
Fabricante de medicamentos: Celgene Corporation (Updated: 2021-02-05)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
OTEZLA® (apremilast) tabletas, para uso oral
Aprobación inicial en EE. UU.: 2014
CAMBIOS MAYORES RECIENTES
INDICACIONES Y USO
OTEZLA, un inhibidor de la fosfodiesterasa 4 (PDE4), está indicado para el tratamiento de:
DOSIS Y ADMINISTRACIÓN
- Para reducir el riesgo de síntomas gastrointestinales, titule a la dosis recomendada de 30 mg dos veces al día de acuerdo con el siguiente programa (2.1)
- Día 1: 10 mg por la mañana
- Día 2: 10 mg por la mañana y 10 mg por la noche
- Día 3: 10 mg por la mañana y 20 mg por la noche
- Día 4: 20 mg por la mañana y 20 mg por la noche
- Día 5: 20 mg por la mañana y 30 mg por la noche
- Día 6 y siguientes: 30 mg dos veces al día
FORMAS Y FUERZAS DE DOSIFICACIÓN
Tabletas: 10 mg, 20 mg, 30 mg (3)
CONTRAINDICACIONES
Hipersensibilidad conocida a apremilast o a cualquier excipiente de la formulación (4)
ADVERTENCIAS Y PRECAUCIONES
- Diarrea, náuseas y vómitos: Considere la reducción de la dosis de OTEZLA o la suspensión si los pacientes desarrollan diarrea grave, náuseas o vómitos (5.1)
- Depresión: Avise a los pacientes, sus cuidadores y familias que estén atentos a la aparición o el empeoramiento de la depresión, los pensamientos suicidas u otros cambios del estado de ánimo y que, si se producen estos cambios, se pongan en contacto con su proveedor de atención médica. Evalúe cuidadosamente los riesgos y beneficios del tratamiento con OTEZLA en pacientes con antecedentes de depresión y/o pensamientos o comportamiento suicida (5.2)
- Pérdida de peso: Controle el peso regularmente. Si se produce una pérdida de peso inexplicable o clínicamente significativa, evalúe la pérdida de peso y considere la interrupción de OTEZLA (5.3)
- Interacciones medicamentosas: No se recomienda el uso con inductores fuertes de la enzima citocromo P450 (por ejemplo, rifampicina, fenobarbital, carbamazepina, fenitoína) porque puede producirse una pérdida de eficacia (5.4, 7.1)
REACCIONES ADVERSAS
- Artritis psoriásica: Las reacciones adversas más comunes (≥5%) son diarrea, náuseas y dolor de cabeza (6.1)
- Psoriasis: Las reacciones adversas más comunes (≥5%) son diarrea, náuseas, infección del tracto respiratorio superior y dolor de cabeza, incluido el dolor de cabeza tensional (6.1)
- Enfermedad de Behçet: Las reacciones adversas más comunes (≥10%) son diarrea, náuseas, dolor de cabeza e infección del tracto respiratorio superior (6.1)
Para informar sobre REACCIONES ADVERSAS SOSPECHOSAS, póngase en contacto con Celgene Corporation al 1-888-423-5436 o con la FDA al 1-800-FDA-1088 o www.fda.gov/medwatch
USO EN POBLACIONES ESPECÍFICAS
Insuficiencia renal grave: Se ha observado un aumento de la exposición sistémica de OTEZLA, se recomienda reducir la dosis a 30 mg una vez al día (2.2, 8.6)
Ver 17 para INFORMACIÓN PARA EL PACIENTE.
Revisado: 7/2019
Tabla de Contenido
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDO*
1 INDICACIONES Y USO
1.1 Artritis Psoriásica
1.2 Psoriasis
1.3 Úlceras Orales Asociadas con la Enfermedad de Behçet
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Dosificación en Artritis Psoriásica, Psoriasis y Enfermedad de Behçet
2.2 Ajuste de la Dosificación en Pacientes con Insuficiencia Renal Grave
3 FORMAS Y FUERZAS DE DOSIFICACIÓN
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Diarrea, Náuseas y Vómitos
5.2 Depresión
5.3 Disminución de Peso
5.4 Interacciones Medicamentosas
6 REACCIONES ADVERSAS
6.1 Experiencia en Ensayos Clínicos
7 INTERACCIONES MEDICAMENTOSAS
7.1 Inductores Fuertes de CYP450
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.4 Uso Pediátrico
8.5 Uso Geriátrico
8.6 Insuficiencia Renal
8.7 Insuficiencia Hepática
10 SOBREDOSIS
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de Acción
12.3 Farmacocinética
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
14 ESTUDIOS CLÍNICOS
14.1 Artritis Psoriásica
14.2 Psoriasis
14.3 Úlceras Orales Asociadas con la Enfermedad de Behçet
16 CÓMO SE SUMINISTRA/ALMACENAMIENTO Y MANEJO
17 INFORMACIÓN PARA EL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información completa de prescripción no están enumeradas.
1 INDICACIONES Y USO
1.1 Artritis Psoriásica
OTEZLA está indicado para el tratamiento de pacientes adultos con artritis psoriásica activa.
1.2 Psoriasis
OTEZLA está indicado para el tratamiento de pacientes con psoriasis en placas de moderada a grave que son candidatos a fototerapia o terapia sistémica.
1.3 Úlceras Orales Asociadas a la Enfermedad de Behçet
OTEZLA está indicado para el tratamiento de pacientes adultos con úlceras orales asociadas a la enfermedad de Behçet.
2 DOSIS Y ADMINISTRACIÓN
2.1 Dosage in Psoriatic Arthritis, Psoriasis, and Behçet’s Disease
La dosificación inicial recomendada de OTEZLA del día 1 al día 5 se muestra en la Tabla 1. Después de la dosificación gradual de 5 días, la dosis de mantenimiento recomendada es de 30 mg dos veces al día por vía oral a partir del día 6. Esta dosificación gradual está destinada a reducir los síntomas gastrointestinales asociados con la terapia inicial.
OTEZLA puede administrarse con o sin alimentos. No triture, divida ni mastique las tabletas.
| Día 1 | Día 2 | Día 3 | Día 4 | Día 5 | Día 6 & thereafter |
|||||
| AM | AM | PM | AM | PM | AM | PM | AM | PM | AM | PM |
| 10 mg | 10 mg | 10 mg | 10 mg | 20 mg | 20 mg | 20 mg | 20 mg | 30 mg | 30 mg | 30 mg |
2.2 Dosage Adjustment in Patients with Severe Renal Impairment
La dosis de OTEZLA debe reducirse a 30 mg una vez al día en pacientes con insuficiencia renal grave (depuración de creatinina (CLcr) de menos de 30 ml por minuto estimada mediante la ecuación de Cockcroft-Gault) [ver Uso en poblaciones específicas (8.6) y Farmacología clínica (12.3)]. Para la dosificación gradual inicial en este grupo, se recomienda que OTEZLA se dosifique gradualmente utilizando solo el programa AM que se indica en la Tabla 1 y que se omitan las dosis PM.
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
OTEZLA está disponible como tabletas recubiertas con película de forma de diamante en las siguientes concentraciones:
- Tableta rosa de 10 mg grabada con “APR” en un lado y “10” en el otro lado
- Tableta marrón de 20 mg grabada con “APR” en un lado y “20” en el otro lado
- Tableta beige de 30 mg grabada con “APR” en un lado y “30” en el otro lado.
4 CONTRAINDICACIONES
OTEZLA está contraindicado en pacientes con hipersensibilidad conocida a apremilast o a cualquiera de los excipientes de la formulación [ver Reacciones adversas (6.1)].
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Diarrea, náuseas y vómitos
Se han notificado casos de diarrea grave, náuseas y vómitos asociados al uso de OTEZLA después de su comercialización. La mayoría de los eventos ocurrieron en las primeras semanas de tratamiento. En algunos casos, los pacientes fueron hospitalizados. Los pacientes de 65 años o más y los pacientes que toman medicamentos que pueden provocar depleción de volumen o hipotensión pueden tener un mayor riesgo de complicaciones por diarrea grave, náuseas o vómitos. Controle a los pacientes que sean más susceptibles a las complicaciones de la diarrea o los vómitos. Los pacientes que redujeron la dosis o suspendieron OTEZLA generalmente mejoraron rápidamente. Considere la reducción de la dosis de OTEZLA o su suspensión si los pacientes desarrollan diarrea grave, náuseas o vómitos.
5.2 Depresión
El tratamiento con OTEZLA se asocia con un aumento en las reacciones adversas de depresión. Antes de usar OTEZLA en pacientes con antecedentes de depresión y/o pensamientos o comportamiento suicidas, los prescriptores deben evaluar cuidadosamente los riesgos y beneficios del tratamiento con OTEZLA en estos pacientes. Se debe aconsejar a los pacientes, sus cuidadores y familias sobre la necesidad de estar atentos a la aparición o el empeoramiento de la depresión, los pensamientos suicidas u otros cambios del estado de ánimo, y si ocurren estos cambios, deben comunicarse con su proveedor de atención médica. Los prescriptores deben evaluar cuidadosamente los riesgos y beneficios de continuar el tratamiento con OTEZLA si ocurren estos eventos.
Artritis psoriásica: Durante el período controlado con placebo de 0 a 16 semanas de los 3 ensayos clínicos controlados, el 1,0% (10/998) de los sujetos tratados con OTEZLA informaron depresión o estado de ánimo depresivo en comparación con el 0,8% (4/495) tratados con placebo. Durante los ensayos clínicos, el 0,3% (4/1441) de los sujetos tratados con OTEZLA suspendieron el tratamiento debido a depresión o estado de ánimo depresivo en comparación con ninguno en los sujetos tratados con placebo (0/495). La depresión se informó como grave en el 0,2% (3/1441) de los sujetos expuestos a OTEZLA, en comparación con ninguno en los sujetos tratados con placebo (0/495). Se han observado casos de ideación y comportamiento suicidas en el 0,2% (3/1441) de los sujetos mientras recibían OTEZLA, en comparación con ninguno en los sujetos tratados con placebo (0/495). En los ensayos clínicos, 2 sujetos que recibieron placebo se suicidaron en comparación con ninguno en los sujetos tratados con OTEZLA.
Psoriasis: Durante el período controlado con placebo de 0 a 16 semanas de los 3 ensayos clínicos controlados, el 1,3% (12/920) de los sujetos tratados con OTEZLA informaron depresión en comparación con el 0,4% (2/506) tratados con placebo. Durante los ensayos clínicos, el 0,1% (1/1308) de los sujetos tratados con OTEZLA suspendieron el tratamiento debido a depresión en comparación con ninguno en los sujetos tratados con placebo (0/506). La depresión se informó como grave en el 0,1% (1/1308) de los sujetos expuestos a OTEZLA, en comparación con ninguno en los sujetos tratados con placebo (0/506). Se han observado casos de comportamiento suicida en el 0,1% (1/1308) de los sujetos mientras recibían OTEZLA, en comparación con el 0,2% (1/506) en los sujetos tratados con placebo. En los ensayos clínicos, un sujeto tratado con OTEZLA intentó suicidarse mientras que uno que recibió placebo se suicidó.
Enfermedad de Behçet: Durante el período controlado con placebo del estudio de fase 3, el 1% (1/104) de los pacientes tratados con OTEZLA informaron depresión/estado de ánimo depresivo en comparación con el 1% (1/103) tratados con placebo. Ninguno de estos informes de depresión fue grave o condujo a la interrupción del estudio. No se informaron casos de ideación o comportamiento suicidas durante el período controlado con placebo del estudio de fase 3 en pacientes tratados con OTEZLA (0/104) o tratados con placebo (0/103).
5.3 Disminución de peso
Durante el período controlado de los estudios en artritis psoriásica (PsA), se informó una disminución de peso entre el 5% y el 10% del peso corporal en el 10% (49/497) de los sujetos tratados con OTEZLA 30 mg dos veces al día en comparación con el 3,3% (16/495) tratados con placebo.
Durante el período controlado de los ensayos en psoriasis, la disminución de peso entre el 5% y el 10% del peso corporal ocurrió en el 12% (96/784) de los sujetos tratados con OTEZLA en comparación con el 5% (19/382) tratados con placebo. La disminución de peso de ≥10% del peso corporal ocurrió en el 2% (16/784) de los sujetos tratados con OTEZLA 30 mg dos veces al día en comparación con el 1% (3/382) de los sujetos tratados con placebo.
Durante el período controlado del estudio de fase 3 en la enfermedad de Behçet, se informó una disminución de peso >5% del peso corporal en el 4,9% (5/103) de los sujetos tratados con OTEZLA 30 mg dos veces al día en comparación con el 3,9% (4/102) de los pacientes tratados con placebo.
Los pacientes tratados con OTEZLA deben tener su peso controlado regularmente. Si se produce una pérdida de peso inexplicable o clínicamente significativa, se debe evaluar la pérdida de peso y se debe considerar la interrupción de OTEZLA [ver Reacciones adversas (6.1)].
5.4 Interacciones medicamentosas
La coadministración de un inductor potente de la enzima citocromo P450, rifampicina, provocó una reducción de la exposición sistémica de apremilast, lo que puede provocar una pérdida de eficacia de OTEZLA. Por lo tanto, no se recomienda el uso de inductores de la enzima citocromo P450 (por ejemplo, rifampicina, fenobarbital, carbamazepina, fenitoína) con OTEZLA [ver Interacciones medicamentosas (7.1) y Farmacología clínica (12.3)].
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas se describen en otras partes de la etiqueta:
- Diarrea, náuseas y vómitos [ver Advertencias y precauciones (5.1)]
- Depresión [ver Advertencias y precauciones (5.2)]
- Pérdida de peso [ver Advertencias y precauciones (5.3)]
- Interacciones medicamentosas [ver Advertencias y precauciones (5.4)]
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se llevan a cabo en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un medicamento no se pueden comparar directamente con las tasas en los ensayos clínicos de otro medicamento y pueden no reflejar las tasas observadas en la práctica clínica.
Ensayos clínicos de artritis psoriásica
OTEZLA se evaluó en 3 ensayos multicéntricos, aleatorizados, doble ciego, controlados con placebo [Estudios PsA-1, PsA-2 y PsA-3] de diseño similar en pacientes adultos con artritis psoriásica activa [ver Estudios clínicos (14.1)]. En los 3 estudios, hubo 1493 pacientes aleatorizados por igual a placebo, OTEZLA 20 mg dos veces al día u OTEZLA 30 mg dos veces al día. Se utilizó la titulación durante los primeros 5 días [ver Dosificación y administración (2.1)]. Los pacientes con placebo cuyos recuentos de articulaciones sensibles e inflamadas no habían mejorado al menos un 20% fueron re-aleatorizados 1:1 de forma ciega a OTEZLA 20 mg dos veces al día o 30 mg dos veces al día en la semana 16, mientras que los pacientes con OTEZLA permanecieron en su tratamiento inicial. Los pacientes tenían edades comprendidas entre los 18 y los 83 años, con una mediana de edad general de 51 años.
La mayoría de las reacciones adversas más comunes que se presentan en Tabla 2 ocurrieron dentro de las primeras 2 semanas de tratamiento y tendieron a resolverse con el tiempo con la dosificación continua. La diarrea, el dolor de cabeza y las náuseas fueron las reacciones adversas más comúnmente reportadas. Las reacciones adversas más comunes que llevaron a la interrupción del tratamiento para los pacientes que tomaban OTEZLA fueron náuseas (1.8%), diarrea (1.8%) y dolor de cabeza (1.2%). La proporción de pacientes con artritis psoriásica que interrumpieron el tratamiento debido a cualquier reacción adversa fue del 4.6% para los pacientes que tomaron OTEZLA 30 mg dos veces al día y del 1.2% para los pacientes tratados con placebo.
| a De las reacciones adversas gastrointestinales reportadas, 1 sujeto experimentó una reacción adversa grave de náuseas y vómitos en OTEZLA 30 mg dos veces al día; 1 sujeto tratado con OTEZLA 20 mg dos veces al día experimentó una reacción adversa grave de diarrea; 1 paciente tratado con OTEZLA 30 mg dos veces al día experimentó una reacción adversa grave de dolor de cabeza. | ||||
| b De las reacciones adversas a los medicamentos reportadas, ninguna fue grave. | ||||
| c n (%) indica el número de pacientes y el porcentaje. | ||||
| Placebo | OTEZLA 30 mg BID | |||
| Término preferido | Día 1 a 5 (N=495) n (%)c |
Día 6 a Día 112 (N=490) n (%) |
Día 1 a 5 (N=497) n (%) |
Día 6 a Día 112 (N=493) n (%) |
| Diarreaa | 6 (1.2) | 8 (1.6) | 46 (9.3) | 38 (7.7) |
| Náuseasa | 7 (1.4) | 15 (3.1) | 37 (7.4) | 44 (8.9) |
| Dolor de cabezaa | 9 (1.8) | 11 (2.2) | 24 (4.8) | 29 (5.9) |
| Infección de las vías respiratorias superiores infecciónb |
3 (0.6) | 9 (1.8) | 3 (0.6) | 19 (3.9) |
| Vómitosa | 2 (0.4) | 2 (0.4) | 4 (0.8) | 16 (3.2) |
| Nasofaringitisb | 1 (0.2) | 8 (1.6) | 1 (0.2) | 13 (2.6) |
| Dolor abdominal superiorb | 0 (0.0) | 1 (0.2) | 3 (0.6) | 10 (2.0) |
Otras reacciones adversas notificadas en pacientes tratados con OTEZLA en estudios clínicos, incluidos los estudios de extensión:
Trastornos del sistema inmunitario: Hipersensibilidad
Pruebas: Disminución del peso
Trastornos gastrointestinales: Defecación frecuente, enfermedad por reflujo gastroesofágico, dispepsia
Trastornos del metabolismo y la nutrición: Disminución del apetito*
Trastornos del sistema nervioso: Migraña
Trastornos respiratorios, torácicos y mediastínicos: Tos
Trastornos de la piel y del tejido subcutáneo: Erupción cutánea
*1 paciente tratado con OTEZLA 30 mg dos veces al día experimentó una reacción adversa grave.
Ensayos clínicos de psoriasis
La seguridad de OTEZLA se evaluó en 1426 sujetos en 3 ensayos aleatorizados, doble ciego, controlados con placebo en sujetos adultos con psoriasis en placas de moderada a grave que eran candidatos a fototerapia o terapia sistémica. Los sujetos se asignaron aleatoriamente para recibir OTEZLA 30 mg dos veces al día o placebo dos veces al día. Se utilizó la titulación durante los primeros 5 días [ver Posología y forma de administración (2.1)]. Los sujetos tenían edades comprendidas entre los 18 y los 83 años, con una edad media general de 46 años.
La diarrea, las náuseas y la infección de las vías respiratorias altas fueron las reacciones adversas notificadas con mayor frecuencia. Las reacciones adversas más frecuentes que llevaron a la interrupción del tratamiento en los sujetos que tomaban OTEZLA fueron las náuseas (1,6%), la diarrea (1,0%) y la cefalea (0,8%). La proporción de sujetos con psoriasis que interrumpieron el tratamiento debido a cualquier reacción adversa fue del 6,1% para los sujetos tratados con OTEZLA 30 mg dos veces al día y del 4,1% para los sujetos tratados con placebo.
| *Dos sujetos tratados con OTEZLA experimentaron una reacción adversa grave de dolor abdominal. | ||
| Término preferido | Placebo (N=506) n (%) |
OTEZLA 30 mg BID (N=920) n (%) |
| Diarrea | 32 (6) | 160 (17) |
| Náuseas | 35 (7) | 155 (17) |
| Infección de las vías respiratorias altas | 31 (6) | 84 (9) |
| Cefalea tensional | 21 (4) | 75 (8) |
| Cefalea | 19 (4) | 55 (6) |
| Dolor abdominal* | 11 (2) | 39 (4) |
| Vómitos | 8 (2) | 35 (4) |
| Fatiga | 9 (2) | 29 (3) |
| Dispepsia | 6 (1) | 29 (3) |
| Disminución del apetito | 5 (1) | 26 (3) |
| Insomnio | 4 (1) | 21 (2) |
| Dolor de espalda | 4 (1) | 20 (2) |
| Migraña | 5 (1) | 19 (2) |
| Defecaciones frecuentes | 1 (0) | 17 (2) |
| Depresión | 2 (0) | 12 (1) |
| Bronquitis | 2 (0) | 12 (1) |
| Absceso dental | 0 (0) | 10 (1) |
| Foliculitis | 0 (0) | 9 (1) |
| Cefalea sinusal | 0 (0) | 9 (1) |
El empeoramiento grave de la psoriasis (rebote) se produjo en el 0,3% (4/1184) de los sujetos tras la interrupción del tratamiento con OTEZLA.
Ensayos clínicos de la enfermedad de Behçet
OTEZLA se evaluó en un estudio de fase 3, multicéntrico, aleatorizado y controlado con placebo (BCT-002) en pacientes adultos con enfermedad de Behçet (EB) con úlceras orales activas. Un total de 207 pacientes fueron aleatorizados para recibir OTEZLA 30 mg dos veces al día o placebo dos veces al día. Se utilizó la titulación durante los primeros 5 días [ver Posología y forma de administración (2.1)]. Después de la semana 12, todos los pacientes recibieron tratamiento con OTEZLA 30 mg dos veces al día. Los pacientes tenían edades comprendidas entre los 19 y los 72 años, con una edad media de 40 años.
La diarrea, las náuseas, el dolor de cabeza y la infección de las vías respiratorias altas fueron las reacciones adversas más frecuentes. La proporción de pacientes con EB que interrumpieron el tratamiento debido a cualquier reacción adversa durante el período controlado con placebo del estudio fue del 2,9% para los pacientes tratados con OTEZLA 30 mg dos veces al día y del 4,9% para los pacientes tratados con placebo.
| Término preferido | Placebo (N=103) n (%) |
OTEZLA 30 mg dos veces al día (N=104) n (%) |
|---|---|---|
|
||
| Diarrea* | 21 (20.4) | 43 (41.3) |
| Náuseas* | 11 (10.7) | 20 (19.2) |
| Dolor de cabeza | 11 (10.7) | 15 (14.4) |
| Infección de las vías respiratorias altas | 5 (4.9) | 12 (11.5) |
| Dolor abdominal superior | 2 (1.9) | 9 (8.7) |
| Vómitos* | 2 (1.9) | 9 (8.7) |
| Dolor de espalda | 6 (5.8) | 8 (7.7) |
| Infección viral de las vías respiratorias altas | 5 (4.9) | 7 (6.7) |
| Artralgia | 3 (2.9) | 6 (5.8) |
7 INTERACCIONES MEDICAMENTOSAS
7.1 Inductores fuertes del CYP450
La exposición al apremilast disminuye cuando se administra OTEZLA junto con inductores fuertes del CYP450 (como la rifampicina) y puede resultar en pérdida de eficacia [consulte Advertencias y precauciones (5.3) y Farmacología clínica (12.3)].
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Registro de Exposición al Embarazo
Existe un registro de exposición al embarazo que monitorea los resultados del embarazo en mujeres expuestas a OTEZLA durante el embarazo. La información sobre el registro se puede obtener llamando al 1-877-311-8972 o visitando https://mothertobaby.org/ongoing-study/otezla/.
Resumen de Riesgos
Los datos de farmacovigilancia disponibles con el uso de OTEZLA en mujeres embarazadas no han establecido un riesgo asociado a los medicamentos de defectos de nacimiento importantes, aborto espontáneo o resultados adversos maternos o fetales, pero estos datos son extremadamente limitados. Con base en los hallazgos de estudios de reproducción animal, OTEZLA puede aumentar el riesgo de pérdida fetal. En estudios de desarrollo embrio-fetal animal, la administración de apremilast a monos cynomolgus embarazadas durante la organogénesis resultó en aumentos relacionados con la dosis en el aborto / muerte embrio-fetal a exposiciones de dosis 2.1 veces la dosis terapéutica humana máxima recomendada (MRHD) y sin efectos adversos a una exposición de 1.4 veces la MRHD. Cuando se administró a ratones embarazadas, durante la organogénesis no hubo malformaciones inducidas por apremilast hasta exposiciones 4.0 veces la MRHD (ver Datos). Avise a las mujeres embarazadas sobre el riesgo potencial de pérdida fetal. Considere la planificación y prevención del embarazo para las mujeres en edad fértil.
El riesgo de fondo estimado de defectos de nacimiento importantes y aborto espontáneo para las poblaciones indicadas es desconocido. Todos los embarazos tienen un riesgo de fondo de defectos de nacimiento, pérdida u otros resultados adversos. En la población general de EE. UU., el riesgo de fondo estimado de defectos de nacimiento importantes y aborto espontáneo en embarazos clínicamente reconocidos es del 2-4% y del 15-20%, respectivamente.
Datos
Datos de animales
En un estudio de desarrollo embrio-fetal, se administró apremilast a monos cynomolgus embarazadas a dosis de 20, 50, 200 o 1000 mg / kg / día durante el período de organogénesis (días de gestación 20 a 50). Hubo un aumento relacionado con la dosis en abortos espontáneos, con la mayoría de los abortos ocurriendo durante las semanas 3 a 4 de dosificación en el primer trimestre, a dosis aproximadamente 2.1 veces la MRHD y mayor (sobre una base de área bajo la curva [AUC] a dosis ≥50 mg / kg / día). No se observaron efectos abortivos a una dosis aproximadamente 1.4 veces la MRHD (sobre una base de AUC a una dosis de 20 mg / kg / día). Aunque no hubo evidencia de un efecto teratógeno a dosis de 20 mg / kg / día y mayores cuando se examinó al día 100, los fetos abortados no se examinaron.
En un estudio de desarrollo embrio-fetal en ratones, se administró apremilast a dosis de 250, 500 o 750 mg / kg / día a las madres durante la organogénesis (día de gestación 6 a 15). En un estudio combinado de fertilidad y desarrollo embrio-fetal en ratones, se administró apremilast a dosis de 10, 20, 40 u 80 mg / kg / día comenzando 15 días antes de la cohabitación y continuando hasta el día 15 de gestación. No se observaron hallazgos teratógenos atribuidos a apremilast en ninguno de los estudios; sin embargo, hubo un aumento en la pérdida postimplantación a dosis correspondientes a una exposición sistémica de 2.3 veces la MRHD y mayor (≥20 mg / kg / día). A dosis de ≥20 mg / kg / día, las variaciones esqueléticas incluyeron sitios de osificación incompletos de tarsos, cráneo, esternón y vértebras. No se observaron efectos a una dosis aproximadamente 1.3 veces la MRHD (10 mg / kg / día).
Apremilast se distribuyó a través de la placenta al compartimento fetal en ratones y monos.
En un estudio pre y posnatal en ratones, se administró apremilast a ratones hembra embarazadas a dosis de 10, 80 o 300 mg / kg / día desde el día 6 de gestación hasta el día 20 de lactancia, con el destete en el día 21. La distocia, la reducción de la viabilidad y la reducción del peso al nacer ocurrieron a dosis correspondientes a ≥4.0 veces la MRHD (sobre una base de AUC a dosis ≥80 mg / kg / día). No se produjeron efectos adversos a una dosis 1.3 veces la MRHD (10 mg / kg / día). No hubo evidencia de deterioro funcional del desarrollo físico, comportamiento, capacidad de aprendizaje, competencia inmunitaria o fertilidad en la descendencia a dosis de hasta 7.5 veces la MRHD (sobre una base de AUC a una dosis de 300 mg / kg / día).
8.2 Lactancia
Resumen de Riesgos
No hay datos sobre la presencia de apremilast en la leche materna, los efectos en el lactante amamantado o los efectos en la producción de leche. Sin embargo, se detectó apremilast en la leche de ratones lactantes. Cuando un medicamento está presente en la leche animal, es probable que el medicamento esté presente en la leche materna. Los beneficios para el desarrollo y la salud de la lactancia materna deben considerarse junto con la necesidad clínica de la madre de OTEZLA y cualquier efecto adverso potencial en el lactante amamantado de OTEZLA o de la condición materna subyacente.
8.4 Uso pediátrico
No se ha establecido la seguridad y eficacia de OTEZLA en pacientes pediátricos menores de 18 años.
8.5 Uso geriátrico
De los 1493 sujetos que se inscribieron en los estudios PsA-1, PsA-2 y PsA-3, un total de 146 sujetos con artritis psoriásica tenían 65 años o más, incluidos 19 sujetos de 75 años o más. No se observaron diferencias generales en el perfil de seguridad de los sujetos de edad avanzada ≥65 años y los sujetos adultos jóvenes <65 años en los estudios clínicos.
De los 1257 sujetos que se inscribieron en dos ensayos controlados con placebo para la psoriasis (PSOR 1 y PSOR 2), un total de 108 sujetos con psoriasis tenían 65 años o más, incluidos 9 sujetos que tenían 75 años o más. No se observaron diferencias generales en la eficacia y seguridad en los sujetos de edad avanzada ≥65 años y los sujetos adultos jóvenes <65 años en los ensayos clínicos.
8.6 Insuficiencia renal
La farmacocinética de apremilast se caracterizó en sujetos con insuficiencia renal leve, moderada y grave, definida por un aclaramiento de creatinina de 60-89, 30-59 y menos de 30 mL por minuto, respectivamente, mediante la ecuación de Cockcroft–Gault. Si bien no es necesario ajustar la dosis en pacientes con insuficiencia renal leve o moderada, la dosis de OTEZLA debe reducirse a 30 mg una vez al día en pacientes con insuficiencia renal grave [ver Dosis y administración (2.2) y Farmacología clínica (12.3)].
8.7 Insuficiencia hepática
La farmacocinética de apremilast se caracterizó en sujetos con insuficiencia hepática moderada (Child Pugh B) y grave (Child Pugh C). No es necesario ajustar la dosis en estos pacientes.
10 SOBREDOSIS
En caso de sobredosis, los pacientes deben buscar ayuda médica inmediata. Los pacientes deben ser manejados mediante atención sintomática y de apoyo en caso de sobredosis.
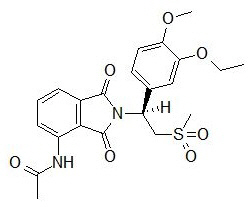
11 DESCRIPCIÓN
El ingrediente activo en las tabletas de OTEZLA es apremilast. Apremilast es un inhibidor de la fosfodiesterasa 4 (PDE4). Apremilast se conoce químicamente como N-[2-[(1S)-1-(3-etoxi-4-metoxifenil)-2-(metilsulfonil)etil]-2,3-dihidro-1,3-dioxo-1H-isoindol-4-il]acetamida. Su fórmula empírica es C22H24N2O7S y su peso molecular es 460.5.
La estructura química es:
Las tabletas de OTEZLA se suministran en concentraciones de 10, 20 y 30 mg para administración oral. Cada tableta contiene apremilast como ingrediente activo y los siguientes ingredientes inactivos: lactosa monohidratada, celulosa microcristalina, croscarmelosa sódica, estearato de magnesio, alcohol polivinílico, dióxido de titanio, polietilenglicol, talco, óxido de hierro rojo, óxido de hierro amarillo (solo 20 y 30 mg) y óxido de hierro negro (solo 30 mg).
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
Apremilast es un inhibidor oral de molécula pequeña de la fosfodiesterasa 4 (PDE4) específica para el monofosfato cíclico de adenosina (AMPc). La inhibición de la PDE4 da como resultado un aumento de los niveles intracelulares de AMPc. El mecanismo o mecanismos específicos por los que apremilast ejerce su acción terapéutica no están bien definidos.
12.3 Farmacocinética
Absorción
Apremilast, cuando se administra por vía oral, se absorbe con una biodisponibilidad absoluta de ~73%, con concentraciones plasmáticas máximas (Cmax) que se producen a un tiempo medio (tmax) de ~2,5 horas. La coadministración con alimentos no altera la extensión de la absorción de apremilast.
Distribución
La unión a proteínas plasmáticas humanas de apremilast es aproximadamente del 68%. El volumen de distribución aparente medio (Vd) es de 87 L.
Metabolismo
Después de la administración oral en humanos, apremilast es un componente circulante principal (45%) seguido del metabolito inactivo M12 (39%), un conjugado glucurónido de apremilast O-desmetilado. Se metaboliza ampliamente en humanos, con hasta 23 metabolitos identificados en plasma, orina y heces. Apremilast se metaboliza tanto por metabolismo oxidativo del citocromo (CYP) con posterior glucuronidación como por hidrólisis no mediada por CYP. In vitro, el metabolismo de CYP de apremilast está mediado principalmente por CYP3A4, con contribuciones menores de CYP1A2 y CYP2A6.
Eliminación
El aclaramiento plasmático de apremilast es de aproximadamente 10 L/h en sujetos sanos, con una vida media de eliminación terminal de aproximadamente 6-9 horas. Después de la administración oral de apremilast radiomarcado, aproximadamente el 58% y el 39% de la radiactividad se recupera en orina y heces, respectivamente, con aproximadamente el 3% y el 7% de la dosis radiactiva recuperada como apremilast en orina y heces, respectivamente.
Poblaciones específicas
Insuficiencia hepática: La farmacocinética de apremilast no se ve afectada por la insuficiencia hepática moderada o grave.
Insuficiencia renal: La farmacocinética de apremilast no se ve afectada por la insuficiencia renal leve o moderada. En 8 sujetos con insuficiencia renal grave a los que se administró una dosis única de 30 mg de apremilast, el AUC y la Cmax de apremilast aumentaron aproximadamente un 88% y un 42%, respectivamente [ver Dosis y administración (2.2) y Uso en poblaciones específicas (8.6)].
Edad: Se estudió una dosis única oral de 30 mg de apremilast en adultos jóvenes y sujetos sanos de edad avanzada. La exposición a apremilast en sujetos de edad avanzada (de 65 a 85 años) fue aproximadamente un 13% más alta en AUC y aproximadamente un 6% más alta en Cmax que en sujetos jóvenes (de 18 a 55 años) [ver Uso en poblaciones específicas (8.5)].
Género: En estudios farmacocinéticos en voluntarios sanos, la extensión de la exposición en mujeres fue aproximadamente un 31% más alta y la Cmax fue aproximadamente un 8% más alta que en los sujetos masculinos.
Raza y origen étnico: La farmacocinética de apremilast en sujetos masculinos sanos chinos y japoneses es comparable a la de los sujetos masculinos sanos caucásicos. Además, la exposición a apremilast es similar entre los caucásicos hispanos, los caucásicos no hispanos y los afroamericanos.
Interacciones medicamentosas
Datos in vitro: Apremilast no es un inhibidor de CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 o CYP3A4 y no es un inductor de CYP1A2, CYP2B6, CYP2C9, CYP2C19 o CYP3A4. Apremilast es un sustrato, pero no un inhibidor de la glicoproteína P (P-gp) y no es un sustrato o un inhibidor del transportador de aniones orgánicos (OAT)1 y OAT3, el transportador de cationes orgánicos (OCT)2, la polipéptido transportador de aniones orgánicos (OATP)1B1 y OATP1B3, o la proteína de resistencia al cáncer de mama (BCRP).
Se realizaron estudios de interacción medicamentosa con apremilast y sustratos de CYP3A4 (anticonceptivo oral que contiene etinilestradiol y norgestimato), inhibidor de CYP3A y P-gp (ketoconazol), inductor de CYP450 (rifampicina) y fármaco que se administra con frecuencia en esta población de pacientes (metotrexato).
No se observaron interacciones farmacocinéticas significativas cuando se administró apremilast oral de 30 mg con anticonceptivos orales, ketoconazol o metotrexato. La coadministración del inductor de CYP450 rifampicina (600 mg una vez al día durante 15 días) con una dosis única oral de 30 mg de apremilast dio como resultado una reducción del AUC y la Cmax de apremilast en un 72% y un 43%, respectivamente [ver Advertencias y precauciones (5.3) e Interacciones medicamentosas (7.1)].
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
Se realizaron estudios a largo plazo en ratones y ratas con apremilast para evaluar su potencial carcinogénico. No se observó evidencia de tumores inducidos por apremilast en ratones a dosis orales de hasta 8.8 veces la Dosis Humana Máxima Recomendada (MRHD) en base al AUC (1000 mg/kg/día) o en ratas a dosis orales de hasta aproximadamente 0.08 y 1.1 veces la MRHD, (20 mg/kg/día en machos y 3 mg/kg/día en hembras, respectivamente).
Apremilast dio negativo en la prueba de Ames, en la prueba de aberración cromosómica in vitro de linfocitos de sangre periférica humana y en la prueba de micronúcleos in vivo en ratones.
En un estudio de fertilidad en ratones machos, apremilast a dosis orales de hasta aproximadamente 3 veces la MRHD basada en AUC (hasta 50 mg/kg/día) no produjo efectos sobre la fertilidad masculina. En un estudio de fertilidad en ratonas, apremilast se administró a dosis orales de 10, 20, 40 o 80 mg/kg/día. A dosis ≥1.8 veces la MRHD (≥20 mg/kg/día), los ciclos estrales se prolongaron, debido al alargamiento del diestro que resultó en un intervalo más largo hasta el apareamiento. Las ratonas que quedaron embarazadas a dosis de 20 mg/kg/día y mayores también tuvieron un aumento de la incidencia de pérdidas postimplantatorias tempranas. No hubo efecto de apremilast aproximadamente 1.0 veces la MRHD (10 mg/kg/día).
14 ESTUDIOS CLÍNICOS
14.1 Artritis psoriásica
Se evaluó la seguridad y eficacia de OTEZLA en 3 ensayos multicéntricos, aleatorizados, doble ciego y controlados con placebo (Estudios PsA-1, PsA-2 y PsA-3) de diseño similar. Se aleatorizó a un total de 1493 pacientes adultos con artritis psoriásica (PsA) activa (≥3 articulaciones inflamadas y ≥3 articulaciones sensibles) a pesar del tratamiento previo o actual con fármacos antirreumáticos modificadores de la enfermedad (DMARD). Los pacientes incluidos en estos estudios tenían un diagnóstico de PsA desde hacía al menos 6 meses. En el Estudio PsA-3 se requería una lesión cutánea psoriásica calificada de al menos 2 cm de diámetro. Se permitía el tratamiento previo con un fármaco biológico, incluidos los inhibidores del TNF (hasta el 10% podían ser fracasos terapéuticos con inhibidores del TNF). En los 3 estudios, los pacientes fueron asignados aleatoriamente a placebo (n=496), OTEZLA 20 mg (n=500) u OTEZLA 30 mg (n=497) administrados por vía oral dos veces al día. Se utilizó un ajuste de dosis durante los primeros 5 días [ver Posología y administración (2.1)]. Durante el ensayo, se permitió a los pacientes recibir dosis estables de metotrexato [MTX (≤25 mg/semana)], sulfasalazina [SSZ (≤2 g/día)], leflunomida [LEF (≤20 mg/día)], dosis bajas de corticosteroides orales (equivalentes a ≤10 mg de prednisona al día) y/o antiinflamatorios no esteroideos (AINE) concomitantes. Las asignaciones de tratamiento se estratificaron en función del uso de DMARD de moléculas pequeñas al inicio del estudio en los Estudios PsA-1, PsA-2 y PsA-3. En el estudio PsA-3 se realizó una estratificación adicional de BSA >3% con psoriasis. Se excluyó a los pacientes que eran fracasos terapéuticos de >3 agentes para la PsA (moléculas pequeñas o productos biológicos), o >1 bloqueador del TNF biológico.
La variable principal de eficacia fue el porcentaje de pacientes que lograron una respuesta ACR 20 del Colegio Estadounidense de Reumatología (ACR) en la Semana 16. Los datos de eficacia controlados con placebo se recogieron y analizaron hasta la Semana 24. Los pacientes cuyo recuento de articulaciones sensibles e inflamadas no había mejorado al menos un 20% se consideraron no respondedores en la Semana 16. Los pacientes que no respondieron al placebo fueron reasignados aleatoriamente 1:1 de forma ciega a OTEZLA 20 mg dos veces al día o 30 mg dos veces al día siguiendo el esquema de ajuste de dosis [ver Posología y administración (2.1)]. Los pacientes tratados con OTEZLA continuaron con su tratamiento inicial. En la Semana 24, todos los pacientes que aún recibían placebo fueron reasignados aleatoriamente a 20 mg dos veces al día o 30 mg dos veces al día.
En los 3 estudios se incluyeron pacientes con subtipos de PsA, como poliartritis simétrica (62,0%), oligoartritis asimétrica (27,0%), artritis de las articulaciones interfalángicas distales (IFD) (6,0%), artritis mutilante (3,0%) y espondilitis predominante (2,1%). La mediana de duración de la enfermedad PsA fue de 5 años. Los pacientes recibieron tratamiento concomitante con al menos un DMARD (65,0%), MTX (55,0%), SSZ (9,0%), LEF (7,0%), dosis bajas de corticosteroides orales (14,0%) y AINE (71,0%). El tratamiento previo solo con DMARD de moléculas pequeñas se notificó en el 76,0% de los pacientes y el tratamiento previo con DMARD biológicos se notificó en el 22,0% de los pacientes, lo que incluye al 9,0% que habían fracasado al tratamiento previo con DMARD biológicos.
Respuesta clínica en pacientes con artritis psoriásica
El porcentaje de pacientes que lograron respuestas ACR 20, 50 y 70 en los Estudios PsA-1, PsA-2 y PsA-3 se presenta en la Tabla 5 a continuación. OTEZLA ± DMARD, en comparación con placebo ± DMARD, dio lugar a una mayor mejoría de los signos y síntomas de la artritis psoriásica, como demuestra la proporción de pacientes con una respuesta ACR 20 en la Semana 16.
| a N es el número de pacientes aleatorizados y tratados. b Diferencia estadísticamente significativa con respecto al placebo (p<0,05). |
||||||
| PsA-1 | PsA-2 | PsA-3 | ||||
| Na | Placebo ± DMARDs N=168 |
OTEZLA 30 mg dos veces al día ± DMARDs N=168 |
Placebo ± DMARDs N=159 |
OTEZLA 30 mg dos veces al día ± DMARDs N=162 |
Placebo ± DMARDs N=169 |
OTEZLA 30 mg dos veces al día ± DMARDs N=167 |
| ACR 20 Semana 16 |
19% | 38% b | 19% | 32% b | 18% | 41% b |
| ACR 50 Semana 16 |
6% | 16% | 5% | 11% | 8% | 15% |
| ACR 70 Semana 16 |
1% | 4% | 1% | 1% | 2% | 4% |
OTEZLA 30 mg dos veces al día resultó en una mejoría para cada componente ACR, en comparación con el placebo en la Semana 16 en el Estudio PsA-1 (Tabla 6). Se observaron resultados consistentes en los Estudios PsA-2 y PsA-3.
| Los cambios medios con respecto al valor inicial son medias de mínimos cuadrados de análisis de covarianza. a Escala 0-78. b Escala 0-76. c VAS=Escala Analógica Visual; 0=mejor, 100=peor. d HAQ-DI=Índice de Discapacidad del Cuestionario de Evaluación de la Salud; 0=mejor, 3=peor; mide la capacidad del sujeto para realizar lo siguiente: vestirse/arreglarse, levantarse, comer, caminar, alcanzar, agarrar, mantener la higiene y mantener la actividad diaria. e CRP=proteína C reactiva; Rango de referencia 0-0.5 mg/dL. * N refleja los pacientes aleatorizados; el número real de pacientes evaluables para cada criterio de valoración puede variar según el punto temporal. |
||
| Placebo (N*=168) |
OTEZLA 30 mg dos veces al día (N*=168) |
|
| Número de articulaciones sensiblesa Tamaño de la muestra Línea base Cambio medio en la semana 16 |
166 23 -2 |
164 23 -7 |
| Número de articulaciones inflamadasb Tamaño de la muestra Línea base Cambio medio en la semana 16 |
166 13 -2 |
164 13 -5 |
| Evaluación del dolor por parte del pacientec Tamaño de la muestra Línea base Cambio medio en la semana 16 |
165 61 -6 |
159 58 -14 |
| Evaluación global de la actividad de la enfermedad por parte del pacientec Tamaño de la muestra Línea base Cambio medio en la semana 16 |
165 59 -3 |
159 56 -10 |
| Evaluación global de la actividad de la enfermedad por parte del médicoc Tamaño de la muestra Línea base Cambio medio en la semana 16 |
158 55 -8 |
159 56 -19 |
| Puntuación HAQ-DId Tamaño de la muestra Línea base Cambio medio en la semana 16 |
165 1.2 -0.09 |
159 1.2 -0.2 |
| CRPe Tamaño de la muestra Línea base Cambio medio en la semana 16 |
166 1.1 0.1 |
167 0.8 -0.1 |
El tratamiento con OTEZLA produjo una mejoría en la dactilitis y la entesitis en pacientes con dactilitis o entesitis preexistentes.
Respuesta de la función física
OTEZLA 30 mg dos veces al día demostró una mejoría mayor en comparación con el placebo en el cambio medio con respecto al valor inicial de la puntuación del Índice de Discapacidad del Cuestionario de Evaluación de la Salud (HAQ-DI) en la Semana 16 [-0,244 frente a -0,086, respectivamente; el IC del 95% para la diferencia fue (-0,26, -0,06)] en el Estudio PsA-1. Las proporciones de respondedores al HAQ-DI (≥0,3 de mejoría con respecto al valor inicial) en la Semana 16 para el grupo de OTEZLA 30 mg dos veces al día fueron del 38%, en comparación con el 27% para el grupo de placebo en el Estudio PsA-1. Se observaron resultados consistentes en los Estudios PsA-2 y PsA-3.
14.2 Psoriasis
Dos ensayos multicéntricos, aleatorizados, doble ciego y controlados con placebo (Estudios PSOR-1 y PSOR-2) reclutaron a un total de 1257 sujetos de 18 años o más con psoriasis en placas de moderada a grave [implicación de la superficie corporal (BSA) del ≥10%, Evaluación Global Estática del Médico (sPGA) del ≥3 (enfermedad moderada o grave), puntuación del Índice de Gravedad y Área de la Psoriasis (PASI) ≥12, candidatos a fototerapia o terapia sistémica]. Se permitió a los sujetos utilizar corticosteroides tópicos de baja potencia en la cara, las axilas y la ingle. Los sujetos con psoriasis del cuero cabelludo podían utilizar champú de alquitrán de hulla y/o preparados tópicos con ácido salicílico en las lesiones del cuero cabelludo.
En el estudio PSOR-1 se reclutó a 844 sujetos y en el estudio PSOR-2 a 413 sujetos. En ambos estudios, los sujetos fueron aleatorizados 2:1 a OTEZLA 30 mg dos veces al día o placebo durante 16 semanas. En ambos estudios se evaluó la proporción de sujetos que alcanzaron PASI-75 en la Semana 16 y la proporción de sujetos que alcanzaron una puntuación en la sPGA de aclaramiento (0) o casi aclaramiento (1) en la Semana 16. En ambos estudios, la edad de los sujetos osciló entre 18 y 83 años, con una mediana de edad global de 46 años. La afectación media de la BSA al inicio del estudio fue del 25,19% (mediana de 21,0%), la puntuación media del PASI al inicio del estudio fue de 19,07 (mediana de 16,80) y la proporción de sujetos con una puntuación de la sPGA de 3 (moderada) y 4 (grave) al inicio del estudio fue del 70,0% y el 29,8%, respectivamente. Aproximadamente el 30% de todos los sujetos habían recibido fototerapia previa y el 54% habían recibido terapia sistémica convencional y/o biológica previa para el tratamiento de la psoriasis, con un 37% que había recibido terapia sistémica convencional previa y un 30% que había recibido terapia biológica previa. Aproximadamente un tercio de los sujetos no habían recibido fototerapia, ni terapia sistémica convencional ni biológica previa. Un total del 18% de los sujetos tenían antecedentes de artritis psoriásica.
Respuesta clínica en sujetos con psoriasis en placas
La proporción de sujetos que lograron respuestas PASI -75 y una puntuación sPGA de aclaramiento (0) o casi aclaramiento (1) se presentan en la Tabla 7.
| a N es el número de pacientes aleatorizados y tratados. b PASI=Índice de gravedad y área de la psoriasis. c sPGA=Evaluación global estática del médico. |
||||
| Estudio PSOR-1 | Estudio PSOR-2 | |||
| Placebo | OTEZLA 30 mg dos veces al día |
Placebo | OTEZLA 30 mg dos veces al día |
|
| Na | N=282 | N=562 | N=137 | N=274 |
| PASIb-75, n (%) | 15 (5.3) | 186 (33.1) | 8 (5.8) | 79 (28.8) |
| sPGAc de aclaramiento o casi aclaramiento, n (%) |
11 (3.9) | 122 (21.7) | 6 (4.4) | 56 (20.4) |
La mediana del tiempo hasta la pérdida de la respuesta PASI-75 entre los sujetos reasignados aleatoriamente a placebo en la Semana 32 durante la Fase de Retirada del Tratamiento Aleatorizado fue de 5,1 semanas.
14.3 Úlceras bucales asociadas a la enfermedad de Behçet
En un ensayo multicéntrico, aleatorizado y controlado con placebo (BCT-002) se reclutó a un total de 207 pacientes adultos con EB con úlceras bucales activas. Los pacientes habían sido tratados previamente con al menos un medicamento no biológico para la EB y eran candidatos a terapia sistémica. Los pacientes cumplían los criterios del Grupo de Estudio Internacional (ISG, por sus siglas en inglés) para la EB. Los pacientes tenían al menos 2 úlceras bucales en el momento de la selección y al menos 2 úlceras bucales en el momento de la aleatorización y sin afectación activa de órganos importantes en ese momento. No se permitió el tratamiento concomitante para la EB.
Los pacientes fueron aleatorizados 1:1 para recibir OTEZLA 30 mg dos veces al día (n=104) o placebo (n=103) durante 12 semanas. Después de la semana 12, todos los pacientes recibieron OTEZLA 30 mg dos veces al día.
La eficacia se evaluó en función del número y el dolor de las úlceras bucales.
La edad de los pacientes oscilaba entre 19 y 72 años, con una media de 40 años. La duración media de la EB fue de 6,84 años. Todos los sujetos tenían antecedentes de úlceras bucales recurrentes que estaban activas en ese momento. Los sujetos tenían antecedentes de lesiones cutáneas (98,6%), úlceras genitales (90,3%), manifestaciones musculoesqueléticas (72,5%), manifestaciones oculares (17,4%), sistema nervioso central (9,7%), manifestaciones gastrointestinales (GI) (9,2%) y afectación vascular (1,4%). El recuento medio de úlceras bucales al inicio del estudio fue de 4,2 y 3,9 en los grupos de OTEZLA y placebo, respectivamente.
Medidas de las úlceras bucales
Las mejoras en las medidas de las úlceras bucales en la semana 12 se presentan en la Tabla 8.
| Criterio de valoración | Placebo N=103 |
OTEZLA 30 mg dos veces al día N=104 |
Diferencia de tratamiento† (IC del 95%‡) |
|---|---|---|---|
|
|||
| Cambio§ con respecto al valor inicial en el dolor de las úlceras bucales medido mediante la EAV¶ en la semana 12 | -18,7 | – 42,7 | -24,1 (-32,4, -15,7) |
| Proporción# de sujetos que lograron una respuesta completa de las úlceras bucales (sin úlceras bucales) en la semana 12 | 22,3% | 52,9% | 30,6%Þ (18,1%, 43,1%) |
| Proporción# de sujetos que lograron una respuesta completa de las úlceras bucales (sin úlceras bucales) en la semana 6, y que permanecieron sin úlceras bucales durante al menos 6 semanas adicionales durante la fase de tratamiento controlado con placebo de 12 semanas | 4,9% | 29,8% | 25,1%Þ (15,5%, 34,6%) |
| Promedio diarioß,à de úlceras bucales durante la fase de tratamiento controlado con placebo de 12 semanas | 2,6 | 1,5 | -1,1 (-1,6, -0,7) |
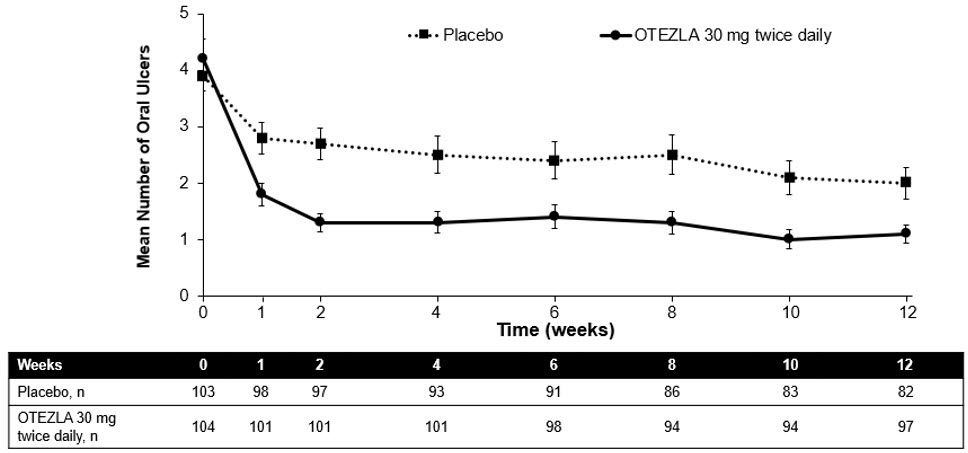
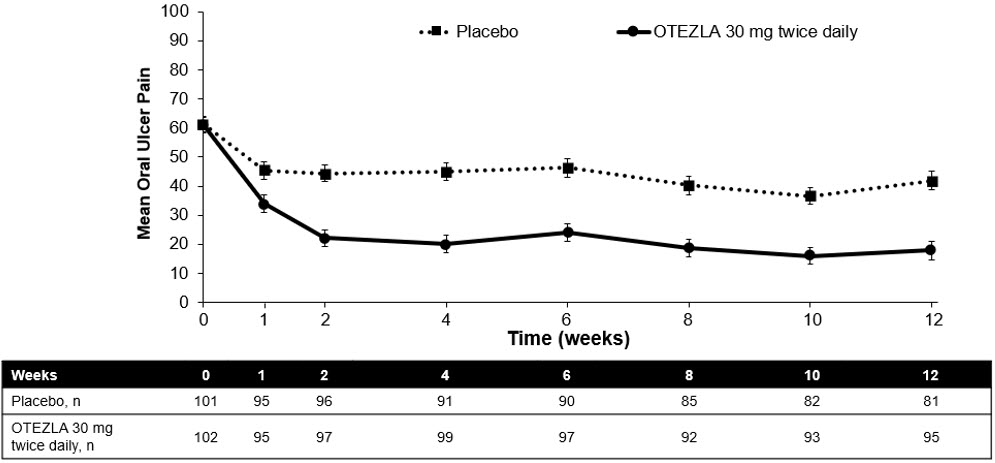
La Figura 1 muestra el número medio de úlceras bucales para cada grupo de tratamiento en cada visita, mientras que la Figura 2 muestra el dolor medio de las úlceras bucales en una escala analógica visual para cada grupo de tratamiento en cada visita.
| ITT = intención de tratar; SE = error estándar. |
| Figura 1: Media (± SE) del número de úlceras bucales por punto temporal hasta la semana 12 (población ITT) |
 |
| ITT=intent-to-treat; SE=standard error. El dolor de la úlcera oral se evaluó en una escala analógica visual de 100 mm con 0 = sin dolor y 100 = el peor dolor posible. Las puntuaciones medias iniciales de dolor en la escala analógica visual fueron de 61,2 y 60,8 en el grupo de tratamiento con OTEZLA 30 mg dos veces al día y en el grupo de tratamiento con placebo, respectivamente. |
| Figura 2: Dolor de la úlcera oral promedio (± SE) en una escala analógica visual por punto de tiempo hasta la semana 12 (población con ITT) |
 |
16 FORMA DE SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
OTEZLA está disponible en tabletas recubiertas con película en forma de diamante en las siguientes concentraciones de dosis: tableta rosa de 10 mg grabada con “APR” en un lado y “10” en el otro lado; tableta marrón de 20 mg grabada con “APR” en un lado y “20” en el otro lado; tableta beige de 30 mg grabada con “APR” en un lado y “30” en el otro lado.
Las tabletas se suministran en las siguientes concentraciones y configuraciones de envase:
| Configuración del envase |
Concentración de la tableta |
Número NDC |
| Frascos de 60 | 30 mg | 59572-631-06 |
| Paquete de inicio de dos semanas | Envase de dosis progresiva blíster de 13 tabletas que contiene: (4) tabletas de 10 mg, (4) de 20 mg, y (5) de 30 mg con (14) tabletas adicionales de 30 mg |
59572-630-27 |
| Caja de 28 unidades | Dos blísteres de 30 mg que contienen (14) tabletas de 30 mg |
59572-631-28 |
| Paquete de inicio de 28 días | Envase de dosis progresiva blíster de 13 tabletas que contiene: (4) tabletas de 10 mg, (4) de 20 mg, y (5) de 30 mg con (42) tabletas adicionales de 30 mg |
59572-632-55 |
Almacenamiento y manipulación
Conservar las tabletas por debajo de 30 °C (86 °F).
17 INFORMACIÓN PARA EL PACIENTE
-
Diarrea, náuseas y vómitos
Indique a los pacientes que se comuniquen con su proveedor de atención médica si experimentan diarrea, náuseas o vómitos intensos. Los médicos que recetan el medicamento deben advertir a los pacientes sobre las posibles complicaciones de la diarrea, las náuseas o los vómitos intensos. Considere la posibilidad de reducir la dosis de OTEZLA o suspenderla si los pacientes presentan diarrea, náuseas o vómitos intensos [consulte Advertencias y precauciones (5.1)]. -
Depresión
Antes de usar OTEZLA en pacientes con antecedentes de depresión o pensamientos y comportamiento suicidas, los médicos que recetan el medicamento deben sopesar cuidadosamente los riesgos y beneficios del tratamiento con OTEZLA en esos pacientes. Se debe advertir a los pacientes, a sus cuidadores y a sus familias sobre la necesidad de estar atentos a la aparición o el agravamiento de la depresión, los pensamientos suicidas u otros cambios en el estado de ánimo, y si se producen esos cambios, que se pongan en contacto con su proveedor de atención médica. Los médicos que recetan el medicamento deben evaluar cuidadosamente los riesgos y beneficios de continuar el tratamiento con OTEZLA si se producen estos eventos [consulte Advertencias y precauciones (5.2)]. -
Disminución de peso
Los pacientes tratados con OTEZLA deben controlarse el peso con regularidad. Si se produce una pérdida de peso inexplicable o clínicamente significativa, esta debe evaluarse y debe considerarse la posibilidad de interrumpir el tratamiento con OTEZLA [consulte Advertencias y precauciones (5.3)]. -
Interacciones con otros medicamentos
No se recomienda el uso de inductores potentes de la enzima del citocromo P450 (p. ej., rifampicina, fenobarbital, carbamazepina, fenitoína) con OTEZLA [consulte Advertencias y precauciones (5.4), Interacciones con otros medicamentos (7.1) y Farmacología clínica (12.3)]. - Indique a los pacientes que tomen OTEZLA solo según las indicaciones.
- Aconseje a los pacientes que OTEZLA puede tomarse con o sin alimentos.
- Aconseje a los pacientes que no deben triturar, partir ni masticar los comprimidos.
- Informe a los pacientes sobre los efectos secundarios asociados con OTEZLA [consulte Reacciones adversas (6.1)].
-
Embarazo
Informe a las pacientes que existe un registro de embarazos para las mujeres embarazadas que han tomado OTEZLA durante el embarazo. Aconseje a las pacientes que se pongan en contacto con el registro llamando al 1-877-311-8972 para inscribirse o que visiten https://mothertobaby.org/ongoing-study/otezla/ [consulte Uso en poblaciones específicas (8.1)]. Advierta a las mujeres embarazadas y a las mujeres en edad fértil sobre el riesgo potencial para el feto. Aconseje a las mujeres que informen a su médico si están embarazadas o sospechan que pueden estarlo.
Fabricado para: Celgene Corporation
Summit, NJ 07901
OTEZLA® es una marca registrada de Celgene Corporation.
Pat. http://www.celgene.com/therapies
© 2014-2019 Celgene Corporation. Todos los derechos reservados.
APRPI.007 07/19
PANEL DE VISUALIZACIÓN PRINCIPAL
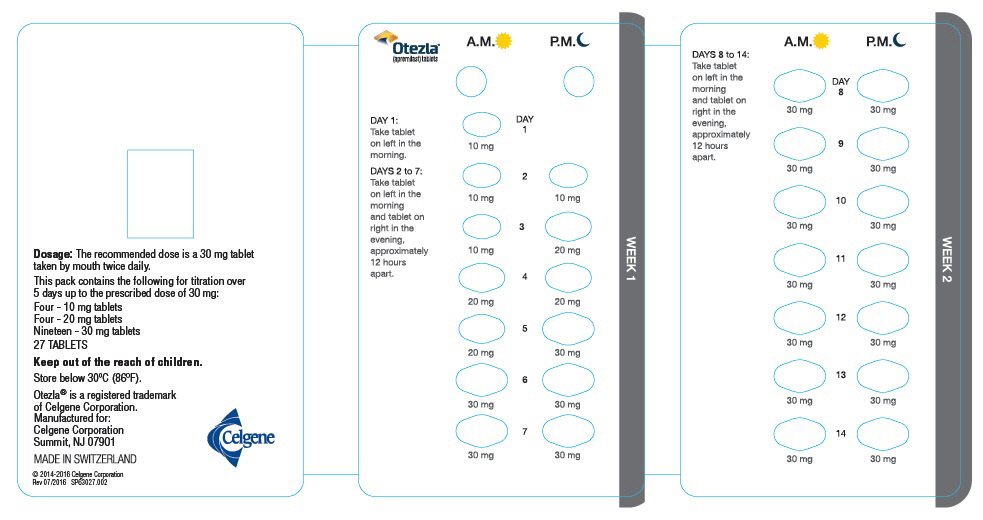
PRINCIPAL DISPLAY PANEL – NDC: 59572-630-27 – Two-Week Starter Pack Wallet Label (Outside)

PANEL DE VISUALIZACIÓN PRINCIPAL
PANEL PRINCIPAL DE EXHIBICIÓN – NDC: 59572-630-27 – Etiqueta de la Cartera del Paquete Inicial de Dos Semanas (Interior)
PANEL DE VISUALIZACIÓN PRINCIPAL
PRINCIPAL DISPLAY PANEL – NDC: 59572-630-99 – Sample Two-Week Starter Pack Wallet Label (Outside)

PANEL DE VISUALIZACIÓN PRINCIPAL
PRINCIPAL DISPLAY PANEL – NDC: 59572-630-99 – Sample Two-Week Starter Pack Wallet Label (Inside)
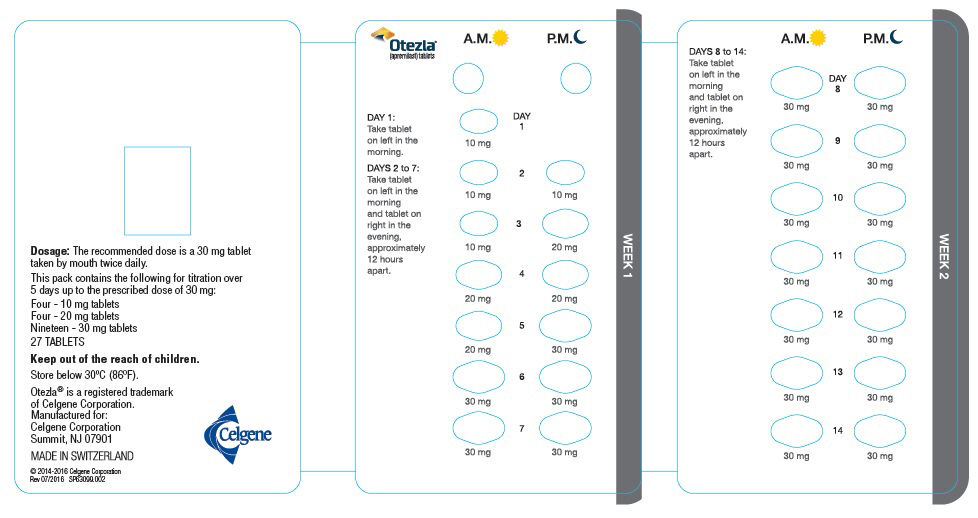
PANEL PRINCIPAL DE VISUALIZACIÓN – Paquete de inicio de muestra – NDC: 59572-630-98
NDC 59572-630-98
Paquete de inicio
Rx solamente
MUESTRA – NO PARA LA VENTA
Cada paquete contiene lo siguiente para la titulación durante
5 días hasta la dosis prescrita de 30 mg:
Cuatro – tabletas de 10 mg
Cuatro – tabletas de 20 mg
Diecinueve – tabletas de 30 mg
Cinco paquetes de inicio, cada uno con 27 TABLETAS
Celgene
Otezla®
(apremilast) tabletas

PANEL PRINCIPAL DE VISUALIZACIÓN – Paquete de inicio de muestra de tableta de 27 – NDC: 59572-630-97
NDC 59572-630-97
Rx Only
Otezla®
(apremilast) tablets
Paquete de inicio
MUESTRA – PROHIBIDA SU VENTA
Este paquete contiene lo siguiente
para la dosificación durante 5 días hasta
la dosis prescrita de 30 mg:
Cuatro comprimidos de 10 mg
Cuatro comprimidos de 20 mg
Diecinueve comprimidos de 30 mg
27 COMPRIMIDOS
Celgene

PANEL DE VISUALIZACIÓN PRINCIPAL
PANEL DE PRESENTACIÓN PRINCIPAL – NDC: 59572-631-06 – Etiqueta de frasco de 60 unidades de 30 mg

PANEL DE VISUALIZACIÓN PRINCIPAL
PANEL DE EXPOSICIÓN PRINCIPAL – NDC: 59572-631-28 – 30 mg, 28 unidades en lámina de blíster

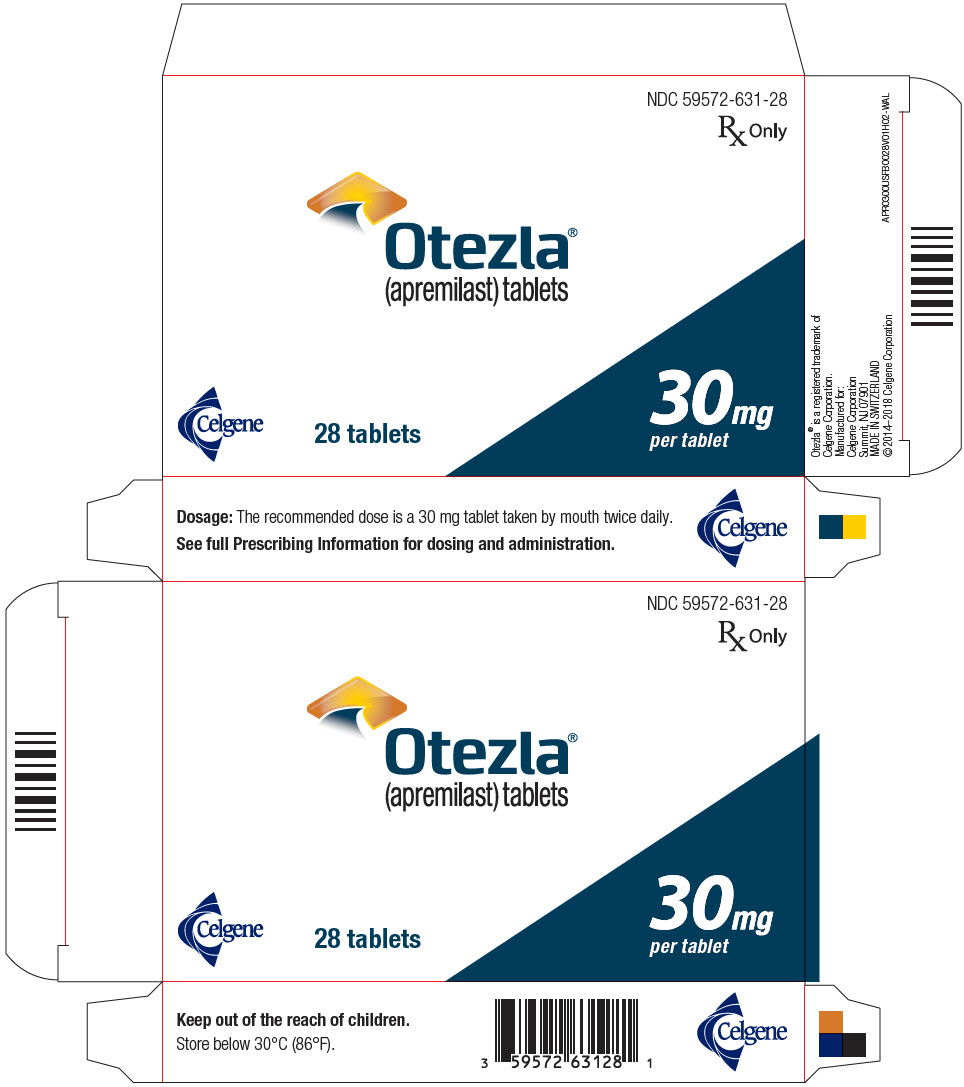
PANEL DE MOSTRACIÓN PRINCIPAL – Blíster de 30 mg en tableta Cartón – NDC: 59572-631-28
NDC 59572-631-28
Sólo receta médica
Otezla®
(apremilast) tabletas
30 mg
por tableta
Celgene
28 tabletas
PANEL DE DESPLIEGUE PRINCIPAL – Caja de blíster con comprimidos de 30 mg
NDC 59572-631-28
Rx Sólo
Otezla®
(apremilast) tabletas
30mg
por tableta
Celgene
28 tabletas
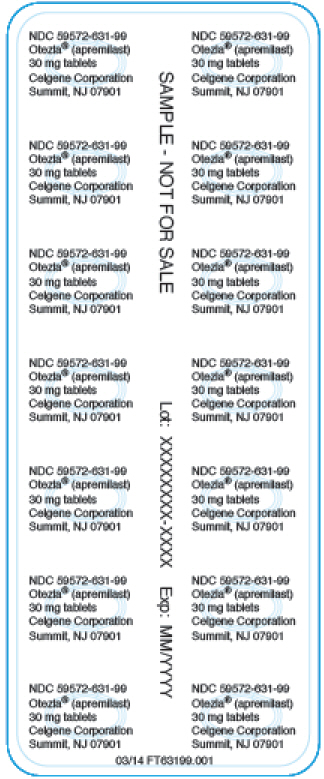
Panel de visualización principal – NDC: 59572-631-99 – Muestra de 30 mg 28 conteo Lámina de blíster
NDC 59572-631-99
Otezla® (apremilast)
30 mg comprimidos
Celgene Corporation
Summit, NJ 07901
MUESTRA – NO DE VENTA
Lote: XXXXXXXX-XXXX
Exp: MM/YYYY
03/14 FT63199.001
PANEL DE VISUALIZACIÓN PRINCIPAL
PANEL PRINCIPAL DE VISUALIZACIÓN – NDC: 59572-631-99 – Etiqueta de la caja de muestra de 30 mg, 28 unidades
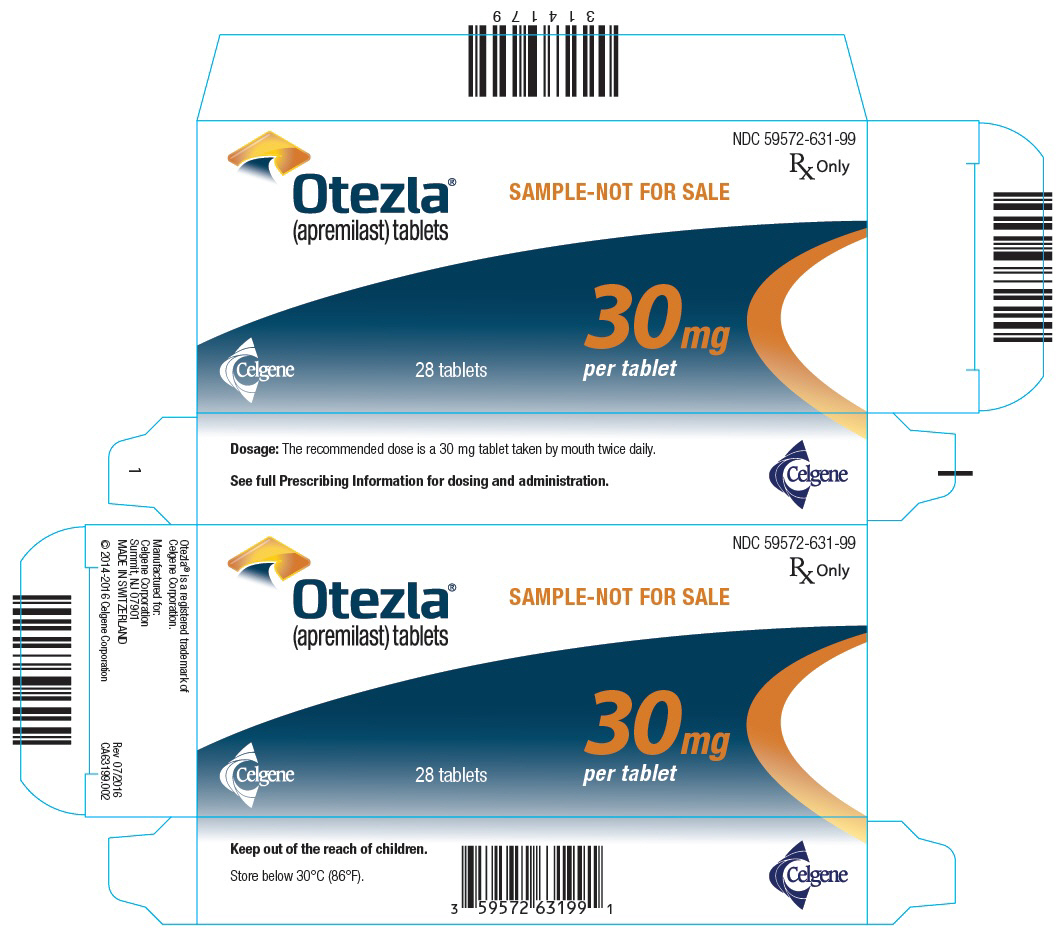
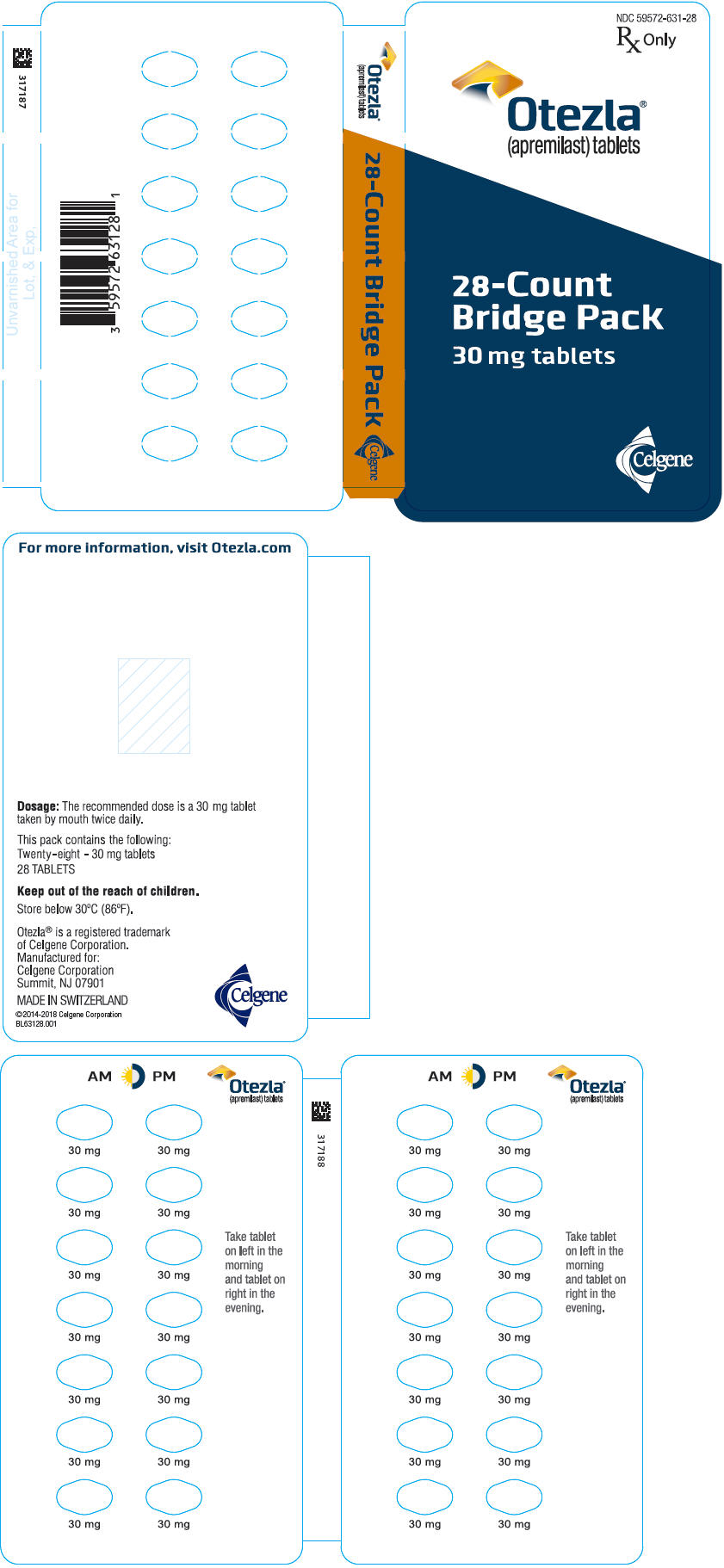
PANEL PRINCIPAL DE PRESENTACIÓN – Tableta de 30 mg Muestra Paquete Puente – NDC: 59572-631-97
NDC 59572-631-97
Sólo con receta
Otezla®
(apremilast) comprimidos
28 unidades
Bolsa puente de muestra
MUESTRA – NO SE VENDE
Comprimidos de 30 mg
Celgene

Panel principal de presentación -砖块 de muestra de tableta de 30 mg, paquete de cartón puente – NDC: 59572-631-97
NDC 59572-631-97
Sólo receta médica
Otezla®
(apremilast) comprimidos
MUESTRA – NO VENTA
Celgene
28 comprimidos
30mg
por comprimido

PANEL PRINCIPAL DE VISUALIZACIÓN – Caja de blíster de 55 tabletas
Paquete de inicio de 28 días
Starter Pack
Este paquete contiene lo siguiente
para la titulación durante 5 días hasta la
dosis prescrita de 30 mg:
Cuatro – tabletas de 10 mg
Cuatro – tabletas de 20 mg
Cuarenta y siete – tabletas de 30 mg
55 TABLETAS
Celgene

PANEL PRINCIPAL DE VISUALIZACIÓN – 55 Tabletas en blíster
NDC 59572-632-55
Rx Only
Otezla®
(apremilast) tablets
Paquete de inicio de 28 días
Starter Pack
Este paquete contiene lo siguiente
para la titulación durante 5 días hasta la
dosis prescrita de 30 mg:
Cuatro – 10 mg tabletas
Cuatro – 20 mg tabletas
Cuarenta y siete – 30 mg tabletas
55 TABLETAS
Celgene
