Fabricante de medicamentos: Aurobindo Pharma Limited (Updated: 2024-10-11)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
Tabletas de TERIFLUNOMIDA, para uso oral
Aprobación inicial en EE. UU.: 2012
ADVERTENCIA: HEPATOTOXICIDAD y TOXICIDAD EMBRIOFETAL
Consulte la información completa de prescripción para obtener la advertencia completa en recuadro.
• Hepatotoxicidad
Se ha notificado lesión hepática clínicamente significativa y potencialmente mortal, incluida la insuficiencia hepática aguda que requiere trasplante, en pacientes tratados con tabletas de teriflunomida en el entorno poscomercialización (5.1). El uso concomitante de tabletas de teriflunomida con otros medicamentos hepatotóxicos puede aumentar el riesgo de lesión hepática grave. Obtenga los niveles de transaminasas y bilirrubina dentro de los 6 meses anteriores al inicio de la teriflunomida y controle los niveles de ALT al menos mensualmente durante seis meses (5.1). Si se sospecha lesión hepática inducida por fármacos, suspenda la teriflunomida e inicie el procedimiento de eliminación acelerada (5.3).
• Toxicidad embrio-fetal
Se produjo teratogenicidad y embrioletalidad en animales a los que se administró teriflunomida (5.2, 8.1). Excluya el embarazo antes de iniciar el tratamiento con teriflunomida (4, 5.2, 8.1, 8.3). Aconseje el uso de métodos anticonceptivos eficaces en mujeres en edad fértil durante el tratamiento y durante un procedimiento de eliminación acelerada del fármaco (4, 5.2, 5.3, 8.1, 8.3). Suspenda la teriflunomida y utilice un procedimiento de eliminación acelerada del fármaco si la paciente queda embarazada (5.2, 5.3, 8.1).
INDICACIONES Y USO
Las tabletas de teriflunomida son un inhibidor de la síntesis de pirimidina indicado para el tratamiento de las formas recurrentes de esclerosis múltiple (EM), que incluyen el síndrome clínicamente aislado, la enfermedad remitente-recurrente y la enfermedad progresiva secundaria activa, en adultos. (1)
DOSIFICACIÓN Y ADMINISTRACIÓN
7 mg o 14 mg por vía oral una vez al día, con o sin alimentos. (2)
FORMAS Y FUERZAS DE DOSIFICACIÓN
Tabletas recubiertas con película de 7 mg y 14 mg (3)
CONTRAINDICACIONES
ADVERTENCIAS Y PRECAUCIONES
- La eliminación de la teriflunomida se puede acelerar mediante la administración de colestiramina o carbón activado durante 11 días. (5.3)
- La teriflunomida puede disminuir los glóbulos blancos. Debe estar disponible un CBC reciente antes de comenzar la teriflunomida. Controle los signos y síntomas de infección. Considere suspender el tratamiento con teriflunomida en caso de infección grave. No comience la teriflunomida en pacientes con infecciones activas. (5.4)
- Suspenda la teriflunomida si el paciente presenta anafilaxia, angioedema, síndrome de Stevens-Johnson, necrólisis epidérmica tóxica, reacción medicamentosa con eosinofilia y síntomas sistémicos; inicie la eliminación rápida. (5.3, 5.5, 5.6, 5.7)
- Si el paciente desarrolla síntomas compatibles con neuropatía periférica, evalúe al paciente y considere la posibilidad de suspender la teriflunomida. (5.8)
- La teriflunomida puede aumentar la presión arterial. Mida la presión arterial al inicio del tratamiento y controle la presión arterial durante el tratamiento. (5.9)
REACCIONES ADVERSAS
Reacciones adversas más comunes (≥10% y ≥2% mayores que el placebo): dolor de cabeza, diarrea, náuseas, alopecia, aumento de la ALT. (6)
Para informar sobre REACCIONES ADVERSAS SOSPECHOSAS, comuníquese con Aurobindo Pharma USA, Inc. al 1-866-850-2876 o con la FDA al 1-800-FDA-1088 o www.fda.gov/medwatch.
INTERACCIONES MEDICAMENTOSAS
- Fármacos metabolizados por los transportadores CYP2C8 y OAT3: Controle a los pacientes porque la teriflunomida puede aumentar la exposición a estos fármacos. (7)
- La teriflunomida puede aumentar la exposición al etinilestradiol y la levonorgestrel. Elija un anticonceptivo oral adecuado. (7)
- Fármacos metabolizados por CYP1A2: Controle a los pacientes porque la teriflunomida puede disminuir la exposición a estos fármacos. (7)
- Warfarina: Controle el INR ya que la teriflunomida puede disminuir el INR. (7)
- Fármacos metabolizados por los transportadores BCRP y OATP1B1/B3: Controle a los pacientes porque la teriflunomida puede aumentar la exposición a estos fármacos. (7)
- Rosuvastatina: La dosis de rosuvastatina no debe exceder los 10 mg una vez al día en pacientes que toman teriflunomida. (7)
Consulte 17 para obtener INFORMACIÓN PARA EL PACIENTE Y Guía de medicamentos.
Revisado: 10/2024
Tabla de Contenido
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDO*
ADVERTENCIA: HEPATOTOXICIDAD Y TOXICIDAD EMBRIOFETAL
1 INDICACIONES Y USO
2 DOSIFICACIÓN Y ADMINISTRACIÓN
3 FORMAS Y FUERZAS DE DOSIFICACIÓN
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Hepatotoxicidad
5.2 Toxicidad embrio-fetal
5.3 Procedimiento para la eliminación acelerada de teriflunomida
5.4 Efectos en la médula ósea/Potencial inmunosupresor/Infecciones
5.5 Reacciones de hipersensibilidad
5.6 Reacciones cutáneas graves
5.7 Reacción medicamentosa con eosinofilia y síntomas sistémicos
5.8 Neuropatía periférica
5.9 Aumento de la presión arterial
5.10 Efectos respiratorios
5.11 Pancreatitis en pacientes pediátricos
5.12 Uso concomitante con terapias inmunosupresoras o inmunomoduladoras
6 REACCIONES ADVERSAS
6.1 Experiencia en ensayos clínicos
6.2 Experiencia postcomercialización
7 INTERACCIONES MEDICAMENTOSAS
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.3 Mujeres y hombres en edad fértil
8.4 Uso pediátrico
8.5 Uso geriátrico
8.6 Insuficiencia hepática
8.7 Insuficiencia renal
10 SOBREDOSIS
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
12.2 Farmacodinamia
12.3 Farmacocinética
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, mutagénesis, deterioro de la fertilidad
14 ESTUDIOS CLÍNICOS
16 FORMA DE SUMINISTRO/ALMACENAMIENTO Y MANEJO
17 INFORMACIÓN PARA EL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información completa de prescripción no están enumeradas.
ADVERTENCIA EN EL RECUADRO
ADVERTENCIA: HEPATOTOXICIDAD Y TOXICIDAD EMBRIOFETAL
• Hepatotoxicidad
Se ha notificado lesión hepática clínicamente significativa y potencialmente mortal, incluida la insuficiencia hepática aguda que requiere trasplante, en pacientes tratados con teriflunomida en el entorno postcomercialización [ver Advertencias y precauciones (5.1)]. El uso concomitante de teriflunomida con otros fármacos hepatotóxicos puede aumentar el riesgo de lesión hepática grave.
Obtenga los niveles de transaminasas y bilirrubina dentro de los 6 meses previos al inicio del tratamiento con teriflunomida. Controle los niveles de ALT al menos mensualmente durante seis meses después de comenzar el tratamiento con teriflunomida [ver Advertencias y precauciones (5.1)]. Si se sospecha una lesión hepática inducida por fármacos, suspenda la teriflunomida e inicie un procedimiento de eliminación acelerada con colestiramina o carbón [ver Advertencias y precauciones (5.3)]. La teriflunomida está contraindicada en pacientes con insuficiencia hepática grave [ver Contraindicaciones (4)]. Los pacientes con enfermedad hepática preexistente pueden tener un mayor riesgo de desarrollar niveles elevados de transaminasas séricas cuando toman teriflunomida.
• Toxicidad embrio-fetal
La teriflunomida está contraindicada para su uso en mujeres embarazadas y en mujeres en edad fértil que no utilizan métodos anticonceptivos eficaces debido al potencial de daño fetal. La teratogenicidad y la embrioletalidad se produjeron en animales a exposiciones plasmáticas de teriflunomida inferiores a las de los humanos. Excluya el embarazo antes de comenzar el tratamiento con teriflunomida en mujeres en edad fértil. Aconseje a las mujeres en edad fértil que utilicen métodos anticonceptivos eficaces durante el tratamiento con teriflunomida y durante un procedimiento de eliminación acelerada del fármaco después del tratamiento con teriflunomida. Suspenda la teriflunomida y utilice un procedimiento de eliminación acelerada del fármaco si la paciente queda embarazada [ver Contraindicaciones (4), Advertencias y precauciones (5.2, 5.3), Uso en poblaciones específicas (8.1, 8.3), y Farmacología clínica (12.3)].
1 INDICACIONES Y USO
Las tabletas de teriflunomida están indicadas para el tratamiento de las formas recurrentes de esclerosis múltiple (EM), que incluyen el síndrome clínicamente aislado, la enfermedad remitente-recurrente y la enfermedad progresiva secundaria activa, en adultos.
2 DOSIS Y ADMINISTRACIÓN
La dosis recomendada de tabletas de teriflunomida es de 7 mg o 14 mg por vía oral una vez al día.
Las tabletas de teriflunomida se pueden tomar con o sin alimentos.
Monitoreo para evaluar la seguridad
- Obtenga los niveles de transaminasas y bilirrubina dentro de los 6 meses anteriores al inicio del tratamiento con tabletas de teriflunomida. Monitoree los niveles de ALT al menos mensualmente durante seis meses después de comenzar el tratamiento con tabletas de teriflunomida [ver Advertencias y precauciones (5.1)].
- Obtenga un hemograma completo (CBC) dentro de los 6 meses anteriores al inicio del tratamiento con tabletas de teriflunomida. El monitoreo adicional debe basarse en los signos y síntomas de infección [ver Advertencias y precauciones (5.4)].
- Antes de iniciar el tratamiento con tabletas de teriflunomida, examine a los pacientes para detectar infección latente por tuberculosis con una prueba cutánea de tuberculina o una prueba de sangre para la infección por Mycobacterium tuberculosis [ver Advertencias y precauciones (5.4)].
- Excluye el embarazo antes de iniciar el tratamiento con teriflunomida en mujeres en edad fértil [ver Advertencias y precauciones (5.2)].
- Revise la presión arterial antes de comenzar el tratamiento con tabletas de teriflunomida y periódicamente después [ver Advertencias y precauciones (5.9)].
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
Las tabletas de teriflunomida están disponibles en tabletas de 7 mg y 14 mg.
La tableta de 7 mg es de color verde claro a verde, redonda, recubierta con película y con la marca “N” en un lado y “7” en el otro.
La tableta de 14 mg es de color azul claro a azul, redonda, recubierta con película y con la marca “N” en un lado y “14” en el otro.
4 CONTRAINDICACIONES
Las tabletas de teriflunomida están contraindicadas en/con:
- Pacientes con insuficiencia hepática grave [ver Advertencias y precauciones (5.1)].
- Mujeres embarazadas y mujeres en edad fértil que no utilizan métodos anticonceptivos eficaces. Las tabletas de teriflunomida pueden causar daño fetal [ver Advertencias y precauciones (5.2, 5.3) y Uso en poblaciones específicas (8.1)].
- Pacientes con antecedentes de reacción de hipersensibilidad a teriflunomida, leflunomida o a cualquiera de los ingredientes inactivos en las tabletas de teriflunomida. Las reacciones han incluido anafilaxia, angioedema y reacciones cutáneas graves [ver Advertencias y precauciones (5.5)].
- Administración conjunta con leflunomida [ver Farmacología clínica (12.3)].
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Hepatotoxicidad
Se ha informado de lesión hepática clínicamente significativa y potencialmente mortal, incluyendo insuficiencia hepática aguda que requiere trasplante, en pacientes tratados con teriflunomida en la fase posterior a la comercialización. Los pacientes con enfermedad hepática preexistente y los pacientes que toman otros medicamentos hepatotóxicos pueden tener un mayor riesgo de desarrollar lesión hepática al tomar teriflunomida. La lesión hepática clínicamente significativa puede ocurrir en cualquier momento durante el tratamiento con teriflunomida.
Los pacientes con enfermedad hepática aguda o crónica preexistente, o aquellos con alanina aminotransferasa (ALT) sérica mayor de dos veces el límite superior de lo normal (LSN) antes de iniciar el tratamiento, normalmente no deben ser tratados con teriflunomida. La teriflunomida está contraindicada en pacientes con insuficiencia hepática grave [ver Contraindicaciones (4)].
En ensayos controlados con placebo en pacientes adultos, se observó ALT mayor de tres veces el LSN en 61/1045 (5.8%) y 62/1002 (6.2%) de los pacientes que recibieron teriflunomida 7 mg y 14 mg, respectivamente, y 38/997 (3.8%) de los pacientes que recibieron placebo, durante el período de tratamiento. Estas elevaciones ocurrieron principalmente dentro del primer año de tratamiento. La mitad de los casos volvieron a la normalidad sin suspender el medicamento. En los ensayos clínicos, si la elevación de la ALT era mayor de tres veces el LSN en dos pruebas consecutivas, se suspendió la teriflunomida y los pacientes se sometieron a un procedimiento de eliminación acelerada [ver Advertencias y precauciones (5.3)]. De los pacientes que se sometieron a la interrupción y eliminación acelerada en ensayos controlados, la mitad volvió a valores normales o casi normales en 2 meses.
Un paciente en los ensayos controlados en pacientes adultos desarrolló ALT 32 veces el LSN e ictericia 5 meses después del inicio del tratamiento con teriflunomida 14 mg. El paciente fue hospitalizado durante 5 semanas y se recuperó después de la plasmaféresis y el procedimiento de eliminación acelerada con colestiramina. No se pudo descartar la lesión hepática inducida por teriflunomida en este paciente.
Obtener los niveles séricos de transaminasas y bilirrubina dentro de los 6 meses previos al inicio del tratamiento con teriflunomida. Controlar los niveles de ALT al menos mensualmente durante seis meses después de comenzar con teriflunomida. Considerar un control adicional cuando se administra teriflunomida con otros medicamentos potencialmente hepatotóxicos.
Considerar la interrupción de teriflunomida si se confirma un aumento de las transaminasas séricas (mayor de tres veces el LSN). Controlar las transaminasas séricas y la bilirrubina durante el tratamiento con teriflunomida, particularmente en pacientes que desarrollan síntomas sugestivos de disfunción hepática, como náuseas inexplicables, vómitos, dolor abdominal, fatiga, anorexia o ictericia y/u orina oscura. Si se sospecha que la lesión hepática es inducida por teriflunomida, interrumpir la teriflunomida e iniciar un procedimiento de eliminación acelerada [ver Advertencias y precauciones (5.3)] y controlar las pruebas hepáticas semanalmente hasta que se normalicen. Si es poco probable que la lesión hepática sea inducida por teriflunomida porque se ha encontrado alguna otra causa probable, se puede considerar la reanudación del tratamiento con teriflunomida.
5.2 Toxicidad embriofetal
La teriflunomida puede causar daño fetal cuando se administra a una mujer embarazada. Se observó teratogenicidad y letalidad embriofetal en estudios de reproducción animal en múltiples especies animales con exposiciones plasmáticas a teriflunomida similares o inferiores a las de los humanos con la dosis máxima recomendada en humanos (DMRH) de 14 mg/día [ver Uso en poblaciones específicas (8.1)].
La teriflunomida está contraindicada para su uso en mujeres embarazadas y en mujeres en edad fértil que no utilizan anticonceptivos eficaces [ver Contraindicaciones (4)]. Descartar el embarazo antes de iniciar el tratamiento con teriflunomida en mujeres en edad fértil [ver Posología y administración (2)]. Aconsejar a las mujeres en edad fértil que utilicen anticonceptivos eficaces durante el tratamiento con teriflunomida y durante un procedimiento de eliminación acelerada del fármaco después del tratamiento con teriflunomida [ver Uso en poblaciones específicas (8.3)]. Si una mujer queda embarazada mientras toma teriflunomida, interrumpir el tratamiento con teriflunomida, informar a la paciente sobre el riesgo potencial para el feto y realizar un procedimiento de eliminación acelerada del fármaco para lograr una concentración plasmática de teriflunomida inferior a 0.02 mg/L [ver Advertencias y precauciones (5.3)].
Al suspender la teriflunomida, se recomienda que todas las mujeres en edad fértil se sometan a un procedimiento de eliminación acelerada del fármaco. Las mujeres que reciben tratamiento con teriflunomida y desean quedar embarazadas deben suspender la teriflunomida y someterse a un procedimiento de eliminación acelerada del fármaco, que incluye la verificación de que las concentraciones plasmáticas de teriflunomida sean inferiores a 0.02 mg/L (0.02 mcg/mL). Los hombres que desean tener un hijo también deben suspender el uso de teriflunomida y someterse a un procedimiento de eliminación acelerada o esperar hasta que se verifique que la concentración plasmática de teriflunomida sea inferior a 0.02 mg/L (0.02 mcg/mL) [ver Uso en poblaciones específicas (8.3)]. Según los datos en animales, se espera que las concentraciones plasmáticas de teriflunomida en humanos inferiores a 0.02 mg/L (0.02 mcg/mL) tengan un riesgo embriofetal mínimo [ver Contraindicaciones (4), Advertencias y precauciones (5.3) y Uso en poblaciones específicas (8.1)].
5.3 Procedimiento para la eliminación acelerada de teriflunomida
La teriflunomida se elimina lentamente del plasma [see Clinical Pharmacology (12.3)]. Sin un procedimiento de eliminación acelerada, se tarda un promedio de 8 meses en alcanzar concentraciones plasmáticas inferiores a 0.02 mg/L, aunque debido a las variaciones individuales en la eliminación del fármaco, puede tardar hasta 2 años. Se puede utilizar un procedimiento de eliminación acelerada en cualquier momento después de la interrupción de la teriflunomida. La eliminación se puede acelerar mediante cualquiera de los siguientes procedimientos:
- Administración de 8 g de colestiramina cada 8 horas durante 11 días. Si no se tolera bien la colestiramina 8 g tres veces al día, se puede utilizar colestiramina 4 g tres veces al día.
- Administración de 50 g de carbón activado en polvo por vía oral cada 12 horas durante 11 días.
Si alguno de los procedimientos de eliminación se tolera mal, los días de tratamiento no necesitan ser consecutivos a menos que sea necesario reducir rápidamente la concentración plasmática de teriflunomida.
Al final de los 11 días, ambos regímenes aceleraron con éxito la eliminación de teriflunomida, lo que condujo a una disminución de más del 98% en las concentraciones plasmáticas de teriflunomida.
El uso del procedimiento de eliminación acelerada puede resultar en el retorno de la actividad de la enfermedad si el paciente había estado respondiendo al tratamiento con teriflunomida.
5.4 Efectos sobre la médula ósea/Potencial de inmunosupresión/Infecciones
Efectos sobre la médula ósea
En los ensayos controlados con placebo en pacientes adultos con 7 mg y 14 mg de teriflunomida, se observó una disminución media en comparación con el valor inicial en el recuento de glóbulos blancos (GB) de aproximadamente el 15% (principalmente neutrófilos y linfocitos) y en el recuento de plaquetas de aproximadamente el 10%. La disminución en el recuento medio de GB ocurrió durante las primeras 6 semanas y el recuento de GB se mantuvo bajo durante el tratamiento. En estudios controlados con placebo en pacientes adultos, se observó un recuento de neutrófilos <1.5 x 109/L en el 12% y el 16% de los pacientes que recibieron teriflunomida 7 mg y 14 mg, respectivamente, en comparación con el 7% de los pacientes que recibieron placebo; se observó un recuento de linfocitos <0.8 x 109/L en el 10% y el 12% de los pacientes que recibieron teriflunomida 7 mg y 14 mg, respectivamente, en comparación con el 6% de los pacientes que recibieron placebo. No se informaron casos de pancitopenia grave en los ensayos clínicos previos a la comercialización de teriflunomida, pero se han informado casos raros de pancitopenia y agranulocitosis en la fase posterior a la comercialización con leflunomida. Se esperaría un riesgo similar para la teriflunomida [see Clinical Pharmacology (12.3)]. Se han notificado casos de trombocitopenia con teriflunomida, incluidos casos raros con recuentos de plaquetas inferiores a 50 000/mm3, en la fase posterior a la comercialización. Obtenga un hemograma completo (CBC) dentro de los 6 meses previos al inicio del tratamiento con teriflunomida. El seguimiento adicional debe basarse en signos y síntomas que sugieran supresión de la médula ósea.
Riesgo de infección/Detección de tuberculosis
Los pacientes con infecciones agudas o crónicas activas no deben comenzar el tratamiento hasta que se resuelvan las infecciones. Si un paciente desarrolla una infección grave, considere suspender el tratamiento con teriflunomida y utilizar un procedimiento de eliminación acelerada. Reevalúe los beneficios y riesgos antes de reanudar la terapia. Indique a los pacientes que reciben teriflunomida que informen los síntomas de infecciones a un médico.
No se recomienda la teriflunomida para pacientes con inmunodeficiencia grave, enfermedad de la médula ósea o infecciones graves no controladas. Los medicamentos como la teriflunomida que tienen potencial de inmunosupresión pueden hacer que los pacientes sean más susceptibles a las infecciones, incluidas las infecciones oportunistas.
En estudios controlados con placebo de teriflunomida en pacientes adultos, no se observó un aumento general en el riesgo de infecciones graves con teriflunomida 7 mg (2.2%) o 14 mg (2.7%) en comparación con placebo (2.2%).
Sin embargo, se produjo un caso mortal de sepsis por neumonía por klebsiella en un paciente que tomaba teriflunomida 14 mg durante 1.7 años. Se han notificado infecciones mortales en la fase posterior a la comercialización en pacientes que reciben leflunomida, especialmente neumonía por Pneumocystis jirovecii y aspergilosis. La mayoría de los informes se vieron confundidos por la terapia inmunosupresora concomitante y/o una enfermedad comórbida que, además de la enfermedad reumatoide, puede predisponer a los pacientes a la infección. En estudios clínicos con teriflunomida, se ha observado reactivación de la hepatitis por citomegalovirus.
En estudios clínicos con teriflunomida en pacientes adultos, se han observado casos de tuberculosis. Antes de iniciar la teriflunomida, examine a los pacientes para detectar infección tuberculosa latente con una prueba cutánea de tuberculina o con un análisis de sangre para detectar infección por mycobacterium tuberculosis. La teriflunomida no se ha estudiado en pacientes con una prueba de tuberculosis positiva, y se desconoce la seguridad de la teriflunomida en personas con infección tuberculosa latente. Para los pacientes que den positivo en la prueba de detección de tuberculosis, trate según la práctica médica estándar antes de la terapia con teriflunomida.
Vacunación
No hay datos clínicos disponibles sobre la eficacia y seguridad de las vacunas vivas en pacientes que toman teriflunomida. No se recomienda la vacunación con vacunas vivas. Se debe considerar la larga vida media de la teriflunomida al contemplar la administración de una vacuna viva después de suspender la teriflunomida.
Malignidad
El riesgo de malignidad, en particular los trastornos linfoproliferativos, aumenta con el uso de algunos medicamentos inmunosupresores. Existe un potencial de inmunosupresión con teriflunomida. No se informó un aumento aparente en la incidencia de neoplasias malignas y trastornos linfoproliferativos en los ensayos clínicos de teriflunomida, pero se necesitarían estudios más amplios y a más largo plazo para determinar si existe un mayor riesgo de neoplasias malignas o trastornos linfoproliferativos con teriflunomida.
5.5 Reacciones de hipersensibilidad
La teriflunomida puede causar anafilaxia y reacciones alérgicas graves [see Contraindications (4)]. Los signos y síntomas han incluido disnea, urticaria y angioedema, incluidos labios, ojos, garganta y lengua.
Informe a los pacientes sobre los signos y síntomas de anafilaxia y angioedema.
5.6 Reacciones cutáneas graves
Se han notificado casos de reacciones cutáneas graves, a veces mortales, como el síndrome de Stevens-Johnson (SSJ), la necrólisis epidérmica tóxica (NET) y la reacción farmacológica con eosinofilia y síntomas sistémicos (DRESS) [ver Advertencias y precauciones (5.7)], con teriflunomida. Se notificaron desenlaces fatales en un caso de NET y un caso de DRESS.
Informe a los pacientes sobre los signos y síntomas que pueden indicar una reacción cutánea grave. Indique a los pacientes que suspendan la teriflunomida y busquen atención médica inmediata si se presentan estos signos y síntomas. A menos que la reacción claramente no esté relacionada con el medicamento, suspenda la teriflunomida y comience un procedimiento de eliminación acelerada inmediatamente [ver Advertencias y precauciones (5.3)]. En tales casos, los pacientes no deben volver a exponerse a la teriflunomida [ver Contraindicaciones (4)].
5.7 Reacción farmacológica con eosinofilia y síntomas sistémicos
Se ha producido una reacción farmacológica con eosinofilia y síntomas sistémicos (DRESS), también conocida como hipersensibilidad multiorgánica, con teriflunomida. Se ha notificado un caso mortal de DRESS que se produjo en estrecha asociación temporal (34 días) con el inicio del tratamiento con teriflunomida en la fase posterior a la comercialización. El DRESS típicamente, aunque no exclusivamente, se presenta con fiebre, erupción cutánea, linfadenopatía y/o hinchazón facial, en asociación con la afectación de otros sistemas orgánicos, como hepatitis, nefritis, anomalías hematológicas, miocarditis o miositis, que a veces se asemeja a una infección viral aguda. A menudo se presenta eosinofilia. Este trastorno es variable en su expresión y pueden estar implicados otros sistemas orgánicos no mencionados aquí. Es importante tener en cuenta que las manifestaciones tempranas de hipersensibilidad (p. ej., fiebre, linfadenopatía) pueden estar presentes aunque no haya erupción cutánea evidente. Si se presentan tales signos o síntomas, el paciente debe ser evaluado inmediatamente.
Suspenda la teriflunomida, a menos que se establezca una etiología alternativa para los signos o síntomas, y comience un procedimiento de eliminación acelerada inmediatamente [ver Advertencias y precauciones (5.3)]. En tales casos, los pacientes no deben volver a exponerse a la teriflunomida [ver Contraindicaciones (4)].
5.8 Neuropatía periférica
En estudios controlados con placebo en pacientes adultos, la neuropatía periférica, que incluye tanto la polineuropatía como la mononeuropatía (p. ej., síndrome del túnel carpiano), se produjo con mayor frecuencia en pacientes que tomaban teriflunomida que en pacientes que tomaban placebo. La incidencia de neuropatía periférica confirmada por estudios de conducción nerviosa fue del 1,4 % (13 pacientes) y del 1,9 % (17 pacientes) de los pacientes que recibieron 7 mg y 14 mg de teriflunomida, respectivamente, en comparación con el 0,4 % que recibió placebo (4 pacientes). El tratamiento se suspendió en el 0,7 % (8 pacientes) con neuropatía periférica confirmada (3 pacientes que recibieron teriflunomida 7 mg y 5 pacientes que recibieron teriflunomida 14 mg). Cinco de ellos se recuperaron después de la suspensión del tratamiento. No todos los casos de neuropatía periférica se resolvieron con la continuación del tratamiento. La neuropatía periférica también se produjo en pacientes que recibieron leflunomida.
Una edad superior a 60 años, medicamentos neurotóxicos concomitantes y diabetes pueden aumentar el riesgo de neuropatía periférica. Si un paciente que toma teriflunomida desarrolla síntomas compatibles con neuropatía periférica, como entumecimiento bilateral u hormigueo en las manos o los pies, considere suspender el tratamiento con teriflunomida y realizar un procedimiento de eliminación acelerada [ver Advertencias y precauciones (5.3)].
5.9 Aumento de la presión arterial
En estudios controlados con placebo en pacientes adultos, el cambio medio desde el inicio hasta el final del estudio en la presión arterial sistólica fue de +2,3 mmHg y +2,7 mmHg para teriflunomida 7 mg y 14 mg, respectivamente, y de -0,6 mmHg para placebo. El cambio desde el inicio en la presión arterial diastólica fue de +1,4 mmHg y +1,9 mmHg para teriflunomida 7 mg y 14 mg, respectivamente, y de -0,3 mmHg para placebo. La hipertensión fue una reacción adversa en el 3,1 % y el 4,3 % de los pacientes tratados con 7 mg o 14 mg de teriflunomida en comparación con el 1,8 % para placebo. Controle la presión arterial antes de iniciar el tratamiento con teriflunomida y periódicamente a partir de entonces. La presión arterial elevada debe manejarse adecuadamente durante el tratamiento con teriflunomida.
5.10 Efectos respiratorios
Se ha notificado enfermedad pulmonar intersticial, incluida neumonitis intersticial aguda, con teriflunomida en la fase posterior a la comercialización.
Se ha notificado enfermedad pulmonar intersticial y empeoramiento de la enfermedad pulmonar intersticial preexistente durante el tratamiento con leflunomida. La enfermedad pulmonar intersticial puede ser mortal y puede ocurrir de forma aguda en cualquier momento durante el tratamiento con una presentación clínica variable. La aparición de nuevos síntomas pulmonares o el empeoramiento de los mismos, como tos y disnea, con o sin fiebre asociada, pueden ser motivo para la suspensión del tratamiento y para una mayor investigación, según corresponda. Si es necesaria la suspensión del medicamento, considere la posibilidad de iniciar un procedimiento de eliminación acelerada [ver Advertencias y precauciones (5.3)].
5.11 Pancreatitis en pacientes pediátricos
La teriflunomida no está aprobada para su uso en pacientes pediátricos. En el ensayo clínico pediátrico, se observaron casos de pancreatitis en el 1,8 % (2/109) de los pacientes que recibieron teriflunomida; uno de estos casos fue grave [ver Uso en poblaciones específicas (8.4)]. Si se sospecha pancreatitis, suspenda la teriflunomida e inicie un procedimiento de eliminación acelerada [ver Advertencias y precauciones (5.3)].
5.12 Uso concomitante con terapias inmunosupresoras o inmunomoduladoras
No se ha evaluado la administración conjunta con terapias antineoplásicas o inmunosupresoras utilizadas para el tratamiento de la esclerosis múltiple. Los estudios de seguridad en los que se administró teriflunomida concomitantemente con otras terapias inmunomoduladoras durante un máximo de un año (interferón beta, acetato de glatiramer) no revelaron ningún problema de seguridad específico. No se ha establecido la seguridad a largo plazo de estas combinaciones en el tratamiento de la esclerosis múltiple.
En cualquier situación en la que se tome la decisión de cambiar de teriflunomida a otro agente con un potencial conocido de supresión hematológica, sería prudente monitorizar la toxicidad hematológica, ya que habrá una superposición de la exposición sistémica a ambos compuestos. El uso de un procedimiento de eliminación acelerada puede disminuir este riesgo, pero también puede resultar en el retorno de la actividad de la enfermedad si el paciente había estado respondiendo al tratamiento con teriflunomida [see Warnings and Precautions (5.3)].
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas graves se describen en otras partes de la información de prescripción:
- Hepatotoxicity [see Contraindications (4) and Warnings and Precautions (5.1)]
- Bone Marrow Effects/Immunosuppression Potential/Infections [see Warnings and Precautions (5.4)]
- Hypersensitivity Reactions [see Contraindications (4) and Warnings and Precautions (5.5)]
- Serious Skin Reactions [see Warnings and Precautions (5.6)]
- Drug Reaction with Eosinophilia and Systemic Symptoms [see Warnings and Precautions (5.7)]
- Peripheral Neuropathy [see Warnings and Precautions (5.8)]
- Increased Blood Pressure [see Warnings and Precautions (5.9)]
- Respiratory Effects [see Warnings and Precautions (5.10)]
- Pancreatitis in Pediatric Patients [see Warnings and Precautions (5.11)]
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se realizan bajo condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un medicamento no pueden compararse directamente con las tasas en los ensayos clínicos de otro medicamento y es posible que no reflejen las tasas observadas en la práctica clínica.
Un total de 2047 pacientes que recibieron teriflunomida (7 mg o 14 mg una vez al día) constituyeron la población de seguridad en el análisis conjunto de estudios controlados con placebo en pacientes con formas recurrentes de esclerosis múltiple; de estos, el 71% eran mujeres. La edad promedio fue de 37 años.
La Tabla 1 enumera las reacciones adversas en los ensayos controlados con placebo con tasas que fueron al menos del 2% para los pacientes con teriflunomida y también al menos un 2% por encima de la tasa en los pacientes con placebo. Las más comunes fueron dolor de cabeza, un aumento de ALT, diarrea, alopecia y náuseas. La reacción adversa más comúnmente asociada con la interrupción fue un aumento de ALT (3.3%, 2.6% y 2.3% de todos los pacientes en los brazos de tratamiento con teriflunomida 7 mg, teriflunomida 14 mg y placebo, respectivamente).
| Reacción adversa |
Teriflunomida 7 mg (N=1,045) |
Teriflunomida 14 mg (N=1,002) |
Placebo (N=997) |
| Dolor de cabeza | 18% | 16% | 15% |
| Aumento de alanina aminotransferasa | 13% | 15% | 9% |
| Diarrea | 13% | 14% | 8% |
| Alopecia | 10% | 13% | 5% |
| Náuseas | 8% | 11% | 7% |
| Paresthesia | 8% | 9% | 7% |
| Arthralgia | 8% | 6% | 5% |
| Neutropenia | 4% | 6% | 2% |
| Hipertensión | 3% | 4% | 2% |
Muertes cardiovasculares
Se reportaron cuatro muertes cardiovasculares, incluyendo tres muertes súbitas y un infarto de miocardio en un paciente con antecedentes de hiperlipidemia e hipertensión, entre aproximadamente 2600 pacientes expuestos a teriflunomida en la base de datos previa a la comercialización. Estas muertes cardiovasculares ocurrieron durante estudios de extensión no controlados, de uno a nueve años después del inicio del tratamiento. No se ha establecido una relación entre teriflunomida y la muerte cardiovascular.
Insuficiencia renal aguda
En estudios controlados con placebo, los valores de creatinina aumentaron más del 100 % sobre el valor inicial en 8/1045 (0,8 %) pacientes en el grupo de teriflunomida de 7 mg y en 6/1002 (0,6 %) pacientes en el grupo de teriflunomida de 14 mg frente a 4/997 (0,4 %) pacientes en el grupo placebo. Estas elevaciones fueron transitorias. Algunas elevaciones estuvieron acompañadas de hiperpotasemia. La teriflunomida puede causar nefropatía aguda por ácido úrico con insuficiencia renal aguda transitoria porque la teriflunomida aumenta la depuración renal de ácido úrico.
Hipofosfatemia
En ensayos clínicos, el 18 % de los pacientes tratados con teriflunomida tuvieron hipofosfatemia con niveles séricos de fósforo de al menos 0,6 mmol/L, en comparación con el 7 % de los pacientes tratados con placebo; el 4 % de los pacientes tratados con teriflunomida tuvieron hipofosfatemia con niveles séricos de fósforo de al menos 0,3 mmol/L pero menos de 0,6 mmol/L, en comparación con el 0,8 % de los pacientes tratados con placebo. Ningún paciente en ningún grupo de tratamiento tuvo un fósforo sérico por debajo de 0,3 mmol/L.
6.2 Experiencia posterior a la comercialización
Las siguientes reacciones adversas se han identificado durante el uso posterior a la aprobación de teriflunomida. Debido a que estas reacciones se notifican voluntariamente a partir de una población de tamaño incierto, no siempre es posible estimar de forma fiable su frecuencia o establecer una relación causal con la exposición al fármaco.
- Trastornos de la sangre y del sistema linfático: Trombocitopenia [ver Advertencias y precauciones (5.4)]
- Trastornos gastrointestinales: Pancreatitis, colitis
- Trastornos hepatobiliares: Lesión hepática inducida por fármacos (DILI) [ver Advertencias y precauciones (5.1)]
- Trastornos del sistema inmunitario: Reacciones de hipersensibilidad, algunas de las cuales fueron graves, como anafilaxia y angioedema [ver Advertencias y precauciones (5.5)]
- Trastornos respiratorios, torácicos y mediastínicos: Enfermedad pulmonar intersticial [ver Advertencias y precauciones (5.10)]
- Trastornos de la piel y del tejido subcutáneo: Reacciones cutáneas graves, como necrólisis epidérmica tóxica y síndrome de Stevens-Johnson [ver Advertencias y precauciones (5.6)]; reacción farmacológica con eosinofilia y síntomas sistémicos (DRESS) [ver Advertencias y precauciones (5.7)]; psoriasis o empeoramiento de la psoriasis (incluida la psoriasis pustulosa y la psoriasis ungueal); trastornos de las uñas
7 INTERACCIONES MEDICAMENTOSAS
Efecto de Teriflunomida en Sustratos de CYP2C8
Teriflunomida es un inhibidor de CYP2C8 in vivo. En pacientes que toman teriflunomida, la exposición a medicamentos metabolizados por CYP2C8 (por ejemplo, paclitaxel, pioglitazona, repaglinida, rosiglitazona) puede aumentar. Monitoree a estos pacientes y ajuste la dosis del medicamento(s) concomitante(s) metabolizado(s) por CYP2C8 según sea necesario [ver Farmacología Clínica (12.3)].
Efecto de Teriflunomida en Warfarina
La administración conjunta de teriflunomida con warfarina requiere una estrecha vigilancia de la relación internacional normalizada (INR) porque la teriflunomida puede disminuir el pico de INR en aproximadamente un 25%.
Efecto de Teriflunomida en Anticonceptivos Orales
Teriflunomida puede aumentar las exposiciones sistémicas de etinilestradiol y levonorgestrel. Se debe considerar el tipo o la dosis de los anticonceptivos utilizados en combinación con teriflunomida [ver Farmacología Clínica (12.3)].
Efecto de Teriflunomida en Sustratos de CYP1A2
Teriflunomida puede ser un inductor débil de CYP1A2 in vivo. En pacientes que toman teriflunomida, la exposición a medicamentos metabolizados por CYP1A2 (por ejemplo, alosetrón, duloxetina, teofilina, tizanidina) puede reducirse. Monitoree a estos pacientes y ajuste la dosis del medicamento(s) concomitante(s) metabolizado(s) por CYP1A2 según sea necesario [ver Farmacología Clínica (12.3)].
Efecto de Teriflunomida en Sustratos del Transportador de Aniones Orgánicos 3 (OAT3)
Teriflunomida inhibe la actividad de OAT3 in vivo. En pacientes que toman teriflunomida, la exposición a medicamentos que son sustratos de OAT3 (por ejemplo, cefaclor, cimetidina, ciprofloxacina, penicilina G, ketoprofeno, furosemida, metotrexato, zidovudina) puede aumentar. Monitoree a estos pacientes y ajuste la dosis del medicamento(s) concomitante(s) que son sustratos de OAT3 según sea necesario [ver Farmacología Clínica (12.3)].
Efecto de Teriflunomida en Sustratos de BCRP y Polipéptido de Transporte de Aniones Orgánicos B1 y B3 (OATP1B1/1B3)
Teriflunomida inhibe la actividad de BCRP y OATP1B1/1B3 in vivo. Para un paciente que toma teriflunomida, la dosis de rosuvastatina no debe exceder los 10 mg una vez al día. Para otros sustratos de BCRP (por ejemplo, mitoxantrona) y medicamentos de la familia OATP (por ejemplo, metotrexato, rifampicina), especialmente inhibidores de la HMG-Co reductasa (por ejemplo, atorvastatina, nateglinida, pravastatina, repaglinida y simvastatina), considere reducir la dosis de estos medicamentos y monitorear a los pacientes de cerca para detectar signos y síntomas de exposiciones aumentadas a los medicamentos mientras los pacientes están tomando teriflunomida [ver Farmacología Clínica (12.3)].
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de Riesgos
Teriflunomida está contraindicada para su uso en mujeres embarazadas y mujeres en edad fértil que no utilizan métodos anticonceptivos eficaces debido al potencial de daño fetal basado en datos de animales [ver Contraindicaciones (4) y Advertencias y precauciones (5.2)].
En estudios de reproducción en animales en ratas y conejos, la administración oral de teriflunomida durante la organogénesis causó teratogenicidad y embrioletalidad a exposiciones plasmáticas (AUC) inferiores a las de la dosis máxima recomendada en humanos (MRHD) de 14 mg/día [ver Datos]. Los datos humanos disponibles de registros de embarazo, ensayos clínicos, casos de farmacovigilancia y literatura publicada son demasiado limitados para sacar conclusiones, pero no indican claramente un aumento de los defectos de nacimiento o los abortos espontáneos asociados con la exposición inadvertida a teriflunomida en el primer trimestre temprano cuando se sigue un procedimiento de eliminación acelerada [ver Consideraciones clínicas y Datos]. No hay datos humanos relacionados con exposiciones más adelante en el primer trimestre o más allá.
En la población general de EE. UU., el riesgo de fondo estimado de defectos de nacimiento mayores y abortos espontáneos en embarazos clínicamente reconocidos es del 2% al 4% y del 15% al 20%, respectivamente. Se desconoce el riesgo de fondo de defectos de nacimiento mayores y abortos espontáneos en la población indicada.
Consideraciones clínicas
Reacciones adversas fetales/neonatales
Reducir la concentración plasmática de teriflunomida mediante la instauración de un procedimiento de eliminación acelerada del fármaco tan pronto como se detecte el embarazo puede disminuir el riesgo para el feto de teriflunomida. El procedimiento de eliminación acelerada del fármaco incluye la verificación de que la concentración plasmática de teriflunomida es inferior a 0,02 mg/L [ver Advertencias y precauciones (5.3) y Farmacología clínica (12.3)].
Datos
Datos humanos
Los datos humanos disponibles son limitados. Los datos informados prospectivamente (de ensayos clínicos e informes poscomercialización) de >150 embarazos en pacientes tratados con teriflunomida y >300 embarazos en pacientes tratados con leflunomida no han demostrado un aumento de la tasa de malformaciones congénitas o abortos espontáneos tras la exposición a teriflunomida en el primer trimestre temprano cuando se sigue un procedimiento de eliminación acelerada. No se han observado patrones específicos de malformaciones congénitas mayores en humanos. Las limitaciones de estos datos incluyen un número inadecuado de embarazos informados para sacar conclusiones, la corta duración de la exposición al fármaco en los embarazos informados, lo que impide una evaluación completa de los riesgos fetales, la notificación incompleta y la incapacidad de controlar los factores de confusión (como la enfermedad materna subyacente y el uso de medicamentos concomitantes).
Datos de animales
Cuando se administró teriflunomida (dosis orales de 1, 3 o 10 mg/kg/día) a ratas embarazadas durante todo el período de organogénesis, se observaron altas incidencias de malformación fetal (principalmente craneofacial, y defectos esqueléticos axiales y apendiculares) y muerte fetal a dosis no asociadas con toxicidad materna. Se observaron efectos adversos en el desarrollo fetal tras la dosificación en varias etapas durante la organogénesis. La exposición plasmática materna en el nivel sin efecto (1 mg/kg/día) para la toxicidad del desarrollo fetal en ratas fue inferior a la de los humanos a la dosis máxima recomendada en humanos (MRHD, 14 mg/día).
La administración de teriflunomida (dosis orales de 1, 3.5 o 12 mg/kg/día) a conejas embarazadas durante toda la organogénesis dio como resultado altas incidencias de malformación fetal (principalmente craneofacial, y defectos esqueléticos axiales y apendiculares) y muerte fetal a dosis asociadas con toxicidad materna mínima. La exposición plasmática materna a la dosis sin efecto (1 mg/kg/día) para la toxicidad del desarrollo fetal en conejos fue inferior a la de los humanos a la MRHD.
En estudios en los que se administró teriflunomida (dosis orales de 0,05, 0,1, 0,3, 0,6 o 1 mg/kg/día) a ratas durante la gestación y la lactancia, se observó un crecimiento disminuido, anomalías oculares y cutáneas, y altas incidencias de malformación (defectos de las extremidades) y muerte postnatal en la descendencia a dosis no asociadas con toxicidad materna. La exposición plasmática materna a la dosis sin efecto para la toxicidad del desarrollo prenatal y postnatal en ratas (0,10 mg/kg/día) fue inferior a la de los humanos a la MRHD.
En estudios de reproducción en animales de leflunomida, se observaron efectos embrioletales y teratógenos en ratas y conejas embarazadas a concentraciones plasmáticas de teriflunomida clínicamente relevantes (AUC) o inferiores. En estudios de reproducción publicados en ratones embarazadas, la leflunomida fue embrioletal y aumentó la incidencia de malformaciones (craneofacial, esquelética axial, cardíaca y de grandes vasos). La suplementación con uridina exógena redujo los efectos teratógenos en ratones embarazadas, lo que sugiere que el modo de acción (inhibición de la enzima mitocondrial dihidroorotato deshidrogenasa) es el mismo para la eficacia terapéutica y la toxicidad del desarrollo.
A las dosis recomendadas en humanos, teriflunomida y leflunomida dan como resultado un rango similar de concentraciones plasmáticas de teriflunomida.
8.2 Lactancia
Resumen de Riesgos
No hay datos sobre la presencia de teriflunomida en la leche materna, los efectos en el lactante amamantado o los efectos en la producción de leche. Se detectó teriflunomida en la leche de rata tras una sola dosis oral. Debido al potencial de reacciones adversas en un lactante amamantado por teriflunomida, las mujeres no deben amamantar durante el tratamiento con teriflunomida.
8.3 Mujeres y hombres en edad fértil
Prueba de embarazo
Excluir el embarazo antes de iniciar el tratamiento con teriflunomida en mujeres en edad fértil. Aconsejar a las mujeres que notifiquen inmediatamente a su médico si se produce o se sospecha un embarazo durante el tratamiento [ver Advertencias y precauciones (5.2, 5.3) y Uso en poblaciones específicas (8.1)].
Contracepción
Mujeres
Las mujeres en edad fértil deben utilizar métodos anticonceptivos eficaces mientras toman teriflunomida. Si se interrumpe el tratamiento con teriflunomida, se debe continuar con el uso de anticonceptivos hasta que se verifique que las concentraciones plasmáticas de teriflunomida son inferiores a 0,02 mg/L (0,02 mcg/mL), el nivel que se espera que tenga un riesgo fetal mínimo, según los datos en animales.
Las mujeres en edad fértil que deseen quedar embarazadas deben interrumpir el tratamiento con teriflunomida y someterse a un procedimiento de eliminación acelerada. Se debe utilizar un método anticonceptivo eficaz hasta que se verifique que las concentraciones plasmáticas de teriflunomida son inferiores a 0,02 mg/L (0,02 mcg/mL) [ver Advertencias y precauciones (5.2, 5.3) y Uso en poblaciones específicas (8.1)].
Hombres
La teriflunomida se detecta en el semen humano. No se han realizado estudios en animales para evaluar específicamente el riesgo de toxicidad fetal mediada por el hombre. Para minimizar cualquier posible riesgo, los hombres que no deseen tener un hijo y sus parejas femeninas deben utilizar métodos anticonceptivos eficaces. Los hombres que deseen tener un hijo deben interrumpir el uso de teriflunomida y someterse a un procedimiento de eliminación acelerada o esperar hasta que se verifique que la concentración plasmática de teriflunomida es inferior a 0,02 mg/L (0,02 mcg/mL) [ver Advertencias y precauciones (5.3)].
8.4 Uso pediátrico
No se ha establecido la seguridad y eficacia en pacientes pediátricos. La eficacia de la teriflunomida para el tratamiento de la forma recurrente de la esclerosis múltiple en pacientes pediátricos (de 10 a 17 años de edad) no se estableció en un estudio clínico adecuado y bien controlado en 166 pacientes (109 pacientes recibieron dosis diarias de teriflunomida y 57 pacientes recibieron placebo) durante un máximo de 96 semanas.
Se ha notificado pancreatitis en adultos en el contexto de la poscomercialización, pero parece ocurrir con mayor frecuencia en la población pediátrica. En este estudio pediátrico, se notificaron casos de pancreatitis en el 1,8% (2/109) de los pacientes que recibieron teriflunomida en comparación con ningún paciente en el grupo placebo. Todos los pacientes del ensayo pediátrico se recuperaron o se estaban recuperando después de la interrupción del tratamiento y el procedimiento de eliminación acelerada [ver Advertencias y precauciones (5.11)].
Además, se notificó una concentración elevada o anormal de creatina fosfoquinasa en sangre en el 6,4% de los pacientes pediátricos que recibieron teriflunomida en comparación con ningún paciente en el grupo placebo.
Datos de toxicidad en animales jóvenes
La administración oral de teriflunomida (0, 0,3, 3 o 6 mg/kg/día) a ratas jóvenes en los días posnatales 21 a 70 dio como resultado la supresión de la función inmunitaria (respuesta de anticuerpos dependiente de células T) en las dosis medias y altas, y efectos adversos en los órganos reproductores masculinos (reducción del recuento de espermatozoides) y alteración de la función neuroconductual (aumento de la actividad locomotora) en la dosis alta. En la dosis sin efecto (0,3 mg/kg/día) para la toxicidad del desarrollo en ratas jóvenes, las exposiciones plasmáticas fueron inferiores a las de los pacientes pediátricos en las dosis de teriflunomida probadas en el estudio clínico.
8.5 Uso en geriatría
Los estudios clínicos de teriflunomida no incluyeron pacientes mayores de 65 años.
8.6 Insuficiencia hepática
No es necesario ajustar la dosis en pacientes con insuficiencia hepática leve o moderada. No se ha evaluado la farmacocinética de la teriflunomida en la insuficiencia hepática grave.
La teriflunomida está contraindicada en pacientes con insuficiencia hepática grave [ver Contraindicaciones (4), Advertencias y precauciones (5.1), y Farmacología clínica (12.3)].
8.7 Insuficiencia renal
No es necesario ajustar la dosis en pacientes con insuficiencia renal leve, moderada y grave [ver Farmacología clínica (12.3)].
10 SOBREDOSIS
No hay experiencia con sobredosis o intoxicación por teriflunomida en humanos. Teriflunomida 70 mg diarios hasta 14 días fue bien tolerado por sujetos sanos.
En caso de sobredosis o toxicidad clínicamente significativa, se recomienda colestiramina o carbón activado para acelerar la eliminación [ver Advertencias y precauciones (5.3)].
11 DESCRIPCIÓN
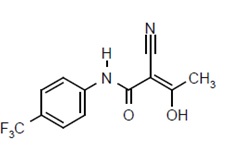
Teriflunomida es un inhibidor oral de novo de la síntesis de pirimidina de la enzima DHO-DH, con el nombre químico ácido (Z)-2-ciano-3-hidroxi-but-2-enoico-(4-trifluorometilfenil)-amida. Su peso molecular es 270.21, y la fórmula molecular es C12H9F3N2O2 con la siguiente estructura química:
Teriflunomida es un polvo blanco a casi blanco que es escasamente soluble en acetona y ligeramente soluble en acetonitrilo.
Teriflunomida se formula como comprimidos recubiertos con película para administración oral. Los comprimidos de teriflunomida contienen 7 mg o 14 mg de teriflunomida y los siguientes ingredientes inactivos: dióxido de silicio coloidal, almidón de maíz, FD & C azul # 2/ lago de aluminio de índigo carmín, hidroxipropilcelulosa, hipromelosa, monohidrato de lactosa, estearato de magnesio, celulosa microcristalina, polietilenglicol, glicolato de almidón sódico, talco y dióxido de titanio. Además, 7 mg contiene óxido de hierro amarillo.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
La teriflunomida, un agente inmunomodulador con propiedades antiinflamatorias, inhibe la dihidroorotato deshidrogenasa, una enzima mitocondrial involucrada en la síntesis de novo de pirimidinas. El mecanismo exacto por el cual la teriflunomida ejerce su efecto terapéutico en la esclerosis múltiple es desconocido, pero puede involucrar una reducción en el número de linfocitos activados en el SNC.
12.2 Farmacodinamia
Potencial para prolongar el intervalo QT
En un estudio QT exhaustivo controlado con placebo realizado en sujetos adultos sanos, no hubo evidencia de que la teriflunomida causara una prolongación del intervalo QT de importancia clínica (es decir, el límite superior del intervalo de confianza del 90% para el mayor QTc ajustado por placebo y corregido con respecto al basal fue inferior a 10 ms).
12.3 Farmacocinética
La teriflunomida es el principal metabolito activo de la leflunomida y es responsable de la actividad de la leflunomida in vivo. A las dosis recomendadas, la teriflunomida y la leflunomida dan lugar a un rango similar de concentraciones plasmáticas de teriflunomida.
Basado en un análisis poblacional de la teriflunomida en voluntarios adultos sanos y pacientes adultos con EM, la mediana t1/2 fue de aproximadamente 18 y 19 días después de dosis repetidas de 7 mg y 14 mg, respectivamente. Se tarda aproximadamente 3 meses, respectivamente, para alcanzar las concentraciones en estado de equilibrio. La razón de acumulación estimada del AUC es de aproximadamente 30 después de dosis repetidas de 7 o 14 mg.
Absorción
La mediana del tiempo para alcanzar las concentraciones plasmáticas máximas es de entre 1 a 4 horas después de la administración oral de teriflunomida.
Los alimentos no tienen un efecto clínicamente relevante en la farmacocinética de la teriflunomida.
Distribución
La teriflunomida se une ampliamente a las proteínas plasmáticas (> 99%) y se distribuye principalmente en el plasma. El volumen de distribución es de 11 L después de una administración intravenosa (IV) única.
Metabolismo
La teriflunomida es la principal fracción circulante detectada en el plasma. La vía de biotransformación primaria a metabolitos menores de la teriflunomida es la hidrólisis, siendo la oxidación una vía menor. Las vías secundarias involucran la oxidación, la N-acetilación y la conjugación con sulfato.
Eliminación
La teriflunomida se elimina principalmente a través de la excreción biliar directa del fármaco sin cambios, así como la excreción renal de los metabolitos. Durante más de 21 días, el 60,1% de la dosis administrada se excreta a través de las heces (37,5%) y la orina (22,6%). Después de un procedimiento de eliminación acelerada con colestiramina, se recuperó un adicional del 23,1% (principalmente en las heces). Después de una administración IV única, la depuración corporal total de la teriflunomida es de 30,5 mL/h.
Estudios de interacción de fármacos
La teriflunomida no se metaboliza por las enzimas del Citocromo P450 o la flavina monoamino oxidasa.
El efecto potencial de la teriflunomida en otros fármacos
Sustratos de CYP2C8
Hubo un aumento en la Cmáx y el AUC medios de la repaglinida (1,7- y 2,4 veces, respectivamente) después de dosis repetidas de teriflunomida y una dosis única de 0,25 mg de repaglinida, lo que sugiere que la teriflunomida es un inhibidor de CYP2C8 in vivo. La magnitud de la interacción podría ser mayor con la dosis recomendada de repaglinida [véase Interacciones de fármacos (7)].
Sustratos de CYP1A2
Las dosis repetidas de teriflunomida disminuyeron la Cmáx y el AUC medios de la cafeína en un 18% y un 55%, respectivamente, lo que sugiere que la teriflunomida puede ser un inductor débil de CYP1A2 in vivo [véase Interacciones de fármacos (7)].
Sustratos de OAT3
Hubo un aumento en la Cmáx y el AUC medios del cefaclor (1,43- y 1,54 veces, respectivamente) después de dosis repetidas de teriflunomida, lo que sugiere que la teriflunomida es un inhibidor del transportador de aniones orgánicos 3 (OAT3) in vivo [véase Interacciones de fármacos (7)].
Sustratos de BCRP y OATP1B1/1B3
Hubo un aumento en la Cmáx y el AUC medios de la rosuvastatina (2,65- y 2,51 veces, respectivamente) después de dosis repetidas de teriflunomida, lo que sugiere que la teriflunomida es un inhibidor del transportador BCRP y del polipéptido transportador de aniones orgánicos 1B1 y 1B3 (OATP1B1/1B3) [véase Interacciones de fármacos (7)].
Anticonceptivos orales
Hubo un aumento en la Cmáx y el AUC0-24 medios del etinilestradiol (1,58- y 1,54 veces, respectivamente) y de la levonorgestrel Cmáx y AUC0-24 (1,33- y 1,41 veces, respectivamente) después de dosis repetidas de teriflunomida [véase Interacciones de fármacos (7)].
La teriflunomida no afectó la farmacocinética de la bupropión (un sustrato de CYP2B6), el midazolam (un sustrato de CYP3A4), la S-warfarina (un sustrato de CYP2C9), el omeprazol (un sustrato de CYP2C19) y el metoprolol (un sustrato de CYP2D6).
El efecto potencial de otros fármacos en la teriflunomida
Inductores potentes de CYP y transportadores: La rifampicina no afectó la farmacocinética de la teriflunomida.
Poblaciones específicas
Insuficiencia hepática
La insuficiencia hepática leve y moderada no tuvo impacto en la farmacocinética de la teriflunomida. La farmacocinética de la teriflunomida en la insuficiencia hepática grave no ha sido evaluada [véase Contraindicaciones (4), Advertencias y Precauciones (5.1) y Uso en poblaciones específicas (8.6)].
Insuficiencia renal
La insuficiencia renal grave no tuvo impacto en la farmacocinética de la teriflunomida [véase Uso en poblaciones específicas (8.7)].
Género
En un análisis poblacional, la tasa de depuración de la teriflunomida es un 23% menor en las mujeres que en los hombres.
Raza
El efecto de la raza en la farmacocinética de la teriflunomida no puede ser evaluado adecuadamente debido al bajo número de pacientes que se identificaron como negros o afroamericanos, asiáticos u otras razas en los ensayos clínicos.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
Carcinogénesis
No se observó evidencia de carcinogenicidad en bioensayos de carcinogenicidad de por vida en ratones y ratas. En ratones, la teriflunomida se administró por vía oral en dosis de hasta 12 mg/kg/día durante un máximo de 95 a 104 semanas; las exposiciones plasmáticas a la teriflunomida (AUC) en la dosis más alta probada son aproximadamente 3 veces mayores que en humanos a la dosis humana máxima recomendada (MRHD, 14 mg/día). En ratas, la teriflunomida se administró por vía oral en dosis de hasta 4 mg/kg/día durante un máximo de 97 a 104 semanas; las AUC plasmáticas de teriflunomida en las dosis más altas probadas son menores que en humanos a la MRHD.
Mutagénesis
La teriflunomida fue negativa en la prueba de mutación inversa bacteriana in vitro (Ames), la prueba HPRT in vitro y en las pruebas de micronúcleos y aberraciones cromosómicas in vivo. La teriflunomida fue positiva en una prueba de aberración cromosómica in vitro en linfocitos humanos, con y sin activación metabólica. La adición de uridina (para complementar el grupo de pirimidinas) redujo la magnitud del efecto clastogénico; sin embargo, la teriflunomida fue positiva en la prueba de aberración cromosómica in vitro, incluso en presencia de uridina.
La 4-trifluorometilanilina (4-TFMA), un metabolito menor de la teriflunomida, fue positiva en la prueba de mutación inversa bacteriana in vitro (Ames), la prueba HPRT in vitro y la prueba de aberración cromosómica in vitro en células de mamíferos. La 4-TFMA fue negativa en las pruebas de micronúcleos y aberraciones cromosómicas in vivo.
Deterioro de la Fertilidad
La administración oral de teriflunomida (0, 1, 3, 10 mg/kg/día) a ratas macho antes y durante el apareamiento (con hembras no tratadas) no produjo efectos adversos sobre la fertilidad; sin embargo, se observó una reducción en el recuento de espermatozoides epididimarios en las dosis media y alta probadas. La dosis sin efecto para la toxicidad reproductiva en ratas macho (1 mg/kg) es menor que la MRHD en base a mg/m2.
La administración oral de teriflunomida (0, 0,84, 2,6, 8,6 mg/kg/día) a ratas hembra, antes y durante el apareamiento (con machos no tratados) y continuando hasta el día 6 de gestación, produjo embrioletalidad, reducción del peso corporal fetal y/o malformaciones en todas las dosis probadas. Debido a la marcada embrioletalidad en la dosis más alta probada, no hubo fetos disponibles para la evaluación. La dosis más baja probada es menor que la MRHD en base a mg/m2.
14 ESTUDIOS CLÍNICOS
Cuatro ensayos clínicos aleatorizados, controlados, doble ciego establecieron la eficacia de la teriflunomida en pacientes con formas recurrentes de esclerosis múltiple.
El estudio 1 fue un ensayo clínico doble ciego, controlado con placebo que evaluó dosis diarias de teriflunomida 7 mg y teriflunomida 14 mg durante un máximo de 26 meses en pacientes con formas recurrentes de esclerosis múltiple. Los pacientes debían tener un diagnóstico de esclerosis múltiple que mostrara un curso clínico recurrente, con o sin progresión, y haber experimentado al menos un brote durante el año anterior al ensayo o al menos dos brotes durante los dos años anteriores al ensayo. Los pacientes no debían haber recibido interferón-beta durante al menos cuatro meses, ni ningún otro medicamento para la esclerosis múltiple durante al menos seis meses antes de ingresar al estudio, ni se permitieron estos medicamentos durante el estudio. Las evaluaciones neurológicas debían realizarse en la selección, cada 12 semanas hasta la semana 108, y después de los brotes sospechosos. La resonancia magnética debía realizarse en la selección y en la semana 24, 48, 72 y 108. El criterio de valoración principal fue la tasa de recaídas anualizada (ARR).
En el estudio 1, 1.088 pacientes fueron aleatorizados para recibir teriflunomida 7 mg (n=366), teriflunomida 14 mg (n=359) o placebo (n=363). Al ingresar, los pacientes tenían una puntuación de la Escala de Estado de Discapacidad Expandida (EDSS) ≤5.5. Los pacientes tenían una edad media de 38 años, una duración media de la enfermedad de 5 años y una EDSS media al inicio del estudio de 2.7. Un total del 91% de los pacientes tenían esclerosis múltiple remitente recurrente y el 9% tenía una forma progresiva de esclerosis múltiple con recaídas. La duración media del tratamiento fue de 635, 627 y 631 días para teriflunomida 7 mg, teriflunomida 14 mg y placebo, respectivamente. El porcentaje de pacientes que completaron el período de tratamiento del estudio fue del 75%, 73% y 71% para teriflunomida 7 mg, teriflunomida 14 mg y placebo, respectivamente.
Hubo una reducción estadísticamente significativa en la ARR para los pacientes que recibieron teriflunomida 7 mg o teriflunomida 14 mg, en comparación con los pacientes que recibieron placebo (ver Tabla 2). Se observó una reducción consistente de la ARR en los subgrupos definidos por sexo, grupo de edad, terapia previa para la esclerosis múltiple y actividad de la enfermedad al inicio del estudio.
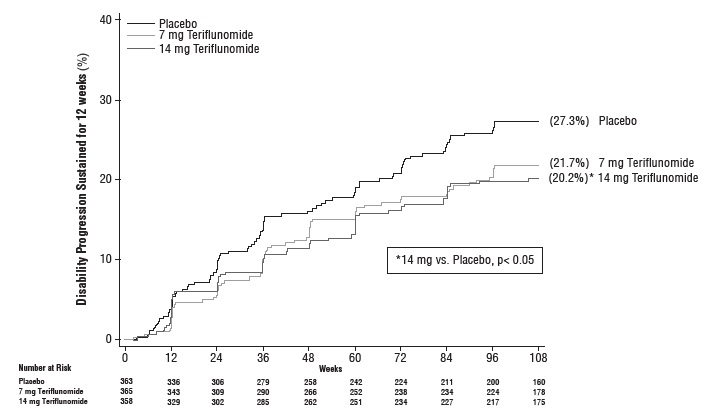
Hubo una reducción estadísticamente significativa en el riesgo relativo de progresión de la discapacidad en la semana 108 sostenida durante 12 semanas (medida por un aumento de al menos 1 punto desde la línea de base EDSS ≤5.5 o un aumento de 0.5 puntos para aquellos con una línea de base EDSS >5.5) en el grupo de teriflunomida 14 mg en comparación con el placebo (ver Tabla 2 y Figura 1).
El efecto de la teriflunomida en varias variables de resonancia magnética (RM), incluido el volumen total de la lesión de las lesiones T2 e hipointensas T1, se evaluó en el estudio 1. El cambio en el volumen total de la lesión desde la línea de base fue significativamente menor en los grupos de teriflunomida 7 mg y teriflunomida 14 mg que en el grupo placebo. Los pacientes en ambos grupos de teriflunomida tuvieron significativamente menos lesiones que realzan con gadolinio por exploración ponderada en T1 que los del grupo placebo (ver Tabla 2).
| Teriflunomida 7 mg N=365 |
Teriflunomida 14 mg N=358 |
Placebo N=363 |
|
|---|---|---|---|
| * Volumen total de la lesión: suma del volumen de la lesión T2 e hipointensa T1 en mL † Valores p basados en datos transformados por raíz cúbica para el volumen total de la lesión |
|||
| Criterios de valoración clínicos |
|||
| Tasa de recaídas anualizada | 0.370 (p=0.0002) |
0.369 (p=0.0005) |
0.539 |
| Reducción del riesgo relativo | 31% | 31% | – |
| Porcentaje de pacientes que permanecen libres de recaídas en la semana 108 | 53.7% | 56.5% | 45.6% |
| Porcentaje de progresión de la discapacidad en la semana 108 | 21.7% (p=0.084) |
20.2% (p=0.028) |
27.3% |
| Razón de riesgo | 0.76 | 0.70 | – |
| Criterios de valoración de RM |
|||
| Cambio mediano desde el inicio en el volumen total de la lesión* (mL) en la semana 108 | 0.755 (p=0.0317) † |
0.345 (p=0.0003)† |
1.127 |
| Número medio de lesiones T1 con realce de Gd por exploración | 0.570 (p<0.0001) |
0.261 (p<0.0001) |
1.331 |
Figura 1: Gráfica de Kaplan-Meier del tiempo hasta la progresión de la discapacidad sostenida durante 12 semanas (Estudio 1)
El estudio 2 fue un ensayo clínico doble ciego, controlado con placebo que evaluó dosis diarias de teriflunomida 7 mg y teriflunomida 14 mg durante un máximo de 40 meses en pacientes con formas recurrentes de esclerosis múltiple. Los pacientes debían tener un diagnóstico de esclerosis múltiple que mostrara un curso clínico recurrente y haber experimentado al menos un recaída durante el año anterior al ensayo, o al menos dos recaídas durante los dos años anteriores al ensayo. Los pacientes debían no haber recibido ningún medicamento para la esclerosis múltiple durante al menos tres meses antes de ingresar al ensayo, ni se permitieron estos medicamentos durante el ensayo. Las evaluaciones neurológicas debían realizarse en la selección, cada 12 semanas hasta la finalización, y después de cada recaída sospechosa. El punto final primario fue la ARR.
Un total de 1.165 pacientes recibieron teriflunomida 7 mg (n=407), teriflunomida 14 mg (n=370), o placebo (n=388). Los pacientes tenían una edad media de 38 años, una duración media de la enfermedad de 5 años y una EDSS media al inicio de 2,7. Un total del 98% de los pacientes tenían esclerosis múltiple remitente recurrente, y el 2% tenía una forma progresiva de esclerosis múltiple con recaídas. La duración media del tratamiento fue de 552, 567 y 571 días para teriflunomida 7 mg, teriflunomida 14 mg y placebo, respectivamente. El porcentaje de pacientes que completaron el período de tratamiento del estudio fue del 67%, 66% y 68% para teriflunomida 7 mg, teriflunomida 14 mg y placebo, respectivamente.
Hubo una reducción estadísticamente significativa en la ARR para los pacientes que recibieron teriflunomida 7 mg o teriflunomida 14 mg en comparación con los pacientes que recibieron placebo (ver Tabla 3). Hubo una reducción constante de la ARR observada en subgrupos definidos por sexo, grupo de edad, terapia previa para la esclerosis múltiple y actividad de la enfermedad al inicio.
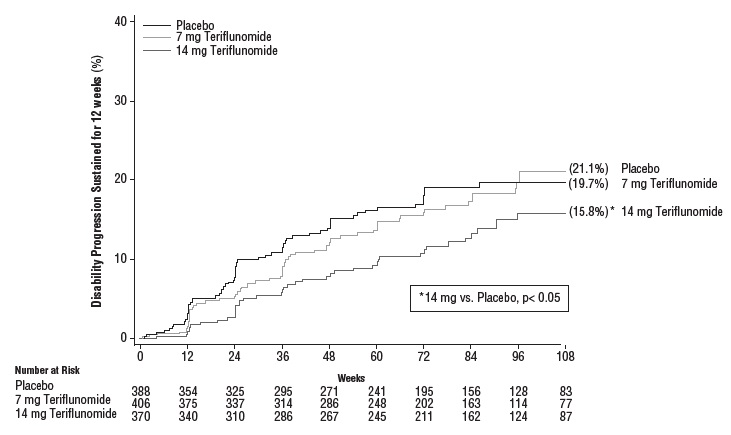
Hubo una reducción estadísticamente significativa en el riesgo relativo de progresión de la discapacidad en la semana 108 sostenida durante 12 semanas (medida por al menos un aumento de 1 punto desde el inicio de la EDSS ≤5,5 o un aumento de 0,5 puntos para aquellos con una EDSS inicial >5,5) en el grupo de teriflunomida 14 mg en comparación con placebo (ver Tabla 3 y Figura 2).
| Teriflunomida 7 mg N=407 |
Teriflunomida 14 mg N=370 |
Placebo N=388 |
|
|---|---|---|---|
| Puntos finales clínicos |
|||
| Tasa anualizada de recaídas | 0.389 (p=0.0183) |
0.319 (p=0.0001) |
0.501 |
| Reducción del riesgo relativo | 22% | 36% | – |
| Porcentaje de pacientes que permanecen libres de recaídas en la semana 108 | 58.2% | 57.1% | 46.8% |
| Porcentaje de progresión de la discapacidad en la semana 108 | 21.1% (p=0.762) |
15.8% (p=0.044) |
19.7% |
| Razón de riesgo | 0.96 | 0.69 | – |
Figura 2: Gráfica de Kaplan-Meier del tiempo hasta la progresión de la discapacidad sostenida durante 12 semanas (Estudio 2)
El Estudio 3 fue un ensayo clínico doble ciego controlado con placebo que evaluó dosis diarias de teriflunomida 7 mg y teriflunomida 14 mg durante un máximo de 108 semanas en pacientes con esclerosis múltiple remitente. Los pacientes debían haber tenido un primer evento clínico consistente con desmielinización aguda que ocurriera dentro de los 90 días de la aleatorización con 2 o más lesiones T2 de al menos 3 mm de diámetro que fueran características de la esclerosis múltiple. Un total de 614 pacientes recibieron teriflunomida 7 mg (n=203), teriflunomida 14 mg (n=214) o placebo (n=197). Los pacientes tenían una edad media de 32 años, EDSS al inicio del estudio de 1,7 y una duración media de la enfermedad de dos meses. La proporción de pacientes libres de recaída fue mayor en los grupos de teriflunomida 7 mg (70,5%, p<0,05) y teriflunomida 14 mg (72,2%, p<0,05) que en el grupo placebo (61,7%).
El efecto de la teriflunomida sobre la actividad de la RMN también se demostró en el Estudio 4, un ensayo clínico aleatorizado, doble ciego, controlado con placebo en pacientes con esclerosis múltiple con recaída. En el Estudio 4, la RMN debía realizarse al inicio del estudio, a las 6 semanas, a las 12 semanas, a las 18 semanas, a las 24 semanas, a las 30 semanas y a las 36 semanas después del inicio del tratamiento. Un total de 179 pacientes fueron aleatorizados a teriflunomida 7 mg (n=61), teriflunomida 14 mg (n=57) o placebo (n=61). Las características demográficas al inicio del estudio fueron consistentes entre los grupos de tratamiento. El criterio de valoración principal fue el número medio de lesiones activas únicas por exploración de RMN durante el tratamiento. El número medio de lesiones activas únicas por exploración de RMN cerebral durante el período de tratamiento de 36 semanas fue menor en los pacientes tratados con teriflunomida 7 mg (1,06) y teriflunomida 14 mg (0,98) en comparación con el placebo (2,69), siendo la diferencia estadísticamente significativa para ambos (p=0,0234 y p=0,0052, respectivamente).
16 FORMA DE SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
Teriflunomide está disponible en tabletas de 7 mg y 14 mg.
La tableta de 7 mg es de color verde claro a verde, redonda, recubierta con película, con “N” grabado en un lado y con “7” en el otro lado. Cada tableta contiene 7 mg de teriflunomida.
Cajas de 28 tabletas que contienen 2 blísteres de 14 tabletas cada uno NDC 59651-054-28
Botellas de 30 NDC 59651-054-30
La tableta de 14 mg es de color azul claro a azul, redonda, recubierta con película, con “N” grabado en un lado y con “14” en el otro lado. Cada tableta contiene 14 mg de teriflunomida.
Cajas de 28 tabletas que contienen 2 blísteres de 14 tabletas cada uno NDC 59651-055-28
Botellas de 30 NDC 59651-055-30
Almacenar a 20o a 25oC (68o a 77oF) [ver Temperatura ambiente controlada USP].
17 INFORMACIÓN PARA EL PACIENTE
Aconseje al paciente que lea el etiquetado del paciente aprobado por la FDA (Guía de medicamentos).
Se requiere una Guía de medicamentos para su distribución con teriflunomida.
Hepatotoxicidad
Informe a los pacientes que la teriflunomida puede causar daño hepático, que puede ser potencialmente mortal, y que sus enzimas hepáticas se controlarán antes de comenzar la teriflunomida y al menos mensualmente durante 6 meses después de comenzar la teriflunomida [ver Dosificación y administración (2) y Advertencias y precauciones (5.1)]. Aconseje a los pacientes que deben comunicarse con su médico si tienen náuseas, vómitos, dolor abdominal, fatiga, anorexia o ictericia y/o orina oscura inexplicables.
Toxicidad embrio-fetal
- Aconseje a las mujeres en edad fértil
- del riesgo potencial de daño fetal si se toma teriflunomida durante el embarazo
- que notifiquen a su proveedor de atención médica de inmediato si se produce o se sospecha un embarazo
- que usen métodos anticonceptivos efectivos durante el tratamiento con teriflunomida y hasta que se verifique que la concentración plasmática de teriflunomida es inferior a 0,02 mg/L [ver Advertencias y precauciones (5.2, 5.3), Uso en poblaciones específicas (8.1, 8.3), Farmacología clínica (12.3)].
- Indique a los hombres que toman teriflunomida y que no desean engendrar un hijo que usen métodos anticonceptivos efectivos para minimizar cualquier riesgo posible para el feto; sus parejas femeninas también deben usar métodos anticonceptivos efectivos.
- Aconseje a los hombres que desean engendrar un hijo que interrumpan el uso de teriflunomida y se sometan a un procedimiento de eliminación acelerada.
Disponibilidad de un procedimiento de eliminación acelerada
Aconseje a los pacientes que la teriflunomida puede permanecer en la sangre hasta 2 años después de la última dosis y que se puede utilizar un procedimiento de eliminación acelerada si es necesario [ver Advertencias y precauciones (5.3)].
Riesgo de infecciones
Informe a los pacientes que pueden desarrollar una disminución en el recuento de glóbulos blancos y que se controlará su recuento sanguíneo antes de comenzar la teriflunomida.
Informe a los pacientes que pueden tener más probabilidades de contraer infecciones cuando toman teriflunomida y que deben comunicarse con su médico si desarrollan síntomas de infección, particularmente en caso de fiebre [ver Advertencias y precauciones (5.4)].
Aconseje a los pacientes que se debe evitar el uso de algunas vacunas durante el tratamiento con teriflunomida y durante al menos 6 meses después de la interrupción.
Reacciones de hipersensibilidad
Aconseje a los pacientes que interrumpan la teriflunomida y busquen atención médica inmediata si se presenta algún signo o síntoma de una reacción de hipersensibilidad [ver Contraindicaciones (4) y Advertencias y precauciones (5.5)]. Los signos y síntomas pueden incluir disnea, urticaria, angioedema que afecta los labios, los ojos, la garganta o la lengua, o erupción cutánea.
Reacciones cutáneas graves
Aconseje a los pacientes que interrumpan la teriflunomida y busquen atención médica inmediata si se presenta algún signo de una reacción cutánea grave, como SJS o TEN [ver Advertencias y precauciones (5.6)]. Los signos y síntomas pueden incluir erupción cutánea, llagas en la boca, ampollas o descamación de la piel.
DRESS/Hipersensibilidad multiorgánica
Indique a los pacientes y cuidadores que la fiebre o la erupción cutánea asociada con signos de afectación de otros sistemas orgánicos (por ejemplo, linfadenopatía, disfunción hepática) puede estar relacionada con el medicamento y debe informarse a su proveedor de atención médica de inmediato. La teriflunomida debe interrumpirse de inmediato si se sospecha una reacción de hipersensibilidad grave [ver Advertencias y precauciones (5.7)].
Neuropatía periférica
Informe a los pacientes que pueden desarrollar neuropatía periférica. Aconseje a los pacientes que deben comunicarse con su médico si desarrollan síntomas de neuropatía periférica, como entumecimiento u hormigueo de las manos o los pies [ver Advertencias y precauciones (5.8)].
Presión arterial aumentada
Informe a los pacientes que la teriflunomida puede aumentar la presión arterial [ver Advertencias y precauciones (5.9)].
Lactancia
Aconseje a las mujeres que no deben amamantar durante el tratamiento con teriflunomida [ver Uso en poblaciones específicas (8.2)].
Dispense con la Guía de medicamentos disponible en: www.aurobindousa.com/medication-guides
Distribuido por:
Aurobindo Pharma USA, Inc.
279 Princeton-Hightstown Road
East Windsor, NJ 08520
Fabricado por:
Aurobindo Pharma Limited
Hyderabad-500 032, India
Revisado: 10/2024
Dispense con la Guía de medicamentos disponible en: www.aurobindousa.com/medication-guides
Guía de medicación
| Guía de Medicamentos Teriflunomida (ter″ i floo′ noe mide) Tabletas, para uso oral |
| Lea esta Guía de Medicamentos antes de empezar a usar las tabletas de teriflunomida y cada vez que renueve su receta. Puede haber nueva información. Esta información no reemplaza el hablar con su médico sobre su condición médica o su tratamiento. |
| ¿Cuál es la información más importante que debo saber sobre las tabletas de teriflunomida? Las tabletas de teriflunomida pueden causar efectos secundarios graves, que incluyen:
Llame a su médico de inmediato si tiene alguno de los siguientes síntomas de problemas hepáticos:
|
¿Qué son las tabletas de teriflunomida?
|
| ¿Quién no debe tomar tabletas de teriflunomida? No tome tabletas de teriflunomida si usted:
|
| ¿Qué debo informarle a mi médico antes de tomar tabletas de teriflunomida? Antes de tomar tabletas de teriflunomida, informe a su médico sobre todas sus afecciones médicas, incluso si usted:
|
¿Cómo debo tomar las tabletas de teriflunomida?
|
| ¿Cuáles son los posibles efectos secundarios de las tabletas de teriflunomida? Las tabletas de teriflunomida pueden causar efectos secundarios graves, que incluyen:
Si tiene fiebre o sarpullido con alguno de los síntomas anteriores, deje de tomar las tabletas de teriflunomida y llame a su médico de inmediato.
Informe a su médico si tiene entumecimiento u hormigueo en las manos o los pies que es diferente de su EM.
Los efectos secundarios más comunes de las tabletas de teriflunomida incluyen:
|
¿Cómo debo almacenar las tabletas de teriflunomida?
|
| Información general sobre el uso seguro y eficaz de las tabletas de teriflunomida. Los medicamentos a veces se recetan para fines distintos a los que se enumeran en una Guía del Medicamento. No use las tabletas de teriflunomida para una afección para la que no fue recetada. No le dé las tabletas de teriflunomida a otras personas, incluso si tienen los mismos síntomas que usted. Podrían dañarlas. Puede pedirle a su médico o farmacéutico información sobre las tabletas de teriflunomida que esté escrita para profesionales de la salud. |
| ¿Cuáles son los ingredientes de las tabletas de teriflunomida? Ingrediente activo: teriflunomide Ingredientes inactivos en tabletas de 7 mg y 14 mg: dióxido de silicio coloidal, almidón de maíz, FD & C azul # 2/laca de aluminio índigo carmín, hidroxipropilcelulosa, hipromelosa, lactosa monohidrato, estearato de magnesio, celulosa microcristalina, polietilenglicol, glicolato de almidón sódico, talco y dióxido de titanio. Además, 7 mg contienen óxido de hierro amarillo. Para obtener más información, llame a Aurobindo Pharma USA, Inc. al 1-866-850-2876. Distribuido por: Fabricado por: |
Esta Guía del Medicamento ha sido aprobada por la Administración de Alimentos y Medicamentos de los EE. UU.
Revisado: 10/2024
ETIQUETA DEL ENVASE – PANEL DE VISUALIZACIÓN PRINCIPAL – 7 mg – (Frasco de 30 Tabletas)
NDC 59651-054-30
Rx only
Teriflunomide Tablets
7 mg
PHARMACIST: Dispense the Medication Guide provided
separately to each patient.
AUROBINDO 30 Tablets

ETIQUETA DEL ENVASE – PANEL PRINCIPAL DE VISUALIZACIÓN – 7 mg – 28 Comprimidos (2 blísteres de 14 comprimidos cada uno)
NDC 59651-054-28
Rx only
Teriflunomide Tablets
7 mg
FARMACÉUTICO: Dispensar la Guía del Medicamento que se proporciona
por separado a cada paciente.
28 Tabletas (2 blísteres de 14 tabletas cada uno)
AUROBINDO
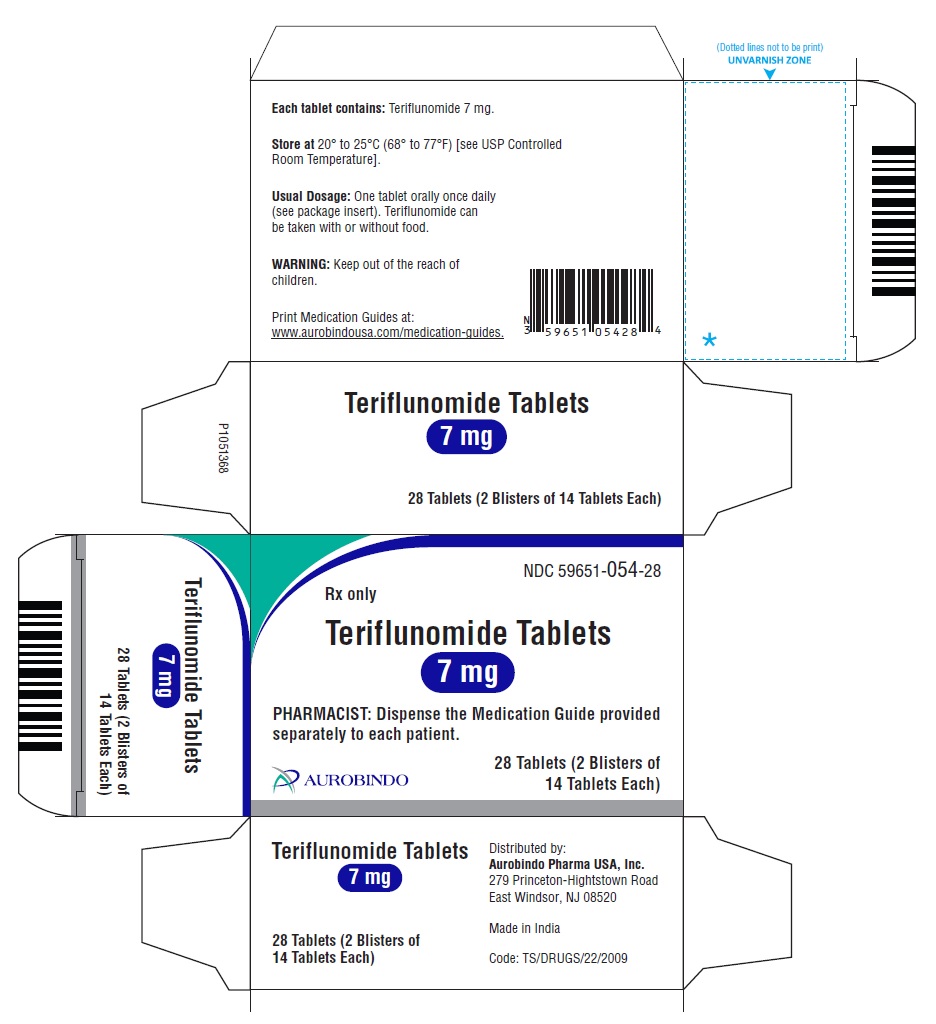
ETIQUETA DEL ENVASE – PANEL DE VISUALIZACIÓN PRINCIPAL – 14 mg – (Frasco de 30 Tabletas)
NDC 59651-055-30
Rx only
Teriflunomide Tablets
14 mg
FARMACÉUTICO: Entregue la Guía del Medicamento que se proporciona
por separado a cada paciente.
AUROBINDO 30 Tabletas

ETIQUETA DEL ENVASE – PANEL DE VISUALIZACIÓN PRINCIPAL – 14 mg – 28 Tabletas (2 Blísteres de 14 Tabletas Cada Uno)
NDC 59651-055-28
Rx only
Teriflunomide Tablets
14 mg
PHARMACIST: Dispense the Medication Guide provided
separately to each patient.
28 Tablets (2 Blisters of 14 Tablets Each)
AUROBINDO