
Fabricante de medicamentos: Janssen Biotech, Inc. (Updated: 2025-02-07)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
SIMPONI ARIA
®(golimumab) inyección, para uso intravenoso
Aprobación inicial en EE. UU.: 2009
ADVERTENCIA: INFECCIONES GRAVES y NEOPLASIAS
Consulte la información completa de prescripción para ver la advertencia completa en recuadro.
-
Se han producido infecciones graves que han requerido hospitalización o han causado la muerte, incluidas tuberculosis (TB), sepsis bacteriana, infecciones fúngicas invasivas (como histoplasmosis) y otras infecciones oportunistas en pacientes que recibieron SIMPONI ARIA (
5.1).
-
Interrumpa el tratamiento con SIMPONI ARIA si un paciente desarrolla una infección grave o sepsis (
5.1).
-
Realice una prueba de TB latente; si el resultado es positivo, inicie el tratamiento para la TB antes de comenzar el tratamiento con SIMPONI ARIA (
5.1).
-
Controle a todos los pacientes para detectar TB activa durante el tratamiento, incluso si la prueba inicial de TB latente es negativa (
5.1).
-
Se han notificado linfoma y otras neoplasias, algunas mortales, en niños y adolescentes tratados con bloqueadores del TNF, de los cuales SIMPONI ARIA es un miembro (
5.2).
INDICACIONES Y USO
SIMPONI ARIA es un bloqueador del factor de necrosis tumoral (TNF) indicado para el tratamiento de:
- Pacientes adultos con artritis reumatoide (AR) moderada a gravemente activa en combinación con metotrexato (
1.1)
- Artritis psoriásica (APs) activa en pacientes de 2 años de edad o mayores (
1.2)
- Pacientes adultos con espondilitis anquilosante (EA) activa (
1.3)
- Artritis idiopática juvenil (AIJ) poliarticular activa en pacientes de 2 años de edad o mayores (
1.4)
POSOLOGÍA Y ADMINISTRACIÓN
- Pacientes adultos con artritis reumatoide, artritis psoriásica y espondilitis anquilosante:
- 2 mg/kg de infusión intravenosa durante 30 minutos en las semanas 0 y 4, y cada 8 semanas a partir de entonces (
2.1)
- 2 mg/kg de infusión intravenosa durante 30 minutos en las semanas 0 y 4, y cada 8 semanas a partir de entonces (
- Pacientes pediátricos con artritis idiopática juvenil poliarticular y artritis psoriásica:
- 80 mg/m
2infusión intravenosa durante 30 minutos en las semanas 0 y 4, y cada 8 semanas a partir de entonces (
2.2)
- 80 mg/m
- Se requiere la dilución de la solución de SIMPONI ARIA suministrada con inyección de cloruro de sodio al 0,9%, USP antes de la administración. Como alternativa, también se puede utilizar inyección de cloruro de sodio al 0,45%, USP (
2.4)
FORMAS Y CONCENTRACIONES FARMACÉUTICAS
- Inyección: solución de 50 mg/4 mL (12,5 mg/mL) en un vial de dosis única (
3)
CONTRAINDICACIONES
- Ninguna (
4)
ADVERTENCIAS Y PRECAUCIONES
- Infecciones graves: No inicie el tratamiento con SIMPONI ARIA durante una infección activa. Si se desarrolla una infección, controle cuidadosamente y suspenda SIMPONI ARIA si la infección se agrava (
5.1).
- Infecciones fúngicas invasivas: En pacientes que desarrollan una enfermedad sistémica con SIMPONI ARIA, considere la terapia antifúngica empírica para aquellos que residen o viajan a regiones donde las micosis son endémicas (
5.1).
- Reactivación de la hepatitis B: Controle a los portadores del VHB durante y varios meses después del tratamiento. Si se produce una reactivación, suspenda SIMPONI ARIA e inicie un tratamiento antiviral (
5.1).
- Neoplasias: Se han observado más casos de linfoma entre los pacientes que recibieron bloqueadores del TNF en comparación con los pacientes de los grupos de control. Se han observado casos de otras neoplasias entre los pacientes que recibieron bloqueadores del TNF (
5.2).
- Insuficiencia cardíaca congestiva: Puede producirse un empeoramiento o una aparición nueva. Suspenda SIMPONI ARIA si aparecen síntomas nuevos o que empeoran (
5.3).
- Trastornos desmielinizantes: Puede producirse un empeoramiento o una aparición nueva (
5.4).
- Síndrome similar al lupus: Suspenda SIMPONI ARIA si aparecen síntomas (
5.5).
- Reacciones de hipersensibilidad: Pueden producirse reacciones de hipersensibilidad sistémicas graves, incluida la anafilaxia (
5.11).
REACCIONES ADVERSAS
Las reacciones adversas más comunes (incidencia ≥ 3%) son: infección de las vías respiratorias superiores, aumento de la alanina aminotransferasa, infección viral, aumento de la aspartato aminotransferasa, disminución del recuento de neutrófilos, bronquitis, hipertensión y erupción cutánea (
6.1).
Para notificar SOSPECHAS DE REACCIONES ADVERSAS, póngase en contacto con Janssen Biotech, Inc. al 1-800-JANSSEN (1-800-526-7736) o con la FDA al 1-800-FDA-1088 o
www.fda.gov/medwatch.
INTERACCIONES MEDICAMENTOSAS
Consulte el apartado 17 para obtener información sobre el ASESORAMIENTO AL PACIENTE y la Guía de medicamentos.
Revisado: 7/2023
Tabla de contenido
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDO*
ADVERTENCIA: INFECCIONES GRAVES y MALIGNIDAD
1 INDICACIONES Y USO
1.1 Artritis Reumatoide (AR)
1.2 Artritis Psoriásica (APs)
1.3 Espondilitis Anquilosante (EA)
1.4 Artritis Idiopática Juvenil Poliarticular (AIJp)
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Dosis en Adultos con Artritis Reumatoide, Artritis Psoriásica y Espondilitis Anquilosante
2.2 Dosis en Pacientes Pediátricos con Artritis Idiopática Juvenil Poliarticular y Artritis Psoriásica
2.3 Evaluación de Tuberculosis y Hepatitis B Antes de la Dosificación
2.4 Instrucciones Importantes de Administración
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Infecciones Graves
5.2 Tumores Malignos
5.3 Insuficiencia Cardíaca Congestiva
5.4 Trastornos Desmielinizantes
5.5 Autoinmunidad
5.6 Uso con Abatacept
5.7 Uso con Anakinra
5.8 Cambio Entre Fármacos Biológicos Antirreumáticos Modificadores de la Enfermedad (FARME)
5.9 Citopenias Hematológicas
5.10 Vacunas/Agentes Infecciosos Terapéuticos
5.11 Reacciones de Hipersensibilidad
6 REACCIONES ADVERSAS
6.1 Experiencia en Ensayos Clínicos
6.2 Inmunogenicidad
6.3 Experiencia Poscomercialización
7 INTERACCIONES MEDICAMENTOSAS
7.1 Metotrexato
7.2 Productos Biológicos para AR, APs, EA y AIJp
7.3 Vacunas Vivas/Agentes Infecciosos Terapéuticos
7.4 Sustratos del Citocromo P450
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.4 Uso Pediátrico
8.5 Uso Geriátrico
10 SOBREDOSIS
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de Acción
12.2 Farmacodinamia
12.3 Farmacocinética
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
14 ESTUDIOS CLÍNICOS
14.1 Artritis Reumatoide
14.2 Artritis Psoriásica
14.3 Espondilitis Anquilosante
14.4 Artritis Idiopática Juvenil Poliarticular (AIJp)
16 CÓMO SE SUMINISTRA/ALMACENAMIENTO Y MANIPULACIÓN
17 INFORMACIÓN DE ASESORAMIENTO AL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información completa de prescripción no se enumeran.
ADVERTENCIA RECUADRO
ADVERTENCIA: INFECCIONES GRAVES Y NEOPLASIAS
INFECCIONES GRAVES
Los pacientes tratados con SIMPONI ARIA presentan un mayor riesgo de desarrollar infecciones graves que pueden provocar hospitalización o la muerte
[ver
Advertencias y precauciones (5.1)]
. La mayoría de los pacientes que desarrollaron estas infecciones estaban tomando inmunosupresores concomitantes como metotrexato o corticosteroides.
Interrumpa el tratamiento con SIMPONI ARIA si un paciente desarrolla una infección grave.
Las infecciones notificadas con bloqueadores del TNF, de los que SIMPONI ARIA es miembro, incluyen:
- Tuberculosis activa, incluida la reactivación de la tuberculosis latente. Los pacientes con tuberculosis con frecuencia han presentado enfermedad diseminada o extrapulmonar. Realice pruebas a los pacientes para detectar tuberculosis latente antes del uso de SIMPONI ARIA y durante el tratamiento. Inicie el tratamiento para la tuberculosis latente antes del uso de SIMPONI ARIA.
- Infecciones fúngicas invasoras, incluidas histoplasmosis, coccidioidomicosis, candidiasis, aspergilosis, blastomicosis y neumocistosis. Los pacientes con histoplasmosis u otras infecciones fúngicas invasoras pueden presentar enfermedad diseminada en lugar de localizada. Las pruebas de antígeno y anticuerpos para la histoplasmosis pueden ser negativas en algunos pacientes con infección activa. Considere la terapia antifúngica empírica en pacientes con riesgo de infecciones fúngicas invasoras que desarrollan una enfermedad sistémica grave.
- Infecciones bacterianas, víricas y otras infecciones debidas a patógenos oportunistas, incluidos Legionella y Listeria.
Considere los riesgos y beneficios del tratamiento con SIMPONI ARIA antes de iniciar el tratamiento en pacientes con infección crónica o recurrente.
Controle estrechamente a los pacientes para detectar el desarrollo de signos y síntomas de infección durante y después del tratamiento con SIMPONI ARIA, incluido el posible desarrollo de tuberculosis en pacientes que dieron negativo en la prueba de infección tuberculosa latente antes de iniciar el tratamiento
[ver
Advertencias y precauciones (5.1)]
.
NEOPLASIAS
Se han notificado linfoma y otras neoplasias, algunas mortales, en niños y adolescentes tratados con bloqueadores del TNF, de los que SIMPONI ARIA es miembro
[ver
Advertencias y precauciones (5.2)]
.
1 INDICACIONES Y USO
1.1 Artritis Reumatoide (AR)
SIMPONI ARIA, en combinación con metotrexato (MTX), está indicado para el tratamiento de pacientes adultos con artritis reumatoide activa de moderada a grave.
1.2 Artritis Psoriásica (APs)
SIMPONI ARIA está indicado para el tratamiento de la artritis psoriásica activa en pacientes de 2 años de edad y mayores.
1.3 Espondilitis Anquilosante (EA)
SIMPONI ARIA está indicado para el tratamiento de pacientes adultos con espondilitis anquilosante activa.
1.4 Artritis Idiopática Juvenil Poliarticular (AIJp)
SIMPONI ARIA está indicado para el tratamiento de la artritis idiopática juvenil poliarticular (AIJp) activa en pacientes de 2 años de edad y mayores.
2 DOSIS Y ADMINISTRACIÓN
2.1 Dosificación en adultos con artritis reumatoide, artritis psoriásica y espondilitis anquilosante
El régimen de dosificación de SIMPONI ARIA es de 2 mg por kg administrado como infusión intravenosa durante 30 minutos en las semanas 0 y 4, y cada 8 semanas a partir de entonces. Siga las instrucciones de dilución y administración para SIMPONI ARIA
[ver
Dosificación y administración (2.4)]
.
Para pacientes con artritis reumatoide (AR), SIMPONI ARIA debe administrarse en combinación con metotrexato.
No se ha establecido la eficacia y seguridad del cambio entre las formulaciones intravenosa y subcutánea y las vías de administración.
2.2 Dosificación en pacientes pediátricos con artritis idiopática juvenil poliarticular y artritis psoriásica
El régimen de dosificación de SIMPONI ARIA, basado en el área de superficie corporal (ASC), es de 80 mg/m
2administrado como infusión intravenosa durante 30 minutos en las semanas 0 y 4, y cada 8 semanas a partir de entonces. Siga las instrucciones de dilución y administración para SIMPONI ARIA
[ver
Dosificación y administración (2.4)]
.
2.3 Evaluación de tuberculosis y hepatitis B antes de la dosificación
Antes de iniciar SIMPONI ARIA y periódicamente durante el tratamiento, evalúe a los pacientes para detectar tuberculosis activa y realice pruebas de infección latente
[ver
Advertencias y precauciones (5.1)]
. Antes de iniciar SIMPONI ARIA, realice pruebas a los pacientes para detectar infección viral por hepatitis B
[ver
Advertencias y precauciones (5.1)]
.
2.4 Instrucciones importantes de administración
La solución de SIMPONI ARIA para infusión intravenosa debe ser diluida por un profesional sanitario utilizando una técnica aséptica de la siguiente manera:
- Calcule la dosis y el número de viales de SIMPONI ARIA necesarios en función de la dosis recomendada para adultos de 2 mg/kg y el peso del paciente para AR, AP y EA. Calcule la dosis y el número de viales de SIMPONI ARIA necesarios en función de la dosis pediátrica recomendada de 80 mg/m
2y el área de superficie corporal (ASC) del paciente, para AIJ poliarticular y pacientes pediátricos con AP. Cada vial de 4 mL de SIMPONI ARIA contiene 50 mg de golimumab.
- Compruebe que la solución en cada vial sea incolora a amarillo claro. La solución puede desarrollar algunas partículas traslúcidas finas, ya que el golimumab es una proteína. No lo use si hay partículas opacas, decoloración u otras partículas extrañas presentes.
- Diluya el volumen total de la solución de SIMPONI ARIA con inyección de cloruro de sodio al 0,9%, USP hasta un volumen final de 100 mL. Por ejemplo, esto se puede lograr extrayendo un volumen de la inyección de cloruro de sodio al 0,9%, USP de la bolsa o frasco de infusión de 100 mL igual al volumen total de SIMPONI ARIA. Añada lentamente el volumen total de la solución de SIMPONI ARIA a la bolsa o frasco de infusión de 100 mL. Mezcle suavemente. Deseche cualquier solución no utilizada que quede en los viales. Alternativamente, SIMPONI ARIA puede diluirse utilizando el mismo método descrito anteriormente con inyección de cloruro de sodio al 0,45%, USP.
- Antes de la infusión, inspeccione visualmente la solución de SIMPONI ARIA diluida para detectar la presencia de partículas o decoloración. No lo use si están presentes.
- Utilice únicamente un equipo de infusión con un filtro en línea, estéril, no pirogénico y de baja unión a proteínas (tamaño de poro de 0,22 micrómetros o menos).
- No infunda SIMPONI ARIA de forma concomitante en la misma vía intravenosa con otros agentes. No se han realizado estudios de compatibilidad fisicoquímica para evaluar el uso de SIMPONI ARIA con otros agentes intravenosos en la misma vía intravenosa.
- Infunda la solución diluida durante 30 minutos.
- Una vez diluida, la solución de infusión puede almacenarse hasta 4 horas a temperatura ambiente.
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
Inyección: solución incolora a amarillo claro de 50 mg/4 mL (12.5 mg/mL) en vial de dosis única.
4 CONTRAINDICACIONES
Ninguna.
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Infecciones Graves
Los pacientes tratados con SIMPONI ARIA presentan un mayor riesgo de desarrollar infecciones graves que afectan a diversos sistemas y sitios orgánicos, lo que puede provocar hospitalización o la muerte.
Se han notificado infecciones oportunistas debidas a organismos bacterianos, micobacterianos, fúngicos invasivos, víricos o parasitarios, incluyendo aspergilosis, blastomicosis, candidiasis, coccidioidomicosis, histoplasmosis, legionelosis, listeriosis, neumocistosis y tuberculosis con los bloqueadores del TNF. Los pacientes con frecuencia han presentado una enfermedad diseminada en lugar de localizada. El uso concomitante de un bloqueador del TNF y abatacept o anakinra se asoció con un mayor riesgo de infecciones graves; por lo tanto, no se recomienda el uso concomitante de SIMPONI ARIA y estos productos biológicos
[ver
Advertencias y precauciones (5.6),
5.7)y
Interacciones medicamentosas (7.2)]
.
No se debe iniciar el tratamiento con SIMPONI ARIA en pacientes con una infección activa, incluidas las infecciones localizadas clínicamente importantes. Los pacientes mayores de 65 años, los pacientes con afecciones comórbidas y/o los pacientes que toman inmunosupresores concomitantes como corticosteroides o metotrexato pueden tener un mayor riesgo de infección. Considere los riesgos y beneficios del tratamiento antes de iniciar SIMPONI ARIA en pacientes:
- con infección crónica o recurrente;
- que han estado expuestos a la tuberculosis;
- con antecedentes de una infección oportunista;
- que han residido o viajado a zonas de tuberculosis endémica o micosis endémicas, como histoplasmosis, coccidioidomicosis o blastomicosis; o
- con afecciones subyacentes que puedan predisponerlos a la infección.
Monitorización
Controle estrechamente a los pacientes para detectar el desarrollo de signos y síntomas de infección durante y después del tratamiento con SIMPONI ARIA. Suspenda SIMPONI ARIA si un paciente desarrolla una infección grave, una infección oportunista o sepsis. En los pacientes que desarrollan una nueva infección durante el tratamiento con SIMPONI ARIA, realice una evaluación diagnóstica rápida y completa adecuada para un paciente inmunodeprimido e inicie una terapia antimicrobiana adecuada y controlelos estrechamente.
Tuberculosis
Se han observado casos de reactivación de la tuberculosis o nuevas infecciones tuberculosas en pacientes que reciben bloqueadores del TNF, incluidos los pacientes que han recibido previamente tratamiento para la tuberculosis latente o activa. Evalúe a los pacientes para detectar factores de riesgo de tuberculosis y realice pruebas de infección latente antes de iniciar SIMPONI ARIA y periódicamente durante el tratamiento.
Se ha demostrado que el tratamiento de la infección tuberculosa latente antes del tratamiento con bloqueadores del TNF reduce el riesgo de reactivación de la tuberculosis durante el tratamiento. Antes de iniciar SIMPONI ARIA, evalúe si es necesario el tratamiento para la tuberculosis latente; una induración de 5 mm o más es una prueba cutánea de tuberculina positiva, incluso para pacientes previamente vacunados con Bacilo de Calmette-Guérin (BCG).
Considere la terapia antituberculosa antes de iniciar SIMPONI ARIA en pacientes con antecedentes de tuberculosis latente o activa en los que no se puede confirmar un tratamiento adecuado, y en pacientes con una prueba negativa para tuberculosis latente pero que presentan factores de riesgo de infección tuberculosa. Se recomienda consultar con un médico con experiencia en el tratamiento de la tuberculosis para ayudar a decidir si es adecuado iniciar la terapia antituberculosa para un paciente individual.
Se han producido casos de tuberculosis activa en pacientes tratados con la formulación subcutánea de golimumab durante y después del tratamiento de la tuberculosis latente. Controle a los pacientes para detectar el desarrollo de signos y síntomas de tuberculosis, incluidos los pacientes que dieron negativo en la prueba de infección tuberculosa latente antes de iniciar el tratamiento, los pacientes que están en tratamiento para la tuberculosis latente o los pacientes que fueron tratados previamente para la infección tuberculosa.
Considere la tuberculosis en el diagnóstico diferencial en pacientes que desarrollan una nueva infección durante el tratamiento con SIMPONI ARIA, especialmente en pacientes que han viajado previamente o recientemente a países con una alta prevalencia de tuberculosis, o que han tenido contacto cercano con una persona con tuberculosis activa.
Infecciones Fúngicas Invasivas
Si los pacientes desarrollan una enfermedad sistémica grave y residen o viajan a regiones donde las micosis son endémicas, considere la infección fúngica invasiva en el diagnóstico diferencial. Considere la terapia antifúngica empírica adecuada y tenga en cuenta tanto el riesgo de infección fúngica grave como los riesgos de la terapia antifúngica mientras se realiza una evaluación diagnóstica. Las pruebas de antígeno y anticuerpos para la histoplasmosis pueden ser negativas en algunos pacientes con infección activa. Para ayudar en el manejo de estos pacientes, considere la consulta con un médico con experiencia en el diagnóstico y tratamiento de infecciones fúngicas invasivas.
Reactivación del Virus de la Hepatitis B
El uso de bloqueadores del TNF, de los cuales SIMPONI ARIA es un miembro, se ha asociado con la reactivación del virus de la hepatitis B (VHB) en pacientes que son portadores crónicos de hepatitis B (es decir, antígeno de superficie positivo). En algunos casos, la reactivación del VHB que ocurre junto con la terapia con bloqueadores del TNF ha sido fatal. La mayoría de estos informes se han producido en pacientes que recibieron inmunosupresores concomitantes.
Todos los pacientes deben ser analizados para detectar infección por VHB antes de iniciar el tratamiento con bloqueadores del TNF. Para los pacientes que den positivo en la prueba de antígeno de superficie de la hepatitis B, se recomienda consultar con un médico con experiencia en el tratamiento de la hepatitis B antes de iniciar el tratamiento con bloqueadores del TNF. Los riesgos y beneficios del tratamiento deben considerarse antes de prescribir bloqueadores del TNF, incluido SIMPONI ARIA, a pacientes portadores del VHB. No se dispone de datos suficientes sobre si el tratamiento antiviral puede reducir el riesgo de reactivación del VHB en portadores del VHB tratados con bloqueadores del TNF. Los pacientes portadores del VHB que requieran tratamiento con bloqueadores del TNF deben ser controlados estrechamente para detectar signos clínicos y de laboratorio de infección activa por VHB durante todo el tratamiento y durante varios meses después de la finalización del tratamiento.
En los pacientes que desarrollen reactivación del VHB, se deben suspender los bloqueadores del TNF e iniciar un tratamiento antiviral con tratamiento de apoyo adecuado. No se conoce la seguridad de reanudar los bloqueadores del TNF después de que se haya controlado la reactivación del VHB. Por lo tanto, los médicos deben actuar con precaución al considerar la reanudación de los bloqueadores del TNF en esta situación y controlar estrechamente a los pacientes.
5.2 Neoplasias malignas
Neoplasias malignas en pacientes pediátricos
Se han notificado neoplasias malignas, algunas mortales, entre niños, adolescentes y adultos jóvenes que recibieron tratamiento con agentes bloqueadores del TNF (inicio del tratamiento ≤ 18 años de edad), incluido golimumab. Aproximadamente la mitad de los casos fueron linfomas, incluidos el linfoma de Hodgkin y el linfoma no Hodgkin. Los demás casos representaron una variedad de neoplasias malignas, incluidas neoplasias malignas raras que suelen asociarse con inmunosupresión, y neoplasias malignas que normalmente no se observan en niños y adolescentes. Las neoplasias malignas se produjeron después de una mediana de 30 meses (rango de 1 a 84 meses) después de la primera dosis de terapia con bloqueadores del TNF. La mayoría de los pacientes recibían inmunosupresores concomitantes. La mayoría de los casos se notificaron después de la comercialización y provienen de diversas fuentes, incluidos registros e informes espontáneos posteriores a la comercialización.
Neoplasias malignas en pacientes adultos
Los riesgos y beneficios del tratamiento con bloqueadores del TNF, incluido SIMPONI ARIA, deben considerarse antes de iniciar el tratamiento en pacientes con una neoplasia maligna conocida que no sea un cáncer de piel no melanoma (CPNM) tratado con éxito o cuando se considere continuar con un bloqueador del TNF en pacientes que desarrollan una neoplasia maligna.
En las partes controladas de los ensayos clínicos de bloqueadores del TNF, incluida la formulación subcutánea de golimumab, se han observado más casos de linfoma entre los pacientes que recibieron tratamiento anti-TNF en comparación con los pacientes de los grupos de control. Los pacientes con AR y otras enfermedades inflamatorias crónicas, particularmente los pacientes con enfermedad altamente activa y/o exposición crónica a terapias inmunosupresoras, pueden tener un riesgo mayor (hasta varias veces) que la población general de desarrollar linfoma, incluso en ausencia de terapia con bloqueadores del TNF. Se han notificado casos de leucemia aguda y crónica con el uso de bloqueadores del TNF, incluido SIMPONI ARIA, en la artritis reumatoide y otras indicaciones. Incluso en ausencia de terapia con bloqueadores del TNF, los pacientes con artritis reumatoide pueden tener un riesgo mayor (aproximadamente el doble) que la población general de desarrollar leucemia.
Se han notificado casos raros posteriores a la comercialización de linfoma de células T hepatosplénico (LCTH) en pacientes tratados con agentes bloqueadores del TNF. Este tipo raro de linfoma de células T tiene un curso de enfermedad muy agresivo y suele ser mortal. Casi todos los casos notificados asociados con bloqueadores del TNF se han producido en pacientes con enfermedad de Crohn o colitis ulcerosa. La mayoría fueron varones adolescentes y adultos jóvenes. Casi todos estos pacientes habían recibido tratamiento con azatioprina (AZA) o 6-mercaptopurina (6-MP) concomitantemente con un bloqueador del TNF en o antes del diagnóstico. No se puede excluir un riesgo de desarrollo de linfoma de células T hepatosplénico en pacientes tratados con bloqueadores del TNF.
Se han notificado melanoma y carcinoma de células de Merkel en pacientes tratados con agentes bloqueadores del TNF, incluido SIMPONI ARIA. Se recomienda un examen periódico de la piel para todos los pacientes, particularmente aquellos con factores de riesgo de cáncer de piel.
En ensayos controlados de otros bloqueadores del TNF en pacientes con mayor riesgo de neoplasias malignas (por ejemplo, pacientes con enfermedad pulmonar obstructiva crónica [EPOC], pacientes con granulomatosis de Wegener tratados con ciclofosfamida concomitante), una mayor proporción de neoplasias malignas se produjo en el grupo de bloqueadores del TNF en comparación con el grupo controlado. En un ensayo clínico exploratorio que evaluó el uso de la formulación subcutánea de golimumab en pacientes con asma persistente grave, más pacientes tratados con golimumab informaron de neoplasias malignas en comparación con los pacientes del grupo de control. Se desconoce la importancia de este hallazgo.
Durante la parte controlada del ensayo de fase 3 en AR para SIMPONI ARIA, la incidencia de neoplasias malignas distintas del linfoma y el CPNM por cada 100 pacientes-año de seguimiento fue de 0,56 (IC del 95%: 0,01, 3,11) en el grupo de SIMPONI ARIA en comparación con una incidencia de 0 (IC del 95%: 0,00, 3,79) en el grupo placebo.
5.3 Insuficiencia cardíaca congestiva
Se han notificado casos de empeoramiento de la insuficiencia cardíaca congestiva (ICC) y de aparición de nueva ICC con bloqueadores del TNF, incluido SIMPONI ARIA. Algunos casos tuvieron un desenlace mortal. En varios ensayos exploratorios de otros bloqueadores del TNF en el tratamiento de la ICC, hubo mayores proporciones de pacientes tratados con bloqueadores del TNF que tuvieron exacerbaciones de la ICC que requirieron hospitalización o mayor mortalidad. SIMPONI ARIA no se ha estudiado en pacientes con antecedentes de ICC y SIMPONI ARIA debe utilizarse con precaución en pacientes con ICC. Si se decide administrar SIMPONI ARIA a pacientes con ICC, estos pacientes deben ser controlados estrechamente durante el tratamiento, y SIMPONI ARIA debe suspenderse si aparecen síntomas nuevos o que empeoran de ICC.
5.4 Trastornos desmielinizantes
El uso de bloqueadores del TNF, incluido SIMPONI ARIA, se ha asociado con casos poco frecuentes de aparición de nuevos trastornos desmielinizantes del sistema nervioso central (SNC) o exacerbación de los mismos, incluida la esclerosis múltiple (EM), y trastornos desmielinizantes periféricos, incluido el síndrome de Guillain-Barré. En raras ocasiones, se han notificado casos de desmielinización central, EM, neuritis óptica y polineuropatía desmielinizante periférica en pacientes tratados con golimumab. Los médicos deben tener precaución al considerar el uso de bloqueadores del TNF, incluido SIMPONI ARIA, en pacientes con trastornos desmielinizantes del sistema nervioso central o periférico. Debe considerarse la interrupción de SIMPONI ARIA si se desarrollan estos trastornos.
5.5 Autoinmunidad
El tratamiento con bloqueadores del TNF, incluido SIMPONI ARIA, puede provocar la formación de anticuerpos antinucleares (AAN). En raras ocasiones, el tratamiento con bloqueadores del TNF puede provocar el desarrollo de un síndrome similar al lupus
[ver
. Si un paciente desarrolla síntomas que sugieran un síndrome similar al lupus tras el tratamiento con SIMPONI ARIA, debe interrumpirse el tratamiento.
5.6 Uso con abatacept
En ensayos controlados, la administración concomitante de otro bloqueador del TNF y abatacept se asoció con una mayor proporción de infecciones graves que el uso de un bloqueador del TNF solo; y la terapia combinada, en comparación con el uso de un bloqueador del TNF solo, no ha demostrado una mejora del beneficio clínico en el tratamiento de la AR. Por lo tanto, no se recomienda la combinación de bloqueadores del TNF, incluido SIMPONI ARIA, y abatacept
[ver
Interacciones medicamentosas (7.2)]
.
5.7 Uso con anakinra
La administración concomitante de anakinra (un antagonista de la interleucina-1) y otro bloqueador del TNF se asoció con una mayor proporción de infecciones graves y neutropenia y sin beneficios adicionales en comparación con el bloqueador del TNF solo. Por lo tanto, no se recomienda la combinación de anakinra con bloqueadores del TNF, incluido SIMPONI ARIA
[ver
Interacciones medicamentosas (7.2)]
.
5.8 Cambio entre fármacos antirreumáticos modificadores de la enfermedad biológicos (DMARD)
Se debe tener precaución al cambiar de un producto biológico a otro, ya que la actividad biológica superpuesta puede aumentar aún más el riesgo de infección.
5.9 Citopenias hematológicas
Se han notificado casos de pancitopenia, leucopenia, neutropenia, agranulocitosis, anemia aplásica y trombocitopenia en pacientes que reciben golimumab. Se debe tener precaución al usar bloqueadores del TNF, incluido SIMPONI ARIA, en pacientes que tienen o han tenido citopenias significativas.
5.10 Vacunaciones/Agentes infecciosos terapéuticos
Vacunas vivas
Evite las vacunas vivas en pacientes tratados con SIMPONI ARIA. En pacientes que reciben terapia anti-TNF, hay datos limitados disponibles sobre la respuesta a la vacunación con vacunas vivas, o sobre la transmisión secundaria de la infección por vacunas vivas. El uso de vacunas vivas podría provocar infecciones clínicas, incluidas infecciones diseminadas.
No se recomienda la administración de vacunas vivas a lactantes expuestos a SIMPONI ARIA
in uterodurante 6 meses después de la última infusión de SIMPONI ARIA de la madre durante el embarazo
[ver
Interacciones medicamentosas (7.3)y
Uso en poblaciones específicas (8.1)]
.
Siempre que sea posible, actualice las inmunizaciones antes de iniciar el tratamiento con SIMPONI ARIA siguiendo las pautas de inmunización actuales para pacientes que reciben agentes inmunosupresores. Aconseje a los pacientes que hablen con el médico antes de buscar cualquier inmunización.
Agentes infecciosos terapéuticos
Otros usos de agentes infecciosos terapéuticos como bacterias atenuadas vivas (p. ej., instilación vesical de BCG para el tratamiento del cáncer) podrían provocar infecciones clínicas, incluidas infecciones diseminadas. Se recomienda que no se administren agentes infecciosos terapéuticos de forma concomitante con SIMPONI ARIA.
5.11 Reacciones de hipersensibilidad
En la experiencia postcomercialización, se han notificado reacciones de hipersensibilidad sistémicas graves (incluida la anafilaxia) tras la administración de las formulaciones subcutánea e intravenosa de golimumab, incluido SIMPONI ARIA. Se notificaron reacciones de hipersensibilidad, incluidas urticaria, prurito, disnea y náuseas, durante la infusión y, por lo general, en el plazo de una hora después de la infusión. Algunas de estas reacciones se produjeron después de la primera administración de golimumab. Si se produce una reacción alérgica grave o anafiláctica, debe interrumpirse inmediatamente la administración de SIMPONI ARIA e instaurarse el tratamiento adecuado.
6 REACCIONES ADVERSAS
Las reacciones adversas más graves fueron:
- Infecciones graves
[ver
Advertencias y precauciones (5.1)]
- Tumores malignos
[ver
Advertencias y precauciones (5.2)]
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se realizan bajo condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un medicamento no se pueden comparar directamente con las tasas en los ensayos clínicos de otro medicamento y es posible que no reflejen las tasas observadas en la práctica clínica.
Los datos de seguridad que se describen a continuación se basan en un ensayo de Fase 3 aleatorizado, doble ciego y controlado en pacientes con AR que recibieron SIMPONI ARIA por infusión intravenosa (Ensayo AR). El protocolo incluyó disposiciones para que los pacientes que tomaban placebo recibieran tratamiento con SIMPONI ARIA en la Semana 16 o la Semana 24, ya sea por respuesta del paciente (basada en la actividad incontrolada de la enfermedad) o por diseño, de modo que los eventos adversos no siempre se pueden atribuir inequívocamente a un tratamiento determinado. Las comparaciones entre el placebo y SIMPONI ARIA se basaron en las primeras 24 semanas de exposición.
El Ensayo AR incluyó a 197 pacientes tratados con control y 463 pacientes tratados con SIMPONI ARIA (que incluye a los pacientes tratados con control que cambiaron a SIMPONI ARIA en la Semana 16). La proporción de pacientes que interrumpieron el tratamiento debido a reacciones adversas en la fase controlada del Ensayo AR hasta la Semana 24 fue del 3.5% para los pacientes tratados con SIMPONI ARIA y del 0.5% para los pacientes tratados con placebo. La infección de las vías respiratorias superiores fue la reacción adversa más común reportada en el ensayo hasta la Semana 24, y ocurrió en el 6.5% de los pacientes tratados con SIMPONI ARIA en comparación con el 7.6% de los pacientes tratados con control, respectivamente.
Infecciones
Las infecciones graves observadas en pacientes tratados con SIMPONI ARIA incluyeron sepsis, neumonía, celulitis, abscesos, infecciones oportunistas, tuberculosis (TB) e infecciones fúngicas invasivas. Los casos de TB incluyeron TB pulmonar y extrapulmonar. La mayoría de los casos de TB ocurrieron en países con una alta tasa de incidencia de TB
[ver
Advertencias y precauciones (5.1)]
.
En la fase controlada del Ensayo AR hasta la Semana 24, se observaron infecciones en el 27% de los pacientes tratados con SIMPONI ARIA en comparación con el 24% de los pacientes tratados con control, y se observaron infecciones graves en el 0.9% de los pacientes tratados con SIMPONI ARIA y el 0.0% de los pacientes tratados con control. Hasta la Semana 24, la incidencia de infecciones graves por 100 pacientes-año de seguimiento fue de 2.2 (IC del 95%: 0.61, 5.71) para el grupo de SIMPONI ARIA y 0 (0.00, 3.79) para el grupo de placebo. En las partes controladas y no controladas del Ensayo AR, 958 pacientes-año de seguimiento total con una mediana de seguimiento de aproximadamente 92 semanas, la incidencia por 100 pacientes-año de todas las infecciones graves fue de 4.07 (IC del 95%: 2.90, 5.57) en pacientes que recibieron SIMPONI ARIA
[ver
Advertencias y precauciones (5.1)]
. En las partes controladas y no controladas del Ensayo AR, en pacientes tratados con SIMPONI ARIA, la incidencia de TB activa por 100 pacientes-año fue de 0.31 (IC del 95%: 0.06; 0.92) y la incidencia de otras infecciones oportunistas por 100 pacientes-año fue de 0.42 (IC del 95%: 0.11, 1.07).
Tumores malignos
Se reportó un caso de neoplasia maligna diferente a linfoma y NMSC con SIMPONI ARIA hasta la Semana 24 durante la fase controlada del Ensayo AR. En las partes controladas y no controladas hasta aproximadamente 92 semanas, la incidencia de neoplasias malignas por 100 pacientes-año, distintas del linfoma y el NMSC, en pacientes tratados con SIMPONI ARIA fue de 0.31 (IC del 95%: 0.06, 0.92) y la incidencia de NMSC fue de 0.1 (IC del 95%: 0.00, 0.58).
Elevaciones de las enzimas hepáticas
Ha habido informes de reacciones hepáticas graves, incluida insuficiencia hepática aguda, en pacientes que reciben bloqueadores del TNF.
En la fase controlada del Ensayo AR, hasta la Semana 24, se produjeron elevaciones de ALT ≥ 5 × LSN en el 0.8% de los pacientes tratados con SIMPONI ARIA y el 0% de los pacientes tratados con control, y se produjeron elevaciones de ALT ≥ 3 × LSN en el 2.3% de los pacientes tratados con SIMPONI ARIA y el 2.5% de los pacientes tratados con control.
En la fase controlada del Ensayo APs, hasta la Semana 24, se produjeron elevaciones de ALT ≥ 5 × LSN en el 1.7% de los pacientes tratados con SIMPONI ARIA y <1% de los pacientes tratados con placebo, y se produjeron elevaciones de ALT ≥ 3 × LSN a < 5 × LSN en el 2.9% de los pacientes tratados con SIMPONI ARIA y <1% de los pacientes tratados con placebo.
Dado que muchos de los pacientes en los ensayos de Fase 3 también tomaban medicamentos que causan elevaciones de las enzimas hepáticas (p. ej., medicamentos antiinflamatorios no esteroideos [AINE], MTX o profilaxis con isoniazida), la relación entre SIMPONI ARIA y la elevación de las enzimas hepáticas no está clara.
Trastornos autoinmunes y autoanticuerpos
En la Semana 20 del Ensayo AR, el 17% de los pacientes tratados con SIMPONI ARIA y el 13% de los pacientes de control fueron nuevos positivos para anticuerpos antinucleares (ANA). De estos pacientes, un paciente tratado con SIMPONI ARIA y ningún paciente tratado con control tuvieron anticuerpos anti-dsDNA recientemente positivos
[ver
Advertencias y precauciones (5.5)]
.
Reacciones a la administración
En la fase controlada del Ensayo AR hasta la Semana 24, el 1.1% de las infusiones de SIMPONI ARIA se asociaron con una reacción a la infusión en comparación con el 0.2% de las infusiones en el grupo de control. La reacción a la infusión más común en los pacientes tratados con SIMPONI ARIA fue el sarpullido. No se reportaron reacciones graves a la infusión.
Otras reacciones adversas
La Tabla 1 resume las reacciones adversas a medicamentos que ocurrieron a una tasa de al menos el 1% en el grupo de SIMPONI ARIA + MTX con una mayor incidencia que en el grupo de placebo + MTX durante el período controlado del Ensayo AR hasta la Semana 24.
| Placebo + MTX | SIMPONI ARIA + MTX | |
|---|---|---|
| Pacientes tratados | 197 | 463 |
| Reacción adversa | ||
| Infecciones e infestaciones | ||
| Infección del tracto respiratorio superior (como infección del tracto respiratorio superior, nasofaringitis, faringitis, laringitis y rinitis) | 12% | 13% |
| Infecciones víricas (como gripe y herpes) | 3% | 4% |
| Infecciones bacterianas | 0% | 1% |
| Bronquitis | 1% | 3% |
| Trastornos vasculares | ||
| Hipertensión | 2% | 3% |
| Trastornos de la piel y del tejido subcutáneo | ||
| Erupción | 1% | 3% |
| Trastornos generales y alteraciones en el lugar de administración | ||
| Pirexia | 1% | 2% |
| Trastornos sanguíneos y del sistema linfático | ||
| Leucopenia | 0% | 1% |
Otras reacciones adversas a medicamentos menos frecuentes en ensayos clínicos
Las reacciones adversas a medicamentos que no aparecen en la Tabla 1 o que ocurrieron < 1% en pacientes tratados con SIMPONI ARIA durante el ensayo RA hasta la semana 24 y que no aparecen en la sección de Advertencias y precauciones incluyeron los siguientes eventos, clasificados por clase de órgano del sistema:
Infecciones e infestaciones:Infección micótica superficial, sinusitis, absceso, infección del tracto respiratorio inferior (neumonía), pielonefritis
Pruebas:Aumento de la alanino aminotransferasa (ALT), aumento de la aspartato aminotransferasa (AST), disminución del recuento de neutrófilos
Trastornos del sistema nervioso:Mareo, parestesia
Trastornos gastrointestinales:Estreñimiento
Artritis psoriásica
El ensayo PsA evaluó a 480 pacientes
[ver
. Las reacciones adversas fueron similares a las observadas en pacientes con AR, con la excepción de la psoriasis (aparición o empeoramiento nuevos, palmar/plantar y pustular), que se produjo en <1% de los pacientes tratados con SIMPONI ARIA. La incidencia de las reacciones adversas notificadas en el ensayo PsA fue similar a la del ensayo RA, con las excepciones de una mayor incidencia en SIMPONI ARIA para el aumento de ALT (7,9% frente al 2,1% en placebo), el aumento de AST (5,4% frente al 2,1% en placebo) y la disminución del recuento de neutrófilos (4,6% frente al 2,1% en placebo).
Espondilitis anquilosante
El ensayo AS evaluó a 208 pacientes
[ver
. Las reacciones adversas fueron similares a las notificadas en pacientes con AR, con la excepción de la mayor incidencia de aumento de ALT, que se produjo en el 2,9% de los pacientes tratados con SIMPONI ARIA en comparación con ninguno de los pacientes tratados con placebo.
Pacientes pediátricos con artritis idiopática juvenil poliarticular y artritis psoriásica
El ensayo pJIA evaluó a 127 pacientes con JIA con poliartritis activa
[ver
Uso en poblaciones específicas (8.4) y
Las reacciones adversas observadas fueron consistentes con el perfil de seguridad establecido de SIMPONI ARIA en pacientes adultos con AR y PsA.
6.2 Inmunogenicidad
Al igual que con todas las proteínas terapéuticas, existe la posibilidad de inmunogenicidad. La detección de la formación de anticuerpos depende en gran medida de la sensibilidad y especificidad del ensayo. Además, la incidencia observada de positividad de anticuerpos (incluidos los anticuerpos neutralizantes) en un ensayo puede verse influenciada por varios factores, como la metodología del ensayo, el manejo de las muestras, el momento de la toma de muestras, los medicamentos concomitantes y la enfermedad subyacente. Por estas razones, la comparación de la incidencia de anticuerpos a golimumab en los ensayos descritos a continuación con la incidencia de anticuerpos en otros ensayos o con otros productos puede ser engañosa.
Utilizando un método de inmunoensayo enzimático (EIA), se detectaron anticuerpos contra golimumab en 13 (3%) pacientes tratados con golimumab después de la administración IV de SIMPONI ARIA en combinación con MTX hasta la semana 24 del ensayo RA, de los cuales todos fueron anticuerpos neutralizantes.
Se desarrolló y validó un método de inmunoensayo enzimático tolerante a fármacos (EIA tolerante a fármacos) para detectar anticuerpos contra golimumab. Este método es aproximadamente 16 veces más sensible que el método EIA original con menos interferencia de golimumab en suero. En aproximadamente 6 meses, la incidencia de anticuerpos contra golimumab con el método EIA tolerante a fármacos para los ensayos RA, PsA, AS y pJIA fue del 21%, 19%, 19% y 31%, respectivamente. Donde se probó, aproximadamente un tercio a la mitad fueron neutralizantes.
Los pacientes con AR, PsA, AS y pJIA que desarrollaron anticuerpos contra golimumab generalmente tuvieron concentraciones séricas en estado estacionario de golimumab más bajas en el valle
[ver
6.3 Experiencia postcomercialización
Las siguientes reacciones adversas se han identificado durante el uso posterior a la aprobación de golimumab. Debido a que estas reacciones se notifican voluntariamente a partir de una población de tamaño incierto, no siempre es posible estimar de forma fiable su frecuencia o establecer una relación causal con la exposición a golimumab:
Trastornos generales y afecciones en el lugar de administración:Reacciones relacionadas con la infusión
[ver
Advertencias y precauciones (5.11)]
Neoplasias benignas y malignas: Melanoma, carcinoma de células de Merkel
[ver
Advertencias y precauciones (5.2)]
Trastornos del sistema inmunitario: Reacciones de hipersensibilidad sistémicas graves (incluida la reacción anafiláctica)
[ver
Advertencias y precauciones (5.11)]
, sarcoidosis
Trastornos respiratorios, torácicos y mediastínicos:Enfermedad pulmonar intersticial
Trastornos de la piel y del tejido subcutáneo:Exfoliación de la piel, reacciones liquenoides, reacciones cutáneas ampollosas
7 INTERACCIONES MEDICAMENTOSAS
7.1 Metotrexato
SIMPONI ARIA debe utilizarse con MTX para el tratamiento de la AR
[véase
. Tras la administración IV, la administración concomitante de metotrexato disminuye la depuración de SIMPONI ARIA aproximadamente un 9% según el análisis farmacocinético (PK) poblacional. Además, la administración concomitante de metotrexato disminuye la depuración de SIMPONI ARIA al reducir el desarrollo de anticuerpos contra el golimumab.
7.2 Productos biológicos para AR, PsA, AS y pJIA
Se ha observado un mayor riesgo de infecciones graves en estudios clínicos de AR con otros bloqueadores del TNF utilizados en combinación con anakinra o abatacept, sin beneficio añadido; por lo tanto, no se recomienda el uso de SIMPONI ARIA con otros productos biológicos, incluidos abatacept o anakinra
[véase
Advertencias y precauciones (5.6y
5.7)]
. También se ha observado una mayor tasa de infecciones graves en pacientes con AR tratados con rituximab que recibieron posteriormente tratamiento con un bloqueador del TNF. No se recomienda el uso concomitante de SIMPONI ARIA con biológicos aprobados para tratar la AR, la PsA, la AS y la pJIA debido a la posibilidad de un mayor riesgo de infección.
7.3 Vacunas vivas/Agentes infecciosos terapéuticos
No deben administrarse vacunas vivas de forma concomitante con SIMPONI ARIA
[véase
Advertencias y precauciones (5.10)]
.
No deben administrarse agentes infecciosos terapéuticos de forma concomitante con SIMPONI ARIA
[véase
Advertencias y precauciones (5.10)]
.
Los lactantes nacidos de mujeres tratadas con SIMPONI ARIA durante el embarazo pueden tener un mayor riesgo de infección hasta por 6 meses. No se recomienda la administración de vacunas vivas a los lactantes expuestos a SIMPONI ARIA
in uterodurante 6 meses después de la última infusión de SIMPONI ARIA de la madre durante el embarazo
[véase
Advertencias y precauciones (5.10),
Uso en poblaciones específicas (8.1)]
.
7.4 Sustratos del citocromo P450
La formación de enzimas CYP450 puede verse suprimida por el aumento de los niveles de citocinas (p. ej., TNFα) durante la inflamación crónica. Por lo tanto, se espera que para una molécula que antagoniza la actividad de las citocinas, como el golimumab, la formación de enzimas CYP450 pueda normalizarse. Al iniciar o suspender SIMPONI ARIA en pacientes tratados con sustratos de CYP450 con un índice terapéutico estrecho, se recomienda controlar el efecto (p. ej., warfarina) o la concentración del fármaco (p. ej., ciclosporina o teofilina) y ajustar la dosis individual del producto farmacéutico según sea necesario.
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de Riesgo
No hay ensayos adecuados y bien controlados de SIMPONI ARIA en mujeres embarazadas. Los anticuerpos monoclonales, como el golimumab, se transportan a través de la placenta durante el tercer trimestre del embarazo y pueden afectar la respuesta inmunitaria en el lactante expuesto
in utero. Existen consideraciones clínicas para el uso de SIMPONI ARIA en mujeres embarazadas
[ver
. En un estudio de reproducción animal, el golimumab administrado por vía subcutánea a monos preñados, durante el período de organogénesis, en dosis que produjeron exposiciones aproximadamente 200 veces la dosis máxima recomendada para humanos (MRHD), no tuvo efectos fetales adversos.
Todos los embarazos tienen un riesgo de base de defectos de nacimiento, pérdida u otros resultados adversos. Se desconoce el riesgo de base estimado de defectos de nacimiento importantes y aborto espontáneo para las poblaciones indicadas. En la población general de EE. UU., los riesgos de base estimados de defectos de nacimiento importantes y aborto espontáneo en embarazos clínicamente reconocidos son del 2 al 4 % y del 15 al 20 %, respectivamente.
Consideraciones Clínicas
Reacciones adversas fetales/neonatales
El golimumab atraviesa la placenta durante el embarazo. Otro anticuerpo monoclonal bloqueante del TNF administrado durante el embarazo se detectó hasta por 6 meses en el suero de los lactantes. En consecuencia, estos lactantes pueden tener un mayor riesgo de infección. No se recomienda la administración de vacunas vivas a los lactantes expuestos a SIMPONI ARIA
in uterodurante 6 meses después de la última infusión de SIMPONI ARIA de la madre durante el embarazo
[ver
Advertencias y precauciones (5.10) y
Interacciones medicamentosas (7.3)]
.
Datos
Datos en humanos
Los datos limitados sobre el uso de SIMPONI ARIA en mujeres embarazadas de estudios observacionales, informes de casos publicados y vigilancia postcomercialización son insuficientes para informar un riesgo asociado al medicamento.
Datos en animales
En un estudio de toxicología del desarrollo embriofetal en el que se trató a monos cynomolgus preñados con golimumab durante el período de organogénesis desde los días de gestación (GD) 20 a 51, las exposiciones hasta 200 veces mayores que la exposición a la MRHD (sobre la base del área bajo la curva (AUC) con dosis subcutáneas maternas de hasta 50 mg/kg dos veces por semana) no produjeron evidencia de malformaciones fetales o embriotoxicidad. No hubo evidencia de toxicidad materna. Las muestras de sangre del cordón umbilical recolectadas al final del segundo trimestre mostraron que los fetos estuvieron expuestos al golimumab durante la gestación.
En un estudio de desarrollo pre y postnatal en el que se trató a monos cynomolgus preñados con golimumab desde el día de gestación 50 hasta el día posparto 33, las concentraciones máximas de fármaco hasta 33 veces mayores que las encontradas con la MRHD (sobre la base de la concentración máxima en sangre (C
max) en estado estacionario con dosis subcutáneas maternas de hasta 50 mg/kg dos veces por semana) no se asociaron con ninguna evidencia de defectos del desarrollo en los lactantes. No hubo evidencia de toxicidad materna. El golimumab estuvo presente en el suero fetal al final del segundo trimestre y en el suero neonatal desde el momento del nacimiento y hasta 6 meses después del parto.
8.2 Lactancia
Resumen de Riesgo
No hay información sobre la presencia de SIMPONI ARIA en la leche materna, los efectos en los lactantes amamantados o los efectos en la producción de leche. Se sabe que la IgG materna está presente en la leche materna. Si el golimumab se transfiere a la leche materna, se desconocen los efectos de la exposición local en el tracto gastrointestinal y la posible exposición sistémica limitada en el lactante al golimumab. Se deben considerar los beneficios para el desarrollo y la salud de la lactancia materna junto con la necesidad clínica de la madre de SIMPONI ARIA y cualquier posible efecto adverso en los lactantes amamantados por SIMPONI ARIA o por la enfermedad materna subyacente.
Datos
Datos en animales
En el estudio de desarrollo pre y postnatal en monos cynomolgus en el que se administró golimumab por vía subcutánea durante el embarazo y la lactancia, se detectó golimumab en la leche materna en concentraciones aproximadamente 400 veces menores que las concentraciones séricas maternas.
8.4 Uso pediátrico
La seguridad y eficacia de SIMPONI ARIA para la artritis idiopática juvenil poliarticular activa y la PsA se han establecido en pacientes pediátricos de 2 años o más.
El uso de SIMPONI ARIA en estos grupos de edad se basa en evidencia de estudios adecuados y bien controlados de SIMPONI ARIA en adultos con AR y PsA, datos farmacocinéticos de pacientes adultos con AR y PsA y pacientes pediátricos con AIJ con poliartritis activa, y datos de seguridad de un estudio clínico en 127 pacientes pediátricos de 2 a < 18 años de edad con AIJ con poliartritis activa. Las concentraciones previas a la dosis (valle) observadas son generalmente comparables entre adultos con AR y PsA y pacientes pediátricos con AIJ con poliartritis activa, y se espera que la exposición farmacocinética sea comparable entre los pacientes adultos con PsA y los pacientes pediátricos con PsA
[ver
.
Se han notificado neoplasias malignas, algunas mortales, entre niños, adolescentes y adultos jóvenes que recibieron tratamiento con golimumab y otros agentes bloqueantes del TNF
[ver
Advertencias y precauciones (5.2)].
No se ha establecido la seguridad y eficacia en pacientes pediátricos menores de 2 años en AIJp o en PsA. No se ha establecido la seguridad y eficacia de SIMPONI ARIA en pacientes pediátricos con afecciones distintas de AIJp y PsA.
8.5 Uso en geriátricos
En el ensayo RA, el número de pacientes de 65 años o más fue demasiado pequeño para realizar comparaciones con pacientes más jóvenes tratados con SIMPONI ARIA. Debido a que hay una mayor incidencia de infecciones en la población geriátrica en general, se debe tener precaución al tratar a pacientes geriátricos con SIMPONI ARIA.
10 SOBREDOSIS
En un estudio clínico, 5 pacientes recibieron infusiones únicas de hasta 1000 mg de SIMPONI ARIA sin reacciones adversas graves u otras reacciones significativas.
11 DESCRIPCIÓN
Golimumab es un anticuerpo monoclonal IgG1қ humano específico para el factor de necrosis tumoral alfa (TNFα) humano que exhibe múltiples glicoformas con masas moleculares de aproximadamente 150 a 151 kilodaltons. Golimumab se creó utilizando ratones genéticamente modificados inmunizados con TNF humano, lo que resultó en un anticuerpo con regiones variables y constantes de anticuerpos derivadas de humanos. Golimumab se produce mediante una línea celular recombinante cultivada por perfusión continua y se purifica mediante una serie de pasos que incluyen medidas para inactivar y eliminar virus.
La inyección de SIMPONI ARIA
®(golimumab) es una solución estéril del anticuerpo golimumab suministrada en un vial de vidrio de 4 ml para infusión intravenosa.
SIMPONI ARIA es una solución incolora a amarillo claro, sin conservantes, con un pH de aproximadamente 5,5. SIMPONI ARIA no está hecho con látex de caucho natural. Cada vial de 4 ml de SIMPONI ARIA contiene 50 mg de golimumab, L-histidina (1,14 mg), monohidrocloruro de L-histidina monohidrato (6,42 mg), polisorbato 80 (0,6 mg), sorbitol (180 mg) y agua para inyección.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
Golimumab es un anticuerpo monoclonal humano que se une a las formas bioactivas solubles y transmembrana del TNFα humano. Esta interacción impide la unión del TNFα a sus receptores, inhibiendo así la actividad biológica del TNFα (una proteína citocina). No hubo evidencia de unión del anticuerpo golimumab a otros ligandos de la superfamilia del TNF; en particular, el anticuerpo golimumab no se unió ni neutralizó la linfotoxina humana. Golimumab no lisó los monocitos humanos que expresaban TNF transmembrana en presencia de complemento o células efectoras.
Se ha implicado a los niveles elevados de TNFα en sangre, sinovia y articulaciones en la fisiopatología de varias enfermedades inflamatorias crónicas como la artritis reumatoide, la artritis psoriásica y la espondilitis anquilosante. El TNFα es un mediador importante de la inflamación articular que es característica de estas enfermedades. Golimumab moduló los efectos biológicos in vitro mediados por el TNF en varios bioensayos, incluida la expresión de proteínas de adhesión responsables de la infiltración de leucocitos (E-selectina, ICAM-1 y VCAM-1) y la secreción de citocinas proinflamatorias (IL-6, IL-8, G-CSF y GM-CSF). Se desconoce la relevancia clínica de estos hallazgos.
12.2 Farmacodinamia
Después del tratamiento con SIMPONI ARIA en pacientes con AR, se observaron disminuciones desde el valor basal en el inhibidor tisular de la metaloproteinasa-1 (TIMP-1), la metaloproteinasa de matriz-1 (MMP-1), la metaloproteinasa de matriz-3 (MMP-3), la resistina, la interleucina-6 (IL-6), la proteína inflamatoria de macrófagos-1 (MIP-1b), el factor de crecimiento endotelial vascular (VEGF), el amiloide sérico A (SAA), S100A12 y la proteína C reactiva de alta sensibilidad (hsCRP). Por el contrario, se observaron aumentos desde el valor basal en la fosfatasa ácida resistente al tartrato (TRAP-5b). Se desconoce la relevancia clínica de esta información.
12.3 Farmacocinética
Golimumab exhibió una farmacocinética aproximadamente proporcional a la dosis en pacientes con AR en el rango de dosis de 0,1 a 10,0 mg/kg después de una dosis intravenosa única.
Absorción
Después de una administración intravenosa única de 2 mg/kg de SIMPONI ARIA, se observó una Cmax media de 44,4 ± 11,3 mcg/mL en pacientes con AR. No hay datos que comparen directamente la administración intravenosa de 2 mg/kg y la administración subcutánea de 50 mg.
Distribución
Después de una administración intravenosa única de 2 mg/kg de SIMPONI ARIA, el volumen de distribución medio se estimó en 115 ± 19 mL/kg en sujetos sanos y en 151 ± 61 mL/kg en pacientes con AR. El volumen de distribución de golimumab puede indicar que golimumab se distribuye principalmente en el sistema circulatorio con una distribución extravascular limitada.
Eliminación
Después de una administración intravenosa única de 2 mg/kg de SIMPONI ARIA, el aclaramiento sistémico de golimumab se estimó en 6,9 ± 2,0 mL/día/kg en sujetos sanos y en 7,6 ± 2,0 mL/día/kg en pacientes con AR. La semivida terminal media se estimó en 12 ± 3 días en sujetos sanos y la semivida terminal media en pacientes con AR fue de 14 ± 4 días.
Dosis múltiples
Cuando se administraron 2 mg/kg de SIMPONI ARIA por vía intravenosa a pacientes con AR en las semanas 0, 4 y cada 8 semanas a partir de entonces, las concentraciones séricas alcanzaron el estado estacionario en la semana 12. Con el uso concomitante de MTX, el tratamiento con 2 mg/kg de golimumab cada 8 semanas dio como resultado una concentración sérica media en el valle en estado estacionario de aproximadamente 0,4 ± 0,4 mcg/mL en pacientes con AR activa. La concentración sérica media en el valle en estado estacionario en pacientes con Artritis Psoriásica fue de 0,7 ± 0,6 mcg/mL. La concentración sérica media en el valle en estado estacionario en pacientes con EA fue de 0,8 ± 0,6 mcg/mL.
Los pacientes con AR, Artritis Psoriásica y EA que desarrollaron anticuerpos contra golimumab generalmente tuvieron concentraciones séricas en el valle en estado estacionario más bajas de golimumab [véase Reacciones adversas (6.2)].
Poblaciones específicas
No se realizó ningún estudio formal del efecto del deterioro renal o hepático en la farmacocinética de golimumab.
Peso corporal
Después de la administración intravenosa, los pacientes con mayor peso corporal tendieron a tener concentraciones séricas de golimumab ligeramente más altas que los pacientes con pesos corporales más bajos cuando golimumab se administró en base a mg/kg (peso corporal). Sin embargo, según el análisis farmacocinético poblacional, no hubo diferencias clínicamente relevantes en la exposición a golimumab después de la administración intravenosa de 2 mg/kg de SIMPONI ARIA en pacientes adultos en un rango de diferentes pesos corporales.
Pediatría
Cuando se administraron 80 mg/m2 de SIMPONI ARIA por vía intravenosa a pacientes con ARJ con poliartritis activa en las semanas 0, 4 y cada 8 semanas a partir de entonces, las concentraciones séricas alcanzaron el estado estacionario en la semana 12. Con el uso concomitante de MTX, el tratamiento con 80 mg/m2 de SIMPONI ARIA dio como resultado una concentración sérica media en el valle en estado estacionario de golimumab de aproximadamente 0,5 ± 0,4 mcg/mL y un AUC media en estado estacionario de 425 ± 125 mcg∙día/mL en pacientes con ARJ con poliartritis activa. En general, las concentraciones en el valle de golimumab en estado estacionario observadas en pacientes con ARJ con poliartritis activa se encontraban dentro del rango de las observadas para la AR y la Artritis Psoriásica adultas después de la administración de SIMPONI ARIA.
De acuerdo con los datos intravenosos en pacientes adultos con AR, los análisis farmacocinéticos poblacionales para SIMPONI ARIA intravenoso en pARJ revelaron que no hubo diferencias clínicamente relevantes en la exposición a golimumab después de la administración intravenosa de 80 mg/m2 de SIMPONI ARIA en pacientes pediátricos en un rango de edad y diferentes pesos corporales. El efecto de la respuesta inmunitaria en el aclaramiento de golimumab en pacientes con ARJ con poliartritis activa fue comparable al de los adultos con AR.
Estudios de interacción medicamentosa
No se han realizado estudios específicos de interacción medicamentosa con SIMPONI ARIA.
El análisis farmacocinético poblacional indicó que el uso concomitante de MTX, AINE, corticosteroides orales o sulfasalazina (SSZ) no influyó significativamente en el aclaramiento de golimumab después de la administración IV.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogenicidad, Mutagénesis, Deterioro de la Fertilidad
No se han realizado estudios a largo plazo en animales con golimumab para evaluar su potencial carcinogénico. No se han realizado estudios de mutagenicidad con golimumab. Un estudio de fertilidad realizado en ratones utilizando un anticuerpo anti-TNFα análogo administrado por vía intravenosa en dosis de hasta 40 mg/kg una vez por semana no mostró deterioro de la fertilidad.
14 ESTUDIOS CLÍNICOS
14.1 Artritis Reumatoide
La eficacia y seguridad de SIMPONI ARIA se evaluaron en un ensayo controlado, doble ciego, aleatorizado, multicéntrico (Ensayo AR, NCT00973479) en 592 pacientes ≥ 18 años de edad con AR de moderada a severamente activa a pesar del tratamiento concomitante con MTX y que no habían sido tratados previamente con un bloqueador de TNF biológico. Los pacientes fueron diagnosticados de acuerdo con los criterios del Colegio Americano de Reumatología (ACR), al menos 3 meses antes de la administración del agente del estudio y se les requirió tener al menos 6 articulaciones inflamadas y 6 sensibles. Los pacientes fueron aleatorizados para recibir SIMPONI ARIA 2 mg/kg (N=395) o placebo (N=197) durante una infusión intravenosa de 30 minutos en las Semanas 0, 4 y cada 8 semanas a partir de entonces, además de su dosis semanal de mantenimiento de MTX (15–25 mg). Todos los pacientes que recibieron placebo + MTX recibieron SIMPONI ARIA + MTX después de la Semana 24, pero el ensayo permaneció ciego hasta que todos los pacientes completaron 108 semanas de tratamiento. Se recopilaron y analizaron datos de eficacia hasta la Semana 52. Se permitió a los pacientes continuar con dosis estables concomitantes de corticosteroides en dosis bajas (equivalente a ≤ 10 mg de prednisona al día) y/o AINE. Se prohibió el uso de otros FARME, incluidos agentes citotóxicos u otros productos biológicos.
El criterio de valoración principal en el Ensayo AR fue el porcentaje de pacientes que lograron una respuesta ACR 20 en la Semana 14. En el Ensayo AR, la mayoría de los sujetos eran mujeres (82%) y caucásicos (80%) con una mediana de edad de 52 años y un peso medio de 70 kg. La mediana de duración de la enfermedad fue de 4,7 años, y el 50% de los pacientes utilizaron al menos un FARME diferente al MTX en el pasado. Al inicio del estudio, el 81% de los pacientes recibieron AINE concomitantes y el 81% de los pacientes recibieron corticosteroides en dosis bajas (equivalente a ≤ 10 mg de prednisona al día). La mediana del DAS28-PCR basal fue de 5,9 y la mediana de la puntuación de van der Heijde-Sharp al inicio del estudio fue de 28,5.
Respuesta Clínica
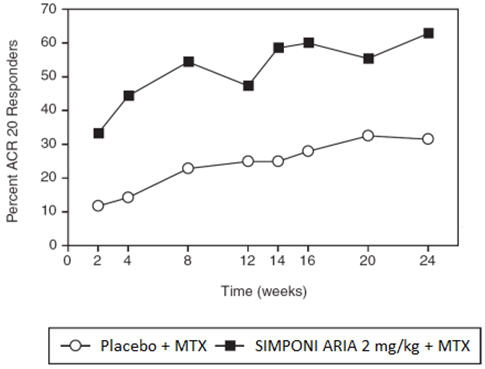
Un mayor porcentaje de pacientes tratados con la combinación de SIMPONI ARIA + MTX lograron ACR 20 en la Semana 14 y ACR 50 en la Semana 24 en comparación con los pacientes tratados con placebo + MTX, como se muestra en la Tabla 2. El porcentaje de pacientes que lograron respuestas ACR 20 por visita para el Ensayo AR se muestra en la Figura 1.
| Ensayo AR AR activa, a pesar de MTX |
|||
|---|---|---|---|
| Placebo + MTX | SIMPONI ARIA + MTX | 95% CI | |
| N | 197 | 395 | |
| ACR 20 | |||
| Semana 14 | 25% | 59% | 25.9, 41.4 |
| Semana 24 | 32% | 63% | 23.3, 39.4 |
| ACR 50 | |||
| Semana 14 | 9% | 30% | 15.3, 27.2 |
| Semana 24 | 13% | 35% | 15.1, 28.4 |
| ACR 70 | |||
| Semana 14 | 3% | 12% | 5.3, 13.4 |
| Semana 24 | 4% | 18% | 8.8, 18.1 |
 |
||
| El análisis se basa en la población por intención de tratar. Se realizó la última observación trasladada para los datos faltantes. Los pacientes que interrumpieron el tratamiento debido a la falta de eficacia se contaron como no respondedores, al igual que los pacientes que comenzaron a tomar medicamentos prohibidos o no lograron al menos una mejora del 10% en el recuento de articulaciones en la Semana 16. |
La mejoría en todos los componentes de los criterios de respuesta ACR para el grupo de SIMPONI ARIA + MTX fue mayor en comparación con el grupo de placebo + MTX en el Ensayo AR como se muestra en la Tabla 3.
| Ensayo AR AR activa, a pesar del MTX |
||
|---|---|---|
| Placebo + MTX | SIMPONI ARIA + MTX | |
| Nota: Todos los valores son medias. | ||
|
||
| N | 197 | 395 |
| Número de articulaciones inflamadas (0–66) | ||
| Línea base | 15 | 15 |
| Semana 14 | 11 | 6 |
| Número de articulaciones sensibles (0–68) | ||
| Línea base | 26 | 26 |
| Semana 14 | 20 | 13 |
| Evaluación del dolor por parte del paciente (0–10) | ||
| Línea base | 6.5 | 6.5 |
| Semana 14 | 5.6 | 3.9 |
| Evaluación global de la actividad de la enfermedad por parte del paciente (0–10) | ||
| Línea base | 6.5 | 6.5 |
| Semana 14 | 5.5 | 4.0 |
| Evaluación global de la actividad de la enfermedad por parte del médico (0–10) | ||
| Línea base | 6.3 | 6.2 |
| Semana 14 | 4.9 | 3.1 |
| Puntuación HAQ (0–3) | ||
| Línea base | 1.6 | 1.6 |
| Semana 14 | 1.4 | 1.1 |
| CRP (mg/dL) (0–1) | ||
| Línea base | 2.2 | 2.8 |
| Semana 14 | 1.8 | 0.9 |
En la semana 14, una mayor proporción de pacientes tratados con SIMPONI ARIA + MTX lograron un nivel bajo de actividad de la enfermedad según la medición de un DAS28-CRP inferior a 2,6 en comparación con el grupo de placebo + MTX (15 % en comparación con el 5 %; IC del 95 % para la diferencia [6,3 %, 15,5 %]).
Respuesta radiográfica
En el ensayo RA, el daño estructural de las articulaciones se evaluó radiográficamente y se expresó como un cambio en la puntuación de Sharp modificada por van der Heijde (vdH-S) y sus componentes, la puntuación de erosión y la puntuación de estrechamiento del espacio articular (JSN), en la semana 24 en comparación con el valor inicial. El grupo de tratamiento con SIMPONI ARIA + MTX inhibió la progresión del daño estructural en comparación con el placebo + MTX, según la evaluación de la puntuación total de vdH-S, como se muestra en la Tabla 4.
| Placebo + MTX (N=197) |
SIMPONI ARIA + MTX (N=395) |
|
|---|---|---|
| Media | Media | |
| Cambiar la puntuación total de vdH-S | 1.1 | 0.03 |
| Cambiar la puntuación de erosión | 0.5 | -0.1 |
| Cambiar la puntuación de JSN | 0.6 | 0.1 |
En la semana 24, una mayor proporción de pacientes en el grupo de SIMPONI ARIA + MTX (71%) no tuvo progresión del daño estructural (cambio en la puntuación total de vdH-S ≤ 0), en comparación con el 57% de los pacientes en el grupo de placebo + MTX. En la semana 52, el cambio medio desde el inicio en la puntuación total de vdH-S fue de 1,2 en los pacientes originalmente aleatorizados a placebo + MTX que cambiaron a SIMPONI ARIA + MTX en la semana 16 o 24, y de 0,1 en los pacientes originalmente aleatorizados a SIMPONI ARIA + MTX que permanecieron en tratamiento activo.
Respuesta de la función física en pacientes con AR
La función física se evaluó mediante el índice de discapacidad del Cuestionario de Evaluación de la Salud (HAQ-DI). En la semana 14, el grupo de SIMPONI ARIA + MTX mostró una mayor mejoría media en el HAQ-DI en comparación con el placebo + MTX (0,5 en comparación con 0,2; IC del 95% para la diferencia [0,2, 0,4]).
Otros resultados relacionados con la salud
El estado de salud general se evaluó mediante la Encuesta de Salud de Formato Corto de 36 ítems (SF-36). En el ensayo RA, los pacientes que recibieron SIMPONI ARIA + MTX demostraron una mayor mejoría desde el inicio en comparación con el placebo + MTX en el resumen del componente físico (PCS), las puntuaciones del resumen del componente mental (MCS) y en los 8 dominios del SF-36.
La fatiga se evaluó mediante la puntuación de la Evaluación Funcional de la Terapia de Enfermedades Crónicas-Fatiga (FACIT-F) en el Ensayo RA. El tratamiento con SIMPONI ARIA resultó en una mejoría de la fatiga medida por FACIT-F.
14.2 Artritis psoriásica
La eficacia y seguridad de SIMPONI ARIA se evaluaron en un ensayo multicéntrico, aleatorizado, doble ciego, controlado con placebo en 480 pacientes ≥ 18 años de edad con artritis psoriásica activa a pesar del tratamiento con AINE o DMARD (Ensayo PsA, NCT02181673). No se permitió el tratamiento previo con un biológico. Los pacientes de este ensayo tenían un diagnóstico de APs durante al menos seis meses y presentaban síntomas de enfermedad activa [≥5 articulaciones inflamadas y ≥5 articulaciones sensibles y un nivel de PCR de ≥ 0,6 mg/dL]. Los pacientes fueron aleatorizados para recibir SIMPONI ARIA 2 mg/kg (N=241) o placebo (N=239) en forma de infusión intravenosa de 30 minutos en las semanas 0, 4, 12 y 20. Todos los pacientes que recibieron placebo recibieron SIMPONI ARIA en la semana 24, la semana 28 y cada 8 semanas a partir de entonces hasta la semana 52. Los pacientes del grupo de tratamiento con SIMPONI ARIA continuaron recibiendo infusiones de SIMPONI ARIA en la semana 28 y cada 8 semanas hasta la semana 52.
Se permitió a los pacientes continuar con dosis estables de MTX, AINE y corticosteroides orales en dosis bajas (equivalentes a ≤ 10 mg de prednisona al día) durante el ensayo. Se prohibió el uso de otros DMARD, incluidos los agentes citotóxicos u otros biológicos.
El criterio de valoración principal fue el porcentaje de pacientes que lograron una respuesta ACR 20 en la semana 14.
Se incluyeron pacientes con cada subtipo de APs, incluyendo artritis poliarticular con ausencia de nódulos reumatoides (44%), artritis periférica asimétrica (19%), afectación de las articulaciones interfalángicas distales (8,1%), espondilitis con artritis periférica (25%) y artritis mutilante (4,8%). La mediana de la duración de la enfermedad APs fue de 3,5 años, el 86% de los pacientes había utilizado previamente MTX y el 35% de los pacientes recibió al menos otro DMARD en el pasado. Al inicio del estudio, el 76% y el 54% de los pacientes presentaban entesitis y dactilitis, respectivamente. La mediana de la puntuación total de vdH-S modificada al inicio del estudio fue de 15,5. Durante el ensayo, los medicamentos concomitantes utilizados incluyeron MTX (70%), corticosteroides orales (28%) y AINE (71%).
Respuesta clínica
En el Ensayo PsA, el tratamiento con SIMPONI ARIA, en comparación con el placebo, resultó en una mejoría significativa de los signos y síntomas, como lo demuestra el porcentaje de pacientes con una respuesta ACR 20 en la semana 14 (véase
Tabla 5). Se observaron respuestas ACR 20 similares en la semana 24 en pacientes con diferentes subtipos de APs. Las respuestas ACR 20 observadas en los grupos tratados con SIMPONI ARIA fueron similares en los pacientes que recibían o no MTX concomitante.
| Placebo | SIMPONI ARIA | Diferencia con respecto al placebo (IC del 95%) |
|
|---|---|---|---|
| (N
*=239) |
(N
*=241) |
||
| Nota: El análisis se basa en la población por intención de tratar. Se realizó la última observación trasladada para los datos parcialmente faltantes y la imputación de no respondedores para los datos completamente faltantes. Los pacientes que interrumpieron el tratamiento debido a la falta de eficacia fueron imputados como no respondedores, al igual que los pacientes que comenzaron a tomar medicación prohibida, aumentaron los corticosteroides o el MTX, o no lograron al menos una mejoría del 5% en el recuento articular en la semana 16 y recibieron una intervención con medicación concomitante (corticosteroides, MTX o AINE). | |||
| Respuesta ACR 20 | |||
| Semana 14 | 22% | 75% | 53%
†
|
| Semana 24 | 24% | 77% | 53% (45, 60) |
| Respuesta ACR 50 | |||
| Semana 14 | 6.3% | 44% | 37% (30, 44) |
| Semana 24 | 6.3% | 54% | 47% (40, 54) |
| Respuesta ACR 70 | |||
| Semana 14 | 2.1% | 25% | 22% (17, 28) |
| Semana 24 | 3.3% | 33% | 29% (23, 36) |
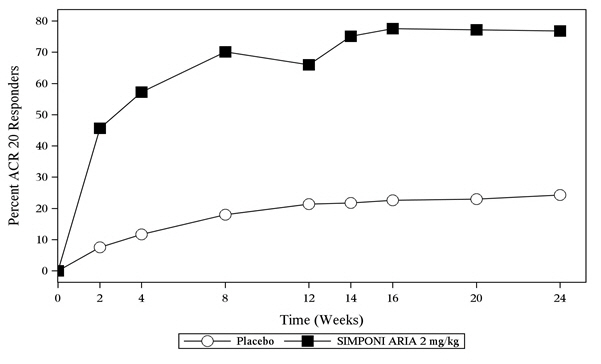
El porcentaje de pacientes que lograron respuestas ACR20 por visita hasta la semana 24 para el ensayo PsA se muestra en la Figura 2.
| Figura 2: Ensayo PsA – Porcentaje de pacientes que lograron una respuesta ACR20 hasta la semana 24 | |
|
|
|
| El análisis se basa en la población por intención de tratar. Se realizó la última observación trasladada para datos parcialmente faltantes e imputación de no respondedores para datos completamente faltantes. Los pacientes que interrumpieron el tratamiento debido a la falta de eficacia se imputaron como no respondedores, al igual que los pacientes que comenzaron a tomar medicamentos prohibidos, aumentaron los corticosteroides o MTX, o no lograron al menos una mejora del 5% en el recuento de articulaciones en la semana 16 y recibieron una intervención con medicación concomitante (corticosteroides, MTX o AINE). | |
La Tabla 6 muestra la mejora en los componentes individuales de los criterios de respuesta ACR para los grupos de SIMPONI ARIA y placebo en el Ensayo PsA.
| Placebo N *=239 |
SIMPONI ARIA N *=241 |
|||
|---|---|---|---|---|
| Línea de base | Cambio en la semana 14 respecto a la línea de base | Línea de base | Cambio en la semana 14 respecto a la línea de base | |
| Nota: Todos los valores son medias. | ||||
| Componentes de ACR | ||||
| N.º de articulaciones inflamadas (0–66) | 14 | -2.9 | 14 | -11 |
| Número de articulaciones sensibles (0–68) | 26 | -4.2 | 25 | -15 |
| Evaluación del dolor por parte del paciente (0–100 mm) | 64 | -11 | 63 | -31 |
| Evaluación global del paciente (0–100 mm) | 63 | -11 | 65 | -32 |
| Evaluación global del médico (0–100 mm) | 64 | -13 | 62 | -39 |
| Índice de discapacidad (HAQ) (0–3) | 1.3 | -0.13 | 1.3 | -0.60 |
| hsCRP (mg/L) | 20 | -2.9 | 19 | -16 |
Los pacientes con entesitis al inicio del estudio fueron evaluados para determinar la mejoría media utilizando el Índice de Entesitis de Leeds (LEI) en una escala de 0 a 6. Los pacientes tratados con SIMPONI ARIA mostraron una mejoría significativamente mayor en la entesitis, con una reducción media de 1,8 en comparación con una reducción media de 0,8 en los pacientes tratados con placebo en la semana 14. Los pacientes con dactilitis al inicio del estudio fueron evaluados para determinar la mejoría media en una escala de 0 a 60. Los pacientes tratados con SIMPONI ARIA mostraron una mejoría significativamente mayor, con una reducción media de 7,8 en comparación con una reducción media de 2,8 en los pacientes tratados con placebo en la semana 14.
Respuesta radiográfica
En el ensayo PsA, el daño estructural de las articulaciones se evaluó radiográficamente y se expresó como un cambio con respecto al valor inicial en la semana 24 en la puntuación total vdH-S modificada y sus componentes, la puntuación de erosión y la puntuación JSN. SIMPONI ARIA inhibió la progresión del daño estructural en comparación con el placebo, según la evaluación de la puntuación total vdH-S modificada, como se muestra en la Tabla 7.
| Placebo N *=237 |
SIMPONI ARIA N *=237 |
Diferencia con respecto al placebo (95% CI) |
|
|---|---|---|---|
| Media | Media | ||
| Nota: Todos los valores son medias. | |||
|
|||
| Cambio en la puntuación total vdH-S modificada | 2.0 | -0.4 | -2.3 (-2.9, -1.7) |
En la semana 24, una mayor proporción de pacientes en el grupo de SIMPONI ARIA (72%) no tuvo progresión del daño estructural (cambio en la puntuación total vdH-S modificada ≤ 0), en comparación con el 43% de los pacientes en el grupo placebo.
Función Física y Respuestas
La mejora en la función física evaluada por el Índice de Discapacidad del Cuestionario de Evaluación de la Salud (HAQ-DI) demostró que la proporción de pacientes que lograron una mejora clínicamente significativa de ≥ 0.3 en la puntuación HAQ-DI desde el inicio fue mayor en el grupo tratado con SIMPONI ARIA en comparación con el placebo en la semana 14 (69% en comparación con 32%).
Otros Resultados Relacionados con la Salud
El estado de salud general se evaluó mediante la Encuesta de Salud de Formato Corto de 36 ítems (SF-36). En el Ensayo PsA, los pacientes que recibieron SIMPONI ARIA demostraron una mayor mejora desde el inicio en comparación con el placebo en el resumen del componente físico, las puntuaciones del resumen del componente mental y en los 8 dominios del SF-36.
La fatiga se evaluó mediante la puntuación de la Evaluación Funcional de la Terapia de Enfermedades Crónicas-Fatiga (FACIT-F) en el Ensayo PsA. El tratamiento con SIMPONI ARIA resultó en una mejora en la fatiga medida por FACIT-F.
Tratamiento de Pacientes Pediátricos
La eficacia de SIMPONI ARIA en pacientes pediátricos con PsA se basa en la exposición farmacocinética y la extrapolación de la eficacia establecida de SIMPONI ARIA en pacientes adultos con PsA en el Ensayo PsA
[ver
Uso en Poblaciones Específicas (8.4),
14.3 Espondilitis Anquilosante
La eficacia y seguridad de SIMPONI ARIA se evaluaron en un ensayo multicéntrico, aleatorizado, doble ciego, controlado con placebo (Ensayo AS, NCT02186873) en 208 pacientes ≥ 18 años de edad con espondilitis anquilosante (EA) activa y respuesta inadecuada o intolerancia a los AINE. Los pacientes tenían un diagnóstico de EA definitiva durante al menos 3 meses según los criterios modificados de Nueva York. Los pacientes tenían síntomas de enfermedad activa [Índice de Actividad de la Enfermedad de Bath AS (BASDAI) ≥ 4, EAV para el dolor de espalda total de ≥ 4, en escalas de 0 a 10 cm (0 a 100 mm) y un nivel de PCRhs de ≥ 0.3 mg/dL (3 mg/L)]. Los pacientes fueron aleatorizados para recibir SIMPONI ARIA 2 mg/kg (N=105) o placebo (N=103) como una infusión intravenosa de 30 minutos en las Semanas 0, 4 y 12. Todos los pacientes con placebo recibieron SIMPONI ARIA en la Semana 16, Semana 20 y cada 8 semanas a partir de entonces hasta la Semana 52. Los pacientes en el grupo de tratamiento con SIMPONI ARIA continuaron recibiendo infusiones de SIMPONI ARIA en la Semana 20 y cada 8 semanas hasta la Semana 52. Se permitió a los pacientes continuar con dosis estables de MTX, SSZ, hidroxicloroquina (HCQ), corticosteroides orales en dosis bajas (equivalente a ≤ 10 mg de prednisona por día) y/o AINE concomitantes durante el ensayo. Se prohibió el uso de otros FARME, incluidos agentes citotóxicos u otros productos biológicos.
El criterio de valoración principal fue el porcentaje de pacientes que lograron una respuesta ASAS (Assessment in Ankylosing Spondylitis) 20 en la semana 16.
En el Ensayo AS, la mediana de la duración de la enfermedad AS fue de 2.8 años, la mediana de la duración del dolor lumbar inflamatorio fue de 8 años, el 90% fueron HLA-B27 positivos, el 8.2% se sometió a una cirugía o procedimiento articular previo, el 5.8% tenía anquilosis completa de la columna, el 14% había recibido terapia previa con un bloqueador del TNF biológico (que no sea golimumab) y la suspendió por razones distintas a la falta de eficacia dentro de las primeras 16 semanas de tratamiento (fracaso primario), y el 76% recibió al menos un FARME en el pasado. Durante el ensayo, el uso de medicamentos concomitantes fue AINE (88%), SSZ (38%), corticosteroides (26%), MTX (18%) y HCQ (0.5%).
Respuesta Clínica
En el Ensayo AS, el tratamiento con SIMPONI ARIA, en comparación con el placebo, resultó en una mejora significativa en los signos y síntomas, como lo demuestra el porcentaje de pacientes con una respuesta ASAS 20 en la Semana 16 (ver
Tabla 8).
| Placebo N *=103 |
SIMPONI ARIA N *=105 |
Diferencia de Tratamiento (95% CI) |
|
|---|---|---|---|
| Nota: El análisis se basa en la población por intención de tratar. Se realizó la última observación trasladada para datos parcialmente faltantes e imputación de no respondedores para datos completamente faltantes. | |||
| Respondedores | |||
| ASAS 20 | 26% | 73% | 47%
†
|
| ASAS 40 | 8.7% | 48% | 39% (28, 50) |
El porcentaje de pacientes que lograron respuestas ASAS 20 por visita hasta la semana 16 para el Ensayo AS se muestra en la Figura 3.
| Figura 3: Ensayo AS – Porcentaje de pacientes que logran una respuesta ASAS 20 hasta la semana 16 | |
|
|
|
| El análisis se basa en la población por intención de tratar. Se realizó la última observación trasladada para datos parcialmente faltantes e imputación de no respondedores para datos completamente faltantes. | |
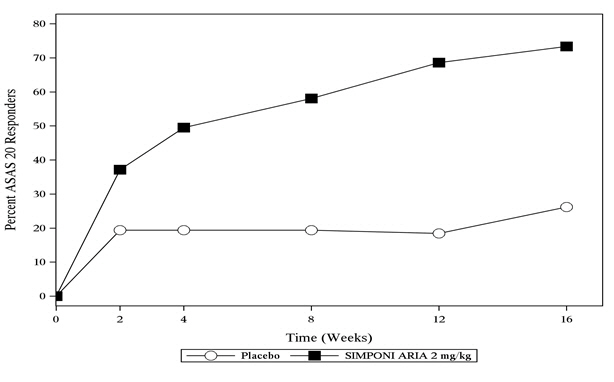
La Tabla 9 muestra la mejora en los componentes de los criterios de respuesta ASAS y otras medidas de la actividad de la enfermedad para los grupos de SIMPONI ARIA y placebo en el Ensayo AS.
| Placebo N *=103 |
SIMPONI ARIA N *=105 |
|||
|---|---|---|---|---|
| Línea base | Cambio en la semana 16 respecto a la línea base | Línea base | Cambio en la semana 16 respecto a la línea base | |
| Nota: Todos los valores son medias. | ||||
|
||||
| Criterios de respuesta ASAS 20 | ||||
| Evaluación global del paciente de la actividad de la enfermedad (0–100 mm) | 71 | -8.3 | 73 | -34 |
| Dolor de espalda total (0–100 mm) | 73 | -12 | 72 | -32 |
| BASFI (0–10) | 6.1 | -0.5 | 6.3 | -2.4 |
| Inflamación (0–10) | 7.4 | -1.1 | 7.3 | -3.6 |
| Puntuación BASDAI | 7.1 | -1.1 | 7.1 | -3.1 |
| BASMI | 5.0 | -0.1 | 5.0 | -0.4 |
| hsCRP (mg/L) | 19 | -2.3 | 20 | -17 |
En la semana 16, un mayor porcentaje de pacientes tratados con SIMPONI ARIA logró un bajo nivel de actividad de la enfermedad (<2 [en una escala de 0 a 10 cm] en los cuatro dominios ASAS) en comparación con los pacientes tratados con placebo (16.2% vs. 3.9%).
Otros Resultados Relacionados con la Salud
El estado de salud general se evaluó mediante la Encuesta de Salud de Formato Corto de 36 ítems (SF-36). En el Ensayo AS, los pacientes que recibieron SIMPONI ARIA demostraron una mayor mejoría desde el inicio en comparación con el placebo en las puntuaciones resumidas de los componentes físico y mental y en los 8 dominios del SF-36.
Los pacientes tratados con SIMPONI ARIA mostraron una mejoría significativa en comparación con los pacientes tratados con placebo en la calidad de vida relacionada con la salud, según la evaluación del cuestionario de Calidad de Vida de la Espondilitis Anquilosante (ASQoL).
14.4 Artritis Idiopática Juvenil Poliarticular (AIJp)
La eficacia de SIMPONI ARIA en pacientes pediátricos con AIJp se basa en la exposición farmacocinética y la extrapolación de la eficacia establecida de SIMPONI ARIA en pacientes con AR. La eficacia de SIMPONI ARIA también se evaluó en un estudio multicéntrico, abierto, de un solo brazo en 127 niños (de 2 a < 18 años de edad) con AIJ con poliartritis activa a pesar del tratamiento con MTX durante al menos 2 meses (Ensayo AIJp, NCT02277444). Los subtipos de pacientes con AIJ poliarticular al inicio del estudio incluyeron: factor reumatoide negativo (43%), factor reumatoide positivo (35%), artritis relacionada con entesitis (9%), oligoarticular extendida (6%), artritis psoriásica juvenil (4%) y AIJ sistémica sin manifestaciones sistémicas (3%). Todos los pacientes recibieron SIMPONI ARIA 80 mg/m
2como infusión intravenosa en la semana 0, 4 y cada 8 semanas hasta la semana 52. Los pacientes continuaron con dosis estables de MTX semanalmente hasta la semana 28; después de la semana 28, se permitieron cambios en la dosis de MTX. La eficacia se evaluó como criterios de valoración de apoyo hasta la semana 52. La eficacia fue generalmente consistente con las respuestas en pacientes con AR.
16 SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
SIMPONI ARIA
®(golimumab) Injection es una solución incolora a amarillo claro disponible en envases de 1 vial NDC 57894-350-01.
Almacenamiento y manipulación
Refrigere SIMPONI ARIA a 36ºF a 46ºF (2ºC a 8ºC) y protéjalo de la luz. Mantenga el producto en su caja original para protegerlo de la luz hasta el momento de su uso. No congelar. No agitar.
Si es necesario, SIMPONI ARIA puede almacenarse a temperatura ambiente hasta 77ºF (25ºC) durante un período máximo único de 30 días en su caja original para protegerlo de la luz. Una vez que SIMPONI ARIA se haya almacenado a temperatura ambiente, no devuelva el producto al refrigerador. Si no se usa dentro de los 30 días a temperatura ambiente, deseche SIMPONI ARIA.
17 INFORMACIÓN DE ASESORAMIENTO AL PACIENTE
Ver
la información para el paciente aprobada por la FDA (Guía del medicamento).
Informe a los pacientes sobre los posibles beneficios y riesgos de SIMPONI ARIA. Indique a los pacientes que lean la Guía del medicamento antes de comenzar el tratamiento con SIMPONI ARIA y que la lean cada vez que se renueve la receta.
Infecciones
Informe a los pacientes que SIMPONI ARIA puede disminuir la capacidad de su sistema inmunitario para combatir infecciones. Indique al paciente la importancia de ponerse en contacto con su médico si desarrolla algún síntoma de infección, incluidas la tuberculosis, las infecciones fúngicas invasivas y la reactivación de la hepatitis B.
Tumores malignos
Se debe informar a los pacientes sobre el riesgo de linfoma y otros tumores malignos mientras reciben SIMPONI ARIA. Se recomienda un examen periódico de la piel para todos los pacientes, particularmente aquellos con factores de riesgo de cáncer de piel.
Otras afecciones médicas
Aconseje a los pacientes que informen sobre cualquier signo de nuevas afecciones médicas o que empeoren, como insuficiencia cardíaca congestiva, trastornos desmielinizantes, enfermedades autoinmunitarias, enfermedad hepática, citopenias o psoriasis.
Vacunaciones
Informe a los pacientes que, dado que SIMPONI ARIA puede disminuir la capacidad de su sistema inmunitario para combatir infecciones, deben evitar las vacunas vivas. Informe a las pacientes embarazadas que reciben SIMPONI ARIA que sus bebés no deben recibir vacunas vivas durante los 6 meses posteriores a la última infusión de SIMPONI ARIA durante el embarazo. Aconseje a los pacientes y a los bebés de mujeres que recibieron SIMPONI ARIA durante el embarazo que consulten a un médico antes de recibir cualquier inmunización.
SECCIÓN NO CLASIFICADA DE SPL
Fabricado por:
Janssen Biotech, Inc.
Horsham, PA 19044
Licencia de EE. UU. n.° 1864
© 2017, 2020 Janssen Pharmaceutical Companies
GUÍA DEL MEDICAMENTO
| GUÍA DEL MEDICAMENTO SIMPONI ARIA ®( SIM-po-nee AHR-ee-uh ) |
|||
| ¿Cuál es la información más importante que debo saber sobre SIMPONI ARIA? | |||
| SIMPONI ARIA es un medicamento que afecta su sistema inmunitario. SIMPONI ARIA puede disminuir la capacidad de su sistema inmunitario para combatir infecciones. Algunas personas han tenido infecciones graves mientras recibían SIMPONI ARIA, incluyendo tuberculosis (TB), e infecciones causadas por bacterias, hongos o virus que se propagan por todo el cuerpo. Algunas personas han muerto a causa de estas infecciones graves. | |||
|
|||
| No debe comenzar a recibir SIMPONI ARIA si tiene algún tipo de infección, a menos que su médico se lo indique. | |||
| Antes de recibir SIMPONI ARIA, informe a su médico si: | |||
|
|||
|
|
||
|
|||
| Después de recibir SIMPONI ARIA, llame a su médico de inmediato si tiene algún síntoma de infección. SIMPONI ARIA puede aumentar la probabilidad de que contraiga infecciones o empeorar cualquier infección que tenga. | |||
| Cáncer | |||
|
|||
| ¿Qué es SIMPONI ARIA? | |||
SIMPONI ARIA es un medicamento recetado llamado bloqueador del TNF. SIMPONI ARIA se usa para tratar:
|
|||
| No se sabe si SIMPONI ARIA es seguro y eficaz en niños con APs y AIJp menores de 2 años de edad o en niños con afecciones distintas de APs y AIJp. | |||
| ¿Qué debo decirle a mi médico antes de comenzar el tratamiento con SIMPONI ARIA? | |||
| Ver ”
¿Cuál es la información más importante que debo saber sobre SIMPONI ARIA?“. |
|||
| Antes de comenzar con SIMPONI ARIA, informe a su médico sobre todas sus afecciones médicas, incluso si: | |||
|
|||
| Informe a su médico sobre todos los medicamentos que toma, incluidos los medicamentos recetados y de venta libre, las vitaminas y los suplementos herbales. | |||
| Especialmente, informe a su médico si: | |||
|
|||
| Pregunte a su médico o farmacéutico por una lista de estos medicamentos si no está seguro. | |||
| Lleve una lista de todos sus medicamentos con usted para mostrársela a su médico y farmacéutico cada vez que obtenga un nuevo medicamento. | |||
| ¿Cómo debo recibir SIMPONI ARIA? | |||
|
|||
|
|||
|
|||
| ¿Cuáles son los posibles efectos secundarios de SIMPONI ARIA? | |||
| SIMPONI ARIA puede causar efectos secundarios graves, que incluyen: | |||
| Ver ”
¿Cuál es la información más importante que debo saber sobre SIMPONI ARIA?” |
|||
|
|||
|
|
||
|
|||
|
|
||
| Infección por hepatitis B en personas que portan el virus en la sangre.Si usted es portador del virus de la hepatitis B (un virus que afecta el hígado), el virus puede activarse mientras usa SIMPONI ARIA. Su médico debe realizar análisis de sangre antes de comenzar el tratamiento con SIMPONI ARIA y mientras recibe SIMPONI ARIA. | |||
|
|||
|
|
||
| Insuficiencia cardíaca, incluyendo nueva insuficiencia cardíaca o empeoramiento de la insuficiencia cardíaca que ya padece, puede ocurrir en personas que usan medicamentos bloqueadores del TNF, incluyendo SIMPONI ARIA.Si desarrolla insuficiencia cardíaca nueva o que empeora con SIMPONI ARIA, es posible que necesite ser tratado en un hospital, y puede resultar en la muerte. | |||
|
|||
| Problemas del sistema nervioso.En raras ocasiones, las personas que reciben medicamentos bloqueadores del TNF, incluido SIMPONI ARIA, tienen problemas del sistema nervioso como esclerosis múltiple o síndrome de Guillain-Barré. Informe a su médico de inmediato si presenta alguno de estos síntomas: | |||
|
|
||
| Problemas del sistema inmunitario.En raras ocasiones, las personas que reciben medicamentos bloqueadores del TNF han desarrollado síntomas similares a los síntomas del lupus. Informe a su médico si tiene alguno de estos síntomas: | |||
|
|
||
| Problemas hepáticos.Pueden ocurrir problemas hepáticos en personas que reciben medicamentos bloqueadores del TNF, incluido SIMPONI ARIA. Estos problemas pueden provocar insuficiencia hepática y la muerte. Llame a su médico de inmediato si tiene alguno de estos síntomas: | |||
|
|
||
| Problemas sanguíneos.Se han observado recuentos sanguíneos bajos con SIMPONI ARIA. Es posible que su cuerpo no produzca suficientes células sanguíneas que ayudan a combatir infecciones o a detener el sangrado. Los síntomas incluyen fiebre, moretones o sangrado con facilidad, o palidez. Su médico controlará sus recuentos sanguíneos antes y durante el tratamiento con SIMPONI ARIA. | |||
| Reacciones alérgicas.Pueden ocurrir reacciones alérgicas en personas que reciben medicamentos bloqueadores del TNF, incluido SIMPONI ARIA. Algunas reacciones pueden ser graves y potencialmente mortales. Algunas de estas reacciones pueden ocurrir después de recibir su primera dosis de SIMPONI ARIA. Llame a su médico de inmediato si tiene alguno de estos síntomas de una reacción alérgica: | |||
|
|
||
| Los efectos secundarios más comunes de SIMPONI ARIA incluyen: | |||
|
|||
| Estos no son todos los posibles efectos secundarios de SIMPONI ARIA. | |||
| Informe a su médico sobre cualquier efecto secundario que le moleste o que no desaparezca. Llame a su médico para obtener asesoramiento médico sobre los efectos secundarios. Puede informar los efectos secundarios a la FDA al 1-800-FDA-1088. | |||
| Información general sobre el uso seguro y eficaz de SIMPONI ARIA. | |||
| A veces se recetan medicamentos para fines distintos a los enumerados en una Guía del Medicamento. | |||
| Puede pedirle a su médico o farmacéutico información sobre SIMPONI ARIA que esté escrita para profesionales de la salud. | |||
| ¿Cuáles son los ingredientes de SIMPONI ARIA? | |||
| Ingrediente activo: golimumab. | |||
| Ingredientes inactivos: L-histidina, monohidrocloruro de L-histidina monohidrato, polisorbato 80, sorbitol y agua para inyección. SIMPONI ARIA no contiene conservantes y no está hecho con látex de caucho natural. | |||
| Fabricado por: Janssen Biotech, Inc. Horsham, PA 19044 US License No. 1864 © 2017, 2020 Janssen Pharmaceutical Companies | |||
| Para obtener más información, visite www.SIMPONIARIA.com o llame al 1-800-JANSSEN (1-800-526-7736). | |||
| Esta Guía del Medicamento ha sido aprobada por la Administración de Alimentos y Medicamentos de los EE. UU. | Revisado: 9/2020 | ||
PANEL PRINCIPAL DE EXHIBICIÓN – Caja de vial de 4 mL
NDC 57894-350-01
Simponi ARIA
®
golimumab
50 mg/4 mL
(12.5 mg/mL)
Para infusión intravenosa
Vial de dosis única.
Deseche la porción no utilizada.
Rx solamente
Proporcione la Guía de medicamentos adjunta
a cada paciente.
