Fabricante de medicamentos: Vertex Pharmaceuticals Incorporated (Updated: 2024-12-26)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
Estos aspectos destacados no incluyen toda la información necesaria para usar TRIKAFTA de forma segura y eficaz. Consulte la información de prescripción completa para TRIKAFTA.
TRIKAFTA® (elexacaftor, tezacaftor e ivacaftor comprimidos; ivacaftor comprimidos), co-envasados para uso oral
TRIKAFTA® (elexacaftor, tezacaftor e ivacaftor gránulos orales; ivacaftor gránulos orales), co-envasados
Aprobación inicial en EE. UU.: 2019
ADVERTENCIA: DAÑO HEPÁTICO INDUCIDO POR MEDICAMENTOS E INSUFICIENCIA HEPÁTICA
Consulte la información de prescripción completa para ver la advertencia completa en recuadro.
-
TRIKAFTA puede causar daño hepático grave y potencialmente mortal inducido por medicamentos. Se ha informado de insuficiencia hepática que ha provocado trasplante y muerte. (5.1, 6)
-
Evalúe las pruebas de función hepática (ALT, AST, fosfatasa alcalina, bilirrubina) en todos los pacientes antes de iniciar TRIKAFTA. (2.1, 5.1)
-
Controle las pruebas de función hepática (ALT, AST, fosfatasa alcalina, bilirrubina) cada mes durante los primeros 6 meses de tratamiento, luego cada 3 meses durante los siguientes 12 meses y, posteriormente, al menos una vez al año. (2.1, 5.1)
-
Interrumpa TRIKAFTA en caso de elevaciones significativas en las pruebas de función hepática o signos o síntomas de daño hepático. Siga a los pacientes de cerca con monitoreo clínico y de laboratorio hasta que las anomalías se resuelvan. (5.1)
-
Reanude TRIKAFTA si las anomalías se resuelven y solo si se espera que el beneficio supere el riesgo. (5.1)
-
TRIKAFTA no debe usarse en pacientes con insuficiencia hepática grave (Child-Pugh Clase C). No se recomienda el uso de TRIKAFTA en pacientes con insuficiencia hepática moderada (Child-Pugh Clase B). (2.3, 5.1, 8.7, 12.3)
CAMBIOS RECIENTES IMPORTANTES
| Recuadro de advertencia |
12/2024 |
| Indicaciones y uso (1) |
12/2024 |
| Dosis y administración (2.1) |
12/2024 |
| Advertencias y precauciones, daño hepático inducido por medicamentos e insuficiencia hepática (5.1) |
12/2024 |
INDICACIONES Y USO
TRIKAFTA es una combinación de ivacaftor, un potenciador de CFTR, tezacaftor y elexacaftor indicado para el tratamiento de la fibrosis quística (FQ) en pacientes de 2 años de edad y mayores que tienen al menos una mutación F508del en el gen CFTR o una mutación en el gen CFTR que responde según datos clínicos o in vitro.
Si se desconoce el genotipo del paciente, se debe utilizar una prueba de mutación de FQ aprobada por la FDA para confirmar la presencia de al menos una mutación indicada. (1)
DOSIS Y ADMINISTRACIÓN
Antes de iniciar TRIKAFTA, obtenga pruebas de función hepática (ALT, AST, fosfatasa alcalina y bilirrubina) en todos los pacientes. Controle las pruebas de función hepática cada mes durante los primeros 6 meses de tratamiento, luego cada 3 meses durante los siguientes 12 meses y, posteriormente, al menos una vez al año. (2.1, 5.1)
| Dosis recomendada para pacientes adultos y pediátricos de 2 años de edad y mayores (con alimentos que contienen grasa (2.2, 12.3)) |
| Edad |
Peso |
Dosis matutina |
Dosis vespertina |
| 2 a menos de 6 años |
Menos de 14 kg |
Un paquete que contiene elexacaftor 80 mg/tezacaftor 40 mg/ivacaftor 60 mg gránulos orales |
Un paquete que contiene ivacaftor 59.5 mg gránulos orales |
| 14 kg o más |
Un paquete que contiene elexacaftor 100 mg/tezacaftor 50 mg/ivacaftor 75 mg gránulos orales |
Un paquete que contiene ivacaftor 75 mg gránulos orales |
| 6 a menos de 12 años |
Menos de 30 kg |
Dos comprimidos, cada uno con elexacaftor 50 mg/tezacaftor 25 mg/ivacaftor 37.5 mg |
Un comprimido de ivacaftor 75 mg |
| 30 kg o más |
Dos comprimidos, cada uno con elexacaftor 100 mg/tezacaftor 50 mg/ivacaftor 75 mg |
Un comprimido de ivacaftor 150 mg |
| 12 años y mayores |
– |
Dos comprimidos, cada uno con elexacaftor 100 mg/tezacaftor 50 mg/ivacaftor 75 mg |
Un comprimido de ivacaftor 150 mg |
- No debe usarse en pacientes con insuficiencia hepática grave. No se recomienda su uso en pacientes con insuficiencia hepática moderada a menos que el beneficio supere el riesgo. Reducir la dosis si se usa en pacientes con insuficiencia hepática moderada. Se deben monitorear de cerca las pruebas de función hepática. (2.3, 5.1, 6, 8.7, 12.3)
- Consulte la información de prescripción completa para las modificaciones de la dosis debido a las interacciones medicamentosas con TRIKAFTA. (2.4, 5.4, 7.1, 12.3)
FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
Tabletas:
- Combinación de dosis fija que contiene elexacaftor 50 mg, tezacaftor 25 mg e ivacaftor 37.5 mg envasados conjuntamente con ivacaftor 75 mg;
- Combinación de dosis fija que contiene elexacaftor 100 mg, tezacaftor 50 mg e ivacaftor 75 mg envasados conjuntamente con ivacaftor 150 mg. (3)
Gránulos orales:
- Paquetes de dosis unitaria de elexacaftor 100 mg, tezacaftor 50 mg e ivacaftor 75 mg envasados conjuntamente con paquetes de dosis unitaria de ivacaftor 75 mg;
- Paquetes de dosis unitaria de elexacaftor 80 mg, tezacaftor 40 mg e ivacaftor 60 mg envasados conjuntamente con paquetes de dosis unitaria de ivacaftor 59.5 mg. (3)
ADVERTENCIAS Y PRECAUCIONES
-
Lesión hepática inducida por fármacos e insuficiencia hepática: TRIKAFTA puede causar lesión hepática inducida por fármacos grave y potencialmente mortal. Evalúe las pruebas de función hepática (ALT, AST, fosfatasa alcalina, bilirrubina) en todos los pacientes antes de iniciar y durante el tratamiento con TRIKAFTA. Interrumpa TRIKAFTA en caso de elevaciones significativas en las pruebas de función hepática o signos o síntomas de lesión hepática. TRIKAFTA no debe usarse en pacientes con insuficiencia hepática grave (Child-Pugh Clase C). No se recomienda TRIKAFTA en pacientes con insuficiencia hepática moderada (Child-Pugh Clase B). (2.1, 2.3, 5.1, 6, 8.7, 12.3)
-
Reacciones de hipersensibilidad: Se ha informado de angioedema y anafilaxia con TRIKAFTA en la fase posterior a la comercialización. Inicie la terapia adecuada en caso de una reacción de hipersensibilidad. (5.2)
-
Uso con inductores de CYP3A: El uso concomitante con inductores fuertes de CYP3A (p. ej., rifampicina, hierba de San Juan) disminuye significativamente la exposición a ivacaftor y se espera que disminuya la exposición a elexacaftor y tezacaftor, lo que puede reducir la eficacia de TRIKAFTA. Por lo tanto, no se recomienda el uso concomitante. (5.3, 7.1, 12.3)
-
Cataratas: Se han notificado opacidades/cataratas del cristalino no congénitas en pacientes pediátricos tratados con regímenes que contienen ivacaftor. Se recomiendan exámenes iniciales y de seguimiento en pacientes pediátricos que inician el tratamiento con TRIKAFTA. (5.5, 8.4)
REACCIONES ADVERSAS
Las reacciones adversas más comunes a TRIKAFTA (≥5% de los pacientes y con una frecuencia mayor que el placebo en ≥1%) fueron dolor de cabeza, infección de las vías respiratorias superiores, dolor abdominal, diarrea, erupción cutánea, aumento de la alanina aminotransferasa, congestión nasal, aumento de la creatina fosfoquinasa en sangre, aumento de la aspartato aminotransferasa, rinorrea, rinitis, influenza, sinusitis, aumento de la bilirrubina en sangre y estreñimiento. (6.1)
Para informar REACCIONES ADVERSAS SOSPECHOSAS, comuníquese con Vertex Pharmaceuticals Incorporated al 1-877-634-8789 o con la FDA al 1-800-FDA-1088 o www.fda.gov/medwatch.
INTERACCIONES CON LA DROGA
- Inductores fuertes de CYP3A: Evite el uso concomitante. (5.3, 7.1, 12.3)
- Inhibidores fuertes o moderados de CYP3A: Reduzca la dosis de TRIKAFTA cuando se use concomitantemente. Evite los alimentos o bebidas que contengan pomelo. (2.4, 5.4, 7.1, 12.3)
Consulte 17 para obtener INFORMACIÓN DE ASESORAMIENTO AL PACIENTE y la Guía del Medicamento.
Revisado: 12/2024
Tabla de contenido
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDO*
ADVERTENCIA EN EL RECUADRO
ADVERTENCIA: LESIÓN HEPÁTICA Y FALLO HEPÁTICO INDUCIDOS POR FÁRMACOS
TRIKAFTA puede causar lesión hepática inducida por fármacos grave y potencialmente mortal. Se han notificado casos de insuficiencia hepática que llevaron a un trasplante y a la muerte en pacientes con y sin antecedentes de enfermedad hepática que tomaban TRIKAFTA, tanto en ensayos clínicos como en la fase posterior a la comercialización [véase Reacciones adversas (6)]. Se ha notificado lesión hepática en el primer mes de tratamiento y hasta 15 meses después del inicio del tratamiento con TRIKAFTA.
Evalúe las pruebas de función hepática (ALT, AST, fosfatasa alcalina y bilirrubina) en todos los pacientes antes de iniciar el tratamiento con TRIKAFTA. Evalúe las pruebas de función hepática cada mes durante los primeros 6 meses de tratamiento, luego cada 3 meses durante los siguientes 12 meses y, posteriormente, al menos una vez al año. Considere un control más frecuente para los pacientes con antecedentes de enfermedad hepática o elevaciones de las pruebas de función hepática en el momento basal [véase Posología y administración (2.1), Advertencias y precauciones (5.1), Reacciones adversas (6) y Uso en poblaciones específicas (8.7)].
Interrumpa el tratamiento con TRIKAFTA en caso de elevaciones significativas de las pruebas de función hepática o en caso de signos o síntomas de lesión hepática. Considere la derivación a un hepatólogo. Realice un seguimiento estrecho de los pacientes mediante monitorización clínica y de laboratorio hasta que las anomalías se resuelvan. Si las anomalías se resuelven, reanude el tratamiento solo si se espera que el beneficio supere el riesgo. Se recomienda una monitorización más estrecha después de reanudar el tratamiento con TRIKAFTA [véase Advertencias y precauciones (5.1)].
TRIKAFTA no debe utilizarse en pacientes con insuficiencia hepática grave (Child-Pugh clase C). No se recomienda el uso de TRIKAFTA en pacientes con insuficiencia hepática moderada (Child-Pugh clase B). Si se utiliza, hágalo con precaución a una dosis reducida y controle estrechamente a los pacientes [véase Posología y administración (2.3), Advertencias y precauciones (5.1), Reacciones adversas (6), Uso en poblaciones específicas (8.7) y Farmacología clínica (12.3)].
1 INDICACIONES Y USO
TRIKAFTA está indicado para el tratamiento de la fibrosis quística (FQ) en pacientes de 2 años de edad o mayores que tengan al menos una mutación F508del en el gen del regulador de la conductancia transmembrana de la fibrosis quística (CFTR) o una mutación en el gen CFTR que sea sensible según datos clínicos y/o in vitro (véase Tabla 6) [véase Farmacología Clínica (12.1)].
Si se desconoce el genotipo del paciente, se debe utilizar una prueba de mutación de FQ aprobada por la FDA para confirmar la presencia de al menos una mutación indicada [véase Farmacología Clínica (12.1)].
2 DOSIS Y ADMINISTRACIÓN
2.1 Pruebas de laboratorio recomendadas antes de iniciar TRIKAFTA y durante el tratamiento
Antes de iniciar el tratamiento con TRIKAFTA, obtenga pruebas de función hepática (ALT, AST, fosfatasa alcalina y bilirrubina) para todos los pacientes. Controle las pruebas de función hepática mensualmente durante los primeros 6 meses de tratamiento, luego cada 3 meses durante los siguientes 12 meses y, posteriormente, al menos una vez al año. Considere un control más frecuente para pacientes con antecedentes de enfermedad hepática o elevaciones en las pruebas de función hepática al inicio del tratamiento [ver Advertencias y precauciones (5.1) y Uso en poblaciones específicas (8.7)].
2.2 Dosis recomendada en adultos y pacientes pediátricos de 2 años o más
La dosis recomendada para adultos y pacientes pediátricos de 2 años o más se proporciona en la Tabla 1. Administre los comprimidos de TRIKAFTA (trague los comprimidos enteros) o los gránulos orales por vía oral con alimentos que contengan grasa, por la mañana y por la noche, con aproximadamente 12 horas de diferencia. Ejemplos de comidas o refrigerios que contienen grasa son aquellos preparados con mantequilla o aceites, o aquellos que contienen huevos, mantequilla de cacahuete, quesos, frutos secos, leche entera o carnes [ver Farmacología clínica (12.3)].
Administre cada dosis de gránulos orales de TRIKAFTA inmediatamente antes o después de la ingestión de alimentos que contengan grasa. Mezcle todo el contenido de cada sobre de gránulos orales con una cucharadita (5 mL) de alimento blando o líquido apropiado para la edad que esté a temperatura ambiente o inferior. Algunos ejemplos de alimentos blandos o líquidos incluyen puré de frutas o verduras, yogur, puré de manzana, agua, leche o zumo. Una vez mezclado, el producto debe consumirse completamente en una hora.
2.3 Dosis recomendada para pacientes con insuficiencia hepática
2.4 Modificación de la dosis para pacientes que toman medicamentos que son inhibidores de CYP3A
La Tabla 3 describe la modificación de la dosis recomendada para TRIKAFTA cuando se usa concomitantemente con inhibidores de CYP3A fuertes (por ejemplo, ketoconazol, itraconazol, posaconazol, voriconazol, telitromicina y claritromicina) o moderados (por ejemplo, fluconazol, eritromicina). Administrar TRIKAFTA por vía oral con alimentos que contengan grasa [ver Dosificación y administración (2.2)]. Evitar alimentos o bebidas que contengan toronja durante el tratamiento con TRIKAFTA [ver Advertencias y precauciones (5.4), Interacciones medicamentosas (7.1) y Farmacología clínica (12.3)].
2.5 Recomendaciones sobre dosis olvidada(s)
Si han transcurrido 6 horas o menos desde la dosis matutina o vespertina olvidada, el paciente debe tomar la dosis olvidada lo antes posible y continuar con el horario original.
Si han transcurrido más de 6 horas desde:
- la dosis matutina olvidada, el paciente debe tomar la dosis olvidada lo antes posible y no debe tomar la dosis vespertina. La siguiente dosis matutina programada debe tomarse a la hora habitual.
- la dosis vespertina olvidada, el paciente no debe tomar la dosis olvidada. La siguiente dosis matutina programada debe tomarse a la hora habitual.
Las dosis matutina y vespertina no deben tomarse al mismo tiempo.
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
Tabletas:
Combinación de dosis fija que contiene elexacaftor 50 mg, tezacaftor 25 mg e ivacaftor 37.5 mg, envasadas conjuntamente con ivacaftor 75 mg:
- Las tabletas de elexacaftor, tezacaftor e ivacaftor son de color naranja claro, oblongas y llevan grabado “T50” en un lado y lisas en el otro.
- Las tabletas de ivacaftor son de color azul claro, oblongas y llevan impresa “V 75” con tinta negra en un lado y lisas en el otro.
Combinación de dosis fija que contiene elexacaftor 100 mg, tezacaftor 50 mg e ivacaftor 75 mg, envasadas conjuntamente con ivacaftor 150 mg:
- Las tabletas de elexacaftor, tezacaftor e ivacaftor son de color naranja, oblongas y llevan grabado “T100” en un lado y lisas en el otro.
- Las tabletas de ivacaftor son de color azul claro, oblongas y llevan impresa “V 150” con tinta negra en un lado y lisas en el otro.
Gránulos orales:
Gránulos orales de combinación de dosis fija que contienen elexacaftor 100 mg, tezacaftor 50 mg e ivacaftor 75 mg, envasados conjuntamente con gránulos orales de ivacaftor 75 mg:
- Los gránulos orales de elexacaftor, tezacaftor e ivacaftor son de color blanco a blanquecino, endulzados, sin sabor, de aproximadamente 2 mm de diámetro, contenidos en un sobre unidosis blanco y naranja.
- Los gránulos orales de ivacaftor son de color blanco a blanquecino, endulzados, sin sabor, de aproximadamente 2 mm de diámetro, contenidos en un sobre unidosis blanco y rosa.
Gránulos orales de combinación de dosis fija que contienen elexacaftor 80 mg, tezacaftor 40 mg e ivacaftor 60 mg, envasados conjuntamente con gránulos orales de ivacaftor 59.5 mg:
- Los gránulos orales de elexacaftor, tezacaftor e ivacaftor son de color blanco a blanquecino, endulzados, sin sabor, de aproximadamente 2 mm de diámetro, contenidos en un sobre unidosis blanco y azul.
- Los gránulos orales de ivacaftor son de color blanco a blanquecino, endulzados, sin sabor, de aproximadamente 2 mm de diámetro, contenidos en un sobre unidosis blanco y verde.
4 CONTRAINDICACIONES
Ninguna.
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Lesión Hepática inducida por fármacos e insuficiencia hepática
TRIKAFTA puede causar lesión hepática inducida por fármacos, grave y potencialmente mortal. Se han notificado casos de insuficiencia hepática que llevaron a un trasplante y a la muerte en pacientes con y sin antecedentes de enfermedad hepática que tomaban TRIKAFTA, tanto en ensayos clínicos como en la fase posterior a la comercialización [véase Reacciones adversas (6)]. Se ha notificado lesión hepática en el primer mes de tratamiento y hasta 15 meses después del inicio del tratamiento con TRIKAFTA.
Evalúe las pruebas de función hepática (ALT, AST, fosfatasa alcalina y bilirrubina) en todos los pacientes antes de iniciar el tratamiento con TRIKAFTA. Evalúe las pruebas de función hepática cada mes durante los primeros 6 meses de tratamiento, luego cada 3 meses durante los siguientes 12 meses y, posteriormente, al menos una vez al año. Considere un control más frecuente para los pacientes con antecedentes de enfermedad hepática o elevaciones de las pruebas de función hepática en el momento basal [véase Posología y administración (2.1), Reacciones adversas (6) y Uso en poblaciones específicas (8.7)].
Interrumpa el tratamiento con TRIKAFTA en caso de signos o síntomas de lesión hepática. Estos pueden incluir:
-
Elevaciones significativas de las pruebas de función hepática (p. ej., ALT o AST >5 × el límite superior de la normalidad (LSN) o ALT o AST >3 × LSN con bilirrubina >2 × LSN)
-
Síntomas clínicos que sugieran lesión hepática (p. ej., ictericia, dolor en el cuadrante superior derecho, náuseas, vómitos, alteración del estado mental, ascitis).
Considere la derivación a un hepatólogo y realice un seguimiento estrecho de los pacientes con control clínico y de laboratorio hasta que las anomalías se resuelvan. Si las anomalías se resuelven y si se espera que el beneficio supere el riesgo, reanude el tratamiento con TRIKAFTA con un control estrecho.
TRIKAFTA no debe utilizarse en pacientes con insuficiencia hepática grave (Child-Pugh clase C). TRIKAFTA no se recomienda en pacientes con insuficiencia hepática moderada (Child-Pugh clase B) y solo debe considerarse cuando exista una clara necesidad médica y el beneficio supere el riesgo. Si se utiliza, hágalo con precaución a una dosis reducida y controle estrechamente a los pacientes [véase Posología y administración (2.3), Uso en poblaciones específicas (8.7) y Farmacología clínica (12.3)].
5.2 Reacciones de hipersensibilidad, incluida la anafilaxia
Se han notificado reacciones de hipersensibilidad, incluidos casos de angioedema y anafilaxia, en la fase posterior a la comercialización [véase Reacciones adversas (6.2)]. Si aparecen signos o síntomas de reacciones de hipersensibilidad graves durante el tratamiento, interrumpa el tratamiento con TRIKAFTA e instaurar el tratamiento adecuado. Considere los beneficios y los riesgos para cada paciente para determinar si se debe reanudar el tratamiento con TRIKAFTA.
5.3 Uso concomitante con inductores del CYP3A
La exposición a ivacaftor disminuye significativamente y se espera que la exposición a elexacaftor y tezacaftor disminuya con el uso concomitante de inductores potentes del CYP3A, lo que puede reducir la eficacia terapéutica de TRIKAFTA. Por lo tanto, no se recomienda el uso concomitante con inductores potentes del CYP3A [véase Interacciones medicamentosas (7.1) y Farmacología clínica (12.3)].
5.5 Cataratas
Se han notificado casos de opacidades del cristalino no congénitas en pacientes pediátricos tratados con regímenes que contienen ivacaftor. Aunque en algunos casos existían otros factores de riesgo (como el uso de corticosteroides, la exposición a la radiación), no se puede excluir un posible riesgo atribuible al tratamiento con ivacaftor. Se recomiendan exploraciones oftalmológicas iniciales y de seguimiento en pacientes pediátricos que inicien el tratamiento con TRIKAFTA [véase Uso en poblaciones específicas (8.4)].
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas clínicamente significativas se describen con mayor detalle en otras secciones del etiquetado:
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se realizan en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no se pueden comparar directamente con las tasas en los ensayos clínicos de otro fármaco y pueden no reflejar las tasas observadas en la práctica clínica.
Pacientes con fibrosis quística con al menos una mutación F508del
El perfil de seguridad de TRIKAFTA en pacientes con fibrosis quística con al menos una mutación F508del se basa en datos de 510 pacientes de 12 años o más en dos ensayos doble ciego controlados con placebo de 24 semanas y 4 semanas de duración del tratamiento (Ensayos 1 y 2, respectivamente). Los pacientes elegibles también pudieron participar en un estudio de seguridad de extensión abierta (hasta 96 semanas de TRIKAFTA). En los dos ensayos controlados, un total de 257 pacientes de 12 años o más recibieron al menos una dosis de TRIKAFTA.
En el Ensayo 1, la proporción de pacientes que interrumpieron el fármaco del estudio prematuramente debido a eventos adversos fue del 1% para los pacientes tratados con TRIKAFTA y del 0% para los pacientes tratados con placebo.
En el Ensayo 1, las reacciones adversas graves que ocurrieron con mayor frecuencia en los pacientes tratados con TRIKAFTA en comparación con el placebo fueron erupción cutánea (1% vs <1%) e influenza (1% vs 0%). No hubo muertes.
La Tabla 4 muestra las reacciones adversas que ocurrieron en ≥5% de los pacientes tratados con TRIKAFTA y superiores al placebo en ≥1% en el ensayo de 24 semanas, controlado con placebo y de grupos paralelos (Ensayo 1).
Otras reacciones adversas que ocurrieron en pacientes tratados con TRIKAFTA con una frecuencia del 2% al <5% y superior al placebo en ≥1% incluyen las siguientes: flatulencia, distensión abdominal, conjuntivitis, faringitis, infección del tracto respiratorio, amigdalitis, infección del tracto urinario, aumento de la proteína C reactiva, hipoglucemia, mareos, dismenorrea, acné, eccema y prurito.
Además, también se han realizado los siguientes ensayos clínicos [ver Uso en poblaciones específicas (8.4), Farmacología clínica (12.3) y Estudios clínicos (14)]:
- un ensayo abierto de 24 semanas en 66 pacientes con CF de 6 a menos de 12 años de edad que eran homocigotos para la mutación F508del o heterocigotos para la mutación F508del, y una mutación en el segundo alelo que da como resultado una proteína CFTR no funcional o una proteína CFTR que no responde a ivacaftor y tezacaftor/ivacaftor (Ensayo 3).
- un ensayo abierto de 24 semanas en 75 pacientes con CF de 2 a menos de 6 años de edad. Los pacientes que tenían al menos una mutación F508del o una mutación conocida por responder a TRIKAFTA fueron elegibles para el estudio (Ensayo 4).
El perfil de seguridad para los pacientes con CF inscritos en los Ensayos 2, 3 y 4 fue consistente con el observado en el Ensayo 1.
Pacientes con fibrosis quística con al menos una mutación no-F508del que califica
La seguridad de TRIKAFTA en pacientes con CF con al menos una mutación no-F508del se basa en datos de 307 pacientes de 6 años o más con al menos una mutación CFTR no-F508del que califica y que respondió a TRIKAFTA. El Ensayo 5 fue un ensayo aleatorizado, doble ciego, controlado con placebo con una duración del tratamiento de 24 semanas en el que 205 pacientes recibieron al menos una dosis de TRIKAFTA. Los pacientes elegibles también pudieron participar en un estudio de seguridad de extensión abierta.
En el Ensayo 5, la proporción de pacientes que interrumpieron el fármaco del estudio prematuramente debido a reacciones adversas fue del 2% para los pacientes tratados con TRIKAFTA y del 0% para los pacientes tratados con placebo.
La Tabla 5 muestra las reacciones adversas que ocurrieron en ≥5% de los pacientes tratados con TRIKAFTA y superiores al placebo en ≥1% en el ensayo de 24 semanas, controlado con placebo y de grupos paralelos (Ensayo 5).
Reacciones adversas específicas
Elevaciones de las pruebas de función hepática
En el Ensayo 1, la incidencia de transaminasas máximas (ALT o AST) >8, >5 o >3 × LSN fue del 1 %, 2 % y 8 % en pacientes tratados con TRIKAFTA y del 1 %, 1 % y 5 % en pacientes tratados con placebo. La incidencia de reacciones adversas de elevaciones de transaminasas (AST y/o ALT) fue del 11 % en pacientes tratados con TRIKAFTA y del 4 % en pacientes tratados con placebo.
En el Ensayo 1, la incidencia de elevación máxima de bilirrubina total >2 × LSN fue del 4 % en pacientes tratados con TRIKAFTA y <1 % en pacientes tratados con placebo. Las elevaciones máximas de bilirrubina indirecta y directa >1,5 × LSN se produjeron en el 11 % y el 3 % de los pacientes tratados con TRIKAFTA, respectivamente. Ningún paciente tratado con TRIKAFTA presentó una elevación máxima de bilirrubina directa >2 × LSN.
Durante el Ensayo 3, en pacientes de 6 a menos de 12 años, la incidencia de transaminasas máximas (ALT o AST) >8, >5 y >3 × LSN fue del 0 %, 1,5 % y 10,6 %, respectivamente. Ningún paciente tratado con TRIKAFTA presentó una elevación de transaminasas >3 × LSN asociada con una elevación de bilirrubina total >2 × LSN o interrumpió el tratamiento debido a elevaciones de transaminasas.
Durante el Ensayo 4 en pacientes de 2 a menos de 6 años, la incidencia de transaminasas máximas (ALT o AST) >8, >5 y >3 × LSN fue del 1,3 %, 2,7 % y 8,0 %, respectivamente. Ningún paciente tratado con TRIKAFTA presentó una elevación de transaminasas >3 × LSN asociada con una elevación de bilirrubina total >2 × LSN. Un paciente requirió la interrupción del tratamiento durante el Ensayo 4 y posteriormente interrumpió el tratamiento con TRIKAFTA durante la extensión de etiqueta abierta debido a elevaciones de transaminasas.
En el Ensayo 5, la incidencia de transaminasas máximas (ALT o AST) >8, >5 y >3 × LSN fue del 2,0 %, 2,0 % y 6,3 %, respectivamente, y provocó la interrupción del tratamiento en el 0,5 % y las interrupciones del tratamiento en el 1,5 % de los pacientes tratados con TRIKAFTA. No hubo elevaciones de transaminasas >3 × LSN en los pacientes tratados con placebo.
Erupción cutánea
En el Ensayo 1, la incidencia general de erupción cutánea fue del 10 % en pacientes tratados con TRIKAFTA y del 5 % en pacientes tratados con placebo (véase Tabla 4). La incidencia de erupción cutánea fue mayor en las pacientes mujeres tratadas con TRIKAFTA (16 %) que en los pacientes varones tratados con TRIKAFTA (5 %).
En el Ensayo 5, la incidencia general de erupción cutánea fue del 23 % en pacientes tratados con TRIKAFTA y del 2 % en pacientes tratados con placebo (véase Tabla 5). La incidencia de erupción cutánea fue mayor en las pacientes mujeres tratadas con TRIKAFTA (27 %) que en los pacientes varones tratados con TRIKAFTA (20 %).
No se puede descartar la influencia de los anticonceptivos hormonales en la aparición de erupciones cutáneas [véase Interacciones medicamentosas (7.3)].
Aumento de la creatina fosfocinasa
En el Ensayo 1, la incidencia de elevación máxima de creatina fosfocinasa >5 × LSN fue del 10 % en pacientes tratados con TRIKAFTA y del 5 % en pacientes tratados con placebo. Entre los pacientes tratados con TRIKAFTA con elevación de creatina fosfocinasa >5 × LSN, el 14 % (3/21) requirió la interrupción del tratamiento y ninguno interrumpió el tratamiento.
En el Ensayo 5, la incidencia de elevación máxima de creatina fosfocinasa >5 × LSN fue del 5,4 % (11/205) en pacientes tratados con TRIKAFTA y del 1 % (1/102) en pacientes tratados con placebo. La incidencia de elevación máxima de creatina fosfocinasa >10 × LSN fue del 2,4 % (5/205) en pacientes tratados con TRIKAFTA y del 1 % (1/102) en pacientes tratados con placebo. No hubo interrupciones ni suspensiones entre los pacientes tratados con TRIKAFTA con elevación de creatina fosfocinasa >5 × LSN. Entre los pacientes tratados con TRIKAFTA con elevación de creatina fosfocinasa >10 × LSN, dos pacientes, que habían realizado ejercicio en las 72 horas anteriores, presentaron rabdomiólisis sin evidencia de afectación renal, lo que provocó la interrupción del tratamiento en 1 paciente.
Aumento de la presión arterial
En el Ensayo 1, el aumento máximo con respecto a los valores basales en la presión arterial sistólica y diastólica media fue de 3,5 mmHg y 1,9 mmHg, respectivamente, para los pacientes tratados con TRIKAFTA (valor basal: 113 mmHg sistólica y 69 mmHg diastólica) y de 0,9 mmHg y 0,5 mmHg, respectivamente, para los pacientes tratados con placebo (valor basal: 114 mmHg sistólica y 70 mmHg diastólica).
La proporción de pacientes que presentaron una presión arterial sistólica >140 mmHg y un aumento de 10 mmHg con respecto a los valores basales en al menos dos ocasiones fue del 4 % en los pacientes tratados con TRIKAFTA y del 1 % en los pacientes tratados con placebo. La proporción de pacientes que presentaron una presión arterial diastólica >90 mmHg y un aumento de 5 mmHg con respecto a los valores basales en al menos dos ocasiones fue del 1 % en los pacientes tratados con TRIKAFTA y del 2 % en los pacientes tratados con placebo.
Con la excepción de las diferencias de sexo en la erupción cutánea, el perfil de seguridad de TRIKAFTA fue generalmente similar en todos los subgrupos de pacientes, incluido el análisis por edad, sexo, porcentaje predicho basal de FEV1 (ppFEV1) y regiones geográficas.
6.2 Experiencia postcomercialización
Las siguientes reacciones adversas se han identificado durante el uso posterior a la aprobación de TRIKAFTA. Debido a que estas reacciones se notifican voluntariamente a partir de una población de tamaño incierto, no siempre es posible estimar su frecuencia de forma fiable o establecer una relación causal con la exposición al fármaco.
Hepatobiliar: lesión hepática, insuficiencia hepática mortal, trasplante de hígado
Trastornos del sistema inmunitario: anafilaxia, angioedema
7 INTERACCIONES MEDICAMENTOSAS
7.1 Efecto de Otros Medicamentos y la Toronja/Pomelo sobre TRIKAFTA
Inductores Fuertes del CYP3A
No se recomienda el uso concomitante de TRIKAFTA con inductores fuertes del CYP3A. Elexacaftor, tezacaftor e ivacaftor son sustratos del CYP3A (ivacaftor es un sustrato sensible del CYP3A). El uso concomitante de inductores del CYP3A puede resultar en exposiciones reducidas y, por lo tanto, en una eficacia reducida de TRIKAFTA [ver Advertencias y precauciones (5.3)]. El uso concomitante de ivacaftor con rifampicina, un inductor fuerte del CYP3A, disminuyó significativamente el área bajo la curva (AUC) de ivacaftor en un 89%. Se espera que las exposiciones a elexacaftor y tezacaftor disminuyan durante el uso concomitante con inductores fuertes del CYP3A [ver Farmacología Clínica (12.3)].
Ejemplos de inductores fuertes del CYP3A incluyen:
- rifampicina, rifabutina, fenobarbital, carbamazepina, fenitoína y hierba de San Juan (Hypericum perforatum)
Inhibidores Fuertes o Moderados del CYP3A
La dosis de TRIKAFTA debe reducirse cuando se usa concomitantemente con inhibidores fuertes del CYP3A [ver Dosis y Administración (2.4) y Advertencias y precauciones (5.4)]. El uso concomitante con itraconazol, un inhibidor fuerte del CYP3A, aumentó el AUC de elexacaftor en 2.8 veces y el AUC de tezacaftor en 4.0 a 4.5 veces. Cuando se usa concomitantemente con itraconazol y ketoconazol, el AUC de ivacaftor aumentó en 15.6 veces y 8.5 veces, respectivamente [ver Farmacología Clínica (12.3)].
Ejemplos de inhibidores fuertes del CYP3A incluyen:
- ketoconazol, itraconazol, posaconazol y voriconazol
- telitromicina y claritromicina
La dosis de TRIKAFTA debe reducirse cuando se usa concomitantemente con inhibidores moderados del CYP3A [ver Dosis y Administración (2.4) y Advertencias y precauciones (5.4)]. Las simulaciones indicaron que el uso concomitante con inhibidores moderados del CYP3A puede aumentar el AUC de elexacaftor y tezacaftor en aproximadamente 1.9 a 2.3 veces y 2.1 veces, respectivamente. El uso concomitante de fluconazol aumentó el AUC de ivacaftor en 2.9 veces [ver Farmacología Clínica (12.3)].
Ejemplos de inhibidores moderados del CYP3A incluyen:
Toronja/Pomelo
El uso concomitante de TRIKAFTA con jugo de toronja/pomelo, que contiene uno o más componentes que inhiben moderadamente el CYP3A, puede aumentar la exposición a elexacaftor, tezacaftor e ivacaftor; por lo tanto, se deben evitar los alimentos o bebidas que contengan toronja/pomelo durante el tratamiento con TRIKAFTA [ver Dosis y Administración (2.4)].
7.2 Efecto de TRIKAFTA sobre Otros Medicamentos
Sustratos del CYP2C9
Ivacaftor puede inhibir el CYP2C9; por lo tanto, se recomienda la monitorización del índice internacional normalizado (INR) durante el uso concomitante de TRIKAFTA con warfarina. Otros medicamentos para los cuales la exposición puede aumentar con TRIKAFTA incluyen glimepirida y glipizida; estos medicamentos deben usarse con precaución [ver Farmacología Clínica (12.3)].
Transportadores
El uso concomitante de ivacaftor o tezacaftor/ivacaftor con digoxina, un sustrato sensible de la P-gp, aumentó el AUC de la digoxina en 1.3 veces, lo que concuerda con una inhibición débil de la P-gp por ivacaftor. La administración de TRIKAFTA puede aumentar la exposición sistémica de los medicamentos que son sustratos sensibles de la P-gp, lo que puede aumentar o prolongar su efecto terapéutico y las reacciones adversas. Cuando se usa concomitantemente con digoxina u otros sustratos de la P-gp con un índice terapéutico estrecho, como ciclosporina, everolimus, sirolimus y tacrolimus, se debe tener precaución y una monitorización adecuada [ver Farmacología Clínica (12.3)].
Elexacaftor y M23-ELX inhiben la captación por OATP1B1 y OATP1B3 in vitro. El uso concomitante de TRIKAFTA puede aumentar las exposiciones de los medicamentos que son sustratos de estos transportadores, como estatinas, gliburida, nateglinida y repaglinida. Cuando se usa concomitantemente con sustratos de OATP1B1 o OATP1B3, se debe tener precaución y una monitorización adecuada [ver Farmacología Clínica (12.3)]. La bilirrubina es un sustrato de OATP1B1 y OATP1B3.
7.3 Medicamentos sin Interacciones Clínicamente Significativas con TRIKAFTA
Ciprofloxacino
Ciprofloxacino no tuvo un efecto clínicamente relevante sobre la exposición a tezacaftor o ivacaftor y no se espera que afecte la exposición a elexacaftor. Por lo tanto, no es necesario ajustar la dosis durante la administración concomitante de TRIKAFTA con ciprofloxacino [ver Farmacología Clínica (12.3)].
Anticonceptivos Hormonales
TRIKAFTA se ha estudiado con etinilestradiol/levonorgestrel y se encontró que no tiene un efecto clínicamente relevante sobre las exposiciones del anticonceptivo oral. No se espera que TRIKAFTA tenga un impacto en la eficacia de los anticonceptivos orales.
Los anticonceptivos hormonales pueden desempeñar un papel en la aparición de erupciones cutáneas y no se pueden excluir [ver Reacciones Adversas (6.1)]. Para los pacientes con FQ que toman anticonceptivos hormonales y desarrollan erupciones cutáneas, considere interrumpir TRIKAFTA y los anticonceptivos hormonales. Después de la resolución de la erupción cutánea, considere reanudar TRIKAFTA sin los anticonceptivos hormonales. Si la erupción no reaparece, se puede considerar la reanudación de los anticonceptivos hormonales.
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de Riesgo
Existen datos humanos limitados e incompletos de ensayos clínicos sobre el uso de TRIKAFTA o sus componentes individuales, elexacaftor, tezacaftor e ivacaftor, en mujeres embarazadas para informar sobre un riesgo asociado al fármaco. Aunque no hay estudios de reproducción animal con la administración concomitante de elexacaftor, tezacaftor e ivacaftor, se realizaron estudios de reproducción y desarrollo por separado con cada componente activo de TRIKAFTA en ratas y conejas embarazadas.
En estudios de desarrollo embriofetal (EFD) en animales, la administración oral de elexacaftor a ratas y conejas embarazadas durante la organogénesis no demostró efectos adversos en el desarrollo a dosis que produjeron exposiciones maternas hasta aproximadamente 2 veces la exposición a la dosis máxima recomendada en humanos (MRHD) en ratas y 4 veces la MRHD en conejas [basado en AUC sumadas de elexacaftor y su metabolito (para rata) y AUC de elexacaftor (para coneja)]. La administración oral de tezacaftor a ratas y conejas embarazadas durante la organogénesis no demostró efectos adversos en el desarrollo a dosis que produjeron exposiciones maternas hasta aproximadamente 3 veces la exposición a la MRHD en ratas y 0,2 veces la MRHD en conejas (basado en AUC sumadas de tezacaftor y M1-TEZ). La administración oral de ivacaftor a ratas y conejas embarazadas durante la organogénesis no demostró efectos adversos en el desarrollo a dosis que produjeron exposiciones maternas hasta aproximadamente 5 y 14 veces la exposición a la MRHD, respectivamente [basado en AUC sumadas de ivacaftor y sus metabolitos (para rata) y AUC de ivacaftor (para coneja)]. No se observaron efectos adversos en el desarrollo después de la administración oral de elexacaftor, tezacaftor o ivacaftor a ratas embarazadas desde el período de organogénesis hasta la lactancia a dosis que produjeron exposiciones maternas aproximadamente 1 vez, aproximadamente 1 vez y 3 veces las exposiciones a la MRHD, respectivamente [basado en AUC sumadas de compuesto principal y metabolito(s)] (ver Datos).
Se desconoce el riesgo de fondo de defectos congénitos importantes y aborto espontáneo para la población indicada. En la población general de EE. UU., el riesgo de fondo estimado de defectos congénitos importantes y aborto espontáneo en embarazos clínicamente reconocidos es del 2% al 4% y del 15% al 20%, respectivamente.
Datos
Datos en animales
Elexacaftor
En un estudio de EFD, se administraron a ratas embarazadas dosis orales de elexacaftor a 15, 25 y 40 mg/kg/día durante el período de organogénesis desde el día 6 hasta el 17 de gestación. El elexacaftor no causó resultados de desarrollo adversos a exposiciones de hasta 9 veces la MRHD (basado en AUC sumadas para elexacaftor y su metabolito a dosis maternas de hasta 40 mg/kg/día). Se observaron pesos fetales medios más bajos a dosis ≥25 mg/kg/día que produjeron exposiciones maternas ≥4 veces la MRHD. Se observó toxicidad materna a 40 mg/kg/día (9 veces la MRHD). En un estudio de EFD, se administraron a conejas embarazadas dosis orales de elexacaftor a 50, 100 o 125 mg/kg/día durante el período de organogénesis desde el día 7 hasta el 20 de gestación. El elexacaftor no fue teratógeno a exposiciones de hasta 4 veces la MRHD (basado en el AUC de elexacaftor a dosis maternas de hasta 125 mg/kg/día). Se observó toxicidad materna a 125 mg/kg/día (4 veces la MRHD). En un estudio de desarrollo pre y postnatal (PPND), se administraron a ratas embarazadas dosis orales de elexacaftor a 5, 7,5 y 10 mg/kg/día desde el día 6 de gestación hasta el día 18 de lactancia. El elexacaftor no causó resultados de desarrollo adversos en las crías a dosis maternas de hasta 10 mg/kg/día (aproximadamente 1 vez la MRHD basada en AUC sumadas de elexacaftor y su metabolito). Se observó transferencia placentaria de elexacaftor en ratas embarazadas.
Tezacaftor
En un estudio de EFD, se administró tezacaftor a ratas embarazadas a dosis orales de 25, 50 o 100 mg/kg/día durante el período de organogénesis desde el día 6 hasta el 17 de gestación. El tezacaftor no causó efectos adversos en el desarrollo a exposiciones de hasta 3 veces la MRHD (basado en AUC sumadas de tezacaftor y M1-TEZ). Se observó toxicidad materna en ratas a más de 50 mg/kg/día (aproximadamente mayor o igual a 1 vez la MRHD). En un estudio de EFD, se administró tezacaftor a conejas embarazadas a dosis orales de 10, 25 o 50 mg/kg/día durante el período de organogénesis desde el día 7 hasta el 20 de gestación. El tezacaftor no afectó los resultados del desarrollo fetal a exposiciones de hasta 0,2 veces la MRHD (basado en AUC sumadas de tezacaftor y M1-TEZ). Se observaron pesos fetales más bajos en conejas a una dosis tóxica para la madre que produjo exposiciones aproximadamente 1 vez la MRHD (basado en AUC sumadas de tezacaftor y M1-TEZ a una dosis materna de 50 mg/kg/día). En un estudio de PPND, se administró tezacaftor a ratas embarazadas a dosis orales de 25, 50 o 100 mg/kg/día desde el día 6 de gestación hasta el día 18 de lactancia. El tezacaftor no tuvo efectos adversos en el desarrollo de las crías a una exposición de aproximadamente 1 vez la MRHD (basado en AUC sumadas para tezacaftor y M1-TEZ a una dosis materna de 25 mg/kg/día). Se produjeron disminuciones en el peso corporal fetal y retrasos en el desarrollo temprano en el desprendimiento del pabellón auricular, la apertura de los ojos y el reflejo de enderezamiento a una dosis tóxica para la madre (basado en la pérdida de peso materna) que produjo exposiciones aproximadamente 2 veces la exposición a la MRHD (basado en AUC sumadas para tezacaftor y M1-TEZ). Se observó transferencia placentaria de tezacaftor en ratas embarazadas.
Ivacaftor
En un estudio de EFD, se administraron ratas preñadas ivacaftor en dosis orales de 50, 100 o 200 mg/kg/día durante el período de organogénesis desde los días 7 a 17 de gestación. Ivacaftor no afectó la supervivencia fetal en exposiciones hasta 5 veces la MRHD (basado en la suma de las AUC de ivacaftor y sus metabolitos en dosis orales maternas de hasta 200 mg/kg/día). Se observó toxicidad materna a 100 y 200 mg/kg/día (3 y 5 veces la exposición a la MRHD) y se asoció con una disminución en el peso corporal fetal a una dosis materna de 200 mg/kg/día (5 veces la MRHD). En un estudio de EFD, se administraron conejas preñadas ivacaftor en dosis orales de 25, 50 o 100 mg/kg/día durante el período de organogénesis desde los días 7 a 19 de gestación. Ivacaftor no afectó el desarrollo o la supervivencia fetal en exposiciones de hasta 14 veces la MRHD (sobre la base del AUC de ivacaftor en dosis orales maternas de hasta 100 mg/kg/día). Se observó toxicidad materna (es decir, muerte, disminución del consumo de alimentos, disminución del peso corporal medio y del aumento de peso corporal, disminución del estado clínico, abortos) en dosis iguales o superiores a 50 mg/kg/día (aproximadamente 5 veces la MRHD). En un estudio de PPND, se administraron ratas preñadas ivacaftor en dosis orales de 50, 100 o 200 mg/kg/día desde el día 7 de gestación hasta el día 20 de lactancia. Ivacaftor no tuvo efectos en el parto ni en el crecimiento y desarrollo de la descendencia en exposiciones de hasta 3 veces la MRHD (basado en la suma de las AUC de ivacaftor y sus metabolitos en dosis orales maternas de hasta 100 mg/kg/día). Se observó una disminución del peso corporal fetal a una dosis tóxica para la madre que produjo exposiciones 5 veces superiores a la MRHD (basado en la suma de las AUC de ivacaftor y sus metabolitos). Se observó transferencia placentaria de ivacaftor en ratas y conejas preñadas.
8.2 Lactancia
Resumen de Riesgo
No hay información sobre la presencia de elexacaftor, tezacaftor o ivacaftor en la leche materna, los efectos en el lactante o los efectos sobre la producción de leche. Elexacaftor, tezacaftor e ivacaftor se excretan en la leche de ratas lactantes (ver Datos). Se deben considerar los beneficios para el desarrollo y la salud de la lactancia materna junto con la necesidad clínica de la madre de TRIKAFTA y cualquier posible efecto adverso en el niño amamantado por TRIKAFTA o por la enfermedad materna subyacente.
Datos
Elexacaftor: La excreción láctea de elexacaftor en ratas se demostró después de una dosis oral única (10 mg/kg) de 14C-elexacaftor administrada de 6 a 10 días después del parto a madres lactantes. La exposición a 14C-elexacaftor en la leche fue aproximadamente 0,4 veces el valor observado en el plasma (basado en AUC0-72h).
Tezacaftor: La excreción láctea de tezacaftor en ratas se demostró después de una dosis oral única (30 mg/kg) de 14C-tezacaftor administrada de 6 a 10 días después del parto a madres lactantes. La exposición a 14C-tezacaftor en la leche fue aproximadamente 3 veces mayor que en el plasma (basado en AUC0-72h).
Ivacaftor: La excreción láctea de ivacaftor en ratas se demostró después de una dosis oral única (100 mg/kg) de 14C-ivacaftor administrada de 9 a 10 días después del parto a madres lactantes. La exposición a 14C-ivacaftor en la leche fue aproximadamente 1,5 veces mayor que en el plasma (basado en AUC0-24h).
8.4 Uso Pediátrico
La seguridad y eficacia de TRIKAFTA para el tratamiento de la fibrosis quística se han establecido en pacientes pediátricos de 2 a menos de 18 años que tienen al menos una mutación F508del en el gen CFTR o una mutación en el gen CFTR que responde según datos clínicos y/o in vitro. El uso de TRIKAFTA para esta indicación en pacientes pediátricos de 12 años o más se basó en la evidencia de dos estudios adecuados y bien controlados (Estudios 1 y 2) en pacientes con fibrosis quística de 12 años o más [ver Reacciones adversas (6.1) y Estudios clínicos (14)].
El uso de TRIKAFTA para esta indicación en pacientes pediátricos de 2 a menos de 12 años se basa en lo siguiente:
- Ensayo 1, 56 pacientes pediátricos de 12 a menos de 18 años que tenían una mutación F508del en un alelo y una mutación en el segundo alelo que resulta en ninguna proteína CFTR o una proteína CFTR que no responde a ivacaftor y tezacaftor/ivacaftor [ver Reacciones adversas (6) y Estudios clínicos (14)].
- Ensayo 2, 16 pacientes pediátricos de 12 a menos de 18 años que eran homocigotos para la mutación F508del [ver Reacciones adversas (6) y Estudios clínicos (14)].
- Ensayo 3, 66 pacientes pediátricos de 6 a menos de 12 años que eran homocigotos para la mutación F508del o heterocigotos para la mutación F508del con una mutación en el segundo alelo que resulta en ninguna proteína CFTR o una proteína CFTR que no responde a ivacaftor y tezacaftor/ivacaftor [ver Reacciones adversas (6) y Farmacología clínica (12.3)].
- Ensayo 4, 75 pacientes pediátricos de 2 a menos de 6 años que tenían al menos una mutación F508del o una mutación conocida por responder a TRIKAFTA [ver Reacciones adversas (6) y Farmacología clínica (12.3)].
- Ensayo 5, 64 pacientes pediátricos de 6 a menos de 18 años que tenían al menos una mutación TRIKAFTA-responsiva no-F508del calificadora y no tenían una mutación excluyente [ver Reacciones adversas (6) y Estudios clínicos (14.2)].
La eficacia de TRIKAFTA en pacientes de 2 a menos de 12 años se extrapoló de pacientes de 12 años o más con el apoyo de análisis farmacocinéticos poblacionales que muestran niveles de exposición a elexacaftor, tezacaftor e ivacaftor en pacientes de 2 a menos de 12 años dentro del rango de exposiciones observadas en pacientes de 12 años o más [ver Farmacología clínica (12.3)]. La seguridad de TRIKAFTA en pacientes de 6 a menos de 12 años se derivó de un ensayo clínico abierto de 24 semanas en 66 pacientes de 6 a menos de 12 años (edad media al inicio 9,3 años) a los que se administró una dosis total de elexacaftor 100 mg/tezacaftor 50 mg/ivacaftor 75 mg por la mañana e ivacaftor 75 mg por la noche (para pacientes que pesan menos de 30 kg) o una dosis total de elexacaftor 200 mg/tezacaftor 100 mg/ivacaftor 150 mg por la mañana e ivacaftor 150 mg por la noche (para pacientes que pesan 30 kg o más) (Ensayo 3). La seguridad de TRIKAFTA en pacientes de 2 a menos de 6 años se derivó de un ensayo clínico abierto de 24 semanas en 75 pacientes de 2 a menos de 6 años (edad media al inicio 4,1 años) a los que se administró una dosis total de elexacaftor 80 mg/tezacaftor 40 mg/ivacaftor 60 mg por la mañana e ivacaftor 59,5 mg por la noche (para pacientes que pesan de 10 kg a menos de 14 kg) o una dosis total de elexacaftor 100 mg/tezacaftor 50 mg/ivacaftor 75 mg por la mañana e ivacaftor 75 mg por la noche (para pacientes que pesan 14 kg o más) (Ensayo 4). El perfil de seguridad de los pacientes en estos ensayos fue similar al observado en el Ensayo 1 [ver Reacciones adversas (6)].
No se ha establecido la seguridad y eficacia de TRIKAFTA en pacientes con CF menores de 2 años de edad.
Datos de toxicidad en animales jóvenes
Se observaron hallazgos de cataratas en ratas jóvenes a las que se administraron dosis de ivacaftor de 10 mg/kg/día o más (0,21 veces la dosis máxima recomendada para humanos (MRHD) basada en la exposición sistémica a ivacaftor y sus metabolitos) desde el día postnatal 7 hasta el 35. Este hallazgo no se ha observado en animales mayores [ver Advertencias y precauciones (5.5)].
Se realizaron estudios con tezacaftor en ratas jóvenes a partir del día postnatal (PND) 21 y hasta los PND 35 a 49. Se observaron hallazgos de convulsiones y muerte en ratas jóvenes que recibieron una dosis de tezacaftor de 100 mg/kg/día (aproximadamente equivalente a 1,9 veces la MRHD basada en la suma de las AUC de tezacaftor y su metabolito, M1-TEZ). Se identificó una dosis sin efecto a los 30 mg/kg/día (aproximadamente equivalente a 0,8 veces la MRHD basada en la suma de las AUC de tezacaftor y su metabolito, M1-TEZ). Los hallazgos fueron relacionados con la dosis y generalmente más graves cuando la administración de tezacaftor se inició antes en el período postnatal (PND 7, que sería aproximadamente equivalente a un recién nacido humano). Tezacaftor y su metabolito, M1-TEZ, son sustratos de la P-glicoproteína. Los niveles más bajos de actividad de la P-glicoproteína en el cerebro de las ratas más jóvenes dieron como resultado niveles más altos de tezacaftor y M1-TEZ en el cerebro. Estos hallazgos no son relevantes para la población pediátrica indicada, de 2 años de edad o más, para quienes los niveles de actividad de la P-glicoproteína son equivalentes a los niveles observados en adultos.
8.5 Uso en geriátricos
Los estudios clínicos de TRIKAFTA no incluyeron pacientes de 65 años o más.
8.6 Insuficiencia renal
TRIKAFTA no se ha estudiado en pacientes con insuficiencia renal grave o enfermedad renal terminal. No se recomienda ningún ajuste de dosis en pacientes con insuficiencia renal leve (eGFR 60 a <90 mL/min/1,73 m2) o moderada (eGFR 30 a <60 mL/min/1,73 m2). Usar con precaución en pacientes con insuficiencia renal grave (eGFR <30 mL/min/1,73 m2) o enfermedad renal terminal [ver Farmacología clínica (12.3)].
8.7 Insuficiencia hepática
-
Insuficiencia hepática grave (Child-Pugh clase C): No debe utilizarse. TRIKAFTA no se ha estudiado en pacientes con insuficiencia hepática grave (Child-Pugh clase C), pero se espera que la exposición sea mayor que en pacientes con insuficiencia hepática moderada [véase Posología y administración (2.3), Advertencias y precauciones (5.1), Reacciones adversas (6) y Farmacología clínica (12.3)].
-
Insuficiencia hepática moderada (Child-Pugh clase B): No se recomienda el tratamiento. El uso de TRIKAFTA en pacientes con insuficiencia hepática moderada solo debe considerarse cuando exista una clara necesidad médica y el beneficio supere el riesgo. Si se utiliza en pacientes con insuficiencia hepática moderada, TRIKAFTA debe utilizarse a una dosis reducida. Las pruebas de función hepática deben controlarse estrechamente [véase Posología y administración (2.1, 2.3) y Advertencias y precauciones (5.1)].
En un estudio clínico de 11 sujetos con insuficiencia hepática moderada, un sujeto presentó elevaciones de bilirrubina total y directa >2 × ULN, y un segundo sujeto presentó una elevación de bilirrubina directa >4.5 × ULN [véase Farmacología clínica (12.3)].
-
Insuficiencia hepática leve (Child-Pugh clase A): No se recomienda ningún ajuste de dosis. Las pruebas de función hepática deben controlarse estrechamente [véase Posología y administración (2.1) y Advertencias y precauciones (5.1)].
8.8 Pacientes con disfunción pulmonar grave
El ensayo 1 incluyó un total de 18 pacientes que recibieron TRIKAFTA con ppFEV1 <40 al inicio del estudio. La seguridad y la eficacia en este subgrupo fueron comparables a las observadas en la población general.
10 SOBREDOSIS
El tratamiento de la sobredosis consiste en medidas generales de apoyo, incluyendo la monitorización de las constantes vitales y la observación del estado clínico del paciente.
11 DESCRIPCIÓN
TRIKAFTA es una combinación de comprimidos o gránulos de elexacaftor, tezacaftor e ivacaftor en dosis fija, e ivacaftor en comprimidos o gránulos. Tanto los comprimidos como los gránulos son para administración oral.
Los comprimidos de combinación de dosis fija de elexacaftor, tezacaftor e ivacaftor están disponibles como: comprimido recubierto con película de color naranja, oblongo, que contiene 100 mg de elexacaftor, 50 mg de tezacaftor, 75 mg de ivacaftor, o comprimido recubierto con película de color naranja claro, oblongo, que contiene 50 mg de elexacaftor, 25 mg de tezacaftor, 37.5 mg de ivacaftor. El comprimido de combinación de dosis fija contiene los siguientes excipientes inactivos: croscarmelosa sódica, hipromelosa, hipromelosa acetato succinato, estearato de magnesio, celulosa microcristalina y laurilsulfato de sodio. La cubierta pelicular del comprimido contiene hidroxipropilcelulosa, hipromelosa, óxido de hierro rojo, óxido de hierro amarillo, talco y dióxido de titanio.
El comprimido de ivacaftor está disponible como un comprimido recubierto con película de color azul claro, oblongo, que contiene 150 mg o 75 mg de ivacaftor y los siguientes excipientes inactivos: dióxido de silicio coloidal, croscarmelosa sódica, hipromelosa acetato succinato, lactosa monohidrato, estearato de magnesio, celulosa microcristalina y laurilsulfato de sodio. La cubierta pelicular del comprimido contiene cera de carnauba, FD&C Azul #2, PEG 3350, alcohol polivinílico, talco y dióxido de titanio. La tinta de impresión contiene hidróxido de amonio, óxido de hierro negro, propilenglicol y goma laca.
Los gránulos orales de combinación de dosis fija de elexacaftor, tezacaftor e ivacaftor son gránulos blancos a blanquecinos, endulzados, sin sabor, de aproximadamente 2 mm de diámetro, envasados en sobres unidosis. Cada sobre unidosis contiene 100 mg de elexacaftor, 50 mg de tezacaftor, 75 mg de ivacaftor o 80 mg de elexacaftor, 40 mg de tezacaftor, 60 mg de ivacaftor y los siguientes excipientes inactivos: dióxido de silicio coloidal, croscarmelosa sódica, hipromelosa, hipromelosa acetato succinato, lactosa monohidrato, estearato de magnesio, manitol, laurilsulfato de sodio y sucralosa.
Los gránulos orales de ivacaftor son gránulos blancos a blanquecinos, endulzados, sin sabor, de aproximadamente 2 mm de diámetro, envasados en sobres unidosis. Cada sobre unidosis contiene 75 mg o 59,5 mg de ivacaftor y los siguientes excipientes inactivos: dióxido de silicio coloidal, croscarmelosa sódica, hipromelosa acetato succinato, lactosa monohidrato, estearato de magnesio, manitol, laurilsulfato de sodio y sucralosa.
Los principios activos de TRIKAFTA se describen a continuación.
Elexacaftor
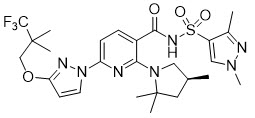
El elexacaftor es un sólido blanco que es prácticamente insoluble en agua (<1 mg/mL). Su nombre químico es N-(1,3-dimetil-1H-pirazolo-4-sulfonil)-6-[3-(3,3,3-trifluoro-2,2-dimetilpropoxi)-1H-pirazolo-1-il]-2-[(4S)-2,2,4-trimetilpirrolidin-1-il]piridina-3-carboxamida. Su fórmula molecular es C26H34N7O4SF3 y su peso molecular es 597,66. El elexacaftor tiene la siguiente fórmula estructural:
Tezacaftor
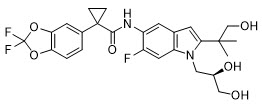
El tezacaftor es un sólido blanco a blanquecino que es prácticamente insoluble en agua (<5 microgramos/mL). Su nombre químico es 1-(2,2-difluoro-2H-1,3-benzodioxol-5-il)-N-{1-[(2R)-2,3-dihidroxopropil]-6-fluoro-2-(1-hidroxi-2-metilpropan-2-il)-1H-indol-5-il}ciclopropano-1-carboxamida. Su fórmula molecular es C26H27N2F3O6 y su peso molecular es 520,50. El tezacaftor tiene la siguiente fórmula estructural:
Ivacaftor
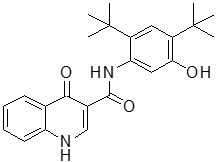
El ivacaftor es un sólido cristalino blanco a blanquecino que es prácticamente insoluble en agua (<0,05 microgramos/mL). Farmacológicamente es un potenciador de CFTR. Su nombre químico es N-(2,4-di-terc-butil-5-hidroxifenil)-1,4-dihidro-4-oxoquinolina-3-carboxamida. Su fórmula molecular es C24H28N2O3 y su peso molecular es 392,49. El ivacaftor tiene la siguiente fórmula estructural:
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de Acción
Elexacaftor y tezacaftor se unen a diferentes sitios en la proteína CFTR y tienen un efecto aditivo al facilitar el procesamiento celular y el tráfico de formas mutantes selectas de CFTR (incluida F508del-CFTR) para aumentar la cantidad de proteína CFTR que llega a la superficie celular en comparación con cualquiera de las moléculas por separado. Ivacaftor potencia la probabilidad de apertura del canal (o activación) de la proteína CFTR en la superficie celular.
El efecto combinado de elexacaftor, tezacaftor e ivacaftor es una mayor cantidad y función de CFTR en la superficie celular, lo que resulta en una mayor actividad de CFTR, medida tanto por el transporte de cloruro mediado por CFTR in vitro como por el cloruro en el sudor en pacientes con FQ.
Ensayo de transporte de cloruro CFTR en células tiroideas de rata Fischer que expresan la proteína CFTR mutante
Los efectos de elexacaftor/tezacaftor/ivacaftor sobre el transporte de cloruro para las proteínas CFTR mutantes se determinaron en estudios de electrofisiología de cámara Ussing utilizando un panel de líneas celulares de tiroides de rata Fischer (FRT) que expresan de forma estable proteínas CFTR mutantes individuales. Elexacaftor/tezacaftor/ivacaftor aumentó el transporte de cloruro en células FRT que expresan mutaciones CFTR, como se identifica en la Tabla 6.
El umbral que debe superar el aumento inducido por el tratamiento en el transporte de cloruro para que la proteína CFTR mutante se considere sensible es ≥10% del valor normal sobre el valor basal. Este umbral se utilizó porque se espera que prediga el beneficio clínico. Para mutaciones individuales, la magnitud del cambio neto sobre el valor basal en el transporte de cloruro mediado por CFTR in vitro no se correlaciona con la magnitud de la respuesta clínica.
Ensayo de transporte de cloruro CFTR en células epiteliales bronquiales humanas que expresan la proteína CFTR mutante
Las células epiteliales bronquiales humanas (HBE) homocigotas y heterocigotas N1303K mostraron un mayor transporte de cloruro en presencia de elexacaftor/tezacaftor/ivacaftor que las células HBE F508del/F508del tratadas con tezacaftor/ivacaftor (que ha mostrado beneficio clínico en personas homocigotas para F508del).
Selección de pacientes
Seleccione pacientes de 2 años de edad y mayores para el tratamiento de la FQ con TRIKAFTA según la presencia de al menos una mutación F508del u otra mutación sensible en el gen CFTR (consulte la Tabla 6) [ver Indicaciones y uso (1)].
La Tabla 6 enumera las mutaciones CFTR que responden a TRIKAFTA según la respuesta clínica y/o los datos in vitro en células FRT o HBE o según la extrapolación de la eficacia.
12.2 Farmacodinamia
Evaluación de cloruro en sudor
En el Ensayo 1 (pacientes con una mutación F508del en un alelo y una mutación en el segundo alelo que resulta en ausencia de proteína CFTR o en una proteína CFTR que no responde a ivacaftor y tezacaftor/ivacaftor), se observó una reducción en el cloruro en sudor desde el inicio en la Semana 4 y se mantuvo durante el período de tratamiento de 24 semanas [ver Estudios Clínicos (14.1)]. En el Ensayo 2 (pacientes homocigotos para la mutación F508del), se observó una reducción en el cloruro en sudor desde el inicio en la Semana 4 [ver Estudios Clínicos (14.2)]. En el Ensayo 3 (pacientes de 6 a menos de 12 años que son homocigotos para la mutación F508del o heterocigotos para la mutación F508del y una mutación en el segundo alelo que resulta en ausencia de proteína CFTR o en una proteína CFTR que no responde a ivacaftor y tezacaftor/ivacaftor), el cambio medio absoluto en el cloruro en sudor desde el inicio hasta la Semana 24 fue de -60.9 mmol/L (IC del 95%: -63.7, -58.2). En el Ensayo 4 (pacientes de 2 a menos de 6 años que tenían al menos una mutación F508del o una mutación que se sabe que responde a TRIKAFTA), el cambio medio absoluto en el cloruro en sudor desde el inicio hasta la Semana 24 fue de -57.9 mmol/L (IC del 95%: -61.3, -54.6). En el Ensayo 5 (pacientes de 6 años o mayores con al menos una mutación CFTR que no sea F508del y que responda a elexacaftor/tezacaftor/ivacaftor), el cambio medio absoluto en el cloruro en sudor desde el inicio hasta la Semana 24 en comparación con el placebo fue de -28.3 mmol/L (IC del 95%: -32.1, -24.5).
Electrofisiología cardíaca
A dosis de hasta 2 veces la dosis máxima recomendada de elexacaftor y 3 veces la dosis máxima recomendada de tezacaftor e ivacaftor, el intervalo QT/QTc en sujetos sanos no se prolongó en ninguna medida clínicamente relevante.
12.3 Farmacocinética
La farmacocinética de elexacaftor, tezacaftor e ivacaftor es similar entre los sujetos adultos sanos y los pacientes con FQ. Los parámetros farmacocinéticos de elexacaftor, tezacaftor e ivacaftor en pacientes con FQ de 12 años o mayores se muestran en la Tabla 7.
Poblaciones Específicas
Pacientes Pediátricos de 2 a Menos de 12 Años de Edad
Las exposiciones a elexacaftor, tezacaftor e ivacaftor observadas en pacientes de 2 a menos de 12 años, según lo determinado mediante el análisis PK poblacional, se presentan por grupo de edad y dosis administrada en la Tabla 8. Las exposiciones a elexacaftor, tezacaftor e ivacaftor en esta población de pacientes están dentro del rango observado en pacientes de 12 años o mayores.
Pacientes pediátricos de 12 a menos de 18 años de edad
Las siguientes conclusiones sobre las exposiciones entre adultos y la población pediátrica se basan en análisis farmacocinéticos (PK) poblacionales. Después de la administración oral de TRIKAFTA a pacientes de 12 a menos de 18 años de edad (elexacaftor 200 mg qd/tezacaftor 100 mg qd/ivacaftor 150 mg q12h), el AUCss medio (±DE) fue de 147 (36,8) mcg∙h/mL, 88,8 (21,8) mcg∙h/mL y 10,6 (3,35) mcg∙h/mL, respectivamente para elexacaftor, tezacaftor e ivacaftor, similar al AUCss en pacientes adultos.
Pacientes con insuficiencia renal
La excreción renal de elexacaftor, tezacaftor e ivacaftor es mínima. Elexacaftor solo o en combinación con tezacaftor e ivacaftor no se ha estudiado en sujetos con insuficiencia renal grave (TFG <30 mL/min/1,73 m2) o enfermedad renal en etapa terminal. Según los análisis de PK poblacionales, la depuración de elexacaftor y tezacaftor fue similar en sujetos con insuficiencia renal leve (TFG de 60 a <90 mL/min/1,73 m2) o moderada (TFG de 30 a <60 mL/min/1,73 m2) en relación con los pacientes con función renal normal [ver Uso en poblaciones específicas (8.6)].
Pacientes con insuficiencia hepática
Elexacaftor solo o en combinación con tezacaftor e ivacaftor no se ha estudiado en sujetos con insuficiencia hepática grave (Child-Pugh Clase C, puntuación 10-15). En un estudio clínico, después de dosis múltiples de elexacaftor, tezacaftor e ivacaftor durante 10 días, los sujetos con función hepática moderadamente deteriorada (Child-Pugh Clase B, puntuación 7-9) tuvieron un AUC un 25 % mayor y una Cmáx un 12 % mayor para elexacaftor, un AUC un 73 % mayor y una Cmáx un 70 % mayor para M23-ELX, un AUC un 36 % mayor y una Cmáx un 24 % mayor para la combinación de elexacaftor y M23-ELX, un AUC un 20 % mayor pero una Cmáx similar para tezacaftor y un AUC 1,5 veces mayor y una Cmáx un 10 % mayor para ivacaftor en comparación con sujetos sanos con características demográficas similares [ver Posología y administración (2.3), Advertencias y precauciones (5.1), Reacciones adversas (6) y Uso en poblaciones específicas (8.7)].
Tezacaftor e Ivacaftor
Después de dosis múltiples de tezacaftor e ivacaftor durante 10 días, los sujetos con función hepática moderadamente deteriorada tuvieron un AUC aproximadamente un 36 % mayor y una Cmáx un 10 % mayor para tezacaftor y un AUC 1,5 veces mayor pero una Cmáx similar para ivacaftor en comparación con sujetos sanos con características demográficas similares.
Ivacaftor
En un estudio con ivacaftor solo, los sujetos con insuficiencia hepática moderada tuvieron una Cmáx de ivacaftor similar, pero un AUC0-∞ de ivacaftor aproximadamente 2,0 veces mayor en comparación con sujetos sanos con características demográficas similares.
Pacientes masculinos y femeninos
Según el análisis farmacocinético poblacional, las exposiciones de elexacaftor, tezacaftor e ivacaftor son similares en hombres y mujeres.
Estudios de interacción farmacológica
Se realizaron estudios de interacción farmacológica con elexacaftor, tezacaftor y/o ivacaftor y otros fármacos que probablemente se administren conjuntamente o fármacos comúnmente utilizados como sondas para estudios de interacción farmacocinética [ver Interacciones farmacológicas (7)].
Posibilidad de que Elexacaftor, Tezacaftor y/o Ivacaftor afecten a otros fármacos
Según los resultados in vitro, elexacaftor y tezacaftor tienen un bajo potencial para inhibir CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 y CYP3A4, mientras que ivacaftor tiene el potencial de inhibir CYP2C8, CYP2C9 y CYP3A. Sin embargo, los estudios clínicos demostraron que el régimen de combinación de tezacaftor/ivacaftor no es un inhibidor de CYP3A e ivacaftor no es un inhibidor de CYP2C8 o CYP2D6.
Según los resultados in vitro, no es probable que elexacaftor, tezacaftor e ivacaftor induzcan CYP3A, CYP1A2 y CYP2B6.
Según los resultados in vitro, elexacaftor y tezacaftor tienen un bajo potencial para inhibir el transportador P-gp, mientras que ivacaftor tiene el potencial de inhibir P-gp. La coadministración de tezacaftor/ivacaftor con digoxina, un sustrato sensible a la P-gp, aumentó la exposición a la digoxina en 1,3 veces en un estudio clínico. Según los resultados in vitro, elexacaftor y M23-ELX pueden inhibir la captación de OATP1B1 y OATP1B3. Tezacaftor tiene un bajo potencial para inhibir BCRP, OCT2, OAT1 u OAT3. Ivacaftor no es un inhibidor de los transportadores OCT1, OCT2, OAT1 u OAT3.
Los efectos de elexacaftor, tezacaftor y/o ivacaftor sobre la exposición a fármacos coadministrados se muestran en la Tabla 9 [ver Interacciones farmacológicas (7)].
Potencial de otros medicamentos para afectar a Elexacaftor, Tezacaftor y/o Ivacaftor
Los estudios in vitro mostraron que elexacaftor, tezacaftor e ivacaftor son metabolizados por el CYP3A. La exposición a elexacaftor, tezacaftor e ivacaftor puede reducirse por la administración concomitante de inductores de CYP3A y aumentarse por la administración concomitante de inhibidores de CYP3A.
Los estudios in vitro mostraron que elexacaftor y tezacaftor son sustratos del transportador de eflujo P-gp, pero ivacaftor no lo es. Elexacaftor e ivacaftor no son sustratos de OATP1B1 u OATP1B3; tezacaftor es un sustrato de OATP1B1, pero no de OATP1B3. Tezacaftor es un sustrato de BCRP.
Los efectos de los medicamentos coadministrados sobre la exposición a elexacaftor, tezacaftor y/o ivacaftor se muestran en la Tabla 10 [ver Dosis y Administración (2.4) y Interacciones Medicamentosas (7)].
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogenicidad, Mutagénesis, Deterioro de la Fertilidad
No se realizaron estudios de carcinogenicidad, mutagénesis o deterioro de la fertilidad con la combinación de elexacaftor, tezacaftor e ivacaftor; sin embargo, a continuación se describen estudios separados de elexacaftor, tezacaftor e ivacaftor.
Elexacaftor
Un estudio de 6 meses en ratones transgénicos Tg.rasH2 no mostró evidencia de tumorigenicidad a una dosis de 50 mg/kg/día, la dosis más alta probada.
Se realizó un estudio de dos años en ratas para evaluar el potencial carcinogénico de elexacaftor. No se observó evidencia de tumorigenicidad en ratas a dosis orales de elexacaftor de hasta 10 mg/kg/día (aproximadamente 2 y 5 veces la MRHD basada en el AUC sumado de elexacaftor y su metabolito en ratas macho y hembra, respectivamente).
Elexacaftor resultó negativo para genotoxicidad en los siguientes ensayos: prueba de Ames para mutación genética bacteriana, ensayo de micronúcleos de células de mamíferos in vitro en células TK6 y prueba de micronúcleos in vivo en ratones.
Elexacaftor no causó toxicidad en el sistema reproductivo en ratas macho a 55 mg/kg/día y ratas hembra a 25 mg/kg/día, equivalente a aproximadamente 6 y 4 veces la MRHD, respectivamente (basado en el AUC sumado de elexacaftor y su metabolito). Elexacaftor no causó toxicidad embrionaria a 35 mg/kg/día, que fue la dosis más alta probada, equivalente a aproximadamente 7 veces la MRHD (basado en el AUC sumado de elexacaftor y su metabolito). Se observaron índices más bajos de fertilidad masculina y femenina, cópula masculina y concepción femenina en machos a 75 mg/kg/día y hembras a 35 mg/kg/día, equivalente a aproximadamente 6 y 7 veces, respectivamente, la MRHD (basado en el AUC sumado de elexacaftor y su metabolito).
Tezacaftor
Se realizaron un estudio de dos años en ratas Sprague-Dawley y un estudio de 6 meses en ratones transgénicos Tg.rasH2 para evaluar el potencial carcinogénico de tezacaftor. No se observó evidencia de tumorigenicidad por tezacaftor en ratas macho y hembra a dosis orales de hasta 50 y 75 mg/kg/día (aproximadamente 1 y 2 veces la MRHD basada en el AUC sumado de tezacaftor y sus metabolitos en machos y hembras, respectivamente). No se observó evidencia de tumorigenicidad en ratones transgénicos Tg.rasH2 machos y hembras a dosis de tezacaftor de hasta 500 mg/kg/día.
Tezacaftor resultó negativo para genotoxicidad en los siguientes ensayos: prueba de Ames para mutación genética bacteriana, ensayo de aberración cromosómica in vitro en células de ovario de hámster chino y prueba de micronúcleos in vivo en ratones.
No hubo efectos sobre la fertilidad masculina o femenina y el desarrollo embrionario temprano en ratas a dosis orales de tezacaftor de hasta 100 mg/kg/día (aproximadamente 3 veces la MRHD basada en el AUC sumado de tezacaftor y M1-TEZ).
Ivacaftor
Se realizaron estudios de dos años en ratones CD-1 y ratas Sprague-Dawley para evaluar el potencial carcinogénico de ivacaftor. No se observó evidencia de tumorigenicidad por ivacaftor en ratones o ratas a dosis orales de hasta 200 mg/kg/día y 50 mg/kg/día, respectivamente (aproximadamente equivalente a 2 y 7 veces la MRHD, respectivamente, basado en el AUC sumado de ivacaftor y sus metabolitos).
Ivacaftor resultó negativo para genotoxicidad en los siguientes ensayos: prueba de Ames para mutación genética bacteriana, ensayo de aberración cromosómica in vitro en células de ovario de hámster chino y prueba de micronúcleos in vivo en ratones.
Ivacaftor deterioró los índices de fertilidad y rendimiento reproductivo en ratas macho y hembra a 200 mg/kg/día (aproximadamente 7 y 5 veces, respectivamente, la MRHD basada en el AUC sumado de ivacaftor y sus metabolitos). Se observaron aumentos en el diestro prolongado en hembras a 200 mg/kg/día. Ivacaftor también aumentó el número de hembras con todos los embriones no viables y disminuyó los cuerpos lúteos, las implantaciones y los embriones viables en ratas a 200 mg/kg/día (aproximadamente 5 veces la MRHD basada en el AUC sumado de ivacaftor y sus metabolitos) cuando las madres fueron dosificadas antes y durante el embarazo temprano. Estos deterioros de la fertilidad y el rendimiento reproductivo en ratas macho y hembra a 200 mg/kg/día se atribuyeron a toxicidad severa.
14 ESTUDIOS CLÍNICOS
14.1 Estudios clínicos en pacientes con fibrosis quística con al menos una mutación F508del
La eficacia de TRIKAFTA en pacientes de 12 años o más con fibrosis quística (FQ) con al menos una mutación F508del se evaluó en dos ensayos aleatorizados, doble ciego y controlados (Ensayos 1 y 2).
El Ensayo 1 (NCT03525444) fue un estudio de 24 semanas, aleatorizado, doble ciego y controlado con placebo en pacientes que tenían una mutación F508del en un alelo y una mutación en el segundo alelo que da como resultado ninguna proteína CFTR o una proteína CFTR que no responde a ivacaftor y tezacaftor/ivacaftor. Se planeó un análisis intermedio cuando al menos 140 pacientes completaron la Semana 4 y al menos 100 pacientes completaron la Semana 12.
El Ensayo 2 (NCT03525548) fue un estudio de 4 semanas, aleatorizado, doble ciego y controlado con activo en pacientes homocigotos para la mutación F508del. Los pacientes recibieron tezacaftor 100 mg qd/ivacaftor 150 mg q12h durante un período de inclusión abierto de 4 semanas y luego fueron aleatorizados y dosificados para recibir TRIKAFTA o tezacaftor 100 mg qd/ivacaftor 150 mg q12h durante un período de tratamiento doble ciego de 4 semanas.
Los pacientes de los Ensayos 1 y 2 tenían un diagnóstico confirmado de FQ y al menos una mutación F508del. Los pacientes interrumpieron cualquier terapia previa con moduladores de CFTR, pero continuaron con sus otras terapias estándar para la FQ (por ejemplo, broncodilatadores, antibióticos inhalados, dornaza alfa y solución salina hipertónica). Los pacientes tenían un ppFEV1 en la evaluación inicial entre 40-90%. Los pacientes con antecedentes de colonización con organismos asociados con una disminución más rápida del estado pulmonar, incluyendo pero no limitado a Burkholderia cenocepacia, Burkholderia dolosa o Mycobacterium abscessus, o que tenían una prueba de función hepática anormal en la evaluación inicial (ALT, AST, ALP o GGT ≥3 × ULN, o bilirrubina total ≥2 × ULN), fueron excluidos de los ensayos. Los pacientes de los Ensayos 1 y 2 fueron elegibles para pasar a un estudio de extensión de etiqueta abierta.
Ensayo 1
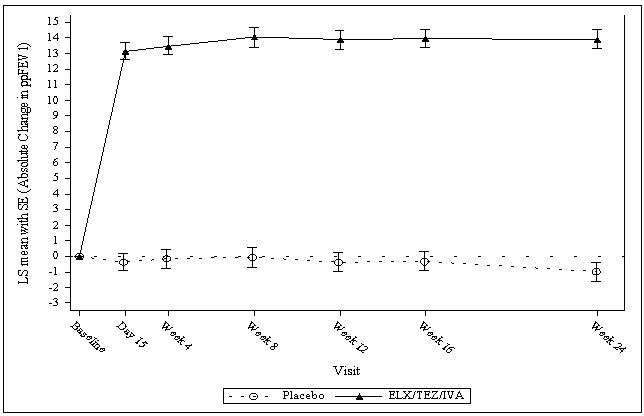
El Ensayo 1 evaluó a 403 pacientes (200 TRIKAFTA, 203 placebo) con FQ de 12 años o más (edad media 26,2 años). El ppFEV1 medio al inicio del estudio fue del 61,4% (rango: 32,3%, 97,1%). El criterio de valoración principal evaluado en el momento del análisis intermedio fue el cambio absoluto medio en el ppFEV1 desde el inicio del estudio en la Semana 4. El análisis final evaluó todos los criterios de valoración secundarios clave en los 403 pacientes que completaron la participación en el estudio de 24 semanas, incluyendo el cambio absoluto en el ppFEV1 desde el inicio del estudio hasta la Semana 24; el cambio absoluto en el cloruro del sudor desde el inicio del estudio en la Semana 4 y hasta la Semana 24; el número de exacerbaciones pulmonares hasta la Semana 24; el cambio absoluto en el IMC desde el inicio del estudio en la Semana 24, y el cambio absoluto en la puntuación del dominio respiratorio del CFQ-R (una medida de los síntomas respiratorios relevantes para los pacientes con FQ, como la tos, la producción de esputo y la dificultad para respirar) desde el inicio del estudio en la Semana 4 y hasta la Semana 24.
De los 403 pacientes incluidos en el análisis intermedio, la diferencia de tratamiento entre TRIKAFTA y placebo para el cambio absoluto medio desde el inicio del estudio en el ppFEV1 en la Semana 4 fue de 13,8 puntos porcentuales (IC del 95%: 12,1, 15,4; P<0,0001).
La diferencia de tratamiento entre TRIKAFTA y placebo para el cambio absoluto medio en el ppFEV1 desde el inicio del estudio hasta la Semana 24 fue de 14,3 puntos porcentuales (IC del 95%: 12,7, 15,8; P<0,0001). Se observó una mejoría media en el ppFEV1 en la primera evaluación el día 15 y se mantuvo durante el período de tratamiento de 24 semanas (véase la Figura 1). Se observaron mejoras en el ppFEV1 independientemente de la edad, el ppFEV1 basal, el sexo y la región geográfica. Véase la Tabla 11 para un resumen de los resultados principales y secundarios clave en el Ensayo 1.