Fabricante de medicamentos: Mylan Pharmaceuticals Inc. (Updated: 2024-07-26)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
Tabletas de SORAFENIB, para uso oral
Aprobación inicial en EE. UU.: 2005
INDICACIONES Y USO
Las tabletas de sorafenib son un inhibidor de la cinasa indicado para el tratamiento de
DOSIFICACIÓN Y ADMINISTRACIÓN
- •
- La dosis recomendada es de 400 mg por vía oral dos veces al día sin alimentos. (2.1)
FORMAS Y FUERZAS DE DOSIFICACIÓN
Tabletas: 200 mg (3)
CONTRAINDICACIONES
- •
- Las tabletas de sorafenib están contraindicadas en pacientes con hipersensibilidad grave conocida a sorafenib o cualquier otro componente de las tabletas de sorafenib. (4)
- •
- Las tabletas de sorafenib en combinación con carboplatino y paclitaxel están contraindicadas en pacientes con cáncer de pulmón de células escamosas. (4)
ADVERTENCIAS Y PRECAUCIONES
- •
- Eventos cardiovasculares: Considere la interrupción temporal o permanente de las tabletas de sorafenib. (2.2, 5.1)
- •
- Hemorragia: Interrumpa las tabletas de sorafenib si es necesario. (5.2)
- •
- Hipertensión: Controle la presión arterial semanalmente durante las primeras 6 semanas y periódicamente a partir de entonces. Considere la interrupción temporal o permanente para la hipertensión grave o persistente a pesar del tratamiento antihipertensivo. (5.3)
- •
- Toxicidades dermatológicas: Interrupción y/o reducción de la dosis. Interrupción para reacciones graves o persistentes, o si se sospecha síndrome de Stevens-Johnson y necrólisis epidérmica tóxica. (5.4)
- •
- Perforación gastrointestinal: Interrupción de las tabletas de sorafenib. (5.5)
- •
- Riesgo de deterioro de la cicatrización de heridas: Suspenda las tabletas de sorafenib durante al menos 10 días antes de la cirugía electiva. No administre durante al menos 2 semanas después de una cirugía mayor y hasta que la cicatrización de la herida sea adecuada. No se ha establecido la seguridad de la reanudación de las tabletas de sorafenib después de la resolución de las complicaciones de la cicatrización de la herida. (5.7)
- •
- Prolongación del QT: Controle los electrocardiogramas y los electrolitos en pacientes con riesgo aumentado de arritmias ventriculares. Corrija los electrolitos. Interrupción si el QTc es mayor de 500 mseg o aumenta más de 60 mseg con respecto al valor inicial. (2.2, 5.9, 12.2)
- •
- Lesión hepática inducida por fármacos: Controle las pruebas de función hepática regularmente; suspenda para elevaciones inexplicables de las transaminasas. (5.10)
- •
- Toxicidad embrio-fetal: Sorafenib puede causar daño fetal. Avise a las pacientes del riesgo potencial para un feto y de que usen métodos anticonceptivos eficaces. (5.11, 8.1, 8.3)
- •
- Deterioro de la supresión de la hormona estimulante del tiroides (TSH) en el CDT: Controle la TSH mensualmente y ajuste la terapia de reemplazo tiroideo en pacientes con cáncer de tiroides. (5.12)
REACCIONES ADVERSAS
Las reacciones adversas más comunes (≥ 20%) son diarrea, fatiga, infección, alopecia, reacción cutánea mano-pie, erupción cutánea, pérdida de peso, disminución del apetito, náuseas, dolores gastrointestinales y abdominales, hipertensión y hemorragia. (6)
Para informar sobre REACCIONES ADVERSAS SOSPECHOSAS, póngase en contacto con Mylan al 1-877-446-3679 (1-877-4-INFO-RX) o con la FDA al 1-800-FDA-1088 o www.fda.gov/medwatch.
INTERACCIONES MEDICAMENTOSAS
- •
- Inductores fuertes del CYP3A: Evite los inductores fuertes del CYP3A4. (7.1)
Ver 17 para INFORMACIÓN DE ASESORAMIENTO AL PACIENTE y etiquetado del paciente aprobado por la FDA.
Revisado: 7/2024
Tabla de Contenido
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDO*
1 INDICACIONES Y USO
1.1 Carcinoma Hepatocelular
1.2 Carcinoma de Células Renales
1.3 Carcinoma Tiroideo Diferenciado
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Dosis Recomendada
2.2 Modificaciones de la Dosis para Reacciones Adversas
3 FORMAS Y FUERZAS DE DOSIFICACIÓN
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Eventos Cardiovasculares
5.2 Hemorragia
5.3 Hipertensión
5.4 Toxicidades Dermatológicas
5.5 Perforación Gastrointestinal
5.6 Aumento del Riesgo de Sangrado con el Uso Concomitante de Warfarina
5.7 Riesgo de Deterioro de la Cicatrización de Heridas
5.8 Aumento de la Mortalidad Observada con Sorafenib Administrado en Combinación con Carboplatino/Paclitaxel y Gemcitabina/Cisplatino en Cáncer de Pulmón de Células Escamosas
5.9 Prolongación del Intervalo QT
5.10 Lesión Hepática Inducida por Fármacos
5.11 Toxicidad Embriofetal
5.12 Deterioro de la Supresión de la Hormona Estimulante de la Tiroides en el Carcinoma Tiroideo Diferenciado
6 REACCIONES ADVERSAS
6.1 Experiencia en Ensayos Clínicos
6.2 Experiencia Postcomercialización
7 INTERACCIONES MEDICAMENTOSAS
7.1 Efecto de Otros Fármacos en Sorafenib
7.2 Uso Concomitante de Warfarina
7.3 Fármacos que Prolongan el Intervalo QT
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.3 Mujeres y Hombres en Potencial Reproductivo
8.4 Uso Pediátrico
8.5 Uso Geriátrico
8.6 Insuficiencia Renal
8.7 Insuficiencia Hepática
10 SOBREDOSIS
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de Acción
12.2 Farmacodinamia
12.3 Farmacocinética
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
14 ESTUDIOS CLÍNICOS
14.1 Carcinoma Hepatocelular
14.2 Carcinoma de Células Renales
14.3 Carcinoma Tiroideo Diferenciado
16 FORMA DE SUMINISTRO/ALMACENAMIENTO Y MANEJO
17 INFORMACIÓN PARA EL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información completa de prescripción no están enumeradas.
1 INDICACIONES Y USO
1.1 Carcinoma Hepatocelular
Las tabletas de Sorafenib están indicadas para el tratamiento de pacientes con carcinoma hepatocelular (HCC) irresecable.
1.2 Carcinoma de Células Renales
Las tabletas de Sorafenib están indicadas para el tratamiento de pacientes con carcinoma de células renales (RCC) avanzado.
1.3 Carcinoma Diferenciado de Tiroides
Las tabletas de Sorafenib están indicadas para el tratamiento de pacientes con carcinoma diferenciado de tiroides (DTC) localmente recurrente o metastásico, progresivo, que es refractario al tratamiento con yodo radiactivo.
2 DOSIS Y ADMINISTRACIÓN
2.1 Dosis Recomendada
La dosis recomendada de tabletas de sorafenib es de 400 mg por vía oral dos veces al día sin alimentos (al menos 1 hora antes o 2 horas después de una comida) hasta que el paciente deje de beneficiarse clínicamente de la terapia o hasta que se presente una toxicidad inaceptable.
2.2 Modificaciones de la Dosis por Reacciones Adversas
Recommended Dosage Modifications
Las modificaciones de dosis recomendadas para las reacciones adversas se proporcionan en las Tablas 1, 2 y 3.
|
Dose Reduction |
Hepatocellular Carcinoma and Renal Cell Carcinoma |
Differentiated Thyroid Carcinoma |
|
Primera Reducción de Dosis |
400 mg por vía oral una vez al día |
400 mg por vía oral por la mañana y 200 mg por vía oral por la noche con un intervalo de aproximadamente 12 horas O 200 mg por vía oral por la mañana y 400 mg por vía oral por la noche con un intervalo de aproximadamente 12 horas |
|
Segunda Reducción de Dosis |
200 mg por vía oral una vez al día O 400 en días alternos |
200 mg por vía oral dos veces al día |
|
Tercera Reducción de Dosis |
Ninguna |
200 mg por vía oral una vez al día |
|
||
|
Reacción adversa |
Severidad* |
Modificación de la dosis de tabletas de Sorafenib |
|
Eventos cardiovasculares [ver Advertencias y precauciones (5.1)] |
||
|
Isquemia e/o infarto cardíaco |
Grado 2 y superior |
Suspender permanentemente. |
|
Insuficiencia cardíaca congestiva |
Grado 3 |
Interrumpir† hasta Grado 1 o menos, reanudar con una dosis reducida en 1 nivel de dosis.‡ |
|
Grado 4 |
Suspender permanentemente. |
|
|
Hemorragia [ver Advertencias y precauciones (5.2)] |
Grado 2 y superior que requiere intervención médica |
Suspender permanentemente. |
|
Hipertensión [ver Advertencias y precauciones (5.3)] |
Grado 2 (sintomático/persistente) O Aumento sintomático de Grado 2 en más de 20 mm Hg (diastólica) o mayor de 140/90 mm Hg si previamente estaba dentro de los límites normales O Grado 3 |
Interrumpir hasta que los síntomas se resuelvan y la presión arterial diastólica sea inferior a 90 mm Hg, luego reanudar con una dosis reducida en 1 nivel de dosis.‡
Si es necesario, reduzca otro nivel de dosis.‡ |
|
Grado 4 |
Suspender permanentemente. |
|
|
Perforación gastrointestinal [ver Advertencias y precauciones (5.5)] |
Cualquier grado |
Suspender permanentemente. |
|
Prolongación del intervalo QT [ver Advertencias y precauciones (5.9)] |
Mayor de 500 milisegundos O Aumento desde el valor inicial de 60 milisegundos o mayor |
Interrumpir y corregir las anomalías electrolíticas (magnesio, potasio, calcio). Utilizar criterio médico antes de reiniciar. |
|
Lesión hepática inducida por fármacos [ver Advertencias y precauciones (5.10)] |
ALT de Grado 3 o superior en ausencia de otra causa§ O AST/ALT mayor de 3 x límite superior normal (LSN) con bilirrubina mayor de 2 x LSN en ausencia de otra causa§ |
Suspender permanentemente. |
|
Toxicidades no hematológicas [ver Reacciones adversas (6.1)] |
Grado 2 |
Continuar el tratamiento con una dosis reducida en 1 nivel de dosis. |
|
Grado 3 |
||
|
1ra aparición |
Interrumpir hasta el Grado 2 o menor, luego reanudar con una dosis reducida en 1 nivel de dosis. |
|
|
Sin mejora dentro de los 7 días O 2da o 3ra aparición |
Interrumpir hasta el Grado 2 o menor, luego reanudar con una dosis reducida en 2 niveles de dosis. |
|
|
4ta aparición |
Interrumpir hasta el Grado 2 o menor, luego reanudar con una dosis reducida en 2 niveles de dosis para HCC y RCC o 3 niveles de dosis para DTC. |
|
|
Grado 4 |
Interrumpir permanentemente. |
|
|
Grado de Toxicidad Dermatológica |
Aparición |
Modificación de la dosis de tabletas de Sorafenib |
|
|
Carcinoma Hepatocelular y Renal |
Carcinoma Diferenciado de Tiroides |
||
|
Grado 2: Eritema doloroso e hinchazón de las manos o los pies y/o molestias que afectan las actividades normales del paciente |
1ra aparición |
Continuar con las tabletas de sorafenib y considerar la terapia tópica para el alivio sintomático. Si no hay mejoría dentro de los 7 días, ver a continuación. |
Disminuir las tabletas de sorafenib a 600 mg al día. Si no hay mejoría dentro de los 7 días, ver a continuación. |
|
Sin mejora dentro de los 7 días con la dosis reducida O 2da y 3ra aparición |
Interrumpir las tabletas de sorafenib hasta que se resuelva o mejore al Grado 0 a 1. |
Interrumpir las tabletas de sorafenib hasta que se resuelva completamente o mejore al Grado 1. |
|
|
Al reanudar el tratamiento, disminuir la dosis en 1 nivel de dosis. |
Al reanudar el tratamiento, disminuir la dosis en 1 nivel de dosis para la 2da aparición y 2 niveles de dosis para la 3ra aparición. |
||
|
4ta aparición |
Interrumpir el tratamiento con tabletas de sorafenib. |
||
|
Grado 3: Descamación húmeda, ulceración, ampollas o dolor intenso en las manos o los pies, lo que resulta en la incapacidad para trabajar o realizar las actividades de la vida diaria |
1ra aparición |
Interrumpir las tabletas de sorafenib hasta que se resuelva o mejore al Grado 0 a 1. |
Interrumpir las tabletas de sorafenib hasta que se resuelva completamente o mejore al Grado 1. |
|
Al reanudar el tratamiento, disminuir la dosis en 1 nivel de dosis. |
Al reanudar el tratamiento, disminuir la dosis en 1 nivel de dosis. |
||
|
2da aparición |
Interrumpir las tabletas de sorafenib hasta que se resuelva o mejore al Grado 0 a 1. |
Interrumpir las tabletas de sorafenib hasta que se resuelva completamente o mejore al Grado 1. |
|
|
Al reanudar el tratamiento, disminuir la dosis en 1 nivel de dosis. |
Al reanudar el tratamiento, disminuir la dosis en 2 niveles de dosis. |
||
|
3ra aparición |
Interrumpir el tratamiento con tabletas de sorafenib. |
||
Después de la mejora de la toxicidad dermatológica de Grado 2 o 3 al Grado 0 o 1 durante al menos 28 días con una dosis reducida de tabletas de sorafenib, la dosis de tabletas de sorafenib puede aumentarse 1 nivel de dosis a partir de la dosis reducida. Se espera que aproximadamente el 50% de los pacientes que requieren una reducción de la dosis por toxicidad dermatológica cumplan con estos criterios para la reanudación de la dosis más alta y se espera que aproximadamente el 50% de los pacientes que reanudan la dosis anterior toleren la dosis más alta (es decir, mantengan el nivel de dosis más alto sin toxicidad dermatológica recurrente de Grado 2 o superior).
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
Las tabletas de Sorafenib, USP están disponibles en presentación de 200 mg de sorafenib, equivalente a 274 mg de sorafenib tosilato, USP.
- •
- Las tabletas de 200 mg son de color durazno, recubiertas con película, redondas, sin ranura, con 200 grabado en un lado de la tableta y NAT en el otro lado.
4 CONTRAINDICACIONES
- •
- Las tabletas de sorafenib están contraindicadas en pacientes con hipersensibilidad grave conocida a sorafenib o cualquier otro componente de las tabletas de sorafenib.
- •
- Las tabletas de sorafenib en combinación con carboplatino y paclitaxel están contraindicadas en pacientes con cáncer de pulmón de células escamosas [ver Advertencias y precauciones (5.8)].
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Eventos Cardiovasculares
En el estudio SHARP (HCC), la incidencia de isquemia/infarto cardíaco fue del 2,7% en los pacientes tratados con sorafenib en comparación con el 1,3% en los que recibieron placebo; en el estudio TARGET (RCC), la incidencia de isquemia/infarto cardíaco fue mayor en el grupo tratado con sorafenib (2,9%) en comparación con los pacientes que recibieron placebo (0,4%), y en el estudio DECISION (DTC), la incidencia de isquemia/infarto cardíaco fue del 1,9% en el grupo tratado con sorafenib en comparación con el 0% en los pacientes que recibieron placebo. Los pacientes con enfermedad coronaria inestable o infarto de miocardio reciente fueron excluidos de este estudio. En múltiples ensayos clínicos, se ha informado insuficiencia cardíaca congestiva en el 1,9% de los pacientes tratados con sorafenib (N = 2276) [ver Reacciones adversas (6.2)].
Considere la interrupción temporal o permanente de sorafenib en pacientes que desarrollan eventos cardiovasculares [ver Dosis y administración (2.2)].
5.2 Hemorragia
Puede ocurrir un mayor riesgo de sangrado después de la administración de sorafenib. En el estudio SHARP (HCC), las tasas de sangrado por varices esofágicas (2,4% y 4%) y de sangrado con desenlace fatal desde cualquier sitio (2,4% y 4%) fueron similares en los pacientes tratados con sorafenib y en los que recibieron placebo, respectivamente. En el estudio TARGET (RCC), se informó sangrado en el 15,3% de los pacientes en el grupo tratado con sorafenib y en el 8,2% de los pacientes que recibieron placebo. La incidencia de sangrado de grado 3 y 4 fue del 2% y el 0%, respectivamente, en los pacientes tratados con sorafenib, y del 1,3% y el 0,2%, respectivamente, en los que recibieron placebo. Hubo una hemorragia fatal en cada grupo de tratamiento en el estudio TARGET (RCC). En el estudio DECISION (DTC), se informó sangrado en el 17,4% de los pacientes tratados con sorafenib y en el 9,6% de los que recibieron placebo; sin embargo, la incidencia de sangrado de grado 3 fue similar (1% y 1,4%) en los pacientes tratados con sorafenib y en los que recibieron placebo.
Si algún sangrado requiere intervención médica, considere la interrupción permanente de sorafenib [ver Dosis y administración (2.2)]. Debido al riesgo potencial de sangrado, trate la infiltración traqueal, bronquial y esofágica con terapia local antes de administrar tabletas de sorafenib en pacientes con DTC.
5.3 Hipertensión
En el estudio SHARP (HCC), se informó hipertensión en el 9,4% de los pacientes tratados con sorafenib y en el 4,3% de los pacientes que recibieron placebo. En el estudio TARGET (RCC), se informó hipertensión en el 16,9% de los pacientes tratados con sorafenib y en el 1,8% de los pacientes que recibieron placebo. En el estudio DECISION (DTC), se informó hipertensión en el 40,6% de los pacientes tratados con sorafenib y en el 12,4% de los pacientes que recibieron placebo. La hipertensión fue generalmente leve a moderada, ocurrió al principio del tratamiento y se controló con terapia antihipertensiva estándar. La interrupción permanente debido a la hipertensión ocurrió en 1 de 297 pacientes tratados con sorafenib en el estudio SHARP (HCC), 1 de 451 pacientes tratados con sorafenib en el estudio TARGET (RCC) y 1 de 207 pacientes tratados con sorafenib en el estudio DECISION (DTC).
Controle la presión arterial semanalmente durante las primeras 6 semanas de sorafenib. A partir de entonces, controle la presión arterial y trate la hipertensión, si es necesario, de acuerdo con la práctica médica estándar. En casos de hipertensión grave o persistente a pesar de la institución de terapia antihipertensiva, considere la interrupción temporal o permanente de las tabletas de sorafenib [ver Dosis y administración (2.2)].
5.4 Toxicidades Dermatológicas
La reacción cutánea mano-pie y la erupción representan las reacciones adversas más comunes atribuidas a sorafenib. La erupción y la reacción cutánea mano-pie suelen ser de grado 1 y 2 y generalmente aparecen durante las primeras seis semanas de tratamiento con sorafenib. La interrupción permanente de la terapia debido a la reacción cutánea mano-pie ocurrió en 4 (1,3%) de 297 pacientes tratados con sorafenib con HCC, 3 (0,7%) de 451 pacientes tratados con sorafenib con RCC y 11 (5,3%) de 207 pacientes tratados con sorafenib con DTC.
El manejo de las toxicidades dermatológicas puede incluir terapias tópicas para el alivio sintomático, la interrupción temporal del tratamiento y/o la reducción de la dosis de sorafenib, o en casos graves o persistentes, la interrupción permanente de sorafenib [ver Dosis y administración (2.2)].
Ha habido informes de toxicidades dermatológicas graves, incluido el síndrome de Stevens-Johnson (SJS) y la necrólisis epidérmica tóxica (TEN). Estos casos pueden ser potencialmente mortales. Suspenda sorafenib si se sospecha SJS o TEN.
5.5 Perforación Gastrointestinal
Se ha informado perforación gastrointestinal en menos del 1% de los pacientes que toman sorafenib. En algunos casos, esto no estuvo asociado con un tumor intraabdominal aparente. En caso de perforación gastrointestinal, suspenda permanentemente sorafenib.
5.6 Mayor riesgo de sangrado con el uso concomitante de warfarina
Se ha informado sangrado infrecuente o elevaciones en la Razón Internacional Normalizada (INR) en algunos pacientes que toman warfarina mientras están en sorafenib. Controle a los pacientes que toman warfarina concomitante regularmente para detectar cambios en el tiempo de protrombina (PT), INR o episodios de sangrado clínico.
5.7 Riesgo de cicatrización de heridas deteriorada
La cicatrización de heridas deteriorada puede ocurrir en pacientes que reciben medicamentos que inhiben la vía de señalización VEGF. Por lo tanto, sorafenib tiene el potencial de afectar negativamente la cicatrización de heridas.
Retenga sorafenib durante al menos 10 días antes de la cirugía electiva. No administre durante al menos 2 semanas después de una cirugía mayor y hasta que la cicatrización de la herida sea adecuada. No se ha establecido la seguridad de la reanudación de sorafenib después de la resolución de las complicaciones de la cicatrización de la herida.
5.8 Aumento de la Mortalidad Observada con Sorafenib Administrado en Combinación con Carboplatino/Paclitaxel y Gemcitabina/Cisplatino en Cáncer de Pulmón de Células Escamosas
En un análisis de subgrupos de dos ensayos controlados aleatorizados en pacientes con cáncer de pulmón de células no pequeñas en estadio IIIB-IV que no habían recibido quimioterapia, los pacientes con carcinoma de células escamosas experimentaron una mayor mortalidad con la adición de sorafenib en comparación con aquellos tratados solo con carboplatino/paclitaxel (HR 1,81; IC del 95% 1,19, 2,74) y gemcitabina/cisplatino solo (HR 1,22; IC del 95% 0,82, 1,80). El uso de sorafenib en combinación con carboplatino/paclitaxel está contraindicado en pacientes con cáncer de pulmón de células escamosas. No se recomienda sorafenib en combinación con gemcitabina/cisplatino en pacientes con cáncer de pulmón de células escamosas. No se ha establecido la seguridad y eficacia de sorafenib en pacientes con cáncer de pulmón de células no pequeñas.
5.9 Prolongación del Intervalo QT
Sorafenib puede prolongar el intervalo QT/QTc. La prolongación del intervalo QT/QTc aumenta el riesgo de arritmias ventriculares.
Evite sorafenib en pacientes con síndrome de QT largo congénito. Controle los electrolitos y los electrocardiogramas en pacientes con insuficiencia cardíaca congestiva, bradiarritmias, medicamentos que se sabe que prolongan el intervalo QT, incluidos los antiarrítmicos de clase Ia y III. Corrija las anomalías electrolíticas (magnesio, potasio, calcio). Interrumpa sorafenib si el intervalo QTc es mayor de 500 milisegundos o para un aumento desde el valor inicial de 60 milisegundos o más [ver Farmacología Clínica (12.2)].
5.10 Lesión Hepática Inducida por Medicamentos
La hepatitis inducida por sorafenib se caracteriza por un patrón hepatocelular de daño hepático con aumentos significativos de las transaminasas que pueden provocar insuficiencia hepática y muerte. También pueden ocurrir aumentos en la bilirrubina y el INR. La incidencia de lesión hepática inducida por medicamentos grave, definida como niveles elevados de transaminasas por encima de 20 veces el límite superior de lo normal o elevaciones de transaminasas con secuelas clínicas significativas (por ejemplo, INR elevado, ascitis, fatal o trasplante), fue de dos de 3.357 pacientes (0,06%) en una base de datos global de monoterapia.
Controle las pruebas de función hepática regularmente. En caso de aumento significativo de las transaminasas sin explicación alternativa, como hepatitis viral o progresión de la malignidad subyacente, suspenda sorafenib [ver Dosificación y Administración (2.2)].
5.11 Toxicidad Embriofetal
Con base en su mecanismo de acción y los hallazgos en animales, sorafenib puede causar daño fetal cuando se administra a una mujer embarazada. Sorafenib causó toxicidades embriofetales en animales a exposiciones maternas que fueron significativamente más bajas que las exposiciones humanas a la dosis recomendada de 400 mg dos veces al día. Avise a las mujeres embarazadas y a las mujeres en edad fértil sobre el riesgo potencial para el feto. Avise a las mujeres en edad fértil que usen métodos anticonceptivos efectivos durante el tratamiento y durante 6 meses después de la última dosis de sorafenib. Avise a los pacientes masculinos con parejas femeninas en edad fértil y parejas embarazadas que usen métodos anticonceptivos efectivos durante el tratamiento y durante 3 meses después de la última dosis de sorafenib [ver Uso en Poblaciones Específicas (8.1, 8.3)].
5.12 Deterioro de la Supresión de la Hormona Estimulante de la Tiroides en el Carcinoma Tiroideo Diferenciado
Sorafenib deteriora la supresión tiroidea exógena. En el estudio DECISION (DTC), el 99% de los pacientes tenían un nivel basal de hormona estimulante de la tiroides (TSH) inferior a 0,5 mU/L. Se observó una elevación del nivel de TSH por encima de 0,5 mU/L en el 41% de los pacientes tratados con sorafenib en comparación con el 16% de los que recibieron placebo. Para los pacientes con supresión de TSH deteriorada mientras reciben sorafenib, la TSH máxima mediana fue de 1,6 mU/L y el 25% tuvo niveles de TSH superiores a 4,4 mU/L.
Controle los niveles de TSH mensualmente y ajuste la medicación de reemplazo tiroideo según sea necesario en pacientes con DTC.
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas clínicamente significativas se discuten en otra parte de la etiqueta:
- •
- Eventos cardiovasculares [ver Advertencias y precauciones (5.1)]
- •
- Hemorragia [ver Advertencias y precauciones (5.2)]
- •
- Hipertensión [ver Advertencias y precauciones (5.3)]
- •
- Toxicidades dermatológicas [ver Advertencias y precauciones (5.4)]
- •
- Perforación gastrointestinal [ver Advertencias y precauciones (5.5)]
- •
- Prolongación del intervalo QT [ver Advertencias y precauciones (5.9) y Farmacología clínica (12.2)]
- •
- Lesión hepática inducida por fármacos [ver Advertencias y precauciones (5.10)]
- •
- Deterioro de la supresión de TSH en DTC [ver Advertencias y precauciones (5.12)]
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se llevan a cabo en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no se pueden comparar directamente con las tasas en los ensayos clínicos de otro fármaco y pueden no reflejar las tasas observadas en la práctica.
Los datos descritos reflejan la exposición a sorafenib en 955 pacientes que participaron en estudios controlados con placebo en carcinoma hepatocelular (N = 297), carcinoma de células renales avanzado (N = 451) o carcinoma diferenciado de tiroides (N = 207). Las reacciones adversas más comunes (≥ 20%), que se consideraron relacionadas con sorafenib, en pacientes con HCC, RCC o DTC son diarrea, fatiga, infección, alopecia, reacción cutánea mano-pie, erupción cutánea, pérdida de peso, disminución del apetito, náuseas, dolores gastrointestinales y abdominales, hipertensión y hemorragia.
Carcinoma hepatocelular
La Tabla 4 muestra el porcentaje de pacientes en el estudio SHARP (HCC) que experimentaron reacciones adversas que se informaron en al menos el 10% de los pacientes y a una tasa más alta en el grupo tratado con sorafenib que en aquellos que recibieron placebo.
|
||||||
|
Tabletas de sorafenib N = 297 |
Placebo N = 302 |
|||||
|
Reacción adversa* |
Todos los grados % |
Grado 3 % |
Grado 4 % |
Todos los grados % |
Grado 3 % |
Grado 4 % |
|
Cualquier reacción adversa |
98 |
39 |
6 |
96 |
24 |
8 |
|
Gastrointestinal |
||||||
|
Diarrea |
55 |
10 |
< 1 |
25 |
2 |
0 |
|
Anorexia |
29 |
3 |
0 |
18 |
3 |
< 1 |
|
Náuseas |
24 |
1 |
0 |
20 |
3 |
0 |
|
Vómitos |
15 |
2 |
0 |
11 |
2 |
0 |
|
Estreñimiento |
14 |
0 |
0 |
10 |
0 |
0 |
|
Síntomas constitucionales |
||||||
|
Fatiga |
46 |
9 |
1 |
45 |
12 |
2 |
|
Pérdida de peso |
30 |
2 |
0 |
10 |
1 |
0 |
|
Dolor |
||||||
|
Dolor abdominal |
31 |
9 |
0 |
26 |
5 |
1 |
|
Dermatología/piel |
||||||
|
Reacción cutánea mano-pie |
21 |
8 |
0 |
3 |
< 1 |
0 |
|
Erupción/descamación |
19 |
1 |
0 |
14 |
0 |
0 |
|
Alopecia |
14 |
0 |
0 |
2 |
0 |
0 |
|
Prurito |
14 |
< 1 |
0 |
11 |
< 1 |
0 |
|
Piel seca |
10 |
0 |
0 |
6 |
0 |
0 |
|
Hepatobiliar/páncreas |
||||||
|
Disfunción hepática |
11 |
2 |
1 |
8 |
2 |
1 |
Se informó hipertensión en el 9% de los pacientes tratados con sorafenib y en el 4% de los que recibieron placebo. Se informó hipertensión de grado 3 en el 4% de los pacientes tratados con sorafenib y en el 1% de los que recibieron placebo.
Se informó hemorragia/sangrado en el 18% de los que recibieron sorafenib y en el 20% de los pacientes que recibieron placebo. Las tasas de sangrado de grado 3 y 4 también fueron más altas en los pacientes que recibieron placebo (grado 3: 3% sorafenib y 5% placebo y grado 4: 2% sorafenib y 4% placebo).
Se informó sangrado por varices esofágicas en el 2,4% de los pacientes tratados con sorafenib y en el 4% de los pacientes que recibieron placebo.
Se informó insuficiencia renal en < 1% de los pacientes tratados con sorafenib y en el 3% de los pacientes que recibieron placebo. Se informó pancreatitis clínica en 1 de 297 pacientes tratados con sorafenib (grado 2).
La tasa de reacciones adversas (incluidas las asociadas con la enfermedad progresiva) que provocaron la interrupción permanente fue similar tanto en los pacientes tratados con sorafenib como en los que recibieron placebo (32% de los pacientes tratados con sorafenib y 35% de los pacientes que recibieron placebo).
Las anomalías en las pruebas de laboratorio informadas en SHARP se presentan en la Tabla 5.
| NR = no reportado | ||||
|
||||
|
Parámetro de laboratorio* |
Tabletas de Sorafenib N = 297 |
Placebo N = 302 |
||
|
Todos los grados (%) |
Grado 3 o 4 (%) |
Todos los grados (%) |
Grado 3 o 4 (%) |
|
|
Hipoalbuminemia |
59 |
0 |
47 |
0 |
|
Liasa elevada |
40 |
9 |
37 |
9 |
|
Linfopenia |
47 |
NR |
42 |
NR |
|
Trombocitopenia |
46 |
4 |
41 |
< 1 |
|
INR elevado |
42 |
4 |
34 |
2 |
|
Hipofosfatemia |
35 |
11 |
11 |
2 |
|
Amilasa elevada |
34 |
2 |
29 |
2 |
|
Hipocalcemia |
27 |
2.4 |
15 |
1 |
|
Hipopotasemia |
10 |
< 1 |
6 |
< 1 |
Carcinoma de células renales
La Tabla 6 muestra el porcentaje de pacientes en el estudio TARGET (RCC) que experimentaron reacciones adversas que se informaron en al menos el 10% de los pacientes y a una tasa más alta en el brazo de pacientes tratados con sorafenib que en aquellos que recibieron placebo.
La tasa de reacciones adversas (incluidas las asociadas con la enfermedad progresiva) que provocaron la interrupción permanente fue similar tanto en los pacientes tratados con sorafenib como en los pacientes que recibieron placebo (10% y 8%, respectivamente).
Se informó pancreatitis clínica en 3 de 451 pacientes tratados con sorafenib (un Grado 2 y dos Grado 4).
|
||||||
|
Tabletas de Sorafenib N = 451 |
Placebo N = 451 |
|||||
|
Reacción adversa* |
Todos los grados % |
Grado 3 % |
Grado 4 % |
Todos los grados % |
Grado 3 % |
Grado 4 % |
|
Cualquier reacción adversa |
95 |
31 |
7 |
86 |
22 |
6 |
|
Síntomas gastrointestinales |
||||||
|
Diarrea |
43 |
2 |
0 |
13 |
< 1 |
0 |
|
Náuseas |
23 |
< 1 |
0 |
19 |
< 1 |
0 |
|
Anorexia |
16 |
< 1 |
0 |
13 |
1 |
0 |
|
Vómitos |
16 |
< 1 |
0 |
12 |
1 |
0 |
|
Constipation |
15 |
< 1 |
0 |
11 |
< 1 |
0 |
|
Dermatology/skin |
||||||
|
Rash/desquamation |
40 |
< 1 |
0 |
16 |
< 1 |
0 |
|
Hand-foot skin reaction |
30 |
6 |
0 |
7 |
0 |
0 |
|
Alopecia |
27 |
< 1 |
0 |
3 |
0 |
0 |
|
Pruritus |
19 |
< 1 |
0 |
6 |
0 |
0 |
|
Dry skin |
11 |
0 |
0 |
4 |
0 |
0 |
|
Constitutional symptoms |
||||||
|
Fatigue |
37 |
5 |
< 1 |
28 |
3 |
< 1 |
|
Weight loss |
10 |
< 1 |
0 |
6 |
0 |
0 |
|
Cardiovascular, General |
||||||
|
Hipertensión |
17 |
3 |
< 1 |
2 |
< 1 |
0 |
|
Hemorragia/sangrado |
||||||
|
Hemorragia – todos los sitios |
15 |
2 |
0 |
8 |
1 |
< 1 |
|
Pulmonar |
||||||
|
Disnea |
14 |
3 |
< 1 |
12 |
2 |
< 1 |
|
Neurología |
||||||
|
Neuropatía-sensorial |
13 |
< 1 |
0 |
6 |
< 1 |
0 |
|
Dolor |
||||||
|
Dolor, abdomen |
11 |
2 |
0 |
9 |
2 |
0 |
|
Dolor, cefalea |
10 |
< 1 |
0 |
6 |
< 1 |
0 |
|
Dolor, articular |
10 |
2 |
0 |
6 |
< 1 |
0 |
Las anormalidades en las pruebas de laboratorio reportadas en TARGET se presentan en la Tabla 7.
|
||||
|
Parámetro de laboratorio* |
Tabletas de Sorafenib N = 451 |
Placebo N = 451 |
||
|
Todos los grados (%) |
Grado 3 o 4 (%) |
Todos los grados (%) |
Grado 3 o 4 (%) |
|
|
Hipo fosfatemia |
45 |
13 |
11 |
3 |
|
Anemia |
44 |
2 |
49 |
4 |
|
Liasa elevada |
41 |
12 |
30 |
7 |
|
Amilasa elevada |
30 |
1 |
23 |
3 |
|
Linfopenia |
23 |
13 |
13 |
7 |
|
Neutropenia |
18 |
5 |
10 |
2 |
|
Trombocitopenia |
12 |
1 |
5 |
0 |
|
Hipocalcemia |
12 |
2 |
8 |
<1 |
|
Hipokalemia |
5 |
1 |
<1 |
<1 |
Carcinoma diferenciado de tiroides
La seguridad de sorafenib se evaluó en DECISION en 416 pacientes con carcinoma diferenciado de tiroides (CDT) localmente recurrente o metastásico, progresivo y refractario al tratamiento con yodo radiactivo (RAI) aleatorizados para recibir 400 mg dos veces al día de sorafenib (n = 207) o placebo coincidente (n = 209) hasta la progresión de la enfermedad o toxicidad intolerable en un ensayo doble ciego [ver Estudios clínicos (14.3)]. Los datos descritos a continuación reflejan una exposición media a sorafenib durante 46 semanas (rango de 0,3 a 135). La población expuesta a sorafenib fue 50% masculina y tenía una edad media de 63 años.
Se requirieron interrupciones de la dosis por reacciones adversas en el 66% de los pacientes que recibieron sorafenib y se requirieron reducciones de la dosis en el 64% de los pacientes. Las reacciones adversas que provocaron la interrupción del tratamiento se informaron en el 14% de los pacientes tratados con sorafenib en comparación con el 1,4% de los pacientes que recibieron placebo.
La tabla 8 muestra el porcentaje de pacientes con CDT que experimentaron reacciones adversas a una tasa más alta en los pacientes tratados con sorafenib que en los pacientes que recibieron placebo en la fase doble ciego del estudio DECISION. Las reacciones adversas de grado 3 ocurrieron en el 53% de los pacientes tratados con sorafenib en comparación con el 23% de los pacientes que recibieron placebo. Las reacciones adversas de grado 4 ocurrieron en el 12% de los pacientes tratados con sorafenib en comparación con el 7% de los pacientes que recibieron placebo.
|
||||
|
Reacción adversa |
Tabletas de sorafenib N = 207 |
Placebo N = 209 |
||
|
Todos los grados (%) |
Grados 3 y 4 (%) |
Todos los grados (%) |
Grados 3 y 4 (%) |
|
|
Trastornos de la piel y del tejido subcutáneo |
||||
|
PPES† |
69 |
19 |
8 |
0 |
|
Alopecia |
67 |
0 |
8 |
0 |
|
Erupción |
35 |
5 |
7 |
0 |
|
Prurito |
20 |
0.5 |
11 |
0 |
|
Piel seca |
13 |
0.5 |
5 |
0 |
|
Eritema |
10 |
0 |
0.5 |
0 |
|
Hiperqueratosis |
7 |
0 |
0 |
0 |
|
Trastornos gastrointestinales |
||||
|
Diarrea |
68 |
6 |
15 |
1 |
|
Estomatitis‡ |
24 |
2 |
3 |
0 |
|
Náuseas |
21 |
0 |
12 |
0 |
|
Dolor abdominal§ |
20 |
1 |
7 |
1 |
|
Estreñimiento |
16 |
0 |
8 |
0.5 |
|
Dolor oral¶ |
14 |
0.5 |
6 |
0 |
|
Vómitos |
11 |
0 |
3 |
0 |
|
Pruebas de laboratorio |
||||
|
Pérdida de peso |
49 |
6 |
14 |
1 |
|
Trastornos generales y condiciones del lugar de administración |
||||
|
Fatiga |
41 |
5 |
20 |
1 |
|
Astenia |
12 |
0 |
7 |
0 |
|
Pirexia |
11 |
1 |
5 |
0 |
|
Trastornos vasculares |
||||
|
Hipertensión# |
41 |
10 |
12 |
2 |
|
Trastornos del metabolismo y la nutrición |
||||
|
Disminución del apetito |
30 |
2 |
5 |
0 |
|
Trastornos del sistema nervioso |
||||
|
Dolor de cabeza |
17 |
0 |
6 |
0 |
|
Disgeusia |
6 |
0 |
0 |
0 |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
||||
|
Dolor en las extremidades |
15 |
1 |
7 |
0 |
|
Espasmos musculares |
10 |
0 |
3 |
0 |
|
Trastornos respiratorios, torácicos y mediastínicos |
||||
|
Disfonía |
13 |
0.5 |
3 |
0 |
|
Epistaxis |
7 |
0 |
1 |
0 |
|
Neoplasias benignas, malignas y no especificadas |
||||
|
Carcinoma de células escamosas de la piel |
3 |
3 |
0 |
0 |
El aumento relativo de las siguientes anormalidades de laboratorio observadas en pacientes tratados con sorafenib en comparación con los pacientes que recibieron placebo en el estudio DECISION es similar al observado en los estudios de RCC y HCC: lipasa, amilasa, hipokalemia, hipofosfatemia, neutropenia, linfopenia, anemia y trombocitopenia. La hipocalcemia fue más frecuente y más grave en pacientes con DTC, especialmente aquellos con antecedentes de hipoparatiroidismo, en comparación con los pacientes con RCC o HCC. Otras anormalidades en las pruebas de laboratorio reportadas en DECISION se presentan en la Tabla 9
|
||||
|
Parámetro de laboratorio* |
Tabletas de Sorafenib N = 207 |
Placebo N = 209 |
||
|
Todos los grados (%) |
Grado 3 o 4 (%) |
Todos los grados (%) |
Grado 3 o 4 (%) |
|
|
ALT elevada |
59 |
4 |
24 |
0 |
|
AST elevada |
54 |
2 |
15 |
0 |
|
Hipocalcemia |
36 |
10 |
11 |
3 |
Datos adicionales de múltiples ensayos clínicos
Las siguientes reacciones adversas relacionadas con el fármaco y las anomalías de laboratorio se informaron en ensayos clínicos de sorafenib (muy frecuentes 10% o más, frecuentes 1 a menos del 10%, poco frecuentes 0.1% a menos del 1%, raras menos del 0.1%):
Cardiovasculares: Frecuentes: insuficiencia cardíaca congestiva*†, isquemia y/o infarto de miocardio Poco frecuentes: crisis hipertensiva* Raras: prolongación del QT*
Dermatológicas: Muy frecuentes: eritema Frecuentes: dermatitis exfoliativa, acné, rubor, foliculitis, hiperqueratosis Poco frecuentes: eczema, eritema multiforme
Digestivas: Muy frecuentes: aumento de la lipasa, aumento de la amilasa Frecuentes: mucositis, estomatitis (incluida la boca seca y la glositis), dispepsia, disfagia, reflujo gastrointestinal Poco frecuentes: pancreatitis, gastritis, perforaciones gastrointestinales*, colecistitis, colangitis
Tenga en cuenta que las elevaciones en la lipasa son muy comunes (41%, ver más abajo); no se debe hacer un diagnóstico de pancreatitis únicamente en base a valores de laboratorio anormales
Trastornos generales: Muy frecuentes: infección, hemorragia (incluida la gastrointestinal* y la del tracto respiratorio* y casos poco frecuentes de hemorragia cerebral*), astenia, dolor (incluido el dolor de boca, hueso y tumor), pirexia, disminución del apetito Frecuentes: enfermedad similar a la gripe
Hematología: Muy frecuentes: leucopenia, linfopenia Frecuentes: anemia, neutropenia, trombocitopenia Poco frecuentes: INR anormal
Trastornos hepatobiliares: Raras: lesión hepática inducida por fármacos (incluida la insuficiencia hepática y la muerte)
Hipersensibilidad: Poco frecuentes: reacciones de hipersensibilidad (incluidas reacciones cutáneas y urticaria), reacción anafiláctica
Metabolismo y nutrición: Muy frecuentes: hipofosfatemia Frecuentes: aumentos transitorios de las transaminasas, hipocalcemia, hipopotasemia, hiponatremia, hipotiroidismo Poco frecuentes: deshidratación, aumentos transitorios de la fosfatasa alcalina, aumento de la bilirrubina (incluida la ictericia), hipertiroidismo
Musculoesquelético: Muy frecuentes: artralgia Frecuentes: mialgia, espasmos musculares
Sistema nervioso y psiquiátrico: Frecuentes: depresión, disgeusia Poco frecuentes: tinnitus, leucoencefalopatía posterior reversible*
Renal y genitourinario: Frecuentes: insuficiencia renal, proteinuria Raras: síndrome nefrótico
Reproductivo: Frecuentes: disfunción eréctil Poco frecuentes: ginecomastia
Respiratorio: Frecuentes: rinorrea Poco frecuentes: eventos similares a la enfermedad pulmonar intersticial (incluye informes de neumonitis, neumonitis por radiación, dificultad respiratoria aguda, neumonía intersticial, pulmonitis e inflamación pulmonar)
Además, las siguientes reacciones adversas médicamente significativas fueron poco frecuentes durante los ensayos clínicos de sorafenib: ataque isquémico transitorio, arritmia y tromboembolismo. Para estas reacciones adversas, no se ha establecido la relación causal con sorafenib.
* las reacciones adversas pueden tener un resultado potencialmente mortal o fatal.
† reportado en el 1.9% de los pacientes tratados con sorafenib (N = 2276).
6.2 Experiencia postcomercialización
Las siguientes reacciones adversas se han identificado durante el uso posterior a la aprobación de sorafenib. Debido a que estas reacciones se informan voluntariamente de una población de tamaño incierto, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal con la exposición al fármaco.
Trastornos de la sangre y del sistema linfático: Microangiopatía trombótica (TMA)
Dermatológicas: Síndrome de Stevens-Johnson y necrólisis epidérmica tóxica (TEN)
Hipersensibilidad: Angioedema
Musculoesquelético: Rabdomiólisis, osteonecrosis de la mandíbula
Respiratorio: Eventos similares a la enfermedad pulmonar intersticial (que pueden tener un resultado potencialmente mortal o fatal)
Vascular: Aneurisma arterial (incluido el aórtico), disecciones y rotura
7 INTERACCIONES MEDICAMENTOSAS
7.1 Efecto de Otros Medicamentos en Sorafenib
Inductores Fuertes de CYP3A4
El uso concomitante de sorafenib con rifampicina, un inductor fuerte de CYP3A4, disminuyó el AUC medio de sorafenib, lo que puede disminuir la actividad antitumoral [ver Farmacología Clínica (12.3)]. Evite el uso concomitante de sorafenib con inductores fuertes de CYP3A4, cuando sea posible, porque estos medicamentos pueden disminuir la exposición sistémica a sorafenib.
Neomicina
El uso concomitante de sorafenib con neomicina disminuyó el AUC medio de sorafenib, lo que puede disminuir la actividad antitumoral. Evite el uso concomitante de sorafenib con neomicina. Los efectos de otros antibióticos sobre la farmacocinética de sorafenib no se han estudiado [ver Farmacología Clínica (12.3)].
7.2 Uso Concomitante de Warfarina
El uso concomitante de sorafenib y warfarina puede aumentar el riesgo de sangrado o aumentar el INR. Controle el INR y los episodios de sangrado clínico en pacientes que toman warfarina mientras reciben sorafenib [ver Advertencias y Precauciones (5.6)].
7.3 Medicamentos que Prolongan el Intervalo QT
Sorafenib se asocia con la prolongación del intervalo QTc. Evite la administración conjunta de sorafenib con medicamentos con un potencial conocido para prolongar el intervalo QT/QTc [ver Advertencias y Precauciones (5.9), Farmacología Clínica (12.2)].
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de Riesgos
Con base en los hallazgos de estudios en animales y su mecanismo de acción [ver Farmacología Clínica (12.1)], sorafenib puede causar daño fetal cuando se administra a una mujer embarazada. No hay datos disponibles en mujeres embarazadas para informar un riesgo asociado con el medicamento. En estudios de reproducción en animales, la administración oral de sorafenib a ratas y conejas embarazadas durante el período de organogénesis resultó en toxicidades embrio-fetales a exposiciones maternas que fueron significativamente más bajas que las exposiciones humanas a la dosis recomendada de 400 mg dos veces al día (ver Datos). Advierta a las mujeres embarazadas y a las mujeres en edad fértil sobre el riesgo potencial para el feto.
El riesgo de fondo de defectos de nacimiento mayores y aborto espontáneo para la población indicada es desconocido. En la población general de los EE. UU., el riesgo de fondo estimado de defectos de nacimiento mayores y aborto espontáneo en embarazos clínicamente reconocidos es del 2% al 4% y del 15% al 20%, respectivamente.
Datos
Datos de animales
En estudios de reproducción en animales, sorafenib fue teratógeno e indujo toxicidad embrio-fetal (incluida una mayor pérdida postimplantación, resorciones, retardos esqueléticos y peso fetal retardado) cuando se administró por vía oral a ratas y conejas embarazadas durante el período de organogénesis. Los efectos ocurrieron a dosis considerablemente inferiores a la dosis humana recomendada de 400 mg dos veces al día (aproximadamente 500 mg/m2/día en base al área de superficie corporal). Se observaron efectos adversos en el desarrollo intrauterino a dosis > 0,2 mg/kg/día (1,2 mg/m2/día) en ratas y ≥ 0,3 mg/kg/día (≥ 3,6 mg/m2/día) en conejos. Estas dosis dan como resultado exposiciones (AUC) que son aproximadamente 0,008 veces el AUC en pacientes a la dosis recomendada.
8.2 Lactancia
Resumen de Riesgos
No hay datos sobre la presencia de sorafenib o sus metabolitos en la leche materna, o sus efectos en el niño amamantado o en la producción de leche. Sorafenib estuvo presente en la leche de ratas lactantes (ver Datos). Debido al potencial de reacciones adversas graves en un niño amamantado por sorafenib, se debe aconsejar a las mujeres que no amamanten durante el tratamiento con sorafenib y durante 2 semanas después de la última dosis.
8.3 Mujeres y Hombres en Edad Fértil
Sorafenib puede causar daño fetal cuando se administra a una mujer embarazada [ver Uso en Poblaciones Específicas (8.1)].
Prueba de Embarazo
Verifique el estado de embarazo de las mujeres en edad fértil antes de iniciar el tratamiento con sorafenib.
Anticoncepción
Mujeres
Aconseje a las mujeres en edad fértil que usen métodos anticonceptivos efectivos durante el tratamiento y durante 6 meses después de la última dosis de sorafenib.
Hombres
Con base en la genotoxicidad y los hallazgos en estudios de reproducción en animales, se debe aconsejar a los hombres con parejas femeninas en edad fértil y parejas embarazadas que usen métodos anticonceptivos efectivos durante el tratamiento con sorafenib y durante 3 meses después de la última dosis de sorafenib [ver Uso en Poblaciones Específicas (8.1), Toxicología No Clínica (13.1)].
Infertilidad
Hombres
Con base en los hallazgos en estudios en animales, sorafenib puede afectar la fertilidad en hombres en edad fértil [ver Toxicología No Clínica (13.1)].
8.4 Uso en poblaciones específicas
No se ha establecido la seguridad y eficacia de sorafenib en pacientes pediátricos.
Datos de toxicidad en animales jóvenes
La dosificación repetida de sorafenib en perros jóvenes y en crecimiento resultó en un engrosamiento irregular de la placa de crecimiento femoral a dosis diarias de sorafenib ≥ 600 mg/m2 (aproximadamente 0.3 veces el AUC a la dosis humana recomendada), hipocelularidad de la médula ósea adyacente a la placa de crecimiento a 200 mg/m2/día (aproximadamente 0.1 veces el AUC a la dosis humana recomendada), y alteraciones de la composición de la dentina a 600 mg/m2/día. No se observaron efectos similares en perros adultos cuando se administraron durante 4 semanas o menos.
8.5 Uso geriátrico
En total, el 59% de los pacientes con HCC tratados con sorafenib tenían 65 años o más y el 19% tenían 75 años o más. En total, el 32% de los pacientes con RCC tratados con sorafenib tenían 65 años o más y el 4% tenían 75 años o más. No se observaron diferencias en la seguridad o eficacia entre los pacientes mayores y los más jóvenes, y otras experiencias clínicas reportadas no han identificado diferencias en las respuestas entre los pacientes ancianos y los más jóvenes, pero no se puede descartar una mayor sensibilidad de algunos individuos mayores.
8.6 Insuficiencia renal
No es necesario ajustar la dosis para pacientes con insuficiencia renal leve, moderada o grave que no estén en diálisis. No se ha estudiado la farmacocinética de sorafenib en pacientes que están en diálisis [ver Farmacología clínica (12.3)].
8.7 Insuficiencia hepática
No es necesario ajustar la dosis para pacientes con insuficiencia hepática leve o moderada. No se ha estudiado la farmacocinética de sorafenib en pacientes con insuficiencia hepática grave (Child-Pugh C) [ver Farmacología clínica (12.3)].
10 SOBREDOSIS
Las reacciones adversas observadas a una dosis de 800 mg dos veces al día (2 veces la dosis recomendada) fueron principalmente diarrea y dermatológicas. No hay información disponible sobre los síntomas de sobredosis aguda en animales debido a la saturación de la absorción en estudios de toxicidad aguda oral realizados en animales.
En caso de sospecha de sobredosis, suspenda sorafenib e instituya cuidados de apoyo.
11 DESCRIPCIÓN
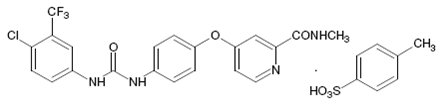
Sorafenib, un inhibidor de la quinasa, es la sal tosilato de sorafenib. El tosilato de sorafenib, USP tiene el nombre químico 4-[4-({[4-cloro-3-(trifluorometil)fenil]carbamoil}amino)fenoxi]-N-metilpiridina-2-carboxamida, tosilato. La fórmula molecular del tosilato de sorafenib, USP es C21H16ClF3N4O3•C7H8O3S y el peso molecular del tosilato de sorafenib, USP es de 637.03 g/mol. Su fórmula estructural es:
El tosilato de sorafenib, USP es un polvo cristalino de color crema a amarillo. El tosilato de sorafenib, USP es prácticamente insoluble en medios acuosos, ligeramente soluble en etanol y soluble en PEG 400.
Los comprimidos de sorafenib, USP para uso oral se suministran como comprimidos recubiertos con película que contienen 200 mg de sorafenib equivalente a 274 mg de tosilato de sorafenib, USP y los siguientes ingredientes inactivos: carboximetilcelulosa cálcica, croscarmelosa sódica, hipromelosa, estearato de magnesio, celulosa microcristalina, polietilenglicol, óxido de hierro rojo, lauril sulfato de sodio y dióxido de titanio.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
Sorafenib es un inhibidor de la quinasa que disminuye la proliferación de células tumorales in vitro. Se demostró que sorafenib inhibe múltiples quinasas intracelulares (c-CRAF, BRAF y BRAF mutante) y de la superficie celular (KIT, FLT-3, RET, RET/PTC, VEGFR-1, VEGFR-2, VEGFR-3 y PDGFR-ß). Se cree que varias de estas quinasas están involucradas en la señalización, angiogénesis y apoptosis de las células tumorales. Sorafenib inhibió el crecimiento tumoral de xenoinjertos de tumores humanos de HCC, RCC y DTC en ratones inmunocomprometidos. Se observaron reducciones en la angiogénesis tumoral en modelos de HCC y RCC tras el tratamiento con sorafenib, y se observaron aumentos en la apoptosis tumoral en modelos de HCC, RCC y DTC.
12.2 Farmacodinámica
Electrofisiología cardíaca
El efecto de sorafenib 400 mg dos veces al día sobre el intervalo QTc se evaluó en un ensayo multicéntrico, abierto, no aleatorizado en 53 pacientes con cáncer avanzado. No se detectaron grandes cambios en los intervalos QTc medios (es decir, > 20 ms) desde el inicio en el ensayo. Después de un ciclo de tratamiento de 28 días, el mayor cambio medio en el intervalo QTc de 8.5 ms (límite superior del intervalo de confianza del 90 % bilateral, 13.3 ms) se observó a las 6 horas posteriores a la dosis el día 1 del ciclo 2 [see Advertencias y precauciones (5.9), Interacciones medicamentosas (7.3)].
12.3 Farmacocinética
Dosis múltiples de sorafenib durante 7 días resultaron en una acumulación de 2.5 a 7 veces en comparación con una dosis única. Las concentraciones plasmáticas de sorafenib en estado estacionario se alcanzaron en 7 días, con una proporción pico-valle de concentraciones medias de menos de 2.
Las concentraciones en estado estacionario de sorafenib después de la administración de sorafenib 400 mg dos veces al día se evaluaron en pacientes con DTC, RCC y HCC. Los pacientes con DTC tienen concentraciones medias en estado estacionario que son 1.8 veces más altas que los pacientes con HCC y 2.3 veces más altas que aquellos con RCC. Se desconoce la razón del aumento de las concentraciones de sorafenib en pacientes con DTC.
La Cmáx y el AUC medios aumentaron menos que proporcionalmente más allá de las dosis orales de 400 mg administradas dos veces al día.
Absorción
Después de la administración de tabletas de sorafenib, la biodisponibilidad relativa media fue del 38-49% en comparación con una solución oral. Después de la administración oral, sorafenib alcanzó niveles plasmáticos máximos en aproximadamente 3 horas.
Eliminación
La vida media de eliminación promedio de sorafenib fue de aproximadamente 25 a 48 horas.
Metabolismo
Sorafenib se somete a metabolismo oxidativo por el CYP3A4 hepático, así como a glucuronidación por UGT1A9.
Excreción
Sorafenib representó aproximadamente del 70 al 85 % de los analitos circulantes en plasma en estado estacionario. Se han identificado ocho metabolitos de sorafenib, de los cuales 5 se han detectado en plasma. El principal metabolito circulante de sorafenib, el N-óxido de piridina que comprende aproximadamente del 9 al 16 % de los analitos circulantes en estado estacionario, mostró una potencia in vitro similar a la de sorafenib.
Después de la administración oral de una dosis de 100 mg de una formulación en solución de sorafenib, el 96 % de la dosis se recuperó en 14 días, con el 77 % de la dosis excretada en las heces y el 19 % de la dosis excretada en la orina como metabolitos glucuronidados. El sorafenib sin cambios, que representa el 51 % de la dosis, se encontró en las heces pero no en la orina.
Poblaciones específicas
Un estudio de la farmacocinética de sorafenib indicó que el AUC medio de sorafenib en asiáticos (N = 78) fue un 30 % menor que en blancos (N = 40). El sexo y la edad no tienen un efecto clínicamente significativo sobre la farmacocinética de sorafenib.
Pacientes con insuficiencia renal
La insuficiencia renal leve (CLcr 50-80 ml/min), moderada (CLcr 30 – < 50 ml/min) y grave (CLcr < 30 ml/min) no afecta la farmacocinética de sorafenib [see Uso en poblaciones específicas (8.6)].
Pacientes con insuficiencia hepática
La insuficiencia hepática leve (Child-Pugh A) y moderada (Child-Pugh B) no afecta la farmacocinética de sorafenib [ver Uso en poblaciones específicas (8.7)].
Estudios de interacciones medicamentosas
Efecto de los inhibidores potentes del CYP3A4 sobre sorafenib
El ketoconazol, un potente inhibidor del CYP3A4 y la glicoproteína P, administrado en una dosis de 400 mg una vez al día durante 7 días no alteró el AUC medio de una dosis oral única de 50 mg de sorafenib en sujetos sanos.
Efecto de los inductores potentes del CYP3A4 sobre sorafenib
El uso concomitante de sorafenib con rifampicina administrada en una dosis de 600 mg una vez al día durante 5 días con una dosis oral única de 400 mg de sorafenib en voluntarios sanos resultó en una disminución del 37% en el AUC medio de sorafenib.
Efecto de la neomicina sobre sorafenib
La neomicina administrada como una dosis oral de 1 g tres veces al día durante 5 días disminuyó el AUC medio de sorafenib en un 54% en sujetos sanos a los que se les administró una dosis oral única de 400 mg de sorafenib.
Efecto de sorafenib sobre otros medicamentos
Sorafenib 400 mg dos veces al día durante 28 días no aumentó la exposición sistémica de midazolam (sustrato del CYP3A4), dextrometorfano (sustrato del CYP2D6) y omeprazol (sustrato del CYP2C19) administrados concomitantemente [ver Farmacología clínica (12.3)].
Medicamentos que aumentan el pH gástrico
La solubilidad acuosa de sorafenib depende del pH, y un pH más alto resulta en una menor solubilidad. Sin embargo, el omeprazol, un inhibidor de la bomba de protones, administrado en una dosis de 40 mg una vez al día durante 5 días, no produjo un cambio clínicamente significativo en la exposición a una dosis única de sorafenib.
Estudios in vitro
Sorafenib inhibió competitivamente el CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 y CYP3A4 in vitro. Sin embargo, sorafenib 400 mg dos veces al día durante 28 días con sustratos de CYP3A4, CYP2D6 y CYP2C19 no aumentó la exposición sistémica de estos sustratos [ver Interacciones medicamentosas (7.3)].
Sorafenib no aumentó las actividades de CYP1A2 y CYP3A4, lo que sugiere que es poco probable que sorafenib induzca CYP1A2 o CYP3A4 en humanos.
Sorafenib inhibe la glucuronidación por UGT1A1 y UGT1A9 in vitro. Sorafenib podría aumentar la exposición sistémica de los medicamentos administrados concomitantemente que son sustratos de UGT1A1 o UGT1A9.
Sorafenib inhibió la glicoproteína P in vitro. Sorafenib podría aumentar las concentraciones de los medicamentos administrados concomitantemente que son sustratos de la glicoproteína P.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
No se han realizado estudios de carcinogenicidad con sorafenib. Sorafenib fue clastogénico cuando se probó en un ensayo de células de mamíferos in vitro (ovario de hámster chino) en presencia de activación metabólica. Sorafenib no fue mutagénico en el ensayo de células bacterianas de Ames in vitro o clastogénico en un ensayo de micronúcleos de ratón in vivo . Un intermedio en el proceso de fabricación, que también está presente en la sustancia farmacéutica final (< 0,15%), fue positivo para mutagénesis en un ensayo de células bacterianas in vitro (prueba de Ames) cuando se probó de forma independiente.
No se han realizado estudios específicos con sorafenib en animales para evaluar el efecto sobre la fertilidad. Sin embargo, los resultados de los estudios de toxicidad de dosis repetidas sugieren que existe un potencial para que sorafenib afecte la función reproductiva y la fertilidad. Se observaron múltiples efectos adversos en los órganos reproductivos masculinos y femeninos, siendo la rata más susceptible que los ratones o los perros. Los cambios típicos en las ratas consistieron en atrofia o degeneración testicular, degeneración del epidídimo, próstata y vesículas seminales, necrosis central de los cuerpos lúteos y desarrollo folicular detenido. Los efectos relacionados con sorafenib en los órganos reproductivos de las ratas se manifestaron a dosis orales diarias ≥ 5 mg/kg (30 mg/m2). Esta dosis da como resultado una exposición (AUC) que es aproximadamente 0,5 veces el AUC en pacientes a la dosis humana recomendada. Los perros mostraron degeneración tubular en los testículos a 30 mg/kg/día (600 mg/m2/día). Esta dosis da como resultado una exposición que es aproximadamente 0,3 veces el AUC a la dosis humana recomendada. Se observó oligospermia en perros a 60 mg/kg/día (1200 mg/m2/día) de sorafenib.
14 ESTUDIOS CLÍNICOS
14.1 Carcinoma Hepatocelular
El estudio SHARP (HCC) (NCT00105443) fue un ensayo internacional, multicéntrico, aleatorizado, doble ciego, controlado con placebo en pacientes con carcinoma hepatocelular irresecable. La supervivencia general fue el criterio de valoración principal. Se aleatorizaron un total de 602 pacientes; 299 a sorafenib 400 mg dos veces al día y 303 a placebo coincidente. Los 602 sujetos aleatorizados se incluyeron en la población ITT para los análisis de eficacia.
Las características demográficas y de la enfermedad de referencia fueron similares entre los brazos de sorafenib y placebo con respecto a la edad, el sexo, la raza, el estado de rendimiento, la etiología (incluidas la hepatitis B, la hepatitis C y la enfermedad hepática alcohólica), el estadio TNM (estadio I: < 1% vs. < 1%; estadio II: 10,4% vs. 8,3%; estadio III: 37,8% vs. 43,6%; estadio IV: 50,8% vs. 46,9%), ausencia de invasión vascular macroscópica y diseminación tumoral extrahepática (30,1% vs. 30,0%) y estadio del Cáncer Hepático de la Clínica de Barcelona (estadio B: 18,1% vs. 16,8%; estadio C: 81,6% vs. 83,2%; estadio D: < 1% vs. 0%). La insuficiencia hepática según la puntuación de Child-Pugh fue comparable entre los brazos de sorafenib y placebo (Clase A: 95% vs. 98%; B: 5% vs. 2%). Solo se incluyó un paciente con clase C de Child-Pugh. Los tratamientos previos incluyeron procedimientos de resección quirúrgica (19,1% vs. 20,5%), terapias locoregionales (incluida la ablación por radiofrecuencia, la inyección de etanol percutáneo y la quimioembolización transarterial; 38,8% vs. 40,6%), radioterapia (4,3% vs. 5,0%) y terapia sistémica (3,0% vs. 5,0%).
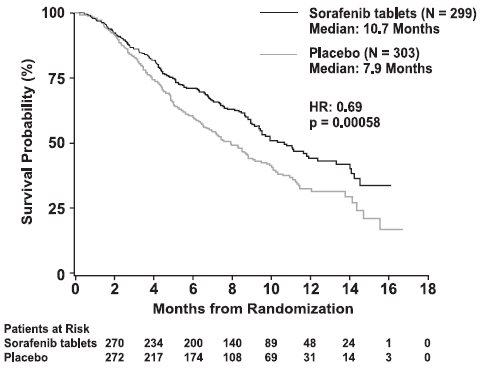
El ensayo se detuvo por eficacia después de un análisis intermedio preespecificado de segundo nivel para la supervivencia que mostró una ventaja estadísticamente significativa para sorafenib sobre placebo para la supervivencia general (HR: 0,69, p = 0,00058) (ver Tabla 10 y Figura 1). Esta ventaja fue consistente en todos los subconjuntos analizados.
El análisis final del tiempo hasta la progresión tumoral (TTP) basado en datos de un punto de tiempo anterior (por revisión radiológica independiente) también fue significativamente más largo en el brazo de sorafenib (HR: 0,58, p = 0,000007) (ver Tabla 10).
| CI = Intervalo de confianza | ||
|
||
|
Parámetro de eficacia |
Tabletas de Sorafenib (N = 299) |
Placebo (N = 303) |
|
Supervivencia general |
||
|
Número de eventos |
143 |
178 |
|
Mediana, meses |
10,7 |
7,9 |
|
(95% CI) |
(9,4, 13,3) |
(6,8, 9,1) |
|
Razón de riesgo* (95% CI) |
0,69 (0,55, 0,87) |
|
|
Valor p (prueba de log-rank†) |
0,00058 |
|
|
Tiempo hasta la progresión‡ |
||
|
Número de eventos |
107 |
156 |
|
Mediana, meses |
5,5 |
2,8 |
|
(95% CI) |
(4,1, 6,9) |
(2,7, 3,9) |
|
Razón de riesgo* (95% CI) |
0,58 (0,45, 0,74) |
|
|
Valor p (prueba de log-rank†) |
0,000007 |
|
14.2 Carcinoma de células renales
La seguridad y eficacia de sorafenib en el tratamiento del carcinoma de células renales (CCR) avanzado se estudiaron en los siguientes dos ensayos clínicos controlados aleatorizados.
TARGET:TARGET (NCT00073307) fue un ensayo internacional, multicéntrico, aleatorizado, doble ciego, controlado con placebo en pacientes con carcinoma de células renales avanzado que habían recibido una terapia sistémica previa. Los criterios de valoración primarios del estudio incluyeron la supervivencia general y la supervivencia libre de progresión (SLP). La tasa de respuesta tumoral fue un criterio de valoración secundario. El análisis de SLP incluyó 769 pacientes, según el protocolo, estratificados por categoría de riesgo pronóstico de MSKCC (Memorial Sloan Kettering Cancer Center) (baja o intermedia) y país y aleatorizados a sorafenib 400 mg dos veces al día (N = 384) o a placebo (N = 385).
La tabla 11 resume las características demográficas y de la enfermedad de la población del estudio analizada. Las características demográficas y de la enfermedad de referencia estaban bien equilibradas para ambos grupos de tratamiento. El tiempo medio desde el diagnóstico inicial de CCR hasta la aleatorización fue de 1,6 y 1,9 años para los brazos de sorafenib y placebo, respectivamente.
|
||||
|
Características |
Tabletas de sorafenib N = 384 |
Placebo N = 385 |
||
|
N |
(%) |
N |
(%) |
|
|
Género |
||||
|
Masculino |
267 |
(70) |
287 |
(75) |
|
Femenino |
116 |
(30) |
98 |
(25) |
|
Raza |
||||
|
Blanca |
276 |
(72) |
278 |
(73) |
|
Negra/Asiática/Hispana/Otra |
11 |
(3) |
10 |
(2) |
|
No reportado* |
97 |
(25) |
97 |
(25) |
|
Grupo de edad |
||||
|
< 65 años |
255 |
(67) |
280 |
(73) |
|
≥ 65 años |
127 |
(33) |
103 |
(27) |
|
ECOG performance status at baseline |
||||
|
0 |
184 |
(48) |
180 |
(47) |
|
1 |
191 |
(50) |
201 |
(52) |
|
2 |
6 |
(2) |
1 |
(< 1) |
|
No reportado |
3 |
(< 1) |
3 |
(< 1) |
|
MSKCC prognostic risk category |
||||
|
Bajo |
200 |
(52) |
194 |
(50) |
|
Intermedio |
184 |
(48) |
191 |
(50) |
|
Prior IL-2 and/or interferon |
||||
|
Sí |
319 |
(83) |
313 |
(81) |
|
No |
65 |
(17) |
72 |
(19) |
La supervivencia libre de progresión, definida como el tiempo desde la aleatorización hasta la progresión o la muerte por cualquier causa, lo que ocurriera primero, se evaluó mediante una revisión radiológica independiente ciega utilizando los criterios RECIST. La figura 2 muestra las curvas de Kaplan-Meier para la SLP. El análisis de la SLP se basó en una prueba de Log-Rank bilateral estratificada por categoría de riesgo pronóstico de MSKCC y país.
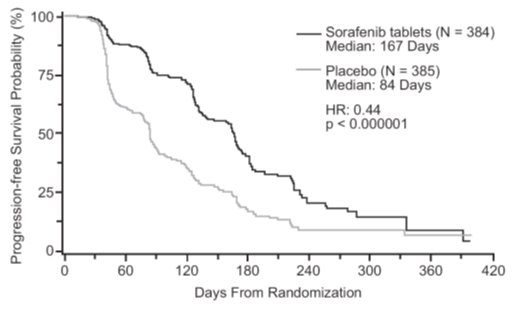
NOTA: HR es del modelo de regresión de Cox con las siguientes covariables: categoría de riesgo pronóstico de MSKCC y país. El valor p es de la prueba de Log-Rank bilateral estratificada por categoría de riesgo pronóstico de MSKCC y país.
La SLP mediana para los pacientes aleatorizados a sorafenib fue de 167 días en comparación con 84 días para los pacientes aleatorizados a placebo. La razón de riesgo estimada (riesgo inmediato de progresión o muerte con sorafenib en comparación con placebo) fue de 0,44 (IC del 95%: 0,35, 0,55).
Se examinó una serie de subconjuntos de pacientes en análisis univariados exploratorios de la SLP. Los subconjuntos incluyeron edad superior o inferior a 65 años, ECOG PS 0 o 1, categoría de riesgo pronóstico de MSKCC, si la terapia previa fue para la enfermedad metastásica progresiva o para un entorno de enfermedad anterior y el tiempo desde el diagnóstico de menos o más de 1,5 años. El efecto de sorafenib en la SLP fue consistente en todos estos subconjuntos, incluidos los pacientes que no habían recibido terapia previa con IL-2 o interferón (N = 137; 65 pacientes que recibieron sorafenib y 72 placebo), para quienes la SLP mediana fue de 172 días en el brazo de sorafenib en comparación con 85 días en el brazo de placebo.
La respuesta tumoral se determinó mediante una revisión radiológica independiente de acuerdo con los criterios RECIST. En general, de los 672 pacientes que fueron evaluables para la respuesta, 7 (2%) pacientes en el brazo de sorafenib y ningún (0%) paciente en el brazo de placebo tuvieron una respuesta parcial confirmada. Por lo tanto, la ganancia en la SLP refleja principalmente la población de enfermedad estable.
En el momento de un análisis de supervivencia interino planificado, basado en 220 muertes, la supervivencia general fue más larga para aquellos aleatorizados a sorafenib en comparación con placebo con una razón de riesgo de 0,72. Este análisis no cumplió con los criterios preespecificados para la significancia estadística. Se planean análisis adicionales a medida que maduren los datos de supervivencia.
BAY43-9006: BAY43-9006 (NCT00101413) fue un ensayo de discontinuación aleatorizado en pacientes con malignidades metastásicas, incluido el RCC. El criterio de valoración principal fue el porcentaje de pacientes aleatorizados que permanecieron libres de progresión a las 24 semanas. Todos los pacientes recibieron sorafenib durante las primeras 12 semanas. La evaluación radiológica se repitió en la semana 12. Los pacientes con un cambio < 25% en las mediciones bidimensionales del tumor desde el inicio se aleatorizaron a sorafenib o placebo durante 12 semanas más. Los pacientes que fueron aleatorizados a placebo pudieron cambiar a sorafenib de etiqueta abierta al progresar. Los pacientes con una reducción del tumor ≥ 25% continuaron con sorafenib, mientras que los pacientes con un crecimiento del tumor ≥ 25% interrumpieron el tratamiento.
Se inscribieron un total de 202 pacientes con RCC avanzado en BAY43-9006, incluidos pacientes que no habían recibido terapia previa y pacientes con histología tumoral diferente al carcinoma de células claras. Después de las primeras 12 semanas de sorafenib, 79 pacientes con RCC continuaron con sorafenib de etiqueta abierta, y 65 pacientes fueron aleatorizados a sorafenib o placebo. Después de 12 semanas adicionales, en la semana 24, para los 65 pacientes aleatorizados, la tasa de progresión libre fue significativamente mayor en los pacientes aleatorizados a sorafenib (16/32, 50%) que en los pacientes aleatorizados a placebo (6/33, 18%) (p = 0,0077). La supervivencia libre de progresión fue significativamente más larga en el brazo de sorafenib (163 días) que en aquellos aleatorizados a placebo (41 días) (p = 0,0001, HR = 0,29).
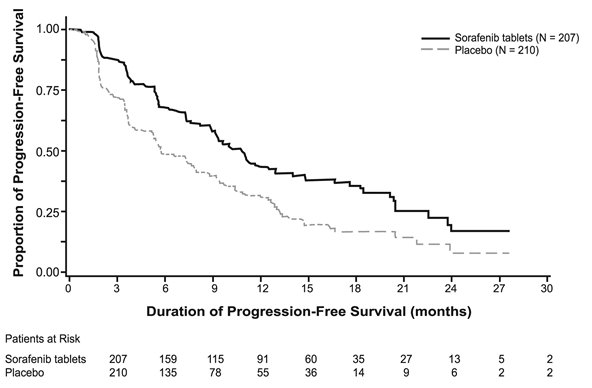
14.3 Carcinoma diferenciado de tiroides
La seguridad y eficacia de sorafenib se evaluaron en un ensayo multicéntrico, aleatorizado (1:1), doble ciego, controlado con placebo (DECISION; NCT00984282) realizado en 417 pacientes con carcinoma diferenciado de tiroides (CDT) localmente recurrente o metastásico, progresivo, refractario al tratamiento con yodo radiactivo (RAI). La aleatorización se estratificó por edad (< 60 años frente a ≥ 60 años) y región geográfica (América del Norte, Europa y Asia). Los 417 sujetos se incluyeron en la población ITT para los análisis de eficacia.
Todos los pacientes debían tener una enfermedad en progresión activa, definida como progresión dentro de los 14 meses posteriores a la inscripción. La enfermedad refractaria al RAI se definió en función de cuatro criterios que no eran mutuamente excluyentes. Todos los tratamientos con RAI y las exploraciones de diagnóstico debían realizarse en condiciones de una dieta baja en yodo y una estimulación adecuada de la TSH. A continuación, se presentan los criterios de refractariedad al RAI y la proporción de pacientes en el estudio que cumplieron cada uno de ellos: una lesión diana sin captación de yodo en la exploración con RAI (68%); tumores con captación de yodo y progresión después del tratamiento con RAI dentro de los 16 meses posteriores a la inscripción (12%); tumores con captación de yodo y múltiples tratamientos con RAI con el último tratamiento realizado más de 16 meses antes de la inscripción, y progresión de la enfermedad después de cada uno de los dos tratamientos con RAI administrados dentro de los 16 meses uno del otro (7%); dosis acumulada de RAI ≥ 600 mCi administrada (34%). La principal medida de resultado de eficacia fue la supervivencia libre de progresión (SLP) determinada por una revisión radiológica independiente ciega utilizando una versión modificada de los Criterios de Evaluación de la Respuesta en Tumores Sólidos v. 1.0 (RECIST). RECIST se modificó mediante la inclusión de la progresión clínica de las lesiones óseas basada en la necesidad de radiación de haz externo (4,4% de los eventos de progresión). Las medidas de resultado de eficacia adicionales incluyeron la supervivencia general (SG), la tasa de respuesta tumoral y la duración de la respuesta.
Los pacientes fueron aleatorizados para recibir sorafenib 400 mg dos veces al día (n = 207) o placebo (n = 210). De los 417 pacientes aleatorizados, el 48% eran hombres, la edad media fue de 63 años, el 61% tenía 60 años o más, el 60% eran blancos, el 62% tenía un estado de rendimiento ECOG de 0 y el 99% se había sometido a tiroidectomía. Los diagnósticos histológicos fueron carcinoma papilar en el 57%, carcinoma folicular (incluido el de células de Hürthle) en el 25%, y carcinoma pobremente diferenciado en el 10%, y otros en el 8% de la población del estudio. Las metástasis estaban presentes en el 96% de los pacientes: pulmones en el 86%, ganglios linfáticos en el 51% y huesos en el 27%. La actividad acumulada media de RAI administrada antes de la entrada al estudio fue de 400 mCi.
Se demostró una prolongación estadísticamente significativa de la PFS para los pacientes tratados con sorafenib en comparación con los que recibieron placebo (Figura 3); no se observó una diferencia estadísticamente significativa en el análisis final de supervivencia general (OS) (Tabla 12). El cambio a sorafenib de etiqueta abierta ocurrió en 161 (77%) pacientes aleatorizados a placebo después de la progresión de la enfermedad determinada por el investigador.
| NR = No Alcanzado, CI = Intervalo de confianza, NE = No Estimable | ||
|
||
|
Tabletas de Sorafenib N = 207 |
Placebo N = 210 |
|
|
Supervivencia libre de progresión* |
||
|
Número de muertes o progresión |
113 (55%) |
136 (65%) |
|
PFS mediana en meses (IC del 95%) |
10.8 (9.1, 12.9) |
5.8 (5.3, 7.8) |
|
Hazard Ratio (IC del 95%) |
0.59 (0.46, 0.76) |
|
|
Valor p† |
< 0.001 |
|
|
Supervivencia general‡ |
||
|
Número de muertes |
103 (49.8%) |
109 (51.9%) |
|
OS mediana en meses (IC del 95%) |
42.8 (34.6, 52.6) |
39.4 (32.7, 51.4) |
|
Hazard Ratio (IC del 95%) |
0.92 (0.71, 1.21) |
|
|
Valor p† |
0.570 |
|
|
Respuesta objetiva |
||
|
Número de respondedores objetivos§ |
24 (12%) |
1 (0.5%) |
|
(IC del 95%) |
(7.6%, 16.8%) |
(0.01%, 2.7%) |
|
Duración mediana de la respuesta en meses (IC del 95%) |
10.2 (7.4, 16.6) |
NE |
16 FORMA DE SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
Las tabletas de Sorafenib, USP están disponibles en presentaciones que contienen 200 mg de sorafenib, equivalente a 274 mg de sorafenib tosilato, USP.
Las tabletas de 200 mg son de color melocotón, recubiertas con película, redondas, sin ranurar y con la marca 200 en un lado de la tableta y NAT en el otro lado. Están disponibles de la siguiente manera:
NDC 0378-1201-78
frascos de 120 tabletas
Almacenar a 20° a 25°C (68° a 77°F). [Ver Temperatura ambiente controlada USP.] Almacenar en un lugar seco.
Dispensar en un recipiente hermético, resistente a la luz, según se define en la USP, utilizando un cierre a prueba de niños.
17 INFORMACIÓN PARA EL PACIENTE
Aconseje al paciente que lea el etiquetado del paciente aprobado por la FDA (Información para el paciente).
Eventos cardiovasculares: Hable con los pacientes sobre la isquemia cardíaca y/o el infarto y la insuficiencia cardíaca congestiva, que se han notificado durante el tratamiento con sorafenib, y que deben informar inmediatamente cualquier episodio de dolor en el pecho u otros síntomas de isquemia cardíaca o insuficiencia cardíaca congestiva [ver Advertencias y precauciones (5.1)].
Hemorragia: Informe a los pacientes que sorafenib puede aumentar el riesgo de hemorragia y que deben informar rápidamente cualquier episodio de hemorragia [ver Advertencias y precauciones (5.2)].
Informe a los pacientes que se han notificado hemorragias o elevaciones en la Razón Internacional Normalizada (INR) en algunos pacientes que toman warfarina mientras están en tratamiento con sorafenib y que su INR debe controlarse regularmente [ver Advertencias y precauciones (5.6)].
Hipertensión: Informe a los pacientes que la hipertensión puede desarrollarse durante el tratamiento con sorafenib, especialmente durante las primeras seis semanas de terapia, y que la presión arterial debe controlarse regularmente durante el tratamiento [ver Advertencias y precauciones (5.3)].
Reacciones cutáneas: Avise a los pacientes de la posible aparición de reacción cutánea mano-pie y erupción durante el tratamiento con sorafenib y las medidas de contraataque apropiadas [ver Advertencias y precauciones (5.4)].
Perforación gastrointestinal: Avise a los pacientes que se han notificado casos de perforación gastrointestinal en pacientes que toman sorafenib [ver Advertencias y precauciones (5.5)].
Riesgo de cicatrización de heridas deteriorada: Avise a los pacientes que sorafenib puede deteriorar la cicatrización de heridas. Avise a los pacientes que informen a su médico de cualquier procedimiento quirúrgico planificado [ver Advertencias y precauciones (5.7)].
Prolongación del intervalo QT: Informe a los pacientes con antecedentes de intervalo QT prolongado que sorafenib puede empeorar la condición [ver Advertencias y precauciones (5.9) y Farmacología clínica (12.2)].
Lesión hepática inducida por fármacos: Informe a los pacientes que sorafenib puede causar hepatitis, lo que puede provocar insuficiencia hepática y muerte. Avise a los pacientes que las pruebas de función hepática deben controlarse regularmente durante el tratamiento y que deben informar los signos y síntomas de hepatitis [ver Advertencias y precauciones (5.10)].
Toxicidad embrio-fetal: Avise a las mujeres que informen a su médico si están embarazadas o quedan embarazadas. Informe a las pacientes mujeres del riesgo para un feto y la posible pérdida del embarazo [ver Uso en poblaciones específicas (8.1)]. Avise a las mujeres en edad fértil que utilicen métodos anticonceptivos eficaces durante el tratamiento con sorafenib y durante 6 meses después de la última dosis. Avise a los pacientes varones con parejas femeninas en edad fértil o que estén embarazadas que utilicen métodos anticonceptivos eficaces durante el tratamiento con sorafenib y durante 3 meses después de recibir la última dosis de sorafenib [ver Advertencias y precauciones (5.11), Uso en poblaciones específicas (8.1, 8.3)].
Lactancia: Avise a los pacientes que no deben amamantar mientras toman sorafenib y durante 2 semanas después de recibir la última dosis de sorafenib [ver Uso en poblaciones específicas (8.2)].
Dosis olvidadas: Indique a los pacientes que si se olvida una dosis de sorafenib, deben tomar la siguiente dosis a la hora programada regularmente, y no duplicar la dosis. Indique a los pacientes que se pongan en contacto con su médico inmediatamente si toman demasiado sorafenib.
INFORMACIÓN PARA EL PACIENTE
|
Sorafenib Tablets, USP (soe raf’ e nib) |
|
|
¿Qué son las tabletas de sorafenib? Las tabletas de sorafenib son un medicamento recetado que se usa para tratar:
No se sabe si las tabletas de sorafenib son seguras y efectivas en niños. |
|
|
No tome tabletas de sorafenib si usted:
|
|
|
Antes de tomar tabletas de sorafenib, informe a su proveedor de atención médica sobre todas sus afecciones médicas, incluso si usted:
Informe a su proveedor de atención médica sobre todos los medicamentos que toma, incluidos los medicamentos recetados y de venta libre, las vitaminas y los suplementos herbales. Especialmente informe a su proveedor de atención médica si toma el medicamento warfarina. |
|
|
¿Cómo debo tomar tabletas de sorafenib?
|
|
|
¿Cuáles son los posibles efectos secundarios de las tabletas de sorafenib? Las tabletas de sorafenib pueden causar efectos secundarios graves, que incluyen:
|
|
|
|
|
|
|
|
Los efectos secundarios más comunes de las tabletas de sorafenib incluyen: |
|
|
|
|
Las tabletas de sorafenib pueden causar problemas de fertilidad en los hombres. Esto puede afectar su capacidad para engendrar un hijo. Hable con su proveedor de atención médica si esto le preocupa.
Estos no son todos los posibles efectos secundarios de las tabletas de sorafenib. Llame a su médico para obtener asesoramiento médico sobre los efectos secundarios. Puede informar los efectos secundarios a la FDA al 1-800-FDA-1088. |
|
|
¿Cómo debo almacenar las tabletas de sorafenib?
Mantenga las tabletas de sorafenib y todos los medicamentos fuera del alcance de los niños. |
|
|
Información general sobre el uso seguro y eficaz de las tabletas de sorafenib Los medicamentos a veces se recetan para fines distintos de los que se enumeran en un folleto de información para el paciente. No use tabletas de sorafenib para una condición para la que no fueron recetadas. No le dé tabletas de sorafenib a otras personas, incluso si tienen los mismos síntomas que usted. Pueden hacerles daño. Puede pedirle a su proveedor de atención médica o farmacéutico información sobre las tabletas de sorafenib que esté escrita para profesionales de la salud. |
|
|
¿Cuáles son los ingredientes de las tabletas de sorafenib? Ingrediente activo: tosilato de sorafenib Ingredientes inactivos: celulosa microcristalina, estearato de magnesio, hipromelosa, laurilsulfato de sodio, dióxido de titanio, croscarmelosa sódica, polietilenglicol, óxido de hierro rojo, carboximetilcelulosa cálcica.
Fabricado para: Mylan Pharmaceuticals Inc., Morgantown, WV 26505 U.S.A. Para obtener más información, llame a Mylan al 1-877-446-3679 (1-877-4-INFO-RX). |
|
Esta Información para el Paciente ha sido aprobada por la Administración de Alimentos y Medicamentos de los Estados Unidos.
Fabricado para:
Mylan Pharmaceuticals Inc.
Morgantown, WV 26505 U.S.A.
Fabricado por:
Natco Pharma Limited
Plot No.: 89 & 90, JNPC SEZ
Ramky Pharma City
Parawada, Visakhapatnam
Andhra Pradesh, India – 531019
40500541
Revisado: 7/2024
NPLV:SORA:R3
PANEL PRINCIPAL DE VISUALIZACIÓN – 200 mg
NDC 0378-1201-78
Sorafenib
Tabletas, USP
200 mg
Rx only 120 Tablets
Cada tableta recubierta con película contiene
200 mg de sorafenib equivalente a
274 mg de sorafenib tosilato, USP.
Almacenar a 20° a 25°C (68° a 77°F).
[Ver USP Controlled Room
Temperature.] Almacenar en un lugar seco.
Dosis habitual: Ver información de
prescripción adjunta.
Dispensar en un recipiente hermético, resistente a la luz
como se define en la USP
utilizando un cierre a prueba de niños.
Mantener el recipiente bien cerrado.
Mantener este y todos los medicamentos
fuera del alcance de los niños.
Fabricado para:
Mylan Pharmaceuticals Inc.
Morgantown, WV 26505 U.S.A.
Fabricado por:
Natco Pharma Limited
Visakhapatnam – 531019
AP, India
Mylan.com
M.L.: 18/VSP/AP/2018/F/G
40100226
RNPLV1201DA