Fabricante de medicamentos: E.R. Squibb & Sons, L.L.C. (Updated: 2024-03-06)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
OPDIVO® (nivolumab) inyección, para uso intravenoso
Aprobación inicial en EE. UU.: 2014
INDICACIONES Y USO
OPDIVO es un anticuerpo bloqueador del receptor 1 de muerte programada (PD-1) indicado para el tratamiento de:
Melanoma
- •
- pacientes adultos y pediátricos (de 12 años o más) con melanoma irresecable o metastásico, como agente único o en combinación con ipilimumab. (1.1)
- •
- para el tratamiento adyuvante de pacientes adultos y pediátricos de 12 años o más con melanoma completamente resecado en estadio IIB, estadio IIC, estadio III o estadio IV. (1.2)
Cáncer de pulmón de células no pequeñas (NSCLC)
- •
- pacientes adultos con cáncer de pulmón de células no pequeñas resecable (tumores ≥4 cm o ganglios positivos) en el contexto neoadyuvante, en combinación con quimioterapia de doblete de platino. (1.3)
- •
- pacientes adultos con cáncer de pulmón de células no pequeñas metastásico que expresa PD-L1 (≥1%) según lo determinado por una prueba aprobada por la FDA, sin aberraciones genómicas tumorales EGFR o ALK, como tratamiento de primera línea en combinación con ipilimumab. (1.4)
- •
- pacientes adultos con cáncer de pulmón de células no pequeñas metastásico o recurrente sin aberraciones genómicas tumorales EGFR o ALK como tratamiento de primera línea, en combinación con ipilimumab y 2 ciclos de quimioterapia de doblete de platino. (1.4)
- •
- pacientes adultos con cáncer de pulmón de células no pequeñas metastásico y progresión durante o después de la quimioterapia basada en platino. Los pacientes con aberraciones genómicas tumorales EGFR o ALK deben haber presentado progresión de la enfermedad durante la terapia aprobada por la FDA para estas aberraciones antes de recibir OPDIVO. (1.4)
Mesotelioma pleural maligno
- •
- pacientes adultos con mesotelioma pleural maligno irresecable, como tratamiento de primera línea en combinación con ipilimumab. (1.5)
Cáncer de células renales (RCC)
- •
- pacientes adultos con cáncer de células renales avanzado de riesgo intermedio o pobre, como tratamiento de primera línea en combinación con ipilimumab. (1.6)
- •
- pacientes adultos con cáncer de células renales avanzado, como tratamiento de primera línea en combinación con cabozantinib. (1.6)
- •
- pacientes adultos con cáncer de células renales avanzado que han recibido terapia antiangiogénica previa. (1.6)
Linfoma de Hodgkin clásico (cHL)
- •
- pacientes adultos con linfoma de Hodgkin clásico que ha recaído o progresado después dea: (1.7)
- •
- trasplante autólogo de células madre hematopoyéticas (HSCT) y brentuximab vedotin, o
- •
- 3 o más líneas de terapia sistémica que incluyen HSCT autólogo.
Carcinoma de células escamosas de cabeza y cuello (SCCHN)
- •
- pacientes adultos con carcinoma de células escamosas de cabeza y cuello recurrente o metastásico con progresión de la enfermedad durante o después de una terapia basada en platino. (1.8)
Carcinoma urotelial
- •
- tratamiento adyuvante de pacientes adultos con carcinoma urotelial (UC) que tienen un alto riesgo de recurrencia después de someterse a una resección radical de UC. (1.9)
- •
- pacientes adultos con carcinoma urotelial irresecable o metastásico, como tratamiento de primera línea en combinación con cisplatino y gemcitabina. (1.9)
- •
- pacientes adultos con carcinoma urotelial localmente avanzado o metastásico que:
- •
- tienen progresión de la enfermedad durante o después de la quimioterapia que contiene platino.
- •
- tienen progresión de la enfermedad dentro de los 12 meses del tratamiento neoadyuvante o adyuvante con quimioterapia que contiene platino. (1.9)
Cáncer colorrectal
- •
- pacientes adultos y pediátricos (de 12 años o más) con cáncer colorrectal metastásico de alta inestabilidad de microsatélites (MSI-H) o deficiencia de reparación de desajuste (dMMR) que ha progresado después del tratamiento con una fluoropirimidina, oxaliplatino e irinotecán, como agente único o en combinación con ipilimumab.a (1.10)
Carcinoma hepatocelular (HCC)
- •
- pacientes adultos con carcinoma hepatocelular que han sido tratados previamente con sorafenib en combinación con ipilimumab.a (1.11)
Cáncer de esófago
- •
- pacientes adultos con cáncer de esófago o unión gastroesofágica completamente resecado con enfermedad patológica residual, que han recibido quimiorradioterapia (CRT) neoadyuvante. (1.12)
- •
- pacientes adultos con carcinoma de células escamosas de esófago avanzado o metastásico irresecable como tratamiento de primera línea en combinación con quimioterapia que contiene fluoropirimidina y platino. (1.12)
- •
- pacientes adultos con carcinoma de células escamosas de esófago avanzado o metastásico irresecable como tratamiento de primera línea en combinación con ipilimumab. (1.12)
- •
- pacientes adultos con carcinoma de células escamosas de esófago (ESCC) avanzado, recurrente o metastásico irresecable después de quimioterapia previa basada en fluoropirimidina y platino. (1.12)
Cáncer gástrico, cáncer de unión gastroesofágica y adenocarcinoma de esófago
- •
- pacientes adultos con cáncer gástrico avanzado o metastásico, cáncer de la unión gastroesofágica y adenocarcinoma de esófago en combinación con quimioterapia que contiene fluoropirimidina y platino. (1.13)
a Esta indicación está aprobada bajo aprobación acelerada basada en la tasa de respuesta general y la duración de la respuesta. La aprobación continua para esta indicación puede estar sujeta a la verificación y descripción del beneficio clínico en ensayos confirmatorios.
DOSIFICACIÓN Y ADMINISTRACIÓN
- •
- Administrar por infusión intravenosa después de la dilución, según la velocidad de infusión recomendada para cada indicación. (2)
- •
- Melanoma irresecable o metastásico
- •
- Pacientes adultos y pediátricos que pesan 40 kg o más: 240 mg cada 2 semanas o 480 mg cada 4 semanas. (2.2)
- •
- Pacientes pediátricos que pesan menos de 40 kg: 3 mg/kg cada 2 semanas o 6 mg/kg cada 4 semanas. (2.2)
- •
- Pacientes adultos y pediátricos que pesan 40 kg o más: 1 mg/kg seguido de ipilimumab 3 mg/kg el mismo día cada 3 semanas durante 4 dosis, luego 240 mg cada 2 semanas o 480 mg cada 4 semanas. (2.2)
- •
- Pacientes pediátricos que pesan menos de 40 kg: 1 mg/kg seguido de ipilimumab 3 mg/kg el mismo día cada 3 semanas durante 4 dosis, luego 3 mg/kg cada 2 semanas o 6 mg/kg cada 4 semanas. (2.2)
- •
- Tratamiento adyuvante del melanoma
- •
- Tratamiento neoadyuvante del cáncer de pulmón de células no pequeñas resecable (tumores ≥4 cm o ganglios positivos)
- •
- 360 mg con quimioterapia de doblete de platino el mismo día cada 3 semanas durante 3 ciclos. (2.2)
- •
- Cáncer de pulmón de células no pequeñas metastásico
- •
- Mesotelioma pleural maligno
- •
- 360 mg cada 3 semanas con ipilimumab 1 mg/kg cada 6 semanas. (2.2)
- •
- Carcinoma de células renales avanzado
- •
- 3 mg/kg seguido de ipilimumab 1 mg/kg el mismo día cada 3 semanas durante 4 dosis, luego 240 mg cada 2 semanas o 480 mg cada 4 semanas. (2.2)
- •
- 240 mg cada 2 semanas o 480 mg cada 4 semanas administrados en combinación con cabozantinib 40 mg una vez al día sin alimentos. (2.2)
- •
- 240 mg cada 2 semanas o 480 mg cada 4 semanas. (2.2)
- •
- Linfoma de Hodgkin clásico
- •
- 240 mg cada 2 semanas o 480 mg cada 4 semanas. (2.2)
- •
- Carcinoma de células escamosas de cabeza y cuello recurrente o metastásico
- •
- 240 mg cada 2 semanas o 480 mg cada 4 semanas. (2.2)
- •
- Tratamiento adyuvante del carcinoma urotelial
- •
- 240 mg cada 2 semanas o 480 mg cada 4 semanas. (2.2)
- •
- Carcinoma urotelial metastásico o irresecable de primera línea
- •
- 360 mg cada 3 semanas con cisplatino y gemcitabina el mismo día durante un máximo de 6 ciclos, luego 240 mg cada 2 semanas o 480 mg cada 4 semanas. (2.2)
- •
- Carcinoma urotelial localmente avanzado o metastásico previamente tratado
- •
- 240 mg cada 2 semanas o 480 mg cada 4 semanas. (2.2)
- •
- Cáncer colorrectal metastásico de alta inestabilidad de microsatélites (MSI-H) o deficiencia de reparación de desajuste (dMMR)
- •
- Pacientes adultos y pediátricos que pesan 40 kg o más: 240 mg cada 2 semanas o 480 mg cada 4 semanas. (2.2)
- •
- Pacientes pediátricos que pesan menos de 40 kg: 3 mg/kg cada 2 semanas. (2.2)
- •
- Pacientes adultos y pediátricos que pesan 40 kg o más: 3 mg/kg seguido de ipilimumab 1 mg/kg el mismo día cada 3 semanas durante 4 dosis, luego 240 mg cada 2 semanas o 480 mg cada 4 semanas. (2.2)
- •
- Carcinoma hepatocelular
- •
- 1 mg/kg seguido de ipilimumab 3 mg/kg el mismo día cada 3 semanas durante 4 dosis, luego 240 mg cada 2 semanas o 480 mg cada 4 semanas. (2.2)
- •
- Tratamiento adyuvante del cáncer esofágico o gastroesofágico resecado
- •
- 240 mg cada 2 semanas o 480 mg cada 4 semanas durante una duración total del tratamiento de 1 año. (2.2)
- •
- Carcinoma de células escamosas esofágico
- •
- Cáncer gástrico, cáncer de la unión gastroesofágica y adenocarcinoma esofágico (GC, GEJC o EAC)
- •
- Consulte la información completa de prescripción para obtener instrucciones de preparación y administración y modificaciones de la dosis para reacciones adversas.
FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
- •
- Inyección: 40 mg/4 mL (10 mg/mL), 100 mg/10 mL (10 mg/mL), 120 mg/12 mL (10 mg/mL) y 240 mg/24 mL (10 mg/mL) solución en un vial de dosis única. (3)
CONTRAINDICACIONES
- •
- Ninguna. (4)
ADVERTENCIAS Y PRECAUCIONES
- •
-
Reacciones adversas inmunomediadas: (5.1)
- •
- Las reacciones adversas inmunomediadas, que pueden ser graves o fatales, pueden ocurrir en cualquier sistema de órganos o tejido, incluyendo las siguientes: neumonitis inmunomediada, colitis inmunomediada, hepatitis inmunomediada y hepatotoxicidad, endocrinopatías inmunomediadas, reacciones adversas dermatológicas inmunomediadas y nefritis inmunomediada y disfunción renal.
- •
- Monitorear para la identificación temprana y el manejo. Evaluar las enzimas hepáticas, la creatinina y la función tiroidea al inicio y periódicamente durante el tratamiento.
- •
- Suspender o suspender permanentemente según la gravedad y el tipo de reacción. (2.3)
- •
- Reacciones relacionadas con la infusión: Interromper, disminuir la velocidad de infusión o suspender permanentemente OPDIVO según la gravedad de la reacción. (5.2)
- •
- Complicaciones del trasplante de células madre hematopoyéticas alogénico (HSCT): Pueden ocurrir complicaciones fatales y otras graves en pacientes que reciben HSCT alogénico antes o después de ser tratados con un anticuerpo bloqueador de PD-1/PD-L1. (5.3)
- •
- Toxicidad embrio-fetal: Puede causar daño fetal. Advertir a las mujeres en edad fértil sobre el riesgo potencial para el feto y el uso de métodos anticonceptivos efectivos. (5.4, 8.1, 8.3)
- •
- No se recomienda el tratamiento de pacientes con mieloma múltiple con un anticuerpo bloqueador de PD-1 o PD-L1 en combinación con un análogo de talidomida más dexametasona fuera de ensayos clínicos controlados. (5.5)
REACCIONES ADVERSAS
Las reacciones adversas más comunes (incidencia ≥20%) en pacientes fueron:
- •
- Como agente único: fatiga, erupción cutánea, dolor musculoesquelético, prurito, diarrea, náuseas, astenia, tos, disnea, estreñimiento, disminución del apetito, dolor de espalda, artralgia, infección del tracto respiratorio superior, pirexia, cefalea, dolor abdominal, vómitos e infección del tracto urinario. (6.1)
- •
- En combinación con ipilimumab: fatiga, diarrea, erupción cutánea, prurito, náuseas, dolor musculoesquelético, pirexia, tos, disminución del apetito, vómitos, dolor abdominal, disnea, infección del tracto respiratorio superior, artralgia, cefalea, hipotiroidismo, estreñimiento, disminución de peso y mareos. (6.1)
- •
- En combinación con quimioterapia de doblete de platino: náuseas, fatiga, dolor musculoesquelético, estreñimiento, disminución del apetito, erupción cutánea, vómitos y neuropatía periférica. (6.1)
- •
- En combinación con ipilimumab y quimioterapia de doblete de platino: fatiga, dolor musculoesquelético, náuseas, diarrea, erupción cutánea, disminución del apetito, estreñimiento y prurito. (6.1)
- •
- En combinación con cabozantinib: diarrea, fatiga, hepatotoxicidad, síndrome de eritrodisestesia palmar-plantar, estomatitis, erupción cutánea, hipertensión, hipotiroidismo, dolor musculoesquelético, disminución del apetito, náuseas, disgeusia, dolor abdominal, tos e infección del tracto respiratorio superior. (6.1)
- •
- En combinación con quimioterapia que contiene fluoropirimidina y platino: náuseas, neuropatía periférica, disminución del apetito, fatiga, estreñimiento, estomatitis, diarrea, vómitos, dolor abdominal y dolor musculoesquelético. (6.1)
Para reportar REACCIONES ADVERSAS SOSPECHOSAS, comuníquese con Bristol-Myers Squibb al 1-800-721-5072 o con la FDA al 1-800-FDA-1088 o www.fda.gov/medwatch.
Ver 17 para INFORMACIÓN PARA EL PACIENTE y Guía de Medicamentos.
Revisado: 3/2024
Tabla de contenido
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDO*
1 INDICACIONES Y USO
1.1 Melanoma irresecable o metastásico
1.2 Tratamiento adyuvante del melanoma
1.3 Tratamiento neoadyuvante del cáncer de pulmón de células no pequeñas resecable
1.4 Cáncer de pulmón de células no pequeñas metastásico
1.5 Mesotelioma pleural maligno
1.6 Carcinoma de células renales avanzado
1.7 Linfoma de Hodgkin clásico
1.8 Carcinoma de células escamosas de cabeza y cuello
1.9 Carcinoma urotelial
1.10 Cáncer colorrectal metastásico con alta inestabilidad de microsatélites o deficiencia de reparación de desajuste
1.11 Carcinoma hepatocelular
1.12 Cáncer de esófago
1.13 Cáncer gástrico, cáncer de la unión gastroesofágica y adenocarcinoma de esófago
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Selección del paciente
2.2 Dosis recomendada
2.3 Modificaciones de la dosis
2.4 Preparación y administración
3 FORMAS Y FUERZAS DE DOSIFICACIÓN
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Reacciones adversas inmunomediadas graves y fatales
5.2 Reacciones relacionadas con la infusión
5.3 Complicaciones del trasplante alogénico de células madre hematopoyéticas
5.4 Toxicidad embrio-fetal
5.5 Aumento de la mortalidad en pacientes con mieloma múltiple cuando OPDIVO se agrega a un análogo de la talidomida y dexametasona
6 REACCIONES ADVERSAS
6.1 Experiencia en ensayos clínicos
6.2 Experiencia postcomercialización
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.3 Mujeres y hombres en edad fértil
8.4 Uso pediátrico
8.5 Uso geriátrico
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
12.2 Farmacodinamia
12.3 Farmacocinética
12.6 Inmunogenicidad
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, mutagénesis, deterioro de la fertilidad
13.2 Toxicología y/o farmacología animal
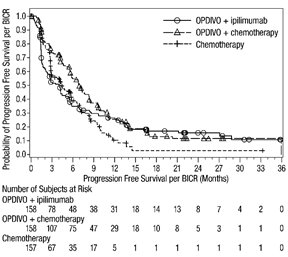
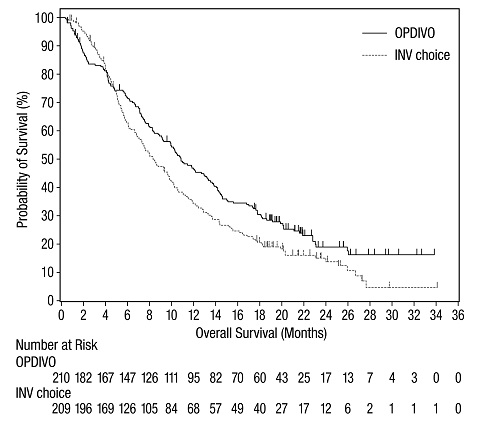
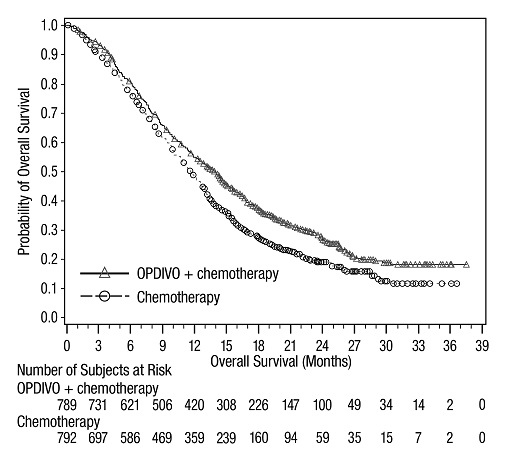
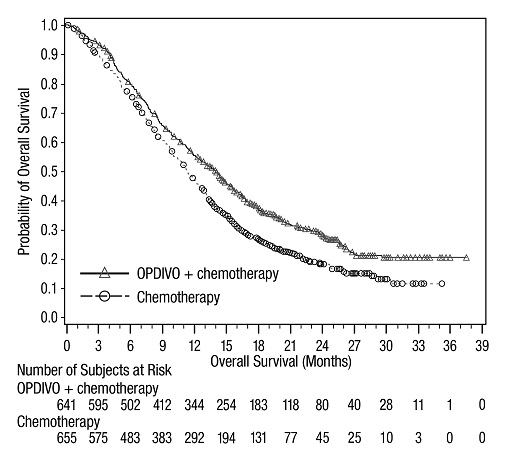
14 ESTUDIOS CLÍNICOS
14.1 Melanoma irresecable o metastásico
14.2 Tratamiento adyuvante del melanoma
14.3 Tratamiento neoadyuvante del cáncer de pulmón de células no pequeñas resecable (tumores ≥4 cm o ganglios positivos)
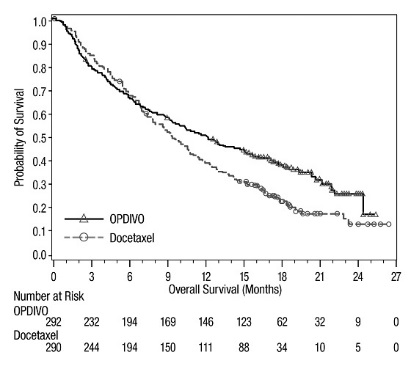
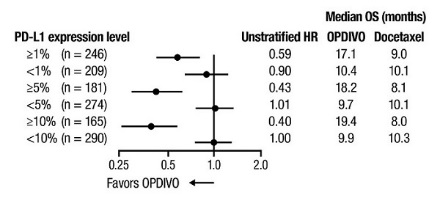
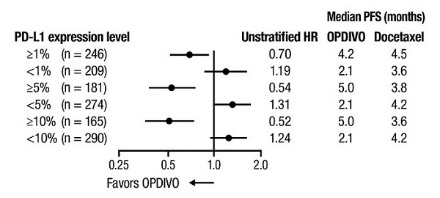
14.4 Cáncer de pulmón de células no pequeñas metastásico
14.5 Mesotelioma pleural maligno
14.6 Carcinoma de células renales avanzado
14.7 Linfoma de Hodgkin clásico
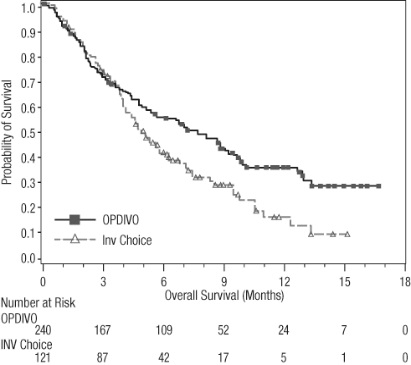
14.8 Carcinoma de células escamosas de cabeza y cuello recurrente o metastásico
14.9 Carcinoma urotelial
14.10 Cáncer colorrectal metastásico con alta inestabilidad de microsatélites o deficiencia de reparación de desajuste
14.11 Carcinoma hepatocelular
14.12 Cáncer de esófago
14.13 Cáncer gástrico, cáncer de la unión gastroesofágica y adenocarcinoma de esófago
16 CÓMO SE SUMINISTRA/ALMACENAMIENTO Y MANEJO
17 INFORMACIÓN PARA EL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información completa de prescripción no están enumeradas.
1 INDICACIONES Y USO
1.1 Melanoma Irresecable o Metastásico
OPDIVO, como agente único o en combinación con ipilimumab, está indicado para el tratamiento de adultos y pacientes pediátricos de 12 años o más con melanoma irresecable o metastásico.
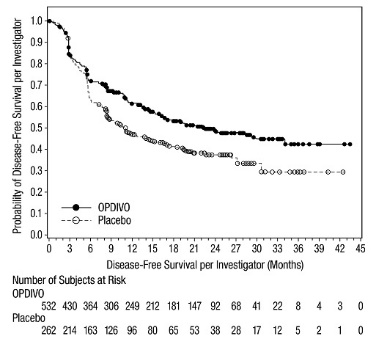
1.2 Tratamiento Adyuvante del Melanoma
OPDIVO está indicado para el tratamiento adyuvante de adultos y pacientes pediátricos de 12 años o más con melanoma completamente resecado en estadio IIB, estadio IIC, estadio III o estadio IV.
1.3 Tratamiento Neoadyuvante del Cáncer de Pulmón de Células No Pequeñas Resecable
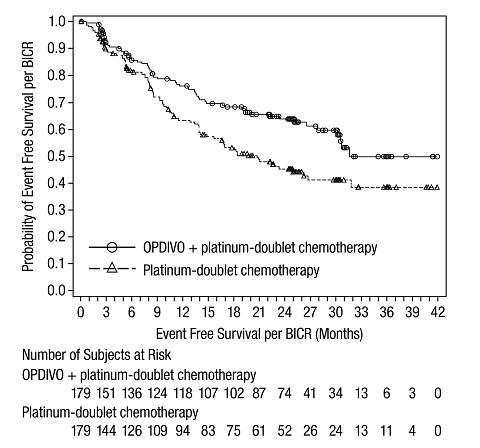
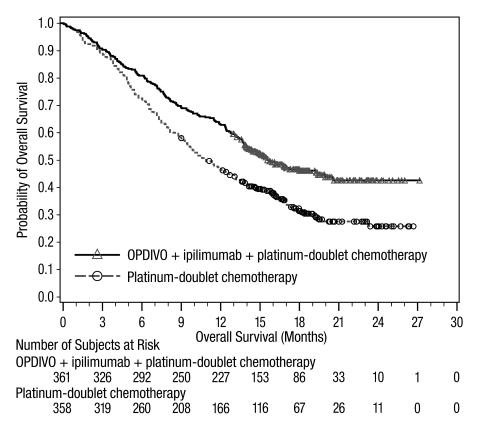
OPDIVO, en combinación con quimioterapia de doblete de platino, está indicado como tratamiento neoadyuvante de pacientes adultos con cáncer de pulmón de células no pequeñas (NSCLC) resecable (tumores ≥4 cm o ganglios positivos).
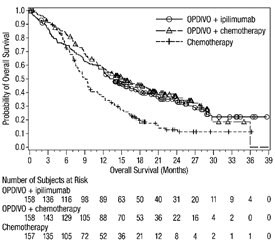
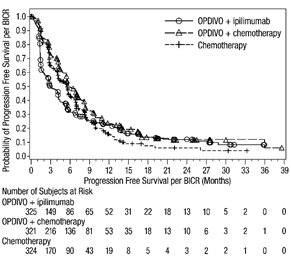
1.4 Cáncer de Pulmón de Células No Pequeñas Metastásico
- •
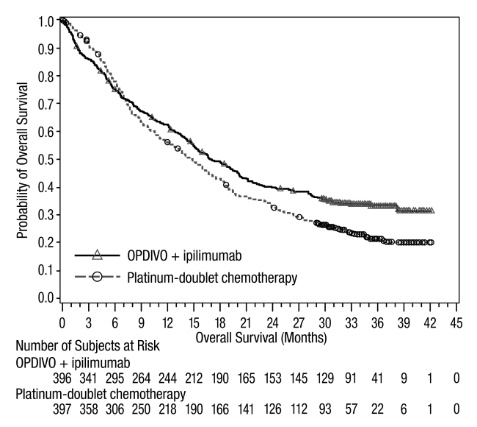
- OPDIVO, en combinación con ipilimumab, está indicado para el tratamiento de primera línea de pacientes adultos con NSCLC metastásico cuyos tumores expresan PD-L1 (≥1%) según lo determinado por una prueba aprobada por la FDA [ver Dosificación y administración (2.1)], sin aberraciones genómicas tumorales EGFR o ALK.
- •
- OPDIVO, en combinación con ipilimumab y 2 ciclos de quimioterapia de doblete de platino, está indicado para el tratamiento de primera línea de pacientes adultos con NSCLC metastásico o recurrente, sin aberraciones genómicas tumorales EGFR o ALK.
- •
- OPDIVO está indicado para el tratamiento de pacientes adultos con NSCLC metastásico con progresión en o después de la quimioterapia basada en platino. Los pacientes con aberraciones genómicas tumorales EGFR o ALK deben tener progresión de la enfermedad en la terapia aprobada por la FDA para estas aberraciones antes de recibir OPDIVO.
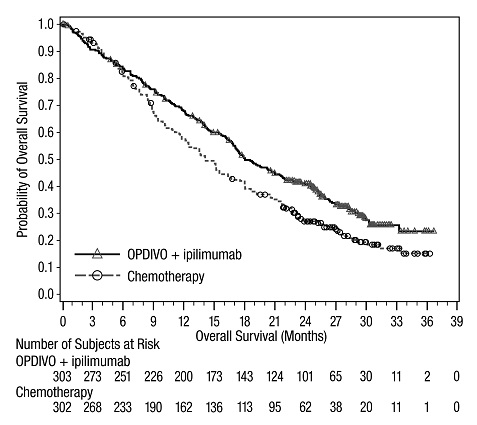
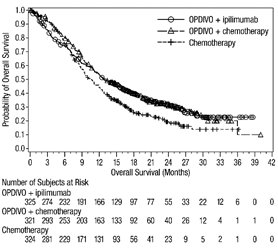
1.5 Mesotelioma Pleural Maligno
OPDIVO, en combinación con ipilimumab, está indicado para el tratamiento de primera línea de pacientes adultos con mesotelioma pleural maligno irresecable.
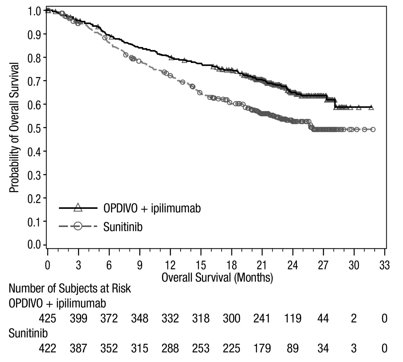
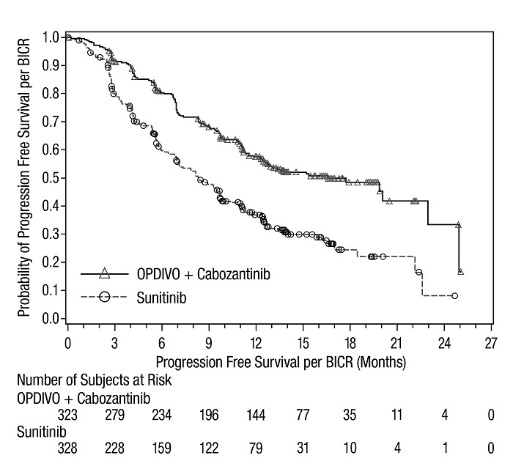
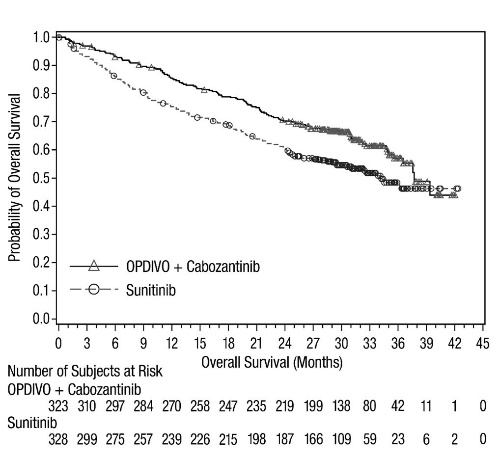
1.6 Carcinoma de Células Renales Avanzado
- •
- OPDIVO, en combinación con ipilimumab, está indicado para el tratamiento de primera línea de pacientes adultos con RCC avanzado de riesgo intermedio o pobre.
- •
- OPDIVO, en combinación con cabozantinib, está indicado para el tratamiento de primera línea de pacientes adultos con RCC avanzado.
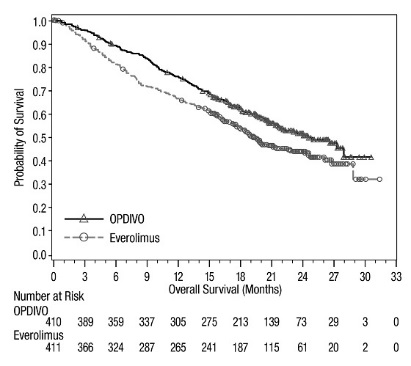
- •
- OPDIVO como agente único está indicado para el tratamiento de pacientes adultos con carcinoma de células renales (RCC) avanzado que han recibido terapia antiangiogénica previa.
1.7 Linfoma de Hodgkin Clásico
OPDIVO está indicado para el tratamiento de pacientes adultos con linfoma de Hodgkin clásico (cHL) que ha recaído o progresado después de:
- •
- trasplante autólogo de células madre hematopoyéticas (HSCT) y brentuximab vedotin, o
- •
- 3 o más líneas de terapia sistémica que incluyen HSCT autólogo.
Esta indicación está aprobada bajo aprobación acelerada en base a la tasa de respuesta general [ver Estudios clínicos (14.7)]. La aprobación continua para esta indicación puede estar supeditada a la verificación y descripción del beneficio clínico en ensayos confirmatorios.
1.8 Carcinoma de Células Escamosas de Cabeza y Cuello
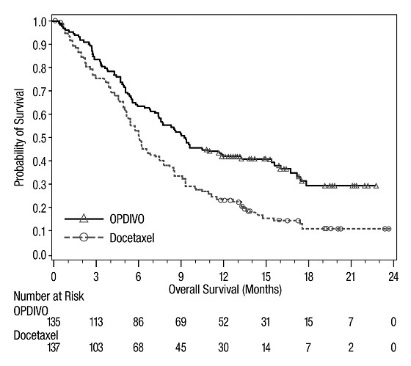
OPDIVO está indicado para el tratamiento de pacientes adultos con carcinoma de células escamosas de cabeza y cuello (SCCHN) recurrente o metastásico con progresión de la enfermedad en o después de la terapia basada en platino.
1.9 Carcinoma Urotel
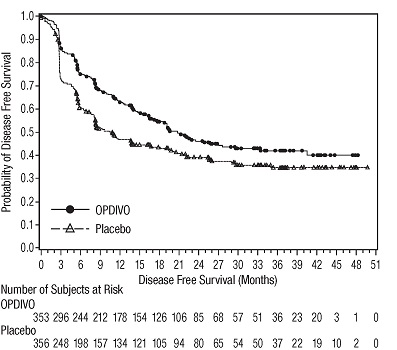
OPDIVO está indicado para el tratamiento adyuvante de pacientes adultos con carcinoma urotel (UC) que tienen un alto riesgo de recurrencia después de someterse a una resección radical de UC [ver Estudios clínicos (14.9)].
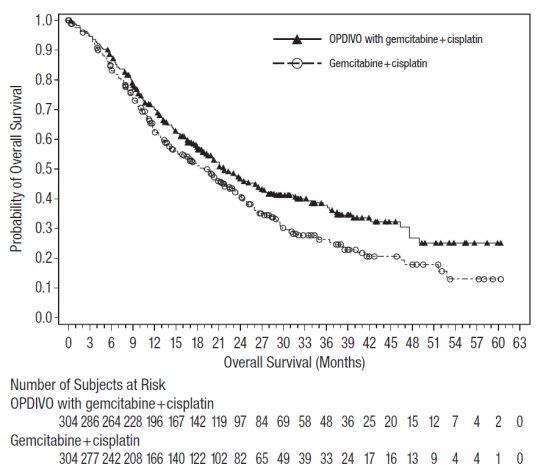
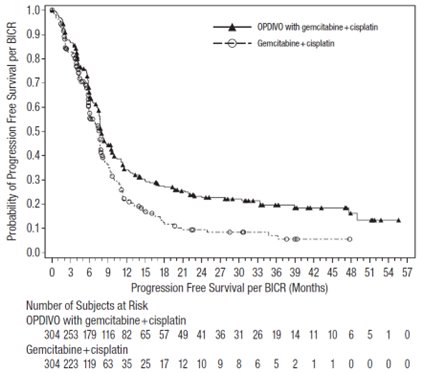
OPDIVO, en combinación con cisplatino y gemcitabina, está indicado para el tratamiento de primera línea de pacientes adultos con carcinoma urotel irresecable o metastásico.
OPDIVO está indicado para el tratamiento de pacientes adultos con carcinoma urotel localmente avanzado o metastásico que:
- •
- tienen progresión de la enfermedad durante o después de la quimioterapia que contiene platino.
- •
- tienen progresión de la enfermedad dentro de los 12 meses del tratamiento neoadyuvante o adyuvante con quimioterapia que contiene platino.
1.10 Cáncer Colorrectal Metastásico de Instabilidad de Microsatélites Alta o Deficiencia de Reparación de Desajuste
OPDIVO, como agente único o en combinación con ipilimumab, está indicado para el tratamiento de adultos y pacientes pediátricos de 12 años o más con cáncer colorrectal (CRC) metastásico de instabilidad de microsatélites alta (MSI-H) o deficiencia de reparación de desajuste (dMMR) que ha progresado después del tratamiento con una fluoropirimidina, oxaliplatino e irinotecán.
Esta indicación está aprobada bajo aprobación acelerada en base a la tasa de respuesta general y la duración de la respuesta [ver Estudios clínicos (14.10)]. La aprobación continua para esta indicación puede estar supeditada a la verificación y descripción del beneficio clínico en ensayos confirmatorios.
1.11 Carcinoma Hepatocelular
OPDIVO, en combinación con ipilimumab, está indicado para el tratamiento de pacientes adultos con carcinoma hepatocelular (HCC) que han sido tratados previamente con sorafenib. Esta indicación está aprobada bajo aprobación acelerada en base a la tasa de respuesta general y la duración de la respuesta [ver Estudios Clínicos (14.11)]. La aprobación continua para esta indicación puede estar sujeta a la verificación y descripción del beneficio clínico en los ensayos confirmatorios.
1.12 Cáncer Esofágico
- •
- OPDIVO está indicado para el tratamiento adyuvante del cáncer esofágico o de la unión gastroesofágica completamente resecado con enfermedad patológica residual en pacientes adultos que han recibido quimiorradioterapia neoadyuvante (CRT).
- •
- OPDIVO, en combinación con quimioterapia que contiene fluoropirimidina y platino, está indicado para el tratamiento de primera línea de pacientes adultos con carcinoma de células escamosas esofágico (ESCC) avanzado o metastásico irresecable.
- •
- OPDIVO, en combinación con ipilimumab, está indicado para el tratamiento de primera línea de pacientes adultos con carcinoma de células escamosas esofágico (ESCC) avanzado o metastásico irresecable.
- •
- OPDIVO está indicado para el tratamiento de pacientes adultos con carcinoma de células escamosas esofágico (ESCC) avanzado, recurrente o metastásico irresecable después de la quimioterapia previa basada en fluoropirimidina y platino.
1.13 Cáncer Gástrico, Cáncer de la Unión Gastroesofágica y Adenocarcinoma Esofágico
OPDIVO, en combinación con quimioterapia que contiene fluoropirimidina y platino, está indicado para el tratamiento de pacientes adultos con cáncer gástrico avanzado o metastásico, cáncer de la unión gastroesofágica y adenocarcinoma esofágico.
2 DOSIS Y ADMINISTRACIÓN
2.1 Selección del Paciente
Seleccione pacientes con NSCLC metastásico para tratamiento con OPDIVO en combinación con ipilimumab en función de la expresión de PD-L1 [ver Estudios Clínicos (14.4)].
La información sobre las pruebas aprobadas por la FDA para la determinación de la expresión de PD-L1 en NSCLC está disponible en: http://www.fda.gov/CompanionDiagnostics.
2.2 Dosis Recomendada
Las dosis recomendadas de OPDIVO como agente único se presentan en la Tabla 1.
| * Infusión intravenosa de 30 minutos. | ||
|
Indicación |
Dosis recomendada de OPDIVO |
Duración de la terapia |
|
Cáncer de pulmón de células no pequeñas metastásico |
240 mg cada 2 semanas* o 480 mg cada 4 semanas* |
Hasta la progresión de la enfermedad o la toxicidad inaceptable |
|
Carcinoma de células renales avanzado |
||
|
Linfoma de Hodgkin clásico |
||
|
Carcinoma de células escamosas de cabeza y cuello |
||
|
Carcinoma urotelial localmente avanzado o metastásico |
||
|
Carcinoma de células escamosas de esófago |
||
|
Melanoma irresecable o metastásico |
Pacientes adultos y pacientes pediátricos de 12 años o más y con un peso de 40 kg o más: |
Hasta la progresión de la enfermedad o la toxicidad inaceptable |
|
Pacientes pediátricos de 12 años o más y con un peso inferior a 40 kg: o 6 mg/kg cada 4 semanas* |
||
|
Tratamiento adyuvante del melanoma |
Pacientes adultos y pacientes pediátricos de 12 años o más y con un peso de 40 kg o más: o 480 mg cada 4 semanas* |
Hasta la recurrencia de la enfermedad o la toxicidad inaceptable durante un máximo de 1 año |
|
Pacientes pediátricos de 12 años o más y con un peso inferior a 40 kg: o 6 mg/kg cada 4 semanas* |
||
|
Tratamiento adyuvante del carcinoma urotelial (UC) |
240 mg cada 2 semanas* o 480 mg cada 4 semanas* |
Hasta la recurrencia de la enfermedad o la toxicidad inaceptable durante un máximo de 1 año |
|
Cáncer colorrectal metastásico de alta inestabilidad de microsatélites (MSI-H) o deficiencia de reparación de desajuste (dMMR) |
Pacientes adultos y pacientes pediátricos de 12 años o más y con un peso de 40 kg o más: o 480 mg cada 4 semanas* |
Hasta la progresión de la enfermedad o la toxicidad inaceptable |
|
Pacientes pediátricos de 12 años o más y con un peso inferior a 40 kg: |
||
|
Tratamiento adyuvante del cáncer de esófago o de la unión gastroesofágica resecado |
240 mg cada 2 semanas* o 480 mg cada 4 semanas* |
Hasta la progresión de la enfermedad o la toxicidad inaceptable durante una duración total del tratamiento de 1 año |
Las dosis recomendadas de OPDIVO en combinación con otros agentes terapéuticos se presentan en la Tabla 2. Consulte la Información de Prescripción respectiva para cada agente terapéutico administrado en combinación con OPDIVO para obtener la información de dosificación recomendada, según corresponda.
| * Infusión intravenosa de 30 minutos el mismo día. | ||
|
Indicación |
Dosis recomendada de OPDIVO |
Duración de la terapia |
|
Melanoma irresecable o metastásico |
1 mg/kg cada 3 semanas* |
En combinación con ipilimumab por un máximo de 4 dosis o hasta que se produzca una toxicidad inaceptable, lo que ocurra primero |
|
Pacientes adultos y pacientes pediátricos de 12 años o más y con un peso de 40 kg o más: 240 mg cada 2 semanas* |
Después de completar 4 dosis de terapia combinada, administrar como agente único hasta la progresión de la enfermedad o la toxicidad inaceptable |
|
|
Pacientes pediátricos de 12 años o más y con un peso inferior a 40 kg:
3 mg/kg cada 2 semanas* |
||
|
Tratamiento neoadyuvante del cáncer de pulmón de células no pequeñas resecable |
360 mg cada 3 semanas* |
En combinación con quimioterapia de doblete de platino durante 3 ciclos |
|
Cáncer de pulmón de células no pequeñas metastásico que expresa PD-L1 |
360 mg cada 3 semanas* |
En combinación con ipilimumab hasta la progresión de la enfermedad, la toxicidad inaceptable o hasta 2 años en pacientes sin progresión de la enfermedad |
|
Cáncer de pulmón de células no pequeñas metastásico o recurrente |
360 mg cada 3 semanas* |
En combinación con ipilimumab hasta la progresión de la enfermedad, la toxicidad inaceptable o hasta 2 años en pacientes sin progresión de la enfermedad |
|
2 ciclos de quimioterapia de doblete de platino basada en la histología |
||
|
Mesotelioma pleural maligno |
360 mg cada 3 semanas* |
En combinación con ipilimumab hasta la progresión de la enfermedad, la toxicidad inaceptable o hasta 2 años en pacientes sin progresión de la enfermedad |
|
Carcinoma de células renales avanzado |
3 mg/kg cada 3 semanas* |
En combinación con ipilimumab |
|
240 mg cada 2 semanas*
o Administrar OPDIVO en combinación con cabozantinib 40 mg por vía oral una vez al día sin alimentos |
OPDIVO: Hasta la progresión de la enfermedad, la toxicidad inaceptable o hasta 2 años |
|
|
Cabozantinib: Hasta la progresión de la enfermedad o la toxicidad inaceptable |
||
|
240 mg cada 2 semanas* 480 mg cada 4 semanas* |
Después de completar 4 dosis de terapia combinada con ipilimumab, administrar como agente único hasta la progresión de la enfermedad o la toxicidad inaceptable |
|
|
Carcinoma urotelial irresecable o metastásico de primera línea |
360 mg cada 3 semanas* |
En combinación con cisplatino y gemcitabina |
|
240 mg cada 2 semanas* |
Después de completar hasta 6 ciclos de terapia combinada, administrar como agente único hasta la progresión de la enfermedad, toxicidad inaceptable o hasta 2 años desde la primera dosis |
|
|
Cáncer colorrectal metastásico con inestabilidad de microsatélites alta (MSI-H) o deficiencia de reparación de desajuste (dMMR) |
3 mg/kg cada 3 semanas* |
En combinación con ipilimumab |
|
Pacientes adultos y pacientes pediátricos de 12 años o más y con un peso de 40 kg o más: |
Después de completar 4 dosis de terapia combinada, administrar como agente único hasta la progresión de la enfermedad o toxicidad inaceptable |
|
|
Pacientes pediátricos de 12 años o más y con un peso inferior a 40 kg: |
||
|
Carcinoma hepatocelular |
1 mg/kg cada 3 semanas* |
En combinación con ipilimumab |
|
240 mg cada 2 semanas* |
Después de completar 4 dosis de terapia combinada, administrar como agente único hasta la progresión de la enfermedad o toxicidad inaceptable |
|
|
Carcinoma de células escamosas de esófago |
240 mg cada 2 semanas* 480 mg cada 4 semanas* |
OPDIVO: Hasta la progresión de la enfermedad, toxicidad inaceptable o hasta 2 años |
|
Quimioterapia: Hasta la progresión de la enfermedad o toxicidad inaceptable |
||
|
3 mg/kg cada 2 semanas* |
En combinación con ipilimumab hasta la progresión de la enfermedad, toxicidad inaceptable o hasta 2 años |
|
|
Cáncer gástrico, cáncer de la unión gastroesofágica y adenocarcinoma de esófago |
240 mg cada 2 semanas* o 360 mg cada 3 semanas* |
Hasta la progresión de la enfermedad, toxicidad inaceptable o hasta 2 años |
2.3 Modificaciones de la dosis
No se recomienda la reducción de la dosis de OPDIVO. En general, suspenda OPDIVO para reacciones adversas inmunomediadas graves (Grado 3). Suspenda permanentemente OPDIVO para reacciones adversas inmunomediadas potencialmente mortales (Grado 4), reacciones inmunomediadas graves recurrentes (Grado 3) que requieran tratamiento inmunosupresor sistémico, o la incapacidad de reducir la dosis de corticosteroides a 10 mg o menos de prednisona o equivalente por día dentro de las 12 semanas de iniciar los esteroides.
Las modificaciones de la dosis para OPDIVO o OPDIVO en combinación para reacciones adversas que requieren un manejo diferente a estas pautas generales se resumen en la Tabla 3 y la Tabla 4.
Cuando OPDIVO se administra en combinación con ipilimumab, suspenda o suspenda permanentemente tanto ipilimumab como OPDIVO para una reacción adversa que cumpla con estas pautas de modificación de la dosis.
| a Reanudar en pacientes con resolución completa o parcial (Grado 0 a 1) después de la reducción gradual de los corticosteroides. Suspenda permanentemente si no hay resolución completa o parcial dentro de las 12 semanas de la última dosis o incapacidad de reducir la prednisona a 10 mg por día (o equivalente) o menos dentro de las 12 semanas de iniciar los esteroides. b Si AST y ALT son menores o iguales a ULN al inicio, suspenda o suspenda permanentemente OPDIVO según las recomendaciones para la hepatitis sin afectación hepática. c Dependiendo de la gravedad clínica, considere suspender la endocrinopatía de Grado 2 hasta que los síntomas mejoren con la terapia de reemplazo hormonal. Reanudar una vez que los síntomas agudos se hayan resuelto. ALT = alanina aminotransferasa, AST = aspartato aminotransferasa, DRESS = Erupción medicamentosa con eosinofilia y síntomas sistémicos, SJS = Síndrome de Stevens-Johnson, TEN = Necrólisis epidérmica tóxica, ULN = Límite superior normal |
||
|
Reacción adversa |
Severidad |
Modificación de la dosis |
|
Reacciones adversas inmunomediadas [ver Advertencias y precauciones (5.1)] |
||
|
Neumonitis |
Grado 2 |
Suspendaa |
|
Grados 3 o 4 |
Suspenda permanentemente |
|
|
Colitis
Para la colitis en pacientes tratados con terapia combinada con ipilimumab, consulte la Tabla 4. |
Grado 2 o 3 |
Suspendaa |
|
Grado 4 |
Suspenda permanentemente |
|
|
Hepatitis sin afectación tumoral del hígado
Para las elevaciones de las enzimas hepáticas en pacientes tratados con terapia combinada con ipilimumab, consulte la Tabla 4. |
Aumentos de AST/ALT a >3 y ≤8 veces ULN o Aumentos de bilirrubina total a >1.5 y ≤3 veces ULN. |
Suspendaa |
|
Aumentos de AST o ALT a >8 veces ULN o Aumentos de bilirrubina total a >3 veces ULN. |
Suspenda permanentemente |
|
|
Hepatitis con afectación tumoral del hígadob
Para las elevaciones de las enzimas hepáticas en pacientes tratados con terapia combinada con ipilimumab, consulte la Tabla 4. |
AST/ALT basal es >1 y ≤3 veces ULN y aumenta a >5 y ≤10 veces ULN o AST/ALT basal es >3 y ≤5 veces ULN y aumenta a >8 y ≤10 veces ULN. |
Suspendaa |
|
AST/ALT aumenta a >10 veces ULN o Aumentos de bilirrubina total a >3 veces ULN. |
Suspenda permanentemente |
|
|
Endocrinopatíasc |
Grado 3 o 4 |
Suspenda hasta que sea clínicamente estable o suspenda permanentemente dependiendo de la gravedad |
|
Nefritis con disfunción renal |
Aumento de la creatinina en sangre de Grado 2 o 3 |
Suspendaa |
|
Aumento de la creatinina en sangre de Grado 4 |
Suspenda permanentemente |
|
|
Condiciones dermatológicas exfoliativas |
SJS, TEN o DRESS sospechosos |
Suspenda |
|
SJS, TEN o DRESS confirmado |
Suspenda permanentemente |
|
|
Miocarditis |
Grados 2, 3 o 4 |
Suspenda permanentemente |
|
Toxicidades neurológicas |
Grado 2 |
Retengaa |
|
Grado 3 o 4 |
Suspenda permanentemente |
|
|
Otras reacciones adversas |
||
|
Reacciones relacionadas con la infusión |
Grado 1 o 2 |
Interrumpa o disminuya la velocidad de la infusión |
|
Grado 3 o 4 |
Suspenda permanentemente |
|
| a Reanudar en pacientes con resolución completa o parcial (Grado 0 a 1) después de la reducción gradual de los corticosteroides. Suspender permanentemente si no hay resolución completa o parcial dentro de las 12 semanas de la última dosis o si no se puede reducir la prednisona a 10 mg por día (o equivalente) o menos dentro de las 12 semanas de iniciar los esteroides. b Si AST y ALT son menores o iguales a ULN al inicio, suspender o suspender permanentemente OPDIVO en combinación con ipilimumab según las recomendaciones para la hepatitis sin afectación hepática. c Considere la terapia con corticosteroides para las reacciones adversas hepáticas si OPDIVO se suspende o se suspende cuando se administra en combinación con cabozantinib. d Después de la recuperación, se puede considerar un nuevo desafío con uno o ambos de OPDIVO y cabozantinib. Si se vuelve a desafiar con cabozantinib con o sin OPDIVO, consulte la información de prescripción de cabozantinib. |
|||
|
Tratamiento |
Reacción adversa |
Severidad |
Modificación de la dosis |
|
OPDIVO en combinación con ipilimumab |
Colitis |
Grado 2 |
Suspendera |
|
Grado 3 o 4 |
Suspender permanentemente |
||
|
Hepatitis sin afectación tumoral del hígado |
Aumentos de AST/ALT a >3 veces ULN y ≤5 veces ULN |
Suspendera |
|
|
AST o ALT >5 veces ULN |
Suspender permanentemente |
||
|
Hepatitis con afectación tumoral del hígadob/HCC |
El AST/ALT basal es >1 y ≤3 veces ULN y aumenta a >5 y ≤10 veces ULN |
Suspendera |
|
|
AST/ALT aumenta a >10 veces ULN |
Suspender permanentemente |
||
|
OPDIVO en combinación con cabozantinib |
Elevaciones de las enzimas hepáticas |
ALT o AST >3 veces ULN pero ≤10 veces ULN con bilirrubina total concurrente <2 veces ULN |
Suspenderc tanto OPDIVO como cabozantinib hasta que las reacciones adversas se recuperend a los Grados 0-1 |
|
ALT o AST >10 veces ULN o >3 veces ULN con bilirrubina total concurrente ≥2 veces ULN |
Suspender permanentementec tanto OPDIVO como cabozantinib |
||
2.4 Preparación y Administración
Inspeccione visualmente para detectar partículas y decoloración. OPDIVO es una solución transparente a opalescente, incolora a amarillo pálido. Deseche si está turbia, descolorida o contiene partículas extrañas distintas de unas pocas partículas proteicas translúcidas a blancas. No agitar.
Preparación
- •
- Retire el volumen requerido de OPDIVO y transfiéralo a un contenedor intravenoso.
- •
- Diluyar OPDIVO con Inyección de Cloruro de Sodio al 0,9%, USP o Inyección de Dextrosa al 5%, USP para preparar una infusión con una concentración final que oscile entre 1 mg/mL y 10 mg/mL. El volumen total de infusión no debe exceder los 160 mL.
• Para pacientes adultos y pediátricos con un peso corporal de 40 kg o más, no exceda un volumen total de infusión de 160 mL.
• Para pacientes adultos y pediátricos con un peso corporal inferior a 40 kg, no exceda un volumen total de infusión de 4 mL/kg de peso corporal. - •
- Mezcle la solución diluida mediante inversión suave. No agitar.
- •
- Deseche los viales parcialmente utilizados o los viales vacíos de OPDIVO.
- •
- El producto no contiene conservante.
- •
- Después de la preparación, almacene la solución diluida de la siguiente manera:
• a temperatura ambiente y luz ambiente durante no más de 8 horas desde el momento de la preparación hasta el final de la infusión. Deseche la solución diluida si no se utiliza dentro de las 8 horas posteriores a la preparación; o
• bajo refrigeración a 2°C a 8°C (36°F a 46°F) y protegido de la luz durante no más de 7 días desde el momento de la preparación hasta el final de la infusión. Deseche la solución diluida si no se utiliza dentro de los 7 días posteriores a la preparación. - •
- No congelar.
Administración
- •
- Administre la infusión, después de la dilución, durante 30 minutos a través de una línea intravenosa que contenga un filtro en línea estéril, no pirogénico, de baja unión a proteínas (tamaño de poro de 0,2 micrómetros a 1,2 micrómetros).
- •
- Administre OPDIVO en combinación con otros agentes terapéuticos de la siguiente manera:
- •
- Con ipilimumab: administre OPDIVO primero seguido de ipilimumab el mismo día.
- •
- Con quimioterapia de doblete de platino: administre OPDIVO primero seguido de quimioterapia de doblete de platino el mismo día
- •
- Con ipilimumab y quimioterapia de doblete de platino: administre OPDIVO primero seguido de ipilimumab y luego quimioterapia de doblete de platino el mismo día.
- •
- Con quimioterapia que contiene fluoropirimidina y platino: administre OPDIVO primero seguido de quimioterapia que contiene fluoropirimidina y platino el mismo día.
- •
- Utilice bolsas de infusión y filtros separados para cada infusión.
- •
- Enjuague la línea intravenosa al final de la infusión.
- •
- No administre otros medicamentos a través de la misma línea intravenosa.
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
Inyección: 40 mg/4 mL (10 mg/mL), 100 mg/10 mL (10 mg/mL), 120 mg/12 mL (10 mg/mL) y 240 mg/24 mL (10 mg/mL) solución clara u opalescente, incolora a amarillo pálido en un frasco para una dosis única.
4 CONTRAINDICACIONES
Ninguna.
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Reacciones adversas inmunitarias graves y mortales
OPDIVO es un anticuerpo monoclonal que pertenece a una clase de fármacos que se unen al receptor de muerte programada 1 (PD-1) o al ligando 1 de PD (PD-L1), bloqueando la vía PD-1/PD-L1, eliminando así la inhibición de la respuesta inmunitaria, potencialmente rompiendo la tolerancia periférica e induciendo reacciones adversas inmunitarias. Las reacciones adversas inmunitarias importantes que se enumeran en Advertencias y precauciones pueden no incluir todas las posibles reacciones inmunitarias graves y mortales.
Las reacciones adversas inmunitarias, que pueden ser graves o mortales, pueden ocurrir en cualquier sistema orgánico o tejido. Las reacciones adversas inmunitarias pueden ocurrir en cualquier momento después de comenzar el tratamiento con un anticuerpo bloqueador de PD-1/PD-L1. Si bien las reacciones adversas inmunitarias generalmente se manifiestan durante el tratamiento con anticuerpos bloqueadores de PD-1/PD-L1, las reacciones adversas inmunitarias también pueden manifestarse después de la interrupción de los anticuerpos bloqueadores de PD-1/PD-L1.
La identificación temprana y el manejo de las reacciones adversas inmunitarias son esenciales para garantizar el uso seguro de los anticuerpos bloqueadores de PD-1/PD-L1. Monitoree a los pacientes de cerca en busca de síntomas y signos que puedan ser manifestaciones clínicas de reacciones adversas inmunitarias subyacentes. Evalúe las enzimas hepáticas, la creatinina y la función tiroidea al inicio y periódicamente durante el tratamiento. En casos de sospecha de reacciones adversas inmunitarias, inicie una evaluación adecuada para excluir etiologías alternativas, incluida la infección. Instituya el manejo médico de inmediato, incluida la consulta de especialistas según corresponda.
Suspenda o suspenda permanentemente OPDIVO según la gravedad [ver Dosificación y administración (2.3)]. En general, si OPDIVO requiere interrupción o suspensión, administre terapia con corticosteroides sistémicos (1 a 2 mg/kg/día de prednisona o equivalente) hasta que mejore a Grado 1 o menos. Una vez que mejore a Grado 1 o menos, inicie la reducción gradual de los corticosteroides y continúe reduciéndolos gradualmente durante al menos 1 mes. Considere la administración de otros inmunosupresores sistémicos en pacientes cuyas reacciones adversas inmunitarias no se controlan con terapia con corticosteroides.
Las pautas de manejo de la toxicidad para las reacciones adversas que no necesariamente requieren esteroides sistémicos (por ejemplo, endocrinopatías y reacciones dermatológicas) se analizan a continuación.
Neumonitis inmunitaria
OPDIVO puede causar neumonitis inmunitaria, que se define como la necesidad de usar esteroides y no hay una etiología alternativa clara. En pacientes tratados con otros anticuerpos bloqueadores de PD-1/PD-L1, la incidencia de neumonitis es mayor en pacientes que han recibido radiación torácica previa.
OPDIVO como agente único
La neumonitis inmunitaria ocurrió en el 3.1% (61/1994) de los pacientes que recibieron OPDIVO como agente único, incluidas reacciones adversas de Grado 4 (<0.1%), Grado 3 (0.9%) y Grado 2 (2.1%). La neumonitis provocó la suspensión permanente de OPDIVO en el 1.1% y la suspensión de OPDIVO en el 0.8% de los pacientes.
Se requirieron corticosteroides sistémicos en el 100% (61/61) de los pacientes con neumonitis. La neumonitis se resolvió en el 84% de los 61 pacientes. De los 15 pacientes en quienes se suspendió OPDIVO por neumonitis, 14 reiniciaron OPDIVO después de la mejora de los síntomas; de estos, 4 (29%) tuvieron recurrencia de neumonitis.
OPDIVO con Ipilimumab
OPDIVO 3 mg/kg con Ipilimumab 1 mg/kg: En NSCLC, la neumonitis inmunitaria ocurrió en el 9% (50/576) de los pacientes que recibieron OPDIVO 3 mg/kg cada 2 semanas con ipilimumab 1 mg/kg cada 6 semanas, incluidas neumonitis inmunitarias de Grado 4 (0.5%), Grado 3 (3.5%) y Grado 2 (4.0%). Cuatro pacientes (0.7%) murieron debido a neumonitis. La neumonitis inmunitaria provocó la suspensión permanente de OPDIVO con ipilimumab en el 5% de los pacientes y la suspensión de OPDIVO con ipilimumab en el 3.6% de los pacientes.
Se requirieron corticosteroides sistémicos en el 100% de los pacientes con neumonitis. La neumonitis se resolvió en el 72% de los pacientes. Aproximadamente el 13% (2/16) de los pacientes tuvieron recurrencia de neumonitis después de la reiniciación de OPDIVO con ipilimumab.
Colitis inmunitaria
OPDIVO puede causar colitis inmunitaria, definida como la necesidad de usar corticosteroides y no hay una etiología alternativa clara. Un síntoma común incluido en la definición de colitis fue la diarrea. Se ha informado infección/reactivación por citomegalovirus (CMV) en pacientes con colitis inmunitaria refractaria a los corticosteroides. En casos de colitis refractaria a los corticosteroides, considere repetir la evaluación infecciosa para excluir etiologías alternativas.
OPDIVO como agente único
La colitis inmunitaria ocurrió en el 2.9% (58/1994) de los pacientes que recibieron OPDIVO como agente único, incluidas reacciones adversas de Grado 3 (1.7%) y Grado 2 (1%). La colitis provocó la suspensión permanente de OPDIVO en el 0.7% y la suspensión de OPDIVO en el 0.9% de los pacientes.
Se requirieron corticosteroides sistémicos en el 100% (58/58) de los pacientes con colitis. Cuatro pacientes requirieron la adición de infliximab a corticosteroides de alta dosis. La colitis se resolvió en el 86% de los 58 pacientes. De los 18 pacientes en quienes se suspendió OPDIVO por colitis, 16 reiniciaron OPDIVO después de la mejora de los síntomas; de estos, 12 (75%) tuvieron recurrencia de colitis.
OPDIVO con Ipilimumab
OPDIVO 1 mg/kg con Ipilimumab 3 mg/kg: La colitis inmunitaria ocurrió en el 25% (115/456) de los pacientes con melanoma o HCC que recibieron OPDIVO 1 mg/kg con ipilimumab 3 mg/kg cada 3 semanas, incluidas reacciones adversas de Grado 4 (0.4%), Grado 3 (14%) y Grado 2 (8%). La colitis provocó la suspensión permanente de OPDIVO con ipilimumab en el 14% y la suspensión de OPDIVO con ipilimumab en el 4.4% de los pacientes.
Se requirieron corticosteroides sistémicos en el 100% (115/115) de los pacientes con colitis. Aproximadamente el 23% de los pacientes requirieron la adición de infliximab a corticosteroides de alta dosis. La colitis se resolvió en el 93% de los 115 pacientes. De los 20 pacientes en quienes se suspendió OPDIVO con ipilimumab por colitis, 16 reiniciaron el tratamiento después de la mejora de los síntomas; de estos, 9 (56%) tuvieron recurrencia de colitis.
OPDIVO 3 mg/kg con Ipilimumab 1 mg/kg: La colitis inmunitaria ocurrió en el 9% (60/666) de los pacientes con RCC o CRC que recibieron OPDIVO 3 mg/kg con ipilimumab 1 mg/kg cada 3 semanas, incluidas reacciones adversas de Grado 3 (4.4%) y Grado 2 (3.7%). La colitis provocó la suspensión permanente de OPDIVO con ipilimumab en el 3.2% y la suspensión de OPDIVO con ipilimumab en el 2.7% de los pacientes con RCC o CRC.
Se requirieron corticosteroides sistémicos en el 100% (60/60) de los pacientes con colitis. Aproximadamente el 23% de los pacientes con colitis inmunomediada requirieron la adición de infliximab a corticosteroides de alta dosis. La colitis se resolvió en el 95% de los 60 pacientes. De los 18 pacientes en quienes se suspendió OPDIVO con ipilimumab por colitis, 16 reiniciaron el tratamiento después de la mejoría de los síntomas; de estos, 10 (63%) tuvieron recurrencia de la colitis.
Hepatitis inmunomediada y hepatotoxicidad
OPDIVO puede causar hepatitis inmunomediada, definida como la necesidad de usar corticosteroides y sin una etiología alternativa clara.
OPDIVO como agente único
La hepatitis inmunomediada ocurrió en el 1.8% (35/1994) de los pacientes que recibieron OPDIVO como agente único, incluidas reacciones adversas de Grado 4 (0.2%), Grado 3 (1.3%) y Grado 2 (0.4%). La hepatitis provocó la interrupción permanente de OPDIVO en el 0.7% y la suspensión de OPDIVO en el 0.6% de los pacientes.
Se requirieron corticosteroides sistémicos en el 100% (35/35) de los pacientes con hepatitis. Dos pacientes requirieron la adición de ácido micofenólico a corticosteroides de alta dosis. La hepatitis se resolvió en el 91% de los 35 pacientes. De los 12 pacientes en quienes se suspendió OPDIVO por hepatitis, 11 reiniciaron OPDIVO después de la mejoría de los síntomas; de estos, 9 (82%) tuvieron recurrencia de la hepatitis.
OPDIVO con Ipilimumab
OPDIVO 1 mg/kg con Ipilimumab 3 mg/kg: La hepatitis inmunomediada ocurrió en el 15% (70/456) de los pacientes con melanoma o HCC que recibieron OPDIVO 1 mg/kg con ipilimumab 3 mg/kg cada 3 semanas, incluidas reacciones adversas de Grado 4 (2.4%), Grado 3 (11%) y Grado 2 (1.8%). La hepatitis inmunomediada provocó la interrupción permanente de OPDIVO con ipilimumab en el 8% o la suspensión de OPDIVO con ipilimumab en el 3.5% de los pacientes.
Se requirieron corticosteroides sistémicos en el 100% (70/70) de los pacientes con hepatitis. Aproximadamente el 9% de los pacientes con hepatitis inmunomediada requirieron la adición de ácido micofenólico a corticosteroides de alta dosis. La hepatitis se resolvió en el 91% de los 70 pacientes. De los 16 pacientes en quienes se suspendió OPDIVO con ipilimumab por hepatitis, 14 reiniciaron el tratamiento después de la mejoría de los síntomas; de estos, 8 (57%) tuvieron recurrencia de la hepatitis.
OPDIVO 3 mg/kg con Ipilimumab 1 mg/kg: La hepatitis inmunomediada ocurrió en el 7% (48/666) de los pacientes con RCC o CRC que recibieron OPDIVO 3 mg/kg con ipilimumab 1 mg/kg cada 3 semanas, incluidas reacciones adversas de Grado 4 (1.2%), Grado 3 (4.9%) y Grado 2 (0.4%). La hepatitis inmunomediada provocó la interrupción permanente de OPDIVO con ipilimumab en el 3.6% y la suspensión de OPDIVO con ipilimumab en el 2.6% de los pacientes con RCC o CRC.
Se requirieron corticosteroides sistémicos en el 100% (48/48) de los pacientes con hepatitis. Aproximadamente el 19% de los pacientes con hepatitis inmunomediada requirieron la adición de ácido micofenólico a corticosteroides de alta dosis. La hepatitis se resolvió en el 88% de los 48 pacientes. De los 17 pacientes en quienes se suspendió OPDIVO con ipilimumab por hepatitis, 14 reiniciaron el tratamiento después de la mejoría de los síntomas; de estos, 10 (71%) tuvieron recurrencia de la hepatitis.
OPDIVO con Cabozantinib
OPDIVO en combinación con cabozantinib puede causar toxicidad hepática con mayores frecuencias de elevaciones de ALT y AST de Grado 3 y 4 en comparación con OPDIVO solo. Monitorear las enzimas hepáticas antes de iniciar y periódicamente durante el tratamiento. Considere un monitoreo más frecuente de las enzimas hepáticas en comparación con cuando los medicamentos se administran como agentes únicos. Para las enzimas hepáticas elevadas, interrumpa OPDIVO y cabozantinib y considere administrar corticosteroides [ver Dosificación y administración (2.3)].
Con la combinación de OPDIVO y cabozantinib, se observaron ALT o AST aumentadas de Grados 3 y 4 en el 11% de los pacientes [ver Reacciones adversas (6.1)]. Se informó ALT o AST >3 veces el LSN (Grado ≥2) en 83 pacientes, de los cuales 23 (28%) recibieron corticosteroides sistémicos; ALT o AST se resolvieron a Grados 0-1 en 74 (89%). Entre los 44 pacientes con ALT o AST aumentada de Grado ≥2 que fueron retados con OPDIVO (n=11) o cabozantinib (n=9) administrados como agente único o con ambos (n=24), se observó recurrencia de ALT o AST aumentada de Grado ≥2 en 2 pacientes que recibieron OPDIVO, 2 pacientes que recibieron cabozantinib y 7 pacientes que recibieron tanto OPDIVO como cabozantinib.
Endocrinopatías inmunomediadas
Insuficiencia suprarrenal
OPDIVO puede causar insuficiencia suprarrenal primaria o secundaria. Para la insuficiencia suprarrenal de grado 2 o superior, inicie el tratamiento sintomático, incluida la terapia de reemplazo hormonal según esté clínicamente indicado. Suspenda OPDIVO dependiendo de la gravedad [ver Dosificación y administración (2.3)].
OPDIVO como agente único
La insuficiencia suprarrenal ocurrió en el 1% (20/1994) de los pacientes que recibieron OPDIVO como agente único, incluidas reacciones adversas de Grado 3 (0.4%) y Grado 2 (0.6%). La insuficiencia suprarrenal provocó la interrupción permanente de OPDIVO en el 0.1% y la suspensión de OPDIVO en el 0.4% de los pacientes.
Aproximadamente el 85% de los pacientes con insuficiencia suprarrenal recibieron terapia de reemplazo hormonal. Se requirieron corticosteroides sistémicos en el 90% (18/20) de los pacientes con insuficiencia suprarrenal. La insuficiencia suprarrenal se resolvió en el 35% de los 20 pacientes. De los 8 pacientes en quienes se suspendió OPDIVO por insuficiencia suprarrenal, 4 reiniciaron OPDIVO después de la mejoría de los síntomas y todos requirieron terapia de reemplazo hormonal para su insuficiencia suprarrenal en curso.
OPDIVO con Ipilimumab
OPDIVO 1 mg/kg con Ipilimumab 3 mg/kg: La insuficiencia suprarrenal ocurrió en el 8% (35/456) de los pacientes con melanoma o HCC que recibieron OPDIVO 1 mg/kg con ipilimumab 3 mg/kg cada 3 semanas, incluidas reacciones adversas de Grado 4 (0.2%), Grado 3 (2.4%) y Grado 2 (4.2%). La insuficiencia suprarrenal provocó la interrupción permanente de OPDIVO con ipilimumab en el 0.4% y la suspensión de OPDIVO con ipilimumab en el 2.0% de los pacientes.
Aproximadamente el 71% (25/35) de los pacientes con insuficiencia suprarrenal recibieron terapia de reemplazo hormonal, incluidos corticosteroides sistémicos. La insuficiencia suprarrenal se resolvió en el 37% de los 35 pacientes. De los 9 pacientes en quienes se suspendió OPDIVO con ipilimumab por insuficiencia suprarrenal, 7 reiniciaron el tratamiento después de la mejoría de los síntomas y todos requirieron terapia de reemplazo hormonal para su insuficiencia suprarrenal en curso.
OPDIVO 3 mg/kg con Ipilimumab 1 mg/kg: La insuficiencia suprarrenal ocurrió en el 7% (48/666) de los pacientes con CCR o CRC que recibieron OPDIVO 3 mg/kg con ipilimumab 1 mg/kg cada 3 semanas, incluidas reacciones adversas de Grado 4 (0.3%), Grado 3 (2.5%) y Grado 2 (4.1%). La insuficiencia suprarrenal condujo a la interrupción permanente de OPDIVO con ipilimumab en el 1.2% y a la suspensión de OPDIVO con ipilimumab en el 2.1% de los pacientes con CCR o CRC.
Aproximadamente el 94% (45/48) de los pacientes con insuficiencia suprarrenal recibieron terapia de reemplazo hormonal, incluidos corticosteroides sistémicos. La insuficiencia suprarrenal se resolvió en el 29% de los 48 pacientes. De los 14 pacientes en quienes se suspendió OPDIVO con ipilimumab por insuficiencia suprarrenal, 11 reiniciaron el tratamiento después de la mejora de los síntomas; de estos, todos recibieron terapia de reemplazo hormonal y 2 (18%) tuvieron recurrencia de la insuficiencia suprarrenal.
OPDIVO con Cabozantinib
La insuficiencia suprarrenal ocurrió en el 4.7% (15/320) de los pacientes con CCR que recibieron OPDIVO con cabozantinib, incluidas reacciones adversas de Grado 3 (2.2%) y Grado 2 (1.9%). La insuficiencia suprarrenal condujo a la interrupción permanente de OPDIVO y cabozantinib en el 0.9% y a la suspensión de OPDIVO y cabozantinib en el 2.8% de los pacientes con CCR.
Aproximadamente el 80% (12/15) de los pacientes con insuficiencia suprarrenal recibieron terapia de reemplazo hormonal, incluidos corticosteroides sistémicos. La insuficiencia suprarrenal se resolvió en el 27% (n=4) de los 15 pacientes. De los 9 pacientes en quienes se suspendió OPDIVO con cabozantinib por insuficiencia suprarrenal, 6 reiniciaron el tratamiento después de la mejora de los síntomas; de estos, todos (n=6) recibieron terapia de reemplazo hormonal y 2 tuvieron recurrencia de la insuficiencia suprarrenal.
Hipofisitis
OPDIVO puede causar hipofisitis mediada por el sistema inmunitario. La hipofisitis puede presentarse con síntomas agudos asociados con el efecto de masa, como dolor de cabeza, fotofobia o defectos del campo visual. La hipofisitis puede causar hipopituitarismo. Inicie el reemplazo hormonal según esté clínicamente indicado. Suspenda o interrumpa permanentemente OPDIVO según la gravedad [ver Dosificación y administración (2.3)].
OPDIVO como agente único
La hipofisitis ocurrió en el 0.6% (12/1994) de los pacientes que recibieron OPDIVO como agente único, incluidas reacciones adversas de Grado 3 (0.2%) y Grado 2 (0.3%). La hipofisitis condujo a la interrupción permanente de OPDIVO en <0.1% y a la suspensión de OPDIVO en el 0.2% de los pacientes.
Aproximadamente el 67% (8/12) de los pacientes con hipofisitis recibieron terapia de reemplazo hormonal, incluidos corticosteroides sistémicos. La hipofisitis se resolvió en el 42% de los 12 pacientes. De los 3 pacientes en quienes se suspendió OPDIVO por hipofisitis, 2 reiniciaron OPDIVO después de la mejora de los síntomas; de estos, ninguno tuvo recurrencia de la hipofisitis.
OPDIVO con Ipilimumab
OPDIVO 1 mg/kg con Ipilimumab 3 mg/kg: La hipofisitis ocurrió en el 9% (42/456) de los pacientes con melanoma o HCC que recibieron OPDIVO 1 mg/kg con ipilimumab 3 mg/kg cada 3 semanas, incluidas reacciones adversas de Grado 3 (2.4%) y Grado 2 (6%). La hipofisitis condujo a la interrupción permanente de OPDIVO con ipilimumab en el 0.9% y a la suspensión de OPDIVO con ipilimumab en el 4.2% de los pacientes.
Aproximadamente el 86% de los pacientes con hipofisitis recibieron terapia de reemplazo hormonal. Los corticosteroides sistémicos fueron necesarios en el 88% (37/42) de los pacientes con hipofisitis. La hipofisitis se resolvió en el 38% de los 42 pacientes. De los 19 pacientes en quienes se suspendió OPDIVO con ipilimumab por hipofisitis, 9 reiniciaron el tratamiento después de la mejora de los síntomas; de estos, 1 (11%) tuvo recurrencia de la hipofisitis.
OPDIVO 3 mg/kg con Ipilimumab 1 mg/kg: La hipofisitis ocurrió en el 4.4% (29/666) de los pacientes con CCR o CRC que recibieron OPDIVO 3 mg/kg con ipilimumab 1 mg/kg cada 3 semanas, incluidas reacciones adversas de Grado 4 (0.3%), Grado 3 (2.4%) y Grado 2 (0.9%). La hipofisitis condujo a la interrupción permanente de OPDIVO con ipilimumab en el 1.2% y a la suspensión de OPDIVO con ipilimumab en el 2.1% de los pacientes con CCR o CRC.
Aproximadamente el 72% (21/29) de los pacientes con hipofisitis recibieron terapia de reemplazo hormonal, incluidos corticosteroides sistémicos. La hipofisitis se resolvió en el 59% de los 29 pacientes. De los 14 pacientes en quienes se suspendió OPDIVO con ipilimumab por hipofisitis, 11 reiniciaron el tratamiento después de la mejora de los síntomas; de estos, 2 (18%) tuvieron recurrencia de la hipofisitis.
Trastornos de la tiroides
OPDIVO puede causar trastornos de la tiroides mediados por el sistema inmunitario. La tiroiditis puede presentarse con o sin endocrinopatía. El hipotiroidismo puede seguir al hipertiroidismo. Inicie el reemplazo hormonal o el manejo médico según esté clínicamente indicado. Suspenda o interrumpa permanentemente OPDIVO según la gravedad [ver Dosificación y administración (2.3)].
Tiroiditis
OPDIVO como agente único
La tiroiditis ocurrió en el 0.6% (12/1994) de los pacientes que recibieron OPDIVO como agente único, incluidas reacciones adversas de Grado 2 (0.2%). La tiroiditis condujo a la interrupción permanente de OPDIVO en ningún paciente y a la suspensión de OPDIVO en el 0.2% de los pacientes.
Los corticosteroides sistémicos fueron necesarios en el 17% (2/12) de los pacientes con tiroiditis. La tiroiditis se resolvió en el 58% de los 12 pacientes. De los 3 pacientes en quienes se suspendió OPDIVO por tiroiditis, 1 reinició OPDIVO después de la mejora de los síntomas sin recurrencia de la tiroiditis.
Hipertiroidismo
OPDIVO como agente único
El hipertiroidismo ocurrió en el 2.7% (54/1994) de los pacientes que recibieron OPDIVO como agente único, incluidas reacciones adversas de Grado 3 (<0.1%) y Grado 2 (1.2%). El hipertiroidismo condujo a la interrupción permanente de OPDIVO en ningún paciente y a la suspensión de OPDIVO en el 0.4% de los pacientes.
Aproximadamente el 19% de los pacientes con hipertiroidismo recibieron metimazol, el 7% recibieron carbimazol y el 4% recibieron propiltiouracilo. Los corticosteroides sistémicos fueron necesarios en el 9% (5/54) de los pacientes. El hipertiroidismo se resolvió en el 76% de los 54 pacientes. De los 7 pacientes en quienes se suspendió OPDIVO por hipertiroidismo, 4 reiniciaron OPDIVO después de la mejora de los síntomas; de estos, ninguno tuvo recurrencia del hipertiroidismo.
OPDIVO con Ipilimumab
OPDIVO 1 mg/kg con Ipilimumab 3 mg/kg: El hipertiroidismo ocurrió en el 9% (42/456) de los pacientes con melanoma o HCC que recibieron OPDIVO 1 mg/kg con ipilimumab 3 mg/kg cada 3 semanas, incluidas reacciones adversas de Grado 3 (0.9%) y Grado 2 (4.2%). El hipertiroidismo condujo a la interrupción permanente de OPDIVO con ipilimumab en ningún paciente y a la suspensión de OPDIVO con ipilimumab en el 2.4% de los pacientes.
Aproximadamente el 26% de los pacientes con hipertiroidismo recibieron metimazol y el 21% recibieron carbimazol. Se requirieron corticosteroides sistémicos en el 17% (7/42) de los pacientes. El hipertiroidismo se resolvió en el 91% de los 42 pacientes. De los 11 pacientes en quienes se suspendió OPDIVO con ipilimumab por hipertiroidismo, 8 reiniciaron el tratamiento después de la mejora de los síntomas; de estos, 1 (13%) tuvo recurrencia del hipertiroidismo.
OPDIVO 3 mg/kg con Ipilimumab 1 mg/kg: El hipertiroidismo ocurrió en el 12% (80/666) de los pacientes con CCR o CRC que recibieron OPDIVO 3 mg/kg con ipilimumab 1 mg/kg cada 3 semanas, incluidas reacciones adversas de Grado 3 (0.6%) y Grado 2 (4.5%). El hipertiroidismo condujo a la interrupción permanente de OPDIVO con ipilimumab en ningún paciente y a la suspensión de OPDIVO con ipilimumab en el 2.3% de los pacientes con CCR o CRC.
De los 80 pacientes con CCR o CRC que desarrollaron hipertiroidismo, aproximadamente el 16% recibió metimazol y el 3% recibió carbimazol. Se requirieron corticosteroides sistémicos en el 20% (16/80) de los pacientes con hipertiroidismo. El hipertiroidismo se resolvió en el 85% de los 80 pacientes. De los 15 pacientes en quienes se suspendió OPDIVO con ipilimumab por hipertiroidismo, 11 reiniciaron el tratamiento después de la mejora de los síntomas; de estos, 3 (27%) tuvieron recurrencia del hipertiroidismo.
Hipotiroidismo
OPDIVO como agente único
El hipotiroidismo ocurrió en el 8% (163/1994) de los pacientes que recibieron OPDIVO como agente único, incluidas reacciones adversas de Grado 3 (0.2%) y Grado 2 (4.8%). El hipotiroidismo condujo a la interrupción permanente de OPDIVO en ningún paciente y a la suspensión de OPDIVO en el 0.5% de los pacientes.
Aproximadamente el 79% de los pacientes con hipotiroidismo recibieron levotiroxina. Se requirieron corticosteroides sistémicos en el 3.1% (5/163) de los pacientes con hipotiroidismo. El hipotiroidismo se resolvió en el 35% de los 163 pacientes. De los 9 pacientes en quienes se suspendió OPDIVO por hipotiroidismo, 3 reiniciaron OPDIVO después de la mejora de los síntomas; de estos, 1 (33%) tuvo recurrencia del hipotiroidismo.
OPDIVO con Ipilimumab
OPDIVO 1 mg/kg con Ipilimumab 3 mg/kg: El hipotiroidismo ocurrió en el 20% (91/456) de los pacientes con melanoma o HCC que recibieron OPDIVO 1 mg/kg con ipilimumab 3 mg/kg cada 3 semanas, incluidas reacciones adversas de Grado 3 (0.4%) y Grado 2 (11%). El hipotiroidismo condujo a la interrupción permanente de OPDIVO con ipilimumab en el 0.9% y a la suspensión de OPDIVO con ipilimumab en el 0.9% de los pacientes.
Aproximadamente el 89% de los pacientes con hipotiroidismo recibieron levotiroxina. Se requirieron corticosteroides sistémicos en el 2.2% (2/91) de los pacientes con hipotiroidismo. El hipotiroidismo se resolvió en el 41% de los 91 pacientes. De los 4 pacientes en quienes se suspendió OPDIVO con ipilimumab por hipotiroidismo, 2 reiniciaron el tratamiento después de la mejora de los síntomas; de estos, ninguno tuvo recurrencia del hipotiroidismo.
OPDIVO 3 mg/kg con Ipilimumab 1 mg/kg: El hipotiroidismo ocurrió en el 18% (122/666) de los pacientes con CCR o CRC que recibieron OPDIVO 3 mg/kg e ipilimumab 1 mg/kg cada 3 semanas, incluidas reacciones adversas de Grado 3 (0.6%) y Grado 2 (11%). El hipotiroidismo condujo a la interrupción permanente de OPDIVO con ipilimumab en el 0.2% y a la suspensión de OPDIVO con ipilimumab en el 1.4% de los pacientes con CCR o CRC.
De los 122 pacientes con CCR o CRC que desarrollaron hipotiroidismo, aproximadamente el 82% recibió levotiroxina. Se requirieron corticosteroides sistémicos en el 7% (9/122) de los pacientes con hipotiroidismo. El hipotiroidismo se resolvió en el 27% de los 122 pacientes. De los 9 pacientes en quienes se suspendió OPDIVO con ipilimumab por hipotiroidismo, 5 reiniciaron el tratamiento después de la mejora de los síntomas; de estos, 1 (20%) tuvo recurrencia del hipotiroidismo.
Diabetes mellitus tipo 1, que puede presentarse con cetoacidosis diabética
Controle a los pacientes para detectar hiperglucemia u otros signos y síntomas de diabetes. Inicie el tratamiento con insulina según esté clínicamente indicado. Suspenda OPDIVO según la gravedad [ver Dosificación y administración (2.3)].
OPDIVO como agente único
La diabetes ocurrió en el 0.9% (17/1994) de los pacientes que recibieron OPDIVO como agente único, incluidas reacciones adversas de Grado 3 (0.4%) y Grado 2 (0.3%), y dos casos de cetoacidosis diabética. La diabetes condujo a la interrupción permanente de OPDIVO en ningún paciente y a la suspensión de OPDIVO en el 0.1% de los pacientes.
Ningún paciente (0/17) con diabetes requirió corticosteroides sistémicos. La diabetes se resolvió en el 29% de los 17 pacientes. De los 2 pacientes en quienes se suspendió OPDIVO por diabetes, ambos reiniciaron OPDIVO después de la mejora de los síntomas; de estos, ninguno tuvo recurrencia de la diabetes.
Nefritis inmunomediada con disfunción renal
OPDIVO puede causar nefritis inmunomediada, que se define como la necesidad de usar esteroides y sin una etiología alternativa clara.
OPDIVO como agente único
La nefritis inmunomediada y la disfunción renal ocurrieron en el 1.2% (23/1994) de los pacientes que recibieron OPDIVO como agente único, incluidas reacciones adversas de Grado 4 (<0.1%), Grado 3 (0.5%) y Grado 2 (0.6%). La nefritis inmunomediada y la disfunción renal condujeron a la interrupción permanente de OPDIVO en el 0.3% y a la suspensión de OPDIVO en el 0.4% de los pacientes.
Se requirieron corticosteroides sistémicos en el 100% (23/23) de los pacientes con nefritis y disfunción renal. La nefritis y la disfunción renal se resolvieron en el 78% de los 23 pacientes. De los 7 pacientes en quienes se suspendió OPDIVO por nefritis o disfunción renal, 7 reiniciaron OPDIVO después de la mejora de los síntomas; de estos, 1 (14%) tuvo recurrencia de nefritis o disfunción renal.
Reacciones adversas dermatológicas inmunomediadas
OPDIVO puede causar erupción o dermatitis inmunomediada, definida como la necesidad de usar esteroides y sin una etiología alternativa clara. La dermatitis exfoliativa, incluido el síndrome de Stevens-Johnson, la necrólisis epidérmica tóxica (NET) y el DRESS (erupción medicamentosa con eosinofilia y síntomas sistémicos) ha ocurrido con anticuerpos bloqueadores de PD-1/PD-L1. Los emolientes tópicos y/o los corticosteroides tópicos pueden ser adecuados para tratar erupciones no exfoliativas leves a moderadas. Suspenda o interrumpa permanentemente OPDIVO según la gravedad [ver Dosificación y administración (2.3)].
OPDIVO como agente único
La erupción inmunomediada ocurrió en el 9% (171/1994) de los pacientes, incluidas reacciones adversas de Grado 3 (1.1%) y Grado 2 (2.2%). La erupción inmunomediada condujo a la interrupción permanente de OPDIVO en el 0.3% y a la suspensión de OPDIVO en el 0.5% de los pacientes.
Se requirieron corticosteroides sistémicos en el 100% (171/171) de los pacientes con erupción cutánea inmunomediada. La erupción se resolvió en el 72% de los 171 pacientes. De los 10 pacientes en quienes se suspendió OPDIVO por erupción cutánea inmunomediada, 9 reiniciaron OPDIVO después de la mejora de los síntomas; de estos, 3 (33%) tuvieron recurrencia de erupción cutánea inmunomediada.
OPDIVO con Ipilimumab
OPDIVO 1 mg/kg con Ipilimumab 3 mg/kg: La erupción cutánea inmunomediada ocurrió en el 28% (127/456) de los pacientes con melanoma o HCC que recibieron OPDIVO 1 mg/kg con ipilimumab 3 mg/kg cada 3 semanas, incluidas reacciones adversas de Grado 3 (4,8%) y Grado 2 (10%). La erupción cutánea inmunomediada condujo a la interrupción permanente de OPDIVO con ipilimumab en el 0,4% y a la suspensión de OPDIVO con ipilimumab en el 3,9% de los pacientes.
Se requirieron corticosteroides sistémicos en el 100% (127/127) de los pacientes con erupción cutánea inmunomediada. La erupción se resolvió en el 84% de los 127 pacientes. De los 18 pacientes en quienes se suspendió OPDIVO con ipilimumab por erupción cutánea inmunomediada, 15 reiniciaron el tratamiento después de la mejora de los síntomas; de estos, 8 (53%) tuvieron recurrencia de erupción cutánea inmunomediada.
OPDIVO 3 mg/kg con Ipilimumab 1 mg/kg: La erupción cutánea inmunomediada ocurrió en el 16% (108/666) de los pacientes con RCC o CRC que recibieron OPDIVO 3 mg/kg con ipilimumab 1 mg/kg cada 3 semanas, incluidas reacciones adversas de Grado 3 (3,5%) y Grado 2 (4,2%). La erupción cutánea inmunomediada condujo a la interrupción permanente de OPDIVO con ipilimumab en el 0,5% de los pacientes y a la suspensión de OPDIVO con ipilimumab en el 2,0% de los pacientes con RCC o CRC.
Se requirieron corticosteroides sistémicos en el 100% (108/108) de los pacientes con erupción cutánea inmunomediada. La erupción se resolvió en el 75% de los 108 pacientes. De los 13 pacientes en quienes se suspendió OPDIVO con ipilimumab por erupción cutánea inmunomediada, 11 reiniciaron el tratamiento después de la mejora de los síntomas; de estos, 5 (46%) tuvieron recurrencia de erupción cutánea inmunomediada.
Otras reacciones adversas inmunomediadas
Las siguientes reacciones adversas inmunomediadas clínicamente significativas ocurrieron con una incidencia de <1% (a menos que se indique lo contrario) en pacientes que recibieron OPDIVO o OPDIVO en combinación con ipilimumab, o se informaron con el uso de otros anticuerpos bloqueadores de PD-1/PD-L1. Se han informado casos graves o fatales para algunas de estas reacciones adversas.
Cardiovascular: Miocarditis, pericarditis, vasculitis
Sistema nervioso: Meningitis, encefalitis, mielitis y desmielinización, síndrome miasténico/miastenia gravis (incluida la exacerbación), síndrome de Guillain-Barré, paresia nerviosa, neuropatía autoinmune
Ocular: Uveítis, iritis y otras toxicidades inflamatorias oculares pueden ocurrir. Algunos casos pueden estar asociados con desprendimiento de retina. Pueden ocurrir diversos grados de discapacidad visual, incluida la ceguera. Si se produce uveítis en combinación con otras reacciones adversas inmunomediadas, considere un síndrome similar a Vogt-Koyanagi-Harada, ya que esto puede requerir tratamiento con esteroides sistémicos para reducir el riesgo de pérdida permanente de la visión
Gastrointestinal: Pancreatitis que incluye aumentos en los niveles séricos de amilasa y lipasa, gastritis, duodenitis
Musculoesquelético y tejido conectivo: Miositis/polimiositis, rabdomiólisis y secuelas asociadas que incluyen insuficiencia renal, artritis, polimialgia reumática
Endocrino: Hipoparatiroidismo
Otros (hematológicos/inmunológicos): Anemia hemolítica, anemia aplásica, linfohistiocitosis hemofagocítica, síndrome de respuesta inflamatoria sistémica, linfadenitis necrotizante histiocítica (linfadenitis de Kikuchi), sarcoidosis, púrpura trombocitopénica inmunitaria, rechazo de trasplante de órgano sólido, rechazo de otro trasplante (incluido el trasplante de injerto corneal)
5.2 Reacciones relacionadas con la infusión
OPDIVO puede causar reacciones graves relacionadas con la infusión, que se han informado en <1,0% de los pacientes en ensayos clínicos. Suspenda OPDIVO en pacientes con reacciones relacionadas con la infusión graves o potencialmente mortales. Interrumpa o disminuya la velocidad de infusión en pacientes con reacciones relacionadas con la infusión leves o moderadas [ver Dosificación y administración (2.3)].
OPDIVO como agente único
En pacientes que recibieron OPDIVO como una infusión intravenosa de 60 minutos, las reacciones relacionadas con la infusión ocurrieron en el 6,4% (127/1994) de los pacientes.
En un ensayo que evaluó la farmacocinética y la seguridad de una infusión más rápida, en la que los pacientes recibieron OPDIVO como una infusión intravenosa de 60 minutos o una infusión intravenosa de 30 minutos, las reacciones relacionadas con la infusión ocurrieron en el 2,2% (8/368) y el 2,7% (10/369) de los pacientes, respectivamente. Además, el 0,5% (2/368) y el 1,4% (5/369) de los pacientes, respectivamente, experimentaron reacciones adversas dentro de las 48 horas posteriores a la infusión que llevaron a un retraso en la dosis, una interrupción permanente o la suspensión de OPDIVO.
OPDIVO con Ipilimumab
OPDIVO 1 mg/kg con Ipilimumab 3 mg/kg
Las reacciones relacionadas con la infusión ocurrieron en el 2,5% (10/407) de los pacientes con melanoma y en el 8% (4/49) de los pacientes con HCC que recibieron OPDIVO 1 mg/kg con ipilimumab 3 mg/kg cada 3 semanas.
OPDIVO 3 mg/kg con Ipilimumab 1 mg/kg
Las reacciones relacionadas con la infusión ocurrieron en el 5,1% (28/547) de los pacientes con RCC y el 4,2% (5/119) de los pacientes con CRC que recibieron OPDIVO 3 mg/kg con ipilimumab 1 mg/kg cada 3 semanas, respectivamente. Las reacciones relacionadas con la infusión ocurrieron en el 12% (37/300) de los pacientes con mesotelioma pleural maligno que recibieron OPDIVO 3 mg/kg cada 2 semanas con ipilimumab 1 mg/kg cada 6 semanas.
5.3 Complicaciones del Trasplante Alogénico de Células Madre Hematopoyéticas
Pueden ocurrir complicaciones fatales y otras graves en pacientes que reciben trasplante alogénico de células madre hematopoyéticas (HSCT) antes o después de ser tratados con un anticuerpo bloqueador del receptor PD-1. Las complicaciones relacionadas con el trasplante incluyen enfermedad de injerto contra huésped (GVHD) hiperaguda, GVHD aguda, GVHD crónica, enfermedad veno-oclusiva hepática (VOD) después del acondicionamiento de intensidad reducida y síndrome febril que requiere esteroides (sin una causa infecciosa identificada) [ver Reacciones adversas (6.1)]. Estas complicaciones pueden ocurrir a pesar de la terapia de intervención entre el bloqueo de PD-1 y el HSCT alogénico.
Controle de cerca a los pacientes en busca de evidencia de complicaciones relacionadas con el trasplante e intervenga de inmediato. Considere el beneficio versus los riesgos del tratamiento con un anticuerpo bloqueador del receptor PD-1 antes o después de un HSCT alogénico.
5.4 Toxicidad Embriofetal
Con base en su mecanismo de acción y datos de estudios en animales, OPDIVO puede causar daño fetal cuando se administra a una mujer embarazada. En estudios de reproducción en animales, la administración de nivolumab a monos cynomolgus desde el inicio de la organogénesis hasta el parto resultó en un aumento del aborto y la muerte infantil prematura. Advierta a las mujeres embarazadas sobre el riesgo potencial para un feto. Avise a las mujeres en edad fértil que usen métodos anticonceptivos efectivos durante el tratamiento con OPDIVO y durante 5 meses después de la última dosis [ver Uso en poblaciones específicas (8.1, 8.3)].
5.5 Aumento de la Mortalidad en Pacientes con Mieloma Múltiple Cuando OPDIVO Se Agrega a un Análogo de Talidomida y Dexametasona
En ensayos clínicos aleatorizados en pacientes con mieloma múltiple, la adición de un anticuerpo bloqueador de PD-1, incluido OPDIVO, a un análogo de talidomida más dexametasona, un uso para el cual no está indicado ningún anticuerpo bloqueador de PD-1 o PD-L1, resultó en un aumento de la mortalidad. No se recomienda el tratamiento de pacientes con mieloma múltiple con un anticuerpo bloqueador de PD-1 o PD-L1 en combinación con un análogo de talidomida más dexametasona fuera de los ensayos clínicos controlados.
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas clínicamente significativas se describen en otra parte del etiquetado.
- •
- Reacciones adversas inmunomediadas graves y mortales [ver Advertencias y precauciones (5.1)]
- •
- Reacciones relacionadas con la infusión [ver Advertencias y precauciones (5.2)]
- •
- Complicaciones del alo-TPH [ver Advertencias y precauciones (5.3)]
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se realizan en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un medicamento no pueden compararse directamente con las tasas en los ensayos clínicos de otro medicamento y pueden no reflejar las tasas observadas en la práctica.
Los datos de ADVERTENCIAS Y PRECAUCIONES reflejan la exposición a OPDIVO como agente único en 1994 pacientes incluidos en CHECKMATE-037, CHECKMATE-017, CHECKMATE-057, CHECKMATE-066, CHECKMATE-025, CHECKMATE-067, CHECKMATE-205, CHECKMATE-039 o un ensayo de un solo brazo en NSCLC (n=117); OPDIVO 1 mg/kg con ipilimumab 3 mg/kg en pacientes incluidos en CHECKMATE-067 (n=313), CHECKMATE-040 (n=49) u otro ensayo aleatorizado (n=94); OPDIVO 3 mg/kg administrado con ipilimumab 1 mg/kg (n=666) en pacientes incluidos en CHECKMATE-214 o CHECKMATE-142; OPDIVO 3 mg/kg cada 2 semanas con ipilimumab 1 mg/kg cada 6 semanas en pacientes incluidos en CHECKMATE-227 (n=576) o CHECKMATE-743 (n=300); OPDIVO 360 mg con ipilimumab 1 mg/kg y 2 ciclos de quimioterapia con doblete de platino en CHECKMATE-9LA (n=361); y OPDIVO 240 mg con cabozantinib 40 mg en pacientes incluidos en CHECKMATE-9ER (n=320).
Melanoma irresecable o metastásico
Melanoma metastásico previamente tratado
Se evaluó la seguridad de OPDIVO en CHECKMATE-037, un ensayo aleatorizado, abierto, en 370 pacientes con melanoma irresecable o metastásico [ver Estudios clínicos (14.1)]. Los pacientes tenían progresión documentada de la enfermedad después del tratamiento con ipilimumab y, si eran positivos para la mutación BRAF V600, un inhibidor de BRAF. El ensayo excluyó a los pacientes con enfermedad autoinmune, reacciones adversas de grado 4 relacionadas con ipilimumab previas (excepto endocrinopatías) o reacciones adversas de grado 3 relacionadas con ipilimumab que no se hubieran resuelto o estuvieran controladas de forma inadecuada en las 12 semanas posteriores al evento inicial, pacientes con una condición que requiriera tratamiento sistémico crónico con corticosteroides (>10 mg de equivalente de prednisona al día) u otros medicamentos inmunosupresores, una prueba positiva para hepatitis B o C, y antecedentes de VIH. Los pacientes recibieron OPDIVO 3 mg/kg por infusión intravenosa durante 60 minutos cada 2 semanas (n=268) o la elección del investigador de quimioterapia (n=102): dacarbazina 1000 mg/m2 por vía intravenosa cada 3 semanas o carboplatino AUC 6 mg/mL/min y paclitaxel 175 mg/m2 por vía intravenosa cada 3 semanas. La mediana de duración de la exposición fue de 5.3 meses (rango: 1 día a 13.8+ meses) en pacientes tratados con OPDIVO y fue de 2 meses (rango: 1 día a 9.6+ meses) en pacientes tratados con quimioterapia. En este ensayo en curso, el 24% de los pacientes recibieron OPDIVO durante >6 meses y el 3% de los pacientes recibieron OPDIVO durante >1 año.
Las características de la población en el grupo de OPDIVO y el grupo de quimioterapia fueron similares: 66% hombres, mediana de edad 59.5 años, 98% blancos, estado funcional inicial del Eastern Cooperative Oncology Group (ECOG) 0 (59%) o 1 (41%), 74% con enfermedad en estadio M1c, 73% con melanoma cutáneo, 11% con melanoma mucoso, 73% recibieron dos o más terapias previas para la enfermedad avanzada o metastásica, y el 18% tenía metástasis cerebral. Hubo más pacientes en el grupo de OPDIVO con lactato deshidrogenasa (LDH) elevada al inicio del estudio (51% frente a 38%).
Se produjeron reacciones adversas graves en el 41% de los pacientes que recibieron OPDIVO. Se interrumpió el tratamiento con OPDIVO debido a reacciones adversas en el 9% de los pacientes. El veintiséis por ciento de los pacientes que recibieron OPDIVO tuvieron una interrupción de la dosis debido a una reacción adversa. Se produjeron reacciones adversas de grado 3 y 4 en el 42% de los pacientes que recibieron OPDIVO. Las reacciones adversas de grado 3 y 4 más frecuentes notificadas en el 2% a <5% de los pacientes que recibieron OPDIVO fueron dolor abdominal, hiponatremia, aumento de la aspartato aminotransferasa y aumento de la lipasa. La reacción adversa más común (notificada en ≥20% de los pacientes) fue el exantema.
Las tablas 5 y 6 resumen las reacciones adversas y las anomalías de laboratorio, respectivamente, en CHECKMATE-037.
| La toxicidad se clasificó según NCI CTCAE v4. a Incluye erupción maculopapular, erupción eritematosa, erupción pruriginosa, erupción folicular, erupción macular, erupción papular, erupción pustular, erupción vesicular y dermatitis acneiforme. b Incluye rinitis, faringitis y nasofaringitis. |
||||
|
Reacción adversa |
OPDIVO |
Quimioterapia |
||
|
Todos los grados (%) |
Grados 3-4 (%) |
Todos los grados (%) |
Grados 3-4 (%) |
|
|
Piel y tejido subcutáneo |
||||
|
Erupción cutáneaa |
21 |
0.4 |
7 |
0 |
|
Prurito |
19 |
0 |
3.9 |
0 |
|
Respiratorias, torácicas y mediastínicas |
||||
|
Tos |
17 |
0 |
6 |
0 |
|
Infecciones |
||||
|
Infección de las vías respiratorias superioresb |
11 |
0 |
2 |
0 |
|
Generales |
||||
|
Edema periférico |
10 |
0 |
5 |
0 |
Las reacciones adversas clínicamente importantes en <10% de los pacientes que recibieron OPDIVO fueron:
Trastornos cardíacos: arritmia ventricular
Trastornos oculares: iridociclitis
Trastornos generales y afecciones en el sitio de administración: reacciones relacionadas con la infusión
Investigaciones: aumento de la amilasa, aumento de la lipasa
Trastornos del sistema nervioso: mareos, neuropatía periférica y sensorial
Trastornos de la piel y del tejido subcutáneo: dermatitis exfoliativa, eritema multiforme, vitiligo, psoriasis
| a La incidencia de cada prueba se basa en el número de pacientes que tenían disponible tanto la medición de laboratorio inicial como al menos una medición de laboratorio durante el estudio: grupo de OPDIVO (rango: 252 a 256 pacientes) y grupo de quimioterapia (rango: 94 a 96 pacientes). | ||||
|
Anomalía de laboratorio |
OPDIVO |
Quimioterapia |
||
|
Todos los grados (%) |
Grados 3-4 (%) |
Todos los grados (%) |
Grados 3-4 (%) |
|
|
Aumento de AST |
28 |
2.4 |
12 |
1 |
|
Hiponatremia |
25 |
5 |
18 |
1.1 |
|
Aumento de la fosfatasa alcalina |
22 |
2.4 |
13 |
1.1 |
|
Aumento de ALT |
16 |
1.6 |
5 |
0 |
|
Hiperpotasemia |
15 |
2 |
6 |
0 |
Melanoma metastásico no tratado previamente
CHECKMATE-066
La seguridad de OPDIVO también se evaluó en CHECKMATE-066, un ensayo aleatorizado, doble ciego y con control activo en 411 pacientes con melanoma irresecable o metastásico con BRAF V600 de tipo natural no tratados previamente [ver Estudios clínicos (14.1)]. Se excluyeron del ensayo los pacientes con enfermedad autoinmunitaria y los pacientes que requerían tratamiento sistémico crónico con corticosteroides (>10 mg de equivalente de prednisona al día) u otros medicamentos inmunosupresores. Los pacientes recibieron OPDIVO 3 mg/kg mediante infusión intravenosa durante 60 minutos cada 2 semanas (n=206) o 1000 mg/m2 de dacarbazina por vía intravenosa cada 3 semanas (n=205). La mediana de duración de la exposición fue de 6.5 meses (rango: 1 día a 16.6 meses) en los pacientes tratados con OPDIVO. En este ensayo, el 47 % de los pacientes recibieron OPDIVO durante >6 meses y el 12 % de los pacientes recibieron OPDIVO durante >1 año.
Las características de la población del ensayo en el grupo de OPDIVO y en el grupo de dacarbazina fueron: 59 % hombres, mediana de edad de 65 años, 99.5 % blancos, 61 % con enfermedad en estadio M1c, 74 % con melanoma cutáneo, 11 % con melanoma mucoso, 4 % con metástasis cerebral y 37 % con LDH elevada al inicio del estudio. Hubo más pacientes en el grupo de OPDIVO con un estado funcional ECOG de 0 (71 % frente a 59 %).
Se produjeron reacciones adversas graves en el 36 % de los pacientes que recibieron OPDIVO. Las reacciones adversas provocaron la interrupción permanente de OPDIVO en el 7 % de los pacientes y la interrupción de la dosis en el 26 % de los pacientes; ningún tipo de reacción adversa representó la mayoría de las interrupciones de OPDIVO. Se produjeron reacciones adversas de grado 3 y 4 en el 41 % de los pacientes que recibieron OPDIVO.
Las reacciones adversas de grado 3 y 4 más frecuentes notificadas en ≥2 % de los pacientes que recibieron OPDIVO fueron aumento de la gamma-glutamil transferasa (3.9 %) y diarrea (3.4 %). Las reacciones adversas más comunes (notificadas en ≥20 % de los pacientes y con una incidencia mayor que en el grupo de dacarbazina) fueron fatiga, dolor musculoesquelético, erupción cutánea y prurito.
Las Tablas 7 y 8 resumen las reacciones adversas y las anomalías de laboratorio seleccionadas, respectivamente, en CHECKMATE-066.
| La toxicidad se clasificó según los criterios comunes de toxicidad del NCI v4. a Incluye edema periorbitario, edema facial, edema generalizado, edema gravitacional, edema localizado, edema periférico, edema pulmonar y linfedema. b Incluye dolor de espalda, dolor óseo, dolor torácico musculoesquelético, molestias musculoesqueléticas, mialgia, dolor de cuello, dolor en las extremidades, dolor en la mandíbula y dolor espinal. c Incluye erupción maculopapular, erupción eritematosa, erupción pruriginosa, erupción folicular, erupción macular, erupción papular, erupción pustular, erupción vesicular, dermatitis, dermatitis alérgica, dermatitis exfoliativa, dermatitis acneiforme, erupción medicamentosa y reacción cutánea. d Incluye rinitis, rinitis vírica, faringitis y nasofaringitis. |
||||
|
Reacción adversa |
OPDIVO |
Dacarbazina |
||
|
Todos los grados (%) |
Grados 3-4 (%) |
Todos los grados (%) |
Grados 3-4 (%) |
|
|
Generales |
||||
|
Fatiga |
49 |
1.9 |
39 |
3.4 |
|
Edemaa |
12 |
1.5 |
4.9 |
0 |
|
Musculoesqueléticas y del tejido conjuntivo |
||||
|
Dolor musculoesqueléticob |
32 |
2.9 |
25 |
2.4 |
|
De la piel y del tejido subcutáneo |
||||
|
Erupción cutáneac |
28 |
1.5 |
12 |
0 |
|
Prurito |
23 |
0.5 |
12 |
0 |
|
Vitiligo |
11 |
0 |
0.5 |
0 |
|
Eritema |
10 |
0 |
2.9 |
0 |
|
Infecciones |
||||
|
Infección de las vías respiratorias superioresd |
17 |
0 |
6 |
0 |
Las reacciones adversas clínicamente importantes en <10% de los pacientes que recibieron OPDIVO fueron:
Trastornos del sistema nervioso: neuropatía periférica
| a La incidencia de cada prueba se basa en el número de pacientes que tenían disponible tanto la medición de laboratorio inicial como al menos una en el estudio: grupo de OPDIVO (rango: 194 a 197 pacientes) y grupo de dacarbazina (rango: 186 a 193 pacientes). | ||||
|
Anormalidad de laboratorio |
OPDIVO |
Dacarbazina |
||
|
Todos los grados (%) |
Grados 3-4 (%) |
Todos los grados (%) |
Grados 3-4 (%) |
|
|
Aumento de ALT |
25 |
3 |
19 |
0.5 |
|
Aumento de AST |
24 |
3.6 |
19 |
0.5 |
|
Aumento de la fosfatasa alcalina |
21 |
2.6 |
14 |
1.6 |
|
Aumento de la bilirrubina |
13 |
3.1 |
6 |
0 |
CHECKMATE-067
Se evaluó la seguridad de OPDIVO, administrado con ipilimumab o como agente único, en CHECKMATE-067, un ensayo aleatorizado (1:1:1), doble ciego en 937 pacientes con melanoma no resecable o metastásico no tratado previamente [ver Estudios clínicos (14.1)]. Se excluyeron del ensayo pacientes con enfermedad autoinmune, una afección médica que requiriera tratamiento sistémico con corticosteroides (más de 10 mg de equivalente de prednisona al día) u otra medicación inmunosupresora en los 14 días anteriores al inicio del tratamiento del estudio, un resultado positivo en la prueba de hepatitis B o C, o antecedentes de VIH.
Los pacientes fueron aleatorizados para recibir:
- •
- OPDIVO 1 mg/kg durante 60 minutos con ipilimumab 3 mg/kg mediante infusión intravenosa cada 3 semanas durante 4 dosis seguido de OPDIVO como agente único a una dosis de 3 mg/kg mediante infusión intravenosa durante 60 minutos cada 2 semanas (grupo de OPDIVO e ipilimumab; n=313), o
- •
- OPDIVO 3 mg/kg mediante infusión intravenosa durante 60 minutos cada 2 semanas (grupo de OPDIVO; n=313), o
- •
- Ipilimumab 3 mg/kg mediante infusión intravenosa cada 3 semanas hasta 4 dosis (grupo de ipilimumab; n=311).
La mediana de duración de la exposición a OPDIVO fue de 2.8 meses (rango: 1 día a 36.4 meses) para el grupo de OPDIVO e ipilimumab y de 6.6 meses (rango: 1 día a 36.0 meses) para el grupo de OPDIVO. En el grupo de OPDIVO e ipilimumab, el 39% estuvo expuesto a OPDIVO durante ≥6 meses y el 30% estuvo expuesto durante >1 año. En el grupo de OPDIVO, el 53% estuvo expuesto durante ≥6 meses y el 40% durante >1 año.
Las características de la población fueron: 65% hombres, mediana de edad 61 años, 97% blancos, estado funcional ECOG inicial 0 (73%) o 1 (27%), 93% con enfermedad en estadio IV según la American Joint Committee on Cancer (AJCC), 58% con enfermedad en estadio M1c; 36% con LDH elevada al inicio, 4% con antecedentes de metástasis cerebral y 22% había recibido tratamiento adyuvante.
Las reacciones adversas graves (74% y 44%), las reacciones adversas que provocaron la interrupción permanente (47% y 18%) o los retrasos en la administración (58% y 36%) y las reacciones adversas de grado 3 o 4 (72% y 51%) se produjeron con mayor frecuencia en el grupo de OPDIVO e ipilimumab en comparación con el grupo de OPDIVO.
Las reacciones adversas graves más frecuentes (≥10%) en el grupo de OPDIVO e ipilimumab y en el grupo de OPDIVO, respectivamente, fueron diarrea (13% y 2.2%), colitis (10% y 1.9%) y pirexia (10% y 1.0%). Las reacciones adversas que provocaron la interrupción de ambos fármacos en el grupo de OPDIVO e ipilimumab y de OPDIVO en el grupo de OPDIVO, respectivamente, fueron colitis (10% y 0.6%), diarrea (8% y 2.2%), aumento de la ALT (4.8% y 1.0%), aumento de la AST (4.5% y 0.6%) y neumonitis (1.9% y 0.3%).
Las reacciones adversas más comunes (≥20%) en el grupo de OPDIVO e ipilimumab fueron fatiga, diarrea, erupción cutánea, náuseas, pirexia, prurito, dolor musculoesquelético, vómitos, disminución del apetito, tos, dolor de cabeza, disnea, infección de las vías respiratorias superiores, artralgia y aumento de las transaminasas. Las reacciones adversas más comunes (≥20%) en el grupo de OPDIVO fueron fatiga, erupción cutánea, dolor musculoesquelético, diarrea, náuseas, tos, prurito, infección de las vías respiratorias superiores, disminución del apetito, dolor de cabeza, estreñimiento, artralgia y vómitos.
Las Tablas 9 y 10 resumen la incidencia de reacciones adversas y anomalías de laboratorio, respectivamente, en CHECKMATE-067.
| La toxicidad se clasificó según los criterios comunes de toxicidad del National Cancer Institute versión 4 (NCI CTCAE v4). a Incluye astenia y fatiga. b Incluye erupción pustulosa, dermatitis, dermatitis acneiforme, dermatitis alérgica, dermatitis atópica, dermatitis ampollosa, dermatitis exfoliativa, dermatitis psoriasiforme, erupción medicamentosa, erupción exfoliativa, erupción eritematosa, erupción generalizada, erupción macular, erupción maculopapular, erupción morbiliforme, erupción papular, erupción papuloescamosa y erupción pruriginosa. c Incluye dolor de espalda, dolor óseo, dolor torácico musculoesquelético, molestias musculoesqueléticas, mialgia, dolor de cuello, dolor en las extremidades y dolor espinal. d Incluye infección de las vías respiratorias superiores, nasofaringitis, faringitis y rinitis. e Incluye hipertensión y aumento de la presión arterial. |
||||||
|
Reacción adversa |
OPDIVO e ipilimumab |
OPDIVO |
Ipilimumab |
|||
|
Todos los grados (%) |
Grados 3-4 (%) |
Todos los grados (%) |
Grados 3-4 (%) |
Todos los grados (%) |
Grados 3-4 (%) |
|
|
Generales |
||||||
|
Fatigaa |
62 |
7 |
59 |
1.6 |
51 |
4.2 |
|
Pyrexia |
40 |
1.6 |
16 |
0 |
18 |
0.6 |
|
Gastrointestinales |
||||||
|
Diarrea |
54 |
11 |
36 |
5 |
47 |
7 |
|
Náuseas |
44 |
3.8 |
30 |
0.6 |
31 |
1.9 |
|
Vómitos |
31 |
3.8 |
20 |
1 |
17 |
1.6 |
|
Piel y tejido subcutáneo |
||||||
|
Erupción cutáneab |
53 |
6 |
40 |
1.9 |
42 |
3.5 |
|
Vitiligo |
9 |
0 |
10 |
0.3 |
5 |
0 |
|
Musculoesquelético y tejido conectivo |
||||||
|
Dolor musculoesqueléticoc |
32 |
2.6 |
42 |
3.8 |
36 |
1.9 |
|
Artralgia |
21 |
0.3 |
21 |
1 |
16 |
0.3 |
|
Metabolismo y nutrición |
||||||
|
Disminución del apetito |
29 |
1.9 |
22 |
0 |
24 |
1.3 |
|
Respiratorias, torácicas y mediastínicas |
||||||
|
Tos/tos productiva |
27 |
0.3 |
28 |
0.6 |
22 |
0 |
|
Disnea/disnea de esfuerzo |
24 |
2.9 |
18 |
1.3 |
17 |
0.6 |
|
Infecciones |
||||||
|
Infección de las vías respiratorias superioresd |
23 |
0 |
22 |
0.3 |
17 |
0 |
|
Endocrino |
||||||
|
Hipotiroidismo |
19 |
0.6 |
11 |
0 |
5 |
0 |
|
Hipertiroidismo |
11 |
1.3 |
6 |
0 |
1 |
0 |
|
Investigaciones |
||||||
|
Pérdida de peso |
12 |
0 |
7 |
0 |
7 |
0.3 |
|
Vascular |
||||||
|
Hipertensióne |
7 |
2.2 |
11 |
5 |
9 |
2.3 |
Las reacciones adversas clínicamente importantes en <10% de los pacientes que recibieron OPDIVO con ipilimumab u OPDIVO como agente único fueron:
Trastornos gastrointestinales: estomatitis, perforación intestinal
Trastornos de la piel y del tejido subcutáneo: vitiligo
Trastornos musculoesqueléticos y del tejido conjuntivo: miopatía, síndrome de Sjogren, espondiloartropatía, miositis (incluida la polimiositis)
Trastornos del sistema nervioso: neuritis, parálisis del nervio peroneo
| a La incidencia de cada prueba se basa en el número de pacientes que tenían disponible tanto la medición de laboratorio inicial como al menos una medición de laboratorio durante el estudio: OPDIVO e ipilimumab (rango: 75 a 297); OPDIVO (rango: 81 a 306); ipilimumab (rango: 61 a 301). | ||||||
|
Anormalidad de laboratorio |
OPDIVO e ipilimumab |
OPDIVO |
Ipilimumab |
|||
|
Todos los grados (%) |
Grados 3-4 (%) |
Todos los grados (%) |
Grados 3-4 (%) |
Todos los grados (%) |
Grados 3-4 (%) |
|
|
Química |
||||||
|
Aumento de ALT |
55 |
16 |
25 |
3 |
29 |
2.7 |
|
Hiperglucemia |
53 |
5.3 |
46 |
7 |
26 |
0 |
|
Aumento de AST |
52 |
13 |
29 |
3.7 |
29 |
1.7 |
|
Hiponatremia |
45 |
10 |
22 |
3.3 |
26 |
7 |
|
Aumento de la lipasa |
43 |
22 |
32 |
12 |
24 |
7 |
|
Aumento de la fosfatasa |
41 |
6 |
27 |
2 |
23 |
2 |
|
Hipocalcemia |
31 |
1.1 |
15 |
0.7 |
20 |
0.7 |
|
Aumento de la amilasa |
27 |
10 |
19 |
2.7 |
15 |
1.6 |
|
Increased creatinine |
26 |
2.7 |
19 |
0.7 |
17 |
1.3 |
|
Hematología |
||||||
|
Anemia |
52 |
2.7 |
41 |
2.6 |
41 |
6 |
|
Linfopenia |
39 |
5 |
41 |
4.9 |
29 |
4 |
Tratamiento adyuvante del melanoma
CHECKMATE-76K
La seguridad de OPDIVO como agente único se evaluó en CHECKMATE-76K, un ensayo aleatorizado (2:1), doble ciego, en 788 pacientes con melanoma en estadio IIB/C completamente resecado que recibieron OPDIVO 480 mg por infusión intravenosa durante 30 minutos cada 4 semanas (n=524) o placebo por infusión intravenosa durante 30 minutos cada 4 semanas (n=264) durante un máximo de 1 año [ver Estudios clínicos (14.2)]. La mediana de duración de la exposición fue de 11 meses en los pacientes tratados con OPDIVO y de 11 meses en los pacientes tratados con placebo.
Se produjeron reacciones adversas graves en el 18% de los pacientes tratados con OPDIVO. Se produjo una reacción adversa mortal en 1 (0,2%) paciente (insuficiencia cardíaca y lesión renal aguda). La interrupción permanente de OPDIVO debido a una reacción adversa se produjo en el 17% de los pacientes. Las reacciones adversas que provocaron la interrupción permanente de OPDIVO en >1% de los pacientes incluyeron diarrea (1,1%), artralgia (1,7%) y erupción cutánea (1,7%).
Se produjeron interrupciones de la dosis de OPDIVO debido a una reacción adversa en el 25% de los pacientes. Las reacciones adversas que requirieron la interrupción de la dosis en >1% de los pacientes incluyeron infección por COVID-19, reacción relacionada con la infusión, diarrea, artralgia y aumento de la ALT.
Las reacciones adversas más frecuentes (notificadas en ≥20% de los pacientes) fueron fatiga, dolor musculoesquelético, erupción cutánea, diarrea y prurito.
En las Tablas 11 y 12 se resumen las reacciones adversas y las anomalías de laboratorio, respectivamente, en CHECKMATE-76K.
| La toxicidad se clasificó según los criterios comunes de toxicidad del NCI v5. a Incluye astenia. b Incluye artralgia, artritis, dolor de espalda, dolor óseo, dolor torácico musculoesquelético, molestias musculoesqueléticas, rigidez musculoesquelética, mialgia, dolor de cuello, dolor torácico no cardíaco, dolor espinal, dolor en las extremidades. c Incluye dermatitis, dermatitis acneiforme, eccema dishidrótico, eccema, eccema asteatótico, erupción cutánea en los párpados, erupción cutánea genital, penfigoide, erupción cutánea en el pene, erupción cutánea eritematosa, erupción cutánea folicular, erupción cutánea macular, erupción cutánea maculopapular, erupción cutánea papular, erupción cutánea pruriginosa, erupción cutánea pustulosa, erupción cutánea vesicular, exfoliación cutánea, erupción cutánea tóxica. d Incluye colitis autoinmune, colitis, diarrea, enteritis, enterocolitis e Incluye hipotiroidismo autoinmune, aumento de la hormona estimulante de la tiroides en sangre. f Incluye cefalea en racimos, migraña. |
||||
|
Reacción adversa |
OPDIVO |
Placebo |
||
|
Todos los grados (%) |
Grados 3-4 (%) |
Todos los grados (%) |
Grados 3-4 (%) |
|
|
Generales |
||||
|
Fatigaa |
36 |
0.4 |
34 |
0.4 |
|
Musculoesqueléticas y tejido conectivo |
||||
|
Dolor musculoesqueléticob |
30 |
0.4 |
26 |
0.4 |
|
Piel y tejido subcutáneo |
||||
|
Erupción cutáneac |
28 |
1.1 |
15 |
0.4 |
|
Prurito |
20 |
0.2 |
11 |
0 |
|
Gastrointestinales |
||||
|
Diarread |
23 |
1.3 |
16 |
0 |
|
Náuseas |
14 |
0 |
11 |
0 |
|
Endocrino |
||||
|
Hipotiroidismoe |
14 |
0 |
2.3 |
0 |
|
Sistema nervioso |
||||
|
Dolor de cabezaf |
12 |
0.2 |
14 |
0.8 |
| a La incidencia de cada prueba se basa en el número de pacientes que tenían disponible tanto la medición de laboratorio inicial como al menos una medición de laboratorio durante el estudio: grupo de OPDIVO (rango: 262 a 513 pacientes) y grupo de placebo (rango: 138 a 261 pacientes). | ||||
|
Anormalidad de laboratorio |
OPDIVO |
Placebo |
||
|
Todos los grados (%) |
Grados 3-4 (%) |
Todos los grados (%) |
Grados 3-4 (%) |
|
|
Hematología |
||||
|
Anemia |
19 |
0 |
14 |
0 |
|
Linfopenia |
17 |
1.1 |
17 |
1.7 |
|
Neutropenia |
10 |
0 |
10 |
0.4 |
|
Química |
||||
|
AST aumentado |
25 |
2.2 |
16 |
0.4 |
|
Lipasa aumentada |
22 |
2.9 |
21 |
2.3 |
|
ALT aumentada |
20 |
2.1 |
15 |
0.4 |
|
Amilasa aumentada |
17 |
0.4 |
9 |
0 |
|
Creatinina aumentada |
15 |
0.4 |
13 |
0 |
|
Sodio disminuido |
13 |
0.6 |
11 |
0.4 |
|
Potasio aumentado |
13 |
1 |
15 |
1.1 |
CHECKMATE-238
La seguridad de OPDIVO como agente único se evaluó en CHECKMATE-238, un ensayo aleatorizado (1:1), doble ciego, en 905 pacientes con melanoma en estadio IIIB/C o en estadio IV completamente resecado que recibieron OPDIVO 3 mg/kg por infusión intravenosa durante 60 minutos cada 2 semanas (n=452) o ipilimumab 10 mg/kg por infusión intravenosa cada 3 semanas durante 4 dosis y luego cada 12 semanas a partir de la semana 24 durante un máximo de 1 año (n=453) [ver Estudios clínicos (14.2)]. La mediana de duración de la exposición fue de 11.5 meses en los pacientes tratados con OPDIVO y de 2.7 meses en los pacientes tratados con ipilimumab. En este ensayo en curso, el 74% de los pacientes recibieron OPDIVO durante >6 meses.
Se produjeron reacciones adversas graves en el 18% de los pacientes tratados con OPDIVO. Se interrumpió el tratamiento del estudio debido a reacciones adversas en el 9% de los pacientes tratados con OPDIVO y en el 42% de los pacientes tratados con ipilimumab. El veintiocho por ciento de los pacientes tratados con OPDIVO tuvieron al menos una omisión de la dosis debido a una reacción adversa. Se produjeron reacciones adversas de grado 3 o 4 en el 25% de los pacientes tratados con OPDIVO.
Las reacciones adversas de grado 3 y 4 más frecuentes notificadas en ≥2% de los pacientes tratados con OPDIVO fueron diarrea y aumento de la lipasa y la amilasa. Las reacciones adversas más comunes (al menos el 20%) fueron fatiga, diarrea, erupción cutánea, dolor musculoesquelético, prurito, dolor de cabeza, náuseas, infección de las vías respiratorias superiores y dolor abdominal. Las reacciones adversas inmunomediadas más comunes fueron erupción cutánea (16%), diarrea/colitis (6%) y hepatitis (3%).
En las tablas 13 y 14 se resumen las reacciones adversas y las anomalías de laboratorio, respectivamente, en CHECKMATE-238.
| La toxicidad se clasificó según los NCI CTCAE v4. a Incluye astenia. b Incluye molestias abdominales, dolor abdominal bajo, dolor abdominal alto y sensibilidad abdominal. c Incluye dermatitis descrita como acneiforme, alérgica, ampollosa o exfoliativa y erupción cutánea descrita como generalizada, eritematosa, macular, papular, maculopapular, pruriginosa, pustulosa, vesicular o en mariposa, y erupción medicamentosa. d Incluye dolor de espalda, dolor óseo, dolor torácico musculoesquelético, molestias musculoesqueléticas, mialgia, dolor de cuello, dolor espinal y dolor en las extremidades. e Incluye mareos posturales y vértigo. f Incluye infección de las vías respiratorias superiores, incluyendo infección viral de las vías respiratorias, infección de las vías respiratorias inferiores, rinitis, faringitis y nasofaringitis. g Incluye hipotiroidismo secundario e hipotiroidismo autoinmune. |
||||
|
Reacción adversa |
OPDIVO |
Ipilimumab 10 mg/kg |
||
|
Todos los grados (%) |
Grados 3-4 (%) |
Todos los grados (%) |
Grados 3-4 (%) |
|
|
Generales |
||||
|
Fatigaa |
57 |
0.9 |
55 |
2.4 |
|
Gastrointestinales |
||||
|
Diarrea |
37 |
2.4 |
55 |
11 |
|
Náuseas |
23 |
0.2 |
28 |
0 |
|
Dolor abdominalb |
21 |
0.2 |
23 |
0.9 |
|
Estreñimiento |
10 |
0 |
9 |
0 |
|
Piel y tejido subcutáneo |
||||
|
Erupción cutáneac |
35 |
1.1 |
47 |
5.3 |
|
Prurito |
28 |
0 |
37 |
1.1 |
|
Musculoesquelético y tejido conjuntivo |
||||
|
Dolor |
32 |
0.4 |
27 |
0.4 |
|
Artralgia |
19 |
0.4 |
13 |
0.4 |
|
Sistema nervioso |
||||
|
Dolor de cabeza |
23 |
0.4 |
31 |
2.0 |
|
Mareoe |
11 |
0 |
8 |
0 |
|
Infecciones |
||||
|
Infección de las vías |
22 |
0 |
15 |
0.2 |
|
Respiratorio, torácico y mediastínico |
||||
|
Tos/tos |
19 |
0 |
19 |
0 |
|
Disnea/disnea de |
10 |
0.4 |
10 |
0.2 |
|
Endocrino |
||||
|
Hipotiroidismog |
12 |
0.2 |
7.5 |
0.4 |
| a La incidencia de cada prueba se basa en el número de pacientes que tenían disponible tanto el valor inicial como al menos una medición de laboratorio durante el estudio: grupo de OPDIVO (rango: 400 a 447 pacientes) y grupo de ipilimumab 10 mg/kg (rango: 392 a 443 pacientes). | ||||
|
Anormalidad de laboratorio |
OPDIVO |
Ipilimumab 10 mg/kg |
||
|
Todos los grados (%) |
Grados 3-4 (%) |
Todos los grados (%) |
Grados 3-4 (%) |
|
|
Hematología |
||||
|
Linfopenia |
27 |
0.4 |
12 |
0.9 |
|
Anemia |
26 |
0 |
34 |
0.5 |
|
Leucopenia |
14 |
0 |
2.7 |
0.2 |
|
Neutropenia |
13 |
0 |
6 |
0.5 |
|
Química |
||||
|
Aumento de la lipasa |
25 |
7 |
23 |
9 |
|
Aumento de ALT |
25 |
1.8 |
40 |
12 |
|
Aumento de AST |
24 |
1.3 |
33 |
9 |
|
Aumento de la amilasa |
17 |
3.3 |
13 |
3.1 |
|
Hiponatremia |
16 |
1.1 |
22 |
3.2 |
|
Hiperpotasemia |
12 |
0.2 |
9 |
0.5 |
|
Increased Creatinine |
12 |
0 |
13 |
0 |
|
Hipocalcemia |
10 |
0.7 |
16 |
0.5 |
Tratamiento neoadyuvante del cáncer de pulmón de célula no pequeña resecable (tumores ≥ 4 cm o con afectación ganglionar positiva)
La seguridad de OPDIVO en combinación con la quimioterapia de platino doble se evaluó en CHECKMATE-816, un ensayo aleatorizado, de etiqueta abierta, multicéntrico en pacientes con NSCLC resecable [véase Estudios clínicos (14.3)]. Los pacientes recibieron bien OPDIVO 360 mg administrado en combinación con quimioterapia de platino doble administrada cada 3 semanas durante 3 ciclos; o quimioterapia de platino doble administrada cada 3 semanas durante 3 ciclos.
La mediana de edad de los pacientes que recibieron OPDIVO en combinación con quimioterapia de platino doble o quimioterapia de platino doble fue de 65 años (rango: 34 – 84); 72% varones; 47% blancos, 50% asiáticos y 2% negros/afroamericanos.
Se produjeron reacciones adversas graves en el 30% de los pacientes que fueron tratados con OPDIVO en combinación con quimioterapia de platino doble. Las reacciones adversas graves en > 2% incluyeron neumonía y vómitos. No se produjeron reacciones adversas fatales en los pacientes que recibieron OPDIVO en combinación con quimioterapia de platino doble.
El tratamiento del estudio con OPDIVO en combinación con quimioterapia de platino doble se suspendió definitivamente por reacciones adversas en el 10% de los pacientes y el 30% tuvieron al menos un tratamiento suspendido por una reacción adversa. Las reacciones adversas más comunes (≥ 1%) que dieron lugar a la suspensión permanente de OPDIVO en combinación con quimioterapia de platino doble fueron reacción anafiláctica (1,7%), lesión renal aguda (1,1%), erupción cutánea (1,1%) y fatiga (1,1%).
Las reacciones adversas más comunes (> 20%) fueron náuseas, estreñimiento, fatiga, pérdida de apetito y erupción cutánea. Las anomalías de laboratorio más comunes de Grado 3 o 4 (≥ 2%) fueron neutropenia, hiperglucemia, leucopenia, linfopenia, aumento de amilasa, anemia, trombocitopenia e hiponatremia.
Las Tablas 15 y 16 resumen las reacciones adversas seleccionadas y las anomalías de laboratorio, respectivamente, en CHECKMATE-816.
| La toxicidad se graduó según NCI CTCAE v4. a Incluye fatiga y astenia b Incluye erupción cutánea, dermatitis, dermatitis acneiforme, dermatitis atópica, dermatitis ampollosa, erupción por fármaco, erupción maculopapular y erupción con prurito. c Incluye neuropatía periférica, disestesia, hipoestesia, neuropatía motora periférica, neuropatía sensitiva periférica. |
||||
|
Reacción adversa |
OPDIVO y quimioterapia de platino doble |
Quimioterapia de platino doble |
||
|
Todos los grados (%) |
Grados 3 o 4 (%) |
Todos los grados (%) |
Grados 3 o 4 (%) |
|
|
Gastrointestinal |
||||
|
Náuseas |
38 |
0,6 |
45 |
1,1 |
|
Estreñimiento |
34 |
0 |
32 |
1,1 |
|
Vómitos |
11 |
1,1 |
13 |
0,6 |
|
General |
||||
|
Fatigaa |
26 |
2,3 |
23 |
1,1 |
|
Malestar |
15 |
0,6 |
14 |
0,6 |
|
Metabolismo y nutrición |
||||
|
Disminución del apetito |
20 |
1.1 |
23 |
2.3 |
|
Piel y tejido subcutáneo |
||||
|
Erupción cutáneab |
20 |
2.3 |
7 |
0 |
|
Alopecia |
11 |
0 |
15 |
0 |
|
Sistema nervioso |
||||
|
Peripheral neuropathyc |
13 |
0 |
6 |
0 |
| a La incidencia de cada prueba se basa en el número de pacientes que tenían disponible tanto la medición de laboratorio inicial como al menos una medición de laboratorio durante el estudio: grupo de OPDIVO y quimioterapia con doblete de platino (rango: 73 a 171 pacientes) y grupo de quimioterapia con doblete de platino (rango: 68 a 171 pacientes). | ||||
|
Anomalía de laboratorio |
OPDIVO y quimioterapia con doblete de platinoa |
Quimioterapia con doblete de platinoa |
||
|
Todos los grados (%) |
Grados 3 o 4 (%) |
Todos los grados (%) |
Grados 3 o 4 (%) |
|
|
Hematología |
||||
|
Anemia |
63 |
3.5 |
70 |
6 |
|
Neutropenia |
58 |
22 |
58 |
27 |
|
Leucopenia |
53 |
5 |
51 |
11 |
|
Linfopenia |
38 |
4.7 |
31 |
1.8 |
|
Trombocitopenia |
24 |
2.9 |
22 |
3 |
|
Química |
||||
|
Hiperglucemia |
37 |
6 |
35 |
2.9 |
|
Hipomagnesemia |
25 |
1.2 |
29 |
1.2 |
|
Hiponatremia |
25 |
2.4 |
28 |
1.8 |
|
Aumento de la amilasa |
23 |
3.6 |
13 |
1.8 |
|
Aumento de la ALT |
23 |
0 |
20 |
1.2 |
Cáncer de pulmón no microcítico metastásico
Tratamiento de primera línea del CPNM metastásico: en combinación con ipilimumab
La seguridad de OPDIVO en combinación con ipilimumab se evaluó en CHECKMATE-227, un ensayo aleatorizado, multicéntrico, de múltiples cohortes y abierto en pacientes con CPNM metastásico o recurrente no tratado previamente sin aberraciones genómicas tumorales en EGFR o ALK [ver Estudios clínicos (14.4)]. Se excluyeron del ensayo pacientes con metástasis cerebrales no tratadas, meningitis carcinomatosa, enfermedad autoinmune activa o afecciones médicas que requirieran inmunosupresión sistémica. Los pacientes recibieron OPDIVO 3 mg/kg mediante infusión intravenosa durante 30 minutos cada 2 semanas e ipilimumab 1 mg/kg mediante infusión intravenosa durante 30 minutos cada 6 semanas o quimioterapia con un doblete de platino cada 3 semanas durante 4 ciclos. La mediana de duración del tratamiento en pacientes tratados con OPDIVO e ipilimumab fue de 4.2 meses (rango: 1 día a 25.5 meses): el 39 % de los pacientes recibieron OPDIVO e ipilimumab durante >6 meses y el 23 % de los pacientes recibieron OPDIVO e ipilimumab durante >1 año. Las características de la población fueron: mediana de edad de 64 años (rango: 26 a 87); el 48 % tenía ≥65 años, el 76 % eran de raza blanca y el 67 % eran hombres. El estado funcional ECOG inicial fue de 0 (35 %) o 1 (65 %), el 85 % eran fumadores o exfumadores, el 11 % tenía metástasis cerebrales, el 28 % tenía histología escamosa y el 72 % tenía histología no escamosa.
Se produjeron reacciones adversas graves en el 58 % de los pacientes. Se interrumpió la administración de OPDIVO e ipilimumab debido a reacciones adversas en el 24 % de los pacientes y al 53 % se le suspendió al menos una dosis debido a una reacción adversa.
Las reacciones adversas graves más frecuentes (≥2 %) fueron neumonía, diarrea/colitis, neumonitis, hepatitis, embolia pulmonar, insuficiencia suprarrenal e hipophisitis. Se produjeron reacciones adversas mortales en el 1.7 % de los pacientes; estas incluyeron episodios de neumonitis (4 pacientes), miocarditis, lesión renal aguda, shock, hiperglucemia, fallo multiorgánico e insuficiencia renal. Las reacciones adversas más frecuentes (≥20 %) fueron fatiga, erupción cutánea, disminución del apetito, dolor musculoesquelético, diarrea/colitis, disnea, tos, hepatitis, náuseas y prurito.
Las Tablas 17 y 18 resumen las reacciones adversas y las anomalías de laboratorio seleccionadas, respectivamente, en CHECKMATE-227.
| a Incluye fatiga y astenia. b Incluye edema de párpado, edema facial, edema generalizado, edema localizado, edema, edema periférico y edema periorbital. c Incluye dermatitis autoinmune, dermatitis, dermatitis acneiforme, dermatitis alérgica, dermatitis atópica, dermatitis ampollosa, dermatitis de contacto, dermatitis exfoliativa, dermatitis psoriasiforme, dermatitis granulomatosa, erupción cutánea generalizada, erupción medicamentosa, eccema dishidrótico, eccema, erupción exfoliativa, erupción nodular, erupción cutánea, erupción eritematosa, erupción macular, erupción maculopapular, erupción papular, erupción pruriginosa, erupción pustulosa, erupción cutánea tóxica. d Incluye prurito y prurito generalizado. e Incluye dolor de espalda, dolor óseo, dolor torácico musculoesquelético, molestias musculoesqueléticas, dolor musculoesquelético, mialgia y dolor en las extremidades. f Incluye colitis, colitis microscópica, colitis ulcerosa, diarrea, enteritis infecciosa, enterocolitis, enterocolitis infecciosa y enterocolitis vírica. g Incluye molestias abdominales, dolor abdominal, dolor abdominal bajo, dolor abdominal alto y sensibilidad abdominal. h Incluye disnea y disnea de esfuerzo. i Incluye tos y tos productiva. j Incluye aumento de la alanina aminotransferasa, aumento de la aspartato aminotransferasa, hepatitis autoinmune, aumento de la bilirrubina en sangre, aumento de las enzimas hepáticas, insuficiencia hepática, función hepática anormal, hepatitis, hepatitis E, lesión hepatocelular, hepatotoxicidad, hiperbilirrubinemia, hepatitis mediada por el sistema inmunitario, prueba de función hepática anormal, aumento de la prueba de función hepática, aumento de las transaminasas. k Incluye tiroiditis autoinmune, aumento de la hormona estimulante de la tiroides en sangre, hipotiroidismo, hipotiroidismo primario, tiroiditis y disminución de la triyodotironina libre. l Contiene disminución de la hormona estimulante de la tiroides en sangre, hipertiroidismo y aumento de la triyodotironina libre. m Incluye infección de las vías respiratorias inferiores, infección bacteriana de las vías respiratorias inferiores, infección pulmonar, neumonía, neumonía adenoviral, neumonía por aspiración, neumonía bacteriana, neumonía por Klebsiella, neumonía gripal, neumonía vírica, neumonía atípica, neumonía organizada. |
||||
|
Reacción adversa |
OPDIVO e ipilimumab |
Quimioterapia con doblete de platino |
||
|
Todos los grados |
Grados 3-4 |
Todos los grados |
Grados 3-4 |
|
|
Generales |
||||
|
Fatigaa |
44 |
6 |
42 |
4.4 |
|
Pirexia |
18 |
0.5 |
11 |
0.4 |
|
Edemab |
14 |
0.2 |
12 |
0.5 |
|
Piel y Tejido Subcutáneo |
||||
|
Erupción cutáneac |
34 |
4.7 |
10 |
0.4 |
|
Pruritod |
21 |
0.5 |
3.3 |
0 |
|
Metabolismo y Nutrición |
||||
|
Disminución del apetito |
31 |
2.3 |
26 |
1.4 |
|
Musculoesquelético y Tejido Conectivo |
||||
|
Dolor musculoesqueléticoe |
27 |
1.9 |
16 |
0.7 |
|
Artralgia |
13 |
0.9 |
2.5 |
0.2 |
|
Gastrointestinal |
||||
|
Diarrea/colitisf |
26 |
3.6 |
16 |
0.9 |
|
Náuseas |
21 |
1 |
42 |
2.5 |
|
Estreñimiento |
18 |
0.3 |
27 |
0.5 |
|
Vómitos |
13 |
1 |
18 |
2.3 |
|
Dolor abdominalg |
10 |
0.2 |
9 |
0.7 |
|
Respiratorio, Torácico y Mediastínico |
||||
|
Disneah |
26 |
4.3 |
16 |
2.1 |
|
Tosi |
23 |
0.2 |
13 |
0 |
|
Hepatobiliary |
||||
|
Hepatitisj |
21 |
9 |
10 |
1.2 |
|
Endocrino |
||||
|
Hipotiroidismok |
16 |
0.5 |
1.2 |
0 |
|
Hipertiroidismol |
10 |
0 |
0.5 |
0 |
|
Infecciones e infestaciones |
||||
|
Neumoníam |
13 |
7 |
8 |
4 |
|
Sistema nervioso |
||||
|
Dolor de cabeza |
11 |
0.5 |
6 |
0 |
Otras reacciones adversas clínicamente importantes en CHECKMATE-227 fueron:
Piel y tejido subcutáneo: urticaria, alopecia, eritema multiforme, vitiligo
Gastrointestinales: estomatitis, pancreatitis, gastritis
Musculoesqueléticas y del tejido conjuntivo: artritis, polimialgia reumática, rabdomiólisis
Sistema nervioso: neuropatía periférica, encefalitis autoinmune
Sangre y sistema linfático: eosinofilia
Trastornos oculares: visión borrosa, uveítis
Cardíacas: fibrilación auricular, miocarditis
| a La incidencia de cada prueba se basa en el número de pacientes que tenían disponible tanto el valor inicial como al menos una medición de laboratorio durante el estudio: grupo de OPDIVO e ipilimumab (rango: 494 a 556 pacientes) y grupo de quimioterapia (rango: 469 a 542 pacientes). | ||||
|
Anomalía de laboratorio |
OPDIVO e ipilimumab |
Quimioterapia con doblete de platino |
||
|
Grados 1-4 |
Grados 3-4 |
Grados 1-4 |
Grados 3-4 |
|
|
Hematología |
||||
|
Anemia |
46 |
3.6 |
78 |
14 |
|
Linfopenia |
46 |
5 |
60 |
15 |
|
Química |
||||
|
Hiponatremia |
41 |
12 |
26 |
4.9 |
|
Aumento de AST |
39 |
5 |
26 |
0.4 |
|
Aumento de ALT |
36 |
7 |
27 |
0.7 |
|
Aumento de lipasa |
35 |
14 |
14 |
3.4 |
|
Aumento de fosfatasa |
34 |
3.8 |
20 |
0.2 |
|
Aumento de amilasa |
28 |
9 |
18 |
1.9 |
|
Hipocalcemia |
28 |
1.7 |
17 |
1.3 |
|
Hyperkalemia |
27 |
3.4 |
22 |
0.4 |
|
Aumento de la creatinina |
22 |
0.9 |
17 |
0.2 |
Tratamiento de primera línea del CPNM metastásico o recurrente: en combinación con ipilimumab y quimioterapia con doblete de platino
La seguridad de OPDIVO en combinación con ipilimumab y quimioterapia con doblete de platino se evaluó en CHECKMATE-9LA [ver Estudios clínicos (14.4)]. Los pacientes recibieron 360 mg de OPDIVO administrados cada 3 semanas en combinación con 1 mg/kg de ipilimumab administrado cada 6 semanas y quimioterapia con doblete de platino administrada cada 3 semanas durante 2 ciclos; o quimioterapia con doblete de platino administrada cada 3 semanas durante 4 ciclos. La mediana de duración del tratamiento con OPDIVO en combinación con ipilimumab y quimioterapia con doblete de platino fue de 6 meses (rango: 1 día a 19 meses): el 50 % de los pacientes recibieron OPDIVO e ipilimumab durante >6 meses y el 13 % de los pacientes recibieron OPDIVO e ipilimumab durante >1 año.
Se produjeron reacciones adversas graves en el 57 % de los pacientes que recibieron tratamiento con OPDIVO en combinación con ipilimumab y quimioterapia con doblete de platino. Las reacciones adversas graves más frecuentes (>2 %) fueron neumonía, diarrea, neutropenia febril, anemia, lesión renal aguda, dolor musculoesquelético, disnea, neumonitis e insuficiencia respiratoria. Se produjeron reacciones adversas mortales en 7 (2 %) pacientes, entre las que se incluyeron toxicidad hepática, insuficiencia renal aguda, sepsis, neumonitis, diarrea con hipopotasemia y hemoptisis masiva en el contexto de trombocitopenia.
El tratamiento en estudio con OPDIVO en combinación con ipilimumab y quimioterapia con doblete de platino se suspendió de forma permanente debido a reacciones adversas en el 24 % de los pacientes y al 56 % se le interrumpió al menos un tratamiento debido a una reacción adversa. Las reacciones adversas más frecuentes (>20 %) fueron fatiga, dolor musculoesquelético, náuseas, diarrea, erupción cutánea, disminución del apetito, estreñimiento y prurito.
En las Tablas 19 y 20 se resumen las reacciones adversas y las anomalías de laboratorio seleccionadas, respectivamente, en CHECKMATE-9LA.
| La toxicidad se clasificó según los criterios comunes de toxicidad del NCI v4. a Incluye fatiga y astenia b Incluye mialgia, dolor de espalda, dolor en las extremidades, dolor musculoesquelético, dolor óseo, dolor en el costado, espasmos musculares, dolor torácico musculoesquelético, trastorno musculoesquelético, osteítis, rigidez musculoesquelética, dolor torácico no cardíaco, artralgia, artritis, artropatía, derrame articular, artropatía psoriásica, sinovitis c Incluye colitis, colitis ulcerosa, diarrea y enterocolitis d Incluye molestias abdominales, dolor abdominal, dolor abdominal bajo, dolor abdominal alto y dolor gastrointestinal e Incluye acné, dermatitis, dermatitis acneiforme, dermatitis alérgica, dermatitis atópica, dermatitis ampollosa, dermatitis exfoliativa generalizada, eccema, queratodermia blenorrágica, síndrome de eritrodisestesia palmo-plantar, erupción cutánea, erupción cutánea eritematosa, erupción cutánea generalizada, erupción cutánea macular, erupción cutánea maculopapular, erupción cutánea morbiliforme, erupción cutánea papular, erupción cutánea pruriginosa, exfoliación cutánea, reacción cutánea, toxicidad cutánea, síndrome de Stevens-Johnson, urticaria f Incluye prurito y prurito generalizado g Incluye tos, tos productiva y síndrome de tos de las vías respiratorias altas h Incluye disnea, disnea en reposo y disnea de esfuerzo i Incluye tiroiditis autoinmune, aumento de la hormona estimulante de la tiroides en sangre, hipotiroidismo, tiroiditis y disminución de la triyodotironina libre j Incluye mareos, vértigo y vértigo posicional |
||||
|
Reacción adversa |
OPDIVO e ipilimumab y quimioterapia con doblete de platino |
Quimioterapia con doblete de platino |
||
|
Todos los grados (%) |
Grados 3-4 (%) |
Todos los grados (%) |
Grados 3-4 (%) |
|
|
Generales |
||||
|
Fatigaa |
49 |
5 |
40 |
4.9 |
|
Pirexia |
14 |
0.6 |
10 |
0.6 |
|
Musculoesqueléticos y del tejido conjuntivo |
||||
|
Dolor musculoesqueléticob |
39 |
4.5 |
27 |
2 |
|
Gastrointestinales |
||||
|
Náuseas |
32 |
1.7 |
41 |
0.9 |
|
Diarreac |
31 |
6 |
18 |
1.7 |
|
Estreñimiento |
21 |
0.6 |
23 |
0.6 |
|
Vómitos |
18 |
2 |
17 |
1.4 |
|
Dolor abdominald |
12 |
0.6 |
11 |
0.9 |
|
Piel y tejido subcutáneo |
||||
|
Erupción cutáneae |
30 |
4.7 |
10 |
0.3 |
|
Pruritof |
21 |
0.8 |
2.9 |
0 |
|
Alopecia |
11 |
0.8 |
10 |
0.6 |
|
Metabolismo y nutrición |
||||
|
Disminución del apetito |
28 |
2 |
22 |
1.7 |
|
Respiratorio, torácico y mediastínico |
||||
|
Tosg |
19 |
0.6 |
15 |
0.9 |
|
Disneah |
18 |
4.7 |
14 |
3.2 |
|
Endocrino |
||||
|
Hipotiroidismoi |
19 |
0.3 |
3.4 |
0 |
|
Sistema nervioso |
||||
|
Dolor de cabeza |
11 |
0.6 |
7 |
0 |
|
Mareosj |
11 |
0.6 |
6 |
0 |
| a La incidencia de cada prueba se basa en el número de pacientes que tenían disponible tanto la medición de laboratorio inicial como al menos una medición de laboratorio durante el estudio: grupo de OPDIVO e ipilimumab y quimioterapia con doblete de platino (rango: 197 a 347 pacientes) y grupo de quimioterapia con doblete de platino (rango: 191 a 335 pacientes). | ||||
|
Anomalía de laboratorio |
OPDIVO e ipilimumab y quimioterapia con doblete de platino |
Quimioterapia con doblete de platino |
||
|
Grados 1-4 (%) |
Grados 3-4 (%) |
Grados 1-4 (%) |
Grados 3-4 (%) |
|
|
Hematología |
||||
|
Anemia |
70 |
9 |
74 |
16 |
|
Linfopenia |
41 |
6 |
40 |
11 |
|
Neutropenia |
40 |
15 |
42 |
15 |
|
Leucopenia |
36 |
10 |
40 |
9 |
|
Trombocitopenia |
23 |
4.3 |
24 |
5 |
|
Química |
||||
|
Hiperglucemia |
45 |
7 |
42 |
2.6 |
|
Hiponatremia |
37 |
10 |
27 |
7 |
|
Aumento de ALT |
34 |
4.3 |
24 |
1.2 |
|
Aumento de la lipasa |
31 |
12 |
10 |
2.2 |
|
Aumento de la fosfatasa alcalina |
31 |
1.2 |
26 |
0.3 |
|
Aumento de la amilasa |
30 |
7 |
19 |
1.3 |
|
Aumento de AST |
30 |
3.5 |
22 |
0.3 |
|
Hipomagnesemia |
29 |
1.2 |
33 |
0.6 |
|
Hipocalcemia |
26 |
1.4 |
22 |
1.8 |
|
Aumento de la creatinina |
26 |
1.2 |
23 |
0.6 |
|
Hiperpotasemia |
22 |
1.7 |
21 |
2.1 |
Tratamiento de segunda línea del CPNM metastásico
La seguridad de OPDIVO se evaluó en CHECKMATE-017, un ensayo multicéntrico, abierto y aleatorizado en pacientes con CPNM de células escamosas metastásico y progresión durante o después de un régimen de quimioterapia basado en un doblete de platino previo, y en CHECKMATE-057, un ensayo multicéntrico, abierto y aleatorizado en pacientes con CPNM no escamoso metastásico y progresión durante o después de un régimen de quimioterapia basado en un doblete de platino previo [ver Estudios clínicos (14.4)]. Estos ensayos excluyeron a pacientes con enfermedad autoinmune activa, afecciones médicas que requieren inmunosupresión sistémica o con enfermedad pulmonar intersticial sintomática. Los pacientes recibieron 3 mg/kg de OPDIVO durante 60 minutos mediante infusión intravenosa cada 2 semanas o 75 mg/m2 de docetaxel por vía intravenosa cada 3 semanas. La mediana de duración del tratamiento en los pacientes tratados con OPDIVO en CHECKMATE-017 fue de 3.3 meses (rango: 1 día a 21.7+ meses) y en CHECKMATE-057 fue de 2.6 meses (rango: 0 a 24.0+ meses). En CHECKMATE-017, el 36 % de los pacientes recibieron OPDIVO durante al menos 6 meses y el 18 % de los pacientes recibieron OPDIVO durante al menos 1 año y en CHECKMATE-057, el 30 % de los pacientes recibieron OPDIVO durante >6 meses y el 20 % de los pacientes recibieron OPDIVO durante >1 año.
En ambos ensayos, la mediana de edad de los pacientes tratados con OPDIVO fue de 61 años (rango: 37 a 85); el 38 % tenía ≥65 años, el 61 % eran hombres y el 91 % eran blancos. El diez por ciento de los pacientes tenían metástasis cerebrales y el estado funcional ECOG era 0 (26 %) o 1 (74 %).
En CHECKMATE-057, en el grupo de OPDIVO, siete muertes se debieron a infección, incluido un caso de neumonía por Pneumocystis jirovecii, cuatro se debieron a embolia pulmonar y una muerte se debió a encefalitis límbica. Se produjeron reacciones adversas graves en el 46 % de los pacientes que recibieron OPDIVO. Se interrumpió el tratamiento con OPDIVO en el 11 % de los pacientes y se retrasó en el 28 % de los pacientes debido a una reacción adversa.
Las reacciones adversas graves más frecuentes notificadas en ≥2 % de los pacientes que recibieron OPDIVO fueron neumonía, embolia pulmonar, disnea, pirexia, derrame pleural, neumonitis e insuficiencia respiratoria. En ambos ensayos, las reacciones adversas más comunes (≥20 %) fueron fatiga, dolor musculoesquelético, tos, disnea y disminución del apetito.
Las Tablas 21 y 22 resumen las reacciones adversas y las anomalías de laboratorio seleccionadas, respectivamente, en CHECKMATE-057.
| La toxicidad se clasificó según los criterios comunes de toxicidad del NCI v4. | ||||
|
Reacción adversa |
OPDIVO |
Docetaxel |
||
|
Todos los grados (%) |
Grados 3-4 (%) |
Todos los grados (%) |
Grados 3-4 (%) |
|
|
Respiratorias, torácicas y mediastínicas |
||||
|
Tos |
31 |
0.7 |
24 |
0 |
|
Metabolismo y nutrición |
||||
|
Disminución del apetito |
28 |
1.4 |
23 |
1.5 |
|
Piel y tejido subcutáneo |
||||
|
Prurito |
10 |
0.2 |
2 |
0 |
Otras reacciones adversas clínicamente importantes observadas en pacientes tratados con OPDIVO y que ocurrieron con una incidencia similar en pacientes tratados con docetaxel y que no se enumeran en otra parte de la sección 6 incluyen: fatiga/astenia (48 % todos los grados, 5 % grado 3-4), dolor musculoesquelético (33 % todos los grados), derrame pleural (4,5 % todos los grados), embolia pulmonar (3,3 % todos los grados).
| a La incidencia de cada prueba se basa en el número de pacientes que tenían disponible tanto el valor inicial como al menos una medición de laboratorio durante el estudio: grupo de OPDIVO (rango: 405 a 417 pacientes) y grupo de docetaxel (rango: 372 a 390 pacientes), excepto para la TSH: grupo de OPDIVO n=314 y grupo de docetaxel n=297. b No clasificada según NCI CTCAE v4. |
||||
|
Anormalidad de laboratorio |
OPDIVO |
Docetaxel |
||
|
Todos los grados (%) |
Grados 3-4 (%) |
Todos los grados (%) |
Grados 3-4 (%) |
|
|
Química |
||||
|
Hiponatremia |
35 |
7 |
34 |
4.9 |
|
Aumento de AST |
27 |
1.9 |
13 |
0.8 |
|
Aumento de la fosfatasa alcalina |
26 |
0.7 |
18 |
0.8 |
|
Aumento de ALT |
22 |
1.7 |
17 |
0.5 |
|
Aumento de creatinina |
18 |
0 |
12 |
0.5 |
|
Aumento de TSHb |
14 |
N/A |
6 |
N/A |
Mesotelioma pleural maligno
Se evaluó la seguridad de OPDIVO en combinación con ipilimumab en CHECKMATE-743, un ensayo aleatorizado, abierto en pacientes con mesotelioma pleural maligno no resecable previamente no tratado [ver Estudios clínicos (14.5)]. Los pacientes recibieron OPDIVO 3 mg/kg durante 30 minutos por infusión intravenosa cada 2 semanas e ipilimumab 1 mg/kg durante 30 minutos por infusión intravenosa cada 6 semanas durante un máximo de 2 años; o quimioterapia con doblete de platino durante un máximo de 6 ciclos. La mediana de la duración del tratamiento en pacientes tratados con OPDIVO e ipilimumab fue de 5.6 meses (rango: 0 a 26.2 meses); el 48 % de los pacientes recibieron OPDIVO e ipilimumab durante >6 meses y el 24 % de los pacientes recibieron OPDIVO e ipilimumab durante >1 año.
Se produjeron reacciones adversas graves en el 54 % de los pacientes que fueron tratados con OPDIVO en combinación con ipilimumab. Las reacciones adversas graves más frecuentes (≥2 %) fueron neumonía, pirexia, diarrea, neumonitis, derrame pleural, disnea, lesión renal aguda, reacción relacionada con la infusión, dolor musculoesquelético y embolia pulmonar. Se produjeron reacciones adversas mortales en 4 (1.3 %) pacientes e incluyeron neumonitis, insuficiencia cardíaca aguda, sepsis y encefalitis.
Tanto OPDIVO como ipilimumab se suspendieron permanentemente debido a reacciones adversas en el 23 % de los pacientes y el 52 % tuvo al menos una dosis retenida debido a una reacción adversa.
Las reacciones adversas más comunes (≥20 %) fueron fatiga, dolor musculoesquelético, erupción cutánea, diarrea, disnea, náuseas, disminución del apetito, tos y prurito.
Las Tablas 23 y 24 resumen las reacciones adversas y las anomalías de laboratorio, respectivamente, en CHECKMATE-743.
| a Incluye fatiga y astenia. b Incluye pirexia y fiebre tumoral. c Incluye edema, edema generalizado, edema periférico e hinchazón periférica. d Incluye dolor musculoesquelético, dolor de espalda, dolor óseo, dolor en el flanco, contracciones musculares involuntarias, espasmos musculares, fasciculaciones musculares, dolor torácico musculoesquelético, rigidez musculoesquelética, mialgia, dolor de cuello, dolor torácico no cardíaco, dolor en las extremidades, polimialgia reumática y dolor espinal. e Incluye erupción cutánea, acné, dermatitis acneiforme, dermatitis alérgica, dermatitis atópica, dermatitis autoinmune, dermatitis ampollosa, dermatitis de contacto, dermatitis, erupción medicamentosa, eccema dishidrótico, eccema, erupción eritematosa, erupción exfoliativa, dermatitis exfoliativa generalizada, erupción generalizada, dermatitis granulomatosa, queratodermia blenorrágica, erupción macular, erupción maculopapular, erupción morbiliforme, erupción nodular, erupción papular, dermatitis psoriasiforme, erupción pruriginosa, erupción pustulosa, exfoliación de la piel, reacción cutánea, toxicidad cutánea, síndrome de Stevens-Johnson, erupción cutánea tóxica y urticaria. f Incluye prurito, prurito alérgico y prurito generalizado. g Incluye diarrea, colitis, enteritis, enteritis infecciosa, enterocolitis, enterocolitis infecciosa, colitis microscópica, colitis ulcerosa y enterocolitis viral. h Incluye dolor abdominal, molestias abdominales, sensibilidad abdominal, dolor gastrointestinal, dolor abdominal inferior y dolor abdominal superior. i Incluye disnea, disnea en reposo y disnea de esfuerzo. j Incluye tos, tos productiva y síndrome de tos de las vías respiratorias superiores. k Incluye hipotiroidismo, tiroiditis autoinmune, disminución de la triyodotironina libre, aumento de la hormona estimulante de la tiroides en sangre, hipotiroidismo primario, tiroiditis e hipotiroidismo autoinmune. l Incluye infección de las vías respiratorias superiores, nasofaringitis, faringitis y rinitis. m Incluye neumonía, infección del tracto respiratorio inferior, infección pulmonar, neumonía por aspiración y neumonía por Pneumocystis jirovecii. |
||||
|
Reacción adversa |
OPDIVO e ipilimumab |
Quimioterapia |
||
|
Todos los grados |
Grados 3-4 |
Todos los grados |
Grados 3-4 |
|
|
General |
||||
|
Fatigaa |
43 |
4.3 |
45 |
6 |
|
Pirexiab |
18 |
1.3 |
4.6 |
0.7 |
|
Edemac |
17 |
0 |
8 |
0 |
|
Musculoesquelético y tejido conectivo |
||||
|
Dolor musculoesqueléticod |
38 |
3.3 |
17 |
1.1 |
|
Artralgia |
13 |
1 |
1.1 |
0 |
|
Piel y tejido subcutáneo |
||||
|
Erupción cutáneae |
34 |
2.7 |
11 |
0.4 |
|
Pruritof |
21 |
1 |
1.4 |
0 |
|
Gastrointestinal |
||||
|
Diarreag |
32 |
6 |
12 |
1.1 |
|
Náuseas |
24 |
0.7 |
43 |
2.5 |
|
Estreñimiento |
19 |
0.3 |
30 |
0.7 |
|
Dolor abdominalh |
15 |
1 |
10 |
0.7 |
|
Vómitos |
14 |
0 |
18 |
2.1 |
|
Respiratorio, torácico y mediastínico |
||||
|
Disneai |
27 |
2.3 |
16 |
3.2 |
|
Tosj |
23 |
0.7 |
9 |
0 |
|
Metabolismo y nutrición |
||||
|
Disminución del apetito |
24 |
1 |
25 |
1.4 |
|
Endocrino |
||||
|
Hipotiroidismok |
15 |
0 |
1.4 |
0 |
|
Infecciones e infestaciones |
||||
|
Infección de las vías respiratorias superioresl |
12 |
0.3 |
7 |
0 |
|
Neumoníam |
10 |
4 |
4.2 |
2.1 |
| a La incidencia de cada prueba se basa en el número de pacientes que tenían disponible tanto la medición de laboratorio inicial como al menos una medición de laboratorio durante el estudio: grupo de OPDIVO e ipilimumab (rango: 109 a 297 pacientes) y grupo de quimioterapia (rango: 90 a 276 pacientes). | ||||
|
Anomalía de laboratorio |
OPDIVO e ipilimumab |
Quimioterapia |
||
|
Grados 1-4 |
Grados 3-4 |
Grados 1-4 |
Grados 3-4 |
|
|
Química |
||||
|
Hiperglucemia |
53 |
3.7 |
34 |
1.1 |
|
Aumento de AST |
38 |
7 |
17 |
0 |
|
Aumento de ALT |
37 |
7 |
15 |
0.4 |
|
Aumento de la lipasa |
34 |
13 |
9 |
0.8 |
|
Hiponatremia |
32 |
8 |
21 |
2.9 |
|
Aumento de la fosfatasa alcalina |
31 |
3.1 |
12 |
0 |
|
Hiperpotasemia |
30 |
4.1 |
16 |
0.7 |
|
Hipocalcemia |
28 |
0 |
16 |
0 |
|
Aumento de la amilasa |
26 |
5 |
13 |
0.9 |
|
Aumento de la creatinina |
20 |
0.3 |
20 |
0.4 |
|
Hematología |
||||
|
Lymphopenia |
43 |
8 |
57 |
14 |
|
Anemia |
43 |
2.4 |
75 |
15 |
Carcinoma de células renales avanzado
Carcinoma de células renales de primera línea
CHECKMATE-214
La seguridad de OPDIVO con ipilimumab se evaluó en CHECKMATE-214, un ensayo abierto aleatorizado en 1082 pacientes con CCR avanzado no tratado previamente que recibieron OPDIVO 3 mg/kg durante 60 minutos con ipilimumab 1 mg/kg por vía intravenosa cada 3 semanas durante 4 dosis seguido de OPDIVO como agente único a una dosis de 3 mg/kg por infusión intravenosa cada 2 semanas (n=547) o sunitinib 50 mg por vía oral diariamente durante las primeras 4 semanas de un ciclo de 6 semanas (n=535) [ver Estudios clínicos (14.6)]. La mediana de duración del tratamiento fue de 7.9 meses (rango: 1 día a 21.4+ meses) en pacientes tratados con OPDIVO e ipilimumab y de 7.8 meses (rango: 1 día a 20.2+ meses) en pacientes tratados con sunitinib. En este ensayo, el 57 % de los pacientes del grupo de OPDIVO e ipilimumab estuvieron expuestos al tratamiento durante >6 meses y el 38 % de los pacientes estuvieron expuestos al tratamiento durante >1 año.
Se produjeron reacciones adversas graves en el 59 % de los pacientes que recibieron OPDIVO e ipilimumab. El tratamiento del estudio se interrumpió debido a reacciones adversas en el 31 % de los pacientes tratados con OPDIVO e ipilimumab. El cincuenta y cuatro por ciento (54 %) de los pacientes que recibieron OPDIVO e ipilimumab tuvieron una interrupción de la dosis debido a una reacción adversa.
Las reacciones adversas graves más frecuentes notificadas en ≥2 % de los pacientes tratados con OPDIVO e ipilimumab fueron diarrea, pirexia, neumonía, neumonitis, hipophisitis, lesión renal aguda, disnea, insuficiencia suprarrenal y colitis; en los pacientes tratados con sunitinib, fueron neumonía, derrame pleural y disnea. Las reacciones adversas más comunes (notificadas en ≥20 % de los pacientes) fueron fatiga, erupción cutánea, diarrea, dolor musculoesquelético, prurito, náuseas, tos, pirexia, artralgia y disminución del apetito. Las anomalías de laboratorio más comunes que empeoraron en comparación con el valor inicial en ≥30 % de los pacientes tratados con OPDIVO e ipilimumab incluyen aumento de la lipasa, anemia, aumento de la creatinina, aumento de la ALT, aumento de la AST, hiponatremia, aumento de la amilasa y linfopenia.
Las Tablas 25 y 26 resumen las reacciones adversas y las anomalías de laboratorio, respectivamente, que se produjeron en >15 % de los pacientes tratados con OPDIVO e ipilimumab en CHECKMATE-214.
| La toxicidad se clasificó según los criterios comunes de toxicidad del NCI v4. a Incluye astenia. b Incluye edema periférico, inflamación periférica. c Incluye dermatitis descrita como acneiforme, ampollosa y exfoliativa, erupción medicamentosa, erupción cutánea descrita como exfoliativa, eritematosa, folicular, generalizada, macular, maculopapular, papular, pruriginosa y pustulosa, erupción fija por fármacos. d Incluye dolor de espalda, dolor óseo, dolor torácico musculoesquelético, molestias musculoesqueléticas, mialgia, dolor de cuello, dolor en las extremidades, dolor espinal. |
||||
|
Reacción adversa |
OPDIVO e ipilimumab |
Sunitinib |
||
|
Grados 1-4 (%) |
Grados 3-4 (%) |
Grados 1-4 (%) |
Grados 3-4 (%) |
|
|
Reacción adversa |
99 |
65 |
99 |
76 |
|
Generales |
||||
|
Fatigaa |
58 |
8 |
69 |
13 |
|
Pirexia |
25 |
0.7 |
17 |
0.6 |
|
Edemab |
16 |
0.5 |
17 |
0.6 |
|
Piel y tejido subcutáneo |
||||
|
Erupción cutáneac |
39 |
3.7 |
25 |
1.1 |
|
Pruritus/generalized pruritus |
33 |
0.5 |
11 |
0 |
|
Gastrointestinal |
||||
|
Diarrea |
38 |
4.6 |
58 |
6 |
|
Náuseas |
30 |
2 |
43 |
1.5 |
|
Vómitos |
20 |
0.9 |
28 |
2.1 |
|
Dolor abdominal |
19 |
1.6 |
24 |
1.9 |
|
Estreñimiento |
17 |
0.4 |
18 |
0 |
|
Musculoskeletal and Connective Tissue |
||||
|
Musculoskeletal paind |
37 |
4 |
40 |
2.6 |
|
Artralgia |
23 |
1.3 |
16 |
0 |
|
Respiratory, Thoracic and Mediastinal |
||||
|
Cough/productive cough |
28 |
0.2 |
25 |
0.4 |
|
Disnea/disnea de esfuerzo |
20 |
2.4 |
21 |
2.1 |
|
Metabolism and Nutrition |
||||
|
Disminución del apetito |
21 |
1.8 |
29 |
0.9 |
|
Nervous System |
||||
|
Dolor de cabeza |
19 |
0.9 |
23 |
0.9 |
|
Endocrine |
||||
|
Hipotiroidismo |
18 |
0.4 |
27 |
0.2 |
| a La incidencia de cada prueba se basa en el número de pacientes que tenían disponible tanto el valor inicial como al menos una medición de laboratorio durante el estudio: grupo de OPDIVO e ipilimumab (rango: 490 a 538 pacientes) y grupo de sunitinib (rango: 485 a 523 pacientes). | ||||
|
Anomalía de laboratorio |
OPDIVO e ipilimumab |
Sunitinib |
||
|
Grados 1-4 (%) |
Grados 3-4 (%) |
Grados 1-4 (%) |
Grados 3-4 (%) |
|
|
Química |
||||
|
Aumento de la lipasa |
48 |
20 |
51 |
20 |
|
Aumento de la creatinina |
42 |
2.1 |
46 |
1.7 |
|
Aumento de la ALT |
41 |
7 |
44 |
2.7 |
|
Aumento de la AST |
40 |
4.8 |
60 |
2.1 |
|
Aumento de la amilasa |
39 |
12 |
33 |
7 |
|
Hiponatremia |
39 |
10 |
36 |
7 |
|
Aumento de la fosfatasa alcalina |
29 |
2 |
32 |
1 |
|
Hiperpotasemia |
29 |
2.4 |
28 |
2.9 |
|
Hipocalcemia |
21 |
0.4 |
35 |
0.6 |
|
Hipomagnesemia |
16 |
0.4 |
26 |
1.6 |
|
Hematología |
||||
|
Anemia |
43 |
3 |
64 |
9 |
|
Linfopenia |
36 |
5 |
63 |
14 |
Además, entre los pacientes con TSH ≤LSN al inicio, una menor proporción de pacientes experimentó un aumento de TSH > LSN emergente del tratamiento en el grupo de OPDIVO e ipilimumab en comparación con el grupo de sunitinib (31 % y 61 %, respectivamente).
CHECKMATE-9ER
Se evaluó la seguridad de OPDIVO con cabozantinib en CHECKMATE-9ER, un estudio aleatorizado, abierto, en pacientes con CCR avanzado no tratado previamente. Los pacientes recibieron 240 mg de OPDIVO durante 30 minutos cada 2 semanas con 40 mg de cabozantinib por vía oral una vez al día (n=320) o 50 mg de sunitinib al día, administrados por vía oral durante 4 semanas en tratamiento seguidos de 2 semanas sin tratamiento [ver Estudios clínicos (14.6)]. El tratamiento con cabozantinib se pudo interrumpir o reducir a 20 mg al día o 20 mg en días alternos. La mediana de la duración del tratamiento fue de 14 meses (rango: 0.2 a 27 meses) en los pacientes tratados con OPDIVO y cabozantinib. En este ensayo, el 82 % de los pacientes del grupo de OPDIVO y cabozantinib estuvieron expuestos al tratamiento durante >6 meses y el 60 % de los pacientes estuvieron expuestos al tratamiento durante >1 año.
Se produjeron reacciones adversas graves en el 48 % de los pacientes que recibieron OPDIVO y cabozantinib. Las reacciones adversas graves más frecuentes (≥2 %) fueron diarrea, neumonía, neumonitis, embolia pulmonar, infección del tracto urinario e hiponatremia. Se produjeron perforaciones intestinales mortales en 3 (0.9 %) pacientes.
Se produjeron reacciones adversas que provocaron la interrupción de OPDIVO o cabozantinib en el 20 % de los pacientes: 7 % solo OPDIVO, 8 % solo cabozantinib y 6 % ambos fármacos debido a la misma reacción adversa al mismo tiempo. Se produjo una reacción adversa que provocó la interrupción o reducción de la dosis de OPDIVO o cabozantinib en el 83 % de los pacientes: 3 % solo OPDIVO, 46 % solo cabozantinib y 21 % ambos fármacos debido a la misma reacción adversa al mismo tiempo, y 6 % ambos fármacos de forma secuencial.
Las reacciones adversas más comunes notificadas en ≥20 % de los pacientes tratados con OPDIVO y cabozantinib fueron diarrea, fatiga, hepatotoxicidad, síndrome de eritrodisestesia palmo-plantar, estomatitis, erupción cutánea, hipertensión, hipotiroidismo, dolor musculoesquelético, disminución del apetito, náuseas, disgeusia, dolor abdominal, tos e infección de las vías respiratorias superiores.
Las Tablas 27 y 28 resumen las reacciones adversas y las anomalías de laboratorio, respectivamente, en CHECKMATE-9ER.
| La toxicidad se clasificó según los criterios comunes de toxicidad del NCI v4. a Incluye molestias abdominales, dolor abdominal inferior, dolor abdominal superior. b Incluye enfermedad por reflujo gastroesofágico. c Incluye astenia. d Incluye hepatotoxicidad, aumento de ALT, aumento de AST, aumento de la fosfatasa alcalina en sangre, aumento de la gamma-glutamiltransferasa, hepatitis autoinmune, aumento de la bilirrubina en sangre, lesión hepática inducida por fármacos, aumento de las enzimas hepáticas, hepatitis, hiperbilirrubinemia, aumento de las pruebas de función hepática, pruebas de función hepática anormales, aumento de las transaminasas, insuficiencia hepática. e Incluye inflamación de la mucosa, úlcera aftosa, ulceración bucal. f Incluye dermatitis, dermatitis acneiforme, dermatitis ampollosa, erupción cutánea exfoliativa, erupción eritematosa, erupción folicular, erupción macular, erupción maculopapular, erupción papular, erupción pruriginosa. g Incluye aumento de la presión arterial, aumento de la presión arterial sistólica. h Incluye hipotiroidismo primario. i Incluye dolor de espalda, dolor óseo, dolor torácico musculoesquelético, molestias musculoesqueléticas, mialgia, dolor de cuello, dolor en las extremidades, dolor espinal. j Incluye tos productiva. k Incluye nasofaringitis, faringitis, rinitis. |
||||
|
Reacción adversa |
OPDIVO y cabozantinib |
Sunitinib |
||
|
Grados 1-4 (%) |
Grados 3-4 (%) |
Grados 1-4 (%) |
Grados 3-4 (%) |
|
|
Gastrointestinales |
||||
|
Diarrea |
64 |
7 |
47 |
4.4 |
|
Náuseas |
27 |
0.6 |
31 |
0.3 |
|
Dolor abdominala |
22 |
1.9 |
15 |
0.3 |
|
Vómitos |
17 |
1.9 |
21 |
0.3 |
|
Dispepsiab |
15 |
0 |
22 |
0.3 |
|
Generales |
||||
|
Fatigac |
51 |
8 |
50 |
8 |
|
Hepatobiliares |
||||
|
Hepatotoxicidadd |
44 |
11 |
26 |
5 |
|
Piel y tejido subcutáneo |
||||
|
Síndrome de |
40 |
8 |
41 |
8 |
|
Estomatitise |
37 |
3.4 |
46 |
4.4 |
|
Erupción cutáneaf |
36 |
3.1 |
14 |
0 |
|
Prurito |
19 |
0.3 |
4.4 |
0 |
|
Vasculares |
||||
|
Hipertensióng |
36 |
13 |
39 |
14 |
|
Endocrinas |
||||
|
Hipotiroidismoh |
34 |
0.3 |
30 |
0.3 |
|
Musculoesqueléticas y del tejido conjuntivo |
||||
|
Dolor |
33 |
3.8 |
29 |
3.1 |
|
Artralgia |
18 |
0.3 |
9 |
0.3 |
|
Metabolismo y nutrición |
||||
|
Disminución del apetito |
28 |
1.9 |
20 |
1.3 |
|
Sistema nervioso |
||||
|
Disgeusia |
24 |
0 |
22 |
0 |
|
Dolor de cabeza |
16 |
0 |
12 |
0.6 |
|
Respiratorias, torácicas y mediastínicas |
||||
|
Tosj |
20 |
0.3 |
17 |
0 |
|
Disfonía |
17 |
0.3 |
3.4 |
0 |
|
Infecciones e infestaciones |
||||
|
Infección de las vías respiratorias superioresk |
20 |
0.3 |
8 |
0.3 |
| a La incidencia de cada prueba se basa en el número de pacientes que tenían disponible tanto la medición de laboratorio inicial como al menos una medición de laboratorio durante el estudio: Grupo de OPDIVO y cabozantinib (rango: 170 a 317 pacientes) y grupo de sunitinib (rango: 173 a 311 pacientes). | ||||
|
Anomalía de laboratorio |
OPDIVO y cabozantinib |
Sunitinib |
||
|
Grados 1-4 (%) |
Grados 3-4 (%) |
Grados 1-4 (%) |
Grados 3-4 (%) |
|
|
Química |
||||
|
Aumento de ALT |
79 |
9.8 |
39 |
3.5 |
|
Aumento de AST |
77 |
7.9 |
57 |
2.6 |
|
Hipofosfatemia |
69 |
28 |
48 |
10 |
|
Hipocalcemia |
54 |
1.9 |
24 |
0.6 |
|
Hipomagnesemia |
47 |
1.3 |
25 |
0.3 |
|
Hiperglucemia |
44 |
3.5 |
44 |
1.7 |
|
Hiponatremia |
43 |
11 |
36 |
12 |
|
Aumento de la lipasa |
41 |
14 |
38 |
13 |
|
Aumento de la amilasa |
41 |
10 |
28 |
6 |
|
Aumento de la fosfatasa alcalina |
41 |
2.8 |
37 |
1.6 |
|
Increased creatinine |
39 |
1.3 |
42 |
0.6 |
|
Hiperpotasemia |
35 |
4.7 |
27 |
1 |
|
Hipoglucemia |
26 |
0.8 |
14 |
0.4 |
|
Hematológicas |
||||
|
Linfopenia |
42 |
6.6 |
45 |
10 |
|
Trombocitopenia |
41 |
0.3 |
70 |
9.7 |
|
Anemia |
37 |
2.5 |
61 |
4.8 |
|
Leucopenia |
37 |
0.3 |
66 |
5.1 |
|
Neutropenia |
35 |
3.2 |
67 |
12 |
Carcinoma de células renales previamente tratado
CHECKMATE-025
Se evaluó la seguridad de OPDIVO en CHECKMATE-025, un ensayo abierto aleatorizado en 803 pacientes con CCR avanzado que habían experimentado progresión de la enfermedad durante o después de al menos un régimen de tratamiento antiangiogénico que recibieron OPDIVO 3 mg/kg durante 60 minutos mediante infusión intravenosa cada 2 semanas (n=406) o everolimus 10 mg al día (n=397) [consulte Estudios clínicos (14.6)]. La mediana de la duración del tratamiento fue de 5.5 meses (rango: 1 día a 29.6+ meses) en pacientes tratados con OPDIVO y de 3.7 meses (rango: 6 días a 25.7+ meses) en pacientes tratados con everolimus.
La tasa de muerte durante el tratamiento o dentro de los 30 días posteriores a la última dosis fue del 4.7 % en el grupo de OPDIVO. Se produjeron reacciones adversas graves en el 47 % de los pacientes que recibieron OPDIVO. Se interrumpió el tratamiento del estudio debido a reacciones adversas en el 16 % de los pacientes tratados con OPDIVO. El cuarenta y cuatro por ciento (44 %) de los pacientes que recibieron OPDIVO tuvieron una interrupción de la dosis debido a una reacción adversa.
Las reacciones adversas graves más frecuentes en al menos el 2 % de los pacientes fueron: lesión renal aguda, derrame pleural, neumonía, diarrea e hipercalcemia. Las reacciones adversas más comunes (≥20 %) fueron fatiga, tos, náuseas, erupción cutánea, disnea, diarrea, estreñimiento, disminución del apetito, dolor de espalda y artralgia. Las anomalías de laboratorio más comunes que empeoraron en comparación con el valor inicial en ≥30 % de los pacientes incluyen aumento de la creatinina, linfopenia, anemia, aumento de AST, aumento de la fosfatasa alcalina, hiponatremia, aumento de los triglicéridos e hiperpotasemia. Además, entre los pacientes con TSH < LSN al inicio, una mayor proporción de pacientes experimentó una elevación emergente del tratamiento de TSH > LSN en el grupo de OPDIVO en comparación con el grupo de everolimus (26 % y 14 %, respectivamente).
Las tablas 29 y 30 resumen las reacciones adversas y las anomalías de laboratorio, respectivamente, en CHECKMATE-025.
| La toxicidad se clasificó según NCI CTCAE v4. a Incluye astenia, disminución de la actividad, fatiga y malestar general. b Incluye nasofaringitis, faringitis, rinitis e infección de las vías respiratorias superiores (IVRS) vírica. c Incluye colitis, enterocolitis y gastroenteritis. d Incluye dermatitis, dermatitis acneiforme, erupción eritematosa, erupción generalizada, erupción macular, erupción maculopapular, erupción papular, erupción pruriginosa, eritema multiforme y eritema. |
||||
|
Reacción adversa |
OPDIVO |
Everolimus |
||
|
Grados 1-4 (%) |
Grados 3-4 (%) |
Grados 1-4 (%) |
Grados 3-4 (%) |
|
|
Reacción adversa |
98 |
56 |
96 |
62 |
|
Generales |
||||
|
Fatigaa |
56 |
6 |
57 |
7 |
|
Pirexia |
17 |
0.7 |
20 |
0.8 |
|
Respiratorias, torácicas y mediastínicas |
||||
|
Tos/tos productiva |
34 |
0 |
38 |
0.5 |
|
Disnea/disnea de esfuerzo |
27 |
3 |
31 |
2 |
|
Infección de las vías respiratorias superioresb |
18 |
0 |
11 |
0 |
|
Gastrointestinales |
||||
|
Náuseas |
28 |
0.5 |
29 |
1 |
|
Diarreac |
25 |
2.2 |
32 |
1.8 |
|
Estreñimiento |
23 |
0.5 |
18 |
0.5 |
|
Vómitos |
16 |
0.5 |
16 |
0.5 |
|
Piel y tejido subcutáneo |
||||
|
Erupción cutánead |
28 |
1.5 |
36 |
1 |
|
Prurito/prurito generalizado |
19 |
0 |
14 |
0 |
|
Metabolismo y Nutrición |
||||
|
Disminución del apetito |
23 |
1.2 |
30 |
1.5 |
|
Musculoesquelético y tejido conectivo |
||||
|
Artralgia |
20 |
1 |
14 |
0.5 |
|
Dolor de espalda |
21 |
3.4 |
16 |
2.8 |
Otras reacciones adversas clínicamente importantes en CHECKMATE-025 fueron:
Trastornos generales y afecciones en el sitio de administración: edema/edema periférico
Trastornos gastrointestinales: dolor/molestia abdominal
Trastornos musculoesqueléticos y del tejido conectivo: dolor en las extremidades, dolor musculoesquelético
Trastornos del sistema nervioso: dolor de cabeza/migraña, neuropatía periférica
Investigaciones: pérdida de peso
Trastornos de la piel: eritrodisestesia palmo-plantar
| a La incidencia de cada prueba se basa en el número de pacientes que tenían disponible tanto el valor inicial como al menos una medición de laboratorio durante el estudio: grupo OPDIVO (rango: 259 a 401 pacientes) y grupo everolimus (rango: 257 a 376 pacientes). | ||||
|
Anomalía de laboratorio |
OPDIVO |
Everolimus |
||
|
Grados 1-4 (%) |
Grados 3-4 (%) |
Grados 1-4 (%) |
Grados 3-4 (%) |
|
|
Hematología |
||||
|
Linfopenia |
42 |
6 |
53 |
11 |
|
Anemia |
39 |
8 |
69 |
16 |
|
Química |
||||
|
Aumento de la creatinina |
42 |
2 |
45 |
1.6 |
|
Aumento de AST |
33 |
2.8 |
39 |
1.6 |
|
Aumento de la fosfatasa alcalina |
32 |
2.3 |
32 |
0.8 |
|
Hiponatremia |
32 |
7 |
26 |
6 |
|
Hiperpotasemia |
30 |
4 |
20 |
2.1 |
|
Hipocalcemia |
23 |
0.9 |
26 |
1.3 |
|
Aumento de ALT |
22 |
3.2 |
31 |
0.8 |
|
Hipercalcemia |
19 |
3.2 |
6 |
0.3 |
|
Lípidos |
||||
|
Aumento de triglicéridos |
32 |
1.5 |
67 |
11 |
|
Aumento del colesterol |
21 |
0.3 |
55 |
1.4 |
Linfoma de Hodgkin clásico
Se evaluó la seguridad de OPDIVO en 266 pacientes adultos con LCHc (243 pacientes en los ensayos CHECKMATE-205 y 23 pacientes en el CHECKMATE-039) [ver Estudios clínicos (14.7)]. Los pacientes recibieron 3 mg/kg de OPDIVO en infusión intravenosa durante 60 minutos cada 2 semanas hasta la progresión de la enfermedad, el beneficio clínico máximo o una toxicidad inaceptable.
La mediana de edad fue de 34 años (intervalo: 18 a 72), el 98 % de los pacientes había recibido un TPH autógeno, ninguno había recibido un TPH alogénico y el 74 % había recibido brentuximab vedotin. La mediana del número de regímenes sistémicos previos fue de 4 (intervalo: 2 a 15). Los pacientes recibieron una mediana de 23 dosis (ciclos) de OPDIVO (intervalo: 1 a 48), con una mediana de duración del tratamiento de 11 meses (intervalo: 0 a 23 meses).
Once pacientes fallecieron por causas distintas a la progresión de la enfermedad: 3 por reacciones adversas dentro de los 30 días posteriores a la última dosis de nivolumab, 2 por infección de 8 a 9 meses después de finalizar el tratamiento con nivolumab y 6 por complicaciones del TPH alogénico. Se produjeron reacciones adversas graves en el 26 % de los pacientes. Se produjo un retraso en la dosis debido a una reacción adversa en el 34 % de los pacientes. Se interrumpió el tratamiento con OPDIVO debido a reacciones adversas en el 7 % de los pacientes.
Las reacciones adversas graves más frecuentes notificadas en ≥ 1 % de los pacientes fueron neumonía, reacción relacionada con la infusión, pirexia, colitis o diarrea, derrame pleural, neumonitis y erupción cutánea. Las reacciones adversas más comunes (≥ 20 %) entre todos los pacientes fueron infección de las vías respiratorias superiores, fatiga, tos, diarrea, pirexia, dolor musculoesquelético, erupción cutánea, náuseas y prurito.
Las Tablas 31 y 32 resumen las reacciones adversas y las anomalías de laboratorio, respectivamente, en CHECKMATE-205 y CHECKMATE-039.
| La toxicidad se clasificó según los criterios comunes de toxicidad del National Cancer Institute versión 4 (NCI CTCAE v4). a Incluye eventos que ocurrieron hasta 30 días después de la última dosis de nivolumab, independientemente de la causalidad. Después de una reacción adversa mediada por el sistema inmunitario, se incluyeron las reacciones posteriores a la reexposición a nivolumab si ocurrían hasta 30 días después de finalizar el ciclo inicial de nivolumab. b Incluye nasofaringitis, faringitis, rinitis y sinusitis. c Incluye neumonía bacteriana, neumonía por micoplasma, neumonía por Pneumocystis jirovecii. d Incluye astenia. e Incluye colitis. f Incluye molestias abdominales y dolor en la parte superior del abdomen. g Incluye dolor de espalda, dolor óseo, dolor torácico musculoesquelético, molestias musculoesqueléticas, mialgia, dolor de cuello y dolor en las extremidades. h Incluye dermatitis, dermatitis acneiforme, dermatitis exfoliativa y erupción cutánea descrita como macular, papular, maculopapular, pruriginosa, exfoliativa o acneiforme. i Incluye hiperestesia, hipoestesia, parestesia, disestesia, neuropatía motora periférica, neuropatía sensorial periférica y polineuropatía. Estas cifras son específicas de los eventos emergentes del tratamiento. |
||
|
Reacción adversaa |
OPDIVO (n=266) |
|
|
Todos los grados (%) |
Grados 3-4 (%) |
|
|
Infecciones |
||
|
Infección de las vías respiratorias superioresb |
44 |
0.8 |
|
Neumonía/bronconeumoníac |
13 |
3.8 |
|
Congestión nasal |
11 |
0 |
|
Generales |
||
|
Fatigad |
39 |
1.9 |
|
Pirexia |
29 |
<1 |
|
Respiratorias, torácicas y mediastínicas |
||
|
Tos/tos productiva |
36 |
0 |
|
Disnea/disnea de esfuerzo |
15 |
1.5 |
|
Gastrointestinales |
||
|
Diarreae |
33 |
1.5 |
|
Náuseas |
20 |
0 |
|
Vómitos |
19 |
<1 |
|
Dolor abdominalf |
16 |
<1 |
|
Estreñimiento |
14 |
0.4 |
|
Musculoesquelético y tejido conjuntivo |
||
|
Dolor musculoesqueléticog |
26 |
1.1 |
|
Artralgia |
16 |
<1 |
|
Piel y tejido subcutáneo |
||
|
Erupción cutáneah |
24 |
1.5 |
|
Prurito |
20 |
0 |
|
Sistema nervioso |
||
|
Dolor de cabeza |
17 |
<1 |
|
Neuropatía periféricai |
12 |
<1 |
|
Lesiones, intoxicaciones y complicaciones de procedimientos |
||
|
Reacción relacionada con la infusión |
14 |
<1 |
|
Endocrino |
||
|
Hipotiroidismo/tiroiditis |
12 |
0 |
Información adicional con respecto a las reacciones adversas clínicamente importantes:
Neumonitis inmunomediada: En CHECKMATE-205 y CHECKMATE-039, se produjo neumonitis, incluida la enfermedad pulmonar intersticial, en el 6.0% (16/266) de los pacientes que recibieron OPDIVO. Se produjo neumonitis inmunomediada en el 4.9% (13/266) de los pacientes que recibieron OPDIVO (un Grado 3 y 12 Grado 2). La mediana del tiempo hasta la aparición fue de 4.5 meses (rango: 5 días a 12 meses). Los 13 pacientes recibieron corticosteroides sistémicos, con resolución en 12. Cuatro pacientes suspendieron permanentemente OPDIVO debido a la neumonitis. Ocho pacientes continuaron con OPDIVO (tres después del retraso de la dosis), de los cuales dos tuvieron recurrencia de la neumonitis.
Neuropatía periférica: Se reportó neuropatía periférica emergente del tratamiento en el 12% (31/266) de todos los pacientes que recibieron OPDIVO. Veintiocho pacientes (11%) tuvieron neuropatía periférica de nueva aparición y 3 pacientes tuvieron empeoramiento de la neuropatía desde el inicio. La mediana del tiempo hasta la aparición fue de 50 (rango: 1 a 309) días.
Complicaciones del alo-TPH después de OPDIVO: De 17 pacientes con linfoma de Hodgkin clásico de los ensayos CHECKMATE-205 y CHECKMATE-039 que se sometieron a alo-TPH después del tratamiento con OPDIVO, 6 pacientes (35%) murieron por complicaciones relacionadas con el trasplante. Se produjeron cinco muertes en el contexto de EICH grave (Grado 3 a 4) o refractaria. La EICH hiperaguda ocurrió en 2 pacientes (12%) y se reportó EICH de Grado 3 o superior en 5 pacientes (29%). La EHO hepática ocurrió en 1 paciente, que recibió alo-TPH condicionado de intensidad reducida y murió de EICH e insuficiencia multiorgánica.
La Tabla 32 resume las anomalías de laboratorio en pacientes con linfoma de Hodgkin clásico. Las anomalías de laboratorio emergentes del tratamiento más comunes (≥20%) incluyeron citopenias, anomalías de la función hepática y aumento de la lipasa. Otros hallazgos comunes (≥10%) incluyeron aumento de la creatinina, anomalías electrolíticas y aumento de la amilasa.
| a La incidencia de cada prueba se basa en el número de pacientes que tenían tanto mediciones de laboratorio iniciales como al menos una medición de laboratorio durante el estudio: rango: 203 a 266 pacientes. b Incluye eventos que ocurren hasta 30 días después de la última dosis de nivolumab. Después de una reacción adversa inmunomediada, las reacciones posteriores al retratamiento con nivolumab se incluyeron si ocurrían dentro de los 30 días posteriores a la finalización del ciclo inicial de nivolumab. c Además, en la población de seguridad, se reportó hiperglucemia en ayunas (todos de grado 1-2) en 27 de 69 (39%) pacientes evaluables e hipoglucemia en ayunas (todos de grado 1-2) en 11 de 69 (16%). |
||
|
Anomalía de laboratorio |
OPDIVOa |
|
|
Todos los grados (%)b |
Grados 3-4 (%)b |
|
|
Hematología |
||
|
Leucopenia |
38 |
4.5 |
|
Neutropenia |
37 |
5 |
|
Trombocitopenia |
37 |
3 |
|
Linfopenia |
32 |
11 |
|
Anemia |
26 |
2.6 |
|
Químicac |
||
|
Aumento de AST |
33 |
2.6 |
|
Aumento de ALT |
31 |
3.4 |
|
Aumento de lipasa |
22 |
9 |
|
Aumento de la fosfatasa alcalina |
20 |
1.5 |
|
Hiponatremia |
20 |
1.1 |
|
Hipopotasemia |
16 |
1.9 |
|
Increased creatinine |
16 |
<1 |
|
Hipocalcemia |
15 |
<1 |
|
Hipercalemia |
15 |
1.5 |
|
Hipomagnesemia |
14 |
<1 |
|
Increased amylase |
13 |
1.5 |
|
Increased bilirubin |
11 |
1.5 |
Carcinoma escamoso de células de cabeza y cuello
La seguridad de OPDIVO se evaluó en CHECKMATE-141, un ensayo aleatorizado, controlado activamente, abierto, multicéntrico en pacientes con SCCHN recurrente o metastásico con progresión durante o dentro de los 6 meses de recibir tratamiento previo basado en platino [ver Estudios clínicos (14.8)]. El ensayo excluyó a pacientes con enfermedad autoinmune activa, afecciones médicas que requieran inmunosupresión sistémica o carcinoma metastásico o recurrente de la nasofaringe, carcinoma de células escamosas de histología primaria desconocida, glándulas salivales o histologías no escamosas (por ejemplo, melanoma mucoso). Los pacientes recibieron OPDIVO 3 mg/kg por perfusión intravenosa durante 60 minutos cada 2 semanas (n=236) o la elección del investigador, ya sea cetuximab (dosis inicial de 400 mg/m2 por vía intravenosa seguido de 250 mg/m2 semanales), metotrexato (40 a 60 mg/m2 por vía intravenosa semanalmente) o docetaxel (30 a 40 mg/m2 por vía intravenosa semanalmente). La mediana de duración de la exposición a nivolumab fue de 1,9 meses (rango: 1 día a 16,1+ meses) en los pacientes tratados con OPDIVO. En este ensayo, el 18% de los pacientes recibió OPDIVO durante más de 6 meses y el 2,5% de los pacientes recibió OPDIVO durante más de 1 año.
La edad mediana de todos los pacientes aleatorizados fue de 60 años (rango: 28 a 83); el 28% de los pacientes en el grupo OPDIVO tenían ≥65 años y el 37% en el grupo comparador tenían ≥65 años, el 83% eran varones y el 83% eran blancos, el 12% eran asiáticos y el 4% eran negros. El estado de funcionamiento ECOG de referencia fue 0 (20%) o 1 (78%), el 45% de los pacientes recibió solo una línea previa de terapia sistémica, el 55% restante de los pacientes tuvo dos o más líneas previas de terapia y el 90% recibió radioterapia previa.
Se produjeron reacciones adversas graves en el 49% de los pacientes que recibieron OPDIVO. OPDIVO se suspendió en el 14% de los pacientes y se retrasó en el 24% de los pacientes por una reacción adversa. Las reacciones adversas y las anomalías de laboratorio que se presentaron en pacientes con SCCHN fueron generalmente similares a las que ocurrieron en pacientes con melanoma y NSCLC.
Las reacciones adversas graves más frecuentes reportadas en ≥2% de los pacientes que recibieron OPDIVO fueron neumonía, dificultad respiratoria, insuficiencia respiratoria, infección de las vías respiratorias y sepsis. Las reacciones adversas más comunes que se presentaron en ≥10% de los pacientes tratados con OPDIVO y con una incidencia mayor que la elección del investigador fueron tos y dificultad respiratoria. Las anomalías de laboratorio más comunes que se presentaron en ≥10% de los pacientes tratados con OPDIVO y con una incidencia mayor que la elección del investigador fueron el aumento de la fosfatasa alcalina, el aumento de la amilasa, la hipercalcemia, la hiperpotasemia y el aumento del TSH.
Tratamiento adyuvante de carcinoma urotelial (UC)
La seguridad de OPDIVO se evaluó en CHECKMATE-274, un ensayo aleatorizado, doble ciego, multicéntrico de OPDIVO adyuvante frente a placebo en pacientes adultos que se habían sometido a resección radical de UC que se originaba en la vejiga o en el tracto urinario superior (pelvis renal o uréter) y tenían un riesgo alto de recurrencia [ver Estudios clínicos (14.9)]. Los pacientes recibieron OPDIVO 240 mg por perfusión intravenosa durante 30 minutos cada 2 semanas (n = 351) o placebo (n = 348) hasta la recurrencia o toxicidad inaceptable durante un máximo de 1 año. La mediana de duración del tratamiento con OPDIVO fue de 8,8 meses (rango: 0 a 12,5).
Se produjeron reacciones adversas graves en el 30% de los pacientes con OPDIVO. La reacción adversa grave más frecuente reportada en ≥2% de los pacientes fue la infección del tracto urinario. Ocurrieron reacciones adversas fatales en el 1% de los pacientes; entre estos eventos se incluyeron neumonitis (0,6%). OPDIVO se suspendió por reacciones adversas en el 18% de los pacientes. OPDIVO se retrasó por reacción adversa en el 33% de los pacientes.
Las reacciones adversas más comunes (reportadas en ≥20% de los pacientes) fueron erupción cutánea, fatiga, diarrea, prurito, dolor musculoesquelético y infección del tracto urinario.
Las Tablas 33 y 34 resumen las reacciones adversas y las anomalías de laboratorio, respectivamente, en CHECKMATE-274.
| La toxicidad fue gradada según NCI CTCAE v4. a Incluye acné, ampolla, dermatitis, dermatitis acneiforme, dermatitis alérgica, dermatitis de contacto, eccema, eccema asteatósico, eccema numular, eritema, eritema multiforme, liquen escleroso, queratosis liqueniforme, penfigoide, reacción fototérmica, trastorno de la pigmentación, psoriasis, erupción cutánea, lesión cutánea, reacción cutánea, erupción tóxica cutánea y urticaria. b Incluye colitis, colitis microscópica, diarrea, duodenitis, enteritis e enterocolitis inmunomediada c Incluye dolor abdominal, malestar abdominal, sensibilidad abdominal, dolor abdominal inferior y superior. d Incluye dolor musculoesquelético, dolor de espalda, dolor óseo, dolor musculoesquelético en el pecho, malestar musculoesquelético, mialgia, dolor de cuello, dolor en extremidades y dolor espinal. e Incluye cistitis, infección urinaria por Escherichia coli, pielonefritis, pielonefritis aguda, pielonefritis crónica, uretritis, infección urinaria, infección urinaria bacteriana, infección urinaria estafilocócica y urosepsis. f Incluye infección del tracto respiratorio superior, nasofaringitis, faringitis y rinitis. g Incluye lesión renal aguda, nefritis autoinmune, aumento de la creatinina sérica, disminución de la tasa de filtración glomerular, nefritis inmunomediada, nefritis, insuficiencia renal y deterioro renal. h Incluye tos, tos productiva y síndrome de tos de vías respiratorias superiores. i Incluye dificultad respiratoria y dificultad respiratoria por esfuerzo. j Incluye mareos, mareos posturales y vértigo. k Incluye aumento de la aspartato aminotransferasa, aumento de la alanina aminotransferasa, aumento de la bilirrubina sérica, colangitis, lesión hepática inducida por fármacos, fallo hepático, anormalidad de la función hepática, hepatitis, lesión hepatocelular, hiperbilirrubinemia, aumento de la gamma-glutamil transferasa, lesión hepática y aumento de las transaminasas. |
||||
|
Reacción adversa |
OPDIVO (n=351) |
Placebo (n=348) |
||
|
Todos los grados (%) |
Grados 3-4 (%) |
Todos los grados (%) |
Grados 3-4 (%) |
|
|
Piel y tejido subcutáneo |
||||
|
Rasha |
36 |
1.7 |
19 |
0.3 |
|
Prurito |
30 |
0 |
16 |
0 |
|
General |
||||
|
Fatiga/Astenia |
36 |
1.1 |
32 |
0.3 |
|
Pyrexia |
10 |
0.3 |
10 |
0.3 |
|
Gastrointestinal |
||||
|
Diarrheab |
30 |
2.8 |
27 |
1.7 |
|
Náuseas |
16 |
0.6 |
13 |
0 |
|
Dolor abdominalc |
15 |
0.9 |
15 |
0.6 |
|
Estreñimiento |
13 |
0.3 |
15 |
0.3 |
|
Musculoesquelético y tejido conectivo |
||||
|
Dolor musculoesqueléticod |
28 |
0.6 |
24 |
0.9 |
|
Arthralgia |
11 |
0.3 |
13 |
0 |
|
Infecciones |
||||
|
Infección del tracto urinarioe |
22 |
6 |
23 |
9 |
|
Infección del tracto respiratorio superiorf |
16 |
0.3 |
16 |
0.6 |
|
Endocrino |
||||
|
Hipertiroidismo |
11 |
0 |
1.1 |
0 |
|
Hipotiroidismo |
11 |
0 |
2.3 |
0 |
|
Trastornos renales y urinarios |
||||
|
Disfunción renalg |
17 |
1.7 |
16 |
0.9 |
|
Respiratorios, torácicos y mediastínicos |
||||
|
Tosh |
14 |
0 |
11 |
0 |
|
Disneai |
11 |
0.3 |
6 |
0.3 |
|
Metabolismo y nutrición |
||||
|
Disminución del apetito |
13 |
0.9 |
7 |
0.3 |
|
Trastornos del sistema nervioso |
||||
|
Mareoj |
11 |
0.3 |
9 |
0 |
|
Hepatobiliar |
||||
|
Hepatitisk |
11 |
4 |
8 |
0.6 |
| a La incidencia de cada prueba se basa en el número de pacientes que disponían tanto del valor inicial como de al menos una medición de laboratorio durante el estudio: Grupo OPDIVO (rango: 322 a 348 pacientes) y grupo placebo (rango: 312 a 341 pacientes). | ||||
|
Anormalidad de laboratorio |
OPDIVO (n=351) |
Placebo (n=348) |
||
|
Todos los grados (%) |
Grados 3-4 (%) |
Todos los grados (%) |
Grados 3-4 (%) |
|
|
Química |
||||
|
Aumento de la creatinina |
36 |
1.7 |
36 |
2.6 |
|
Aumento de la amilasa |
34 |
8 |
23 |
3.2 |
|
Aumento de la lipasa |
33 |
12 |
31 |
10 |
|
Hiperpotasemia |
32 |
5 |
30 |
6 |
|
Aumento de la fosfatasa alcalina |
24 |
2.3 |
15 |
0.6 |
|
Aumento de AST |
24 |
3.5 |
16 |
0.9 |
|
Aumento de ALT |
23 |
2.9 |
15 |
0.6 |
|
Hiponatremia |
22 |
4.1 |
17 |
1.8 |
|
Hipocalcemia |
17 |
1.2 |
11 |
0.9 |
|
Hipomagnesemia |
16 |
0 |
9 |
0 |
|
Hipercalcemia |
12 |
0.3 |
8 |
0.3 |
|
Hematología |
||||
|
Linfopenia |
33 |
2.9 |
27 |
1.5 |
|
Anemia |
30 |
1.4 |
28 |
0.9 |
|
Neutropenia |
11 |
0.6 |
10 |
0.3 |
Tratamiento de primera línea del CU irresecable o metastásico
La seguridad de OPDIVO se evaluó en CHECKMATE-901, un ensayo aleatorizado y abierto en pacientes elegibles para cisplatino con CU irresecable o metastásico [ver Estudios clínicos (14.9)]. Los pacientes recibieron 360 mg de OPDIVO con cisplatino y gemcitabina cada 3 semanas durante un máximo de 6 ciclos, seguidos de 480 mg de OPDIVO en monoterapia cada 4 semanas durante un máximo de 2 años (n=304), o quimioterapia con cisplatino y gemcitabina cada 3 semanas durante un máximo de 6 ciclos (n=288). Los pacientes que interrumpieron el cisplatino solo pudieron cambiar a carboplatino.
Entre los pacientes que recibieron OPDIVO con quimioterapia, la mediana de duración de la exposición a OPDIVO fue de 7.4 meses (rango: 0.03 a 47.9 meses). Se produjeron reacciones adversas graves en el 48% de los pacientes que recibieron OPDIVO en combinación con quimioterapia. Las reacciones adversas graves más frecuentes notificadas en ≥2% de los pacientes que recibieron OPDIVO con quimioterapia fueron infección del tracto urinario (4.9%), lesión renal aguda (4.3%), anemia (3%), embolia pulmonar (2.6%), sepsis (2.3%) y disminución del recuento de plaquetas (2.3%). Las reacciones adversas más comunes (notificadas en ≥20% de los pacientes) fueron náuseas, fatiga, dolor musculoesquelético, estreñimiento, disminución del apetito, erupción cutánea, vómitos y neuropatía periférica.
Se produjeron reacciones adversas mortales en el 3.6% de los pacientes que recibieron OPDIVO en combinación con quimioterapia; entre ellas, sepsis (1%).
El tratamiento con OPDIVO y/o quimioterapia se interrumpió en el 30% de los pacientes y se retrasó en el 67% de los pacientes debido a una reacción adversa.
En las Tablas 35 y 36 se resumen las reacciones adversas y las anomalías de laboratorio, respectivamente, en el estudio CHECKMATE-901.
| La toxicidad se clasificó según los criterios comunes de toxicidad del National Cancer Institute versión 4 (NCI CTCAE v4). a Incluye colitis, enterocolitis mediada por el sistema inmunitario. b Incluye dolor en la parte superior del abdomen, dolor en la parte inferior del abdomen, molestias abdominales, molestias epigástricas, dolor gastrointestinal y dolor hepático. c Incluye astenia. d Incluye edema periférico, inflamación, inflamación periférica, edema localizado, inflamación, edema facial, edema testicular, edema gravitacional y edema genital. e Incluye hipertermia, aumento de la temperatura corporal e hiperpirexia. f Incluye dolor de espalda, artralgia, dolor óseo, artritis, dolor torácico musculoesquelético, dolor torácico no cardíaco, mialgia, dolor de cuello, dolor en las extremidades y dolor espinal. g Incluye erupción maculopapular, erupción eritematosa, erupción macular, erupción papular, erupción pustular, dermatitis acneiforme, dermatitis, dermatitis alérgica, dermatitis atópica, erupción exfoliativa, eccema asteatótico, eritema multiforme, síndrome de eritrodisestesia palmo-plantar, eccema, dermatitis exfoliativa generalizada y exfoliación cutánea. h Incluye parestesia, neuropatía sensorial periférica, hipoestesia, disestesia, neuralgia, hiperestesia, neuropatía motora periférica, polineuropatía. i Incluye neuralgia occipital. j Incluye urosepsis, cistitis, pielonefritis, pielonefritis aguda, infección del tracto urinario enterocócica, infección del tracto urinario por Escherichia. k Incluye aumento de la hormona estimulante de la sangre. l Incluye lesión renal aguda, insuficiencia renal, insuficiencia renal, disminución de la tasa de filtración glomerular, anuria, azoemia. |
||||
|
Reacción adversa |
OPDIVO y quimioterapia con doblete de platino |
Quimioterapia con doblete de platino (n=288) |
||
|
Todos los grados (%) |
Grados 3-4 (%) |
Todos los grados (%) |
Grados 3-4 (%) |
|
|
Trastornos gastrointestinales |
||||
|
Náuseas |
52 |
0.3 |
53 |
1 |
|
Estreñimiento |
30 |
0 |
28 |
0.7 |
|
Vómitos |
23 |
1.3 |
19 |
2.1 |
|
Diarreaa |
19 |
2 |
14 |
0 |
|
Dolor abdominalb |
14 |
0.3 |
9 |
0.3 |
|
Generales |
||||
|
Fatigac |
48 |
3.9 |
43 |
4.2 |
|
Edemad |
18 |
0 |
9 |
0.3 |
|
Pirexiae |
14 |
1 |
14 |
0 |
|
Musculoesqueléticas y del tejido conjuntivo |
||||
|
Dolor musculoesqueléticof |
33 |
3 |
21 |
0.3 |
|
Metabolismo y nutrición |
||||
|
Disminución del apetito |
30 |
1.6 |
19 |
1 |
|
Piel y tejido subcutáneo |
||||
|
Erupción cutáneag |
25 |
2.3 |
7 |
0.3 |
|
Prurito |
17 |
0.7 |
3.5 |
0 |
|
Trastornos del sistema nervioso |
||||
|
Neuropatía periféricah |
20 |
0.7 |
14 |
0 |
|
Dolor de cabezai |
11 |
0 |
5 |
0 |
|
Infecciones |
||||
|
Infección del tracto urinarioj |
19 |
8 |
18 |
8 |
|
Trastornos endocrinos |
||||
|
Hipotiroidismok |
17 |
0 |
0.3 |
0 |
|
Trastornos renales y urinarios |
||||
|
Renal dysfunctionl |
14 |
6 |
11 |
1.7 |
|
Hematuria |
11 |
1 |
7 |
1.4 |
|
Investigaciones |
||||
|
Pérdida de peso |
11 |
0.3 |
6 |
0 |
| a La incidencia de cada prueba se basa en el número de pacientes que tenían disponible tanto la medición de laboratorio inicial como al menos una medición de laboratorio durante el estudio: grupo de OPDIVO (rango: 289-301 pacientes) y grupo de quimioterapia (rango: 265-281 pacientes). | ||||
|
Anormalidad de laboratorio |
OPDIVO y quimioterapia con doblete de platino |
Quimioterapia con doblete de platino |
||
|
Grados 1-4 (%) |
Grados 3-4 (%) |
Grados 1-4 (%) |
Grados 3-4 (%) |
|
|
Hematología |
||||
|
Anemia |
88 |
21 |
80 |
21 |
|
Neutropenia |
82 |
35 |
76 |
28 |
|
Linfopenia |
71 |
17 |
56 |
13 |
|
Trombocitopenia |
60 |
13 |
51 |
8 |
|
Química |
||||
|
Aumento de la creatinina |
53 |
2.4 |
42 |
1.1 |
|
Hipomagnesemia |
48 |
3.8 |
39 |
1.5 |
|
Hiponatremia |
43 |
13 |
39 |
8 |
|
Hiperglucemia |
41 |
3.9 |
37 |
3.2 |
|
Hipocalcemia |
36 |
2.1 |
24 |
1.1 |
|
Hiperpotasemia |
33 |
3.0 |
32 |
1.1 |
|
Increased amylase |
32 |
4.2 |
23 |
3.6 |
|
Increased AST |
31 |
2.4 |
17 |
0.7 |
|
Increased ALT |
29 |
2.4 |
19 |
0.7 |
UC avanzado o metastásico previamente tratado
La seguridad de OPDIVO se evaluó en CHECKMATE-275, un ensayo unicéntrico en el que 270 pacientes con UC localmente avanzado o metastásico presentaron progresión de la enfermedad durante o después de la quimioterapia que contenía platino o tuvieron progresión de la enfermedad en los 12 meses posteriores al tratamientooadyuvante o neoadyuvante con quimioterapia que contenía platino [ver Estudios clínicos (14.9)]. Los pacientes recibieron OPDIVO 3 mg/kg en infusión intravenosa durante 60 minutos cada 2 semanas hasta la progresión de la enfermedad o toxicidad inaceptable. La duración media del tratamiento fue de 3,3 meses (rango: 0 a 13,4+). Cuarenta y seis por ciento (46%) de los pacientes tuvieron una interrupción de la dosis por una reacción adversa.
Catorce pacientes (5,2%) murieron por causas diferentes a la progresión de la enfermedad. Esto incluye 4 pacientes (1,5%) que murieron por neumonitis o insuficiencia cardiovascular atribuida al tratamiento con OPDIVO. Ocurrieron reacciones adversas graves en el 54% de los pacientes. OPDIVO se discontinuó por reacciones adversas en el 17% de los pacientes.
Las reacciones adversas graves más frecuentes reportadas en ≥ 2% de los pacientes fueron infección del tracto urinario, sepsis, diarrea, obstrucción intestinal pequeña y deterioro general de la salud física. Las reacciones adversas más comunes (reportadas en ≥ 20% de los pacientes) fueron fatiga, dolor musculoesquelético, náuseas y pérdida de apetito.
Las tablas 37 y 38 resumen las reacciones adversas y las anomalías de laboratorio, respectivamente, en CHECKMATE-275.
| La toxicidad se clasificó según NCI CTCAE v4. a Incluye dolor de espalda, dolor óseo, dolor musculoesquelético torácico, molestia musculoesquelética, mialgia, dolor de cuello, dolor en las extremidades y dolor de la columna vertebral. b Incluye molestia abdominal, dolor abdominal inferior y superior. c Incluye dermatitis, dermatitis acneiforme, dermatitis ampollosa y erupción descrita como generalizada, macular, maculo-papular o pruriginosa. d Incluye tiroiditis autoinmune, disminución del TSH en sangre, aumento del TSH en sangre, hipertiroidismo, hipotiroidismo, tiroiditis, disminución de la tiroxina, aumento de la tiroxina libre, aumento de la tiroxina, aumento de la triyodotironina libre, aumento de la triyodotironina. |
||
|
Reacción adversa |
OPDIVO |
|
|
Todos los grados (%) |
Grados 3-4 (%) |
|
|
Reacción adversa |
99 |
51 |
|
General |
||
|
Astenia / fatiga / malestar |
46 |
7 |
|
Pirexia / fiebre asociada al tumor |
17 |
0,4 |
|
Edema / edema periférico / hinchazón periférica |
13 |
0,4 |
|
Musculoesquelético y tejido conjuntivo |
||
|
Dolor musculoesqueléticoa |
30 |
2,6 |
|
Artralgia |
10 |
0,7 |
|
Metabolismo y nutrición |
||
|
Disminución del apetito |
22 |
2,2 |
|
Gastrointestinal |
||
|
Náuseas |
22 |
0,7 |
|
Diarrea |
17 |
2,6 |
|
Estreñimiento |
16 |
0,4 |
|
Dolor abdominalb |
13 |
1.5 |
|
Vómitos |
12 |
1.9 |
|
Respiratory, Thoracic and Mediastinal |
||
|
Tos/tos productiva |
18 |
0 |
|
Disnea/disnea de esfuerzo |
14 |
3.3 |
|
Infecciones |
||
|
Infección del tracto urinario/escherichia/infección fúngica del tracto urinario |
17 |
7 |
|
Skin and Subcutaneous Tissue |
||
|
Erupción cutáneac |
16 |
1.5 |
|
Prurito |
12 |
0 |
|
Endocrine |
||
|
Trastornos de la tiroidesd |
15 |
0 |
| a La incidencia de cada prueba se basa en el número de pacientes que tenían disponible tanto la medición de laboratorio inicial como al menos una medición de laboratorio durante el estudio: rango: de 84 a 256 pacientes. | ||
|
Anormalidad de laboratorio |
OPDIVOa |
|
|
Todos los grados (%) |
Grados 3-4 (%) |
|
|
Química |
||
|
Hiperglucemia |
42 |
2.4 |
|
Hiponatremia |
41 |
11 |
|
Aumento de la creatinina |
39 |
2 |
|
Aumento de la fosfatasa alcalina |
33 |
5.5 |
|
Hipocalcemia |
26 |
0.8 |
|
Aumento de AST |
24 |
3.5 |
|
Aumento de la lipasa |
20 |
7 |
|
Hiperpotasemia |
19 |
1.2 |
|
Aumento de ALT |
18 |
1.2 |
|
Aumento de la amilasa |
18 |
4.4 |
|
Hipomagnesemia |
16 |
0 |
|
Hematología |
||
|
Linfopenia |
42 |
9 |
|
Anemia |
40 |
7 |
|
Trombocitopenia |
15 |
2.4 |
|
Leucopenia |
11 |
0 |
Cáncer Colorrectal Metastásico con MSI-H o dMMR
Se evaluó la seguridad de OPDIVO administrado como agente único o en combinación con ipilimumab en CHECKMATE-142, un ensayo multicéntrico, no aleatorizado, de cohortes paralelas múltiples, abierto [ver Estudios Clínicos (14.10)]. En CHECKMATE-142, 74 pacientes con CCRm recibieron OPDIVO 3 mg/kg por infusión intravenosa durante 60 minutos cada 2 semanas hasta la progresión de la enfermedad o hasta una toxicidad intolerable y 119 pacientes con CCRm recibieron OPDIVO 3 mg/kg e ipilimumab 1 mg/kg cada 3 semanas durante 4 dosis, luego OPDIVO 3 mg/kg cada 2 semanas hasta la progresión de la enfermedad o hasta una toxicidad inaceptable.
En la cohorte de OPDIVO con ipilimumab, se produjeron reacciones adversas graves en el 47% de los pacientes. Se interrumpió el tratamiento en el 13% de los pacientes y se retrasó en el 45% de los pacientes debido a una reacción adversa. Las reacciones adversas graves más frecuentes notificadas en ≥2% de los pacientes fueron colitis/diarrea, episodios hepáticos, dolor abdominal, lesión renal aguda, pirexia y deshidratación. Las reacciones adversas más comunes (notificadas en ≥20% de los pacientes) fueron fatiga, diarrea, pirexia, dolor musculoesquelético, dolor abdominal, prurito, náuseas, erupción cutánea, disminución del apetito y vómitos.
Las Tablas 39 y 40 resumen las reacciones adversas y las anomalías de laboratorio, respectivamente, en CHECKMATE-142. Según el diseño de CHECKMATE-142, los datos siguientes no pueden utilizarse para identificar diferencias estadísticamente significativas entre las dos cohortes que se resumen a continuación para ninguna reacción adversa.
| La toxicidad se clasificó según NCI CTCAE v4. a Incluye astenia. b Incluye edema periférico e hinchazón periférica. c Incluye dolor abdominal superior, dolor abdominal inferior y molestias abdominales. d Incluye dolor de espalda, dolor en las extremidades, mialgia, dolor de cuello y dolor óseo. e Incluye dermatitis, dermatitis acneiforme y erupción cutánea descrita como maculopapular, eritematosa y generalizada. f Incluye nasofaringitis y rinitis. |
||||
|
Reacción Adversa |
OPDIVO |
OPDIVO e Ipilimumab |
||
|
Todos los Grados (%) |
Grados 3-4 (%) |
Todos los Grados (%) |
Grados 3-4 (%) |
|
|
General |
||||
|
Fatigaa |
54 |
5 |
49 |
6 |
|
Pirexia |
24 |
0 |
36 |
0 |
|
Edemab |
12 |
0 |
7 |
0 |
|
Gastrointestinales |
||||
|
Diarrea |
43 |
2.7 |
45 |
3.4 |
|
Dolor abdominalc |
34 |
2.7 |
30 |
5 |
|
Náuseas |
34 |
1.4 |
26 |
0.8 |
|
Vómitos |
28 |
4.1 |
20 |
1.7 |
|
Estreñimiento |
20 |
0 |
15 |
0 |
|
Musculoesquelético y tejido conectivo |
||||
|
Dolor musculoesqueléticod |
28 |
1.4 |
36 |
3.4 |
|
Artralgia |
19 |
0 |
14 |
0.8 |
|
Respiratorio, torácico y mediastínico |
||||
|
Tos |
26 |
0 |
19 |
0.8 |
|
Disnea |
8 |
1 |
13 |
1.7 |
|
Piel y tejido subcutáneo |
||||
|
Erupción cutáneae |
23 |
1.4 |
25 |
4.2 |
|
Prurito |
19 |
0 |
28 |
1.7 |
|
Piel seca |
7 |
0 |
11 |
0 |
|
Infecciones |
||||
|
Infección de las vías respiratorias superioresf |
20 |
0 |
9 |
0 |
|
Endocrino |
||||
|
Hiperglucemia |
19 |
2.7 |
6 |
1 |
|
Hipotiroidismo |
5 |
0 |
14 |
0.8 |
|
Hipertiroidismo |
4 |
0 |
12 |
0 |
|
Sistema nervioso |
||||
|
Dolor de cabeza |
16 |
0 |
17 |
1.7 |
|
Mareos |
14 |
0 |
11 |
0 |
|
Metabolismo y nutrición |
||||
|
Disminución del apetito |
14 |
1.4 |
20 |
1.7 |
|
Psiquiátrico |
||||
|
Insomnio |
9 |
0 |
13 |
0.8 |
|
Investigaciones |
||||
|
Pérdida de peso |
8 |
0 |
10 |
0 |
Las reacciones adversas clínicamente importantes notificadas en <10 % de los pacientes que recibieron OPDIVO con ipilimumab fueron encefalitis (0,8 %), miositis necrotizante (0,8 %) y uveítis (0,8 %).
| a La incidencia de cada prueba se basa en el número de pacientes que tenían disponible tanto la medición de laboratorio inicial como al menos una medición de laboratorio durante el estudio. El número de pacientes evaluables oscila entre 62 y 71 para la cohorte de OPDIVO y entre 87 y 114 para la cohorte de OPDIVO e ipilimumab. | ||||
|
Anomalía de laboratorio |
OPDIVO |
OPDIVO e ipilimumab |
||
|
Todos los grados (%) |
Grados 3-4 (%) |
Todos los grados (%) |
Grados 3-4 (%) |
|
|
Hematología |
||||
|
Anemia |
50 |
7 |
42 |
9 |
|
Linfopenia |
36 |
7 |
25 |
6 |
|
Neutropenia |
20 |
4.3 |
18 |
0 |
|
Trombocitopenia |
16 |
1.4 |
26 |
0.9 |
|
Química |
||||
|
Aumento de la |
37 |
2.8 |
28 |
5 |
|
Aumento de la lipasa |
33 |
19 |
39 |
12 |
|
Aumento de la ALT |
32 |
2.8 |
33 |
12 |
|
Aumento de la AST |
31 |
1.4 |
40 |
12 |
|
Hiponatremia |
27 |
4.3 |
26 |
5 |
|
Hipocalcemia |
19 |
0 |
16 |
0 |
|
Hipomagnesemia |
17 |
0 |
18 |
0 |
|
Aumento de amilasa |
16 |
4.8 |
36 |
3.4 |
|
Aumento de la bilirrubina |
14 |
4.2 |
21 |
5 |
|
Hipocalemia |
14 |
0 |
15 |
1.8 |
|
Aumento de creatinina |
12 |
0 |
25 |
3.6 |
|
Hipercalemia |
11 |
0 |
23 |
0.9 |
Carcinoma hepatocelular
Se evaluó la seguridad de OPDIVO 1 mg/kg en combinación con ipilimumab 3 mg/kg en un subgrupo compuesto por 49 pacientes con HCC y cirrosis de clase A de Child-Pugh incluidos en la Cohorte 4 de CHECKMATE-040, un ensayo multicéntrico, de múltiples cohortes y abierto [ver Estudios clínicos (14.11)] que progresaron o no toleraron sorafenib. OPDIVO e ipilimumab se administraron cada 3 semanas durante 4 dosis, seguidas de OPDIVO 240 mg como agente único cada 2 semanas hasta la progresión de la enfermedad o la aparición de toxicidad inaceptable. Durante el período de combinación de OPDIVO e ipilimumab, 33 de 49 pacientes (67 %) recibieron las 4 dosis planificadas de OPDIVO e ipilimumab. Durante todo el período de tratamiento, la mediana de la duración de la exposición a OPDIVO fue de 5.1 meses (rango: 0 a más de 35 meses) y a ipilimumab fue de 2.1 meses (rango: 0 a 4.5 meses). El 47 % de los pacientes estuvieron expuestos al tratamiento durante más de 6 meses y el 35 % de los pacientes estuvieron expuestos al tratamiento durante más de 1 año. Se produjeron reacciones adversas graves en el 59 % de los pacientes. Se interrumpió el tratamiento en el 29 % de los pacientes y se retrasó en el 65 % de los pacientes debido a una reacción adversa.
Las reacciones adversas graves más frecuentes (notificadas en ≥4 % de los pacientes) fueron pirexia, diarrea, anemia, aumento de AST, insuficiencia suprarrenal, ascitis, hemorragia por varices esofágicas, hiponatremia, aumento de la bilirrubina en sangre y neumonitis.
Las Tablas 41 y 42 resumen las reacciones adversas y las anomalías de laboratorio, respectivamente, en CHECKMATE-040.
|
Reacción adversa |
OPDIVO e ipilimumab (n=49) |
|
|
Todos los grados (%) |
Grados 3-4 (%) |
|
|
Piel y tejido subcutáneo |
||
|
Erupción cutánea |
53 |
8 |
|
Prurito |
53 |
4 |
|
Musculoesquelético y tejido conjuntivo |
||
|
Dolor musculoesquelético |
41 |
2 |
|
Artralgia |
10 |
0 |
|
Gastrointestinales |
||
|
Diarrea |
39 |
4 |
|
Dolor abdominal |
22 |
6 |
|
Náuseas |
20 |
0 |
|
Ascitis |
14 |
6 |
|
Estreñimiento |
14 |
0 |
|
Sequedad de boca |
12 |
0 |
|
Dispepsia |
12 |
2 |
|
Vómitos |
12 |
2 |
|
Estomatitis |
10 |
0 |
|
Respiratorias, torácicas y mediastínicas |
||
|
Tos |
37 |
0 |
|
Disnea |
14 |
0 |
|
Neumonitis |
10 |
2 |
|
Metabolismo y Nutrición |
||
|
Disminución del apetito |
35 |
2 |
|
General |
||
|
Fatiga |
27 |
2 |
|
Pirexia |
27 |
0 |
|
Malestar |
18 |
2 |
|
Edema |
16 |
2 |
|
Enfermedad tipo influenza |
14 |
0 |
|
Escalofríos |
10 |
0 |
|
Sistema Nervioso |
||
|
Dolor de cabeza |
22 |
0 |
|
Mareo |
20 |
0 |
|
Endocrino |
||
|
Hipotiroidismo |
20 |
0 |
|
Insuficiencia suprarrenal |
18 |
4 |
|
Investigaciones |
||
|
Pérdida de peso |
20 |
0 |
|
Psiquiátrico |
||
|
Insomnio |
18 |
0 |
|
Sangre y Sistema Linfático |
||
|
Anemia |
10 |
4 |
|
Infecciones |
||
|
Influenza |
10 |
2 |
|
Vascular |
||
|
Hipotensión |
10 |
0 |
Las reacciones adversas clínicamente importantes notificadas en <10 % de los pacientes que recibieron OPDIVO con ipilimumab fueron hiperglucemia (8 %), colitis (4 %) y aumento de la creatina fosfocinasa en sangre (2 %).
|
Anomalía de laboratorio |
OPDIVO e ipilimumab (n=47) |
|
|
Todos los grados (%) |
Grados 3-4 (%) |
|
|
Hematología |
||
|
Linfopenia |
53 |
13 |
|
Anemia |
43 |
4.3 |
|
Neutropenia |
43 |
9 |
|
Leucopenia |
40 |
2.1 |
|
Trombocitopenia |
34 |
4.3 |
|
Química |
||
|
Aumento de AST |
66 |
40 |
|
Aumento de ALT |
66 |
21 |
|
Aumento de la bilirrubina |
55 |
11 |
|
Aumento de la lipasa |
51 |
26 |
|
Hiponatremia |
49 |
32 |
|
Hipocalcemia |
47 |
0 |
|
Aumento de la fosfatasa alcalina |
40 |
4.3 |
|
Aumento de la amilasa |
38 |
15 |
|
Hipopotasemia |
26 |
2.1 |
|
Hiperpotasemia |
23 |
4.3 |
|
Aumento de la creatinina |
21 |
0 |
|
Hipomagnesemia |
11 |
0 |
En pacientes que recibieron OPDIVO con ipilimumab, se produjo una exacerbación virológica en 4 de 28 (14 %) pacientes y en 2 de 4 (50 %) pacientes con VHB o VHC activa al inicio, respectivamente. La exacerbación virológica del VHB se definió como un aumento de al menos 1 log en el ADN del VHB para aquellos pacientes con ADN del VHB detectable al inicio. La exacerbación virológica del VHC se definió como un aumento de 1 log en el ARN del VHC desde el inicio.
Cáncer de esófago
Tratamiento adyuvante del cáncer de esófago o de la unión gastroesofágica resecado
Se evaluó la seguridad de OPDIVO en CHECKMATE-577, un ensayo multicéntrico, aleatorizado, controlado con placebo, doble ciego, en 792 pacientes tratados con cáncer de esófago o de la unión gastroesofágica completamente resecado (márgenes negativos) que tenían enfermedad patológica residual después de quimiorradioterapia (QRT) [ver Estudios clínicos (14.12)]. El ensayo excluyó a los pacientes que no recibieron QRT concurrente antes de la cirugía, que tenían enfermedad resecable en estadio IV, enfermedad autoinmune o cualquier afección que requiriera tratamiento sistémico con corticosteroides (>10 mg diarios de prednisona o equivalente) u otros medicamentos inmunosupresores. Los pacientes recibieron 240 mg de OPDIVO o placebo mediante infusión intravenosa durante 30 minutos cada 2 semanas durante 16 semanas, seguidos de 480 mg o placebo mediante infusión intravenosa durante 30 minutos cada 4 semanas a partir de la semana 17. Los pacientes recibieron tratamiento hasta la recurrencia de la enfermedad, la toxicidad inaceptable o hasta una duración total de 1 año. La mediana de duración de la exposición fue de 10.1 meses (rango: <0.1 a 14 meses) en los pacientes tratados con OPDIVO y de 9 meses (rango: <0.1 a 15 meses) en los pacientes tratados con placebo. Entre los pacientes que recibieron OPDIVO, el 61 % estuvo expuesto durante >6 meses y el 54 % estuvo expuesto durante >9 meses.
Se produjeron reacciones adversas graves en el 33 % de los pacientes que recibieron OPDIVO. Una reacción adversa grave notificada en ≥2 % de los pacientes que recibieron OPDIVO fue neumonitis. Se produjo una reacción adversa mortal de infarto de miocardio en un paciente que recibió OPDIVO.
Se interrumpió el tratamiento con OPDIVO en el 12 % de los pacientes y se retrasó en el 28 % de los pacientes debido a una reacción adversa.
Las Tablas 43 y 44 resumen las reacciones adversas y las anomalías de laboratorio, respectivamente, en CHECKMATE-577.
| a Incluye dolor abdominal superior, dolor abdominal inferior y molestias abdominales. b Incluye reflujo gastroesofágico. c Incluye astenia. d Incluye tos productiva. e Incluye disnea de esfuerzo. f Incluye erupción pustulosa, dermatitis, dermatitis acneiforme, dermatitis alérgica, dermatitis ampollosa, erupción exfoliativa, erupción eritematosa, erupción macular, erupción maculopapular, erupción papular, erupción pruriginosa. g Incluye dolor de espalda, dolor óseo, dolor torácico musculoesquelético, molestias musculoesqueléticas, mialgia, mialgia intercostal, dolor de cuello, dolor en las extremidades, dolor espinal. |
||||
|
Reacción adversa |
OPDIVO (n=532) |
Placebo (n=260) |
||
|
Todos los grados (%) |
Grados 3-4 (%) |
Todos los grados (%) |
Grados 3-4 (%) |
|
|
Reacción adversa |
96 |
34 |
93 |
32 |
|
Gastrointestinales |
||||
|
Diarrea |
29 |
0.9 |
29 |
0.8 |
|
Náuseas |
23 |
0.8 |
21 |
0 |
|
Dolor abdominala |
17 |
0.8 |
20 |
1.5 |
|
Vómitos |
15 |
0.6 |
16 |
1.2 |
|
Disfagia |
13 |
0.8 |
17 |
3.5 |
|
Dispepsiab |
12 |
0.2 |
16 |
0.4 |
|
Estreñimiento |
11 |
0 |
12 |
0 |
|
Generales |
||||
|
Fatigac |
34 |
1.3 |
29 |
1.5 |
|
Respiratorias, torácicas y mediastínicas |
||||
|
Tosd |
20 |
0.2 |
21 |
0.4 |
|
Disneae |
12 |
0.8 |
12 |
0.4 |
|
Piel y tejido subcutáneo |
||||
|
Erupción cutáneaf |
21 |
0.9 |
10 |
0.4 |
|
Prurito |
13 |
0.4 |
6 |
0 |
|
Investigaciones |
||||
|
Pérdida de peso |
13 |
0.4 |
9 |
0 |
|
Musculoesqueléticas y del tejido conectivo |
||||
|
Dolor musculoesqueléticog |
21 |
0.6 |
20 |
0.8 |
|
Artralgia |
10 |
0.2 |
8 |
0 |
|
Metabolismo y nutrición |
||||
|
Disminución del apetito |
15 |
0.9 |
10 |
0.8 |
|
Endocrino |
||||
|
Hipotiroidismo |
11 |
0 |
1.5 |
0 |
| a La incidencia de cada prueba se basa en el número de pacientes que tenían disponible tanto el valor inicial como al menos una medición de laboratorio durante el estudio: grupo de OPDIVO (rango: 163 a 526 pacientes) y grupo de placebo (rango: 86 a 256 pacientes). b Incluye aumento de la alanina aminotransferasa, aumento de la aspartato aminotransferasa. |
||||
|
Anormalidad de laboratorio |
OPDIVO (n=532) |
Placebo (n=260) |
||
|
Todos los grados (%) |
Grados 3-4 (%) |
Todos los grados (%) |
Grados 3-4 (%) |
|
|
Química |
||||
|
Aumento de AST |
27 |
2.1 |
22 |
0.8 |
|
Aumento de la fosfatasa alcalina |
25 |
0.8 |
18 |
0.8 |
|
Aumento de la albúmina |
21 |
0.2 |
18 |
0 |
|
Aumento de ALT |
20 |
1.9 |
16 |
1.2 |
|
Aumento de la amilasa |
20 |
3.9 |
13 |
1.3 |
|
Hiponatremia |
19 |
1.7 |
12 |
1.2 |
|
Hiperpotasemia |
17 |
0.8 |
15 |
1.6 |
|
Hipopotasemia |
12 |
1 |
11 |
1.2 |
|
Transaminasas elevadasb |
11 |
1.5 |
6 |
1.2 |
|
Hematología |
||||
|
Linfopenia |
44 |
17 |
35 |
12 |
|
Anemia |
27 |
0.8 |
21 |
0.4 |
|
Neutropenia |
24 |
1.5 |
23 |
0.4 |
Tratamiento de primera línea del CCEE avanzado o metastásico irresecable
La seguridad de OPDIVO en combinación con quimioterapia o en combinación con ipilimumab se evaluó en CHECKMATE-648, un ensayo aleatorizado, controlado con activo, multicéntrico y abierto en pacientes con CCEE avanzado, recurrente o metastásico irresecable no tratado previamente [ver Estudios clínicos (14.12)]. Los pacientes recibieron uno de los siguientes tratamientos:
- •
- OPDIVO 240 mg en los días 1 y 15, 5-FU (fluorouracilo) 800 mg/m2/día por vía intravenosa en los días 1 a 5 (durante 5 días) y cisplatino 80 mg/m2 por vía intravenosa en el día 1 (de un ciclo de 4 semanas).
- •
- OPDIVO 3 mg/kg cada 2 semanas en combinación con ipilimumab 1 mg/kg cada 6 semanas.
- •
- 5-FU (fluorouracilo) 800 mg/m2/día por vía intravenosa en los días 1 a 5 (durante 5 días) y cisplatino 80 mg/m2 por vía intravenosa en el día 1 (de un ciclo de 4 semanas).
Entre los pacientes que recibieron OPDIVO con quimioterapia, la mediana de duración de la exposición fue de 5.7 meses (rango: 0.1 a 30.6 meses). Entre los pacientes que recibieron OPDIVO e ipilimumab, la mediana de duración de la exposición fue de 2.8 meses (rango: 0 a 24 meses).
Se produjeron reacciones adversas graves en el 62% de los pacientes que recibieron OPDIVO en combinación con quimioterapia y en el 69% de los pacientes que recibieron OPDIVO en combinación con ipilimumab. Las reacciones adversas graves más frecuentes notificadas en ≥ 2% de los pacientes que recibieron OPDIVO con quimioterapia fueron neumonía (11%), disfagia (7%), estenosis esofágica (2.9%), lesión renal aguda (2.9%) y pirexia (2.3%). Las reacciones adversas graves más frecuentes notificadas en ≥ 2% de los pacientes que recibieron OPDIVO con ipilimumab fueron neumonía (10%), pirexia (4.3%), neumonitis (4%), neumonía por aspiración (3.7%), disfagia (3.7%), función hepática anormal (2.8%), disminución del apetito (2.8%), insuficiencia suprarrenal (2.5%) y deshidratación (2.5%).
Se produjeron reacciones adversas mortales en 5 (1.6%) pacientes que recibieron OPDIVO en combinación con quimioterapia; estas incluyeron neumonitis, neumatosis intestinal, neumonía y lesión renal aguda y en 5 (1.6%) pacientes que recibieron OPDIVO en combinación con ipilimumab; estas incluyeron neumonitis, enfermedad pulmonar intersticial, embolia pulmonar y síndrome de dificultad respiratoria aguda.
Se interrumpió el tratamiento con OPDIVO y/o quimioterapia en el 39% de los pacientes y se retrasó en el 71% de los pacientes debido a una reacción adversa. Se interrumpió el tratamiento con OPDIVO y/o ipilimumab en el 23% de los pacientes y se retrasó en el 46% de los pacientes debido a una reacción adversa.
Las reacciones adversas más comunes notificadas en ≥ 20% de los pacientes tratados con OPDIVO en combinación con quimioterapia fueron náuseas, disminución del apetito, fatiga, estreñimiento, estomatitis, diarrea y vómitos. Las reacciones adversas más comunes notificadas en ≥ 20% de los pacientes tratados con OPDIVO en combinación con ipilimumab fueron erupción cutánea, fatiga, pirexia, náuseas, diarrea y estreñimiento.
Las Tablas 45 y 46 resumen las reacciones adversas y las anomalías de laboratorio, respectivamente, en CHECKMATE-648.
| La toxicidad se clasificó según los criterios comunes de toxicidad del NCI v4. a Incluye úlcera aftosa, ulceración bucal e inflamación de las mucosas. b Incluye molestias abdominales, dolor abdominal inferior y dolor abdominal superior. c Incluye astenia y malestar general. d Incluye fiebre asociada al tumor. e Incluye inflamación, edema generalizado, edema periférico e inflamación periférica. f Incluye hiperestesia, hipoestesia, neuropatía motora periférica, neuropatía sensitivomotora periférica y neuropatía sensorial periférica. g Incluye dermatitis, dermatitis acneiforme, dermatitis alérgica, dermatitis bullosa, erupción medicamentosa, erupción exfoliativa, erupción eritematosa, erupción folicular, erupción macular, erupción maculopapular, erupción papular y erupción pruriginosa. h Incluye tos productiva. i Incluye neumonía organizada, neumonía bacteriana y neumonía por pseudomonas. j Incluye dolor de espalda, dolor óseo, dolor torácico musculoesquelético, mialgia, dolor de cuello, dolor en las extremidades y dolor espinal. |
||||||
|
Reacción adversa |
OPDIVO con cisplatino y 5‑FU (n=310) |
OPDIVO e ipilimumab (n=322) |
Cisplatino y 5-FU |
|||
|
Todos los grados (%) |
Grados 3-4 (%) |
Todos los grados (%) |
Grados 3-4 (%) |
Todos los grados (%) |
Grados 3-4 (%) |
|
|
Gastrointestinales |
||||||
|
Náuseas |
65 |
4.2 |
22 |
0.6 |
56 |
2.6 |
|
Estreñimiento |
44 |
1.0 |
20 |
0.3 |
43 |
1 |
|
Estomatitisa |
44 |
9 |
11 |
0.6 |
35 |
3 |
|
Diarrea |
29 |
2.9 |
22 |
1.9 |
20 |
2 |
|
Vómitos |
23 |
2.3 |
15 |
1.6 |
19 |
3 |
|
Disfagia |
14 |
7 |
12 |
5 |
12 |
4.9 |
|
Dolor abdominalb |
13 |
1.9 |
10 |
0.9 |
11 |
0.7 |
|
Metabolismo y nutrición |
||||||
|
Disminución del |
51 |
7 |
17 |
4 |
50 |
6 |
|
General |
||||||
|
Fatigac |
47 |
3.5 |
28 |
2.5 |
41 |
4.9 |
|
Pirexiad |
19 |
0.3 |
23 |
0.9 |
12 |
0.3 |
|
Edemae |
16 |
0 |
7 |
0 |
13 |
0 |
|
Sistema nervioso |
||||||
|
Peripheral |
18 |
1.3 |
2.8 |
0 |
13 |
1 |
|
Psiquiátricos |
||||||
|
Insomnio |
16 |
0 |
8 |
0 |
10 |
0.3 |
|
Piel y tejido subcutáneo |
||||||
|
Rashg |
16 |
0.6 |
31 |
3.1 |
7 |
0 |
|
Prurito |
11 |
0 |
17 |
0.9 |
3.6 |
0 |
|
Alopecia |
10 |
0 |
11 |
0 |
||
|
Respiratorias, torácicas y mediastínicas |
||||||
|
Coughh |
16 |
0.3 |
13 |
0.3 |
13 |
0.3 |
|
Infecciones e infestaciones |
||||||
|
Pneumoniai |
13 |
5 |
14 |
8 |
10 |
2.6 |
|
Endocrino |
||||||
|
Hipotiroidismo |
7 |
0 |
14 |
0 |
0.3 |
0 |
|
Investigaciones |
||||||
|
Weight decreased |
12 |
0.6 |
12 |
1.9 |
11 |
1 |
|
Musculoesquelético y tejido conectivo |
||||||
|
Dolor |
11 |
0.3 |
14 |
0.6 |
8 |
0.3 |
| a Cada incidencia de prueba se basa en el número de pacientes que tenían disponible tanto la medición de laboratorio inicial como al menos una medición de laboratorio durante el estudio: grupo de OPDIVO con cisplatino y 5-FU (rango: 60 a 305 pacientes), grupo de OPDIVO e ipilimumab (rango: 59 a 307 pacientes) o grupo de cisplatino y 5-FU (rango: 56 a 283 pacientes). | ||||||
|
Anomalía de laboratorio |
OPDIVO con cisplatino y 5-FU |
OPDIVO e ipilimumab |
Cisplatino y 5-FU |
|||
|
Grados 1-4 (%) |
Grados 3-4 (%) |
Grados 1-4 (%) |
Grados 3-4 (%) |
Grados 1-4 (%) |
Grados 3-4 (%) |
|
|
Hematología |
||||||
|
Anemia |
81 |
21 |
52 |
7 |
66 |
14 |
|
Linfopenia |
67 |
23 |
50 |
13 |
44 |
8 |
|
Neutropenia |
61 |
18 |
13 |
1.3 |
48 |
13 |
|
Leucopenia |
53 |
11 |
39 |
5 |
||
|
Trombocitopenia |
43 |
3.3 |
12 |
1 |
29 |
2.8 |
|
Química |
||||||
|
Hiponatremia |
52 |
15 |
45 |
11 |
40 |
8 |
|
Hipocalcemia |
43 |
3 |
32 |
0 |
23 |
0.7 |
|
Aumento de la creatinina |
41 |
2.3 |
15 |
0.7 |
31 |
0.7 |
|
Hipomagnesemia |
35 |
1.7 |
15 |
0 |
25 |
1.8 |
|
Hiperglucemia |
34 |
0 |
43 |
4.3 |
36 |
0.8 |
|
Hiperpotasemia |
33 |
2.3 |
23 |
1.6 |
24 |
0.7 |
|
Hipopotasemia |
29 |
9 |
19 |
5 |
17 |
6 |
|
Aumento de la fosfatasa alcalina |
26 |
1.3 |
31 |
3.3 |
15 |
0 |
|
Aumento de AST |
23 |
3.3 |
39 |
6 |
11 |
1.4 |
|
Aumento de ALT |
23 |
2.3 |
33 |
6 |
8 |
0.7 |
|
Hipoglucemia |
18 |
0.4 |
15 |
1.2 |
7 |
0 |
|
Hipercalcemia |
11 |
2.6 |
15 |
2 |
8 |
0 |
Carcinoma Escamoso de Esófago (CEE) Avanzado, Recurrente o Metastásico Irresecable Previamente Tratado
Se evaluó la seguridad de OPDIVO en ATTRACTION-3, un ensayo aleatorizado, con control activo, abierto, multicéntrico en 209 pacientes con CEE irresecable avanzado, recurrente o metastásico refractario o intolerante a al menos una quimioterapia basada en fluoropirimidina y platino [ver Estudios Clínicos (14.12)]. El ensayo excluyó a pacientes que eran refractarios o intolerantes a la terapia con taxanos, tenían metástasis cerebrales que eran sintomáticas o requerían tratamiento, tenían enfermedad autoinmune, usaban corticosteroides sistémicos o inmunosupresores, tenían invasión tumoral aparente de órganos adyacentes al tumor esofágico o tenían stents en el esófago o en el tracto respiratorio. Los pacientes recibieron OPDIVO 240 mg por infusión intravenosa durante 30 minutos cada 2 semanas (n=209) o según criterio del investigador: docetaxel 75 mg/m2 por vía intravenosa cada 3 semanas (n=65) o paclitaxel 100 mg/m2 por vía intravenosa una vez a la semana durante 6 semanas seguidas de 1 semana sin tratamiento (n=143). Los pacientes fueron tratados hasta la progresión de la enfermedad o la toxicidad inaceptable. La mediana de duración de la exposición fue de 2.6 meses (rango: 0 a 29.2 meses) en pacientes tratados con OPDIVO y de 2.6 meses (rango: 0 a 21.4 meses) en pacientes tratados con docetaxel o paclitaxel. Entre los pacientes que recibieron OPDIVO, el 26% estuvo expuesto durante >6 meses y el 10% estuvo expuesto durante >1 año.
Se produjeron reacciones adversas graves en el 38% de los pacientes que recibieron OPDIVO. Las reacciones adversas graves notificadas en ≥2% de los pacientes que recibieron OPDIVO fueron neumonía, fístula esofágica, enfermedad pulmonar intersticial y pirexia. Las siguientes reacciones adversas mortales ocurrieron en pacientes que recibieron OPDIVO: enfermedad pulmonar intersticial o neumonitis (1.4%), neumonía (1.0%), shock séptico (0.5%), fístula esofágica (0.5%), hemorragia gastrointestinal (0.5%), embolia pulmonar (0.5%) y muerte súbita (0.5%).
Se interrumpió el tratamiento con OPDIVO en el 13% de los pacientes y se retrasó en el 27% de los pacientes debido a una reacción adversa.
Las Tablas 47 y 48 resumen las reacciones adversas y las anomalías de laboratorio, respectivamente, en ATTRACTION-3.
| La toxicidad se clasificó según NCI CTCAE v4. a Incluye urticaria, erupción por fármacos, eccema, eccema asteatósico, eccema numular, síndrome de eritrodisestesia palmo-plantar, eritema, eritema multiforme, ampolla, exfoliación cutánea, síndrome de Stevens-Johnson, dermatitis, dermatitis descrita como acneiforme, ampollosa o de contacto, y erupción cutánea descrita como maculopapular, generalizada o pustulosa. b Incluye hipofagia y aversión a los alimentos. c Incluye colitis. d Incluye espondilolistesis, periartritis, dolor torácico musculoesquelético, dolor de cuello, artralgia, dolor de espalda, mialgia, dolor en las extremidades, artritis, dolor óseo y periartritis calcárea. e Incluye influenza, enfermedad similar a la influenza, faringitis, nasofaringitis, traqueítis y bronquitis e infección de las vías respiratorias superiores con bronquitis. f Incluye neumonía por aspiración, neumonía bacteriana e infección pulmonar. Dos pacientes (1.0%) murieron de neumonía en el grupo de tratamiento con OPDIVO. Dos pacientes (1.0%) murieron de neumonía en el grupo de tratamiento con quimioterapia; estas muertes ocurrieron solo con paclitaxel. g Incluye tos productiva. h Incluye fiebre asociada al tumor. i Incluye astenia. j Incluye disminución de la hemoglobina y anemia por deficiencia de hierro. k Incluye aumento de la hormona estimulante de la tiroides en sangre. |
||||
|
Reacción Adversa |
OPDIVO (n=209) |
Docetaxel o Paclitaxel (n=208) |
||
|
Todos los Grados (%) |
Grados 3-4 (%) |
Todos los Grados (%) |
Grados 3-4 (%) |
|
|
Piel y Tejido Subcutáneo |
||||
|
Erupción Cutáneaa |
22 |
1.9 |
28 |
1 |
|
Prurito |
12 |
0 |
7 |
0 |
|
Metabolismo y Nutrición |
||||
|
Disminución del apetitob |
21 |
1.9 |
35 |
5 |
|
Gastrointestinales |
||||
|
Diarreac |
18 |
1.9 |
17 |
1.4 |
|
Estreñimiento |
17 |
0 |
19 |
0 |
|
Náuseas |
11 |
0 |
20 |
0.5 |
|
Musculoesquelético y tejido conjuntivo |
||||
|
Dolor |
17 |
0 |
26 |
1.4 |
|
Infecciones |
||||
|
Infección de las vías |
17 |
1 |
14 |
0 |
|
Neumoníaf |
13 |
5 |
19 |
9 |
|
Respiratorias, torácicas y mediastínicas |
||||
|
Tosg |
16 |
0 |
14 |
0.5 |
|
Generales |
||||
|
Pirexiah |
16 |
0.5 |
19 |
0.5 |
|
Fatigai |
12 |
1.4 |
27 |
4.8 |
|
Sangre y sistema linfático |
||||
|
Anemiaj |
13 |
8 |
30 |
13 |
|
Endocrino |
||||
|
Hipotiroidismok |
11 |
0 |
1.4 |
0 |
| a La incidencia de cada prueba se basa en el número de pacientes que tenían disponible tanto la medición de laboratorio inicial como al menos una medición de laboratorio durante el estudio: grupo de OPDIVO (209 pacientes) y grupo de Docetaxel o Paclitaxel (rango: 207 a 208 pacientes). | ||||
|
Anormalidad de laboratorio |
OPDIVO (n=209) |
Docetaxel o Paclitaxel (n=208) |
||
|
Todos los grados (%) |
Grados 3-4 (%) |
Todos los grados (%) |
Grados 3-4 (%) |
|
|
Química |
||||
|
Increased creatinine |
78 |
0.5 |
68 |
0.5 |
|
Hiperglucemia |
52 |
5 |
62 |
5 |
|
Hiponatremia |
42 |
11 |
50 |
12 |
|
Increased AST |
40 |
6 |
30 |
1 |
|
Increased alkaline phosphatase |
33 |
4.8 |
24 |
1.0 |
|
Increased ALT |
31 |
5 |
22 |
1.9 |
|
Hipercalcemia |
22 |
6 |
14 |
2.9 |
|
Hiperpotasemia |
22 |
0.5 |
31 |
1 |
|
Hipoglucemia |
14 |
1.4 |
14 |
0.5 |
|
Hipopotasemia |
11 |
2.9 |
13 |
3.4 |
|
Hematología |
||||
|
Linfopenia |
46 |
19 |
72 |
43 |
|
Anemia |
42 |
9 |
71 |
17 |
|
Leucopenia |
11 |
0.5 |
79 |
45 |
Cáncer gástrico, cáncer de la unión gastroesofágica y adenocarcinoma esofágico
La seguridad de OPDIVO en combinación con quimioterapia se evaluó en CHECKMATE-649, un ensayo aleatorizado, multicéntrico y abierto en pacientes con cáncer gástrico avanzado o metastásico, cáncer de la unión gastroesofágica y adenocarcinoma esofágico no tratados previamente [ver Estudios clínicos (14.13)]. Se excluyeron del ensayo pacientes con receptor 2 del factor de crecimiento epidérmico humano (HER2) positivo conocido o con metástasis del sistema nervioso central (SNC) no tratadas. Los pacientes fueron aleatorizados para recibir OPDIVO en combinación con quimioterapia o quimioterapia. Los pacientes recibieron uno de los siguientes tratamientos:
- •
- OPDIVO 240 mg en combinación con mFOLFOX6 (fluorouracilo, leucovorina y oxaliplatino) cada 2 semanas o mFOLFOX6 cada 2 semanas.
- •
- OPDIVO 360 mg en combinación con CapeOX (capecitabina y oxaliplatino) cada 3 semanas o CapeOX cada 3 semanas.
Los pacientes fueron tratados con OPDIVO en combinación con quimioterapia o quimioterapia hasta la progresión de la enfermedad, toxicidad inaceptable o hasta 2 años. La mediana de duración de la exposición fue de 6.8 meses (rango: 0 a 33.5 meses) en los pacientes tratados con OPDIVO y quimioterapia. Entre los pacientes que recibieron OPDIVO y quimioterapia, el 54% estuvieron expuestos durante >6 meses y el 28% estuvieron expuestos durante >1 año.
Se produjeron reacciones adversas mortales en 16 (2.0%) pacientes que fueron tratados con OPDIVO en combinación con quimioterapia; estas incluyeron neumonitis (4 pacientes), neutropenia febril (2 pacientes), accidente cerebrovascular (2 pacientes), toxicidad gastrointestinal, mucositis intestinal, shock séptico, neumonía, infección, hemorragia gastrointestinal, trombosis de vasos mesentéricos y coagulación intravascular diseminada. Se produjeron reacciones adversas graves en el 52% de los pacientes tratados con OPDIVO en combinación con quimioterapia. Se interrumpió el tratamiento con OPDIVO y/o quimioterapia en el 44% de los pacientes y se suspendió al menos una dosis en el 76% de los pacientes debido a una reacción adversa.
Las reacciones adversas graves más frecuentes notificadas en ≥2% de los pacientes tratados con OPDIVO en combinación con quimioterapia fueron vómitos (3.7%), neumonía (3.6%), anemia (3.6%), pirexia (2.8%), diarrea (2.7%), neutropenia febril (2.6%) y neumonitis (2.4%). Las reacciones adversas más frecuentes notificadas en ≥20% de los pacientes tratados con OPDIVO en combinación con quimioterapia fueron neuropatía periférica, náuseas, fatiga, diarrea, vómitos, disminución del apetito, dolor abdominal, estreñimiento y dolor musculoesquelético.
Las Tablas 49 y 50 resumen las reacciones adversas y las anomalías de laboratorio, respectivamente, en CHECKMATE-649.
| La toxicidad se clasificó según los criterios comunes de toxicidad del NCI v4. a Incluye disestesia, hipoestesia, neuropatía motora periférica, neuropatía sensitivomotora periférica y neuropatía sensorial periférica. b Incluye molestias abdominales, dolor abdominal bajo y dolor abdominal alto. c Incluye úlcera aftosa, ulceración bucal e inflamación de las mucosas. d Incluye astenia. e Incluye fiebre asociada al tumor. f Incluye inflamación, edema generalizado, edema periférico e inflamación periférica. g Incluye disminución de la albúmina en sangre. h Incluye dolor de espalda, dolor óseo, dolor torácico musculoesquelético, molestias musculoesqueléticas, mialgia, dolor de cuello, dolor en las extremidades y dolor de columna. i Incluye dermatitis, dermatitis acneiforme, dermatitis alérgica, dermatitis ampollosa, erupción medicamentosa, erupción exfoliativa, erupción nodular, erupción eritematosa, erupción macular, erupción maculopapular, erupción papular, erupción pruriginosa y erupción vesicular. j Incluye tos productiva. k Incluye nasofaringitis, faringitis y rinitis. |
||||
|
Reacción adversa |
OPDIVO y mFOLFOX6 o CapeOX (n=782) |
mFOLFOX6 o CapeOX |
||
|
Todos los grados (%) |
Grados 3-4 (%) |
Todos los grados (%) |
Grados 3-4 (%) |
|
|
Reacción adversa |
99 |
69 |
98 |
59 |
|
Sistema nervioso |
||||
|
Neuropatía periféricaa |
53 |
7 |
46 |
4.8 |
|
Dolor de cabeza |
11 |
0.8 |
6 |
0.3 |
|
Gastrointestinales |
||||
|
Náuseas |
48 |
3.2 |
44 |
3.7 |
|
Diarrea |
39 |
5 |
34 |
3.7 |
|
Vómitos |
31 |
4.2 |
29 |
4.2 |
|
Dolor abdominalb |
27 |
2.8 |
24 |
2.6 |
|
Estreñimiento |
25 |
0.6 |
21 |
0.4 |
|
Estomatitisc |
17 |
1.8 |
13 |
0.8 |
|
Generales |
||||
|
Fatigad |
44 |
7 |
40 |
5 |
|
Pirexiae |
19 |
1 |
11 |
0.4 |
|
Edemaf |
12 |
0.5 |
8 |
0.1 |
|
Metabolismo y Nutrición |
||||
|
Disminución del apetito |
29 |
3.6 |
26 |
2.5 |
|
Hipoalbuminemiag |
14 |
0.3 |
9 |
0.3 |
|
Investigaciones |
||||
|
Pérdida de peso |
17 |
1.3 |
15 |
0.7 |
|
Aumento de la lipasa |
14 |
7 |
8 |
3.7 |
|
Aumento de la amilasa |
12 |
3.1 |
5 |
0.4 |
|
Musculoesquelético y tejido conjuntivo |
||||
|
Dolor musculoesqueléticoh |
20 |
1.3 |
14 |
2 |
|
Piel y tejido subcutáneo |
||||
|
Erupción cutáneai |
18 |
1.7 |
4.4 |
0.1 |
|
Síndrome de eritrodisestesia |
13 |
1.5 |
12 |
0.8 |
|
Respiratorio, torácico y mediastínico |
||||
|
Tosj |
13 |
0.1 |
9 |
0 |
|
Infecciones e infestaciones |
||||
|
Infección de las vías respiratorias superioresk |
10 |
0.1 |
7 |
0.1 |
|
Anomalía de laboratorio |
OPDIVO y mFOLFOX6 o CapeOX |
mFOLFOX6 o CapeOX |
||
|
Grados 1-4 (%) |
Grados 3-4 (%) |
Grados 1-4 (%) |
Grados 3-4 (%) |
|
|
Hematología |
||||
|
Neutropenia |
73 |
29 |
62 |
23 |
|
Leucopenia |
69 |
12 |
59 |
9 |
|
Trombocitopenia |
68 |
7 |
63 |
4.4 |
|
Anemia |
59 |
14 |
60 |
10 |
|
Linfopenia |
59 |
12 |
49 |
9 |
|
Química |
||||
|
Aumento de AST |
52 |
4.6 |
47 |
1.9 |
|
Hipocalcemia |
42 |
1.6 |
37 |
1 |
|
Hiperglucemia |
41 |
3.9 |
38 |
2.7 |
|
Aumento de ALT |
37 |
3.4 |
30 |
1.9 |
|
Hiponatremia |
34 |
6 |
24 |
5 |
|
Hipopotasemia |
27 |
7 |
24 |
4.8 |
|
Hiperbilirrubinemia |
24 |
2.8 |
21 |
2 |
|
Aumento de la creatinina |
15 |
1 |
9 |
0.5 |
|
Hiperpotasemia |
14 |
1.4 |
11 |
0.7 |
|
Hipoglucemia |
12 |
0.7 |
9 |
0.2 |
|
Hipernatremia |
11 |
0.5 |
7.1 |
0 |
a Cada incidencia de prueba se basa en el número de pacientes que tenían tanto la medición de laboratorio inicial como al menos una en estudio disponible: grupo OPDIVO y mFOLFOX6 o CapeOX (407 a 767 pacientes) o grupo mFOLFOX6 o CapeOX (rango: 405 a 735 pacientes).
6.2 Experiencia posterior a la comercialización
Las siguientes reacciones adversas se han identificado durante el uso posterior a la aprobación de OPDIVO. Debido a que estas reacciones se notifican voluntariamente a partir de una población de tamaño incierto, no siempre es posible estimar con fiabilidad su frecuencia o establecer una relación causal con la exposición al fármaco.
Ojo: síndrome de Vogt-Koyanagi-Harada (VKH)
Complicaciones del tratamiento con OPDIVO después del TCTH alogénico: EICH aguda y crónica grave refractaria al tratamiento
Trastornos de la sangre y del sistema linfático: linfohistiocitosis hemofagocítica (LHH) (incluidos casos mortales), anemia hemolítica autoinmune (incluidos casos mortales)
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de Riesgos
Basado en datos de estudios en animales y su mecanismo de acción [ver Farmacología Clínica (12.1)], OPDIVO puede causar daño fetal cuando se administra a una mujer embarazada. En estudios de reproducción en animales, la administración de nivolumab a monos cynomolgus desde el inicio de la organogénesis hasta el parto resultó en un aumento del aborto y la muerte infantil prematura (ver Datos). Se sabe que la IgG4 humana cruza la barrera placentaria y el nivolumab es una inmunoglobulina G4 (IgG4); por lo tanto, el nivolumab tiene el potencial de transmitirse de la madre al feto en desarrollo. Es probable que los efectos de OPDIVO sean mayores durante el segundo y tercer trimestre del embarazo. No hay datos disponibles sobre el uso de OPDIVO en mujeres embarazadas para evaluar un riesgo asociado al fármaco. Avise a las mujeres embarazadas del riesgo potencial para un feto.
El riesgo de fondo en la población general de EE. UU. de defectos de nacimiento importantes es del 2% al 4% y de aborto espontáneo es del 15% al 20% de los embarazos clínicamente reconocidos.
Datos
Datos de animales
Una función central de la vía PD-1/PD-L1 es preservar el embarazo manteniendo la tolerancia inmunitaria materna al feto. Se ha demostrado que el bloqueo de la señalización de PD-L1 en modelos murinos de embarazo interrumpe la tolerancia al feto y aumenta la pérdida fetal. Los efectos del nivolumab en el desarrollo prenatal y postnatal se evaluaron en monos que recibieron nivolumab dos veces por semana desde el inicio de la organogénesis hasta el parto, a niveles de exposición de entre 9 y 42 veces más altos que los observados en la dosis clínica de 3 mg/kg (basado en AUC). La administración de nivolumab resultó en un aumento no relacionado con la dosis en el aborto espontáneo y un aumento de la muerte neonatal. Basado en su mecanismo de acción, la exposición fetal al nivolumab puede aumentar el riesgo de desarrollar trastornos inmunomediados o alterar la respuesta inmunitaria normal y se han reportado trastornos inmunomediados en ratones knockout de PD-1. En los bebés sobrevivientes (18 de 32 en comparación con 11 de 16 bebés expuestos al vehículo) de monos cynomolgus tratados con nivolumab, no hubo malformaciones aparentes ni efectos en los parámetros neuroconductuales, inmunológicos o de patología clínica durante el período postnatal de 6 meses.
8.2 Lactancia
Resumen de Riesgos
No hay datos sobre la presencia de nivolumab en la leche materna, los efectos en el niño amamantado o los efectos en la producción de leche. Debido al potencial de reacciones adversas graves en el niño amamantado, se recomienda a las mujeres que no amamanten durante el tratamiento y durante 5 meses después de la última dosis de OPDIVO.
8.3 Mujeres y Hombres con Potencial Reproductivo
Prueba de Embarazo
Verifique el estado de embarazo de las mujeres con potencial reproductivo antes de iniciar OPDIVO [ver Uso en Poblaciones Específicas (8.1)].
Anticoncepción
OPDIVO puede causar daño fetal cuando se administra a una mujer embarazada [ver Uso en Poblaciones Específicas (8.1)]. Avise a las mujeres con potencial reproductivo que usen métodos anticonceptivos efectivos durante el tratamiento con OPDIVO y durante 5 meses después de la última dosis.
8.4 Uso Pediátrico
La seguridad y eficacia de OPDIVO se han establecido en pacientes pediátricos de 12 años o más para las siguientes indicaciones: como agente único y en combinación con ipilimumab para el tratamiento del melanoma irresecable o metastásico, como agente único para el tratamiento adyuvante del melanoma completamente resecado en estadio IIB, estadio IIC, estadio III o estadio IV y, como agente único o en combinación con ipilimumab para el tratamiento de mCRC MSI-H o dMMR que ha progresado después del tratamiento con una fluoropirimidina, oxaliplatino e irinotecán. El uso de OPDIVO para estas indicaciones está respaldado por evidencia de estudios adecuados y bien controlados en adultos con melanoma o mCRC MSI-H o dMMR y datos farmacocinéticos adicionales en pacientes pediátricos. La exposición al nivolumab en pacientes pediátricos de 12 años o más es comparable a la de los adultos y el curso del melanoma y el mCRC MSI-H o dMMR es similar en pacientes pediátricos de 12 años o más al de los adultos para permitir la extrapolación de la seguridad y la eficacia [ver Reacciones Adversas (6.1), Farmacología Clínica (12.3), Estudios Clínicos (14.1, 14.10)].
La seguridad y eficacia de OPDIVO no se han establecido en pacientes pediátricos menores de 12 años con melanoma o mCRC MSI-H o dMMR.
La seguridad y eficacia de OPDIVO no se han establecido en pacientes pediátricos con cáncer de pulmón de células no pequeñas, mesotelioma pleural maligno, carcinoma de células renales avanzado, linfoma de Hodgkin clásico, carcinoma de células escamosas de cabeza y cuello, carcinoma urotelial, carcinoma hepatocelular, cáncer de esófago, cáncer gástrico, cáncer gastroesofágico y adenocarcinoma de esófago.
8.5 Uso Geriátrico
Agente Único
De 3569 pacientes con melanoma, NSCLC, carcinoma de células renales, carcinoma urotelial, ESCC y cáncer de esófago o unión gastroesofágica que se asignaron aleatoriamente a OPDIVO en monoterapia en estudios clínicos, el 41% tenía 65 años o más y el 10% tenía 75 años o más. No se observaron diferencias generales en la seguridad o la eficacia entre los pacientes de edad avanzada y los pacientes más jóvenes [ver Estudios clínicos (14.1, 14.2, 14.4, 14.6, 14.9, 14.12)].
En pacientes con cHL, SCC recurrente de cabeza y cuello, o dMMR o CRC metastásico MSI-H (mCRC) que fueron tratados con OPDIVO en monoterapia en estudios clínicos, no se incluyó un número suficiente de pacientes de 65 años o más para determinar si responden de manera diferente a los pacientes más jóvenes [ver Estudios clínicos (14.7, 14.8, 14.10)].
En combinación con ipilimumab
De los 314 pacientes con melanoma que se asignaron aleatoriamente a OPDIVO en combinación con ipilimumab, el 41% tenía 65 años o más y el 11% tenía 75 años o más. No se informaron diferencias generales en la seguridad o la eficacia entre los pacientes de edad avanzada y los pacientes más jóvenes [ver Estudios clínicos (14.1)].
De los 576 pacientes con NSCLC que se asignaron aleatoriamente a OPDIVO en combinación con ipilimumab, el 48% tenía 65 años o más y el 10% tenía 75 años o más. No se informó ninguna diferencia general en la seguridad entre los pacientes mayores y los pacientes más jóvenes; sin embargo, hubo una tasa de interrupción más alta debido a reacciones adversas en pacientes de 75 años o más (29%) en relación con todos los pacientes que recibieron OPDIVO con ipilimumab (18%). De los 396 pacientes en la población de eficacia primaria (PD-L1 ≥1%) asignados aleatoriamente a OPDIVO en combinación con ipilimumab, la razón de riesgo para la supervivencia general fue de 0,70 (IC del 95%: 0,55, 0,89) en los 199 pacientes menores de 65 años en comparación con 0,91 (IC del 95%: 0,72, 1,15) en los 197 pacientes de 65 años o más [ver Estudios clínicos (14.3)].
De los 303 pacientes con mesotelioma pleural maligno que se asignaron aleatoriamente a OPDIVO en combinación con ipilimumab, el 77% tenía 65 años o más y el 26% tenía 75 años o más. No se informó ninguna diferencia general en la seguridad entre los pacientes mayores y los pacientes más jóvenes; sin embargo, hubo tasas más altas de reacciones adversas graves y de interrupción debido a reacciones adversas en pacientes de 75 años o más (68% y 35%, respectivamente) en relación con todos los pacientes que recibieron OPDIVO con ipilimumab (54% y 28%, respectivamente). Para los pacientes de 75 años o más que recibieron quimioterapia, la tasa de reacciones adversas graves fue del 34% y la tasa de interrupción debido a reacciones adversas fue del 26% en relación con el 28% y el 19%, respectivamente, para todos los pacientes. La razón de riesgo para la supervivencia general fue de 0,76 (IC del 95%: 0,52, 1,11) en los 71 pacientes menores de 65 años en comparación con 0,74 (IC del 95%: 0,59, 0,93) en los 232 pacientes de 65 años o más asignados aleatoriamente a OPDIVO en combinación con ipilimumab [ver Estudios clínicos (14.5)].
De los 550 pacientes con carcinoma de células renales que se asignaron aleatoriamente a OPDIVO en combinación con ipilimumab, el 38% tenía 65 años o más y el 8% tenía 75 años o más. No se informó ninguna diferencia general en la seguridad entre los pacientes de edad avanzada y los pacientes más jóvenes. En pacientes de edad avanzada con riesgo intermedio o pobre, no se informó ninguna diferencia general en la eficacia [ver Estudios clínicos (14.6)].
De los 49 pacientes con carcinoma hepatocelular que fueron tratados con OPDIVO en combinación con ipilimumab, el 29% tenía entre 65 y 74 años de edad y el 8% tenía 75 años o más. Los estudios clínicos de OPDIVO en combinación con ipilimumab no incluyeron un número suficiente de pacientes con carcinoma hepatocelular de 65 años o más para determinar si responden de manera diferente a los pacientes más jóvenes [ver Estudios clínicos (14.11)].
De los 325 pacientes con ESCC que se asignaron aleatoriamente a OPDIVO en combinación con ipilimumab, el 43% tenía 65 años o más y el 7% tenía 75 años o más. No se informó ninguna diferencia general en la seguridad entre los pacientes mayores y los pacientes más jóvenes; sin embargo, hubo una tasa de interrupción más alta debido a reacciones adversas en pacientes de 75 años o más (38%) en relación con todos los pacientes que recibieron OPDIVO con ipilimumab (23%). Para los pacientes de 75 años o más que recibieron quimioterapia, la tasa de interrupción debido a reacciones adversas fue del 33% en relación con el 23% para todos los pacientes [ver Estudios clínicos (14.12)].
En combinación con quimioterapia que contiene platino
De los 179 pacientes con NSCLC que se asignaron aleatoriamente a OPDIVO en combinación con quimioterapia de doblete de platino, el 48% tenía 65 años o más y el 6% tenía 75 años o más. No se informaron diferencias generales en la seguridad o la eficacia entre los pacientes mayores y menores de 65 años [ver Estudios clínicos (14.3)].
De los 1110 pacientes con ESCC, GC, GEJC o EAC que se asignaron aleatoriamente a OPDIVO en combinación con quimioterapia que contiene fluoropirimidina y platino), el 42% tenía 65 años o más y el 10% tenía 75 años o más. No se informó ninguna diferencia general en la seguridad entre los pacientes de edad avanzada y los pacientes más jóvenes [ver Estudios clínicos (14.12, 14.13)].
De los 304 pacientes con UC que fueron tratados con OPDIVO en combinación con gemcitabina y quimioterapia de doblete de platino, el 40% tenía 65 años o más y el 11% tenía 75 años o más. No se observaron diferencias generales en la seguridad o la eficacia entre los pacientes de 65 años o más y los pacientes más jóvenes. Los estudios clínicos de OPDIVO con quimioterapia de doblete de platino no incluyeron un número suficiente de pacientes de 75 años o más para determinar si la seguridad y la eficacia difieren en comparación con los pacientes más jóvenes. [ver Estudios clínicos (14.9)].
En combinación con ipilimumab y quimioterapia de doblete de platino
De los 361 pacientes con CPNM que fueron asignados al azar a OPDIVO en combinación con ipilimumab y quimioterapia de doblete de platino, el 51% tenía 65 años o más y el 10% tenía 75 años o más. No se informó ninguna diferencia general en la seguridad entre los pacientes mayores y los pacientes más jóvenes; sin embargo, hubo una tasa de interrupción más alta debido a reacciones adversas en pacientes de 75 años o más (43%) en relación con todos los pacientes que recibieron OPDIVO con ipilimumab y quimioterapia (24%). Para los pacientes de 75 años o más que recibieron solo quimioterapia, la tasa de interrupción debido a reacciones adversas fue del 16% en relación con todos los pacientes que tuvieron una tasa de interrupción del 13%. Con base en un análisis actualizado para la supervivencia general, de los 361 pacientes asignados al azar a OPDIVO en combinación con ipilimumab y quimioterapia de doblete de platino, la razón de riesgo para la supervivencia general fue de 0,61 (IC del 95%: 0,47, 0,80) en los 176 pacientes menores de 65 años en comparación con 0,73 (IC del 95%: 0,56, 0,95) en los 185 pacientes de 65 años o más [ver Estudios clínicos (14.4)].
En combinación con cabozantinib
De los 320 pacientes con carcinoma de células renales que fueron tratados con OPDIVO en combinación con cabozantinib, el 41% tenía 65 años o más y el 9% tenía 75 años o más. No se informó ninguna diferencia general en la seguridad entre los pacientes de edad avanzada y los pacientes más jóvenes [ver Estudios clínicos (14.6)].
11 DESCRIPCIÓN
Nivolumab es un anticuerpo bloqueador del receptor 1 de muerte programada (PD-1). Nivolumab es una inmunoglobulina IgG4 kappa que tiene una masa molecular calculada de 146 kDa. Se expresa en una línea celular recombinante de ovario de hámster chino (CHO).
OPDIVO es un líquido estéril, sin conservantes, no pirogénico, transparente a opalescente, incoloro a amarillo pálido que puede contener partículas ligeras (pocas).
OPDIVO (nivolumab) inyección para uso intravenoso se suministra en viales de dosis única. Cada mL de solución de OPDIVO contiene nivolumab 10 mg, manitol (30 mg), ácido pentetítico (0,008 mg), polisorbato 80 (0,2 mg), cloruro de sodio (2,92 mg), citrato de sodio dihidratado (5,88 mg) y Agua para inyección, USP. Puede contener ácido clorhídrico y/o hidróxido de sodio para ajustar el pH a 6.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
La unión de los ligandos de PD-1, PD-L1 y PD-L2, al receptor PD-1 que se encuentra en las células T, inhibe la proliferación de las células T y la producción de citoquinas. La regulación positiva de los ligandos de PD-1 ocurre en algunos tumores y la señalización a través de esta vía puede contribuir a la inhibición de la vigilancia inmunitaria activa de los tumores por las células T. Nivolumab es un anticuerpo monoclonal humano de inmunoglobulina G4 (IgG4) que se une al receptor PD-1 y bloquea su interacción con PD-L1 y PD-L2, liberando la inhibición mediada por la vía PD-1 de la respuesta inmunitaria, incluida la respuesta inmunitaria antitumoral. En modelos de tumores de ratón singenéticos, el bloqueo de la actividad de PD-1 resultó en una disminución del crecimiento tumoral.
La inhibición mediada por la combinación de nivolumab (anti-PD-1) e ipilimumab (anti-CTLA-4) da como resultado una función mejorada de las células T que es mayor que los efectos de cualquiera de los anticuerpos solos, y da como resultado respuestas antitumorales mejoradas en el melanoma metastásico y el RCC avanzado. En modelos de tumores singenéticos de ratón, el bloqueo dual de PD-1 y CTLA-4 resultó en una mayor actividad antitumoral.
12.2 Farmacodinamia
No hay relaciones significativas clínicamente entre la exposición y la respuesta para la eficacia o la seguridad de la monoterapia con nivolumab en todos los regímenes de dosificación aprobados, independientemente del tipo de cáncer.
12.3 Farmacocinética
La farmacocinética (PK) de nivolumab se evaluó utilizando un enfoque de PK poblacional tanto para OPDIVO en monoterapia como para OPDIVO con ipilimumab. La PK de nivolumab se estudió en pacientes en un rango de dosis de 0,1 mg/kg a 20 mg/kg administrados como una dosis única o como dosis múltiples de OPDIVO como una infusión intravenosa de 60 minutos cada 2 o 3 semanas. La exposición a nivolumab aumenta proporcionalmente a la dosis en el rango de dosis de 0,1 a 10 mg/kg administrados cada 2 semanas. La exposición predicha de nivolumab después de una infusión de 30 minutos es comparable a la observada con una infusión de 60 minutos. Las concentraciones en estado estacionario de nivolumab se alcanzaron a las 12 semanas cuando se administraron a 3 mg/kg cada 2 semanas, y la acumulación sistémica fue de 3,7 veces.
Distribución
El volumen de distribución medio geométrico en estado estacionario (Vss) y el coeficiente de variación (CV%) es de 6,8 L (27,3%).
Eliminación
El aclaramiento (CL) de nivolumab disminuye con el tiempo, con una reducción media máxima de los valores iniciales (CV%) del 24,5% (47,6%), lo que da como resultado un aclaramiento medio geométrico en estado estacionario (CLss) (CV%) de 8,2 mL/h (53,9%) en pacientes con tumores metastásicos; la disminución en CLss no se considera clínicamente relevante. El aclaramiento de nivolumab no disminuye con el tiempo en pacientes con melanoma completamente resecado, ya que el aclaramiento poblacional medio geométrico es un 24% menor en esta población de pacientes en comparación con los pacientes con melanoma metastásico en estado estacionario.
La vida media de eliminación media geométrica (t1/2) es de 25 días (77,5%).
Poblaciones específicas
Los siguientes factores no tuvieron un efecto clínicamente importante en el aclaramiento de nivolumab: edad (29 a 87 años), peso (35 a 160 kg), sexo, raza, LDH basal, expresión de PD-L1, tipo de tumor sólido, tamaño del tumor, insuficiencia renal (eGFR ≥ 15 mL/min/1,73 m2) e insuficiencia hepática leve (bilirrubina total [TB] menor o igual que el ULN y AST mayor que ULN o TB mayor que 1 a 1,5 veces ULN y cualquier AST) o moderada (TB mayor que 1,5 a 3 veces ULN y cualquier AST). Nivolumab no se ha estudiado en pacientes con insuficiencia hepática grave (TB mayor que 3 veces ULN y cualquier AST).
Pacientes pediátricos
Las exposiciones de nivolumab en pacientes pediátricos de 12 años de edad o mayores son comparables a las de los adultos a la dosis recomendada [ver Dosificación y administración (2.2)].
Estudios de interacción medicamentosa
Cuando se administró OPDIVO 3 mg/kg cada 3 semanas en combinación con ipilimumab 1 mg/kg cada 3 semanas, el CL de nivolumab e ipilimumab no cambió en comparación con nivolumab o ipilimumab administrados solos.
Cuando se administró OPDIVO 1 mg/kg cada 3 semanas en combinación con ipilimumab 3 mg/kg cada 3 semanas, el CL de nivolumab aumentó un 29% en comparación con OPDIVO administrado solo y el CL de ipilimumab no cambió en comparación con ipilimumab administrado solo.
Cuando se administró OPDIVO 3 mg/kg cada 2 semanas en combinación con ipilimumab 1 mg/kg cada 6 semanas, el CL de nivolumab no cambió en comparación con OPDIVO administrado solo y el CL de ipilimumab aumentó un 30% en comparación con ipilimumab administrado solo.
Cuando se administró OPDIVO 360 mg cada 3 semanas en combinación con ipilimumab 1 mg/kg cada 6 semanas y quimioterapia, el CL de nivolumab no cambió en comparación con OPDIVO administrado solo y el CL de ipilimumab aumentó un 22% en comparación con ipilimumab administrado solo.
12.6 Inmunogenicidad
La incidencia observada de anticuerpos antidrogas depende en gran medida de la sensibilidad y la especificidad del ensayo. Las diferencias en los métodos de ensayo excluyen comparaciones significativas de la incidencia de anticuerpos antidrogas en los estudios descritos a continuación con la incidencia de anticuerpos antidrogas en otros estudios, incluidos los de OPDIVO o de otros productos de nivolumab.
Las respuestas de anticuerpos antidrogas y anticuerpos neutralizantes se monitorearon durante todo el período de tratamiento donde se evaluó la relación beneficio-riesgo. La incidencia de anticuerpos antidrogas y anticuerpos neutralizantes se presenta en la Tabla 51.
| a Los detalles de cada régimen de tratamiento se describen en la Sección 14 [ver Estudios clínicos (14)]. b La incidencia de NAb se informa entre el subconjunto de pacientes positivos para ADA. c Incluye melanoma irresecable o metastásico, NSCLC metastásico, RCC avanzado, cHL, SCCHN recurrente o metastásico e indicaciones de UC. ADA = anticuerpos anti-nivolumab emergentes durante el tratamiento, NAb = anticuerpos neutralizantes, HCC = carcinoma hepatocelular, RCC = carcinoma de células renales, CRC = cáncer colorrectal, NSCLC = cáncer de pulmón de células no pequeñas. |
|||
|
Régimen de tratamientoa |
Indicación(es) |
ADA |
NAbb |
|---|---|---|---|
|
Treatment Regimena |
Indication(s) |
ADA |
NAbb |
|
OPDIVO como agente único |
Múltiplec |
11% |
7% |
|
OPDIVO con ipilimumab durante 4 dosis seguido de OPDIVO como agente único |
Melanoma |
38% |
12% |
|
HCC |
56% |
41% |
|
|
RCC y CRC |
26% |
3% |
|
|
OPDIVO con ipilimumab |
Mesotelioma pleural maligno |
26% |
2.9% |
|
NSCLC |
37% |
3.9% |
|
|
OPDIVO con ipilimumab y 2 ciclos de quimioterapia de doblete de platino |
NSCLC |
34% |
8% |
Efectos de los anticuerpos antidrogas
La presencia de anticuerpos anti-nivolumab emergentes del tratamiento aumentó el aclaramiento de nivolumab hasta en un 20% después de la administración de nivolumab como monoterapia o en combinación con ipilimumab. Estos cambios farmacocinéticos asociados a anticuerpos antidrogas no se consideraron clínicamente significativos. No se identificó ningún efecto clínicamente significativo de los anticuerpos antidrogas en la incidencia de reacciones relacionadas con la infusión. Los efectos de los anticuerpos antidrogas en la eficacia no se han caracterizado completamente.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogenicidad, Mutagénesis, Deterioro de la Fertilidad
No se han realizado estudios para evaluar el potencial de nivolumab para la carcinogenicidad o genotoxicidad. No se han realizado estudios de fertilidad con nivolumab. En estudios de toxicología de dosis repetidas de 1 mes y 3 meses en monos, no hubo efectos notables en los órganos reproductores masculinos y femeninos; sin embargo, la mayoría de los animales en estos estudios no eran sexualmente maduros.
13.2 Toxicología y/o Farmacología Animal
En modelos animales, la inhibición de la señalización de PD-1 aumentó la gravedad de algunas infecciones y mejoró las respuestas inflamatorias. Los ratones knockout de PD-1 infectados con M. tuberculosis exhiben una supervivencia marcadamente disminuida en comparación con los controles de tipo salvaje, lo que se correlacionó con una mayor proliferación bacteriana y respuestas inflamatorias en estos animales. Los ratones knockout de PD-1 también han mostrado una disminución de la supervivencia después de la infección con el virus de la coriomeningitis linfocítica.
14 ESTUDIOS CLÍNICOS
14.1 Melanoma no resecable o metastásico
Melanoma metastásico previamente tratado
CHECKMATE-037 (NCT01721746) fue un ensayo multicéntrico, abierto, que aleatorizó (2:1) a pacientes con melanoma no resecable o metastásico para recibir OPDIVO 3 mg/kg por vía intravenosa cada 2 semanas o la quimioterapia de elección del investigador, ya sea dacarbazina como agente único 1000 mg/m2 cada 3 semanas o la combinación de carboplatino AUC 6 por vía intravenosa cada 3 semanas y paclitaxel 175 mg/m2 por vía intravenosa cada 3 semanas. Se requirió que los pacientes tuvieran progresión de la enfermedad durante o después del tratamiento con ipilimumab y, si eran positivos para la mutación BRAF V600, un inhibidor de BRAF. El ensayo excluyó a pacientes con enfermedad autoinmune, afecciones médicas que requieren inmunosupresión sistémica, melanoma ocular, metástasis cerebral activa o antecedentes de reacciones adversas relacionadas con ipilimumab de Grado 4 (excepto endocrinopatías) o reacciones adversas relacionadas con ipilimumab de Grado 3 que no se hubieran resuelto o estuvieran inadecuadamente controladas dentro de las 12 semanas posteriores al evento inicial. Las evaluaciones tumorales se realizaron 9 semanas después de la aleatorización, luego cada 6 semanas durante el primer año y, posteriormente, cada 12 semanas.
Se evaluó la eficacia en un análisis intermedio planificado, no comparativo, de un solo grupo, de los primeros 120 pacientes que recibieron OPDIVO en CHECKMATE-037 y en quienes la duración mínima del seguimiento fue de 6 meses. Las principales medidas de resultado de eficacia en esta población fueron la tasa de respuesta global (ORR) confirmada, medida por una revisión central independiente ciega utilizando los Criterios de Evaluación de Respuesta en Tumores Sólidos (RECIST 1.1) y la duración de la respuesta.
Entre los 120 pacientes tratados con OPDIVO, la mediana de edad fue de 58 años (rango: 25 a 88), el 65 % de los pacientes eran hombres, el 98 % eran blancos y el estado funcional ECOG fue de 0 (58 %) o 1 (42 %). Las características de la enfermedad fueron enfermedad M1c (76 %), mutación BRAF V600 positiva (22 %), LDH elevada (56 %), antecedentes de metástasis cerebrales (18 %) y dos o más terapias sistémicas previas para la enfermedad metastásica (68 %).
La ORR fue del 32 % (intervalo de confianza [IC] del 95 %: 23, 41), que consistió en 4 respuestas completas y 34 respuestas parciales en pacientes tratados con OPDIVO. De los 38 pacientes con respuestas, el 87 % tuvo respuestas continuas con duraciones que van desde 2,6+ a 10+ meses, que incluyeron a 13 pacientes con respuestas continuas de 6 meses o más.
Hubo respuestas en pacientes con y sin melanoma con mutación BRAF V600 positiva. Se aleatorizó a un total de 405 pacientes y la mediana de la SG fue de 15,7 meses (IC del 95 %: 12,9, 19,9) en los pacientes tratados con OPDIVO en comparación con los 14,4 meses (IC del 95 %: 11,7, 18,2) (HR 0,95; IC del 95,54 %: 0,73, 1,24) en los pacientes asignados al tratamiento de elección del investigador. La Figura 1 resume los resultados de la SG.
Figura 1: Supervivencia global – CHECKMATE-037*
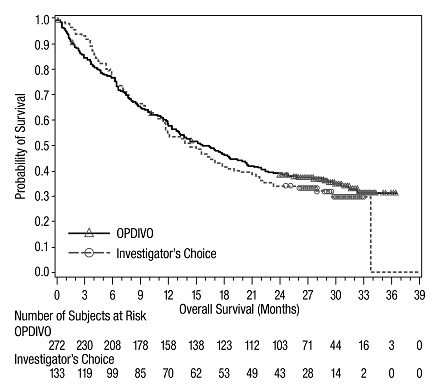
* El análisis primario de SG no se ajustó para tener en cuenta las terapias posteriores, con 54 (40,6 %) pacientes en el grupo de quimioterapia que posteriormente recibieron un tratamiento anti-PD1. La SG puede verse confundida por el abandono, el desequilibrio de las terapias posteriores y las diferencias en los factores basales.
Melanoma metastásico no tratado previamente
CHECKMATE-066
CHECKMATE-066 (NCT01721772) fue un ensayo multicéntrico, doble ciego, aleatorizado (1:1) en 418 pacientes con melanoma irresecable o metastásico con BRAF V600 de tipo salvaje. Los pacientes fueron aleatorizados para recibir OPDIVO 3 mg/kg por infusión intravenosa cada 2 semanas o dacarbazina 1000 mg/m2 por vía intravenosa cada 3 semanas hasta la progresión de la enfermedad o una toxicidad inaceptable. La aleatorización se estratificó según el estado de PD-L1 (≥5 % de tinción de la membrana de células tumorales mediante inmunohistoquímica frente a <5 % o resultado indeterminado) y el estadio M (M0/M1a/M1b frente a M1c). Los criterios de elegibilidad clave incluyeron melanoma cutáneo, mucoso o acral confirmado histológicamente, irresecable o metastásico; sin tratamiento previo para la enfermedad metastásica; finalización del tratamiento adyuvante o neoadyuvante previo al menos 6 semanas antes de la aleatorización; estado funcional ECOG 0 o 1; ausencia de enfermedad autoinmune; y ausencia de metástasis cerebrales o leptomeníngeas activas. El ensayo excluyó a pacientes con melanoma ocular. Las evaluaciones tumorales se realizaron 9 semanas después de la aleatorización, luego cada 6 semanas durante el primer año y luego cada 12 semanas. La principal medida de resultado de eficacia fue la supervivencia global (SG). Las medidas de resultado adicionales incluyeron la supervivencia libre de progresión (SLP) evaluada por el investigador y la ORR según RECIST v1.1.
Las características de la población del ensayo fueron: la mediana de edad fue de 65 años (rango: 18 a 87), el 59 % eran hombres y el 99,5 % eran blancos. Las características de la enfermedad fueron enfermedad en estadio M1c (61 %), melanoma cutáneo (74 %), melanoma mucoso (11 %), nivel elevado de LDH (37 %), expresión de PD-L1 ≥5 % en la membrana de las células tumorales (35 %) y antecedentes de metástasis cerebrales (4 %). Más pacientes en el grupo de OPDIVO tenían un estado funcional ECOG de 0 (71 % frente a 58 %).
CHECKMATE-066 demostró una mejora estadísticamente significativa en la SG para el grupo de OPDIVO en comparación con el grupo de dacarbazina en un análisis intermedio basado en el 47 % del total de eventos planificados para la SG. En el momento del análisis, el 88 % (63/72) de los pacientes tratados con OPDIVO tenían respuestas continuas, lo que incluía a 43 pacientes con respuesta continua de 6 meses o más. Los resultados de eficacia se muestran en la Tabla 52 y la Figura 2.
| a No alcanzado b Basado en un modelo de riesgos proporcionales estratificado. c Basado en la prueba de rango logarítmico estratificado. d El valor p se compara con el alfa asignado de 0,0021 para este análisis intermedio. |
||
|
OPDIVO |
Dacarbazina |
|
|
Supervivencia global |
||
|
Muertes (%) |
50 (24) |
96 (46) |
|
Mediana (meses) (IC del 95%) |
NRa |
10.8 (9.3, 12.1) |
|
Razón de riesgo (IC del 95%)b |
0.42 (0.30, 0.60) |
|
|
Valor pc,d |
<0.0001 |
|
|
Supervivencia sin progresión |
||
|
Progresión de la enfermedad o muerte (%) |
108 (51) |
163 (78) |
|
Mediana (meses) (IC del 95%) |
5.1 (3.5, 10.8) |
2.2 (2.1, 2.4) |
|
Razón de riesgo (IC del 95%)b |
0.43 (0.34, 0.56) |
|
|
Valor pc,d |
<0.0001 |
|
|
Tasa de respuesta global |
34% |
9% |
|
(IC del 95%) |
(28, 41) |
(5, 13) |
|
Tasa de respuesta completa |
4% |
1% |
|
Tasa de respuesta parcial |
30% |
8% |
Figura 2: Supervivencia general – CHECKMATE-066
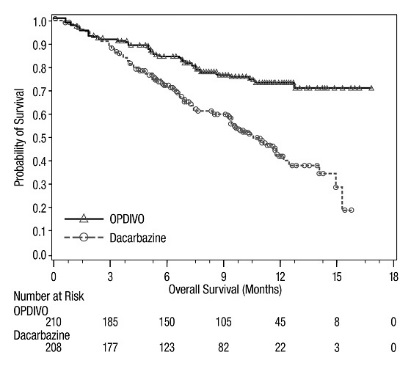
CHECKMATE-067
CHECKMATE-067 (NCT01844505) fue un ensayo multicéntrico, aleatorizado (1:1:1), doble ciego en 945 pacientes con melanoma no resecable o metastásico previamente no tratado a uno de los siguientes brazos: OPDIVO e ipilimumab, OPDIVO o ipilimumab. Los pacientes debían haber completado el tratamiento adyuvante o neoadyuvante al menos 6 semanas antes de la aleatorización y no haber recibido tratamiento previo con anticuerpos anti-CTLA-4 ni evidencia de metástasis cerebral activa, melanoma ocular, enfermedad autoinmune o afecciones médicas que requirieran inmunosupresión sistémica.
Los pacientes fueron aleatorizados para recibir:
- •
- OPDIVO 1 mg/kg con ipilimumab 3 mg/kg por vía intravenosa cada 3 semanas durante 4 dosis, seguido de OPDIVO como agente único a una dosis de 3 mg/kg por infusión intravenosa cada 2 semanas (brazo de OPDIVO e ipilimumab),
- •
- OPDIVO 3 mg/kg por infusión intravenosa cada 2 semanas (brazo de OPDIVO), o
- •
- Ipilimumab 3 mg/kg por vía intravenosa cada 3 semanas durante 4 dosis, seguido de placebo cada 2 semanas (brazo de ipilimumab).
La aleatorización se estratificó por expresión de PD-L1 (≥5% vs. <5% de expresión de la membrana de células tumorales) según lo determinado por un ensayo clínico, estado de mutación BRAF V600 y estadio M según el sistema de estadificación AJCC (M0, M1a, M1b vs. M1c). Las evaluaciones tumorales se realizaron 12 semanas después de la aleatorización, luego cada 6 semanas durante el primer año y cada 12 semanas a partir de entonces. Las principales medidas de resultado de eficacia fueron la PFS evaluada por el investigador según RECIST v1.1 y la OS. Las medidas de resultado de eficacia adicionales fueron la ORR confirmada y la duración de la respuesta.
Las características de la población del ensayo fueron: mediana de edad 61 años (rango: 18 a 90); 65% hombres; 97% blancos; puntuación de rendimiento ECOG 0 (73%) o 1 (27%). Las características de la enfermedad fueron: enfermedad en estadio IV del AJCC (93%); enfermedad M1c (58%); LDH elevada (36%); antecedentes de metástasis cerebrales (4%); melanoma con mutación BRAF V600 positivo (32%); expresión de PD-L1 ≥5% en la membrana de las células tumorales según lo determinado por el ensayo clínico (46%); y terapia adyuvante previa (22%).
CHECKMATE-067 demostró mejoras estadísticamente significativas en la OS y la PFS para los pacientes aleatorizados a cualquiera de los brazos que contenían OPDIVO en comparación con el brazo de ipilimumab. El ensayo no se diseñó para evaluar si la adición de ipilimumab a OPDIVO mejora la PFS o la OS en comparación con OPDIVO como agente único. Los resultados de eficacia se muestran en la Tabla 53 y la Figura 3.
| a Los resultados de la OS se basan en el análisis final de la OS con 28 meses de seguimiento mínimo; los resultados de la PFS (criterio de valoración principal) y la ORR (criterio de valoración secundario) se basaron en el análisis primario con 9 meses de seguimiento mínimo. b Basado en un modelo de riesgos proporcionales estratificado. c Basado en la prueba de rango logarítmico estratificado. d Si el máximo de los dos valores p de la OS es inferior a 0,04 (un nivel de significación asignado por el procedimiento de Hochberg), entonces ambos valores p se consideran significativos. e El valor p se compara con 0,005 del alfa asignado para las comparaciones finales del tratamiento de la PFS. f Basado en la prueba de Cochran-Mantel-Haenszel estratificada. + Observación censurada |
|||
|
OPDIVO e Ipilimumab |
OPDIVO |
Ipilimumab |
|
|
Supervivencia generala |
|||
|
Muertes (%) |
128 (41) |
142 (45) |
197 (63) |
|
Hazard ratiob (vs. ipilimumab) |
0.55 |
0.63 |
|
|
p-valuec, d |
<0.0001 |
<0.0001 |
|
|
Supervivencia libre de progresióna |
|||
|
Progresión de la enfermedad o muerte |
151 (48%) |
174 (55%) |
234 (74%) |
|
Mediana (meses) |
11.5 |
6.9 |
2.9 |
|
Hazard ratiob (frente a ipilimumab) |
0.42 |
0.57 |
|
|
Valor pc, e |
<0.0001 |
<0.0001 |
|
|
Tasa de respuesta global confirmadaa |
50% |
40% |
14% |
|
(IC del 95%) |
(44, 55) |
(34, 46) |
(10, 18) |
|
Valor pf |
<0.0001 |
<0.0001 |
|
|
Respuesta completa |
8.9% |
8.5% |
1.9% |
|
Respuesta parcial |
41% |
31% |
12% |
|
Duración de la respuesta |
|||
|
Proporción ≥6 meses de duración |
76% |
74% |
63% |
|
Rango (meses) |
1.2+ a 15.8+ |
1.3+ a 14.6+ |
1.0+ a 13.8+ |
Figura 3: Supervivencia global – CHECKMATE-067
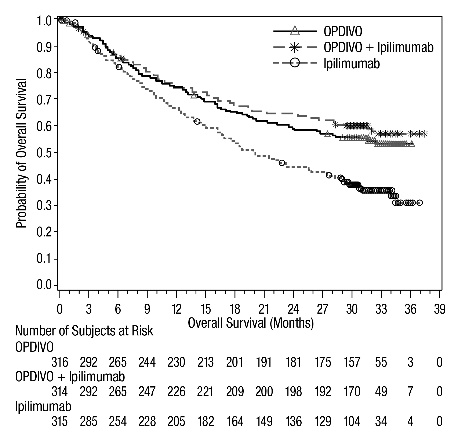
Con un seguimiento mínimo de 48 meses, la mediana de la supervivencia global no se alcanzó (IC del 95%: 38,2, NR) en el brazo de OPDIVO e ipilimumab. La mediana de la supervivencia global fue de 36,9 meses (IC del 95%: 28,3, NR) en el brazo de OPDIVO y de 19,9 meses (IC del 95%: 16,9, 24,6) en el brazo de ipilimumab.
Con un seguimiento mínimo de 28 meses, la mediana de la PFS fue de 11,7 meses (IC del 95%: 8,9, 21,9) en el brazo de OPDIVO e ipilimumab, de 6,9 meses (IC del 95%: 4,3, 9,5) en el brazo de OPDIVO y de 2,9 meses (IC del 95%: 2,8, 3,2) en el brazo de ipilimumab. Con un seguimiento mínimo de 28 meses, la proporción de respuestas que duraron ≥ 24 meses fue del 55% en el brazo de OPDIVO e ipilimumab, del 56% en el brazo de OPDIVO y del 39% en el brazo de ipilimumab.
14.2 Tratamiento adyuvante del melanoma
CHECKMATE-76K
CHECKMATE-76K (NCT04099251) fue un ensayo aleatorizado, doble ciego en 790 pacientes con melanoma en estadio IIB/C completamente resecado. Los pacientes se aleatorizaron (2:1) para recibir OPDIVO 480 mg o placebo por infusión intravenosa cada 4 semanas durante hasta 1 año o hasta la recurrencia de la enfermedad o toxicidad inaceptable. La inscripción requirió la resección completa del melanoma primario con márgenes negativos y un nodo linfático centinela negativo dentro de las 12 semanas previas a la aleatorización, y un estado de rendimiento ECOG de 0 o 1. El ensayo excluyó a pacientes con melanoma ocular/uveal o mucoso, enfermedad autoinmune, cualquier afección que requiriera tratamiento sistémico con corticosteroides (≥ 10 mg de prednisona diaria o equivalente) u otros medicamentos inmunosupresores, así como pacientes con tratamiento previo para el melanoma excepto cirugía. La aleatorización se estratificó por la edición 8ª del sistema de estadificación AJCC (T3b vs. T4a vs. T4b). El principal resultado de eficacia fue la supervivencia libre de recurrencia (RFS) definida como el tiempo entre la fecha de aleatorización y la fecha de la primera recurrencia (local, regional o a distancia), un nuevo melanoma primario o muerte, por cualquier causa, lo que ocurriera primero y según lo evaluado por el investigador. Las evaluaciones tumorales se llevaron a cabo cada 26 semanas durante los años 1-3 y cada 52 semanas a partir de entonces hasta el año 5.
Las características de la población del ensayo fueron: mediana de edad 62 años (rango: 19 a 92), el 61% eran hombres, el 98% eran blancos, el 0,4% negros o afroamericanos, el 0,1% asiáticos y el 1,1% raza desconocida, el 2,2% hispanos o latinos, el 58% no hispanos o latinos, el 40% etnia desconocida y el 94% tenía un estado de rendimiento ECOG de 0. El 61% tenían melanoma en estadio IIB y el 39% en estadio IIC.
CHECKMATE-76K demostró una mejora estadísticamente significativa en la RFS de los pacientes aleatorizados al brazo de OPDIVO en comparación con el brazo de placebo. Los resultados de eficacia se muestran en la Tabla 54 y la Figura 4.
| a No alcanzado. b Basado en estimaciones de Kaplan-Meier. c El ratio de riesgo es de OPDIVO sobre placebo basado en un modelo de riesgo proporcional de Cox estratificado. d Basado en una prueba de log-rank estratificada bilateral. Límite de significación estadística: valor p < 0,033. |
||
|
OPDIVO |
Placebo |
|
|
Supervivencia libre de recurrencia |
||
|
Número de eventos, n (%) |
66 (13%) |
69 (26%) |
|
Mediana (meses) b |
NRa |
NRa |
|
Ratio de riesgoc |
0,42 |
|
Figura 4: Supervivencia libre de recurrencia – CHECKMATE-76K
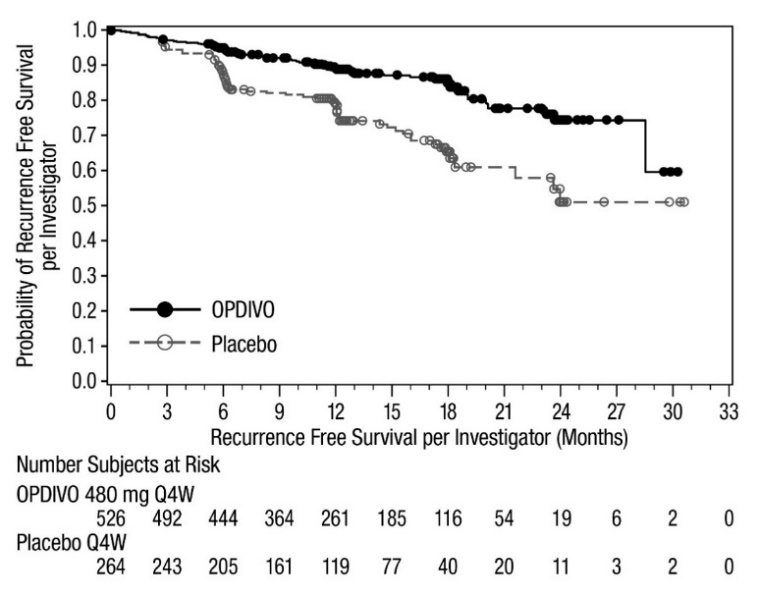
CHECKMATE-238
CHECKMATE-238 (NCT02388906) fue un ensayo aleatorizado, doble ciego en 906 pacientes con melanoma en estadio IIIB/C o estadio IV completamente resecado. Los pacientes fueron aleatorizados (1:1) para recibir OPDIVO 3 mg/kg por infusión intravenosa cada 2 semanas o ipilimumab 10 mg/kg por vía intravenosa cada 3 semanas durante 4 dosis y luego cada 12 semanas comenzando en la semana 24 por hasta 1 año. La inscripción requirió la resección completa del melanoma con márgenes negativos para la enfermedad dentro de las 12 semanas previas a la aleatorización. El ensayo excluyó a pacientes con antecedentes de melanoma ocular/uveal, enfermedad autoinmune y cualquier condición que requiriera tratamiento sistémico con corticosteroides (≥10 mg diarios de prednisona o equivalente) u otros medicamentos inmunosupresores, así como pacientes con terapia previa para melanoma, excepto cirugía, radioterapia adyuvante después de la resección neuroquirúrgica de lesiones del sistema nervioso central e interferón adyuvante previo completado ≥6 meses antes de la aleatorización. La aleatorización se estratificó por estado de PD-L1 (positivo [basado en un nivel del 5%] frente a negativo/indeterminado) y estadio AJCC (estadio IIIB/C frente a estadio IV M1a-M1b frente a estadio IV M1c). La medida de resultado de eficacia principal fue la supervivencia libre de recurrencia (SLR) definida como el tiempo entre la fecha de aleatorización y la fecha de la primera recurrencia (metástasis local, regional o distante), nuevo melanoma primario o muerte, por cualquier causa, lo que ocurriera primero y según la evaluación del investigador. Los pacientes se sometieron a imágenes para detectar recurrencia tumoral cada 12 semanas durante los primeros 2 años y luego cada 6 meses a partir de entonces.
Las características de la población del ensayo fueron: mediana de edad de 55 años (rango: 18 a 86), 58% hombres, 95% blancos y 90% con un estado funcional ECOG de 0. Las características de la enfermedad fueron estadio AJCC IIIB (34%), estadio IIIC (47%), estadio IV (19%), M1a-b (14%), mutación BRAF V600 positiva (42%), BRAF de tipo salvaje (45%), LDH elevada (8%), expresión de la membrana celular tumoral PD-L1 ≥5% determinada por ensayo clínico (34%), ganglios linfáticos macroscópicos (48%) y ulceración tumoral (32%).
CHECKMATE-238 demostró una mejora estadísticamente significativa en la SLR para los pacientes aleatorizados al grupo de OPDIVO en comparación con el grupo de ipilimumab 10 mg/kg. Los resultados de eficacia se muestran en la Tabla 55 y la Figura 5.
| a No alcanzado. b Basado en un modelo de riesgos proporcionales estratificado. c Basado en una prueba de rango logarítmico estratificado. d El valor p se compara con 0.0244 del alfa asignado para este análisis. e En el momento del análisis final de la SG, se observaron menos eventos de supervivencia general de lo que se había previsto originalmente (aproximadamente 302). |
||
|
OPDIVO |
Ipilimumab 10 mg/kg |
|
|
Supervivencia libre de recurrencia |
||
|
Número de eventos, n (%) |
154 (34%) |
206 (45%) |
|
Mediana (meses) |
NAa |
NAa |
|
Razón de riesgob |
0.65 |
|
|
Supervivencia global |
||
|
Número de eventos, n (%)e |
100 (22%) |
111 (25%) |
|
Mediana (meses) |
NAa |
NAa |
|
Razón de riesgob |
0.87 |
|
Figura 5: Supervivencia libre de recurrencia – CHECKMATE-238
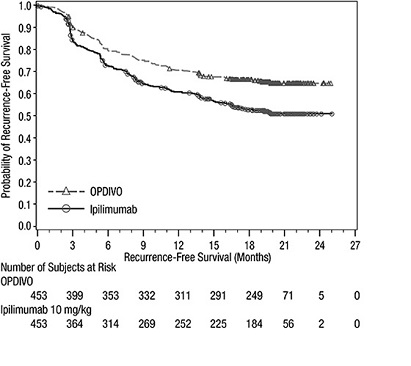
14.3 Tratamiento neoadyuvante del cáncer de pulmón de células no pequeñas resecable (tumor ≥ 4 cm o ganglio positivo)
CHECKMATE-816 (NCT02998528) fue un ensayo aleatorio, de etiqueta abierta en pacientes con NSCLC resecable. El ensayo incluyó a pacientes con NSCLC resecable, histológicamente confirmado en estadio IB (≥ 4 cm), II o IIIA (según los criterios de estadificación de la 7ª edición del Comité Conjunto Americano sobre Cáncer/Unión Internacional contra el Cáncer [AJCC/UICC]), estado funcional ECOG de 0 o 1 y enfermedad medible (según la versión 1.1 del RECIST). Se excluyeron del estudio pacientes con NSCLC irresecable o metastásico, mutaciones conocidas de EGFR o translocaciones de ALK, neuropatía periférica de grado 2 o mayor, enfermedad autoinmune activa o condiciones médicas que requiriesen inmunosupresión sistémica.
Los pacientes fueron aleatorizados para recibir bien:
- •
- OPDIVO 360 mg administrado por vía intravenosa durante 30 minutos y quimioterapia con platino doble administrada por vía intravenosa cada 3 semanas durante un máximo de 3 ciclos, o
- •
- Quimioterapia con platino doble administrada cada 3 semanas durante un máximo de 3 ciclos.
La quimioterapia con platino doble consistió en paclitaxel 175 mg/m2 o 200 mg/m2 y carboplatino AUC 5 o AUC 6 (cualquier histología); pemetrexed 500 mg/m2 y cisplatino 75 mg/m2 (histología no escamosa); o gemcitabina 1000 mg/m2 o 1250 mg/m2 y cisplatino 75 mg/m2 (histología escamosa). En el brazo de la quimioterapia con platino doble, dos opciones de régimen de tratamiento adicionales incluyeron vinorelbine 25 mg/m2 o 30 mg/m2 y cisplatino 75 mg/m2; o docetaxel 60 mg/m2 o 75 mg/m2 y cisplatino 75 mg/m2 (cualquier histología).
Los factores de estratificación para la aleatorización fueron el nivel de expresióntumoral PD-L1 (≥ 1% frente a < 1% o no cuantificable), el estadio de la enfermedad (IB/II frente a IIIA) y el sexo (masculino frente a femenino). Las evaluaciones del tumor se realizaron en el punto basal, en un plazo de 14 días de la cirugía, cada 12 semanas después de la cirugía durante 2 años, luego cada 6 meses durante 3 años y cada año durante 5 años hasta la recurrencia o progresión de la enfermedad. Las principales medidas de resultado de eficacia fueron la supervivencia libre de eventos (EFS) basada en la evaluación central independiente ciega (BICR) y la respuesta patológica completa (pCR) evaluada mediante revisión patológica independiente ciega (BIPR). Entre las medidas de resultado de eficacia adicionales se incluyó la OS.
Un total de 358 pacientes fueron aleatorizados a recibir bien OPDIVO en combinación con quimioterapia con platino doble (n = 179) o quimioterapia con platino doble (n = 179). La edad mediana fue de 65 años (rango: 34 a 84) con 51% de los pacientes ≥ 65 años y 7% ≥ 75 años, 50% eran asiáticos, 47% eran blancos, 2% eran negros y 71% eran hombres. El estado funcional ECOG basal fue 0 (67%) o 1 (33%); 50% tenían tumores con expresiónde PD-L1 ≥ 1%; 35% tenían estadio IB/II y 64% tenían estadio IIIA; 51% tenían tumores con histología escamosa y 49% tenían tumores con histología no escamosa; y 89% eran fumadores antiguos/actuales.
El 83% de los pacientes en el brazo de OPDIVO en combinación con quimioterapia con platino doble tuvieron cirugía definitiva en comparación con el 75% de los pacientes en el brazo de quimioterapia con platino doble.
El estudio demostró mejoras estadísticamente significativas en la EFS y la pCR. Los resultados de eficacia se presentan en la Tabla 56 y la Figura 6.
| El seguimiento mínimo para la EFS fue de 21 meses. a Estimación de Kaplan-Meier. b Basado en un modelo de riesgo proporcional de Cox estratificado. c Basado en una prueba de log-rank estratificada. Límite para la significancia estadística: valor p < 0.0262. d Basado en el método de Clopper y Pearson. e Diferencia ajustada por estratos basada en el método de ponderación de Cochran-Mantel-Haenszel. f De la prueba CMH estratificada. |
||
|
OPDIVO y quimioterapia con platino doble |
Quimioterapia con platino doble |
|
|
Supervivencia libre de eventos (EFS) por BICR |
||
|
Eventos (%) |
64 (35.8) |
87 (48.6) |
|
Mediana (meses)a (95% IC) |
31.6 (30.2, NR) |
20.8 (14.0, 26.7) |
|
Ratio de riesgosb (95% IC) |
0.63 (0.45, 0.87) |
|
|
Valor p de la prueba log-rank estratificadac |
0.0052 |
|
|
Respuesta patológica completa (pCR) por BIPR |
||
|
Número de pacientes con pCR |
43 |
4 |