Fabricante de medicamentos: Novartis Pharmaceuticals Corporation (Updated: 2024-12-18)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
TASIGNA® (nilotinib) cápsulas, para uso oral
Aprobación inicial en EE. UU.: 2007
ADVERTENCIA: PROLONGACIÓN DEL QT Y MUERTES SÚBITAS
Consulte la información de prescripción completa para ver el recuadro de advertencia completo.
- Tasigna prolonga el intervalo QT. Antes de la administración de Tasigna y periódicamente, controle la hipopotasemia o la hipomagnesemia y corrija las deficiencias. (5.2) Obtenga ECG para controlar el QTc al inicio, siete días después del inicio y periódicamente a partir de entonces, y después de cualquier ajuste de dosis. (5.2, 5.3, 5.7, 5.12)
- Se han reportado muertes súbitas en pacientes que reciben Tasigna. (5.3) No administre Tasigna a pacientes con hipopotasemia, hipomagnesemia o síndrome de QT largo. (4, 5.2)
- Evite el uso concomitante de medicamentos que se sabe que prolongan el intervalo QT y los inhibidores potentes del CYP3A4. (7.1, 7.2)
- Evite los alimentos 2 horas antes y 1 hora después de tomar la dosis. (2.1)
INDICACIONES Y USO
Tasigna es un inhibidor de la quinasa indicado para el tratamiento de:
- Pacientes adultos y pediátricos mayores o iguales a 1 año de edad con leucemia mieloide crónica cromosoma Filadelfia positivo (Ph+ CML) recién diagnosticada en fase crónica. (1.1)
- Pacientes adultos con LMC Ph+ en fase crónica (FC) y fase acelerada (FA) resistentes o intolerantes a la terapia previa que incluyó imatinib. (1.2)
- Pacientes pediátricos mayores o iguales a 1 año de edad con LMC Ph+ en FC y FA resistentes o intolerantes a la terapia previa con inhibidores de la tirosina quinasa (TKI). (1.3)
DOSIFICACIÓN Y ADMINISTRACIÓN
- Dosis recomendada para adultos: LMC Ph+ FC recién diagnosticada: 300 mg por vía oral dos veces al día. LMC Ph+ FC y FA resistente o intolerante: 400 mg por vía oral dos veces al día. (2.1)
- Dosis pediátrica recomendada: LMC Ph+ FC recién diagnosticada o LMC Ph+ FC y FA resistente o intolerante a la terapia previa con TKI: 230 mg/m2 por vía oral dos veces al día, redondeado a la dosis de 50 mg más cercana (hasta una dosis única máxima de 400 mg). (2.1)
- Consulte la sección Dosis y administración para obtener instrucciones completas de dosificación e instrucciones de reducción de dosis por toxicidad. (2.1)
- Reduzca la dosis inicial en pacientes con insuficiencia hepática basal. (2.7)
- Los pacientes adultos recién diagnosticados elegibles con LMC Ph+ FC que han recibido Tasigna durante un mínimo de 3 años y han logrado una respuesta molecular sostenida (RM4.5) y los pacientes con LMC Ph+ FC resistentes o intolerantes a imatinib que han recibido Tasigna durante al menos 3 años y han logrado una respuesta molecular sostenida (RM4.5) pueden ser considerados para la interrupción del tratamiento. (2.2, 2.3, 5.16)
FORMAS FARMACÉUTICAS Y CONCENTRACIONES
Cápsulas: 50 mg, 150 mg y 200 mg (3)
CONTRAINDICACIONES
Tasigna está contraindicado en pacientes con hipopotasemia, hipomagnesemia o síndrome de QT largo. (4)
ADVERTENCIAS Y PRECAUCIONES
- Mielosupresión: Controle el hemograma completo (CBC) durante el tratamiento y maneje mediante la interrupción del tratamiento o la reducción de la dosis. (5.1)
- Eventos oclusivos vasculares cardíacos y arteriales: Evalúe el estado cardiovascular, controle y maneje los factores de riesgo cardiovascular durante el tratamiento con Tasigna. (5.4)
- Pancreatitis y lipasa sérica elevada: Controle la lipasa sérica; si las elevaciones están acompañadas de síntomas abdominales, interrumpa las dosis y considere los diagnósticos apropiados para excluir la pancreatitis. (5.5)
- Hepatotoxicidad: Controle las pruebas de función hepática mensualmente o según esté clínicamente indicado. (5.6)
- Anomalías electrolíticas: Tasigna puede causar hipofosfatemia, hipopotasemia, hiperpotasemia, hipocalcemia e hiponatremia. Corrija las anomalías electrolíticas antes de iniciar Tasigna y controle periódicamente durante el tratamiento. (5.7)
- Síndrome de lisis tumoral: Mantenga una hidratación adecuada y corrija los niveles de ácido úrico antes de iniciar el tratamiento con Tasigna. (5.8)
- Hemorragia: Puede producirse hemorragia desde cualquier lugar. Aconseje a los pacientes que informen sobre los signos y síntomas de sangrado y que lo controlen médicamente según sea necesario. (5.9)
- Retención de líquidos: Controle a los pacientes para detectar un aumento de peso rápido inesperado, hinchazón y dificultad para respirar. Maneje médicamente. (5.13)
- Efectos sobre el crecimiento y el desarrollo en pacientes pediátricos: Se ha informado de retraso del crecimiento en pacientes pediátricos tratados con Tasigna. Controle el crecimiento y el desarrollo en pacientes pediátricos. (5.14)
- Toxicidad embriofetal: Aconseje a las mujeres en edad fértil sobre el riesgo potencial para el feto y que utilicen métodos anticonceptivos eficaces. (5.15, 8.1, 8.3)
- Interrupción del tratamiento: Los pacientes deben tener transcripciones típicas de BCR-ABL. Se debe utilizar una prueba autorizada por la FDA con un límite de detección inferior a MR4.5 para determinar la elegibilidad para la interrupción. Los pacientes deben ser controlados con frecuencia mediante la prueba autorizada por la FDA para detectar una posible pérdida de la remisión. (5.16)
REACCIONES ADVERSAS
Las reacciones adversas no hematológicas más frecuentes (≥ 20%) en pacientes adultos y pediátricos fueron náuseas, erupción cutánea, dolor de cabeza, fatiga, prurito, vómitos, diarrea, tos, estreñimiento, artralgia, nasofaringitis, pirexia y sudores nocturnos. Las reacciones adversas hematológicas a los medicamentos incluyen mielosupresión: trombocitopenia, neutropenia y anemia. (6.1)
Para informar de REACCIONES ADVERSAS SOSPECHOSAS, póngase en contacto con Novartis Pharmaceuticals Corporation en el 1-888-669-6682 o con la FDA en el 1-800-FDA-1088 o en www.fda.gov/medwatch.
INTERACCIONES CON OTROS MEDICAMENTOS
- Inhibidores potentes del CYP3A: Evite el uso concomitante con Tasigna, o reduzca la dosis de Tasigna si no se puede evitar la administración conjunta. (7.1)
- Inductores potentes del CYP3A: Evite el uso concomitante con Tasigna. (7.1)
- Inhibidores de la bomba de protones: Utilice antiácidos de acción corta o bloqueadores H2 como alternativa a los inhibidores de la bomba de protones. (7.1)
USO EN POBLACIONES ESPECÍFICAS
- Lactancia: Aconseje a las mujeres que no amamanten. (8.2)
Consulte 17 para obtener INFORMACIÓN DE ASESORAMIENTO AL PACIENTE y la Guía del Medicamento.
Revisado: 2/2024
Tabla de contenido
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDO*
ADVERTENCIA: PROLONGACIÓN DEL INTERVALO QT Y MUERTES SÚBITAS
1
INDICACIONES Y USO
1.1
Pacientes adultos y pediátricos con LMC-FC Ph+ de nuevo diagnóstico
1.2
Pacientes adultos con LMC-FC Ph+ resistente o intolerante y LMC-AF
1.3
Pacientes pediátricos con LMC-FC Ph+ resistente o intolerante y LMC-AF
2
POSOLOGÍA Y ADMINISTRACIÓN
2.1
Posología recomendada
2.2
Interrupción del tratamiento después de una respuesta molecular sostenida (RM4.5) con Tasigna
2.3
Reanudación del tratamiento en pacientes que pierden la respuesta molecular después de la interrupción de la terapia con Tasigna
2.4
Modificación de la dosis para la prolongación del intervalo QT
2.5
Modificaciones de la dosis para la mielosupresión
2.6
Modificaciones de la dosis para anomalías de laboratorio no hematológicas seleccionadas y otras toxicidades
2.7
Modificación de la dosis para insuficiencia hepática
2.8
Modificación de la dosis con inhibidores potentes concomitantes del CYP3A4
3
FORMAS Y CONCENTRACIONES FARMACÉUTICAS
4
CONTRAINDICACIONES
5
ADVERTENCIAS Y PRECAUCIONES
5.1
Mielosupresión
5.2
Prolongación del intervalo QT
5.3
Muertes súbitas
5.4
Eventos oclusivos vasculares cardíacos y arteriales
5.5
Pancreatitis y aumento de la lipasa sérica
5.6
Hepatotoxicidad
5.7
Anomalías electrolíticas
5.8
Síndrome de lisis tumoral
5.9
Hemorragia
5.10
Gastrectomía total
5.11
Lactosa
5.12
Monitorización de las pruebas de laboratorio
5.13
Retención de líquidos
5.14
Efectos sobre el crecimiento y el desarrollo en pacientes pediátricos
5.15
Toxicidad embriofetal
5.16
Monitorización de los niveles de transcripción de BCR-ABL
6
REACCIONES ADVERSAS
6.1
Experiencia en ensayos clínicos
6.2
Experiencia postcomercialización
7
INTERACCIONES MEDICAMENTOSAS
7.1
Efecto de otros medicamentos sobre Tasigna
7.2
Medicamentos que prolongan el intervalo QT
8
USO EN POBLACIONES ESPECÍFICAS
8.1
Embarazo
8.2
Lactancia
8.3
Mujeres y hombres en edad fértil
8.4
Uso pediátrico
8.5
Uso geriátrico
8.6
Trastornos cardíacos
8.7
Insuficiencia hepática
10
SOBREDOSIS
11
DESCRIPCIÓN
12
FARMACOLOGÍA CLÍNICA
12.1
Mecanismo de acción
12.2
Farmacodinamia
12.3
Farmacocinética
12.5
Farmacogenómica
13
TOXICOLOGÍA NO CLÍNICA
13.1
Carcinogénesis, mutagénesis, deterioro de la fertilidad
14
ESTUDIOS CLÍNICOS
14.1
LMC-FC Ph+ de nuevo diagnóstico en adultos
14.2
Pacientes adultos con LMC-FC Ph+ resistente o intolerante y LMC-AF
14.3
Interrupción del tratamiento en pacientes con LMC-FC Ph+ de nuevo diagnóstico que han alcanzado una respuesta molecular sostenida (RM4.5)
14.4
Interrupción del tratamiento en pacientes con LMC-FC Ph+ que han alcanzado una respuesta molecular sostenida (RM4.5) con Tasigna después de la terapia previa con imatinib
14.5
Pacientes pediátricos con LMC-FC Ph+ de nuevo diagnóstico o LMC-FC Ph+ resistente o intolerante
16
PRESENTACIÓN/CONSERVACIÓN Y MANIPULACIÓN
17
INFORMACIÓN PARA EL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información completa de prescripción no figuran en la lista.
ADVERTENCIA EN EL RECUADRO
ADVERTENCIA: PROLONGACIÓN DEL INTERVALO QT Y MUERTES SÚBITAS
- Tasigna prolonga el intervalo QT. Antes de la administración de Tasigna y periódicamente, controlar la hipopotasemia o hipomagnesemia y corregir las deficiencias [ver Advertencias y precauciones (5.2)]. Obtener ECGs para controlar el QTc al inicio, siete días después del inicio, y periódicamente a partir de entonces, y después de cualquier ajuste de dosis [ver Advertencias y precauciones (5.2, 5.3, 5.7, 5.12)].
- Se han notificado muertes súbitas en pacientes que reciben Tasigna [ver Advertencias y precauciones (5.3)]. No administrar Tasigna a pacientes con hipopotasemia, hipomagnesemia o síndrome de QT largo [ver Contraindicaciones (4), Advertencias y precauciones (5.2)].
- Evitar el uso concomitante de medicamentos que se sabe que prolongan el intervalo QT e inhibidores potentes del CYP3A4 [ver Interacciones medicamentosas (7.1, 7.2)].
- Evitar los alimentos 2 horas antes y 1 hora después de tomar la dosis [ver Posología y administración (2.1)].
1 INDICACIONES Y USO
1.1
Pacientes adultos y pediátricos con LMC-CP Ph+ de nuevo diagnóstico
Tasigna está indicado para el tratamiento de pacientes adultos y pediátricos mayores o iguales a 1 año de edad con leucemia mieloide crónica positiva para el cromosoma Filadelfia (LMC Ph+) de novo en fase crónica.
1.2
Pacientes adultos con LMC-CP y LMC-AP Ph+ resistente o intolerante
Tasigna está indicado para el tratamiento de pacientes adultos con leucemia mielógena crónica positiva para el cromosoma Filadelfia (LMC Ph+) en fase crónica y fase acelerada resistente o intolerante a la terapia previa que incluyó imatinib.
1.3
Pacientes pediátricos con LMC-CP y LMC-AP Ph+ resistente o intolerante
Tasigna está indicado para el tratamiento de pacientes pediátricos mayores o iguales a 1 año de edad con leucemia mieloide crónica positiva para el cromosoma Filadelfia (LMC Ph+) en fase crónica y fase acelerada con resistencia o intolerancia a la terapia previa con inhibidor de la tirosina cinasa (TKI).
2 DOSIS Y ADMINISTRACIÓN
2.1
Dosis Recomendada
Administre Tasigna dos veces al día con un intervalo aproximado de 12 horas con el estómago vacío. No se deben consumir alimentos al menos 2 horas antes de tomar la dosis y al menos 1 hora después de tomarla. Indique a los pacientes que traguen las cápsulas enteras con agua [ver Advertencia en recuadro, Farmacología clínica (12.3)].
Para los pacientes que no pueden tragar cápsulas, el contenido de cada cápsula se puede dispersar en 1 cucharadita de puré de manzana. La mezcla debe tomarse inmediatamente (dentro de los 15 minutos) y no debe almacenarse para uso futuro [ver Farmacología clínica (12.3)].
Tasigna se puede administrar en combinación con factores de crecimiento hematopoyéticos, como eritropoyetina o G-CSF si está clínicamente indicado. Tasigna se puede administrar con hidroxiurea o anagrelida si está clínicamente indicado.
Dosis en pacientes adultos con LMC-CP Ph+ de nuevo diagnóstico
La dosis recomendada de Tasigna es de 300 mg por vía oral dos veces al día.
Dosis en pacientes adultos con LMC-CP Ph+ resistente o intolerante y LMC-AP
La dosis recomendada de Tasigna es de 400 mg por vía oral dos veces al día.
Dosis en pacientes pediátricos con LMC-CP Ph+ de nuevo diagnóstico o LMC-CP Ph+ resistente o intolerante y LMC-AP
La dosis recomendada de Tasigna para pacientes pediátricos es de 230 mg/m2 por vía oral dos veces al día, redondeada a la dosis de 50 mg más cercana (hasta una dosis única máxima de 400 mg) (ver Tabla 1). Si es necesario, obtenga la dosis deseada combinando diferentes concentraciones de cápsulas de Tasigna. Continúe el tratamiento mientras se observe un beneficio clínico o hasta que se produzca una toxicidad inaceptable.
| Superficie corporal | Dosis única | Dosis diaria total |
| Hasta 0,32 m2 | 50 mg | 100 mg |
| 0,33 – 0,54 m2 | 100 mg | 200 mg |
| 0,55 – 0,76 m2 | 150 mg | 300 mg |
| 0,77 – 0,97 m2 | 200 mg | 400 mg |
| 0,98 – 1,19 m2 | 250 mg | 500 mg |
| 1,20 – 1,41 m2 | 300 mg | 600 mg |
| 1,42 – 1,63 m2 | 350 mg | 700 mg |
| ≥ 1,64 m2 | 400 mg | 800 mg |
2.2
Interrupción del tratamiento después de una respuesta molecular sostenida (RM4.5) con Tasigna
Selección del paciente
Elegibilidad para la interrupción del tratamiento
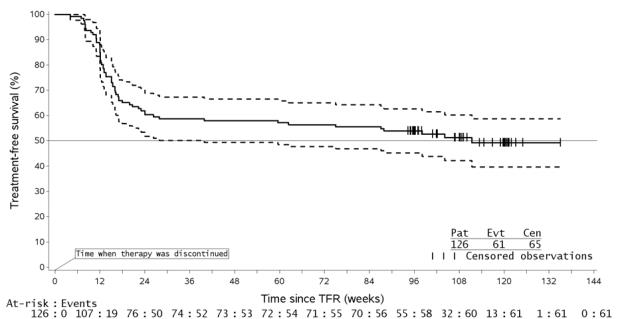
Los pacientes con LMC-FC Ph+ con transcritos BCR-ABL típicos, que han estado tomando Tasigna durante un mínimo de 3 años y han logrado una respuesta molecular sostenida (MR4.5, correspondiente a = BCR-ABL/ABL ≤ 0.0032% IS), pueden ser elegibles para la interrupción del tratamiento [véase Estudios clínicos (14.3, 14.4)]. La información sobre las pruebas autorizadas por la FDA para la detección y cuantificación de los transcritos BCR-ABL para determinar la elegibilidad para la interrupción del tratamiento está disponible en http://www.fda.gov/CompanionDiagnostics.
Los pacientes con transcritos BCR-ABL típicos (e13a2/b2a2 o e14a2/b3a2), que alcanzan los criterios de RM4.5 sostenida, son elegibles para la interrupción de Tasigna. Los pacientes deben continuar siendo monitorizados para detectar una posible pérdida de remisión molecular después de la interrupción del tratamiento. Utilice la misma prueba autorizada por la FDA para monitorizar de forma consistente los niveles de respuesta molecular durante y después del tratamiento.
Considere la interrupción en pacientes con LMC-FC Ph+ de nuevo diagnóstico que tengan:
- han sido tratados con Tasigna durante al menos 3 años
- mantuvieron una respuesta molecular de al menos MR4.0 (correspondiente a = BCR-ABL/ABL ≤ 0.01% IS) durante un año antes de la interrupción de la terapia
- alcanzaron una MR4.5 en la última evaluación realizada inmediatamente antes de la interrupción de la terapia
- se ha confirmado que expresan los transcritos BCR-ABL típicos (e13a2/b2a2 o e14a2/b3a2)
- sin antecedentes de fase acelerada o crisis blástica
- sin antecedentes de intentos previos de interrupción de la remisión sin tratamiento que hayan provocado una recaída.
Considere la interrupción en pacientes con LMC-FC Ph+ resistentes o intolerantes al imatinib que hayan logrado una respuesta molecular sostenida (MR4.5) con Tasigna que tengan:
- han sido tratados con Tasigna durante un mínimo de 3 años
- han sido tratados únicamente con imatinib antes del tratamiento con Tasigna
- alcanzaron una respuesta molecular de MR4.5 (correspondiente a = BCR-ABL/ABL ≤ 0.0032% IS)
- mantuvieron una MR4.5 durante un mínimo de un año inmediatamente antes de la interrupción de la terapia
- se ha confirmado que expresan los transcritos BCR-ABL típicos (e13a2/b2a2 o e14a2/b3a2)
- sin antecedentes de fase acelerada o crisis blástica
- sin antecedentes de intentos previos de interrupción de la remisión sin tratamiento que hayan provocado una recaída.
Controle los niveles de transcritos BCR-ABL y el hemograma completo (HC) con diferencial en pacientes que hayan interrumpido el tratamiento con Tasigna mensualmente durante un año, luego cada 6 semanas durante el segundo año y cada 12 semanas a partir de entonces [véase Advertencias y precauciones (5.16)].
Tras la pérdida de MR4.0 (correspondiente a = BCR-ABL/ABL ≤ 0.01% IS) durante la fase sin tratamiento, controle los niveles de transcritos BCR-ABL cada 2 semanas hasta que los niveles de BCR-ABL se mantengan por debajo de la respuesta molecular mayor [(RMM), correspondiente a MR3.0 o = BCR-ABL/ABL ≤ 0.1% IS] durante 4 mediciones consecutivas. El paciente puede entonces continuar con el programa de monitorización original.
2.3
Reanudación del tratamiento en pacientes que pierden la respuesta molecular después de la interrupción de la terapia con Tasigna
- Los pacientes de nuevo diagnóstico que pierden la RMM deben reiniciar el tratamiento en un plazo de 4 semanas al nivel de dosis anterior a la interrupción de la terapia [véase Advertencias y precauciones (5.16)]. Los pacientes que reanuden la terapia con Tasigna deben tener sus niveles de transcritos BCR-ABL controlados mensualmente hasta que se restablezca la respuesta molecular mayor y cada 12 semanas a partir de entonces.
- Los pacientes resistentes o intolerantes al tratamiento previo que incluyó imatinib con pérdida confirmada de MR4.0 (2 medidas consecutivas separadas por al menos 4 semanas que muestran la pérdida de MR4.0) o pérdida de RMM deben reiniciar el tratamiento en un plazo de 4 semanas al nivel de dosis anterior a la interrupción de la terapia [véase Advertencias y precauciones (5.16)]. Los pacientes que reanuden la terapia con Tasigna deben tener sus niveles de transcritos BCR-ABL controlados mensualmente hasta que se restablezca la respuesta molecular mayor o MR4.0 anterior y cada 12 semanas a partir de entonces.
2.4
Modificación de la dosis por prolongación del intervalo QT
Consulte la Tabla 2 para los ajustes de dosis en caso de prolongación del intervalo QT [véase Advertencias y precauciones (5.2), Farmacología clínica (12.2)].
| Abreviatura: ECG, electrocardiograma. | |
| Grado de prolongación del QTc | Ajuste de la dosis |
| ECG con un QTc superior a 480 mseg |
1. Suspender Tasigna y realizar un análisis del potasio y el magnesio séricos; si están por debajo del límite inferior de la normalidad, corregirlos con suplementos hasta alcanzar los límites normales. Se debe revisar el uso concomitante de medicamentos. 2. Reanudar en un plazo de 2 semanas a la dosis anterior si el QTcF vuelve a ser inferior a 450 mseg y a menos de 20 mseg de la línea de base. 3. Si el QTcF está entre 450 mseg y 480 mseg después de 2 semanas, reducir la dosis a 400 mg una vez al día en adultos y a 230 mg/m2 una vez al día en pacientes pediátricos. 4. Interrumpir Tasigna si, después de reducir la dosis a 400 mg una vez al día en adultos y a 230 mg/m2 una vez al día en pacientes pediátricos, el QTcF vuelve a ser superior a 480 mseg. 5. Se debe repetir un ECG aproximadamente 7 días después de cualquier ajuste de la dosis. |
2.5
Modificaciones de la dosis por mielosupresión
Suspenda o reduzca la dosis de Tasigna para las toxicidades hematológicas (neutropenia, trombocitopenia) que no estén relacionadas con la leucemia subyacente (Tabla 3) [véase Advertencias y precauciones (5.1)].
| Abreviaturas: ANC, recuento absoluto de neutrófilos; LMC Ph+, leucemia mieloide crónica positiva para el cromosoma Filadelfia. | ||
| Diagnóstico | Grado de mielosupresión | Ajuste de dosis |
Pacientes adultos con:
|
ANC menor que 1,0 x 109/L y/o recuento de plaquetas menor que 50 x 109/L | 1. Suspenda Tasigna y controle los recuentos sanguíneos. 2. Reanude dentro de las 2 semanas con la dosis anterior si el ANC es mayor que 1,0 x 109/L y las plaquetas son mayores que 50 x 109/L. 3. Si los recuentos sanguíneos permanecen bajos durante más de 2 semanas, reduzca la dosis a 400 mg una vez al día.
|
Pacientes pediátricos con:
|
ANC menor que 1,0 x 109/L y/o recuento de plaquetas menor que 50 x 109/L | 1. Suspenda Tasigna y controle los recuentos sanguíneos. 2. Reanude dentro de las 2 semanas con la dosis anterior si el ANC es mayor que 1,5 x 109/L y/o las plaquetas son mayores que 75 x 109/L. 3. Si los recuentos sanguíneos permanecen bajos durante más de 2 semanas, puede ser necesaria una reducción de la dosis a 230 mg/m2 una vez al día. 4. Si el evento ocurre después de la reducción de la dosis, considere interrumpir el tratamiento. |
2.6
Modificaciones de la dosis para anomalías de laboratorio no hematológicas seleccionadas y otras toxicidades
Consulte la Tabla 4 para los ajustes de dosis para elevaciones de lipasa, amilasa, bilirrubina y/o transaminasas hepáticas [véase Advertencias y precauciones (5.5, 5.6), Reacciones adversas (6.1)].
| Grado de anomalía de laboratorio no hematológica | Ajuste de dosis |
| Lipasa o amilasa séricas elevadas mayor o igual que Grado 3 | Pacientes adultos: 1. Suspenda Tasigna y controle la lipasa o amilasa séricas. 2. Reanude el tratamiento a 400 mg una vez al día si la lipasa o amilasa séricas vuelven a ser menor o igual que Grado 1. |
| Pacientes pediátricos: 1. Interrupción de Tasigna hasta que el evento regrese a menos de o igual a Grado 1. 2. Reanude el tratamiento a 230 mg/m2 una vez al día si la dosis anterior era 230 mg/m2 dos veces al día; suspenda el tratamiento si la dosis anterior era 230 mg/m2 una vez al día. |
|
| Bilirrubina elevada mayor o igual que Grado 3 en pacientes adultos y mayor o igual que Grado 2 en pacientes pediátricos | Pacientes adultos: 1. Suspenda Tasigna y controle la bilirrubina. 2. Reanude el tratamiento a 400 mg una vez al día si la bilirrubina vuelve a ser menor o igual que Grado 1. |
| Pacientes pediátricos: 1. Interrupción de Tasigna hasta que el evento regrese a menos de o igual a Grado 1. 2. Reanude el tratamiento a 230 mg/m2 una vez al día si la dosis anterior era 230 mg/m2 dos veces al día; suspenda el tratamiento si la dosis anterior era 230 mg/m2 una vez al día y la recuperación a menos de o igual a Grado 1 tarda más de 28 días. |
|
|
Transaminasas hepáticas elevadas mayor o igual que Grado 3 |
Pacientes adultos: 1. Suspenda Tasigna y controle las transaminasas hepáticas. 2. Reanude el tratamiento a 400 mg una vez al día si las transaminasas hepáticas vuelven a ser menor o igual que Grado 1. |
| Pacientes pediátricos: 1. Interrupción de Tasigna hasta que el evento regrese a menos de o igual a Grado 1. 2. Reanude el tratamiento a 230 mg/m2 una vez al día si la dosis anterior era 230 mg/m2 dos veces al día; suspenda el tratamiento si la dosis anterior era 230 mg/m2 una vez al día y la recuperación a menos de o igual a Grado 1 tarda más de 28 días. |
Si se desarrolla toxicidad no hematológica moderada o grave clínicamente significativa (incluida la retención de líquidos médicamente grave), consulte la Tabla 5 para realizar ajustes de la dosis [véase Reacciones adversas (6.1)].
| Abreviaturas: LMC-FA, leucemia mieloide crónica-fase acelerada; LMC-FC, leucemia mieloide crónica-fase crónica; Ph+, cromosoma Filadelfia positivo. | |
| Grado de “otra toxicidad no hematológica” | Ajuste de la dosis |
| Otra toxicidad no hematológica clínicamente moderada o grave | Pacientes adultos: 1. Suspender Tasigna hasta que la toxicidad haya desaparecido. 2. Reanudar el tratamiento a 400 mg una vez al día si la dosis anterior fue de 300 mg dos veces al día en pacientes adultos recién diagnosticados con LMC-FC o 400 mg dos veces al día en pacientes adultos con LMC-FC y LMC-FA resistente o intolerante. 3. Interrumpir el tratamiento si la dosis anterior fue de 400 mg una vez al día en pacientes adultos. 4. Si es clínicamente apropiado, considerar la reescalada de la dosis a 300 mg (LMC-FC Ph+ recién diagnosticada) o 400 mg (LMC-FC y LMC-FA Ph+ resistente o intolerante) dos veces al día. |
| Pacientes pediátricos: 1. Interrumpir Tasigna hasta que la toxicidad haya desaparecido. 2. Reanudar el tratamiento a 230 mg/m2 una vez al día si la dosis anterior fue de 230 mg/m2 dos veces al día; interrumpir el tratamiento si la dosis anterior fue de 230 mg/m2 una vez al día. 3. Si es clínicamente apropiado, considerar la reescalada de la dosis a 230 mg/m2 dos veces al día. |
|
2.7
Modificación de la dosis por insuficiencia hepática
Si es posible, considere terapias alternativas. Si Tasigna debe administrarse a pacientes con insuficiencia hepática, considere la siguiente reducción de la dosis [véase Uso en poblaciones específicas (8.7)]:
| Diagnóstico | Grado de insuficiencia hepática | Ajuste de la dosis |
| LMC Ph+ recién diagnosticada en fase crónica | Leve (Child-Pugh A), Moderada (Child-Pugh B) o Grave (Child-Pugh C) | Reducir la dosis a 200 mg dos veces al día. Aumentar la dosis a 300 mg dos veces al día según la tolerabilidad. |
|
LMC Ph+ resistente o intolerante en fase crónica o fase acelerada |
Leve o Moderada | Reducir la dosis a 300 mg dos veces al día. Aumentar la dosis a 400 mg dos veces al día según la tolerabilidad. |
| Grave | Reducir la dosis a 200 mg dos veces al día. Aumentar la dosis a 300 mg dos veces al día y luego a 400 mg dos veces al día según la tolerabilidad. |
2.8
Modificación de la dosis con inhibidores potentes concomitantes del CYP3A4
Evite el uso concomitante de inhibidores potentes del CYP3A4. Si se requiere tratamiento con alguno de estos agentes, interrumpa el tratamiento con Tasigna. Si los pacientes deben recibir un inhibidor potente del CYP3A4 de forma concomitante, reduzca la dosis a 300 mg una vez al día en pacientes con LMC Ph+ resistente o intolerante o a 200 mg una vez al día en pacientes con LMC-FC Ph+ recién diagnosticada. Sin embargo, no hay datos clínicos con este ajuste de dosis en pacientes que reciben inhibidores potentes del CYP3A4. Si se interrumpe el inhibidor potente, permita un período de lavado antes de ajustar la dosis de Tasigna hacia arriba a la dosis indicada. Para los pacientes que no pueden evitar el uso de inhibidores potentes del CYP3A4, controle estrechamente la prolongación del intervalo QT [véase Advertencia en recuadro, Advertencias y precauciones (5.2), Interacciones medicamentosas (7.1, 7.2), Farmacología clínica (12.3)].
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
Cápsulas:
- 50 mg cápsulas de gelatina dura con cuerpo de color amarillo claro opaco y tapa roja opaca, con impresión radial negra “NVR/ABL.”
- 150 mg cápsulas de gelatina dura opacas de color rojo con impresión axial negra “NVR/BCR.”
- 200 mg cápsulas de gelatina dura opacas de color amarillo claro con impresión axial roja “NVR/TKI.”
4 CONTRAINDICACIONES
Tasigna está contraindicado en pacientes con hipopotasemia, hipomagnesemia o síndrome de QT largo [ver Advertencia en recuadro].
5 ADVERTENCIAS Y PRECAUCIONES
5.1
Mielosupresión
El tratamiento con Tasigna puede causar trombocitopenia, neutropenia y anemia de grado 3/4. Realice hemogramas completos cada 2 semanas durante los primeros 2 meses y luego mensualmente, o según esté clínicamente indicado. La mielosupresión fue generalmente reversible y usualmente se manejó suspendiendo Tasigna temporalmente o reduciendo la dosis [see Dosage and Administration (2.5)].
5.2
Prolongación del QT
Se ha demostrado que Tasigna prolonga la repolarización ventricular cardíaca según lo medido por el intervalo QT en el electrocardiograma de superficie (ECG) de manera dependiente de la concentración [see Adverse Reactions (6.1), Clinical Pharmacology (12.2)]. La prolongación del intervalo QT puede resultar en un tipo de taquicardia ventricular llamada torsade de pointes, que puede resultar en síncope, convulsiones y/o muerte. Se deben realizar electrocardiogramas al inicio, 7 días después del inicio de Tasigna y periódicamente según esté clínicamente indicado y después de los ajustes de dosis [see Dosage and Administration (2.4), Warnings and Precautions (5.12)].
Tasigna no debe usarse en pacientes que tienen hipopotasemia, hipomagnesemia o síndrome de QT largo. Antes de iniciar Tasigna y periódicamente, examine los niveles sanguíneos de electrolitos, calcio y magnesio. La hipopotasemia o la hipomagnesemia deben corregirse antes de iniciar Tasigna y estos electrolitos deben controlarse periódicamente durante el tratamiento [see Warnings and Precautions (5.12)].
Puede ocurrir una prolongación significativa del intervalo QT cuando Tasigna se toma inapropiadamente con alimentos y/o inhibidores potentes del CYP3A4 y/o medicamentos con un potencial conocido de prolongar el QT. Por lo tanto, se debe evitar la administración conjunta con alimentos y se debe evitar el uso concomitante con inhibidores potentes del CYP3A4 y/o medicamentos con un potencial conocido de prolongar el QT [see Dosage and Administration (2.1), Drug Interactions (7.1, 7.2)]. La presencia de hipopotasemia e hipomagnesemia puede prolongar aún más el intervalo QT [see Warnings and Precautions (5.7, 5.12)].
5.3
Muertes Súbita
Se han reportado muertes súbitas en el 0.3% de los pacientes con LMC tratados con Tasigna en estudios clínicos de 5661 pacientes. La aparición relativamente temprana de algunas de estas muertes en relación con el inicio de Tasigna sugiere la posibilidad de que las anomalías de la repolarización ventricular puedan haber contribuido a su aparición.
5.4
Eventos Oclusivos Vasculares Cardíacos y Arteriales
Se informaron eventos cardiovasculares, incluidos eventos oclusivos vasculares arteriales, en un ensayo clínico aleatorizado en pacientes con LMC recién diagnosticada y se observaron en los informes posteriores a la comercialización de pacientes que recibían terapia con Tasigna [see Adverse Reactions (6.1)]. Con un tiempo medio de tratamiento de 60 meses en el ensayo clínico, se produjeron eventos cardiovasculares, incluidos eventos oclusivos vasculares arteriales, en el 9 % y el 15 % de los pacientes en los grupos de Tasigna 300 y 400 mg dos veces al día, respectivamente, y en el 3,2 % en el grupo de imatinib. Estos incluyeron casos de eventos cardiovasculares, incluidos eventos cardíacos relacionados con cardiopatía isquémica (5 % y 9 % en los grupos de Tasigna 300 mg y 400 mg dos veces al día, respectivamente, y 2,5 % en el grupo de imatinib), enfermedad oclusiva arterial periférica (3,6 % y 2,9 % en los grupos de Tasigna 300 mg y 400 mg dos veces al día, respectivamente, y 0 % en el grupo de imatinib) y eventos cerebrovasculares isquémicos (1,4 % y 3,2 % en los grupos de Tasigna 300 mg y 400 mg dos veces al día, respectivamente, y 0,7 % en el grupo de imatinib). Si se presentan signos o síntomas agudos de eventos cardiovasculares, aconseje a los pacientes que busquen atención médica inmediata. El estado cardiovascular de los pacientes debe evaluarse y los factores de riesgo cardiovascular deben controlarse y manejarse activamente durante el tratamiento con Tasigna de acuerdo con las pautas estándar [see Dosage and Administration (2.4)].
5.5
Pancreatitis y Lipasa Sérica Elevada
Tasigna puede causar aumentos en la lipasa sérica [see Adverse Reactions (6.1)]. Los pacientes con antecedentes de pancreatitis pueden tener un mayor riesgo de lipasa sérica elevada. Si las elevaciones de la lipasa se acompañan de síntomas abdominales, interrumpa la dosificación y considere diagnósticos apropiados para descartar pancreatitis [see Dosage and Administration (2.6)]. Examine los niveles de lipasa sérica mensualmente o según esté clínicamente indicado.
5.6
Hepatotoxicidad
Tasigna puede provocar hepatotoxicidad, medida por elevaciones en la bilirrubina, aspartato aminotransferasa (AST), alanina aminotransferasa (ALT) y fosfatasa alcalina. Se informaron elevaciones de grado 3-4 de bilirrubina, AST y ALT con una frecuencia mayor en pacientes pediátricos que en pacientes adultos. Controle las pruebas de función hepática mensualmente o según esté clínicamente indicado [see Warnings and Precautions (5.12)] y después de los ajustes de dosis. [see Dosage and Administration (2.6)].
5.7
Anomalías Electrolíticas
El uso de Tasigna puede causar hipofosfatemia, hipopotasemia, hiperpotasemia, hipocalcemia e hiponatremia. Corrija las anomalías electrolíticas antes de iniciar Tasigna y durante el tratamiento. Controle estos electrolitos periódicamente durante el tratamiento [see Warnings and Precautions (5.12)].
5.8
Síndrome de lisis tumoral
Se han notificado casos de síndrome de lisis tumoral (SLT) en pacientes tratados con Tasigna con LMC resistente o intolerante. En la mayoría de estos casos, se presentó progresión de la enfermedad maligna, recuentos altos de glóbulos blancos (GB) y/o deshidratación. Debido al potencial de SLT, mantenga una hidratación adecuada y corrija los niveles de ácido úrico antes de iniciar el tratamiento con Tasigna.
5.9
Hemorragia
Se han producido eventos hemorrágicos graves, incluyendo eventos mortales, en pacientes con LMC tratados con Tasigna. En un ensayo aleatorizado en pacientes con LMC Ph+ recién diagnosticada en fase crónica que comparaba Tasigna e imatinib, se produjo hemorragia de Grado 3 o 4 en el 1.1% de los pacientes en el grupo de Tasigna 300 mg dos veces al día, en el 1.8% de los pacientes en el grupo de Tasigna 400 mg dos veces al día y en el 0.4% de los pacientes en el grupo de imatinib. Se produjo hemorragia gastrointestinal en el 2.9% y el 5% de los pacientes en los grupos de Tasigna 300 mg dos veces al día y 400 mg dos veces al día, y en el 1.4% de los pacientes en el grupo de imatinib, respectivamente. Los eventos de Grado 3 o 4 se produjeron en el 0.7% y el 1.4% de los pacientes en los grupos de Tasigna 300 mg dos veces al día y 400 mg dos veces al día, respectivamente, y en ningún paciente en el grupo de imatinib. Vigile las señales y los síntomas de sangrado y adminístrele tratamiento médico según sea necesario.
5.10
Gastrectomía total
Dado que la exposición a Tasigna se reduce en pacientes con gastrectomía total, realice un seguimiento más frecuente de estos pacientes. Considere aumentar la dosis o un tratamiento alternativo en pacientes con gastrectomía total [see Clinical Pharmacology (12.3)].
5.11
Lactosa
Dado que las cápsulas contienen lactosa, Tasigna no se recomienda para pacientes con problemas hereditarios raros de intolerancia a la galactosa, deficiencia grave de lactasa con un grado grave de intolerancia a los productos que contienen lactosa o malabsorción de glucosa-galactosa.
5.12
Pruebas de laboratorio de monitorización
Se deben realizar hemogramas completos cada 2 semanas durante los primeros 2 meses y luego mensualmente. Realice paneles químicos, incluidos electrolitos, calcio, magnesio, enzimas hepáticas, perfil lipídico y glucosa antes del tratamiento y periódicamente. Se deben obtener electrocardiogramas al inicio, 7 días después del inicio y periódicamente a partir de entonces, así como después de los ajustes de dosis [see Warnings and Precautions (5.2)]. Monitorice los perfiles lipídicos y la glucosa periódicamente durante el primer año de tratamiento con Tasigna y al menos anualmente durante el tratamiento crónico. Si se necesita tratamiento con cualquier inhibidor de la HMG-CoA reductasa (un agente reductor de lípidos) para tratar las elevaciones de lípidos, evalúe el potencial de una interacción fármaco-fármaco antes de iniciar el tratamiento, ya que ciertos inhibidores de la HMG-CoA reductasa se metabolizan por la vía CYP3A4 [see Drug Interactions (7.1)]. Evalúe los niveles de glucosa antes de iniciar el tratamiento con Tasigna y monitorice durante el tratamiento según esté clínicamente indicado. Si los resultados de las pruebas justifican el tratamiento, los médicos deben seguir sus estándares de práctica y pautas de tratamiento locales.
5.13
Retención de líquidos
En el ensayo aleatorizado en pacientes con LMC Ph+ recién diagnosticada en fase crónica, se produjo retención de líquidos grave (Grado 3 o 4) en el 3.9% y el 2.9% de los pacientes que recibieron Tasigna 300 mg dos veces al día y 400 mg dos veces al día, respectivamente, y en el 2.5% de los pacientes que recibieron imatinib. Se observaron derrames (incluyendo derrame pleural, derrame pericárdico, ascitis) o edema pulmonar en el 2.2% y el 1.1% de los pacientes que recibieron Tasigna 300 mg dos veces al día y 400 mg dos veces al día, respectivamente, y en el 2.1% de los pacientes que recibieron imatinib. Los derrames fueron graves (Grado 3 o 4) en el 0.7% y el 0.4% de los pacientes que recibieron Tasigna 300 mg dos veces al día y 400 mg dos veces al día, respectivamente, y en ningún paciente que recibió imatinib. También se observaron eventos similares en los informes posteriores a la comercialización. Vigile a los pacientes para detectar signos de retención grave de líquidos (p. ej., aumento de peso rápido inesperado o hinchazón) y síntomas de compromiso respiratorio o cardíaco (p. ej., dificultad para respirar) durante el tratamiento con Tasigna; evalúe la etiología y trate a los pacientes en consecuencia.
5.14
Efectos sobre el crecimiento y el desarrollo en pacientes pediátricos
Se ha notificado retraso del crecimiento en pacientes pediátricos con LMC Ph+ en fase crónica tratados con Tasigna. En un ensayo pediátrico con 58 pacientes con LMC Ph+ en fase crónica con una mediana de exposición de 56.7 meses, se observó una deceleración del crecimiento (cruzando al menos dos líneas de percentil de altura principales desde el inicio) en ocho pacientes: cinco (9%) cruzaron dos líneas de percentil principales desde el inicio y tres (5%) cruzaron tres líneas de percentil principales desde el inicio (líneas de percentil: 5th, 10th, 25th, 50th, 75th, 90th y 95th). La deceleración del crecimiento fue más pronunciada en niños menores de 12 años al inicio del estudio. Se notificaron reacciones adversas asociadas con retraso del crecimiento en 3 pacientes (5%). Monitorice el crecimiento y el desarrollo en pacientes pediátricos que reciben tratamiento con Tasigna.
5.15
Toxicidad Embrio-Fetal
Según los hallazgos de estudios en animales y su mecanismo de acción, Tasigna puede causar daño fetal cuando se administra a una mujer embarazada. En estudios de reproducción animal, la administración de nilotinib a ratas y conejas preñadas durante la organogénesis causó resultados adversos en el desarrollo, incluyendo letalidad/efectos fetales embrio-fetales (papila renal pequeña, edema fetal y variaciones esqueléticas) en ratas y aumento de las resorciones de fetos y variaciones esqueléticas fetales en conejos con un área bajo la curva (AUC) materna de aproximadamente 2 y 0.5 veces, respectivamente, el AUC en pacientes que reciben la dosis recomendada. Advierta a las mujeres embarazadas sobre el riesgo potencial para el feto.
Aconseje a las mujeres en edad fértil que utilicen métodos anticonceptivos eficaces durante el tratamiento y durante 14 días después de la última dosis [see Use in Specific Populations (8.1, 8.3), Clinical Pharmacology (12.1)].
5.16
Monitoreo de los Niveles de Transcripción de BCR-ABL
Monitoreo de los Niveles de Transcripción de BCR-ABL en Pacientes que Descontinuaron Tasigna
Monitoree los niveles de transcripción de BCR-ABL en pacientes elegibles para la interrupción del tratamiento utilizando una prueba autorizada por la FDA validada para medir los niveles de respuesta molecular con una sensibilidad de al menos MR4.5 (BCR-ABL/ABL ≤ 0.0032% IS). En pacientes que interrumpen el tratamiento con Tasigna, evalúe los niveles de transcripción de BCR-ABL mensualmente durante un año, luego cada 6 semanas durante el segundo año y cada 12 semanas a partir de entonces durante la interrupción del tratamiento [see Clinical Studies (14.3,14.4), Dosage and Administration (2.2)].
Los pacientes recién diagnosticados deben reiniciar el tratamiento con Tasigna dentro de las 4 semanas posteriores a la pérdida de la respuesta molecular mayor [(MMR), correspondiente a MR3.0 o = BCR-ABL/ABL ≤ 0.1% IS].
Los pacientes resistentes o intolerantes al tratamiento previo que incluyó imatinib deben reiniciar el tratamiento con Tasigna dentro de las 4 semanas posteriores a la pérdida de MMR o la pérdida confirmada de MR4.0 (dos mediciones consecutivas separadas por al menos 4 semanas que muestren pérdida de MR4.0, correspondiente a = BCR-ABL/ABL ≤ 0.01% IS).
Para los pacientes que no logran MMR después de tres meses de reinicio del tratamiento, se deben realizar pruebas de mutación del dominio de la cinasa BCR-ABL.
Monitoreo de los Niveles de Transcripción de BCR-ABL en Pacientes que Han Reiniciado el Tratamiento Después de la Pérdida de la Respuesta Molecular
Monitoree el CBC y las transcripciones de BCR-ABL en pacientes que reinician el tratamiento con Tasigna debido a la pérdida de la cuantificación de la respuesta molecular cada 4 semanas hasta que se restablezca una respuesta molecular mayor, luego cada 12 semanas.
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas clínicamente significativas pueden ocurrir con Tasigna y se discuten con mayor detalle en otras secciones del etiquetado:
- Mielosupresión [ver Advertencias y precauciones (5.1)]
- Prolongación del QT [ver Advertencia en recuadro, Advertencias y precauciones (5.2)]
- Muertes súbitas [ver Advertencia en recuadro, Advertencias y precauciones (5.3)]
- Eventos oclusivos vasculares cardíacos y arteriales [ver Advertencias y precauciones (5.4)]
- Pancreatitis y lipasa sérica elevada [ver Advertencias y precauciones (5.5)]
- Hepatotoxicidad [ver Advertencias y precauciones (5.6)]
- Anormalidades electrolíticas [ver Advertencia en recuadro, Advertencias y precauciones (5.7)]
- Hemorragia [ver Advertencias y precauciones (5.9)]
- Retención de líquidos [ver Advertencias y precauciones (5.13)]
6.1
Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se realizan en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no se pueden comparar directamente con las tasas en los ensayos clínicos de otro fármaco y pueden no reflejar las tasas observadas en la práctica.
En pacientes adultos con LMC Ph+ de novo en fase crónica
Los datos a continuación reflejan la exposición a Tasigna de un ensayo aleatorizado en pacientes con LMC Ph+ de novo en fase crónica tratados con la dosis recomendada de 300 mg dos veces al día (n = 279). El tiempo medio de tratamiento en el grupo de Tasigna 300 mg dos veces al día fue de 61 meses (rango, 0,1 a 71 meses). La intensidad de la dosis real mediana fue de 593 mg/día en el grupo de Tasigna 300 mg dos veces al día.
Las reacciones adversas farmacológicas no hematológicas más comunes (mayores al 10%) fueron erupción cutánea, prurito, cefalea, náuseas, fatiga, alopecia, mialgia y dolor abdominal superior. Estreñimiento, diarrea, piel seca, espasmos musculares, artralgia, dolor abdominal, edema periférico, vómitos y astenia se observaron con menos frecuencia (menos del 10% y mayor del 5%) y han sido de gravedad leve a moderada, manejables y generalmente no requirieron reducción de la dosis.
Se observó un aumento en el QTcF mayor de 60 mseg desde el valor basal en 1 paciente (0,4%) en el grupo de tratamiento de 300 mg dos veces al día . Ningún paciente tuvo un QTcF absoluto mayor de 500 mseg mientras estuvo en el estudio.
Las reacciones adversas hematológicas más comunes (todos los grados) fueron mielosupresión, incluyendo: trombocitopenia (18%), neutropenia (15%) y anemia (8%). Consulte la Tabla 9 para obtener información sobre las anormalidades de laboratorio de grado 3/4.
La interrupción debido a reacciones adversas, independientemente de la relación con el fármaco del estudio, se observó en el 10% de los pacientes.
En pacientes adultos con LMC Ph+ resistente o intolerante a la terapia y LMC-AP
En el ensayo clínico multicéntrico, abierto, de un solo brazo, se trataron un total de 458 pacientes con LMC Ph+ en fase crónica y LMC-AP resistentes o intolerantes a al menos una terapia previa, incluida la imatinib (LMC-CP = 321; LMC-AP = 137) a la dosis recomendada de 400 mg dos veces al día.
La duración media de la exposición en días para los pacientes con LMC-CP y LMC-AP es de 561 (rango, 1 a 1096) y 264 (rango, 2 a 1160), respectivamente. La intensidad de la dosis mediana para pacientes con LMC-CP y LMC-AP es de 789 mg/día (rango, 151 a 1110) y 780 mg/día (rango, 150 a 1149), respectivamente, y correspondió a la dosificación planificada de 400 mg dos veces al día.
La duración acumulativa mediana en días de interrupciones de la dosis para los pacientes con LMC-CP fue de 20 (rango, 1 a 345), y la duración mediana en días de interrupciones de la dosis para los pacientes con LMC-AP fue de 23 (rango, 1 a 234).
En pacientes con LMC-CP, las reacciones adversas farmacológicas no hematológicas más comúnmente notificadas (mayores o iguales al 10%) fueron erupción cutánea, prurito, náuseas, fatiga, cefalea, estreñimiento, diarrea, vómitos y mialgia. Las reacciones adversas graves relacionadas con el fármaco comunes (mayores o iguales al 1% y menores al 10%) fueron trombocitopenia, neutropenia y anemia.
En pacientes con LMC-AP, las reacciones adversas farmacológicas no hematológicas más comúnmente notificadas (mayores o iguales al 10%) fueron erupción cutánea, prurito y fatiga. Las reacciones adversas graves comunes (mayores o iguales al 1% y menores al 10%) fueron trombocitopenia, neutropenia, neutropenia febril, neumonía, leucopenia, hemorragia intracraneal, lipasa elevada y pirexia.
Se notificaron muertes súbitas y prolongación del QT. El cambio máximo medio de QTcF desde el valor basal en estado estacionario fue de 10 mseg. Se observó un aumento en el QTcF mayor de 60 mseg desde el valor basal en el 4,1% de los pacientes y un QTcF mayor de 500 mseg se observó en 4 pacientes (menos del 1%) [ver Advertencia en recuadro, Advertencias y precauciones (5.2, 5.3), Farmacología clínica (12.2)].
Se observó la interrupción debido a reacciones adversas a los medicamentos en el 16% de los pacientes con LMC-CP y el 10% de los pacientes con LMC-AP.
Reacciones adversas más frecuentemente notificadas
Las tablas 7 y 8 muestran el porcentaje de pacientes adultos que experimentaron reacciones adversas no hematológicas (excluyendo las anormalidades de laboratorio), independientemente de la relación con el fármaco del estudio. Se enumeran las reacciones adversas notificadas en más del 10% de los pacientes adultos que recibieron al menos 1 dosis de Tasigna.