
Fabricante de medicamentos: E.R. Squibb & Sons, L.L.C. (Updated: 2024-12-13)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
ORENCIA (abatacept) for injection, for intravenous use
ORENCIA (abatacept) injection, for subcutaneous use
Initial U.S. Approval: 2005
CAMBIOS RECIENTES IMPORTANTES
INDICACIONES Y USO
ORENCIA es un modulador selectivo de la coestimulación de células T indicado para:
- •
- el tratamiento de pacientes adultos con artritis reumatoide (AR) activa de moderada a grave. (1.1)
- •
- el tratamiento de pacientes de 2 años de edad y mayores con artritis idiopática juvenil poliarticular (AIJp) activa de moderada a grave. (1.2)
- •
- el tratamiento de pacientes de 2 años de edad y mayores con artritis psoriásica (APs) activa. (1.3)
- •
- la profilaxis de la enfermedad del injerto contra el huésped aguda (EICHa), en combinación con un inhibidor de la calcineurina y metotrexato, en adultos y pacientes pediátricos de 2 años de edad y mayores que se someten a un trasplante de células madre hematopoyéticas (TCMH) de un donante no emparentado compatible o con un desajuste de 1 alelo. (1.4)
Limitaciones de uso:
No se recomienda el uso concomitante de ORENCIA con otros inmunosupresores [p. ej., fármacos biológicos antirreumáticos modificadores de la enfermedad (FARMEb), inhibidores de la Janus quinasa (JAK)] (1.5, 5.1).
DOSIS Y ADMINISTRACIÓN
Uso intravenoso para AR en adultos (2.1) y APs en adultos (2.3)
- •
- Administrar a las 0, 2 y 4 semanas, y cada 4 semanas a partir de entonces, como una infusión de 30 minutos.
| Peso corporal del paciente | Dosis | Número de viales |
|---|---|---|
|
Menos de 60 kg |
500 mg |
2 |
|
60 a 100 kg |
750 mg |
3 |
|
Más de 100 kg |
1,000 mg |
4 |
Uso subcutáneo para AR en adultos (2.1)
- •
- Antes de la primera dosis subcutánea, se puede administrar una dosis de carga opcional como una única infusión intravenosa según las categorías de peso corporal anteriores.
- •
- Administrar 125 mg mediante inyección subcutánea una vez por semana (dentro de un día de la infusión intravenosa si se administra la infusión).
- •
- Pacientes que cambian del uso intravenoso al uso subcutáneo, administrar la primera dosis subcutánea en lugar de la siguiente dosis intravenosa programada.
Uso intravenoso para AIJp en pacientes pediátricos de ≥6 años de edad (2.2)
- •
- Pacientes pediátricos que pesan <75 kg administrar 10 mg/kg por vía intravenosa y aquellos que pesan ≥75 kg administrar el régimen de dosificación intravenosa para adultos (sin exceder una dosis máxima de 1,000 mg), como una infusión de 30 minutos.
- •
- Subsecuentemente administrar infusiones a las 2 y 4 semanas y cada 4 semanas a partir de entonces.
Uso subcutáneo para AIJp y APs en pacientes pediátricos de ≥2 años de edad (2.2)
- •
- Administrar por vía subcutánea sin una dosis de carga intravenosa
| Peso corporal del paciente pediátrico | Dosis (una vez por semana) |
|---|---|
|
10 kg a menos de 25 kg |
50 mg |
|
25 kg a menos de 50 kg |
87.5 mg |
|
50 kg o más |
125 mg |
Uso subcutáneo para PsA en adultos (2.3)
- •
- Administrar 125 mg por inyección subcutánea una vez por semana sin una dosis de carga intravenosa.
- •
- Pacientes que cambian del uso intravenoso al uso subcutáneo, administrar la primera dosis subcutánea en lugar de la siguiente dosis intravenosa programada.
Uso intravenoso para la profilaxis de la eGvHD (2.4)
- •
- Para pacientes de 6 años o mayores, administrar a una dosis de 10 mg/kg (dosis máxima de 1,000 mg) como una infusión de 60 minutos el día antes del trasplante, seguida de una dosis el día 5, 14 y 28 después del trasplante.
- •
- Para pacientes de 2 a menos de 6 años, administrar una dosis de 15 mg/kg como una infusión de 60 minutos el día antes del trasplante, seguida de una dosis de 12 mg/kg como una infusión de 60 minutos el día 5, 14 y 28 después del trasplante.
Instrucciones de preparación y administración (2.5, 2.6)
- •
- Administrar como una infusión intravenosa de 30 minutos para AR, AIJp y PsA en adultos (2.5).
- •
- Administrar como una infusión intravenosa de 60 minutos para la profilaxis de la eGvHD (2.5).
- •
- Consulte la Información de prescripción completa para obtener instrucciones de preparación y administración para infusión intravenosa y recomendaciones para uso subcutáneo (2.5, 2.6). Prepare ORENCIA utilizando solo la jeringa desechable sin silicona (2.5).
FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
Infusión intravenosa
- •
- Para inyección: 250 mg de polvo liofilizado en un vial de dosis única. (3)
Uso subcutáneo
CONTRAINDICACIONES
- Ninguna. (4)
ADVERTENCIAS Y PRECAUCIONES
- •
- El uso concomitante con un antagonista del TNF puede aumentar el riesgo de infecciones e infecciones graves. (5.1)
- •
- Se han producido hipersensibilidad y anafilaxia. (5.2)
- •
- Infecciones graves reportadas. Los pacientes con antecedentes de infecciones recurrentes o afecciones subyacentes que predisponen a infecciones pueden experimentar más infecciones. Suspenda si se desarrolla una infección grave. (5.3)
- •
- Realice pruebas de detección de infección latente por TB antes de iniciar el tratamiento. Los pacientes con resultados positivos deben ser tratados antes de iniciar ORENCIA. (5.3)
- •
- Realice pruebas de detección de hepatitis viral antes de iniciar ORENCIA. (5.3)
- •
- Actualice las vacunas antes de iniciar ORENCIA. Las vacunas vivas no deben administrarse simultáneamente ni dentro de los 3 meses posteriores a la interrupción. ORENCIA puede disminuir la eficacia de algunas vacunas. (5.4)
- •
- Los pacientes con EPOC pueden desarrollar reacciones adversas respiratorias con mayor frecuencia. (5.5)
- •
- Reactivación del citomegalovirus (CMV) y del virus de Epstein-Barr (VEB) en pacientes tratados para la profilaxis de la eGvHD. (5.7)
REACCIONES ADVERSAS
- •
- Los eventos adversos más comunes (≥10%) en la AR son dolor de cabeza, infección de las vías respiratorias superiores, nasofaringitis y náuseas. (6.1)
- •
- Las reacciones adversas más comunes (≥10%) en la profilaxis de la eGvHD son anemia, hipertensión, reactivación del CMV/infección por CMV, pirexia, neumonía, epistaxis, disminución de linfocitos CD4, hipermagnesemia y lesión renal aguda. (6.1)
Para informar REACCIONES ADVERSAS SOSPECHOSAS, comuníquese con Bristol-Myers Squibb al 1-800-721-5072 o con la FDA al 1-800-FDA-1088 o www.fda.gov/medwatch.
Consulte 17 para obtener INFORMACIÓN DE ASESORAMIENTO AL PACIENTE y el etiquetado del paciente aprobado por la FDA.
Revisado: 5/2024
Tabla de contenido
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDO*
1 INDICACIONES Y USO
1.1 Artritis Reumatoide en Adultos
1.2 Artritis Idiopática Juvenil Poliarticular
1.3 Artritis Psoriásica
1.4 Profilaxis de la Enfermedad de Injerto contra Huésped Aguda
1.5 Limitaciones de Uso
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Dosificación en Artritis Reumatoide en Adultos
2.2 Dosificación en Artritis Idiopática Juvenil Poliarticular
2.3 Dosificación en Artritis Psoriásica
2.4 Dosificación en la Profilaxis de la Enfermedad de Injerto contra Huésped Aguda en Adultos y Pacientes Pediátricos de 2 Años o Mayores
2.5 Instrucciones de Preparación y Administración para Infusión Intravenosa
2.6 Recomendaciones para la Administración Subcutánea
3 PRESENTACIONES Y CONCENTRACIONES
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Mayor Riesgo de Infección con el Uso Concomitante de Antagonistas del TNF, Otra Terapia Biológica para AR/APs o Inhibidores de JAK
5.2 Reacciones de Hipersensibilidad
5.3 Infecciones
5.4 Inmunizaciones
5.5 Mayor Riesgo de Reacciones Adversas Cuando se Usa en Pacientes con Enfermedad Pulmonar Obstructiva Crónica (EPOC)
5.6 Inmunosupresión
5.7 Reactivación del Citomegalovirus (CMV) y del Virus de Epstein-Barr (VEB) en la Profilaxis de la EICH después del Trasplante de Células Madre Hematopoyéticas (TCMH)
6 REACCIONES ADVERSAS
6.1 Experiencia en Ensayos Clínicos
6.2 Inmunogenicidad
6.3 Experiencia Postcomercialización
7 INTERACCIONES MEDICAMENTOSAS
7.1 Inmunosupresores
7.2 Prueba de Glucosa en Sangre
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.4 Uso Pediátrico
8.5 Uso Geriátrico
10 SOBREDOSIFICACIÓN
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de Acción
12.2 Farmacodinamia
12.3 Farmacocinética
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
13.2 Toxicología y/o Farmacología Animal
14 ESTUDIOS CLÍNICOS
14.1 Artritis Reumatoide en Adultos
14.2 Artritis Idiopática Juvenil Poliarticular
14.3 Artritis Psoriásica
14.4 Profilaxis de la Enfermedad de Injerto contra Huésped Aguda
16 CÓMO SE SUMINISTRA/ALMACENAMIENTO Y MANEJO
17 INFORMACIÓN PARA EL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información completa de prescripción no se enumeran.
1 INDICACIONES Y USO
1.1 Artritis Reumatoide en Adultos
ORENCIA® está indicado para el tratamiento de pacientes adultos con artritis reumatoide (AR) moderada a gravemente activa.
1.2 Artritis Idiopática Juvenil Poliarticular
ORENCIA está indicado para el tratamiento de pacientes de 2 años de edad o mayores con artritis idiopática juvenil poliarticular (AIJp) moderada a gravemente activa.
1.3 Artritis Psoriásica
ORENCIA está indicado para el tratamiento de pacientes de 2 años de edad o mayores con artritis psoriásica (APs) activa.
1.4 Profilaxis para la Enfermedad Injerto contra Huésped Aguda
ORENCIA está indicado para la profilaxis de la enfermedad injerto contra huésped aguda (EICHa), en combinación con un inhibidor de la calcineurina y metotrexato, en adultos y pacientes pediátricos de 2 años de edad o mayores sometidos a trasplante de células madre hematopoyéticas (TCMH) de un donante no emparentado compatible o con una discrepancia de 1 alelo.
1.5 Limitaciones de Uso
No se recomienda el uso concomitante de ORENCIA con otros inmunosupresores potentes [p. ej., fármacos antirreumáticos modificadores de la enfermedad biológicos (bDMARD), inhibidores de la Janus quinasa (JAK)].
2 DOSIS Y ADMINISTRACIÓN
2.1 Dosificación en adultos con artritis reumatoide
Para pacientes adultos con AR, administrar como infusión intravenosa o como inyección subcutánea. ORENCIA puede utilizarse como monoterapia o concomitantemente con fármacos antirreumáticos modificadores de la enfermedad (DMARD) distintos de los inhibidores de JAK o bDMARD (p. ej., antagonistas del TNF).
Dosificación intravenosa
Reconstituir el polvo liofilizado de ORENCIA y administrar después de la dilución [véase Dosificación y administración (2.5)] como una infusión intravenosa de 30 minutos utilizando la dosificación basada en el rango de peso recomendada en la Tabla 1. Después de la infusión intravenosa inicial, administrar como infusión intravenosa a las 2 y 4 semanas y cada 4 semanas a partir de entonces.
| Peso corporal del paciente adulto | Dosis | Número de vialesa |
|---|---|---|
| a Cada vial proporciona 250 mg de abatacept para administración. | ||
|
Menos de 60 kg |
500 mg |
2 |
|
60 a 100 kg |
750 mg |
3 |
|
Más de 100 kg |
1000 mg |
4 |
Dosificación subcutánea
Antes de la primera dosis subcutánea, se puede administrar una dosis de carga opcional como una sola infusión intravenosa (según las categorías de peso corporal en la Tabla 1). Si se utiliza una dosis de carga intravenosa, administrar la primera inyección subcutánea dentro de un día de la infusión. Administrar ORENCIA 125 mg en jeringas precargadas o en el autoinyector ORENCIA ClickJect™ mediante inyección subcutánea una vez por semana [véase Dosificación y administración (2.6)].
Para los pacientes que cambian de la terapia intravenosa con ORENCIA a la administración subcutánea, administrar la primera dosis subcutánea en lugar de la siguiente dosis intravenosa programada.
2.2 Dosificación en la artritis idiopática juvenil poliarticular
Para pacientes pediátricos con AIJp, administrar ORENCIA como infusión intravenosa (solo pacientes de 6 años de edad o mayores) o como inyección subcutánea (solo pacientes de 2 años de edad o mayores) [véase Uso en poblaciones específicas (8.4)]. ORENCIA puede utilizarse como monoterapia o concomitantemente con metotrexato.
Dosificación intravenosa
Administrar ORENCIA como una infusión intravenosa de 30 minutos basada en el peso corporal [véase Dosificación y administración (2.5)]:
- •
- Para un peso corporal inferior a 75 kg, administrar una dosis de 10 mg/kg.
- •
- Para un peso corporal de 75 kg o superior, administrar según las recomendaciones de la Tabla 1 (seguir el régimen de dosificación intravenosa para adultos), sin exceder una dosis máxima de 1000 mg.
Después de la infusión intravenosa inicial, administrar infusiones a las 2 y 4 semanas y cada 4 semanas a partir de entonces. Desechar inmediatamente cualquier porción no utilizada en los viales.
Dosificación subcutánea
Administrar ORENCIA para inyección subcutánea, sin dosis de carga intravenosa, utilizando la dosificación basada en el rango de peso según lo recomendado en la Tabla 2 [véase Dosificación y administración (2.6)]. Posteriormente administrar una vez por semana.
| Peso corporal del paciente pediátrico | Dosis (una vez por semana) |
|---|---|
|
10 a menos de 25 kg |
50 mg |
|
25 a menos de 50 kg |
87,5 mg |
|
50 kg o más |
125 mg |
Los pacientes con pJIA pueden autoinyectarse ORENCIA o el cuidador del paciente puede administrar ORENCIA si tanto el profesional sanitario como el padre/tutor legal determinan que es apropiado. No se ha probado la capacidad de los pacientes pediátricos para autoinyectarse con el autoinyector.
2.3 Dosificación en la artritis psoriásica
Pacientes adultos
Para pacientes adultos con artritis psoriásica, administrar como infusión intravenosa o inyección subcutánea.
ORENCIA puede utilizarse con o sin DMARDs no biológicos.
Dosificación intravenosa
Administrar ORENCIA como una infusión intravenosa de 30 minutos utilizando la dosificación basada en el rango de peso especificada en la Tabla 1. Después de la administración intravenosa inicial, administrar una infusión intravenosa a las 2 y 4 semanas y cada 4 semanas a partir de entonces.
Dosificación subcutánea
Administrar 125 mg de ORENCIA por vía subcutánea una vez por semana (no se necesita dosis de carga intravenosa) [ver Dosificación y administración (2.6)].
Para los pacientes que cambian de infusiones intravenosas de ORENCIA a administración subcutánea, administrar la primera dosis subcutánea en lugar de la siguiente dosis intravenosa programada.
Pacientes pediátricos
Administrar ORENCIA como inyección subcutánea en pacientes pediátricos de 2 años de edad o mayores con artritis psoriásica [ver Uso en poblaciones específicas (8.4)]. ORENCIA puede utilizarse como monoterapia o concomitantemente con metotrexato. La administración intravenosa no está aprobada para pacientes pediátricos con artritis psoriásica.
Dosificación subcutánea
Administrar ORENCIA para inyección subcutánea semanalmente, utilizando la dosificación basada en el rango de peso como se recomienda en la Tabla 3 [ver Dosificación y administración (2.6)].
- Tabla 3:Dosis de ORENCIA para administración subcutánea en pacientes de 2 años de edad o mayores con artritis psoriásica
|
Peso corporal del paciente pediátrico |
Dosis (una vez por semana) |
||
|
10 a menos de 25 kg |
50 mg |
||
|
25 a menos de 50 kg |
87,5 mg |
||
|
50 kg o más |
125 mg |
Los pacientes pediátricos con artritis psoriásica pueden autoinyectarse ORENCIA o el cuidador del paciente puede administrar ORENCIA si tanto el profesional sanitario como el padre/tutor legal determinan que es apropiado. No se ha probado la capacidad de los pacientes pediátricos para autoinyectarse con el autoinyector.
2.4 Dosificación en la profilaxis de la enfermedad injerto contra huésped aguda en adultos y pacientes pediátricos de 2 años o más
Tratamiento profiláctico antiviral
Antes de administrar ORENCIA, administre el tratamiento profiláctico antiviral recomendado para la reactivación del virus de Epstein-Barr (VEB) y continúe durante seis meses después del TCH. Además, considere antivirales profilácticos para la infección/reactivación por citomegalovirus (CMV) durante el tratamiento y durante seis meses después del TCH [véase Advertencias y precauciones (5.7)].
Régimen de dosificación intravenosa
Para pacientes de 6 años o más, administre ORENCIA 10 mg/kg (dosis máxima de 1000 mg) como infusión intravenosa durante 60 minutos el día anterior al trasplante (día -1), seguido de la administración en los días 5, 14 y 28 después del trasplante.
Para pacientes de 2 a menos de 6 años, administre ORENCIA 15 mg/kg como infusión intravenosa durante 60 minutos el día anterior al trasplante (día -1), seguido de 12 mg/kg como infusión intravenosa durante 60 minutos en los días 5, 14 y 28 después del trasplante.
2.5 Instrucciones de preparación y administración para infusión intravenosa
Calcule la dosis de ORENCIA, el volumen total de solución reconstituida requerido y el número de viales de ORENCIA necesarios. Para una dosis completa, puede ser necesaria una cantidad inferior al contenido completo de un vial o más de un vial. Utilizando una técnica aséptica, reconstituya, diluya y luego administre ORENCIA de la siguiente manera:
Reconstitución
- 1)
- Utilice el vial solo si hay vacío.
- 2)
- Reconstituya cada vial de polvo liofilizado ORENCIA suministrado (cada vial suministra 250 mg de abatacept) con 10 mL de Agua estéril para inyección, USP (dirija el chorro hacia la pared interior del vial) para obtener una concentración de 25 mg/mL. Utilice únicamente la jeringa sin silicona proporcionada con una aguja de calibre 18 a 21:
- a.
- Si el polvo liofilizado ORENCIA se reconstituye accidentalmente con una jeringa siliconada, la solución puede desarrollar algunas partículas traslúcidas (deseche cualquier solución preparada con jeringas siliconadas).
- b.
- Si la jeringa desechable sin silicona se cae o se contamina, utilice una nueva jeringa desechable sin silicona. Para obtener nuevas jeringas sin silicona, póngase en contacto con Bristol-Myers Squibb en el 1-800-ORENCIA.
- 3)
- Gire suavemente el vial para minimizar la formación de espuma, hasta que el contenido esté completamente disuelto. No agite. Evite la agitación prolongada o vigorosa.
- 4)
- Una vez disuelto completamente el polvo liofilizado, ventile el vial con una aguja para disipar cualquier espuma que pueda estar presente.
- 5)
- Inspeccione visualmente la solución reconstituida (la solución debe ser transparente e incolora a amarillo pálido). No utilice si hay partículas opacas, decoloración u otras partículas extrañas presentes.
- 6)
- Repita los pasos 2) a 5) si se necesitan dos, tres o cuatro viales para una dosis (véase la Tabla 1).
Dilución
- 7)
- Debe diluir aún más la solución reconstituida de ORENCIA a 100 mL de la siguiente manera:
- a.
- De una bolsa o frasco de infusión de 100 mL de inyección de cloruro de sodio al 0,9%, USP, extraiga un volumen igual al volumen de la solución reconstituida de ORENCIA necesaria para la dosis del paciente.
- b.
- Añada lentamente la(s) solución(es) reconstituida(s) de ORENCIA a la bolsa o frasco utilizando la jeringa desechable sin silicona proporcionada con cada vial.
- c.
- Mezcle suavemente. No agite la bolsa o el frasco. La concentración final de abatacept en la bolsa o el frasco dependerá de la cantidad de abatacept añadida, pero no será superior a 10 mg/mL. Deseche inmediatamente cualquier parte no utilizada en el vial de ORENCIA.
Administración
- 8)
- Antes de la administración, inspeccione visualmente la solución diluida de ORENCIA para detectar la presencia de partículas y decoloración. Deseche la solución diluida si observa alguna partícula o decoloración.
- 9)
- Utilizando un equipo de infusión y un filtro estéril, no pirógeno, de baja unión a proteínas (tamaño de poro de 0,2 μm a 1,2 μm), administre toda la solución diluida de ORENCIA durante:
- •
- 30 minutos para AR, AJIA y adultos con APs
- •
- 60 minutos para la profilaxis de la enfermedad injerto contra huésped aguda
- 10)
- Debe completar la infusión de la solución diluida de ORENCIA en las 24 horas siguientes a la reconstitución de los viales de ORENCIA.
No infunda ORENCIA de forma concomitante en la misma vía intravenosa con otros agentes. No se han realizado estudios de compatibilidad física o bioquímica para evaluar la coadministración de ORENCIA con otros fármacos.
Almacenamiento de la solución diluida de ORENCIA
Puede almacenar la solución diluida de ORENCIA a temperatura ambiente o refrigerada a 2ºC a 8ºC (36ºF a 46ºF) hasta 24 horas antes de su uso. Deseche la solución diluida si no se administra en un plazo de 24 horas.
2.6 Recomendaciones para la administración subcutánea
Las jeringas precargadas de ORENCIA y los autoinyectores ClickJect de ORENCIA están destinados a:
- •
- Solo para uso subcutáneo y no está indicado para infusión intravenosa.
- •
- Usar bajo la supervisión de un profesional sanitario.
Después de la capacitación adecuada en la técnica de inyección subcutánea, un paciente o el cuidador del paciente puede administrar una inyección subcutánea de ORENCIA (autoinyector ClickJect o jeringa precargada) si un profesional sanitario determina que es apropiado. Instruya a los pacientes y/o cuidadores a seguir las instrucciones proporcionadas en las Instrucciones de Uso para obtener detalles adicionales sobre la administración. Instrúyalos específicamente a inyectar la cantidad total (que proporciona la dosis adecuada de ORENCIA), a rotar los sitios de inyección y a evitar las inyecciones en áreas donde la piel esté sensible, magullada, roja o dura.
Inspeccione visualmente la presencia de partículas y decoloración antes de la administración. No use jeringas precargadas de ORENCIA o autoinyectores ClickJect de ORENCIA que presenten partículas o decoloración. ORENCIA debe ser transparente a ligeramente opalescente e incolora a amarillo pálido.
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
- •
- Infusion intravenosa
- Para inyección: 250 mg de polvo liofilizado blanco en un vial de dosis única [ver Posología y administración (2.1, 2.2, 2.3, 2.5)].
- •
- Uso subcutáneo
- Inyección: 50 mg/0.4 mL, 87.5 mg/0.7 mL y 125 mg/mL de una solución transparente a ligeramente opalescente, incolora a amarillo pálido en una jeringa precargada de dosis única.
- Inyección: 125 mg/mL de una solución transparente a ligeramente opalescente, incolora a amarillo pálido en un autoinyector ClickJect precargado de dosis única.
4 CONTRAINDICACIONES
Ninguna.
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Mayor Riesgo de Infección con el Uso Concomitante de Antagonistas del TNF, Otra Terapia Biológica para AR/PsA o Inhibidores de JAK
En ensayos clínicos controlados en pacientes adultos con AR, los pacientes que recibieron terapia concomitante con ORENCIA intravenoso y un antagonista del TNF experimentaron más infecciones (63% vs. 43%) e infecciones graves (4.4% vs. 0.8%) en comparación con los pacientes tratados solo con antagonistas del TNF [ver Reacciones adversas (6.1)]. Estos ensayos no demostraron una mejora importante de la eficacia con la administración concomitante de ORENCIA con antagonistas del TNF; por lo tanto, no se recomienda la terapia concomitante con ORENCIA y un antagonista del TNF. Durante la transición de la terapia con antagonistas del TNF a la terapia con ORENCIA, los pacientes deben ser monitoreados para detectar signos de infección. Además, no se recomienda el uso concomitante de ORENCIA con otra terapia biológica para AR/PsA o inhibidores de JAK.
5.2 Reacciones de Hipersensibilidad
En ensayos clínicos de 2688 pacientes adultos con AR tratados con ORENCIA intravenoso, hubo dos casos (<0.1%) de reacciones de anafilaxia. Otras reacciones potencialmente asociadas con hipersensibilidad a los medicamentos, como hipotensión, urticaria y disnea, ocurrieron en menos del 0.9% de los pacientes tratados con ORENCIA. De los 190 pacientes tratados con ORENCIA en ensayos clínicos de ARJIp, hubo un caso de reacción de hipersensibilidad (0.5%) [ver Reacciones adversas (6.1)].
En la experiencia postcomercialización, se han notificado anafilaxia fatal después de la primera infusión de ORENCIA y casos potencialmente mortales de angioedema. El angioedema se ha producido tan pronto como después de la primera dosis de ORENCIA, pero también se ha producido con dosis posteriores. Las reacciones de angioedema se han producido en cuestión de horas después de la administración y, en algunos casos, han tenido un inicio tardío (es decir, días).
Las medidas de apoyo médico apropiadas para el tratamiento de las reacciones de hipersensibilidad deben estar disponibles para su uso inmediato en caso de reacción. Si se produce una reacción anafiláctica u otra reacción alérgica grave, la administración de ORENCIA intravenoso o subcutáneo debe interrumpirse inmediatamente con la terapia adecuada instituida, y el uso de ORENCIA debe suspenderse permanentemente.
5.3 Infecciones
Se han notificado infecciones graves, incluida la sepsis y la neumonía, en pacientes que recibieron ORENCIA (las infecciones graves se notificaron en el 3% y el 1.9% de los pacientes con AR tratados con ORENCIA intravenoso y placebo, respectivamente) [ver Reacciones adversas (6.1)]. Algunas de estas infecciones han sido mortales. Muchas de las infecciones graves se han producido en pacientes con terapia inmunosupresora concomitante que, además de su enfermedad subyacente, podría predisponerlos aún más a la infección. Se ha observado una mayor tasa de infecciones graves en pacientes adultos con AR tratados con antagonistas del TNF y ORENCIA concomitantes en comparación con aquellos tratados con ORENCIA solo [ver Advertencias y precauciones (5.1)].
Los profesionales de la salud deben tener precaución al considerar el uso de ORENCIA en pacientes con antecedentes de infecciones recurrentes, afecciones subyacentes que puedan predisponerlos a infecciones o infecciones crónicas, latentes o localizadas. Los pacientes que desarrollan una nueva infección mientras se someten a tratamiento con ORENCIA deben ser monitoreados de cerca. La administración de ORENCIA debe interrumpirse si un paciente desarrolla una infección grave.
Antes de iniciar ORENCIA, los pacientes deben ser examinados para detectar infección latente por tuberculosis (TB) de acuerdo con las pautas actuales de TB. ORENCIA no se ha estudiado en pacientes con una prueba de TB positiva, y se desconoce la seguridad de ORENCIA en individuos con infección latente por TB. Los pacientes que den positivo en la prueba de detección de TB deben ser tratados según la práctica médica estándar antes de la terapia con ORENCIA.
Las terapias antirreumáticas se han asociado con la reactivación de la hepatitis B. Por lo tanto, se debe realizar una prueba de detección de hepatitis viral de acuerdo con las pautas publicadas antes de comenzar el tratamiento con ORENCIA. En estudios clínicos con ORENCIA, los pacientes que dieron positivo en la prueba de detección de hepatitis fueron excluidos del estudio.
5.4 Inmunizaciones
Antes de iniciar ORENCIA en pacientes pediátricos y adultos, actualice las vacunas de acuerdo con las pautas actuales de vacunación. Los pacientes tratados con ORENCIA pueden recibir vacunas no vivas actuales. Las vacunas vivas no deben administrarse concomitantemente con ORENCIA o dentro de los 3 meses posteriores a la interrupción. No hay datos disponibles sobre la transmisión secundaria de la infección de personas que reciben vacunas vivas a pacientes que reciben ORENCIA. Además, existen consideraciones clínicas para administrar vacunas vivas a bebés que estuvieron expuestos a ORENCIA mientras estaban en el útero [ver Uso en poblaciones específicas (8.1)]. Debido a su mecanismo de acción, ORENCIA puede reducir la eficacia de algunas inmunizaciones.
5.5 Mayor Riesgo de Reacciones Adversas Cuando se Usa en Pacientes con Enfermedad Pulmonar Obstructiva Crónica (EPOC)
En el Estudio V, los pacientes adultos con EPOC tratados con ORENCIA para la AR desarrollaron reacciones adversas con más frecuencia que los tratados con placebo, incluidas exacerbaciones de la EPOC, tos, roncus y disnea. Un mayor porcentaje de pacientes tratados con ORENCIA desarrolló un evento adverso grave en comparación con los pacientes tratados con placebo (27% vs 6%) [ver Estudios clínicos (14.1) y Reacciones adversas (6.1)]. El uso de ORENCIA en pacientes con EPOC debe realizarse con precaución y estos pacientes deben ser monitoreados para detectar el empeoramiento de su estado respiratorio.
5.6 Inmunosupresión
Existe la posibilidad de que los fármacos que inhiben la activación de los linfocitos T, incluido ORENCIA, afecten las defensas del huésped contra infecciones y neoplasias, ya que los linfocitos T median las respuestas inmunitarias celulares. En los ensayos clínicos en pacientes adultos con AR, se observó una mayor tasa de infecciones en los pacientes tratados con ORENCIA en comparación con los pacientes tratados con placebo [ver Advertencias y precauciones (5.3) y Reacciones adversas (6.1)]. No se comprende completamente el impacto del tratamiento con ORENCIA en el desarrollo y el curso de las neoplasias [ver Reacciones adversas (6.1)]. Se han notificado neoplasias, incluido cáncer de piel, en pacientes que recibieron ORENCIA [ver Reacciones adversas (6.3)]. Se recomiendan exámenes periódicos de la piel para todos los pacientes tratados con ORENCIA, particularmente aquellos con factores de riesgo de cáncer de piel.
5.7 Reactivación del citomegalovirus (CMV) y del virus de Epstein-Barr (VEB) en la profilaxis de la enfermedad injerto contra huésped (EICH) después del trasplante de células madre hematopoyéticas (TCMH)
Se produjo un trastorno linfoproliferativo postrasplante (TLP) en pacientes que recibieron ORENCIA para la profilaxis de la EICH durante el TCMH no relacionado. De 116 pacientes que recibieron ORENCIA, 4 pacientes (3,4%) experimentaron TLP. Todos los eventos de TLP se asociaron con la infección por el virus de Epstein-Barr (VEB). Tres de los cuatro pacientes fueron serológicos positivos para VEB al inicio del estudio; un paciente tuvo una serología basal negativa para VEB con serología de VEB del donante desconocida. Tres de los 4 pacientes interrumpieron la profilaxis con aciclovir a los 30 días postrasplante. El rango de tiempo hasta el inicio de los eventos fue de 49 a 89 días postrasplante. Controle a los pacientes para detectar la reactivación del VEB de acuerdo con las prácticas institucionales. Proporcione profilaxis para la infección por VEB durante 6 meses postrasplante para prevenir el TLP asociado con VEB [ver Posología y administración (2.4)].
Se produjo una enfermedad invasiva por citomegalovirus (CMV) en pacientes que recibieron ORENCIA para la profilaxis de la EICH durante el TCMH no relacionado. De 116 pacientes que recibieron ORENCIA, el 7% experimentó enfermedades invasivas por CMV hasta el día 225 postrasplante. Todos los pacientes que experimentaron enfermedad invasiva por CMV fueron serológicos positivos para CMV al inicio del estudio. El tiempo medio hasta el inicio del evento fue de 91 días postrasplante. Las enfermedades invasivas por CMV afectaron predominantemente el tracto gastrointestinal [ver Reacciones adversas (6.1)].
Controle a los pacientes para detectar infección/reactivación por CMV durante 6 meses postrasplante, independientemente de los resultados de la serología pre-trasplante de CMV del donante y del receptor. Considere la profilaxis para la infección/reactivación por CMV [ver Posología y administración (2.4)].
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas clínicamente significativas se describen en otras partes del etiquetado:
- •
- Aumento del riesgo de infección con el uso concomitante con antagonistas del TNF, otra terapia biológica para AR/PsA o inhibidores de JAK [ver Advertencias y precauciones (5.1)]
- •
- Reacciones de hipersensibilidad [ver Advertencias y precauciones (5.2)]
- •
- Infecciones [ver Advertencias y precauciones (5.3)]
- •
- Aumento del riesgo de reacciones adversas cuando se usa en pacientes con enfermedad pulmonar obstructiva crónica (EPOC) [ver Advertencias y precauciones (5.5)]
- •
- Inmunosupresión [ver Advertencias y precauciones (5.6)]
- •
- Reactivación del citomegalovirus (CMV) y del virus de Epstein-Barr (VEB) en la profilaxis de la enfermedad de injerto contra huésped (EICH) después del trasplante de células madre hematopoyéticas (TCMH) [ver Advertencias y precauciones (5.7)]
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se realizan en condiciones ampliamente variables y controladas, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no se pueden comparar directamente con las tasas en los ensayos clínicos de otro fármaco y pueden no predecir las tasas observadas en una población de pacientes más amplia en la práctica clínica.
Reacciones adversas en pacientes adultos con AR
Reacciones adversas en pacientes adultos con AR tratados con ORENCIA intravenoso
Los datos de los estudios controlados con placebo que se describen aquí reflejan la exposición a ORENCIA administrado por vía intravenosa en pacientes con AR activa (1955 pacientes con ORENCIA, 989 con placebo) (Estudios I a VI) [ver Estudios clínicos (14.1)]. Los estudios tuvieron un período doble ciego controlado con placebo de 6 meses (258 pacientes con ORENCIA, 133 con placebo) o 1 año (1697 pacientes con ORENCIA, 856 con placebo). Un subgrupo de estos pacientes recibió terapia concomitante con DMARD biológicos, como un antagonista del TNF (204 pacientes con ORENCIA, 134 con placebo). No se recomienda el uso concomitante de ORENCIA con un antagonista del TNF [ver Indicaciones y uso (1.5)]. La mayoría de los pacientes en los estudios clínicos de AR recibieron uno o más de los siguientes medicamentos concomitantes con ORENCIA: metotrexato, fármacos antiinflamatorios no esteroideos (AINE), corticosteroides, antagonista del TNF, azatioprina, cloroquina, oro, hidroxicloroquina, leflunomida, sulfasalazina y anakinra.
Las reacciones adversas más graves fueron infecciones graves y neoplasias malignas. Los eventos adversos más comúnmente notificados (que ocurrieron en ≥10% de los pacientes tratados con ORENCIA) fueron cefalea, infección del tracto respiratorio superior, nasofaringitis y náuseas.
Las reacciones adversas que con mayor frecuencia dieron lugar a una intervención clínica (interrupción o suspensión de ORENCIA) se debieron a una infección. Las infecciones más frecuentemente notificadas que provocaron la interrupción de la dosis fueron infección del tracto respiratorio superior (1%), bronquitis (0,7%) y herpes zóster (0,7%). Las infecciones más frecuentes que provocaron la suspensión fueron neumonía (0,2%), infección localizada (0,2%) y bronquitis (0,1%).
Reacciones adversas más comunes en pacientes adultos con AR tratados con ORENCIA intravenoso
Las reacciones adversas que ocurrieron en 3% o más de los pacientes y al menos 1% más frecuentemente en pacientes tratados con ORENCIA (intravenoso) durante los estudios de AR controlados con placebo se resumen en la Tabla 4.
| Intravenoso ORENCIA (n=1955)a |
Placebo (n=989)b |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| * Ocurrió en ≥3% de los pacientes y >1% más frecuentemente en pacientes tratados con ORENCIA. a Incluye 204 pacientes con DMARD biológicos concomitantes (adalimumab, anakinra, etanercept o infliximab). b Incluye 134 pacientes con DMARD biológicos concomitantes (adalimumab, anakinra, etanercept o infliximab). |
||||||||||||
|
Cefalea |
18% |
13% |
||||||||||
|
Nasofaringitis |
12% |
9% |
||||||||||
|
Mareo |
9% |
7% |
||||||||||
|
Tos |
8% |
7% |
||||||||||
|
Dolor de espalda |
7% |
6% |
||||||||||
|
Hipertensión |
7% |
4% |
||||||||||
|
Dispepsia |
6% |
4% |
||||||||||
|
Infección del tracto urinario |
6% |
5% |
|
Erupción cutánea |
4% |
3% |
|
Dolor en las extremidades |
3% |
2% |
Infecciones en pacientes adultos con AR tratados con ORENCIA intravenoso
En los ensayos controlados con placebo en pacientes con AR, se notificaron infecciones en el 54% de los pacientes tratados con ORENCIA intravenoso y en el 48% de los pacientes tratados con placebo. Las infecciones más comúnmente notificadas (reportadas en el 5%-13% de los pacientes) fueron infección del tracto respiratorio superior, rinofaringitis, sinusitis, infección del tracto urinario, gripe y bronquitis. Otras infecciones notificadas en menos del 5% de los pacientes con una frecuencia más alta (>0.5%) con ORENCIA en comparación con placebo fueron rinitis, herpes simple y neumonía [ver Advertencias y Precauciones (5.3)].
Se notificaron infecciones graves en el 3% de los pacientes tratados con ORENCIA y en el 1.9% de los pacientes tratados con placebo. Las infecciones graves más comunes (0.2%-0.5%) notificadas con ORENCIA fueron neumonía, celulitis, infección del tracto urinario, bronquitis, diverticulitis y pielonefritis aguda [ver Advertencias y Precauciones (5.3)].
Malignidades en pacientes adultos con AR tratados con ORENCIA intravenoso
En las porciones controladas con placebo de los ensayos clínicos (1955 pacientes tratados por AR con ORENCIA durante una mediana de 12 meses), las frecuencias generales de malignidades fueron similares en los pacientes tratados con ORENCIA y con placebo (1.3% y 1.1%, respectivamente). Sin embargo, se observaron más casos de cáncer de pulmón en los pacientes tratados con ORENCIA (4 casos, 0.2%) que en los pacientes tratados con placebo (0 casos, 0%). En los ensayos clínicos acumulativos de ORENCIA intravenoso en pacientes con AR (controlados con placebo y no controlados, de etiqueta abierta) se observaron un total de 8 casos de cáncer de pulmón (0.21 casos por 100 pacientes-año) y 4 linfomas (0.10 casos por 100 pacientes-año) en 2688 pacientes (3827 pacientes-año). La tasa observada para el linfoma es aproximadamente 3.5 veces mayor que la esperada en una población general emparejada por edad y género basada en la Base de Datos de Vigilancia, Epidemiología y Resultados Finales del Instituto Nacional del Cáncer. Los pacientes con AR, particularmente aquellos con enfermedad altamente activa, tienen un mayor riesgo de desarrollar linfoma. Otras malignidades incluyeron cáncer de piel, mama, conducto biliar, vejiga, cuello uterino, endometrio, linfoma, melanoma, síndrome mielodisplásico, ovario, próstata, riñón, tiroides y útero [ver Advertencias y Precauciones (5.6)]. El papel potencial de ORENCIA en el desarrollo de malignidades en humanos es desconocido.
Reacciones relacionadas con la infusión y reacciones de hipersensibilidad en pacientes adultos con AR tratados con ORENCIA intravenoso
Los eventos agudos relacionados con la infusión (reacciones adversas que ocurren dentro de la primera hora del inicio de la infusión) en los Estudios III, IV y V [ver Estudios Clínicos (14.1)] fueron más comunes en los pacientes tratados con ORENCIA que en los pacientes tratados con placebo (9% para ORENCIA, 6% para placebo). Los eventos más frecuentemente reportados (1%-2%) fueron mareos, dolor de cabeza y hipertensión.
Los eventos agudos relacionados con la infusión que se notificaron en >0.1% y ≤1% de los pacientes tratados con ORENCIA incluyeron síntomas cardiopulmonares, como hipotensión, aumento de la presión arterial y disnea; otros síntomas incluyeron náuseas, rubor, urticaria, tos, hipersensibilidad, prurito, erupción cutánea y sibilancias. La mayoría de estas reacciones fueron leves (68%) a moderadas (28%). Menos del 1% de los pacientes tratados con ORENCIA suspendieron el tratamiento debido a un evento agudo relacionado con la infusión. En los ensayos controlados, 6 pacientes tratados con ORENCIA en comparación con 2 pacientes tratados con placebo suspendieron el tratamiento del estudio debido a eventos agudos relacionados con la infusión.
En los ensayos clínicos de 2688 pacientes adultos con AR tratados con ORENCIA intravenoso, hubo dos casos (<0.1%) de anafilaxia. Otras reacciones potencialmente asociadas con hipersensibilidad a la droga, como hipotensión, urticaria y disnea, cada una ocurrió en menos del 0.9% de los pacientes tratados con ORENCIA y generalmente ocurrieron dentro de las 24 horas de la infusión de ORENCIA. Deben estar disponibles medidas de apoyo médico apropiadas para el tratamiento de las reacciones de hipersensibilidad para su uso inmediato en caso de una reacción [ver Advertencias y Precauciones (5.2)].
Reacciones adversas en pacientes con EPOC tratados por AR con ORENCIA intravenoso
En el Estudio V [ver Estudios Clínicos (14.1)], hubo 37 y 17 pacientes con enfermedad pulmonar obstructiva crónica (EPOC) que fueron tratados por AR con ORENCIA y placebo, respectivamente. Los pacientes con EPOC tratados con ORENCIA para AR desarrollaron eventos adversos con más frecuencia que aquellos tratados con placebo (97% frente a 88%, respectivamente). Los trastornos respiratorios ocurrieron con más frecuencia en los pacientes tratados con ORENCIA en comparación con los pacientes tratados con placebo (43% frente a 24%, respectivamente), incluyendo exacerbación de la EPOC, tos, ronquidos y disnea. Un mayor porcentaje de pacientes tratados con ORENCIA desarrolló un evento adverso grave en comparación con los pacientes tratados con placebo (27% frente a 6%), incluyendo exacerbación de la EPOC [3 de 37 pacientes (8%)] y neumonía [1 de 37 pacientes (3%)] [ver Advertencias y Precauciones (5.5)].
Reacciones adversas en pacientes naïve a metotrexato con AR tratados con ORENCIA intravenoso
El Estudio VI fue un ensayo clínico controlado activo en pacientes naïve a metotrexato [ver Estudios Clínicos (14.1)]. La experiencia de seguridad en estos pacientes fue consistente con la de los pacientes en los Estudios I-V.
Reacciones adversas en pacientes adultos con AR tratados con ORENCIA subcutánea o intravenosa
Los datos que se describen a continuación provienen del Estudio SC-1. El Estudio SC-1 fue un estudio de no inferioridad, aleatorizado, doble ciego, doble ficticio que comparó la seguridad de ORENCIA administrada por vía subcutánea o intravenosa en 1457 pacientes con AR que recibieron metotrexato como tratamiento de fondo y experimentaron una respuesta inadecuada al metotrexato (MTX-IR) [véase Estudios clínicos (14.1)]. El perfil de reacciones adversas en los pacientes tratados con ORENCIA subcutánea fue similar al perfil de reacciones adversas en los pacientes tratados con ORENCIA intravenosa y consistente con la administración de ORENCIA intravenosa en los Estudios I-VI.
Reacciones en el lugar de inyección en pacientes adultos con AR tratados con ORENCIA subcutánea
La frecuencia general de reacciones en el lugar de inyección en el Estudio SC-1 fue del 2,6 % (19/736) y del 2,5 % (18/721) para el grupo de ORENCIA subcutánea y el grupo de placebo subcutáneo (que recibió ORENCIA intravenosa), respectivamente [véase Estudios clínicos (14.1)]. Todas estas reacciones en el lugar de inyección (incluidos hematoma, prurito y eritema) fueron de leves (83 %) a moderadas (17 %) en gravedad, y ninguna requirió la interrupción del medicamento.
Reacciones adversas en pacientes adultos con PsA
Reacciones adversas en pacientes adultos con PsA tratados con ORENCIA intravenosa o subcutánea
La seguridad de ORENCIA se evaluó en 594 pacientes con PsA (341 pacientes con ORENCIA y 253 pacientes con placebo), en dos ensayos aleatorizados, doble ciego y controlados con placebo [véase Estudios clínicos (14.3)]. De los 341 pacientes que recibieron ORENCIA, 128 pacientes recibieron ORENCIA intravenosa (PsA-I) y 213 pacientes recibieron ORENCIA subcutánea (PsA-II). El perfil de seguridad fue comparable entre ORENCIA administrada por vía intravenosa en el Estudio PsA-I y ORENCIA administrada por vía subcutánea en el Estudio PsA-II y también consistente con el perfil de seguridad de ORENCIA en pacientes con AR [véase Advertencias y precauciones (5), Reacciones adversas (6.1)].
Reacciones adversas en pacientes con ARJp tratados con ORENCIA intravenosa
En general, la frecuencia y el tipo de acontecimientos adversos en pacientes pediátricos con ARJp tratados con ORENCIA intravenosa fueron similares a los observados en pacientes adultos con AR tratados con ORENCIA intravenosa [véase Advertencias y precauciones (5) y Reacciones adversas (6)].
El Estudio JIA-1 fue un estudio de tres partes que incluyó una extensión de etiqueta abierta que evaluó la seguridad de ORENCIA intravenosa en 190 pacientes pediátricos de 6 a 17 años de edad con ARJp. La frecuencia general de acontecimientos adversos en el período de 4 meses de introducción, de etiqueta abierta, del estudio fue del 70 %; las infecciones se produjeron con una frecuencia del 36 % [véase Estudios clínicos (14.2)]. Las infecciones más comunes fueron la infección del tracto respiratorio superior y la nasofaringitis. Las infecciones se resolvieron sin secuelas, y los tipos de infecciones fueron consistentes con los que se observan comúnmente en las poblaciones pediátricas ambulatorias. Otros eventos que ocurrieron con una prevalencia de al menos el 5 % fueron dolor de cabeza, náuseas, diarrea, tos, pirexia y dolor abdominal.
Se notificaron un total de 6 acontecimientos adversos graves [leucemia linfocítica aguda, quiste ovárico, infección por varicela, exacerbación de la enfermedad (2) y desgaste articular] durante los primeros 4 meses de tratamiento con ORENCIA intravenosa.
De los 190 pacientes pediátricos con ARJp tratados con ORENCIA intravenosa en ensayos clínicos, hubo un caso de reacción de hipersensibilidad (0,5 %). Durante los períodos A, B y C, las reacciones agudas relacionadas con la infusión se produjeron con una frecuencia del 4 %, 2 % y 3 %, respectivamente, y fueron consistentes con los tipos de eventos notificados en adultos.
Tras el tratamiento continuado en el período de extensión de etiqueta abierta, la frecuencia y el tipo de acontecimientos adversos fueron similares a los observados en pacientes adultos, excepto por un solo paciente diagnosticado con esclerosis múltiple mientras recibía tratamiento de etiqueta abierta.
Reacciones adversas en pacientes con ARJp tratados con ORENCIA subcutánea
El Estudio JIA-2 fue un estudio de etiqueta abierta con un período a corto plazo de 4 meses y un período de extensión a largo plazo que evaluó la seguridad de ORENCIA subcutánea en 205 pacientes pediátricos de 2 a 17 años de edad con ARJp. El perfil de reacciones adversas en pacientes con ARJp tratados con ORENCIA administrada por vía subcutánea en el Estudio JIA-2 fue consistente con el perfil de reacciones adversas en pacientes con ARJp tratados con el Estudio JIA-1 intravenoso.
No hubo casos notificados de reacciones de hipersensibilidad. Las reacciones locales en el lugar de inyección se produjeron con una frecuencia del 4,4 %.
Reacciones adversas en pacientes sometidos a trasplante de células madre hematopoyéticas (TCMH) de donante no relacionado con ORENCIA intravenoso
Los datos descritos aquí proceden de un estudio clínico de ORENCIA (GVHD-1) para la profilaxis de la aGVHD en pacientes de 6 años o más con neoplasias hematológicas sometidos a un TCMH de donante no relacionado, en el que todos los pacientes recibieron un inhibidor de la calcineurina y metotrexato como estándar de atención para la profilaxis de la aGVHD [véase Estudios clínicos (14.4)]. Se estudiaron dos cohortes a 10 mg/kg (dosis máxima de 1000 mg) como infusión intravenosa durante 60 minutos el día anterior al trasplante (día -1), seguida de la administración en los días 5, 14 y 28 después del trasplante:
1) Una cohorte de un solo brazo de pacientes tratados con ORENCIA (n=43) que se sometieron a un TCMH de 7 de 8 antígenos leucocitarios humanos (HLA) coincidentes de donantes no relacionados (cohorte de 7 de 8) y
2) Una cohorte aleatorizada compuesta por pacientes tratados con ORENCIA (n=73) y pacientes tratados con placebo (n=69) que se sometieron a un TCMH de 8 de 8 HLA coincidentes de donantes no relacionados (cohorte de 8 de 8).
De los 116 pacientes que recibieron ORENCIA, 27 (23%) tenían entre 6 y menos de 17 años [véase Uso en poblaciones específicas (8.4)].
A continuación se presenta la información de seguridad desde la fecha de la primera dosis de ORENCIA hasta el día 225 después del trasplante de este estudio. La incidencia de reacciones adversas se determinó en base a datos agrupados de pacientes tratados con ORENCIA de las 2 cohortes del estudio (n=116).
Las reacciones adversas graves notificadas en > 5% de los pacientes que recibieron ORENCIA en combinación con un inhibidor de la calcineurina y metotrexato incluyeron pirexia (20%), neumonía (8%), lesión renal aguda (7%), diarrea (6%), hipoxia (5%) y náuseas (5%).
La interrupción permanente de ORENCIA debido a una reacción adversa se produjo en dos pacientes (1,7%) debido a un caso de neumonía y una reacción alérgica.
Las reacciones adversas más frecuentes (≥10%) en los pacientes tratados con ORENCIA fueron anemia, hipertensión, reactivación/infección por CMV, pirexia, neumonía, epistaxis, disminución de los linfocitos CD4, hipermagnesemia y lesión renal aguda.
La Tabla 5 resume la frecuencia de las reacciones adversas notificadas en el estudio de ORENCIA en GVHD-1.
| Cohorte de 7 de 8 | Cohorte de 8 de 8 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Reacción adversa | ORENCIA (+Inhibidor de la calcineurina y MTX) (N=43) |
ORENCIA (+Inhibidor de la calcineurina y MTX) (N=73) |
Placebo (+Inhibidor de la calcineurina y MTX) (N=69) |
|||||||
|
Todos los grados (%) |
Grado 3 o 4 (%) |
Todos los grados (%) |
Grado 3 o 4 (%) |
Todos los grados (%) |
Grado 3 o 4 (%) |
|||||
|
Trastornos de la sangre y del sistema linfático |
||||||||||
|
Anemia |
56 |
56 |
69 |
69 |
57 |
57 |
||||
|
Disminución de linfocitos CD4 |
14 |
14 |
14 |
14 |
9 |
9 |
||||
|
Trastornos vasculares |
||||||||||
|
Hipertensión |
49 |
49 |
43 |
43 |
38 |
38 |
||||
|
Trastornos generales y condiciones en el sitio de administración |
||||||||||
|
Pirexia |
28 |
9 |
19 |
10 |
20 |
4 |
||||
|
Infecciones e infestaciones |
||||||||||
|
Reactivación/Infección por CMV |
26 |
26 |
32 |
32 |
22 |
22 |
||||
|
Neumonía |
19 |
19 |
12 |
12 |
10 |
9 |
||||
|
Trastornos respiratorios y mediastínicos |
||||||||||
|
Epistaxis |
12 |
12 |
16 |
16 |
10 |
10 |
||||
|
Trastornos renales y urinarios |
||||||||||
|
Insuficiencia renal aguda |
9 |
7 |
15 |
15 |
10 |
10 |
|
Trastornos del metabolismo y la nutrición |
||||||
|
Hipermagnesemia |
5 |
5 |
18 |
18 |
10 |
10 |
En el Estudio GVHD-1, las reacciones adversas clínicamente relevantes en <10% de los pacientes que recibieron ORENCIA en combinación con un inhibidor de la calcineurina y metotrexato incluyeron la reactivación del EBV.
6.2 Inmunogenicidad
Como con todas las proteínas terapéuticas, existe el potencial de inmunogenicidad. La detección de la formación de anticuerpos depende en gran medida de la sensibilidad y especificidad del ensayo. Además, la incidencia observada de positividad de anticuerpos (incluyendo anticuerpos neutralizantes) en un ensayo puede verse influenciada por varios factores, incluyendo la metodología del ensayo, el manejo de la muestra, el momento de la recolección de la muestra, los medicamentos concomitantes y la enfermedad subyacente. Por estas razones, la comparación de la incidencia de anticuerpos en los estudios descritos a continuación con la incidencia de anticuerpos en otros estudios o con otros productos de abatacept puede ser engañosa.
Inmunogenicidad en pacientes adultos con AR tratados con ORENCIA intravenosa
Los anticuerpos dirigidos contra la molécula completa de abatacept o a la porción CTLA-4 de abatacept se evaluaron mediante ensayos ELISA en pacientes con AR durante un máximo de 2 años después del tratamiento repetido con ORENCIA intravenosa. Treinta y cuatro de 1993 (2%) pacientes desarrollaron anticuerpos de unión a la molécula completa de abatacept o a la porción CTLA-4 de abatacept. Debido a que los niveles valle de abatacept pueden interferir con los resultados del ensayo, se realizó un análisis de subconjunto. En el análisis de subconjunto, 9 de 154 (6%) pacientes que habían suspendido el tratamiento intravenoso de ORENCIA durante más de 56 días desarrollaron anticuerpos. Las muestras con actividad de unión confirmada a CTLA-4 se evaluaron para la presencia de anticuerpos neutralizantes en un ensayo de reportero de luciferasa basado en células. Se demostró que 6 de 9 (67%) pacientes evaluables poseían anticuerpos neutralizantes. Sin embargo, el desarrollo de anticuerpos neutralizantes puede estar subreportado debido a la falta de sensibilidad del ensayo.
No se observó correlación entre el desarrollo de anticuerpos anti-abatacept y la respuesta clínica o los eventos adversos.
Inmunogenicidad en pacientes adultos con AR tratados con ORENCIA subcutánea o intravenosa
El Estudio SC-1 comparó la inmunogenicidad a abatacept después de la administración subcutánea o intravenosa de ORENCIA. La frecuencia de inmunogenicidad general a abatacept fue del 1% (8/725) y del 2% (16/710) para los grupos subcutáneo e intravenoso, respectivamente. La tasa es consistente con la experiencia previa, y no hubo correlación de la inmunogenicidad con los efectos en la farmacocinética, la seguridad o la eficacia.
Inmunogenicidad en pacientes adultos con AR tratados con ORENCIA subcutánea en monoterapia
El Estudio SC-2 se llevó a cabo para determinar el efecto del uso de monoterapia subcutánea de ORENCIA en la inmunogenicidad (sin una dosis de carga intravenosa) en 100 pacientes con AR, que no habían recibido previamente ORENCIA u otro CTLA4Ig. Los pacientes en este estudio recibieron ya sea ORENCIA subcutáneo más metotrexato (n = 51) o ORENCIA subcutáneo en monoterapia (n = 49). Ningún paciente en ninguno de los grupos desarrolló anticuerpos anti-abatacept después de 4 meses de tratamiento. La seguridad observada en este estudio fue consistente con la observada en los otros estudios subcutáneos.
Inmunogenicidad en pacientes adultos con AR después del tratamiento, la retirada y el reinicio de ORENCIA subcutánea
El Estudio SC-3 se llevó a cabo para investigar la inmunogenicidad en pacientes adultos con AR después del tratamiento, la retirada (tres meses) y el reinicio del tratamiento subcutáneo de ORENCIA (los pacientes fueron tratados concomitantemente con metotrexato). Ciento sesenta y siete pacientes fueron inscritos en el primer período de tratamiento de 3 meses y los respondedores (n = 120) fueron aleatorizados ya sea a ORENCIA subcutáneo o a placebo para el segundo período de 3 meses (período de retirada). Los pacientes de este período luego recibieron un tratamiento abierto con ORENCIA subcutáneo en el último período de 3 meses del estudio (período 3). Al final del período de retirada, 0/38 (0%) pacientes que continuaron recibiendo ORENCIA subcutáneo desarrollaron anticuerpos anti-abatacept en comparación con 7/73 (10%) de los pacientes a los que se les retiró ORENCIA subcutánea durante este período. La mitad de los pacientes que recibieron placebo subcutáneo durante el período de retirada recibió una única infusión intravenosa de ORENCIA al inicio del período 3 y la mitad recibió placebo intravenoso. Al final del período 3, cuando todos los pacientes nuevamente recibieron ORENCIA subcutánea, las tasas de inmunogenicidad fueron 1/38 (3%) en el grupo que recibió ORENCIA subcutáneo durante todo el estudio, y 2/73 (3%) en el grupo que recibió placebo durante el período de retirada. Al reanudar el tratamiento, no hubo reacciones por inyección y no hubo diferencias en la respuesta al tratamiento en los pacientes que fueron retirados del tratamiento subcutáneo durante hasta 3 meses en comparación con aquellos que permanecieron en el tratamiento subcutáneo (estos resultados ocurrieron en aquellos que recibieron o no recibieron una dosis de carga intravenosa). La seguridad observada en este estudio fue consistente con la observada en los otros estudios.
Inmunogenicidad en pacientes con pJIA tratados con ORENCIA intravenosa
Los anticuerpos dirigidos contra la molécula completa de abatacept o a la porción CTLA-4 de abatacept se evaluaron mediante ensayos ELISA en pacientes con pJIA después del tratamiento repetido con ORENCIA intravenosa durante todo el período de etiqueta abierta. Para los pacientes que fueron retirados del tratamiento durante hasta 6 meses durante el período de doble ciego, la tasa de formación de anticuerpos a la porción CTLA-4 de la molécula fue del 41% (22/54), mientras que para aquellos que permanecieron en el tratamiento la tasa fue del 13% (7/54). Veinte de estos pacientes tenían muestras que podían ser analizadas para anticuerpos con actividad neutralizante; de estos, 8 (40%) pacientes se demostró que poseían anticuerpos neutralizantes.
La presencia de anticuerpos fue generalmente transitoria y los títulos fueron bajos. La presencia de anticuerpos no se asoció con eventos adversos, cambios en la eficacia o un efecto en las concentraciones séricas de abatacept. Para los pacientes que fueron retirados de ORENCIA durante el período doble ciego hasta por 6 meses, no se observaron eventos graves relacionados con la infusión aguda tras la reiniciación del tratamiento con ORENCIA.
Inmunogenicidad en pacientes tratados para la profilaxis de aGVHD con ORENCIA intravenosa
Se evaluó la inmunogenicidad en pacientes sometidos a TCHC. En general, la incidencia de inmunogenicidad y los títulos de anticuerpos asociados fueron bajos a partir del régimen de ORENCIA intravenosa de 4 dosis utilizado en este estudio. De los 114 sujetos evaluables para inmunogenicidad en los grupos de ORENCIA, ninguno fue positivo durante el período de tratamiento con ORENCIA (Día -1 a Día 28 después del trasplante). Durante el período sin tratamiento (Día 29 y hasta el Día 180 después del trasplante); 6 de 91 sujetos evaluables para inmunogenicidad (6,6%) fueron positivos para CTLA4 y posiblemente Ig; 4 de los 6 sujetos positivos presentaron al menos una muestra positiva con actividad de neutralización. En este estudio, los sujetos con inmunogenicidad positiva solo tuvieron muestras positivas de ADA en el Día 180 (período sin tratamiento) y, por lo tanto, debido al momento de la respuesta, no se pudo determinar el impacto en la farmacocinética, la seguridad o la eficacia.
6.3 Experiencia postcomercialización
Se han notificado reacciones adversas durante el uso posterior a la aprobación de ORENCIA. Debido a que estas reacciones se notifican voluntariamente a partir de una población de tamaño incierto, no siempre es posible estimar su frecuencia de manera fiable o establecer una relación causal con ORENCIA. Sobre la base de la experiencia postcomercialización con ORENCIA, se han identificado las siguientes reacciones adversas:
- •
- Vasculitis (incluida la vasculitis cutánea y la vasculitis leucocitoclástica)
- •
- Psoriasis nueva o en empeoramiento
- •
- Cánceres de piel no melanoma (carcinoma basocelular y carcinoma de células escamosas)
- •
- Reacciones de angioedema [ver Advertencias y precauciones (5.2)]
Durante la experiencia postcomercialización con ORENCIA intravenosa, las reacciones a la infusión sistémica fueron similares a las observadas en la experiencia de los ensayos clínicos con ORENCIA intravenosa, con la excepción de un caso de anafilaxia mortal [ver Advertencias y precauciones (5.2)]. Se han producido notificaciones postcomercialización de reacciones a la inyección sistémica (p. ej., prurito, opresión de garganta, disnea) tras el uso de ORENCIA subcutánea.
7 INTERACCIONES MEDICAMENTOSAS
7.1 Inmunosupresores
La administración concomitante de un antagonista del TNF con ORENCIA se ha asociado con un mayor riesgo de infecciones graves y ninguna eficacia adicional significativa sobre el uso de los antagonistas del TNF solos. No se recomienda la terapia concomitante con ORENCIA y antagonistas del TNF [ver Advertencias y precauciones (5.1)].
No hay suficiente experiencia para evaluar la seguridad y eficacia de ORENCIA administrada concurrentemente con otra terapia biológica para la AR, como anakinra, u otra terapia biológica para la PsA, e inhibidores de JAK y, por lo tanto, no se recomienda dicho uso. [ver Advertencias y precauciones (5.1)].
7.2 Análisis de glucosa en sangre
Los productos farmacéuticos parenterales que contienen maltosa pueden interferir con las lecturas de los monitores de glucosa en sangre que utilizan tiras reactivas con glucosa deshidrogenasa pirroloquinolina quinona (GDH-PQQ). Los sistemas de monitorización de glucosa basados en GDH-PQQ pueden reaccionar con la maltosa presente en ORENCIA para administración intravenosa, lo que provoca lecturas falsamente elevadas de glucosa en sangre el día de la infusión. Cuando reciben ORENCIA intravenosa, se debe aconsejar a los pacientes que requieren monitorización de glucosa en sangre que consideren métodos que no reaccionan con la maltosa, como los basados en los métodos de prueba de glucosa deshidrogenasa nicotinamida adenina dinucleótido (GDH-NAD), glucosa oxidasa o glucosa hexocinasa.
ORENCIA para administración subcutánea no contiene maltosa; por lo tanto, los pacientes no necesitan modificar su monitorización de glucosa.
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de riesgos
Los datos sobre el uso de ORENCIA en mujeres embarazadas son insuficientes para informar sobre el riesgo asociado al fármaco. Sin embargo, existen consideraciones clínicas para la administración de vacunas vivas a lactantes que estuvieron expuestos a ORENCIA en el útero (ver Consideraciones clínicas). En estudios de toxicología reproductiva en ratas y conejos, no se observaron malformaciones fetales con la administración intravenosa de ORENCIA durante la organogénesis en dosis que produjeron exposiciones aproximadamente 29 veces la exposición en la dosis humana máxima recomendada (DHMR) de 10 mg/kg/mes en base a AUC. Sin embargo, en un estudio de desarrollo pre y posnatal en ratas, ORENCIA alteró la función inmune en ratas hembras en 11 veces la DHMR en base a AUC.
Consideraciones clínicas
Lactantes y administración de vacunas vivas
No se sabe si el abatacept puede cruzar la placenta hacia el feto cuando una mujer es tratada con ORENCIA durante el embarazo. El abatacept es un agente inmunomodulador. No se sabe si la respuesta inmune de un lactante que estuvo expuesto en el útero al abatacept y posteriormente se le administra una vacuna viva se ve afectada. Los riesgos y beneficios deben ser considerados antes de vacunar a tales lactantes [ver Advertencias y Precauciones (5.4)].
Datos
Datos humanos
No existen estudios adecuados y bien controlados sobre el uso de ORENCIA en mujeres embarazadas. Los datos sobre el uso de ORENCIA en mujeres embarazadas son insuficientes para informar sobre el riesgo asociado al fármaco.
Datos animales
La administración intravenosa de abatacept durante la organogénesis a ratones (10, 55 o 300 mg/kg/día), ratas (10, 45 o 200 mg/kg/día) y conejos (10, 45 o 200 mg/kg cada 3 días) produjo exposiciones en ratas y conejos que fueron aproximadamente 29 veces la DHMR en base a AUC (en dosis maternas de 200 mg/kg/día en ratas y conejos), y no se observó embriotoxicidad ni malformaciones fetales en ninguna especie.
En un estudio de desarrollo pre y posnatal en ratas (10, 45 o 200 mg/kg cada 3 días desde el día 6 de gestación hasta el día 21 de lactancia), se produjeron alteraciones en la función inmune en la descendencia femenina, consistentes en un aumento de 9 veces en la respuesta de anticuerpos dependientes de células T en relación con los controles en el día posnatal (DPN) 56 y tiroiditis en una sola cría femenina en el DPN 112, que ocurrieron en aproximadamente 11 veces la DHMR en base a AUC (en una dosis materna de 200 mg/kg). No se observaron efectos adversos en aproximadamente 3 veces la DHMR (una dosis materna de 45 mg/kg). No se sabe si las perturbaciones inmunológicas en ratas son indicadores relevantes de un riesgo de desarrollo de enfermedades autoinmunes en humanos expuestos en el útero al abatacept. La exposición al abatacept en la rata juvenil, que puede ser más representativa del estado del sistema inmune fetal en el humano, resultó en anormalidades del sistema inmune incluyendo inflamación de la tiroides y el páncreas [ver Toxicología no clínica (13.2)].
8.4 Uso pediátrico
Artritis idiopática juvenil poliarticular
Se ha establecido la seguridad y eficacia de ORENCIA para reducir signos y síntomas en pacientes de 2 años de edad y mayores con artritis idiopática juvenil poliarticular (AIJP) moderada a gravemente activa (ORENCIA puede ser usado como monoterapia o concomitantemente con metotrexato). El uso de ORENCIA para esta indicación está respaldado por la evidencia de los siguientes estudios:
- Uso intravenoso: Un estudio aleatorizado de retirada de eficacia, seguridad y farmacocinética de ORENCIA intravenoso en 190 pacientes pediátricos de 6 a 17 años de edad con Artritis Idiopática Juvenil poliarticular (AIJp) [véase Farmacología Clínica (12.3) y Estudios Clínicos (14.2)]. Dado que los análisis farmacocinéticos (PK) poblacionales (después de la administración intravenosa de ORENCIA) mostraron que el aclaramiento de abatacept aumentó con el peso corporal basal, ORENCIA intravenoso se administra en función del peso o del rango de peso [véase Posología y administración (2.2)]. La administración intravenosa de ORENCIA no se ha estudiado en pacientes menores de 6 años de edad.
- Uso subcutáneo: Un estudio abierto de PK y seguridad de ORENCIA subcutáneo en 205 pacientes pediátricos de 2 a 17 años de edad con AIJp, extrapolación de la eficacia de ORENCIA intravenoso en pacientes con AIJp y ORENCIA subcutáneo en pacientes con AR [véase Farmacología Clínica (12.3) y Estudios Clínicos (14.2)]. Dado que los análisis de PK poblacionales (después de la inyección subcutánea de ORENCIA) en pacientes con AIJp mostraron que había una tendencia hacia un mayor aclaramiento de abatacept con el aumento del peso corporal, la dosis de ORENCIA subcutánea se basa en el rango de peso [véase Posología y administración (2.2)].
No se ha establecido la seguridad y eficacia del uso de ORENCIA en AIJp en pacientes pediátricos menores de dos años de edad.
Profilaxis de la enfermedad aguda del injerto contra el huésped
Se ha establecido la seguridad y eficacia de ORENCIA para la profilaxis de la enfermedad aguda del injerto contra el huésped (EaICH), en combinación con un inhibidor de la calcineurina y metotrexato, en pacientes pediátricos de 2 años de edad o más sometidos a TCHH de un donante no emparentado compatible o con 1 alelo no compatible. El uso de ORENCIA para esta indicación se basa en evidencia de:
- •
- estudios adecuados y bien controlados en adultos y pacientes pediátricos de 6 años o más a los que se administró una dosis de 10 mg/kg por vía intravenosa el día anterior al trasplante, seguida de una dosis de 10 mg/kg por vía intravenosa los días 5, 14 y 28 después del trasplante y
- •
- modelado farmacocinético y simulaciones de la exposición a abatacept en pacientes pediátricos de 2 a menos de 6 años a los que se administró una dosis de 15 mg/kg por vía intravenosa el día anterior al trasplante, seguida de una dosis de 12 mg/kg por vía intravenosa los días 5, 14 y 28 después del trasplante.
Además, el curso de la enfermedad es suficientemente similar en pacientes pediátricos de 2 a menos de 6 años al de los pacientes de 6 años o más para permitir la extrapolación de datos a pacientes pediátricos más jóvenes [véase Farmacología Clínica (12.3) y Estudios Clínicos (14.4)]. No se observaron nuevas señales de seguridad en pacientes pediátricos de 6 años o más en el Estudio GVHD-1.
No se ha establecido la seguridad y eficacia de ORENCIA para esta indicación en pacientes pediátricos menores de 2 años de edad.
Artritis psoriásica
Administración subcutánea
Se ha establecido la seguridad y eficacia de ORENCIA subcutáneo para el tratamiento de la artritis psoriásica en pacientes pediátricos de 2 a 17 años de edad.
El uso de ORENCIA en este grupo de edad se basa en evidencia de estudios adecuados y bien controlados de ORENCIA en adultos con APs, datos farmacocinéticos de pacientes adultos con AR, pacientes adultos con APs y pacientes pediátricos con AIJp, y datos de seguridad de estudios clínicos en pacientes pediátricos de 2 a 17 años de edad con AIJp utilizando la formulación subcutánea.
Las concentraciones previas a la dosis (valle) observadas son generalmente comparables entre adultos con AR y APs y pacientes pediátricos con AIJ con poliartritis activa, y se espera que la exposición a PK sea comparable entre adultos con APs y pacientes pediátricos con APs. [véase Reacciones adversas (6.1), Farmacología Clínica (12.3), y Estudios Clínicos (14.1, 14.2, 14.3)].
No se ha establecido la seguridad y eficacia de ORENCIA subcutáneo en pacientes pediátricos menores de 2 años de edad con artritis psoriásica.
Administración intravenosa
No se ha establecido la seguridad y eficacia de ORENCIA intravenoso en pacientes pediátricos con artritis psoriásica.
Datos de toxicidad en animales jóvenes
Un estudio en animales jóvenes realizado en ratas a las que se administró abatacept de 4 a 94 días de edad (antes de la madurez del sistema inmunitario) mostró un aumento en la incidencia de infecciones que provocaron la muerte en todas las dosis en comparación con los controles. Se observaron alteraciones en los subconjuntos de células T, incluido un aumento de las células T auxiliares y una reducción de las células T reguladoras. Además, se observó la inhibición de las respuestas de anticuerpos dependientes de células T (TDAR). Al seguir a estos animales hasta la edad adulta, se observó inflamación linfocítica de la tiroides y los islotes pancreáticos. En contraste, los estudios en ratones y monos adultos no han demostrado hallazgos similares. Como el sistema inmunitario de la rata no está desarrollado en las primeras semanas después del nacimiento, se desconoce la relevancia de estos resultados para los humanos.
8.5 Uso en geriatría
Artritis reumatoide
Un total de 323 pacientes de 65 años o más, incluyendo 53 pacientes de 75 años o más, recibieron ORENCIA en estudios clínicos. No se observaron diferencias generales en seguridad o eficacia entre pacientes geriátricos (pacientes de 65 años o más) y adultos más jóvenes, y otras experiencias clínicas reportadas no han identificado diferencias en las respuestas entre pacientes geriátricos y adultos más jóvenes, pero no se puede descartar una mayor sensibilidad de algunos pacientes geriátricos. La frecuencia de infecciones graves y malignidad entre los pacientes tratados con ORENCIA mayores de 65 años fue mayor que para aquellos menores de 65 años. Debido a que hay una mayor incidencia de infecciones y malignidades en la población geriátrica en general, se debe tener precaución al tratar a pacientes geriátricos.
Profilaxis de la Enfermedad de Injerto contra Huésped Aguda
De los 116 pacientes en el Estudio GVHD-1 que recibieron ORENCIA a una dosis de 10 mg/kg para la profilaxis de aGVHD, 12 (10%) tenían 65 años o más, y 2 (2%) pacientes tenían 75 años o más [ver Estudios Clínicos (14.4)]. Los estudios clínicos de ORENCIA para aGVHD no incluyeron un número suficiente de pacientes de 65 años o más para determinar si responden de manera diferente a los pacientes adultos más jóvenes.
10 SOBREDOSIS
Se han administrado por vía intravenosa dosis de ORENCIA de hasta 50 mg/kg (5 veces la dosis máxima recomendada en pacientes de 6 años o más y 3,3 veces la dosis máxima recomendada en pacientes de 2 a menos de 6 años) sin efectos tóxicos aparentes. En caso de sobredosis, se recomienda controlar al paciente para detectar cualquier signo o síntoma de reacciones adversas e instaurar el tratamiento sintomático adecuado.
11 DESCRIPCIÓN
El abatacept es un modulador selectivo de la coestimulación de las células T. El abatacept es una proteína de fusión soluble que consiste en el dominio extracelular del antígeno 4 asociado a linfocitos T citotóxicos humanos (CTLA-4) unido a la porción modificada Fc (regiones bisagra, CH2 y CH3) de la inmunoglobulina G1 humana (IgG1). El abatacept se produce mediante tecnología de ADN recombinante en un sistema de expresión celular de mamíferos. El peso molecular aparente del abatacept es de 92 kilodaltons.
ORENCIA (abatacept) para inyección es un polvo liofilizado estéril, blanco, sin conservantes, para reconstitución y dilución antes de la infusión intravenosa. Después de la reconstitución del polvo liofilizado con 10 mL de Agua Estéril para Inyección, USP, la solución reconstituida de ORENCIA es transparente, incolora a amarillo pálido, con una concentración de 25 mg/mL y con un rango de pH de 7.2 a 7.8. Cada vial de dosis única de ORENCIA proporciona 250 mg de abatacept, maltosa (500 mg), fosfato monosódico (17.2 mg) y cloruro de sodio (14.6 mg).
La inyección de ORENCIA (abatacept) es una solución estéril, sin conservantes, transparente a ligeramente opalescente, incolora a amarillo pálido, con un rango de pH de 6.8 a 7.4 para administración subcutánea. La inyección de ORENCIA se suministra como una jeringa precargada de dosis única o como un autoinyector ClickJect de dosis única (ver Tabla 6).
| Presentación | Cantidad y volumen del principio activo | Contenido de ingredientes inactivos |
|---|---|---|
|
Inyección de ORENCIA 50 mg/0.4 mL jeringa precargada |
50 mg de abatacept en |
fosfato disódico anhidro (0.335 mg) fosfato monosódico monohidratado (0.114 mg) poloxámero 188 (3.2 mg) sacarosa (68 mg) c.s. hasta 0.4 mL de Agua para Inyección, USP |
|
Inyección de ORENCIA 87.5 mg/0.7 mL jeringa precargada |
87.5 mg de abatacept en |
fosfato disódico anhidro (0.587 mg) fosfato monosódico monohidratado (0.200 mg) poloxámero 188 (5.6 mg) sacarosa (119 mg) c.s. hasta 0.7 mL de Agua para Inyección, USP |
|
Inyección de ORENCIA 125 mg/mL jeringa precargada y autoinyector ClickJect |
125 mg de abatacept en |
fosfato disódico anhidro (0.838 mg) fosfato monosódico monohidratado (0.286 mg) poloxámero 188 (8 mg) sacarosa (170 mg) c.s. hasta 1 mL de Agua para Inyección, USP |
A diferencia de la formulación liofilizada para uso intravenoso, las soluciones de ORENCIA para administración subcutánea no contienen maltosa.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
El abatacept, un modulador selectivo de la costimulación, inhibe la activación de las células T (linfocitos T) al unirse a CD80 y CD86, bloqueando así la interacción con CD28. Esta interacción proporciona una señal de costimulación necesaria para la activación completa de los linfocitos T. Los linfocitos T activados están implicados en la patogénesis de la AR, la AIJp y la PsA, y se encuentran en el sinovio de pacientes con AR, AIJp y PsA.
In vitro, el abatacept disminuye la proliferación de las células T e inhibe la producción de las citocinas TNF alfa (TNFα), interferón-γ e interleucina-2. En un modelo de artritis inducida por colágeno en ratas, el abatacept suprime la inflamación, disminuye la producción de anticuerpos anti-colágeno y reduce la producción específica de antígeno de interferón-γ. La relación de estos marcadores de respuesta biológica con los mecanismos por los cuales ORENCIA ejerce sus efectos clínicos es desconocida.
12.2 Farmacodinámica
En ensayos clínicos con ORENCIA en dosis aproximadas de 10 mg/kg, se observaron disminuciones en los niveles séricos del receptor de interleucina-2 soluble (sIL-2R), interleucina-6 (IL-6), factor reumatoide (FR), proteína C reactiva (PCR), metaloproteinasa de matriz 3 (MMP3) y TNFα. La relación de estos marcadores de respuesta biológica con los mecanismos por los cuales ORENCIA ejerce sus efectos clínicos es desconocida.
No se han realizado análisis farmacodinámicos formales de marcadores de respuesta biológica en pacientes expuestos a ORENCIA como profilaxis de la EICH-a.
12.3 Farmacocinética
Adultos sanos y adultos con AR – Administración intravenosa
Se estudiaron la farmacocinética del abatacept en sujetos adultos sanos después de una infusión intravenosa única de 10 mg/kg y en pacientes con AR después de múltiples infusiones intravenosas de 10 mg/kg de ORENCIA (ver Tabla 7).
| Parámetro PK | Sujetos Sanos (Después de una dosis única de 10 mg/kg) n = 13 |
Pacientes con AR (Después de múltiples dosis de 10 mg/kga) n = 14 |
|---|---|---|
| a Se administraron múltiples infusiones intravenosas de ORENCIA en los días 1, 15, 30 y mensualmente a partir de entonces. | ||
|
Concentración máxima (Cmax) [mcg/mL] |
292 (175 – 427) |
295 (171 – 398) |
|
Semivida terminal (t1/2) [días] |
16,7 (12 – 23) |
13,1 (8 – 25) |
|
Aclaramiento sistémico (CL) [mL/h/kg] |
0,23 (0,16 – 0,30) |
0,22 (0,13 – 0,47) |
|
Volumen de distribución (Vss) [L/kg] |
0,09 (0,06 – 0,13) |
0,07 (0,02 – 0,13) |
La farmacocinética de abatacept en pacientes con artritis reumatoide (AR) y sujetos sanos parecía ser comparable. En pacientes con AR, después de múltiples infusiones intravenosas, la farmacocinética de abatacept mostró aumentos proporcionales de Cmax y AUC en el rango de dosis de 2 mg/kg a 10 mg/kg. A 10 mg/kg, la concentración en suero pareció alcanzar un estado estable en el día 60 con una concentración valle media (rango) de 24 mcg/mL (1 a 66 mcg/mL). No se produjo acumulación sistémica de abatacept al continuar el tratamiento repetido con 10 mg/kg a intervalos mensuales en pacientes con AR.
Los análisis de farmacocinética poblacional en pacientes con AR revelaron que existía una tendencia hacia una mayor depuración de abatacept con el aumento del peso corporal. La edad y el género (cuando se corrigió por el peso corporal) no afectaron a la depuración. El metotrexato, los AINE, los corticosteroides y los antagonistas del TNF concomitantes no influyeron en la depuración de abatacept.
No se realizaron estudios formales para examinar los efectos de la insuficiencia renal o hepática en la farmacocinética de abatacept.
AR en adultos – Administración subcutánea
Abatacept exhibió farmacocinética lineal tras la administración subcutánea. La media (rango) de Cmin y Cmax en el estado estable observado después de 85 días de tratamiento fue de 32,5 mcg/mL (6,6 a 113,8 mcg/mL) y 48,1 mcg/mL (9,8 a 132,4 mcg/mL), respectivamente. La biodisponibilidad de abatacept tras la administración subcutánea en relación con la administración intravenosa fue del 79%. Las estimaciones medias de la depuración sistémica (0,28 mL/h/kg), el volumen de distribución (0,11 L/kg) y la vida media terminal (14,3 días) fueron comparables entre la administración subcutánea y la intravenosa.
Se llevó a cabo el estudio SC-2 para determinar el efecto del uso de monoterapia subcutánea de ORENCIA en la inmunogenicidad (sin una dosis de carga intravenosa) en 100 pacientes con AR [véase Reacciones adversas (6.3)]. En este estudio, se alcanzó una concentración valle media de 12,6 mcg/mL después de 2 semanas de dosificación.
En consonancia con los datos intravenosos, los análisis de farmacocinética poblacional de ORENCIA subcutánea en pacientes con AR revelaron que existía una tendencia hacia una mayor depuración de abatacept con el aumento del peso corporal [véase Dosificación y administración (2.1)]. La edad y el género (cuando se corrigió por el peso corporal) no afectaron a la depuración aparente. Los medicamentos concomitantes, como el metotrexato, los corticosteroides y los AINE, no influyeron en la depuración aparente de abatacept.
Artritis idiopática juvenil poliarticular – Administración intravenosa
En el estudio JIA-1 en pacientes de 6 a 17 años de edad, las concentraciones medias (rango) en el pico y valle en el estado estable de abatacept en suero fueron de 217 mcg/mL (57 a 700 mcg/mL) y 11,9 mcg/mL (0,15 a 44,6 mcg/mL) [véase Estudios clínicos (14.2)]. Los análisis de farmacocinética poblacional de los datos de concentración en suero mostraron que la depuración de abatacept aumentó con el peso corporal basal [véase Dosificación y administración (2.2)]. La depuración media estimada (rango) de abatacept en los pacientes con artritis idiopática juvenil fue de 0,4 mL/h/kg (0,20 a 1,12 mL/h/kg). Después de tener en cuenta el efecto del peso corporal, la depuración de abatacept no estaba relacionada con la edad y el género. También se demostró que el metotrexato, los corticosteroides y los AINE concomitantes no influían en la depuración de abatacept.
Artritis idiopática juvenil poliarticular – Administración subcutánea
En el estudio JIA-2 en pacientes de 2 a 17 años de edad, el estado estable de abatacept se alcanzó en el día 85 después de la dosificación subcutánea escalada por peso corporal semanal de ORENCIA [véase Estudios clínicos (14.2)]. Se alcanzaron concentraciones valle comparables en todos los escalones de peso y grupos de edad mediante el régimen de dosificación subcutánea escalada por peso corporal. La concentración valle media (rango) de abatacept en el día 113 fue de 44,4 mcg/mL (13,4 a 88,1 mcg/mL), 46,6 mcg/mL (22,4 a 97,0 mcg/mL) y 38,5 mcg/mL (9,3 a 73,2 mcg/mL) en pacientes pediátricos con JIA que pesaban 10 a <25 kg, 25 a <50 kg y ≥50 kg, respectivamente.
En consonancia con los datos intravenosos, los análisis de farmacocinética poblacional de ORENCIA subcutánea en pacientes con JIA revelaron que existía una tendencia hacia una mayor depuración de abatacept con el aumento del peso corporal [véase Dosificación y administración (2.2)]. La edad y el género (cuando se corrigió por el peso corporal) no afectaron a la depuración aparente. Los medicamentos concomitantes, como el metotrexato, los corticosteroides y los AINE, no influyeron en la depuración aparente de abatacept.
Artritis psoriásica en adultos – Administración intravenosa y subcutánea
En el estudio PsA-I, un estudio de rango de dosis, se administró ORENCIA intravenoso a 3 mg/kg, dosificación basada en el rango de peso: 500 mg para pacientes que pesaban menos de 60 kg, 750 mg para pacientes que pesaban 60 a 100 kg y 1.000 mg para pacientes que pesaban más de 100 kg, o dosis de 30 mg/kg en los días 1 y 15 seguidas de dosificación basada en el rango de peso [véase Estudios clínicos (14.3)]. Después de la administración mensual de ORENCIA intravenoso, abatacept mostró farmacocinética lineal en el rango de dosis en este estudio. Con la dosificación basada en el rango de peso (véase arriba), el estado estable de abatacept se alcanzó en el día 57 y la concentración valle geométrica (CV%) media (Cmin) fue de 24,3 mcg/mL (40,8%) en el día 169. En el estudio PsA-II después de la administración subcutánea semanal de ORENCIA a 125 mg, el estado estable de abatacept se alcanzó en el día 57 y la Cmin geométrica (CV%) media fue de 25,6 mcg/mL (47,7%) en el día 169.
De acuerdo con los resultados de la artritis reumatoide, los análisis farmacocinéticos poblacionales de abatacept en pacientes con psoriasis artrítica revelaron una tendencia hacia una mayor depuración (L/h) de abatacept con el aumento del peso corporal [ver Dosis y Administración (2.3)]. Además, en comparación con los pacientes con artritis reumatoide con el mismo peso corporal, la depuración de abatacept en pacientes con psoriasis artrítica fue aproximadamente un 8% menor, lo que resulta en una mayor exposición a abatacept en pacientes con psoriasis artrítica. Sin embargo, esta ligera diferencia en la exposición no se considera clínicamente significativa.
Profilaxis de la enfermedad de injerto contra huésped aguda – Administración intravenosa
| a Cmin observada en el Día 5 del período de tratamiento; n = 18 para la cohorte 7/8; n = 32 para la cohorte 8/8. Cmax, t1/2, CL y Vss se predijeron mediante el modelo después de la primera infusión intravenosa de 10 mg/kg de ORENCIA. |
||
|
Parámetro PK |
Cohorte 7 de 8 |
Cohorte 8 de 8 |
|
Concentración mínima (Cmin) a [mcg/mL] |
59 (26 – 112) |
43 (25 – 73) |
|
Concentración pico (Cmax) [mcg/mL] |
221 (163 – 292) |
172 (107 – 254) |
|
Semivida terminal (t1/2) [días] |
20.6 (6 – 43) |
20.8 (12 – 38) |
|
Depuración sistémica (CL) [mL/h/kg] |
0.26 (0.15 – 0.65) |
0.32 (0.18 – 0.56) |
|
Volumen de distribución (Vss) [L/kg] |
0.13 (0.08 – 0.27) |
0.17 (0.11 – 0.26) |
En un estudio de pacientes que recibieron ORENCIA para la profilaxis de la enfermedad de injerto contra huésped aguda (aGVHD) de 6 años o más, las concentraciones medias geométricas (%CV) en el valle (Cmin) de abatacept en el Día 63 después del trasplante después de 4 dosis utilizando una dosificación basada en el peso de 10 mg/kg (dosis máxima de 1.000 mg) administrada el día anterior al trasplante (Día -1), seguida de una dosis en el Día 5, 14 y 28 después del trasplante, fueron de 22.5 mcg/mL (243.9 %CV) para los receptores de HSCT emparejados 8 de 8 de antígeno leucocitario humano (HLA) de donantes no relacionados (URD), y de 31.1 mcg/mL (114.4 %CV) para los receptores de HSCT 7 de 8 de HLA emparejados de donantes no relacionados (URD), respectivamente.
Los análisis farmacocinéticos poblacionales en pacientes con aGVHD demostraron que los receptores de HSCT 7 de 8 HLA emparejados tenían una depuración un 29% menor en comparación con los receptores de HSCT 8 de 8 HLA emparejados. De acuerdo con datos anteriores, el aumento del peso corporal se asoció con una mayor depuración de abatacept, mientras que la edad (cuando se corrige por el peso corporal) no afectó la depuración aparente. Los medicamentos concomitantes, como el metotrexato y los inhibidores de la calcineurina (por ejemplo, ciclosporina y tacrolimus), no influyeron en la depuración de abatacept.
Basado en la modelización y simulación farmacocinética poblacional con datos de pacientes de 6 años o más, las exposiciones simuladas de abatacept después de la primera y última dosis en sujetos pediátricos de 2 a menos de 6 años de edad que recibieron 15 mg/kg de ORENCIA mediante infusión intravenosa de 60 minutos en el Día -1, seguida de 12 mg/kg mediante infusión intravenosa de 60 minutos en el Día 5, 14 y 28 son comparables a las de pacientes pediátricos de 6 a menos de 17 años de edad y pacientes adultos que recibieron 10 mg/kg mediante infusión intravenosa de 60 minutos en el Día -1, 5, 14 y 28.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
En un estudio de carcinogenicidad en ratones, las inyecciones subcutáneas semanales de 20, 65 o 200 mg/kg de abatacept administradas hasta por 84 semanas en machos y 88 semanas en hembras se asociaron con aumentos en la incidencia de linfomas malignos (todas las dosis) y tumores de glándula mamaria (dosis intermedia y alta en hembras). Los ratones de este estudio fueron infectados con virus de leucemia murina y virus del tumor mamario de ratón. Estos virus están asociados con un aumento en la incidencia de linfomas y tumores de glándula mamaria, respectivamente, en ratones inmunodeprimidos. Las dosis utilizadas en estos estudios produjeron exposiciones 0,8, 2,0 y 3,0 veces más altas, respectivamente, que la exposición asociada con la dosis máxima recomendada en humanos (MRHD) de 10 mg/kg basada en el AUC (área bajo la curva de concentración-tiempo). Se desconoce la relevancia de estos hallazgos para el uso clínico de ORENCIA.
En un estudio de toxicidad de un año en monos cynomolgus, se administró abatacept por vía intravenosa una vez por semana en dosis de hasta 50 mg/kg (produciendo 9 veces la exposición MRHD basada en el AUC). Abatacept no se asoció con ninguna toxicidad significativa relacionada con el fármaco. Los efectos farmacológicos reversibles consistieron en disminuciones mínimas transitorias en la IgG sérica y en un agotamiento linfoide mínimo a severo de los centros germinales en el bazo y/o los ganglios linfáticos. No se observó evidencia de linfomas o cambios morfológicos preneoplásicos, a pesar de la presencia de un virus (linfocriptovirus) conocido por causar estas lesiones en monos inmunodeprimidos dentro del plazo de este estudio. Se desconoce la relevancia de estos hallazgos para el uso clínico de ORENCIA.
No se observó potencial mutagénico de abatacept en las pruebas de mutación inversa bacteriana in vitro (Ames) o en las pruebas de mutación puntual directa de ovario de hámster chino/hipoxantina guanina fosforribosiltransferasa (CHO/HGPRT) con o sin activación metabólica, y no se observaron aberraciones cromosómicas en linfocitos humanos tratados con abatacept con o sin activación metabólica.
Abatacept no tuvo efectos adversos sobre la fertilidad masculina o femenina en ratas a dosis de hasta 200 mg/kg cada tres días (11 veces la exposición MRHD basada en el AUC).
13.2 Toxicología y/o Farmacología Animal
En estudios con ratones y monos adultos, la inhibición de TDAR fue aparente. Sin embargo, no se observaron infección y mortalidad, alteración de las células T ayudantes e inflamación de la tiroides y el páncreas.
14 ESTUDIOS CLÍNICOS
14.1 Artritis reumatoide adulta
Descripción de los estudios clínicos de ORENCIA intravenosa para el tratamiento de pacientes con AR
La eficacia y seguridad de ORENCIA para administración intravenosa se evaluaron en seis estudios aleatorizados, doble ciego, controlados (cinco controlados con placebo y uno controlado con un fármaco activo) en pacientes de ≥18 años de edad con artritis reumatoide (AR) activa diagnosticada de acuerdo con los criterios del Colegio Americano de Reumatología (ACR). Los estudios I, II, III, IV y VI requerían que los pacientes tuvieran al menos 12 articulaciones sensibles y 10 articulaciones hinchadas en la randomización, y el Estudio V no requería un número específico de articulaciones sensibles o hinchadas. El tratamiento con ORENCIA o placebo se administró por vía intravenosa en las semanas 0, 2 y 4 y luego cada 4 semanas a partir de entonces en los Estudios I, II, III, IV y VI.
- •
- El Estudio I (NCT00279760) evaluó ORENCIA como monoterapia en 122 pacientes con AR activa que habían fallado al menos un fármaco antirreumático modificador de la enfermedad (DMARD) no biológico o etanercept.
- •
- En el Estudio II (NCT00162266) y el Estudio III (NCT00048568), se evaluó la eficacia de ORENCIA en pacientes con una respuesta inadecuada al metotrexato (MTX) y que continuaron con su dosis estable de MTX.
- •
- En el Estudio IV (NCT00048581), se evaluó la eficacia de ORENCIA en pacientes con una respuesta inadecuada a un antagonista del factor de necrosis tumoral (TNF), con el antagonista del TNF suspendido antes de la randomización; se permitieron otros DMARD.
- •
- El Estudio V (NCT00048932) evaluó principalmente la seguridad en pacientes con AR activa que requerían una intervención adicional a pesar del tratamiento actual con DMARD; todos los DMARD utilizados en la inscripción se continuaron. Los pacientes en el Estudio V no fueron excluidos por condiciones médicas comórbidas.
- •
- En el Estudio VI (NCT00122382), se evaluó la eficacia y seguridad de ORENCIA en pacientes naïve al metotrexato con AR de menos de 2 años de duración de la enfermedad. En el Estudio VI, los pacientes previamente naïve al metotrexato fueron aleatorizados para recibir ORENCIA más metotrexato o metotrexato más placebo.
Los pacientes del Estudio I fueron aleatorizados para recibir una de tres dosis de ORENCIA (0.5, 2 o 10 mg/kg) o placebo hasta la semana 8. Los pacientes del Estudio II fueron aleatorizados para recibir ORENCIA 2 o 10 mg/kg o placebo durante 12 meses. Los pacientes de los Estudios III, IV, V y VI fueron aleatorizados para recibir una dosis de ORENCIA basada en el rango de peso o placebo durante 12 meses (Estudios III, V y VI) o 6 meses (Estudio IV). La dosis de ORENCIA fue de 500 mg para pacientes que pesaban menos de 60 kg, 750 mg para pacientes que pesaban de 60 a 100 kg y 1,000 mg para pacientes que pesaban más de 100 kg.
Descripción de los estudios clínicos de ORENCIA subcutánea o intravenosa para el tratamiento de pacientes con AR adulta
La eficacia de ORENCIA para administración subcutánea se evaluó en el Estudio SC-1 (NCT00559585), que fue un estudio aleatorizado, doble ciego, doble simulacro, de no inferioridad que comparó ORENCIA administrada subcutáneamente con ORENCIA administrada por vía intravenosa en 1457 pacientes con AR de moderada a gravemente activa, que recibían metotrexato (MTX) de fondo y tenían una respuesta inadecuada al metotrexato (MTX-IR). En el Estudio SC-1, los pacientes fueron aleatorizados con estratificación por peso corporal (<60 kg, 60 a 100 kg, >100 kg) para recibir (1) inyecciones subcutáneas de ORENCIA 125 mg semanales, después de una dosis de carga intravenosa única de ORENCIA basada en el peso corporal o (2) ORENCIA por vía intravenosa en los Días 1, 15, 29 y cada cuatro semanas a partir de entonces. Los sujetos continuaron tomando su dosis actual de MTX desde el día de la randomización.
Respuesta clínica en pacientes con AR adulta
El porcentaje de pacientes tratados con ORENCIA que alcanzaron respuestas ACR 20, 50 y 70 y una respuesta clínica importante en los Estudios I, III, IV y VI se muestra en la Tabla 9. Los pacientes tratados con ORENCIA tuvieron tasas de respuesta ACR 20, 50 y 70 más altas a los 6 meses en comparación con los pacientes tratados con placebo. Las tasas de respuesta ACR en el mes 6 en el grupo de 10 mg/kg del Estudio II fueron similares al grupo de ORENCIA en el Estudio III.
En los Estudios III y IV, se observó una mejora en la tasa de respuesta ACR 20 en comparación con el placebo en algunos pacientes dentro de los 15 días y dentro de los 29 días en comparación con el MTX en el Estudio VI. En los Estudios II, III y VI, las tasas de respuesta ACR se mantuvieron hasta los 12 meses en los pacientes tratados con ORENCIA. Las respuestas ACR se mantuvieron hasta tres años en la extensión de etiqueta abierta del Estudio II. En el Estudio III, los pacientes tratados con ORENCIA experimentaron una mayor mejora que los pacientes tratados con placebo en la rigidez matutina.
En el Estudio VI, una mayor proporción de pacientes tratados con ORENCIA más MTX alcanzó un nivel bajo de actividad de la enfermedad, medido por un DAS28-CRP menor a 2.6 a los 12 meses en comparación con aquellos tratados con MTX más placebo (Tabla 9). De los pacientes tratados con ORENCIA más MTX que alcanzaron un DAS28-CRP menor a 2.6, el 54% no tenía articulaciones activas, el 17% tenía una articulación activa, el 7% tenía dos articulaciones activas y el 22% tenía tres o más articulaciones activas, donde una articulación activa era una articulación que fue calificada como sensible o hinchada o ambas.
En el Estudio SC-1, la medida de resultado principal fue ACR 20 a los 6 meses. El margen de no inferioridad preespecificado fue una diferencia de tratamiento de -7.5%. Como se muestra en la Tabla 10, el estudio demostró la no inferioridad de ORENCIA administrada subcutáneamente en comparación con las infusiones intravenosas de ORENCIA con respecto a las respuestas ACR 20 hasta 6 meses de tratamiento. Las respuestas ACR 50 y 70 también se muestran en la Tabla 9. No se observaron diferencias importantes en las respuestas ACR entre los grupos de tratamiento intravenoso y subcutáneo en los subgrupos basados en las categorías de peso (menos de 60 kg, 60 a 100 kg y más de 100 kg; datos no mostrados).
| Porcentaje de pacientes | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
DMARDs
Metotrexato
(MTX)
Antagonistas del TNF
a MTX
† p<0.01, ORENCIA vs placebo o MTX.
‡ p<0.001, ORENCIA vs placebo o MTX.
§ 95% CI: −4.2, 4.8 (basado en el margen preespecificado para la no inferioridad de −7.5%).
a 10 mg/kg.
b Dosis basada en el rango de peso [ver Dosis y Administración (2.1)].
c La respuesta clínica principal se define como lograr una respuesta ACR 70 durante un período continuo de 6 meses.
d Consulte el texto para obtener una descripción adicional de la actividad articular restante.
e Los datos por protocolo se presentan en la tabla. Para ITT; n=736, 721 para ORENCIA SC e IV, respectivamente.
Tasa de Respuesta
ORNa
n=32
PBO
n=32
ORNb
+MTX
n=424
PBO
+MTX
n=214
ORNb +
DMARDs
n=256
PBO +
DMARDs
n=133
ORNb
+MTX
n=256
PBO
+MTX
n=253
ORNe
SC
+MTX
n=693
ORNe
IV
+MTX
n=678
ACR 20
Mes 3
53%
31%
62%‡
37%
46%‡
18%
64%*
53%
68%
69%
Mes 6
NA
NA
68%‡
40%
50%‡
20%
75%†
62%
76%§
76%
Mes 12
NA
NA
73%‡
40%
NA
NA
76%‡
62%
NA
NA
ACR 50
Mes 3
16%
6%
32%‡
8%
18%†
6%
40%‡
23%
33%
39%
Mes 6
NA
NA
40%‡
17%
20%‡
4%
53%‡
38%
52%
50%
Mes 12
NA
NA
48%‡
18%
NA
NA
57%‡
42%
NA
NA
ACR 70
Mes 3
6%
0
13%‡
3%
6%*
1%
19%†
10%
13%
16%
Mes 6
NA
NA
20%‡
7%
10%†
2%
32%†
20%
26%
25%
Mes 12
NA
NA
29%‡
6%
NA
NA
43%‡
27%
NA
NA
Respuesta
Clínica
Importantec
NA
NA
14%‡
2%
NA
NA
27%‡
12%
NA
NA
DAS28-CRP
<2.6d
Mes 12
NA
NA
NA
NA
NA
NA
41%‡
23%
NA
NA
Los resultados de los componentes de los criterios de respuesta ACR para los Estudios III, IV y SC-1 se muestran en la Tabla 10 (resultados al inicio [BL] y 6 meses [6 M]). En los pacientes tratados con ORENCIA, se observó una mayor mejoría en todos los componentes de los criterios de respuesta ACR durante 6 y 12 meses que en los pacientes tratados con placebo.
| Administración Intravenosa | Administración Subcutánea o Intravenosa | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Respuesta Inadecuada al MTX |
Respuesta Inadecuada a Antagonistas del TNF |
Respuesta Inadecuada a MTX |
||||||||||
| Estudio III | Estudio IV | Estudio SC-1c | ||||||||||
| † p<0.01, ORENCIA (ORN) vs placebo (PBO), basado en el cambio porcentual medio desde el inicio. ‡ p<0.001, ORENCIA vs placebo, basado en el cambio porcentual medio desde el inicio. a Escala analógica visual: 0 = mejor, 100 = peor. b Cuestionario de Evaluación de Salud: 0 = mejor, 3 = peor; 20 preguntas; 8 categorías: vestirse y arreglarse, levantarse, comer, caminar, higiene, alcance, agarre y actividades. c SC-1 es un estudio de no inferioridad. Los datos por protocolo se presentan en la tabla. |
||||||||||||
|
|
ORN |
PBO |
ORN |
PBO |
ORN SC |
ORN IV |
||||||
|
Componente |
BL |
6 M |
BL |
6 M |
BL |
6 M |
BL |
6 M |
BL |
6 M |
BL |
6 M |
|
Número de |
28 |
7‡ |
31 |
14 |
30 |
13‡ |
31 |
24 |
27 |
5 |
27 |
6 |
|
Número de |
19 |
5‡ |
20 |
11 |
21 |
10‡ |
20 |
14 |
18 |
4 |
18 |
3 |
|
Dolora |
67 |
27‡ |
70 |
50 |
73 |
43† |
74 |
64 |
71 |
25 |
70 |
28 |
|
Evaluación global del |
66 |
29‡ |
64 |
48 |
71 |
44‡ |
73 |
63 |
70 |
26 |
68 |
27 |
|
Índice de |
1.75 |
1.13‡ |
1.75 |
1.38 |
1.88 |
1.38‡ |
2.00 |
1.75 |
1.88 |
1.00 |
1.75 |
1.00 |
|
Evaluación global del |
69 |
21‡ |
68 |
40 |
71 |
32‡ |
69 |
54 |
65 |
16 |
65 |
15 |
|
CRP (mg/dL) |
2.2 |
0.9‡ |
2.1 |
1.8 |
3.4 |
1.3‡ |
2.8 |
2.3 |
1.6 |
0.7 |
1.8 |
0.7 |
El porcentaje de pacientes que lograron la respuesta ACR 50 para el Estudio III por visita se muestra en la Figura 1. El curso temporal para el grupo de ORENCIA en el Estudio VI fue similar al del Estudio III.
Figura 1: Porcentaje de pacientes que logran una respuesta ACR 50 por visita* (Estudio III)

*Es posible que los mismos pacientes no hayan respondido en cada punto de tiempo.
El porcentaje de pacientes que lograron la respuesta ACR 50 para el Estudio SC-1 en los brazos de tratamiento subcutáneo (SC) e intravenoso (IV) de ORENCIA en cada visita de tratamiento fue el siguiente: Día 15—SC 3%, IV 5%; Día 29—SC 11%, IV 14%; Día 57—SC 24%, IV 30%; Día 85—SC 33%, IV 38%; Día 113—SC 39%, IV 41%; Día 141—SC 46%, IV 47%; Día 169—SC 51%, IV 50%.
Respuesta radiográfica en pacientes adultos con AR
En el Estudio III y el Estudio VI, el daño estructural articular se evaluó radiográficamente y se expresó como el cambio con respecto al valor inicial en la Puntuación Total de Sharp (TSS) modificada por Genant y sus componentes, la Puntuación de Erosión (ES) y la puntuación de Estrechamiento del Espacio Articular (JSN). ORENCIA/MTX retrasó la progresión del daño estructural en comparación con placebo/MTX después de 12 meses de tratamiento, como se muestra en la Tabla 11.
| Parámetro | ORENCIA/MTX | Placebo/MTX | Diferencias | Valor Pd | |
|---|---|---|---|---|---|
| a Pacientes con una respuesta inadecuada al MTX. b Pacientes sin tratamiento previo con MTX. c Los pacientes recibieron 1 año de placebo/MTX seguido de 1 año de ORENCIA/MTX. d Basado en un modelo ANCOVA no paramétrico. |
|||||
|
Estudio III |
|
|
|
|
|
|
Primer año |
|||||
|
TSS |
1.07 |
2.43 |
1.36 |
<0.01 |
|
|
ES |
0.61 |
1.47 |
0.86 |
<0.01 |
|
|
Puntuación JSN |
0.46 |
0.97 |
0.51 |
<0.01 |
|
|
Segundo año |
|||||
| Respuesta Inadecuada al Metotrexato | ||||
|---|---|---|---|---|
| Estudio II | Estudio III | |||
| *** p<0.001, ORENCIA vs placebo. a 10 mg/kg. b Dosis basada en el rango de peso [ver Dosis y Administración (2.1)]. c Cuestionario de Evaluación de Salud Modificado: 0 = mejor, 3 = peor; 8 preguntas; 8 categorías: vestirse y arreglarse, levantarse, comer, caminar, higiene, alcanzar, agarrar y actividades. d Cuestionario de Evaluación de Salud: 0 = mejor, 3 = peor; 20 preguntas; 8 categorías: vestirse y arreglarse, levantarse, comer, caminar, higiene, alcanzar, agarrar y actividades. |
||||
|
Índice de Discapacidad HAQ |
ORENCIAa |
Placebo |
ORENCIAb |
Placebo |
|
Línea de base (Media) |
0.98c |
0.97c |
1.69d |
1.69d |
|
Mejora media |
|
|
|
|
La calidad de vida relacionada con la salud se evaluó mediante el cuestionario SF-36 a los 6 meses en los Estudios II, III y IV y a los 12 meses en los Estudios II y III. En estos estudios, se observó una mejora en el grupo de ORENCIA en comparación con el grupo placebo en las 8 áreas del SF-36, así como en el Resumen de Componente Físico (PCS) y el Resumen de Componente Mental (MCS).
14.2 Artritis Idiopática Juvenil Poliarticular
Artritis Idiopática Juvenil Poliarticular – Administración Intravenosa
La seguridad y la eficacia de ORENCIA con administración intravenosa se evaluaron en el Estudio JIA-1 (NCT00095173), un estudio en tres partes que incluyó una extensión de etiqueta abierta en pacientes pediátricos con artritis idiopática juvenil poliarticular (pJIA). Se trataron pacientes de 6 a 17 años de edad (n = 190) con pJIA de actividad moderada a grave que habían tenido una respuesta inadecuada a uno o más DMARDs, como MTX o antagonistas del TNF. Los pacientes tenían una duración de la enfermedad de aproximadamente 4 años con enfermedad de actividad moderada a grave al inicio del estudio, según se determinó por los recuentos basales de articulaciones activas (media, 16) y articulaciones con pérdida de movimiento (media, 16); los pacientes tenían niveles elevados de proteína C reactiva (PCR) (media, 3,2 mg/dL) y eritrosedimentación (ESR) (media, 32 mm/h). Los subtipos de JIA de los pacientes inscritos al inicio de la enfermedad incluyeron oligoarticular (16%), poliarticular (64%; 20% eran positivos para el factor reumatoide) y JIA sistémica sin manifestaciones sistémicas (20%). Al inicio del estudio, el 74% de los pacientes recibía MTX (dosis media, 13,2 mg/m2 por semana) y se mantuvo en una dosis estable de MTX (aquellos que no recibían MTX no iniciaron el tratamientoo con MTX durante el estudio).
En el Período A (etiqueta abierta, fase inicial), los pacientes recibieron 10 mg/kg (máximo 1.000 mg por dosis) por vía intravenosa en los días 1, 15, 29 y mensualmente a partir de entonces. La respuesta se evaluó utilizando la definición de mejora ACR Pediatric 30, definida como una mejora ≥30% en al menos 3 de las 6 variables del conjunto central de JIA y un empeoramiento ≥30% en no más de 1 de las 6 variables del conjunto central de JIA. Los pacientes que demostraron una respuesta ACR Pedi 30 al final del Período A fueron aleatorizados en la fase doble ciego (Período B) y recibieron ORENCIA o placebo durante 6 meses o hasta que se presentara un brote de la enfermedad. El brote de la enfermedad se definió como un empeoramiento ≥30% en al menos 3 de las 6 variables del conjunto central de JIA con una mejora ≥30% en no más de 1 de las 6 variables del conjunto central de JIA; se requería un empeoramiento de ≥2 cm en la Evaluación Global del Médico o del Padre/Madre si se usaba como 1 de las 3 variables del conjunto central de JIA utilizadas para definir el brote, y un empeoramiento en ≥2 articulaciones era necesario si el número de articulaciones activas o articulaciones con limitación de movimiento se usaba como 1 de las 3 variables del conjunto central de JIA utilizadas para definir el brote.
Al final del Período A, las respuestas pediátricas ACR 30/50/70 fueron del 65%, 50% y 28%, respectivamente. Las respuestas ACR 30 pediátricas fueron similares en todos los subtipos de JIA estudiados.
Durante la fase de retirada aleatorizada doble ciego (Período B), los pacientes tratados con ORENCIA (intravenosa) tuvieron significativamente menos brotes de la enfermedad en comparación con los pacientes tratados con placebo (20% vs 53%); IC del 95% de la diferencia (15%, 52%). El riesgo de brote de la enfermedad entre los pacientes que continuaron con ORENCIA intravenosa fue menos de un tercio que el de los pacientes que se retiraron del tratamientoo con ORENCIA intravenosa (razón de riesgo = 0,31, IC del 95% [0,16, 0,59]). Entre los pacientes que recibieron ORENCIA intravenosa durante todo el estudio (Período A, Período B y la extensión de etiqueta abierta Período C), la proporción de pacientes respondedores ACR 30/50/70 pediátricos se ha mantenido constante durante 1 año.
Artritis Idiopática Juvenil Poliarticular – Administración Subcutánea
ORENCIA para administración subcutánea sin una dosis de carga intravenosa se evaluó en el Estudio JIA-2 (NCT01844518), un estudio de 2 períodos, de etiqueta abierta que incluyó pacientes pediátricos de 2 a 17 años de edad (n = 205). Los pacientes tenían enfermedad poliarticular activa en el momento del estudio y habían tenido una respuesta inadecuada a al menos un DMARD no biológico o biológico. Los subtipos de pacientes con JIA al inicio del estudio incluyeron poliarticular (79%; 22% eran positivos para el factor reumatoide), oligoarticular extendido y persistente (14%), artritis relacionada con la entesis (1%) y JIA sistémica sin manifestaciones sistémicas (2%). Los pacientes tenían una duración media de la enfermedad de 2,5 años con articulaciones activas (media, 11,9), articulaciones con pérdida de movimiento (media, 10,4) y niveles elevados de proteína C reactiva (PCR) (media, 1,2 mg/dL). Al inicio del estudio, el 80% de los pacientes recibía MTX y se mantuvo en una dosis estable de MTX. Los pacientes recibieron ORENCIA subcutáneo de forma abierta semanalmente mediante un régimen de dosificación basado en el peso. El objetivo principal del estudio fue la evaluación de la farmacocinética para apoyar la extrapolación de la eficacia basada en la exposición a ORENCIA apoyada por la eficacia descriptiva [ver Farmacología Clínica (12.3)].
Las respuestas JIA ACR 30/50/70 evaluadas a los 4 meses en los pacientes de 2 a 17 años tratados con ORENCIA subcutáneo fueron consistentes con los resultados de ORENCIA intravenosa en el Estudio JIA-1.
14.3 Artritis Psoriásica
La eficacia de ORENCIA se evaluó en 594 pacientes adultos (de 18 años o más) con artritis psoriásica (PsA), en dos estudios aleatorizados, doble ciego, controlados con placebo (Estudios PsA-I [NCT00534313] y PsA-II [NCT01860976]). Los pacientes tenían PsA activa (≥3 articulaciones hinchadas y ≥3 articulaciones dolorosas) a pesar del tratamientoo previo con terapia DMARD y tenían una lesión cutánea psoriásica que calificaba de al menos 2 cm de diámetro. En PsA-I y PsA-II, el 37% y el 61% de los pacientes, respectivamente, habían sido tratados previamente con antagonistas del TNF.
Durante el período inicial de 24 semanas, doble ciego del Estudio PsA-I, 170 pacientes fueron aleatorizados para recibir uno de cuatro tratamientos intravenosos en los Días 1, 15, 29 y luego cada 28 días (no hubo escape durante el período de 24 semanas):
- •
- Placebo
- •
- ORENCIA 3 mg/kg
- •
- ORENCIA 500 mg para pacientes que pesan menos de 60 kg, ORENCIA 750 mg para pacientes que pesan entre 60 y 100 kg, y ORENCIA 1,000 mg para pacientes que pesan más de 100 kg (dosificación basada en el rango de peso), o
- •
- ORENCIA 30 mg/kg en los Días 1 y 15 seguido de una dosificación de ORENCIA basada en el rango de peso (es decir, 500 mg para pacientes que pesan menos de 60 kg, 750 mg para pacientes que pesan entre 60 y 100 kg, y 1,000 mg para pacientes que pesan más de 100 kg).
Después del período doble ciego de 24 semanas en el Estudio PsA-I, los pacientes recibieron ORENCIA intravenoso de etiqueta abierta cada 28 días.
Se permitió a los pacientes recibir dosis estables de MTX concomitante, dosis bajas de corticosteroides (equivalente a ≤10 mg de prednisona) y/o AINEs durante el ensayo. Al momento de la inscripción, aproximadamente el 60% de los pacientes estaban recibiendo MTX. Al inicio, la PCR media (DE) para ORENCIA IV fue de 17 mg/L (33.0) y el número medio (DE) de articulaciones sensibles e inflamadas fue de 22.2 (14.3) y 10.9 (7.6), respectivamente.
En PsA-II, 424 pacientes fueron aleatorizados 1:1 para recibir dosis semanales de placebo subcutáneo o ORENCIA 125 mg sin una dosis de carga durante 24 semanas, de forma doble ciego, seguido de ORENCIA subcutáneo de etiqueta abierta 125 mg semanalmente. Se permitió a los pacientes recibir dosis estables de MTX, sulfasalazina, leflunomida, hidroxicloroquina, dosis bajas de corticosteroides (equivalente a ≤10 mg de prednisona) y/o AINEs concomitantes durante el ensayo. En la aleatorización, el 60% de los pacientes estaban recibiendo MTX. Las características basales de la enfermedad incluyeron la presencia de erosión articular en radiografías en el 84% (341/407) con una puntuación media (DE) de erosión de Sharp van der Heijde modificada para PsA (SHS) de 10.8 (24.2), proteína C reactiva sérica elevada (PCR) en el 66% [277/421]) con una media (DE) de 14.1 mg/L (25.9), y enfermedad poliarticular en el 98% (416/424) de los pacientes con un número medio (DE) de articulaciones sensibles e inflamadas de 20.2 (13.3) y 11.6 (7.5), respectivamente. Los pacientes que no habían logrado al menos una mejora del 20% con respecto al valor basal en sus recuentos de articulaciones inflamadas y sensibles para la Semana 16 escaparon a ORENCIA subcutáneo de etiqueta abierta 125 mg semanalmente.
El criterio de valoración principal tanto para PsA-I como para PsA-II fue la proporción de pacientes que lograron una respuesta ACR 20 en la Semana 24 (Día 169).
Respuesta Clínica en Adultos con PsA
Una mayor proporción de pacientes adultos con PsA logró una respuesta ACR 20 después del tratamiento con ORENCIA intravenoso (dosificación basada en el rango de peso como se describe anteriormente) en comparación con el placebo en el Estudio PsA-I y una mayor proporción de pacientes adultos con PsA logró una respuesta ACR 20 después del tratamiento con 125 mg subcutáneos en comparación con el placebo en el Estudio PsA-II en la Semana 24. Se observaron respuestas independientemente del tratamiento previo con antagonistas del TNF e independientemente del tratamiento concomitante con FARME no biológicos. El porcentaje de pacientes que lograron respuestas ACR 20, 50 o 70 en los Estudios PsA-I y PsA-II se presentan en la Tabla 13 a continuación.
| PsA-I | PsA-II | |||
|---|---|---|---|---|
| * p<0.05 versus placebo. a Los pacientes que tuvieron una mejora menor al 20% en el recuento de articulaciones sensibles o inflamadas en la Semana 16 cumplieron con los criterios de escape y se consideraron no respondedores. b Dosificación intravenosa basada en el rango de peso: ORENCIA 500 mg para pacientes que pesan menos de 60 kg, ORENCIA 750 mg para pacientes que pesan entre 60 y 100 kg, y ORENCIA 1,000 mg para pacientes que pesan más de 100 kg. |
||||
|
ORENCIA |
|
ORENCIA 125 mg |
|
|
|
ACR 20 |
47.5%* |
19.0% |
39.4%* |
22.3% |
|
ACR 50 |
25.0% |
2.4% |
19.2% |
12.3% |
|
ACR 70 |
12.5% |
0% |
10.3% |
6.6% |
El porcentaje de pacientes en PsA-II que lograron una respuesta ACR 20 hasta la semana 24 se muestra a continuación en la Figura 2.
Figura 2: Porcentaje de pacientes que logran una respuesta ACR 20a en el estudio PsA-II hasta la semana 24 (día 169)

Los resultados fueron generalmente consistentes entre los componentes de ACR en los estudios PsA-I y PsA-II.
Se observaron mejoras en la entesitis y la dactilitis con el tratamiento con ORENCIA en la semana 24 tanto en PsA-I como en PsA-II.
Respuesta de la función física en adultos con PsA
En el estudio PsA-I, hubo una mayor proporción de pacientes con al menos una disminución de 0.30 desde el inicio en la puntuación del Índice de Discapacidad del Cuestionario de Evaluación de la Salud (HAQ-DI) en la semana 24, con una diferencia estimada para la dosificación de ORENCIA basada en el rango de peso como se describe anteriormente (45%) vs. placebo (19%) de 26.1 (intervalo de confianza del 95%: 6.8, 45.5). En el estudio PsA-II, la proporción de pacientes con al menos una disminución de 0.35 desde el inicio en el HAQ-DI con ORENCIA fue del 31%, en comparación con el 24% con placebo (diferencia estimada: 7%; intervalo de confianza del 95%: -1%, 16%). Hubo un mayor cambio medio ajustado desde el inicio en el HAQ-DI con ORENCIA (-0.33) vs. placebo (-0.20) en la semana 24, con una diferencia estimada de -0.13 (intervalo de confianza del 95%: -0.25, -0.01).
14.4 Profilaxis de la enfermedad aguda de injerto contra huésped
Estudio GVHD-1
La eficacia de ORENCIA, en combinación con un inhibidor de la calcineurina (CNI) y metotrexato (MTX), para la profilaxis de la enfermedad aguda de injerto contra huésped (aGVHD), se evaluó en un estudio clínico multicéntrico de dos cohortes (GVHD-1, NCT01743131) en pacientes de 6 años o mayores que se sometieron a un trasplante de células madre hematopoyéticas (HSCT) de un donante no emparentado compatible o con un desajuste de 1 alelo (URD). Las dos cohortes en GVHD-1 incluyeron:
- 1)
- un estudio abierto, de un solo brazo, de 43 pacientes que se sometieron a un trasplante de HSCT compatible con 7 de 8 antígenos leucocitarios humanos (HLA) (cohorte de 7 de 8); y
- 2)
- un estudio aleatorizado (1:1), doble ciego, controlado con placebo, de pacientes que se sometieron a un trasplante de HSCT compatible con 8 de 8 HLA que recibieron ORENCIA o placebo en combinación con un CNI y MTX (cohorte de 8 de 8).
Tanto en la cohorte de 7/8 como en la de 8/8, ORENCIA se administró a una dosis de 10 mg/kg (dosis máxima de 1000 mg) como infusión intravenosa durante 60 minutos, comenzando el día anterior al trasplante (día -1), seguido de la administración en los días 5, 14 y 28 después del trasplante. Las características demográficas y clínicas basales de las cohortes de 7 de 8 y 8 de 8 se describen a continuación en la Tabla 14.
|
Cohorte de 7 de 8 |
Cohorte de 8 de 8 |
||
|
ORENCIA (+ CNI y MTX) N=43 |
ORENCIA (+ CNI y MTX) N=73 |
Placebo (+CNI y MTX) N=69 |
|
|
Edad – Mediana |
38 |
44 |
40 |
|
Edad – Rango |
6-76 |
6-71 |
7-74 |
|
Sexo – Masculino |
27 (63) |
41 (56) |
37 (54) |
|
Blanco |
31 (72) |
63 (86) |
61 (88) |
|
Negro o afroamericano |
7 (16) |
3 (4.1) |
2 (2.9) |
|
Asiático |
2 (4.7) |
4 (6) |
2 (2.9) |
|
Hispano |
7 (16) |
4 (6) |
2 (2.9) |
|
Malignancy type |
|||
|
Acute Myeloid Leukemia (AML) |
15 (35) |
30 (41) |
22 (32) |
|
Myelodysplastic Syndrome (MDS) |
11 (26) |
15 (21) |
12 (17) |
|
Acute Lymphoblastic Leukemia (ALL) |
8 (19) |
20 (27) |
22 (32) |
|
Leucemia aguda o de linaje ambiguo |
1 (2.3) |
0 |
1 (1.4) |
|
Linfoma de Hodgkin y no Hodgkin |
1 (2.3) |
1 (1.4) |
1 (1.4) |
|
Linfoma linfoblástico agudo en segunda remisión completa o mayor |
1 (2.3) |
4 (6) |
1 (1.4) |
|
Leucemia mielomonocítica crónica |
1 (2.3) |
1 (1.4) |
4 (6) |
|
Leucemia mielógena crónica |
4 (9) |
1 (1.4) |
5 (7) |
|
No reportado |
1 (2.3) |
1 (1.4) |
1 (1.4) |
|
Profilaxis de GVHD |
|||
|
Ciclosporina |
16 (37) |
11 (15) |
11 (16) |
|
Tacrolimus |
27 (63) |
62 (85) |
58 (84) |
|
Tipo de injerto |
|||
|
Médula ósea |
21 (49) |
33 (45) |
26 (38) |
|
Sangre periférica movilizada por citocinas (PBSC) |
22 (51) |
40 (55) |
43 (62) |
|
Régimen de acondicionamiento |
|||
|
TBI y quimioterapia |
11 (26) |
20 (27) |
26 (38) |
|
Busulfan and Cyclophosphamide |
13 (30) |
28 (38) |
21 (30) |
|
Busulfan and Fludarabine |
8 (19) |
7 (10) |
2 (2.9) |
|
Melphalan and Fludarabine |
11 (26) |
18 (25) |
20 (29) |
La eficacia se estableció en base a los resultados de supervivencia general (SG) y supervivencia libre de aGVHD de grado II-IV (SLG) evaluados en el día 180 después del trasplante. ORENCIA + CNI y MTX no mejoraron significativamente la SLG de grado III-IV en comparación con placebo + CNI y MTX en el día 180 después del trasplante. Los resultados de eficacia de la cohorte 8 de 8 de GVHD-1 se muestran en la Tabla 15.
| a La supervivencia libre de aGVHD de grado III-IV se midió desde la fecha del trasplante hasta la aparición de aGVHD de grado III-IV documentada, o la muerte por cualquier causa hasta el día 180 después del trasplante. b La supervivencia libre de aGVHD de grado II-IV se midió desde la fecha del trasplante hasta la aparición de aGVHD de grado II-IV documentada, o la muerte por cualquier causa hasta el día 180 después del trasplante. |
||
|
Endpoint |
ORENCIA (+CNI and MTX) n=73 |
Placebo (+CNI and MTX) n=69 |
|
Gr III-IV aGVHD Free Survivala Rate (95% CI) |
87% (77%, 93%) |
75% (63%, 84%) |
|
Hazard Ratio (95% CI) |
0.55 (0.26, 1.18) |
|
|
Gr II-IV aGVHD Free Survivalb Rate (95% CI) |
50% (38%, 61%) |
32% (21%, 43%) |
|
Hazard Ratio (95% CI) |
0.54 (0.35, 0.83) |
|
|
Overall Survival Rate (95% CI) |
97% (89%, 99%) |
84% (73%, 91%) |
|
Hazard Ratio (95% CI) |
0.33 (0.12, 0.93) |
|
En un análisis exploratorio de la cohorte 7 de 8 de pacientes tratados con ORENCIA (n=43), las tasas de supervivencia libre de GVHD de grado III-IV, supervivencia libre de GVHD de grado II-IV y supervivencia general en el día 180 después del trasplante fueron del 95 % (IC del 95 %: 83 %, 99 %), 53 % (IC del 95 %: 38 %, 67 %) y 98 % (IC del 95 %: 85 %, 100 %), respectivamente.
Study GVHD-2
GVHD-2 (NCT05421299) fue un estudio clínico que utilizó datos del Center for International Blood and Marrow Transplant Research (CIBMTR). El estudio analizó los resultados de ORENCIA en combinación con un CNI y MTX, frente a un CNI y MTX solo, para la profilaxis de la aGVHD, en pacientes de 6 años o mayores que se sometieron a un HSCT de un URD con desajuste de 1 alelo entre 2011 y 2018. El grupo tratado con ORENCIA + CNI y MTX (n=54) incluyó a 42 pacientes de GVHD-1, además de 12 pacientes tratados con ORENCIA fuera de GVHD-1. El grupo comparador (n=162) se seleccionó aleatoriamente en una proporción de 3:1 con respecto al grupo tratado con ORENCIA del registro CIBMTR de pacientes que no habían recibido ORENCIA durante el período de estudio. Los análisis utilizaron la correspondencia de puntuaciones de propensión y la probabilidad inversa de ponderación del tratamiento para ayudar a abordar el impacto del sesgo de selección.
La eficacia se basó en la supervivencia general (SG) en el día 180 después del HSCT. La tasa de SG en el día 180 en el grupo de ORENCIA en combinación con CNI y MTX fue del 98 % (IC del 95 %: 78, 100) y la tasa de SG en el día 180 en el grupo de CNI y MTX fue del 75 % (IC del 95 %: 67, 82).
16 SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
Para Infusión Intravenosa
ORENCIA® (abatacept) para inyección es un polvo liofilizado blanco para infusión intravenosa después de la reconstitución y dilución. Se suministra como un vial de dosis única, envasado individualmente (se puede usar menos del contenido total del vial o más de un vial) con una jeringa desechable sin silicona, que proporciona 250 mg de abatacept:
NDC 0003-2187-10: en presentación tipo concha
NDC 0003-2187-13: en presentación de cartón
Para Uso Subcutáneo
ORENCIA® (abatacept) inyección y ORENCIA® ClickJect (abatacept) inyección son soluciones transparentes a ligeramente opalescentes, incoloras a amarillo pálido para administración subcutánea.
Jeringa Prellenada
ORENCIA (abatacept) inyección, 50 mg/0.4 mL, 87.5 mg/0.7 mL y 125 mg/mL, se suministra como jeringas de vidrio prellenadas desechables de dosis única con protector de aguja pasivo BD UltraSafe™ y extensores de brida.
La jeringa de vidrio Tipo I tiene un tapón recubierto y una aguja de acero inoxidable fija (bisel 5, calibre 29 de pared delgada, aguja de ½ pulgada) cubierta con un protector de aguja rígido. La jeringa prellenada proporciona ORENCIA en los siguientes envases:
NDC 0003-2814-11 (50 mg/0.4 mL): paquete de 4 jeringas con protector de seguridad pasivo para agujas
NDC 0003-2818-11 (87.5 mg/0.7 mL): paquete de 4 jeringas con protector de seguridad pasivo para agujas
NDC 0003-2188-11 (125 mg/mL): paquete de 4 jeringas con protector de seguridad pasivo para agujas
Autoinyector ClickJect
ORENCIA (abatacept) ClickJect, 125 mg/mL, se suministra como un autoinyector prellenado desechable de dosis única. La jeringa de vidrio Tipo I contenida en el autoinyector tiene un tapón recubierto y una aguja de acero inoxidable fija (bisel 5, calibre 27 de pared delgada especial, aguja de ½ pulgada) cubierta con un protector de aguja rígido. El autoinyector proporciona 125 mg de abatacept en 1 mL y se proporciona en el siguiente envase:
NDC 0003-2188-51: paquete de 4 autoinyectores
Almacenamiento
Refrigerar el polvo liofilizado ORENCIA suministrado en un vial a 2°C a 8°C (36°F a 46°F). No usar después de la fecha de caducidad que figura en el vial. Proteger los viales de la luz almacenándolos en el envase original hasta el momento de su uso.
Refrigerar la solución ORENCIA suministrada en una jeringa prellenada o autoinyector ClickJect a 2°C a 8°C (36°F a 46°F). No usar después de la fecha de caducidad que figura en la jeringa prellenada o el autoinyector. Proteger de la luz almacenándolos en el envase original hasta el momento de su uso. No permitir que la jeringa prellenada o el autoinyector se congelen.
17 INFORMACIÓN DE ASESORAMIENTO AL PACIENTE
Aconseje al paciente que lea el etiquetado del paciente aprobado por la FDA (Información para el paciente e Instrucciones de uso).
Mayor riesgo de infección con el uso concomitante con inmunosupresores para la AR
Informe a los pacientes que no se recomienda el uso concomitante con otros inmunosupresores (por ejemplo, DMARD biológicos, inhibidores de JAK) [ver Advertencias y precauciones (5.1) y Interacciones medicamentosas (7.1)].
Reacciones de hipersensibilidad
Indique a los pacientes que informen inmediatamente a su profesional sanitario si experimentan síntomas de una reacción alérgica el día de la administración o el día después de la administración de ORENCIA [ver Advertencias y precauciones (5.2)].
Infecciones
Informe a los pacientes que se han notificado infecciones graves en pacientes que reciben ORENCIA [ver Advertencias y precauciones (5.3)].
Inmunizaciones
Informe a los pacientes que no se deben administrar vacunas vivas de forma concomitante con ORENCIA o en los 3 meses posteriores a su interrupción [ver Advertencias y precauciones (5.4)].
Análisis de glucosa en sangre
ORENCIA para administración intravenosa contiene maltosa y puede proporcionar lecturas falsamente elevadas de glucosa en sangre con ciertos monitores de glucosa en sangre el día de la infusión de ORENCIA. Aconseje a los pacientes tratados con ORENCIA intravenosa que estén utilizando sistemas de monitorización de glucosa basados en GDH-PQQ (por ejemplo, diabéticos) que consideren el uso de otros métodos para la monitorización de la glucosa. Esta recomendación no es aplicable a los pacientes tratados con ORENCIA subcutánea [ver Interacciones medicamentosas (7.2)].
Eliminación de jeringas precargadas y autoinyectores ClickJect
Aconseje a los pacientes que sigan las instrucciones de eliminación que figuran en las Instrucciones de uso. Debe utilizarse un recipiente resistente a las perforaciones para la eliminación de agujas y jeringas. Indique a los pacientes que deberán seguir las directrices de su comunidad para la forma correcta de desechar su recipiente de eliminación de objetos punzantes. Indique a los pacientes que no deben reciclar su recipiente de eliminación de objetos punzantes usado.
SECCIÓN NO CLASIFICADA DE SPL
Bristol-Myers Squibb Company
Princeton, Nueva Jersey 08543 EE. UU.
Número de licencia de EE. UU. 1713
PROSPECTO PARA EL PACIENTE
|
INFORMACIÓN PARA EL PACIENTE
ORENCIA®(oh-REN-see-ah) |
|||||
|
¿Qué es ORENCIA?
ORENCIA también se usa para el tratamiento preventivo de la enfermedad aguda del injerto contra el huésped (EICH), en combinación con un inhibidor de la calcineurina y metotrexato, en:
No se sabe si ORENCIA es seguro y eficaz en niños menores de dos años para el tratamiento de la AIJp. No se sabe si ORENCIA es seguro y eficaz en niños menores de dos años para el tratamiento de la APs. No se sabe si ORENCIA es seguro y eficaz en niños menores de dos años para el tratamiento preventivo de la EICH. |
|||||
|
Antes de recibir o usar ORENCIA, informe a su proveedor de atención médica sobre todas sus afecciones médicas, incluso si:
|
|||||
|
|
||||
|
|||||
|
|||||
|
|||||
|
|||||
|
Informe a su proveedor de atención médica sobre todos los medicamentos que toma, incluidos los medicamentos recetados y de venta libre, las vitaminas y los suplementos herbales. ORENCIA puede afectar la forma en que funcionan otros medicamentos, y otros medicamentos pueden afectar la forma en que funciona ORENCIA causando efectos secundarios graves. Especialmente, informe a su proveedor de atención médica si toma otros medicamentos biológicos que puedan afectar su sistema inmunitario, como: |
|||||
- •
- Enbrel® (etanercept)
- •
- Humira® (adalimumab)
- •
- Remicade® (infliximab)
- •
- Kineret® (anakinra)
- •
- Rituxan® (rituximab)
- •
- Simponi® (golimumab)
- •
- Cimzia® (certolizumab pegol)
- •
- Actemra® (tocilizumab)
Si toma ORENCIA con otros medicamentos biológicos que pueden afectar su sistema inmunitario, puede tener una mayor probabilidad de contraer una infección grave.
Conozca los medicamentos que toma. Lleve una lista de ellos para mostrársela a su proveedor de atención médica y farmacéutico cuando reciba una nueva receta médica.
¿Cómo recibiré o usaré ORENCIA?
Para el tratamiento de AR, ARJp o PsA:
- •
- Puede recibir ORENCIA administrado por un proveedor de atención médica a través de una vena en su brazo (infusión intravenosa). Se tarda unos 30 minutos en administrarle la dosis completa del medicamento. Luego, recibirá ORENCIA 2 semanas y 4 semanas después de la primera dosis y luego cada 4 semanas. La administración intravenosa de ORENCIA no está aprobada para pacientes pediátricos con artritis psoriásica.
- •
- También puede recibir ORENCIA como una inyección debajo de la piel (subcutánea). Para uso doméstico, ORENCIA viene en una jeringa precargada o en un autoinyector ClickJect precargado. Su proveedor de atención médica le recetará el tipo que sea mejor para usted. Si su proveedor de atención médica decide que usted o un cuidador pueden administrarse las inyecciones de jeringas precargadas de ORENCIA o autoinyectores ClickJect de ORENCIA en casa, usted o su cuidador deben recibir capacitación sobre la forma correcta de preparar e inyectar ORENCIA. No intente inyectarse ORENCIA hasta que su proveedor de atención médica le haya mostrado la forma correcta de administrar las inyecciones.
- •
- Su proveedor de atención médica le indicará cuánto ORENCIA usar y cuándo usarlo.
Consulte las instrucciones de uso al final de este folleto de información para el paciente para obtener instrucciones sobre la forma correcta de preparar y administrar sus inyecciones de ORENCIA en casa.
Para el tratamiento preventivo de la aGVHD:
- •
- Recibirá ORENCIA administrado por un proveedor de atención médica a través de una vena en su brazo (infusión intravenosa) durante 60 minutos el día anterior al trasplante (Día -1). Luego, recibirá ORENCIA los días 5, 14 y 28 después del trasplante.
- •
- Su proveedor de atención médica puede administrarle medicamentos antivirales antes, durante y después del trasplante para ayudar a prevenir infecciones por el virus de Epstein-Barr (VEB) y el citomegalovirus (CMV).
¿Cuáles son los posibles efectos secundarios de ORENCIA?
ORENCIA puede causar efectos secundarios graves, que incluyen:
- •
- infecciones. ORENCIA puede aumentar la probabilidad de que contraiga infecciones o empeorar la infección que ya tiene. Algunas personas han muerto a causa de estas infecciones. Llame a su proveedor de atención médica de inmediato si tiene algún síntoma de infección. Los síntomas de una infección pueden incluir:
- o
- fiebre
- o
- sensación de cansancio extremo
- o
- tos
- o
- síntomas similares a la gripe
- o
- piel caliente, roja o dolorosa
- •
- reacciones alérgicas. Las reacciones alérgicas pueden ocurrir en personas que reciben tratamiento con ORENCIA. Llame a su proveedor de atención médica o vaya a la sala de emergencias de inmediato si tiene algún síntoma de una reacción alérgica. Los síntomas de una reacción alérgica pueden incluir:
- o
- ronchas
- o
- cara, párpados, labios o lengua hinchados
- o
- dificultad para respirar
- •
- infección por hepatitis B en personas que portan el virus en la sangre. Si usted es portador del virus de la hepatitis B (un virus que afecta el hígado), el virus puede activarse durante el tratamiento con ORENCIA. Su proveedor de atención médica puede hacerle un análisis de sangre antes de comenzar el tratamiento con ORENCIA.
- •
- vacunaciones. No debe recibir ORENCIA con ciertos tipos de vacunas (vacunas vivas). Puede recibir vacunas no vivas, como las vacunas antineumocócicas y la vacuna inactivada contra la influenza (gripe). ORENCIA también puede hacer que algunas vacunas sean menos efectivas. Hable con su proveedor de atención médica sobre sus planes de vacunación.
- •
- problemas respiratorios en personas con enfermedad pulmonar obstructiva crónica (EPOC). Puede tener ciertos problemas respiratorios con más frecuencia si recibe ORENCIA y tiene EPOC. Los síntomas de los problemas respiratorios incluyen:
- o
- EPOC que empeora
- o
- tos
- o
- dificultad para respirar
- •
- cáncer (tumores malignos). Se han notificado ciertos tipos de cáncer en personas que usan ORENCIA. No se sabe si ORENCIA aumenta la probabilidad de contraer ciertos tipos de cáncer.
- •
- Infecciones por citomegalovirus (CMV) y virus de Epstein-Barr (VEB). Se han producido infecciones por CMV y VEB y la reaparición del CMV y el VEB (reactivación) en personas que recibieron ORENCIA para el tratamiento preventivo de la aGVHD durante el TCH no relacionado. Su proveedor de atención médica lo controlará durante 6 meses después del trasplante y puede tratarlo con medicamentos para ayudar a prevenir la infección por CMV y VEB si es necesario.
Los efectos secundarios más comunes de ORENCIA en personas con AR incluyen:
|
|
||||
|
En niños y adolescentes, otros efectos secundarios pueden incluir: |
|||||
|
|
||||
|
Los efectos secundarios más comunes de ORENCIA en la prevención de la aGVHD incluyen: |
|||||
|
|
||||
|
Estos no son todos los posibles efectos secundarios de ORENCIA. Consulte a su médico para obtener asesoramiento médico sobre los efectos secundarios. Puede informar los efectos secundarios a la FDA al 1-800-FDA-1088. |
|||||
|
¿Cómo debo almacenar ORENCIA?
Mantenga ORENCIA y todos los medicamentos fuera del alcance de los niños. |
|||||
|
Información general sobre el uso seguro y eficaz de ORENCIA. A veces, los medicamentos se recetan para fines distintos de los que figuran en un prospecto de información para el paciente. No use ORENCIA para una afección para la que no se lo recetaron. No le dé ORENCIA a otras personas, incluso si tienen los mismos síntomas que usted. Podría causarles daño. Puede pedirle a su farmacéutico o proveedor de atención médica información sobre ORENCIA que esté escrita para profesionales de la salud. |
|||||
|
¿Cuáles son los ingredientes de ORENCIA? Ingrediente activo: abatacept Ingredientes inactivos intravenosos: maltosa, fosfato monosódico, cloruro de sodio para administración Ingredientes inactivos subcutáneos: sacarosa, poloxámero 188, fosfato monosódico monohidrato, fosfato disódico anhidro, agua para inyección. |
|||||
|
Bristol-Myers Squibb Company Todas las demás marcas comerciales son propiedad de sus respectivos dueños. Para obtener más información, visite www.ORENCIA.com o llame al 1-800-ORENCIA. |
|||||
Esta información para el paciente ha sido aprobada por la Administración de Alimentos y Medicamentos de EE. UU.
Revisado: 5/2024
INSTRUCCIONES DE USO
ORENCIA®(oh-REN-see-ah)
(abatacept)
Jeringa precargada con protector de aguja pasivo BD UltraSafe™




Paso 1: Preparación para una inyección de ORENCIA






Paso 2: Examine la jeringa precargada


Paso 3: Compruebe la dosis en la jeringa precargada

Paso 4: Elija y prepare un sitio de inyección



Paso 5: Inyecte su dosis de ORENCIA



Paso 6: Después de la inyección
|
Cuidado del sitio de inyección:
|
 |
||
|
Desecho de jeringas precargadas usadas:
|
|||
|
|
|
||
|
|||
|
Consulte las Preguntas frecuentes para obtener información adicional sobre el desecho. Si un cuidador le administra la inyección, esta persona también debe tener cuidado al manipular la jeringa para evitar lesiones accidentales por pinchazo de aguja y posiblemente la propagación de infecciones. Mantenga las jeringas precargadas ORENCIA y el contenedor para objetos punzocortantes fuera del alcance de los niños. |
 |
||
|
Cómo almacenar la jeringa precargada ORENCIA
|
|||
|
Ir a la siguiente página |
|||
|
Preguntas frecuentes |
|||
|
P. |
¿Por qué debo dejar que la jeringa precargada se caliente a temperatura ambiente durante 30 minutos antes de inyectarla? |
||
|
R. |
Este paso es principalmente para su comodidad. Nunca intente acelerar el proceso de calentamiento de ninguna manera, como usar el microondas o colocar la jeringa en agua tibia. |
||
|
P. |
¿Es necesario mantener el pellizco de la piel durante todo el tiempo que me inyecto la dosis? |
||
|
R. |
Debe pellizcar la piel durante la inserción de la aguja; sin embargo, para su comodidad, puede soltar el pellizco de la piel mientras administra la inyección. |
||
|
P. |
¿Qué pasa si mi jeringa precargada parece estar rota o dañada? |
||
|
R. |
No use la jeringa precargada. Comuníquese con su proveedor de atención médica o farmacéutico para obtener más instrucciones. |
||
|
P. |
¿Qué pasa si no puedo ver claramente el líquido dentro de la jeringa? |
||
|
R. |
Mire la jeringa de cerca sosteniéndola a la altura de los ojos y hacia la luz. Puede inclinar la jeringa lentamente para obtener una mejor vista del líquido del medicamento. Si todavía tiene problemas, comuníquese con su proveedor de atención médica o farmacéutico para obtener más instrucciones. |
||
|
P. |
¿Es normal sentir un poco de ardor o dolor durante la inyección? |
||
|
A. |
Puede sentir un pinchazo de la aguja. A veces, el medicamento puede causar una ligera irritación cerca del sitio de inyección. Las molestias deben ser leves o moderadas. Si tiene algún efecto secundario, incluyendo dolor, hinchazón o decoloración cerca del sitio de inyección, comuníquese con su proveedor de atención médica. |
||
|
Ir a la siguiente página |
|||
|
Preguntas frecuentes |
|||
|
P. |
¿Cómo debo desechar una jeringa precargada usada? |
||
|
A. |
Coloque la jeringa precargada usada en un contenedor para objetos punzocortantes aprobado por la FDA. Si no tiene uno, puede usar un recipiente doméstico que sea:
|
||
|
P. |
¿Cómo debo mantener mis jeringas precargadas frías mientras viajo? |
||
|
A. |
Guárdelas en una nevera portátil a una temperatura entre 2ºC y 8ºC (36ºF a 46ºF). |
||
|
P. |
¿Puedo llevar mis jeringas precargadas en un avión? |
||
|
A. |
Generalmente, se le permite llevar sus jeringas precargadas consigo en un avión. No las ponga en su equipaje facturado. Debe llevar sus jeringas precargadas consigo en su nevera portátil a una temperatura de 2ºC a 8ºC (36ºF a 46ºF). Mantenga sus jeringas precargadas en la caja original, con sus etiquetas preimpresas originales y protegidas de la luz. |
||
|
P. |
¿Qué sucede si mi jeringa precargada no se mantiene fría durante un período de tiempo prolongado? ¿Es peligroso usarla? |
||
|
A. |
Comuníquese con el 1-800-673-6242 para obtener más información. |
||
|
Ir a la contraportada |
|||
|
Si tiene preguntas o inquietudes sobre su jeringa precargada, comuníquese con su proveedor de atención médica o llame a nuestra línea de ayuda gratuita al 1-800-673-6242. |
|||
Bristol-Myers Squibb Company
Princeton, NJ 08543 EE. UU., Número de licencia de EE. UU. 1713
Estas instrucciones de uso han sido aprobadas por la Administración de Alimentos y Medicamentos de EE. UU.
ORENCIA es una marca registrada de Bristol-Myers Squibb Company.
BD UltraSafe Passive™ es una marca registrada de Becton, Dickinson, and Company.
Revisado 10/2023
INSTRUCCIONES DE USO
ORENCIA® ClickJect™(oh-REN-see-ah)
(abatacept)
Autoinyector precargado








Paso 1: Prepare su autoinyector


Paso 2: Preparación para la inyección



Paso 3: Inyección de la dosis


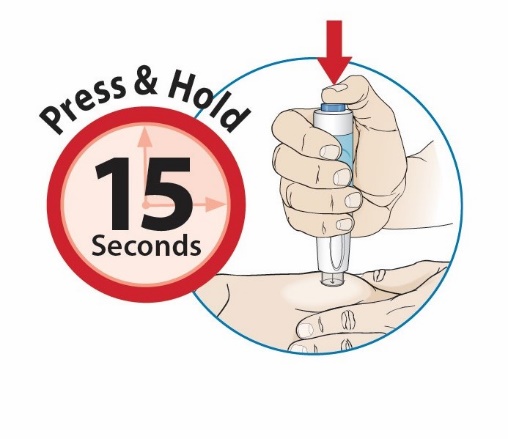

Paso 4: Después de la inyección
|
Cuidado del sitio de inyección:
|
  |
||
|
Desecho de los autoinyectores ClickJect usados:
|
|||
|
|
|
||
|
|||
|
Consulte las Preguntas frecuentes para obtener información adicional sobre el desecho. Si un cuidador le administra la inyección, esta persona también debe manipular el autoinyector con cuidado para evitar lesiones accidentales por pinchazos de aguja y posiblemente la propagación de infecciones. |
 |
||
|
Mantenga el autoinyector y el contenedor para objetos punzocortantes fuera del alcance de los niños. Cómo almacenar el autoinyector ClickJect de ORENCIA
|
|||
|
Continúa en la página siguiente |
|||
|
|
|||
|
Preguntas frecuentes |
|||
|
P. |
¿Por qué debo dejar que el autoinyector se caliente a temperatura ambiente durante 30 minutos antes de inyectarlo? |
||
|
R. |
Este paso es principalmente para su comodidad. Si el medicamento está frío, la inyección puede tardar más de 15 segundos. Nunca intente acelerar el proceso de calentamiento de ninguna manera, como usar el microondas o colocar el autoinyector en agua tibia. |
||
|
P. |
¿Qué sucede si accidentalmente quito la cubierta de la aguja (tapa naranja) antes de estar listo para usar el autoinyector? |
||
|
R. |
Si quita la tapa antes de estar listo para usar el autoinyector, tenga cuidado. No intente volver a colocarla. Use el autoinyector lo antes posible. Mientras se prepara para la inyección, coloque cuidadosamente el autoinyector de lado sobre una superficie limpia y plana. Asegúrese de mantener el autoinyector fuera del alcance de los niños. |
||
|
P. |
¿Qué sucede si el autoinyector parece estar roto o dañado? |
||
|
R. |
No use el autoinyector. Comuníquese con su proveedor de atención médica o farmacéutico para obtener más instrucciones. |
||
|
P. |
¿Qué sucede si no se activó la inyección? |
||
|
A. |
Antes de que se pueda activar la inyección, el dispositivo debe desbloquearse. Para desbloquearlo, presione firmemente el autoinyector contra la piel sin tocar el botón. Cuando se sienta el punto de parada, el dispositivo estará desbloqueado y se podrá activar presionando el botón. |
||
|
P. |
Siento un poco de ardor o dolor durante la inyección. ¿Es esto normal? |
||
|
A. |
Al administrar una inyección, puede sentir un pinchazo de la aguja. A veces, el medicamento puede causar una ligera irritación cerca del sitio de inyección. Si esto ocurre, la molestia debe ser leve a moderada. Si experimenta algún efecto secundario, incluyendo dolor, hinchazón o decoloración cerca del sitio de inyección, comuníquese con su proveedor de atención médica o farmacéutico inmediatamente. Se le recomienda informar los efectos secundarios de los medicamentos recetados a la FDA. Visite www.fda.gov/medwatch o llame al 1-800-FDA-1088. |
||
|
P. |
¿Cómo sé que recibí mi dosis completa? |
||
|
A. |
Antes de levantar el autoinyector del sitio de inyección, verifique que el indicador azul haya dejado de moverse. Luego, antes de desechar el autoinyector, verifique la parte inferior de la ventana transparente para asegurarse de que no quede líquido adentro. Si el medicamento no se ha inyectado completamente, consulte a su proveedor de atención médica o farmacéutico. |
||
|
Continúa en la página siguiente |
|||
|
Preguntas Frecuentes |
|||
|
P. |
¿Cómo debo desechar un autoinyector usado? |
||
|
A. |
Coloque el autoinyector usado en un contenedor para objetos punzocortantes aprobado por la FDA inmediatamente después de su uso.
|
||
|
|||
|
|||
|
P. |
¿Cómo debo mantener mi autoinyector fresco mientras viajo? |
||
|
A. |
Su proveedor de atención médica o farmacéutico puede estar familiarizado con estuches especiales para medicamentos inyectables. Almacene en el refrigerador a una temperatura de 2°C a 8°C (36°F a 46°F). No congele. Proteja de la luz. |
||
|
P. |
¿Puedo llevar mi autoinyector a bordo de una aeronave? |
||
|
A. |
Generalmente, esto está permitido. Asegúrese de empacar su autoinyector en su equipaje de mano y no lo coloque en su equipaje facturado. Debe llevarlo consigo en su nevera de viaje a una temperatura de 2°C a 8°C (36°F a 46°F) hasta que esté listo para usarlo. Los procedimientos de seguridad aeroportuaria y las políticas de las aerolíneas cambian de vez en cuando, por lo que es mejor consultar con las autoridades aeroportuarias y la aerolínea para conocer las reglas especiales. Antes de volar, obtenga una carta de su proveedor de atención médica para explicar que viaja con un medicamento recetado que utiliza un dispositivo con una aguja; si lleva un contenedor para objetos punzocortantes en su equipaje de mano, notifique al agente de control en el aeropuerto. |
||
|
P. |
¿Qué pasa si mi autoinyector no se mantiene fresco durante un período de tiempo prolongado? ¿Es peligroso usarlo? |
||
|
A. |
Comuníquese al 1-800-673-6242 para obtener más detalles. |
||
|
|
|||
|
Si tiene preguntas o inquietudes sobre su autoinyector, comuníquese con un proveedor de atención médica o llame a nuestra línea de ayuda gratuita al 1-800-673-6242. |
|||
Bristol-Myers Squibb Company
Princeton, NJ 08543 EE. UU., Número de licencia de EE. UU. 1713
Estas instrucciones de uso han sido aprobadas por la Administración de Alimentos y Medicamentos de EE. UU.
ORENCIA es una marca registrada y ClickJect es una marca comercial de Bristol-Myers Squibb Company.
Revisado 1/2024
ORENCIA 250 mg/vial para inyección Envase representativo
Consulte la sección Presentación para obtener una lista completa de los envases disponibles de ORENCIA.
Rx solamente
NDC 0003-2187-13
ORENCIA®
(abatacept)
Polvo liofilizado para infusión IV
250 mg/vial
UTILICE ÚNICAMENTE LA JERINGA DESECHABLE SIN SILICONA INCLUIDA EN EL ENVASE PARA LA RECONSTITUCIÓN
Bristol-Myers Squibb Company
Princeton, New Jersey 08543 USA
Licencia de EE. UU. N.º 1713

ORENCIA 50 mg/0.4 mL Presentación representativa del inyectable
Bristol-Myers Squibb
NDC 0003-2814-11
4 Jeringas precargadas de dosis única con protector de aguja pasivo BD UltraSafe™
ORENCIA®
(abatacept)
Solución inyectable
50 mg/0.4mL
Jeringa precargada de dosis única con protector de aguja pasivo BD UltraSafe™
PARA USO SUBCUTÁNEO EXCLUSIVO
ADVERTENCIA: Manténgase fuera del alcance de los niños
Deseche cada jeringa después de su uso
Rx solamente

ORENCIA 87.5 mg/0.7 mL Envase Representativo de la Inyección
Bristol-Myers Squibb
NDC 0003-2818-11
4 Jeringas precargadas de dosis única con protector de aguja pasivo BD UltraSafe™
ORENCIA®
(abatacept)
Solución inyectable
87.5 mg/0.7mL
Jeringa precargada de dosis única con protector de aguja pasivo BD UltraSafe™
SOLO PARA USO SUBCUTÁNEO
ADVERTENCIA: Manténgase fuera del alcance de los niños
Deseche cada jeringa después de su uso
Rx solamente

ORENCIA 125 mg/mL Envase Representativo de la Inyección
Bristol-Myers Squibb
NDC 0003-2188-11
4 Jeringas precargadas de dosis única con protector de aguja pasivo BD UltraSafe™
ORENCIA®
(abatacept)
Solución inyectable
125 mg/mL
Jeringa precargada de dosis única con protector de aguja pasivo BD UltraSafe™
SOLO PARA USO SUBCUTÁNEO
ADVERTENCIA: Manténgase fuera del alcance de los niños
Deseche cada jeringa después de su uso
Rx solamente
ORENCIA 125 mg/mL Envase representativo ClickJect
Bristol-Myers Squibb
NDC 0003-2188-51
4 Autoinyectores prellenados de dosis única
ORENCIA® ClickJect™
(abatacept)
Inyección
125 mg/mL
Autoinyector de dosis única
SOLO PARA USO SUBCUTÁNEO
Refrigerar inmediatamente
ADVERTENCIA: Mantener fuera del alcance de los niños
Deseche cada autoinyector después de su uso
Abrir desde este lado Rx only
