Fabricante de medicamentos: AbbVie Inc. (Updated: 2024-04-26)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
RINVOQ® (upadacitinib) comprimidos de liberación prolongada, para administración oral RINVOQ® LQ (upadacitinib) solución oral
Aprobación inicial en EE. UU.: 2019
ADVERTENCIA: INFECCIONES GRAVES, MORTALIDAD, MALIGNIDAD, EVENTOS CARDIOVASCULARES ADVERSOS MAYORES (MACE) Y TROMBOSIS
Consulte la información completa de prescripción para la advertencia completa en recuadro.
-
Aumento del riesgo de infecciones bacterianas, micóticas, víricas y oportunistas graves que conducen a la hospitalización o la muerte, incluida la tuberculosis (TB). Interrumpa el tratamiento con RINVOQ/RINVOQ LQ si se produce una infección grave hasta que la infección esté controlada. Realice pruebas para detectar TB latente antes y durante la terapia; trate la TB latente antes de su uso. Controle a todos los pacientes para detectar TB activa durante el tratamiento, incluso a los pacientes con prueba inicial negativa, de TB latente. (5.1)
-
Tasa más alta de mortalidad por todas las causas, incluida la muerte cardiovascular súbita con otro inhibidor de la cinasa de Janus (JAK) frente a los bloqueadores del factor de necrosis tumoral (TNF) en pacientes con artritis reumatoide (AR). (5.2)
-
Se han producido malignidades en pacientes tratados con RINVOQ. Tasa más alta de linfomas y cánceres de pulmón con otro inhibidor de JAK frente a los bloqueadores del TNF en pacientes con AR. (5.3)
-
Tasa más alta de MACE (definidos como muerte cardiovascular, infarto de miocardio y accidente cerebrovascular) con otro inhibidor de JAK frente a los bloqueadores del TNF en pacientes con AR. (5.4)
- Se ha producido trombosis en pacientes tratados con RINVOQ. Aumento de la incidencia de embolia pulmonar, trombosis venosa y arterial con otro inhibidor de JAK frente a los bloqueadores del TNF. (5.5)
CAMBIOS RECIENTES IMPORTANTES
INFORMACIÓN IMPORTANTE DE LA RECETA
RINVOQ/RINVOQ LQ es un inhibidor de la cinasa de Janus (JAK).
- RINVOQ está indicado para el tratamiento de adultos con artritis reumatoide activa de moderada a grave que han tenido una respuesta inadecuada o intolerancia a uno o más bloqueadores del TNF. (1.1)
Limitaciones de uso
RINVOQ no se recomienda para su uso en combinación con otros inhibidores de JAK, DMARD biológicos o con inmunosupresores potentes como azatioprina y ciclosporina. (1.1) - RINVOQ/RINVOQ LQ está indicado para el tratamiento de adultos y pacientes pediátricos de 2 años de edad o mayores con artritis psoriásica activa que han tenido una respuesta inadecuada o intolerancia a uno o más bloqueadores del TNF. (1.2)
Limitaciónes de uso
RINVOQ/RINVOQ LQ no se recomienda para su uso en combinación con otros inhibidores de JAK, DMARD biológicos o con inmunosupresores potentes como azatioprina y ciclosporina. (1.2) - RINVOQ está indicado para el tratamiento de adultos y pacientes pediátricos de 12 años de edad o mayores con dermatitis atópica refractaria, de moderada a grave, cuya enfermedad no está adecuadamente controlada con otros productos farmacéuticos sistémicos, incluidos los biológicos, o cuando el uso de esas terapias es desaconsejable. (1.3)
Limitaciones de uso
RINVOQ no se recomienda para su uso en combinación con otros inhibidores de JAK, inmunomoduladores biológicos o con otros inmunosupresores. (1.3) - RINVOQ está indicado para el tratamiento de adultos con colitis ulcerosa activa de moderada a grave que han tenido una respuesta inadecuada o intolerancia a uno o más bloqueadores del TNF. (1.4)
Limitaciones de uso
RINVOQ no se recomienda para su uso en combinación con otros inhibidores de JAK, terapias biológicas para la colitis ulcerosa o con inmunosupresores potentes como azatioprina y ciclosporina. (1.4) - RINVOQ está indicado para el tratamiento de adultos con enfermedad de Crohn activa de moderada a grave que han tenido una respuesta inadecuada o intolerancia a uno o más bloqueadores del TNF. (1.5)
Limitaciones de uso
RINVOQ no se recomienda para su uso en combinación con otros inhibidores de JAK, terapias biológicas para la enfermedad de Crohn o con inmunosupresores potentes como azatioprina y ciclosporina. (1.5) - RINVOQ está indicado para el tratamiento de adultos con espondilitis anquilosante activa que han tenido una respuesta inadecuada o intolerancia a uno o más bloqueadores del TNF. (1.6)
Limitaciones de uso
RINVOQ no se recomienda para su uso en combinación con otros inhibidores de JAK, DMARD biológicos o con inmunosupresores potentes como azatioprina y ciclosporina. (1.6) - RINVOQ está indicado para el tratamiento de adultos con espondiloartritis axial no radiográfica activa con signos objetivos de inflamación que han tenido una respuesta inadecuada o intolerancia a la terapia con bloqueadores del TNF. (1.7)
Limitaciones de uso
RINVOQ no se recomienda para su uso en combinación con otros inhibidores de JAK, DMARD biológicos o con inmunosupresores potentes como azatioprina y ciclosporina. (1.7) - RINVOQ/RINVOQ LQ está indicado para el tratamiento de pacientes de 2 años de edad o mayores con artritis idiopática juvenil poliarticular activa que han tenido una respuesta inadecuada o intolerancia a uno o más bloqueadores del TNF. (1.8)
Limitaciones de uso
RINVOQ/RINVOQ LQ no se recomienda para su uso en combinación con otros inhibidores de JAK, DMARD biológicos o con inmunosupresores potentes como azatioprina y ciclosporina. (1.8)
DOSIFICACIÓN Y ADMINISTRACIÓN
- RINVOQ LQ solución oral no es sustituible con comprimidos de liberación prolongada de RINVOQ (2.2, 2.10).
- Los cambios entre la solución oral de RINVOQ LQ y los comprimidos de liberación prolongada de RINVOQ deben ser realizados por el profesional sanitario.
- Antes del tratamiento, actualice las inmunizaciones y considere evaluar la tuberculosis activa y latente, la hepatitis viral, la función hepática y el estado de embarazo (2.1)
- Evite iniciar o interrumpa RINVOQ/RINVOQ LQ si el recuento absoluto de linfocitos es inferior a 500 células/mm3, el recuento absoluto de neutrófilos es inferior a 1000 células/mm3 o el nivel de hemoglobina es inferior a 8 g/dL. (2.1, 2.13)
Artritis reumatoide, Espondilitis anquilosante y espondiloartritis axial no radiográfica
Artritis psoriásica
-
Pacientes pediátricos de 2 a menos de 18 años de edad que pesen al menos 10 kg: La dosis recomendada se basa en el peso corporal (2.4)
- Adultos: La dosis recomendada de RINVOQ es de 15 mg una vez al día. (2.4)
Dermatitis atópica
-
Pacientes pediátricos de 12 años de edad o mayores que pesen al menos 40 kg y adultos menores de 65 años de edad: Inicie el tratamiento con RINVOQ 15 mg por vía oral una vez al día. Si no se logra una respuesta adecuada, considere aumentar la dosis a 30 mg por vía oral una vez al día. (2.5)
- Adultos de 65 años de edad o mayores: La dosis recomendada de RINVOQ es de 15 mg una vez al día. (2.5)
- Insuficiencia renal grave: La dosis recomendada de RINVOQ es de 15 mg una vez al día. (2.11)
Colitis ulcerosa
- Adultos: La dosis de inducción recomendada de RINVOQ es de 45 mg una vez al día durante 8 semanas. La dosis de mantenimiento recomendada de RINVOQ es de 15 mg una vez al día. Se puede considerar una dosis de mantenimiento de 30 mg una vez al día para pacientes con enfermedad refractaria, grave o extensa. Suspenda RINVOQ si no se logra una respuesta terapéutica adecuada con la dosis de 30 mg. Use la dosis efectiva más baja necesaria para mantener la respuesta. (2.6)
- Consulte la información completa de prescripción para la dosis recomendada en pacientes con insuficiencia renal o hepática y para la modificación de la dosis debido a interacciones medicamentosas. (2.11, 2.12)
Enfermedad de Crohn
- Adultos: La dosis de inducción recomendada de RINVOQ es de 45 mg una vez al día durante 12 semanas. La dosis de mantenimiento recomendada de RINVOQ es de 15 mg una vez al día. Se puede considerar una dosis de mantenimiento de 30 mg una vez al día para pacientes con enfermedad refractaria, grave o extensa. Suspenda RINVOQ si no se logra una respuesta terapéutica adecuada con la dosis de 30 mg. Use la dosis efectiva más baja necesaria para mantener la respuesta. (2.7)
- Consulte la información completa de prescripción para la dosis recomendada en pacientes con insuficiencia renal o hepática y para la modificación de la dosis debido a interacciones medicamentosas. (2.11, 2.12)
Artritis idiopática juvenil poliarticular
- La dosis recomendada se basa en el peso corporal (2.10)
FORMAS Y FUERZAS DE DOSIFICACIÓN
CONTRAINDICACIONES
Hipersensibilidad conocida a upadacitinib o a cualquiera de los excipientes de RINVOQ/RINVOQ LQ. (4, 5.6)
ADVERTENCIAS Y PRECAUCIONES
- Infecciones graves: Evite el uso en pacientes con infección activa grave, incluidas las infecciones localizadas. (5.1)
- Hipersensibilidad: Se han notificado reacciones graves de hipersensibilidad (p. ej., anafilaxia). Suspenda el tratamiento si se produce una reacción grave de hipersensibilidad. (5.6)
- Perforaciones gastrointestinales (GI) : Controle a los pacientes con riesgo de perforaciones GI y evalúe rápidamente a los pacientes con síntomas. (5.7)
- Anormalidades de laboratorio: Se recomienda el control debido a posibles cambios en los linfocitos, neutrófilos, hemoglobina, enzimas hepáticas y lípidos. (5.8)
- Toxicidad embrio-fetal: Puede causar daño fetal según estudios en animales. Avise a las pacientes en edad fértil del riesgo potencial para el feto y de la necesidad de utilizar métodos anticonceptivos eficaces. (5.9, 8.1, 8.3)
- Vacunaciones: Evite el uso con vacunas vivas. (5.10)
- Residuos de medicamentos en las heces: Se ha observado en las heces o en la salida de la ostomía en pacientes con tiempos de tránsito GI acortados. Controle a los pacientes clínicamente y considere un tratamiento alternativo si la respuesta terapéutica es inadecuada. (5.11)
REACCIONES ADVERSAS
- Artritis reumatoide, artritis psoriásica, espondilitis anquilosante y espondiloartritis axial no radiográfica: Las reacciones adversas (≥ 1%) fueron: infecciones del tracto respiratorio superior, herpes zóster, herpes simple, bronquitis, náuseas, tos, pirexia, acné, y dolor de cabeza. (6.1)
- Dermatitis atópica: Las reacciones adversas (≥ 1%) son: infecciones del tracto respiratorio superior, acné, herpes simple, dolor de cabeza, aumento de la creatina fosfoquinasa en sangre, tos, hipersensibilidad, foliculitis, náuseas, dolor abdominal, pirexia, aumento de peso, herpes zóster, gripe, fatiga, neutropenia, mialgia y síntomas gripales. (6.1)
- Colitis ulcerosa: Las reacciones adversas (≥ 5%) notificadas durante la inducción o el mantenimiento son: infecciones del tracto respiratorio superior, aumento de la creatina fosfoquinasa en sangre, acné, neutropenia, elevación de las enzimas hepáticas y erupción cutánea. (6.1)
- Enfermedad de Crohn: Las reacciones adversas (≥ 5%) notificadas durante la inducción o el mantenimiento son: infecciones del tracto respiratorio superior, anemia, pirexia, acné, herpes zóster y dolor de cabeza. (6.1)
Para notificar SOSPECHAS DE REACCIONES ADVERSAS, póngase en contacto con AbbVie Inc. al 1-800-633-9110 o con la FDA al 1-800-FDA-1088 o www.fda.gov/medwatch.
INTERACCIONES MEDICAMENTOSAS
- Inhibidores potentes del CYP3A4: Consulte la información completa de prescripción para la modificación de la dosis en pacientes con dermatitis atópica, colitis ulcerosa y enfermedad de Crohn. (2.12, 7.1)
- Inductores potentes del CYP3A4: No se recomienda la coadministración de RINVOQ/RINVOQ LQ con inductores potentes del CYP3A4. (7.2)
USO EN POBLACIONES ESPECÍFICAS
Ver 17 para INFORMACIÓN PARA EL PACIENTE y Guía de medicamentos.
Revisado: 4/2024
Tabla de Contenido
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDO*
ADVERTENCIA: INFECCIONES GRAVES, MORTALIDAD, MALIGNIDAD, EVENTOS CARDIOVASCULARES ADVERSOS MAYORES Y TROMBOSIS
1 INDICACIONES Y USO
1.1 Artritis Reumatoide
1.2 Artritis Psoriásica
1.3 Dermatitis Atópica
1.4 Colitis Ulcerosa
1.5 Enfermedad de Crohn
1.6 Espondilitis Anquilosante
1.7 Espondiloartritis Axial No Radiográfica
1.8 Artritis Idiopática Juvenil Poliarticular
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Evaluaciones e Inmunizaciones Recomendadas Antes del Inicio del Tratamiento
2.2 Instrucciones Importantes de Administración
2.3 Dosificación Recomendada en Artritis Reumatoide
2.4 Dosificación Recomendada en Artritis Psoriásica
2.5 Dosificación Recomendada en Dermatitis Atópica
2.6 Dosificación Recomendada en Colitis Ulcerosa
2.7 Dosificación Recomendada en Enfermedad de Crohn
2.8 Dosificación Recomendada en Espondilitis Anquilosante
2.9 Recomended Dosificación en Espondiloartritis Axial No Radiográfica
2.10 Dosificación Recomendada en Artritis Idiopática Juvenil Poliarticular
2.11 Dosificación Recomendada en Pacientes con Insuficiencia Renal o Hepática
2.12 Modificaciones de la Dosificación Debido a Interacciones Medicamentosas
2.13 Interrupción de la Dosificación
3 FORMAS Y FUERZAS DE DOSIFICACIÓN
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Infecciones Graves
5.2 Mortalidad
5.3 Malignidad y Trastornos Linfoproliferativos
5.4 Eventos Cardiovasculares Adversos Mayores
5.5 Trombosis
5.6 Reacciones de Hipersensibilidad
5.7 Perforaciones Gastrointestinales
5.8 Anormalidades de Laboratorio
5.9 Toxicidad Embriofetal
5.10 Vacunacións
5.11 Residuo de Medicamentos en las Heces
6 REACCIONES ADVERSAS
6.1 Experiencia en Ensayos Clínicos
7 INTERACCIONES MEDICAMENTOSAS
7.1 Inhibidores Fuertes de CYP3A4
7.2 Inductores Fuertes de CYP3A4
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.3 Mujeres y Hombres en Potencial Reproductivo
8.4 Uso Pediátrico
8.5 Uso Geriátrico
8.6 Insuficiencia Renal
8.7 Insuficiencia Hepática
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de Acción
12.2 Farmacodynamics
12.3 Farmacocinética
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Disminución de la Fertilidad
14 ESTUDIOS CLÍNICOS
14.1 Artritis Reumatoide
14.2 Artritis Psoriásica
14.3 Dermatitis Atópica
14.4 Colitis Ulcerosa
14.5 Enfermedad de Crohn
14.6 Espondilitis Anquilosante
14.7 Espondiloartritis axial no radiográfica
14.8 Artritis Idiopática Juvenil Poliarticular
16 SUMINISTRO / ALMACENAMIENTO Y MANIPULACIÓN
17 INFORMACIÓN PARA LA CONSEJERÍA AL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información completa de prescripción no se enumeran.
ADVERTENCIA RECUADRADA
WARNING: INFECCIONES GRAVES, MORTALIDAD, TUMORES MALIGNOS, EVENTOS CARDIOVASCULARES ADVERSOS MAYORES Y TROMBOSIS
INFECCIONES GRAVES
Los pacientes tratados con RINVOQ/RINVOQ LQ corren mayor riesgo de contraer infecciones graves que pueden provocar hospitalización o la muerte [ver Advertencias y precauciones (5.1), Reacciones adversas (6.1)]. La mayoría de los pacientes que contrajeron estas infecciones estaban tomando inmunodepresores concomitantes como metotrexato o corticosteroides.
Si se produce una infección grave, interrumpa el tratamiento con RINVOQ/RINVOQ LQ hasta que la infección esté controlada.
Las infecciones notificadas incluyen:
-
Tuberculosis activa, que puede presentarse con enfermedad pulmonar o extrapulmonar. Se debe realizar una prueba de detección de tuberculosis latente a los pacientes antes del uso de RINVOQ/RINVOQ LQ y durante el tratamiento. Se debe considerar el tratamiento de la infección latente antes del uso de RINVOQ/RINVOQ LQ.
-
Infecciones fúngicas invasivas, como criptococosis y neumonocistosis.
- Infecciones bacterianas, víricas, incluido el herpes zóster, y otras infecciones por patógenos oportunistas.
Los riesgos y beneficios del tratamiento con RINVOQ/RINVOQ LQ deben considerarse cuidadosamente antes de iniciar el tratamiento en pacientes con infección crónica o recurrente.
Se debe vigilar estrechamente a los pacientes para detectar el desarrollo de signos y síntomas de infección durante y después del tratamiento con RINVOQ/RINVOQ LQ, incluido el posible desarrollo de tuberculosis en pacientes que dieron negativo en la prueba de infección tuberculosa latente antes de iniciar el tratamiento [ver Advertencias y precauciones (5.1)].
MORTALIDAD
En un estudio de seguridad posterior a la comercialización, aleatorizado y amplio en pacientes con artritis reumatoide (AR) de 50 años o mayores con al menos un factor de riesgo cardiovascular que comparó otro inhibidor de la Janus quinasa (JAK) con bloqueadores del factor de necrosis tumoral (TNF), se observó una tasa más alta de mortalidad por todas las causas, incluida la muerte cardiovascular súbita, con el inhibidor de JAK [ver Advertencias y precauciones (5.2)].
TUMORES MALIGNOS
Se han observado linfomas y otros tumores malignos en pacientes tratados con RINVOQ. En pacientes con AR tratados con otro inhibidor de JAK, se observó una tasa más alta de tumores malignos (excluido el cáncer de piel no melanoma [NMSC]) en comparación con los bloqueadores de TNF. Los pacientes que son fumadores actuales o pasados corren un mayor riesgo adicional [ver Advertencias y precauciones (5.3)].
EVENTOS CARDIOVASCULARES ADVERSOS MAYORES
En pacientes con AR de 50 años o mayores con al menos un factor de riesgo cardiovascular tratados con otro inhibidor de JAK, se observó una tasa más alta de eventos cardiovasculares adversos mayores (MACE) (definidos como muerte cardiovascular, infarto de miocardio y accidente cerebrovascular) en comparación con los bloqueadores de TNF. Los pacientes que son fumadores actuales o pasados corren un mayor riesgo adicional. Suspenda el tratamiento con RINVOQ/RINVOQ LQ en pacientes que hayan sufrido un infarto de miocardio o un accidente cerebrovascular [ver Advertencias y precauciones (5.4)].
TROMBOSIS
Se han producido casos de trombosis, como trombosis venosa profunda, embolia pulmonar y trombosis arterial, en pacientes tratados con inhibidores de JAK utilizados para tratar afecciones inflamatorias. Muchos de estos eventos adversos fueron graves y algunos provocaron la muerte. En pacientes con AR de 50 años o mayores con al menos un factor de riesgo cardiovascular tratados con otro inhibidor de JAK, se observó una tasa más alta de trombosis en comparación con los bloqueadores de TNF. Evite el uso de RINVOQ/RINVOQ LQ en pacientes en riesgo. Los pacientes con síntomas de trombosis deben suspender el tratamiento con RINVOQ/RINVOQ LQ y ser evaluados de inmediato [ver Advertencias y precauciones (5.5)].
1 INDICACIONES Y USO
1.1 Artritis Reumatoide
RINVOQ® está indicado para el tratamiento de adultos con artritis reumatoide activa de moderada a grave que han tenido una respuesta inadecuada o intolerancia a uno o más bloqueadores del TNF.
● Limitaciones de uso: No se recomienda el uso de RINVOQ en combinación con otros inhibidores de JAK, fármacos antirreumáticos modificadores de la enfermedad (DMARD) biológicos o con inmunosupresores potentes como azatioprina y ciclosporina.
1.2 Artritis Psoriásica
RINVOQ/RINVOQ LQ está indicado para el tratamiento de adultos y pacientes pediátricos de 2 años de edad o mayores con artritis psoriásica activa que han tenido una respuesta inadecuada o intolerancia a uno o más bloqueadores del TNF.
● Limitaciones de uso: No se recomienda el uso de RINVOQ/RINVOQ LQ en combinación con otros inhibidores de JAK, DMARD biológicos o con inmunosupresores potentes como azatioprina y ciclosporina.
1.3 Dermatitis Atópica
RINVOQ está indicado para el tratamiento de adultos y pacientes pediátricos de 12 años de edad o mayores con dermatitis atópica refractaria, de moderada a grave, cuya enfermedad no está adecuadamente controlada con otros productos farmacéuticos sistémicos, incluidos los biológicos, o cuando el uso de esas terapias es desaconsejable.
● Limitaciones de uso: No se recomienda el uso de RINVOQ en combinación con otros inhibidores de JAK, inmunomoduladores biológicos o con otros inmunosupresores.
1.4 Colitis Ulcerosa
RINVOQ está indicado para el tratamiento de pacientes adultos con colitis ulcerosa activa de moderada a grave que han tenido una respuesta inadecuada o intolerancia a uno o más bloqueadores del TNF.
● Limitaciones de uso: No se recomienda el uso de RINVOQ en combinación con otros inhibidores de JAK, terapias biológicas para la colitis ulcerosa o con inmunosupresores potentes como azatioprina y ciclosporina.
1.5 Enfermedad de Crohn
RINVOQ está indicado para el tratamiento de pacientes adultos con enfermedad de Crohn activa de moderada a grave que han tenido una respuesta inadecuada o intolerancia a uno o más bloqueadores del TNF.
● Limitaciones de uso: No se recomienda el uso de RINVOQ en combinación con otros inhibidores de JAK, terapias biológicas para la enfermedad de Crohn o con inmunosupresores potentes como azatioprina y ciclosporina.
1.6 Espondilitis Anquilosante
RINVOQ está indicado para el tratamiento de adultos con espondilitis anquilosante activa que han tenido una respuesta inadecuada o intolerancia a uno o más bloqueadores del TNF.
● Limitaciones de uso: No se recomienda el uso de RINVOQ en combinación con otros inhibidores de JAK, DMARD biológicos o con inmunosupresores potentes como azatioprina y ciclosporina.
1.7 Espondiloartritis Axial No Radiográfica
RINVOQ está indicado para el tratamiento de adultos con espondiloartritis axial no radiográfica activa con signos objetivos de inflamación que han tenido una respuesta inadecuada o intolerancia a la terapia con bloqueadores del TNF.
● Limitaciones de uso: No se recomienda el uso de RINVOQ en combinación con otros inhibidores de JAK, DMARD biológicos o con inmunosupresores potentes como azatioprina y ciclosporina.
1.8 Artritis Idiopática Juvenil Poliarticular
RINVOQ/RINVOQ LQ está indicado para el tratamiento de pacientes de 2 años de edad o mayores con artritis idiopática juvenil poliarticular (pJIA) activa que han tenido una respuesta inadecuada o intolerancia a uno o más bloqueadores del TNF.
● Limitaciones de uso: No se recomienda el uso de RINVOQ/RINVOQ LQ en combinación con otros inhibidores de JAK, DMARD biológicos o con inmunosupresores potentes como azatioprina y ciclosporina.
2 DOSIS Y ADMINISTRACIÓN
2.1 Evaluaciones y Vacunaciones Recomendadas Antes del Inicio del Tratamiento
Antes de iniciar el tratamiento con RINVOQ/RINVOQ LQ, considere realizar las siguientes evaluaciones:
● Evaluación de la infección por tuberculosis (TB) activa y latente – Si es positiva, trate la TB antes de usar RINVOQ/RINVOQ LQ [ver Advertencias y precauciones (5.1)].
● Detección de hepatitis viral de acuerdo con las pautas clínicas – No se recomienda iniciar RINVOQ/RINVOQ LQ en pacientes con hepatitis B o hepatitis C activa [ver Advertencias y precauciones (5.1)].
● Un hemograma completo – No se recomienda iniciar RINVOQ/RINVOQ LQ en pacientes con un recuento absoluto de linfocitos inferior a 500 células/mm3, recuento absoluto de neutrófilos inferior a 1000 células/mm3, o nivel de hemoglobina inferior a 8 g/dL [ver Dosis y administración (2.13) y Advertencias y precauciones (5.8)].
● Función hepática basal: No se recomienda iniciar RINVOQ/RINVOQ LQ para pacientes con insuficiencia hepática grave (Child-Pugh C) [ver Uso en poblaciones específicas (8.7) y Farmacología clínica (12.3)].
● Estado de embarazo: Verifique el estado de embarazo de las mujeres en edad fértil antes de comenzar el tratamiento [ver Advertencias y precauciones (5.9) y Uso en poblaciones específicas (8.1, 8.3)].
Actualice las inmunizaciones de acuerdo con las pautas de inmunización actuales [ver Advertencias y precauciones (5.10)].
2.2 Instrucciones Importantes de Administración
● La solución oral RINVOQ LQ no es sustituible por las tabletas de liberación prolongada RINVOQ [ver Dosis y administración (2.4, 2.10)].
● Los cambios entre la solución oral RINVOQ LQ y las tabletas de liberación prolongada RINVOQ deben ser realizados por el profesional de la salud.
● RINVOQ/RINVOQ LQ debe tomarse por vía oral con o sin alimentos [ver Farmacología clínica (12.3)].
● Las tabletas de RINVOQ deben tragarse enteras. Las tabletas de RINVOQ no deben dividirse, triturarse ni masticarse.
● RINVOQ LQ debe administrarse utilizando el adaptador de botella de presión y la jeringa oral proporcionados [ver Instrucciones de uso].
● RINVOQ LQ se dosifica dos veces al día [ver Dosis y administración (2.4, 2.10)].
2.3 Dosis Recomendada en Artritis Reumatoide
La dosis recomendada de RINVOQ es de 15 mg una vez al día.
2.4 Dosis Recomendada en Artritis Psoriásica
Pacientes pediátricos de 2 a menos de 18 años de edad
La dosis recomendada se basa en el peso corporal (Tabla 1).
| Peso del paciente | RINVOQ LQ | RINVOQ |
| 10 kg a menos de 20 kg | 3 mg (3 mL de solución oral) dos veces al día | No recomendado |
| 20 kg a menos de 30 kg | 4 mg (4 mL de solución oral) dos veces al día | No recomendado |
| 30 kg y más | 6 mg (6 mL de solución oral) dos veces al día | 15 mg (una tableta de 15 mg) una vez al día |
La solución oral RINVOQ LQ no es sustituible por las tabletas de liberación prolongada de RINVOQ. Los cambios entre la solución oral RINVOQ LQ y las tabletas de liberación prolongada de RINVOQ deben ser realizados por el profesional de la salud.
Adultos de 18 años de edad o mayores
La dosis recomendada de RINVOQ es de 15 mg una vez al día.
2.5 Dosis recomendada en dermatitis atópica
Pacientes pediátricos de 12 años de edad o mayores que pesen al menos 40 kg y adultos menores de 65 años de edad
Inicie el tratamiento con RINVOQ 15 mg una vez al día. Si no se logra una respuesta adecuada, considere aumentar la dosis a 30 mg una vez al día. Suspenda RINVOQ si no se logra una respuesta adecuada con la dosis de 30 mg. Use la dosis efectiva más baja necesaria para mantener la respuesta.
Adultos de 65 años de edad o mayores
La dosis recomendada de RINVOQ es de 15 mg una vez al día.
2.6 Dosis recomendada en colitis ulcerosa
Pacientes adultos: Inducción
La dosis de inducción recomendada de RINVOQ es de 45 mg una vez al día durante 8 semanas.
Pacientes adultos: Mantenimiento
La dosis recomendada de RINVOQ para el tratamiento de mantenimiento es de 15 mg una vez al día. Se puede considerar una dosis de 30 mg una vez al día para pacientes con enfermedad refractaria, grave o extensa. Suspenda RINVOQ si no se logra una respuesta terapéutica adecuada con la dosis de 30 mg. Use la dosis efectiva más baja necesaria para mantener la respuesta.
2.7 Dosis recomendada en enfermedad de Crohn
Pacientes adultos: Inducción
La dosis de inducción recomendada de RINVOQ es de 45 mg una vez al día durante 12 semanas.
Pacientes adultos: Mantenimiento
La dosis recomendada de RINVOQ para el tratamiento de mantenimiento es de 15 mg una vez al día. Se puede considerar una dosis de 30 mg una vez al día para pacientes con enfermedad refractaria, grave o extensa. Suspenda RINVOQ si no se logra una respuesta terapéutica adecuada con la dosis de 30 mg. Use la dosis efectiva más baja necesaria para mantener la respuesta.
2.8 Dosis recomendada en Espondilitis anquilosante
La dosis recomendada de RINVOQ es de 15 mg una vez al día.
2.9 Dosis recomendada en espondiloartritis axial no radiográfica
La dosis recomendada de RINVOQ es de 15 mg una vez al día.
2.10 Dosis recomendada en Artritis idiopática juvenil poliarticular
La dosis recomendada se basa en el peso corporal (Tabla 2).
| Peso del paciente | RINVOQ LQ | RINVOQ |
| 10 kg a menos de 20 kg | 3 mg (3 mL de solución oral) dos veces al día | No recomendado |
| 20 kg a menos de 30 kg | 4 mg (4 mL de solución oral) dos veces al día | No recomendado |
| 30 kg o más | 6 mg (6 mL de solución oral) dos veces al día | 15 mg (una tableta de 15 mg) una vez al día |
La solución oral RINVOQ LQ no es intercambiable con las tabletas de liberación prolongada de RINVOQ. Los cambios entre la solución oral RINVOQ LQ y las tabletas de liberación prolongada de RINVOQ deben ser realizados por el profesional de la salud.
2.11 Dosis recomendada en pacientes con insuficiencia renal o hepática
Insuficiencia renal
Artritis reumatoide, Artritis psoriásica, Espondilitis anquilosante, Espondiloartritis axial no radiográfica, y pJIA:
● No se necesita ajuste de dosis para pacientes con insuficiencia renal leve, moderada o grave.
Dermatitis atópica:
● Para pacientes con insuficiencia renal grave [tasa de filtración glomerular estimada (TFGe) 15 a < 30 mL/min/1.73m2] la dosis recomendada de RINVOQ es de 15 mg una vez al día [ver Uso en poblaciones específicas (8.6)].
● No se necesita ajuste de dosis para pacientes con insuficiencia renal leve o moderada (TFGe ≥ 30 mL/min/1.73m2).
● No se recomienda el uso de RINVOQ en pacientes con enfermedad renal en etapa terminal (TFGe < 15 mL/min/1.73m2) [ver Uso en poblaciones específicas (8.6)].
Colitis ulcerosa:
● Para pacientes con insuficiencia renal grave (TFGe 15 a < 30 mL/min/1.73m2), la dosis recomendada de RINVOQ es:
• Inducción: 30 mg una vez al día durante 8 semanas
• Mantenimiento: 15 mg una vez al día
● No se necesita ajuste de dosis para pacientes con insuficiencia renal leve o moderada (TFGe ≥ 30 mL/min/1.73m2).
● No se recomienda el uso de RINVOQ en pacientes con enfermedad renal en etapa terminal (TFGe < 15 mL/min/1.73m2) [ver Uso en poblaciones específicas (8.6)].
Enfermedad de Crohn:
● Para pacientes con insuficiencia renal grave (TFGe 15 a < 30 mL/min/1.73m2), la dosis recomendada de RINVOQ es:
• Inducción: 30 mg una vez al día durante 12 semanas
• Mantenimiento: 15 mg una vez al día
● No se necesita ajuste de dosis para pacientes con insuficiencia renal leve o moderada (TFGe ≥ 30 mL/min/1.73m2).
● No se recomienda el uso de RINVOQ en pacientes con enfermedad renal en etapa terminal (TFGe < 15 mL/min/1.73m2) [ver Uso en poblaciones específicas (8.6)].
Insuficiencia hepática
No se recomienda el uso de RINVOQ/RINVOQ LQ en pacientes con insuficiencia hepática grave (Child-Pugh C) [ver Uso en poblaciones específicas (8.7)].
Artritis reumatoide, Artritis psoriásica, Dermatitis atópica, Espondilitis anquilosante, Espondiloartritis axial no radiográfica, y pJIA:
No se necesita ajuste de dosis para pacientes con insuficiencia hepática leve o moderada (Child-Pugh A o B).
Colitis ulcerosa:
Para pacientes con insuficiencia hepática leve a moderada (Child-Pugh A o B) la dosis recomendada de RINVOQ es:
- Inducción: 30 mg una vez al día durante 8 semanas
- Mantenimiento: 15 mg una vez al día
Enfermedad de Crohn:
Para pacientes con insuficiencia hepática leve a moderada (Child-Pugh A o B) la dosis recomendada de RINVOQ es:
• Inducción: 30 mg una vez al día durante 12 semanas
• Mantenimiento: 15 mg una vez al día
2.12 Modificaciones de la dosis debido a interacciones medicamentosas
Artritis reumatoide, Artritis psoriásica, Espondilitis anquilosante, Espondiloartritis axial no radiográfica, y pJIA
No se necesita ajuste de dosis en pacientes que reciben inhibidores potentes del CYP3A4 [ver Interacciones medicamentosas (7.1)].
Dermatitis atópica
La dosis recomendada de RINVOQ en pacientes que reciben inhibidores potentes del CYP3A4 es de 15 mg una vez al día [ver Interacciones medicamentosas (7.1)].
Colitis ulcerosa
La dosis recomendada de RINVOQ en pacientes con colitis ulcerosa que reciben inhibidores potentes del CYP3A4 [ver Interacciones medicamentosas (7.1)]:
- Inducción: 30 mg una vez al día durante 8 semanas
- Mantenimiento: 15 mg una vez al día
Enfermedad de Crohn
La dosis recomendada de RINVOQ en pacientes con enfermedad de Crohn que reciben inhibidores potentes del CYP3A4 [ver Interacciones medicamentosas (7.1)]:
• Inducción: 30 mg una vez al día durante 12 semanas
• Mantenimiento: 15 mg una vez al día
2.13 Interrupción de la dosis
Infecciones
Si un paciente desarrolla una infección grave, incluida una infección oportunista grave, interrumpa el tratamiento con RINVOQ/RINVOQ LQ hasta que la infección esté controlada [ver Advertencias y precauciones (5.1)].
Anormalidades de laboratorio
Puede ser necesaria la interrupción de la dosificación para el manejo de las anormalidades de laboratorio como se describe en la Tabla 3 [ver Advertencias y precauciones (5.8)].
| Medida de laboratorio | Acción |
| Recuento absoluto de neutrófilos (RAN) | Interrupción del tratamiento si el RAN es inferior a 1000 células/mm3; el tratamiento puede reanudarse una vez que el RAN vuelva a estar por encima de este valor |
| Recuento absoluto de linfocitos (RAL) | Interrupción del tratamiento si el RAL es inferior a 500 células/mm3; el tratamiento puede reanudarse una vez que el RAL vuelva a estar por encima de este valor |
| Hemoglobina (Hb) | Interrupción del tratamiento si la Hb es inferior a 8 g/dL; el tratamiento puede reanudarse una vez que la Hb vuelva a estar por encima de este valor |
| Transaminasas hepáticas | Interrupción del tratamiento si se sospecha una lesión hepática inducida por fármacos, hasta que se excluya este diagnóstico. |
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
Tabletas de liberación prolongada RINVOQ:
- 15 mg de upadacitinib: púrpura, biconvexa oblonga, con dimensiones de 14 x 8 mm, y con la marca ‘a15’ en un lado.
- 30 mg de upadacitinib: roja, biconvexa oblonga, con dimensiones de 14 x 8 mm, y con la marca ‘a30’ en un lado.
- 45 mg de upadacitinib: amarilla a amarilla moteada, biconvexa oblonga, con dimensiones de 14 x 8 mm, y con la marca ‘a45’ en un lado.
Solución oral RINVOQ LQ:
- 1 mg/mL de upadacitinib; solución transparente, incolora a ligeramente amarilla en frasco de 180 mL.
4 CONTRAINDICACIONES
RINVOQ/RINVOQ LQ está contraindicado en pacientes con hipersensibilidad conocida a upadacitinib o a cualquiera de sus excipientes [ver Advertencias y precauciones (5.6)].
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Infecciones graves
Se han notificado infecciones graves y en ocasiones mortales en pacientes que reciben RINVOQ. Las infecciones graves más frecuentes notificadas con RINVOQ incluyeron neumonía y celulitis [véase Reacciones adversas (6.1)]. Entre las infecciones oportunistas, se notificaron tuberculosis, herpes zóster multidermatomal, candidiasis oral/esofágica y criptococosis con RINVOQ. Se observó una tasa más alta de infecciones graves con RINVOQ 30 mg en comparación con RINVOQ 15 mg.
Evite el uso de RINVOQ/RINVOQ LQ en pacientes con una infección activa grave, incluidas las infecciones localizadas. Considere los riesgos y beneficios del tratamiento antes de iniciar RINVOQ/RINVOQ LQ en pacientes:
- con infección crónica o recurrente
- que han estado expuestos a la tuberculosis
- con antecedentes de una infección grave u oportunista
- que han residido o viajado en áreas de tuberculosis endémica o micosis endémicas; o
- con afecciones subyacentes que pueden predisponerlos a la infección.
Controle de cerca a los pacientes para detectar el desarrollo de signos y síntomas de infección durante y después del tratamiento con RINVOQ/RINVOQ LQ. Interrumpa RINVOQ/RINVOQ LQ si un paciente desarrolla una infección grave u oportunista.
Un paciente que desarrolla una nueva infección durante el tratamiento con RINVOQ/RINVOQ LQ debe someterse a pruebas de diagnóstico rápidas y completas apropiadas para un paciente inmunocomprometido; se debe iniciar la terapia antimicrobiana apropiada, se debe controlar de cerca al paciente y se debe interrumpir RINVOQ/RINVOQ LQ si el paciente no responde a la terapia antimicrobiana. RINVOQ/RINVOQ LQ se puede reanudar una vez que la infección esté controlada.
Tuberculosis
Evalúe y examine a los pacientes para detectar la infección por tuberculosis (TB) latente y activa antes de administrar RINVOQ/RINVOQ LQ. Los pacientes con TB latente deben recibir tratamiento con terapia antimicobacteriana estándar antes de iniciar RINVOQ/RINVOQ LQ. RINVOQ/RINVOQ LQ no debe administrarse a pacientes con TB activa. Considere la terapia anti-TB antes de iniciar RINVOQ/RINVOQ LQ en pacientes con TB latente previamente no tratada o TB activa en quienes no se puede confirmar un curso de tratamiento adecuado, y para pacientes con una prueba negativa para TB latente pero que tienen factores de riesgo para la infección por TB.
Se recomienda consultar con un médico con experiencia en el tratamiento de la TB para ayudar en la decisión sobre si es apropiado iniciar la terapia anti-TB para un paciente individual.
Durante el uso de RINVOQ/RINVOQ LQ, controle a los pacientes para detectar el desarrollo de signos y síntomas de TB, incluidos los pacientes que dieron negativo en la prueba de infección por TB latente antes de iniciar la terapia.
Reactivación viral
Se notificó reactivación viral, incluidos casos de reactivación del virus del herpes (p. ej., herpes zóster) y reactivación del virus de la hepatitis B, en ensayos clínicos con RINVOQ [véase Reacciones adversas (6.1)]. El riesgo de herpes zóster parece ser mayor en pacientes tratados con RINVOQ en Japón. Si un paciente desarrolla herpes zóster, considere interrumpir temporalmente RINVOQ/RINVOQ LQ hasta que el episodio se resuelva.
La detección de hepatitis viral y el control de la reactivación deben realizarse de acuerdo con las pautas clínicas antes de comenzar y durante la terapia con RINVOQ/RINVOQ LQ. Los pacientes que dieron positivo para anticuerpos contra la hepatitis C y ARN del virus de la hepatitis C fueron excluidos de los ensayos clínicos. Los pacientes que dieron positivo para el antígeno de superficie de la hepatitis B o el ADN del virus de la hepatitis B fueron excluidos de los ensayos clínicos. Sin embargo, todavía se notificaron casos de reactivación de la hepatitis B en pacientes inscritos en los ensayos de fase 3 de RINVOQ. Si se detecta ADN del virus de la hepatitis B mientras recibe RINVOQ/RINVOQ LQ, se debe consultar a un especialista en hígado.
5.2 Mortalidad
En un estudio de seguridad poscomercialización grande, aleatorizado, de otro inhibidor de JAK en pacientes con AR de 50 años de edad o más con al menos un factor de riesgo cardiovascular, se observó una tasa más alta de mortalidad por todas las causas, incluida la muerte cardiovascular súbita, en pacientes tratados con el inhibidor de JAK en comparación con los bloqueadores del TNF.
Considere los beneficios y riesgos para el paciente individual antes de iniciar o continuar la terapia con RINVOQ/RINVOQ LQ.
5.3 Malignidad y trastornos linfoproliferativos
Se observaron malignidades, incluidos linfomas, en ensayos clínicos de RINVOQ [véase Reacciones adversas (6.1)].
En un estudio de seguridad poscomercialización grande, aleatorizado, de otro inhibidor de JAK en pacientes con AR, se observó una tasa más alta de malignidades (excluyendo NMSC) en pacientes tratados con el inhibidor de JAK en comparación con los tratados con bloqueadores del TNF. Se observó una tasa más alta de linfomas en pacientes tratados con el inhibidor de JAK en comparación con los tratados con bloqueadores del TNF. Se observó una tasa más alta de cánceres de pulmón en fumadores actuales o pasados tratados con el inhibidor de JAK en comparación con los tratados con bloqueadores del TNF. En este estudio, los fumadores actuales o pasados tuvieron un riesgo adicional aumentado de malignidades generales.
Considere los beneficios y riesgos para el paciente individual antes de iniciar o continuar la terapia con RINVOQ/RINVOQ LQ, particularmente en pacientes con una malignidad conocida (que no sea un NMSC tratado con éxito), pacientes que desarrollan una malignidad cuando están en tratamiento y pacientes que son fumadores actuales o pasados.
Cáncer de piel no melanoma
Se han notificado NMSC en pacientes tratados con RINVOQ. Se recomienda un examen periódico de la piel para los pacientes que tienen un riesgo aumentado de cáncer de piel.
La exposición a la luz solar y los rayos UV debe limitarse usando ropa protectora y usando un protector solar de amplio espectro.
5.4 Eventos cardiovasculares adversos mayores
En un estudio de seguridad poscomercialización grande, aleatorizado, de otro inhibidor de JAK en pacientes con AR de 50 años de edad o más con al menos un factor de riesgo cardiovascular, se observó una tasa más alta de eventos cardiovasculares adversos mayores (MACE) definidos como muerte cardiovascular, infarto de miocardio (IM) no fatal y accidente cerebrovascular no fatal con el inhibidor de JAK en comparación con aquellos tratados con bloqueadores del TNF. Los pacientes que son fumadores actuales o pasados tienen un riesgo adicional aumentado.
Considere los beneficios y los riesgos para el paciente individual antes de iniciar o continuar el tratamiento con RINVOQ/RINVOQ LQ, particularmente en pacientes que son fumadores actuales o pasados y pacientes con otros factores de riesgo cardiovascular. Los pacientes deben ser informados sobre los síntomas de los eventos cardiovasculares graves y los pasos a seguir si ocurren. Suspenda RINVOQ/RINVOQ LQ en pacientes que hayan experimentado un infarto de miocardio o un accidente cerebrovascular.
5.5 Trombosis
Se ha producido trombosis, incluida la trombosis venosa profunda (TVP), la embolia pulmonar (EP) y la trombosis arterial, en pacientes tratados por afecciones inflamatorias con inhibidores de JAK, incluido RINVOQ. Muchos de estos eventos adversos fueron graves y algunos provocaron la muerte.
En un estudio de seguridad poscomercialización grande, aleatorizado, de otro inhibidor de JAK en pacientes con AR de 50 años de edad o más con al menos un factor de riesgo cardiovascular, se observaron tasas más altas de trombosis general, TVP y EP en comparación con aquellos tratados con bloqueadores del TNF.
Si se presentan síntomas de trombosis, los pacientes deben suspender RINVOQ/RINVOQ LQ y ser evaluados de inmediato y tratados de manera adecuada. Evite RINVOQ/RINVOQ LQ en pacientes que puedan tener un mayor riesgo de trombosis.
5.6 Reacciones de hipersensibilidad
Se informaron reacciones de hipersensibilidad graves como anafilaxia y angioedema en pacientes que recibieron RINVOQ en ensayos clínicos. Si se produce una reacción de hipersensibilidad clínicamente significativa, suspenda RINVOQ/RINVOQ LQ e instituya la terapia adecuada [ver Reacciones adversas (6.1)].
5.7 Perforaciones gastrointestinales
Se han informado perforaciones gastrointestinales en ensayos clínicos con RINVOQ [ver Reacciones adversas (6.1)].
Controle a los pacientes tratados con RINVOQ/RINVOQ LQ que puedan tener riesgo de perforación gastrointestinal (por ejemplo, pacientes con antecedentes de diverticulitis y aquellos que toman medicamentos concomitantes, incluidos AINE o corticosteroides). Evalúe de inmediato a los pacientes que presenten dolor abdominal de nueva aparición para la identificación temprana de la perforación gastrointestinal.
5.8 Anormalidades de laboratorio
Neutropenia
El tratamiento con RINVOQ se asoció con una mayor incidencia de neutropenia (ANC menor que 1000 células/mm3).
Evalúe los recuentos de neutrófilos al inicio y posteriormente de acuerdo con la gestión rutinaria del paciente. Evite la iniciación de RINVOQ/RINVOQ LQ e interrumpa el tratamiento con RINVOQ/RINVOQ LQ en pacientes con un recuento bajo de neutrófilos (es decir, ANC menor que 1000 células/mm3) [ver Dosificación y administración (2.1, 2.13)].
Linfopenia
Se informaron ALC menores que 500 células/mm3 en pacientes tratados con RINVOQ en ensayos clínicos.
Evalúe los recuentos de linfocitos al inicio y posteriormente de acuerdo con la gestión rutinaria del paciente. Evite la iniciación de RINVOQ/RINVOQ LQ o interrumpa el tratamiento con RINVOQ/RINVOQ LQ en pacientes con un recuento bajo de linfocitos (es decir, menos de 500 células/mm3) [ver Dosificación y administración (2.1, 2.13)].
Anemia
Se informaron disminuciones en los niveles de hemoglobina a menos de 8 g/dL en pacientes tratados con RINVOQ en ensayos clínicos.
Evalúe la hemoglobina al inicio y posteriormente de acuerdo con la gestión rutinaria del paciente. Evite la iniciación de RINVOQ/RINVOQ LQ o interrumpa el tratamiento con RINVOQ/RINVOQ LQ en pacientes con un nivel bajo de hemoglobina (es decir, menos de 8 g/dL) [ver Dosificación y administración (2.1, 2.13)].
Lípidos
El tratamiento con RINVOQ se asoció con aumentos en los parámetros lipídicos, incluido el colesterol total, el colesterol de lipoproteínas de baja densidad (LDL) y el colesterol de lipoproteínas de alta densidad (HDL) [ver Reacciones adversas (6.1)]. Las elevaciones en el colesterol LDL disminuyeron a los niveles previos al tratamiento en respuesta a la terapia con estatinas. No se ha determinado el efecto de estas elevaciones en los parámetros lipídicos sobre la morbilidad y la mortalidad cardiovascular.
Evalúe los parámetros lipídicos aproximadamente 12 semanas después de iniciar el tratamiento y posteriormente de acuerdo con las pautas clínicas para la hiperlipidemia. Maneje a los pacientes de acuerdo con las pautas clínicas para el manejo de la hiperlipidemia.
Elevaciones de las enzimas hepáticas
El tratamiento con RINVOQ se asoció con una mayor incidencia de elevaciones de las enzimas hepáticas en comparación con el tratamiento con placebo.
Evalúe las enzimas hepáticas al inicio y posteriormente de acuerdo con la gestión rutinaria del paciente. Se recomienda una investigación inmediata de la causa de la elevación de las enzimas hepáticas para identificar posibles casos de lesión hepática inducida por fármacos.
Si se observan aumentos en ALT o AST durante la gestión rutinaria del paciente y se sospecha una lesión hepática inducida por fármacos, se debe interrumpir RINVOQ/RINVOQ LQ hasta que se excluya este diagnóstico.
5.9 Toxicidad embrionaria y fetal
Con base en los hallazgos en estudios con animales, RINVOQ/RINVOQ LQ puede causar daño fetal cuando se administra a una mujer embarazada. La administración de upadacitinib a ratas y conejos durante la organogénesis causó aumentos en las malformaciones fetales. Verifique el estado de embarazo de las pacientes en edad reproductiva antes de comenzar el tratamiento. Avise a las mujeres en edad reproductiva del riesgo potencial para el feto y de que deben usar métodos anticonceptivos eficaces durante el tratamiento con RINVOQ/RINVOQ LQ y durante las 4 semanas posteriores a la finalización de la terapia [ver Uso en poblaciones específicas (8.1, 8.3)].
5.10 Vacunaciónes
Evite el uso de vacunas vivas durante o inmediatamente antes del inicio del tratamiento con RINVOQ/RINVOQ LQ. Antes de iniciar el tratamiento con RINVOQ/RINVOQ LQ, se recomienda que los pacientes se pongan al día con todas las inmunizaciones, incluidas las vacunas profilácticas contra la varicela zóster o el herpes zóster, de acuerdo con las pautas de inmunización actuales.
5.11 Residuo de medicamentos en las heces
Se han notificado casos de residuos de medicamentos en las heces o en la salida de la ostomía en pacientes que toman RINVOQ. La mayoría de los informes describieron condiciones gastrointestinales anatómicas (por ejemplo, ileostomía, colostomía, resección intestinal) o funcionales con tiempos de tránsito gastrointestinal acortados. Indique a los pacientes que se pongan en contacto con su proveedor de atención médica si se observa residuo de medicamentos de forma repetida. Controle a los pacientes clínicamente y considere un tratamiento alternativo si no hay una respuesta terapéutica adecuada.
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas clínicamente significativas se describen en otras partes de la etiqueta:
- Infecciones graves [ver Advertencias y precauciones (5.1)]
- Mortalidad [ver Advertencias y precauciones (5.2)]
- Neoplasia y trastornos linfoproliferativos [ver Advertencias y precauciones (5.3)]
- Eventos cardiovasculares adversos mayores [ver Advertencias y precauciones (5.4)]
- Trombosis [ver Advertencias y precauciones (5.5)]
- Reacciones de hipersensibilidad [ver Advertencias y precauciones (5.6)]
- Perforaciones gastrointestinales [ver Advertencias y precauciones (5.7)]
- Anormalidades de laboratorio [ver Advertencias y precauciones (5.8)]
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se llevan a cabo en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no se pueden comparar directamente con las tasas en los ensayos clínicos de otro fármaco y es posible que no reflejen las tasas observadas en la práctica.
Reacciones adversas en pacientes con artritis reumatoide
Un total de 3833 pacientes adultos con artritis reumatoide fueron tratados con RINVOQ 15 mg o upadacitinib 30 mg comprimidos una vez al día en los ensayos clínicos de fase 3, de los cuales 2806 estuvieron expuestos durante al menos un año.
Los pacientes podían avanzar o cambiar a RINVOQ 15 mg desde placebo, o ser rescatados a RINVOQ desde un comparador activo o placebo desde la semana 12, dependiendo del diseño del ensayo.
Un total de 2630 pacientes recibieron al menos 1 dosis de RINVOQ 15 mg, de los cuales 1860 estuvieron expuestos durante al menos un año. En los ensayos RA-I, RA-II, RA-III y RA-V, 1213 pacientes recibieron al menos 1 dosis de RINVOQ 15 mg, de los cuales 986 pacientes estuvieron expuestos durante al menos un año, y 1203 pacientes recibieron al menos 1 dosis de upadacitinib 30 mg, de los cuales 946 estuvieron expuestos durante al menos un año.
| Reacción adversa | Placebo | RINVOQ 15 mg |
| N = 1042 (%) |
N = 1035 (%) |
|
| Infección de las vías respiratorias superiores (IVRS)* | 9.5 | 13.5 |
| Náuseas | 2.2 | 3.5 |
| Tos | 1.0 | 2.2 |
| Fiebre | 0 | 1.2 |
| *IVRS incluye: sinusitis aguda, laringitis, nasofaringitis, dolor orofaríngeo, faringitis, faringoamigdalitis, rinitis, sinusitis, amigdalitis, infección viral de las vías respiratorias superiores |
||
Otras reacciones adversas notificadas en menos del 1% de los pacientes en el grupo de RINVOQ 15 mg y a una tasa más alta que en el grupo placebo hasta la semana 12 incluyeron neumonía, herpes zóster, herpes simple (incluye herpes oral) y candidiasis oral.
Se presentan cuatro conjuntos de datos integrados en la sección Reacciones adversas específicas:
Ensayos controlados con placebo: Los ensayos RA-III, RA-IV y RA-V se integraron para representar la seguridad hasta las 12/14 semanas para placebo (n=1042) y RINVOQ 15 mg (n=1035). Los ensayos RA-III y RA-V se integraron para representar la seguridad hasta las 12 semanas para placebo (n=390), RINVOQ 15 mg (n=385) y upadacitinib 30 mg (n=384). El ensayo RA-IV no incluyó la dosis de 30 mg y, por lo tanto, los datos de seguridad para upadacitinib 30 mg solo se pueden comparar con las tasas de placebo y RINVOQ 15 mg de la agrupación de los ensayos RA-III y RA-V.
Ensayos controlados con MTX: Los ensayos RA-I y RA-II se integraron para representar la seguridad hasta las 12/14 semanas para MTX (n=530), RINVOQ 15 mg (n=534) y upadacitinib 30 mg (n=529).
Conjunto de datos de exposición de 12 meses: Los ensayos RA-I, II, III y V se integraron para representar la seguridad a largo plazo de RINVOQ 15 mg (n=1213) y upadacitinib 30 mg (n=1203).
Las tasas de incidencia ajustadas por exposición se ajustaron por ensayo para todos los eventos adversos notificados en esta sección.
Reacciones adversas específicas
Infecciones
Ensayos controlados con placebo: En RA-III, RA-IV y RA-V, se notificaron infecciones en 218 pacientes (95,7 por 100 años-paciente) tratados con placebo y 284 pacientes (127,8 por 100 años-paciente) tratados con RINVOQ 15 mg. En RA-III y RA-V, se notificaron infecciones en 99 pacientes (136,5 por 100 años-paciente) tratados con placebo, 118 pacientes (164,5 por 100 años-paciente) tratados con RINVOQ 15 mg y 126 pacientes (180,3 por 100 años-paciente) tratados con upadacitinib 30 mg.
Ensayos controlados con MTX: Se notificaron infecciones en 127 pacientes (119,5 por 100 años-paciente) tratados con monoterapia con MTX, 104 pacientes (91,8 por 100 años-paciente) tratados con monoterapia con RINVOQ 15 mg y 128 pacientes (115,1 por 100 años-paciente) tratados con monoterapia con upadacitinib 30 mg.
Conjunto de datos de exposición de 12 meses: Se notificaron infecciones en 615 pacientes (83,8 por 100 años-paciente) tratados con RINVOQ 15 mg y 674 pacientes (99,7 por 100 años-paciente) tratados con upadacitinib 30 mg.
Infecciones graves
Ensayos controlados con placebo: En RA-III, RA-IV y RA-V, se notificaron infecciones graves en 6 pacientes (2,3 por 100 años-paciente) tratados con placebo y 12 pacientes (4,6 por 100 años-paciente) tratados con RINVOQ 15 mg. En RA-III y RA-V, se notificaron infecciones graves en 1 paciente (1,2 por 100 años-paciente) tratado con placebo, 2 pacientes (2,3 por 100 años-paciente) tratados con RINVOQ 15 mg y 7 pacientes (8,2 por 100 años-paciente) tratados con upadacitinib 30 mg.
Ensayos controlados con MTX: Se notificaron infecciones graves en 2 pacientes (1,6 por 100 años-paciente) tratados con monoterapia con MTX, 3 pacientes (2,4 por 100 años-paciente) tratados con monoterapia con RINVOQ 15 mg y 8 pacientes (6,4 por 100 años-paciente) tratados con monoterapia con upadacitinib 30 mg.
Conjunto de datos de exposición de 12 meses: Se notificaron infecciones graves en 38 pacientes (3,5 por 100 años-paciente) tratados con RINVOQ 15 mg y 59 pacientes (5,6 por 100 años-paciente) tratados con upadacitinib 30 mg.
Las infecciones graves notificadas con mayor frecuencia fueron neumonía y celulitis.
Tuberculosis
Ensayos controlados con placebo y ensayos controlados con MTX: En el período controlado con placebo, no se notificaron casos activos de tuberculosis en los grupos placebo, RINVOQ 15 mg y upadacitinib 30 mg. En el período controlado con MTX, no se notificaron casos activos de tuberculosis en los grupos de monoterapia con MTX, monoterapia con RINVOQ 15 mg y monoterapia con upadacitinib 30 mg.
Conjunto de datos de exposición de 12 meses: Se notificó tuberculosis activa en 2 pacientes tratados con RINVOQ 15 mg y 1 paciente tratado con upadacitinib 30 mg. Se notificaron casos de tuberculosis extrapulmonar.
Infecciones oportunistas (excluyendo tuberculosis)
Ensayos controlados con placebo: En RA-III, RA-IV y RA-V, se notificaron infecciones oportunistas en 3 pacientes (1,2 por 100 años-paciente) tratados con placebo y 5 pacientes (1,9 por 100 años-paciente) tratados con RINVOQ 15 mg. En RA-III y RA-V, se notificaron infecciones oportunistas en 1 paciente (1,2 por 100 años-paciente) tratado con placebo, 2 pacientes (2,3 por 100 años-paciente) tratados con RINVOQ 15 mg y 6 pacientes (7,1 por 100 años-paciente) tratados con upadacitinib 30 mg.
Ensayos controlados con MTX: Se notificaron infecciones oportunistas en 1 paciente (0,8 por 100 años-paciente) tratado con monoterapia con MTX, 0 pacientes tratados con monoterapia con RINVOQ 15 mg y 4 pacientes (3,2 por 100 años-paciente) tratados con monoterapia con upadacitinib 30 mg.
Conjunto de datos de exposición de 12 meses: Se notificaron infecciones oportunistas en 7 pacientes (0,6 por 100 años-paciente) tratados con RINVOQ 15 mg y 15 pacientes (1,4 por 100 años-paciente) tratados con upadacitinib 30 mg.
Neoplasias malignas
Ensayos controlados con placebo: En RA-III, RA-IV y RA-V, se notificaron neoplasias malignas excluyendo NMSC en 1 paciente (0,4 por 100 años-paciente) tratado con placebo y 1 paciente (0,4 por 100 años-paciente) tratado con RINVOQ 15 mg. En RA-III y RA-V, se notificaron neoplasias malignas excluyendo NMSC en 0 pacientes tratados con placebo, 1 paciente (1,1 por 100 años-paciente) tratado con RINVOQ 15 mg y 3 pacientes (3,5 por 100 años-paciente) tratados con upadacitinib 30 mg.
Ensayos controlados con MTX: Se notificaron neoplasias malignas excluyendo NMSC en 1 paciente (0,8 por 100 años-paciente) tratado con monoterapia con MTX, 3 pacientes (2,4 por 100 años-paciente) tratados con monoterapia con RINVOQ 15 mg y 0 pacientes tratados con monoterapia con upadacitinib 30 mg.
Conjunto de datos de exposición de 12 meses: Se notificaron neoplasias malignas excluyendo NMSC en 13 pacientes (1,2 por 100 años-paciente) tratados con RINVOQ 15 mg y 14 pacientes (1,3 por 100 años-paciente) tratados con upadacitinib 30 mg.
Perforaciones gastrointestinales
Ensayos controlados con placebo: No se notificaron perforaciones gastrointestinales (basadas en la revisión médica) en pacientes tratados con placebo, RINVOQ 15 mg y upadacitinib 30 mg.
Ensayos controlados con MTX: No se notificaron casos de perforaciones gastrointestinales en el grupo MTX y RINVOQ 15 mg hasta las 12/14 semanas. Se observaron dos casos de perforaciones gastrointestinales en el grupo upadacitinib 30 mg.
Conjunto de datos de exposición de 12 meses: Se notificaron perforaciones gastrointestinales en 1 paciente tratado con RINVOQ 15 mg y 4 pacientes tratados con upadacitinib 30 mg.
Trombosis
Ensayos controlados con placebo: En RA-IV, se observó trombosis venosa (embolia pulmonar o trombosis venosa profunda) en 1 paciente tratado con placebo y 1 paciente tratado con RINVOQ 15 mg. En RA-V, se observó trombosis venosa en 1 paciente tratado con RINVOQ 15 mg. No se reportaron casos observados de trombosis venosa en RA-III. No se observaron casos de trombosis arterial hasta las 12/14 semanas.
Ensayos controlados con MTX: En RA-II, se observó trombosis venosa en 0 pacientes tratados con monoterapia con MTX, 1 paciente tratado con monoterapia con RINVOQ 15 mg y 0 pacientes tratados con monoterapia con upadacitinib 30 mg hasta la semana 14. En RA-II, no se observaron casos de trombosis arterial hasta las 12/14 semanas. En RA-I, se observó trombosis venosa en 1 paciente tratado con MTX, 0 pacientes tratados con RINVOQ 15 mg y 1 paciente tratado con upadacitinib 30 mg hasta la semana 24. En RA-I, se observó trombosis arterial en 1 paciente tratado con upadacitinib 30 mg hasta la semana 24.
Conjunto de datos de exposición de 12 meses: Se reportaron eventos de trombosis venosa en 5 pacientes (0,5 por 100 años-paciente) tratados con RINVOQ 15 mg y 4 pacientes (0,4 por 100 años-paciente) tratados con upadacitinib 30 mg. Se reportaron eventos de trombosis arterial en 0 pacientes tratados con RINVOQ 15 mg y 2 pacientes (0,2 por 100 años-paciente) tratados con upadacitinib 30 mg.
Anormalidades de laboratorio
Elevaciones de transaminasas hepáticas
En ensayos controlados con placebo (RA-III, RA-IV y RA-V) con DMARDs de fondo, hasta 12/14 semanas, se observaron elevaciones de alanina transaminasa (ALT) y aspartato transaminasa (AST) ≥ 3 x límite superior normal (ULN) en al menos una medición en 2,1% y 1,5% de los pacientes tratados con RINVOQ 15 mg, y en 1,5% y 0,7% de los pacientes tratados con placebo, respectivamente. En RA-III y RA-V, se observaron elevaciones de ALT y AST ≥ 3 x ULN en al menos una medición en 0,8% y 1,0% de los pacientes tratados con RINVOQ 15 mg, 1,0% y 0% de los pacientes tratados con upadacitinib 30 mg y en 1,3% y 1,0% de los pacientes tratados con placebo, respectivamente.
En ensayos controlados con MTX, hasta 12/14 semanas, se observaron elevaciones de ALT y AST ≥ 3 x ULN en al menos una medición en 0,8% y 0,4% de los pacientes tratados con RINVOQ 15 mg, 1,7% y 1,3% de los pacientes tratados con upadacitinib 30 mg y en 1,9% y 0,9% de los pacientes tratados con MTX, respectivamente.
Elevaciones de lípidos
El tratamiento con upadacitinib se asoció con aumentos relacionados con la dosis en el colesterol total, los triglicéridos y el colesterol LDL. Upadacitinib también se asoció con aumentos en el colesterol HDL. Las elevaciones en el colesterol LDL y HDL alcanzaron su punto máximo en la semana 8 y se mantuvieron estables a partir de entonces. En ensayos controlados, hasta 12/14 semanas, los cambios desde el inicio en los parámetros lipídicos en pacientes tratados con RINVOQ 15 mg y upadacitinib 30 mg, respectivamente, se resumen a continuación:
- El colesterol LDL medio aumentó en 14,81 mg/dL y 17,17 mg/dL.
- El colesterol HDL medio aumentó en 8,16 mg/dL y 9,01 mg/dL.
- La relación LDL/HDL media se mantuvo estable.
- Los triglicéridos medios aumentaron en 13,55 mg/dL y 14,44 mg/dL.
Elevaciones de creatina fosfoquinasa
En ensayos controlados con placebo (RA-III, RA-IV y RA-V) con DMARDs de fondo, hasta 12/14 semanas, se observaron aumentos relacionados con la dosis en los valores de creatina fosfoquinasa (CPK). Se reportaron elevaciones de CPK > 5 x ULN en 1,0% y 0,3% de los pacientes durante más de 12/14 semanas en los grupos de RINVOQ 15 mg y placebo, respectivamente. La mayoría de las elevaciones > 5 x ULN fueron transitorias y no requirieron la interrupción del tratamiento. En RA-III y RA-V, se observaron elevaciones de CPK > 5 x ULN en 0,3% de los pacientes tratados con placebo, 1,6% de los pacientes tratados con RINVOQ 15 mg y ninguno en los pacientes tratados con upadacitinib 30 mg.
Neutropenia
En ensayos controlados con placebo (RA-III, RA-IV y RA-V) con DMARDs de fondo, hasta 12/14 semanas, se produjeron disminuciones relacionadas con la dosis en los recuentos de neutrófilos, por debajo de 1000 células/mm3 en al menos una medición en 1,1% y <0,1% de los pacientes en los grupos de RINVOQ 15 mg y placebo, respectivamente. En RA-III y RA-V, las disminuciones en los recuentos de neutrófilos por debajo de 1000 células/mm3 en al menos una medición ocurrieron en 0,3% de los pacientes tratados con placebo, 1,3% de los pacientes tratados con RINVOQ 15 mg y 2,4% de los pacientes tratados con upadacitinib 30 mg. En los ensayos clínicos, el tratamiento se interrumpió en respuesta a un ANC inferior a 1000 células/mm3.
Linfopenia
En ensayos controlados con placebo (RA-III, RA-IV y RA-V) con DMARDs de fondo, hasta 12/14 semanas, se produjeron disminuciones relacionadas con la dosis en los recuentos de linfocitos por debajo de 500 células/mm3 en al menos una medición en 0,9% y 0,7% de los pacientes en los grupos de RINVOQ 15 mg y placebo, respectivamente. En RA-III y RA-V, las disminuciones en los recuentos de linfocitos por debajo de 500 células/mm3 en al menos una medición ocurrieron en 0,5% de los pacientes tratados con placebo, 0,5% de los pacientes tratados con RINVOQ 15 mg y 2,4% de los pacientes tratados con upadacitinib 30 mg.
Anemia
En ensayos controlados con placebo (RA-III, RA-IV y RA-V) con DMARDs de fondo, hasta 12/14 semanas, las disminuciones de hemoglobina por debajo de 8 g/dL en al menos una medición ocurrieron en <0,1% de los pacientes en ambos grupos de RINVOQ 15 mg y placebo. En RA-III y RA-V, se observaron disminuciones de hemoglobina por debajo de 8 g/dL en al menos una medición en 0,3% de los pacientes tratados con placebo, y ninguno en los pacientes tratados con RINVOQ 15 mg y upadacitinib 30 mg.
Reacciones adversas en pacientes con artritis psoriásica
Un total de 1827 pacientes adultos con artritis psoriásica fueron tratados con RINVOQ 15 mg o upadacitinib 30 mg comprimidos una vez al día en ensayos clínicos, lo que representa 1639,2 años-paciente de exposición, de los cuales 722 estuvieron expuestos a upadacitinib durante al menos un año. En los dos ensayos de fase 3, 907 pacientes recibieron al menos 1 dosis de RINVOQ 15 mg, de los cuales 359 estuvieron expuestos durante al menos un año.
Se integraron dos ensayos controlados con placebo (640 pacientes con RINVOQ 15 mg una vez al día y 635 pacientes con placebo) para evaluar la seguridad de RINVOQ 15 mg en comparación con placebo hasta 24 semanas después del inicio del tratamiento.
En general, el perfil de seguridad observado en pacientes con artritis psoriásica activa tratados con RINVOQ 15 mg fue consistente con el perfil de seguridad observado en pacientes con artritis reumatoide. Durante el período controlado con placebo de 24 semanas, las frecuencias de herpes zóster y herpes simple fueron ≥1% (1,1% y 1,4%, respectivamente) con RINVOQ 15 mg y 0,8% y 1,3%, respectivamente, con placebo. También se observó una mayor incidencia de acné y bronquitis en pacientes tratados con RINVOQ 15 mg (1,3% y 3,9%, respectivamente) en comparación con placebo (0,3% y 2,7%, respectivamente).
Reacciones adversas en pacientes con dermatitis atópica
Tres ensayos clínicos aleatorizados, doble ciego, controlados con placebo y multicéntricos de Fase 3 (AD-1, AD-2 y AD-3) y uno de Fase 2b (AD-4) evaluaron la seguridad de RINVOQ en pacientes con dermatitis atópica de moderada a grave. La mayoría de los pacientes eran blancos (68%) y hombres (57%). La edad media fue de 34 años (rango de 12 a 75 años) y el 13% de los pacientes tenían entre 12 y menos de 18 años. En estos 4 ensayos, 2612 pacientes fueron tratados con RINVOQ 15 mg comprimidos o 30 mg comprimidos por vía oral una vez al día, con o sin corticosteroides tópicos (TCS) concomitantes.
En los ensayos clínicos de Fase 3 (AD-1, AD-2 y AD-3), un total de 1239 pacientes recibieron RINVOQ 15 mg, de los cuales 791 estuvieron expuestos durante al menos un año y 1246 pacientes recibieron RINVOQ 30 mg, de los cuales 826 estuvieron expuestos durante al menos un año.
Los ensayos AD-1, AD-2 y AD-4 compararon la seguridad de la monoterapia con RINVOQ con placebo hasta la semana 16. El ensayo AD-3 comparó la seguridad de RINVOQ + TCS con placebo + TCS hasta la semana 16.
Semanas 0 a 16 (Ensayos AD-1 a AD-4)
En los ensayos de RINVOQ con y sin TCS (Ensayos AD-1, 2, 3 y 4) hasta la semana 16, la proporción de pacientes que interrumpieron el tratamiento debido a reacciones adversas en los grupos de RINVOQ 15 mg, 30 mg y placebo fue del 2,3%, 2,9% y 3,8%, respectivamente. La Tabla 5 resume las reacciones adversas que ocurrieron con una frecuencia de al menos el 1% en los grupos de RINVOQ 15 mg o 30 mg durante las primeras 16 semanas de tratamiento.
| Reacción adversa | Placebo | RINVOQ 15 mg |
RINVOQ 30 mg |
| N = 902 (%) |
N = 899 (%) |
N = 906 (%) |
|
| Infección de las vías respiratorias superiores (IVRS)* | 17 | 23 | 25 |
| Acné** | 2 | 10 | 16 |
| Herpes simple*** | 2 | 4 | 8 |
| Dolor de cabeza | 4 | 6 | 6 |
| Aumento de la creatina quinasa en sangre | 2 | 5 | 6 |
| Tos | 1 | 3 | 3 |
| Hipersensibilidad**** | 2 | 2 | 3 |
| Foliculitis | 1 | 2 | 3 |
| Náuseas | 1 | 3 | 3 |
| Dolor abdominal***** | 1 | 3 | 2 |
| Fiebre | 1 | 2 | 2 |
| Aumento de peso | 1 | 2 | 2 |
| Herpes zóster****** | 1 | 2 | 2 |
| Influenza | <1 | 2 | 2 |
| Fatiga | 1 | 1 | 2 |
| Neutropenia | <1 | 1 | 2 |
| Mialgia | 1 | 1 | 2 |
| Enfermedad similar a la gripe | 1 | 1 | 2 |
| * Incluye: laringitis, laringitis viral, nasofaringitis, dolor orofaríngeo, absceso faríngeo, faringitis, faringitis estreptocócica, faringoamigdalitis, infección del tracto respiratorio, infección del tracto respiratorio viral, rinitis, rinolaríngitis, sinusitis, amigdalitis, amigdalitis bacteriana, infección del tracto respiratorio superior, faringitis viral, infección del tracto respiratorio superior viral ** Incluye: acné y dermatitis acneiforme *** Incluye: herpes genital, herpes simple genital, dermatitis herpética, herpes oftálmico, herpes simple, herpes nasal, herpes simple oftálmico, infección por virus del herpes, herpes oral **** Incluye reacción anafiláctica, shock anafiláctico, angioedema, dermatitis exfoliativa generalizada, hipersensibilidad a fármacos, edema de párpados, edema facial, hipersensibilidad, hinchazón periorbitaria, hinchazón faríngea, hinchazón facial, erupción cutánea tóxica, hipersensibilidad tipo I, urticaria ***** Incluye dolor abdominal y dolor abdominal superior ****** Incluye herpes zóster y varicela |
|||
Otras reacciones adversas notificadas en menos del 1% de los pacientes en el grupo de RINVOQ 15 mg y/o 30 mg y a una tasa más alta que en el grupo placebo hasta la semana 16 incluyeron anemia, candidiasis oral, neumonía, cáncer de piel no melanoma y el evento adverso de desprendimiento de retina.
El perfil de seguridad de RINVOQ hasta la semana 52 fue generalmente consistente con el perfil de seguridad observado en la semana 16.
En general, el perfil de seguridad observado en pacientes con AD tratados con RINVOQ fue similar al perfil de seguridad en pacientes con AR. Otras reacciones adversas específicas que se notificaron en pacientes con AD incluyeron eccema herpético/erupción variceliforme de Kaposi.
Eccema Herpético/Erupción Variceliforme de Kaposi
Período controlado con placebo (16 semanas): Se notificó eccema herpético en 4 pacientes (1,6 por 100 años-paciente) tratados con placebo, 6 pacientes (2,2 por 100 años-paciente) tratados con RINVOQ 15 mg y 7 pacientes (2,6 por 100 años-paciente) tratados con RINVOQ 30 mg.
Exposición de 12 meses (semanas 0 a 52): Se notificó eccema herpético en 18 pacientes (1,6 por 100 años-paciente) tratados con RINVOQ 15 mg y 17 pacientes (1,5 por 100 años-paciente) tratados con RINVOQ 30 mg.
Reacciones adversas en pacientes con colitis ulcerosa
RINVOQ se estudió hasta 8 semanas en pacientes con colitis ulcerosa activa de moderada a grave en dos estudios de inducción aleatorizados, doble ciego, controlados con placebo (UC-1, UC-2) y un estudio de búsqueda de dosis aleatorizado, doble ciego, controlado con placebo (UC-4; NCT02819635). La seguridad a largo plazo hasta las 52 semanas se evaluó en pacientes que respondieron a la terapia de inducción en un estudio de mantenimiento aleatorizado, doble ciego, controlado con placebo (UC-3) y un estudio de extensión a largo plazo [ver Estudios clínicos (14.4)].
En los dos estudios de inducción (UC-1, UC-2) y un estudio de búsqueda de dosis (UC-4), se inscribieron 1097 pacientes, de los cuales 719 pacientes recibieron tabletas de RINVOQ 45 mg una vez al día.
En el estudio de mantenimiento (UC-3), se inscribieron 746 pacientes, de los cuales 250 pacientes recibieron tabletas de RINVOQ 15 mg una vez al día y 251 pacientes recibieron tabletas de RINVOQ 30 mg una vez al día.
Las reacciones adversas notificadas en ≥2% de los pacientes en cualquier brazo de tratamiento en los estudios de inducción y mantenimiento se muestran en las Tablas 6 y 7, respectivamente.
| Reacción adversa | Placebo | RINVOQ 45 mg Una vez al día |
| N = 378 (%) |
N = 719 (%) |
|
| Infección del tracto respiratorio superior* | 7 | 9 |
| Acné* | 1 | 6 |
| Creatin fosfoquinasa sanguínea aumentada | 1 | 5 |
| Neutropenia* | <1 | 5 |
| Erupción* | 1 | 4 |
| Enzimas hepáticas elevadas** | 2 | 3 |
| Linfopenia* | 1 | 3 |
| Foliculitis | 1 | 2 |
| Herpes simple* | <1 | 2 |
| * Compuesto de varios términos similares ** Enzimas hepáticas elevadas compuestas de ALT, AST, GGT, ALP, transaminasas hepáticas, enzimas hepáticas, bilirrubina, lesión hepática inducida por fármacos y colestasis. |
||
Otras reacciones adversas notificadas en menos del 2% de los pacientes en el grupo de RINVOQ 45 mg y a una tasa más alta que en el grupo placebo hasta la semana 8 incluyeron herpes zóster y neumonía.
| Reacción adversa | Placebo | RINVOQ 15 mg Una vez al día |
RINVOQ 30 mg Una vez al día |
| N = 245 (%) |
N = 250 (%) |
N = 251 (%) |
|
| Infección de las vías respiratorias altas* | 18 | 16 | 20 |
| Creatina fosfocinasa sanguínea aumentada | 2 | 6 | 8 |
| Neutropenia* | 2 | 3 | 6 |
| Enzimas hepáticas elevadas** | 1 | 6 | 4 |
| Erupción* | 4 | 5 | 5 |
| Herpes zóster | 0 | 4 | 4 |
| Foliculitis | 2 | 2 | 4 |
| Hipercolesterolemia* | 1 | 2 | 4 |
| Influenza | 1 | 3 | 3 |
| Herpes simple* | 1 | 2 | 3 |
| Linfopenia* | 2 | 3 | 2 |
| Hiperlipidemia* | 0 | 2 | 2 |
| 1 Pacientes que fueron respondedores a la terapia de inducción de 8 semanas con RINVOQ 45 mg una vez al día * Compuesto de varios términos similares ** Enzimas hepáticas elevadas compuestas de ALT, AST, GGT, ALP, transaminasas hepáticas, enzimas hepáticas, bilirrubina, lesión hepática inducida por fármacos y colestasis. |
|||
La reacción adversa de cáncer de piel no melanoma se informó en el 1% de los pacientes en el grupo de RINVOQ 30 mg y en ninguno de los pacientes en el grupo de RINVOQ 15 mg o placebo hasta la semana 52.
El perfil de seguridad de RINVOQ en el estudio de extensión a largo plazo fue similar al perfil de seguridad observado en los períodos de inducción y mantenimiento controlados con placebo.
En general, el perfil de seguridad observado en pacientes con colitis ulcerosa tratados con RINVOQ fue generalmente similar al perfil de seguridad en pacientes con AR y AD.
Reacciones adversas específicas
Infecciones graves
Estudios de inducción: En UC-1, UC-2 y UC-4, se informaron infecciones graves en 5 pacientes (8,4 por 100 años-paciente) tratados con placebo y 9 pacientes (8,4 por 100 años-paciente) tratados con RINVOQ 45 mg hasta las 8 semanas.
Estudio de mantenimiento controlado con placebo: En UC-3, se informaron infecciones graves en 8 pacientes (6,3 por 100 años-paciente) tratados con placebo, 8 pacientes (4,5 por 100 años-paciente) tratados con RINVOQ 15 mg y 6 pacientes (3,1 por 100 años-paciente) tratados con RINVOQ 30 mg hasta las 52 semanas.
Anormalidades de laboratorio
Elevaciones de transaminasas hepáticas
En los estudios UC-1, UC-2 y UC-4, se observaron elevaciones de ALT a ≥ 3 x ULN en al menos una medición en el 1,5% de los pacientes tratados con RINVOQ 45 mg y en el 0% de los pacientes tratados con placebo durante 8 semanas. Las elevaciones de AST a ≥ 3 x ULN ocurrieron en el 1,5% de los pacientes tratados con RINVOQ 45 mg y en el 0,3% de los pacientes tratados con placebo. Las elevaciones de ALT a ≥ 5 x ULN ocurrieron en el 0,4% de los pacientes tratados con RINVOQ 45 mg y en el 0% de los pacientes tratados con placebo.
En UC-3, se observaron elevaciones de ALT a ≥ 3 x ULN en al menos una medición en el 4% de los pacientes tratados con RINVOQ 30 mg, el 2% de los pacientes tratados con RINVOQ 15 mg y el 0,8% de los pacientes tratados con placebo durante 52 semanas. Las elevaciones de AST a ≥ 3 x ULN en al menos una medición se observaron en el 2% de los pacientes tratados con RINVOQ 30 mg, el 1,6% de los pacientes tratados con RINVOQ 15 mg y el 0,4% de los pacientes tratados con placebo. Se observaron elevaciones de ALT a ≥ 5 x ULN en el 0,8% de los pacientes tratados con 30 mg, el 0,4% de los pacientes tratados con 15 mg y el 0,4% de los pacientes tratados con placebo.
En general, las anormalidades de laboratorio observadas en pacientes con colitis ulcerosa tratados con RINVOQ fueron similares a las descritas en pacientes con AR.
Reacciones adversas en pacientes con enfermedad de Crohn
RINVOQ se estudió hasta 12 semanas en pacientes con CD de moderada a gravemente activa en dos estudios de inducción aleatorizados, doble ciego y controlados con placebo (CD-1, CD-2). La seguridad a largo plazo hasta las 52 semanas se evaluó en pacientes que respondieron a la terapia de inducción en un estudio de mantenimiento aleatorizado, doble ciego y controlado con placebo (CD-3), con datos adicionales proporcionados de un período de extensión a largo plazo (LTE) [ver Estudios clínicos (14.5)].
En los dos estudios de inducción (CD-1, CD-2), se inscribieron 1021 pacientes, de los cuales 674 pacientes recibieron tabletas de RINVOQ 45 mg una vez al día durante el período controlado con placebo.
En el estudio de mantenimiento (CD-3), se inscribieron 673 pacientes, de los cuales 221 pacientes recibieron tabletas de RINVOQ 15 mg una vez al día y 229 pacientes recibieron tabletas de RINVOQ 30 mg una vez al día durante el período aleatorizado y controlado con placebo.
En general, el perfil de seguridad observado en pacientes con enfermedad de Crohn tratados con RINVOQ fue consistente con el perfil de seguridad conocido para RINVOQ en otras indicaciones.
Las reacciones adversas informadas en ≥2% de los pacientes tratados con RINVOQ y a una tasa más alta que el placebo en los estudios de inducción y mantenimiento se muestran en las Tablas 8 y 9, respectivamente.
| Reacción adversa | Placebo | RINVOQ 45 mg Una vez al día |
| N = 347 (%) |
N = 674 (%) |
|
| Infección del tracto respiratorio superior* | 8 | 13 |
| Anemia* | 6 | 7 |
| Acné* | 2 | 6 |
| Pirexia | 3 | 4 |
| Creatina fosfoquinasa sanguínea aumentada | 1 | 3 |
| Influenza | 1 | 3 |
| Herpes simplex* | 1 | 3 |
| Leucopenia* | 1 | 2 |
| Neutropenia* | <1 | 2 |
| Herpes zóster | 0 | 2 |
| * Compuesto de varios términos similares | ||
Las reacciones adversas notificadas en menos del 2% de los pacientes en el grupo de RINVOQ 45 mg y a una tasa más alta que en el grupo placebo hasta la semana 12 incluyeron foliculitis, hipercolesterolemia, bronquitis, neumonía, candidiasis oral e hiperlipidemia.
| Reacción adversa | Placebo | RINVOQ 15 mg Una vez al día |
RINVOQ 30 mg Una vez al día |
| N = 223 (%) |
N = 221 (%) |
N = 229 (%) |
|
| Infección del tracto respiratorio superior* | 11 | 14 | 12 |
| Pirexia | 2 | 3 | 7 |
| Herpes zóster* | 2 | 3 | 5 |
| Dolor de cabeza* | 1 | 3 | 5 |
| Acné* | 3 | 2 | 5 |
| Gastroenteritis* | 2 | 3 | 3 |
| Fatiga | 2 | 3 | 3 |
| Creatina fosfoquinasa sanguínea aumentada | 1 | 2 | 3 |
| Enzimas hepáticas elevadas2 | <1 | 2 | 3 |
| Leucopenia* | <1 | 1 | 2 |
| Neutropenia* | <1 | 1 | 2 |
| Bronquitis* | 0 | 1 | 2 |
| Neumonía* | 1 | 4 | 1 |
| Tos | 2 | 3 | 1 |
| 1 Pacientes que fueron respondedores a la terapia de inducción de 12 semanas con RINVOQ 45 mg una vez al día. 2 Las enzimas hepáticas elevadas incluyen alanina aminotransferasa aumentada, aspartato aminotransferasa aumentada, fosfatasa alcalina sanguínea aumentada, transaminasas aumentadas, bilirrubina sanguínea aumentada. * Compuesto de varios términos similares |
|||
Las reacciones adversas notificadas en menos del 2% de los pacientes en el grupo de RINVOQ 15 mg o 30 mg y a una tasa más alta que en el grupo placebo hasta la semana 52 incluyeron hiperlipidemia, candidiasis oral e hipercolesterolemia.
El perfil de seguridad de RINVOQ en el estudio de extensión a largo plazo fue similar al perfil de seguridad observado en los períodos de inducción y mantenimiento controlados con placebo.
Reacciones adversas específicas
Infecciones graves
Estudios de inducción: En CD-1 y CD-2, se notificaron infecciones graves en 6 pacientes (8 por 100 años-paciente) tratados con placebo y 13 pacientes (9 por 100 años-paciente) tratados con RINVOQ 45 mg durante las 12 semanas del período controlado con placebo.
Estudio de mantenimiento/LTE: En el período controlado con placebo a largo plazo, se notificaron infecciones graves en 10 pacientes (7 por 100 años-paciente) tratados con placebo, 7 pacientes (4 por 100 años-paciente) tratados con RINVOQ 15 mg y 13 pacientes (6 por 100 años-paciente) tratados con RINVOQ 30 mg.
Perforaciones gastrointestinales
Estudios de inducción: Durante los estudios de inducción en todos los pacientes tratados con RINVOQ 45 mg (N=938), se notificó perforación gastrointestinal en 4 pacientes (2 por 100 años-paciente). En el período de inducción controlado con placebo, en CD-1 y CD-2, se notificó perforación gastrointestinal en ningún paciente tratado con placebo (N=347) y 1 paciente (1 por 100 años-paciente) tratado con RINVOQ 45 mg (N=674) durante las 12 semanas.
Estudio de mantenimiento/LTE: En el período controlado con placebo a largo plazo, se notificó perforación gastrointestinal en 1 paciente (1 por 100 años-paciente) tratado con placebo, 1 paciente (<1 por 100 años-paciente) tratado con RINVOQ 15 mg y 1 paciente (<1 por 100 años-paciente) tratado con RINVOQ 30 mg.
Los pacientes que recibieron placebo o RINVOQ 15 mg para terapia de mantenimiento y perdieron la respuesta fueron tratados con RINVOQ 30 mg de rescate (N=336). Entre estos pacientes, se notificó perforación gastrointestinal en 3 pacientes (1 por 100 años-paciente) durante el tratamiento a largo plazo.
Reacciones adversas en pacientes con Espondilitis anquilosante
Un total de 596 pacientes con espondilitis anquilosante fueron tratados con tabletas de RINVOQ 15 mg en los dos ensayos clínicos que representan 577,3 años-paciente de exposición, de los cuales 220 estuvieron expuestos a RINVOQ 15 mg durante al menos un año.
En general, el perfil de seguridad observado en pacientes con espondilitis anquilosante activa tratados con RINVOQ 15 mg fue consistente con el perfil de seguridad observado en pacientes con artritis reumatoide y artritis psoriásica. Durante el período controlado con placebo de 14 semanas en el ensayo AS-I, la frecuencia de cefalea fue del 5,4% con RINVOQ 15 mg y del 2,1% con placebo. Durante el período controlado con placebo de 14 semanas en el ensayo AS-II, la frecuencia de cefalea fue del 3,3% con RINVOQ 15 mg y del 1,4% con placebo.
Reacciones adversas en pacientes con Espondiloartritis axial no radiográfica
Un total de 187 pacientes con espondiloartritis axial no radiográfica fueron tratados con tabletas de RINVOQ 15 mg en el ensayo clínico que representa 116,6 años-paciente de exposición, de los cuales 31 estuvieron expuestos a RINVOQ 15 mg durante al menos un año.
En general, el perfil de seguridad observado en pacientes con espondiloartritis axial no radiográfica activa tratados con RINVOQ 15 mg fue consistente con el perfil de seguridad observado en pacientes con artritis reumatoide, artritis psoriásica y espondilitis anquilosante.
Reacciones adversas en pacientes con artritis idiopática juvenil poliarticular
Un total de 83 pacientes pediátricos con artritis idiopática juvenil (AIJ) con poliartritis activa fueron tratados con RINVOQ/RINVOQ LQ en el ensayo clínico, que representa 123,7 años-paciente de exposición, de los cuales 48 estuvieron expuestos a RINVOQ/RINVOQ LQ durante al menos un año.
En general, el perfil de seguridad observado en pacientes pediátricos con AIJ con poliartritis activa tratados con RINVOQ/RINVOQ LQ fue consistente con el perfil de seguridad conocido de RINVOQ.
7 INTERACCIONES MEDICAMENTOSAS
7.1 Inhibidores fuertes de CYP3A4
La exposición a upadacitinib aumenta cuando se administra conjuntamente con un inhibidor fuerte de CYP3A4 (como ketoconazol, claritromicina y pomelo), lo que puede aumentar el riesgo de reacciones adversas [ver Farmacología clínica (12.3)]. Controle de cerca a los pacientes con artritis reumatoide, artritis psoriásica, espondilitis anquilosante, espondiloartritis axial no radiográfica o pJIA para detectar reacciones adversas cuando administre conjuntamente RINVOQ/RINVOQ LQ con inhibidores fuertes de CYP3A4. Se debe evitar el consumo de alimentos o bebidas que contengan pomelo durante el tratamiento con RINVOQ/RINVOQ LQ.
Para pacientes con dermatitis atópica, no se recomienda la administración conjunta de RINVOQ 30 mg una vez al día con inhibidores fuertes de CYP3A4.
Para pacientes con colitis ulcerosa o enfermedad de Crohn que toman inhibidores fuertes de CYP3A4, reduzca la dosis de inducción de RINVOQ a 30 mg una vez al día. La dosis de mantenimiento recomendada es de 15 mg una vez al día [ver Dosificación y administración (2.12)].
7.2 Inductores fuertes de CYP3A4
La exposición a upadacitinib disminuye cuando se administra conjuntamente con inductores fuertes de CYP3A4 (como rifampicina), lo que puede conducir a una reducción del efecto terapéutico [ver Farmacología clínica (12.3)]. No se recomienda la administración conjunta de RINVOQ/RINVOQ LQ con inductores fuertes de CYP3A4.
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Programa de Vigilancia del Embarazo
Existe un programa de vigilancia del embarazo para RINVOQ/RINVOQ LQ que monitorea los resultados del embarazo en mujeres expuestas a RINVOQ/RINVOQ LQ. Si la exposición a RINVOQ/RINVOQ LQ ocurre durante el embarazo, los proveedores de atención médica o los pacientes deben reportar el embarazo llamando al 1-800-633-9110.
Resumen de Riesgos
Los datos disponibles de la base de datos de seguridad de farmacovigilancia y los informes de casos posteriores a la comercialización sobre el uso de RINVOQ en mujeres embarazadas no son suficientes para evaluar un riesgo asociado al fármaco para defectos de nacimiento importantes o aborto espontáneo. Con base en estudios en animales, RINVOQ/RINVOQ LQ tiene el potencial de afectar adversamente a un feto en desarrollo. Avise a las pacientes con potencial reproductivo y a las pacientes embarazadas del posible riesgo para el feto.
En estudios de desarrollo embrio-fetal en animales, la administración oral de upadacitinib a ratas y conejas embarazadas a exposiciones iguales o mayores que aproximadamente 1.6 y 15 veces la dosis de la tableta de 15 mg, 0.8 y 7.6 veces la dosis de la tableta de 30 mg, y 0.6 y 5.6 veces la dosis humana máxima recomendada (MRHD) de 45 mg (sobre una base de AUC) resultó en aumentos relacionados con la dosis en malformaciones esqueléticas (solo ratas), una mayor incidencia de malformaciones cardiovasculares (solo conejos), un aumento de la pérdida postimplantación (solo conejos) y una disminución del peso corporal fetal tanto en ratas como en conejos. No se observó toxicidad para el desarrollo en ratas y conejas embarazadas tratadas con upadacitinib oral durante la organogénesis a exposiciones aproximadamente 0.29 y 2.2 veces la dosis de 15 mg, 0.15 veces y 1.1 veces la dosis de 30 mg, y a 0.11 y 0.82 veces la MRHD (sobre una base de AUC). En un estudio de desarrollo pre y postnatal en ratas hembra embarazadas, la administración oral de upadacitinib a exposiciones aproximadamente 3 veces la dosis de 15 mg, 1.4 veces la dosis de 30 mg y la misma que la MRHD (sobre una base de AUC) no resultó en toxicidad materna o de desarrollo (ver Datos).
Los riesgos de fondo de defectos de nacimiento importantes y aborto espontáneo para las poblaciones indicadas son desconocidos. Todos los embarazos tienen un riesgo de fondo de defectos de nacimiento, pérdida u otros resultados adversos. En la población general de EE. UU., el riesgo de fondo estimado de defectos de nacimiento importantes y abortos espontáneos es del 2-4% y del 15-20%, respectivamente.
Consideraciones Clínicas
Riesgo Materno y/o Embrionario/Fetal Asociado a la Enfermedad
Los datos publicados sugieren que el aumento de la actividad de la enfermedad está asociado con el riesgo de desarrollar resultados adversos del embarazo en mujeres con artritis reumatoide o enfermedad inflamatoria intestinal. Los resultados adversos del embarazo incluyen parto prematuro (antes de las 37 semanas de gestación), bebés con bajo peso al nacer (menos de 2500 g) y pequeños para la edad gestacional al nacer.
Datos
Datos de Animales
En un estudio de desarrollo embrio-fetal oral, las ratas embarazadas recibieron upadacitinib en dosis de 5, 25 y 75 mg/kg/día durante el período de organogénesis desde el día 6 hasta el 17 de gestación. El upadacitinib fue teratógeno (malformaciones esqueléticas que consistieron en húmero deforme y escápula doblada) a exposiciones iguales o mayores que aproximadamente 1.7 veces la dosis de la tableta de 15 mg, 0.9 veces la dosis de la tableta de 30 mg y 0.6 veces la MRHD (sobre una base de AUC a dosis orales maternas de 5 mg/kg/día y más alto). Se observaron malformaciones esqueléticas adicionales (extremidades anteriores/posteriores dobladas y defectos de costillas/vértebras) y una disminución del peso corporal fetal en ausencia de toxicidad materna a una exposición aproximadamente 84 veces la dosis de 15 mg, 43 veces la dosis de 30 mg y 31 veces la MRHD (sobre una base de AUC a una dosis oral materna de 75 mg/kg/día).
En un segundo estudio de desarrollo embrio-fetal oral, las ratas embarazadas recibieron upadacitinib en dosis de 1.5 y 4 mg/kg/día durante el período de organogénesis desde el día 6 hasta el 17 de gestación. El upadacitinib fue teratógeno (malformaciones esqueléticas que incluyeron húmero doblado y escápula) a exposiciones aproximadamente 1.6 veces la dosis de 15 mg, 0.8 veces la dosis de 30 mg y 0.6 veces la MRHD (sobre una base de AUC a dosis orales maternas de 4 mg/kg/día). No se observó toxicidad para el desarrollo en ratas a una exposición aproximadamente 0.29 veces la dosis de la tableta de 15 mg, 0.15 veces la dosis de la tableta de 30 mg y 0.11 veces la MRHD (sobre una base de AUC a una dosis oral materna de 1.5 mg/kg/día).
En un estudio de desarrollo embrio-fetal oral, las conejas embarazadas recibieron upadacitinib en dosis de 2.5, 10 y 25 mg/kg/día durante el período de organogénesis desde el día 7 hasta el 19 de gestación. Se observó embrioletalidad, disminución del peso corporal fetal y malformaciones cardiovasculares en presencia de toxicidad materna a una exposición aproximadamente 15 veces la dosis de la tableta de 15 mg, 7.6 veces la dosis de la tableta de 30 mg y 5.6 veces la MRHD (sobre una base de AUC a una dosis oral materna de 25 mg/kg/día). La embrioletalidad consistió en un aumento de la pérdida postimplantación que se debió a incidencias elevadas tanto de resorciones totales como tempranas. No se observó toxicidad para el desarrollo en conejos a una exposición aproximadamente 2.2 veces la dosis de la tableta de 15 mg, 1.1 veces la dosis de la tableta de 30 mg y 0.82 veces la MRHD (sobre una base de AUC a una dosis oral materna de 10 mg/kg/día).
En un estudio de desarrollo pre y postnatal oral, las ratas hembra embarazadas recibieron upadacitinib en dosis de 2.5, 5 y 10 mg/kg/día desde el día 6 de gestación hasta el día 20 de lactancia. No se observó toxicidad materna o de desarrollo ni en las madres ni en las crías, respectivamente, a una exposición aproximadamente 3 veces la dosis de la tableta de 15 mg, 1.4 veces la dosis de la tableta de 30 mg y a aproximadamente la misma exposición que la MRHD (sobre una base de AUC a una dosis oral materna de 10 mg/kg/día).
8.2 Lactancia
Resumen de Riesgos
No hay datos sobre la presencia de upadacitinib en la leche materna, los efectos en el lactante amamantado o los efectos en la producción de leche. Los datos farmacodinámicos/toxicológicos disponibles en animales han demostrado la excreción de upadacitinib en la leche (ver Datos). Cuando un fármaco está presente en la leche animal, es probable que el fármaco esté presente en la leche humana. Debido al potencial de reacciones adversas graves en el lactante amamantado, avise a las pacientes que no se recomienda la lactancia durante el tratamiento con RINVOQ/RINVOQ LQ y durante 6 días (aproximadamente 10 vidas medias) después de la última dosis.
Datos
Se administró una sola dosis oral de 10 mg/kg de upadacitinib radiomarcado a ratas Sprague-Dawley lactantes en los días 7-8 posparto. La exposición al fármaco fue aproximadamente 30 veces mayor en la leche que en el plasma materno según los valores de AUC0-t. Aproximadamente el 97% del material relacionado con el fármaco en la leche fue el fármaco original.
8.3 Mujeres y hombres en edad fértil
Prueba de embarazo
Verifique el estado de embarazo de las mujeres en edad fértil antes de comenzar el tratamiento con RINVOQ/RINVOQ LQ [ver Uso en poblaciones específicas (8.1)].
Anticoncepción
Mujeres
Según estudios en animales, el upadacitinib puede causar daño embrio-fetal cuando se administra a mujeres embarazadas [ver Uso en poblaciones específicas (8.1)]. Aconseje a las pacientes mujeres en edad fértil que utilicen métodos anticonceptivos eficaces durante el tratamiento con RINVOQ/RINVOQ LQ y durante 4 semanas después de la dosis final.
8.4 Uso pediátrico
Espondilitis anquilosante, espondiloartritis axial no radiográfica, colitis ulcerosa y enfermedad de Crohn
No se ha establecido la seguridad y eficacia de RINVOQ/RINVOQ LQ en pacientes pediátricos con espondilitis anquilosante, espondiloartritis axial no radiográfica, colitis ulcerosa o enfermedad de Crohn.
Artritis idiopática juvenil poliarticular y artritis psoriásica
Se ha establecido la seguridad y eficacia de RINVOQ/RINVOQ LQ en pacientes pediátricos de 2 a menos de 18 años de edad con AIJ y artritis psoriásica.
El uso de RINVOQ/RINVOQ LQ en estos grupos de edad está respaldado por evidencia de estudios bien controlados de RINVOQ en adultos con artritis reumatoide y artritis psoriásica, datos farmacocinéticos de pacientes adultos con artritis reumatoide y artritis psoriásica y 51 pacientes pediátricos con AIJ con poliartritis activa, y datos de seguridad de 83 pacientes pediátricos de 2 a < 18 años de edad con AIJ con poliartritis activa. Se predice que las exposiciones plasmáticas de upadacitinib en pacientes pediátricos con AIJ y artritis psoriásica a la dosis recomendada serán comparables a las observadas en adultos con artritis reumatoide y artritis psoriásica, según el modelado farmacocinético poblacional y la simulación [ver Dosificación y administración (2.4, 2.10), Reacciones adversas (6.1), Farmacología clínica (12.3) y Estudios clínicos (14.8)].
No se ha establecido la seguridad y eficacia de RINVOQ/RINVOQ LQ en pacientes pediátricos menores de 2 años de edad con AIJ o artritis psoriásica.
Dermatitis atópica
Se ha establecido la seguridad y eficacia de RINVOQ en pacientes pediátricos de 12 años de edad o mayores que pesen al menos 40 kg con dermatitis atópica. Un total de 344 pacientes pediátricos de 12 a 17 años con dermatitis atópica de moderada a grave fueron aleatorizados en tres ensayos (AD-1, AD-2 y AD-3) para recibir RINVOQ 15 mg (N=114) o 30 mg (N=114) o placebo coincidente (N=116) en monoterapia o en combinación con corticosteroides tópicos. La eficacia fue consistente entre los pacientes pediátricos y los adultos [ver Estudios clínicos (14.3)]. El perfil de reacciones adversas en los pacientes pediátricos fue similar al de los adultos [ver Reacciones adversas (6.1)].
No se ha establecido la seguridad y eficacia de RINVOQ en pacientes pediátricos menores de 12 años de edad con dermatitis atópica.
No se ha establecido la seguridad y eficacia de RINVOQ LQ en pacientes pediátricos con dermatitis atópica.
8.5 Uso geriátrico
Artritis reumatoide y artritis psoriásica
De los 4381 pacientes tratados en los cinco ensayos clínicos, un total de 906 pacientes con artritis reumatoide tenían 65 años de edad o más, incluidos 146 pacientes de 75 años o más. De los 1827 pacientes tratados en los dos ensayos clínicos de fase 3 de artritis psoriásica, un total de 274 pacientes tenían 65 años de edad o más, incluidos 34 pacientes de 75 años o más. No se observaron diferencias en la eficacia entre estos pacientes y los pacientes más jóvenes; sin embargo, hubo una tasa más alta de eventos adversos en general, incluidas las infecciones graves, en pacientes de 65 años de edad o más.
Dermatitis atópica
De los 2583 pacientes tratados en los tres ensayos clínicos de fase 3, un total de 120 pacientes con dermatitis atópica tenían 65 años de edad o más, incluidos 6 pacientes de 75 años de edad. No se observaron diferencias en la eficacia entre estos pacientes y los pacientes más jóvenes; sin embargo, hubo una tasa más alta de infecciones graves y malignidades en aquellos pacientes de 65 años de edad o más en el grupo de dosificación de 30 mg en los ensayos a largo plazo.
Colitis ulcerosa
De los 1097 pacientes tratados en los ensayos clínicos controlados, un total de 95 pacientes con colitis ulcerosa tenían 65 años o más. Los estudios clínicos de RINVOQ no incluyeron un número suficiente de pacientes de 65 años de edad o más con colitis ulcerosa para determinar si responden de manera diferente a los pacientes adultos más jóvenes.
Enfermedad de Crohn
De los 1021 pacientes que fueron tratados en los ensayos clínicos controlados de inducción, un total de 39 pacientes con enfermedad de Crohn tenían 65 años de edad o más, y ningún paciente tenía 75 años de edad o más. Los estudios clínicos de RINVOQ no incluyeron un número suficiente de pacientes de 65 años de edad o más con enfermedad de Crohn para determinar si responden de manera diferente a los pacientes adultos más jóvenes.
Espondilitis anquilosante
De los 607 pacientes tratados en los ensayos clínicos controlados, un total de 32 pacientes con espondilitis anquilosante tenían 65 años o más. Los estudios clínicos de RINVOQ no incluyeron un número suficiente de pacientes de 65 años o más con espondilitis anquilosante para determinar si responden de manera diferente a los pacientes adultos más jóvenes.
Espondilitis axial no radiográfica
De los 313 pacientes tratados en un ensayo clínico de fase 3, un total de 9 pacientes con espondilitis axial no radiográfica tenían 65 años o más. Los estudios clínicos de RINVOQ no incluyeron un número suficiente de pacientes de 65 años o más con espondilitis axial no radiográfica para determinar si responden de manera diferente a los pacientes adultos más jóvenes.
8.6 Insuficiencia renal
Para pacientes con artritis reumatoide, artritis psoriásica, espondilitis anquilosante, espondilitis axial no radiográfica o pJIA, no se necesita ajuste de dosis en pacientes con insuficiencia renal leve (eGFR 60 a < 90 mL/min/1.73 m2), moderada (eGFR 30 a < 60 mL/min/1.73 m2), o grave (eGFR 15 a < 30 mL/min/1.73 m2).
Para pacientes con dermatitis atópica, la dosis máxima recomendada de RINVOQ es de 15 mg una vez al día para pacientes con insuficiencia renal grave. No se necesita ajuste de dosis en pacientes con insuficiencia renal leve o moderada.
Para pacientes con colitis ulcerosa o enfermedad de Crohn, la dosis recomendada de RINVOQ para insuficiencia renal grave es de 30 mg una vez al día para la inducción y 15 mg una vez al día para el mantenimiento. No se necesita ajuste de dosis en pacientes con insuficiencia renal leve o moderada [ver Dosis y administración (2.11)].
RINVOQ/RINVOQ LQ no se ha estudiado en pacientes con enfermedad renal en etapa terminal (eGFR <15 mL/min/1.73m2). No se recomienda el uso en pacientes con dermatitis atópica, colitis ulcerosa o enfermedad de Crohn con enfermedad renal en etapa terminal [ver Farmacología clínica (12.3)].
8.7 Insuficiencia hepática
El uso de RINVOQ/RINVOQ LQ no se ha estudiado en pacientes con insuficiencia hepática grave (Child Pugh C), y por lo tanto no se recomienda [ver Dosis y administración (2.11) y Farmacología clínica (12.3)].
Para pacientes con artritis reumatoide, artritis psoriásica, dermatitis atópica, espondilitis anquilosante, espondilitis axial no radiográfica o pJIA no se necesita ajuste de dosis en pacientes con insuficiencia hepática leve (Child Pugh A) o moderada (Child Pugh B).
Para pacientes con colitis ulcerosa o enfermedad de Crohn, la dosis recomendada de RINVOQ para insuficiencia hepática leve a moderada es de 30 mg una vez al día para la inducción y 15 mg una vez al día para el mantenimiento [ver Dosis y administración (2.11)].
11 DESCRIPCIÓN
RINVOQ y RINVOQ LQ están formuladas con upadacitinib, un inhibidor de JAK.
Upadacitinib tiene el siguiente nombre químico: (3S, 4R) – 3 – Etil – 4 – (3H – imidazo [1,2 – a] pirrolo [2,3 – e] pirazina – 8 – il) – N – (2,2,2 – trifluoroetilo) pirrolidina – 1 – carboxamida hidrato (2: 1).
La potencia de upadacitinib se basa en upadacitinib anhidro. La solubilidad de upadacitinib en agua es de 38 hasta menos de 0.2 mg / mL en un rango de pH de 2 a 9 a 37 oC.
Upadacitinib tiene un peso molecular de 389.38 g / mol y una fórmula molecular de C17H19F3N6O • ½ H2O. La estructura química de upadacitinib es:
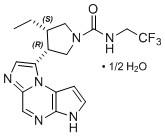
Las tabletas de liberación prolongada RINVOQ 15 mg para administración oral son moradas, oblongas biconvexas, con dimensiones de 14 x 8 mm y están grabadas con ‘a15’ en un lado. Cada tableta contiene los siguientes ingredientes inactivos: dióxido de silicio coloidal, óxido ferroso férrico, hipromelosa, óxido de hierro rojo, estearato de magnesio, manitol, celulosa microcristalina, alcohol polivinílico, polietilenglicol, talco, ácido tartárico y dióxido de titanio.
Las tabletas de liberación prolongada RINVOQ 30 mg para administración oral son rojas, oblongas biconvexas, con dimensiones de 14 x 8 mm y están grabadas con ‘a30’ en un lado. Cada tableta contiene los siguientes ingredientes inactivos: dióxido de silicio coloidal, hipromelosa, óxido de hierro rojo, estearato de magnesio, manitol, celulosa microcristalina, alcohol polivinílico, polietilenglicol, talco, ácido tartárico y dióxido de titanio.
Las tabletas de liberación prolongada RINVOQ 45 mg para administración oral son amarillas a amarillas moteadas, oblongas biconvexas, con dimensiones de 14 x 8 mm y están grabadas con ‘a45’ en un lado. Cada tableta contiene los siguientes ingredientes inactivos: dióxido de silicio coloidal, hipromelosa, óxido de hierro amarillo, óxido de hierro rojo, estearato de magnesio, manitol, celulosa microcristalina, alcohol polivinílico, polietilenglicol, talco, ácido tartárico y dióxido de titanio.
La solución oral RINVOQ LQ para administración oral es una solución clara, incolora a amarillo claro de 1 mg/mL. Cada 1 mL de RINVOQ LQ contiene 1 mg de upadacitinib como base libre (equivalente a 1.02 mg de hemihidrato de upadacitinib) y los siguientes ingredientes inactivos: ácido cítrico anhidro, agua purificada, benzoato de sodio, citrato de sodio dihidrato y sucralosa.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
Upadacitinib es un inhibidor de la Janus quinasa (JAK). Las JAK son enzimas intracelulares que transmiten señales que surgen de las interacciones del receptor de citocinas o factores de crecimiento en la membrana celular para influir en los procesos celulares de hematopoyesis y función de las células inmunitarias. Dentro de la vía de señalización, las JAK fosforilan y activan los transductores de señales y activadores de la transcripción (STAT) que modulan la actividad intracelular, incluida la expresión génica. Upadacitinib modula la vía de señalización en el punto de las JAK, evitando la fosforilación y activación de los STAT.
Las enzimas JAK transmiten la señalización de citocinas a través de su emparejamiento (por ejemplo, JAK1/JAK2, JAK1/JAK3, JAK1/TYK2, JAK2/JAK2, JAK2/TYK2). En un ensayo de enzima aislada sin células, upadacitinib tuvo una mayor potencia inhibitoria en JAK1 y JAK2 en relación con JAK3 y TYK2. En ensayos celulares de leucocitos humanos, upadacitinib inhibió la fosforilación de STAT inducida por citocinas mediada por JAK1 y JAK1/JAK3 de manera más potente que la fosforilación de STAT mediada por JAK2/JAK2. La relevancia de la inhibición de enzimas JAK específicas para la eficacia terapéutica no se conoce actualmente.
12.2 Farmacodinamia
Inhibición de la fosforilación de STAT3 inducida por IL-6 y la fosforilación de STAT5 inducida por IL-7
En voluntarios sanos, la administración de upadacitinib (formulación de liberación inmediata) dio como resultado una inhibición dependiente de la dosis y la concentración de la fosforilación de STAT3 inducida por IL-6 (JAK1/JAK2) y la fosforilación de STAT5 inducida por IL-7 (JAK1/JAK3) en sangre completa. La inhibición máxima se observó 1 hora después de la dosificación, que volvió a casi el nivel basal al final del intervalo de dosificación.
Linfocitos
En pacientes con artritis reumatoide, el tratamiento con upadacitinib se asoció con un pequeño aumento transitorio en el ALC medio desde el inicio hasta la semana 36, que gradualmente volvió a los niveles iniciales o cercanos a ellos con el tratamiento continuo.
Inmunoglobulinas
En pacientes con artritis reumatoide, se observaron pequeñas disminuciones desde el inicio en los niveles medios de IgG e IgM con el tratamiento con upadacitinib en el período controlado; sin embargo, los valores medios al inicio y en todas las visitas estuvieron dentro del rango de referencia normal.
Electrofisiología cardíaca
A 2.5 veces la exposición media de la dosis terapéutica máxima, dosis de 45 mg una vez al día, no hubo ningún efecto clínicamente relevante en el intervalo QTc.
12.3 Farmacocinética
Las exposiciones plasmáticas de upadacitinib son proporcionales a la dosis en el rango de dosis terapéutico. Después de la administración de una dosis única de tabletas de RINVOQ de 15 mg, 30 mg y 45 mg en ayunas en sujetos sanos, la Cmax media fue de 31.6 ng/mL, 71.8 ng/mL y 90.7 ng/mL, respectivamente, y el AUCinf media fue de 265 ng·h/mL, 543 ng·h/mL y 752 ng·h/mL, respectivamente. Las concentraciones plasmáticas en estado estacionario se alcanzan dentro de los 4 días con una acumulación mínima después de la administración una vez al día. Después de la administración de la dosis pediátrica recomendada (Tabla 1, Tabla 2) en pacientes con pJIA y artritis psoriásica, se predice que la Cmax media en estado estacionario será de 47.6 ng/mL y el AUC0-24 media en estado estacionario se predice que será de 342 ng·h/mL. La farmacocinética de upadacitinib es comparable entre pacientes con artritis reumatoide, artritis psoriásica, dermatitis atópica, colitis ulcerosa, enfermedad de Crohn, espondilitis anquilosante y espondiloartritis axial no radiográfica.
Las tabletas de RINVOQ y RINVOQ LQ no son bioequivalentes; por lo tanto, las 2 formas de dosificación no son intercambiables sobre una base de miligramo por miligramo [ver Dosificación y administración (2.2)].
Absorción
Después de la administración oral de tabletas de liberación prolongada de RINVOQ, upadacitinib se absorbe con una Tmax media de 2 a 4 horas. Después de la administración oral de 6 mg de RINVOQ LQ, upadacitinib se absorbe con una Tmax media de 1 hora.
La coadministración de tabletas de RINVOQ con una comida rica en grasas/calorías no tuvo ningún efecto clínicamente relevante en las exposiciones de upadacitinib (aumento del AUCinf en un 29% y la Cmax en un 39% a 60%). No se espera que la coadministración de RINVOQ LQ con alimentos tenga un efecto clínicamente relevante en la exposición de upadacitinib [ver Dosificación y administración (2.2)].
Distribución
Upadacitinib está unido en un 52% a las proteínas plasmáticas. Upadacitinib se reparte de manera similar entre los componentes celulares de la sangre y el plasma con una relación sangre/plasma de 1.0.
Eliminación
Metabolismo
El metabolismo de upadacitinib está mediado principalmente por CYP3A4 con una posible contribución menor de CYP2D6. La actividad farmacológica de upadacitinib se atribuye a la molécula madre. En un estudio radiomarcado en humanos, el upadacitinib sin cambios representó el 79% de la radioactividad total en plasma, mientras que el metabolito principal detectado (producto de la monooxigenación seguido de la glucuronidación) representó el 13% de la radioactividad total en plasma. No se han identificado metabolitos activos para upadacitinib.
Excreción
Después de la administración de una dosis única de solución de liberación inmediata de [14C]-upadacitinib, upadacitinib se eliminó predominantemente como el fármaco madre sin cambios en la orina (24%) y las heces (38%). Aproximadamente el 34% de la dosis de upadacitinib se excretó como metabolitos. La semivida de eliminación terminal media de upadacitinib osciló entre 8 y 14 horas.
Poblaciones específicas
Peso corporal, sexo, y raza
El peso corporal, el sexo, la raza y la etnia no tuvieron un efecto clínicamente significativo en la exposición a upadacitinib en poblaciones de pacientes adultos.
Pacientes pediátricos
En pacientes pediátricos con AJA con poliartritis activa, el aclaramiento de upadacitinib aumentó con el aumento del peso corporal. La edad (en el rango de 2 a < 18 años) no tuvo un efecto adicional en la farmacocinética de upadacitinib después de tener en cuenta el efecto del peso corporal. Se predice que las exposiciones plasmáticas de upadacitinib en pacientes pediátricos con pAJA y artritis psoriásica después de la dosis pediátrica recomendada serán comparables a las observadas en pacientes adultos con artritis reumatoide y artritis psoriásica, respectivamente.
No se observó ninguna diferencia significativa en la exposición sistémica de upadacitinib en pacientes pediátricos con dermatitis atópica de 12 años de edad o más que pesaban al menos 40 kg en comparación con los adultos.
Pacientes geriátricos
No se observaron diferencias clínicamente significativas en la farmacocinética de upadacitinib en pacientes geriátricos (≥ 65 años de edad) en comparación con pacientes adultos más jóvenes.
Pacientes con insuficiencia renal
El AUCinf medio de upadacitinib después de la administración de una dosis única de 15 mg de tabletas de upadacitinib fue un 18%, 33% y 44% más alto en pacientes con insuficiencia renal leve (eGFR de 60 a < 90 mL/min/1,73 m2), moderada (eGFR de 30 a < 60 mL/min/1,73 m2) y grave (eGFR de 15 a < 30 mL/min/1,73 m2), respectivamente, en comparación con los sujetos con función renal normal (eGFR ≥ 90 mL/min/1,73 m2). La Cmax media de upadacitinib fue similar entre los sujetos con función renal normal e insuficiencia renal. En pacientes que reciben RINVOQ/RINVOQ LQ, no se espera que la insuficiencia renal leve y moderada tenga un efecto clínicamente relevante en la exposición a upadacitinib [ver Dosis y administración (2.11) y Uso en poblaciones específicas (8.6)].
Pacientes con insuficiencia hepática
El AUCinf medio de upadacitinib después de la administración de una dosis única de 15 mg de tabletas de upadacitinib fue un 28% y un 24% más alto en pacientes con insuficiencia hepática leve (Child-Pugh A) y moderada (Child-Pugh B), respectivamente, en comparación con los sujetos con función hepática normal. La Cmax media de upadacitinib no cambió en pacientes con insuficiencia hepática leve y fue un 43% más alta en pacientes con insuficiencia hepática moderada en comparación con los sujetos con función hepática normal. Upadacitinib no se estudió en pacientes con insuficiencia hepática grave (Child-Pugh C) [ver Dosis y administración (2.11) y Uso en poblaciones específicas (8.7)].
Estudios de interacción medicamentosa
Posibilidad de que otros medicamentos influyan en la farmacocinética de upadacitinib
Upadacitinib se metaboliza in vitro por CYP3A4 con una contribución menor de CYP2D6. El efecto de los medicamentos coadministrados en las exposiciones plasmáticas de upadacitinib se proporciona en la Tabla 10 [ver Interacciones medicamentosas (7)].
| Co- administrado Medicamento |
Régimen de Co- administrado Medicamento |
Relación (90% IC)a |
|
| Cmax | AUC | ||
| Metotrexato | 10 a 25 mg/semana | 0,97 (0,86-1,09) |
0,99 (0,93- 1,06) |
| Inhibidor potente de CYP3A4: Ketoconazol |
400 mg una vez al día x 6 días |
1,70 (1,55-1,89) |
1,75 (1,62-1,88) |
| Inducidor potente de CYP3A4: Rifampicina |
600 mg una vez al día x 9 días |
0,49 (0,44-0,55) |
0,39 (0,37-0,42) |
| Inhibidor de OATP1B: Rifampicina |
600 mg dosis única | 1,14 (1,02-1,28) |
1,07 (1,01-1,14) |
| IC: Intervalo de confianza a Las relaciones para Cmax y AUC comparan la coadministración del medicamento con upadacitinib frente a la administración de upadacitinib solo. |
|||
No se espera que los medicamentos que modifican el pH (por ejemplo, antiácidos o inhibidores de la bomba de protones) afecten las exposiciones plasmáticas de upadacitinib según las evaluaciones in vitro y los análisis farmacocinéticos poblacionales. El fenotipo metabólico CYP2D6 no tuvo ningún efecto en la farmacocinética de upadacitinib (basado en análisis farmacocinéticos poblacionales), lo que indica que los inhibidores de CYP2D6 no tienen ningún efecto clínicamente relevante en las exposiciones a upadacitinib.
Potencial de Upadacitinib para Influir en la Farmacocinética de Otros Medicamentos
Los estudios in vitro indican que upadacitinib no inhibe la actividad de las enzimas del citocromo P450 (CYP) (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 y CYP3A4) a concentraciones clínicamente relevantes. Los estudios in vitro indican que upadacitinib induce CYP3A4 pero no induce CYP2B6 o CYP1A2 a concentraciones clínicamente relevantes. Los estudios in vitro indican que upadacitinib no inhibe los transportadores P-gp, BCRP, OATP1B1, OATP1B3, OCT1, OCT2, OAT1, OAT3, MATE1 y MATE2K a concentraciones clínicamente relevantes.
Los estudios clínicos indican que upadacitinib no tiene efectos clínicamente relevantes en la farmacocinética de los medicamentos coadministrados. Después de la administración de tabletas de upadacitinib de 30 mg y 45 mg una vez al día, los efectos en cada enzima CYP (CYP1A2, CYP3A, CYP2C9 y CYP2C19) fueron similares entre las dos dosis, excepto por el efecto en CYP2D6. Después de la administración de tabletas de upadacitinib de 30 mg y 45 mg una vez al día, se observó una inducción débil de CYP3A4. Se observó una inhibición débil de CYP2D6 a 45 mg de upadacitinib, pero no a 30 mg. En la Tabla 11 se proporciona un resumen de los resultados de los estudios clínicos que evaluaron el efecto de upadacitinib en otros medicamentos.
| Coadministrado Medicamento o CYP Marcador de Actividad |
Régimen de Dosis Múltiples de Upadacitinib |
Relación (90% IC)a |
|
| Cmax | AUC | ||
| Metotrexato | 6 mg a 24 mg BIDb | 1.03 (0.86-1.23) |
1.14 (0.91-1.43) |
| Sustrato sensible a CYP1A2: Cafeína |
45 mg QDc | 1.05 (0.97-1.14) |
1.04 (0.95-1.13) |
| Sustrato sensible a CYP3A: Midazolam |
30 mg QDc | 0.74 (0.68-0.80) |
0.74 (0.68-0.80) |
| Sustrato sensible a CYP3A: Midazolam |
45 mg QDc | 0.75 (0.69 -0.83) |
0.76 (0.69 -0.83) |
| Sustrato sensible a CYP2D6: Dextrometorfano |
30 mg QDc | 1.09 (0.98-1.21) |
1.07 (0.95-1.22) |
| Sustrato sensible a CYP2D6: Dextrometorfano |
45 mg QDc | 1.30 (1.13-1.50) |
1.35 (1.18-1.54) |
| Sustrato sensible a CYP2C9: S-Warfarina |
45 mg QDc | 1.18 (1.05-1.33) |
1.12 (1.05-1.20) |
| Marcador sensible a CYP2C19: Relación metabólica de 5-OH Omeprazol a Omeprazol |
45 mg QDc | — | 0.96 (0.90-1.02) |
| Sustrato de CYP2B6: Bupropión |
30 mg QDc | 0.87 (0.79-0.96) |
0.92 (0.87-0.98) |
| Rosuvastatina | 30 mg QDc | 0.77 (0.63-0.94) |
0.67 (0.56-0.82) |
| Atorvastatina | 30 mg QDc | 0.88 (0.79-0.97) |
0.77 (0.70-0.85) |
| Etinilestradiol | 30 mg QDc | 0.96 (0.89-1.02) |
1.11 (1.04-1.19) |
| Levonorgestrel | 30 mg QDc | 0.96 (0.87-1.06) |
0.96 (0.85-1.07) |
| CYP: citocromo P450; CI: intervalo de confianza; BID: dos veces al día; QD: una vez al día a Las razones para Cmax y AUC comparan la coadministración del medicamento con upadacitinib frente a la administración del medicamento solo. b Formulación de liberación inmediata c Formulación de tableta de liberación prolongada |
|||
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
Carcinogénesis
El potencial carcinogénico de upadacitinib se evaluó en ratas Sprague-Dawley y ratones Tg.rasH2. No se observó evidencia de tumorigenicidad en ratas machos o hembras que recibieron upadacitinib durante un máximo de 101 semanas a dosis orales de hasta 15 o 20 mg/kg/día, respectivamente (aproximadamente 4 y 10 veces la dosis de la tableta de 15 mg, 2 y 5 veces la dosis de la tableta de 30 mg, y 1.6 y 4 veces la dosis máxima recomendada para humanos (MRHD) de 45 mg en base al AUC, respectivamente). No se observó evidencia de tumorigenicidad en ratones Tg.rasH2 machos o hembras que recibieron upadacitinib durante 26 semanas a dosis orales de hasta 20 mg/kg/día.
Mutagénesis
Upadacitinib dio negativo en las siguientes pruebas de genotoxicidad: la prueba de mutagenicidad bacteriana in vitro (prueba de Ames), la prueba de aberración cromosómica in vitro en linfocitos de sangre periférica humana y la prueba de micronúcleos de médula ósea de rata in vivo.
Deterioro de la Fertilidad
Upadacitinib no tuvo ningún efecto sobre la fertilidad en ratas machos o hembras a dosis orales de hasta 50 mg/kg/día en machos y 75 mg/kg/día en hembras (aproximadamente 42 y 84 veces la dosis de 15 mg, 22 y 43 veces la dosis de 30 mg, y 16 y 31 veces la MRHD, respectivamente, en base al AUC). Sin embargo, el mantenimiento del embarazo se vio afectado negativamente a dosis orales de 25 mg/kg/día y 75 mg/kg/día en base a hallazgos relacionados con la dosis de aumento de las pérdidas postimplantación (aumento de las resorciones) y disminución del número de embriones viables medios por camada (aproximadamente 22 y 84 veces la dosis de la tableta de 15 mg, 11 y 43 veces la dosis de la tableta de 30 mg, y 8 y 31 veces la MRHD en base al AUC, respectivamente). El número de embriones viables no se vio afectado en ratas hembras que recibieron upadacitinib a una dosis oral de 5 mg/kg/día y se aparearon con machos que recibieron la misma dosis (aproximadamente 2 veces la dosis de 15 mg, 0.9 veces la dosis de 30 mg, y 0.6 veces la MRHD en base al AUC).
14 ESTUDIOS CLÍNICOS
14.1 Artritis Reumatoide
Se evaluó la eficacia y seguridad de RINVOQ 15 mg una vez al día en cinco ensayos de fase 3 aleatorizados, doble ciego y multicéntricos en pacientes con artritis reumatoide activa de moderada a grave que cumplían los criterios de clasificación ACR/EULAR 2010. Los pacientes mayores de 18 años eran elegibles para participar. Se requería la presencia de al menos 6 articulaciones sensibles y 6 inflamadas y evidencia de inflamación sistémica basada en la elevación de la PCRhs al inicio del estudio. Aunque se han estudiado otras dosis, la dosis recomendada de RINVOQ es de 15 mg una vez al día.
El ensayo RA-I (NCT02706873) fue un ensayo de monoterapia de 24 semanas en 947 pacientes con artritis reumatoide activa de moderada a grave que eran pacientes sin tratamiento previo con metotrexato (MTX). Los pacientes recibieron 15 mg de RINVOQ o 30 mg de upadacitinib por vía oral una vez al día o MTX como monoterapia. En la semana 26, los pacientes que no respondieron a upadacitinib pudieron ser rescatados con la adición de MTX, mientras que los pacientes con MTX pudieron ser rescatados con la adición de 15 mg de RINVOQ o 30 mg de upadacitinib una vez al día a ciego. El criterio principal de valoración fue la proporción de pacientes que lograron una respuesta ACR50 en la semana 12. Los criterios de valoración secundarios clave incluyeron una puntuación de actividad de la enfermedad (DAS28-PCR) ≤3,2 en la semana 12, DAS28-PCR <2,6 en la semana 24, cambio con respecto al inicio en el HAQ-DI en la semana 12 y cambio con respecto al inicio en la puntuación total de Sharp modificada por van der Heijde (mTSS) en la semana 24.
El ensayo RA-II (NCT02706951) fue un ensayo de monoterapia de 14 semanas en 648 pacientes con artritis reumatoide activa de moderada a grave que tuvieron una respuesta inadecuada al MTX. Los pacientes recibieron 15 mg de RINVOQ o 30 mg de upadacitinib en monoterapia una vez al día o continuaron con su dosis estable de MTX en monoterapia. En la semana 14, los pacientes que fueron aleatorizados a MTX pasaron a recibir 15 mg de RINVOQ o 30 mg de upadacitinib una vez al día en monoterapia de forma ciega en función de la asignación predeterminada al inicio del estudio. El criterio principal de valoración fue la proporción de pacientes que lograron una respuesta ACR20 en la semana 14. Los criterios de valoración secundarios clave incluyeron DAS28-PCR ≤3,2, DAS28-PCR <2,6 y cambio con respecto al inicio en el HAQ-DI en la semana 14.
El ensayo RA-III (NCT02675426) fue un ensayo de 12 semanas en 661 pacientes con artritis reumatoide activa de moderada a grave que tuvieron una respuesta inadecuada a los fármacos antirreumáticos modificadores de la enfermedad convencionales (FAMEc). Los pacientes recibieron 15 mg de RINVOQ o 30 mg de upadacitinib una vez al día o placebo añadido al tratamiento de base con FAMEc. En la semana 12, los pacientes que fueron aleatorizados a placebo pasaron a recibir 15 mg de RINVOQ o 30 mg de upadacitinib una vez al día de forma ciega en función de la asignación predeterminada al inicio del estudio. El criterio principal de valoración fue la proporción de pacientes que lograron una respuesta ACR20 en la semana 12. Los criterios de valoración secundarios clave incluyeron DAS28-PCR ≤3,2, DAS28-PCR<2,6 y cambio con respecto al inicio en el HAQ-DI en la semana 12.
El ensayo RA-IV (NCT02629159) fue un ensayo de 48 semanas en 1629 pacientes con artritis reumatoide activa de moderada a grave que tuvieron una respuesta inadecuada al MTX. Los pacientes recibieron 15 mg de RINVOQ una vez al día, un comparador activo o placebo añadido al MTX de base. A partir de la semana 14, los pacientes que no respondieron a 15 mg de RINVOQ pudieron pasar a recibir el comparador activo de forma ciega, y los pacientes que no respondieron al comparador activo o al placebo pudieron pasar a recibir 15 mg de RINVOQ de forma ciega. En la semana 26, todos los pacientes aleatorizados a placebo pasaron a recibir 15 mg de RINVOQ una vez al día de forma ciega. El criterio principal de valoración fue la proporción de pacientes que lograron una respuesta ACR20 en la semana 12 frente a placebo. Los criterios de valoración secundarios clave frente a placebo incluyeron DAS28-PCR ≤3,2, DAS28-PCR <2,6, cambio con respecto al inicio en el HAQ-DI en la semana 12 y cambio con respecto al inicio en la mTSS en la semana 26.
El ensayo RA-V (NCT02706847) fue un ensayo de 12 semanas en 499 pacientes con artritis reumatoide activa de moderada a grave que tuvieron una respuesta inadecuada o intolerancia a los FAMEb. Los pacientes recibieron 15 mg de RINVOQ o 30 mg de upadacitinib una vez al día o placebo añadido al tratamiento de base con FAMEc. En la semana 12, los pacientes que fueron aleatorizados a placebo pasaron a recibir 15 mg de RINVOQ o 30 mg de upadacitinib una vez al día de forma ciega en función de la asignación predeterminada al inicio del estudio. El criterio principal de valoración fue la proporción de pacientes que lograron una respuesta ACR20 en la semana 12. Los criterios de valoración secundarios clave incluyeron DAS28-PCR ≤3,2 y cambio con respecto al inicio en el HAQ-DI en la semana 12.
Respuesta clínica
Los porcentajes de pacientes tratados con RINVOQ que lograron respuestas ACR20, ACR50 y ACR70, y DAS28(PCR) < 2,6 en todos los ensayos se muestran en la Tabla 12.
Los pacientes tratados con 15 mg de RINVOQ, solos o en combinación con FAMEc, lograron tasas de respuesta ACR más altas en comparación con la monoterapia con MTX o placebo, respectivamente, en el momento de la eficacia primaria (Tabla 12).
En el Ensayo IV, el porcentaje de pacientes que lograron una respuesta ACR20 por visita se muestra en la Figura 1.
En los ensayos RA-III y RA-V, se observaron tasas de respuesta ACR20 más altas en la semana 1 con 15 mg de RINVOQ frente a placebo.
El tratamiento con 15 mg de RINVOQ, solo o en combinación con FAMEc, dio lugar a mayores mejoras en los componentes del ACR en comparación con MTX o placebo en el momento de la eficacia primaria (Tabla 13).
| Ensayo RA-I Pacientes sin tratamiento previo con MTX |
Ensayo RA-II RI-MTX |
Ensayo RA-III RI-FAMEc |
Ensayo RA-IV RI-MTX |
Ensayo RA-V RI-FAMEb |
||||||
| Monoterapia | Monoterapia | FAMEc de base |
MTX de base |
FAMEc de base |
||||||
| MTX | RINVOQ 15 mg % Δ (95% CI) |
MTX | RINVOQ 15 mg % Δ (95% CI) |
PBO | RINVOQ 15 mg % Δ (95% CI) |
PBO | RINVOQ 15 mg % Δ (95% CI) |
PBO | RINVOQ 15 mg % Δ (95% CI) |
|
| N | 314 | 317 | 216 | 217 | 221 | 221 | 651 | 651 | 169 | 164 |
| Semana | ||||||||||
| ACR20 | ||||||||||
| 12a/14b | 54 | 76 22 (14, 29) |
41 | 68 26 (17, 36) |
36 | 64 28 (19, 37) |
36 | 71 34 (29, 39) |
28 | 65 36 (26, 46) |
| 24c/26d | 59 | 79 20 (13, 27) |
36 | 67 32 (27, 37) |
||||||
| ACR50 | ||||||||||
| 12a/14b | 28 | 52 24 (16, 31) |
15 | 42 27 (18, 35) |
15 | 38 23 (15, 31) |
15 | 45 30 (26, 35) |
12 | 34 22 (14, 31) |
| 24c/26d | 33 | 60 27 (19, 34) |
21 | 54 33 (28, 38) |
||||||
| ACR70 | ||||||||||
| 12a/14b | 14 | 32 18 (12, 25) |
3 | 23 20 (14, 26) |
6 | 21 15 (9, 21) |
5 | 25 20 (16, 24) |
7 | 12 5 (-1, 11) |
| 24c/26d | 18 | 44 26 (19, 33) |
10 | 35 25 (21, 29) |
||||||
| DAS28-CRP <2.6 | ||||||||||
| 12a/14b | 14 | 36 22 (15, 28) |
8 | 28 20 (13, 27) |
10 | 31 21 (14, 28) |
6 | 29 23 (19, 27) |
9 | 29 19 (11, 27) |
| 24c/26d | 18 | 48 30 (23, 37) |
9 | 41 32 (27, 36) |
||||||
| Abreviaturas: ACR20 (o 50 o 70) = American College of Rheumatology ≥20% (o ≥50% o ≥70%) de mejoría; bDMARD = fármaco biológico antirreumático modificador de la enfermedad; CRP = proteína C reactiva; DAS28 = Disease Activity Score 28 joints; cDMARDs = fármacos antirreumáticos modificadores de la enfermedad convencionales; MTX = metotrexato; PBO = placebo; IR = respondedor inadecuado Los pacientes que interrumpieron el tratamiento aleatorizado, o tuvieron un cruce entre tratamientos aleatorizados, o les faltaban datos en la semana de evaluación fueron imputados como no respondedores en los análisis. a Ensayo RA-I, Ensayo RA-III, Ensayo RA-IV, Ensayo RA-V b Ensayo RA-II c Ensayo RA-I d Ensayo RA-IV |
||||||||||
| Ensayo RA-I MTX sin tratamiento previo |
Ensayo RA-IIb IR a MTX |
Ensayo RA-III cDMARD-IR |
Ensayo RA-IV IR a MTX |
Ensayo RA-V bDMARD-IR |
||||||
| Monoterapia | Monoterapia | cDMARDs de referencia | MTX de referencia | cDMARDs de referencia | ||||||
| MTX | RINVOQ 15 mg |
MTX | RINVOQ 15 mg |
PBO | RINVOQ 15 mg |
PBO | RINVOQ 15 mg |
PBO | RINVOQ 15 mg |
|
| N | 314 | 317 | 216 | 217 | 221 | 221 | 651 | 651 | 169 | 164 |
| Número de articulaciones sensibles (0-68) | ||||||||||
| Línea base | 26 (16) |
25 (14) |
25 (16) |
24 (15) |
25 (15) |
25 (14) |
26 (14) |
26 (15) |
28 (15) |
28 (16) |
| Semana 12/14 |
13 (15) |
9 (12) |
15 (16) |
10 (13) |
16 (17) |
12 (14) |
16 (15) |
10 (13) |
18 (17) |
11 (14) |
| Número de articulaciones inflamadas (0-66) | ||||||||||
| Línea base | 17 (11) |
17 (10) |
17 (12) |
16 (11) |
15 (9) |
16 (10) |
16 (9) |
17 (10) |
16 (10) |
17 (11) |
| Semana 12/14 |
6 (8) |
5 (7) |
9 (11) |
6 (9) |
9 (10) |
7 (10) |
9 (9) |
5 (7) |
9 (10) |
6 (8) |
| Dolorc | ||||||||||
| Línea base | 66 (21) |
68 (21) |
63 (21) |
62 (23) |
62 (21) |
64 (19) |
65 (21) |
66 (21) |
69 (21) |
68 (20) |
| Semana 12/14 |
41 (25) |
31 (25) |
49 (25) |
36 (27) |
51 (26) |
33 (24) |
49 (25) |
33 (24) |
55 (28) |
41 (28) |
| Evaluación global del pacientec | ||||||||||
| Línea base | 66 (21) |
67 (22) |
60 (22) |
62 (22) |
60 (20) |
63 (22) |
64 (21) |
64 (22) |
66 (23) |
67 (20) |
| Semana 12/14 |
42 (25) |
31 (24) |
48 (26) |
37 (27) |
50 (26) |
32 (24) |
48 (24) |
33 (24) |
54 (28) |
40 (26) |
| Índice de discapacidad (HAQ-DI)d | ||||||||||
| Línea base | 1.60 (0.67) |
1.60 (0.67) |
1.47 (0.66) |
1.47 (0.66) |
1.42 (0.63) |
1.48 (0.61) |
1.61 (0.61) |
1.63 (0.64) |
1.56 (0.60) |
1.67 (0.64) |
| Semana 12/14 |
1.08 (0.72) |
0.76 (0.69) |
1.19 (0.69) |
0.86 (0.67) |
1.13 (0.70) |
0.85 (0.66) |
1.28 (0.67) |
0.98 (0.68) |
1.33 (0.66) |
1.24 (0.77) |
| Evaluación global del médicoc | ||||||||||
| Línea base | 69 (16) |
67 (17) |
62 (17) |
66 (18) |
64 (18) |
64 (16) |
66 (18) |
66 (17) |
67 (17) |
69 (17) |
| Semana 12/14 |
32 (22) |
22 (19) |
37 (24) |
26 (21) |
41 (24) |
26 (21) |
41 (25) |
27 (21) |
39 (25) |
29 (22) |
| CRP (mg/L) | ||||||||||
| Línea base | 21.2 (22.1) |
23.0 (27.4) |
14.5 (17.3) |
14.0 (16.5) |
12.6 (14.0) |
16.6 (19.2) |
18.0 (21.5) |
17.9 (22.5) |
16.3 (21.1) |
16.3 (18.6) |
| Semana 12/14 |
10.9 (14.9) |
4.2 (8.8) |
12.8 (21.4) |
3.7 (7.8) |
13.1 (15.5) |
4.6 (9.6) |
16.2 (19.8) |
5.5 (10.9) |
13.9 (17.3) |
5.0 (14.0) |
| Abbreviations: ACR = American College of Rheumatology; bDMARD = biologic disease-modifying anti-rheumatic drug; CRP = c-reactive protein; cDMARDs = conventional disease-modifying anti-rheumatic drugs; HAQ-DI = Health Assessment Questionnaire Disability Index; IR = inadequate responder; MTX = methotrexate; PBO = placebo
a Los datos mostrados son la media (desviación estándar). |
||||||||||
Figura 1. Porcentaje de pacientes que logran ACR20 en el ensayo RA-IV
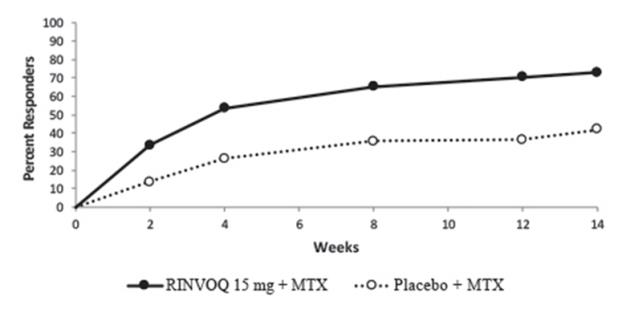
Abreviaturas: ACR20 = mejoría ≥20 % del Colegio Estadounidense de Reumatología; MTX = metotrexato
Los pacientes que interrumpieron el tratamiento aleatorizado, o a quienes les faltaban los resultados del ACR20, o que se perdieron durante el seguimiento o se retiraron del ensayo se imputaron como no respondedores.
En los ensayos RA-I y RA-IV, una mayor proporción de pacientes tratados con RINVOQ 15 mg solo o en combinación con MTX, lograron un DAS28-PCR < 2.6 en comparación con MTX o placebo en el momento de la eficacia primaria (Tabla 14).
| Ensayo RA-I MTX-naïve |
||
| Monoterapia | ||
| DAS28-PCR inferior a 2.6 | MTX N = 314 |
RINVOQ 15 mg N = 317 |
| Proporción de respondedores en la semana 12 (n) | 14% (43) | 36% (113) |
| De los respondedores, proporción con 0 articulaciones activas (n) | 51% (22) | 45% (51) |
| De los respondedores, proporción con 1 articulación activa (n) | 35% (15) | 23% (26) |
| De los respondedores, proporción con 2 articulaciones activas (n) | 9% (4) | 17% (19) |
| De los respondedores, proporción con 3 o más articulaciones activas (n) | 5% (2) | 15% (17) |
| Ensayo RA-IV MTX-IR |
||
| MTX de base | ||
| DAS28-PCR inferior a 2.6 | PBO N = 651 |
RINVOQ 15 mg N = 651 |
| Proporción de respondedores en la semana 12 (n) | 6% (40) | 29% (187) |
| De los respondedores, proporción con 0 articulaciones activas (n) | 60% (24) | 48% (89) |
| De los respondedores, proporción con 1 articulación activa (n) | 20% (8) | 23% (43) |
| De los respondedores, proporción con 2 articulaciones activas (n) | 15% (6) | 13% (25) |
| De los respondedores, proporción con 3 o más articulaciones activas (n) | 5% (2) | 16% (30) |
| Abreviaturas: CRP = proteína C reactiva; DAS28 = puntuación de actividad de la enfermedad de 28 articulaciones; MTX = metotrexato; PBO = placebo; IR = respondedor inadecuado | ||
Radiographic Response
La inhibición de la progresión del daño estructural de las articulaciones se evaluó utilizando la puntuación total de Sharp modificada (mTSS) y sus componentes, la puntuación de erosión y la puntuación de estrechamiento del espacio articular, en la semana 26 en el ensayo RA-IV y en la semana 24 en el ensayo RA-I. También se evaluó la proporción de pacientes sin progresión radiográfica (cambio en la mTSS desde el inicio ≤ 0).
En el ensayo RA-IV, el tratamiento con RINVOQ 15 mg inhibió la progresión del daño estructural de las articulaciones en comparación con el placebo en combinación con cDMARDs en la semana 26 (Tabla 15). Los análisis de las puntuaciones de erosión y estrechamiento del espacio articular fueron consistentes con los resultados generales.
En el grupo de placebo más MTX, el 76 % de los pacientes no experimentaron progresión radiográfica en la semana 26 en comparación con el 83 % de los pacientes tratados con RINVOQ 15 mg.
En el ensayo RA-I, el tratamiento con RINVOQ 15 mg en monoterapia inhibió la progresión del daño estructural de las articulaciones en comparación con la monoterapia con MTX en la semana 24 (Tabla 15). Los análisis de las puntuaciones de erosión y estrechamiento del espacio articular fueron consistentes con los resultados generales.
En el grupo de monoterapia con MTX, el 78 % de los pacientes no experimentaron progresión radiográfica en la semana 24 en comparación con el 87 % de los pacientes tratados con RINVOQ 15 mg en monoterapia.
| Ensayo RA-IV MTX-IR |
|||
| Antecedentes de MTX | |||
| mTSS | PBO (N=651) Media (DE) |
RINVOQ 15 mg (N=651) Media (DE) |
Diferencia estimada frente a PBO en la semana 26 (IC del 95 %)a |
| Inicial | 35.9 (52) | 34.0 (50) | |
| Semana 26b | 0.78 (0.1) | 0.15 (0.1) | -0.63 (-0.92, -0.34) |
| Ensayo RA-I MTX-naïve |
|||
| Monoterapia | |||
| MTX (N=309) Media (DE) |
RINVOQ 15 mg (N=309) Media (DE) |
Diferencia estimada frente a MTX en la semana 24 (IC del 95 %)c |
|
| Inicial | 13.3 (31) | 18.1 (38) | |
| Semana 24d | 0.67 (2.8) | 0.14 (1.4) | -0.53 (-0.85, -0.20) |
| Abreviaturas: mTSS = puntuación total de Sharp modificada, MTX = metotrexato; PBO = placebo; DE = desviación estándar; IR = respondedores inadecuados; bDMARDs = fármacos biológicos antirreumáticos modificadores de la enfermedad; LS = mínimos cuadrados; IC = intervalos de confianza a Medias de LS e IC del 95 % basadas en un modelo de coeficiente aleatorio ajustado al valor de mTSS ajustando por tiempo, grupo de tratamiento, uso previo de bDMARDs, interacción grupo de tratamiento por tiempo, con pendientes aleatorias e intersección aleatoria. b Se presentan la tasa lineal estimada de progresión estructural en la semana 26 y los errores estándar. c Medias de LS e IC del 95 % basadas en un modelo de regresión lineal ajustado al cambio con respecto al valor inicial en la mTSS ajustando por grupo de tratamiento, mTSS inicial y región geográfica. d Se presentan la media del cambio con respecto al valor inicial y la desviación estándar. |
|||
Respuesta de la función física
El tratamiento con RINVOQ 15 mg, solo o en combinación con cDMARD, resultó en una mayor mejoría en la función física en la Semana 12/14 en comparación con todos los comparadores, según lo medido por HAQ-DI.
Otros resultados relacionados con la salud
En todos los ensayos, excepto en el ensayo RA-V, los pacientes que recibieron RINVOQ 15 mg tuvieron una mayor mejoría con respecto al valor inicial en la puntuación del resumen del componente físico (PCS), las puntuaciones del resumen del componente mental (MCS) y en los 8 dominios de la Encuesta de salud de formato corto (SF-36) en comparación con el placebo en combinación con cDMARD o monoterapia con MTX en la Semana 12/14.
La fatiga se evaluó mediante la puntuación de la Evaluación funcional de la terapia de enfermedades crónicas: fatiga (FACIT-F) en los ensayos RA-I, RA-III y RA-IV. Se observó una mejoría en la fatiga en la Semana 12 en pacientes tratados con RINVOQ 15 mg en comparación con los pacientes que recibieron placebo en combinación con cDMARD o monoterapia con MTX.
14.2 Artritis psoriásica
La eficacia y seguridad de RINVOQ 15 mg una vez al día se evaluaron en dos ensayos de fase 3, aleatorizados, doble ciego, multicéntricos y controlados con placebo en pacientes de 18 años o mayores con artritis psoriásica de moderada a gravemente activa. Todos los pacientes tenían artritis psoriásica activa durante al menos 6 meses según los Criterios de clasificación para la artritis psoriásica (CASPAR, por sus siglas en inglés), al menos 3 articulaciones sensibles y al menos 3 articulaciones inflamadas, y psoriasis en placas activa o antecedentes de psoriasis en placas. Aunque se ha estudiado otra dosis, la dosis recomendada de RINVOQ es de 15 mg una vez al día para la artritis psoriásica.
El ensayo PsA-I (NCT03104400) fue un ensayo de 24 semanas en 1705 pacientes con artritis psoriásica de moderada a gravemente activa que tuvieron una respuesta inadecuada o intolerancia a al menos un DMARD no biológico. Los pacientes recibieron RINVOQ 15 mg o upadacitinib 30 mg una vez al día, adalimumab o placebo, solos o en combinación con DMARD no biológicos de fondo. En la semana 24, todos los pacientes asignados aleatoriamente a placebo cambiaron a RINVOQ 15 mg o upadacitinib 30 mg una vez al día de forma ciega. El criterio de valoración principal fue la proporción de pacientes que lograron una respuesta ACR20 en la semana 12.
El ensayo PsA-II (NCT03104374) fue un ensayo de 24 semanas en 642 pacientes con artritis psoriásica de moderada a gravemente activa que tuvieron una respuesta inadecuada o intolerancia a al menos un DMARD biológico. Los pacientes recibieron RINVOQ 15 mg o upadacitinib 30 mg una vez al día o placebo, solos o en combinación con DMARD no biológicos de fondo. En la semana 24, todos los pacientes asignados aleatoriamente a placebo cambiaron a RINVOQ 15 mg o upadacitinib 30 mg una vez al día de forma ciega. El criterio de valoración principal fue la proporción de pacientes que lograron una respuesta ACR20 en la semana 12.
Respuesta clínica
En ambos ensayos, los pacientes tratados con RINVOQ 15 mg lograron respuestas ACR20 significativamente más altas en comparación con el placebo en la semana 12 (Tabla 16, Figura 2). Una mayor proporción de pacientes tratados con RINVOQ 15 mg lograron respuestas ACR50 y ACR70 en la semana 12 en comparación con el placebo.
El tratamiento con RINVOQ 15 mg resultó en mejoras en los componentes del ACR en comparación con el placebo en el momento de la eficacia primaria (Tabla 17).
| Ensayo | Ensayo PsA-I DMARD no biológico-RI |
Ensayo PsA-II bDMARD-RI |
||
| Grupo de tratamiento | PBO
% |
RINVOQ 15 mg % Δ (IC del 95%) |
PBO
% |
RINVOQ 15 mg % Δ (IC del 95%) |
| N | 423 | 429 | 212 | 211 |
| ACR20 | ||||
| Semana 12 | 36 | 71 35 (28, 41) |
24 | 57 33 (24, 42) |
| ACR50 | ||||
| Semana 12 | 13 | 38 24 (19, 30) |
5 | 32 27 (20, 34) |
| ACR70 | ||||
| Semana 12 | 2 | 16 13 (10, 17) |
1 | 9 8 (4, 12) |
| Abreviaturas: ACR20 (o 50 o 70) = ≥20% (o ≥50% o ≥70%) de mejoría del Colegio Estadounidense de Reumatología, bDMARD = fármaco antirreumático modificador de la enfermedad biológico; RI = respondedor inadecuado; PBO = placebo Los pacientes que interrumpieron el tratamiento aleatorizado o a los que les faltaban datos en la semana de evaluación se imputaron como no respondedores en los análisis. |
||||
| Ensayo | Ensayo PsA-I DMARD-IR no biológicos |
Ensayo PsA-II bDMARD-IR |
||
| Grupo de tratamiento | PBO
(N=423) |
RINVOQ 15 mg (N=429) |
PBO
(N=212) |
RINVOQ 15 mg (N=211) |
| Número de articulaciones sensibles/dolorosas (0-68) | ||||
| Línea base | 20.0 (14.3) | 20.4 (14.7) | 25.3 (17.6) | 24.9 (17.3) |
| Semana 12 | 12.5 (13.3) | 8.8 (12.5) | 19.3 (18.5) | 12.6 (15.6) |
| Número de articulaciones inflamadas (0-66) | ||||
| Línea base | 11.0 (8.2) | 11.6 (9.3) | 12.0 (8.9) | 11.3 (8.2) |
| Semana 12 | 5.6 (7.2) | 3.5 (6.0) | 7.3 (9.4) | 4.4 (5.7) |
| Evaluación del dolor por parte del pacienteb | ||||
| Línea base | 6.1 (2.1) | 6.2 (2.1) | 6.6 (2.1) | 6.4 (2.1) |
| Semana 12 | 5.1 (2.3) | 3.8 (2.4) | 5.9 (2.3) | 4.4 (2.5) |
| Evaluación global del pacienteb | ||||
| Línea base | 6.3 (2.0) | 6.6 (2.0) | 6.8 (2.0) | 6.8 (1.9) |
| Semana 12 | 5.2 (2.2) | 3.8 (2.3) | 6.1 (2.3) | 4.5 (2.5) |
| Índice de discapacidad (HAQ-DI)c | ||||
| Línea base | 1.1 (0.6) | 1.2 (0.7) | 1.2 (0.7) | 1.1 (0.6) |
| Semana 12 | 1.0 (0.7) | 0.7 (0.6) | 1.1 (0.6) | 0.8 (0.7) |
| Evaluación global del médicob | ||||
| Línea base | 6.5 (1.6) | 6.7 (1.6) | 6.5 (1.8) | 6.5 (1.8) |
| Semana 12 | 4.3 (2.2) | 3.1 (2.0) | 5.0 (2.2) | 3.4 (2.1) |
| hsCRP (mg/L) | ||||
| Línea base | 11.5 (15.8) | 11.0 (14.9) | 10.4 (18.5) | 11.2 (18.6) |
| Semana 12 | 10.1 (15.2) | 4.2 (9.9) | 9.4 (13.4) | 4.3 (7.9) |
| Abreviaturas: ACR = Colegio Estadounidense de Reumatología; hsCRP = proteína C reactiva de alta sensibilidad; HAQ-DI = Cuestionario de Evaluación de la Salud – Índice de Discapacidad; IR = respondedor inadecuado; PBO = placebo a Los datos mostrados son la media (desviación estándar). b Escala de calificación numérica (NRS): 0 = mejor, 10 = peor c Cuestionario de Evaluación de la Salud – Índice de Discapacidad: 0 = mejor, 3 = peor; 20 preguntas; 8 categorías: vestirse y asearse, levantarse, comer, caminar, higiene, alcanzar, agarrar y actividades. |
||||
El porcentaje de pacientes que lograron una respuesta ACR20 por visita se muestra en la Figura 2.
Figura 2. Porcentaje de pacientes que logran ACR20 en el ensayo PsA-II
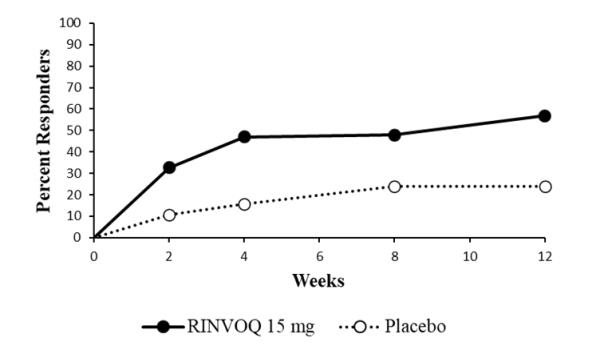
Abreviaturas: ACR20 = Colegio Estadounidense de Reumatología ≥20% de mejoría. Los pacientes que suspendieron el tratamiento aleatorizado, o a quienes les faltaban los resultados del ACR20, o que se perdieron durante el seguimiento o se retiraron del ensayo se imputaron como no respondedores.
El tratamiento con RINVOQ 15 mg produjo una mejoría en la dactilitis y la entesitis en pacientes con dactilitis o entesitis preexistentes.
El tratamiento con RINVOQ 15 mg produjo una mejoría en las manifestaciones cutáneas en pacientes con PsA. Sin embargo, RINVOQ no se ha estudiado ni está indicado para el tratamiento de la psoriasis en placas.
Respuesta de la función física
En ambos ensayos, los pacientes tratados con RINVOQ 15 mg mostraron una mejoría significativa en la función física desde el inicio en comparación con el placebo, según la evaluación del HAQ-DI en la semana 12 (Tabla 16). La diferencia media (IC del 95%) con respecto al placebo en el cambio del HAQ-DI con respecto al inicio en la semana 12 fue de -0,28 (-0,35, -0,22) en el ensayo PsA-I y de -0,21 (-0,30, -0,12) en el ensayo PsA-II.
La proporción de respondedores al HAQ-DI (≥ 0,35 de mejoría con respecto al inicio en la puntuación del HAQ-DI) en la semana 12 en el ensayo PsA-I y el ensayo PsA-II fue del 58% y el 45%, respectivamente, en los pacientes que recibieron RINVOQ 15 mg y del 33% y el 27%, respectivamente, en los pacientes que recibieron placebo.
Respuesta radiográfica
En el ensayo PsA-I, la inhibición de la progresión del daño estructural se evaluó radiográficamente y se expresó como el cambio con respecto al inicio en la puntuación total de Sharp modificada (mTSS) y sus componentes, la puntuación de erosión y la puntuación de estrechamiento del espacio articular, en la semana 24.
El tratamiento con RINVOQ 15 mg inhibió la progresión del daño articular estructural en comparación con el placebo en la semana 24 (Tabla 18). Los análisis de las puntuaciones de erosión y estrechamiento del espacio articular fueron consistentes con los resultados generales. La proporción de pacientes sin progresión radiográfica (cambio en la mTSS ≤ 0) en la semana 24 fue del 93% en los pacientes que recibieron RINVOQ 15 mg y del 89% en los pacientes que recibieron placebo.
| PBO (N=392) Media (DE) |
RINVOQ 15 mg (N=407) Media (DE) |
Diferencia estimada frente a PBO en la semana 24 (IC del 95%)a |
|
| mTSS | |||
| Línea base | 13.32 (31.2) | 13.14 (42.4) | |
| Semana 24b | 0.23 (0.07) | -0.02 (0.04) | -0.25 (-0.41, -0.09) |
| Abreviaturas: IC = intervalos de confianza; LS = mínimos cuadrados; mTSS = puntuación total de Sharp modificada; PBO = placebo; DE = desviación estándar a Medias de LS e IC del 95% basadas en un modelo de coeficientes aleatorios ajustado al valor de la mTSS ajustando por tiempo, grupo de tratamiento, uso actual de DMARD (sí/no), interacción grupo de tratamiento por tiempo, con pendientes aleatorias e intersección aleatoria. b Se presentan la tasa lineal estimada de progresión estructural en la semana 24 y los errores estándar. |
|||
Otros resultados relacionados con la salud
La calidad de vida relacionada con la salud se evaluó mediante el cuestionario SF-36. En ambos ensayos, los pacientes que recibieron RINVOQ 15 mg experimentaron una mejoría significativamente mayor con respecto al valor inicial en la puntuación resumida del componente físico en comparación con el placebo en la semana 12. También se observó una mayor mejoría en la puntuación resumida del componente mental y en los 8 dominios del SF-36 en comparación con el placebo.
Los pacientes que recibieron RINVOQ 15 mg mostraron una mayor mejoría con respecto al valor inicial en la fatiga, medida por la puntuación FACIT-F, en la semana 12 en comparación con el placebo en ambos ensayos.
14.3 Dermatitis atópica
La eficacia de RINVOQ 15 mg y 30 mg una vez al día se evaluó en tres ensayos de fase 3 aleatorizados, doble ciego y multicéntricos (AD-1, AD-2, AD-3; NCT03569293, NCT03607422 y NCT03568318, respectivamente) en un total de 2584 pacientes (de 12 años de edad o mayores). RINVOQ se evaluó en 344 pacientes pediátricos y 2240 pacientes adultos con dermatitis atópica (DA) de moderada a grave no controlada adecuadamente con medicación(es) tópica(s).
La gravedad de la enfermedad al inicio del estudio se definió mediante una puntuación de la Evaluación Global del Investigador validada (vIGA-AD) ≥ 3 en la evaluación general de la DA en una escala de gravedad de 0 a 4, una puntuación del Índice de Gravedad y Área del Eccema (EASI) ≥ 16, una afectación de la superficie corporal (SC) mínima ≥ 10 % y una puntuación media semanal de la Escala Numérica de Valoración del Prurito Peor (NRS) ≥ 4. En general, el 57 % de los pacientes eran hombres y el 69 % eran de raza blanca. La media de edad al inicio del estudio era de 34 años (entre 12 y 75 años) y el 13 % de los pacientes tenían entre 12 y menos de 18 años. Al inicio del estudio, el 49 % de los pacientes tenían una puntuación vIGA-AD de 3 (DA moderada) y el 51 % de los pacientes tenían una puntuación vIGA-AD de 4 (DA grave). La puntuación media del EASI al inicio del estudio fue de 29 y la puntuación media semanal del NRS del Prurito Peor al inicio del estudio fue de 7. Aproximadamente el 52 % de los pacientes habían estado expuestos previamente al tratamiento sistémico de la DA.
En los tres ensayos, los pacientes recibieron dosis orales de RINVOQ una vez al día de 15 mg, 30 mg o placebo correspondiente durante 16 semanas. En el ensayo AD-3, los pacientes también recibieron RINVOQ o placebo con corticosteroides tópicos (CET) concomitantes durante 16 semanas.
En los tres ensayos se evaluaron las variables principales de eficacia de la proporción de pacientes con una puntuación vIGA-AD de 0 (limpia) o 1 (casi limpia) con una mejoría de al menos 2 puntos y la proporción de pacientes con EASI-75 (mejoría de al menos el 75 % en la puntuación EASI con respecto al valor inicial) en la semana 16. Las variables secundarias de eficacia incluyeron EASI-90 y EASI-100 en la semana 16, y la proporción de pacientes con reducción del picor (≥ 4 puntos de mejoría con respecto al valor inicial en el NRS del Prurito Peor) en las semanas 1, 4 y 16. En los ensayos AD-1 y AD-2, la proporción de pacientes con reducción del dolor (≥ 4 puntos de mejoría en el NRS del Dolor Cutáneo de la Escala de Síntomas de la Dermatitis Atópica [ADerm-SS]) desde el inicio hasta la semana 16 fue una variable secundaria de eficacia.
Respuesta clínica
Ensayos de monoterapia (AD-1 y AD-2)
Los resultados de los ensayos de monoterapia con RINVOQ (AD-1 y AD-2) se presentan en la Tabla 19. La Figura 3 presenta la proporción de pacientes con ≥ 4 puntos de mejoría en el NRS del Prurito Peor en las semanas 1, 4 y 16 para los ensayos AD-1 y AD-2.
| Ensayo AD-1 | Ensayo AD-2 | |||||
| PBO | RINVOQ 15 mg |
RINVOQ 30 mg |
PBO | RINVOQ 15 mg |
RINVOQ 30 mg |
|
| Número de pacientes aleatorizados |
281 | 281 | 285 | 278 | 276 | 282 |
| vIGA-AD 0/1a,b | 8% | 48% | 62% | 5% | 39% | 52% |
| Diferencia con respecto a PBO (IC del 95%) |
40% (33%, 46%) |
54% (47%, 60%) |
34% (28%, 40%) |
47% (41%, 54%) |
||
| EASI-75a | 16% | 70% | 80% | 13% | 60% | 73% |
| Diferencia con respecto a PBO (IC del 95%) |
53% (46%, 60%) |
63% (57%, 70%) |
47% (40%, 54%) |
60% (53%, 66%) |
||
| EASI-90a | 8% | 53% | 66% | 5% | 42% | 58% |
| Diferencia con respecto a PBO (IC del 95%) |
45% (39%, 52%) |
58% (51%, 64%) |
37% (31%, 43%) |
53% (47%, 59%) |
||
| EASI-100a | 2% | 17% | 27% | 1% | 14% | 19% |
| Diferencia con respecto a PBO (IC del 95%) |
15% (10%, 20%) |
25% (20%, 31%) |
13% (9%, 18%) |
18% (13%, 23%) |
||
| Número de pacientes con una puntuación inicial en la escala NRS de prurito intenso Prurito ≥ 4 | 272 | 274 | 280 | 274 | 270 | 280 |
| ≥ 4 puntos de mejora en la escala NRS de prurito intensoc | 12% | 52% | 60% | 9% | 42% | 60% |
| Diferencia con respecto a PBO (IC del 95%) |
40% (33%, 48%) |
48% (41%, 55%) |
33% (26%, 39%) |
50% (44%, 57%) |
||
| Número de pacientes con una puntuación inicial en la escala NRS de dolor cutáneo de ADerm-SS NRS ≥ 4 | 233 | 237 | 249 | 247 | 237 | 238 |
| ≥ 4 puntos de mejora en la escala NRS de dolor cutáneo de ADerm-SSd | 15% | 54% | 63% | 13% | 49% | 65% |
| Diferencia con respecto a PBO (IC del 95%) |
39% (31%, 47%) |
49% (41%, 56%) |
36% (28%, 43%) |
52% (44%, 59%) |
||
| Abreviaturas: ADerm-SS = Escala de síntomas de la dermatitis atópica; PBO = placebo a Según el número de pacientes aleatorizados b Se definió como respondedor a un paciente con vIGA-AD de 0 o 1 (“limpio” o “casi limpio”) con una reducción de ≥ 2 puntos en una escala ordinal de 0 a 4 c Según el número de pacientes cuya puntuación inicial en la escala NRS de prurito intenso es ≥ 4 d Según el número de pacientes cuya puntuación inicial en la escala NRS de dolor cutáneo de ADerm-SS es ≥ 4 |
||||||
Figura 3. Proporción de pacientes con DA de moderada a grave con una mejoría de ≥4 puntos en la escala NRS del peor prurito en los ensayos de monoterapia
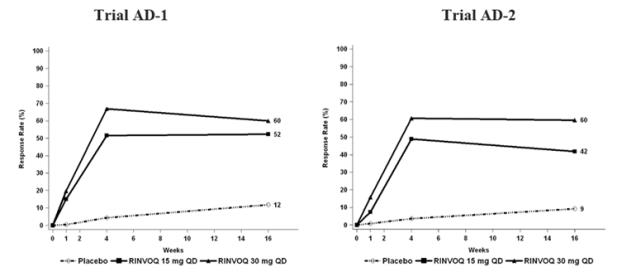
El examen de la edad, el sexo, la raza, el peso y el tratamiento sistémico previo con inmunosupresores no identificó diferencias en la respuesta a RINVOQ entre estos subgrupos en los ensayos AD-1 y AD-2.
Ensayo con TCS concomitante (AD-3)
Los resultados del ensayo de RINVOQ con TCS concomitante (AD-3) se presentan en la Tabla 20. La Figura 4 presenta la proporción de pacientes con una mejoría de ≥ 4 puntos en la escala NRS del peor prurito en las semanas 1, 4 y 16 para el ensayo AD-3.
| Ensayo AD-3 | |||
| PBO + TCS | RINVOQ 15 mg + TCS |
RINVOQ 30 mg + TCS |
|
| Número de pacientes aleatorizados |
304 | 300 | 297 |
| vIGA-AD 0/1a,b | 11% | 40% | 59% |
| Diferencia con respecto a PBO (IC del 95%) |
29% (22%, 35%) |
48% (41%, 54%) |
|
| EASI-75a, | 26% | 65% | 77% |
| Diferencia con respecto a PBO (IC del 95%) |
38% (31%, 45%) |
51% (44%, 57%) |
|
| EASI-90a | 13% | 43% | 63% |
| Diferencia con respecto a PBO (IC del 95%) |
30% (23%, 36%) |
50% (43%, 56%) |
|
| EASI-100a | 1% | 12% | 23% |
| Diferencia con respecto a PBO (IC del 95%) |
11% (7%, 14%) |
21% (16%, 26%) |
|
| Número de pacientes con una puntuación inicial en la escala NRS del peor prurito ≥ 4 | 294 | 288 | 291 |
| ≥ 4 puntos de mejora en la escala NRS del peor pruritoc | 15% | 52% | 64% |
| Diferencia con respecto a PBO (IC del 95%) |
37% (30%, 44%) |
49% (42%, 56%) |
|
| Abreviaturas: PBO = placebo a Basado en el número de pacientes aleatorizados b Se definió como respondedor a un paciente con un vIGA-AD de 0 o 1 (“limpio” o “casi limpio”) con una reducción de ≥ 2 puntos en una escala ordinal de 0-4 c Basado en el número de pacientes cuya puntuación inicial en la escala NRS del peor prurito es ≥ 4 |
|||
Figura 4. Proporción de pacientes con DA de moderada a grave con una mejoría de ≥4 puntos en la calificación numérica del prurito más intenso (Worst Pruritus NRS) en el ensayo con TCS concomitantes
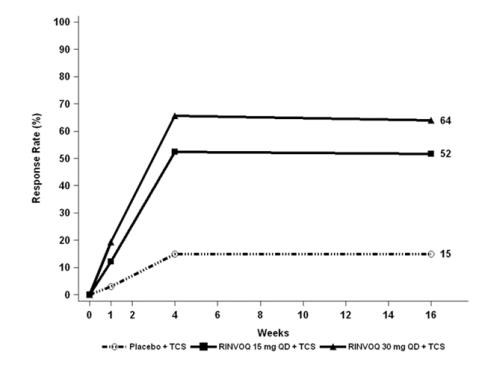
El examen de la edad, el sexo, la raza, el peso y el tratamiento sistémico previo con inmunodepresores no identificó diferencias en la respuesta a RINVOQ entre estos subgrupos en el ensayo AD-3.
Población de pacientes pediátricos
Los resultados de eficacia de los ensayos de RINVOQ en monoterapia (AD-1 y AD-2) y del ensayo de RINVOQ con TCS concomitantes (AD-3) en la semana 16 para pacientes pediátricos de 12 años o mayores se presentan en la Tabla 21 y la Tabla 22, respectivamente.
| Ensayo AD-1 | Ensayo AD-2 | |||||
| PBO | RINVOQ 15 mg |
RINVOQ 30 mg |
PBO | RINVOQ 15 mg |
RINVOQ 30 mg |
|
| Número de pacientes pediátricos aleatorizados | 40 | 42 | 42 | 36 | 33 | 35 |
| vIGA-AD 0/1a,b | 8% | 38% | 69% | 3% | 42% | 62% |
| Diferencia con respecto a PBO (IC del 95%) |
31% (14%, 47%) |
62% (45%, 78%) |
40% (22%, 57%) |
60% (42%, 77%) |
||
| EASI-75a | 8% | 71% | 83% | 14% | 67% | 74% |
| Diferencia con respecto a PBO (IC del 95%) |
63% (47%, 79%) |
75% (61%, 89%) |
53% (33%, 72%) |
61% (42%, 79%) |
||
| Número de pacientes pediátricos con una puntuación inicial en la calificación numérica del prurito más intenso (Worst Pruritus NRS) ≥ 4 | 39 | 40 | 42 | 36 | 30 | 34 |
| ≥ 4 puntos de mejora en la calificación numérica del prurito más intenso (Worst Pruritus NRS)c | 15% | 45% | 55% | 3% | 33% | 50% |
| Diferencia con respecto a PBO (IC del 95%) |
30% (10%, 49%) |
39% (21%, 58%) |
31% (13%, 48%) |
47% (30%, 65%) |
||
| Abreviaturas: PBO = placebo a Según el número de pacientes pediátricos aleatorizados b Se definió como respondedor a un paciente con un vIGA-AD de 0 o 1 (“limpio” o “casi limpio”) con una reducción de ≥ 2 puntos en una escala ordinal de 0 a 4 c Según el número de pacientes pediátricos cuya calificación numérica del prurito más intenso (Worst Pruritus NRS) inicial es ≥ 4 |
||||||
| Ensayo AD-3 | |||
| PBO + TCS | RINVOQ 15 mg + TCS |
RINVOQ 30 mg + TCS |
|
| Número de pacientes pediátricos aleatorizados | 40 | 39 | 37 |
| vIGA-AD 0/1a,b | 8% | 31% | 65% |
| Diferencia con respecto a PBO (IC del 95%) |
23% (7%, 40%) |
57% (40%, 75%) |
|
| EASI-75a | 30% | 56% | 76% |
| Diferencia con respecto a PBO (IC del 95%) |
26% (5%, 47%) |
46% (26%, 65%) |
|
| Número de pacientes pediátricos con una puntuación inicial en la escala NRS del peor prurito ≥ 4 | 38 | 36 | 33 |
| ≥ 4 puntos de mejora en la escala NRS del peor pruritoc | 13% | 42% | 55% |
| Diferencia con respecto a PBO (IC del 95%) |
29% (9%, 48%) |
41% (21%, 61%) |
|
| Abreviaturas: PBO = placebo a Según el número de pacientes pediátricos aleatorizados b Se definió como respondedor a un paciente con un vIGA-AD de 0 o 1 (“limpia” o “casi limpia”) con una reducción de ≥ 2 puntos en una escala ordinal de 0 a 4 c Según el número de pacientes pediátricos cuya puntuación inicial en la escala NRS del peor prurito es ≥ 4 |
|||
14.4 Colitis ulcerosa
Ensayos de inducción (Estudio UC-1 y Estudio UC-2)
En dos ensayos de inducción idénticos (UC-1; NCT02819635 y UC-2; NCT03653026), se aleatorizó a los pacientes en una proporción 2:1 para que recibieran 45 mg de RINVOQ una vez al día o placebo durante 8 semanas. Se analizó un total de 988 pacientes en los dos ensayos. Estos ensayos incluyeron a pacientes adultos con colitis ulcerosa activa de moderada a grave que tuvieron una respuesta inadecuada, pérdida de la respuesta o intolerancia a los aminosalicilatos orales, corticosteroides, inmunosupresores o terapia biológica. Los pacientes incluidos podían usar dosis estables de aminosalicilatos orales, metotrexato, antibióticos relacionados con la colitis ulcerosa o corticosteroides orales (hasta 30 mg/día de prednisona o equivalente). Al inicio, el 38 % de los pacientes recibían corticosteroides y el 68 % de los pacientes recibían aminosalicilatos. Se prohibieron las terapias biológicas concomitantes, la azatioprina, la 6-mercaptopurina y los corticosteroides intravenosos o rectales. Un total del 51 % de los pacientes habían fracasado previamente al tratamiento o eran intolerantes a al menos una terapia biológica. RINVOQ está indicado para pacientes que tienen una respuesta inadecuada o intolerancia a uno o más bloqueadores del TNF [ver Indicaciones y uso (1.4)].
La actividad de la enfermedad se evaluó mediante la puntuación de Mayo modificada (mMS), una puntuación de Mayo de 3 componentes (0-9) que consta de las siguientes subpuntuaciones (de 0 a 3 para cada subpuntuación): frecuencia de deposiciones (SFS), sangrado rectal (RBS) y hallazgos en la puntuación de endoscopia leída centralmente (ES). Una ES de 2 se definió por eritema marcado, falta de patrón vascular, cualquier friabilidad o erosiones, y una puntuación de 3 se definió por sangrado espontáneo y ulceración. Los pacientes incluidos tenían una mMS entre 5 y 9 con una ES de 2 o 3; al inicio, la mediana de la mMS fue de 7, con un 61 % de los pacientes con una mMS inicial de 5 a 7 y un 39 % con una mMS de 8 a 9.
Al inicio, el 39 % y el 37 % de los pacientes recibían corticosteroides, el 1 % y el 1 % de los pacientes recibían metotrexato, y el 68 % y el 69 % de los pacientes recibían aminosalicilatos en los estudios UC-1 y UC-2, respectivamente. La actividad de la enfermedad de los pacientes fue moderada (mMS ≤7) en el 61 % y el 60 % de los pacientes y grave (mMS >7) en el 39 % y el 40 % de los pacientes en los estudios UC-1 y UC-2, respectivamente.
El criterio de valoración principal fue la remisión clínica definida mediante la mMS en la semana 8. Los criterios de valoración secundarios incluyeron la respuesta clínica, la mejoría endoscópica y la mejoría histológica de la mucosa endoscópica (ver Tabla 23 y Tabla 24).
| Estudio UC-1 | |||
| Criterio de valoración | Placebo | RINVOQ 45 mg una vez al día |
Diferencia de tratamiento frente a placebo (IC del 95 %) |
| Remisión clínicaa | |||
| Población total | N=154 5 % |
N=319 26 % |
22 %b (16, 27) |
| Fracaso biológico previoc | N=78 < 1 % |
N=168 18 % |
|
| Sin fracaso biológico previo | N=76 9 % |
N=151 35 % |
|
| Respuesta clínicad | |||
| Población total | N=154 27 % |
N=319 73 % |
46 %b (38, 54) |
| Fracaso biológico previoc | N=78 13 % |
N=168 64 % |
|
| Sin fracaso biológico previo | N=76 42 % |
N=151 82 % |
|
| Mejoría endoscópicae | |||
| Población total | N=154 7 % |
N=319 36 % |
29 %b (23, 36) |
| Fracaso biológico previoc | N=78 2 % |
N=168 27 % |
|
| Sin fracaso biológico previo | N=76 13 % |
N=151 47 % |
|
| Mejoría histológica de la mucosa endoscópicaf | |||
| Población total | N=154 7 % |
N=319 30 % |
24 %b (17, 30) |
| Prior biologic failurec | N=78 1% |
N=168 23% |
|
| Sin fallo biológico previo | N=76 12% |
N=151 38% |
|
| a Según la mMS: SFS ≤1 y no mayor que el valor inicial, RBS = 0, ES de ≤ 1 sin friabilidad b p <0.001, diferencia de tratamiento ajustada (IC del 95%) basada en el método de Cochran-Mantel-Haenszel ajustado para los factores de estratificación de aleatorización c El fallo biológico previo incluye una respuesta inadecuada, la pérdida de respuesta o la intolerancia a uno o más tratamientos biológicos para la colitis ulcerosa. d Según la mMS: disminución ≥ 2 puntos y ≥ 30% con respecto al valor inicial y una disminución de la RBS ≥ 1 con respecto al valor inicial o una RBS absoluta ≤1 e ES ≤ 1 sin friabilidad f ES ≤1 sin friabilidad y puntuación de Geboes ≤ 3,1 (lo que indica infiltración de neutrófilos en <5% de las criptas, sin destrucción de las criptas y sin erosiones, ulceraciones ni tejido de granulación) |
|||
| Estudio UC-2 | |||
| Criterio de valoración | Placebo | RINVOQ 45 mg una vez al día |
Diferencia de tratamiento frente a placebo (95% CI) |
| Remisión clínicaa | |||
| Población total | N=174 4% |
N=341 33% |
29%b (23, 35) |
| Fallo biológico previoc | N=89 2% |
N=173 30% |
|
| Sin fallo biológico previo | N=85 6% |
N=168 38% |
|
| Respuesta clínicad | |||
| Población total | N=174 25% |
N=341 74% |
49%b (42, 57) |
| Fallo biológico previoc | N=89 19% |
N=173 69% |
|
| Sin fallo biológico previo | N=85 32% |
N=168 80% |
|
| Mejoría endoscópicae | |||
| Población total | N=174 8% |
N=341 44% |
35%b (29, 42) |
| Fallo biológico previoc | N=89 5% |
N=173 37% |
|
| Sin fallo biológico previo | N=85 12% |
N=168 51% |
|
| Mejoría histológica endoscópica de la mucosaf | |||
| Población total | N=174 6% |
N=341 37% |
30%b (24, 36) |
| Fallo biológico previoc | N=89 5% |
N=173 31% |
|
| Sin fallo biológico previo | N=85 7% |
N=168 43% |
|
| a Según la mMS: SFS ≤ 1 y no superior al valor inicial, RBS = 0, ES de ≤ 1 sin friabilidad b p <0,001, diferencia de tratamiento ajustada (IC del 95%) basada en el método de Cochran-Mantel-Haenszel ajustado para los factores de estratificación de aleatorización c El fallo biológico previo incluye una respuesta inadecuada, la pérdida de respuesta o la intolerancia a uno o más tratamientos biológicos para la colitis ulcerosa. d Según la mMS: disminución ≥ 2 puntos y ≥ 30% con respecto al valor inicial y una disminución de la RBS ≥ 1 con respecto al valor inicial o una RBS absoluta ≤1 e ES ≤ 1 sin friabilidad f ES ≤1 sin friabilidad y puntuación de Geboes ≤ 3,1 (lo que indica infiltración de neutrófilos en <5% de las criptas, sin destrucción de las criptas y sin erosiones, úlceras o tejido de granulación) |
|||
Los estudios UC-1 y UC-2 no se diseñaron para evaluar la relación entre la mejoría histológica endoscópica de la mucosa en la semana 8 y la progresión de la enfermedad y los resultados a largo plazo.
Puntuaciones parciales de sangrado rectal y frecuencia de deposiciones
El inicio de la respuesta clínica se evaluó mediante la SFS y la RBS (puntuación parcial modificada de Mayo [pmMS]). La respuesta inicial se definió como una disminución de ≥1 punto y ≥30 % con respecto al valor inicial en la pmMS y una disminución de la RBS ≥1 o una RBS absoluta ≤1. El inicio de la respuesta se produjo ya en la semana 2 en una mayor proporción de pacientes tratados con RINVOQ 45 mg una vez al día en comparación con el placebo.
Evaluación endoscópica e histológica
La normalización del aspecto endoscópico de la mucosa (remisión endoscópica) se definió como una ES de 0. En la semana 8, una mayor proporción de pacientes tratados con RINVOQ 45 mg una vez al día en comparación con el placebo logró la remisión endoscópica (UC-1: 14 % frente a 1 %, UC-2: 18 % frente a 2 %). La remisión endoscópica con una puntuación histológica de Geboes < 2,0 (lo que indica que no hay neutrófilos en las criptas ni en la lámina propia y que no hay aumento de eosinófilos, ni destrucción de las criptas, ni erosiones, úlceras o tejido de granulación) se logró en una mayor proporción de pacientes tratados con RINVOQ 45 mg una vez al día en comparación con el placebo en la semana 8 (UC-1: 11 % frente a 1 %, UC-2: 13 % frente a 2 %).
Dolor abdominal y urgencia para defecar
Una mayor proporción de pacientes tratados con RINVOQ 45 mg una vez al día en comparación con el placebo no presentó dolor abdominal (UC-1: 47 % frente a 23 %, UC-2: 54 % frente a 24 %) ni urgencia para defecar (UC-1: 48 % frente a 21 %, UC-2: 54 % frente a 26 %) en la semana 8.
Fatiga
En los estudios UC-1 y UC-2, los pacientes tratados con RINVOQ experimentaron una mejoría clínicamente significativa de la fatiga, según la evaluación del cambio con respecto al valor inicial en la puntuación del FACIT-F, en la semana 8, en comparación con los pacientes tratados con placebo. No se ha establecido el efecto de RINVOQ para mejorar la fatiga después de 8 semanas de inducción.
Estudio de mantenimiento UC-3
En el estudio UC-3 (NCT02819635), un total de 451 pacientes que recibieron RINVOQ 45 mg una vez al día en el estudio UC-1, UC-2 o UC-4 y lograron una respuesta clínica fueron reasignados aleatoriamente para recibir RINVOQ 15 mg, 30 mg o placebo una vez al día durante un máximo de 52 semanas.
El criterio de valoración principal fue la remisión clínica definida mediante la mMS en la semana 52. Los criterios de valoración secundarios incluyeron la remisión clínica sin corticosteroides, la mejoría endoscópica y la mejoría histológica endoscópica de la mucosa (véase la Tabla 25).
| Criterio de valoración | Placebo |
RINVOQ 15 mg Una vez al día |
Diferencia de tratamiento 15 mg frente a placebo (IC del 95 %) |
RINVOQ 30 mg Una vez al día |
Diferencia de tratamiento 30 mg frente a placebo (IC del 95 %) |
| Remisión clínicaa | |||||
| Población total | N=149 12% |
N=148 42% |
31%b (22, 40) |
N=154 52% |
39%b (30, 48) |
| Fallo terapéutico previo con productos biológicosc | N=81 7% |
N=71 41% |
N=73 49% |
||
| Sin fallo terapéutico previo con productos biológicos | N=68 18% |
N=77 44% |
N=81 54% |
||
| Remisión clínica sin corticosteroidesd | |||||
| Población total | N=54 22% |
N=47 57% |
35%b (18, 53) |
N=58 68% |
45%b (29, 62) |
| Fallo terapéutico previo con productos biológicosc | N=22 14% |
N=17 71% |
N=20 73% |
||
| Sin fallo terapéutico previo con productos biológicos | N=32 28% |
N=30 49% |
N=38 65% |
||
| Mejoría endoscópicae | |||||
| Población total | N=149 14% |
N=148 49% |
34%b (25, 44) |
N=154 62% |
46%b (37, 56) |
| Prior biologic failurec | N=81 8% |
N=71 43% |
N=73 56% |
||
| Sin fallo biológico previo | N=68 22% |
N=77 54% |
N=81 67% |
||
| Mejoría Mucosa Endoscópica Histológicaf | |||||
| Población total | N=149 12% |
N=148 35% |
24%b (15, 33) |
N=154 50% |
37%b (28, 47) |
| Prior biologic failurec | N=81 5% |
N=71 33% |
N=73 48% |
||
| Sin fallo biológico previo | N=68 20% |
N=77 37% |
N=81 52% |
||
| a Per mMS: SFS ≤1 and not greater than baseline, RBS = 0, ES ≤1 without friability b p <0.001, adjusted treatment difference (95% CI) based on Cochran-Mantel-Haenszel method adjusted for randomization stratification factors c El fallo biológico previo incluye una respuesta inadecuada, pérdida de respuesta o intolerancia a uno o más tratamientos biológicos para la colitis ulcerosa. d Clinical remission per mMS at Week 52 and corticosteroid free for ≥90 days immediately preceding Week 52 among patients who achieved clinical remission at the end of the induction treatment e ES ≤ 1 without friability f ES ≤1 without friability and Geboes score ≤ 3.1 (indicating neutrophil infiltration in <5% of crypts, no crypt destruction and no erosions, ulcerations or granulation tissue) |
|||||
La relación entre la mejora de la mucosa histológica endoscópica en la semana 52 y la progresión de la enfermedad y los resultados a largo plazo después de la semana 52 no se evaluó en el Estudio UC-3.
Evaluación endoscópica y histológica
La normalización de la apariencia endoscópica de la mucosa (remisión endoscópica) se definió como ES de 0. En UC-3, una mayor proporción de pacientes tratados con RINVOQ 15 mg y 30 mg una vez al día en comparación con placebo lograron remisión endoscópica en la semana 52 (24% y 26% vs 6%). La remisión endoscópica con un puntaje histológico de Geboes < 2.0 se logró en una mayor proporción de pacientes tratados con RINVOQ 15 mg y 30 mg una vez al día en comparación con placebo en la semana 52 (18% y 19% vs 5%).
Dolor abdominal y urgencia intestinal
En la semana 52, una mayor proporción de pacientes tratados con RINVOQ 15 mg y 30 mg una vez al día en comparación con placebo no tuvieron dolor abdominal (46%, 55% y 21%, respectivamente) y no tuvieron urgencia intestinal (56%, 64% y 17%, respectivamente).
14.5 Enfermedad de Crohn
Ensayos de inducción (Estudios CD-1 y CD-2)
En dos ensayos de inducción, CD-1 (NCT03345836) y CD-2 (NCT03345849), los pacientes fueron aleatorizados en una proporción 2:1 para recibir RINVOQ 45 mg o placebo una vez al día durante 12 semanas. La eficacia se evaluó en una población de 857 pacientes (419 pacientes en CD-1 y 438 pacientes en CD-2) con enfermedad de Crohn (CD) moderada a gravemente activa, con un puntaje de índice de actividad de la enfermedad de Crohn (CDAI) basal de al menos 220 y un puntaje endoscópico simple revisado centralmente para la enfermedad de Crohn (SES-CD) de ≥ 6, o ≥ 4 para la enfermedad ileal aislada, excluyendo el componente de estrechamiento. En CD-1, todos los pacientes tuvieron una respuesta inadecuada o fueron intolerantes al tratamiento con una o más terapias biológicas (fracaso biológico previo). En CD-2, el 45% (197/438) de los pacientes tuvieron una respuesta inadecuada o fueron intolerantes al tratamiento con una o más terapias biológicas (fracaso biológico previo). Los pacientes inscritos en ambos estudios pudieron usar dosis estables de antibióticos, aminosalicilatos o metotrexato relacionados con la CD. Se permitieron corticoides concomitantes (hasta 30 mg/día de prednisona o equivalente) en la inscripción; el tapering se inició en la Semana 4.
En CD-1, los pacientes tuvieron una media de edad de 37 años (rango 18 a 74 años); el 46% eran mujeres; y el 72% se identificaron como blancos, el 21% como asiáticos, el 6% como negros o afroamericanos, el 0.5% como indios americanos o nativos de Alaska, y el 0.5% como grupos raciales múltiples. En CD-2, los pacientes tuvieron una media de edad de 40 años (rango de 18 a 74 años); el 45% eran mujeres; el 74% se identificaron como blancos, el 20% como asiáticos, el 4% como negros o afroamericanos, y el 2% como grupos raciales múltiples.
Al inicio, el 36% y el 37% de los pacientes recibieron corticoides, el 7% y el 3% de los pacientes recibieron metotrexato, el 15% y el 24% de los pacientes recibieron aminosalicilatos, y el 2% y el 1% de los pacientes recibieron antibióticos relacionados con la CD en CD-1 y CD-2, respectivamente.
Los puntos finales co-primarios fueron la proporción de pacientes que lograron remisión clínica (por CDAI) en la semana 12 y la proporción de pacientes que lograron respuesta endoscópica (por SES-CD) en la semana 12. Los puntos finales de eficacia secundarios incluyeron respuesta clínica, remisión libre deocorticoides y remisión endoscópica (véase Tabla 26 y Tabla 27).
| CD-1 | |||
| Punto final | Placebo |
RINVOQ 45 mg Una Vez al Día |
Diferencia de Tratamiento vs Placebo (95% CI) |
| Puntos Finales Co-Primarios en la Semana 12 | |||
| Remisión clínicaa | N = 146 18% |
N = 273 36% |
17% (9, 25)* |
| Respuesta endoscópicab | N = 146 3% |
N = 273 34% |
30% (24, 36)* |
| Puntos Finales Adicionales en la Semana 12 | |||
| Respuesta clínica (CR-100)c | N = 146 31% |
N = 273 54% |
22% (13, 31)* |
| Remisión clínica libre de corticosteroides en pacientes que tenían corticosteroides al inicioa,d | N = 53 11% |
N = 96 30% |
17% (5, 29)** |
| Remisión endoscópicae | N = 146 3% |
N = 273 19% |
15% (10, 21)* |
| * p < 0.001, diferencia de tratamiento ajustada (IC del 95%) basada en el método de Cochran-Mantel-Haenszel ajustado por factores de estratificación de aleatorización ** p < 0.01, diferencia de tratamiento ajustada (IC del 95%) basada en el método de Cochran-Mantel-Haenszel ajustado por factores de estratificación de aleatorización a CDAI < 150 b Disminución en el SES-CD > 50% desde el inicio del estudio de inducción (o para pacientes con un SES-CD de 4 al inicio del estudio de inducción, al menos una reducción de 2 puntos desde el inicio del estudio de inducción) c Disminución de al menos 100 puntos en el CDAI desde el inicio d Interrupción de corticosteroides y logro de la remisión clínica entre pacientes con corticosteroides al inicio del estudio e SES-CD ≤ 4 y ninguna subpuntuación individual >1 en ninguna variable individual |
|||
| CD-2 | |||
| Criterio de valoración | Placebo |
RINVOQ 45 mg una vez al día |
Diferencia de tratamiento frente a placebo (IC del 95%) |
| Criterios de valoración coprimarios en la semana 12 | |||
| Remisión clínicaa | |||
| Población total | N=143 23% |
N=295 46% |
24% (15, 32)* |
| Fallo biológico previo | N=62 7% |
N=135 42% |
|
| Sin fallo biológico previo | N=81 35% |
N=160 50% |
|
| Respuesta endoscópicab | |||
| Población total | N=143 13% |
N=295 46% |
33% (26, 41)* |
| Fallo biológico previo | N=62 10% |
N=135 39% |
|
| Sin fallo biológico previo | N=81 15% |
N=160 51% |
|
| Criterios de valoración adicionales en la semana 12 | |||
| Respuesta clínica (CR-100)c | |||
| Población total | N=143 40% |
N=295 64% |
24% (15, 33)* |
| Fallo biológico previo | N=62 25% |
N=135 66% |
|
| Sin fallo biológico previo | N=81 52% |
N=160 62% |
|
| Remisión clínica libre de corticosteroides en pacientes que recibían CS al inicioa,d | |||
| Población total | N=54 13% |
N=108 40% |
27% (14, 39)* |
| Fallo biológico previo | N=29 7% |
N=60 35% |
|
| Sin fallo biológico previo | N=25 20% |
N=48 46% |
|
| Remisión endoscópicae |
|||
| Población total | N=143 8% |
N=295 30% |
22% (16, 29)* |
| Fallo biológico previo | N=62 3% |
N=135 22% |
|
| Sin fallo biológico previo | N=81 11% |
N=160 37% |
|
| * p < 0,001, diferencia de tratamiento ajustada (IC del 95%) basada en el método de Cochran-Mantel-Haenszel ajustado para los factores de estratificación de aleatorización a CDAI < 150 b Disminución en el SES-CD > 50% con respecto al inicio del estudio de inducción (o para pacientes con un SES-CD de 4 al inicio del estudio de inducción, al menos una reducción de 2 puntos con respecto al inicio del estudio de inducción) c Disminución de al menos 100 puntos en el CDAI con respecto al inicio d Interrupción de los corticosteroides y logro de la remisión clínica entre los pacientes que recibían corticosteroides al inicio e SES-CD ≤ 4 y ninguna subpuntuación individual >1 en ninguna variable individual |
|||
El inicio de la respuesta clínica según el CDAI se observó ya a las dos semanas en los estudios CD-1 y CD-2, con una mayor proporción de pacientes que lograron una respuesta clínica en la semana 2 en los pacientes tratados con RINVOQ en comparación con el placebo.
En los estudios CD-1 y CD-2, se observaron reducciones en la frecuencia de las deposiciones y el dolor abdominal en una mayor proporción de pacientes tratados con el régimen de inducción de RINVOQ 45 mg en comparación con el placebo en la semana 12.
En los estudios CD-1 y CD-2, los pacientes tratados con RINVOQ experimentaron una mejoría clínicamente significativa en la fatiga, evaluada mediante el cambio con respecto al valor inicial en la puntuación FACIT-F, en la semana 12, en comparación con los pacientes tratados con placebo. No se ha establecido el efecto de RINVOQ para mejorar la fatiga después de 12 semanas de inducción.
Estudio de mantenimiento (CD-3)
El análisis de eficacia para CD-3 (NCT03345823) evaluó a 343 pacientes que respondieron a 12 semanas de tratamiento de inducción con 45 mg de RINVOQ una vez al día. Los pacientes fueron re-aleatorizados para recibir un régimen de mantenimiento de 15 mg o 30 mg de RINVOQ una vez al día o placebo durante 52 semanas, lo que representa un total de al menos 64 semanas de tratamiento.
Los criterios de valoración co-primarios de remisión clínica (según el CDAI) y respuesta endoscópica (según el SES-CD) se evaluaron en la semana 52. Los criterios de valoración secundarios de eficacia incluyeron la remisión clínica libre de corticosteroides, el mantenimiento de la remisión clínica, la remisión endoscópica y el logro de la remisión clínica y endoscópica, en la semana 52 (véase la Tabla 28).
| Criterio de valoración |
Placebo+ | RINVOQ 15 mg Una vez al día |
RINVOQ 30 mg una vez al día |
Diferencia de tratamiento 15 mg frente a placebo (IC del 95%) |
Diferencia de tratamiento 30 mg frente a placebo (IC del 95%) |
| Criterios de valoración co-primarios | |||||
| Remisión clínicaa | |||||
| Población total | N=111 14% |
N=113 42% |
N=119 55% |
29% (18, 39)* |
40% (29, 51)* |
| Fallo biológico previo | N=87 13% |
N=85 35% |
N=90 54% |
||
| Sin fallo biológico previo |
N=24 21% |
N=28 61% |
N=29 55% |
||
| Respuesta endoscópicab | |||||
| Población total | N=111 7% |
N=113 28% |
N=119 41% |
22% (13, 32)* |
34% (25, 44)* |
| Fallo biológico previo | N=87 5% |
N=85 22% |
N=90 42% |
||
| Sin fallo biológico previo |
N=24 17% |
N=28 45% |
N=29 38% |
||
| Criterios de valoración adicionales | |||||
| Remisión clínica libre de corticosteroidesc |
|||||
| Población total | N=111 14% |
N=113 42% |
N=119 53% |
29% (18, 39)* |
38% (27, 49)* |
| Fallo biológico previo | N=87 13% |
N=85 35% |
N=90 52% |
||
| Sin fallo biológico previo |
N=24 21% |
N=28 61% |
N=29 55% |
||
| Mantenimiento de la remisión clínicad | |||||
| Población total | N=73 22% |
N=72 51% |
N=79 67% |
32% (18, 46)* |
43% (29, 57)* |
| Fracaso biológico previo | N=56 20% |
N=50 46% |
N=55 69% |
||
| Sin fracaso biológico previo |
N=17 29% |
N=22 64% |
N=24 63% |
||
| Remisión endoscópicae | |||||
| Población total | N=111 5% |
N=113 19% |
N=119 30% |
14% (6, 22)* |
25% (15, 34)* |
| Fracaso biológico previo | N=87 3% |
N=85 14% |
N=90 30% |
||
| Sin fracaso biológico previo |
N=24 13% |
N=28 32% |
N=29 31% |
||
| Remisión clínica y endoscópica | |||||
| Población total | N=111 4% |
N=113 16% |
N=119 26% |
13% (6, 21)* |
22% (14, 31)* |
| Fracaso biológico previo | N=87 2% |
N=85 13% |
N=90 26% |
||
| Sin fracaso biológico previo |
N=24 8% |
N=28 25% |
N=29 28% |
||
| + El grupo placebo consistió en pacientes que lograron una respuesta clínica según CDAI con RINVOQ 45 mg al final del estudio de inducción y fueron asignados al azar para recibir placebo al comienzo del tratamiento de mantenimiento. * p < 0.001, diferencia de tratamiento ajustada (IC del 95%) según el método de Cochran-Mantel-Haenszel ajustado por factores de estratificación de aleatorización a CDAI < 150 b Disminución en SES-CD > 50% desde el inicio del estudio de inducción (o para pacientes con un SES-CD de 4 al inicio del estudio de inducción, al menos una reducción de 2 puntos desde el inicio del estudio de inducción) c Sin corticosteroides durante 90 días antes de la semana 52 y logro de la remisión clínica. Entre el subconjunto de pacientes que recibían corticosteroides al inicio de la inducción, el 48 % (N=44) en el grupo de RINVOQ 15 mg, el 44 % (N=45) en el grupo de RINVOQ 30 mg y el 7 % (N=46) en el grupo placebo no recibieron corticosteroides durante 90 días antes de la semana 52 y estaban en remisión clínica. d Definido como el logro de la remisión clínica en la semana 52 en pacientes que lograron la remisión clínica al inicio del estudio de mantenimiento. e SES-CD ≤ 4 y ninguna subpuntuación > 1 en ninguna variable individual |
|||||
En la semana 52, se observaron reducciones en la frecuencia de las heces y el dolor abdominal en una mayor proporción de pacientes tratados con RINVOQ 15 mg y 30 mg en comparación con el placebo.
14.6 Espondilitis anquilosante
La eficacia y seguridad de RINVOQ 15 mg una vez al día se evaluaron en dos ensayos aleatorizados, doble ciego, multicéntricos y controlados con placebo en pacientes de 18 años o mayores con espondilitis anquilosante activa según el Índice de Actividad de la Enfermedad de Bath para la Espondilitis Anquilosante (BASDAI) ≥4 y una puntuación en la Evaluación del Paciente del Dolor Lumbar Total ≥4.
El ensayo AS-I (NCT03178487) fue un ensayo de 14 semanas en 187 pacientes con espondilitis anquilosante con una respuesta inadecuada a al menos dos fármacos antiinflamatorios no esteroideos (AINE) o intolerancia o contraindicación para los AINE y que no habían estado expuestos previamente a fármacos antirreumáticos modificadores de la enfermedad biológicos (fAMEb). Al inicio, los pacientes presentaban síntomas de espondilitis anquilosante desde hacía una media de 14,4 años y aproximadamente el 16% de los pacientes recibían un fAMEc concomitante. Los pacientes recibieron RINVOQ 15 mg una vez al día o placebo. En la semana 14, todos los pacientes aleatorizados a placebo pasaron a recibir RINVOQ 15 mg una vez al día. El criterio de valoración principal fue la proporción de pacientes que lograron una respuesta Assessment of SpondyloArthritis international Society 40 (ASAS40) en la semana 14.
El ensayo AS-II (NCT 04169373) fue un ensayo de 14 semanas en 420 pacientes con espondilitis anquilosante con una respuesta inadecuada a 1 o 2 fAMEb. Al inicio, los pacientes presentaban síntomas de espondilitis anquilosante desde hacía una media de 12,8 años y aproximadamente el 31% de los pacientes recibían un fAMEc concomitante. Los pacientes recibieron RINVOQ 15 mg una vez al día o placebo. En la semana 14, todos los pacientes aleatorizados a placebo pasaron a recibir RINVOQ 15 mg una vez al día. El criterio de valoración principal fue la proporción de pacientes que lograron una respuesta Assessment of SpondyloArthritis international Society 40 (ASAS40) en la semana 14.
Respuesta clínica
En ambos ensayos, una proporción significativamente mayor de pacientes tratados con RINVOQ 15 mg lograron una respuesta ASAS40 en comparación con el placebo en la semana 14 (Tabla 29, Figura 5).
El examen del sexo, el índice de masa corporal (IMC) basal y la PCRhs basal no identificó diferencias en la respuesta a RINVOQ entre estos subgrupos en la semana 14.
| Ensayo AS-I No tratados previamente con fAMEb |
Ensayo AS-II RI a fAMEb |
|||||
| PBO (N=94) |
RINVOQ 15 mg (N=93) |
Diferencia con respecto a PBO (IC del 95%) |
PBO (N=209) |
RINVOQ 15 mg (N=211) |
Diferencia con respecto a PBO (IC del 95%) |
|
| ASAS40a (%) | 25.5 | 50.5 | 25 (12, 38) | 18.2 | 44.5 | 26 (18, 35) |
| ASAS20a (%) | 39.4 | 63.4 | 24 (10, 38) | 38.3 | 65.4 | 27 (18, 36) |
| Abreviaturas: ASAS20 (o 40) = Assessment of SpondyloArthritis international Society mejora ≥20% (o ≥40%); fAMEb = fármaco antirreumático modificador de la enfermedad biológico; RI = respondedores inadecuados; PBO = placebo a Una respuesta ASAS20 (ASAS40) se define como una mejora ≥20% (≥40%) y una mejora absoluta con respecto al valor inicial de ≥1 (≥2) unidad(es) (rango de 0 a 10) en ≥3 de 4 dominios (Global del Paciente, Dolor Lumbar Total, Función e Inflamación), y sin empeoramiento en el posible dominio restante (definido como un empeoramiento ≥20% y ≥1 unidad para ASAS20 o definido como un empeoramiento de > 0 unidades para ASAS40). Para las variables de tipo binario, los resultados de la semana 14 se basan en la imputación de no respondedores (ensayo AS-I) y en la imputación de no respondedores junto con la imputación múltiple (ensayo AS-II). |
||||||
El tratamiento con RINVOQ 15 mg dio lugar a mejoras en los componentes individuales de los criterios de respuesta ASAS40 en comparación con el placebo (Tabla 30).
| Ensayo AS-I bDMARD-naïve |
Ensayo AS-II bDMARD-IR |
|||
| Grupo de tratamiento | PBO | RINVOQ 15 mg |
PBO | RINVOQ 15 mg |
| N | 94 | 93 | 209 | 211 |
| Evaluación global de la actividad de la enfermedad por parte del pacienteb | ||||
| Línea base | 6.8 (1.66) | 6.6 (1.81) | 7.2 (1.40) | 7.4 (1.48) |
| Semana 14 | 5.4 (1.97) | 3.8 (2.44) | 5.9 (2.13) | 4.3 (2.36) |
| Dolor de espalda totalb | ||||
| Línea base | 6.7 (1.78) | 6.8 (1.77) | 7.4 (1.43) | 7.5 (1.48) |
| Semana 14 | 5.0 (2.27) | 3.7 (2.39) | 5.9 (2.09) | 4.4 (2.48) |
| BASFIb | ||||
| Línea base | 5.54 (2.17) | 5.35 (2.36) | 6.18 (1.87) | 6.28 (2.03) |
| Semana 14 | 4.21 (2.26) | 3.14 (2.37) | 5.09 (2.21) | 3.98 (2.45) |
| Inflamaciónc | ||||
| Línea base | 6.66 (1.90) | 6.51 (1.99) | 6.75 (1.55) | 6.88 (1.84) |
| Semana 14 | 4.61 (2.13) | 3.40 (2.16) | 5.11 (2.30) | 3.87 (2.50) |
| hsCRP (mg/L) | ||||
| Línea base | 11.02 (10.85) | 8.90 (12.42) | 14.71 (17.54) | 15.30 (20.53) |
| Semana 14 | 11.72 (15.93) | 2.23 (3.56) | 15.31 (17.55) | 3.82 (8.26) |
| Abreviaturas: ASAS = Assessment of SpondyloArthritis international Society; BASFI = Índice funcional de Bath para la espondilitis anquilosante; bDMARD = fármaco biológico antirreumático modificador de la enfermedad; hsCRP = proteína C reactiva de alta sensibilidad; IR = respondedor inadecuado; PBO = placebo; BASDAI = Índice de actividad de la enfermedad de Bath para la espondilitis anquilosante Los resultados se basan en datos observados de pacientes que tienen observación basal. a Los datos mostrados son la media (desviación estándar). b Escala de calificación numérica (NRS): 0 = mejor, 10 = peor c media de las preguntas 5 y 6 del BASDAI que evalúan la gravedad y la duración de la rigidez matutina: 0 = mejor, 10 = peor |
||||
Figura 5. Porcentaje de pacientes que lograron ASAS40 en el ensayo AS-II*
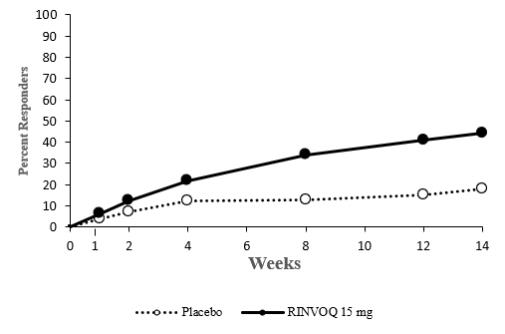
*Los resultados se basan en la imputación de no respondedores junto con la imputación múltiple.
Otros resultados relacionados con la salud
En el ensayo AS-II, los pacientes tratados con RINVOQ 15 mg mostraron mejoras significativas en la calidad de vida relacionada con la salud, medida por la calidad de vida de la espondilitis anquilosante (ASQoL), en comparación con el placebo en la semana 14. En el ensayo AS-I, también se observó una mejora en la ASQoL en comparación con el placebo.
Entesitis
En el ensayo AS-II, los pacientes con entesitis preexistente tratados con RINVOQ 15 mg mostraron una mejora significativa en la entesitis en comparación con el placebo, medida por el cambio desde el inicio en la puntuación de entesitis de espondilitis anquilosante de Maastricht (MASES) en la semana 14. En el ensayo AS-I, también se observó una mejora en la MASES en comparación con el placebo.
Movilidad espinal
En el ensayo AS-II, los pacientes tratados con RINVOQ 15 mg mostraron una mejora significativa en la movilidad espinal en comparación con el placebo, medida por el cambio desde el inicio en el índice de metrología de la espondilitis anquilosante de Bath (BASMI) en la semana 14. En el ensayo AS-I, también se observó una mejora en el BASMI en comparación con el placebo.
14.7 Espondiloartritis axial no radiográfica
La eficacia y seguridad de RINVOQ 15 mg una vez al día se evaluaron en un ensayo aleatorizado, doble ciego, multicéntrico, controlado con placebo en pacientes de 18 años de edad o mayores con espondiloartritis axial no radiográfica activa. El ensayo nr-axSpA (NCT04169373) fue un ensayo controlado con placebo de 52 semanas en 314 pacientes (de los cuales 313 pacientes recibieron tratamiento del estudio) con espondiloartritis axial no radiográfica activa con una respuesta inadecuada a al menos dos fármacos antiinflamatorios no esteroideos (AINE) o intolerancia o contraindicación para los AINE. Los pacientes debían haber tenido signos objetivos de inflamación indicados por una proteína C reactiva (PCR) elevada (definida como > límite superior de lo normal) o sacroilitis en la resonancia magnética (RM), y ninguna evidencia radiográfica definitiva de daño estructural en las articulaciones sacroilíacas. Los pacientes tenían enfermedad activa según la definición del Índice de Actividad de la Enfermedad de Espondilitis Anquilosante de Bath (BASDAI) ≥4, y una puntuación en la Evaluación del Paciente del Dolor Lumbar Total ≥ 4 basada en una escala numérica de calificación (NRS) de 0 a 10 en las visitas de selección y de referencia. Al inicio, aproximadamente el 29.1% de los pacientes estaban tomando un cDMARD concomitante. El 32.9% de los pacientes tuvieron una respuesta inadecuada o intolerancia al tratamiento con bDMARD. Los pacientes recibieron RINVOQ 15 mg una vez al día o placebo. El criterio de valoración principal fue la proporción de pacientes que lograron una respuesta Assessment of SpondyloArthritis international Society 40 (ASAS40) en la semana 14.
Respuesta clínica
En el ensayo nr-axSpA, una proporción significativamente mayor de pacientes tratados con RINVOQ 15 mg lograron una respuesta ASAS40 en comparación con el placebo en la semana 14 (Tabla 31, Figura 6).
El examen del sexo, el IMC inicial, la duración de los síntomas de la espondiloartritis axial no radiográfica, la PCRhs inicial, la sacroilitis por RM y el uso previo de bDMARD no identificó diferencias en la respuesta a RINVOQ entre estos subgrupos en la semana 14.
| PBO (N=157) |
RINVOQ 15 mg (N=156) |
Diferencia con respecto al placebo (IC del 95%) | |
| ASAS40a (%) | 22.3 | 44.9 | 22.5 (12.4, 32.5) |
| ASAS20a (%) | 43.3 | 66.7 | 23.2 (12.6, 33.8) |
| Abreviaturas: ASAS20 (o 40) = Assessment of SpondyloArthritis international Society ≥20% (o ≥40%) de mejora; PBO = placebo a Una respuesta ASAS20 (ASAS40) se define como una mejora ≥20% (≥40%) y una mejora absoluta con respecto al valor inicial de ≥1 (≥2) unidad(es) (rango de 0 a 10) en ≥3 de 4 dominios (Global del paciente, Dolor lumbar total, Función e Inflamación), y sin empeoramiento en el posible dominio restante (definido como un empeoramiento ≥20% y ≥1 unidad para ASAS20 o definido como un empeoramiento de > 0 unidades para ASAS40). Para los criterios de valoración binarios, los resultados se basan en la imputación de no respondedores junto con la imputación múltiple. |
|||
El tratamiento con RINVOQ 15 mg produjo mejoras en los componentes individuales de los criterios de respuesta ASAS40 en comparación con el placebo (Tabla 32).
| Grupo de tratamiento | PBO (N=157) |
RINVOQ 15 mg (N=156) |
| Evaluación global de la actividad de la enfermedad por parte del pacienteb | ||
| Línea base | 7.30 (1.38) | 6.99 (1.62) |
| Semana 14 | 5.35 (2.31) | 4.16 (2.38) |
| Dolor de espalda totalb | ||
| Línea base | 7.29 (1.39) | 7.23 (1.55) |
| Semana 14 | 5.27 (2.36) | 4.29 (2.49) |
| BASFIb | ||
| Línea base | 5.99 (2.14) | 5.89 (2.08) |
| Semana 14 | 4.47 (2.42) | 3.33 (2.39) |
| Inflamaciónc | ||
| Línea base | 6.68 (1.67) | 6.60 (1.83) |
| Semana 14 | 4.69 (2.36) | 3.48 (2.51) |
| hsCRP (mg/L) | ||
| Línea base | 8.75 (12.91) | 9.93 (16.17) |
| Semana 14 | 7.25 (10.61) | 2.84 (4.90) |
| Abreviaturas: ASAS = Sociedad Internacional de Evaluación de la Espondiloartritis; BASFI = Índice Funcional de Bath para la Espondilitis Anquilosante; hsCRP = proteína C reactiva de alta sensibilidad; PBO = placebo a Los datos mostrados son la media (desviación estándar). b Escala numérica de calificación (NRS): 0 = mejor, 10 = peor c Media de las preguntas 5 y 6 del Índice de Actividad de la Enfermedad de Bath para la Espondilitis Anquilosante (BASDAI) que evalúan la gravedad y la duración de la rigidez matutina: 0 = mejor, 10 = peor |
||
Figura 6. Porcentaje de pacientes que logran ASAS40*
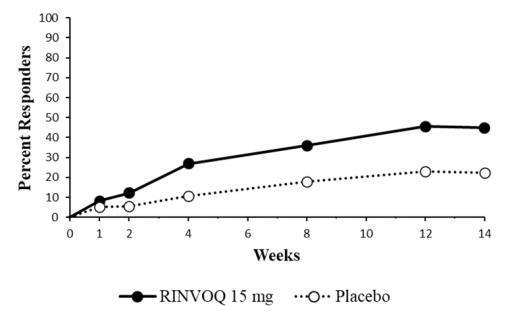
*Los resultados se basan en la imputación de no respondedores junto con la imputación múltiple.
Otros resultados relacionados con la salud
Los pacientes tratados con RINVOQ 15 mg mostraron mejoras significativas en la calidad de vida relacionada con la salud, medida por la calidad de vida en la espondilitis anquilosante (ASQoL), en comparación con el placebo en la semana 14.
14.8 Artritis idiopática juvenil poliarticular
La eficacia de RINVOQ/RINVOQ LQ en pacientes pediátricos con AIJ con poliartritis activa se basa en la extrapolación de la eficacia establecida de RINVOQ en pacientes con artritis reumatoide, ajustada por exposición. La seguridad y eficacia de RINVOQ/RINVOQ LQ también se evaluaron en un estudio multicéntrico, abierto, de un solo brazo, en 83 niños (de 2 a < 18 años de edad) con AIJ con poliartritis activa (NCT03725007). Los subtipos de pacientes con AIJp al inicio del estudio incluyeron poliarticular con factor reumatoide negativo (68.7%), poliarticular con factor reumatoide positivo (15.7%), oligoarticular extendida (13.3%) y AIJ sistémica sin manifestaciones sistémicas (2.4%). Todos los pacientes recibieron dosis de RINVOQ LQ o comprimidos de RINVOQ en función del peso durante un máximo de 156 semanas. Se permitió el ingreso al estudio a los pacientes tratados con una dosis estable de MTX; se permitieron cambios en la dosis de MTX durante el estudio. La eficacia se evaluó como criterios de valoración de apoyo hasta la semana 48. La eficacia fue generalmente consistente con las respuestas en pacientes con artritis reumatoide.
16 FORMA DE SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
Cómo se suministra
Las tabletas de liberación prolongada de RINVOQ se suministran como:
- 15 mg: púrpura, oblonga biconvexa, con dimensiones de 14 x 8 mm, y con la marca ‘a15’ en un lado.
30 tabletas en un frasco; NDC: 0074-2306-30 - 30 mg: roja, oblonga biconvexa, con dimensiones de 14 x 8 mm, y con la marca ‘a30’ en un lado.
30 tabletas en un frasco; NDC: 0074-2310-30 - 45 mg: amarilla a amarilla moteada, oblonga biconvexa, con dimensiones de 14 x 8 mm, y con la marca ‘a45’ en un lado.
28 tabletas en un frasco; NDC: 0074-1043-28
La solución oral de RINVOQ LQ se suministra como:
- Una solución oral de 1 mg/mL en frascos de HDPE con tapa a prueba de niños. Cada frasco contiene un volumen etiquetado de 180 mL de solución transparente, incolora a ligeramente amarilla. El frasco se empaqueta en una caja con un adaptador de frasco de presión y una jeringa oral de 10 mL; NDC: 0074-2320-01
Almacenamiento y manipulación
Tabletas de liberación prolongada de RINVOQ
- Almacenar a 2˚C a 25˚C (36˚F a 77˚F).
- Almacenar en el frasco original para protegerlo de la humedad.
Solución oral de RINVOQ LQ
- Almacenar entre 2°C y 30°C (36°F y 86°F).
- Deseche la solución oral restante 60 días después de abrir el frasco.
17 INFORMACIÓN PARA EL PACIENTE
Aconseje al paciente y al cuidador que lean el etiquetado del paciente aprobado por la FDA (Guía de medicamentos y Instrucciones de uso).
Infecciones graves
Informe a los pacientes que es más probable que desarrollen infecciones cuando toman RINVOQ/RINVOQ LQ. Indique a los pacientes que se pongan en contacto con su médico inmediatamente durante el tratamiento si desarrollan algún signo o síntoma de infección [ver Advertencias y precauciones (5.1)].
Aconseje a los pacientes que el riesgo de herpes zóster está aumentado en los pacientes que toman RINVOQ/RINVOQ LQ y en algunos casos puede ser grave [ver Advertencias y precauciones (5.1)].
Neoplasias
Informe a los pacientes que RINVOQ/RINVOQ LQ puede aumentar su riesgo de ciertos cánceres y que se deben realizar exámenes periódicos de la piel mientras se usa RINVOQ/RINVOQ LQ.
Aconseje a los pacientes que la exposición a la luz solar y a los rayos UV debe limitarse usando ropa protectora y un protector solar de amplio espectro [ver Advertencias y precauciones (5.3)].
Eventos cardiovasculares adversos mayores
Informe a los pacientes que RINVOQ/RINVOQ LQ puede aumentar su riesgo de eventos cardiovasculares adversos mayores (MACE), incluido el infarto de miocardio, el accidente cerebrovascular y la muerte cardiovascular. Indique a todos los pacientes, especialmente a los fumadores actuales o pasados o a los pacientes con otros factores de riesgo cardiovascular, que estén atentos al desarrollo de signos y síntomas de eventos cardiovasculares [ver Advertencias y precauciones (5.4)].
Trombosis
Informe a los pacientes que se han notificado eventos de trombosis venosa profunda y embolia pulmonar en los ensayos clínicos con RINVOQ. Indique a los pacientes que busquen atención médica inmediata si desarrollan algún signo o síntoma de una TVP o EP [ver Advertencias y precauciones (5.5)].
Reacciones de hipersensibilidad
Aconseje a los pacientes que interrumpan el tratamiento con RINVOQ/RINVOQ LQ y busquen atención médica inmediata si desarrollan algún signo o síntoma de reacciones alérgicas [ver Advertencias y precauciones (5.6)].
Perforaciones gastrointestinales
Informe a los pacientes que se han notificado perforaciones gastrointestinales en los ensayos clínicos con RINVOQ y que los factores de riesgo incluyen el uso de AINE, corticosteroides o antecedentes de diverticulitis. Indique a los pacientes que busquen atención médica inmediatamente si experimentan dolor abdominal de nueva aparición, fiebre, escalofríos, náuseas o vómitos [ver Advertencias y Precauciones (5.7)].
Desprendimiento de retina
Informe a los pacientes que se ha notificado desprendimiento de retina en los ensayos clínicos con RINVOQ. Aconseje a los pacientes que informen inmediatamente a su médico si desarrollan algún cambio repentino en la visión mientras reciben RINVOQ/RINVOQ LQ [ver Reacciones adversas (6.1)].
Anormalidades de laboratorio
Informe a los pacientes que RINVOQ/RINVOQ LQ puede afectar ciertas pruebas de laboratorio y que se requieren análisis de sangre antes y durante el tratamiento con RINVOQ/RINVOQ LQ [ver Advertencias y precauciones (5.8)].
Vacunaciones
Aconseje a los pacientes que eviten el uso de vacunas vivas con RINVOQ/RINVOQ LQ. Indique a los pacientes que informen a su médico que están tomando RINVOQ/RINVOQ LQ antes de una posible vacunación [ver Advertencias y precauciones (5.10)].
Toxicidad embrio-fetal
Aconseje a las mujeres embarazadas y a las mujeres en edad fértil que la exposición a RINVOQ/RINVOQ LQ durante el embarazo puede provocar daños fetales. Aconseje a las mujeres que informen a su médico de un embarazo conocido o sospechado [ver Advertencias y precauciones (5.9) y Uso en poblaciones específicas (8.1)].
Aconseje a las mujeres en edad fértil que se debe utilizar una contracepción eficaz durante el tratamiento y durante las 4 semanas posteriores a la última dosis de RINVOQ/RINVOQ LQ [ver Uso en poblaciones específicas (8.3)].
Aconseje a las mujeres expuestas a RINVOQ/RINVOQ LQ durante el embarazo que existe un programa de vigilancia del embarazo que monitoriza los resultados del embarazo [ver Uso en poblaciones específicas (8.1)].
Lactancia
Aconseje a las mujeres que no deben amamantar durante el tratamiento con RINVOQ/RINVOQ LQ y durante los 6 días posteriores a la última dosis [ver Uso en poblaciones específicas (8.2)].
Administración
Aconseje a los pacientes que las tabletas de RINVOQ no son sustituibles por RINVOQ LQ [ver Posología y administración (2.2)].
Aconseje a los pacientes que no mastiquen, trituren ni dividan las tabletas de RINVOQ [ver Posología y administración (2.2)].
Para RINVOQ LQ, indique a los pacientes y a los cuidadores que lean y sigan las Instrucciones de uso para la preparación, administración, almacenamiento y eliminación adecuadas [Ver Posología y administración (2.1)].
Avise a los pacientes que eviten los alimentos o bebidas que contengan pomelo durante el tratamiento con RINVOQ/RINVOQ LQ [ver Interacciones medicamentosas (7.1)].
Residuos de medicamentos en las heces
Indique a los pacientes que notifiquen a su profesional sanitario si observan repetidamente residuos de medicamentos (por ejemplo, comprimidos de RINVOQ intactos o fragmentos) en las heces o en la salida de la ostomía [ver Advertencias y precauciones (5.11)].
Fabricado por: AbbVie Inc., North Chicago, IL 60064, USA
RINVOQ® es una marca registrada de AbbVie Biotechnology Ltd.
©2019-2024 AbbVie Inc.
20080260 Abril 2024
Guía de medicación
| GUÍA DEL MEDICAMENTO | ||||||
| RINVOQ® (RIN-VOKE) (upadacitinib) comprimidos de liberación prolongada, para administración oral |
RINVOQ® LQ (RIN-VOKE EL-CUE) (upadacitinib) solución oral |
|||||
| ¿Cuál es la información más importante que debo saber sobre RINVOQ/RINVOQ LQ? RINVOQ/RINVOQ LQ puede causar efectos secundarios graves, que incluyen: 1. Infecciones graves. RINVOQ/RINVOQ LQ es un medicamento que afecta su sistema inmunitario. RINVOQ/RINVOQ LQ puede disminuir la capacidad de su sistema inmunitario para combatir infecciones. Algunas personas han tenido infecciones graves mientras tomaban RINVOQ, incluida la tuberculosis (TB), e infecciones causadas por bacterias, hongos o virus que pueden propagarse por todo el cuerpo. Algunas personas han muerto a causa de estas infecciones.
|
||||||
|
|
|
||||
2. Mayor riesgo de muerte en personas de 50 años o más que tienen al menos 1 factor de riesgo de enfermedad cardíaca (cardiovascular) y están tomando un medicamento de la clase de medicamentos llamados inhibidores de la Janus quinasa (JAK)s. RINVOQ/RINVOQ LQ es un medicamento inhibidor de JAK.
3. Cáncer y problemas del sistema inmunitario.
RINVOQ/RINVOQ LQ puede aumentar su riesgo de ciertos tipos de cáncer al cambiar la forma en que funciona su sistema inmunitario.
El linfoma y otros tipos de cáncer, incluidos los cánceres de piel, pueden ocurrir en personas que toman RINVOQ/RINVOQ LQ. Las personas que toman un medicamento de la clase de medicamentos llamados inhibidores de la Janus quinasa (JAK) tienen un mayor riesgo de ciertos tipos de cáncer, incluido el linfoma y el cáncer de pulmón, especialmente si usted es un fumador actual o pasado.
Informe a su proveedor de atención médica si alguna vez ha tenido algún tipo de cáncer. Siga los consejos de su proveedor de atención médica sobre cómo revisarse la piel para detectar cáncer de piel durante el tratamiento con RINVOQ/RINVOQ LQ. Limite la cantidad de tiempo que pasa al sol. Evite el uso de camas de bronceado o lámparas solares. Use ropa protectora cuando esté al sol y use un protector solar con un factor de protección alto (FPS 30 o superior). Esto es especialmente importante si su piel es muy clara o si tiene antecedentes familiares de cáncer de piel.
4. Mayor riesgo de eventos cardiovasculares mayores como ataque cardíaco, accidente cerebrovascular o muerte en personas de 50 años o más que tienen al menos 1 factor de riesgo de enfermedad cardíaca (cardiovascular) y están tomando un medicamento de la clase de medicamentos llamados inhibidores de JAKs, especialmente si usted es un fumador actual o pasado.
Busque ayuda de emergencia de inmediato si tiene algún síntoma de ataque cardíaco o accidente cerebrovascular mientras toma RINVOQ/RINVOQ LQ, que incluyen:
- molestia en el centro del pecho que dura más de unos minutos, o que desaparece y vuelve
- opresión, dolor, presión o pesadez intensos en el pecho, la garganta, el cuello o la mandíbula
- dolor o molestia en los brazos, la espalda, el cuello, la mandíbula o el estómago
- falta de aliento con o sin molestia en el pecho
- sudoración fría
- náuseas o vómitos
- sensación de mareo
- debilidad en una parte o en un lado del cuerpo
- habla arrastrada
5. Coágulos de sangre (trombosis).
Los coágulos de sangre en las venas de las piernas (trombosis venosa profunda, TVP) o los pulmones (embolia pulmonar, EP) y las arterias (trombosis arterial) pueden ocurrir en algunas personas que toman RINVOQ/RINVOQ LQ. Esto puede ser potencialmente mortal y causar la muerte. Los coágulos de sangre en las venas de las piernas (TVP) y los pulmones (EP) han ocurrido con más frecuencia en personas de 50 años o más y con al menos 1 factor de riesgo de enfermedad cardíaca (cardiovascular) que toman un medicamento de la clase de medicamentos llamados inhibidores de la Janus quinasa (JAK).
- Informe a su proveedor de atención médica si ha tenido coágulos de sangre en las venas de las piernas o los pulmones en el pasado.
- Busque atención médica de inmediato si tiene signos y síntomas de coágulos de sangre durante el tratamiento con RINVOQ/RINVOQ LQ, que incluyen:
○ dolor o sensibilidad en una o ambas piernas
○ falta de aliento o dificultad para respirar
- Informe a su proveedor de atención médica si ha tenido diverticulitis (inflamación en partes del intestino grueso) o úlceras en el estómago o los intestinos. Algunas personas que toman RINVOQ/RINVOQ LQ pueden tener desgarros en el estómago o los intestinos. Esto ocurre con mayor frecuencia en personas que toman medicamentos antiinflamatorios no esteroideos (AINE) o corticosteroides.
- Busque atención médica de inmediato si tiene dolor en el área del estómago, fiebre, escalofríos, náuseas o vómitos.
Su proveedor de atención médica debe realizar análisis de sangre antes de comenzar a tomar RINVOQ/RINVOQ LQ y mientras toma RINVOQ/RINVOQ LQ para verificar lo siguiente:
- bajo recuento de neutrófilos y linfocitos. Los neutrófilos y los linfocitos son tipos de glóbulos blancos que ayudan al cuerpo a combatir las infecciones.
- bajo recuento de glóbulos rojos. Los glóbulos rojos transportan oxígeno. Un bajo recuento de glóbulos rojos significa que puede tener anemia, lo que puede hacer que se sienta débil y cansado.
- aumento de los niveles de colesterol. Su proveedor de atención médica debe realizar análisis de sangre para verificar sus niveles de colesterol aproximadamente 12 semanas después de comenzar a tomar RINVOQ/RINVOQ LQ, y según sea necesario.
- elevación de las enzimas hepáticas. Las enzimas hepáticas ayudan a determinar si su hígado está funcionando normalmente. Las enzimas hepáticas elevadas pueden indicar que su proveedor de atención médica necesita realizar pruebas adicionales en su hígado.
No debe tomar RINVOQ/RINVOQ LQ si su recuento de neutrófilos, recuento de linfocitos o recuento de glóbulos rojos es demasiado bajo o si sus pruebas de hígado son demasiado altas. Su proveedor de atención médica puede detener su tratamiento con RINVOQ/RINVOQ LQ por un período de tiempo si es necesario debido a cambios en estos resultados de análisis de sangre.
Vea “¿Cuáles son los posibles efectos secundarios de RINVOQ/RINVOQ LQ?” para obtener más información sobre los efectos secundarios.
RINVOQ/RINVOQ LQ es un medicamento recetado que es un inhibidor de la cinasa de Janus (JAK).
- RINVOQ se usa para tratar a adultos con artritis reumatoide moderada a grave cuando se han usado 1 o más medicamentos llamados bloqueadores del factor de necrosis tumoral (TNF), y no funcionaron bien o no se pudieron tolerar.
- RINVOQ se usa para tratar a adultos con artritis psoriásica activa cuando se han usado 1 o más medicamentos llamados bloqueadores del factor de necrosis tumoral (TNF), y no funcionaron bien o no se pudieron tolerar.
- RINVOQ se usa para tratar a adultos y niños de 12 años de edad o mayores con eccema moderado a grave (dermatitis atópica) que no respondió al tratamiento previo y su eccema no está bien controlado con otras píldoras o inyecciones, incluidas las medicinas biológicas, o el uso de otras píldoras o inyecciones no es recomendado.
- RINVOQ se usa para tratar a adultos con colitis ulcerosa moderada a grave cuando se han usado 1 o más medicamentos llamados bloqueadores del factor de necrosis tumoral (TNF), y no funcionaron bien o no se pudieron tolerar.
- RINVOQ se usa para tratar a adultos con enfermedad de Crohn moderada a grave cuando se han usado 1 o más medicamentos llamados bloqueadores del factor de necrosis tumoral (TNF), y no funcionaron bien o no se pudieron tolerar.
- RINVOQ se usa para tratar a adultos con espondilitis anquilosante activa cuando se han usado 1 o más medicamentos llamados bloqueadores del factor de necrosis tumoral (TNF), y no funcionaron bien o no se pudieron tolerar.
- RINVOQ se usa para tratar a adultos con espondiloartritis axial no radiográfica activa con signos objetivos de inflamación cuando se ha usado un medicamento bloqueador del factor de necrosis tumoral (TNF), y no funcionó bien o no se pudo tolerar.
- RINVOQ/RINVOQ LQ se usa para tratar a niños de 2 años de edad o mayores con artritis idiopática juvenil poliarticular activa (pJIA) cuando se han usado 1 o más medicamentos llamados bloqueadores del factor de necrosis tumoral (TNF), y no funcionaron bien o no se pudieron tolerar.
- RINVOQ/RINVOQ LQ se usa para tratar a niños de 2 a menos de 18 años de edad con artritis psoriásica activa cuando se han usado 1 o más medicamentos llamados bloqueadores del factor de necrosis tumoral (TNF), y no funcionaron bien o no se pudieron tolerar.
No se sabe si RINVOQ/RINVOQ LQ es seguro y eficaz en niños con espondilitis anquilosante, espondiloartritis axial no radiográfica, colitis ulcerosa o enfermedad de Crohn.
No se sabe si RINVOQ es seguro y eficaz en niños menores de 12 años de edad con dermatitis atópica.
No se sabe si RINVOQ LQ es seguro y eficaz en niños con dermatitis atópica.
No se sabe si RINVOQ/RINVOQ LQ es seguro y eficaz en niños menores de 2 años de edad con pJIA o artritis psoriásica.
Antes de tomar RINVOQ/RINVOQ LQ, informe a su proveedor de atención médica sobre todas sus afecciones médicas, incluso si:
- Vea “¿Cuál es la información más importante que debo saber sobre RINVOQ/RINVOQ LQ?”
- tiene una infección.
- es un fumador actual o pasado.
- ha tenido un ataque cardíaco, otros problemas cardíacos o un derrame cerebral.
- tiene problemas hepáticos.
- tiene problemas renales.
- tiene dolor abdominal (abdominal) inexplicable, tiene antecedentes de diverticulitis o úlceras en el estómago o los intestinos, o está tomando AINE.
- tiene recuentos bajos de glóbulos rojos o blancos.
- recibió recientemente o está programado para recibir una inmunización (vacuna). Las personas que toman RINVOQ/RINVOQ LQ no deben recibir vacunas vivas.
- está embarazada o planea quedar embarazada. Según estudios en animales, RINVOQ/RINVOQ LQ puede dañar a su bebé nonato.
Mujeres que pueden quedar embarazadas:
- Su proveedor de atención médica verificará si está embarazada o no antes de comenzar el tratamiento con RINVOQ/RINVOQ LQ.
- Debe usar un método anticonceptivo eficaz (anticoncepción) para evitar quedar embarazada durante el tratamiento con RINVOQ/RINVOQ LQ y durante 4 semanas después de su última dosis de RINVOQ/RINVOQ LQ.
- Informe a su proveedor de atención médica si cree que está embarazada o queda embarazada durante el tratamiento con RINVOQ/RINVOQ LQ.
- Existe un programa de vigilancia del embarazo para RINVOQ/RINVOQ LQ. El propósito del programa es recopilar información sobre la salud de usted y su bebé. Si queda embarazada mientras toma RINVOQ/RINVOQ LQ, se le recomienda que informe el embarazo llamando al 1-800-633-9110.
- está amamantando o planea amamantar. RINVOQ/RINVOQ LQ puede pasar a la leche materna. Usted y su proveedor de atención médica deben decidir si tomará RINVOQ/RINVOQ LQ o amamantará. No amamante durante el tratamiento con RINVOQ/RINVOQ LQ y durante 6 días después de su última dosis de RINVOQ/RINVOQ LQ.
Especialmente informe a su proveedor de atención médica si toma:
- medicamentos para infecciones fúngicas (como ketoconazol, itraconazol, posaconazol o voriconazol) o claritromicina (para infecciones bacterianas) ya que estos medicamentos pueden aumentar la cantidad de RINVOQ/RINVOQ LQ en su sangre.
- rifampicina (para infecciones bacterianas) o fenitoína (para trastornos neurológicos) ya que estos medicamentos pueden disminuir el efecto de RINVOQ/RINVOQ LQ.
- medicamentos que afectan su sistema inmunológico (como azatioprina y ciclosporina) ya que estos medicamentos pueden aumentar su riesgo de infección.
Pregúntele a su proveedor de atención médica o farmacéutico si no está seguro de si está tomando alguno de estos medicamentos.
Conozca los medicamentos que toma. Mantenga una lista de ellos para mostrársela a su proveedor de atención médica y farmacéutico cuando obtenga un nuevo medicamento.
- Tome RINVOQ/RINVOQ LQ exactamente como su proveedor de atención médica le indique que lo use.
- Tome RINVOQ LQ 2 veces al día con o sin alimentos.
- Tome las tabletas de RINVOQ 1 vez al día con o sin alimentos.
- Trague las tabletas de RINVOQ enteras. No divida, triture ni mastique las tabletas.
- RINVOQ LQ no es lo mismo que las tabletas de RINVOQ. No cambie entre RINVOQ LQ y las tabletas de RINVOQ a menos que el cambio haya sido realizado por su proveedor de atención médica.
- Si toma demasiado RINVOQ/RINVOQ LQ, llame a su proveedor de atención médica o a la línea de ayuda para envenenamiento al 1-800-222-1222, o vaya a la sala de emergencias del hospital más cercano de inmediato.
¿Qué debo evitar mientras tomo RINVOQ/RINVOQ LQ?
Evite los alimentos o bebidas que contengan toronja durante el tratamiento con RINVOQ/RINVOQ LQ. Comer toronja o beber jugo de toronja puede aumentar el riesgo de efectos secundarios.
¿Cuáles son los posibles efectos secundarios de RINVOQ/RINVOQ LQ?
RINVOQ/RINVOQ LQ puede causar efectos secundarios graves, que incluyen:
- Ver “¿Cuál es la información más importante que debo saber sobre RINVOQ/RINVOQ LQ?”
Los efectos secundarios más comunes de RINVOQ/RINVOQ LQ en personas tratadas para la artritis reumatoide, la artritis psoriásica, la espondilitis anquilosante, la espondiloartritis axial no radiográfica, y la pJIA incluyen:
• herpes zóster (culebrilla)
• infecciones por virus del herpes simple, incluidas las úlceras bucales
• bronquitis
• tos
• fiebre
• acné
• dolor de cabeza
• acné
• infecciones por virus del herpes simple, incluidas las úlceras bucales
• dolor de cabeza
• aumento de los niveles sanguíneos de creatina fosfoquinasa
• tos
• reacciones alérgicas
• inflamación de los folículos pilosos
• náuseas
• fiebre
• aumento de peso
• herpes zóster (culebrilla)
• gripe
• cansancio
• bajo recuento de glóbulos blancos (neutropenia)
• dolor muscular
• enfermedad similar a la gripe
• acné
• infecciones por virus del herpes simple, incluidas las úlceras bucales
• inflamación de los folículos pilosos
• erupción cutánea
• gripe
• aumento de los niveles de colesterol en sangre
• aumento de los niveles sanguíneos de creatina fosfoquinasa
• aumento de los niveles de enzimas hepáticas
• bajo número de ciertos tipos de glóbulos blancos (neutropenia, linfopenia)
• bronquitis
• neumonía
• gripe
• acné
• infecciones por virus del herpes simple, incluidas las úlceras bucales
• cansancio
• tos
• herpes zóster (culebrilla)
• dolor de cabeza
• aumento de los niveles sanguíneos de creatina fosfoquinasa
• aumento de los niveles de enzimas hepáticas
• bajo número de glóbulos rojos (anemia)
• bajo número de glóbulos blancos (neutropenia, leucopenia)
• infección del estómago y el intestino (gastroenteritis)
Algunas personas que toman RINVOQ pueden ver residuos de medicamentos (una tableta entera o pedazos de tabletas) en sus heces. Si esto sucede, llame a su proveedor de atención médica.
Estos no son todos los posibles efectos secundarios de RINVOQ/RINVOQ LQ.
Llame a su médico para obtener asesoramiento médico sobre los efectos secundarios. Puede informar los efectos secundarios a la FDA al 1-800-FDA-1088.
¿Cómo debo almacenar RINVOQ/RINVOQ LQ?
- Almacene las tabletas de RINVOQ entre 36°F y 77°F (2°C y 25°C).
- Almacene las tabletas de RINVOQ en el frasco original para protegerlas de la humedad.
- Almacene RINVOQ LQ entre 36°F y 86°F (2°C y 30°C).
- Deseche (descarte) el RINVOQ LQ restante 60 días después de abrir el frasco.
- Mantenga RINVOQ/RINVOQ LQ y todos los medicamentos fuera del alcance de los niños.
Los medicamentos a veces se recetan para fines distintos de los que se enumeran en una Guía de medicamentos. No use RINVOQ/RINVOQ LQ para una condición para la que no fue recetado.
No le dé RINVOQ/RINVOQ LQ a otras personas, incluso si tienen los mismos síntomas que usted. Puede hacerles daño.
Puede pedirle a su farmacéutico o proveedor de atención médica información sobre RINVOQ/RINVOQ LQ que esté escrita para profesionales de la salud.
¿Cuáles son los ingredientes de las tabletas de RINVOQ 15 mg?
Ingrediente activo: upadacitinib
Ingredientes inactivos: dióxido de silicio coloidal, óxido ferroso férrico, hipromelosa, óxido de hierro rojo, estearato de magnesio, manitol, celulosa microcristalina, alcohol polivinílico, polietilenglicol, talco, ácido tartárico y dióxido de titanio.
¿Cuáles son los ingredientes de las tabletas de RINVOQ 30 mg?
Ingrediente activo: upadacitinib
Ingredientes inactivos: dióxido de silicio coloidal, hipromelosa, óxido de hierro rojo, estearato de magnesio, manitol, celulosa microcristalina, alcohol polivinílico, polietilenglicol, talco, ácido tartárico y dióxido de titanio.
¿Cuáles son los ingredientes de las tabletas de RINVOQ 45 mg?
Ingrediente activo: upadacitinib
Ingredientes inactivos: dióxido de silicio coloidal, hipromelosa, óxido de hierro amarillo y óxido de hierro rojo, estearato de magnesio, manitol, celulosa microcristalina, alcohol polivinílico, polietilenglicol, talco, ácido tartárico y dióxido de titanio.
¿Cuáles son los ingredientes de RINVOQ LQ?
Ingrediente activo: upadacitinib
Ingredientes inactivos: ácido cítrico anhidro, agua purificada, benzoato de sodio, citrato de sodio dihidratado y sucralosa.
Fabricado por: AbbVie Inc., North Chicago, IL 60064, EE. UU.
RINVOQ® es una marca registrada de AbbVie Biotechnology Ltd.
©2019-2024 AbbVie Inc.
Para obtener más información, llame al 1-800 2-RINVOQ (1-800-274-6867) o visite www.RINVOQ.com.
20081173
INSTRUCCIONES DE USO
INSTRUCCIONES DE USO
RINVOQ® LQ [RIN-VOKE EL-CUE]
(upadacitinib)
solución oral
Estas Instrucciones de uso contienen información sobre cómo preparar y administrar una dosis de solución oral RINVOQ LQ.
Lea estas Instrucciones de uso antes de administrar RINVOQ LQ a su hijo y cada vez que reciba una nueva receta. Puede haber nueva información. Esta información no reemplaza hablar con su proveedor de atención médica sobre la condición médica o el tratamiento de su hijo.
Información importante que necesita saber antes de administrar RINVOQ LQ
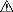
Mantenga el frasco y los suministros fuera de la vista y el alcance de los niños.
Utilice únicamente la jeringa proporcionada. No comparta la jeringa con otras personas ni la use con otros medicamentos.
Llame a su proveedor de atención médica o al 1-800-2-RINVOQ o al 1-800-274-6867 si necesita ayuda o tiene alguna pregunta sobre cómo administrar RINVOQ LQ de la manera correcta.
Comuníquese con su proveedor de atención médica si su hijo toma demasiada solución oral o no recibe la dosis completa.
- Use RINVOQ LQ dentro de los 60 días posteriores a la apertura del frasco. Para ayudarlo a recordar, escriba la fecha en que abrió el frasco en la caja.
- No abra un frasco nuevo de RINVOQ LQ hasta que haya terminado el frasco anterior. Use una jeringa nueva cuando abra un frasco nuevo.
- La solución oral RINVOQ LQ es transparente e incolora a ligeramente amarilla.
- Guarde estas instrucciones y la caja en la que vino su solución oral RINVOQ LQ y los suministros para uso futuro.
Suministros en cada caja
Figura A
Eliminación de RINVOQ LQ
Deseche (tire) la jeringa y el frasco cuando haya terminado el frasco o 60 días después de la apertura.
- Pregúntele a su farmacéutico cómo desechar (tirar) adecuadamente cualquier medicamento no utilizado.
- Enjuague la jeringa y luego colóquela en la basura doméstica.
Preparación de RINVOQ LQ
1. Verifique la dosis prescrita.
a. Verifique la dosis prescrita de su hijo en mililitros (mL) y encuentre esta marca de mL en la jeringa.
b. Utilice únicamente la jeringa proporcionada para administrar la dosis prescrita.
2. Verifique la fecha de vencimiento.
a. Verifique el frasco y asegúrese de que la fecha de vencimiento no haya pasado (vea Figura B).
No use RINVOQ LQ después de la fecha de vencimiento impresa en la caja y el frasco después de “EXP”.
Figura B
3. Verifique los suministros.
a. Verifique los suministros y asegúrese de que no estén dañados (vea Figura C).
b. Utilice únicamente la jeringa si está limpia y seca.
c. Asegúrese de que el émbolo esté completamente dentro de la jeringa.
No use los suministros si están húmedos, dañados o parecen estar manipulados.
Figura C
4. Abra el frasco.
a. Presione hacia abajo y gire la tapa para quitarla del frasco (vea Figura D). No tire la tapa.
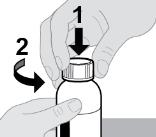
Figura D
5. Inserte el adaptador (solo la primera vez).
a. Mientras sostiene el frasco firmemente, use su pulgar para empujar el adaptador hasta el borde del frasco (vea Figura E).
Nota: Es posible que deba aplicar presión al adaptador.
No retire el adaptador después de insertarlo.
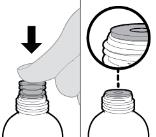
Figura E
Medición de la dosis
6. Inserte la jeringa en el frasco y luego gírelo boca abajo.
a. Inserte la punta de la jeringa en el adaptador.
b. Con la jeringa conectada al frasco, gire el frasco boca abajo (vea Figura F).
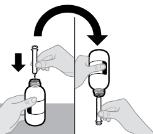
Figura F
7. Extraiga la solución oral en la jeringa.
a. Tire lentamente del émbolo hacia abajo (vea Figura G).
b. Verifique la jeringa para detectar burbujas de aire.
Nota: Es posible que sienta presión al tirar del émbolo.
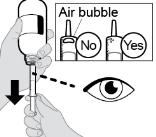
Figura G
8. Elimine las burbujas de aire grandes (vea la Figura H).
a. Mientras sostiene el frasco, agite los lados de la jeringa para enviar las burbujas de aire grandes a la punta.
b. Con la jeringa conectada al frasco, mueva el émbolo hacia arriba y hacia abajo para devolver las burbujas de aire al frasco.
c. Repita el Paso 8 hasta que desaparezcan las burbujas de aire grandes.
Nota: Las burbujas de aire pequeñas son normales.
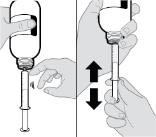
Figura H
9. Mida la dosis.
a. Después de eliminar las burbujas de aire grandes, mueva el émbolo hasta que quede a la altura de la marca de la dosis (vea la Figura I).
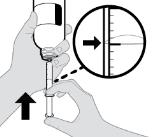
Figura I
10. Gire el frasco hacia arriba y luego retire la jeringa.
a. Con la jeringa conectada al frasco, gire el frasco hacia arriba.
b. Sostenga la jeringa por el medio y retírela con cuidado del frasco (vea la Figura J).
No toque el émbolo para evitar que la solución oral salga accidentalmente de la jeringa antes de que esté listo para administrar el medicamento.
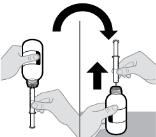
Figura J
Administración de RINVOQ LQ
11. Compruebe la dosis.
a. Compruebe que la jeringa tenga la dosis correcta de solución oral (vea la Figura K).
b. Compruebe la jeringa para ver si hay burbujas de aire grandes.
c. Si la dosis no es correcta o ve burbujas de aire grandes, vuelva al Paso 6.
Figura K
12. Administre la solución oral.
a. Coloque la jeringa contra la parte interna de la mejilla del niño.
b. Empuje el émbolo para administrar toda la dosis en la boca del niño (vea la Figura L).
c. Dele al niño un vaso de agua.
Nota: La solución oral debe administrarse dentro de la hora siguiente a llenar la jeringa.
Figura L
Almacenamiento de RINVOQ LQ
13. Cierre y guarde el frasco.
a. Con el adaptador aún insertado en el frasco, vuelva a atornillar la tapa al frasco para sellarlo (vea la Figura M).
b. Guarde el frasco en posición vertical en la caja entre usos.
c. Guarde el frasco en la caja entre 36°F y 86°F (2°C y 30°C) en un lugar fresco y oscuro.
d. Mantenga el frasco, los suministros y todos los medicamentos fuera del alcance y la vista de los niños.
Figura M
14. Enjuague y guarde la jeringa.
a. Retire el émbolo de la jeringa y luego enjuague ambas partes con agua (vea la Figura N).
No use jabón ni coloque la jeringa en el lavavajillas para limpiarla.
b. Deje que las partes separadas se sequen al aire sobre una superficie limpia.
c. Guarde la jeringa en un lugar limpio y seco.
Figura N
15. Cuando haya terminado el frasco, consulte “Eliminación de RINVOQ LQ”.

Fabricado por: AbbVie Inc., North Chicago, IL 60064, EE. UU.
RINVOQ® es una marca registrada de AbbVie Biotechnology Ltd.
©2019-2024 AbbVie Inc.
Estas Instrucciones de uso han sido aprobadas por la Administración de Alimentos y Medicamentos de los EE. UU.
Emitido: abril de 2024
20081174 R1
PANEL DE VISUALIZACIÓN PRINCIPAL
NDC 0074-2306-30
RINVOQ™
upadacitinib
Tabletas de liberación prolongada 15 mg
Distribuir en el embalaje original
VOLTEE LA TAPA PARA CORTAR LA Lámina
DESPEGUE EL REVÉS PARA LAS INSTRUCCIONES
30 Tabletas
Sólo receta médica
abbvie

PANEL DE VISUALIZACIÓN PRINCIPAL
NDC 0074-2310-30
RINVOQ®
upadacitinib
Comprimidos de liberación prolongada
30 mg
Dispensar en el envase original
GIRE LA TAPA PARA CORTAR LA Lámina
DESPEGUE PARA VER LAS INSTRUCCIONES
30 Comprimidos
Sólo con receta médica
abbvie
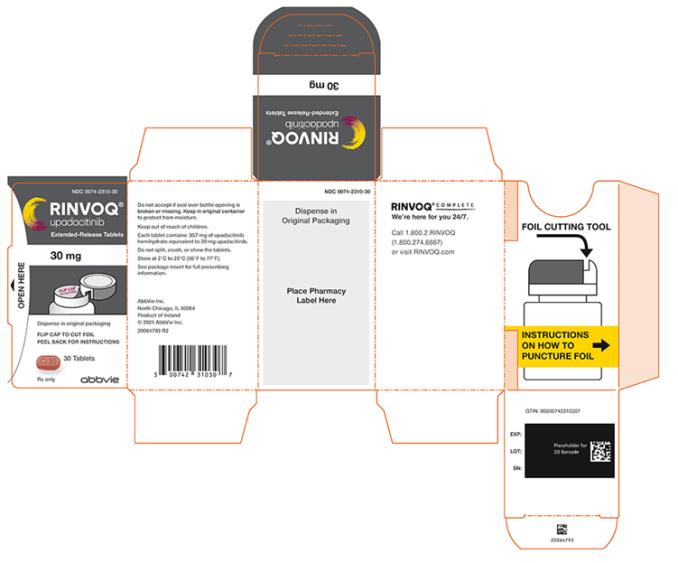
PANEL DE VISUALIZACIÓN PRINCIPAL
NDC 0074-1043-28
RINVOQ®
upadacitinib
Comprimidos de liberación prolongada
45 mg
Dispensar con la Guía de Medicamento
VOLTEE LA TAPA PARA CORTAR LA Lámina DE FOIL
DESPEGUE PARA OBTENER LAS INSTRUCCIONES
28 Comprimidos
Sólo con receta
abbvie
PANEL DE VISUALIZACIÓN PRINCIPAL
NDC 0074-2320-01
RINVOQ®LQ
upadacitinib
Solución oral
1 mg/mL
RINVOQ LQ y RINVOQ
no son sustituibles.
Inserte el adaptador de la botella
en el primer uso.
No retire
tras la inserción.
180 mL por botella
Sólo con receta
abbvie