
Fabricante de medicamentos: Clovis Oncology, Inc. (Updated: 2024-02-21)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
RUBRACA
®(rucaparib) tabletas, para uso oral
Aprobación inicial en EE. UU.: 2016
CAMBIOS RECIENTES IMPORTANTES
| Indicaciones y uso (
1.1) |
12/2022 |
| Indicaciones y uso, tratamiento de
BRCA-cáncer de ovario mutado después de 2 o más quimioterapias ( 1.1) Eliminado |
06/2022 |
| Dosificación y administración (
2.1) |
12/2022 |
| Dosificación y administración, tratamiento de
BRCA-cáncer de ovario mutado después de 2 o más quimioterapias ( 2.1) Eliminado |
06/2022 |
| Advertencias y precauciones (
5.1) |
12/2022 |
INDICACIONES Y USO
RUBRACA es un inhibidor de la poli (ADP-ribosa) polimerasa (PARP) indicado para:
Cáncer de ovario
- para el tratamiento de mantenimiento de pacientes adultos con una mutación
BRCAdeleteria (germinal y/o somática) asociada a cáncer epitelial recurrente de ovario, trompa de Falopio o peritoneo primario que se encuentran en respuesta completa o parcial a la quimioterapia basada en platino. (
1.1)
Cáncer de próstata
- para el tratamiento de pacientes adultos con una mutación
BRCAdeleteria (germinal y/o somática) asociada a cáncer de próstata metastásico resistente a la castración (mCRPC) que han sido tratados con terapia dirigida al receptor de andrógenos y quimioterapia basada en taxanos. Seleccione pacientes para terapia en función de un diagnóstico complementario aprobado por la FDA para RUBRACA. (
1.2,
2.1)
Esta indicación está aprobada bajo aprobación acelerada en función de la tasa de respuesta objetiva y la duración de la respuesta. La aprobación continua para esta indicación puede estar supeditada a la verificación y descripción del beneficio clínico en ensayos confirmatorios. (
1.2)
DOSIFICACIÓN Y ADMINISTRACIÓN
- La dosis recomendada es de 600 mg por vía oral dos veces al día con o sin alimentos. (
2.2)
- Continúe el tratamiento hasta la progresión de la enfermedad o la toxicidad inaceptable. (
2.2)
- Para las reacciones adversas, considere la interrupción del tratamiento o la reducción de la dosis. (
2.3)
- Los pacientes que reciben RUBRACA para mCRPC también deben recibir un análogo de la hormona liberadora de gonadotropina (GnRH) de forma concurrente o deben haberse sometido a una orquiectomía bilateral. (
2.2)
FORMAS Y FUERZAS DE DOSIFICACIÓN
Tabletas: 200 mg, 250 mg y 300 mg (
3)
CONTRAINDICACIONES
Ninguna. (
4)
ADVERTENCIAS Y PRECAUCIONES
- Síndrome mielodisplásico/leucemia mieloide aguda (SMD/LMA): Se produjo SMD/LMA en pacientes expuestos a RUBRACA, y algunos casos fueron fatales. Controle a los pacientes para detectar toxicidad hematológica al inicio y mensualmente a partir de entonces. Interrumpa o reduzca la dosis en función de la gravedad de la reacción. Suspenda si se confirma SMD/LMA. (
2.3,
5.1)
- Toxicidad embrio-fetal: RUBRACA puede causar daño fetal. Avise del riesgo potencial para un feto y del uso de métodos anticonceptivos eficaces. (
5.2,
8.1,
8.3)
REACCIONES ADVERSAS
- Las reacciones adversas más comunes (≥ 10%) entre los pacientes con cáncer de ovario fueron náuseas, fatiga (incluida la astenia), anemia, aumento de AST/ALT, vómitos, diarrea, disminución del apetito, trombocitopenia, disgeusia, neutropenia, aumento de la creatinina en sangre, disnea, mareos, dispepsia, reacción de fotosensibilidad y leucopenia. (
6.1)
- Las reacciones adversas más comunes (≥ 20%) entre los pacientes con
BRCA-mCRPC mutado fueron fatiga (incluida la astenia), náuseas, anemia, aumento de ALT/AST, disminución del apetito, erupción cutánea, estreñimiento, trombocitopenia, vómitos, diarrea. (
6.1)
Para informar sobre SOSPECHAS DE REACCIONES ADVERSAS, póngase en contacto con Clovis Oncology, Inc. al 1-844-258-7662 o con la FDA al 1-800-FDA-1088 owww.fda.gov/medwatch.
Consulte el punto 17 para obtener INFORMACIÓN PARA EL PACIENTE y el etiquetado del paciente aprobado por la FDA.
Revisado: 12/2022
Tabla de Contenido
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDO*
1 INDICACIONES Y USO
1.1 Tratamiento de mantenimiento del cáncer de ovario recurrente con mutación en
BRCA
1.2 Cáncer de próstata metastásico resistente a la castración con mutación en
BRCA
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Selección del paciente
2.2 Dosis recomendada
2.3 Modificaciones de la dosis para reacciones adversas
3 FORMAS Y FUERZAS DE DOSIFICACIÓN
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Síndrome mielodisplásico/Leucemia mieloide aguda
5.2 Toxicidad embrionaria-fetal
6 REACCIONES ADVERSAS
6.1 Experiencia en ensayos clínicos
7 INTERACCIONES MEDICAMENTOSAS
7.1 Efecto de Rubraca sobre otros medicamentos
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.3 Mujeres y hombres en edad fértil
8.4 Uso pediátrico
8.5 Uso geriátrico
8.6 Insuficiencia renal
8.7 Insuficiencia hepática
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
12.2 Farmacodinamia
12.3 Farmacocinética
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, mutagénesis, deterioro de la fertilidad
14 ESTUDIOS CLÍNICOS
14.1 Tratamiento de mantenimiento del cáncer de ovario recurrente con mutación en
BRCA
14.2 Cáncer de próstata metastásico resistente a la castración con mutación en
BRCA
16 FORMA DE SUMINISTRO/ALMACENAMIENTO Y MANEJO
17 INFORMACIÓN PARA EL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información completa de prescripción no están enumeradas.
1 INDICACIONES Y USO
1.1 Tratamiento de mantenimiento del cáncer de ovario recurrente con mutación
BRCA
Rubraca está indicado para el tratamiento de mantenimiento de pacientes adultos con una mutación
BRCAdeleteria (germinal y/o somática) asociada a cáncer epitelial de ovario, trompa de Falopio o peritoneal primario recurrente que se encuentran en respuesta completa o parcial a la quimioterapia a base de platino.
1.2
BRCA-mutación metastásica de cáncer de próstata resistente a la castración
Rubraca está indicado para el tratamiento de pacientes adultos con una mutación
BRCAdeleteria (germinal y/o somática) asociada a cáncer de próstata metastásico resistente a la castración (mCRPC) que han sido tratados con terapia dirigida al receptor de andrógenos y quimioterapia a base de taxanos. Seleccione pacientes para terapia en función de un diagnóstico complementario aprobado por la FDA para Rubraca
[ver Dosificación y administración (
2.1)].
Esta indicación está aprobada bajo aprobación acelerada en base a la tasa de respuesta objetiva y la duración de la respuesta
[ver Estudios clínicos (
14.2)]
. La aprobación continua para esta indicación puede estar supeditada a la verificación y descripción del beneficio clínico en ensayos confirmatorios.
2 DOSIS Y ADMINISTRACIÓN
2.1 Selección del Paciente
Tratamiento de mantenimiento delBRCA-cáncer de ovario recurrente mutado
Seleccione pacientes para el tratamiento de mantenimiento del cáncer de ovario recurrente con Rubraca en función de la presencia de una mutación
BRCAdeleteria (germinal y/o somática) [
ver Estudios Clínicos (
14.1)
].
Actualmente no hay disponible una prueba aprobada por la FDA para la detección de mutaciones
BRCAdeleterias germinales y/o somáticas.
Tratamiento deBRCA-mCRPC mutado después de la terapia dirigida al receptor de andrógenos y la quimioterapia
Seleccione pacientes para el tratamiento de mCRPC con Rubraca en función de la presencia de una mutación
BRCAdeleteria (germinal y/o somática) en muestras de plasma
[ver Estudios Clínicos (
14.2)].
Un resultado negativo de una muestra de plasma no significa que el tumor del paciente sea negativo para
BRCAmutaciones. Si la muestra de plasma tiene un resultado negativo, considere realizar pruebas genómicas adicionales utilizando muestras de tumor según lo indicado clínicamente.
La información sobre las pruebas aprobadas por la FDA para la detección de una mutación
BRCAen pacientes con cáncer de ovario o con cáncer de próstata está disponible en: http://www.fda.gov/CompanionDiagnostics.
2.2 Dosis Recomendada
La dosis recomendada de Rubraca es de 600 mg (dos comprimidos de 300 mg) administrados por vía oral dos veces al día con o sin alimentos, para una dosis diaria total de 1.200 mg.
Continúe el tratamiento hasta la progresión de la enfermedad o la toxicidad inaceptable.
Si un paciente olvida una dosis de Rubraca, indíquele que tome la siguiente dosis a la hora programada. Las dosis vomitadas no deben reemplazarse.
Los pacientes que reciben Rubraca para mCRPC también deben recibir un análogo de la hormona liberadora de gonadotropina (GnRH) de forma concurrente o deben haberse sometido a una orquiectomía bilateral.
2.3 Modificaciones de la Dosis para Reacciones Adversas
Para controlar las reacciones adversas, considere la interrupción del tratamiento o la reducción de la dosis. Las modificaciones de la dosis de Rubraca recomendadas para las reacciones adversas se indican en
| Reducción de la Dosis | Dosis |
| Dosis Inicial | 600 mg dos veces al día (dos comprimidos de 300 mg) |
| Primera Reducción de la Dosis | 500 mg dos veces al día (dos comprimidos de 250 mg) |
| Segunda Reducción de la Dosis | 400 mg dos veces al día (dos comprimidos de 200 mg) |
| Tercera Reducción de la Dosis | 300 mg dos veces al día (un comprimido de 300 mg) |
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
- Tabletas (200 mg): azul, redonda, de liberación inmediata, recubierta con película, con la marca “C2”.
- Tabletas (250 mg): blanca, diamante, de liberación inmediata, recubierta con película, con la marca “C25”.
- Tabletas (300 mg): amarilla, ovalada, de liberación inmediata, recubierta con película, con la marca “C3”.
4 CONTRAINDICACIONES
Ninguna.
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Síndrome mielodisplásico/leucemia mieloide aguda
El síndrome mielodisplásico (SMD)/leucemia mieloide aguda (LMA) se produce en pacientes tratados con Rubraca y son reacciones adversas potencialmente mortales. En 1594 pacientes con cáncer de ovario tratados
[ver Reacciones adversas (
6.1)]
, se produjo SMD/LMA en 32 pacientes (2%), incluidos aquellos en seguimiento a largo plazo. De estos, 14 se produjeron durante el tratamiento o durante el seguimiento de seguridad de 28 días (0.9%). La duración del tratamiento con Rubraca antes del diagnóstico de SMD/LMA osciló entre < 2 meses y aproximadamente 72 meses. Los casos fueron típicos de SMD secundario/LMA relacionada con la terapia del cáncer; en todos los casos, los pacientes habían recibido regímenes de quimioterapia previos con platino y/u otros agentes que dañan el ADN.
En ARIEL3, de los pacientes con una mutación germinal y/o somática en
BRCA tratados con Rubraca, se produjo SMD/LMA en 9 de 129 (7%) pacientes tratados con Rubraca y en 4 de 66 (6%) pacientes tratados con placebo. La duración del tratamiento con Rubraca en pacientes que desarrollaron SMD secundario/LMA relacionada con la terapia del cáncer varió de 1.2 a 4.7 años.
En TRITON2, no se observó SMD/LMA en pacientes con mCRPC (n=209) independientemente de la mutación de deficiencia de recombinación homóloga (HRD)
[ver Reacciones adversas (
6.1)].
No inicie el tratamiento con Rubraca hasta que los pacientes se hayan recuperado de la toxicidad hematológica causada por la quimioterapia previa (≤ Grado 1). Controle los hemogramas completos para detectar citopenia al inicio y mensualmente a partir de entonces para detectar cambios clínicamente significativos durante el tratamiento. En caso de toxicidades hematológicas prolongadas (> 4 semanas), interrumpa el tratamiento con Rubraca o reduzca la dosis de acuerdo con
Tabla 1[ver Posología y administración (
2.3)]
y controle los hemogramas semanalmente hasta la recuperación. Si los niveles no se han recuperado a Grado 1 o inferior después de 4 semanas o si se sospecha SMD/LMA, remita al paciente a un hematólogo para que le realice más pruebas, incluido un análisis de médula ósea y una muestra de sangre para el análisis citogenético. Si se confirma el SMD/LMA, interrumpa el tratamiento con Rubraca.
5.2 Toxicidad embriofetal
Rubraca puede causar daño fetal cuando se administra a una mujer embarazada según su mecanismo de acción y los hallazgos de estudios en animales. En un estudio de reproducción animal, la administración de rucaparib a ratas preñadas durante el período de organogénesis provocó la muerte embriofetal a exposiciones que fueron 0.04 veces el AUC
0-24hen pacientes que recibieron la dosis humana recomendada de 600 mg dos veces al día. Informe a las mujeres embarazadas sobre el riesgo potencial para el feto. Aconseje a las mujeres con posibilidad de embarazo que utilicen métodos anticonceptivos eficaces durante el tratamiento y durante los 6 meses siguientes a la última dosis de Rubraca
[ver Uso en poblaciones específicas (
8.1,
8.3) y Farmacología clínica (
12.1)].
Según los hallazgos de los estudios de toxicidad genética y reproducción animal, se debe aconsejar a los pacientes varones con parejas femeninas con posibilidad de embarazo o que estén embarazadas que utilicen métodos anticonceptivos eficaces durante el tratamiento y durante los 3 meses siguientes a la última dosis de Rubraca
[ver Uso en poblaciones específicas (
8.1,
8.3)].
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas graves se discuten en otra parte de la etiqueta:
- Síndrome mielodisplásico/Leucemia mieloide aguda
[ver Advertencias y precauciones (
5.1)].
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se llevan a cabo en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no se pueden comparar directamente con las tasas en los ensayos clínicos de otro fármaco y pueden no reflejar las tasas observadas en la práctica.
La población de seguridad agrupada para pacientes con cáncer de ovario en la sección ADVERTENCIAS Y PRECAUCIONES refleja la exposición a Rubraca a 600 mg por vía oral dos veces al día en 1594 pacientes tratados en ensayos clínicos, incluido ARIEL3. En este grupo, el 57% de los pacientes estuvieron expuestos durante 6 meses o más y el 33% estuvieron expuestos durante más de un año.
En la población de seguridad agrupada para pacientes con cáncer de ovario, las reacciones adversas más comunes en ≥ 10% de los pacientes fueron náuseas (68%), astenia/fatiga (65%), anemia/disminución de la hemoglobina (45%), aumento de AST/ALT (39%), vómitos (36%), diarrea (29%), disminución del apetito (27%), trombocitopenia/disminución del recuento plaquetario (25%), disgeusia (24%), neutropenia/disminución del recuento de neutrófilos (21%), aumento de la creatinina en sangre (17%), disnea (16%), mareos (14%), dispepsia (11%), reacción de fotosensibilidad (10%) y leucopenia/disminución del recuento de glóbulos blancos (10%).
Tratamiento de mantenimiento deBRCA-cáncer de ovario recurrente mutado
La seguridad de Rubraca para el tratamiento de mantenimiento de pacientes con
BRCA-cáncer epitelial de ovario, trompa de Falopio o peritoneo primario recurrente mutado se investigó en ARIEL3, un estudio aleatorizado (2:1), doble ciego, controlado con placebo en el que 195 pacientes con una mutación
BRCAdeleteria recibieron Rubraca 600 mg por vía oral dos veces al día (n=129) o placebo (n=66) hasta la progresión de la enfermedad o la toxicidad inaceptable. La duración media del tratamiento del estudio fue de 13,6 meses (rango: < 1 mes a 39 meses) para los pacientes que recibieron Rubraca y 5,5 meses para los pacientes que recibieron placebo.
Las interrupciones de la dosis debido a una reacción adversa de cualquier grado ocurrieron en el 67% de los pacientes que recibieron Rubraca y el 14% de los que recibieron placebo; las reducciones de la dosis debido a una reacción adversa ocurrieron en el 57% de los pacientes con Rubraca y el 6% de los pacientes con placebo. Las reacciones adversas más frecuentes que llevaron a la interrupción o reducción de la dosis de Rubraca fueron trombocitopenia (25%), anemia (19%), aumento de AST/ALT (16%), fatiga/astenia (14%) y náuseas (10%). La interrupción debido a reacciones adversas ocurrió en el 15% de los pacientes con Rubraca y el 5% de los pacientes con placebo. Las reacciones adversas específicas que con mayor frecuencia llevaron a la interrupción en los pacientes tratados con Rubraca fueron trombocitopenia (4%), náuseas (3%) y anemia (2%).
Tabla 2describe las reacciones adversas que ocurren en ≥20% de los pacientes; mientras que
Tabla 3describe las anormalidades de laboratorio que ocurren en ≥25% de los pacientes que ocurren en ARIEL3.
|
aCriterios de terminología común del Instituto Nacional del Cáncer para eventos adversos (NCI CTCAE versión 4.03) |
||||
|
bConsiste en términos relacionados agrupados que reflejan el concepto médico de la reacción adversa |
||||
| Reacciones adversas | Rubraca N=129 |
Placebo N=66 |
||
| Grados 1-4a % |
Grados 3-4 % |
Grados 1-4a % |
Grados 3-4 % |
|
| Trastornos gastrointestinales | ||||
| Náuseas | 79 | 2 | 29 | 0 |
| Dolor/distensión abdominal
b |
48 | 3 | 49 | 2 |
| Estreñimiento | 39 | 4 | 36 | 2 |
| Vómitos | 37 | 4 | 14 | 0 |
| Diarrea | 34 | 2 | 18 | 0 |
| Estomatitis
b |
28 | 0.8 | 12 | 0 |
| Trastornos generales y condiciones del lugar de administración | ||||
| Fatiga/astenia | 74 | 9 | 52 | 5 |
| Trastornos de la piel y del tejido subcutáneo | ||||
| Erupción cutánea
b |
45 | 0 | 23 | 0 |
| Trastornos del sistema nervioso | ||||
| Disgeusia | 33 | 0 | 6 | 0 |
| Dolor de cabeza | 22 | 0 | 15 | 2 |
| Investigaciones | ||||
| Elevación de AST/ALT | 33 | 16 | 3 | 0 |
| Trastornos de la sangre y del sistema linfático | ||||
| Anemia | 41 | 26 | 6 | 0 |
| Trombocitopenia | 35 | 6 | 3 | 0 |
| Neutropenia | 22 | 8 | 6 | 0 |
| Trastornos respiratorios, torácicos y mediastínicos | ||||
| Nasofaringitis/Infección del tracto respiratorio superior
b |
29 | 0 | 20 | 2 |
| Trastornos del metabolismo y la nutrición | ||||
| Disminución del apetito | 23 | 2 | 14 | 0 |
Las reacciones adversas que ocurren en < 20% de los pacientes tratados con Rubraca incluyen insomnio (19%), disnea (17%), mareos (15%), pirexia (15%), dispepsia (12%), edema periférico (12%) y depresión (11%).
|
aSe permitió que los pacientes ingresaran a los estudios clínicos con valores de laboratorio de CTCAE Grado 1. |
||||
|
bNCI CTCAE versión 4.03. |
||||
| Parámetro de laboratorioa | Rubraca N=129 |
Placebo N=66 |
||
| Grados 1-4b % |
Grados 3-4 % |
Grados 1-4b % |
Grados 3-4 % |
|
| Química | ||||
| Aumento en creatinina | 96 | 0 | 89 | 0 |
| Aumento en ALT | 67 | 11 | 6 | 0 |
| Aumento en AST | 59 | 1 | 6 | 0 |
| Aumento en colesterol | 39 | 3 | 20 | 0 |
| Aumento en Fosfatasa Alcalina | 39 | 0 | 3 | 0 |
| Hematología | ||||
| Disminución en hemoglobina | 61 | 18 | 14 | 2 |
| Disminución en plaquetas | 47 | 2 | 8 | 0 |
| Disminución en leucocitos | 39 | 3 | 23 | 0 |
| Disminución en neutrófilos | 38 | 6 | 18 | 2 |
| Disminución en linfocitos | 33 | 7 | 14 | 2 |
Tratamiento deBRCA-mCRPC mutado después de la terapia dirigida al receptor de andrógenos y la quimioterapia
La seguridad de Rubraca 600 mg dos veces al día se evaluó en un ensayo de un solo brazo (TRITON2)
[ver Estudios clínicos (
14.2)]
. TRITON2 reclutó a 209 pacientes con mCRPC HRD-positivo, incluidos 115 con
BRCA-mCRPC mutado. Entre los pacientes con
BRCA-mCRPC mutado, la duración media del tratamiento con Rubraca fue de 6,5 meses (rango de 0,5 a 26,7).
Hubo 2 (1,7%) pacientes con reacciones adversas que llevaron a la muerte, una atribuida a síndrome de dificultad respiratoria aguda y neumonía.
Las interrupciones de la dosis debido a una reacción adversa ocurrieron en el 57% de los pacientes que recibieron Rubraca. Las reacciones adversas que requirieron interrupción de la dosis en más del 3% de los pacientes incluyeron anemia, trombocitopenia, astenia/fatiga, náuseas, vómitos, neutropenia, aumento de ALT/AST, aumento de creatinina, disminución del apetito, lesión renal aguda e hipofosfatemia.
Las reducciones de dosis debido a una reacción adversa ocurrieron en el 41% de los pacientes que recibieron Rubraca. Las reacciones adversas que requirieron reducción de la dosis en más del 3% de los pacientes fueron anemia (14%), astenia/fatiga (10%), trombocitopenia (7%), náuseas (6%), disminución del apetito (4%) y erupción cutánea (3%).
La interrupción debido a reacciones adversas ocurrió en el 8% de los pacientes que recibieron Rubraca. Ninguna de las reacciones adversas que llevaron a la interrupción de Rubraca (prolongación del QT en el ECG, síndrome de dificultad respiratoria aguda, anemia, trastorno del equilibrio, insuficiencia cardíaca, disminución del apetito/fatiga/pérdida de peso, leucopenia/neutropenia, aumento de ALT/AST y neumonía) ocurrió en más de un paciente (<1%).
5resumen las reacciones adversas y las anormalidades de laboratorio, respectivamente, en pacientes con
BRCA–mCRPC mutado en TRITON2.
| Reacción adversa | Rubraca N = 115 |
|
|---|---|---|
| Grados 1-4
a |
Grados 3-4 (%) |
|
|
aNCI CTCAE versión 4.03. |
||
|
bIncluye recuento de plaquetas disminuido |
||
|
cIncluye ampolla, ampolla de sangre, dermatitis, dermatitis de contacto, eczema, erupción cutánea genital, síndrome de eritrodisestesia palmar-plantar, reacción de fotosensibilidad, psoriasis, erupción cutánea, erupción cutánea maculopapular, erupción cutánea pruriginosa, exfoliación cutánea, lesión cutánea, urticaria |
||
| Trastornos generales y condiciones del lugar de administración | ||
| Astenia/Fatiga | 62 | 9 |
| Trastornos gastrointestinales | ||
| Náuseas | 52 | 3 |
| Estreñimiento | 27 | 1 |
| Vómitos | 22 | 1 |
| Diarrea | 20 | 0 |
| Trastornos de la sangre y del sistema linfático | ||
| Anemia | 43 | 25 |
| Trombocitopenia
b |
25 | 10 |
| Trastornos del metabolismo y la nutrición | ||
| Disminución del apetito | 28 | 2 |
| Trastornos de la piel y del tejido subcutáneo | ||
| Erupción cutánea
c |
27 | 2 |
| Investigaciones | ||
| Aumento de ALT/AST | 33 | 5 |
Otras reacciones adversas clínicamente relevantes que ocurrieron en menos del 20% de los pacientes incluyeron disnea, mareos, hemorragia, infección del tracto urinario, disgeusia, dispepsia, hipersensibilidad (incluidos sofocos, asma, sensación de ahogo, hinchazón periorbitaria, hinchazón de la cara y sibilancias), neumonía, sepsis, eventos cardiovasculares isquémicos, insuficiencia renal, tromboembolismo venoso y estomatitis.
|
aEl denominador para cada parámetro de laboratorio se basa en el número de pacientes con un valor de laboratorio de referencia y posterior al tratamiento disponible para 111 a 115 pacientes. |
||
|
bNCI CTCAE versión 5.0; la disminución en el fosfato se clasifica utilizando NCI CTACE versión 4.03 |
||
|
cLa elevación de ALT o AST de grado 3-4 provocó la interrupción del fármaco en 4 pacientes, de los cuales 1 tuvo una reducción de la dosis tras el reto. |
||
| Parámetro de laboratorio | Rubraca N = 115 a |
|
| Grados 1-4
b
|
Grados 3-4 (%) |
|
| Química clínica | ||
| Aumento de ALT
c |
69 | 5 |
| Disminución de fosfato | 68 | 15 |
| Aumento de la fosfatasa alcalina | 44 | 2 |
| Aumento de creatinina | 43 | 2 |
| Aumento de triglicéridos | 42 | 5 |
| Disminución de sodio | 38 | 3 |
| Hematología | ||
| Disminución de leucocitos | 69 | 5 |
| Disminución del recuento absoluto de neutrófilos | 62 | 10 |
| Disminución de hemoglobina | 59 | 25 |
| Disminución de linfocitos | 42 | 17 |
| Disminución de plaquetas | 40 | 10 |
7 INTERACCIONES MEDICAMENTOSAS
7.1 Efecto de Rubraca en otros medicamentos
Ciertos sustratos de CYP1A2, CYP3A, CYP2C9 o CYP2C19
La administración concomitante de Rubraca con sustratos de CYP1A2, CYP3A, CYP2C9 o CYP2C19 puede aumentar la exposición sistémica de estos sustratos
[ver Farmacología clínica (
12.3)]
, lo que puede aumentar la frecuencia o gravedad de las reacciones adversas de estos sustratos. Si la administración concomitante es inevitable entre Rubraca y los sustratos de estas enzimas donde los cambios mínimos de concentración pueden provocar reacciones adversas graves, disminuya la dosis del sustrato de acuerdo con la información de prescripción aprobada.
Si no se puede evitar la administración concomitante con warfarina (un sustrato de CYP2C9), considere aumentar la frecuencia del control de la relación internacional normalizada (INR).
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de Riesgos
Basándose en los hallazgos de estudios en animales y su mecanismo de acción, Rubraca puede causar daño fetal cuando se administra a mujeres embarazadas. No hay datos disponibles en mujeres embarazadas para informar el riesgo asociado al fármaco. En un estudio de reproducción en animales, la administración de rucaparib a ratas embarazadas durante la organogénesis provocó la muerte embrio-fetal a exposiciones maternas que fueron 0.04 veces el AUC
0-24hen pacientes que recibieron la dosis recomendada de 600 mg dos veces al día
[ver Datos]. Informar a las mujeres embarazadas del riesgo potencial para el feto.
El riesgo de fondo de defectos de nacimiento mayores y aborto espontáneo para la población indicada es desconocido. En la población general de los EE. UU., el riesgo de fondo estimado de defectos de nacimiento mayores y aborto espontáneo en embarazos clínicamente reconocidos es del 2% al 4% y del 15% al 20%, respectivamente.
Datos en Animales
En un estudio de desarrollo embrio-fetal de búsqueda de rango de dosis, ratas embarazadas recibieron dosis orales de 50, 150, 500 o 1000 mg/kg/día de rucaparib durante el período de organogénesis. Se observó pérdida postimplantación (100% de resorciones tempranas) en todos los animales a dosis mayores o iguales a 50 mg/kg/día (con exposiciones sistémicas maternas aproximadamente 0.04 veces la exposición humana a la dosis recomendada basada en AUC
0-24h).
8.2 Lactancia
Resumen de Riesgos
No hay información sobre la presencia de rucaparib en la leche materna, ni sobre sus efectos en la producción de leche o en el niño amamantado. Debido al potencial de reacciones adversas graves en los niños amamantados por Rubraca, se recomienda a las mujeres lactantes que no amamanten durante el tratamiento con Rubraca y durante 2 semanas después de la última dosis.
8.3 Mujeres y Hombres en Edad Reproductiva
Rubraca puede causar daño fetal cuando se administra a una mujer embarazada
[ver Uso en Poblaciones Específicas (
8.1)]
.
Prueba de Embarazo
Se recomienda realizar una prueba de embarazo a las mujeres en edad reproductiva antes de iniciar el tratamiento con Rubraca.
Mujeres
Se recomienda a las mujeres en edad reproductiva que utilicen métodos anticonceptivos eficaces durante el tratamiento y durante 6 meses después de la última dosis de Rubraca
[ver Uso en Poblaciones Específicas (
8.1)]
.
Hombres
Basándose en los hallazgos en estudios de toxicidad genética y reproducción en animales, se recomienda a los pacientes masculinos con parejas femeninas en edad reproductiva o que estén embarazadas que utilicen métodos anticonceptivos eficaces durante el tratamiento y durante 3 meses después de la última dosis de Rubraca. Se recomienda a los pacientes masculinos que no donen esperma durante la terapia y durante 3 meses después de la última dosis de Rubraca
[ver Uso en Poblaciones Específicas (
8.1) y Toxicología No Clínica (
13.1)].
8.4 Uso Pediátrico
No se ha establecido la seguridad y eficacia de Rubraca en pacientes pediátricos.
8.5 Uso Geriátrico
De los 937 pacientes con cáncer de ovario que recibieron Rubraca en ensayos clínicos, incluido ARIEL3, el 41% tenía 65 años o más y el 10% tenía 75 años o más. No se observaron diferencias importantes en la seguridad entre los pacientes más jóvenes y los más mayores con cáncer de ovario.
De los 209 pacientes con mCRPC que recibieron Rubraca en TRITON2, el 77% tenía 65 años o más y el 33% tenía 75 años o más. No se observaron diferencias importantes en la seguridad entre los pacientes más jóvenes y los más mayores con mCRPC.
8.6 Insuficiencia Renal
No se recomienda ningún ajuste de dosis para pacientes con insuficiencia renal leve a moderada (aclaramiento de creatinina [CLcr] entre 30 y 89 mL/min, según lo estimado por el método de Cockcroft-Gault)
[ver Farmacología Clínica (
12.3)].
Rubraca no se ha estudiado en pacientes con CLcr < 30 mL/min o pacientes en diálisis.
8.7 Insuficiencia Hepática
No se recomienda ningún ajuste de dosis para pacientes con insuficiencia hepática leve a moderada (bilirrubina total ≤ 3 x límite superior de lo normal [ULN] o AST > ULN)
[ver Farmacología Clínica (
12.3)]
. Rubraca no se ha estudiado en pacientes con insuficiencia hepática grave (bilirrubina total > 3 x ULN y cualquier AST).
11 DESCRIPCIÓN
Rucaparib es un inhibidor de la enzima poli(ADP-ribosa) polimerasa (PARP) de mamíferos. El nombre químico es 8-fluoro-2-{4-[(metilamino)metil]fenil}-1,3,4,5-tetrahidro-6H-azepino[5,4,3-cd]indol-6-ona ((1S,4R)-7,7-dimetil-2-oxobiciclo[2.2.1]hept-1-il)metanosulfonato de ácido. La fórmula química del camsilato de rucaparib es C
19H
18FN
3O•C
10H
16O
4S y la masa molecular relativa es 555,67 Daltons.
La estructura química del camsilato de rucaparib se muestra a continuación:
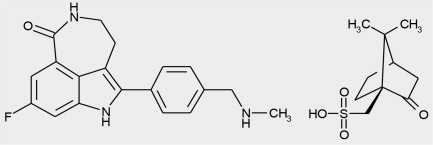
El camsilato de rucaparib es un polvo de blanco a amarillo pálido; formulado en una tableta para uso oral. Rucaparib muestra una baja solubilidad independiente del pH de aproximadamente 1 mg/mL en todo el rango de pH fisiológico.
Las tabletas de Rubraca (rucaparib) contienen camsilato de rucaparib como ingrediente activo. Cada tableta de 200 mg contiene 344 mg de camsilato de rucaparib equivalente a 200 mg de rucaparib base libre. Cada tableta de 250 mg contiene 430 mg de camsilato de rucaparib equivalente a 250 mg de rucaparib base libre. Cada tableta de 300 mg contiene 516 mg de camsilato de rucaparib equivalente a 300 mg de rucaparib base libre.
Los ingredientes inactivos en las tabletas de Rubraca incluyen: celulosa microcristalina, glicolato de almidón sódico, dióxido de silicio coloidal y estearato de magnesio. El recubrimiento de película azul cosmético para tabletas de 200 mg, el recubrimiento de película blanco cosmético para tabletas de 250 mg y el recubrimiento de película amarillo cosmético para tabletas de 300 mg es Opadry II que contiene alcohol polivinílico, dióxido de titanio, polietilenglicol/macrogol y talco. El recubrimiento se colorea de azul utilizando lago de aluminio azul brillante y lago de aluminio índigo carmín, o amarillo utilizando óxido de hierro amarillo.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
El Rucaparib es un inhibidor de las enzimas poli (ADP-ribosa) polimerasa (PARP), incluidas PARP-1, PARP-2 y PARP-3, que desempeñan un papel en la reparación del ADN.
Los estudios in vitro han demostrado que la citotoxicidad inducida por el Rucaparib puede implicar la inhibición de la actividad enzimática de PARP y una mayor formación de complejos PARP-ADN que provocan daños en el ADN, apoptosis y muerte de las células cancerosas. Se observó un aumento de la citotoxicidad inducida por el Rucaparib y de la actividad antitumoral en líneas celulares tumorales con deficiencias en
BRCA1/2 y otros genes de reparación del ADN. Se ha demostrado que el Rucaparib disminuye el crecimiento tumoral en modelos de xenoinjerto de ratón de cáncer humano con o sin deficiencias en
BRCA.
12.2 Farmacodinamia
La relación exposición-respuesta y el curso temporal de la respuesta farmacodinámica para la seguridad y la eficacia del Rucaparib no se han caracterizado completamente.
Electrofisiología cardíaca
Se observó una relación positiva entre la concentración y el QTc en pacientes a los que se administraron dosis continuas de Rubraca que oscilaron entre 40 mg una vez al día (0,03 veces la dosis recomendada aprobada) y 840 mg dos veces al día (1,4 veces la dosis recomendada aprobada). El aumento medio (intervalo de confianza [IC] del 90%) del QTcF respecto al valor basal en estado estacionario con 600 mg de Rubraca dos veces al día fue de 14,9 mseg (IC del 90%: 11,1 a 18,7 mseg).
12.3 Farmacocinética
El AUC y la C
máx. del Rucaparib mostraron una farmacocinética lineal en un intervalo de dosis de 240 mg a 840 mg dos veces al día (de 0,4 a 1,4 veces la dosis recomendada aprobada). La C
máx. media (coeficiente de variación [CV]) del Rucaparib en estado estacionario es de 1940 ng/ml (54%) y el AUC
0-12 h es de 16 900 h×ng/ml (54%) con la dosis recomendada aprobada. La relación media de acumulación del AUC es de 3,5 a 6,2 veces.
Absorción
La mediana de la t
máx. (mín., máx.) en estado estacionario es de 1,9 horas (0 a 5,98) con la dosis recomendada aprobada. La biodisponibilidad absoluta media (mín., máx.) es del 36% (30% a 45%).
Efecto de los alimentos
Tras una comida rica en grasas (aproximadamente de 800 a 1000 calorías, incluidas aproximadamente 250 calorías procedentes de hidratos de carbono, aproximadamente de 500 a 600 calorías procedentes de grasas y aproximadamente 150 calorías procedentes de proteínas), la C
máx. aumentó un 20%, el AUC
0-24 h aumentó un 38% y la t
máx. se retrasó 2,5 horas, en comparación con las condiciones de ayunas
[véase Posología y administración (
2.2)]
.
Distribución
El volumen de distribución aparente medio es de 2300 l (21%).
El Rucaparib se une en un 70% a las proteínas plasmáticas humanas in vitro. El Rucaparib se distribuyó preferentemente en los eritrocitos con una relación de concentración sangre-plasma de 1,8.
Eliminación
El aclaramiento total aparente medio en estado estacionario es de 44,2 l/h (45%) y la semivida de eliminación terminal media es de 26 horas (39%).
Metabolismo
In vitro, el Rucaparib es metabolizado principalmente por el CYP2D6 y, en menor medida, por el CYP1A2 y el CYP3A4. Además de la oxidación basada en el CYP, el Rucaparib también sufre N-desmetilación, N-metilación y glucuronidación.
Excreción
Tras una dosis oral única de Rucaparib radiomarcado, el Rucaparib inalterado representó el 64% de la radiactividad. El Rucaparib representó el 45% y el 95% de la radiactividad en la orina y las heces, respectivamente.
Poblaciones específicas
La edad (de 20 a 86 años), la raza (blanca, negra y asiática), el sexo, el peso corporal (de 41 a 171 kg), la insuficiencia renal leve o moderada (CLcr ≥ 30 ml/min), la insuficiencia hepática leve (bilirrubina total < LSN y AST > LSN o bilirrubina total de 1 a 1,5 × LSN y cualquier AST) y los polimorfismos del genotipo CYP2D6 o CYP1A2 no tuvieron un efecto clínicamente significativo en la farmacocinética del Rucaparib. No se ha estudiado el efecto de la insuficiencia renal grave (CLcr de 15 a 29 ml/min), la enfermedad renal terminal (CLcr < 15 ml/min) o la insuficiencia hepática grave (bilirrubina total > 3 × LSN y cualquier AST).
Insuficiencia hepática
La insuficiencia hepática moderada (bilirrubina total > 1,5 a 3 × LSN y cualquier AST) aumentó el AUC del Rucaparib en un 45%, pero no tuvo ningún efecto sobre la C
máx. en comparación con los pacientes con función hepática normal.
Efecto de otros fármacos sobre el Rucaparib
La administración concomitante de Rubraca con un inhibidor de la bomba de protones no tuvo un efecto clínicamente significativo sobre las concentraciones de Rucaparib en estado estacionario.
Efecto del Rucaparib sobre otros fármacos
La administración concomitante de Rubraca con rosuvastatina (sustrato de la BCRP) no tuvo un efecto clínicamente significativo sobre las concentraciones de rosuvastatina.
La administración conjunta de Rubraca con los siguientes sustratos aumentó la C
max de cada sustrato coadministrado en ≤ 1,1 veces y aumentó el AUC de cada sustrato de la siguiente manera:
- Cafeína (CYP1A2): 2,6 veces
- Midazolam (CYP3A4): 1,4 veces
- Warfarina (CYP2C9): 1,5 veces
- Omeprazol (CYP2C19): 1,6 veces
- Digoxina (glucoproteína P): 1,2 veces
La administración concomitante de Rubraca con un anticonceptivo oral que contiene etinilestradiol y levonorgestrel (sustratos de CYP3A): aumentó el AUC de etinilestradiol en 1,4 veces y el AUC de levonorgestrel en 1,6 veces, pero no tuvo un efecto clínicamente significativo en su C
max.
Estudios in vitro
Enzimas del citocromo P450 (CYP): Rucaparib inhibió CYP2C8 y CYP2D6 e indujo CYP1A2.
Enzimas UDP-glucuronosiltransferasa (UGT): Rucaparib inhibió UGT1A1.
Sistemas de transporte: Rucaparib es un sustrato de P-gp y BCRP. Rucaparib no es un sustrato de OATP1B1, OATP1B3, OAT1, OAT3 u OCT2.
Rucaparib inhibió OATP1B1, OATP1B3, OAT1, OAT3, MATE1, MATE2-K, OCT1, OCT2 y MRP4. Rucaparib no inhibió MRP2, MRP3 ni BSEP.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogenicidad, Mutagénesis, Deterioro de la Fertilidad
No se han realizado estudios de carcinogenicidad con rucaparib.
Rucaparib fue clastogénico en un ensayo de aberración cromosómica
in vitroen linfocitos humanos cultivados. La respuesta clastogénica en células estimuladas mitóticamente se anticipó en base al mecanismo de acción de rucaparib y indica una posible genotoxicidad en humanos. Rucaparib no fue mutagénico en una prueba de mutación inversa bacteriana (Ames).
No se han realizado estudios de fertilidad con rucaparib. En estudios de toxicología general de dosis repetidas de 3 meses, rucaparib no tuvo efectos sobre los órganos reproductivos masculinos y femeninos a dosis de hasta 100 mg/kg/día y 20 mg/kg/día en ratas y perros, respectivamente. Estos niveles de dosis dieron como resultado exposiciones sistémicas de aproximadamente 0,3 y 0,09 veces la exposición humana (AUC
0-24h), respectivamente, a la dosis recomendada.
14 ESTUDIOS CLÍNICOS
14.1 Tratamiento de mantenimiento del cáncer de ovario recurrente con mutación en
BRCA
La eficacia de Rubraca se investigó en ARIEL3 (NCT01968213), un ensayo clínico multicéntrico, doble ciego, en el que 564 pacientes con cáncer epitelial de ovario, trompa de Falopio o peritoneal primario recurrente que estaban en respuesta a la quimioterapia a base de platino se asignaron aleatoriamente (2:1) para recibir comprimidos de Rubraca 600 mg por vía oral dos veces al día (n=375) o placebo (n=189). El tratamiento continuó hasta la progresión de la enfermedad o la toxicidad inaceptable. Todos los pacientes habían logrado una respuesta (completa o parcial) a su quimioterapia más reciente a base de platino. La aleatorización se estratificó por la mejor respuesta al último platino (completa o parcial), el tiempo hasta la progresión después de la terapia con platino penúltima (6 a ≤ 12 meses y > 12 meses) y el estado del biomarcador tumoral.
Las muestras de tejido tumoral se analizaron utilizando un ensayo de ensayo clínico (CTA) (N=564) y una prueba de tejido de investigación de Foundation Medicine (n=518). De las muestras evaluadas con ambas pruebas, el estado de mutación tumoral
BRCA(t
BRCA) se confirmó para el 99% (177/178) de los pacientes con t
BRCA-positivo determinados por el CTA. Las muestras de sangre para el 94% (186/196) de los pacientes con t
BRCAse evaluaron utilizando una prueba central de línea germinal de sangre
BRCA. Con base en estos resultados, el 70% (130/186) de los pacientes con t
BRCAtenían una mutación germinal
BRCAy el 30% (56/186) tenían una mutación somática
BRCA. Los resultados de eficacia se basan en el subgrupo t
BRCA(germinal o somático).
El principal resultado de eficacia fue la supervivencia libre de progresión (SLP) evaluada por el investigador, evaluada de acuerdo con los Criterios de Evaluación de Respuesta en Tumores Sólidos (RECIST), versión 1.1 (v1.1). La supervivencia general (SG) fue una medida de resultado adicional.
De los 564 pacientes inscritos, 196 pacientes (35%) tenían una mutación t
BRCA. Entre los pacientes que tenían una mutación
tBRCA, la edad media fue de 58 años (rango: 42 a 81) para los pacientes que recibieron Rubraca y 59 años (rango: 36 a 84) para los que recibieron placebo; la mayoría eran blancos (84%); y el 100% tenía un estado de rendimiento ECOG de 0 o 1. Todos los pacientes habían recibido al menos dos quimioterapias previas a base de platino (rango: 2 a 5). Un total del 33% de los pacientes estaban en respuesta completa (RC) a su terapia más reciente. El intervalo libre de progresión al platino penúltimo fue de 6 a 12 meses en el 41% de los pacientes y > 12 meses en el 59%. Se informó terapia previa con bevacizumab para el 22% de los pacientes que recibieron Rubraca y el 17% de los pacientes que recibieron placebo. La enfermedad medible estaba presente en la línea de base en el 32% de los pacientes.
ARIEL3 demostró una mejora estadísticamente significativa en la SLP para los pacientes asignados aleatoriamente a Rubraca en comparación con el placebo en pacientes que tenían una mutación t
BRCA. Los resultados de una revisión radiológica independiente ciega fueron consistentes.
Los resultados de eficacia para pacientes con una mutación t
BRCAse resumen en
|
at BRCAincluye a todos los pacientes con una mutación germinal o somática BRCAdeleteria, según lo determinado por el CTA. |
||
| Rubraca N=130 |
Placebo N=66 |
|
| Supervivencia libre de progresión | ||
| Número de eventos, n (%) | 67 (52%) | 56 (85%) |
| Mediana en meses (IC del 95%) | 16.6 (13.4-22.9) | 5.4 (3.4-6.7) |
| HR (IC del 95%) | 0.23 (0.16, 0.34) | |
| Valor p | < 0.0001 | |
Figura 1. Curvas de Kaplan-Meier de Supervivencia Libre de Progreso en ARIEL3 evaluada por el Investigador: tBRCAGrupo
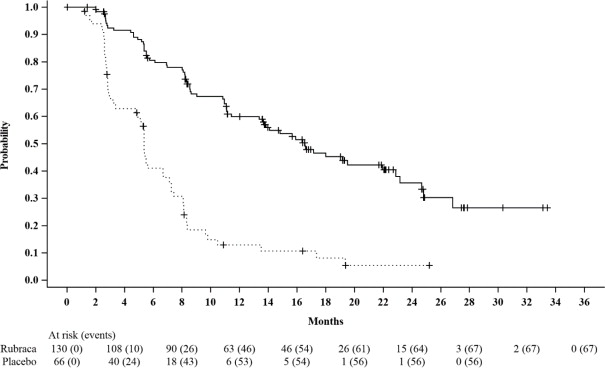
Se realizó un análisis final de SO después de observarse 130 eventos. Los resultados exploratorios de SO mostraron una HR de 0,83 (95% IC: 0,58, 1,19) en el subgrupo t
BRCAcon una mediana de SO de 45,9 meses (95% IC: 37,7, 59,6) para los pacientes tratados con Rubraca y 47,8 meses (95% IC: 43,2, 55,8) para los pacientes con placebo.
14.2
BRCA-mutado cáncer de próstata metastásico resistente a la castración
La eficacia de Rubraca se investigó en TRITON2 (NCT02952534), un ensayo clínico multicéntrico, de un solo brazo en curso en pacientes con
BRCA-mutado mCRPC que habían sido tratados con terapia dirigida al receptor de andrógeno y quimioterapia basada en taxanos. Se inscribieron 115 pacientes con mutaciones germinales o somáticas
BRCAen TRITON2, de los cuales 62 pacientes tenían enfermedad mensurable en el punto basal según la revisión radiológica independiente (IRR). Los pacientes recibieron Rubraca 600 mg por vía oral dos veces al día hasta la progresión de la enfermedad o toxicidad inaceptable. Los pacientes también recibieron un análogo de GnRH concomitante o tenían orquiectomía bilateral previa. La tasa de respuesta objetiva (ORR) y la duración de la respuesta (DOR) se evaluaron en los pacientes con enfermedad mensurable mediante IRR ciega y por el investigador de acuerdo con los criterios modificados RECIST v1.1 / Grupo de Trabajo sobre Cáncer de Próstata 3 (PCWG3).
Para los 62 pacientes con enfermedad mensurable en el punto basal, la edad media era de 73 años (rango 52 a 88); la mayoría eran blancos (73%) y el 10% eran negros; y el 98% de los pacientes tenían un estado funcional ECOG de 0 o 1. Todos los pacientes habían recibido al menos un tratamiento previo dirigido al receptor de andrógeno, el 34% habían recibido 2 tratamientos previos dirigidos al receptor de andrógeno y el 2% habían recibido 3 tratamientos previos dirigidos al receptor de andrógeno, y todos los pacientes también habían recibido la quimioterapia con taxanos previa. El 18% de los pacientes tenían metástasis pulmonares y el 21% tenían metástasis hepáticas en el punto basal. El 24% tenían metástasis solo en los ganglios linfáticos. El 40% tenían 10 o más lesiones óseas en el punto basal.
Todos los 62 pacientes tenían una mutación somática o germinal perjudicial
BRCAdetectada a partir de plasma central (26%), tejido central (32%) o local (42%) pruebas. De los 62 pacientes, el 66% tenían una mutación somática
BRCA, el 34% tenían una mutación germinal
BRCA, el 85% tenían una
BRCA2mutación y el 15% tenían una
BRCA1mutación.
Los principales resultados de eficacia del estudio fueron la ORR confirmada por IRR utilizando los criterios modificados RECIST v1.1 / PCWG3 y la DOR. Los resultados de eficacia de TRITON2 se proporcionan en
Tabla 7. La ORR por IRR fue similar en pacientes con mutación germinal en comparación con la mutación somática
BRCA.
|
NE = no evaluable |
|
|
aDefinida según los criterios modificados RECIST v1.1 y sin progresión ósea confirmada según PCWG3. |
|
|
bEl rango de la DOR fue de 1.7-24+ meses. Quince de los 27 (56%) pacientes con una respuesta objetiva confirmada tenían una DOR de ≥ 6 meses. |
|
| Rubraca (N = 62) |
|
| Tasa de respuesta objetiva confirmada (95% IC)
a |
44% (31, 57) |
| Mediana de la DOR en meses (95% IC)
b |
NE (6.4, NE) |
16 FORMA DE SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
Presentación
Rubraca está disponible en tabletas de 200 mg, 250 mg y 300 mg.
Tabletas de 200 mg:
- Azul, redonda y con “C2” grabado en un lado
- Se suministra en frascos de 60 tabletas (NDC: 69660-201-91)
Tabletas de 250 mg:
- Blanca, diamante y con “C25” grabado en un lado
- Se suministra en frascos de 60 tabletas (NDC: 69660-202-91)
Tabletas de 300 mg:
- Amarilla, ovalada y con “C3” grabado en un lado
- Se suministra en frascos de 60 tabletas (NDC: 69660-203-91)
Almacenamiento
Almacenar a 20°C a 25°C (68°F a 77°F); se permiten excursiones a 15°C a 30°C (59°F a 86°F) [
ver USP Controlled Room Temperature].
17 INFORMACIÓN PARA EL PACIENTE
Aconseje al paciente que lea el etiquetado del paciente aprobado por la FDA (
Información para el paciente).
MDS/AML:Aconseje a los pacientes que se pongan en contacto con su proveedor de atención médica si experimentan debilidad, sensación de cansancio, fiebre, pérdida de peso, infecciones frecuentes, hematomas, sangrado fácil, falta de aliento, sangre en la orina o las heces, y/o hallazgos de laboratorio de recuentos bajos de células sanguíneas, o la necesidad de transfusiones de sangre. Estos pueden ser signos de toxicidad hematológica o un problema más grave de la médula ósea llamado ‘síndrome mielodisplásico’ (MDS) o ‘leucemia mieloide aguda’ (AML) que se han reportado en pacientes tratados con Rubraca
[ver Advertencias y precauciones (
5.1)]
.
Toxicidad embrio-fetal:Aconseje a las mujeres que informen a su proveedor de atención médica si están embarazadas o quedan embarazadas. Informe a las pacientes del riesgo para el feto y la posible pérdida del embarazo
[ver Uso en poblaciones específicas (
8.1)]
. Aconseje a las mujeres en edad fértil que usen métodos anticonceptivos efectivos durante el tratamiento y durante 6 meses después de recibir la última dosis de Rubraca. Aconseje a los pacientes masculinos con parejas femeninas en edad fértil o que estén embarazadas que usen métodos anticonceptivos efectivos durante el tratamiento y durante 3 meses después de recibir la última dosis de Rubraca. Aconseje a los pacientes masculinos que no donen esperma durante la terapia y durante 3 meses después de la última dosis de Rubraca
[ver Advertencias y precauciones (
5.2) y Uso en poblaciones específicas (
8.1,
8.3)]
.
Fotosensibilidad:Aconseje a los pacientes que usen protección solar adecuada debido a la mayor susceptibilidad a las quemaduras solares mientras toman Rubraca
[ver Reacciones adversas a los medicamentos (
6.1)]
.
Lactancia:Aconseje a las mujeres que no amamanten durante el tratamiento y durante 2 semanas después de la última dosis de Rubraca
[ver Uso en poblaciones específicas (
8.2)]
.
Instrucciones de dosificación:Indique a los pacientes que tomen Rubraca por vía oral dos veces al día con o sin alimentos. Las dosis deben tomarse aproximadamente 12 horas separadas. Aconseje a los pacientes que si se olvida una dosis de Rubraca o si el paciente vomita después de tomar una dosis de Rubraca, los pacientes no deben tomar una dosis adicional, sino que deben tomar la siguiente dosis a la hora habitual
[ver Dosificación y administración (
2.2)]
.
Distribuido por:
Clovis Oncology, Inc.
Boulder, CO 80301
1-844-258-7662
Rubraca es una marca registrada de Clovis Oncology, Inc.
INSERTO PARA EL PACIENTE
|
Esta información para el paciente ha sido aprobada por la Administración de Alimentos y Medicamentos de los Estados Unidos. |
Revisado: Diciembre de 2022 |
|
| INFORMACIÓN PARA EL PACIENTE Rubraca®(roo-brah’-kah) (rucaparib) tabletas |
||
|
¿Cuál es la información más importante que debo saber sobre Rubraca? Rubraca puede causar efectos secundarios graves, que incluyen: |
||
|
|
|
Su proveedor de atención médica le hará análisis de sangre para verificar sus recuentos de células sanguíneas:
Ver “¿Cuáles son los posibles efectos secundarios de Rubraca?”para obtener más información sobre los efectos secundarios. |
||
| ¿Qué es Rubraca? Rubraca es un medicamento recetado que se usa en adultos para:
No se sabe si Rubraca es seguro y eficaz en niños. |
||
Antes de tomar Rubraca, informe a su proveedor de atención médica sobre todas sus afecciones médicas, incluso si:
Informe a su proveedor de atención médica sobre todos los medicamentos que toma,incluidos los medicamentos recetados y de venta libre, las vitaminas y los suplementos herbales. |
||
¿Cómo debo tomar Rubraca?
|
||
| ¿Qué debo evitar mientras tomo Rubraca? Evite pasar tiempo bajo el sol. Rubraca puede hacer que su piel sea sensible al sol (fotosensibilidad). Puede quemarse más fácilmente durante el tratamiento con Rubraca. Debe usar un sombrero y ropa que cubra su piel y usar protector solar para ayudar a protegerse de las quemaduras solares si tiene que estar bajo el sol. |
||
|
¿Cuáles son los posibles efectos secundarios de Rubraca? Rubraca puede causar efectos secundarios graves. Los efectos secundarios más comunes de Rubraca en personas con cáncer de ovario incluyen: |
||
|
|
|
| Los efectos secundarios más comunes de Rubraca en personas con cáncer de próstata incluyen: |
||
|
|
|
| Estos no son todos los posibles efectos secundarios de Rubraca. Para obtener más información, consulte a su médico o farmacéutico. Llame a su médico para obtener asesoramiento médico sobre los efectos secundarios. Puede informar los efectos secundarios a la FDA al 1-800-FDA-1088. |
||
¿Cómo debo almacenar Rubraca?
Mantenga Rubraca y todos los medicamentos fuera del alcance de los niños. |
||
| Información general sobre el uso seguro y eficaz de Rubraca. Los medicamentos a veces se recetan para fines distintos de los que se enumeran en un folleto de información para el paciente. No use Rubraca para una afección para la que no fue recetado. No se lo dé a otras personas, incluso si tienen los mismos síntomas que usted. Puede hacerles daño. Puede pedirle a su médico o farmacéutico más información sobre Rubraca. |
||
| ¿Cuáles son los ingredientes de Rubraca? Ingrediente activo:rucaparib Ingredientes inactivos:celulosa microcristalina, glicolato de almidón sódico, dióxido de silicio coloidal y estearato de magnesio. El recubrimiento de la película contiene alcohol polivinílico, dióxido de titanio, polietilenglicol/macrogol y talco. El recubrimiento de la película azul contiene lago azul brillante de aluminio y lago carmín índigo de aluminio. El recubrimiento de la película amarilla contiene óxido de hierro amarillo. Distribuido por: Clovis Oncology, Inc. Boulder, Colorado 80301 Para obtener más información, visite www.Rubraca.com o llame al 1-844-258-7662. |
||
PANEL DE VISUALIZACIÓN PRINCIPAL
Panel de visualización principal – Etiqueta del frasco de tabletas de Rubraca 200 mg
NDC 69660-
201-91
Rubraca®
(rucaparib)tabletas
200 mg*
60 tabletas
Cada tableta contiene 344 mg de rucaparib camsilato equivalente a 200 mg de rucaparib
Rx solamente
Mantener fuera del alcance de los niños

PANEL DE VISUALIZACIÓN PRINCIPAL
Panel de visualización principal – Etiqueta del frasco de tabletas de Rubraca 250 mg
NDC 69660-
202-91
Rubraca®
(rucaparib)tabletas
250 mg*
60 tabletas
Cada tableta contiene 430 mg de rucaparib camsilato equivalente a 250 mg de rucaparib
Rx solamente
Mantener fuera del alcance de los niños

PANEL DE VISUALIZACIÓN PRINCIPAL
Panel de visualización principal – Etiqueta del frasco de tabletas de Rubraca 300 mg
NDC 69660-
203-91
Rubraca®
(rucaparib)tabletas
300 mg*
60 tabletas
Cada tableta contiene 516 mg de rucaparib camsilato equivalente a 300 mg de rucaparib
Rx solamente
Mantener fuera del alcance de los niños
