres componentes:
el anticuerpo monoclonal humanizado, hRS7 IgG1κ (también llamado sacituzumab), que se une a Trop-2 (el antígeno-2 de la superficie celular del trofoblasto);
el fármaco SN-38, un inhibidor de la topoisomerasa;
un enlace hidrolizable (llamado CL2A), que une el anticuerpo monoclonal humanizado a SN-38.
El anticuerpo monoclonal recombinante se produce mediante células de mamíferos (mieloma murino), mientras que los componentes de molécula pequeña SN-38 y CL2A se producen mediante síntesis química. Sacituzumab govitecan-hziy contiene un promedio de 7 a 8 moléculas de SN-38 por molécula de anticuerpo. Sacituzumab govitecan-hziy tiene un peso molecular de aproximadamente 160 kilodaltons. Sacituzumab govitecan-hziy tiene la siguiente estructura química.
TRODELVY (sacituzumab govitecan-hziy) para inyección es un polvo liofilizado estéril, sin conservantes, de blanco a amarillo claro para uso intravenoso en un vial de dosis única de vidrio transparente de 50 mL, con un tapón de goma y sellado con una tapa abatible de aluminio.
Cada vial de dosis única de TRODELVY proporciona 180 mg de sacituzumab govitecan-hziy, 71,7 mg de ácido 2-(N-morfolino) etanosulfónico (MES), 1,8 mg de polisorbato 80 y 153,99 mg de trehalosa. La reconstitución con 20 mL de inyección de cloruro de sodio al 0,9%, USP, da como resultado una concentración de 10 mg/mL con un pH de 6,5.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
Sacituzumab govitecan-hziy es un conjugado anticuerpo-fármaco dirigido a Trop-2. Sacituzumab es un anticuerpo humanizado que reconoce Trop-2. La molécula pequeña, SN-38, es un inhibidor de la topoisomerasa I, que se une covalentemente al anticuerpo mediante un enlazador. Los datos farmacológicos sugieren que sacituzumab govitecan-hziy se une a las células cancerosas que expresan Trop-2 y se internaliza con la posterior liberación de SN-38 a través de la hidrólisis del enlazador. El SN-38 interactúa con la topoisomerasa I y previene la religación de las roturas de una sola cadena inducidas por la topoisomerasa I. El daño resultante en el ADN conduce a la apoptosis y la muerte celular. Sacituzumab govitecan-hziy disminuyó el crecimiento tumoral en modelos de xenoinjerto de ratón de cáncer de mama triple negativo.
12.2 Farmacodinamia
Las relaciones exposición-respuesta de TRODELVY y el curso temporal farmacodinámico para la eficacia no se han caracterizado completamente.
Electrofisiología cardíaca
El cambio medio máximo desde el valor inicial fue de 9,7 mseg (el límite superior del intervalo de confianza del 90 % bilateral es de 16,8 mseg) a la dosis recomendada. Se observó una relación exposición-respuesta positiva entre los aumentos del QTc y las concentraciones de SN-38.
12.3 Farmacocinética
La farmacocinética sérica de sacituzumab govitecan-hziy y SN-38 se evaluó en pacientes con cáncer de mama metastásico (mBC) que recibieron sacituzumab govitecan-hziy como agente único a una dosis de 10 mg/kg. Los parámetros farmacocinéticos de sacituzumab govitecan-hziy y SN-38 libre se presentan en la Tabla 8.
Tabla 8: Resumen de los Parámetros Farmacocinéticos (PK) Medios (CV%) de Sacituzumab Govitecan-hziy y SN-38 Libre*
Sacituzumab govitecan-hziy (N=693)
SN-38 Libre (N=681)
Cmáx: concentración sérica máxima de 0 a 168 horas después de la primera dosis
AUC0–168: área bajo la curva de concentración sérica hasta 168 horas después de la primera dosis
Parámetros estimados con base en análisis farmacocinéticos (PK) poblacionales
Cmáx [ng/mL]
239000 (11%)
98.0 (45%)
AUC0–168 [ng*h/mL]
5640000 (22%)
3696 (56%)
Distribución
Basado en el análisis farmacocinético poblacional, el volumen de distribución en estado estable de sacituzumab govetican-hziy es de 3.6L.
Eliminación
La mediana de la vida media de eliminación (t1/2) de sacituzumab govitecan-hziy y SN-38 libre en pacientes con cáncer de mama triple negativo metastásico fue de 23.4 y 17.6 horas, respectivamente. Basado en el análisis farmacocinético poblacional, la depuración media estimada (%CV) de sacituzumab govitecan-hziy es de 0.13 L/h (12%).
Metabolismo
No se han realizado estudios de metabolismo con sacituzumab govitecan-hziy. SN-38 (la fracción de molécula pequeña de sacituzumab govitecan-hziy) se metaboliza a través de UGT1A1. El metabolito glucurónido de SN-38 (SN-38G) fue detectable en el suero de los pacientes.
Poblaciones Específicas
Los análisis farmacocinéticos en pacientes tratados con TRODELVY no identificaron un efecto de la edad (27 a 88 años), raza (blanca, negra o asiática) o insuficiencia renal leve a moderada (CLcr 30 a 89 mL/min) en la farmacocinética de sacituzumab govitecan-hziy. Se sabe que la eliminación renal contribuye mínimamente a la excreción de SN-38, la fracción de molécula pequeña de sacituzumab govitecan-hziy. No hay datos sobre la farmacocinética de sacituzumab govitecan-hziy en pacientes con insuficiencia renal grave (CLcr 15 a 29 mL/min) o enfermedad renal en etapa terminal (CLcr < 15 mL/min).
Pacientes con Insuficiencia Hepática
La exposición a sacituzumab govitecan-hziy es similar en pacientes con insuficiencia hepática leve (bilirrubina total ≤ LSN con AST > LSN, o bilirrubina >1.0 a ≤ 1.5 LSN con cualquier AST; n=257) a pacientes con función hepática normal (bilirrubina total o AST < LSN; n=526).
Se desconocen las exposiciones a sacituzumab govitecan-hziy y SN-38 libre en pacientes con insuficiencia hepática moderada (bilirrubina total > 1.5 a 3.0 × LSN) o grave (bilirrubina total > 3.0 × LSN).
Estudios de Interacción Farmacológica
No se realizaron estudios de interacción fármaco-fármaco con sacituzumab govitecan-hziy o sus componentes. Los inhibidores o inductores de UGT1A1 pueden aumentar o disminuir la exposición al SN-38, respectivamente [see Drug Interactions (7)].
12.5 Farmacogenómica
El SN-38 se metaboliza a través de UGT1A1 [see Clinical Pharmacology (12.3)]. Las variantes genéticas del gen UGT1A1, como el alelo UGT1A1*28, conducen a una reducción de la actividad de la enzima UGT1A1. Las personas que son homocigotas o heterocigotas para el alelo UGT1A1*28 tienen un mayor riesgo de neutropenia, neutropenia febril y anemia por TRODELVY en comparación con las personas que son de tipo salvaje (*1/*1) [see Warnings and Precautions (5.5)]. Aproximadamente el 20% de la población negra o afroamericana, el 10% de la población blanca y el 2% de la población del este asiático son homocigotos para el alelo UGT1A1*28 (*28/*28). Aproximadamente el 40% de la población negra o afroamericana, el 50% de la población blanca y el 25% de la población del este asiático son heterocigotos para el alelo UGT1A1*28 (*1/*28). Alelos de función disminuida distintos de UGT1A1*28 pueden estar presentes en ciertas poblaciones.
12.6 Inmunogenicidad
La detección de la formación de anticuerpos depende en gran medida de la sensibilidad y especificidad del ensayo. Las diferencias en los métodos de ensayo impiden comparaciones significativas de la incidencia de anticuerpos contra el fármaco en los estudios descritos a continuación con la incidencia de anticuerpos contra el fármaco en otros estudios, incluidos los de TRODELVY.
Durante el período de tratamiento medio de 4 meses en estudios clínicos en pacientes tratados con TRODELVY, 9 (1.1%) de 785 pacientes desarrollaron anticuerpos contra sacituzumab govitecan; 6 de estos pacientes (0.8% de todos los pacientes tratados con TRODELVY) tenían anticuerpos neutralizantes contra sacituzumab govitecan. Debido a la baja incidencia de anticuerpos contra el fármaco, se desconoce el efecto de estos anticuerpos sobre la farmacocinética, la farmacodinámica, la seguridad o la eficacia de sacituzumab govitecan.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogenicidad, Mutagénesis, Deterioro de la Fertilidad
No se han realizado estudios de carcinogenicidad con sacituzumab govitecan-hziy.
SN-38 fue clastogénico en una prueba de micronúcleos de células de mamíferos in vitro en células de ovario de hámster chino y no fue mutagénico en una prueba de reversión de mutación bacteriana in vitro (Ames).
No se han realizado estudios de fertilidad con sacituzumab govitecan-hziy. En un estudio de toxicidad por dosis repetidas en monos cynomolgus, la administración intravenosa de sacituzumab govitecan-hziy en el Día 1 y el Día 4 provocó atrofia endometrial, hemorragia uterina, aumento de la atresia folicular del ovario y atrofia de las células epiteliales vaginales en dosis ≥ 60 mg/kg (≥6 veces la dosis recomendada en humanos de 10 mg/kg según el peso corporal).
14 ESTUDIOS CLÍNICOS
14.1 Cáncer de mama triple negativo localmente avanzado o metastásico
ASCENT
La eficacia se evaluó en un estudio multicéntrico, abierto y aleatorizado (ASCENT; NCT02574455) realizado en 529 pacientes con cáncer de mama triple negativo (mTNBC) localmente avanzado o metastásico irresecable que habían recaído después de al menos dos quimioterapias previas para el cáncer de mama (una de las cuales podría ser en el contexto neoadyuvante o adyuvante, siempre que la progresión se produjera en un plazo de 12 meses). Todas las pacientes recibieron tratamiento previo con taxanos en el contexto adyuvante, neoadyuvante o en estadio avanzado, a menos que hubiera una contraindicación o intolerancia a los taxanos durante o al final del primer ciclo de taxanos. Se requirió una resonancia magnética (RM) para determinar las metástasis cerebrales antes de la inclusión en el estudio para las pacientes con metástasis cerebrales conocidas o sospechosas. Se permitió la inclusión de pacientes con metástasis cerebrales hasta un máximo predefinido del 15 % de los pacientes en el estudio ASCENT. Se excluyeron las pacientes con enfermedad de Gilbert conocida o enfermedad exclusivamente ósea.
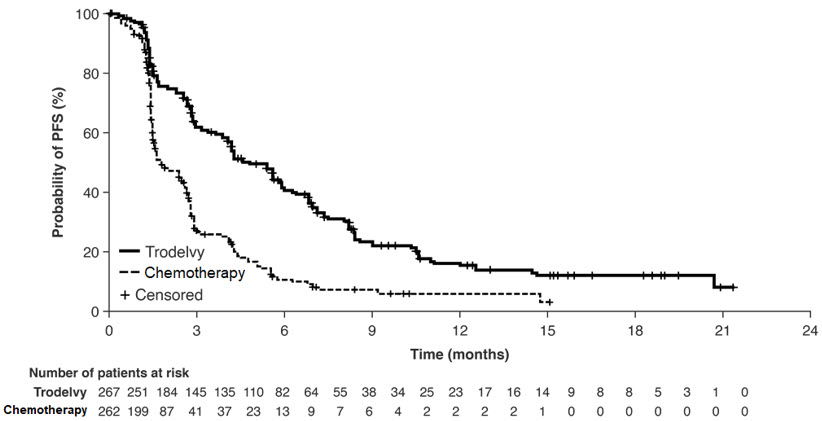
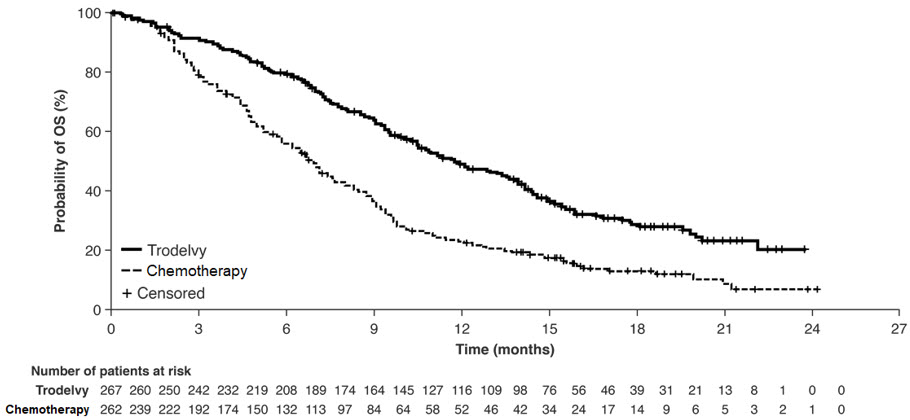
Las pacientes se asignaron aleatoriamente (1:1) para recibir TRODELVY 10 mg/kg como infusión intravenosa los días 1 y 8 de un ciclo de 21 días (n=267) o la quimioterapia de un solo agente a elección del médico (n=262). La quimioterapia de un solo agente fue determinada por el investigador antes de la aleatorización entre una de las siguientes opciones: eribulina (n=139), capecitabina (n=33), gemcitabina (n=38) o vinorelbina (n=52).
Las pacientes fueron tratadas hasta la progresión de la enfermedad o la toxicidad inaceptable. El principal resultado de eficacia fue la supervivencia libre de progresión (SLP) en pacientes sin metástasis cerebrales al inicio (es decir, BMNeg), medida mediante una revisión centralizada, independiente y ciega, evaluada utilizando los Criterios de Evaluación de la Respuesta en Tumores Sólidos (RECIST) versión 1.1. Las medidas de eficacia adicionales incluyeron la SLP para la población completa (todas las pacientes con y sin metástasis cerebrales) y la supervivencia general (SG).
La mediana de edad de las pacientes en la población completa (n = 529) fue de 54 años (rango: 27 a 82 años); el 99,6 % eran mujeres; el 79 % eran blancas, el 12 % eran negras/afroamericanas; y el 81 % de las pacientes tenían menos de 65 años de edad. Todas las pacientes tenían un estado de rendimiento ECOG de 0 (43 %) o 1 (57 %). El 42 % de las pacientes tenían metástasis hepáticas, el 9 % tenían un estado mutacional BRCA1/BRCA2 positivo y el 70 % tenían TNBC en el diagnóstico. El 12 % tenían metástasis cerebrales basales previamente tratadas y estables (n=61; 32 en el brazo de TRODELVY y 29 en el brazo de quimioterapia de un solo agente). En general, el 29 % de las pacientes habían recibido tratamiento previo con PD-1/PD-L1. El trece por ciento de las pacientes del grupo TRODELVY en la población completa recibieron solo 1 línea previa de terapia sistémica en el contexto metastásico.
Los resultados de eficacia se resumen en la Tabla 9 y se muestran en la Figura 1 y la Figura 2. Los resultados de eficacia para el subgrupo de pacientes que habían recibido solo 1 línea previa de terapia sistémica en el contexto metastásico (además de tener recurrencia o progresión de la enfermedad dentro de los 12 meses de la terapia sistémica neoadyuvante/adyuvante) fueron consistentes con los de las pacientes que habían recibido al menos dos líneas previas en el contexto metastásico.
La SLP se define como el tiempo transcurrido desde la fecha de aleatorización hasta la fecha de la primera progresión radiológica de la enfermedad o la muerte por cualquier causa, lo que ocurra primero.
Prueba de log-rank estratificada ajustada por factores de estratificación: número de quimioterapias previas, presencia de metástasis cerebrales conocidas al inicio del estudio y región.
Figura 1: Gráfico de Kaplan-Meier de la PFS según BICR (todos los pacientes aleatorizados) en ASCENT
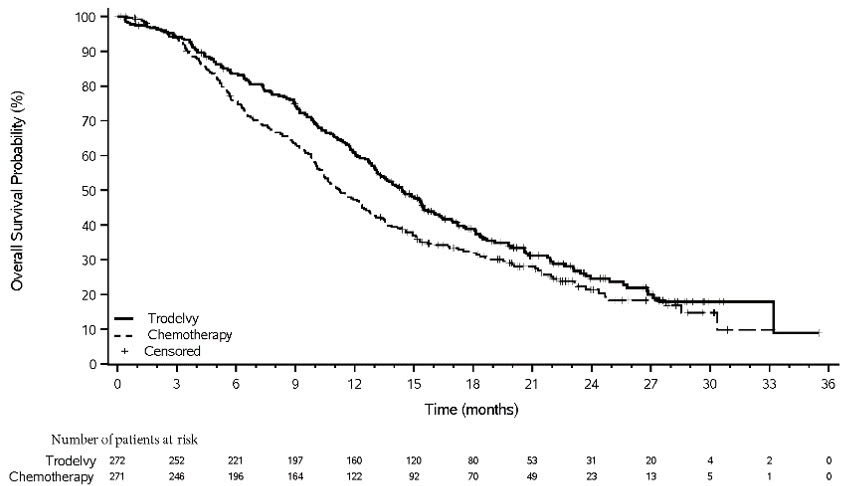
Figura 2: Gráfico de Kaplan-Meier de la OS (todos los pacientes aleatorizados) en ASCENT
Un análisis exploratorio de la PFS en pacientes con metástasis cerebrales estables previamente tratadas mostró una HR estratificada de 0,65 (IC del 95%: 0,35, 1,22). La PFS mediana en el brazo TRODELVY fue de 2,8 meses (IC del 95%: 1,5, 3,9) y la PFS mediana con quimioterapia en monoterapia fue de 1,6 meses (IC del 95%: 1,3, 2,9). El análisis exploratorio de la OS en la misma población mostró una HR estratificada de 0,87 (IC del 95%: 0,47, 1,63). La OS mediana en el brazo TRODELVY fue de 6,8 meses (IC del 95%: 4,7, 14,1) y la OS mediana con quimioterapia en monoterapia fue de 7,4 meses (IC del 95%: 4,7, 11,1).
IMMU-132-01
La eficacia de TRODELVY se evaluó en un estudio multicéntrico, de un solo brazo (NCT01631552) en el que se inscribieron 108 pacientes con cáncer de mama triple negativo metastásico (mTNBC) que habían recibido al menos dos terapias anticancerosas previas para la enfermedad metastásica. Los pacientes con enfermedad voluminosa, definida como una masa >7 cm, no fueron elegibles. Los pacientes con metástasis cerebrales tratadas que no recibían dosis altas de esteroides (> 20 mg de prednisona o equivalente) durante al menos cuatro semanas fueron elegibles. Se excluyeron los pacientes con enfermedad de Gilbert conocida.
Los pacientes recibieron TRODELVY 10 mg/kg por vía intravenosa los días 1 y 8 de un ciclo de tratamiento de 21 días. Los pacientes fueron tratados con TRODELVY hasta la progresión de la enfermedad o la intolerancia a la terapia. Se obtuvieron imágenes tumorales cada 8 semanas, con exploraciones de TC/RM de confirmación obtenidas 4-6 semanas después de una respuesta parcial o completa inicial, hasta la progresión que requirió la interrupción del tratamiento. Las principales medidas de resultado de eficacia fueron la tasa de respuesta general (ORR) evaluada por el investigador utilizando RECIST 1.1 y la duración de la respuesta.
La mediana de edad fue de 55 años (rango: 31 a 80 años); el 87% de los pacientes tenían menos de 65 años. La mayoría de los pacientes fueron mujeres (99%) y blancas (76%). Al inicio del estudio, todos los pacientes tenían un estado de rendimiento ECOG de 0 (29%) o 1 (71%). El setenta y seis por ciento tenía enfermedad visceral, el 42% tenía metástasis hepáticas, el 56% tenía metástasis pulmonares/pleurales y el 2% tenía metástasis cerebrales. Doce pacientes (11%) tenían enfermedad en estadio IV en el momento del diagnóstico inicial.
El número medio de terapias sistémicas previas recibidas en el contexto metastásico fue de 3 (rango: 2 a 10). Las quimioterapias previas en el contexto metastásico incluyeron carboplatino o cisplatino (69%), gemcitabina (55%), paclitaxel o docetaxel (53%), capecitabina (51%), eribulina (45%), doxorrubicina (24%), vinorelbina (16%), ciclofosfamida (19%) e ixabepilona (8%).
En general, el 98% de los pacientes habían recibido taxanos previos y el 86% habían recibido antraciclinas previas en el contexto (neo)adyuvante o metastásico.
El Cuadro 10 resume los resultados de eficacia.
Cuadro 10: Resultados de eficacia para pacientes con mTNBC en IMMU-132-01
14.2 Cáncer de mama localmente avanzado o metastásico HR-positivo, HER2-negativo
Estudio TROPiCS-02
La eficacia de TRODELVY se evaluó en un estudio multicéntrico, abierto y aleatorizado (TROPiCS-02; NCT03901339) realizado en 543 pacientes con cáncer de mama HR-positivo, HER2-negativo (IHC 0, IHC 1+ o IHC 2+/ISH–) localmente avanzado o metastásico irresecable cuya enfermedad había progresado después de lo siguiente en cualquier contexto: un inhibidor de CDK 4/6, terapia endocrina y un taxano; las pacientes recibieron al menos dos quimioterapias previas en el contexto metastásico (una de las cuales podría ser en el contexto neoadyuvante o adyuvante si se producía una recurrencia en un plazo de 12 meses).
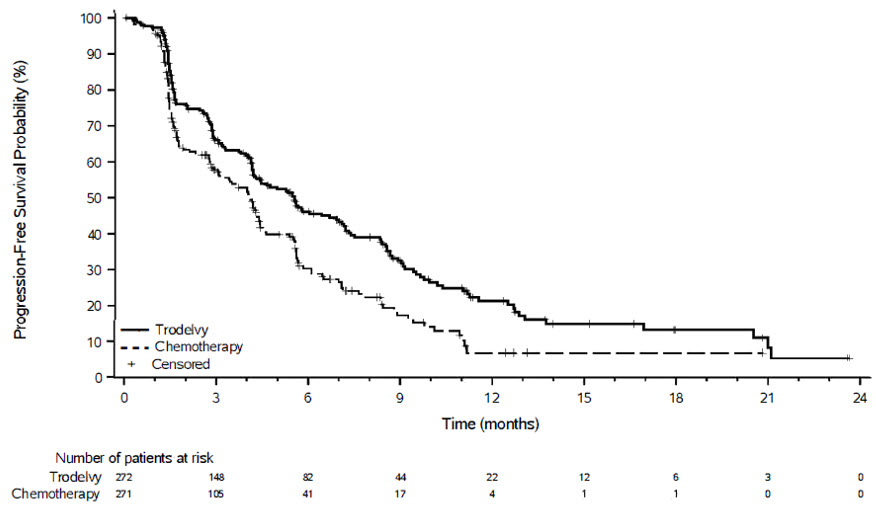
Las pacientes se asignaron aleatoriamente (1:1) para recibir TRODELVY 10 mg/kg como infusión intravenosa los días 1 y 8 de un ciclo de 21 días (n=272) o quimioterapia en monoterapia (n=271). La quimioterapia en monoterapia fue determinada por el investigador antes de la aleatorización a partir de una de las siguientes opciones: eribulina (n=130), vinorelbina (n=63), gemcitabina (n=56) o capecitabina (n=22). La aleatorización se estratificó según los siguientes factores: regímenes de quimioterapia previos para la enfermedad metastásica (2 frente a 3–4), metástasis visceral (Sí o No) y terapia endocrina en el contexto metastásico durante al menos 6 meses (Sí o No).
Las pacientes fueron tratadas hasta la progresión de la enfermedad o la toxicidad inaceptable. La administración de TRODELVY se permitió más allá de la progresión de la enfermedad definida por RECIST si la paciente era clínicamente estable y el investigador consideraba que estaba obteniendo un beneficio clínico. La principal medida de resultado de eficacia fue la PFS determinada por BICR según RECIST v1.1. Las medidas de eficacia adicionales incluyeron la OS, la ORR por BICR y la DOR por BICR.
La mediana de edad de las pacientes en la población del estudio fue de 56 años (rango: 27–86 años), el 26% de las pacientes tenían 65 años o más. La mayoría de las pacientes fueron mujeres (99%); el 67% eran blancas, el 4% eran negras y el 3% eran asiáticas, y el 26% eran de raza desconocida. Las pacientes recibieron una mediana de 7 (rango: 3 a 17) regímenes sistémicos previos en cualquier contexto y 3 (rango: 0 a 8) regímenes de quimioterapia sistémica previos en el contexto metastásico. Aproximadamente el 42% de las pacientes tuvieron 2 regímenes de quimioterapia previos para el tratamiento de la enfermedad metastásica en comparación con el 58% de las pacientes que tuvieron de 3 a 4 regímenes de quimioterapia previos y todas las pacientes tuvieron un estado de rendimiento ECOG de 0 (45%) o 1 (55%). El noventa y cinco por ciento de las pacientes presentaron metástasis viscerales. La mayoría de las pacientes recibieron terapia endocrina en el contexto metastásico durante ≥ 6 meses (86%).
TRODELVY demostró una mejora estadísticamente significativa en la PFS y la OS en comparación con la quimioterapia en monoterapia.
Los resultados de eficacia se resumen en la Tabla 11 y las Figuras 3 y 4.
La PFS se define como el tiempo transcurrido desde la fecha de aleatorización hasta la fecha de la primera progresión radiológica de la enfermedad o la muerte por cualquier causa, lo que ocurra primero.
Prueba de log-rank estratificada ajustada por factores de estratificación: regímenes de quimioterapia previos para la enfermedad metastásica (2 frente a 3–4), metástasis visceral (S/N) y terapia endocrina en el contexto metastásico durante al menos 6 meses (Sí o No). BICR = Revisión central independiente ciega; IC = Intervalo de confianza
TRODELVY (sacituzumab govitecan-hziy) para inyección es un polvo liofilizado estéril, de blanco a amarillo pálido, en un vial de dosis única. Cada vial de TRODELVY se envasa individualmente en una caja de cartón:
NDC 55135-132-01 contiene un vial de 180 mg
Almacene los viales en un refrigerador a 2°C a 8°C (36°F a 46°F) en la caja original para protegerlos de la luz hasta el momento de la reconstitución. No congelar.
TRODELVY es un fármaco peligroso. Siga los procedimientos de manipulación y eliminación especiales aplicables1.
17 INFORMACIÓN DE ASESORAMIENTO AL PACIENTE
Aconseje al paciente que lea el etiquetado del paciente aprobado por la FDA (Información para el paciente)
Neutropenia
Informe a los pacientes sobre el riesgo de neutropenia. Indique a los pacientes que se pongan en contacto inmediatamente con su proveedor de atención médica si experimentan fiebre, escalofríos u otros signos de infección [ver Advertencias y precauciones (5.1)].
Diarrea
Informe a los pacientes sobre el riesgo de diarrea. Indique a los pacientes que se pongan en contacto inmediatamente con su proveedor de atención médica si experimentan diarrea por primera vez durante el tratamiento; heces negras o con sangre; síntomas de deshidratación como mareos, aturdimiento o desmayo; incapacidad para tomar líquidos por vía oral debido a náuseas o vómitos; o incapacidad para controlar la diarrea en 24 horas [ver Advertencias y precauciones (5.2)].
Hipersensibilidad y reacciones relacionadas con la infusión
Informe a los pacientes sobre el riesgo de reacciones graves a la infusión y anafilaxia. Indique a los pacientes que se pongan en contacto inmediatamente con su proveedor de atención médica si experimentan hinchazón facial, de labios, lengua o garganta, urticaria, dificultad para respirar, mareos, aturdimiento, escalofríos, rigidez, sibilancias, prurito, rubor, erupción cutánea, hipotensión o fiebre que ocurran durante o dentro de las 24 horas posteriores a la infusión [ver Advertencias y precauciones (5.3)].
Náuseas/Vómitos
Informe a los pacientes sobre el riesgo de náuseas y vómitos. También se recomienda la premedicación según las pautas establecidas con un régimen de dos o tres medicamentos para la prevención de las náuseas y los vómitos inducidos por quimioterapia (CINV). También se pueden emplear antieméticos adicionales, sedantes y otras medidas de apoyo según lo indicado clínicamente. Todos los pacientes deben recibir medicamentos para llevar a casa para prevenir y tratar las náuseas y los vómitos tardíos, con instrucciones claras. Indique a los pacientes que se pongan en contacto inmediatamente con su proveedor de atención médica si experimentan náuseas o vómitos incontrolables [ver Advertencias y precauciones (5.4)].
Toxicidad embriofetal
Aconseje a las pacientes que se pongan en contacto con su proveedor de atención médica si están embarazadas o quedan embarazadas. Informe a las pacientes sobre el riesgo para el feto y la posible pérdida del embarazo [ver Uso en poblaciones específicas (8.1)].
Anticoncepción
Aconseje a las pacientes con potencial reproductivo que utilicen métodos anticonceptivos eficaces durante el tratamiento y durante 6 meses después de la última dosis de TRODELVY [ver Uso en poblaciones específicas (8.3)].
Aconseje a los pacientes varones con parejas femeninas con potencial reproductivo que utilicen métodos anticonceptivos eficaces durante el tratamiento y durante 3 meses después de la última dosis de TRODELVY [ver Uso en poblaciones específicas (8.3)].
Lactancia
Aconseje a las mujeres que no deben amamantar durante el tratamiento y durante 1 mes después de la última dosis de TRODELVY [ver Uso en poblaciones específicas (8.2)].
Gilead Sciences, Inc. 333 Lakeside Dr. Foster City, CA 94404, USA
Licencia de EE. UU. N.º 2258
PROSPECTO PARA EL PACIENTE
La información para el paciente ha sido aprobada por la Administración de Alimentos y Medicamentos de los EE. UU.
Revisado: 11/2024
Información para el paciente
TRODELVY® (troh-DELL-vee) (sacituzumab govitecan-hziy) para inyección, para uso intravenoso
¿Cuál es la información más importante que debo saber sobre TRODELVY? TRODELVY puede causar efectos secundarios graves, incluyendo:
Recuento bajo de glóbulos blancos (neutropenia). Los recuentos bajos de glóbulos blancos son comunes con TRODELVY y a veces pueden ser graves y provocar infecciones que pueden poner en peligro la vida o causar la muerte. Su proveedor de atención médica debe controlar sus recuentos de células sanguíneas durante el tratamiento con TRODELVY. Si su recuento de glóbulos blancos es demasiado bajo, su proveedor de atención médica puede necesitar reducir su dosis de TRODELVY, administrarle un medicamento para ayudar a prevenir el recuento bajo de células sanguíneas con dosis futuras de TRODELVY o, en algunos casos, suspender TRODELVY. Su proveedor de atención médica puede necesitar administrarle medicamentos antibióticos si desarrolla fiebre mientras su recuento de glóbulos blancos es bajo. Llame a su proveedor de atención médica de inmediato si desarrolla alguno de los siguientes signos de infección durante el tratamiento con TRODELVY:
fiebre
escalofríos
tos
falta de aliento
ardor o dolor al orinar
Diarrea grave. La diarrea es común con TRODELVY y también puede ser grave. La diarrea grave puede provocar la pérdida de demasiado líquido corporal (deshidratación) y problemas renales. Su proveedor de atención médica debe vigilarle la diarrea y administrarle medicamentos según sea necesario para ayudar a controlar su diarrea. Si pierde demasiado líquido corporal, su proveedor de atención médica puede necesitar administrarle líquidos y electrolitos para reponer las sales corporales. Si desarrolla diarrea durante el tratamiento con TRODELVY, su proveedor de atención médica debe verificar si la diarrea puede ser causada por una infección. Su proveedor de atención médica puede disminuir su dosis o suspender TRODELVY si su diarrea es grave y no se puede controlar con medicamentos antidiarreicos. Llame a su proveedor de atención médica de inmediato:
la primera vez que tenga diarrea durante el tratamiento con TRODELVY
si tiene heces negras o con sangre
si tiene síntomas de pérdida de demasiado líquido corporal y sales corporales, como mareos, aturdimiento o desmayo
si no puede tomar líquidos por vía oral debido a náuseas o vómitos
si no puede controlar su diarrea en 24 horas
¿Qué es TRODELVY?
TRODELVY es un medicamento recetado que se usa para tratar a adultos con:
un tipo de cáncer de mama llamado cáncer de mama triple negativo (TNBC), que es receptor hormonal de estrógeno y progesterona (HR)-negativo y receptor del factor de crecimiento epidérmico humano 2 (HER2)-negativo. TRODELVY se puede usar:
cuando su cáncer de mama se ha diseminado a otras partes del cuerpo (metástasis) o no se puede extirpar con cirugía, y
si previamente recibió dos o más tratamientos previos, incluyendo al menos un tratamiento para la enfermedad metastásica.
un tipo de cáncer de mama que es HR-positivo y HER2-negativo. TRODELVY se puede usar:
cuando su cáncer de mama se ha diseminado a otras partes del cuerpo o no se puede extirpar con cirugía, y
si previamente recibió terapia endocrina y al menos dos tratamientos adicionales para la enfermedad metastásica.
No se sabe si TRODELVY es seguro y eficaz en personas con problemas hepáticos moderados o graves. No se sabe si TRODELVY es seguro y eficaz en niños.
No reciba TRODELVY si ha tenido una reacción alérgica grave a TRODELVY. Consulte a su proveedor de atención médica si no está seguro.
Antes de recibir TRODELVY, informe a su proveedor de atención médica sobre todas sus afecciones médicas, incluso si:
le han dicho que porta un gen para la uridina difosfato-glucuronosil transferasa 1A1 (UGT1A1)*28. Las personas que portan este gen tienen un mayor riesgo de sufrir efectos secundarios con TRODELVY, especialmente recuentos bajos de glóbulos blancos, fiebre mientras su recuento de glóbulos blancos es bajo y recuentos bajos de glóbulos rojos. Ver “¿Cuál es la información más importante que debo saber sobre TRODELVY?“
tiene problemas hepáticos.
está embarazada o planea quedar embarazada. TRODELVY puede dañar a su bebé nonato. Su proveedor de atención médica debe verificar si está embarazada antes de comenzar a recibir TRODELVY.
Las mujeres que pueden quedar embarazadas deben usar un método anticonceptivo eficaz durante el tratamiento y durante 6 meses después de su última dosis de TRODELVY. Hable con su proveedor de atención médica sobre las opciones de anticoncepción que pueden ser adecuadas para usted durante este tiempo. Informe a su proveedor de atención médica de inmediato si queda embarazada durante el tratamiento con TRODELVY.
Los hombres con una pareja femenina que puede quedar embarazada deben usar un método anticonceptivo eficaz durante el tratamiento y durante 3 meses después de su última dosis de TRODELVY.
está amamantando o planea amamantar. No se sabe si TRODELVY pasa a la leche materna y puede dañar a su bebé. No amamante durante el tratamiento y durante 1 mes después de su última dosis de TRODELVY.
Informe a su proveedor de atención médica sobre todos los medicamentos que toma, incluidos los medicamentos recetados y de venta libre, las vitaminas y los suplementos herbales. Ciertos medicamentos pueden afectar la forma en que funciona TRODELVY.
¿Cómo recibiré TRODELVY?
Su proveedor de atención médica le administrará TRODELVY en una vena a través de una vía intravenosa (IV).
TRODELVY se administra 1 vez por semana, el día 1 y el día 8 de un ciclo de tratamiento de 21 días.
Recibirá la primera dosis de TRODELVY durante 3 horas. Si tolera bien la primera dosis, las dosis posteriores se pueden administrar en 1 a 2 horas.
Antes de cada dosis de TRODELVY, recibirá medicamentos para ayudar a prevenir reacciones relacionadas con la infusión, náuseas y vómitos.
Lo controlarán para detectar efectos secundarios durante y al menos 30 minutos después de recibir cada infusión de TRODELVY.
Su proveedor de atención médica puede disminuir o detener temporalmente la infusión de TRODELVY si tiene una reacción relacionada con la infusión, o detener permanentemente TRODELVY si tiene una reacción relacionada con la infusión que ponga en peligro su vida.
Su proveedor de atención médica decidirá cuánto tiempo continuará recibiendo TRODELVY.
¿Cuáles son los posibles efectos secundarios de TRODELVY?
TRODELVY puede causar efectos secundarios graves, que incluyen:
Reacciones alérgicas y relacionadas con la infusión. Pueden producirse reacciones alérgicas graves durante el tratamiento con TRODELVY, incluidas reacciones alérgicas que ponen en peligro la vida, y reacciones relacionadas con la infusión. Informe a su proveedor de atención médica o enfermero de inmediato si presenta alguno de los siguientes síntomas de una reacción alérgica o relacionada con la infusión durante la infusión de TRODELVY o dentro de las 24 horas posteriores a la administración de una dosis de TRODELVY:
hinchazón de la cara, los labios, la lengua o la garganta
ronchas
sarpullido, picazón o enrojecimiento de la piel
fiebre
dificultad para respirar o sibilancias
mareos, aturdimiento, sensación de desmayo o desmayo
escalofríos o escalofríos intensos (rígidos)
Náuseas y vómitos. Las náuseas y los vómitos son comunes con TRODELVY y, a veces, pueden ser graves. Antes de cada dosis de TRODELVY, recibirá medicamentos para ayudar a prevenir las náuseas y los vómitos. Se le deben administrar medicamentos para llevar a casa, junto con instrucciones sobre cómo tomarlos para ayudar a prevenir y tratar las náuseas y los vómitos después de recibir TRODELVY. Llame a su proveedor de atención médica de inmediato si tiene náuseas o vómitos que no se controlan con los medicamentos recetados. Su proveedor de atención médica puede decidir disminuir su dosis o suspender TRODELVY si sus náuseas y vómitos son graves y no se pueden controlar con medicamentos antináuseas.
Los efectos secundarios más comunes de TRODELVY incluyen:
disminución del recuento de glóbulos blancos (leucocitos y linfocitos) y glóbulos rojos
sensación de cansancio o debilidad
pérdida de cabello
estreñimiento
aumento de los niveles de azúcar en la sangre
disminución de los niveles de proteínas (albúmina) en la sangre
disminución del apetito
cambios en la prueba de función renal
aumento de los niveles de la enzima llamada fosfatasa alcalina en la sangre (prueba para problemas hepáticos u óseos)
disminución de los niveles de magnesio, potasio y sodio en la sangre
TRODELVY puede causar problemas de fertilidad en las mujeres, lo que podría afectar su capacidad para tener un bebé. Hable con su proveedor de atención médica si la fertilidad es una preocupación para usted. Estos no son todos los posibles efectos secundarios de TRODELVY. Llame a su médico para obtener asesoramiento médico sobre los efectos secundarios. Puede informar los efectos secundarios a la FDA al 1-800-FDA-1088.
Información general sobre el uso seguro y eficaz de TRODELVY.
Los medicamentos a veces se recetan para fines distintos de los que se enumeran en un folleto de información para el paciente. Puede pedirle a su farmacéutico o proveedor de atención médica información sobre TRODELVY que esté escrita para profesionales de la salud.
¿Cuáles son los ingredientes de TRODELVY? Ingrediente activo: sacituzumab govitecan-hziy Ingredientes inactivos: ácido 2-(N-morfolino) etanosulfónico (MES), polisorbato 80 y trehalosa Fabricado por: Gilead Sciences, Inc., 333 Lakeside Dr., Foster City, CA 94404, EE. UU. Licencia de EE. UU. No. 2258 761115-GS-009 Para obtener más información sobre TRODELVY, visite www.TRODELVY.com o llame al 1-888-983-4668.
PRINCIPAL PANEL DE PRESENTACIÓN – Etiqueta del vial de 180 mg
NDC 55135-132-01
Rx only
TRODELVY®
sacituzumab govitecan-hziy Para inyección
180 mg por vial
Sólo para infusión intravenosa
Advertencia: Medicamento peligroso
Vial de dosis única Deseche la porción no utilizada
90370103
PANEL PRINCIPAL DE EXHIBICIÓN – Caja de vial de 180 mg
NDC 55135-132-01
Rx only
TRODELVY®
sacituzumab govitecan-hziy Para inyección
180 mg por vial
Sólo para infusión intravenosa
Advertencia: Medicamento Peligroso
Reconstituir y diluir inmediatamente antes de usar
Vial de dosis única Desechar la porción no utilizada
Fabricante de medicamentos: Jazz Pharmaceuticals, Inc. (Updated: 2023-04-25) Tags: AR ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓNEstos puntos destacados no incluyen toda la información necesaria para usar XYREM de forma segura y eficaz. Consulte la información de prescripción completa para XYREM. XYREM® (oxibato de sodio) solución oral, CIIIAprobación…
Fabricante de medicamentos: Merck Sharp & Dohme LLC (Updated: 2024-07-05) Tags: NS5ANS3/NS4AHepatitis C crónicaHepatitis C ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓNEstos aspectos destacados no incluyen toda la información necesaria para usar ZEPATIER de forma segura y eficaz. Consulte la información completa de prescripción de ZEPATIER. ZEPATIER® (elbasvir…
Fabricante de medicamentos: Eli Lilly and Company (Updated: 2024-11-13) Tags: GLP1R ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓNEstos aspectos destacados no incluyen toda la información necesaria para usar TALTZ de manera segura y eficaz. Consulte la información de prescripción completa para TALTZ.TALTZ (ixekizumab) inyección, para uso subcutáneoAprobación inicial…