
Fabricante de medicamentos: Amgen Inc (Updated: 2024-03-08)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
PROLIA® (denosumab) inyección, para uso subcutáneo
Aprobación inicial en EE. UU.: 2010
ADVERTENCIA: HIPOCALCEMIA GRAVE EN PACIENTES CON ENFERMEDAD RENAL AVANZADA
Consulte la información completa de prescripción para obtener la advertencia completa en recuadro.
- Los pacientes con enfermedad renal crónica avanzada tienen un mayor riesgo de hipocalcemia grave después de la administración de Prolia. Se han notificado casos graves de hipocalcemia que provocaron hospitalización, eventos potencialmente mortales y casos mortales. (5.1)
- La presencia de trastorno mineral óseo en la enfermedad renal crónica (CKD-MBD) aumenta considerablemente el riesgo de hipocalcemia. (5.1)
- Antes de iniciar Prolia en pacientes con enfermedad renal crónica avanzada, evalúe la presencia de CKD-MBD. El tratamiento con Prolia en estos pacientes debe ser supervisado por un profesional sanitario con experiencia en el diagnóstico y el manejo de CKD-MBD. (2.2), (5.1)
CAMBIOS RECIENTES IMPORTANTES
INFORMACIÓN IMPORTANTE DE LA RECETA
Prolia es un inhibidor del ligando RANK (RANKL) indicado para el tratamiento:
- de mujeres posmenopáusicas con osteoporosis con alto riesgo de fractura (1.1)
- para aumentar la masa ósea en hombres con osteoporosis con alto riesgo de fractura (1.2)
- de la osteoporosis inducida por glucocorticoides en hombres y mujeres con alto riesgo de fractura (1.3)
- para aumentar la masa ósea en hombres con alto riesgo de fractura que reciben terapia de privación de andrógenos para el cáncer de próstata no metastásico (1.4)
- para aumentar la masa ósea en mujeres con alto riesgo de fractura que reciben terapia adyuvante con inhibidores de la aromatasa para el cáncer de mama (1.5)
DOSIFICACIÓN Y ADMINISTRACIÓN
- Se debe descartar el embarazo antes de administrar Prolia. (2.1)
- Antes de iniciar Prolia en pacientes con enfermedad renal crónica avanzada, incluidos los pacientes en diálisis, evaluar la presencia de trastorno mineral y óseo de la enfermedad renal crónica con hormona paratiroidea intacta, calcio sérico, vitamina D 25(OH) y vitamina D 1,25(OH)2. (2.2, 5.1, 8.6)
- Prolia debe ser administrado por un profesional de la salud. (2.3)
- Administrar 60 mg cada 6 meses como una inyección subcutánea en la parte superior del brazo, la parte superior del muslo o el abdomen. (2.3)
- Instruya a los pacientes para que tomen 1000 mg de calcio al día y al menos 400 UI de vitamina D al día. (2.3)
FORMAS Y FUERZAS DE DOSIFICACIÓN
- Inyección: Jeringa precargada de dosis única que contiene 60 mg en una solución de 1 mL (3)
CONTRAINDICACIONES
ADVERTENCIAS Y PRECAUCIONES
- Mismo ingrediente activo: Los pacientes que reciben Prolia no deben recibir XGEVA®. (5.2)
- Puede ocurrir hipersensibilidad, incluidas reacciones anafilácticas. Suspenda permanentemente si ocurre una reacción clínicamente significativa. (5.3)
- Hipocalcemia: La hipocalcemia preexistente debe corregirse antes de iniciar Prolia. Puede empeorar, especialmente en pacientes con insuficiencia renal. Suplementar adecuadamente a todos los pacientes con calcio y vitamina D. El uso concomitante de medicamentos calcimiméticos también puede aumentar el riesgo de hipocalcemia. Evaluar la presencia de trastorno mineral óseo de la enfermedad renal crónica. Monitorear el calcio sérico. (5.1)
- Osteonecrosis de la mandíbula: Se ha informado con Prolia. Monitorear los síntomas. (5.4)
- Fracturas femorales atípicas: Se han informado. Evaluar a los pacientes con dolor en el muslo o la ingle para descartar una fractura femoral. (5.5)
- Se han informado múltiples fracturas vertebrales después de la interrupción de Prolia. Los pacientes deben ser transferidos a otro agente antirresortivo si se interrumpe Prolia. (5.6)
- Infecciones graves, incluidas las infecciones cutáneas: Pueden ocurrir, incluidas las que conducen a la hospitalización. Avise a los pacientes que busquen atención médica inmediata si desarrollan signos o síntomas de infección, incluida la celulitis. (5.7)
- Reacciones dermatológicas: Se han informado dermatitis, erupciones cutáneas y eccema. Considere suspender Prolia si se desarrollan síntomas graves. (5.8)
- Puede ocurrir dolor severo en los huesos, las articulaciones y los músculos. Suspenda el uso si se desarrollan síntomas graves. (5.9)
- Supresión de la renovación ósea: Se ha demostrado una supresión significativa. Monitorear las consecuencias de la sobre supresión ósea. (5.10)
REACCIONES ADVERSAS
- Osteoporosis posmenopáusica: Las reacciones adversas más comunes (> 5% y más comunes que el placebo) fueron: dolor de espalda, dolor en las extremidades, hipercolesterolemia, dolor musculoesquelético y cistitis. Se ha informado pancreatitis en ensayos clínicos. (6.1)
- Osteoporosis masculina: Las reacciones adversas más comunes (> 5% y más comunes que el placebo) fueron: dolor de espalda, artralgia y nasofaringitis. (6.1)
- Osteoporosis inducida por glucocorticoides: Las reacciones adversas más comunes (> 3% y más comunes que el grupo de control activo) fueron: dolor de espalda, hipertensión, bronquitis y dolor de cabeza. (6.1)
- Pérdida ósea debido a la ablación hormonal para el cáncer: Las reacciones adversas más comunes (≥ 10% y más comunes que el placebo) fueron: artralgia y dolor de espalda. También se ha informado dolor en las extremidades y dolor musculoesquelético en ensayos clínicos. (6.1)
Para informar las REACCIONES ADVERSAS SOSPECHOSAS, comuníquese con Amgen Inc. al 1-800-77-AMGEN (1-800-772-6436) o la FDA al 1-800-FDA-1088 o www.fda.gov/medwatch.
USO EN POBLACIONES ESPECÍFICAS
- Mujeres embarazadas y mujeres en edad fértil: Prolia puede causar daño fetal cuando se administra a mujeres embarazadas. Aconseje a las mujeres en edad fértil que utilicen métodos anticonceptivos efectivos durante el tratamiento y durante al menos 5 meses después de la última dosis de Prolia. (8.1, 8.3)
- Pacientes pediátricos: Prolia no está aprobado para su uso en pacientes pediátricos. (8.4)
- Insuficiencia renal: No es necesario ajustar la dosis en pacientes con insuficiencia renal. Los pacientes con enfermedad renal crónica avanzada (eGFR < 30 mL/min/1.73 m2), incluidos los pacientes dependientes de diálisis, tienen un mayor riesgo de hipocalcemia grave. La presencia de enfermedad ósea mineral crónica subyacente relacionada con la enfermedad renal crónica aumenta significativamente el riesgo de hipocalcemia. (5.1, 8.6)
Ver 17 para INFORMACIÓN PARA EL PACIENTE y Guía de Medicamentos.
Revisado: 3/2024
Tabla de Contenido
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDO*
ADVERTENCIA: HIPOCALCEMIA GRAVE EN PACIENTES CON ENFERMEDAD RENAL AVANZADA
1 INDICACIONES Y USO
1.1 Tratamiento de mujeres posmenopáusicas con osteoporosis con alto riesgo de fractura
1.2 Tratamiento para aumentar la masa ósea en hombres con osteoporosis
1.3 Tratamiento de la osteoporosis inducida por glucocorticoides
1.4 Tratamiento de la pérdida ósea en hombres que reciben terapia de privación de andrógenos para el cáncer de próstata
1.5 Tratamiento de la pérdida ósea en mujeres que reciben terapia adyuvante con inhibidores de la aromatasa para el cáncer de mama
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Prueba de embarazo antes de iniciar Prolia
2.2 Pruebas de laboratorio en pacientes con enfermedad renal crónica avanzada antes de iniciar Prolia
2.3 Dosis recomendada
2.4 Preparación y administración
3 FORMAS Y FUERZAS DE DOSIFICACIÓN
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Hipocalcemia grave y cambios en el metabolismo mineral
5.2 Productos farmacéuticos con el mismo ingrediente activo
5.3 Hipersensibilidad
5.4 Osteonecrosis de la mandíbula
5.5 Fracturas femorales atípicas subtrocantéricas y diafisarias
5.6 Fracturas vertebrales múltiples (MVF) después de la interrupción del tratamiento con Prolia
5.7 Infecciones graves
5.8 Reacciones adversas dermatológicas
5.9 Dolor musculoesquelético
5.10 Supresión del recambio óseo
5.11 Hipercalcemia en pacientes pediátricos con osteogénesis imperfecta
6 REACCIONES ADVERSAS
6.1 Experiencia en ensayos clínicos
6.2 Experiencia postcomercialización
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.3 Mujeres y hombres en edad fértil
8.4 Uso pediátrico
8.5 Uso geriátrico
8.6 Insuficiencia renal
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
12.2 Farmacodinamia
12.3 Farmacocinética
12.6 Inmunogenicidad
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, mutagénesis, deterioro de la fertilidad
13.2 Toxicología y/o farmacología animal
14 ESTUDIOS CLÍNICOS
14.1 Tratamiento de mujeres posmenopáusicas con osteoporosis
14.2 Tratamiento para aumentar la masa ósea en hombres con osteoporosis
14.3 Tratamiento de la osteoporosis inducida por glucocorticoides
14.4 Tratamiento de la pérdida ósea en hombres con cáncer de próstata
14.5 Tratamiento de la pérdida ósea en mujeres con cáncer de mama
16 CÓMO SE SUMINISTRA/ALMACENAMIENTO Y MANEJO
17 INFORMACIÓN PARA EL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información completa de prescripción no están enumeradas.
ADVERTENCIA RECUADRADA
ADVERTENCIA: HIPOCALCEMIA GRAVE EN PACIENTES CON ENFERMEDAD RENAL AVANZADA
- Los pacientes con enfermedad renal crónica avanzada (eGFR < 30 mL/min/1.73 m2), incluidos los pacientes dependientes de diálisis, tienen un mayor riesgo de hipocalcemia grave después de la administración de Prolia. Se han notificado casos graves de hipocalcemia que provocaron hospitalización, eventos potencialmente mortales y casos fatales [ver Advertencias y precauciones (5.1)].
- La presencia de trastorno óseo mineral de la enfermedad renal crónica (CKD-MBD) aumenta considerablemente el riesgo de hipocalcemia en estos pacientes [ver Advertencias y precauciones (5.1)].
- Antes de iniciar Prolia en pacientes con enfermedad renal crónica avanzada, evalúe la presencia de CKD-MBD. El tratamiento con Prolia en estos pacientes debe ser supervisado por un profesional sanitario con experiencia en el diagnóstico y manejo de CKD-MBD [ver Dosificación y administración (2.2) y Advertencias y precauciones (5.1)].
1 INDICACIONES Y USO
1.1 Tratamiento de mujeres posmenopáusicas con osteoporosis en alto riesgo de fractura
Prolia está indicado para el tratamiento de mujeres posmenopáusicas con osteoporosis en alto riesgo de fractura, definido como un historial de fractura osteoporótica, o múltiples factores de riesgo de fractura; o pacientes que han fallado o son intolerantes a otra terapia disponible para la osteoporosis. En mujeres posmenopáusicas con osteoporosis, Prolia reduce la incidencia de fracturas vertebrales, no vertebrales y de cadera [ver Estudios clínicos (14.1)].
1.2 Tratamiento para aumentar la masa ósea en hombres con osteoporosis
Prolia está indicado para el tratamiento para aumentar la masa ósea en hombres con osteoporosis en alto riesgo de fractura, definido como un historial de fractura osteoporótica, o múltiples factores de riesgo de fractura; o pacientes que han fallado o son intolerantes a otra terapia disponible para la osteoporosis [ver Estudios clínicos (14.2)].
1.3 Tratamiento de la osteoporosis inducida por glucocorticoides
Prolia está indicado para el tratamiento de la osteoporosis inducida por glucocorticoides en hombres y mujeres con alto riesgo de fractura que están iniciando o continuando con glucocorticoides sistémicos en una dosis diaria equivalente a 7,5 mg o más de prednisona y se espera que permanezcan con glucocorticoides durante al menos 6 meses. El alto riesgo de fractura se define como un historial de fractura osteoporótica, múltiples factores de riesgo de fractura, o pacientes que han fallado o son intolerantes a otra terapia disponible para la osteoporosis [ver Estudios clínicos (14.3)].
1.4 Tratamiento de la pérdida ósea en hombres que reciben terapia de privación de andrógenos para el cáncer de próstata
Prolia está indicado como tratamiento para aumentar la masa ósea en hombres con alto riesgo de fractura que reciben terapia de privación de andrógenos (TDA) para el cáncer de próstata no metastásico. En estos pacientes, Prolia también redujo la incidencia de fracturas vertebrales [ver Estudios clínicos (14.4)].
1.5 Tratamiento de la pérdida ósea en mujeres que reciben terapia adyuvante con inhibidores de la aromatasa para el cáncer de mama
Prolia está indicado como tratamiento para aumentar la masa ósea en mujeres con alto riesgo de fractura que reciben terapia adyuvante con inhibidores de la aromatasa para el cáncer de mama [ver Estudios clínicos (14.5)].
2 DOSIS Y ADMINISTRACIÓN
2.1 Prueba de Embarazo Antes de Iniciar Prolia
Se debe descartar el embarazo antes de administrar Prolia. Realice una prueba de embarazo en todas las mujeres en edad fértil antes de administrar Prolia. Con base en los hallazgos en animales, Prolia puede causar daño fetal cuando se administra a mujeres embarazadas [ver Uso en Poblaciones Específicas (8.1, 8.3)].
2.2 Pruebas de Laboratorio en Pacientes con Enfermedad Renal Crónica Avanzada Antes de Iniciar Prolia
En pacientes con enfermedad renal crónica avanzada [es decir, tasa de filtración glomerular estimada (eGFR) < 30 mL/min/1.73 m2], incluidos los pacientes dependientes de diálisis, evalúe la presencia de trastorno mineral y óseo de la enfermedad renal crónica (CKD-MBD) con hormona paratiroidea intacta (iPTH), calcio sérico, 25(OH) vitamina D y 1,25 (OH)2 vitamina D antes de tomar decisiones con respecto al tratamiento con Prolia. Considere también evaluar el estado de recambio óseo (marcadores séricos de recambio óseo o biopsia ósea) para evaluar la enfermedad ósea subyacente que puede estar presente [ver Advertencias y Precauciones (5.1)].
2.3 Dosis Recomendada
Prolia debe ser administrado por un profesional de la salud.
La dosis recomendada de Prolia es de 60 mg administrada como una sola inyección subcutánea una vez cada 6 meses. Administre Prolia mediante inyección subcutánea en la parte superior del brazo, la parte superior del muslo o el abdomen. Todos los pacientes deben recibir 1000 mg de calcio al día y al menos 400 UI de vitamina D al día [ver Advertencias y Precauciones (5.1)].
Si se omite una dosis de Prolia, administre la inyección tan pronto como el paciente esté disponible. Posteriormente, programe las inyecciones cada 6 meses a partir de la fecha de la última inyección.
2.4 Preparación y Administración
Los productos farmacéuticos parenterales deben inspeccionarse visualmente para detectar partículas y decoloración antes de la administración siempre que la solución y el contenedor lo permitan. Prolia es una solución transparente, incolora a amarillo pálido que puede contener trazas de partículas proteicas traslúcidas a blancas. No lo use si la solución está decolorada o turbia o si la solución contiene muchas partículas o materia particulada extraña.
Antes de la administración, Prolia se puede sacar del refrigerador y llevar a temperatura ambiente (hasta 25 °C/77 °F) colocándolo en el contenedor original. Esto generalmente toma de 15 a 30 minutos. No caliente Prolia de ninguna otra manera [ver Cómo se suministra/Almacenamiento y manipulación (16)].
Instrucciones para la Administración de la Jeringa Prellenada de Prolia con Protector de Seguridad de la Aguja
IMPORTANTE: Para minimizar las punciones accidentales con agujas, la jeringa prellenada de dosis única de Prolia tendrá un protector de seguridad verde; active manualmente el protector de seguridad después de administrar la inyección.
NO deslice el protector de seguridad verde hacia adelante sobre la aguja antes de administrar la inyección; se bloqueará en su lugar y evitará la inyección.

Active el protector de seguridad verde (deslice sobre la aguja) después de la inyección.
Paso 1: Retire la Tapa Gris de la Aguja
| Retire la tapa de la aguja. |  |
Paso 2: Administre la Inyección Subcutánea
| Elija un sitio de inyección. Los sitios de inyección recomendados para Prolia incluyen: la parte superior del brazo O la parte superior del muslo O el abdomen. | 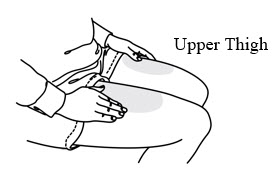 |
|
 |
| Inserte la aguja e inyecte todo el líquido por vía subcutánea. No administre en el músculo o vaso sanguíneo. |
 |
NO vuelva a colocar la tapa gris de la aguja en la aguja.
Paso 3: Deslice Inmediatamente el Protector de Seguridad Verde sobre la Aguja
Con la aguja apuntando hacia afuera.
Sostenga la jeringa prellenada por el agarre de dedo transparente con una mano. Luego, con la otra mano, agarre la base del protector de seguridad verde y deslícelo suavemente hacia la aguja hasta que el protector de seguridad verde se bloquee de forma segura en su lugar y/o escuche un “clic”. NO agarre el protector de seguridad verde con demasiada fuerza; se moverá fácilmente si lo sostiene y lo desliza suavemente.
| Sostenga el agarre de dedo transparente. |  |
|
| Deslice suavemente el protector de seguridad verde sobre la aguja y bloquéelo de forma segura en su lugar. No agarre el protector de seguridad verde con demasiada fuerza al deslizarlo sobre la aguja. | 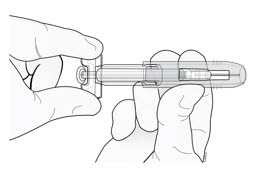 |
Deseche inmediatamente la jeringa y la tapa de la aguja en el contenedor de objetos punzantes más cercano. NO vuelva a colocar la tapa de la aguja en la jeringa usada.
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
- Inyección: 1 mL de una solución de denosumab de 60 mg/mL transparente, incolora a amarillo pálido en una jeringa precargada de dosis única.
4 CONTRAINDICACIONES
Prolia está contraindicado en:
- Pacientes con hipocalcemia: La hipocalcemia preexistente debe corregirse antes de iniciar el tratamiento con Prolia [ver Advertencias y precauciones (5.1)].
- Mujeres embarazadas: Prolia puede causar daño fetal cuando se administra a una mujer embarazada. En mujeres en edad fértil, se debe realizar una prueba de embarazo antes de iniciar el tratamiento con Prolia [ver Uso en poblaciones específicas (8.1)].
- Pacientes con hipersensibilidad a Prolia: Prolia está contraindicado en pacientes con antecedentes de hipersensibilidad sistémica a cualquier componente del producto. Las reacciones han incluido anafilaxia, hinchazón facial y urticaria [ver Advertencias y precauciones (5.3), Reacciones adversas (6.2)].
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Hipocalcemia grave y cambios en el metabolismo mineral
Prolia puede causar hipocalcemia grave y se han notificado casos mortales. La hipocalcemia preexistente debe corregirse antes de iniciar el tratamiento con Prolia. Suplementar adecuadamente a todos los pacientes con calcio y vitamina D [ver Dosificación y administración (2.1), Contraindicaciones (4) y Reacciones adversas (6.1)].
En pacientes sin enfermedad renal crónica avanzada que están predispuestos a la hipocalcemia y alteraciones del metabolismo mineral (por ejemplo, antecedentes de hipoparatiroidismo, cirugía de tiroides, cirugía de paratiroides, síndromes de malabsorción, extirpación del intestino delgado, tratamiento con otros medicamentos que reducen el calcio), evaluar el calcio sérico y los niveles minerales (fósforo y magnesio) de 10 a 14 días después de la inyección de Prolia. En algunos casos de poscomercialización, la hipocalcemia persistió durante semanas o meses y requirió un control frecuente y reemplazo de calcio intravenoso y/u oral, con o sin vitamina D.
Pacientes con enfermedad renal crónica avanzada
Los pacientes con enfermedad renal crónica avanzada [es decir, eGFR < 30 mL/min/1,73 m2] incluidos los pacientes dependientes de diálisis tienen un mayor riesgo de hipocalcemia grave después de la administración de Prolia. Se han notificado casos graves de hipocalcemia que provocaron hospitalización, eventos potencialmente mortales y casos mortales. La presencia de enfermedad renal crónica-trastorno mineral óseo subyacente (CKD-MBD, osteodisplasia renal) aumenta considerablemente el riesgo de hipocalcemia. El uso concomitante de medicamentos calcimiméticos también puede empeorar el riesgo de hipocalcemia.
Para minimizar el riesgo de hipocalcemia en pacientes con enfermedad renal crónica avanzada, evaluar la presencia de trastorno mineral óseo de la enfermedad renal crónica con hormona paratiroidea intacta (iPTH), calcio sérico, 25(OH) vitamina D y 1,25(OH)2 vitamina D antes de tomar decisiones sobre el tratamiento con Prolia. Considere también evaluar el estado de recambio óseo (marcadores séricos de recambio óseo o biopsia ósea) para evaluar la enfermedad ósea subyacente que puede estar presente. Monitorear el calcio sérico semanalmente durante el primer mes después de la administración de Prolia y mensualmente a partir de entonces. Instruya a todos los pacientes con enfermedad renal crónica avanzada, incluidos los que son dependientes de diálisis, sobre los síntomas de la hipocalcemia y la importancia de mantener los niveles de calcio sérico con una suplementación adecuada de calcio y vitamina D activada. El tratamiento con Prolia en estos pacientes debe ser supervisado por un profesional de la salud que tenga experiencia en el diagnóstico y el manejo de CKD-MBD.
5.2 Productos farmacéuticos con el mismo ingrediente activo
Prolia contiene el mismo ingrediente activo (denosumab) que se encuentra en Xgeva. Los pacientes que reciben Prolia no deben recibir Xgeva.
5.3 Hipersensibilidad
Se ha notificado hipersensibilidad clínicamente significativa, incluida la anafilaxia, con Prolia. Los síntomas han incluido hipotensión, disnea, opresión en la garganta, edema facial y de las vías respiratorias superiores, prurito y urticaria. Si se produce una reacción alérgica anafiláctica u otra reacción alérgica clínicamente significativa, inicie la terapia adecuada y suspenda el uso posterior de Prolia [ver Contraindicaciones (4), Reacciones adversas (6.2)].
5.4 Osteonecrosis de la mandíbula
La osteonecrosis de la mandíbula (ONJ), que puede ocurrir espontáneamente, generalmente se asocia con la extracción de dientes y/o infección local con curación retardada. Se ha notificado ONJ en pacientes que reciben denosumab [ver Reacciones adversas (6.1)]. El médico que prescribe debe realizar un examen oral de rutina antes de iniciar el tratamiento con Prolia. Se recomienda un examen dental con odontología preventiva adecuada antes del tratamiento con Prolia en pacientes con factores de riesgo para ONJ como procedimientos dentales invasivos (por ejemplo, extracción de dientes, implantes dentales, cirugía oral), diagnóstico de cáncer, terapias concomitantes (por ejemplo, quimioterapia, corticosteroides, inhibidores de la angiogénesis), higiene oral deficiente y trastornos concomitantes (por ejemplo, enfermedad periodontal y/u otra enfermedad dental preexistente, anemia, coagulopatía, infección, prótesis dentales mal ajustadas). Se deben mantener buenas prácticas de higiene oral durante el tratamiento con Prolia. La administración concomitante de medicamentos asociados con ONJ puede aumentar el riesgo de desarrollar ONJ. El riesgo de ONJ puede aumentar con la duración de la exposición a Prolia.
Para los pacientes que requieren procedimientos dentales invasivos, el juicio clínico del médico tratante y/o cirujano oral debe guiar el plan de manejo de cada paciente en función de la evaluación individual de beneficio-riesgo.
Los pacientes que se sospecha que tienen o que desarrollan ONJ mientras están en Prolia deben recibir atención de un dentista o un cirujano oral. En estos pacientes, la cirugía dental extensa para tratar la ONJ puede exacerbar la afección. Se debe considerar la interrupción de la terapia con Prolia en función de la evaluación individual de beneficio-riesgo.
5.5 Fracturas femorales atípicas subtrocantéricas y diafisarias
Se han notificado fracturas atípicas de baja energía o de bajo trauma del eje en pacientes que reciben Prolia [ver Reacciones adversas (6.1)]. Estas fracturas pueden ocurrir en cualquier parte del eje femoral desde justo debajo del trocánter menor hasta por encima del ensanchamiento supracondíleo y tienen una orientación transversal u oblicua corta sin evidencia de conminución. No se ha establecido la causalidad, ya que estas fracturas también ocurren en pacientes osteoporóticos que no han sido tratados con agentes antirresortivos.
Las fracturas femorales atípicas ocurren con mayor frecuencia con un trauma mínimo o nulo en el área afectada. Pueden ser bilaterales y muchos pacientes informan dolor prodromal en el área afectada, que generalmente se presenta como dolor sordo y punzante en el muslo, semanas o meses antes de que ocurra una fractura completa. Varios informes señalan que los pacientes también estaban recibiendo tratamiento con glucocorticoides (por ejemplo, prednisona) en el momento de la fractura.
Durante el tratamiento con Prolia, se debe aconsejar a los pacientes que informen sobre cualquier dolor nuevo o inusual en el muslo, la cadera o la ingle. Cualquier paciente que presente dolor en el muslo o la ingle debe sospecharse que tiene una fractura atípica y debe ser evaluado para descartar una fractura incompleta del fémur. El paciente que presenta una fractura atípica del fémur también debe ser evaluado para detectar síntomas y signos de fractura en la extremidad contralateral. Se debe considerar la interrupción del tratamiento con Prolia, en función de una evaluación beneficio-riesgo, de forma individual.
5.6 Fracturas Vertebrales Múltiples (MVF) Tras la Interrupción del Tratamiento con Prolia
Tras la interrupción del tratamiento con Prolia, el riesgo de fractura aumenta, incluido el riesgo de fracturas vertebrales múltiples. El tratamiento con Prolia provoca una supresión significativa del recambio óseo y la interrupción del tratamiento con Prolia provoca un aumento del recambio óseo por encima de los valores previos al tratamiento 9 meses después de la última dosis de Prolia. El recambio óseo vuelve entonces a los valores previos al tratamiento 24 meses después de la última dosis de Prolia. Además, la densidad mineral ósea (DMO) vuelve a los valores previos al tratamiento en un plazo de 18 meses después de la última inyección [ver Farmacología Clínica (12.2), Estudios Clínicos (14.1)].
Se produjeron nuevas fracturas vertebrales tan pronto como a los 7 meses (de media 19 meses) después de la última dosis de Prolia. La fractura vertebral previa fue un predictor de fracturas vertebrales múltiples después de la interrupción de Prolia. Evalúe el beneficio-riesgo individual antes de iniciar el tratamiento con Prolia.
Si se interrumpe el tratamiento con Prolia, los pacientes deben ser trasladados a un tratamiento antirresortivo alternativo [ver Reacciones Adversas (6.1)].
5.7 Infecciones Graves
En un ensayo clínico con más de 7800 mujeres con osteoporosis posmenopáusica, se notificaron infecciones graves que llevaron a la hospitalización con más frecuencia en el grupo de Prolia que en el grupo de placebo [ver Reacciones Adversas (6.1)]. Las infecciones cutáneas graves, así como las infecciones del abdomen, el tracto urinario y el oído, fueron más frecuentes en los pacientes tratados con Prolia. También se notificó endocarditis con más frecuencia en los pacientes tratados con Prolia. La incidencia de infecciones oportunistas fue similar entre los grupos de placebo y Prolia, y la incidencia general de infecciones fue similar entre los grupos de tratamiento. Aconseje a los pacientes que busquen atención médica inmediata si desarrollan signos o síntomas de infección grave, incluida la celulitis.
Los pacientes que reciben agentes inmunosupresores concomitantes o que tienen un sistema inmunitario deteriorado pueden tener un mayor riesgo de infecciones graves. Considere el perfil beneficio-riesgo en estos pacientes antes de tratarlos con Prolia. En los pacientes que desarrollan infecciones graves mientras están en tratamiento con Prolia, los prescriptores deben evaluar la necesidad de continuar con el tratamiento con Prolia.
5.8 Reacciones Adversas Dermatológicas
En un gran ensayo clínico con más de 7800 mujeres con osteoporosis posmenopáusica, los eventos adversos epidérmicos y dérmicos como la dermatitis, el eczema y las erupciones cutáneas se produjeron a una tasa significativamente mayor en el grupo de Prolia en comparación con el grupo de placebo. La mayoría de estos eventos no fueron específicos del lugar de la inyección [ver Reacciones Adversas (6.1)]. Considere la posibilidad de interrumpir el tratamiento con Prolia si se desarrollan síntomas graves.
5.9 Dolor Musculoesquelético
En la experiencia postcomercialización, se ha notificado dolor óseo, articular y/o muscular grave y ocasionalmente incapacitante en pacientes que toman Prolia [ver Reacciones Adversas (6.2)]. El tiempo de aparición de los síntomas varió de un día a varios meses después de iniciar el tratamiento con Prolia. Considere la posibilidad de interrumpir el uso si se desarrollan síntomas graves [ver Información para el Paciente (17)].
5.10 Supresión del Recambio Óseo
En los ensayos clínicos en mujeres con osteoporosis posmenopáusica, el tratamiento con Prolia provocó una supresión significativa de la remodelación ósea, como lo demuestran los marcadores del recambio óseo y la histomorfometría ósea [ver Farmacología Clínica (12.2), Estudios Clínicos (14.1)]. Se desconoce la importancia de estos hallazgos y el efecto del tratamiento a largo plazo con Prolia. Las consecuencias a largo plazo del grado de supresión de la remodelación ósea observado con Prolia pueden contribuir a resultados adversos como la osteonecrosis de la mandíbula, las fracturas atípicas y la curación retardada de las fracturas. Controle a los pacientes para detectar estas consecuencias.
5.11 Hipercalcemia en Pacientes Pediátricos con Osteogénesis Imperfecta
Prolia no está aprobado para su uso en pacientes pediátricos. Se ha notificado hipercalcemia en pacientes pediátricos con osteogénesis imperfecta tratados con productos de denosumab, incluido Prolia. Algunos casos requirieron hospitalización [ver Uso en Poblaciones Específicas (8.4)].
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas graves se discuten a continuación y también en otras partes del etiquetado:
- Hipocalcemia [véase Advertencias y Precauciones (5.1)]
- Infecciones graves [véase Advertencias y Precauciones (5.7)]
- Reacciones adversas dermatológicas [véase Advertencias y Precauciones (5.8)]
- Osteonecrosis de la mandíbula [véase Advertencias y Precauciones (5.4)]
- Fracturas femorales atípicas subtrocantéreas y diafisarias [véase Advertencias y Precauciones (5.5)]
- Fracturas vertebrales múltiples (FVM) tras la suspensión del tratamiento con Prolia [véase Advertencias y Precauciones (5.6)]
Las reacciones adversas más comunes notificadas con Prolia en pacientes con osteoporosis posmenopáusica son dolor de espalda, dolor en extremidades, dolor musculoesquelético, hipercolesterolemia y cistitis.
Las reacciones adversas más comunes notificadas con Prolia en hombres con osteoporosis son dolor de espalda, artralgia y nasofaringitis.
Las reacciones adversas más comunes notificadas con Prolia en pacientes con osteoporosis inducida por glucocorticoides son dolor de espalda, hipertensión, bronquitis y cefalea.
Las reacciones adversas más comunes (incidencia por paciente ≥ 10%) notificadas con Prolia en pacientes con pérdida ósea que reciben terapia de privación de andrógenos para el cáncer de próstata o terapia de inhibidor de la aromatasa adyuvante para el cáncer de mama son artralgia y dolor de espalda. También se han notificado dolor en extremidades y dolor musculoesquelético en ensayos clínicos.
Las reacciones adversas más comunes que llevaron a la suspensión de Prolia en pacientes con osteoporosis posmenopáusica son dolor de espalda y estreñimiento.
6.1 Experiencia en ensayos clínicos
Debido a que los estudios clínicos se realizan en condiciones muy variadas, las tasas de reacciones adversas observadas en los estudios clínicos de un fármaco no pueden compararse directamente con las tasas en los estudios clínicos de otro fármaco y pueden no reflejar las tasas observadas en la práctica clínica.
Tratamiento de mujeres posmenopáusicas con osteoporosis
La seguridad de Prolia en el tratamiento de la osteoporosis posmenopáusica se evaluó en un estudio multinacional, aleatorizado, doble ciego, controlado con placebo, de 3 años, de 7 808 mujeres posmenopáusicas de 60 a 91 años de edad. Un total de 3 876 mujeres fueron expuestas a placebo y 3 886 mujeres fueron expuestas a Prolia administrada por vía subcutánea una vez cada 6 meses como una dosis única de 60 mg. A todas las mujeres se les indicó que tomaran al menos 1 000 mg de calcio y 400 UI de suplementación de vitamina D al día.
La incidencia de mortalidad por todas las causas fue del 2,3% (n = 90) en el grupo de placebo y del 1,8% (n = 70) en el grupo de Prolia. La incidencia de eventos adversos graves no fatales fue del 24,2% en el grupo de placebo y del 25,0% en el grupo de Prolia. El porcentaje de pacientes que abandonaron el estudio debido a eventos adversos fue del 2,1% y del 2,4% para los grupos de placebo y Prolia, respectivamente. Las reacciones adversas más comunes notificadas con Prolia en pacientes con osteoporosis posmenopáusica son dolor de espalda, dolor en extremidades, dolor musculoesquelético, hipercolesterolemia y cistitis.
Las reacciones adversas notificadas en ≥ 2% de las mujeres posmenopáusicas con osteoporosis y con mayor frecuencia en las mujeres tratadas con Prolia que en las tratadas con placebo se muestran en la tabla siguiente.
| Término preferido | Prolia (N = 3886) n (%) |
Placebo (N = 3876) n (%) |
|---|---|---|
| Dolor de espalda | 1347 (34.7) | 1340 (34.6) |
| Dolor en extremidades | 453 (11.7) | 430 (11.1) |
| Dolor musculoesquelético | 297 (7.6) | 291 (7.5) |
| Hipercolesterolemia | 280 (7.2) | 236 (6.1) |
| Cistitis | 228 (5.9) | 225 (5.8) |
| Vértigo | 195 (5.0) | 187 (4.8) |
| Infección de las vías respiratorias superiores | 190 (4.9) | 167 (4.3) |
| Edema periférico | 189 (4.9) | 155 (4.0) |
| Ciática | 178 (4.6) | 149 (3.8) |
| Dolor óseo | 142 (3.7) | 117 (3.0) |
| Dolor abdominal superior | 129 (3.3) | 111 (2.9) |
| Anemia | 129 (3.3) | 107 (2.8) |
| Insomnio | 126 (3.2) | 122 (3.1) |
| Mialgia | 114 (2.9) | 94 (2.4) |
| Angina de pecho | 101 (2.6) | 87 (2.2) |
| Erupción cutánea | 96 (2.5) | 79 (2.0) |
| Faringitis | 91 (2.3) | 78 (2.0) |
| Astenia | 90 (2.3) | 73 (1.9) |
| Prurito | 87 (2.2) | 82 (2.1) |
| Flatulencia | 84 (2.2) | 53 (1.4) |
| Osteoartritis espinal | 82 (2.1) | 64 (1.7) |
| Enfermedad por reflujo gastroesofágico | 80 (2.1) | 66 (1.7) |
| Herpes zóster | 79 (2.0) | 72 (1.9) |
Hipocalcemia
Se reportaron disminuciones en los niveles de calcio sérico a menos de 8.5 mg/dL en cualquier visita en el 0.4% de las mujeres en el grupo placebo y en el 1.7% de las mujeres en el grupo Prolia. El punto más bajo en el nivel de calcio sérico ocurrió aproximadamente a los 10 días después de la dosificación de Prolia en sujetos con función renal normal.
En estudios clínicos, los sujetos con función renal deteriorada tuvieron más probabilidades de tener mayores reducciones en los niveles de calcio sérico en comparación con los sujetos con función renal normal. En un estudio de 55 sujetos con diferentes grados de función renal, se observaron niveles de calcio sérico < 7.5 mg/dL o hipocalcemia sintomática en 5 sujetos. Estos incluyeron ningún sujeto en el grupo de función renal normal, el 10% de los sujetos en el grupo de aclaramiento de creatinina de 50 a 80 mL/min, el 29% de los sujetos en el grupo de aclaramiento de creatinina < 30 mL/min y el 29% de los sujetos en el grupo de hemodiálisis. Estos sujetos no recibieron suplementos de calcio y vitamina D. En un estudio de 4550 mujeres posmenopáusicas con osteoporosis, el cambio medio desde el inicio en el nivel de calcio sérico 10 días después de la dosificación de Prolia fue del -5.5% en sujetos con aclaramiento de creatinina < 30 mL/min frente al -3.1% en sujetos con aclaramiento de creatinina ≥ 30 mL/min.
Infecciones graves
El activador del receptor del ligando del factor nuclear kappa-B (RANKL) se expresa en linfocitos T y B activados y en los ganglios linfáticos. Por lo tanto, un inhibidor de RANKL como Prolia puede aumentar el riesgo de infección.
En el estudio clínico de 7808 mujeres posmenopáusicas con osteoporosis, la incidencia de infecciones que provocaron la muerte fue del 0.2% tanto en los grupos de tratamiento con placebo como con Prolia. Sin embargo, la incidencia de infecciones graves no mortales fue del 3.3% en el grupo placebo y del 4.0% en los grupos Prolia. Se reportaron hospitalizaciones debido a infecciones graves en el abdomen (0.7% placebo frente a 0.9% Prolia), tracto urinario (0.5% placebo frente a 0.7% Prolia) y oído (0.0% placebo frente a 0.1% Prolia). Se reportó endocarditis en ningún paciente con placebo y 3 pacientes que recibieron Prolia.
Se reportaron infecciones de la piel, incluida la erisipela y la celulitis, que llevaron a la hospitalización con más frecuencia en pacientes tratados con Prolia (< 0.1% placebo frente a 0.4% Prolia).
La incidencia de infecciones oportunistas fue similar a la reportada con placebo.
Reacciones adversas dermatológicas
Un número significativamente mayor de pacientes tratados con Prolia desarrollaron eventos adversos epidérmicos y dérmicos (como dermatitis, eccema y erupciones), con estos eventos reportados en el 8.2% del grupo placebo y el 10.8% de los grupos Prolia (p < 0.0001). La mayoría de estos eventos no fueron específicos del sitio de inyección [ver Advertencias y precauciones (5.8)].
Osteonecrosis de la mandíbula
Se ha reportado ONJ en el programa de ensayos clínicos de osteoporosis en pacientes tratados con Prolia [ver Advertencias y precauciones (5.4)].
Fracturas femorales atípicas subtrocantéricas y diafisarias
En el programa de ensayos clínicos de osteoporosis, se reportaron fracturas femorales atípicas en pacientes tratados con Prolia. La duración de la exposición a Prolia hasta el momento del diagnóstico de fractura femoral atípica fue tan temprana como 2 años y medio [ver Advertencias y precauciones (5.5)].
Fracturas vertebrales múltiples (MVF) después de la interrupción del tratamiento con Prolia
En el programa de ensayos clínicos de osteoporosis, se reportaron fracturas vertebrales múltiples en pacientes después de la interrupción de Prolia. En el ensayo de fase 3 en mujeres con osteoporosis posmenopáusica, el 6% de las mujeres que interrumpieron Prolia y permanecieron en el estudio desarrollaron nuevas fracturas vertebrales, y el 3% de las mujeres que interrumpieron Prolia y permanecieron en el estudio desarrollaron múltiples nuevas fracturas vertebrales. El tiempo medio hasta el inicio de las fracturas vertebrales múltiples fue de 17 meses (rango: 7-43 meses) después de la última inyección de Prolia. La fractura vertebral previa fue un predictor de fracturas vertebrales múltiples después de la interrupción [ver Advertencias y precauciones (5.6)].
Pancreatitis
Se reportó pancreatitis en 4 pacientes (0.1%) en el grupo placebo y 8 pacientes (0.2%) en los grupos Prolia. De estos informes, 1 paciente en el grupo placebo y los 8 pacientes en el grupo Prolia tuvieron eventos graves, incluida una muerte en el grupo Prolia. Varios pacientes tenían antecedentes de pancreatitis. El tiempo desde la administración del producto hasta la aparición del evento fue variable.
Nuevas neoplasias malignas
La incidencia general de nuevas neoplasias malignas fue del 4.3% en el grupo placebo y del 4.8% en los grupos Prolia. Se reportaron nuevas neoplasias malignas relacionadas con la mama (0.7% placebo frente a 0.9% Prolia), el sistema reproductivo (0.2% placebo frente a 0.5% Prolia) y el sistema gastrointestinal (0.6% placebo frente a 0.9% Prolia). No se ha establecido una relación causal con la exposición al fármaco.
Tratamiento para aumentar la masa ósea en hombres con osteoporosis
La seguridad de Prolia en el tratamiento de hombres con osteoporosis se evaluó en un estudio aleatorizado, doble ciego, controlado con placebo de 1 año. Un total de 120 hombres fueron expuestos a placebo y 120 hombres fueron expuestos a Prolia administrado por vía subcutánea una vez cada 6 meses como una sola dosis de 60 mg. Todos los hombres recibieron instrucciones de tomar al menos 1000 mg de calcio y 800 UI de suplementos de vitamina D por día.
La incidencia de mortalidad por todas las causas fue del 0.8% (n = 1) en el grupo placebo y del 0.8% (n = 1) en el grupo Prolia. La incidencia de eventos adversos graves no mortales fue del 7.5% en el grupo placebo y del 8.3% en el grupo Prolia. El porcentaje de pacientes que se retiraron del estudio debido a eventos adversos fue del 0% y del 2.5% para los grupos placebo y Prolia, respectivamente.
Las reacciones adversas reportadas en ≥ 5% de los hombres con osteoporosis y con más frecuencia con Prolia que en los pacientes tratados con placebo fueron: dolor de espalda (6.7% placebo frente a 8.3% Prolia), artralgia (5.8% placebo frente a 6.7% Prolia) y nasofaringitis (5.8% placebo frente a 6.7% Prolia).
Infecciones graves
Se informó infección grave en 1 paciente (0.8%) en el grupo placebo y en ningún paciente en el grupo Prolia.
Reacciones adversas dermatológicas
Se informaron eventos adversos epidérmicos y dérmicos (como dermatitis, eczema y erupciones) en 4 pacientes (3.3%) en el grupo placebo y 5 pacientes (4.2%) en el grupo Prolia.
Tratamiento de la osteoporosis inducida por glucocorticoides
La seguridad de Prolia en el tratamiento de la osteoporosis inducida por glucocorticoides se evaluó en el análisis primario de 1 año de un estudio aleatorizado, multicéntrico, doble ciego, de grupos paralelos, controlado con activo de 2 años de duración en 795 pacientes (30% hombres y 70% mujeres) de 20 a 94 años (edad media de 63 años) tratados con más de o igual a 7.5 mg/día de prednisona oral (o equivalente). Un total de 384 pacientes fueron expuestos a 5 mg de bisfosfonato oral diario (control activo) y 394 pacientes fueron expuestos a Prolia administrado una vez cada 6 meses como una dosis subcutánea de 60 mg. Se indicó a todos los pacientes que tomaran al menos 1000 mg de calcio y 800 UI de suplementos de vitamina D por día.
La incidencia de mortalidad por todas las causas fue del 0.5% (n = 2) en el grupo control activo y del 1.5% (n = 6) en el grupo Prolia. La incidencia de eventos adversos graves fue del 17% en el grupo control activo y del 16% en el grupo Prolia. El porcentaje de pacientes que se retiraron del estudio debido a eventos adversos fue del 3.6% y del 3.8% para los grupos control activo y Prolia, respectivamente.
Las reacciones adversas notificadas en ≥ 2% de los pacientes con osteoporosis inducida por glucocorticoides y con mayor frecuencia con Prolia que en los pacientes tratados con control activo se muestran en la tabla siguiente.
| Término preferido | Prolia (N = 394) n (%) |
Bisfosfonato oral diario (Control activo) (N = 384) n (%) |
|---|---|---|
|
||
| Dolor de espalda | 18 (4.6) | 17 (4.4) |
| Hipertensión | 15 (3.8) | 13 (3.4) |
| Bronquitis | 15 (3.8) | 11 (2.9) |
| Dolor de cabeza | 14 (3.6) | 7 (1.8) |
| Dispepsia | 12 (3.0) | 10 (2.6) |
| Infección del tracto urinario | 12 (3.0) | 8 (2.1) |
| Dolor abdominal superior | 12 (3.0) | 7 (1.8) |
| Infección de las vías respiratorias superiores | 11 (2.8) | 10 (2.6) |
| Estreñimiento | 11 (2.8) | 6 (1.6) |
| Vómitos | 10 (2.5) | 6 (1.6) |
| Mareos | 9 (2.3) | 8 (2.1) |
| Caída | 8 (2.0) | 7 (1.8) |
| Polimialgia reumática* | 8 (2.0) | 1 (0.3) |
Fracturas femorales atípicas subtrocantéricas y diafisarias
Se reportaron fracturas femorales atípicas en 1 paciente tratado con Prolia. La duración de la exposición a Prolia hasta el momento del diagnóstico de fractura femoral atípica fue de 8.0 meses [ver Advertencias y precauciones (5.5)].
Tratamiento de la pérdida ósea en pacientes que reciben terapia de privación de andrógenos para el cáncer de próstata o terapia adyuvante con inhibidores de la aromatasa para el cáncer de mama
La seguridad de Prolia en el tratamiento de la pérdida ósea en hombres con cáncer de próstata no metastásico que reciben terapia de privación de andrógenos (TDA) se evaluó en un estudio multinacional de 3 años, aleatorizado, doble ciego, controlado con placebo, de 1468 hombres de 48 a 97 años. Un total de 725 hombres fueron expuestos a placebo y 731 hombres fueron expuestos a Prolia administrado una vez cada 6 meses como una sola dosis subcutánea de 60 mg. Todos los hombres recibieron instrucciones de tomar al menos 1000 mg de calcio y 400 UI de vitamina D como suplemento por día.
La incidencia de eventos adversos graves fue del 30.6% en el grupo placebo y del 34.6% en el grupo Prolia. El porcentaje de pacientes que se retiraron del estudio debido a eventos adversos fue del 6.1% y del 7.0% para los grupos placebo y Prolia, respectivamente.
La seguridad de Prolia en el tratamiento de la pérdida ósea en mujeres con cáncer de mama no metastásico que reciben terapia con inhibidores de la aromatasa (IA) se evaluó en un estudio multinacional de 2 años, aleatorizado, doble ciego, controlado con placebo, de 252 mujeres posmenopáusicas de 35 a 84 años. Un total de 120 mujeres fueron expuestas a placebo y 129 mujeres fueron expuestas a Prolia administrado una vez cada 6 meses como una sola dosis subcutánea de 60 mg. Todas las mujeres recibieron instrucciones de tomar al menos 1000 mg de calcio y 400 UI de vitamina D como suplemento por día.
La incidencia de eventos adversos graves fue del 9.2% en el grupo placebo y del 14.7% en el grupo Prolia. El porcentaje de pacientes que se retiraron del estudio debido a eventos adversos fue del 4.2% y del 0.8% para los grupos placebo y Prolia, respectivamente.
Las reacciones adversas reportadas en ≥ 10% de los pacientes tratados con Prolia que recibieron TDA para el cáncer de próstata o terapia adyuvante con IA para el cáncer de mama, y con mayor frecuencia que en los pacientes tratados con placebo fueron: artralgia (13.0% placebo vs. 14.3% Prolia) y dolor de espalda (10.5% placebo vs. 11.5% Prolia). También se ha reportado dolor en las extremidades (7.7% placebo vs. 9.9% Prolia) y dolor musculoesquelético (3.8% placebo vs. 6.0% Prolia) en ensayos clínicos. Además, en hombres tratados con Prolia con cáncer de próstata no metastásico que recibieron TDA, se observó una mayor incidencia de cataratas (1.2% placebo vs. 4.7% Prolia). La hipocalcemia (calcio sérico < 8.4 mg/dL) se reportó solo en pacientes tratados con Prolia (2.4% vs. 0.0%) en la visita del mes 1.
6.2 Experiencia postcomercialización
Debido a que las reacciones postcomercialización se reportan voluntariamente de una población de tamaño incierto, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal con la exposición al fármaco.
Las siguientes reacciones adversas se han identificado durante el uso posterior a la aprobación de Prolia:
- Reacciones de hipersensibilidad relacionadas con el fármaco: anafilaxia, erupción cutánea, urticaria, hinchazón facial y eritema
- Hipocalcemia: hipocalcemia sintomática grave que resulta en hospitalización, eventos que ponen en peligro la vida y casos fatales
- Dolor musculoesquelético, incluidos casos graves
- Hormona paratiroidea (PTH): Elevación marcada de la PTH sérica en pacientes con insuficiencia renal grave (aclaramiento de creatinina < 30 mL/min) o que reciben diálisis
- Múltiples fracturas vertebrales después de la interrupción de Prolia
- Erupciones medicamentosas liquenoides cutáneas y mucosas (por ejemplo, reacciones similares al liquen plano)
- Alopecia
- Vasculitis (por ejemplo, vasculitis positiva para ANCA, vasculitis leucocitoclástica)
- Síndrome de reacción medicamentosa con eosinofilia y síntomas sistémicos (DRESS)
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de Riesgos
Prolia está contraindicado para su uso en mujeres embarazadas porque puede causar daño al feto. No hay datos suficientes sobre el uso de denosumab en mujeres embarazadas para informar sobre ningún riesgo asociado con el fármaco para los resultados del desarrollo adverso. La exposición a denosumab in útero de monos cynomolgus tratados mensualmente con denosumab durante todo el embarazo a una dosis 50 veces mayor que la dosis humana recomendada en función del peso corporal resultó en un aumento de la pérdida fetal, muertes fetales y mortalidad postnatal, y ausencia de ganglios linfáticos, crecimiento óseo anormal y disminución del crecimiento neonatal [ver Datos].
Datos
Datos en animales
Los efectos de denosumab sobre el desarrollo prenatal se han estudiado tanto en monos cynomolgus como en ratones genéticamente modificados en los que la expresión del ligando RANK (RANKL) se desactivó mediante la eliminación de genes (un “ratón knockout”). En monos cynomolgus tratados por vía subcutánea con denosumab durante todo el embarazo a partir del día 20 de gestación y a una dosis farmacológicamente activa 50 veces mayor que la dosis humana recomendada en función del peso corporal, se observó un aumento de la pérdida fetal durante la gestación, muertes fetales y mortalidad postnatal. Otros hallazgos en la descendencia incluyeron la ausencia de ganglios linfáticos axilares, inguinales, mandibulares y mesentéricos; crecimiento óseo anormal, reducción de la resistencia ósea, reducción de la hematopoyesis, displasia dental y desalineación dental; y disminución del crecimiento neonatal. Al nacer y hasta el mes de edad, los bebés tenían niveles sanguíneos medibles de denosumab (22-621% de los niveles maternos).
Tras un período de recuperación desde el nacimiento hasta los 6 meses de edad, los efectos sobre la calidad y la resistencia ósea volvieron a la normalidad; no hubo efectos adversos sobre la erupción dental, aunque la displasia dental seguía siendo evidente; los ganglios linfáticos axilares e inguinales permanecieron ausentes, mientras que los ganglios linfáticos mandibulares y mesentéricos estaban presentes, aunque pequeños; y se observó una mineralización mínima a moderada en múltiples tejidos en un animal de recuperación. No hubo evidencia de daño materno antes del trabajo de parto; los efectos adversos maternos ocurrieron con poca frecuencia durante el trabajo de parto. El desarrollo de la glándula mamaria materna fue normal. No se estableció un NOAEL (nivel sin efecto adverso observable) fetal para este estudio porque solo se evaluó una dosis de 50 mg/kg. La histopatología de la glándula mamaria a los 6 meses de edad fue normal en la descendencia femenina expuesta a denosumab in útero; sin embargo, el desarrollo y la lactancia no se han evaluado completamente.
En ratones knockout RANKL, la ausencia de RANKL (el objetivo de denosumab) también causó agenesia de los ganglios linfáticos fetales y condujo a un deterioro postnatal de la dentición y el crecimiento óseo. Las ratonas embarazadas knockout RANKL mostraron una maduración alterada de la glándula mamaria materna, lo que condujo a una lactancia deteriorada [ver Uso en poblaciones específicas (8.2), Toxicología no clínica (13.2)].
Se desconoce la dosis sin efecto para la teratogenicidad inducida por denosumab. Sin embargo, se identificó una Cmax de 22,9 ng/mL en monos cynomolgus como un nivel en el que no se observaron efectos biológicos (NOEL) de denosumab (sin inhibición de RANKL) [ver Farmacología clínica (12.3)].
8.2 Lactancia
Resumen de Riesgos
No hay información sobre la presencia de denosumab en la leche materna, los efectos sobre el lactante amamantado o los efectos sobre la producción de leche. Se detectó denosumab en la leche materna de monos cynomolgus hasta 1 mes después de la última dosis de denosumab (≤ 0,5% de la relación leche:suero) y el desarrollo de la glándula mamaria materna fue normal, sin lactancia deteriorada. Sin embargo, las ratonas embarazadas knockout RANKL mostraron una maduración alterada de la glándula mamaria materna, lo que condujo a una lactancia deteriorada [ver Uso en poblaciones específicas (8.1), Toxicología no clínica (13.2)].
8.3 Mujeres y hombres en edad fértil
Según los hallazgos en animales, Prolia puede causar daño fetal cuando se administra a una mujer embarazada [ver Uso en poblaciones específicas (8.1)].
Prueba de embarazo
Verifique el estado de embarazo de las mujeres en edad fértil antes de iniciar el tratamiento con Prolia.
Anticoncepción
Mujeres
Aconseje a las mujeres en edad fértil que utilicen métodos anticonceptivos eficaces durante la terapia y durante al menos 5 meses después de la última dosis de Prolia.
Hombres
Denosumab estuvo presente en bajas concentraciones (aproximadamente el 2% de la exposición sérica) en el líquido seminal de los sujetos masculinos que recibieron Prolia. Tras el coito vaginal, la cantidad máxima de denosumab administrada a una pareja femenina daría como resultado exposiciones aproximadamente 11000 veces más bajas que la dosis subcutánea prescrita de 60 mg y al menos 38 veces más bajas que el NOEL en monos.
Por lo tanto, el uso de condón masculino no sería necesario, ya que es poco probable que una pareja femenina o un feto se expongan a concentraciones farmacológicamente relevantes de denosumab a través del líquido seminal [ver Farmacología clínica (12.3)].
8.4 Uso pediátrico
No se ha establecido la seguridad y eficacia de Prolia en pacientes pediátricos.
En un estudio multicéntrico, abierto, realizado en 153 pacientes pediátricos con osteogénesis imperfecta, de 2 a 17 años, que evaluaba la reducción del riesgo de fracturas, no se estableció la eficacia.
Se ha informado hipercalcemia en pacientes pediátricos con osteogénesis imperfecta tratados con productos de denosumab, incluido Prolia. Algunos casos requirieron hospitalización y se complicaron con lesión renal aguda [ver Advertencias y precauciones (5.11)]. Los estudios clínicos en pacientes pediátricos con osteogénesis imperfecta se terminaron antes de tiempo debido a la aparición de eventos potencialmente mortales y hospitalizaciones debido a hipercalcemia.
Con base en los resultados de estudios en animales, Prolia puede afectar negativamente el crecimiento de los huesos largos y la dentición en pacientes pediátricos menores de 4 años.
Datos de toxicidad en animales jóvenes
El tratamiento con Prolia puede afectar el crecimiento de los huesos largos en niños con placas de crecimiento abiertas y puede inhibir la erupción de la dentición. En ratas neonatales, la inhibición de RANKL (el objetivo de la terapia con Prolia) con un constructo de osteoprotegerina unida a Fc (OPG-Fc) a dosis ≤ 10 mg/kg se asoció con la inhibición del crecimiento óseo y la erupción de los dientes. Los primates adolescentes tratados con denosumab a dosis 10 y 50 veces (10 y 50 mg/kg de dosis) más altas que la dosis humana recomendada de 60 mg administrada cada 6 meses, según el peso corporal (mg/kg), tuvieron placas de crecimiento anormales, consideradas consistentes con la actividad farmacológica de denosumab [ver Toxicología no clínica (13.2)].
Los monos cynomolgus expuestos in útero a denosumab exhibieron anormalidades óseas, ausencia de ganglios linfáticos axilares, inguinales, mandibulares y mesentéricos, hematopoyesis reducida, desalineación de los dientes y disminución del crecimiento neonatal. Algunas anormalidades óseas se recuperaron una vez que la exposición cesó después del nacimiento; sin embargo, los ganglios linfáticos axilares e inguinales permanecieron ausentes 6 meses después del nacimiento [ver Uso en poblaciones específicas (8.1)].
8.5 Uso en geriatría
Del número total de pacientes en estudios clínicos de Prolia, 9943 pacientes (76%) tenían ≥ 65 años, mientras que 3576 (27%) tenían ≥ 75 años. De los pacientes en el estudio de osteoporosis en hombres, 133 pacientes (55%) tenían ≥ 65 años, mientras que 39 pacientes (16%) tenían ≥ 75 años. De los pacientes en el estudio de osteoporosis inducida por glucocorticoides, 355 pacientes (47%) tenían ≥ 65 años, mientras que 132 pacientes (17%) tenían ≥ 75 años. No se observaron diferencias generales en la seguridad o la eficacia entre estos pacientes y los pacientes más jóvenes, y otras experiencias clínicas informadas no han identificado diferencias en las respuestas entre los pacientes de edad avanzada y los pacientes más jóvenes, pero no se puede descartar una mayor sensibilidad de algunos individuos mayores.
8.6 Insuficiencia renal
No es necesario ajustar la dosis en pacientes con insuficiencia renal.
Se ha informado hipocalcemia grave que resultó en hospitalización, eventos potencialmente mortales y casos fatales después de la comercialización. En estudios clínicos, los pacientes con enfermedad renal crónica avanzada (es decir, eGFR < 30 mL/min/1,73 m2), incluidos los pacientes dependientes de diálisis, tenían un mayor riesgo de desarrollar hipocalcemia. La presencia de enfermedad renal crónica-trastorno mineral óseo subyacente (CKD-MBD, osteodístrofia renal) aumenta marcadamente el riesgo de hipocalcemia. El uso concomitante de medicamentos calcimiméticos también puede empeorar el riesgo de hipocalcemia. Considere los beneficios y los riesgos para el paciente cuando administre Prolia a pacientes con enfermedad renal crónica avanzada. Monitoree los niveles de calcio y minerales (fósforo y magnesio). La ingesta adecuada de calcio y vitamina D es importante en pacientes con enfermedad renal crónica avanzada, incluidos los pacientes dependientes de diálisis [ver Dosificación y administración (2.2),Advertencias y precauciones (5.1), Reacciones adversas (6.1) y Farmacología clínica (12.3)].
11 DESCRIPCIÓN
Denosumab es un anticuerpo monoclonal humano IgG2 con afinidad y especificidad para RANKL humano (receptor activador del ligando del factor nuclear kappa-B). Denosumab tiene un peso molecular aproximado de 147 kDa y se produce en células de mamíferos (ovario de hámster chino) genéticamente modificadas.
Prolia (denosumab) inyección es una solución estéril, sin conservantes, transparente, incolora a amarillo pálido para uso subcutáneo.
Cada jeringa precargada de dosis única de 1 mL de Prolia contiene 60 mg de denosumab (solución de 60 mg/mL), 4,7% de sorbitol, 17 mM de acetato, 0,01% de polisorbato 20, Agua para inyección (USP) e hidróxido de sodio a un pH de 5,2.
12 FARMACOLOGÍA CLÍNICA
12.1 Mechanism of Action
Prolia se une a RANKL, una proteína transmembrana o soluble esencial para la formación, la función y la supervivencia de los osteoclastos, las células responsables de la resorción ósea. Prolia evita que RANKL active su receptor, RANK, en la superficie de los osteoclastos y sus precursores. La prevención de la interacción RANKL/RANK inhibe la formación, la función y la supervivencia de los osteoclastos, lo que disminuye la resorción ósea y aumenta la masa ósea y la resistencia tanto en el hueso cortical como en el trabecular.
12.2 Pharmacodynamics
En estudios clínicos, el tratamiento con 60 mg de Prolia produjo una reducción en el marcador de resorción ósea del telopéptido C sérico de tipo 1 (CTX) de aproximadamente el 85 % a los 3 días, con reducciones máximas que ocurrieron al mes. Los niveles de CTX estuvieron por debajo del límite de cuantificación del ensayo (0.049 ng/mL) en el 39 % al 68 % de los pacientes 1 a 3 meses después de la administración de Prolia. Al final de cada intervalo de dosificación, las reducciones de CTX se atenuaron parcialmente de una reducción máxima de ≥ 87 % a ≥ 45 % (rango: 45 % a 80 %), a medida que los niveles séricos de denosumab disminuyeron, lo que refleja la reversibilidad de los efectos de Prolia sobre la remodelación ósea. Estos efectos se mantuvieron con el tratamiento continuo. Al reiniciarse, el grado de inhibición de CTX por Prolia fue similar al observado en pacientes que iniciaron el tratamiento con Prolia.
De acuerdo con el acoplamiento fisiológico de la formación y resorción ósea en la remodelación esquelética, se observaron reducciones posteriores en los marcadores de formación ósea (es decir, osteocalcina y péptido N-terminal del procolágeno tipo 1 [P1NP]) a partir de 1 mes después de la primera dosis de Prolia. Después de la interrupción del tratamiento con Prolia, los marcadores de resorción ósea aumentaron a niveles del 40 % al 60 % por encima de los valores previos al tratamiento, pero volvieron a los niveles basales en un plazo de 12 meses.
12.3 Pharmacokinetics
En un estudio realizado en voluntarios sanos de sexo masculino y femenino (n = 73, rango de edad: 18 a 64 años) después de una dosis única de Prolia administrada por vía subcutánea de 60 mg, el área media bajo la curva de concentración-tiempo hasta las 16 semanas (AUC0-16 semanas) de denosumab fue de 316 mcg∙día/mL (DE = 101 mcg∙día/mL). La concentración máxima media de denosumab (Cmáx) fue de 6.75 mcg/mL (DE = 1.89 mcg/mL). No se observa acumulación ni cambio en la farmacocinética de denosumab con el tiempo con dosis múltiples de 60 mg administradas por vía subcutánea una vez cada 6 meses.
Absorción
Después de la administración subcutánea, la mediana del tiempo hasta la concentración máxima de denosumab (Tmáx) fue de 10 días (rango: 3 a 21 días).
Eliminación
Las concentraciones séricas de denosumab disminuyeron durante un período de 4 a 5 meses con una vida media de 25.4 días (DE = 8.5 días; n = 46).
Se realizó un análisis farmacocinético poblacional para evaluar los efectos de las características demográficas. Este análisis no mostró diferencias notables en la farmacocinética con la edad (en mujeres posmenopáusicas), la raza o el peso corporal (36 a 140 kg).
Estudio farmacocinético del líquido seminal
Se midieron las concentraciones séricas y en líquido seminal de denosumab en 12 voluntarios sanos de sexo masculino (rango de edad: 43-65 años). Después de una única administración subcutánea de 60 mg de denosumab, los valores medios (± DE) de Cmáx en las muestras de suero y líquido seminal fueron de 6170 (± 2070) y 100 (± 81.9) ng/mL, respectivamente, lo que dio como resultado una concentración máxima en líquido seminal de aproximadamente el 2 % de los niveles séricos. Los valores medios (rango) de Tmáx en las muestras de suero y líquido seminal fueron de 8.0 (7.9 a 21) y 21 (8.0 a 49) días, respectivamente. Entre los sujetos, la concentración más alta de denosumab en el líquido seminal fue de 301 ng/mL a los 22 días posteriores a la dosis. En el primer día de medición (10 días después de la dosis), nueve de los once sujetos tenían concentraciones cuantificables en el semen. En el último día de medición (106 días después de la dosis), cinco sujetos todavía tenían concentraciones cuantificables de denosumab en el líquido seminal, con una concentración media (± DE) en el líquido seminal de 21.1 (± 36.5) ng/mL en todos los sujetos (n = 12).
Interacciones farmacológicas
En un estudio de 19 mujeres posmenopáusicas con baja DMO y artritis reumatoide tratadas con etanercept (50 mg de inyección subcutánea una vez a la semana), se administró una dosis única de denosumab (60 mg de inyección subcutánea) 7 días después de la dosis previa de etanercept. No se observaron cambios clínicamente significativos en la farmacocinética de etanercept.
Sustratos del citocromo P450
En un estudio de 17 mujeres posmenopáusicas con osteoporosis, se administró midazolam (2 mg por vía oral) 2 semanas después de una dosis única de denosumab (60 mg de inyección subcutánea), que se aproxima a la Tmáx de denosumab. Denosumab no afectó a la farmacocinética de midazolam, que se metaboliza por el citocromo P450 3A4 (CYP3A4). Esto indica que denosumab no debe alterar la farmacocinética de los fármacos metabolizados por CYP3A4 en mujeres posmenopáusicas con osteoporosis.
Poblaciones específicas
Sexo: Los perfiles medios de concentración-tiempo séricos de denosumab observados en un estudio realizado en hombres sanos ≥ 50 años fueron similares a los observados en un estudio realizado en mujeres posmenopáusicas utilizando el mismo régimen de dosificación.
Edad: La farmacocinética de denosumab no se vio afectada por la edad en todas las poblaciones estudiadas cuyas edades oscilaron entre 28 y 87 años.
12.6 Inmunogenicidad
La incidencia observada de anticuerpos contra el fármaco depende en gran medida de la sensibilidad y especificidad del ensayo. Las diferencias en los métodos de ensayo impiden realizar comparaciones significativas de la incidencia de anticuerpos contra el fármaco en los estudios que se describen a continuación con la incidencia de anticuerpos contra el fármaco en otros estudios, incluidos los de Prolia o productos de denosumab.
Utilizando un inmunoensayo de puente electroquimioluminiscente, menos del 1% (55 de 8113) de los pacientes tratados con Prolia durante un máximo de 5 años dieron positivo en las pruebas de anticuerpos de unión (incluidos los anticuerpos preexistentes, transitorios y en desarrollo). Ninguno de los pacientes dio positivo en las pruebas de anticuerpos neutralizantes, según se evaluó mediante un ensayo biológico in vitro quimioluminiscente basado en células.
No se identificó ningún efecto clínicamente significativo de los anticuerpos contra el fármaco sobre la farmacocinética, la farmacodinámica, la seguridad o la eficacia de denosumab.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity
El potencial carcinogénico de denosumab no se ha evaluado en estudios a largo plazo en animales.
Impairment of Fertility
Denosumab no tuvo ningún efecto sobre la fertilidad femenina ni sobre los órganos reproductores masculinos en monos a dosis que fueron de 13 a 50 veces más altas que la dosis humana recomendada de 60 mg administrada por vía subcutánea una vez cada 6 meses, en función del peso corporal (mg/kg).
13.2 Animal Toxicology and/or Pharmacology
Denosumab es un inhibidor de la resorción ósea osteoclastica a través de la inhibición de RANKL.
En monos ovariectomizados, el tratamiento una vez al mes con denosumab suprimió el recambio óseo y aumentó la BMD y la resistencia del hueso trabecular y cortical a dosis 50 veces más altas que la dosis humana recomendada de 60 mg administrada una vez cada 6 meses, en función del peso corporal (mg/kg). El tejido óseo era normal sin evidencia de defectos de mineralización, acumulación de osteoide o hueso entrelazado.
Debido a que la actividad biológica de denosumab en animales es específica de los primates no humanos, la evaluación de ratones genéticamente modificados (“knockout”) o el uso de otros inhibidores biológicos de la vía RANK/RANKL, a saber, OPG-Fc, proporcionó información adicional sobre las propiedades farmacodinámicas de denosumab. Los ratones knockout RANK/RANKL exhibieron ausencia de formación de ganglios linfáticos, así como ausencia de lactancia debido a la inhibición de la maduración de la glándula mamaria (desarrollo de la glándula lobuloalveolar durante el embarazo). Los ratones knockout RANK/RANKL neonatales exhibieron un crecimiento óseo reducido y falta de erupción dental. Un estudio corroborativo en ratas de 2 semanas de edad a las que se administró el inhibidor de RANKL OPG-Fc también mostró un crecimiento óseo reducido, placas de crecimiento alteradas e inhibición de la erupción dental. Estos cambios fueron parcialmente reversibles en este modelo cuando se suspendió la dosificación con los inhibidores de RANKL.
14 ESTUDIOS CLÍNICOS
14.1 Tratamiento de mujeres posmenopáusicas con osteoporosis
La eficacia y seguridad de Prolia en el tratamiento de la osteoporosis posmenopáusica se demostró en un ensayo de 3 años, aleatorizado, doble ciego, controlado con placebo. Las mujeres inscritas tenían una puntuación T de BMD basal entre -2,5 y -4,0 en la columna lumbar o la cadera total. Las mujeres con otras enfermedades (como artritis reumatoide, osteogénesis imperfecta y enfermedad de Paget) o que recibían terapias que afectan los huesos fueron excluidas de este estudio. Las 7808 mujeres inscritas tenían entre 60 y 91 años, con una edad media de 72 años. En general, la puntuación T de BMD de la columna lumbar basal media fue de -2,8, y el 23% de las mujeres tenían una fractura vertebral al inicio del estudio. Las mujeres fueron asignadas aleatoriamente para recibir inyecciones subcutáneas de placebo (N = 3906) o Prolia 60 mg (N = 3902) una vez cada 6 meses. Todas las mujeres recibieron al menos 1000 mg de calcio y 400 UI de vitamina D como suplemento diario.
La variable principal de eficacia fue la incidencia de nuevas fracturas vertebrales morfométricas (diagnosticadas radiológicamente) a los 3 años. Las fracturas vertebrales se diagnosticaron en base a radiografías laterales de la columna vertebral (T4-L4) utilizando un método de puntuación semicuantitativa. Las variables secundarias de eficacia incluyeron la incidencia de fractura de cadera y fractura no vertebral, evaluadas a los 3 años.
Efecto sobre las fracturas vertebrales
Prolia redujo significativamente la incidencia de nuevas fracturas vertebrales morfométricas a los 1, 2 y 3 años (p < 0,0001), como se muestra en la Tabla 3. La incidencia de nuevas fracturas vertebrales al año 3 fue del 7,2% en las mujeres tratadas con placebo en comparación con el 2,3% para las mujeres tratadas con Prolia. La reducción del riesgo absoluto fue del 4,8% y la reducción del riesgo relativo fue del 68% para las nuevas fracturas vertebrales morfométricas al año 3.
| Proporción de mujeres con fractura (%)* | Reducción del riesgo absoluto (%)† (IC del 95%) |
Reducción del riesgo relativo (%)† (IC del 95%) |
||
|---|---|---|---|---|
| Placebo N = 3691 (%) |
Prolia N = 3702 (%) |
|||
| 0-1 año | 2,2 | 0,9 | 1,4 (0,8, 1,9) | 61 (42, 74) |
| 0-2 años | 5,0 | 1,4 | 3,5 (2,7, 4,3) | 71 (61, 79) |
| 0-3 años | 7,2 | 2,3 | 4,8 (3,9, 5,8) | 68 (59, 74) |
Prolia fue eficaz para reducir el riesgo de nuevas fracturas vertebrales morfométricas independientemente de la edad, la tasa basal de recambio óseo, la BMD basal, la historia basal de fractura o el uso previo de un fármaco para la osteoporosis.
Efecto sobre las fracturas de cadera
La incidencia de fractura de cadera fue del 1,2% para las mujeres tratadas con placebo en comparación con el 0,7% para las mujeres tratadas con Prolia al año 3. La reducción del riesgo absoluto ajustado por edad de las fracturas de cadera fue del 0,3% con una reducción del riesgo relativo del 40% a los 3 años (p = 0,04) ( ver Figura 1).
| Figura 1. Incidencia acumulada de fracturas de cadera durante 3 años |
 |
| N = número de sujetos aleatorizados |
Efecto en las fracturas no vertebrales
El tratamiento con Prolia dio como resultado una reducción significativa en la incidencia de fracturas no vertebrales (ver Tabla 4).
| Proporción de mujeres con fractura (%)* | Reducción del riesgo absoluto (%) (IC del 95%) |
Reducción del riesgo relativo (%) (IC del 95%) |
||
|---|---|---|---|---|
| Placebo N = 3906 (%) |
Prolia N = 3902 (%) |
|||
| Fractura no vertebral† | 8.0 | 6.5 | 1.5 (0.3, 2.7) | 20 (5, 33)‡ |
Efecto en la densidad mineral ósea (DMO)
El tratamiento con Prolia aumentó significativamente la DMO en todos los sitios anatómicos medidos a los 3 años. Las diferencias de tratamiento en la DMO a los 3 años fueron del 8,8% en la columna lumbar, del 6,4% en la cadera total y del 5,2% en el cuello femoral. Se observaron efectos consistentes en la DMO en la columna lumbar, independientemente de la edad basal, la raza, el peso/índice de masa corporal (IMC), la DMO basal y el nivel de recambio óseo.
Después de la interrupción de Prolia, la DMO volvió a niveles aproximadamente basales en un plazo de 12 meses.
Histología ósea e histomorfometría
Se obtuvieron un total de 115 especímenes de biopsia ósea de cresta ilíaca de 92 mujeres posmenopáusicas con osteoporosis en el mes 24 y/o en el mes 36 (53 especímenes en el grupo de Prolia, 62 especímenes en el grupo de placebo). De las biopsias obtenidas, 115 (100%) fueron adecuadas para la histología cualitativa y 7 (6%) fueron adecuadas para la evaluación histomorfométrica cuantitativa completa.
Las evaluaciones de histología cualitativa mostraron una arquitectura y calidad normales sin evidencia de defectos de mineralización, hueso tejido o fibrosis de la médula ósea en pacientes tratados con Prolia.
La presencia de doble etiquetado con tetraciclina en un espécimen de biopsia proporciona una indicación de la remodelación ósea activa, mientras que la ausencia de etiquetado con tetraciclina sugiere una supresión de la formación ósea. En los pacientes tratados con Prolia, el 35% no tenía etiquetado con tetraciclina presente en la biopsia del mes 24 y el 38% no tenía etiquetado con tetraciclina presente en la biopsia del mes 36, mientras que el 100% de los pacientes tratados con placebo tenían doble etiquetado presente en ambos puntos de tiempo. En comparación con el placebo, el tratamiento con Prolia dio como resultado una frecuencia de activación prácticamente ausente y una reducción notable de las tasas de formación ósea. Sin embargo, se desconocen las consecuencias a largo plazo de este grado de supresión de la remodelación ósea.
14.2 Tratamiento para aumentar la masa ósea en hombres con osteoporosis
La eficacia y seguridad de Prolia en el tratamiento para aumentar la masa ósea en hombres con osteoporosis se demostró en un ensayo de 1 año, aleatorizado, doble ciego, controlado con placebo. Los hombres inscritos tenían una puntuación T de DMO basal entre -2,0 y -3,5 en la columna lumbar o el cuello femoral. También se inscribieron hombres con una puntuación T de DMO entre -1,0 y -3,5 en la columna lumbar o el cuello femoral si tenían antecedentes de fractura por fragilidad previa. Los hombres con otras enfermedades (como artritis reumatoide, osteogénesis imperfecta y enfermedad de Paget) o que estaban en terapias que pueden afectar los huesos fueron excluidos de este estudio. Los 242 hombres inscritos en el estudio tenían edades comprendidas entre los 31 y los 84 años, con una edad media de 65 años. Los hombres fueron aleatorizados para recibir inyecciones SC de placebo (n = 121) o Prolia 60 mg (n = 121) una vez cada 6 meses. Todos los hombres recibieron al menos 1000 mg de calcio y al menos 800 UI de vitamina D como suplemento diario.
Efecto en la densidad mineral ósea (DMO)
La variable principal de eficacia fue el cambio porcentual en la DMO de la columna lumbar desde el inicio hasta el año 1. Las variables secundarias de eficacia incluyeron el cambio porcentual en la DMO de la cadera total y el cuello femoral desde el inicio hasta el año 1.
El tratamiento con Prolia aumentó significativamente la DMO al año 1. Las diferencias de tratamiento en la DMO al año 1 fueron del 4,8% (+0,9% placebo, +5,7% Prolia; (IC del 95%: 4,0, 5,6); p < 0,0001) en la columna lumbar, del 2,0% (+0,3% placebo, +2,4% Prolia) en la cadera total y del 2,2% (0,0% placebo, +2,1% Prolia) en el cuello femoral. Se observaron efectos consistentes en la DMO en la columna lumbar independientemente de la edad basal, la raza, la DMO, las concentraciones de testosterona y el nivel de recambio óseo.
Histología ósea e histomorfometría
Se obtuvieron un total de 29 especímenes de biopsia ósea de cresta ilíaca de hombres con osteoporosis a los 12 meses (17 especímenes en el grupo de Prolia, 12 especímenes en el grupo de placebo). De las biopsias obtenidas, 29 (100%) fueron adecuadas para la histología cualitativa y, en los pacientes con Prolia, 6 (35%) fueron adecuadas para la evaluación histomorfométrica cuantitativa completa. Las evaluaciones de histología cualitativa mostraron una arquitectura y calidad normales sin evidencia de defectos de mineralización, hueso tejido o fibrosis de la médula ósea en pacientes tratados con Prolia. La presencia de doble etiquetado con tetraciclina en un espécimen de biopsia proporciona una indicación de la remodelación ósea activa, mientras que la ausencia de etiquetado con tetraciclina sugiere una supresión de la formación ósea. En los pacientes tratados con Prolia, el 6% no tenía etiquetado con tetraciclina presente en la biopsia del mes 12, mientras que el 100% de los pacientes tratados con placebo tenían doble etiquetado presente. En comparación con el placebo, el tratamiento con Prolia dio como resultado una reducción notable de las tasas de formación ósea. Sin embargo, se desconocen las consecuencias a largo plazo de este grado de supresión de la remodelación ósea.
14.3 Tratamiento de la osteoporosis inducida por glucocorticoides
La eficacia y seguridad de Prolia en el tratamiento de pacientes con osteoporosis inducida por glucocorticoides se evaluó en el análisis primario de 12 meses de un estudio de 2 años, aleatorizado, multicéntrico, doble ciego, de grupos paralelos, controlado con activo (NCT 01575873) de 795 pacientes (70% mujeres y 30% hombres) de 20 a 94 años (edad media de 63 años) tratados con más de o igual a 7,5 mg/día de prednisona oral (o equivalente) durante < 3 meses antes de la inscripción en el estudio y que planeaban continuar el tratamiento durante un total de al menos 6 meses (subpoblación de inicio de glucocorticoides; n = 290) o ≥ 3 meses antes de la inscripción en el estudio y que planeaban continuar el tratamiento durante un total de al menos 6 meses (subpoblación de continuación de glucocorticoides, n = 505). Los pacientes inscritos < 50 años de edad debían tener antecedentes de fractura osteoporótica. Los pacientes inscritos ≥ 50 años de edad que estaban en la subpoblación de continuación de glucocorticoides debían tener una puntuación T de DMO basal de ≤ -2,0 en la columna lumbar, la cadera total o el cuello femoral; o una puntuación T de DMO ≤ -1,0 en la columna lumbar, la cadera total o el cuello femoral y antecedentes de fractura osteoporótica.
Los pacientes fueron aleatorizados (1:1) para recibir un bisfosfonato oral diario (control activo, risedronato 5 mg una vez al día) (n = 397) o Prolia 60 mg por vía subcutánea una vez cada 6 meses (n = 398) durante un año. La aleatorización se estratificó por sexo dentro de cada subpoblación. Los pacientes recibieron al menos 1000 mg de calcio y 800 UI de vitamina D como suplemento diario.
Efecto en la densidad mineral ósea (DMO)
En la subpoblación que inicia con glucocorticoides, Prolia aumentó significativamente la BMD de la columna lumbar en comparación con el control activo a un año (Control activo 0.8%, Prolia 3.8%) con una diferencia de tratamiento del 2.9% (p < 0.001). En la subpoblación que continúa con glucocorticoides, Prolia aumentó significativamente la BMD de la columna lumbar en comparación con el control activo a un año (Control activo 2.3%, Prolia 4.4%) con una diferencia de tratamiento del 2.2% (p < 0.001). Se observaron efectos consistentes en la BMD de la columna lumbar independientemente del sexo; raza; región geográfica; estado menopáusico; y edad basal, puntuación T de la BMD de la columna lumbar y dosis de glucocorticoides dentro de cada subpoblación.
Histología ósea
Se obtuvieron muestras de biopsia ósea de 17 pacientes (11 en el grupo de tratamiento con control activo y 6 en el grupo de tratamiento con Prolia) en el mes 12. De las biopsias obtenidas, 17 (100%) fueron adecuadas para la histología cualitativa. Las evaluaciones cualitativas mostraron hueso de arquitectura y calidad normales sin defectos de mineralización o anormalidad de la médula ósea. La presencia de doble etiquetado con tetraciclina en una muestra de biopsia proporciona una indicación de la remodelación ósea activa, mientras que la ausencia de etiquetado con tetraciclina sugiere una supresión de la formación ósea. En los pacientes tratados con control activo, el 100% de las biopsias tenían etiquetado con tetraciclina. En los pacientes tratados con Prolia, 1 (33%) tenía etiquetado con tetraciclina y 2 (67%) no tenían etiquetado con tetraciclina presente en la biopsia del mes 12–. La evaluación de la histomorfometría cuantitativa completa, incluidas las tasas de remodelación ósea, no fue posible en la población con osteoporosis inducida por glucocorticoides tratada con Prolia. Se desconocen las consecuencias a largo plazo de este grado de supresión de la remodelación ósea en pacientes tratados con glucocorticoides.
14.4 Tratamiento de la pérdida ósea en hombres con cáncer de próstata
La eficacia y seguridad de Prolia en el tratamiento de la pérdida ósea en hombres con cáncer de próstata no metastásico que reciben terapia de privación de andrógenos (TDA) se demostraron en un estudio multinacional de 3 años, aleatorizado (1:1), doble ciego, controlado con placebo. Los hombres menores de 70 años tenían una puntuación T de la BMD en la columna lumbar, la cadera total o el cuello femoral entre -1.0 y -4.0, o un historial de fractura osteoporótica. La puntuación T de la BMD de la columna lumbar media basal fue de -0.4, y el 22% de los hombres tenían una fractura vertebral al inicio del estudio. Los 1468 hombres inscritos tenían edades comprendidas entre los 48 y los 97 años (mediana de 76 años). Los hombres fueron aleatorizados para recibir inyecciones subcutáneas de placebo (n = 734) o Prolia 60 mg (n = 734) una vez cada 6 meses durante un total de 6 dosis. La aleatorización se estratificó por edad (< 70 años frente a ≥ 70 años) y duración de la TDA al inicio del ensayo (≤ 6 meses frente a > 6 meses). El 79% de los pacientes recibieron TDA durante más de 6 meses al inicio del estudio. Todos los hombres recibieron al menos 1000 mg de calcio y 400 UI de vitamina D como suplemento diario.
Efecto en la densidad mineral ósea (BMD)
La variable principal de eficacia fue el cambio porcentual en la BMD de la columna lumbar desde el inicio hasta el mes 24. Una variable secundaria clave adicional fue la incidencia de nuevas fracturas vertebrales hasta el mes 36 diagnosticadas en base a la evaluación radiográfica por dos radiólogos independientes. La BMD de la columna lumbar fue mayor a los 2 años en los pacientes tratados con Prolia en comparación con los pacientes tratados con placebo [-1.0% placebo, +5.6% Prolia; diferencia de tratamiento 6.7% (IC del 95%: 6.2, 7.1); p < 0.0001].
Con aproximadamente el 62% de los pacientes seguidos durante 3 años, las diferencias de tratamiento en la BMD a los 3 años fueron del 7.9% (-1.2% placebo, +6.8% Prolia) en la columna lumbar, 5.7% (-2.6% placebo, +3.2% Prolia) en la cadera total y 4.9% (-1.8% placebo, +3.0% Prolia) en el cuello femoral. Se observaron efectos consistentes en la BMD en la columna lumbar en subgrupos relevantes definidos por la edad basal, la BMD y el historial basal de fractura vertebral.
Efecto en las fracturas vertebrales
Prolia redujo significativamente la incidencia de nuevas fracturas vertebrales a los 3 años (p = 0.0125), como se muestra en la Tabla 5.
| Proporción de hombres con fractura (%)* | Reducción del riesgo absoluto (%)† (IC del 95%) |
Reducción del riesgo relativo (%)† (IC del 95%) |
||
|---|---|---|---|---|
| Placebo N = 673 (%) |
Prolia N = 679 (%) |
|||
| 0-1 año | 1.9 | 0.3 | 1.6 (0.5, 2.8) | 85 (33, 97) |
| 0-2 años | 3.3 | 1.0 | 2.2 (0.7, 3.8) | 69 (27, 86) |
| 0-3 años | 3.9 | 1.5 | 2.4 (0.7, 4.1) | 62 (22, 81) |
14.5 Tratamiento de la pérdida ósea en mujeres con cáncer de mama
La eficacia y seguridad de Prolia en el tratamiento de la pérdida ósea en mujeres que reciben terapia adyuvante con inhibidores de la aromatasa (IA) para el cáncer de mama se evaluó en un estudio multinacional de 2 años, aleatorizado (1:1), doble ciego, controlado con placebo. Las mujeres tenían puntuaciones T de densidad mineral ósea (DMO) basal entre -1,0 y -2,5 en la columna lumbar, la cadera total o el cuello femoral, y no habían experimentado fracturas después de los 25 años. La puntuación T de DMO de la columna lumbar basal media fue de -1,1, y el 2,0% de las mujeres tenían una fractura vertebral en el momento basal. Las 252 mujeres inscritas tenían edades comprendidas entre los 35 y los 84 años (mediana de 59 años). Las mujeres fueron asignadas aleatoriamente para recibir inyecciones subcutáneas de placebo (n = 125) o Prolia 60 mg (n = 127) una vez cada 6 meses durante un total de 4 dosis. La aleatorización se estratificó por la duración de la terapia adyuvante con IA en el momento de la entrada al ensayo (≤ 6 meses frente a > 6 meses). El sesenta y dos por ciento de las pacientes recibieron terapia adyuvante con IA durante más de 6 meses en el momento de la entrada al estudio. Todas las mujeres recibieron al menos 1000 mg de calcio y 400 UI de vitamina D como suplemento diario.
Efecto sobre la densidad mineral ósea (DMO)
La variable principal de eficacia fue el cambio porcentual en la DMO de la columna lumbar desde el momento basal hasta el mes 12. La DMO de la columna lumbar fue mayor a los 12 meses en las pacientes tratadas con Prolia en comparación con las pacientes tratadas con placebo [-0,7% placebo, +4,8% Prolia; diferencia de tratamiento del 5,5% (IC del 95%: 4,8, 6,3); p < 0,0001].
Con aproximadamente el 81% de las pacientes seguidas durante 2 años, las diferencias de tratamiento en la DMO a los 2 años fueron del 7,6% (-1,4% placebo, +6,2% Prolia) en la columna lumbar, del 4,7% (-1,0% placebo, +3,8% Prolia) en la cadera total y del 3,6% (-0,8% placebo, +2,8% Prolia) en el cuello femoral.
16 FORMA DE SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
Forma de suministro
La inyección de Prolia es una solución transparente, incolora a amarillo pálido que se suministra en una jeringa precargada de dosis única con un protector de seguridad. La jeringa precargada no está hecha con látex de caucho natural.
| 60 mg/mL en una jeringa precargada de dosis única | 1 por caja | NDC 55513-710-01 NDC 55513-710-21 |
Almacenamiento y manipulación
Almacene Prolia en un refrigerador a 2 °C a 8 °C (36 °F a 46 °F) en el envase original. No congelar. Antes de la administración, se puede permitir que Prolia alcance la temperatura ambiente (hasta 25 °C/77 °F) en el envase original. Una vez retirado del refrigerador, Prolia no debe exponerse a temperaturas superiores a 25 °C/77 °F y debe utilizarse en un plazo de 14 días. Si no se utiliza en los 14 días, Prolia debe desecharse. No utilice Prolia después de la fecha de caducidad impresa en la etiqueta. Proteja Prolia de la luz solar directa y del calor.
Evite agitar vigorosamente Prolia.
17 INFORMACIÓN PARA EL PACIENTE
Aconseje al paciente que lea la información para pacientes aprobada por la FDA (Guía del Medicamento).
Hipocalcemia
Aconseje al paciente que se suplemente adecuadamente con calcio y vitamina D e instrúyalo sobre la importancia de mantener los niveles séricos de calcio mientras recibe Prolia [ver Advertencias y precauciones (5.1), Uso en poblaciones específicas (8.6)]. Aconseje a los pacientes que busquen atención médica inmediata si desarrollan signos o síntomas de hipocalcemia.
Hipocalcemia grave en pacientes con enfermedad renal crónica avanzada
Aconseje a los pacientes con enfermedad renal crónica avanzada, incluidos aquellos que son dependientes de diálisis, sobre los síntomas de la hipocalcemia y la importancia de mantener los niveles séricos de calcio con una suplementación adecuada de calcio y vitamina D activada. Aconseje a estos pacientes que se midan el calcio sérico semanalmente durante el primer mes después de la administración de Prolia y mensualmente a partir de entonces [ver Posología y administración (2.2), Advertencias y precauciones (5.1), Uso en poblaciones específicas (8.6)]
Medicamentos con el mismo ingrediente activo
Informe a los pacientes que denosumab también se comercializa como Xgeva, y si están tomando Prolia, no deben recibir Xgeva [ver Advertencias y precauciones (5.2)].
Hipersensibilidad
Aconseje a los pacientes que busquen atención médica inmediata si se presentan signos o síntomas de reacciones de hipersensibilidad. Aconseje a los pacientes que hayan tenido signos o síntomas de reacciones de hipersensibilidad sistémica que no deben recibir denosumab (Prolia o Xgeva) [ver Advertencias y precauciones (5.3), Contraindicaciones (4)].
Osteonecrosis de la mandíbula
Aconseje a los pacientes que mantengan una buena higiene bucal durante el tratamiento con Prolia y que informen a su dentista antes de los procedimientos dentales que están recibiendo Prolia. Los pacientes deben informar a su médico o dentista si experimentan dolor persistente y/o curación lenta de la boca o la mandíbula después de una cirugía dental [ver Advertencias y precauciones (5.4)].
Fracturas femorales atípicas subtrocantéricas y diafisarias
Aconseje a los pacientes que informen sobre dolor nuevo o inusual en el muslo, la cadera o la ingle [ver Advertencias y precauciones (5.5)].
Fracturas vertebrales múltiples (FVM) después de la interrupción del tratamiento con Prolia
Aconseje a los pacientes que no interrumpan el tratamiento con Prolia sin hablar con su médico [ver Advertencias y precauciones (5.6)].
Infecciones graves
Aconseje a los pacientes que busquen atención médica inmediata si desarrollan signos o síntomas de infecciones, incluida la celulitis [ver Advertencias y precauciones (5.7)].
Reacciones adversas dermatológicas
Aconseje a los pacientes que busquen atención médica inmediata si desarrollan signos o síntomas de reacciones dermatológicas (como dermatitis, erupciones cutáneas y eccema) [ver Advertencias y precauciones (5.8)].
Dolor musculoesquelético
Informe a los pacientes que se ha notificado dolor óseo, articular y/o muscular intenso en pacientes que toman Prolia. Los pacientes deben informar de los síntomas graves si los desarrollan [ver Advertencias y precauciones (5.9)].
Embarazo/Lactancia
Aconseje a las mujeres en edad fértil que utilicen un método anticonceptivo eficaz para evitar el embarazo durante el tratamiento y durante al menos 5 meses después de la última dosis de Prolia. Aconseje a la paciente que se ponga en contacto con su médico inmediatamente si se produce un embarazo durante este tiempo. Aconseje a las pacientes que no tomen Prolia durante el embarazo o la lactancia. Si una paciente desea empezar a amamantar después del tratamiento, aconséjele que hable del momento adecuado con su médico [ver Contraindicaciones (4), Uso en poblaciones específicas (8.1)].
Calendario de administración
Aconseje a los pacientes que si se olvida una dosis de Prolia, la inyección debe administrarse tan pronto como sea conveniente. A partir de entonces, programe las inyecciones cada 6 meses a partir de la fecha de la última inyección.
SECCIÓN NO CLASIFICADA SPL
Prolia® (denosumab)
Fabricado por:
Amgen Inc.
One Amgen Center Drive
Thousand Oaks, California 91320-1799
Licencia de EE. UU. No. 1080
Patente: http://pat.amgen.com/prolia/
© 2010-2024 Amgen Inc. Todos los derechos reservados.
1xxxxxx – v26
Guía de medicación
| Esta Guía de Medicamentos ha sido aprobada por la Administración de Alimentos y Medicamentos de los Estados Unidos. | Revisada: 01/2024 | ||
| Guía de Medicamentos Prolia® (PRÓ-lee-a) (denosumab) Inyección, para uso subcutáneo |
|||
| ¿Cuál es la información más importante que debo saber sobre Prolia? Si recibe Prolia, no debe recibir XGEVA®. Prolia contiene el mismo medicamento que Xgeva (denosumab). Prolia puede causar efectos secundarios graves, incluyendo:
|
|||
|
|
||
|
|||
|
|
||
Llame a su médico inmediatamente si presenta cualquiera de estos efectos secundarios. |
|||
| ¿Qué es Prolia? Prolia es un medicamento recetado que se usa para:
No se sabe si Prolia es seguro y eficaz en niños. Prolia no está aprobado para su uso en niños. |
|||
No tome Prolia si:
|
|||
Antes de tomar Prolia, informe a su médico sobre todas sus afecciones médicas, incluso si:
Informe a su médico sobre todos los medicamentos que toma, incluidos los medicamentos recetados y de venta libre, las vitaminas y los suplementos herbales. |
|||
¿Cómo recibiré Prolia?
|
|||
| ¿Cuáles son los posibles efectos secundarios de Prolia? Prolia puede causar efectos secundarios graves.
Los efectos secundarios más comunes de Prolia en mujeres que están siendo tratadas para la osteoporosis después de la menopausia son: |
|||
|
|
||
| Los efectos secundarios más comunes de Prolia en hombres con osteoporosis son: | |||
|
|
||
| Los efectos secundarios más comunes de Prolia en pacientes con osteoporosis inducida por glucocorticoides son: | |||
|
|
||
| Los efectos secundarios más comunes de Prolia en pacientes que reciben ciertos tratamientos para el cáncer de próstata o de mama son: | |||
|
|
||
| Informe a su médico si tiene algún efecto secundario que le moleste o que no desaparezca. Estos no son todos los posibles efectos secundarios de Prolia. Llame a su médico para obtener asesoramiento médico sobre los efectos secundarios. Puede informar los efectos secundarios a la FDA al 1-800-FDA-1088. |
|||
¿Cómo debo almacenar Prolia si necesito recogerlo en una farmacia?
Mantenga Prolia y todos los medicamentos fuera del alcance de los niños. |
|||
| Información general sobre el uso seguro y eficaz de Prolia. Los medicamentos a veces se recetan para fines distintos de los que se enumeran en una Guía de medicamentos. No use Prolia para una condición para la que no fue recetado. No le dé Prolia a otras personas, incluso si tienen los mismos síntomas que usted. Puede hacerles daño. Puede preguntarle a su médico o farmacéutico por información sobre Prolia que esté escrita para profesionales de la salud. |
|||
| ¿Cuáles son los ingredientes de Prolia? Ingrediente activo: denosumab Ingredientes inactivos: sorbitol, acetato, polisorbato 20, Agua para inyección (USP) e hidróxido de sodio Amgen Inc. One Amgen Center Drive Thousand Oaks, California 91320-1799 1xxxxxx – v17 Para obtener más información, visite www.Prolia.com o llame a Amgen al 1-800-772-6436. |
|||
PANEL DE MOSTRACIÓN PRINCIPAL – Cartón de jeringas de 60 mg/mL
1 x 60 mg Jeringa precargada de dosis única
NDC 55513-710-01
AMGEN®
prolia®
(denosumab)
60
mg/mL
60 mg/mL
Inyección – Solo para uso subcutáneo.
Jeringa precargada de dosis única. Deseche la porción no utilizada.
Solución estéril – Sin conservante.
Solo con receta médica
Refrigerar a 2° a 8°C (36° a 46°F). No congelar. Evite agitar excesivamente. Proteger de la luz y el calor directos.
Fabricado por: Amgen Inc., Thousand Oaks, CA 91320-1799
Producto de Singapur

PANEL DE MOSTRACIÓN PRINCIPAL – Cartón de jeringas de 60 mg/mL
1 x 60 mg Jeringa precargada de dosis única
NDC 55513-710-21
AMGEN®
prolia®
(denosumab)
60
mg/mL
60 mg/mL
Inyección – Solo para uso subcutáneo.
Jeringa precargada de dosis única. Deseche la porción no utilizada.
Solución estéril – Sin conservante.
Solo con receta médica
Refrigerar a 2° a 8°C (36° a 46°F). No congelar. Evite agitar excesivamente. Proteger de la luz y el calor directos.
Fabricado por: Amgen Inc., Thousand Oaks, CA 91320-1799
Producto de Singapur
