Fabricante de medicamentos: Novartis Pharmaceuticals Corporation (Updated: 2025-01-03)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
PROMACTA® (eltrombopag) tabletas, para uso oral
PROMACTA® (eltrombopag) para suspensión oral
Aprobación inicial en EE. UU.: 2008
ADVERTENCIA: RIESGO DE DESCOMPENSACIÓN HEPÁTICA EN PACIENTES CON HEPATITIS C CRÓNICA y RIESGO DE HEPATOTOXICIDAD
Consulte la información de prescripción completa para ver la advertencia completa en recuadro.
En pacientes con hepatitis C crónica, PROMACTA en combinación con interferón y ribavirina puede aumentar el riesgo de descompensación hepática. (5.1)
PROMACTA puede aumentar el riesgo de hepatotoxicidad grave y potencialmente mortal. Controle la función hepática y suspenda la dosificación según lo recomendado. (5.2)
INDICACIONES Y USO
PROMACTA es un agonista del receptor de trombopoyetina indicado:
- para el tratamiento de la trombocitopenia en pacientes adultos y pediátricos de 1 año o mayores con trombocitopenia inmunitaria (PTI) persistente o crónica que han tenido una respuesta insuficiente a corticosteroides, inmunoglobulinas o esplenectomía. PROMACTA debe usarse solo en pacientes con PTI cuyo grado de trombocitopenia y condición clínica aumentan el riesgo de sangrado. (1.1)
- para el tratamiento de la trombocitopenia en pacientes con hepatitis C crónica para permitir el inicio y el mantenimiento de la terapia basada en interferón. PROMACTA debe usarse solo en pacientes con hepatitis C crónica cuyo grado de trombocitopenia impide el inicio de la terapia basada en interferón o limita la capacidad de mantener la terapia basada en interferón. (1.2)
- en combinación con la terapia inmunosupresora estándar para el tratamiento de primera línea de pacientes adultos y pediátricos de 2 años o mayores con anemia aplásica grave. (1.3)
- para el tratamiento de pacientes con anemia aplásica grave que han tenido una respuesta insuficiente a la terapia inmunosupresora. (1.3)
Limitaciones de uso:
DOSIFICACIÓN Y ADMINISTRACIÓN
- Tome PROMACTA sin alimentos o con una comida baja en calcio (≤ 50 mg). Tome PROMACTA al menos 2 horas antes o 4 horas después de cualquier medicamento o producto que contenga cationes polivalentes, como antiácidos, alimentos ricos en calcio y suplementos minerales. (2.4, 7.1, 12.3)
- PTI persistente o crónica: Inicie PROMACTA con 50 mg una vez al día para la mayoría de los pacientes adultos y pediátricos de 6 años o mayores, y con 25 mg una vez al día para pacientes pediátricos de 1 a 5 años. Se necesitan reducciones de dosis para pacientes con insuficiencia hepática y algunos pacientes de ascendencia del este/sudeste asiático. Ajuste para mantener un recuento de plaquetas mayor o igual a 50 x 109/L. No exceda los 75 mg por día. (2.1, 8.6, 8.7)
- Trombocitopenia asociada a hepatitis C crónica: Inicie PROMACTA con 25 mg una vez al día para todos los pacientes. Ajuste para lograr el recuento de plaquetas objetivo requerido para iniciar la terapia antiviral. No exceda una dosis diaria de 100 mg. (2.2)
- Anemia aplásica grave de primera línea: Inicie PROMACTA una vez al día con 2.5 mg/kg (en pacientes pediátricos de 2 a 5 años), 75 mg (pacientes pediátricos de 6 a 11 años) o 150 mg para pacientes de 12 años o mayores concomitantemente con la terapia inmunosupresora estándar. Reduzca la dosis inicial en pacientes de ascendencia del este/sudeste asiático. Modifique la dosis por toxicidad o recuentos elevados de plaquetas. (2.3, 8.7)
- Anemia aplásica grave refractaria: Inicie PROMACTA con 50 mg una vez al día. Reduzca la dosis inicial en pacientes con insuficiencia hepática o pacientes de ascendencia del este/sudeste asiático. Ajuste para mantener un recuento de plaquetas mayor a 50 x 109/L. No exceda los 150 mg por día. (2.3, 8.6, 8.7)
FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
CONTRAINDICACIONES
Ninguna. (4)
ADVERTENCIAS Y PRECAUCIONES
- Hepatotoxicidad: Monitorizar la función hepática antes y durante el tratamiento. (5.2)
- Mayor riesgo de muerte y progresión de síndromes mielodisplásicos a leucemia mieloide aguda. (5.3)
- Complicaciones trombóticas/tromboembólicas: Se ha notificado trombosis de la vena porta en pacientes con enfermedad hepática crónica que reciben PROMACTA. Controlar el recuento de plaquetas con regularidad. (5.4)
REACCIONES ADVERSAS
En todas las indicaciones, las reacciones adversas más comunes (≥ 20% en cualquier indicación) fueron: anemia, náuseas, pirexia, aumento de la alanina aminotransferasa, tos, fatiga, dolor de cabeza y diarrea. (6.1)
Para informar de REACCIONES ADVERSAS SOSPECHOSAS, póngase en contacto con Novartis Pharmaceuticals Corporation en el teléfono 1-888-669-6682 o con la FDA en el teléfono 1-800-FDA-1088 o en www.fda.gov/medwatch.
USO EN POBLACIONES ESPECÍFICAS
- Lactancia: Aconsejar a las mujeres que no amamanten durante el tratamiento. (8.2)
Consulte 17 para obtener INFORMACIÓN DE ASESORAMIENTO AL PACIENTE y la Guía del Medicamento.
Revisado: 3/2023
Tabla de contenido
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDO*
ADVERTENCIA: RIESGO DE DESCOMPENSACIÓN HEPÁTICA EN PACIENTES CON HEPATITIS C CRÓNICA Y RIESGO DE HEPATOTOXICIDAD
1 INDICACIONES Y USO
1.1 Tratamiento de la Trombocitopenia en Pacientes con Trombocitopenia Inmune Persistente o Crónica
1.2 Tratamiento de la Trombocitopenia en Pacientes con Infección por Hepatitis C
1.3 Tratamiento de la Anemia Aplásica Grave
1.4 Limitaciones de Uso
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Trombocitopenia Inmune Persistente o Crónica
2.2 Trombocitopenia Asociada a Hepatitis C Crónica
2.3 Anemia Aplásica Grave
2.4 Administración
3 FORMAS Y CONCENTRACIONES FARMACÉUTICAS
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Descompensación Hepática en Pacientes con Hepatitis C Crónica
5.2 Hepatotoxicidad
5.3 Mayor Riesgo de Muerte y Progresión de Síndromes Mielodisplásicos a Leucemia Mieloide Aguda
5.4 Complicaciones Trombóticas/Tromboembólicas
5.5 Cataratas
6 REACCIONES ADVERSAS
6.1 Experiencia en Ensayos Clínicos
6.2 Experiencia Postcomercialización
7 INTERACCIONES MEDICAMENTOSAS
7.1 Cationes Polivalentes (Quelación)
7.2 Transportadores
7.3 Inhibidores de la Proteasa
7.4 Terapia con Peginterferón Alfa-2a/b
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.3 Mujeres y Hombres en Edad Reproductiva
8.4 Uso Pediátrico
8.5 Uso Geriátrico
8.6 Insuficiencia Hepática
8.7 Etnicidad
10 SOBREDOSIFICACIÓN
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de Acción
12.2 Farmacodinamia
12.3 Farmacocinética
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
13.2 Farmacología y/o Toxicología Animal
14 ESTUDIOS CLÍNICOS
14.1 ITP Persistente o Crónica
14.2 Trombocitopenia Asociada a Hepatitis C Crónica
14.3 Anemia Aplásica Grave
16 PRESENTACIÓN/ALMACENAMIENTO Y MANEJO
16.1 Comprimidos
16.2 Para Suspensión Oral
17 INFORMACIÓN PARA EL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información completa de prescripción no se enumeran.
ADVERTENCIA EN EL RECUADRO
ADVERTENCIA: RIESGO DE DESCOMPENSACIÓN HEPÁTICA EN PACIENTES CON HEPATITIS C CRÓNICA Y RIESGO DE HEPATOTOXICIDAD
En pacientes con hepatitis C crónica, PROMACTA en combinación con interferón y ribavirina puede aumentar el riesgo de descompensación hepática [ver Advertencias y precauciones (5.1)].
PROMACTA puede aumentar el riesgo de hepatotoxicidad grave y potencialmente mortal. Controle la función hepática e interrumpa la administración según las recomendaciones [ver Advertencias y precauciones (5.2)].
1 INDICACIONES Y USO
1.1 Tratamiento de la trombocitopenia en pacientes con trombocitopenia inmune persistente o crónica
PROMACTA está indicado para el tratamiento de la trombocitopenia en pacientes adultos y pediátricos de 1 año o más con trombocitopenia inmune (ITP) persistente o crónica que han tenido una respuesta insuficiente a los corticosteroides, inmunoglobulinas o esplenectomía. PROMACTA solo debe usarse en pacientes con ITP cuyo grado de trombocitopenia y condición clínica aumenten el riesgo de hemorragia.
1.2 Tratamiento de la trombocitopenia en pacientes con infección por hepatitis C
PROMACTA está indicado para el tratamiento de la trombocitopenia en pacientes con hepatitis C crónica para permitir el inicio y el mantenimiento de la terapia basada en interferón. PROMACTA solo debe usarse en pacientes con hepatitis C crónica cuyo grado de trombocitopenia impida el inicio de la terapia basada en interferón o limite la capacidad de mantener la terapia basada en interferón.
1.3 Tratamiento de la anemia aplásica grave
- PROMACTA está indicado en combinación con terapia inmunosupresora estándar (IST) para el tratamiento de primera línea de pacientes adultos y pediátricos de 2 años o más con anemia aplásica grave.
- PROMACTA está indicado para el tratamiento de pacientes con anemia aplásica grave que han tenido una respuesta insuficiente a la terapia inmunosupresora.
1.4 Limitaciones de uso
- PROMACTA no está indicado para el tratamiento de pacientes con síndromes mielodisplásicos (SMD) [ver Advertencias y precauciones (5.3)].
- No se ha establecido la seguridad y eficacia en combinación con agentes antivirales de acción directa utilizados sin interferón para el tratamiento de la infección por hepatitis C crónica.
2 DOSIS Y ADMINISTRACIÓN
2.1 Trombocitopenia Inmune Persistente o Crónica
Use la dosis más baja de PROMACTA para lograr y mantener un recuento plaquetario mayor o igual a 50 x 109/L según sea necesario para reducir el riesgo de hemorragia. Los ajustes de la dosis se basan en la respuesta del recuento plaquetario. No use PROMACTA para normalizar los recuentos plaquetarios [véase Advertencias y precauciones (5.4)]. En los ensayos clínicos, los recuentos plaquetarios generalmente aumentaron entre 1 y 2 semanas después de comenzar el tratamiento con PROMACTA y disminuyeron entre 1 y 2 semanas después de suspender PROMACTA [véase Estudios clínicos (14.1)].
Régimen de dosis inicial:
Pacientes adultos y pediátricos de 6 años o más con ITP: Inicie PROMACTA a una dosis de 50 mg una vez al día, excepto en pacientes de ascendencia del este o sudeste asiático o que tengan insuficiencia hepática leve a grave (Child-Pugh clase A, B, C).
Para pacientes de ascendencia del este o sudeste asiático con ITP, inicie PROMACTA a una dosis reducida de 25 mg una vez al día [véase Uso en poblaciones específicas (8.7), Farmacología clínica (12.3)].
Para pacientes con ITP e insuficiencia hepática leve, moderada o grave (Child-Pugh clase A, B, C), inicie PROMACTA a una dosis reducida de 25 mg una vez al día [véase Uso en poblaciones específicas (8.6), Farmacología clínica (12.3)].
Para pacientes de ascendencia del este o sudeste asiático con ITP e insuficiencia hepática (Child-Pugh clase A, B, C), considere iniciar PROMACTA a una dosis reducida de 12.5 mg una vez al día [véase Farmacología clínica (12.3)].
Pacientes pediátricos con ITP de 1 a 5 años: Inicie PROMACTA a una dosis de 25 mg una vez al día [véase Uso en poblaciones específicas (8.7), Farmacología clínica (12.3)].
Monitorización y ajuste de la dosis: Después de iniciar el tratamiento con PROMACTA, ajuste la dosis para lograr y mantener un recuento plaquetario mayor o igual a 50 x 109/L según sea necesario para reducir el riesgo de hemorragia. No exceda una dosis de 75 mg al día. Controle regularmente la hematología clínica y las pruebas de función hepática durante todo el tratamiento con PROMACTA y modifique el régimen de dosificación de PROMACTA según los recuentos plaquetarios como se describe en la Tabla 1. Durante el tratamiento con PROMACTA, evalúe los hemogramas completos (CBC) con diferenciales, incluidos los recuentos plaquetarios, semanalmente hasta que se haya logrado un recuento plaquetario estable. Obtenga hemogramas completos con diferenciales, incluidos los recuentos plaquetarios, mensualmente a partir de entonces.
Cuando se cambie entre la suspensión oral y la tableta, evalúe los recuentos plaquetarios semanalmente durante 2 semanas y luego siga el control mensual estándar.
|
Resultado del recuento plaquetario |
Ajuste de la dosis o respuesta |
|
< 50 x 109/L después de al menos 2 semanas de PROMACTA |
Aumente la dosis diaria en 25 mg hasta un máximo de 75 mg/día. Para los pacientes que toman 12.5 mg una vez al día, aumente la dosis a 25 mg diarios antes de aumentar la cantidad de la dosis en 25 mg. |
|
≥ 200 x 109/L a ≤ 400 x 109/L en cualquier momento |
Disminuya la dosis diaria en 25 mg. Espere 2 semanas para evaluar los efectos de este ajuste de dosis y de cualquier ajuste posterior. Para los pacientes que toman 25 mg una vez al día, disminuya la dosis a 12.5 mg una vez al día. |
|
> 400 x 109/L |
Suspenda PROMACTA; aumente la frecuencia del control plaquetario a dos veces por semana. Una vez que el recuento plaquetario sea < 150 x 109/L, reinicie el tratamiento con una dosis diaria reducida en 25 mg. Para los pacientes que toman 25 mg una vez al día, reinicie el tratamiento con una dosis diaria de 12.5 mg. |
|
> 400 x 109/L después de 2 semanas de tratamiento con la dosis más baja de PROMACTA |
Suspenda PROMACTA. |
En pacientes con PTI y insuficiencia hepática (Child-Pugh clase A, B, C), después de iniciar PROMACTA o después de cualquier aumento posterior de la dosis, espere 3 semanas antes de aumentar la dosis.
Modifique el régimen de dosificación de los medicamentos concomitantes para la PTI, según sea médicamente apropiado, para evitar aumentos excesivos en el recuento de plaquetas durante el tratamiento con PROMACTA. No administre más de una dosis de PROMACTA dentro de un período de 24 horas.
Suspensión: Suspenda PROMACTA si el recuento de plaquetas no aumenta a un nivel suficiente para evitar hemorragias clínicamente importantes después de 4 semanas de tratamiento con PROMACTA a la dosis diaria máxima de 75 mg. Las respuestas excesivas del recuento de plaquetas, como se describe en la Tabla 1, o las anomalías importantes en las pruebas de función hepática también requieren la suspensión de PROMACTA [ver Advertencias y precauciones (5.2)]. Obtenga hemogramas completos con diferenciales, incluyendo recuentos de plaquetas, semanalmente durante al menos 4 semanas después de la suspensión de PROMACTA.
2.2 Trombocitopenia asociada a hepatitis C crónica
Use la dosis más baja de PROMACTA para lograr y mantener un recuento de plaquetas necesario para iniciar y mantener la terapia antiviral con interferón pegilado y ribavirina. Los ajustes de dosis se basan en la respuesta del recuento de plaquetas. No use PROMACTA para normalizar los recuentos de plaquetas [ver Advertencias y precauciones (5.4)]. En los ensayos clínicos, los recuentos de plaquetas generalmente comenzaron a aumentar en la primera semana de tratamiento con PROMACTA [ver Estudios clínicos (14.2)].
Régimen de dosis inicial: Inicie PROMACTA a una dosis de 25 mg una vez al día.
Monitorización y ajuste de la dosis: Ajuste la dosis de PROMACTA en incrementos de 25 mg cada 2 semanas según sea necesario para lograr el recuento de plaquetas objetivo requerido para iniciar la terapia antiviral. Controle los recuentos de plaquetas semanalmente antes de comenzar la terapia antiviral.
Durante la terapia antiviral, ajuste la dosis de PROMACTA para evitar reducciones de la dosis de peginterferón. Controle los hemogramas completos con diferenciales, incluyendo recuentos de plaquetas, semanalmente durante la terapia antiviral hasta que se logre un recuento de plaquetas estable. Controle los recuentos de plaquetas mensualmente a partir de entonces. No exceda una dosis de 100 mg diarios. Controle la hematología clínica y las pruebas de función hepática regularmente durante todo el tratamiento con PROMACTA.
Para obtener instrucciones específicas sobre la dosificación de peginterferón o ribavirina, consulte la información de prescripción correspondiente.
|
Resultado del recuento de plaquetas |
Ajuste de dosis o respuesta |
|
< 50 x 109/L después de al menos 2 semanas de PROMACTA |
Aumente la dosis diaria en 25 mg hasta un máximo de 100 mg/día. |
|
≥ 200 x 109/L a ≤ 400 x 109/L en cualquier momento |
Disminuya la dosis diaria en 25 mg. Espere 2 semanas para evaluar los efectos de este y cualquier ajuste posterior de la dosis. |
|
> 400 x 109/L |
Suspenda PROMACTA; aumente la frecuencia del control de plaquetas a dos veces por semana. Una vez que el recuento de plaquetas sea < 150 x 109/L, reinicie el tratamiento con una dosis diaria reducida en 25 mg. Para pacientes que toman 25 mg una vez al día, reinicie el tratamiento con una dosis diaria de 12,5 mg. |
|
> 400 x 109/L después de 2 semanas de terapia con la dosis más baja de PROMACTA |
Suspenda PROMACTA. |
Suspensión: La información de prescripción para el interferón pegilado y la ribavirina incluye recomendaciones para la suspensión del tratamiento antiviral por inutilidad del tratamiento. Consulte la información de prescripción de interferón pegilado y ribavirina para obtener recomendaciones sobre la suspensión del tratamiento antiviral por inutilidad del tratamiento.
PROMACTA debe suspenderse cuando se suspenda la terapia antiviral. Las respuestas excesivas del recuento de plaquetas, como se describe en la Tabla 2, o las anomalías importantes en las pruebas de función hepática también requieren la suspensión de PROMACTA [ver Advertencias y precauciones (5.2)].
2.3 Anemia aplásica grave
Anemia aplásica grave de primera línea
Inicie PROMACTA concurrentemente con la terapia inmunosupresora estándar [ver Estudios clínicos (14.3)].
Régimen de dosis inicial
El régimen de dosis inicial recomendado se muestra en la Tabla 3. No exceda la dosis inicial de PROMACTA.
|
Edad |
Régimen de dosis |
|
Pacientes de 12 años o más |
150 mg una vez al día durante 6 meses |
|
Pacientes pediátricos de 6 a 11 años |
75 mg una vez al día durante 6 meses |
|
Pacientes pediátricos de 2 a 5 años |
2,5 mg/kg una vez al día durante 6 meses |
Para pacientes con anemia aplásica severa de ascendencia del este/sudeste asiático o aquellos con insuficiencia hepática leve, moderada o severa (Child-Pugh clase A, B, C), disminuya la dosis inicial de PROMACTA en un 50% como se indica en la Tabla 4 [ver Uso en Poblaciones Específicas (8.6, 8.7), Farmacología Clínica (12.3)].
Si los niveles basales de alanino aminotransferasa (ALT) o aspartato aminotransferasa (AST) son > 6 x límite superior de lo normal (LSN), no inicie PROMACTA hasta que los niveles de transaminasas sean < 5 x LSN. Determine la dosis inicial para estos pacientes según la Tabla 3 o la Tabla 4.
|
Edad |
Régimen de dosis |
|
Pacientes de 12 años o más |
75 mg una vez al día durante 6 meses |
|
Pacientes pediátricos de 6 a 11 años |
37.5 mg una vez al día durante 6 meses |
|
Pacientes pediátricos de 2 a 5 años |
1.25 mg/kg una vez al día durante 6 meses |
Monitorización y ajuste de la dosis de PROMACTA: Realice análisis hematológicos clínicos y pruebas hepáticas regularmente durante todo el tratamiento con PROMACTA [ver Advertencias y precauciones (5.2)].
Modifique el régimen de dosificación de PROMACTA según el recuento de plaquetas como se describe en la Tabla 5.
|
Resultado del recuento de plaquetas |
Ajuste de dosis o respuesta |
|
> 200 x 109/L a ≤ 400 x 109/L |
Disminuya la dosis diaria en 25 mg cada 2 semanas hasta la dosis más baja que mantenga el recuento de plaquetas ≥ 50 x 109/L. |
|
> 400 x 109/L |
Suspenda PROMACTA durante una semana. Una vez que el recuento de plaquetas sea < 200 x 109/L, reinicie PROMACTA a una dosis diaria reducida en 25 mg (o 12.5 mg en pacientes pediátricos menores de 12 años). |
La Tabla 6 resume las recomendaciones para la interrupción, reducción o suspensión de la dosis de PROMACTA en el manejo de los niveles elevados de transaminasas hepáticas y los eventos tromboembólicos.
| Abreviaturas: ALT, alanino aminotransferasa; AST, aspartato aminotransferasa; LSN, límite superior de lo normal. | |
|
Evento |
Recomendación |
|
Elevaciones de ALT o AST |
Aumento de ALT o AST > 6 x LSN
Aumento de ALT o AST > 6 x LSN después de reiniciar PROMACTA
Si ALT o AST vuelve a ser > 6 x LSN con la dosis reducida En pacientes pediátricos menores de 12 años, reduzca la dosis diaria al menos en un 15% a la dosis más cercana que se pueda administrar. |
|
Eventos tromboembólicos (p. ej., trombosis venosa profunda, embolia pulmonar, accidente cerebrovascular, infarto de miocardio) |
Suspenda PROMACTA pero permanezca con globulina antitimocítica equina (h-ATG) y ciclosporina. |
La duración total del tratamiento con PROMACTA es de 6 meses.
Anemia aplásica refractaria grave
Use la dosis más baja de PROMACTA para lograr y mantener una respuesta hematológica. Los ajustes de dosis se basan en el recuento de plaquetas. La respuesta hematológica requiere titulación de la dosis, generalmente hasta 150 mg, y puede tardar hasta 16 semanas después de comenzar el tratamiento con PROMACTA [véase Estudios clínicos (14.3)].
Régimen de dosis inicial: Inicie el tratamiento con PROMACTA a una dosis de 50 mg una vez al día.
Para pacientes con anemia aplásica grave de ascendencia del este/sudeste asiático o aquellos con insuficiencia hepática leve, moderada o grave (clase A, B, C de Child-Pugh), inicie PROMACTA a una dosis reducida de 25 mg una vez al día [véase Uso en poblaciones específicas (8.6, 8.7), Farmacología clínica (12.3)].
Monitorización y ajuste de la dosis: Ajuste la dosis de PROMACTA en incrementos de 50 mg cada 2 semanas según sea necesario para lograr el recuento de plaquetas objetivo mayor o igual a 50 x 109/L según sea necesario. No exceda una dosis de 150 mg diarios. Controle la hematología clínica y las pruebas de función hepática regularmente durante todo el tratamiento con PROMACTA y modifique el régimen de dosificación de PROMACTA según los recuentos de plaquetas como se describe en la Tabla 7.
|
Resultado del recuento de plaquetas |
Ajuste de dosis o respuesta |
|
< 50 x 109/L después de al menos 2 semanas de tratamiento con PROMACTA |
Aumente la dosis diaria en 50 mg hasta un máximo de 150 mg/día. Para pacientes que toman 25 mg una vez al día, aumente la dosis a 50 mg diarios antes de aumentar la cantidad de la dosis en 50 mg. |
|
≥ 200 x 109/L a ≤ 400 x 109/L en cualquier momento |
Disminuya la dosis diaria en 50 mg. Espere 2 semanas para evaluar los efectos de este ajuste de dosis y de los ajustes posteriores. |
|
> 400 x 109/L |
Suspenda PROMACTA durante 1 semana. Una vez que el recuento de plaquetas sea < 150 x 109/L, reinicie el tratamiento con una dosis reducida en 50 mg. |
|
> 400 x 109/L después de 2 semanas de tratamiento con la dosis más baja de PROMACTA |
Suspenda PROMACTA. |
Para los pacientes que logran una respuesta trilineal, incluida la independencia de transfusiones, que dure al menos 8 semanas: la dosis de PROMACTA puede reducirse en un 50% [véase Estudios clínicos (14.3)]. Si los recuentos permanecen estables después de 8 semanas con la dosis reducida, suspenda PROMACTA y controle los recuentos sanguíneos. Si el recuento de plaquetas desciende a menos de 30 x 109/L, la hemoglobina a menos de 9 g/dL o el recuento absoluto de neutrófilos (RAN) a menos de 0,5 x 109/L, se puede reiniciar el tratamiento con PROMACTA a la dosis eficaz anterior.
Suspensión del tratamiento: Si no se ha producido una respuesta hematológica después de 16 semanas de tratamiento con PROMACTA, suspenda el tratamiento. Si se observan nuevas anomalías citogenéticas, considere la suspensión de PROMACTA [véase Reacciones adversas (6.1)]. Las respuestas excesivas en el recuento de plaquetas (como se describe en la Tabla 7) o las anomalías importantes en las pruebas de función hepática también requieren la suspensión de PROMACTA [véase Advertencias y precauciones (5.2)].
2.4 Administración
Administración de comprimidos y suspensión oral: Tome PROMACTA sin alimentos o con una comida baja en calcio (≤ 50 mg). Tome PROMACTA al menos 2 horas antes o 4 horas después de otros medicamentos (p. ej., antiácidos), alimentos ricos en calcio (que contengan > 50 mg de calcio, p. ej., productos lácteos, zumos enriquecidos con calcio y ciertas frutas y verduras) o suplementos que contengan cationes polivalentes, como hierro, calcio, aluminio, magnesio, selenio y zinc [véase Interacciones medicamentosas (7.1), Farmacología clínica (12.3)].
No parta, mastique ni triture los comprimidos y no los mezcle con alimentos ni líquidos.
Preparación de la suspensión oral: Antes de usar la suspensión oral, asegúrese de que los pacientes o cuidadores reciban capacitación sobre la dosificación, preparación y administración adecuadas de PROMACTA para suspensión oral.
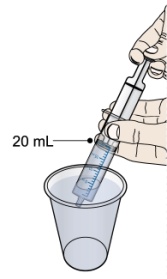
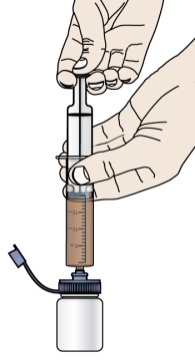
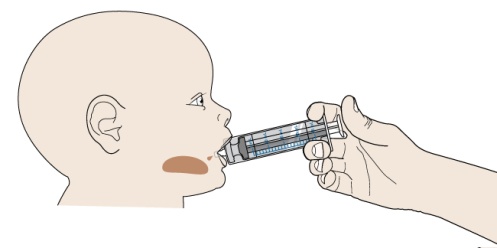
Administre la suspensión oral inmediatamente después de la preparación. Deseche cualquier suspensión que no se administre dentro de los 30 minutos posteriores a la preparación.
Prepare la suspensión solo con agua. NOTA: No utilice agua caliente para preparar la suspensión.
Para obtener más información sobre la preparación y administración de la suspensión, incluida la duración recomendada de uso de cada jeringa dosificadora oral, [véase Instrucciones de uso].
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
Comprimidos
- Comprimidos de 12.5 mg —– comprimidos redondos, biconvexos, blancos, recubiertos con película, con “GS MZ1” y 12.5 grabados en un lado. Cada comprimido, para administración oral, contiene eltrombopag olamine, equivalente a 12.5 mg de eltrombopag ácido libre.
- Comprimidos de 25 mg —– comprimidos redondos, biconvexos, anaranjados, recubiertos con película, con “GS NX3” y 25 grabados en un lado. Cada comprimido, para administración oral, contiene eltrombopag olamine, equivalente a 25 mg de eltrombopag ácido libre.
- Comprimidos de 50 mg —– comprimidos redondos, biconvexos, azules, recubiertos con película, con “GS UFU” y 50 grabados en un lado. Cada comprimido, para administración oral, contiene eltrombopag olamine, equivalente a 50 mg de eltrombopag ácido libre.
- Comprimidos de 75 mg —– comprimidos redondos, biconvexos, rosados, recubiertos con película, con “GS FFS” y 75 grabados en un lado. Cada comprimido, para administración oral, contiene eltrombopag olamine, equivalente a 75 mg de eltrombopag ácido libre.
Para suspensión oral
- Sobre de 12.5 mg —– contiene un polvo de color marrón rojizo a amarillo para reconstitución.
- Sobre de 25 mg —– contiene un polvo de color marrón rojizo a amarillo para reconstitución.
4 CONTRAINDICACIONES
Ninguna.
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Descompensación hepática en pacientes con hepatitis C crónica
En pacientes con hepatitis C crónica, PROMACTA en combinación con interferón y ribavirina puede aumentar el riesgo de descompensación hepática. En dos ensayos clínicos controlados en pacientes con hepatitis C crónica y trombocitopenia, la ascitis y la encefalopatía ocurrieron con mayor frecuencia en el brazo que recibió tratamiento con PROMACTA más antivirales (7%) que en el brazo con placebo más antivirales (4%). Los pacientes con niveles bajos de albúmina (menos de 3.5 g/dL) o una puntuación de Modelo para la Enfermedad Hepática en Estadio Terminal (MELD) mayor o igual a 10 al inicio del tratamiento tuvieron un mayor riesgo de descompensación hepática en el brazo que recibió tratamiento con PROMACTA más antivirales. Suspenda PROMACTA si se interrumpe el tratamiento antiviral.
5.2 Hepatotoxicidad
PROMACTA puede aumentar el riesgo de hepatotoxicidad grave y potencialmente mortal [ver Reacciones adversas (6.1)]. Un paciente (< 1%) con PTI tratado con PROMACTA en ensayos clínicos experimentó una lesión hepática inducida por fármacos. Once pacientes (1%) con hepatitis C crónica tratados con PROMACTA en ensayos clínicos experimentaron una lesión hepática inducida por fármacos.
Tratamiento de la PTI, la trombocitopenia asociada a la hepatitis C crónica y la anemia aplásica refractaria grave
Mida los niveles séricos de ALT, AST y bilirrubina antes de iniciar el tratamiento con PROMACTA, cada 2 semanas durante la fase de ajuste de la dosis y mensualmente después del establecimiento de una dosis estable. PROMACTA inhibe la UDP-glucuronosiltransferasa (UGT)1A1 y la polipéptido transportador de aniones orgánicos (OATP)1B1, lo que puede provocar hiperbilirrubinemia indirecta. Si la bilirrubina está elevada, realice una fraccionación. Evalúe las pruebas hepáticas séricas anormales con pruebas repetidas dentro de los 3 a 5 días. Si se confirman las anomalías, controle las pruebas hepáticas séricas semanalmente hasta que se resuelvan o se estabilicen. Suspenda PROMACTA si los niveles de ALT aumentan a más del triple del límite superior de la normalidad (LSN) en pacientes con función hepática normal o a más del triple del valor basal (o más del quíntuple del LSN, el que sea menor) en pacientes con elevaciones previas al tratamiento de las transaminasas y son:
- progresivamente crecientes, o
- persistentes durante más de 4 semanas, o
- acompañadas de un aumento de la bilirrubina directa, o
- acompañadas de síntomas clínicos de lesión hepática o evidencia de descompensación hepática.
Si se considera que el beneficio potencial de reiniciar el tratamiento con PROMACTA supera el riesgo de hepatotoxicidad, entonces considere la posibilidad de reintroducir PROMACTA con precaución y mida las pruebas hepáticas séricas semanalmente durante la fase de ajuste de la dosis. La hepatotoxicidad puede reaparecer si se reinicia el tratamiento con PROMACTA. Si las anomalías de las pruebas hepáticas persisten, empeoran o reaparecen, suspenda permanentemente PROMACTA.
Tratamiento de primera línea de la anemia aplásica grave
Mida los niveles de ALT, AST y bilirrubina antes de iniciar el tratamiento con PROMACTA, cada dos días mientras esté hospitalizado para la terapia con h-ATG, y luego cada 2 semanas durante el tratamiento. Durante el tratamiento, maneje los aumentos en los niveles de ALT o AST como se recomienda en la Tabla 6.
5.3 Aumento del riesgo de muerte y progresión de los síndromes mielodisplásicos a leucemia mieloide aguda
Un ensayo aleatorizado, doble ciego, controlado con placebo, multicéntrico en pacientes con síndrome mielodisplásico (SMD) de riesgo intermedio-1, intermedio-2 o alto según el Sistema Internacional de Puntuación Pronóstica (IPSS) con trombocitopenia, que recibieron azacitidina en combinación con PROMACTA (n = 179) o placebo (n = 177) se interrumpió debido a la falta de eficacia y razones de seguridad, incluido el aumento de la progresión a leucemia mieloide aguda (LMA). Los pacientes recibieron PROMACTA o placebo a una dosis inicial de 200 mg una vez al día, hasta un máximo de 300 mg una vez al día, en combinación con azacitidina durante al menos seis ciclos. La incidencia de muerte (supervivencia general) fue del 32% (57/179) en el brazo de PROMACTA frente al 29% (51/177) en el brazo de placebo (HR [IC del 95%] = 1,42 [0,97, 2,08], mostrando un aumento del riesgo relativo de muerte en este ensayo del 42% en el brazo de PROMACTA). La incidencia de progresión a LMA fue del 12% (21/179) en el brazo de PROMACTA frente al 6% (10/177) en el brazo de placebo (HR [IC del 95%] = 2,66 [1,31, 5,41], mostrando un aumento del riesgo relativo de progresión a LMA en este ensayo del 166% en el brazo de PROMACTA).
5.4 Complicaciones trombóticas/tromboembólicas
Las complicaciones trombóticas/tromboembólicas pueden ser consecuencia del aumento del recuento plaquetario con PROMACTA. Las complicaciones trombóticas/tromboembólicas notificadas incluyeron eventos venosos y arteriales y se observaron con recuentos plaquetarios bajos y normales.
Considere la posibilidad de un mayor riesgo de tromboembolismo al administrar PROMACTA a pacientes con factores de riesgo conocidos para tromboembolismo (p. ej., Factor V de Leiden, deficiencia de ATIII, síndrome antifosfolípido, enfermedad hepática crónica). Para minimizar el riesgo de complicaciones trombóticas/tromboembólicas, no utilice PROMACTA en un intento de normalizar el recuento plaquetario. Siga las pautas de ajuste de la dosis para lograr y mantener los recuentos plaquetarios objetivo [ver Posología y administración (2.1, 2.2, 2.3)].
En dos ensayos clínicos controlados en pacientes con hepatitis C crónica y trombocitopenia, el 3% (31/955) tratados con PROMACTA experimentaron un evento trombótico en comparación con el 1% (5/484) con placebo. La mayoría de los eventos fueron del sistema venoso portal (1% en pacientes tratados con PROMACTA frente a menos del 1% con placebo).
En un ensayo controlado en pacientes con enfermedad hepática crónica y trombocitopenia no relacionada con ITP que se sometieron a procedimientos invasivos electivos (N = 292), el riesgo de eventos trombóticos aumentó en los pacientes tratados con 75 mg de PROMACTA una vez al día. Se informaron siete complicaciones trombóticas (seis pacientes) en el grupo que recibió PROMACTA y tres complicaciones trombóticas en el grupo placebo (dos pacientes). Todas las complicaciones trombóticas informadas en el grupo que recibió PROMACTA fueron trombosis de la vena porta (TVP). Los síntomas de la TVP incluyeron dolor abdominal, náuseas, vómitos y diarrea. Cinco de los seis pacientes del grupo que recibió PROMACTA experimentaron una complicación trombótica dentro de los 30 días posteriores a la finalización del tratamiento con PROMACTA y con un recuento de plaquetas superior a 200 x 109/L. El riesgo de trombosis venosa portal aumentó en pacientes trombocitopénicos con enfermedad hepática crónica tratados con 75 mg de PROMACTA una vez al día durante 2 semanas en preparación para procedimientos invasivos.
5.5 Cataratas
En los tres ensayos clínicos controlados en adultos con ITP persistente o crónica, se desarrollaron o empeoraron cataratas en 15 (7%) pacientes que recibieron 50 mg de PROMACTA diarios y en 8 (7%) pacientes del grupo placebo. En el ensayo de extensión, las cataratas se desarrollaron o empeoraron en el 11% de los pacientes que se sometieron a un examen ocular antes del tratamiento con PROMACTA. En los dos ensayos clínicos controlados en pacientes con hepatitis C crónica y trombocitopenia, las cataratas se desarrollaron o empeoraron en el 8% de los pacientes tratados con PROMACTA y en el 5% de los pacientes tratados con placebo.
Se observaron cataratas en estudios de toxicología de eltrombopag en roedores [ver Toxicología no clínica (13.2)]. Realice un examen ocular basal antes de la administración de PROMACTA y, durante el tratamiento con PROMACTA, controle regularmente a los pacientes para detectar signos y síntomas de cataratas.
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas clínicamente significativas asociadas con PROMACTA se describen en otras secciones.
- Descompensación hepática en pacientes con hepatitis C crónica [ver Advertencias y precauciones (5.1)]
- Hepatotoxicidad [ver Advertencias y precauciones (5.2)]
- Aumento del riesgo de muerte y progresión de síndromes mielodisplásicos a leucemia mieloide aguda [ver Advertencias y precauciones (5.3)]
- Complicaciones trombóticas/tromboembólicas [ver Advertencias y precauciones (5.4)]
- Cataratas [ver Advertencias y precauciones (5.5)]
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se realizan en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no se pueden comparar directamente con las tasas en los ensayos clínicos de otro fármaco y pueden no reflejar las tasas observadas en la práctica.
Trombocitopenia inmune persistente o crónica
Adultos: En los ensayos clínicos, la hemorragia fue la reacción adversa grave más común y la mayoría de las reacciones hemorrágicas se produjeron después de la interrupción de PROMACTA. Otras reacciones adversas graves incluyeron complicaciones trombóticas/tromboembólicas [ver Advertencias y precauciones (5.4)]. Los datos que se describen a continuación reflejan la exposición de PROMACTA a pacientes con ITP persistente o crónica de 18 a 85 años de edad, de los cuales el 66% eran mujeres, en tres ensayos controlados con placebo y un ensayo de extensión abierto [ver Estudios clínicos (14.1)]. PROMACTA se administró a 330 pacientes durante al menos 6 meses y a 218 pacientes durante al menos 1 año.
La Tabla 8 presenta las reacciones adversas a los medicamentos más comunes (experimentadas por más del 3% de los pacientes que recibieron PROMACTA) de los tres ensayos controlados con placebo, con una mayor incidencia en PROMACTA versus placebo.
| Abreviaturas: ALT, alanina aminotransferasa; AST, aspartato aminotransferasa. aIncluye ITUs de infección del tracto urinario, cistitis, infección bacteriana del tracto urinario y bacteriuria. |
||
|
Reacción adversa |
PROMACTA 50 mg n = 241 (%) |
Placebo n = 128 (%) |
|
Náuseas |
9 |
3 |
|
Diarrea |
9 |
7 |
|
Infección del tracto respiratorio superior |
7 |
6 |
|
Vómitos |
6 |
< 1 |
|
Infección del tracto urinarioa |
5 |
4 |
|
Aumento de ALT |
5 |
3 |
|
Mialgia |
5 |
2 |
|
Dolor orofaríngeo |
4 |
3 |
|
Aumento de AST |
4 |
2 |
|
Faringitis |
4 |
2 |
|
Dolor de espalda |
3 |
2 |
|
Influenza |
3 |
2 |
|
Parestesia |
3 |
2 |
|
Erupción |
3 |
2 |
En los tres ensayos clínicos controlados de ITP persistente o crónica, la alopecia, el dolor musculoesquelético, el aumento de la fosfatasa alcalina en sangre y la boca seca fueron las reacciones adversas notificadas en el 2% de los pacientes tratados con PROMACTA y en ningún paciente que recibió placebo.
Entre 302 pacientes con ITP persistente o crónica que recibieron PROMACTA en el ensayo de extensión de un solo brazo, las reacciones adversas se produjeron con un patrón similar al observado en los ensayos controlados con placebo. La Tabla 9 presenta las reacciones adversas relacionadas con el tratamiento más comunes (experimentadas por más del 3% de los pacientes que recibieron PROMACTA) del ensayo de extensión.
| Abreviaturas: ALT, alanina aminotransferasa; AST, aspartato aminotransferasa. | |
|
Reacción adversa |
PROMACTA 50 mg n = 302 (%) |
|
Dolor de cabeza |
10 |
|
ALT aumentado |
5 |
|
AST aumentado |
5 |
|
Catarata |
5 |
|
Fatiga |
5 |
|
Bilirrubina en sangre aumentada |
4 |
|
Náuseas |
4 |
|
Hiperbilirrubinemia |
3 |
|
Diarrea |
3 |
En los tres ensayos controlados de ITP persistente o crónica, se notificaron anomalías en las pruebas de función hepática en suero (principalmente de grado 2 o menos en gravedad) en el 11 % y el 7 % de los pacientes tratados con PROMACTA y placebo, respectivamente. Cuatro pacientes (1 %) tratados con PROMACTA y tres pacientes del grupo placebo (2 %) interrumpieron el tratamiento debido a anomalías en los parámetros de laboratorio hepatobiliares. Diecisiete de los pacientes tratados con PROMACTA en los ensayos controlados con anomalías en los parámetros de laboratorio hepatobiliares volvieron a exponerse a PROMACTA en el ensayo de extensión. Ocho de estos pacientes volvieron a experimentar anomalías en las pruebas de función hepática (inferiores o iguales al grado 3), lo que provocó la interrupción de PROMACTA en un paciente. En el ensayo de extensión de ITP persistente o crónica, seis pacientes adicionales interrumpieron el tratamiento con PROMACTA debido a anomalías en las pruebas de función hepática (inferiores o iguales al grado 3).
En los tres ensayos controlados de ITP persistente o crónica, se desarrollaron o empeoraron cataratas en el 7 % de los pacientes tratados con PROMACTA y en el 7 % de los pacientes del grupo placebo. Todos los pacientes tenían factores de riesgo preexistentes documentados para la cataractogénesis, incluido el uso de corticosteroides. En el ensayo de extensión, se desarrollaron o empeoraron cataratas en el 11 % de los pacientes que se sometieron a un examen ocular antes del tratamiento con PROMACTA. El setenta y dos por ciento de los pacientes tenían factores de riesgo preexistentes, incluido el uso de corticosteroides.
La seguridad de PROMACTA también se evaluó en todos los pacientes tratados en 7 ensayos clínicos de ITP persistente o crónica en adultos (N = 763 pacientes tratados con PROMACTA y 179 pacientes tratados con placebo). Se notificaron acontecimientos tromboembólicos en el 6 % de los pacientes tratados con PROMACTA frente al 0 % de los pacientes tratados con placebo, y se notificó microangiopatía trombótica con insuficiencia renal aguda en < 1 % de los pacientes tratados con PROMACTA frente al 0 % de los pacientes tratados con placebo.
En un ensayo controlado con placebo de PROMACTA en pacientes con enfermedad hepática crónica y trombocitopenia no relacionada con ITP, seis pacientes tratados con PROMACTA y un paciente del grupo placebo desarrollaron trombosis de la vena porta [véase Advertencias y precauciones (5.4)].
Pacientes pediátricos: Los datos que se describen a continuación reflejan una exposición mediana a PROMACTA de 91 días para 107 pacientes pediátricos (de 1 a 17 años) con ITP persistente o crónica, de los cuales el 53 % eran mujeres, en la fase aleatorizada de dos ensayos controlados con placebo.
La tabla 10 presenta las reacciones adversas a los medicamentos más frecuentes (experimentadas por más del 3 % de los pacientes pediátricos de 1 año o más que recibieron PROMACTA) en los dos ensayos controlados con placebo, con una mayor incidencia para PROMACTA frente a placebo.
| Abreviaturas: ALT, alanina aminotransferasa; AST, aspartato aminotransferasa. aIncluye reacciones adversas o anomalías de laboratorio > 3 x LSN. |
||
|
PROMACTA |
Placebo |
|
|
n = 107 |
n = 50 |
|
|
Reacción adversa |
(%) |
(%) |
|
Infección de las vías respiratorias altas |
17 |
6 |
|
Nasofaringitis |
12 |
4 |
|
Tos |
9 |
0 |
|
Diarrea |
9 |
2 |
|
Pirexia |
9 |
8 |
|
Dolor abdominal |
8 |
4 |
|
Dolor orofaríngeo |
8 |
2 |
|
Dolor de muelas |
6 |
0 |
|
ALT aumentadaa |
6 |
0 |
|
Erupción |
5 |
2 |
|
AST aumentada |
4 |
0 |
|
Rinorrea |
4 |
0 |
En los dos ensayos clínicos controlados de ITP persistente o crónica, se desarrollaron o empeoraron cataratas en 2 (1%) pacientes tratados con PROMACTA. Ambos pacientes habían recibido corticosteroides orales crónicos, un factor de riesgo para la cataractogénesis.
Trombocitopenia crónica asociada a la hepatitis C: En los dos ensayos controlados con placebo, 955 pacientes con trombocitopenia crónica asociada a la hepatitis C recibieron PROMACTA. La Tabla 11 presenta las reacciones adversas a los medicamentos más comunes (experimentadas por más del 10% de los pacientes que recibieron PROMACTA en comparación con el placebo).
| aIncluye TP de insomnio, insomnio inicial y sueño de mala calidad. | ||
|
Reacción adversa |
PROMACTA + Peginterferón/Ribavirina n = 955 (%) |
Placebo + Peginterferón/Ribavirina n = 484 (%) |
|
Anemia |
40 |
35 |
|
Pirexia |
30 |
24 |
|
Fatiga |
28 |
23 |
|
Cefalea |
21 |
20 |
|
Náuseas |
19 |
14 |
|
Diarrea |
19 |
11 |
|
Disminución del apetito |
18 |
14 |
|
Enfermedad tipo influenza |
18 |
16 |
|
Insomnioa |
16 |
15 |
|
Astenia |
16 |
13 |
|
Tos |
15 |
12 |
|
Prurito |
15 |
13 |
|
Escalofríos |
14 |
9 |
|
Mialgia |
12 |
10 |
|
Alopecia |
10 |
6 |
|
Edema periférico |
10 |
5 |
Se notificó erupción cutánea en el 9 % y el 7 % de los pacientes que recibieron PROMACTA y placebo, respectivamente.
En los dos ensayos clínicos controlados en pacientes con hepatitis C crónica, se notificó hiperbilirrubinemia en el 8 % de los pacientes que recibieron PROMACTA en comparación con el 3 % para placebo. Se notificó bilirrubina total mayor o igual a 1,5 x LSN en el 76 % y el 50 % de los pacientes que recibieron PROMACTA y placebo, respectivamente. Se notificó ALT o AST mayor o igual a 3 x LSN en el 34 % y el 38 % de los pacientes para PROMACTA y placebo, respectivamente.
En los dos ensayos clínicos controlados en pacientes con hepatitis C crónica, se desarrollaron o empeoraron cataratas en el 8 % de los pacientes tratados con PROMACTA y el 5 % de los pacientes tratados con placebo.
La seguridad de PROMACTA también se evaluó en todos los pacientes tratados con PROMACTA en los dos ensayos controlados, incluidos los pacientes que inicialmente recibieron PROMACTA en la fase de tratamiento preantiviral del ensayo y luego fueron aleatorizados al brazo de placebo (N = 1520 pacientes tratados con PROMACTA). Se notificó insuficiencia hepática en el 0,8 % de los pacientes tratados con PROMACTA y el 0,4 % de los pacientes tratados con placebo.
Anemia aplásica grave
Tratamiento de primera línea de la anemia aplásica grave
La seguridad de PROMACTA se estableció sobre la base de un ensayo de un solo brazo de 153 pacientes con anemia aplásica grave que no habían recibido terapia inmunosupresora definitiva previa. En este ensayo, PROMACTA se administró en combinación con globulina antitimocítica equina (h-ATG) y ciclosporina [véase Estudios clínicos (14.3)]. Entre los 153 pacientes que recibieron la dosis en este ensayo, 92 pacientes fueron evaluables para la seguridad del uso concomitante de PROMACTA, h-ATG y ciclosporina a la dosis y el régimen recomendados.
En esta cohorte, PROMACTA se administró en dosis de hasta 150 mg una vez al día del día 1 al mes 6 (D1-M6) en combinación con h-ATG del día 1 al 4 y ciclosporina durante 6 meses, seguida de una dosis baja de ciclosporina (dosis de mantenimiento) durante 18 meses adicionales para los pacientes que lograron una respuesta hematológica a los 6 meses. La duración media de la exposición a PROMACTA en esta cohorte fue de 183 días, con el 70 % de los pacientes expuestos durante > 24 semanas.
La Tabla 12 presenta las reacciones adversas más comunes (experimentadas por más del 5 % de los pacientes) asociadas con PROMACTA en la cohorte D1-M6.
| Abreviaturas: ALT, alanina aminotransferasa; AST, aspartato aminotransferasa. | |
|
Reacción adversa |
PROMACTA |
|
Aumento de ALT |
29 |
|
Aumento de AST |
17 |
|
Aumento de bilirrubina en sangre |
17 |
|
Erupción cutánea |
8 |
|
Decoloración de la piel, incluida la hiperpigmentación |
5 |
En el cohorte PROMACTA D1-M6, se informó con mayor frecuencia un aumento de ALT (29%), un aumento de AST (17%) y un aumento de bilirrubina en sangre (17%) que en pacientes con anemia aplásica severa refractaria (ver Tabla 13).
Las anomalías nuevas o que empeoran en los análisis de laboratorio de la función hepática (CTCAE Grado 3 y Grado 4) en el cohorte PROMACTA D1-M6 fueron 15% y 2% para AST, 26% y 4% para ALT, y 12% y 1% para bilirrubina, respectivamente.
En este ensayo clínico abierto de un solo brazo, se informaron ALT o AST > 3 x ULN con bilirrubina total > 1.5 x ULN y ALT o AST > 3 x ULN con bilirrubina total > 2 x ULN en 44% y 32% de los pacientes, respectivamente, en el cohorte PROMACTA D1-M6.
Pacientes pediátricos
Un total de 34 pacientes pediátricos (2 pacientes de 2 a 5 años de edad, 12 pacientes de 6 a 11 años de edad y 20 pacientes de 12 a 16 años de edad) fueron incluidos en este ensayo de un solo brazo, de los cuales 26 pacientes pediátricos fueron incluidos en el cohorte PROMACTA D1-M6. En este cohorte, las reacciones adversas graves más frecuentes (experimentadas por ≥ 10% de los pacientes) fueron infección del tracto respiratorio superior (12% en pacientes de 2 a 16 años de edad en comparación con 5% en pacientes de 17 años de edad o más, respectivamente) y erupción cutánea (12% en comparación con 2%). Las reacciones adversas más comunes (experimentadas por ≥ 10% de los pacientes) asociadas con PROMACTA fueron aumento de ALT (23% en pacientes de 2 a 16 años de edad en comparación con 32% en pacientes de 17 años de edad o más, respectivamente), aumento de bilirrubina en sangre (12% en comparación con 20%), aumento de AST (12% en comparación con 20%) y erupción cutánea (12% en comparación con 6%).
Anomalías citogenéticas
En este ensayo, se evaluaron aspirados de médula ósea de los pacientes para detectar anomalías citogenéticas. Siete pacientes en el cohorte PROMACTA D1-M6 presentaron una nueva anomalía citogenética, de las cuales 4 presentaron pérdida del cromosoma 7; estas 4 ocurrieron dentro de los 6,1 meses. En todos los cohortes, la evolución citogenética clonal ocurrió en 15 de 153 (10%) pacientes. De los 15 pacientes que experimentaron una anomalía citogenética: 7 pacientes presentaron pérdida del cromosoma 7, 6 de las cuales ocurrieron dentro de los 6,1 meses; 4 pacientes presentaron aberraciones cromosómicas de significado incierto; 3 pacientes presentaron una deleción del cromosoma 13; y 1 paciente tuvo una evaluación de médula ósea de seguimiento a los 5 años con características de displasia con hipercelularidad preocupante para el posible desarrollo de SMD. No está claro si estos hallazgos ocurrieron debido a la enfermedad subyacente, la terapia inmunosupresora o el tratamiento con PROMACTA.
Anemia aplásica severa refractaria
En el ensayo abierto de un solo brazo, 43 pacientes con anemia aplásica severa refractaria recibieron PROMACTA. Once pacientes (26%) fueron tratados durante más de 6 meses y 7 pacientes (16%) fueron tratados durante más de 1 año. Las reacciones adversas más comunes (mayores o iguales al 20%) fueron náuseas, fatiga, tos, diarrea y dolor de cabeza.
|
Reacción adversa |
PROMACTA n = 43 (%) |
|
Náuseas |
33 |
|
Fatiga |
28 |
|
Tos |
23 |
|
Diarrea |
21 |
|
Dolor de cabeza |
21 |
|
Dolor en extremidades |
19 |
|
Pirexia |
14 |
|
Mareo |
14 |
|
Dolor orofaríngeo |
14 |
|
Dolor abdominal |
12 |
|
Espasmos musculares |
12 |
|
Aumento de transaminasas |
12 |
|
Artralgia |
12 |
|
Rinorrea |
12 |
Se notificaron erupción e hiperbilirrubinemia en el 7 % de los pacientes; se notificó catarata en el 2 % de los pacientes.
En este ensayo, se notificaron ALT o AST concomitantes superiores a 3 x LSN con bilirrubina total superior a 1,5 x LSN en el 5 % de los pacientes. La bilirrubina total superior a 1,5 x LSN se produjo en el 14 % de los pacientes.
En este ensayo, se evaluaron aspirados de médula ósea de los pacientes para detectar anomalías citogenéticas. Ocho pacientes presentaron una nueva anomalía citogenética notificada durante el tratamiento, incluidos 5 pacientes que presentaron cambios complejos en el cromosoma 7.
6.2 Experiencia postcomercialización
Las siguientes reacciones adversas se han identificado durante el uso de PROMACTA después de la aprobación. Debido a que estas reacciones se notifican voluntariamente a partir de una población de tamaño incierto, no siempre es posible estimar de forma fiable la frecuencia o establecer una relación causal con la exposición al fármaco.
Trastornos de la piel y del tejido subcutáneo: Decoloración de la piel, incluida la hiperpigmentación y el amarillamiento de la piel.
7 INTERACCIONES MEDICAMENTOSAS
7.1 Cationes Polivalentes (Quelación)
Eltrombopag quelata cationes polivalentes (como hierro, calcio, aluminio, magnesio, selenio y zinc) presentes en alimentos, suplementos minerales y antiácidos.
Tome PROMACTA al menos 2 horas antes o 4 horas después de cualquier medicamento o producto que contenga cationes polivalentes, como antiácidos, productos lácteos y suplementos minerales, para evitar una reducción significativa en la absorción de PROMACTA debido a la quelación [ver Dosis y Administración (2.4), Farmacología Clínica (12.3)].
7.2 Transportadores
Use precaución cuando se administre concomitantemente PROMACTA y fármacos que son sustratos de OATP1B1 (p. ej., atorvastatina, bosentán, ezetimiba, fluvastatina, gliburida, olmesartán, pitavastatina, pravastatina, rosuvastatina, repaglinida, rifampicina, ácido simvastatina, SN-38 [metabolito activo del irinotecán], valsartán) o de la proteína de resistencia al cáncer de mama (BCRP) (p. ej., imatinib, irinotecán, lapatinib, metotrexato, mitoxantrona, rosuvastatina, sulfasalazina, topotecan). Controle estrechamente a los pacientes para detectar signos y síntomas de exposición excesiva a los fármacos que son sustratos de OATP1B1 o BCRP y considere la reducción de la dosis de estos fármacos, si procede. En ensayos clínicos con PROMACTA, se recomendó una reducción de la dosis de rosuvastatina del 50%.
7.3 Inhibidores de la Proteasa
Inhibidores de la Proteasa del VIH: No se recomienda ningún ajuste de dosis cuando PROMACTA se coadministra con lopinavir/ritonavir (LPV/RTV). No se han evaluado las interacciones medicamentosas con otros inhibidores de la proteasa del VIH.
Inhibidores de la Proteasa del Virus de la Hepatitis C: No se recomiendan ajustes de dosis cuando PROMACTA se coadministra con boceprevir o telaprevir. No se han evaluado las interacciones medicamentosas con otros inhibidores de la proteasa del virus de la hepatitis C (VHC).
7.4 Terapia con Peginterferón Alfa-2a/b
No se recomiendan ajustes de dosis cuando PROMACTA se coadministra con peginterferón alfa-2a (PEGASYS®) o -2b (PEGINTRON®).
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de riesgos
Los datos disponibles de un pequeño número de informes de casos publicados y la experiencia posterior a la comercialización con el uso de PROMACTA en mujeres embarazadas son insuficientes para evaluar cualquier riesgo asociado con el medicamento de defectos congénitos importantes, aborto espontáneo o resultados adversos maternos o fetales. En estudios de reproducción animal y toxicidad en el desarrollo, la administración oral de eltrombopag a ratas preñadas durante la organogénesis provocó embrioletalidad y reducción del peso fetal a dosis tóxicas para la madre. Estos efectos se observaron a dosis que resultaron en exposiciones que fueron seis veces la exposición clínica humana según el área bajo la curva (AUC) en pacientes con PTI persistente o crónica a 75 mg/día, y tres veces el AUC en pacientes con hepatitis C crónica a 100 mg/día (ver Datos).
Se desconoce el riesgo de fondo estimado de defectos congénitos importantes y aborto espontáneo para la población indicada. Todos los embarazos tienen un riesgo de fondo de defectos de nacimiento, pérdida u otros resultados adversos. En la población general de EE. UU., el riesgo de fondo estimado de defectos congénitos importantes y de aborto espontáneo en embarazos clínicamente reconocidos es del 2% al 4% y del 15% al 20%, respectivamente.
Datos
Datos en animales
En un estudio de desarrollo embrionario temprano, ratas hembra recibieron eltrombopag oral en dosis de 10, 20 o 60 mg/kg/día (0.8, 2 y 6 veces, respectivamente, la exposición clínica humana según el AUC en pacientes con PTI a 75 mg/día y 0.3, 1 y 3 veces, respectivamente, la exposición clínica humana según el AUC en pacientes con hepatitis C crónica a 100 mg/día). Se observó un aumento de la pérdida pre y postimplantación y una reducción del peso fetal a la dosis más alta, que también causó toxicidad materna.
En un estudio de desarrollo embriofetal, se administró eltrombopag por vía oral a ratas preñadas durante el período de organogénesis en dosis de 10, 20 o 60 mg/kg/día (0.8, 2 y 6 veces, respectivamente, la exposición clínica humana según el AUC en pacientes con PTI a 75 mg/día y 0.3, 1 y 3 veces, respectivamente, la exposición clínica humana según el AUC en pacientes con hepatitis C crónica a 100 mg/día). Se observaron disminuciones en el peso fetal (6% a 7%) y un ligero aumento en la presencia de costillas cervicales a la dosis más alta, que también causó toxicidad materna. Sin embargo, no se observó evidencia de malformaciones estructurales importantes.
En un estudio de desarrollo embriofetal, se administró eltrombopag por vía oral a conejas preñadas durante el período de organogénesis en dosis de 30, 80 o 150 mg/kg/día (0.04, 0.3 y 0.5 veces, respectivamente, la exposición clínica humana según el AUC en pacientes con PTI a 75 mg/día y 0.02, 0.1 y 0.3 veces, respectivamente, la exposición clínica humana según el AUC en pacientes con hepatitis C crónica a 100 mg/día). No se observó evidencia de fetotoxicidad, embrioletalidad o teratogenicidad.
En un estudio de toxicidad para el desarrollo pre y posnatal en ratas preñadas (F0), se administró eltrombopag oral desde el día 6 de gestación hasta el día 20 de lactancia. No se observaron efectos adversos sobre la función reproductiva materna ni sobre el desarrollo de las crías (F1) a dosis de hasta 20 mg/kg/día (2 veces la exposición clínica humana según el AUC en pacientes con PTI a 75 mg/día y similar a la exposición clínica humana según el AUC en pacientes con hepatitis C crónica a 100 mg/día). Se detectó eltrombopag en el plasma de las crías (F1). Las concentraciones plasmáticas en las crías aumentaron con la dosis después de la administración del fármaco a las madres F0.
8.2 Lactancia
Resumen de riesgos
No hay datos sobre la presencia de eltrombopag o sus metabolitos en la leche humana, los efectos en el lactante o los efectos sobre la producción de leche. Sin embargo, se detectó eltrombopag en las crías de ratas lactantes 10 días después del parto, lo que sugiere la posibilidad de transferencia durante la lactancia. Debido al potencial de reacciones adversas graves en un lactante por PROMACTA, no se recomienda la lactancia materna durante el tratamiento.
8.3 Mujeres y hombres con potencial reproductivo
Anticoncepción
Según los estudios de reproducción animal, PROMACTA puede causar daño fetal cuando se administra a una mujer embarazada. Las mujeres sexualmente activas en edad fértil deben usar anticonceptivos efectivos (métodos que resulten en tasas de embarazo inferiores al 1%) cuando usen PROMACTA durante el tratamiento y durante al menos 7 días después de suspender el tratamiento con PROMACTA.
8.4 Uso pediátrico
Se ha establecido la seguridad y eficacia de PROMACTA en pacientes pediátricos de 1 año o mayores con PTI persistente o crónica y en pacientes pediátricos de 2 años o mayores con anemia aplásica grave sin tratamiento previo con IST (en combinación con h-ATG y ciclosporina). No se ha establecido la seguridad y eficacia en pacientes pediátricos menores de 1 año con PTI. No se ha establecido la seguridad y eficacia en pacientes pediátricos con trombocitopenia asociada con hepatitis C crónica y anemia aplásica grave refractaria.
La seguridad y la eficacia de PROMACTA en pacientes pediátricos de 1 año o mayores con PTI persistente o crónica se evaluaron en dos ensayos doble ciego controlados con placebo [ver Reacciones adversas (6.1), Estudios clínicos (14.1)]. La farmacocinética de eltrombopag se ha evaluado en 168 pacientes pediátricos de 1 año o mayores con PTI con dosis una vez al día [ver Farmacología clínica (12.3)]. Consulte Posología y administración (2.1) para obtener recomendaciones de dosificación para pacientes pediátricos de 1 año o mayores.
La seguridad y eficacia de PROMACTA en combinación con h-ATG y ciclosporina para el tratamiento de primera línea de la anemia aplásica grave en pacientes pediátricos de 2 años o mayores se evaluaron en un ensayo abierto de un solo brazo [ver Reacciones adversas (6.1), Estudios clínicos (14.3)]. Se evaluó un total de 26 pacientes pediátricos (de 2 a < 17 años); 12 niños (de 2 a < 12 años) y 14 adolescentes (de 12 a < 17 años). Consulte Dosificación y administración (2.3) para obtener recomendaciones de dosificación para pacientes pediátricos de 2 años o mayores. La seguridad y eficacia de PROMACTA en combinación con h-ATG y ciclosporina en pacientes pediátricos menores de 2 años para el tratamiento de primera línea de la anemia aplásica grave aún no se han establecido. En pacientes de 2 a 16 años de edad, el 69 % de los pacientes experimentaron eventos adversos graves en comparación con el 42 % en pacientes de 17 años o mayores. Entre los 12 pacientes de 2 a 11 años de edad en la cohorte D1-M6 de PROMACTA que alcanzaron la evaluación a los 6 meses o se retiraron antes, la tasa de respuesta completa al mes 6 fue del 8 % frente al 46 % en pacientes de 12 a 16 años y el 50 % en pacientes de 17 años o mayores.
8.5 Uso geriátrico
De los 106 pacientes en dos ensayos clínicos aleatorizados de PROMACTA 50 mg en PTI persistente o crónica, el 22% tenía 65 años o más, mientras que el 9% tenía 75 años o más. De los 1439 pacientes en dos ensayos clínicos aleatorizados de PROMACTA en pacientes con hepatitis C crónica y trombocitopenia, el 7% tenía 65 años o más, mientras que < 1% tenía 75 años o más. De los 196 pacientes que recibieron PROMACTA para el tratamiento de la anemia aplásica grave, el 18% tenía 65 años o más, mientras que el 3% tenía 75 años o más. No se observaron diferencias generales en la seguridad o la eficacia entre estos pacientes y los pacientes más jóvenes.
8.6 Insuficiencia hepática
Pacientes con PTI persistente o crónica y anemia aplásica grave
Reduzca la dosis inicial de PROMACTA en pacientes con PTI persistente o crónica (solo pacientes adultos y pediátricos de 6 años o mayores) o anemia aplásica grave refractaria que también tienen insuficiencia hepática (clase A, B, C de Child-Pugh) [ver Dosificación y administración (2.1, 2.3), Advertencias y precauciones (5.2), Farmacología clínica (12.3)].
En un ensayo clínico en pacientes con anemia aplásica grave que no habían recibido terapia inmunosupresora definitiva previa, los pacientes con ALT o AST iniciales > 5 x LSN no fueron elegibles para participar. Si un paciente con insuficiencia hepática (clase A, B, C de Child-Pugh) inicia el tratamiento con PROMACTA para el tratamiento de primera línea de la anemia aplásica grave, reduzca la dosis inicial [ver Dosificación y administración (2.3), Advertencias y precauciones (5.2), Farmacología clínica (12.3)].
Pacientes con hepatitis C crónica
No se recomienda ajustar la dosis en pacientes con hepatitis C crónica e insuficiencia hepática [ver Farmacología clínica (12.3)].
8.7 Origen étnico
Reduzca la dosis inicial de PROMACTA para pacientes de ascendencia del este/sudeste asiático con PTI (solo pacientes adultos y pediátricos de 6 años o mayores) o anemia aplásica grave [ver Dosificación y administración (2.1, 2.3), Farmacología clínica (12.3)]. No se recomienda una reducción en la dosis inicial de PROMACTA en pacientes de ascendencia del este/sudeste asiático con hepatitis C crónica [ver Farmacología clínica (12.3)].
10 SOBREDOSIS
En caso de sobredosis, el recuento de plaquetas puede aumentar excesivamente y provocar complicaciones trombóticas/tromboembólicas.
En un informe, un sujeto que ingirió 5000 mg de PROMACTA experimentó un aumento del recuento de plaquetas hasta un máximo de 929 x 109/L a los 13 días de la ingestión. El paciente también experimentó erupción cutánea, bradicardia, elevaciones de ALT/AST y fatiga. El paciente recibió tratamiento con lavado gástrico, lactulosa oral, líquidos intravenosos, omeprazol, atropina, furosemida, calcio, dexametasona y plasmaféresis; sin embargo, el recuento anormal de plaquetas y las anomalías en las pruebas hepáticas persistieron durante 3 semanas. Después de 2 meses de seguimiento, todos los eventos se habían resuelto sin secuelas.
En caso de sobredosis, considere la administración oral de una preparación que contenga cationes metálicos, como preparaciones de calcio, aluminio o magnesio, para quelar el eltrombopag y así limitar la absorción. Controle estrechamente el recuento de plaquetas. Reinicie el tratamiento con PROMACTA de acuerdo con las recomendaciones de dosificación y administración [ver Dosificación y administración (2.1, 2.2)].
11 DESCRIPCIÓN
Los comprimidos de PROMACTA (eltrombopag) contienen eltrombopag olamine, un agonista del receptor de trombopoyetina (TPO) de molécula pequeña para administración oral.
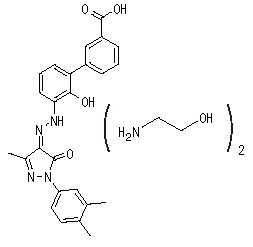
El eltrombopag olamine es una biphenyl hidrazona. El nombre químico del eltrombopag olamine es ácido 3′-{(2Z)-2-[1-(3,4-dimetilfenil)-3-metil-5-oxo-1,5-dihidro-4H-pirazol-4-ilideno]hidrazino}-2′-hidroxi-3-bifenilcarboxílico – 2-aminoetanol (1:2). Tiene la fórmula molecular C25H22N4O4 • 2(C2H7NO). El peso molecular es 564.65 g/mol para eltrombopag olamine y 442.5 g/mol para eltrombopag ácido libre. El eltrombopag olamine tiene la siguiente fórmula estructural:
El eltrombopag olamine es prácticamente insoluble en tampón acuoso en un rango de pH de 1 a 7.4, y es poco soluble en agua.
Los comprimidos de PROMACTA (eltrombopag) contienen eltrombopag olamine en una cantidad equivalente a 12.5 mg, 25 mg, 50 mg o 75 mg de eltrombopag ácido libre. Los ingredientes inactivos de los comprimidos de PROMACTA son:
Núcleo del comprimido: estearato de magnesio, manitol, celulosa microcristalina, povidona y glicolato de almidón sódico.
Recubrimiento: FD&C Azul No. 2 laca de aluminio (comprimido de 50 mg), FD&C Amarillo No. 6 laca de aluminio (comprimido de 25 mg), hipromelosa, óxido de hierro negro y óxido de hierro rojo (comprimido de 75 mg), polietilenglicol 400, polisorbato 80 (comprimido de 12.5 mg) o dióxido de titanio.
PROMACTA (eltrombopag) para suspensión oral en sobres contiene un polvo de color marrón rojizo a amarillo que produce una suspensión marrón rojiza cuando se reconstituye con agua. Cada sobre proporciona eltrombopag olamine equivalente a 12.5 mg o 25 mg de eltrombopag ácido libre. Los ingredientes inactivos de PROMACTA para suspensión oral son manitol, sucralosa y goma xantana.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
Eltrombopag es un agonista del receptor de TPO que interactúa con el dominio transmembrana del receptor de TPO humano (también conocido como cMpl) e inicia cascadas de señalización que inducen la proliferación y diferenciación de megacariocitos, lo que lleva a un aumento en la producción de plaquetas.
12.2 Farmacodinamia
En los ensayos clínicos, el tratamiento con PROMACTA produjo aumentos en el recuento de plaquetas dependientes de la dosis después de la administración repetida (diaria). El aumento en el recuento de plaquetas alcanzó un máximo aproximadamente dos semanas después del inicio de la administración y volvió a la línea de base aproximadamente dos semanas después de la última dosis de PROMACTA.
Electrofisiología cardíaca
En dosis de hasta 150 mg (la dosis máxima recomendada) al día durante 5 días, PROMACTA no prolongó el intervalo QT/QTc en ningún grado relevante.
12.3 Farmacocinética
Eltrombopag demostró un aumento proporcional a la dosis en la exposición entre dosis de 50 a 150 mg/día en sujetos adultos sanos. El AUC de Eltrombopag fue aproximadamente 1,7 veces mayor en pacientes con ITP persistente o crónica y aproximadamente 2,8 veces mayor en pacientes con VHC en comparación con sujetos sanos. Se alcanzó el estado estacionario después de aproximadamente 1 semana de tratamiento diario, con una razón de acumulación geométrica media de 1,56 (intervalo de confianza del 90% 1,20, 1,63) a 75 mg/día. El AUC de Eltrombopag fue aproximadamente 3,2 veces mayor en pacientes con anemia aplásica severa definitiva sin tratamiento inmunosupresor previo en comparación con sujetos sanos, lo que sugiere una mayor exposición relativa en comparación con sujetos sanos o pacientes con ITP y una exposición similar en comparación con pacientes con hepatitis C crónica. La suspensión oral de Eltrombopag proporcionó un 22% más de AUC0-INF en plasma que la formulación en comprimidos.
Absorción
Eltrombopag se absorbe con una concentración máxima que se produce de 2 a 6 horas después de la administración oral. Se estimó que la absorción oral del material relacionado con el fármaco después de la administración de una dosis única de 75 mg en solución fue de al menos el 52%.
Efecto de los alimentos
Un desayuno estándar alto en grasas (876 calorías, 52 g de grasa, 71 g de carbohidratos, 34 g de proteína y 427 mg de calcio) disminuyó significativamente el AUC0-INF de eltrombopag en plasma en aproximadamente un 59% y la Cmax en un 65%, y retrasó la Tmax en 1 hora. La disminución de la exposición se debe principalmente al alto contenido de calcio.
Una comida baja en calcio (≤ 50 mg de calcio) no afectó significativamente la exposición plasmática a eltrombopag, independientemente del contenido calórico y de grasa.
El efecto de la administración de una dosis única de 25 mg de eltrombopag en suspensión oral con una comida alta en calcio, moderada en grasas y moderada en calorías sobre el AUC0-INF y la Cmax en sujetos adultos sanos se presenta en la Tabla 14.
| a372 calorías, 9 g de grasa y 448 mg de calcio. | ||
| Momento de la dosis de eltrombopag en suspensión oral | Media (IC del 90%) de reducción en el AUC0-INF de eltrombopag en plasma | Media (IC del 90%) de reducción en la Cmax de eltrombopag en plasma |
| Con una comida alta en calcio, moderada en grasas y moderada en calorías | 75% (71%, 88%) | 79% (76%, 82%) |
| 2 horas después de la comida alta en calcio, moderada en grasas y moderada en calorías | 47% (40%, 53%) | 48% (40%, 54%) |
| 2 horas antes de la comida alta en calcio, moderada en grasas y moderada en calorías | 20% (9%, 29%) | 14% (2%, 25%) |
Distribución
La concentración de eltrombopag en las células sanguíneas es aproximadamente del 50% al 79% de las concentraciones plasmáticas según un estudio con radiomarcado. In vitro los estudios sugieren que el eltrombopag se une en gran medida a las proteínas plasmáticas humanas (más del 99%). El eltrombopag es un sustrato de BCRP, pero no es un sustrato de P-glucoproteína (P-gp) ni de OATP1B1.
Eliminación
La semivida de eliminación plasmática del eltrombopag es de aproximadamente 21 a 32 horas en sujetos sanos y de 26 a 35 horas en pacientes con ITP.
Metabolismo: El eltrombopag absorbido se metaboliza ampliamente, predominantemente a través de vías, incluyendo la escisión, la oxidación y la conjugación con ácido glucurónico, glutatión o cisteína. In vitro los estudios sugieren que CYP1A2 y CYP2C8 son responsables del metabolismo oxidativo del eltrombopag. UGT1A1 y UGT1A3 son responsables de la glucuronidación del eltrombopag.
Excreción: La vía principal de excreción del eltrombopag es a través de las heces (59%), y el 31% de la dosis se encuentra en la orina. El eltrombopag inalterado en las heces representa aproximadamente el 20% de la dosis; el eltrombopag inalterado no es detectable en la orina.
Poblaciones específicas
Etnia
Las concentraciones de eltrombopag en pacientes de ascendencia asiática oriental/sudoriental con ITP o hepatitis C crónica fueron del 50% al 55% más altas en comparación con los sujetos no asiáticos [véase Dosis y administración (2.1, 2.3)].
La exposición al eltrombopag en sujetos afroamericanos sanos fue aproximadamente un 40% mayor que la observada en sujetos caucásicos en un ensayo de farmacología clínica y similar en otros tres ensayos de farmacología clínica. No se ha establecido el efecto de la etnia afroamericana en la exposición y la seguridad y eficacia relacionadas del eltrombopag.
Insuficiencia hepática
Después de una dosis única de PROMACTA (50 mg), el AUC0-INF plasmático de eltrombopag fue un 41% mayor en pacientes con insuficiencia hepática leve (clase A de Child-Pugh) en comparación con los sujetos con función hepática normal. El AUC0-INF plasmático de eltrombopag fue aproximadamente el doble de alto en pacientes con insuficiencia hepática moderada (clase B de Child-Pugh) y grave (clase C de Child-Pugh) en comparación con los sujetos con función hepática normal. La semivida del eltrombopag se prolongó el doble en estos pacientes. Este ensayo clínico no evaluó los efectos de la unión a proteínas.
Enfermedad hepática crónica
Después de dosis repetidas de eltrombopag en pacientes con trombocitopenia y enfermedad hepática crónica, la insuficiencia hepática leve provocó un aumento del 87% al 110% del AUC(0-τ) plasmático de eltrombopag y la insuficiencia hepática moderada provocó un aumento de aproximadamente el 141% al 240% de los valores de AUC(0-τ) plasmático de eltrombopag en comparación con los pacientes con función hepática normal. La semivida del eltrombopag se prolongó 3 veces en pacientes con insuficiencia hepática leve y 4 veces en pacientes con insuficiencia hepática moderada. Este ensayo clínico no evaluó los efectos de la unión a proteínas.
Hepatitis C crónica
Los pacientes con hepatitis C crónica tratados con PROMACTA tuvieron valores de AUC(0-τ) plasmáticos más altos en comparación con los sujetos sanos, y el AUC(0-τ) aumentó con el aumento de la puntuación de Child-Pugh. Los pacientes con hepatitis C crónica e insuficiencia hepática leve tuvieron un AUC(0-τ) plasmático aproximadamente del 100% al 144% más alto en comparación con los sujetos sanos. Este ensayo clínico no evaluó los efectos de la unión a proteínas.
Insuficiencia renal
Después de una dosis única de PROMACTA (50 mg), el AUC0-INF plasmático total promedio fue del 32% al 36% menor en sujetos con insuficiencia renal leve (aclaramiento de creatinina estimado (CLCr) mediante la ecuación de Cockcroft-Gault: 50 a 80 mL/min) a moderada (CLCr de 30 a 49 mL/min) y un 60% menor en sujetos con insuficiencia renal grave (CLCr inferior a 30 mL/min) en comparación con los sujetos sanos. No se ha evaluado el efecto de la insuficiencia renal en la exposición al eltrombopag libre (activo).
Pacientes pediátricos
La farmacocinética del eltrombopag se ha evaluado en 168 pacientes pediátricos de 1 año o más con ITP con dosis diarias en dos ensayos. El aclaramiento plasmático aparente del eltrombopag después de la administración oral (CL/F) aumentó con el aumento del peso corporal. Los pacientes pediátricos de Asia oriental/sudoriental con ITP tuvieron valores de AUC(0-τ) plasmáticos de eltrombopag aproximadamente un 43% más altos en comparación con los pacientes no asiáticos.
El AUC(0-τ) y la Cmax plasmáticos de eltrombopag en pacientes pediátricos de 12 a 17 años fueron similares a los observados en adultos. Los parámetros farmacocinéticos del eltrombopag en pacientes pediátricos con ITP se muestran en la Tabla 15.
| aParámetros farmacocinéticos presentados como media geométrica (IC del 95%). bBasado en estimaciones post-hoc de farmacocinética poblacional. |
||||
|
Cmaxb |
AUC(0-τ)b |
|||
|
Edad |
(mcg/mL) |
(mcg·hr/mL) |
||
|
Adultos (n = 108) |
7.03 |
101 |
||
|
(6.44, 7.68) |
(91.4, 113) |
|
|
12 a 17 años (n = 62) |
6.80 |
103 |
|
(6.17, 7.50) |
(91.1, 116) |
|
|
6 a 11 años (n = 68) |
10.3 |
153 |
|
(9.42, 11.2) |
(137, 170) |
|
|
1 a 5 años (n = 38) |
11.6 |
162 |
|
(10.4, 12.9) |
(139, 187) |
Estudios de Interacción Medicamentosa
Estudios Clínicos
Efecto de los Medicamentos sobre el Eltrombopag
Efecto de los Antiácidos que Contienen Cationes Polivalentes sobre el Eltrombopag:
La administración conjunta de una dosis única de PROMACTA (75 mg) con un antiácido que contiene cationes polivalentes (1524 mg de hidróxido de aluminio, 1425 mg de carbonato de magnesio y alginato de sodio) disminuyó el AUC0-INF y la Cmax del eltrombopag en plasma aproximadamente en un 70 %. No se conoce la contribución del alginato de sodio a esta interacción.
Efecto de los Inhibidores de la Proteasa del VIH sobre el Eltrombopag:
La administración conjunta de lopinavir 400 mg/ritonavir 100 mg (dos veces al día) en dosis repetidas con una dosis única de PROMACTA (100 mg) disminuyó el AUC0-INF del eltrombopag en plasma en un 17 %.
Efecto de los Inhibidores de la Proteasa del VHC sobre el Eltrombopag:
La administración conjunta de telaprevir (750 mg cada 8 horas) o boceprevir (800 mg cada 8 horas) en dosis repetidas con una dosis única de PROMACTA (200 mg) a sujetos adultos sanos en un ensayo clínico no alteró el AUC0-INF o la Cmax del eltrombopag en plasma en una medida significativa.
Efecto de la Ciclosporina sobre el Eltrombopag:
La administración conjunta de una dosis única de PROMACTA (50 mg) con una dosis única de un inhibidor de OATP y BCRP, ciclosporina (200 mg o 600 mg), disminuyó el AUC0-INF del eltrombopag en plasma en un 18 % a un 24 % y la Cmax en un 25 % a un 39 %.
Efecto del Interferón Peglilado alfa-2a + Ribavirina y del Interferón Peglilado alfa-2b + Ribavirina sobre el Eltrombopag:
La presencia de terapia con interferón peglilado alfa + ribavirina no afectó significativamente la depuración del eltrombopag.
Efecto del Eltrombopag sobre Otros Medicamentos
Efecto del Eltrombopag sobre los Sustratos de las Enzimas del Citocromo P450:
La administración conjunta de dosis múltiples de PROMACTA (75 mg una vez al día durante 7 días) no produjo la inhibición o inducción del metabolismo de una combinación de sustratos de prueba para CYP1A2 (cafeína), CYP2C19 (omeprazol), CYP2C9 (flurbiprofeno) o CYP3A4 (midazolam) en humanos.
Efecto del Eltrombopag sobre la Rosuvastatina:
La administración conjunta de dosis múltiples de PROMACTA (75 mg una vez al día durante 5 días) con una dosis única de rosuvastatina (sustrato de OATP1B1 y BCRP; 10 mg) aumentó el AUC0-INF de la rosuvastatina en plasma en un 55 % y la Cmax en un 103 %.
Efecto del Eltrombopag sobre los Inhibidores de la Proteasa del VHC:
La administración conjunta de telaprevir (750 mg cada 8 horas) o boceprevir (800 mg cada 8 horas) en dosis repetidas con una dosis única de PROMACTA (200 mg) a sujetos adultos sanos en un ensayo clínico no alteró el AUC0-INF o la Cmax del telaprevir o boceprevir en plasma en una medida significativa.
In vitro Estudios
Efecto del Eltrombopag sobre las Enzimas Metabólicas
El eltrombopag ha demostrado el potencial de inhibir CYP2C8, CYP2C9, UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A9, UGT2B7 y UGT2B15.
Efecto del Eltrombopag sobre los Transportadores
El eltrombopag ha demostrado el potencial de inhibir OATP1B1 y BCRP.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogenicidad, Mutagénesis, Deterioro de la Fertilidad
Eltrombopag no estimula la producción de plaquetas en ratas, ratones o perros debido a la especificidad única del receptor TPO. Los datos de estos animales no modelan completamente los efectos en humanos.
Eltrombopag no fue carcinogénico en ratones a dosis de hasta 75 mg/kg/día ni en ratas a dosis de hasta 40 mg/kg/día (exposiciones de hasta 4 veces la exposición clínica humana basada en el AUC en pacientes con ITP a 75 mg/día y 2 veces la exposición clínica humana basada en el AUC en pacientes con hepatitis C crónica a 100 mg/día).
Eltrombopag no fue mutagénico ni clastogénico en una prueba de mutación bacteriana o en dos ensayos in vivo en ratas (micronúcleo y síntesis de ADN no programada, 10 veces la exposición clínica humana basada en Cmax en pacientes con ITP a 75 mg/día y 7 veces la exposición clínica humana basada en Cmax en pacientes con hepatitis C crónica a 100 mg/día). En el ensayo de linfoma de ratón in vitro, eltrombopag fue marginalmente positivo (menos de 3 veces el aumento en la frecuencia de mutación).
Eltrombopag no afectó la fertilidad femenina en ratas a dosis de hasta 20 mg/kg/día (2 veces la exposición clínica humana basada en el AUC en pacientes con ITP a 75 mg/día y similar a la exposición clínica humana basada en el AUC en pacientes con hepatitis C crónica a 100 mg/día). Eltrombopag no afectó la fertilidad masculina en ratas a dosis de hasta 40 mg/kg/día, la dosis más alta probada (3 veces la exposición clínica humana basada en el AUC en pacientes con ITP a 75 mg/día y 2 veces la exposición clínica humana basada en el AUC en pacientes con hepatitis C crónica a 100 mg/día).
13.2 Farmacología y/o Toxicología Animal
Se detectaron cataratas relacionadas con el tratamiento en roedores de una manera dependiente de la dosis y el tiempo. A más de o igual a 6 veces la exposición clínica humana basada en el AUC en pacientes con ITP a 75 mg/día y 3 veces la exposición clínica humana basada en el AUC en pacientes con hepatitis C crónica a 100 mg/día, se observaron cataratas en ratones después de 6 semanas y en ratas después de 28 semanas de dosificación. [ver Advertencias y precauciones (5.5)].
Se observó toxicidad tubular renal en estudios de hasta 14 días de duración en ratones y ratas a exposiciones que generalmente se asociaron con morbilidad y mortalidad. También se observó toxicidad tubular en un estudio de carcinogenicidad oral de 2 años en ratones a dosis de 25, 75 y 150 mg/kg/día. La exposición a la dosis más baja fue 1,2 veces la exposición clínica humana basada en el AUC en pacientes con ITP a 75 mg/día y 0,6 veces la exposición clínica humana basada en el AUC en pacientes con hepatitis C crónica a 100 mg/día. No se observaron efectos similares en ratones después de 13 semanas a exposiciones mayores que las asociadas con cambios renales en el estudio de 2 años, lo que sugiere que este efecto es dependiente tanto de la dosis como del tiempo.
14 ESTUDIOS CLÍNICOS
14.1 Púrpura trombocitopénica inmunitaria (PTI) persistente o crónica
Adultos: La eficacia y seguridad de PROMACTA en pacientes adultos con PTI persistente o crónica se evaluaron en tres ensayos aleatorizados, doble ciego, controlados con placebo y en un ensayo de extensión abierto.
En el Estudio TRA100773B y el Estudio TRA100773A (denominados Estudio 773B y Estudio 773A, respectivamente [NCT00102739]), los pacientes que habían completado al menos un tratamiento previo para la PTI y que tenían un recuento de plaquetas inferior a 30 x 109/L se asignaron aleatoriamente para recibir PROMACTA o placebo diariamente durante un máximo de 6 semanas, seguidas de 6 semanas sin tratamiento. Durante los ensayos, se interrumpió el tratamiento con PROMACTA o placebo si el recuento de plaquetas superaba los 200 x 109/L.
La mediana de edad de los pacientes fue de 50 años y el 60% eran mujeres. Aproximadamente el 70% de los pacientes habían recibido al menos 2 tratamientos previos para la PTI (principalmente corticosteroides, inmunoglobulinas, rituximab, terapias citotóxicas, danazol y azatioprina) y el 40% de los pacientes se habían sometido a una esplenectomía. Las medianas de recuentos de plaquetas basales (aproximadamente 18 x 109/L) fueron similares en todos los grupos de tratamiento.
En el Estudio 773B se asignaron aleatoriamente 114 pacientes (2:1) a PROMACTA 50 mg o placebo. De los 60 pacientes con tiempo transcurrido desde el diagnóstico documentado, aproximadamente el 17% cumplía la definición de PTI persistente con un tiempo transcurrido desde el diagnóstico de 3 a 12 meses. En el Estudio 773A se asignaron aleatoriamente 117 pacientes (1:1:1:1) entre placebo o 1 de 3 regímenes de dosis de PROMACTA, 30 mg, 50 mg o 75 mg, cada uno administrado diariamente. De los 51 pacientes con tiempo transcurrido desde el diagnóstico documentado, aproximadamente el 14% cumplía la definición de PTI persistente.
La eficacia de PROMACTA en este ensayo se evaluó mediante la tasa de respuesta, definida como un cambio de un recuento de plaquetas basal de menos de 30 x 109/L a mayor o igual a 50 x 109/L en cualquier momento durante el período de tratamiento (Tabla 16).
| ap-valor < 0,001 para PROMACTA versus placebo. | ||
|
Estudio |
PROMACTA 50 mg diarios |
Placebo |
|
773B |
43/73 (59%)a |
6/37 (16%) |
|
773A |
19/27 (70%)a |
3/27 (11%) |
La respuesta del recuento plaquetario a PROMACTA fue similar entre los pacientes que se habían sometido o no a una esplenectomía. En general, los aumentos en los recuentos plaquetarios se detectaron 1 semana después del inicio de PROMACTA y la respuesta máxima se observó después de 2 semanas de terapia. En los grupos de placebo y dosis de 50 mg de PROMACTA, el fármaco del ensayo se suspendió debido a un aumento en el recuento de plaquetas a más de 200 x 109/L en el 3% y el 27% de los pacientes, respectivamente. La duración media del tratamiento con la dosis de 50 mg de PROMACTA fue de 43 días en el Estudio 773B y de 42 días en el Estudio 773A.
De 7 pacientes que se sometieron a pruebas hemostáticas, se requirieron medicamentos adicionales para la ITP en 3 de 3 pacientes del grupo placebo y 0 de 4 pacientes tratados con PROMACTA. Los procedimientos quirúrgicos representaron la mayoría de los desafíos hemostáticos. Se produjo una hemorragia que requirió transfusión en un paciente del grupo placebo y en ningún paciente tratado con PROMACTA.
En el estudio RAISE (NCT00370331), 197 pacientes fueron aleatorizados (2:1) para recibir PROMACTA 50 mg una vez al día (n = 135) o placebo (n = 62) durante 6 meses, tiempo durante el cual la dosis de PROMACTA podría ajustarse según los recuentos plaquetarios individuales. De 145 pacientes con tiempo documentado desde el diagnóstico, el 19% cumplió con la definición de ITP persistente. Se permitió a los pacientes reducir gradualmente o suspender los medicamentos concomitantes para la ITP después de haber sido tratados con PROMACTA durante 6 semanas. Los pacientes pudieron recibir tratamientos de rescate en cualquier momento durante el ensayo según lo indicado clínicamente.
Las edades medias de los pacientes tratados con PROMACTA y placebo fueron de 47 años y 52,5 años, respectivamente. Aproximadamente la mitad de los pacientes tratados con PROMACTA y placebo (47% y 50%, respectivamente) estaban recibiendo medicación concomitante para la ITP (principalmente corticosteroides) en la aleatorización y tenían recuentos plaquetarios basales inferiores o iguales a 15 x 109/L (50% y 48%, respectivamente). Un porcentaje similar de pacientes tratados con PROMACTA y placebo (37% y 34%, respectivamente) se habían sometido a una esplenectomía previa.
La eficacia de PROMACTA en este ensayo se evaluó mediante las probabilidades de lograr un recuento plaquetario mayor o igual a 50 x 109/L y menor o igual a 400 x 109/L para los pacientes que recibieron PROMACTA en relación con el placebo y se basó en los perfiles de respuesta del paciente durante el período de tratamiento de 6 meses. En 134 pacientes que completaron 26 semanas de tratamiento, se logró una respuesta plaquetaria sostenida (recuento plaquetario mayor o igual a 50 x 109/L y menor o igual a 400 x 109/L durante 6 de las últimas 8 semanas del período de tratamiento de 26 semanas en ausencia de medicación de rescate en ningún momento) en el 60% de los pacientes tratados con PROMACTA, en comparación con el 10% de los pacientes tratados con placebo (pacientes sometidos a esplenectomía: PROMACTA 51%, placebo 8%; pacientes no sometidos a esplenectomía: PROMACTA 66%, placebo 11%). La proporción de respondedores en el grupo de pacientes tratados con PROMACTA osciló entre el 37% y el 56% en comparación con el 7% y el 19% en el grupo de tratamiento con placebo para todas las visitas durante el tratamiento. Los pacientes tratados con PROMACTA tuvieron significativamente más probabilidades de lograr un recuento plaquetario entre 50 x 109/L y 400 x 109/L durante todo el período de tratamiento de 6 meses en comparación con aquellos pacientes tratados con placebo.
Los resultados del tratamiento se presentan en la Tabla 17 para todos los pacientes inscritos en el ensayo.
|
Resultado |
PROMACTA n = 135 |
Placebo n = 62 |
|
Número medio de semanas con recuentos plaquetarios ≥ 50 x 109/L |
11.3 |
2.4 |
|
Requiriendo terapia de rescate, n (%) |
24 (18) |
25 (40) |
Entre 94 pacientes que recibían otra terapia para la PTI al inicio del estudio, 37 (59%) de 63 pacientes tratados con PROMACTA y 10 (32%) de 31 pacientes del grupo placebo interrumpieron la terapia concomitante en algún momento del ensayo.
En el estudio EXTEND (NCT00351468), los pacientes que completaron cualquier ensayo clínico previo con PROMACTA se inscribieron en un ensayo abierto, de un solo brazo, en el que se intentó disminuir la dosis o eliminar la necesidad de cualquier medicamento concomitante para la PTI. Se administró PROMACTA a 302 pacientes en EXTEND; 218 pacientes completaron 1 año, 180 pacientes completaron 2 años, 107 pacientes completaron 3 años, 75 pacientes completaron 4 años, 34 pacientes completaron 5 años y 18 pacientes completaron 6 años de terapia. La mediana del recuento plaquetario basal fue de 19 x 109/L antes de la administración de PROMACTA. Las medianas de recuentos plaquetarios a los 1, 2, 3, 4, 5, 6 y 7 años de estudio fueron 85 x 109/L, 85 x 109/L, 105 x 109/L, 64 x 109/L, 75 x 109/L, 119 x 109/L y 76 x 109/L, respectivamente.
Pacientes pediátricos: La eficacia y seguridad de PROMACTA en pacientes pediátricos de 1 año o más con PTI persistente o crónica se evaluaron en dos ensayos doble ciego controlados con placebo. Los ensayos difirieron en el tiempo transcurrido desde el diagnóstico de PTI: al menos 6 meses versus al menos 12 meses. Durante los ensayos, las dosis podían aumentarse cada 2 semanas hasta un máximo de 75 mg una vez al día. La dosis de PROMACTA se redujo si el recuento plaquetario superaba los 200 x 109/L y se interrumpió y redujo si superaba los 400 x 109/L.
En el estudio PETIT2 (NCT01520909), pacientes refractarios o recidivantes a al menos una terapia previa para la PTI con un recuento plaquetario inferior a 30 x 109/L (n = 92) se estratificaron por edad y se asignaron al azar (2:1) a PROMACTA (n = 63) o placebo (n = 29). La dosis inicial para pacientes de 6 a 17 años fue de 50 mg una vez al día para aquellos con al menos 27 kg y de 37,5 mg una vez al día para aquellos con menos de 27 kg, administrada en comprimidos orales. Se utilizó una dosis reducida de 25 mg una vez al día para pacientes de Asia oriental/sudoriental de 6 a 17 años, independientemente del peso. La dosis inicial para pacientes de 1 a 5 años fue de 1,2 mg/kg una vez al día (0,8 mg/kg una vez al día para pacientes de Asia oriental/sudoriental) administrada como suspensión oral.
El período aleatorizado, doble ciego de 13 semanas fue seguido por un período abierto de 24 semanas en el que los pacientes de ambos brazos fueron elegibles para recibir PROMACTA.
La mediana de edad de los pacientes fue de 9 años y el 48% eran mujeres. Aproximadamente el 62% de los pacientes tenían un recuento plaquetario basal menor o igual a 15 x 109/L, una característica que fue similar entre los brazos de tratamiento. El porcentaje de pacientes con al menos 2 terapias previas para la PTI (predominantemente corticosteroides e inmunoglobulinas) fue del 73% en el grupo tratado con PROMACTA y del 90% en el grupo tratado con placebo. Cuatro pacientes del grupo tratado con PROMACTA se habían sometido a una esplenectomía.
La eficacia de PROMACTA en este ensayo se evaluó mediante la proporción de sujetos con PROMACTA que alcanzaron recuentos plaquetarios ≥ 50 x 109/L (en ausencia de terapia de rescate) durante al menos 6 de 8 semanas entre las semanas 5 y 12 del período aleatorizado, doble ciego (Tabla 18).
| ap-valor = < 0,001 para PROMACTA versus placebo. | ||
|
Grupo de edad |
PROMACTA |
Placebo |
|
General |
26/63 (41%)a |
1/29 (3%) |
|
12 a 17 años |
10/24 (42%) |
1/10 (10%) |
|
6 a 11 años |
11/25 (44%) |
0/13 (0%) |
|
1 a 5 años |
5/14 (36%) |
0/6 (0%) |
Más pacientes pediátricos tratados con PROMACTA (75%) en comparación con placebo (21%) tuvieron al menos un recuento de plaquetas mayor o igual a 50 x 109/L durante las primeras 12 semanas de tratamiento aleatorizado en ausencia de terapia de rescate. Menos pacientes pediátricos tratados con PROMACTA requirieron tratamiento de rescate durante el período aleatorizado y doble ciego en comparación con los pacientes tratados con placebo (19% [12/63] versus 24% [7/29]). En los pacientes que lograron una respuesta plaquetaria (≥ 50 x 109/L sin rescate) durante 6 de 8 semanas (entre las semanas 5 y 12), el 62% (16/26) tuvo una respuesta inicial en las primeras 2 semanas después de comenzar el tratamiento con PROMACTA.
Se permitió a los pacientes reducir o suspender el tratamiento basal para la PTI solo durante la fase abierta del ensayo. De 15 pacientes que recibieron otro tratamiento para la PTI al inicio del estudio, el 53% (8/15) redujo (n = 1) o suspendió (n = 7) el tratamiento concomitante, principalmente corticosteroides, sin necesidad de terapia de rescate.
En el estudio PETIT (NCT00908037), los pacientes refractarios o recidivantes a al menos un tratamiento previo para la PTI con un recuento de plaquetas inferior a 30 x 109/L (n = 67) se estratificaron por edad y se asignaron al azar (2:1) a PROMACTA (n = 45) o placebo (n = 22). Aproximadamente el 15% de los pacientes cumplieron con la definición de PTI persistente. La dosis inicial para pacientes de 12 a 17 años fue de 37,5 mg una vez al día, independientemente del peso o la raza. La dosis inicial para pacientes de 6 a 11 años fue de 50 mg una vez al día para aquellos con un peso mayor o igual a 27 kg y de 25 mg una vez al día para aquellos con un peso inferior a 27 kg, administrada en comprimidos orales. Se utilizaron dosis reducidas de 25 mg (para aquellos con un peso mayor o igual a 27 kg) y 12,5 mg (para aquellos con un peso inferior a 27 kg), cada una una vez al día, para pacientes del este/sudeste asiático en este rango de edad. La dosis inicial para pacientes de 1 a 5 años fue de 1,5 mg/kg una vez al día (0,8 mg/kg una vez al día para pacientes del este/sudeste asiático) administrada como suspensión oral.
El período de 7 semanas, aleatorizado y doble ciego, fue seguido por un período abierto de hasta 24 semanas en el que los pacientes de ambos brazos pudieron recibir PROMACTA.
La mediana de edad de los pacientes fue de 10 años y el 60% eran mujeres. Aproximadamente el 51% de los pacientes tenían un recuento de plaquetas basal menor o igual a 15 x 109/L. El porcentaje de pacientes con al menos 2 tratamientos previos para la PTI (principalmente corticosteroides e inmunoglobulinas) fue del 84% en el grupo tratado con PROMACTA y del 86% en el grupo tratado con placebo. Cinco pacientes del grupo tratado con PROMACTA se habían sometido a una esplenectomía.
La eficacia de PROMACTA en este ensayo se evaluó mediante la proporción de pacientes que alcanzaron recuentos de plaquetas mayores o iguales a 50 x 109/L (en ausencia de terapia de rescate) al menos una vez entre las semanas 1 y 6 del período aleatorizado y doble ciego (Tabla 19). La respuesta plaquetaria a PROMACTA fue consistente en todos los grupos de edad.
| ap-valor = 0,011 para PROMACTA versus placebo. | ||
|
Grupo de edad |
PROMACTA |
Placebo |
|
Total |
28/45 (62%)a |
7/22 (32%) |
|
12 a 17 años |
10/16 (62%) |
0/8 (0%) |
|
6 a 11 años |
12/19 (63%) |
3/9 (33%) |
|
1 a 5 años |
6/10 (60%) |
4/5 (80%) |
Menos pacientes pediátricos tratados con PROMACTA requirieron tratamiento de rescate durante el período aleatorizado y doble ciego en comparación con los pacientes tratados con placebo (13% [6/45] versus 50% [11/22]).
Se permitió a los pacientes reducir o suspender el tratamiento basal para la PTI solo durante la fase abierta del ensayo. De los 13 pacientes que recibieron otro tratamiento para la PTI al inicio del estudio, el 46% (6/13) redujo (n = 3) o suspendió (n = 3) el tratamiento concomitante, principalmente corticosteroides, sin necesidad de terapia de rescate.
14.2 Trombocitopenia asociada a hepatitis C crónica
La eficacia y seguridad de PROMACTA para el tratamiento de la trombocitopenia en pacientes adultos con hepatitis C crónica se evaluó en dos ensayos aleatorizados, doble ciego y controlados con placebo. El estudio ENABLE1 (NCT00516321) utilizó peginterferón alfa-2a (PEGASYS®) más ribavirina para el tratamiento antiviral y el estudio ENABLE2 (NCT00529568) utilizó peginterferón alfa-2b (PEGINTRON®) más ribavirina. En ambos ensayos, se incluyeron pacientes con un recuento plaquetario inferior a 75 x 109/L y se estratificaron por recuento plaquetario, ARN del VHC en la evaluación y genotipo del VHC. Se excluyó a los pacientes si tenían evidencia de enfermedad hepática descompensada con una puntuación de Child-Pugh superior a 6 (clase B y C), antecedentes de ascitis o encefalopatía hepática. La mediana de edad de los pacientes en ambos ensayos fue de 52 años, el 63% eran hombres y el 74% eran caucásicos. El sesenta y nueve por ciento de los pacientes tenían los genotipos 1, 4, 6 del VHC, y el resto los genotipos 2 y 3. Aproximadamente el 30% de los pacientes habían sido tratados previamente con interferón y ribavirina. La mayoría de los pacientes (90%) tenían fibrosis puente y cirrosis, como lo indica las pruebas no invasivas. Una proporción similar (95%) de pacientes en ambos grupos de tratamiento tenían clase A de Child-Pugh (puntuación de 5 a 6) al inicio del estudio. Una proporción similar de pacientes (2%) en ambos grupos de tratamiento tenían una razón internacional normalizada (INR) basal superior a 1,7. Los recuentos plaquetarios basales medianos (aproximadamente 60 x 109/L) fueron similares en ambos grupos de tratamiento. Los ensayos consistieron en 2 fases: una fase de tratamiento preantiviral y una fase de tratamiento antiviral. En la fase de tratamiento preantiviral, los pacientes recibieron PROMACTA en abierto para aumentar el recuento plaquetario a un umbral superior o igual a 90 x 109/L para ENABLE1 y superior o igual a 100 x 109/L para ENABLE2. PROMACTA se administró a una dosis inicial de 25 mg una vez al día durante 2 semanas y se aumentó en incrementos de 25 mg durante períodos de 2 a 3 semanas para lograr el recuento plaquetario óptimo para iniciar la terapia antiviral. El tiempo máximo que los pacientes pudieron recibir PROMACTA en abierto fue de 9 semanas. Si se alcanzaban los recuentos plaquetarios umbral, los pacientes se aleatorizaban (2:1) a la misma dosis de PROMACTA al final de la fase de pretratamiento o a placebo. PROMACTA se administró en combinación con interferón pegilado y ribavirina según su información de prescripción respectiva durante un máximo de 48 semanas.
La eficacia de PROMACTA para ambos ensayos se evaluó mediante la respuesta virológica sostenida (RVS), definida como el porcentaje de pacientes con ARN del VHC indetectable a las 24 semanas después de la finalización del tratamiento antiviral. El tiempo medio para alcanzar el recuento plaquetario objetivo superior o igual a 90 x 109/L fue de aproximadamente 2 semanas. El noventa y cinco por ciento de los pacientes pudieron iniciar la terapia antiviral.
En ambos ensayos, una proporción significativamente mayor de pacientes tratados con PROMACTA lograron la RVS (ver Tabla 20). La mejora en la proporción de pacientes que lograron la RVS fue consistente en todos los subgrupos según el recuento plaquetario basal (inferior a 50 x 109/L versus superior o igual a 50 x 109/L). En pacientes con altas cargas virales basales (superior o igual a 800 000), la tasa de RVS fue del 18% (82/452) para PROMACTA versus el 8% (20/239) para placebo.
| Abreviatura: VHC, virus de la hepatitis C. aPROMACTA administrado en combinación con peginterferón alfa-2a (180 mcg una vez por semana durante 48 semanas para los genotipos 1/4/6; 24 semanas para el genotipo 2 o 3) más ribavirina (800 a 1200 mg diarios en 2 dosis divididas por vía oral). bPROMACTA administrado en combinación con peginterferón alfa-2b (1,5 mcg/kg una vez por semana durante 48 semanas para los genotipos 1/4/6; 24 semanas para el genotipo 2 o 3) más ribavirina (800 a 1400 mg diarios en 2 dosis divididas por vía oral). cEl recuento plaquetario objetivo fue ≥ 90 x 109/L para ENABLE1 y ≥ 100 x 109/L para ENABLE2. dp-valor < 0,05 para PROMACTA versus placebo. |
||||
|
ENABLE1a |
ENABLE2b |
|||
|
Fase de tratamiento preantiviral |
n = 715 |
n = 805 |
||
|
% de pacientes que alcanzaron los recuentos plaquetarios objetivo e iniciaron la terapia antiviralc |
95% |
94% |
||
|
Fase de tratamiento antiviral |
PROMACTA n = 450 % |
Placebo n = 232 % |
PROMACTA n = 506 % |
Placebo n = 253 % |
|
SVR globald |
23 |
14 |
19 |
13 |
|
Genotipo 2,3 de VHC |
35 |
24 |
34 |
25 |
|
Genotipo 1, 4, 6 de VHC |
18 |
10 |
13 |
7 |
La mayoría de los pacientes tratados con PROMACTA (76%) mantuvieron un recuento plaquetario mayor o igual a 50 x 109/L en comparación con el 19% para el placebo. Una mayor proporción de pacientes con PROMACTA no requirieron ninguna reducción de la dosis antiviral en comparación con el placebo (45% versus 27%).
14.3 Anemia aplásica grave
Tratamiento de primera línea de la anemia aplásica grave
Se investigó PROMACTA en combinación con h-ATG y ciclosporina en un ensayo de cohorte secuencial, de un solo brazo, monocéntrico y abierto (Estudio ETB115AUS01T, denominado Estudio US01T [NCT01623167]) en pacientes con anemia aplásica grave que no habían recibido terapia inmunosupresora previa (IST) con ningún ATG, alemtuzumab o ciclofosfamida en dosis altas. Un total de 153 pacientes recibieron PROMACTA en el Estudio US01T en tres cohortes secuenciales y una extensión de la tercera cohorte. Las múltiples cohortes recibieron la misma dosis inicial de PROMACTA, pero difirieron en el día de inicio del tratamiento y su duración. La dosis inicial de PROMACTA para pacientes de 12 años o más fue de 150 mg una vez al día (se administró una dosis reducida de 75 mg para los pacientes del este/sudeste asiático), 75 mg una vez al día para pacientes pediátricos de 6 a 11 años (se administró una dosis reducida de 37,5 mg para los pacientes del este/sudeste asiático) y 2,5 mg/kg una vez al día para pacientes pediátricos de 2 a 5 años (se administró una dosis reducida de 1,25 mg/kg para los pacientes del este/sudeste asiático).
- Cohorte 1 (n = 30): PROMACTA del día 14 al mes 6 (D14-M6) más h-ATG y ciclosporina
- Cohorte 2 (n = 31): PROMACTA del día 14 al mes 3 (D14-M3) más h-ATG y ciclosporina
- Cohorte 3 + cohorte de extensión [cohorte PROMACTA D1-M6] (n = 92): PROMACTA del día 1 al mes 6 (D1-M6) más h-ATG y ciclosporina (con todos los pacientes elegibles para recibir una dosis baja de ciclosporina (dosis de mantenimiento) si lograron una respuesta hematológica a los 6 meses)
Las reducciones de la dosis de PROMACTA se realizaron para recuentos plaquetarios elevados y deterioro hepático. La Tabla 21 incluye las dosis de h-ATG y ciclosporina administradas en combinación con PROMACTA en el Estudio US01T.
Los datos de la cohorte 3 + cohorte de extensión respaldan la eficacia de PROMACTA para el tratamiento de primera línea de pacientes con anemia aplásica grave (Tabla 22). Los resultados presentados en esta sección representan los hallazgos de la cohorte 3 y la cohorte de extensión (n = 92).
| aLa dosis de ciclosporina se ajustó para lograr los niveles valle objetivo recomendados anteriormente; consulte la información de prescripción de ciclosporina correspondiente. bCalculado como el punto medio entre el peso corporal ideal y el peso corporal real. |
|
|
Agente |
Dosis administrada en el ensayo pivotal |
|
Globulina antitimocítica equina (h-ATG) |
40 mg/kg/día, según el peso corporal real, administrado por vía intravenosa los días 1 a 4 del período de tratamiento de 6 meses |
|
Ciclosporinaa |
Pacientes de 12 años o más (dosis diaria total de 6 mg/kg/día) 3 mg/kg, según el peso corporal real, por vía oral cada 12 horas durante 6 meses, a partir del día 1 Pacientes > 20 años de edad con un índice de masa corporal > 35 o pacientes de 12 a 20 años de edad con un índice de masa corporal > percentil 95th: 3 mg/kg, según el peso corporal ajustadob, por vía oral cada 12 horas durante 6 meses, a partir del día 1 Pacientes de 2 a 11 años de edad (dosis diaria total de 12 mg/kg/día) 6 mg/kg, según el peso corporal real, por vía oral cada 12 horas durante 6 meses, a partir del día 1 Pacientes de 2 a 11 años de edad con un índice de masa corporal > percentil 95th: 6 mg/kg, según el peso corporal ajustadob, por vía oral cada 12 horas durante 6 meses, a partir del día 1 |
|
Ciclosporina |
Para pacientes que logran una respuesta hematológica a los 6 meses 2 mg/kg/día administrados por vía oral a una dosis fija durante 18 meses adicionales |
En la cohorte PROMACTA D1-M6, la mediana de edad fue de 28 años (rango, 5 a 82 años), con un 16 % y un 28 % de pacientes ≥ 65 años y < 17 años de edad, respectivamente. El 46 % de los pacientes eran hombres y la mayoría eran blancos (62 %). Los pacientes con un peso ≤ 12 kg o con ALT o AST > 5 veces el límite superior de la normalidad fueron excluidos del ensayo.
La eficacia de PROMACTA en combinación con h-ATG y ciclosporina se estableció sobre la base de la respuesta hematológica completa a los 6 meses. Se definió una respuesta completa como parámetros hematológicos que cumplían los 3 valores siguientes en 2 recuentos sanguíneos seriados consecutivos con al menos una semana de diferencia: recuento absoluto de neutrófilos (RAN) > 1000/mcL, recuento de plaquetas > 100 x 109/L y hemoglobina > 10 g/dL. Se definió una respuesta parcial como recuentos sanguíneos que ya no cumplían los criterios estándar para pancitopenia grave en anemia aplásica grave, equivalente a 2 de los valores siguientes en 2 recuentos sanguíneos seriados consecutivos con al menos una semana de diferencia: RAN > 500/mcL, recuento de plaquetas > 20 x 109/L o recuento de reticulocitos > 60 000/mcL. La tasa de respuesta global se define como el número de respuestas parciales más respuestas completas.
| Abreviatura: NE, no estimable. aEl número de pacientes que alcanzaron la evaluación de 6 meses o se retiraron antes es el denominador para el cálculo del porcentaje. bNúmero de respondedores en cualquier momento. |
|
|
PROMACTA D1-M6 + h-ATG + ciclosporina |
|
|
Mes 6, na Respuesta global, n (%) [IC del 95 %] Respuesta completa, n (%) [IC del 95 %] |
87 |
|
Duración mediana de la respuesta global, nb |
70 |
|
Meses (IC del 95 %) |
24,3 (21,4, NE) |
|
Duración mediana de la respuesta completa, nb |
46 |
|
Meses (IC del 95 %) |
24,3 (23,0, NE) |
Las tasas generales y completas de respuesta hematológica al año 1 (n = 78) son del 56,4 % y del 38,5 %, y al año 2 (n = 62) son del 38,7 % y del 30,6 %, respectivamente.
Pacientes pediátricos
Treinta y cuatro pacientes de 2 a 16 años de edad fueron incluidos en el Estudio US01T. En la cohorte D1-M6, 7 y 17 de 25 pacientes pediátricos lograron una respuesta completa y general, respectivamente, a los 6 meses.
Anemia aplásica grave refractaria
PROMACTA se estudió en un ensayo de un solo brazo, monocéntrico y abierto (Estudio ETB115AUS28T, denominado Estudio US28T [NCT00922883]) en 43 pacientes con anemia aplásica grave que habían tenido una respuesta insuficiente a al menos una terapia inmunosupresora previa y que tenían un recuento de plaquetas inferior o igual a 30 x 109/L. PROMACTA se administró a una dosis inicial de 50 mg una vez al día durante 2 semanas y se aumentó en períodos de 2 semanas hasta una dosis máxima de 150 mg una vez al día. La eficacia de PROMACTA en el estudio se evaluó mediante la respuesta hematológica evaluada después de 12 semanas de tratamiento. La respuesta hematológica se definió como el cumplimiento de uno o más de los siguientes criterios: 1) aumento del recuento de plaquetas a 20 x 109/L por encima del valor basal, o recuentos de plaquetas estables con independencia de las transfusiones durante un mínimo de 8 semanas; 2) aumento de la hemoglobina en más de 1,5 g/dL, o una reducción en más de o igual a 4 unidades de transfusiones de glóbulos rojos (GR) durante 8 semanas consecutivas; 3) aumento del ANC del 100 % o un aumento del ANC superior a 0,5 x 109/L. PROMACTA se suspendió después de 16 semanas si no se observó ninguna respuesta hematológica. Los pacientes que respondieron continuaron el tratamiento en una fase de extensión del ensayo.
La población tratada tenía una mediana de edad de 45 años (rango, 17 a 77 años) y el 56 % eran hombres. Al inicio del estudio, la mediana del recuento de plaquetas fue de 20 x 109/L, la hemoglobina fue de 8,4 g/dL, el ANC fue de 0,58 x 109/L y el recuento absoluto de reticulocitos fue de 24,3 x 109/L. El ochenta y seis por ciento de los pacientes dependían de transfusiones de glóbulos rojos (GR) y el 91 % dependían de transfusiones de plaquetas. La mayoría de los pacientes (84 %) recibieron al menos 2 terapias inmunosupresoras previas. Tres pacientes presentaron anomalías citogenéticas al inicio del estudio.
La Tabla 23 presenta los resultados de eficacia.
| aIncluye uni y multi-linaje. bNR = no alcanzado debido a pocos eventos (recaída). |
|
|
Resultado |
PROMACTA n = 43 |
|
Tasa de respuestaa, n (%) IC del 95 % (%) |
17 (40) (25, 56) |
|
Mediana de la duración de la respuesta en meses (IC del 95 %) |
NRb (3,0, NRb) |
En los 17 respondedores, el período sin transfusiones de plaquetas osciló entre 8 y 1096 días con una mediana de 200 días, y el período sin transfusiones de GR osciló entre 15 y 1082 días con una mediana de 208 días.
En la fase de extensión, 8 pacientes lograron una respuesta multi-linaje; 4 de estos pacientes posteriormente redujeron gradualmente el tratamiento con PROMACTA y mantuvieron la respuesta (seguimiento mediano: 8,1 meses, rango, 7,2 a 10,6 meses).
16 SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
16.1 Tabletas
- Las tabletas de 12.5 mg son tabletas redondas, biconvexas, blancas, recubiertas con película, grabadas con “GS MZ1” y 12.5 en una cara y están disponibles en frascos de 30: NDC 0078-0684-15
- Las tabletas de 25 mg son tabletas redondas, biconvexas, anaranjadas, recubiertas con película, grabadas con “GS NX3” y 25 en una cara y están disponibles en frascos de 30: NDC 0078-0685-15
- Las tabletas de 50 mg son tabletas redondas, biconvexas, azules, recubiertas con película, grabadas con “GS UFU” y 50 en una cara y están disponibles:
- Frascos de 14 NDC 0078-0686-55
- Frascos de 30 NDC 0078-0686-15
- Las tabletas de 75 mg son tabletas redondas, biconvexas, rosas, recubiertas con película, grabadas con “GS FFS” y 75 en una cara y están disponibles en frascos de 30: NDC 0078-0687-15
Almacenar a temperatura ambiente entre 20°C y 25°C (68°F y 77°F); se permiten excursiones entre 15°C y 30°C (59°F y 86°F) [ver USP Controlled Room Temperature]. Dispensar en el frasco original.
16.2 Para Suspensión Oral
- La suspensión oral de 12.5 mg es un polvo de color marrón rojizo a amarillo en paquetes de dosis unitaria, empaquetado conjuntamente en un kit con un recipiente de reconstitución de 40 cc, un cierre roscado con capacidad para puerto de jeringa y 30 jeringas orales de un solo uso.
Cada kit (NDC 0078-0972-61) contiene 30 paquetes: NDC 0078-0972-19
- La suspensión oral de 25 mg es un polvo de color marrón rojizo a amarillo en paquetes de dosis unitaria, empaquetado conjuntamente en un kit con un recipiente de reconstitución de 40 cc, un cierre roscado con capacidad para puerto de jeringa y 30 jeringas orales de un solo uso.
Cada kit (NDC 0078-0697-61) contiene 30 paquetes: NDC 0078-0697-19
Almacenar a temperatura ambiente entre 20°C y 25°C (68°F y 77°F); se permiten excursiones entre 15°C y 30°C (59°F y 86°F) [ver USP Controlled Room Temperature]. Después de la reconstitución, el producto debe administrarse inmediatamente, pero puede almacenarse durante un período máximo de 30 minutos entre 20°C y 25°C (68°F y 77°F); se permiten excursiones entre 15°C y 30°C (59°F y 86°F) [ver USP Controlled Room Temperature]. Deseche la mezcla si no se utiliza en 30 minutos.
17 INFORMACIÓN DE ASESORAMIENTO AL PACIENTE
Aconseje al paciente o cuidador que lea el etiquetado del paciente aprobado por la FDA (Guía de medicamentos e Instrucciones de uso).
Antes del tratamiento, los pacientes deben comprender completamente y estar informados de los siguientes riesgos y consideraciones para PROMACTA:
Riesgos
Hepatotoxicidad
- La terapia con PROMACTA puede asociarse con anormalidades en las pruebas de laboratorio hepatobiliares [ver Advertencias y precauciones (5.2)].
- Avise a los pacientes con hepatitis C crónica y cirrosis de que pueden tener riesgo de descompensación hepática al recibir PROMACTA con terapia de alfa interferón [ver Advertencias y precauciones (5.1)].
- Avise a los pacientes que deben informar de inmediato a su proveedor de atención médica cualquiera de los siguientes signos y síntomas de problemas hepáticos [ver Advertencias y precauciones (5.2)].
- coloración amarillenta de la piel o el blanco de los ojos (ictericia)
- oscurecimiento inusual de la orina
- cansancio inusual
- dolor en la zona superior derecha del estómago
- confusión
- hinchazón de la zona del estómago (abdomen)
Riesgo de sangrado tras la interrupción de PROMACTA
- Avise a los pacientes que la trombocitopenia y el riesgo de sangrado pueden reaparecer al suspender PROMACTA, particularmente si se suspende PROMACTA mientras el paciente está tomando anticoagulantes o agentes antiplaquetarios. Avise a los pacientes que durante el tratamiento con PROMACTA, deben seguir evitando situaciones o medicamentos que puedan aumentar el riesgo de sangrado.
Complicaciones trombóticas/tromboembólicas
- Avise a los pacientes que una cantidad excesiva de PROMACTA puede provocar un recuento excesivo de plaquetas y un riesgo de complicaciones trombóticas/tromboembólicas [ver Advertencias y precauciones (5.4)].
Cataratas
- Avise a los pacientes que se realicen un examen ocular basal antes de la administración de PROMACTA y que se les controle la presencia de signos y síntomas de cataratas durante el tratamiento [ver Advertencias y precauciones (5.5)].
Interacciones medicamentosas
- Avise a los pacientes que tomen PROMACTA al menos 2 horas antes o 4 horas después de los alimentos ricos en calcio, suplementos minerales y antiácidos que contengan cationes polivalentes, como hierro, calcio, aluminio, magnesio, selenio y zinc [ver Posología y administración (2.4), Interacciones medicamentosas (7.1)].
Lactancia
- Avise a las mujeres que no deben amamantar durante el tratamiento con PROMACTA [ver Uso en poblaciones específicas (8.2)].
Administración de PROMACTA
- Para pacientes con ITP persistente o crónica, la terapia con PROMACTA se administra para lograr y mantener un recuento de plaquetas superior o igual a 50 x 109/L según sea necesario para reducir el riesgo de sangrado [ver Indicaciones y uso (1.1)].
- Para pacientes con hepatitis C crónica, la terapia con PROMACTA se administra para lograr y mantener un recuento de plaquetas necesario para iniciar y mantener la terapia antiviral con interferón pegilado y ribavirina [ver Indicaciones y uso (1.2)].
- Avise a los pacientes que tomen PROMACTA sin comida o con una comida baja en calcio (≤ 50 mg) y al menos 2 horas antes o 4 horas después de otros medicamentos (p. ej., antiácidos) y alimentos ricos en calcio [ver Posología y administración (2.4)].
- Antes de usar la suspensión oral, asegúrese de que los pacientes o cuidadores reciban capacitación sobre la dosificación, preparación y administración adecuadas [ver Posología y administración (2.4)].
- Informe a los pacientes o cuidadores cuántos sobres deben administrar para obtener la dosis completa [ver Instrucciones de uso].
- Informe a los pacientes o cuidadores que deben usar una jeringa dosificadora oral nueva para preparar cada dosis de suspensión oral de PROMACTA [ver Instrucciones de uso].
Las siguientes son marcas comerciales registradas de sus respectivos propietarios: PEGASYS/Hoffmann-La Roche Inc.; PEGINTRON/Schering Corporation.
Distribuido por:
Novartis Pharmaceuticals Corporation
East Hanover, New Jersey 07936
© Novartis
T2023-13
Guía de medicación
| Esta Guía del Medicamento ha sido aprobada por la Administración de Alimentos y Medicamentos de los EE. UU. | Revisado: marzo de 2023 |
| GUÍA DEL MEDICAMENTO | |
| PROMACTA® (pro-MAC-ta) (eltrombopag) comprimidos |
PROMACTA® (pro-MAC-ta) (eltrombopag) para suspensión oral |
| ¿Cuál es la información más importante que debo saber sobre PROMACTA?
PROMACTA puede causar efectos secundarios graves, incluyendo: Problemas hepáticos:
Informe a su proveedor de atención médica de inmediato si tiene alguno de estos signos y síntomas de problemas hepáticos: |
|
|
|
| Consulte “¿Cuáles son los posibles efectos secundarios de PROMACTA?” para conocer otros efectos secundarios de PROMACTA. | |
| ¿Qué es PROMACTA? PROMACTA es un medicamento recetado que se usa para tratar a adultos y niños de 1 año de edad o más con recuentos bajos de plaquetas sanguíneas debido a trombocitopenia inmunitaria persistente o crónica (ITP), cuando otros medicamentos para tratar la ITP o la cirugía para extirpar el bazo no han funcionado lo suficientemente bien. PROMACTA también se usa para tratar a personas con:
PROMACTA se usa para intentar aumentar el recuento de plaquetas a fin de reducir el riesgo de sangrado. PROMACTA no se usa para normalizar el recuento de plaquetas. PROMACTA no se debe usar en personas con una afección precancerosa llamada síndrome mielodisplásico (SMD), ni en personas con recuentos bajos de plaquetas causados por ciertas otras afecciones o enfermedades médicas. No se sabe si PROMACTA es seguro y eficaz cuando se usa con otros medicamentos antivirales para tratar la hepatitis C crónica. No se sabe si PROMACTA es seguro y eficaz en niños:
|
|
Antes de tomar PROMACTA, informe a su proveedor de atención médica sobre todas sus afecciones médicas, incluso si:
Especialmente, informe a su proveedor de atención médica si toma:
Ciertos medicamentos pueden impedir que PROMACTA funcione correctamente. Tome PROMACTA al menos 2 horas antes o 4 horas después de tomar estos productos:
Pregunte a su proveedor de atención médica si no está seguro de si su medicamento es uno de los que se mencionan anteriormente. |
|
¿Cómo debo tomar PROMACTA?
|
|
| ¿Qué debo evitar mientras tomo PROMACTA?
Evite situaciones y medicamentos que puedan aumentar su riesgo de hemorragia. |
|
| ¿Cuáles son los posibles efectos secundarios de PROMACTA?
PROMACTA puede causar efectos secundarios graves, que incluyen:
Los efectos secundarios más comunes de PROMACTA en adultos y niños incluyen: |
|
|
|
| Las pruebas de laboratorio pueden mostrar cambios anormales en las células de la médula ósea. Informe a su proveedor de atención médica si tiene algún efecto secundario que le moleste o que no desaparezca. Estos no son todos los posibles efectos secundarios de PROMACTA. Para obtener más información, consulte a su proveedor de atención médica o farmacéutico. Llame a su médico para obtener asesoramiento médico sobre los efectos secundarios. Puede informar los efectos secundarios a la FDA al 1-800-FDA-1088. |
|
| ¿Cómo debo almacenar los comprimidos de PROMACTA y PROMACTA para suspensión oral?
Comprimidos:
Para suspensión oral:
Mantenga PROMACTA y todos los medicamentos fuera del alcance de los niños. |
|
| Información general sobre el uso seguro y eficaz de PROMACTA
A veces, los medicamentos se recetan para fines distintos de los que figuran en una Guía de medicamentos. No use PROMACTA para una afección para la que no se lo recetaron. No le dé PROMACTA a otras personas, incluso si tienen los mismos síntomas que usted. Puede dañarlos. Puede pedirle a su proveedor de atención médica o farmacéutico información sobre PROMACTA que esté escrita para profesionales de la salud. |
|
| ¿Cuáles son los ingredientes de PROMACTA? Tabletas Ingrediente activo: eltrombopag olamine Ingredientes inactivos:
Para suspensión oral Distribuido por: Novartis Pharmaceuticals Corporation, East Hanover, Nueva Jersey 07936 |
|
T2023-14
INSTRUCCIONES DE USO
PROMACTA® [pro-MAC-ta]
(eltrombopag)
para suspensión oral
Lea todas las instrucciones de uso y siga los pasos a continuación para mezclar y administrar una dosis de PROMACTA para suspensión oral.
Información importante que debe conocer antes de tomar PROMACTA para suspensión oral:
- No tome PROMACTA para suspensión oral ni se lo dé a otra persona hasta que le hayan mostrado cómo mezclar y administrar correctamente una dosis de PROMACTA para suspensión oral. Su proveedor de atención médica o enfermero le mostrará cómo mezclar y administrar correctamente una dosis de PROMACTA para suspensión oral.
- PROMACTA para suspensión oral debe mezclarse únicamente con agua fría o fresca. No utilice agua caliente para preparar la suspensión oral.
- Administre la dosis de suspensión inmediatamente después de mezclarla con agua. Si el medicamento no se administra dentro de los 30 minutos, deberá mezclar una nueva dosis. Deseche la mezcla sin usar en la basura. No la vierta por el desagüe.
- Si PROMACTA para suspensión oral entra en contacto con su piel, lávese la piel inmediatamente con agua y jabón. Llame a su proveedor de atención médica si tiene una reacción cutánea o si tiene alguna pregunta. Si derrama algún polvo o líquido, siga las instrucciones de limpieza en el paso 12.
- Comuníquese con su proveedor de atención médica o farmacéutico si tiene alguna pregunta sobre cómo mezclar o administrar PROMACTA a su hijo, o si daña o pierde alguno de los suministros de su kit.
- No reutilice la jeringa oral dosificadora. Utilice una jeringa oral dosificadora de un solo uso nueva para preparar cada dosis de PROMACTA para suspensión oral.
- Después de haber usado los 30 paquetes, deseche todos los suministros restantes (frasco mezclador, tapa con tapón y jeringa oral dosificadora) en la basura.
Cada kit de PROMACTA para suspensión oral contiene los siguientes suministros:



Necesitará lo siguiente para administrar una dosis de PROMACTA en suspensión oral.
Del kit:
- número prescrito de sobres
- 1 frasco mezclador reutilizable con tapa y tapón. Nota: Debido a su pequeño tamaño, la tapa puede representar un peligro de asfixia para los niños pequeños.
- 1 jeringa dosificadora oral de 20 ml de un solo uso (Use una jeringa dosificadora oral nueva (de un solo uso) para preparar cada dosis de PROMACTA en suspensión oral)
No incluido en el kit:
- 1 vaso o taza limpia llena de agua potable
- tijeras para cortar el sobre
- toallas de papel o paño desechable
- guantes desechables (opcional)