
Fabricante de medicamentos: Janssen Biotech, Inc. (Updated: 2024-11-26)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
®de forma segura y eficaz. Consulte la información completa de prescripción de STELARA
®.
STELARA
®(ustequinumab) inyección, para uso subcutáneo o intravenoso
Aprobación inicial en EE. UU.: 2009
INDICACIONES Y USO
STELARA
®es un antagonista humano de la interleucina-12 y -23 indicado para el tratamiento de:
Pacientes adultos con:
-
psoriasis en placas de moderada a grave (PsO)que son candidatos a fototerapia o terapia sistémica. (
1.1)
-
artritis psoriásica (PsA) activa. (
1.2)
-
enfermedad de Crohn (EC) de moderada a grave actividad. (
1.3)
-
colitis ulcerosa de moderada a grave actividad.(
1.4)
Pacientes pediátricos de 6 años o más con:
POSOLOGÍA Y ADMINISTRACIÓN
Psoriasis Dosis subcutánea recomendada para adultos (
2.1)
:
| Rango de peso (kilogramos) | Dosis |
|---|---|
| menor o igual a 100 kg | 45 mg administrados por vía subcutánea inicialmente y 4 semanas después, seguidos de 45 mg administrados por vía subcutánea cada 12 semanas |
| mayor de 100 kg | 90 mg administrados por vía subcutánea inicialmente y 4 semanas después, seguidos de 90 mg administrados por vía subcutánea cada 12 semanas |
Psoriasis Pacientes pediátricos (6 a 17 años) Dosis subcutánea recomendada (
2.1)
:
Se recomienda la dosificación basada en el peso en la dosis inicial, 4 semanas después y luego cada 12 semanas a partir de entonces.
| Rango de peso (kilogramos) | Dosis |
|---|---|
| menos de 60 kg | 0,75 mg/kg |
| 60 kg a 100 kg | 45 mg |
| mayor de 100 kg | 90 mg |
Artritis psoriásica Dosis subcutánea recomendada para adultos (
2.2):
- La dosis recomendada es de 45 mg administrados por vía subcutánea inicialmente y 4 semanas después, seguidos de 45 mg administrados por vía subcutánea cada 12 semanas.
- Para pacientes con psoriasis en placas de moderada a grave coexistente que pesan más de 100 kg, la dosis recomendada es de 90 mg administrados por vía subcutánea inicialmente y 4 semanas después, seguidos de 90 mg administrados por vía subcutánea cada 12 semanas.
Artritis psoriásica Pediátrica (6 a 17 años) Dosis subcutánea recomendada (
2.2):
Se recomienda la dosificación basada en el peso en la dosis inicial, 4 semanas después y luego cada 12 semanas a partir de entonces.
| Rango de peso (kilogramos) | Dosis |
|---|---|
| menos de 60 kg | 0,75 mg/kg |
| 60 kg o más | 45 mg |
| mayor de 100 kg con psoriasis en placas de moderada a grave coexistente | 90 mg |
Enfermedad de Crohn y Colitis ulcerosa Dosis intravenosa inicial recomendada para adultos (
2.3)
:
Una sola infusión intravenosa utilizando una dosificación basada en el peso:
| Rango de peso (kilogramos) | Dosis recomendada |
|---|---|
| hasta 55 kg | 260 mg (2 viales) |
| mayor de 55 kg a 85 kg | 390 mg (3 viales) |
| mayor de 85 kg | 520 mg (4 viales) |
Dosis recomendada subcutánea para el mantenimiento en adultos con enfermedad de Crohn y colitis ulcerosa (
2.3)
:
Una dosis subcutánea de 90 mg 8 semanas después de la dosis intravenosa inicial, y luego cada 8 semanas a partir de entonces.
FORMAS Y CONCENTRACIONES FARMACÉUTICAS
CONTRAINDICACIONES
Hipersensibilidad clínicamente significativa a ustequinumab o a cualquiera de los excipientes de STELARA
®. (
4)
ADVERTENCIAS Y PRECAUCIONES
-
Infecciones: Se han producido infecciones graves. Evite iniciar el tratamiento con STELARA
®durante cualquier infección activa clínicamente importante. Si se desarrolla una infección grave o clínicamente significativa, suspenda STELARA
®hasta que la infección desaparezca. (
5.1)
-
Riesgo teórico de infecciones particulares: Se han notificado infecciones graves por micobacterias, salmonela y vacunas con bacilo de Calmette-Guérin (BCG) en pacientes con deficiencia genética de IL-12/IL-23. Considere las pruebas de diagnóstico para estas infecciones según lo dicten las circunstancias clínicas. (
5.2)
-
Tuberculosis (TB): Evalúe a los pacientes para detectar tuberculosis antes de iniciar el tratamiento con STELARA
®. Inicie el tratamiento de la tuberculosis latente antes de administrar STELARA
®. (
5.3)
-
Neoplasias malignas: STELARA
®puede aumentar el riesgo de neoplasias malignas. No se ha evaluado la seguridad de STELARA
®en pacientes con antecedentes de neoplasia maligna conocida o desconocida. (
5.4)
-
Reacciones de hipersensibilidad: Si se produce una reacción de hipersensibilidad anafiláctica u otra clínicamente significativa, instaurar el tratamiento adecuado y suspender STELARA
®.(
5.5)
-
Síndrome de encefalopatía posterior reversible (PRES): Si se sospecha PRES, trate con prontitud y suspenda STELARA
®. (
5.6)
-
Inmunizaciones:Evite el uso de vacunas vivas en pacientes durante el tratamiento con STELARA
®. (
5.7)
-
Neumonía no infecciosa: Se han notificado casos de neumonía intersticial, neumonía eosinofílica y neumonía organizada criptogénica durante el uso posterior a la aprobación de STELARA
®. Si se confirma el diagnóstico, suspenda STELARA
®e instituya el tratamiento adecuado. (
5.8)
REACCIONES ADVERSAS
Las reacciones adversas más comunes son:
-
Psoriasis (≥3%):nasofaringitis, infección del tracto respiratorio superior, cefalea y fatiga. (
6.1)
-
Enfermedad de Crohn, inducción (≥3%):vómitos. (
6.1)
-
Enfermedad de Crohn, mantenimiento (≥3%):nasofaringitis, eritema en el sitio de inyección, candidiasis vulvovaginal/infección micótica, bronquitis, prurito, infección del tracto urinario y sinusitis. (
6.1)
-
Colitis ulcerosa, inducción (≥3%):nasofaringitis (
6.1)
-
Colitis ulcerosa, mantenimiento (≥3%):nasofaringitis, cefalea, dolor abdominal, gripe, fiebre, diarrea, sinusitis, fatiga y náuseas (
6.1)
Para notificar SOSPECHAS DE REACCIONES ADVERSAS, póngase en contacto con Janssen Biotech, Inc. al 1-800-JANSSEN (1-800-526-7736) o con la FDA al 1-800-FDA-1088 o
www.fda.gov/medwatch.
Ver 17 para información para el paciente y guía de medicamentos.
Revisado: 11/2024
Tabla de contenido
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDO*
1 INDICACIONES Y USO
1.1 Psoriasis en Placas (PsO)
1.2 Artritis Psoriásica (PsA)
1.3 Enfermedad de Crohn (EC)
1.4 Colitis Ulcerosa
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Dosis Recomendada en Psoriasis en Placas
2.2 Dosis Recomendada en Artritis Psoriásica
2.3 Dosis Recomendada en Enfermedad de Crohn y Colitis Ulcerosa
2.4 Consideraciones Generales para la Administración
2.5 Instrucciones para la Administración de STELARA
®Jeringas Prellenadas Equipadas con Protector de Seguridad para la Aguja
2.6 Preparación y Administración de STELARA
®Vial de 130 mg/26 mL (5 mg/mL) para Infusión Intravenosa (Enfermedad de Crohn y Colitis Ulcerosa)
3 FORMAS Y CONCENTRACIONES FARMACÉUTICAS
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Infecciones
5.2 Riesgo Teórico de Vulnerabilidad a Infecciones Particulares
5.3 Evaluación Previa al Tratamiento para Tuberculosis
5.4 Neoplasias Malignas
5.5 Reacciones de Hipersensibilidad
5.6 Síndrome de Encefalopatía Posterior Reversible (PRES)
5.7 Inmunizaciones
5.8 Neumonía No Infecciosa
6 REACCIONES ADVERSAS
6.1 Experiencia en Ensayos Clínicos
6.2 Inmunogenicidad
6.3 Experiencia Postcomercialización
7 INTERACCIONES MEDICAMENTOSAS
7.1 Terapias Concomitantes
7.2 Sustratos del CYP450
7.3 Inmunoterapia con Alérgenos
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.4 Uso Pediátrico
8.5 Uso Geriátrico
10 SOBREDOSIFICACIÓN
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de Acción
12.2 Farmacodinamia
12.3 Farmacocinética
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
13.2 Toxicología y/o Farmacología Animal
14 ESTUDIOS CLÍNICOS
14.1 Psoriasis en Placas en Adultos
14.2 Psoriasis en Placas en Pediatría
14.3 Artritis Psoriásica
14.4 Enfermedad de Crohn
14.5 Colitis Ulcerosa
15 REFERENCIAS
16 PRESENTACIÓN/ALMACENAMIENTO Y MANEJO
17 INFORMACIÓN PARA EL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información completa de prescripción no se enumeran.
1 INDICACIONES Y USO
1.1 Psoriasis en placas (PsO)
STELARA
® está indicado para el tratamiento de adultos y pacientes pediátricos de 6 años o más con psoriasis en placas de moderada a grave que son candidatos a fototerapia o terapia sistémica.
1.2 Artritis psoriásica (PsA)
STELARA
® está indicado para el tratamiento de adultos y pacientes pediátricos de 6 años o más con artritis psoriásica activa.
1.3 Enfermedad de Crohn (EC)
STELARA
® está indicado para el tratamiento de pacientes adultos con enfermedad de Crohn moderada a gravemente activa.
1.4 Colitis ulcerosa
STELARA
® está indicado para el tratamiento de pacientes adultos con colitis ulcerosa moderada a gravemente activa.
2 DOSIS Y ADMINISTRACIÓN
2.1 Dosis recomendada en psoriasis en placas
Régimen de dosificación subcutánea para adultos
- Para pacientes con un peso de 100 kg o menos, la dosis recomendada es de 45 mg inicialmente y 4 semanas después, seguida de 45 mg cada 12 semanas.
- Para pacientes con un peso superior a 100 kg, la dosis recomendada es de 90 mg inicialmente y 4 semanas después, seguida de 90 mg cada 12 semanas.
En sujetos con un peso superior a 100 kg, también se demostró que 45 mg eran eficaces. Sin embargo, 90 mg resultaron en una mayor eficacia en estos sujetos
[ver
.
Régimen de dosificación subcutánea pediátrica
Administrar STELARA
®por vía subcutánea en las semanas 0 y 4, y luego cada 12 semanas a partir de entonces.
La dosis recomendada de STELARA
®para pacientes pediátricos (6–17 años) con psoriasis en placas según el peso corporal se muestra a continuación (Tabla 1).
| Peso corporal del paciente en el momento de la administración | Dosis recomendada |
|---|---|
| menos de 60 kg | 0,75 mg/kg |
| 60 kg a 100 kg | 45 mg |
| más de 100 kg | 90 mg |
Para pacientes pediátricos que pesan menos de 60 kg, el volumen de administración para la dosis recomendada (0,75 mg/kg) se muestra en la Tabla 2; extraiga el volumen adecuado del vial de dosis única.
| Peso corporal (kg) en el momento de la administración | Dosis (mg) | Volumen de inyección (mL) |
|---|---|---|
|
||
| 15 | 11.3 | 0.12 |
| 16 | 12.0 | 0.13 |
| 17 | 12.8 | 0.14 |
| 18 | 13.5 | 0.15 |
| 19 | 14.3 | 0.16 |
| 20 | 15.0 | 0.17 |
| 21 | 15.8 | 0.17 |
| 22 | 16.5 | 0.18 |
| 23 | 17.3 | 0.19 |
| 24 | 18.0 | 0.20 |
| 25 | 18.8 | 0.21 |
| 26 | 19.5 | 0.22 |
| 27 | 20.3 | 0.22 |
| 28 | 21.0 | 0.23 |
| 29 | 21.8 | 0.24 |
| 30 | 22.5 | 0.25 |
| 31 | 23.3 | 0.26 |
| 32 | 24 | 0.27 |
| 33 | 24.8 | 0.27 |
| 34 | 25.5 | 0.28 |
| 35 | 26.3 | 0.29 |
| 36 | 27 | 0.3 |
| 37 | 27.8 | 0.31 |
| 38 | 28.5 | 0.32 |
| 39 | 29.3 | 0.32 |
| 40 | 30 | 0.33 |
| 41 | 30.8 | 0.34 |
| 42 | 31.5 | 0.35 |
| 43 | 32.3 | 0.36 |
| 44 | 33 | 0.37 |
| 45 | 33.8 | 0.37 |
| 46 | 34.5 | 0.38 |
| 47 | 35.3 | 0.39 |
| 48 | 36 | 0.4 |
| 49 | 36.8 | 0.41 |
| 50 | 37.5 | 0.42 |
| 51 | 38.3 | 0.42 |
| 52 | 39 | 0.43 |
| 53 | 39.8 | 0.44 |
| 54 | 40.5 | 0.45 |
| 55 | 41.3 | 0.46 |
| 56 | 42 | 0.46 |
| 57 | 42.8 | 0.47 |
| 58 | 43.5 | 0.48 |
| 59 | 44.3 | 0.49 |
2.2 Dosis recomendada en la artritis psoriásica
Régimen de dosificación subcutánea para adultos
- La dosis recomendada es de 45 mg inicialmente y 4 semanas después, seguida de 45 mg cada 12 semanas.
- Para pacientes con psoriasis en placas de moderada a grave coexistente que pesen más de 100 kg, la dosis recomendada es de 90 mg inicialmente y 4 semanas después, seguida de 90 mg cada 12 semanas.
Régimen de dosificación subcutánea pediátrica
Administrar STELARA
®por vía subcutánea en las semanas 0 y 4, y luego cada 12 semanas a partir de entonces.
La dosis recomendada de STELARA
®para pacientes pediátricos (de 6 a 17 años) con artritis psoriásica, según el peso corporal, se muestra a continuación (Tabla 3).
| Peso corporal del paciente en el momento de la administración | Dosis recomendada |
|---|---|
|
|
| menos de 60 kg | 0,75 mg/kg |
| 60 kg o más | 45 mg |
| más de 100 kg con psoriasis en placas de moderada a grave coexistente | 90 mg |
2.3 Dosis recomendada en la enfermedad de Crohn y la colitis ulcerosa
Régimen de dosificación intravenosa de inducción para adultos
Una dosis única de infusión intravenosa de STELARA
®utilizando el régimen de dosificación basado en el peso especificado en la Tabla 4
[véase
Instrucciones para la dilución del vial de STELARA® 130 mg para infusión intravenosa (2.6)]
.
| Peso corporal del paciente en el momento de la administración | Dosis | Número de viales de STELARA
®130 mg/26 mL (5 mg/mL) |
|---|---|---|
| 55 kg o menos | 260 mg | 2 |
| más de 55 kg a 85 kg | 390 mg | 3 |
| más de 85 kg | 520 mg | 4 |
2.4 Consideraciones generales para la administración
- STELARA
®está destinado a usarse bajo la guía y supervisión de un proveedor de atención médica. STELARA
®solo debe administrarse a pacientes que serán monitoreados de cerca y que tendrán visitas de seguimiento regulares con un proveedor de atención médica. Un proveedor de atención médica debe determinar la dosis apropiada utilizando el peso actual del paciente en el momento de la administración. En pacientes pediátricos, se recomienda que STELARA
®sea administrado por un proveedor de atención médica. Si un proveedor de atención médica determina que es apropiado, un paciente puede autoinyectarse o un cuidador puede inyectar STELARA
®después de una capacitación adecuada en la técnica de inyección subcutánea. Instruya a los pacientes a que sigan las instrucciones proporcionadas en la Guía de medicamentos
[ver
.
- La cubierta de la aguja de la jeringa precargada contiene caucho natural seco (un derivado del látex). La cubierta de la aguja no debe ser manipulada por personas sensibles al látex.
- Se recomienda que cada inyección se administre en una ubicación anatómica diferente (como la parte superior de los brazos, las regiones glúteas, los muslos o cualquier cuadrante del abdomen) a la inyección anterior, y no en áreas donde la piel esté sensible, magullada, eritematosa o indurada. Cuando se utiliza el vial de dosis única, se recomienda una jeringa de 1 mL con una aguja de calibre 27, ½ pulgada.
- Antes de la administración, inspeccione visualmente STELARA
®para detectar partículas y decoloración. STELARA
®es una solución incolora a ligeramente amarilla y puede contener algunas partículas traslúcidas o blancas pequeñas. No use STELARA
®si está decolorado o turbio, o si hay otras partículas presentes. STELARA
®no contiene conservantes; por lo tanto, deseche cualquier producto sin usar que quede en el vial y/o la jeringa.
2.5 Instrucciones para la administración de jeringas precargadas STELARA
®
Equipadas con protector de seguridad para agujas
Consulte el diagrama a continuación para obtener las instrucciones proporcionadas.
Para evitar la activación prematura del protector de seguridad de la aguja, no toque los CLIP DE ACTIVACIÓN DEL PROTECTOR DE LA AGUJA en ningún momento durante el uso.
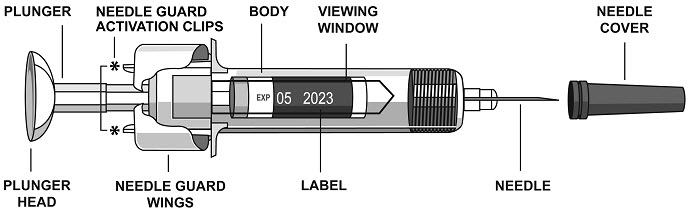
- Sujete el CUERPO y retire la CUBIERTA DE LA AGUJA.
No sujete el ÉMBOLO ni la CABEZA DEL ÉMBOLO mientras retira la CUBIERTA DE LA AGUJA, ya que el émbolo podría moverse. No utilice la jeringa precargada si se cae sin la CUBIERTA DE LA AGUJA en su lugar.
- Inyecte STELARA
®por vía subcutánea según lo recomendado
[ver
Dosificación y administración (2.1,
2.2,
2.3)]
.
- Inyecte todo el medicamento presionando el ÉMBOLO hasta que la CABEZA DEL ÉMBOLO esté completamente entre las alas del protector de la aguja.
Es necesario inyectar todo el contenido de la jeringa precargada para activar el protector de la aguja.
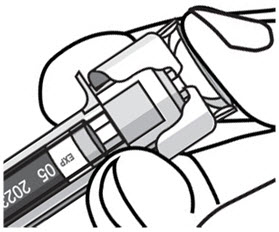
- Después de la inyección, mantenga la presión sobre la CABEZA DEL ÉMBOLO y retire la aguja de la piel. Retire lentamente el pulgar de la CABEZA DEL ÉMBOLO para permitir que la jeringa vacía se mueva hacia arriba hasta que toda la aguja esté cubierta por el protector de la aguja, como se muestra en la ilustración a continuación:
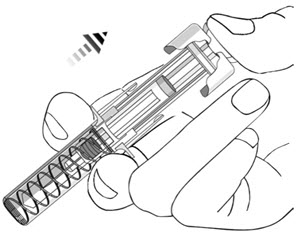
- Las jeringas usadas deben colocarse en un recipiente resistente a las perforaciones.
2.6 Preparación y administración de STELARA
®
Vial de 130 mg/26 mL (5 mg/mL) para infusión intravenosa (enfermedad de Crohn y colitis ulcerosa)
STELARA
®la solución para infusión intravenosa debe ser diluida, preparada e infundida por un profesional de la salud utilizando una técnica aséptica.
- Calcule la dosis y el número de viales de STELARA
®necesarios en función del peso del paciente (Tabla 4). Cada vial de 26 mL de STELARA
®contiene 130 mg de ustequinumab.
- Extraiga y luego deseche un volumen de la inyección de cloruro de sodio al 0,9%, USP de la bolsa de infusión de 250 mL igual al volumen de STELARA
®que se va a añadir (deseche 26 mL de cloruro de sodio por cada vial de STELARA
®necesario; para 2 viales, deseche 52 mL; para 3 viales, deseche 78 mL; para 4 viales, deseche 104 mL). Como alternativa, se puede utilizar una bolsa de infusión de 250 mL que contenga inyección de cloruro de sodio al 0,45%, USP.
- Extraiga 26 mL de STELARA
®de cada vial necesario y añádalos a la bolsa de infusión de 250 mL. El volumen final en la bolsa de infusión debe ser de 250 mL. Mezcle suavemente.
- Inspeccione visualmente la solución diluida antes de la infusión. No la utilice si observa partículas visibles opacas, decoloración o partículas extrañas.
- Infunda la solución diluida durante un período de al menos una hora. Una vez diluida, la infusión debe administrarse completamente dentro de las ocho horas posteriores a la dilución en la bolsa de infusión.
- Utilice únicamente un equipo de infusión con un filtro en línea, estéril, no pirogénico y de baja unión a proteínas (tamaño de poro de 0,2 micrómetros).
- No infunda STELARA
®de forma concomitante en la misma vía intravenosa con otros agentes.
- STELARA
®no contiene conservantes. Cada vial es para una sola dosis. Deseche cualquier solución restante. Deseche cualquier medicamento no utilizado de acuerdo con los requisitos locales.
Conservación
Si es necesario, la solución de infusión diluida puede conservarse a temperatura ambiente hasta 25 °C (77 °F) durante un máximo de 7 horas. El tiempo de almacenamiento a temperatura ambiente comienza una vez que se ha preparado la solución diluida. La infusión debe completarse dentro de las 8 horas posteriores a la dilución en la bolsa de infusión (tiempo acumulado después de la preparación, incluido el almacenamiento y el período de infusión). No congelar. Deseche cualquier parte no utilizada de la solución de infusión.
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
STELARA
®(ustekinumab) es una solución incolora a amarillo claro y puede contener algunas partículas traslúcidas o blancas pequeñas.
Inyección Subcutánea
- Inyección: solución de 45 mg/0.5 mL o 90 mg/mL en una jeringa precargada de dosis única
- Inyección: solución de 45 mg/0.5 mL en un vial de dosis única
4 CONTRAINDICACIONES
STELARA
®está contraindicado en pacientes con hipersensibilidad clínicamente significativa a ustequinumab o a cualquiera de los excipientes de STELARA
®[ver
Advertencias y precauciones (5.5)].
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Infecciones
STELARA
®puede aumentar el riesgo de infecciones y la reactivación de infecciones latentes. Se observaron infecciones bacterianas, micobacterianas, fúngicas y víricas graves en pacientes que recibieron STELARA
®[ver
6.3)]
.
Las infecciones graves que requirieron hospitalización, u otras infecciones clínicamente significativas, notificadas en los ensayos clínicos incluyeron las siguientes:
- Psoriasis en placas: diverticulitis, celulitis, neumonía, apendicitis, colecistitis, sepsis, osteomielitis, infecciones víricas, gastroenteritis e infecciones del tracto urinario.
- Artritis psoriásica: colecistitis.
- Enfermedad de Crohn: absceso anal, gastroenteritis, herpes zóster oftálmico, neumonía y meningitis por listeria.
- Colitis ulcerosa: gastroenteritis, herpes zóster oftálmico, neumonía y listeriosis.
Evite iniciar el tratamiento con STELARA
®en pacientes con cualquier infección activa clínicamente importante hasta que la infección desaparezca o se trate adecuadamente. Considere los riesgos y beneficios del tratamiento antes de iniciar el uso de STELARA
®en pacientes con una infección crónica o un historial de infección recurrente.
Indique a los pacientes que busquen consejo médico si aparecen signos o síntomas que sugieran una infección durante el tratamiento con STELARA
®y suspenda STELARA
®en caso de infecciones graves o clínicamente significativas hasta que la infección desaparezca o se trate adecuadamente.
5.2 Riesgo teórico de vulnerabilidad a infecciones particulares
Las personas genéticamente deficientes en IL-12/IL-23 son particularmente vulnerables a las infecciones diseminadas por micobacterias (incluidas las micobacterias no tuberculosas y ambientales), salmonella (incluidas las cepas no tifoideas) y vacunas con bacilo de Calmette-Guérin (BCG). Se han notificado infecciones graves y resultados fatales en estos pacientes.
No se sabe si los pacientes con bloqueo farmacológico de IL-12/IL-23 por el tratamiento con STELARA
®pueden ser susceptibles a este tipo de infecciones. Considere las pruebas de diagnóstico adecuadas (p. ej., cultivo de tejidos, cultivo de heces, según lo dicten las circunstancias clínicas).
5.3 Evaluación previa al tratamiento para la tuberculosis
Evalúe a los pacientes para detectar una infección por tuberculosis antes de iniciar el tratamiento con STELARA
®.
Evite administrar STELARA
®a pacientes con infección activa por tuberculosis. Inicie el tratamiento de la tuberculosis latente antes de administrar STELARA
®. Considere la terapia antituberculosa antes de iniciar STELARA
®en pacientes con antecedentes de tuberculosis latente o activa en los que no se pueda confirmar un tratamiento adecuado. Supervise estrechamente a los pacientes que reciben STELARA
®para detectar signos y síntomas de tuberculosis activa durante y después del tratamiento.
5.4 Neoplasias malignas
STELARA
®es un inmunosupresor y puede aumentar el riesgo de malignidad. Se notificaron neoplasias malignas entre los sujetos que recibieron STELARA
®en los ensayos clínicos
[ver
. En modelos de roedores, la inhibición de IL-12/IL-23p40 aumentó el riesgo de malignidad
[ver
.
La seguridad de STELARA
®no se ha evaluado en pacientes con antecedentes de neoplasia maligna o que tengan una neoplasia maligna conocida.
Ha habido notificaciones posteriores a la comercialización de la aparición rápida de múltiples carcinomas de células escamosas cutáneos en pacientes que recibieron STELARA
®que tenían factores de riesgo preexistentes para desarrollar cáncer de piel no melanoma. Supervise a todos los pacientes que reciben STELARA
®para detectar la aparición de cáncer de piel no melanoma. Supervise estrechamente a los pacientes mayores de 60 años, a aquellos con antecedentes médicos de terapia inmunosupresora prolongada y a aquellos con antecedentes de tratamiento con PUVA
[ver
.
5.5 Reacciones de hipersensibilidad
Se han notificado reacciones de hipersensibilidad, incluida la anafilaxia y el angioedema, con STELARA
®[ver
6.3)]
. Si se produce una reacción de hipersensibilidad anafiláctica u otra clínicamente significativa, instaurar el tratamiento adecuado y suspender STELARA
®.
5.6 Síndrome de encefalopatía posterior reversible (PRES)
Se notificaron dos casos de síndrome de encefalopatía posterior reversible (PRES), también conocido como síndrome de leucoencefalopatía posterior reversible (RPLS), en los ensayos clínicos. También se han notificado casos en la experiencia posterior a la comercialización en pacientes con psoriasis, artritis psoriásica y enfermedad de Crohn. La presentación clínica incluyó cefaleas, convulsiones, confusión, trastornos visuales y cambios en las imágenes compatibles con PRES unos días a varios meses después del inicio de ustequinumab. Algunos casos notificaron una latencia de un año o más. Los pacientes se recuperaron con cuidados de apoyo tras la retirada de ustequinumab.
Supervise a todos los pacientes tratados con STELARA
®para detectar signos y síntomas de PRES. Si se sospecha PRES, administre rápidamente el tratamiento adecuado y suspenda STELARA
®.
5.7 Inmunizaciones
Antes de iniciar el tratamiento con STELARA
®, los pacientes deben recibir todas las inmunizaciones apropiadas para su edad según lo recomendado por las guías de inmunización actuales. Los pacientes tratados con STELARA
® deben evitar recibir vacunas vivas. Evite administrar vacunas BCG durante el tratamiento con STELARA
® o durante un año antes de iniciar el tratamiento o un año después de la interrupción del tratamiento. Se recomienda precaución al administrar vacunas vivas a los contactos domésticos de pacientes que reciben STELARA
® debido al riesgo potencial de diseminación por parte del contacto doméstico y transmisión al paciente.
Las vacunas no vivas recibidas durante un ciclo de tratamiento con STELARA
® pueden no provocar una respuesta inmunitaria suficiente para prevenir la enfermedad.
5.8 Neumonía no infecciosa
Se han notificado casos de neumonía intersticial, neumonía eosinofílica y neumonía organizada criptogénica durante el uso posterior a la aprobación de STELARA
®. Las manifestaciones clínicas incluyeron tos, disnea e infiltrados intersticiales después de una a tres dosis. Los resultados graves incluyeron insuficiencia respiratoria y hospitalización prolongada. Los pacientes mejoraron con la interrupción del tratamiento y, en ciertos casos, con la administración de corticosteroides. Si se confirma el diagnóstico, suspenda STELARA
®e instituya el tratamiento adecuado
[ver
Experiencia postcomercialización (6.3)]
.
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas graves se describen en otras partes de la etiqueta:
- Infecciones
[ver
Advertencias y precauciones (5.1)]
- Neoplasias malignas
[ver
Advertencias y precauciones (5.4)]
- Reacciones de hipersensibilidad
[ver
Advertencias y precauciones (5.5)]
- Síndrome de encefalopatía posterior reversible (PRES)
[ver
Advertencias y precauciones (5.6)]
- Neumonía no infecciosa
[ver
Advertencias y precauciones (5.8)]
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se realizan en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no se pueden comparar directamente con las tasas en los ensayos clínicos de otro fármaco y pueden no reflejar las tasas observadas en la práctica.
Sujetos adultos con psoriasis en placas
Los datos de seguridad reflejan la exposición a STELARA
®en 3117 sujetos adultos con psoriasis en placas, incluidos 2414 expuestos durante al menos 6 meses, 1855 expuestos durante al menos un año, 1653 expuestos durante al menos dos años, 1569 expuestos durante al menos tres años, 1482 expuestos durante al menos cuatro años y 838 expuestos durante al menos cinco años.
La Tabla 5 resume las reacciones adversas que ocurrieron a una tasa de al menos el 1% con tasas más altas en los grupos de STELARA
®durante el período controlado con placebo de Ps STUDY 1 y Ps STUDY 2
[ver
.
| STELARA
® |
|||
|---|---|---|---|
| Placebo | 45 mg | 90 mg | |
| Sujetos tratados | 665 | 664 | 666 |
| Nasofaringitis | 51 (8%) | 56 (8%) | 49 (7%) |
| Infección de las vías respiratorias superiores | 30 (5%) | 36 (5%) | 28 (4%) |
| Cefalea | 23 (3%) | 33 (5%) | 32 (5%) |
| Fatiga | 14 (2%) | 18 (3%) | 17 (3%) |
| Dorsalgia | 8 (1%) | 9 (1%) | 14 (2%) |
| Mareo | 8 (1%) | 8 (1%) | 14 (2%) |
| Dolor faringolaríngeo | 7 (1%) | 9 (1%) | 12 (2%) |
| Prurito | 9 (1%) | 10 (2%) | 9 (1%) |
| Eritema en el sitio de inyección | 3 (<1%) | 6 (1%) | 13 (2%) |
| Mialgia | 4 (1%) | 7 (1%) | 8 (1%) |
| Depresión | 3 (<1%) | 8 (1%) | 4 (1%) |
Las reacciones adversas que ocurrieron a tasas inferiores al 1% en el período controlado de los ESTUDIOS de Psoriasis 1 y 2 hasta la semana 12 incluyeron: celulitis, herpes zóster, diverticulitis y ciertas reacciones en el sitio de inyección (dolor, hinchazón, prurito, induración, hemorragia, hematomas e irritación).
Se produjo un caso de PRES durante los ensayos clínicos de psoriasis en placa en adultos
[ver
Advertencias y precauciones (5.6)]
.
Infecciones
En el período controlado con placebo de los ensayos clínicos de sujetos con psoriasis en placa (seguimiento promedio de 12,6 semanas para los sujetos tratados con placebo y 13,4 semanas para los sujetos tratados con STELARA
®), el 27% de los sujetos tratados con STELARA
®reportaron infecciones (1,39 por año-sujeto de seguimiento) en comparación con el 24% de los sujetos tratados con placebo (1,21 por año-sujeto de seguimiento). Las infecciones graves ocurrieron en el 0,3% de los sujetos tratados con STELARA
®(0,01 por año-sujeto de seguimiento) y en el 0,4% de los sujetos tratados con placebo (0,02 por año-sujeto de seguimiento)
[ver
Advertencias y precauciones (5.1)]
.
En las partes controladas y no controladas de los ensayos clínicos de psoriasis en placa (seguimiento mediano de 3,2 años), que representan 8998 años-sujeto de exposición, el 72,3% de los sujetos tratados con STELARA
®reportaron infecciones (0,87 por año-sujeto de seguimiento). Se informaron infecciones graves en el 2,8% de los sujetos (0,01 por año-sujeto de seguimiento).
Neoplasias malignas
En las partes controladas y no controladas de los ensayos clínicos de psoriasis en placa (seguimiento mediano de 3,2 años, que representan 8998 años-sujeto de exposición), el 1,7% de los sujetos tratados con STELARA
®reportaron neoplasias malignas, excluyendo los cánceres de piel no melanoma (0,60 por cien años-sujeto de seguimiento). El cáncer de piel no melanoma se informó en el 1,5% de los sujetos tratados con STELARA
®(0,52 por cien años-sujeto de seguimiento)
[ver
Advertencias y precauciones (5.4)]
. Las neoplasias malignas más frecuentemente observadas, distintas del cáncer de piel no melanoma, durante los ensayos clínicos fueron: próstata, melanoma, colorrectal y mama. Las neoplasias malignas distintas del cáncer de piel no melanoma en los sujetos tratados con STELARA
®durante las partes controladas y no controladas de los ensayos fueron similares en tipo y número a lo que se esperaría en la población general de EE. UU. según la base de datos SEER (ajustada por edad, sexo y raza).
1
Sujetos pediátricos con psoriasis en placa
La seguridad de STELARA
®se evaluó en dos ensayos de sujetos pediátricos con psoriasis en placa de moderada a grave. El ESTUDIO de Psoriasis 3 evaluó la seguridad durante un máximo de 60 semanas en 110 sujetos pediátricos de 12 a 17 años de edad. El ESTUDIO de Psoriasis 4 evaluó la seguridad durante un máximo de 56 semanas en 44 sujetos pediátricos de 6 a 11 años de edad. El perfil de seguridad en sujetos pediátricos fue similar al perfil de seguridad de los ensayos en adultos con psoriasis en placa.
Artritis psoriásica
La seguridad de STELARA
®se evaluó en 927 sujetos en dos ensayos aleatorizados, doble ciego, controlados con placebo en adultos con artritis psoriásica activa (APs). El perfil de seguridad general de STELARA
®en sujetos con APs fue consistente con el perfil de seguridad observado en los ensayos clínicos de psoriasis en adultos. Se observó una mayor incidencia de artralgia, náuseas e infecciones dentales en los sujetos tratados con STELARA
®en comparación con los sujetos tratados con placebo (3% frente a 1% para artralgia y 3% frente a 1% para náuseas; 1% frente a 0,6% para infecciones dentales) en las partes controladas con placebo de los ensayos clínicos de APs.
Enfermedad de Crohn
La seguridad de STELARA
®se evaluó en 1407 sujetos con enfermedad de Crohn de moderada a gravemente activa (Índice de actividad de la enfermedad de Crohn [CDAI] mayor o igual a 220 y menor o igual a 450) en tres ensayos aleatorizados, doble ciego, controlados con placebo, de grupos paralelos y multicéntricos. Estos 1407 sujetos incluyeron 40 sujetos que recibieron una formulación intravenosa de ustequinumab de investigación previa, pero que no se incluyeron en los análisis de eficacia. En los ensayos CD-1 y CD-2 hubo 470 sujetos que recibieron STELARA
®6 mg/kg como una dosis de inducción intravenosa única basada en el peso y 466 que recibieron placebo
[ver
. Los sujetos que fueron respondedores en el ensayo CD-1 o CD-2 se asignaron al azar para recibir un régimen de mantenimiento subcutáneo de 90 mg de STELARA
®cada 8 semanas, o placebo durante 44 semanas en el ensayo CD-3. Los sujetos en estos 3 ensayos pueden haber recibido otras terapias concomitantes, incluidos aminosalicilatos, agentes inmunomoduladores [azatioprina (AZA), 6-mercaptopurina (6-MP), metotrexato (MTX)], corticosteroides orales (prednisona o budesonida) y/o antibióticos para su enfermedad de Crohn
[ver
.
El perfil de seguridad general de STELARA
®fue consistente con el perfil de seguridad observado en los ensayos clínicos de psoriasis en adultos y artritis psoriásica. Las reacciones adversas comunes en los ensayos CD-1 y CD-2 y en el ensayo CD-3 se enumeran en las Tablas 6 y 7, respectivamente.
| Placebo | STELARA
® |
|
|---|---|---|
| N=466 | N=470 | |
| Vómitos | 3% | 4% |
Otras reacciones adversas menos comunes notificadas en sujetos en los ensayos CD-1 y CD-2 incluyeron astenia (1% vs 0,4%), acné (1% vs 0,4%) y prurito (2% vs 0,4%).
| Placebo | STELARA
® |
|
|---|---|---|
| N=133 | N=131 | |
| Nasofaringitis | 8% | 11% |
| Eritema en el sitio de inyección | 0 | 5% |
| Candidiasis vulvovaginal/infección micótica | 1% | 5% |
| Bronquitis | 3% | 5% |
| Prurito | 2% | 4% |
| Infección del tracto urinario | 2% | 4% |
| Sinusitis | 2% | 3% |
Infecciones
En sujetos con enfermedad de Crohn, las infecciones graves u otras clínicamente significativas incluyeron absceso anal, gastroenteritis y neumonía. Además, se notificaron meningitis por listeria y herpes zóster oftálmico en un paciente cada uno
[ver
Advertencias y precauciones (5.1)]
.
Neoplasias malignas
Con hasta un año de tratamiento en los ensayos clínicos de la enfermedad de Crohn, el 0,2% de los sujetos tratados con STELARA
® (0,36 eventos por cien pacientes-año) y el 0,2% de los sujetos tratados con placebo (0,58 eventos por cien pacientes-año) desarrollaron cáncer de piel no melanoma. Las neoplasias malignas distintas de los cánceres de piel no melanoma se produjeron en el 0,2% de los sujetos tratados con STELARA
® (0,27 eventos por cien pacientes-año) y en ninguno de los sujetos tratados con placebo.
Reacciones de hipersensibilidad, incluida la anafilaxia
En los ensayos de EC, dos sujetos informaron reacciones de hipersensibilidad después de la administración de STELARA
®. Un paciente experimentó signos y síntomas compatibles con anafilaxia (opresión en la garganta, dificultad para respirar y rubor) después de una sola administración subcutánea (0,1% de los sujetos que recibieron STELARA
® subcutánea). Además, un sujeto experimentó signos y síntomas compatibles con o relacionados con una reacción de hipersensibilidad (molestias en el pecho, rubor, urticaria y aumento de la temperatura corporal) después de la dosis inicial intravenosa de STELARA
® (0,08% de los sujetos que recibieron STELARA
® intravenosa). Estos sujetos fueron tratados con antihistamínicos orales o corticosteroides y en ambos casos los síntomas se resolvieron en una hora.
Colitis ulcerosa
La seguridad de STELARA
® se evaluó en dos ensayos clínicos aleatorizados, doble ciego y controlados con placebo (UC-1 [inducción IV] y UC-2 [mantenimiento SC]) en 960 sujetos adultos con colitis ulcerosa de moderada a gravemente activa
[ver
. El perfil de seguridad general de STELARA
® en sujetos con colitis ulcerosa fue consistente con el perfil de seguridad observado en todas las indicaciones aprobadas. Las reacciones adversas notificadas en al menos el 3% de los sujetos tratados con STELARA
® y a una tasa mayor que el placebo fueron:
- Inducción (UC-1): nasofaringitis (7% vs 4%).
- Mantenimiento (UC-2): nasofaringitis (24% vs 20%), cefalea (10% vs 4%), dolor abdominal (7% vs 3%), gripe (6% vs 5%), fiebre (5% vs. 4%), diarrea (4% vs 1%), sinusitis (4% vs 1%), fatiga (4% vs 2%) y náuseas (3% vs 2%).
Infecciones
En sujetos con colitis ulcerosa, las infecciones graves u otras clínicamente significativas incluyeron gastroenteritis y neumonía. Además, se notificaron listeriosis y herpes zóster oftálmico en un sujeto cada uno
[ver
Advertencias y precauciones (5.1)]
.
Neoplasias malignas
Con hasta un año de tratamiento en los ensayos clínicos de la colitis ulcerosa, el 0,4% de los sujetos tratados con STELARA
® (0,48 eventos por cien pacientes-año) y el 0,0% de los sujetos tratados con placebo (0,00 eventos por cien pacientes-año) desarrollaron cáncer de piel no melanoma. Las neoplasias malignas distintas de los cánceres de piel no melanoma se produjeron en el 0,5% de los sujetos tratados con STELARA
® (0,64 eventos por cien pacientes-año) y el 0,2% de los sujetos tratados con placebo (0,40 eventos por cien pacientes-año).
6.2 Inmunogenicidad
Al igual que con todas las proteínas terapéuticas, existe la posibilidad de inmunogenicidad. La detección de la formación de anticuerpos depende en gran medida de la sensibilidad y la especificidad del ensayo. Además, la incidencia observada de positividad de anticuerpos (incluidos los anticuerpos neutralizantes) en un ensayo puede verse influenciada por varios factores, incluida la metodología del ensayo, el manejo de las muestras, el momento de la recolección de las muestras, los medicamentos concomitantes y la enfermedad subyacente. Por estas razones, la comparación de la incidencia de anticuerpos a ustequinumab en los ensayos descritos a continuación con la incidencia de anticuerpos a otros productos puede ser engañosa.
Aproximadamente del 6 al 12,4% de los sujetos tratados con STELARA
® en ensayos clínicos de psoriasis en placas y artritis psoriásica desarrollaron anticuerpos contra ustequinumab, que generalmente fueron de bajo título. En los ensayos clínicos de psoriasis en placas, los anticuerpos contra ustequinumab se asociaron con concentraciones séricas de ustequinumab reducidas o indetectables y una eficacia reducida. En los ensayos de psoriasis en placas, la mayoría de los sujetos que dieron positivo para anticuerpos contra ustequinumab tenían anticuerpos neutralizantes.
En los ensayos clínicos de enfermedad de Crohn y colitis ulcerosa, el 2,9% y el 4,6% de los sujetos, respectivamente, desarrollaron anticuerpos contra ustequinumab cuando fueron tratados con STELARA
® durante aproximadamente un año. No se observó ninguna asociación aparente entre el desarrollo de anticuerpos contra ustequinumab y el desarrollo de reacciones en el sitio de inyección.
6.3 Experiencia postcomercialización
Las siguientes reacciones adversas se han notificado durante el uso posterior a la aprobación de STELARA
®. Debido a que estas reacciones se notifican voluntariamente a partir de una población de tamaño incierto, no siempre es posible estimar de forma fiable su frecuencia o establecer una relación causal con la exposición a STELARA
® .
Trastornos del sistema inmunitario: Reacciones de hipersensibilidad graves (incluidas anafilaxia y angioedema), otras reacciones de hipersensibilidad (incluidas erupción cutánea y urticaria)
.
Infecciones e infestaciones: Infección del tracto respiratorio inferior (incluidas infecciones fúngicas oportunistas y tuberculosis).
Trastornos neurológicos: Síndrome de encefalopatía posterior reversible (PRES)
.
Trastornos respiratorios, torácicos y mediastínicos:Neumonía intersticial, neumonía eosinofílica y neumonía organizada criptogénica.
Reacciones cutáneas: Psoriasis pustulosa, psoriasis eritrodérmica, vasculitis por hipersensibilidad.
7 INTERACCIONES MEDICAMENTOSAS
7.1 Terapias Concomitantes
En ensayos de psoriasis en placas, no se ha evaluado la seguridad de STELARA
® en combinación con agentes inmunosupresores o fototerapia. En ensayos de artritis psoriásica, el uso concomitante de MTX no pareció influir en la seguridad o eficacia de STELARA
®. En los ensayos de inducción de la enfermedad de Crohn y la colitis ulcerosa, los inmunomoduladores (6-MP, AZA, MTX) se utilizaron de forma concomitante en aproximadamente el 30% de los sujetos y los corticosteroides se utilizaron de forma concomitante en aproximadamente el 40% y el 50% de los sujetos con enfermedad de Crohn y colitis ulcerosa, respectivamente. El uso de estas terapias concomitantes no pareció influir en la seguridad o eficacia general de STELARA
®.
7.2 Sustratos de CYP450
La formación de enzimas CYP450 puede suprimirse por el aumento de los niveles de ciertas citocinas (p. ej., IL-1, IL-6, TNFα, IFN) durante la inflamación crónica. Por lo tanto, el uso de STELARA
®, un antagonista de IL-12 e IL-23, podría normalizar la formación de enzimas CYP450. Al iniciar o suspender STELARA
® en pacientes que reciben sustratos de CYP450 concomitantes, particularmente aquellos con un índice terapéutico estrecho, considere controlar el efecto terapéutico o la concentración del fármaco y ajustar la dosis individual del sustrato de CYP según sea necesario. Consulte la información de prescripción de sustratos de CYP específicos.
No se observó un efecto de interacción farmacológica mediada por CYP en sujetos con enfermedad de Crohn
[ver
.
7.3 Inmunoterapia con Alérgenos
STELARA
® no se ha evaluado en pacientes que se han sometido a inmunoterapia con alérgenos. STELARA
® puede disminuir el efecto protector de la inmunoterapia con alérgenos (disminuir la tolerancia), lo que puede aumentar el riesgo de una reacción alérgica a una dosis de inmunoterapia con alérgenos. Por lo tanto, se debe tener precaución en pacientes que reciben o que han recibido inmunoterapia con alérgenos, particularmente para la anafilaxia.
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de Riesgo
Los datos limitados de estudios observacionales, informes de casos publicados y vigilancia postcomercialización sobre el uso de STELARA
®durante el embarazo son insuficientes para informar un riesgo asociado al fármaco de defectos congénitos importantes, aborto espontáneo y otros resultados adversos maternos o fetales. El transporte del anticuerpo IgG humano a través de la placenta aumenta a medida que avanza el embarazo y alcanza su punto máximo durante el tercer trimestre; por lo tanto, STELARA
®puede transferirse al feto en desarrollo
[ver
En estudios de toxicidad reproductiva y del desarrollo en animales, no se observaron efectos adversos en el desarrollo de la descendencia después de la administración de ustequinumab a monos preñados con exposiciones superiores a 100 veces la dosis máxima recomendada para humanos (MRHD).
Se desconoce el riesgo de fondo de defectos congénitos importantes y aborto espontáneo para la(s) población(es) indicada(s). Todos los embarazos tienen un riesgo de fondo de defectos congénitos, pérdida u otros resultados adversos. En la población general de EE. UU., el riesgo de fondo estimado de defectos congénitos importantes y aborto espontáneo de embarazos clínicamente reconocidos es del 2% al 4% y del 15% al 20%, respectivamente.
Consideraciones Clínicas
Reacciones adversas fetales/neonatales
Debido a que el ustequinumab puede interferir teóricamente con la respuesta inmunitaria a las infecciones, considere los riesgos y beneficios antes de administrar vacunas vivas a los bebés expuestos a STELARA
®in útero.No hay datos suficientes sobre los niveles séricos de ustequinumab en los bebés expuestos al nacer y la duración de la persistencia del ustequinumab en el suero del bebé después del nacimiento. Aunque se desconoce un plazo específico para retrasar la administración de vacunas atenuadas vivas en bebés expuestos
in útero, considere los riesgos y beneficios de retrasar un mínimo de 6 meses después del nacimiento debido a la eliminación del producto.
Datos
Datos en animales
El ustequinumab se probó en dos estudios de toxicidad del desarrollo embriofetal en monos cynomolgus. No se observaron efectos teratogénicos ni otros efectos adversos en el desarrollo en los fetos de monos preñados a los que se administró ustequinumab por vía subcutánea dos veces por semana o por vía intravenosa semanalmente durante el período de organogénesis. Las concentraciones séricas de ustequinumab en monos preñados fueron superiores a 100 veces la concentración sérica en pacientes tratados por vía subcutánea con 90 mg de ustequinumab semanalmente durante 4 semanas.
En un estudio combinado de toxicidad del desarrollo embriofetal y pre y postnatal, se administraron a monos cynomolgus preñados dosis subcutáneas de ustequinumab dos veces por semana con exposiciones superiores a 100 veces la MRHD desde el comienzo de la organogénesis hasta el día 33 después del parto. Se produjeron muertes neonatales en la descendencia de un mono al que se le administró ustequinumab a 22,5 mg/kg y un mono con una dosis de 45 mg/kg. No se observaron efectos relacionados con el ustequinumab en el desarrollo funcional, morfológico o inmunológico en los recién nacidos desde el nacimiento hasta los seis meses de edad.
8.2 Lactancia
Resumen de Riesgo
Los datos limitados de la literatura publicada sugieren que el ustequinumab está presente en la leche materna humana. No hay datos disponibles sobre los efectos del ustequinumab en la producción de leche. Se desconocen los efectos de la exposición gastrointestinal local y la exposición sistémica limitada en el lactante amamantado al ustequinumab. No se han identificado efectos adversos en el lactante amamantado causalmente relacionados con el ustequinumab en la literatura publicada o la experiencia postcomercialización.
Los beneficios para el desarrollo y la salud de la lactancia materna deben considerarse junto con la necesidad clínica de la madre de STELARA
®y cualquier efecto adverso potencial en el niño amamantado por STELARA
®o por la enfermedad materna subyacente.
8.4 Uso Pediátrico
Psoriasis en Placas
La seguridad y la eficacia de STELARA
®se han establecido para el tratamiento de la psoriasis en placas de moderada a grave en pacientes pediátricos de 6 a 17 años de edad que son candidatos a fototerapia o terapia sistémica.
El uso de STELARA
®en pacientes pediátricos de 12 a menos de 17 años de edad está respaldado por la evidencia de un ensayo multicéntrico, aleatorizado, de 60 semanas (Ps STUDY 3) que incluyó una parte de 12 semanas, doble ciego, controlado con placebo, de grupo paralelo, en 110 sujetos pediátricos de 12 años de edad o más
[ver
.
El uso de STELARA
®en pacientes pediátricos de 6 a 11 años de edad está respaldado por la evidencia de un ensayo de eficacia, seguridad y farmacocinética de un solo brazo, abierto, (Ps STUDY 4) en 44 sujetos
[ver
.
La seguridad y la eficacia de STELARA
®no se han establecido en pacientes pediátricos menores de 6 años de edad con psoriasis en placas.
Artritis Psoriásica
La seguridad y eficacia de STELARA
® se han establecido para el tratamiento de la artritis psoriásica en pacientes pediátricos de 6 a 17 años de edad.
El uso de STELARA
® en estos grupos de edad se apoya en evidencia de ensayos adecuados y bien controlados de STELARA
® en adultos con psoriasis y Artritis Psoriásica (APs), datos farmacocinéticos de sujetos adultos con psoriasis, sujetos adultos con APs y sujetos pediátricos con psoriasis, y datos de seguridad de dos ensayos clínicos en 44 sujetos pediátricos de 6 a 11 años de edad con psoriasis y 110 sujetos pediátricos de 12 a 17 años de edad con psoriasis. Las concentraciones previas a la dosis (mínimas) observadas son generalmente comparables entre los sujetos adultos con psoriasis, los sujetos adultos con APs y los sujetos pediátricos con psoriasis, y se espera que la exposición farmacocinética (PK) sea comparable entre los sujetos adultos y pediátricos con APs
[ver
Farmacología clínica (12.3), y
14.2,
14.3)].
La seguridad y eficacia de STELARA
® no se han establecido en pacientes pediátricos menores de 6 años de edad con artritis psoriásica.
8.5 Uso en geriátricos
De los 6709 sujetos expuestos a STELARA
®, un total de 340 tenían 65 años de edad o más (183 sujetos con psoriasis en placas, 65 sujetos con artritis psoriásica, 58 sujetos con enfermedad de Crohn y 34 sujetos con colitis ulcerosa), y 40 sujetos tenían 75 años de edad o más. Los ensayos clínicos de STELARA
® no incluyeron un número suficiente de sujetos de 65 años de edad o más para determinar si responden de manera diferente a los sujetos adultos más jóvenes.
10 SOBREDOSIS
Se han administrado en ensayos clínicos dosis únicas de hasta 6 mg/kg por vía intravenosa sin toxicidad limitante de la dosis. En caso de sobredosis, controle al paciente para detectar cualquier signo o síntoma de reacciones o efectos adversos e instituya inmediatamente el tratamiento sintomático adecuado. Considere contactar con la línea de ayuda contra envenenamiento (1-800-222-1222) o un toxicólogo médico para obtener recomendaciones adicionales sobre el manejo de la sobredosis.
11 DESCRIPCIÓN
El ustequinumab, un anticuerpo monoclonal IgG1κ humano, es un antagonista humano de la interleucina-12 y -23. Utilizando tecnología de ADN recombinante, el ustequinumab se produce en una línea celular murina (Sp2/0). El proceso de fabricación contiene pasos para la eliminación de virus. El ustekinumab está compuesto por 1326 aminoácidos y tiene una masa molecular estimada que oscila entre 148.079 y 149.690 Daltons.
STELARA
®(ustequinumab) inyección es una solución estéril, sin conservantes, incolora a ligeramente amarilla y puede contener algunas pequeñas partículas traslúcidas o blancas con un pH de 5,7–6,3.
STELARA
®para uso subcutáneo
Disponible como 45 mg de ustequinumab en 0,5 mL y 90 mg de ustekinumab en 1 mL, suministrado como una solución estéril en una jeringa precargada de dosis única con una aguja fija de calibre 27 de ½ pulgada y como 45 mg de ustequinumab en 0,5 mL en un vial de vidrio Tipo I de 2 mL de dosis única con un tapón recubierto. La jeringa está equipada con un protector de aguja pasivo y una cubierta de aguja que contiene caucho natural seco (un derivado del látex).
Cada jeringa precargada o vial de 0,5 mL contiene 45 mg de ustequinumab, L-histidina y monohidrocloruro de L-histidina monohidrato (0,5 mg), Polisorbato 80 (0,02 mg) y sacarosa (38 mg).
Cada jeringa precargada de 1 mL contiene 90 mg de ustequinumab, L-histidina y monohidrocloruro de L-histidina monohidrato (1 mg), Polisorbato 80 (0,04 mg) y sacarosa (76 mg).
STELARA
®para infusión intravenosa
Disponible como 130 mg de ustequinumab en 26 mL, suministrado como un vial de vidrio Tipo I de 30 mL de dosis única con un tapón recubierto.
Cada vial de 26 mL contiene 130 mg de ustequinumab, EDTA disódico dihidrato (0,52 mg), L-histidina (20 mg), monohidrocloruro de L-histidina monohidrato (27 mg), L-metionina (10,4 mg), Polisorbato 80 (10,4 mg) y sacarosa (2210 mg).
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
Ustequinumab es un anticuerpo monoclonal IgG1κ humano que se une con especificidad a la subunidad proteica p40 utilizada por las citocinas IL-12 e IL-23. La IL-12 y la IL-23 son citocinas naturales que participan en las respuestas inflamatorias e inmunitarias, como la activación de células asesinas naturales y la diferenciación y activación de células T CD4+. En
in vitro modelos, se demostró que el ustequinumab interrumpía la señalización mediada por IL-12 e IL-23 y las cascadas de citocinas al interrumpir la interacción de estas citocinas con una cadena de receptor de superficie celular compartida, IL-12Rβ1. Las citocinas IL-12 e IL-23 se han implicado como contribuyentes importantes a la inflamación crónica que es un sello distintivo de la enfermedad de Crohn y la colitis ulcerosa. En modelos animales de colitis, se demostró que la ausencia genética o el bloqueo con anticuerpos de la subunidad p40 de IL-12 e IL-23, el objetivo de ustequinumab, era protector.
12.2 Farmacodinamia
Psoriasis en placas
En un pequeño ensayo exploratorio, se observó una disminución en la expresión del ARNm de sus objetivos moleculares IL-12 e IL-23 en biopsias de piel lesional medidas al inicio del estudio y hasta dos semanas después del tratamiento en sujetos con psoriasis en placas.
Colitis ulcerosa
Tanto en el ensayo UC-1 (inducción) como en el ensayo UC-2 (mantenimiento), se observó una relación positiva entre la exposición y las tasas de remisión clínica, respuesta clínica y mejoría endoscópica. La tasa de respuesta se aproximó a una meseta en las exposiciones a ustequinumab asociadas con el régimen de dosificación recomendado para el tratamiento de mantenimiento
[ver
.
12.3 Farmacocinética
Absorción
En sujetos adultos con psoriasis en placas, el tiempo medio para alcanzar la concentración sérica máxima (T
max) fue de 13,5 días y 7 días, respectivamente, después de una administración subcutánea única de 45 mg (N=22) y 90 mg (N=24) de ustequinumab. En sujetos sanos (N=30), la mediana del valor T
max(8,5 días) después de una administración subcutánea única de 90 mg de ustequinumab fue comparable a la observada en sujetos con psoriasis en placas.
Después de múltiples dosis subcutáneas de STELARA
®en sujetos adultos con psoriasis en placas, se alcanzaron concentraciones séricas en estado estacionario de ustequinumab en la semana 28. Las concentraciones séricas mínimas medias (±DE) en estado estacionario de ustequinumab fueron 0,69 ± 0,69 mcg/mL para sujetos con un peso inferior o igual a 100 kg que recibieron una dosis de 45 mg y 0,74 ± 0,78 mcg/mL para sujetos con un peso superior a 100 kg que recibieron una dosis de 90 mg. No hubo acumulación aparente en la concentración sérica de ustequinumab con el tiempo cuando se administró por vía subcutánea cada 12 semanas.
Después de la dosis de inducción intravenosa recomendada, la concentración sérica máxima media ± DE de ustequinumab fue de 125,2 ± 33,6 mcg/mL en sujetos con enfermedad de Crohn y de 129,1 ± 27,6 mcg/mL en sujetos con colitis ulcerosa. A partir de la semana 8, se administró la dosis de mantenimiento subcutánea recomendada de 90 mg de ustequinumab cada 8 semanas. Se alcanzó la concentración de ustequinumab en estado estacionario al comienzo de la segunda dosis de mantenimiento. No hubo acumulación aparente en la concentración de ustequinumab con el tiempo cuando se administró por vía subcutánea cada 8 semanas. La concentración mínima media ± DE en estado estacionario fue de 2,5 ± 2,1 mcg/mL en sujetos con enfermedad de Crohn y de 3,3 ± 2,3 mcg/mL en sujetos con colitis ulcerosa para 90 mg de ustequinumab administrados cada 8 semanas.
Distribución
Los análisis farmacocinéticos poblacionales mostraron que el volumen de distribución de ustequinumab en el compartimento central fue de 2,7 L (IC del 95%: 2,69, 2,78) en sujetos con enfermedad de Crohn y de 3,0 L (IC del 95%: 2,96, 3,07) en sujetos con colitis ulcerosa. El volumen de distribución total en estado estacionario fue de 4,6 L en sujetos con enfermedad de Crohn y de 4,4 L en sujetos con colitis ulcerosa.
Eliminación
La semivida media (±DE) osciló entre 14,9 ± 4,6 y 45,6 ± 80,2 días en todos los ensayos de psoriasis en placas después de la administración subcutánea. Los análisis farmacocinéticos poblacionales mostraron que el aclaramiento de ustequinumab fue de 0,19 L/día (IC del 95%: 0,185, 0,197) en sujetos con enfermedad de Crohn y de 0,19 L/día (IC del 95%: 0,179, 0,192) en sujetos con colitis ulcerosa, con una semivida terminal mediana estimada de aproximadamente 19 días para ambas poblaciones de EII (enfermedad de Crohn y colitis ulcerosa).
Estos resultados indican que la farmacocinética de ustequinumab fue similar entre los sujetos con enfermedad de Crohn y colitis ulcerosa.
Metabolismo
No se ha caracterizado la vía metabólica de ustequinumab. Como anticuerpo monoclonal IgG1κ humano, se espera que el ustequinumab se degrade en pequeños péptidos y aminoácidos a través de vías catabólicas de la misma manera que la IgG endógena.
Poblaciones específicas
Peso
Cuando se les administró la misma dosis, los sujetos con psoriasis en placas o artritis psoriásica con un peso superior a 100 kg tuvieron concentraciones séricas medianas de ustequinumab más bajas en comparación con los sujetos con un peso de 100 kg o menos. Las concentraciones séricas mínimas medianas de ustequinumab en sujetos de mayor peso (mayor de 100 kg) en el grupo de 90 mg fueron comparables a las de sujetos de menor peso (100 kg o menos) en el grupo de 45 mg.
Edad: Población geriátrica
Se realizó un análisis farmacocinético poblacional (N=106/1937 sujetos con psoriasis en placas de 65 años o más) para evaluar el efecto de la edad en la farmacocinética del ustekinumab. No hubo cambios aparentes en los parámetros farmacocinéticos (aclaramiento y volumen de distribución) en sujetos mayores de 65 años.
Edad: Población pediátrica
Después de múltiples dosis recomendadas de STELARA
®en sujetos pediátricos de 6 a 17 años de edad con psoriasis en placas, se alcanzaron las concentraciones séricas en estado estacionario de ustekinumab en la semana 28. En la semana 28, las concentraciones séricas mínimas en estado estacionario medias ± DE de ustekinumab fueron 0,36 ± 0,26 mcg/mL y 0,54 ± 0,43 mcg/mL, respectivamente, en sujetos pediátricos de 6 a 11 años de edad y en sujetos pediátricos de 12 a 17 años de edad.
En general, las concentraciones mínimas de ustekinumab en estado estacionario observadas en sujetos pediátricos con psoriasis en placas se encontraban dentro del rango de las observadas en sujetos adultos con psoriasis en placas y en sujetos adultos con Artritis Psoriásica después de la administración de STELARA
®.
Estudios de interacción medicamentosa
Los efectos de IL-12 o IL-23 en la regulación de las enzimas CYP450 se evaluaron en un estudio in vitro utilizando hepatocitos humanos, que mostraron que IL-12 y/o IL-23 a niveles de 10 ng/mL no alteraron las actividades de las enzimas CYP450 humanas (CYP1A2, 2B6, 2C9, 2C19, 2D6 o 3A4).
No se observaron cambios clínicamente significativos en la exposición a cafeína (sustrato CYP1A2), warfarina (sustrato CYP2C9), omeprazol (sustrato CYP2C19), dextrometorfano (sustrato CYP2D6) o midazolam (sustrato CYP3A) cuando se usaron concomitantemente con ustekinumab en la dosis recomendada aprobada en sujetos con enfermedad de Crohn
[ver
Interacciones medicamentosas (7.2)]
.
Los análisis farmacocinéticos poblacionales indicaron que el aclaramiento de ustekinumab no se vio afectado por el uso concomitante de MTX, AINE y corticosteroides orales, o por la exposición previa a un bloqueador del TNF en sujetos con artritis psoriásica.
En sujetos con enfermedad de Crohn y colitis ulcerosa, los análisis farmacocinéticos poblacionales no indicaron cambios en el aclaramiento de ustekinumab con el uso concomitante de corticosteroides o inmunomoduladores (AZA, 6-MP o MTX); y las concentraciones séricas de ustekinumab no se vieron afectadas por el uso concomitante de estos medicamentos.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogenicidad, Mutagénesis, Deterioro de la Fertilidad
No se han realizado estudios en animales para evaluar el potencial carcinogénico o mutagénico de STELARA
®. La literatura publicada mostró que la administración de IL-12 murina causó un efecto antitumoral en ratones que contenían tumores trasplantados y los ratones knockout de IL-12/IL-23p40 o los ratones tratados con anticuerpos anti-IL-12/IL-23p40 tuvieron una disminución de la defensa del huésped contra los tumores. Los ratones genéticamente manipulados para ser deficientes en IL-12 e IL-23 o solo en IL-12 desarrollaron cánceres de piel inducidos por UV antes y con más frecuencia en comparación con los ratones de tipo salvaje. Se desconoce la relevancia de estos hallazgos experimentales en modelos de ratón para el riesgo de malignidad en humanos.
No se observaron efectos sobre la fertilidad en monos cynomolgus machos a los que se administró ustekinumab por vía subcutánea en dosis de hasta 45 mg/kg dos veces por semana (45 veces la dosis máxima recomendada en humanos (MRHD) en base a mg/kg) antes y durante el período de apareamiento. Sin embargo, no se evaluaron los resultados de fertilidad y embarazo en hembras apareadas.
No se observaron efectos sobre la fertilidad en ratonas hembras a las que se administró un anticuerpo análogo de IL-12/IL-23p40 por vía subcutánea en dosis de hasta 50 mg/kg, dos veces por semana, antes y durante el embarazo temprano.
13.2 Toxicología y/o Farmacología Animal
En un estudio de toxicología de 26 semanas, uno de cada 10 monos a los que se administró ustekinumab por vía subcutánea a 45 mg/kg dos veces por semana durante 26 semanas tuvo una infección bacteriana.
14 ESTUDIOS CLÍNICOS
14.1 Psoriasis en placa en adultos
Dos ensayos multicéntricos, aleatorizados, doble ciego y controlados con placebo (Estudio Ps 1 y Estudio Ps 2) reclutaron un total de 1996 sujetos de 18 años o más con psoriasis en placa que tenían una afectación de la superficie corporal mínima del 10% y una puntuación del Índice de Área y Gravedad de la Psoriasis (PASI) ≥12, y que eran candidatos a fototerapia o terapia sistémica. Los sujetos con psoriasis guttata, eritrodérmica o pustulosa fueron excluidos de los ensayos.
El Estudio Ps 1 reclutó 766 sujetos y el Estudio Ps 2 reclutó 1230 sujetos. Los ensayos tuvieron el mismo diseño hasta la semana 28. En ambos ensayos, los sujetos fueron aleatorizados en igual proporción a placebo, 45 mg o 90 mg de STELARA
®. Los sujetos aleatorizados a STELARA
® recibieron dosis de 45 mg o 90 mg, independientemente del peso, en las semanas 0, 4 y 16. Los sujetos aleatorizados para recibir placebo en las semanas 0 y 4 pasaron a recibir STELARA
® (45 mg o 90 mg) en las semanas 12 y 16.
En ambos ensayos, los sujetos de todos los grupos de tratamiento tuvieron una puntuación PASI basal mediana que osciló entre aproximadamente 17 y 18. La puntuación PGA basal fue marcada o grave en el 44% de los sujetos del Estudio Ps 1 y el 40% de los sujetos del Estudio Ps 2. Aproximadamente dos tercios de todos los sujetos habían recibido fototerapia previa, el 69% había recibido terapia sistémica convencional o biológica previa para el tratamiento de la psoriasis, con un 56% que recibió terapia sistémica convencional previa y un 43% que recibió terapia biológica previa. Un total del 28% de los sujetos tenían antecedentes de artritis psoriásica.
En ambos ensayos, los criterios de valoración fueron la proporción de sujetos que lograron al menos una reducción del 75% en la puntuación PASI (PASI 75) desde el inicio hasta la semana 12 y el éxito del tratamiento (limpio o mínimo) en la Evaluación Global del Médico (PGA). La PGA es una escala de 6 categorías que va de 0 (limpio) a 5 (grave) que indica la evaluación general del médico de la psoriasis centrándose en el grosor/induración de la placa, el eritema y la descamación.
Respuesta clínica
Los resultados del Estudio Ps 1 y del Estudio Ps 2 se presentan en la Tabla 8 a continuación.
| Estudio Ps 1 | Estudio Ps 2 | |||||
|---|---|---|---|---|---|---|
| STELARA
® |
STELARA
® |
|||||
| Placebo | 45 mg | 90 mg | Placebo | 45 mg | 90 mg | |
| Sujetos aleatorizados | 255 | 255 | 256 | 410 | 409 | 411 |
| Respuesta PASI 75 | 8 (3%) | 171 (67%) | 170 (66%) | 15 (4%) | 273 (67%) | 311 (76%) |
| PGA de Limpio o Mínimo | 10 (4%) | 151 (59%) | 156 (61%) | 18 (4%) | 277 (68%) | 300 (73%) |
El examen de los subgrupos de edad, sexo y raza no identificó diferencias en la respuesta a STELARA
®entre estos subgrupos.
En los sujetos que pesaban 100 kg o menos, las tasas de respuesta fueron comparables con las dosis de 45 mg y 90 mg; sin embargo, en los sujetos que pesaban más de 100 kg, se observaron tasas de respuesta más altas con la dosis de 90 mg en comparación con la dosis de 45 mg (Tabla 9 a continuación).
| Estudio Ps 1 | Estudio Ps 2 | |||||
|---|---|---|---|---|---|---|
| STELARA
® |
STELARA
® |
|||||
| Placebo | 45 mg | 90 mg | Placebo | 45 mg | 90 mg | |
|
||||||
| Sujetos aleatorizados | 255 | 255 | 256 | 410 | 409 | 411 |
| Respuesta PASI 75 | ||||||
| ≤100 kg | 4% | 74% | 65% | 4% | 73% | 78% |
| 6/166 | 124/168 | 107/164 | 12/290 | 218/297 | 225/289 | |
| >100 kg | 2% | 54% | 68% | 3% | 49% | 71% |
| 2/89 | 47/87 | 63/92 | 3/120 | 55/112 | 86/121 | |
| PGA de Despejado o Mínimo | ||||||
| ≤100 kg | 4% | 64% | 63% | 5% | 74% | 75% |
| 7/166 | 108/168 | 103/164 | 14/290 | 220/297 | 216/289 | |
| >100 kg | 3% | 49% | 58% | 3% | 51% | 69% |
| 3/89 | 43/87 | 53/92 | 4/120 | 57/112 | 84/121 | |
Los sujetos en el ESTUDIO 1 de Psoriasis que fueron respondedores PASI 75 en las semanas 28 y 40 fueron re-aleatorizados en la semana 40 a una dosis continua de STELARA
®(STELARA
®en la semana 40) o a la retirada del tratamiento (placebo en la semana 40). En la semana 52, el 89% (144/162) de los sujetos re-aleatorizados a tratamiento con STELARA
®fueron respondedores PASI 75 en comparación con el 63% (100/159) de los sujetos re-aleatorizados a placebo (retirada del tratamiento después de la dosis de la semana 28). El tiempo medio hasta la pérdida de la respuesta PASI 75 entre los sujetos aleatorizados a la retirada del tratamiento fue de 16 semanas.
14.2 Psoriasis en placa pediátrica
Un ensayo multicéntrico, aleatorizado, doble ciego, controlado con placebo (ESTUDIO 3 de Psoriasis) incluyó a 110 sujetos pediátricos de 12 a 17 años de edad con una afectación de la superficie corporal mínima del 10%, una puntuación PASI superior o igual a 12 y una puntuación PGA superior o igual a 3, que eran candidatos a fototerapia o terapia sistémica y cuya enfermedad no estaba adecuadamente controlada con terapia tópica.
Los sujetos fueron aleatorizados para recibir placebo (n = 37), la dosis recomendada de STELARA
®(n = 36) o la mitad de la dosis recomendada de STELARA
®(n = 37) mediante inyección subcutánea en las semanas 0 y 4, seguida de una dosis cada 12 semanas (q12s). La dosis recomendada de STELARA
®fue de 0,75 mg/kg para sujetos con un peso inferior a 60 kg, 45 mg para sujetos con un peso de 60 kg a 100 kg y 90 mg para sujetos con un peso superior a 100 kg. En la semana 12, los sujetos que recibieron placebo pasaron a recibir STELARA
®a la dosis recomendada o a la mitad de la dosis recomendada.
De los sujetos pediátricos, aproximadamente el 63% había tenido exposición previa a fototerapia o terapia sistémica convencional y aproximadamente el 11% había tenido exposición previa a biológicos.
Las variables principales fueron la proporción de sujetos que alcanzaron una puntuación PGA de aclarado (0) o mínima (1), PASI 75 y PASI 90 en la semana 12. Los sujetos fueron seguidos durante un máximo de 60 semanas después de la primera administración del agente del ensayo.
Respuesta clínica
Los resultados de eficacia en la semana 12 para el ESTUDIO 3 de Psoriasis se presentan en la Tabla 10.
| ESTUDIO 3 de Psoriasis | ||
|---|---|---|
| Placebo n (%) |
STELARA
®* |
|
|
||
| N | 37 | 36 |
| PGA | ||
| PGA de aclarado (0) o mínima (1) | 2 (5,4%) | 25 (69,4%) |
| PASI | ||
| Respondedores PASI 75 | 4 (10,8%) | 29 (80,6%) |
| Respondedores PASI 90 | 2 (5,4%) | 22 (61,1%) |
14.3 Artritis Psoriásica
La seguridad y eficacia de STELARA
®se evaluó en 927 sujetos (ESTUDIO PsA 1, n=615; ESTUDIO PsA 2, n=312), en dos ensayos aleatorizados, doble ciego, controlados con placebo en sujetos adultos de 18 años de edad o mayores con PsA activa (≥5 articulaciones inflamadas y ≥5 articulaciones dolorosas) a pesar del tratamiento con antiinflamatorios no esteroideos (AINE) o antirreumáticos modificadores de la enfermedad (ARME). Los sujetos en estos ensayos tenían un diagnóstico de PsA durante al menos 6 meses. Se inscribieron sujetos con cada subtipo de PsA, incluida la artritis poliarticular con ausencia de nódulos reumatoideos (39%), espondilitis con artritis periférica (28%), artritis periférica asimétrica (21%), afectación interfalángica distal (12%) y artritis mutilans (0,5%). Más del 70% y el 40% de los pacientes, respectivamente, tenían entesitis y dactilitis al inicio del estudio.
Los sujetos fueron aleatorizados para recibir tratamiento con STELARA
®45 mg, 90 mg o placebo por vía subcutánea en las semanas 0 y 4, seguido de una dosis cada 12 semanas (q12s). Aproximadamente el 50% de los sujetos continuaron con dosis estables de MTX (≤25 mg/semana). El criterio principal de valoración fue el porcentaje de sujetos que lograron una respuesta ACR 20 en la semana 24.
En el ESTUDIO PsA 1 y el ESTUDIO PsA 2, el 80% y el 86% de los sujetos, respectivamente, habían recibido previamente tratamiento con ARME. En el ESTUDIO PsA 1, no se permitió el tratamiento previo con un agente anti-factor de necrosis tumoral (TNF)-α. En el ESTUDIO PsA 2, el 58% (n=180) de los sujetos habían recibido previamente tratamiento con un bloqueador del TNF, de los cuales más del 70% habían interrumpido su tratamiento con bloqueador del TNF por falta de eficacia o intolerancia en algún momento.
Respuesta Clínica
En ambos ensayos, una mayor proporción de sujetos lograron una respuesta ACR 20, ACR 50 y PASI 75 en los grupos de STELARA
®45 mg y 90 mg en comparación con el placebo en la semana 24 (véase
Tabla 11). Las respuestas ACR 70 también fueron mayores en los grupos de STELARA
®45 mg y 90 mg, aunque la diferencia fue solo numérica (p=NS) en el ESTUDIO 2. Las respuestas fueron consistentes en los sujetos tratados con STELARA
®solo o en combinación con metotrexato. Las respuestas fueron similares en los sujetos independientemente de la exposición previa a TNFα.
| ESTUDIO PsA 1 | ESTUDIO PsA 2 | |||||
|---|---|---|---|---|---|---|
| STELARA
® |
STELARA
® |
|||||
| Placebo | 45 mg | 90 mg | Placebo | 45 mg | 90 mg | |
|
||||||
| Número de sujetos aleatorizados | 206 | 205 | 204 | 104 | 103 | 105 |
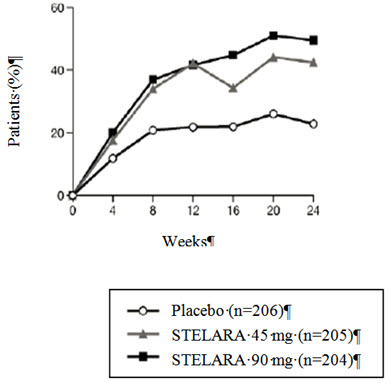
| Respuesta ACR 20, N (%) | 47 (23%) | 87 (42%) | 101 (50%) | 21 (20%) | 45 (44%) | 46 (44%) |
| Respuesta ACR 50, N (%) | 18 (9%) | 51 (25%) | 57 (28%) | 7 (7%) | 18 (17%) | 24 (23%) |
| Respuesta ACR 70, N (%) | 5 (2%) | 25 (12%) | 29 (14%) | 3 (3%) | 7 (7%) | 9 (9%) |
| Número de sujetos con ≥ 3% de SCT | 146 | 145 | 149 | 80 | 80 | 81 |
| Respuesta PASI 75, N (%) | 16 (11%) | 83 (57%) | 93 (62%) | 4 (5%) | 41 (51%) | 45 (56%) |
El porcentaje de sujetos que lograron respuestas ACR 20 por visita se muestra en la Figura 1.
| Figura 1: Porcentaje de sujetos que lograron una respuesta ACR 20 hasta la semana 24 |
|---|
| ESTUDIO PsA 1 |
 |
Los resultados de los componentes de los criterios de respuesta ACR se muestran en la Tabla 12.
| ESTUDIO 1 DE PsA | |||
|---|---|---|---|
| STELARA
® |
|||
| Placebo (N = 206) |
45 mg (N = 205) |
90 mg (N = 204) |
|
|
|||
| Número de articulaciones inflamadas | |||
| Valor inicial | 15 | 12 | 13 |
| Cambio medio en la semana 24 | -3 | -5 | -6 |
| Número de articulaciones doloridas | |||
| Valor inicial | 25 | 22 | 23 |
| Cambio medio en la semana 24 | -4 | -8 | -9 |
| Evaluación del dolor por el sujeto | |||
| Valor inicial | 6.1 | 6.2 | 6.6 |
| Cambio medio en la semana 24 | -0.5 | -2.0 | -2.6 |
| Evaluación global del sujeto | |||
| Valor inicial | 6.1 | 6.3 | 6.4 |
| Cambio medio en la semana 24 | -0.5 | -2.0 | -2.5 |
| Evaluación global del médico | |||
| Valor inicial | 5.8 | 5.7 | 6.1 |
| Cambio medio en la semana 24 | -1.4 | -2.6 | -3.1 |
| Índice de discapacidad (HAQ) | |||
| Valor inicial | 1.2 | 1.2 | 1.2 |
| Cambio medio en la semana 24 | -0.1 | -0.3 | -0.4 |
| PCR (mg/dL) | |||
| Valor inicial | 1.6 | 1.7 | 1.8 |
| Cambio medio en la semana 24 | 0.01 | -0.5 | -0.8 |
Se observó una mejoría en las puntuaciones de entesitis y dactilitis en cada grupo de STELARA
®en comparación con el placebo en la semana 24.
Función física
Los sujetos tratados con STELARA
®mostraron una mejoría en la función física en comparación con los sujetos tratados con placebo, según lo evaluado por HAQ-DI en la semana 24. En ambos ensayos, la proporción de respondedores HAQ-DI (mejora ≥0,3 en la puntuación HAQ-DI) fue mayor en los grupos STELARA
®45 mg y 90 mg en comparación con el placebo en la semana 24.
14.4 Enfermedad de Crohn
STELARA
®se evaluó en tres ensayos clínicos aleatorizados, doble ciego, controlados con placebo en sujetos adultos con enfermedad de Crohn de moderada a gravemente activa (puntuación del Índice de Actividad de la Enfermedad de Crohn [CDAI] de 220 a 450). Hubo dos ensayos de inducción intravenosa de 8 semanas (CD-1 y CD-2) seguidos de un ensayo de mantenimiento de retirada aleatorizado subcutáneo de 44 semanas (CD-3) que representan 52 semanas de terapia. Los sujetos en CD-1 habían fracasado o eran intolerantes al tratamiento con uno o más bloqueadores del TNF, mientras que los sujetos en CD-2 habían fracasado o eran intolerantes al tratamiento con inmunomoduladores o corticosteroides, pero nunca habían fracasado en el tratamiento con un bloqueador del TNF.
Ensayos CD-1 y CD-2
En los ensayos CD-1 y CD-2, se aleatorizaron 1409 sujetos, de los cuales 1368 (CD-1, n=741; CD-2, n=627) se incluyeron en el análisis final de eficacia. Se evaluó la inducción de la respuesta clínica (definida como una reducción en la puntuación CDAI de más de o igual a 100 puntos o una puntuación CDAI de menos de 150) en la semana 6 y la remisión clínica (definida como una puntuación CDAI de menos de 150) en la semana 8. En ambos ensayos, los sujetos fueron aleatorizados para recibir una única administración intravenosa de STELARA
®a aproximadamente 6 mg/kg, placebo (ver
Tabla 4), o 130 mg (una dosis inferior a la recomendada).
En el ensayo CD-1, los sujetos habían fracasado o eran intolerantes al tratamiento previo con un bloqueador del TNF: el 29% de los sujetos tuvieron una respuesta inicial inadecuada (no respondedores primarios), el 69% respondió pero posteriormente perdió la respuesta (no respondedores secundarios) y el 36% fue intolerante a un bloqueador del TNF. De estos sujetos, el 48% fracasó o fue intolerante a un bloqueador del TNF y el 52% había fracasado en 2 o 3 bloqueadores del TNF previos. Al inicio y durante todo el ensayo, aproximadamente el 46% de los sujetos recibieron corticosteroides y el 31% de los sujetos recibieron inmunomoduladores (AZA, 6-MP, MTX). La puntuación CDAI basal mediana fue de 319 en el grupo STELARA
®aproximadamente 6 mg/kg y de 313 en el grupo placebo.
En el ensayo CD-2, los sujetos habían fracasado o eran intolerantes al tratamiento previo con corticosteroides (81% de los sujetos), al menos un inmunomodulador (6-MP, AZA, MTX; 68% de los sujetos), o ambos (49% de los sujetos). Además, el 69% nunca recibió un bloqueador del TNF y el 31% lo recibió previamente pero no había fracasado en un bloqueador del TNF. Al inicio y durante todo el ensayo, aproximadamente el 39% de los sujetos recibieron corticosteroides y el 35% de los sujetos recibieron inmunomoduladores (AZA, 6-MP, MTX). La puntuación CDAI basal mediana fue de 286 en el grupo STELARA
®y de 290 en el grupo placebo.
En estos ensayos de inducción, una mayor proporción de sujetos tratados con STELARA
®(a la dosis recomendada de aproximadamente 6 mg/kg) lograron una respuesta clínica en la semana 6 y una remisión clínica en la semana 8 en comparación con el placebo (ver
Tabla 13 para las tasas de respuesta clínica y remisión). La respuesta clínica y la remisión fueron significativas ya en la semana 3 en los sujetos tratados con STELARA
®y continuaron mejorando hasta la semana 8.
| CD-1 n = 741 |
CD-2 n = 627 |
|||||
|---|---|---|---|---|---|---|
| Placebo N = 247 |
STELARA ®‡ N = 249 |
Diferencia de tratamiento e IC del 95% | Placebo N = 209 |
STELARA ®‡ N = 209 |
Diferencia de tratamiento e IC del 95% | |
| La remisión clínica se define como una puntuación CDAI < 150; La respuesta clínica se define como una reducción en la puntuación CDAI de al menos 100 puntos o estar en remisión clínica: la respuesta de 70 puntos se define como una reducción en la puntuación CDAI de al menos 70 puntos | ||||||
|
||||||
| Respuesta clínica (100 puntos), Semana 6 | 53 (21%) | 84 (34%) § |
12% (4%, 20%) |
60 (29%) | 116 (56%) ¶ |
27% (18%, 36%) |
| Remisión clínica, Semana 8 | 18 (7%) | 52 (21%) ¶ |
14% (8%, 20%) |
41 (20%) | 84 (40%) ¶ |
21% (12%, 29%) |
| Respuesta clínica (100 puntos), Semana 8 | 50 (20%) | 94 (38%) ¶ |
18% (10%, 25%) |
67 (32%) | 121 (58%) ¶ |
26% (17%, 35%) |
| Respuesta de 70 puntos, Semana 6 | 75 (30%) | 109 (44%) § |
13% (5%, 22%) |
81 (39%) | 135 (65%) ¶ |
26% (17%, 35%) |
| Respuesta de 70 puntos, Semana 3 | 67 (27%) | 101 (41%) § |
13% (5%, 22%) |
66 (32%) | 106 (51%) ¶ |
19% (10%, 28%) |
Ensayo CD-3
El ensayo de mantenimiento (CD-3) evaluó a 388 sujetos que lograron una respuesta clínica (reducción ≥100 puntos en la puntuación CDAI) en la semana 8 con cualquiera de las dosis de inducción de STELARA
® en los ensayos CD-1 o CD-2. Los sujetos fueron aleatorizados para recibir un régimen de mantenimiento subcutáneo de 90 mg de STELARA
® cada 8 semanas o placebo durante 44 semanas (véase
Tabla 14).
| Placebo | 90 mg STELARA
® cada 8 semanas |
Diferencia de tratamiento e IC del 95% | |
|---|---|---|---|
| N = 131 | N = 128 | ||
| La remisión clínica se define como una puntuación CDAI < 150; la respuesta clínica se define como una reducción de al menos 100 puntos en el CDAI o estar en remisión clínica | |||
|
|||
| Remisión clínica | 47 (36%) | 68 (53%) | 17% (5%, 29%) |
| Respuesta clínica | 58 (44%) | 76 (59%) | 15% (3%, 27%) |
| Remisión clínica en pacientes en remisión al inicio del tratamiento de mantenimiento | 36/79 (46%) | 52/78 (67%) | 21% (6%, 36%) |
En la semana 44, el 47 % de los sujetos que recibieron STELARA
®estaban libres de corticosteroides y en remisión clínica, en comparación con el 30 % de los sujetos del grupo placebo.
En la semana 0 del ensayo CD-3, 34/56 (61 %) de los sujetos tratados con STELARA
®que previamente habían fracasado o eran intolerantes a las terapias con bloqueadores del TNF estaban en remisión clínica, y 23/56 (41 %) de estos sujetos estaban en remisión clínica en la semana 44. En el brazo placebo, 27/61 (44 %) de los sujetos estaban en remisión clínica en la semana 0, mientras que 16/61 (26 %) de estos sujetos estaban en remisión en la semana 44.
En la semana 0 del ensayo CD-3, 46/72 (64 %) de los sujetos tratados con STELARA
®que previamente habían fracasado en la terapia inmunomoduladora o con corticosteroides (pero no con bloqueadores del TNF) estaban en remisión clínica, y 45/72 (63 %) de estos sujetos estaban en remisión clínica en la semana 44. En el brazo placebo, 50/70 (71 %) de estos sujetos estaban en remisión clínica en la semana 0, mientras que 31/70 (44 %) estaban en remisión en la semana 44. En el subgrupo de estos sujetos que también eran naive a los bloqueadores del TNF, 34/52 (65 %) de los sujetos tratados con STELARA
®estaban en remisión clínica en la semana 44, en comparación con 25/51 (49 %) en el brazo placebo.
Los sujetos que no presentaron respuesta clínica 8 semanas después de la inducción con STELARA
®no se incluyeron en los análisis de eficacia primaria del ensayo CD-3; sin embargo, estos sujetos podían recibir una inyección subcutánea de 90 mg de STELARA
®al entrar en el ensayo CD-3. De estos sujetos, 102/219 (47 %) lograron una respuesta clínica ocho semanas después y fueron seguidos durante la duración del ensayo.
14.5 Colitis ulcerosa
STELARA
®se evaluó en dos ensayos clínicos aleatorizados, doble ciego y controlados con placebo [UC-1 y UC-2 (NCT02407236)] en sujetos adultos con colitis ulcerosa de moderada a gravemente activa que habían tenido una respuesta inadecuada o no habían tolerado un biológico (es decir, bloqueador del TNF y/o vedolizumab), corticosteroides y/o terapia con 6-MP o AZA. El ensayo de inducción intravenosa de 8 semanas (UC-1) fue seguido por el ensayo de mantenimiento de retirada aleatorizada subcutánea de 44 semanas (UC-2) durante un total de 52 semanas de terapia.
La evaluación de la enfermedad se basó en la puntuación de Mayo, que osciló entre 0 y 12 y tiene cuatro subpuntuaciones que se calificaron de 0 (normal) a 3 (más grave): frecuencia de las deposiciones, hemorragia rectal, hallazgos en la endoscopia revisada centralmente y evaluación global del médico. La colitis ulcerosa de moderada a gravemente activa se definió al inicio (semana 0) como una puntuación de Mayo de 6 a 12, incluida una subpuntuación de endoscopia de Mayo ≥2. Una puntuación de endoscopia de 2 se definió por eritema marcado, patrón vascular ausente, friabilidad, erosiones; y una puntuación de 3 se definió por hemorragia espontánea, ulceración. Al inicio, los sujetos tenían una puntuación de Mayo mediana de 9, con el 84 % de los sujetos con enfermedad moderada (puntuación de Mayo 6-10) y el 15 % con enfermedad grave (puntuación de Mayo 11-12).
Los sujetos de estos ensayos pueden haber recibido otras terapias concomitantes, incluidos aminosalicilatos, agentes inmunomoduladores (AZA, 6-MP o MTX) y corticosteroides orales (prednisona).
Ensayo UC-1
En UC-1, 961 sujetos fueron aleatorizados en la semana 0 a una única administración intravenosa de STELARA
®de aproximadamente 6 mg/kg, 130 mg (una dosis inferior a la recomendada) o placebo. Los sujetos inscritos en UC-1 tenían que haber fracasado en la terapia con corticosteroides, inmunomoduladores o al menos un biológico. Un total del 51 % había fracasado en al menos un biológico y el 17 % había fracasado tanto en un bloqueador del TNF como en un bloqueador del receptor de integrina. De la población total, el 46 % había fracasado en los corticosteroides o los inmunomoduladores, pero eran naive a los biológicos, y un 3 % adicional había recibido previamente, pero no había fracasado, un biológico. Al inicio de la inducción y durante todo el ensayo, aproximadamente el 52 % de los sujetos recibían corticosteroides orales, el 28 % de los sujetos recibían inmunomoduladores (AZA, 6-MP o MTX) y el 69 % de los sujetos recibían aminosalicilatos.
El criterio de valoración principal fue la remisión clínica en la semana 8. La remisión clínica con una definición de: subpuntuación de frecuencia de las deposiciones de Mayo de 0 o 1, subpuntuación de hemorragia rectal de Mayo de 0 (sin hemorragia rectal) y subpuntuación de endoscopia de Mayo de 0 o 1 (subpuntuación de endoscopia de Mayo de 0 definida como enfermedad normal o inactiva y subpuntuación de Mayo de 1 definida como presencia de eritema, patrón vascular disminuido y sin friabilidad) se proporciona en la Tabla 15.
Los criterios de valoración secundarios fueron la respuesta clínica, la mejoría endoscópica y la mejoría mucosa histológico-endoscópica. La respuesta clínica con una definición de (≥ 2 puntos y ≥ 30 % de disminución en la puntuación de Mayo modificada, definida como la puntuación de Mayo de 3 componentes sin la evaluación global del médico, con una disminución desde el inicio en la subpuntuación de hemorragia rectal ≥1 o una subpuntuación de hemorragia rectal de 0 o 1), la mejoría endoscópica con una definición de subpuntuación de endoscopia de Mayo de 0 o 1, y la mejoría mucosa histológico-endoscópica con una definición de mejoría endoscópica combinada y mejoría histológica del tejido del colon [infiltración de neutrófilos en <5 % de las criptas, sin destrucción de criptas y sin erosiones, ulceraciones o tejido de granulación]) se proporcionan en la Tabla 15.
En UC-1, una proporción significativamente mayor de sujetos tratados con STELARA
®(a la dosis recomendada de aproximadamente 6 mg/kg) estaban en remisión clínica y respuesta y lograron una mejoría endoscópica y una mejoría mucosa histológico-endoscópica en comparación con el placebo (véase
Tabla 15).
| Criterio de valoración |
Placebo N = 319 |
STELARA
®* |
Diferencia de tratamiento e IC del 97,5 % | ||
|---|---|---|---|---|---|
| N | % | N | % | ||
|---|---|---|---|---|---|
|
|||||
| Remisión clínica | 22 | 7% | 62 | 19% | 12% (7%, 18%) |
| Bio-ingenuo | 14/151 | 9% | 36/147 | 24% | |
| Fallo biológico previo | 7/161 | 4% | 24/166 | 14% | |
| Mejora endoscópica | 40 | 13% | 80 | 25% | 12% (6%, 19%) |
| Bio-ingenuo | 28/151 | 19% | 43/147 | 29% | |
| Fallo biológico previo | 11/161 | 7% | 34/166 | 20% | |
| Respuesta clínica | 99 | 31% | 186 | 58% | 27% (18%, 35%) |
| Bio-ingenuo | 55/151 | 36% | 94/147 | 64% | |
| Fallo biológico previo | 42/161 | 26% | 86/166 | 52% | |
| Mejora de la mucosa histológico-endoscópica | 26 | 8% | 54 | 17% | 9% (3%, 14%) |
| Bio-ingenuo | 19/151 | 13% | 30/147 | 20% | |
| Fallo biológico previo | 6/161 | 4% | 21/166 | 13% | |
No se evaluó la relación entre la mejoría histológica-endoscópica de la mucosa, según se definió en UC-1, en la semana 8 y la progresión de la enfermedad y los resultados a largo plazo durante UC-1.
Subpuntuaciones de sangrado rectal y frecuencia de deposiciones
Se observaron disminuciones en las subpuntuaciones de sangrado rectal y frecuencia de deposiciones ya en la semana 2 en los sujetos tratados con STELARA
®.
Ensayo UC-2
El ensayo de mantenimiento (UC-2) evaluó a 523 sujetos que lograron una respuesta clínica 8 semanas después de la administración intravenosa de la dosis de inducción de STELARA
®en UC-1. Estos sujetos fueron aleatorizados para recibir un régimen de mantenimiento subcutáneo de 90 mg de STELARA
®cada 8 semanas, o cada 12 semanas (una dosis inferior a la recomendada), o placebo durante 44 semanas.
El criterio de valoración principal fue la proporción de sujetos en remisión clínica en la semana 44. Los criterios de valoración secundarios incluyeron la proporción de sujetos que mantuvieron la respuesta clínica en la semana 44, la proporción de sujetos con mejoría endoscópica en la semana 44, la proporción de sujetos con remisión clínica sin corticosteroides en la semana 44 y la proporción de sujetos que mantuvieron la remisión clínica en la semana 44 entre los sujetos que lograron la remisión clínica 8 semanas después de la inducción.
Los resultados de los criterios de valoración principales y secundarios en la semana 44 en los sujetos tratados con STELARA
®a la dosis recomendada (90 mg cada 8 semanas) en comparación con el placebo se muestran en la Tabla 16.
| Criterio de valoración | Placebo
* |
90 mg STELARA
®cada 8 semanas |
Diferencia de tratamiento e IC del 95% | ||
|---|---|---|---|---|---|
| N | % | N | % | ||
|
|||||
| Remisión clínica | 46 | 26% | 79 | 45% | 19% (9%, 28%) |
| Sin tratamiento previo con biológicos | 30/84 | 36% | 39/79 | 49% | |
| Fallo previo de biológicos | 16/88 | 18% | 37/91 | 41% | |
| Mantenimiento de la respuesta clínica en la semana 44 | 84 | 48% | 130 | 74% | 26% (16%, 36%) |
| Bio-naïve | 49/84 | 58% | 62/79 | 78% | |
| Fallo previo con biológico | 35/88 | 40% | 64/91 | 70% | |
| Mejora endoscópica | 47 | 27% | 83 | 47% | 20% (11%, 30%) |
| Bio-naïve | 29/84 | 35% | 42/79 | 53% | |
| Fallo previo con biológico | 18/88 | 20% | 38/91 | 42% | |
| Remisión clínica sin corticosteroides | 45 | 26% | 76 | 43% | 17% (8%, 27%) |
| Bio-naïve | 30/84 | 36% | 38/79 | 48% | |
| Fallo previo con biológico | 15/88 | 17% | 35/91 | 38% | |
| Mantenimiento de la remisión clínica en la semana 44 en sujetos que lograron la remisión clínica 8 semanas después de la inducción | 18/50 | 36% | 27/41 | 66% | 31% (12%, 50%) |
| Bio-naïve | 12/27 | 44% | 14/20 | 70% | |
| Fallo previo con biológico | 6/23 | 26% | 12/18 | 67% | |
Otros criterios de valoración
Sujetos respondedores a la inducción con ustequinumab en la semana 16
Los sujetos que no presentaron respuesta clínica 8 semanas después de la inducción con STELARA
®en UC-1 no se incluyeron en los análisis de eficacia primaria para el ensayo UC-2; sin embargo, estos sujetos fueron elegibles para recibir una inyección subcutánea de 90 mg de STELARA
®en la semana 8. De estos sujetos, 55/101 (54%) lograron una respuesta clínica ocho semanas después (semana 16) y recibieron STELARA
®90 mg por vía subcutánea cada 8 semanas durante el ensayo UC-2. En la semana 44, hubo 97/157 (62%) sujetos que mantuvieron la respuesta clínica y hubo 51/157 (32%) que lograron la remisión clínica.
Mejora mucosa histológica-endoscópica en la semana 44
La proporción de sujetos que lograron una mejora mucosa histológica-endoscópica durante el tratamiento de mantenimiento en UC-2 fue de 75/172 (44%) entre los sujetos tratados con STELARA
®y de 40/172 (23%) en los sujetos tratados con placebo en la semana 44. La relación entre la mejora mucosa histológica-endoscópica, según se definió en UC-2, en la semana 44 y la progresión de la enfermedad o los resultados a largo plazo no se evaluó en UC-2.
Normalización endoscópica
La normalización del aspecto endoscópico de la mucosa se definió como una subpuntuación endoscópica de Mayo de 0. En la semana 8 en UC-1, se logró la normalización endoscópica en 25/322 (8%) de los sujetos tratados con STELARA
®y en 12/319 (4%) de los sujetos del grupo placebo. En la semana 44 de UC-2, se logró la normalización endoscópica en 51/176 (29%) de los sujetos tratados con STELARA
®y en 32/175 (18%) de los sujetos del grupo placebo.
15 REFERENCIAS
- Programa de Vigilancia, Epidemiología y Resultados Finales (SEER) (www.seer.cancer.gov) Base de datos SEER*Stat: Incidencia – SEER 6.6.2 Regs Research Data, Nov 2009 Sub (1973–2007) – Vinculado a atributos del condado – Total de EE. UU., condados de 1969–2007, Instituto Nacional del Cáncer, DCCPS, Programa de Investigación de Vigilancia, Rama de Sistemas de Vigilancia, publicado en abril de 2010, basado en la presentación de noviembre de 2009.
16 SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
STELARA
®(ustekinumab) injection es una solución estéril, sin conservantes, incolora a ligeramente amarilla y puede contener algunas partículas traslúcidas o blancas pequeñas. Se suministra en jeringas precargadas de dosis única, con envase individual, o en viales de dosis única.
Para uso subcutáneo
Jeringas precargadas
- 45 mg/0.5 mL (NDC 57894-060-03)
- 90 mg/mL (NDC 57894-061-03)
Cada jeringa precargada está equipada con una aguja fija de 27 G de ½ pulgada, un protector de seguridad para la aguja y una cubierta de aguja que contiene caucho natural seco.
Vial de dosis única
- 45 mg/0.5 mL (NDC 57894-060-02)
Almacenamiento y estabilidad
Conserve los viales y las jeringas precargadas de STELARA
® refrigerados entre 2°C y 8°C (36°F y 46°F). Almacene los viales de STELARA
® en posición vertical. Mantenga el producto en su envase original para protegerlo de la luz hasta el momento de su uso. No congelar. No agitar.
Si es necesario, las jeringas precargadas individuales se pueden almacenar a temperatura ambiente hasta 30°C (86°F) durante un período máximo único de hasta 30 días en el envase original para protegerlas de la luz. Anote la fecha en que la jeringa precargada se retira por primera vez del refrigerador en el espacio previsto en el envase. Una vez que una jeringa se ha almacenado a temperatura ambiente, no la vuelva a refrigerar. Deseche la jeringa si no se usa dentro de los 30 días de almacenamiento a temperatura ambiente. No use STELARA
® después de la fecha de caducidad que figura en el envase o en la jeringa precargada.
17 INFORMACIÓN DE ASESORAMIENTO AL PACIENTE
Aconseje al paciente y/o cuidador que lean el etiquetado del paciente aprobado por la FDA (Guía de medicamentos e Instrucciones de uso).
Infecciones
Informe a los pacientes que STELARA
®puede disminuir la capacidad de su sistema inmunitario para combatir infecciones y que deben comunicarse con su proveedor de atención médica inmediatamente si desarrollan algún signo o síntoma de infección
[ver
Advertencias y precauciones (5.1)]
.
Neoplasias malignas
Informe a los pacientes sobre el riesgo de desarrollar neoplasias malignas mientras reciben STELARA
®[ver
Advertencias y precauciones (5.4)]
.
Reacciones de hipersensibilidad
- Aconseje a los pacientes que busquen atención médica inmediata si experimentan algún signo o síntoma de reacciones de hipersensibilidad graves y que interrumpan el tratamiento con STELARA
®[ver
Advertencias y precauciones (5.5)].
- Informe a los pacientes que la cubierta de la aguja de la jeringa precargada contiene caucho natural seco (un derivado del látex), que puede causar reacciones alérgicas en individuos sensibles al látex
[ver
Síndrome de encefalopatía posterior reversible (PRES)
Informe a los pacientes que se comuniquen inmediatamente con su proveedor de atención médica si experimentan signos y síntomas de PRES (que pueden incluir dolor de cabeza, convulsiones, confusión o trastornos visuales)
[ver
Advertencias y precauciones (5.6)]
.
Inmunizaciones
Informe a los pacientes que STELARA
®puede interferir con la respuesta habitual a las inmunizaciones y que deben evitar las vacunas vivas
[ver
Advertencias y precauciones (5.7)]
.
Administración
Instruya a los pacientes a seguir las recomendaciones de eliminación de objetos punzocortantes, como se describe en las Instrucciones de uso.
SECCIÓN NO CLASIFICADA DE SPL
Jeringa precargada fabricada por: Janssen Biotech, Inc., Horsham, PA 19044, EE. UU. Licencia nº 1864 en Baxter Pharmaceutical Solutions, Bloomington, IN 47403 y en Cilag AG, Schaffhausen, Suiza
Vial fabricado por: Janssen Biotech, Inc., Horsham, PA 19044, EE. UU. Licencia nº 1864 en Cilag AG, Schaffhausen, Suiza
© 2012, 2016, 2019 Janssen Pharmaceutical Companies
Guía de medicación
| Esta Guía del Medicamento ha sido aprobada por la Administración de Alimentos y Medicamentos de los EE. UU. | Revisado: 03/2024 | ||
| GUÍA DEL MEDICAMENTO STELARA ®(stel ar’ a) (ustekinumab) inyección, para uso subcutáneo o intravenoso |
|||
| ¿Cuál es la información más importante que debo saber sobre STELARA? STELARA es un medicamento que afecta su sistema inmunitario. STELARA puede aumentar su riesgo de sufrir efectos secundarios graves, incluyendo: Infecciones graves.STELARA puede disminuir la capacidad de su sistema inmunitario para combatir infecciones y puede aumentar su riesgo de infecciones. Algunas personas han tenido infecciones graves mientras tomaban STELARA, incluyendo tuberculosis (TB), e infecciones causadas por bacterias, hongos o virus. Algunas personas han tenido que ser hospitalizadas para el tratamiento de su infección.
No debe comenzar a tomar STELARA si tiene algún tipo de infección a menos que su médico diga que está bien. Antes de comenzar con STELARA, informe a su médico si:
|
|||
|
|
||
Después de comenzar con STELARA, llame a su médico de inmediato si tiene algún síntoma de infección (ver arriba). Estos pueden ser signos de infecciones como infecciones pulmonares, o infecciones de la piel o herpes zóster que podrían tener complicaciones graves. STELARA puede hacer que sea más probable que contraiga infecciones o que empeore una infección que ya tenga. Las personas que tienen un problema genético en el que el cuerpo no produce ninguna de las proteínas interleucina 12 (IL-12) e interleucina 23 (IL-23) tienen un mayor riesgo de ciertas infecciones graves. Estas infecciones pueden diseminarse por todo el cuerpo y causar la muerte. Las personas que toman STELARA también pueden ser más propensas a contraer estas infecciones. Cánceres.STELARA puede disminuir la actividad de su sistema inmunitario y aumentar su riesgo de ciertos tipos de cáncer. Informe a su médico si alguna vez ha tenido algún tipo de cáncer. Algunas personas que están recibiendo STELARA y tienen factores de riesgo para el cáncer de piel han desarrollado ciertos tipos de cáncer de piel. Durante su tratamiento con STELARA, informe a su médico si desarrolla algún crecimiento cutáneo nuevo. Síndrome de encefalopatía posterior reversible (PRES).PRES es una afección poco frecuente que afecta al cerebro y puede causar la muerte. Se desconoce la causa del PRES. Si el PRES se detecta y trata precozmente, la mayoría de las personas se recuperan. Informe a su médico de inmediato si tiene algún problema médico nuevo o que empeora, incluyendo: |
|||
|
|
||
| ¿Qué es STELARA? STELARA es un medicamento recetado que se usa para tratar:
No se sabe si STELARA es seguro y eficaz en niños menores de 6 años. |
|||
| No tome STELARA si es alérgico a ustekinumab o a cualquiera de los ingredientes de STELARA. Consulte el final de esta Guía del Medicamento para obtener una lista completa de los ingredientes de STELARA. | |||
|
Antes de recibir STELARA, informe a su médico sobre todas sus afecciones médicas, incluso si:
Informe a su médico sobre todos los medicamentos que toma, incluidos los medicamentos recetados y de venta libre, las vitaminas y los suplementos herbales. Conozca los medicamentos que toma. Lleve una lista de ellos para mostrársela a su médico y farmacéutico cuando obtenga un nuevo medicamento. |
|||
¿Cómo debo usar STELARA?
Lea las instrucciones detalladas de uso al final de esta Guía de medicamentos para obtener instrucciones sobre cómo preparar e inyectar una dosis de STELARA y cómo desechar correctamente las agujas y jeringas usadas. La jeringa, la aguja y el vial nunca deben reutilizarse. Después de perforar el tapón de goma, STELARA puede contaminarse con bacterias dañinas que podrían causar una infección si se reutiliza. Por lo tanto, deseche cualquier porción no utilizada de STELARA. |
|||
|
¿Qué debo evitar mientras uso STELARA? No debe recibir una vacuna viva mientras toma STELARA. Consulte “Antes de recibir STELARA, informe a su médico sobre todas sus afecciones médicas, incluso si:“ |
|||
| ¿Cuáles son los posibles efectos secundarios de STELARA? STELARA puede causar efectos secundarios graves, que incluyen:
|
|||
|
|
||
|
|||
| Efectos secundarios comunes de STELARA incluyen: | |||
|
|
||
| Estos no son todos los posibles efectos secundarios de STELARA. Consulte a su médico para obtener asesoramiento médico sobre los efectos secundarios. Puede informar los efectos secundarios a la FDA al 1-800-FDA-1088. También puede informar los efectos secundarios a Janssen Biotech, Inc. al 1-800 JANSSEN (1-800-526-7736). |
|||
¿Cómo debo almacenar STELARA?
Si es necesario, las jeringas precargadas individuales de STELARA también se pueden almacenar a temperatura ambiente hasta 30°C (86ºF) durante un período máximo único de hasta 30 días en la caja original para protegerlas de la luz. Anote la fecha en que la jeringa precargada se retira por primera vez del refrigerador en la caja en el espacio provisto. Una vez que una jeringa se ha almacenado a temperatura ambiente, no debe volver a colocarse en el refrigerador. Deseche la jeringa si no se usa dentro de los 30 días de almacenamiento a temperatura ambiente. No use STELARA después de la fecha de vencimiento que figura en la caja o en la jeringa precargada. Mantenga STELARA y todos los medicamentos fuera del alcance de los niños. |
|||
| Información general sobre el uso seguro y eficaz de STELARA. Los medicamentos a veces se recetan para fines distintos de los que se enumeran en una Guía de medicamentos. No use STELARA para una afección para la que no se le recetó. No le dé STELARA a otras personas, incluso si tienen los mismos síntomas que usted. Puede dañarlos. Puede pedirle a su médico o farmacéutico información sobre STELARA que esté escrita para profesionales de la salud. |
|||
| ¿Cuáles son los ingredientes de STELARA? Ingrediente activo: ustekinumab Ingredientes inactivos: Jeringa precargada de dosis única para uso subcutáneo contiene L-histidina, monohidrato de monohidrocloruro de L-histidina, polisorbato 80 y sacarosa. Vial de dosis única para uso subcutáneo contiene L-histidina, monohidrato de clorhidrato de L-histidina, polisorbato 80 y sacarosa. Vial de dosis única para infusión intravenosa contiene EDTA sal disódica dihidratada, L-histidina, monohidrato de clorhidrato de L-histidina, L-metionina, polisorbato 80 y sacarosa. Fabricado por: Janssen Biotech, Inc., Horsham, PA 19044, EE. UU. Licencia No. 1864 © 2012, 2016, 2019 Janssen Pharmaceutical Companies Para obtener más información, visite www.stelarainfo.com o llame al 1-800-JANSSEN (1-800-526-7736). |
|||
INSTRUCCIONES DE USO
INSTRUCCIONES DE USO
STELARA (
stel ar’ a
)
(ustekinumab)
inyección, para uso subcutáneo
Instrucciones para inyectar STELARA usando una jeringa precargada.
Lea estas Instrucciones de uso antes de comenzar a usar STELARA. Su médico o enfermero debe mostrarle cómo preparar y administrar su inyección de STELARA de la manera correcta.
Si no puede inyectarse usted mismo:
- pídale ayuda a su médico o enfermero, o
- pídale a alguien que haya sido capacitado por un médico o enfermero que le administre las inyecciones.
No intente inyectarse STELARA usted mismo hasta que su médico, enfermero o profesional de la salud le haya mostrado cómo inyectar STELARA.
Información importante:
- Antes de comenzar, verifique la caja para asegurarse de que sea la dosis correcta. Recibirá 45 mg o 90 mg según lo recetado por su médico.
- Si su dosis es de 45 mg, recibirá una jeringa precargada de 45 mg.
- Si su dosis es de 90 mg, recibirá una jeringa precargada de 90 mg o dos jeringas precargadas de 45 mg.
Si recibe dos jeringas precargadas de 45 mg para una dosis de 90 mg, deberá aplicarse dos inyecciones, una inmediatamente después de la otra.
- Los niños de 12 años o más con psoriasis que pesen 132 libras o más pueden usar una jeringa precargada.
- Verifique la fecha de vencimiento en la jeringa precargada y la caja. Si la fecha de vencimiento ha pasado o si la jeringa precargada se ha mantenido a temperatura ambiente hasta 30 °C (86 °F) durante un período máximo único de 30 días o si la jeringa precargada se ha almacenado a más de 30 °C (86 °F), no la use. Si la fecha de vencimiento ha pasado o si la jeringa precargada se ha almacenado a más de 30 °C (86 °F), llame a su médico o farmacéutico, o llame al 1-800-JANSSEN (1-800-526-7736) para obtener ayuda.
- Asegúrese de que la jeringa no esté dañada.
- La cubierta de la aguja de la jeringa precargada contiene látex. No manipule la cubierta de la aguja de la jeringa precargada STELARA si es alérgico al látex.
- Revise su jeringa precargada para detectar partículas o decoloración. Su jeringa precargada debe verse transparente e incolora a ligeramente amarilla con pocas partículas blancas.
- No use si está congelado, decolorado, turbio o tiene partículas grandes. Obtenga una nueva jeringa precargada.
- No agite la jeringa precargada en ningún momento.Agitar la jeringa precargada puede dañar su medicamento STELARA. Si su jeringa precargada se ha agitado, no la use. Obtenga una nueva jeringa precargada.
- Para reducir el riesgo de pinchazos accidentales con la aguja, cada jeringa precargada tiene un protector de aguja que se activa automáticamente para cubrir la aguja después de que haya administrado su inyección. No tire del émbolo en ningún momento.
Reúna los suministros que necesitará para preparar y administrar su inyección. (Ver
Necesitará:
- toallitas antisépticas
- bolas de algodón o gasas
- vendaje adhesivo
- su dosis prescrita de STELARA
(Ver
- contenedor de eliminación de objetos punzocortantes aprobado por la FDA.
Ver ”
Para evitar la activación temprana del protector de seguridad de la aguja, no toque los CLIP DE ACTIVACIÓN DEL PROTECTOR DE LA AGUJA en ningún momento durante el uso.
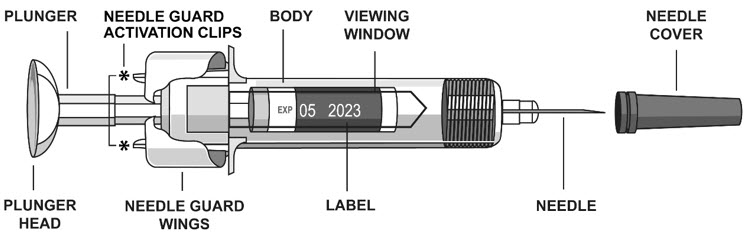
Paso 1: Prepare la inyección.
- Elija una superficie de trabajo limpia, plana y bien iluminada.
- Lávese bien las manos con agua tibia y jabón.
- Sostenga la jeringa precargada con la aguja cubierta apuntando hacia arriba.
Paso 2: Prepare el sitio de inyección
- Elija un sitio de inyección alrededor del área del estómago (abdomen), las nalgas, la parte superior de las piernas (muslos). Si un cuidador le está administrando la inyección, también se puede usar el área externa de la parte superior de los brazos.
(Ver
- Use un sitio de inyección diferente para cada inyección. Noadministre una inyección en un área de la piel que esté sensible, magullada, roja o dura.
- Limpie la piel con una toallita antiséptica donde planea administrarse la inyección.
- Novuelva a tocar esta área antes de administrar la inyección. Deje que su piel se seque antes de inyectar.
- Noabanique ni sople sobre el área limpia.
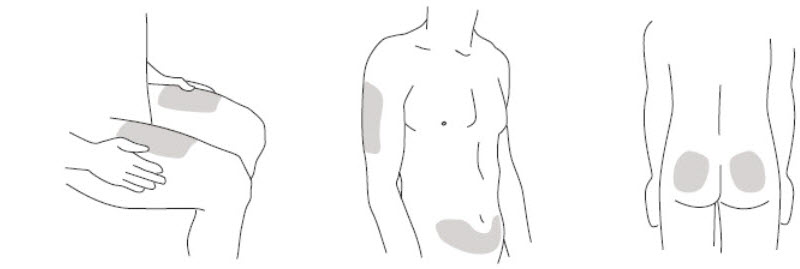
*Las áreas en gris son los sitios de inyección recomendados.
Paso 3: Inyecte STELARA
- Retire la cubierta de la aguja cuando esté listo para inyectar su STELARA.
- Notoque el émbolo mientras retira la cubierta de la aguja.
- Sostenga el cuerpo de la jeringa precargada con una mano y retire la cubierta de la aguja directamente.
(ver
- Tire la cubierta de la aguja a la basura.
- También puede ver una gota de líquido en el extremo de la aguja. Esto es normal.
- Notoque la aguja ni la deje tocar nada.
- Nouse la jeringa precargada si se cae sin la cubierta de la aguja en su lugar. Llame a su médico, enfermero o profesional de la salud para obtener instrucciones.
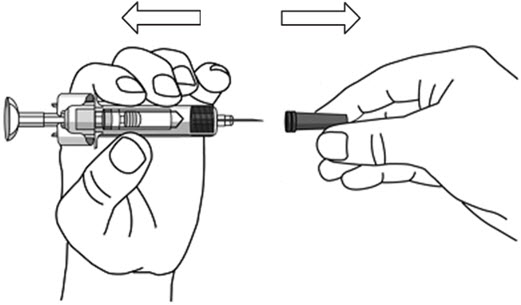
- Sostenga el cuerpo de la jeringa precargada en una mano entre el pulgar y el índice.
(Ver
- Notire de atrás del émbolo en ningún momento.
- Use la otra mano para pellizcar suavemente el área de piel limpia. Mantenga firmemente.
- Use un movimiento rápido, como de dardo, para insertar la aguja en la piel pellizcada en un ángulo de aproximadamente 45 grados.
(Ver
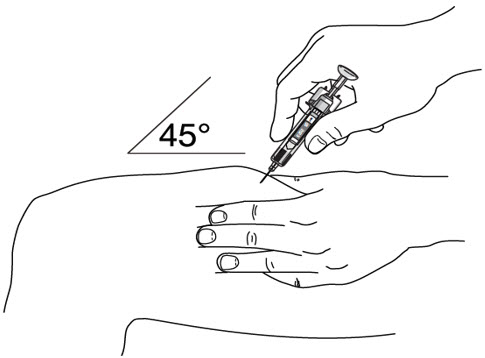
- Inyecte todo el líquido usando el pulgar para empujar el émbolo hasta que la cabeza del émbolo esté completamente entre las alas del protector de la aguja.
(Ver
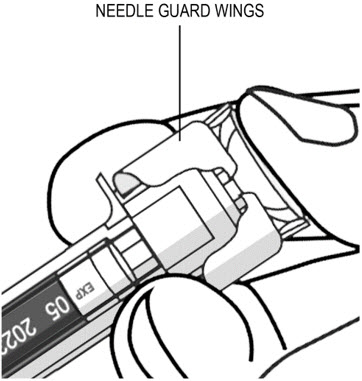
- Cuando el émbolo esté empujado hasta donde pueda llegar, mantenga presión sobre la cabeza del émbolo. Saque la aguja de la piel y suelte la piel.
- Quite lentamente el pulgar de la cabeza del émbolo. Esto permitirá que la jeringa vacía se mueva hacia arriba hasta que toda la aguja esté cubierta por el protector de la aguja.
(Ver
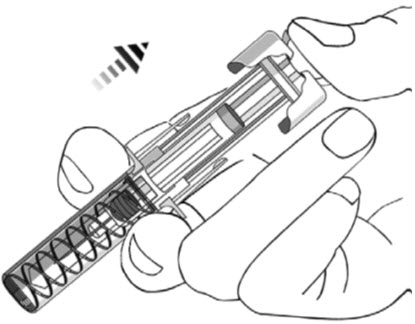
- Cuando se extraiga la aguja de la piel, puede haber un pequeño sangrado en el sitio de inyección. Esto es normal. Puede presionar una bola de algodón o una gasa en el sitio de inyección si es necesario. No frote el sitio de inyección. Puede cubrir el sitio de inyección con una pequeña venda adhesiva, si es necesario.
Si su dosis es de 90 mg, recibirá una jeringa precargada de 90 mg o dos jeringas precargadas de 45 mg. Si recibe dos jeringas precargadas de 45 mg para una dosis de 90 mg, deberá aplicarse una segunda inyección inmediatamente después de la primera. Repita los pasos 1 a 3 para la segunda inyección usando una jeringa nueva. Elija un sitio diferente para la segunda inyección.
- Coloque la jeringa en un contenedor de eliminación de objetos punzocortantes aprobado por la FDA inmediatamente después de su uso.
No tire (deseche) las jeringas sueltas a la basura de su casa.
- Si no tiene un contenedor de eliminación de objetos punzocortantes aprobado por la FDA, puede usar un contenedor doméstico que sea:
- hecho de plástico de alta resistencia.
- se pueda cerrar con una tapa hermética y resistente a las perforaciones, sin que los objetos punzocortantes puedan salir.
- sea vertical y estable durante el uso,
- a prueba de fugas,
- y esté correctamente etiquetado para advertir sobre los residuos peligrosos dentro del contenedor.
- Cuando su contenedor de eliminación de objetos punzocortantes esté casi lleno, deberá seguir las pautas de su comunidad para la forma correcta de desechar su contenedor de eliminación de objetos punzocortantes. Puede haber leyes locales o estatales sobre cómo desechar jeringas y agujas. Para obtener más información sobre la eliminación segura de objetos punzocortantes y para obtener información específica sobre la eliminación de objetos punzocortantes en el estado en el que vive, visite el sitio web de la FDA en: http://www.fda.gov/safesharpsdisposal.
- No deseche su contenedor de eliminación de objetos punzocortantes en la basura de su casa a menos que las pautas de su comunidad lo permitan. No recicle su contenedor de eliminación de objetos punzocortantes.
- Si tiene alguna pregunta, hable con su médico o farmacéutico.
Mantenga STELARA y todos los medicamentos fuera del alcance de los niños.
Jeringa precargada fabricada por:
Janssen Biotech, Inc., Horsham, PA 19044, EE. UU. Licencia No. 1864 en Baxter Pharmaceutical Solutions, Bloomington, IN 47403 y en Cilag AG, Schaffhausen, Suiza
Estas instrucciones de uso han sido aprobadas por la Administración de Alimentos y Medicamentos de EE. UU.
Revisado: 03/2020
© 2012 Janssen Pharmaceutical Companies
INSTRUCCIONES DE USO
INSTRUCCIONES DE USO
STELARA (
stel ar’ a
)
(ustekinumab)
inyección, para uso subcutáneo
Instrucciones para inyectar STELARA a partir de un vial.
Lea estas Instrucciones de uso antes de comenzar a usar STELARA. Su médico o enfermero debe mostrarle cómo preparar, medir su dosis y administrar su inyección de STELARA de la manera correcta.
Si no puede autoadministrarse la inyección:
- pídale ayuda a su médico o enfermero, o
- pídale a alguien que haya sido capacitado por un médico o enfermero que le administre las inyecciones.
No intente inyectarse STELARA usted mismo hasta que su médico, enfermero o profesional de la salud le haya mostrado cómo inyectar STELARA.
Información importante:
- Antes de comenzar, verifique la caja para asegurarse de que sea la dosis correcta. Recibirá 45 mg o 90 mg según lo recetado por su médico.
- Si su dosis es de 45 mg o menos, recibirá un vial de 45 mg.
- Si su dosis es de 90 mg, recibirá dos viales de 45 mg
y deberá administrarse dos inyecciones, una inmediatamente después de la otra.
- Los niños de 12 años o más que pesen menos de 60 kg requieren una dosis inferior a 45 mg.
- Verifique la fecha de vencimiento en el vial y la caja. Si la fecha de vencimiento ha pasado, no lo use. Si la fecha de vencimiento ha pasado, llame a su médico o farmacéutico, o llame al 1-800-JANSSEN (1-800-526-7736) para obtener ayuda.
- Revise el vial para detectar partículas o decoloración. Su vial debe verse transparente e incoloro a ligeramente amarillo con pocas partículas blancas.
- No lo use si está congelado, decolorado, turbio o tiene partículas grandes. Obtenga un vial nuevo.
- No agite el vial en ningún momento.Agitar el vial puede dañar su medicamento STELARA. Si su vial se ha agitado, no lo use. Obtenga un vial nuevo.
- No use un vial de STELARA más de una vez, incluso si queda medicamento en el vial. Después de perforar el tapón de goma, STELARA puede contaminarse con bacterias dañinas que podrían causar una infección si se reutiliza. Por lo tanto, deseche cualquier STELARA sin usar después de administrarse la inyección.
- Deseche (elimine) de forma segura los viales de STELARA después de su uso.
- No reutilice jeringas ni agujas. Consulte ”
Paso 6: Deseche las agujas y las jeringas.”
- Para evitar lesiones por pinchazos con agujas,
norecoloque las agujas.
Reúna los suministros que necesitará para preparar STELARA y administrarse la inyección. (Consulte
Necesitará:
- una jeringa con la aguja puesta, necesitará una receta de su proveedor de atención médica para obtener jeringas con agujas de su farmacia.
- toallitas antisépticas
- bolas de algodón o gasas
- vendaje adhesivo
- su dosis prescrita de STELARA
- contenedor de eliminación de objetos punzocortantes aprobado por la FDA. Consulte ”
Paso 6: Deseche las agujas y las jeringas.”
Figura A
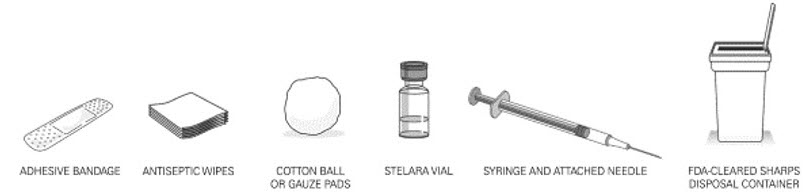
Paso 1: Prepare la inyección.
- Elija una superficie de trabajo limpia, plana y bien iluminada.
- Lávese bien las manos con agua tibia y jabón.
Paso 2: Prepare el sitio de inyección
- Elija un sitio de inyección alrededor del área del estómago (abdomen), las nalgas y la parte superior de las piernas (muslos).
Si un cuidador le está administrando la inyección, también se puede usar el área externa de la parte superior de los brazos.(Consulte
- Use un sitio de inyección diferente para cada inyección. Noadministre una inyección en un área de la piel que esté sensible, magullada, roja o dura.
- Limpie la piel con una toallita antiséptica donde planea administrarse la inyección.
- Novuelva a tocar esta área antes de administrar la inyección. Deje que su piel se seque antes de inyectar.
- Noabanique ni sople sobre el área limpia.
Figura B
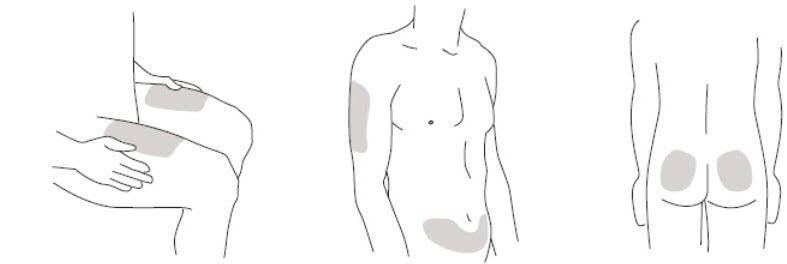
*Las áreas en gris son los sitios de inyección recomendados.
Paso 3: Prepare el vial.
- Retire la tapa de la parte superior del vial. Deseche la tapa, pero no retire el tapón de goma.
(Consulte
Figura C

- Limpie el tapón de goma con un hisopo antiséptico.
(Consulte
Figura D
- No toque el tapón de goma después de limpiarlo.
- Coloque el vial sobre una superficie plana.
Paso 4: Prepare la aguja
- Tome la jeringa con la aguja puesta.
- Retire la tapa que cubre la aguja.
(Consulte
- Deseche la tapa de la aguja. No toque la aguja ni permita que la aguja toque nada.
Figura E
- Tire cuidadosamente del émbolo hasta la línea que coincida con la dosis prescrita por su médico.
- Sujete el vial entre el pulgar y el índice (dedo índice).
- Use su otra mano para empujar la aguja de la jeringa a través del centro del tapón de goma.
(Consulte
Figura F
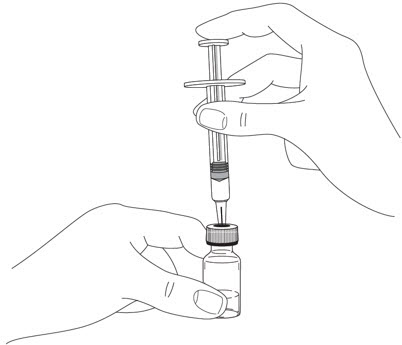
- Presione el émbolo hasta que todo el aire haya salido de la jeringa hacia el vial.
- Gire el vial y la jeringa boca abajo.
(Ver
- Sujete el vial de STELARA con una mano.
- Es importante que la aguja esté siempre dentro del líquido para evitar que se formen burbujas de aire en la jeringa.
- Tire del émbolo de la jeringa con la otra mano.
- Llene la jeringa hasta que la punta negra del émbolo se alinee con la marca que coincida con la dosis prescrita.
Figura G
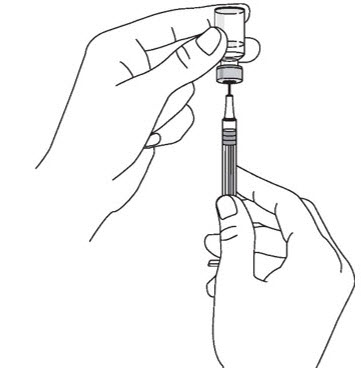
- No retire la aguja del vial.Sujete la jeringa con la aguja apuntando hacia arriba para ver si tiene burbujas de aire en su interior.
- Si hay burbujas de aire, golpee suavemente el lateral de la jeringa hasta que las burbujas de aire suban a la superficie.
(Ver
- Presione lentamente el émbolo hacia arriba hasta que todas las burbujas de aire hayan salido de la jeringa (pero sin que salga líquido).
- Retire la jeringa del vial. No coloque la jeringa boca abajo ni permita que la aguja toque nada.
Figura H
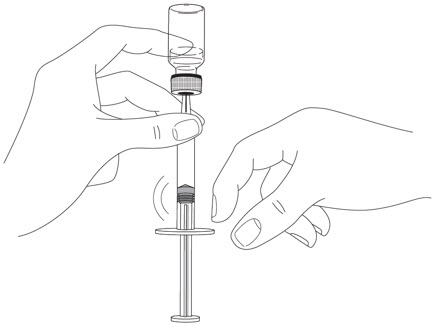
Paso 5: Inyecte STELARA
- Sujete el cuerpo de la jeringa con una mano, entre el pulgar y el índice.
- Notire del émbolo en ningún momento.
- Use la otra mano para pellizcar suavemente el área de piel limpia. Mantenga firmemente.
- Use un movimiento rápido y similar a una flecha para insertar la aguja en la piel pellizcada con un ángulo de aproximadamente 45 grados.
(Ver
Figura I
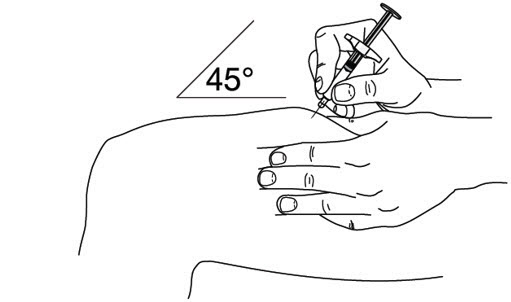
- Empuje el émbolo con el pulgar hasta donde llegue para inyectar todo el líquido. Empújelo lenta y uniformemente, manteniendo la piel suavemente pellizcada.
- Cuando la jeringa esté vacía, retire la aguja de la piel y suelte la piel.
(Ver
Figura J
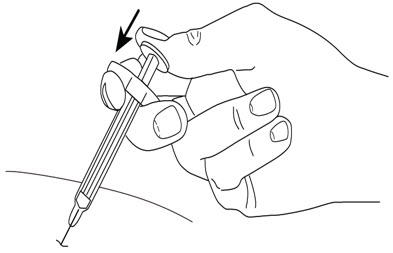
- Cuando se retire la aguja de la piel, puede haber un pequeño sangrado en el lugar de la inyección. Esto es normal. Puede presionar una bola de algodón o una gasa en el lugar de la inyección si es necesario. No frote el lugar de la inyección. Puede cubrir el lugar de la inyección con una pequeña venda adhesiva, si es necesario.
Si su dosis es de 90 mg, recibirá dos viales de 45 mg y deberá aplicarse una segunda inyección inmediatamente después de la primera. Repita los pasos 1 a 5 utilizando una jeringa nueva.Elija un sitio diferente para la segunda inyección.
Paso 6: Deseche las agujas y las jeringas.
- Noreutilice una jeringa o aguja.
- Para evitar lesiones por pinchazos con agujas, no vuelva a colocar la tapa a una aguja.
- Coloque sus agujas y jeringas en un contenedor de eliminación de objetos punzantes aprobado por la FDA inmediatamente después de su uso.
No tire (deseche) las agujas y jeringas sueltas a la basura de su hogar.
- Si no tiene un contenedor de eliminación de objetos punzantes aprobado por la FDA, puede usar un contenedor doméstico que sea:
- hecho de plástico de alta resistencia
- se pueda cerrar con una tapa hermética y resistente a las perforaciones, sin que los objetos punzantes puedan salir
- sea vertical y estable durante su uso
- a prueba de fugas,
- y debidamente etiquetado para advertir sobre los residuos peligrosos dentro del contenedor.
- Cuando su contenedor de eliminación de objetos punzantes esté casi lleno, deberá seguir las pautas de su comunidad para la forma correcta de desechar su contenedor de eliminación de objetos punzantes. Puede haber leyes locales o estatales sobre cómo desechar jeringas y agujas. Para obtener más información sobre la eliminación segura de objetos punzantes y para obtener información específica sobre la eliminación de objetos punzantes en el estado en el que vive, visite el sitio web de la FDA en: http://www.fda.gov/safesharpsdisposal.
- No deseche su contenedor de eliminación de objetos punzantes en la basura de su hogar a menos que las pautas de su comunidad lo permitan. No recicle su contenedor de eliminación de objetos punzantes.
- Tire el vial al contenedor donde puso las jeringas y las agujas.
- Si tiene alguna pregunta, hable con su médico o farmacéutico.
Mantenga STELARA y todos los medicamentos fuera del alcance de los niños.
Vial Fabricado por: Janssen Biotech, Inc., Horsham, PA 19044, EE. UU. Licencia No. 1864 en Cilag AG, Schaffhausen, Suiza
Estas instrucciones de uso han sido aprobadas por la Administración de Alimentos y Medicamentos de EE. UU.
Revisado: 03/2020
© 2012, 2016 Janssen Pharmaceutical Companies
PRINCIPAL PANEL DE PRESENTACIÓN – Vial de 45 mg/0.5 mL en caja
Stelara®
(ustekinumab)
Inyección
45 mg/0.5 mL
Para uso subcutáneo
Contiene una jeringa precargada de 45 mg/0.5 mL
NDC 57894-060-03
Jeringa precargada de dosis única – Deseche la porción no utilizada
La jeringa precargada de 45 mg contiene:
45 mg de ustekinumab, 0.5 mg de L-histidina y L-histidina
monohidrocloruro monohidrato, 0.02 mg de polisorbato 80,
y 38 mg de sacarosa para completar un volumen final de 0.5 mL
Consulte el prospecto para obtener información sobre la dosificación
Rx solamente
ATENCIÓN: Entregue la Guía del medicamento adjunta
a cada paciente.
© 2009 Janssen

PANEL PRINCIPAL DE PRESENTACIÓN – Jeringa de 90 mg/mL, Caja
Stelara®
(ustekinumab)
Inyección
90 mg/mL
Para uso subcutáneo
Contiene una jeringa de 90 mg/mL
NDC 57894-061-03
Jeringa precargada de dosis única – Deseche la porción no utilizada
La jeringa precargada de 90 mg contiene:
90 mg de ustekinumab, 1 mg de L-histidina y L-histidina
monohidrocloruro monohidrato, 0,04 mg de polisorbato 80,
y 76 mg de sacarosa para completar un volumen final de 1 mL
Consulte el prospecto para obtener información sobre la dosificación
Rx solamente
ATENCIÓN: Entregue la Guía del medicamento adjunta
a cada paciente.
© 2009 Janssen
PRINCIPAL PANEL DE PRESENTACIÓN – Vial de 130 mg/26 mL en caja
NDC 57894-054-27
Vial de dosis única
Deseche la porción no utilizada
Stelara®
(ustekinumab)
Solución inyectable
130 mg/26 mL
(5 mg/mL)
Sólo para infusión intravenosa
Debe diluirse
ATENCIÓN: Entregue
la Guía del medicamento adjunta
a cada paciente.
Rx solamente
janssen
