ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
Estos aspectos destacados no incluyen toda la información necesaria para usar OFEV de forma segura y eficaz. Consulte la información completa de prescripción de OFEV.
OFEV® (cápsulas de nintedanib), para uso oral Primera aprobación en EE. UU.: 2014
INDICACIONES Y USO
OFEV es un inhibidor de quinasa indicado en adultos para:
Tratamiento de la fibrosis pulmonar idiopática (FPI) (1.1)
Tratamiento de enfermedades pulmonares intersticiales fibróticas crónicas (ILDs) con un fenotipo progresivo (1.2)
Reducción de la velocidad de disminución de la función pulmonar en pacientes con enfermedad pulmonar intersticial asociada a esclerosis sistémica (SSc-ILD) (1.3)
DOSIFICACIÓN Y ADMINISTRACIÓN
Dosificación recomendada: 150 mg por vía oral dos veces al día, aproximadamente con 12 horas de diferencia, tomados con alimentos. (2.2)
Dosificación recomendada en pacientes con deterioro hepático leve (Child Pugh A): 100 mg por vía oral dos veces al día, aproximadamente con 12 horas de diferencia, tomados con alimentos. (2.3, 8.6)
Considere una reducción temporal de la dosis a 100 mg, interrupción del tratamiento o suspensión para el manejo de reacciones adversas. (2.4, 5.2, 5.3, 6)
Antes del inicio del tratamiento, realice pruebas de función hepática en todos los pacientes y una prueba de embarazo en mujeres en edad fértil. (2.1, 5.2, 5.4)
Deterioro hepático: OFEV no se recomienda para uso en pacientes con deterioro hepático moderado o grave. En pacientes con deterioro hepático leve (Child Pugh A), la dosis recomendada es de 100 mg dos veces al día, aproximadamente con 12 horas de diferencia, tomados con alimentos. Considere la interrupción del tratamiento o la suspensión para el manejo de reacciones adversas en estos pacientes. (2.3, 2.4, 5.1, 8.6, 12.3)
Elevación de enzimas hepáticas y lesión hepática inducida por fármacos: Las elevaciones de ALT, AST y bilirrubina han ocurrido con OFEV, incluyendo casos de lesión hepática inducida por fármacos. En el período postcomercialización, se han reportado casos no graves y graves de lesión hepática inducida por fármacos, incluyendo lesiones hepáticas graves con resultado fatal. La mayoría de los eventos hepáticos ocurren dentro de los primeros tres meses de tratamiento. Los aumentos de enzimas hepáticas y bilirrubina fueron reversibles con la modificación de la dosis o la interrupción en la mayoría de los casos. Monitorice ALT, AST y bilirrubina antes del inicio del tratamiento, a intervalos regulares durante los primeros tres meses de tratamiento y periódicamente después o según lo indique clínicamente. Pueden ser necesarias reducciones temporales de la dosis o suspensiones. (2.1, 2.4, 5.2)
Trastornos gastrointestinales: Diarrea, náuseas y vómitos han ocurrido con OFEV. Trate a los pacientes a los primeros signos con hidratación adecuada y medicamentos antidiarreicos (por ejemplo, loperamida) o antieméticos. Suspenda OFEV si la diarrea, náuseas o vómitos graves persisten a pesar del tratamiento sintomático. (5.3)
Toxicidad embriofetal: Puede causar daño fetal. Advierte a las mujeres en edad fértil del riesgo potencial para el feto y que usen un método anticonceptivo altamente eficaz. Advierte a las mujeres que toman anticonceptivos hormonales orales y que experimentan vómitos, diarrea u otras condiciones en las que la absorción del fármaco puede reducirse que usen un método anticonceptivo altamente eficaz alternativo. (5.4, 8.1, 8.3)
Se han reportado eventos tromboembólicos arteriales. Tenga precaución al tratar a pacientes con mayor riesgo cardiovascular, incluyendo enfermedad coronaria conocida. (5.5)
Se han reportado eventos hemorrágicos. Use OFEV en pacientes con riesgo conocido de hemorragia solo si el beneficio esperado supera el riesgo potencial. (5.6)
Se ha reportado perforación gastrointestinal. Use OFEV con precaución al tratar a pacientes con cirugía abdominal reciente, antecedentes de enfermedad diverticular o que reciben corticosteroides o AINEs concomitantes. Suspenda OFEV en pacientes que desarrollen perforación gastrointestinal. Use OFEV solo en pacientes con riesgo conocido de perforación gastrointestinal si el beneficio esperado supera el riesgo potencial. (5.7)
Se ha reportado proteinuria en el rango nefrótico. Considere la interrupción del tratamiento en pacientes que desarrollen proteinuria nueva o empeoramiento. (5.8)
REACCIONES ADVERSAS
Las reacciones adversas más comunes (≥5%) son: diarrea, náuseas, dolor abdominal, vómitos, elevación de enzimas hepáticas, disminución del apetito, dolor de cabeza, pérdida de peso y hipertensión. (6.1)
Para reportar REACCIONES ADVERSAS SOSPECHOSAS, comuníquese con Boehringer Ingelheim Pharmaceuticals, Inc. al (800) 542 – 6257 o con la FDA al 1 – 800 – FDA – 1088 o www.fda.gov/medwatch.
INTERACCIONES MEDICAMENTOSAS
La administración conjunta de inhibidores de la P-gp y del CYP3A4 puede aumentar la exposición a nintedanib. Monitoree a los pacientes de cerca para determinar la tolerabilidad de OFEV. (7.1)
USO EN POBLACIONES ESPECÍFICAS
Lactancia: No se recomienda la lactancia materna. (8.2)
Insuficiencia renal: No se ha estudiado la seguridad y eficacia de OFEV en pacientes con insuficiencia renal grave y enfermedad renal en etapa terminal. (8.7, 12.3)
Fumadores: Se ha observado una menor exposición en fumadores, lo que puede alterar el perfil de eficacia de OFEV. (8.8)
Consulte 17 para obtener INFORMACIÓN DE ASESORAMIENTO AL PACIENTE y el etiquetado del paciente aprobado por la FDA.
Las secciones o subsecciones omitidas de la información completa de prescripción no están listadas.
1 INDICACIONES Y USO
1.1 Fibrosis Pulmonar Idiopática
OFEV está indicado para el tratamiento de adultos con fibrosis pulmonar idiopática (FPI).
1.2 Enfermedades Pulmonares Intersticiales Fibrosantes Crónicas con un Fenotipo Progresivo
OFEV está indicado para el tratamiento de adultos con enfermedades pulmonares intersticiales fibrosantes crónicas (EPIFC) con un fenotipo progresivo [ver Estudios Clínicos (14.2)].
1.3 Enfermedad Pulmonar Intersticial Asociada a Esclerodermia
OFEV está indicado para ralentizar la tasa de disminución de la función pulmonar en pacientes adultos con enfermedad pulmonar intersticial asociada a esclerodermia (EPIES).
2 DOSIS Y ADMINISTRACIÓN
2.1 Pruebas antes de la administración de OFEV
Realice pruebas de función hepática en todos los pacientes y una prueba de embarazo en mujeres en edad fértil antes de iniciar el tratamiento con OFEV [ver Advertencias y precauciones (5.2, 5.4)].
2.2 Dosis recomendada
La dosis recomendada de OFEV es de 150 mg por vía oral dos veces al día, administrada con un intervalo aproximado de 12 horas.
Información de administración
Las cápsulas de OFEV deben tomarse con alimentos [ver Farmacología clínica (12.3)] y tragarse enteras con líquido. Las cápsulas de OFEV no deben masticarse debido a su sabor amargo.
Las cápsulas de OFEV no deben abrirse ni triturarse. Si se produce contacto con el contenido de la cápsula, lávese las manos inmediata y minuciosamente. Se desconoce el efecto de masticar o triturar la cápsula sobre la farmacocinética de nintedanib.
Información sobre la dosis olvidada
Si se olvida una dosis de OFEV, la siguiente dosis debe tomarse a la hora programada siguiente. Aconseje al paciente que no compense la dosis olvidada. No exceda la dosis máxima diaria recomendada de 300 mg.
2.3 Dosis recomendada para pacientes con insuficiencia hepática
Insuficiencia hepática leve
En pacientes con insuficiencia hepática leve (Child Pugh A), la dosis recomendada de OFEV es de 100 mg por vía oral dos veces al día con un intervalo aproximado de 12 horas, tomada con alimentos [ver Uso en poblaciones específicas (8.6)].
2.4 Modificación de la dosis debido a reacciones adversas
Además del tratamiento sintomático, si corresponde, el manejo de las reacciones adversas de OFEV puede requerir una reducción de la dosis o una interrupción temporal hasta que la reacción adversa específica se resuelva a niveles que permitan continuar con la terapia. El tratamiento con OFEV puede reanudarse con la dosis completa (150 mg dos veces al día) o con la dosis reducida (100 mg dos veces al día), que posteriormente puede aumentarse a la dosis completa. Si un paciente no tolera 100 mg dos veces al día, suspenda el tratamiento con OFEV [ver Advertencias y precauciones (5.2, 5.3, 5.5, 5.7) y Reacciones adversas (6.1)].
Elevación de las enzimas hepáticas
Pueden ser necesarias modificaciones o interrupciones de la dosis para las elevaciones de las enzimas hepáticas. Realice pruebas de función hepática (aspartato aminotransferasa (AST), alanina aminotransferasa (ALT) y bilirrubina) antes de iniciar el tratamiento con OFEV, a intervalos regulares durante los primeros tres meses de tratamiento y, posteriormente, de forma periódica o según esté clínicamente indicado. Mida las pruebas hepáticas con prontitud en pacientes que reporten síntomas que puedan indicar daño hepático, incluyendo fatiga, anorexia, molestias en la parte superior derecha del abdomen, orina oscura o ictericia. Suspenda OFEV en pacientes con AST o ALT mayor a 3 veces el límite superior de lo normal (LSN) con signos o síntomas de daño hepático y para elevaciones de AST o ALT mayores a 5 veces el límite superior de lo normal. Para AST o ALT mayor a 3 veces y menor a 5 veces el LSN sin signos de daño hepático, interrumpa el tratamiento o reduzca OFEV a 100 mg dos veces al día. Una vez que las enzimas hepáticas hayan regresado a los valores basales, el tratamiento con OFEV puede reintroducirse a una dosis reducida (100 mg dos veces al día), que posteriormente puede aumentarse a la dosis completa (150 mg dos veces al día) [ver Advertencias y precauciones (5.2) y Reacciones adversas (6.1)].
En pacientes con insuficiencia hepática leve (Child Pugh A), considere la interrupción o la suspensión del tratamiento para el manejo de las reacciones adversas.
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
Cápsulas:
150 mg, marrón, opaco, oblongo, cápsulas blandas impresas en negro con el símbolo de la compañía Boehringer Ingelheim y “150”.
100 mg, melocotón, opaco, oblongo, cápsulas blandas impresas en negro con el símbolo de la compañía Boehringer Ingelheim y “100”.
5.2 Elevación de las enzimas hepáticas y lesión hepática inducida por fármacos
Se han observado casos de lesión hepática inducida por fármacos (DILI) con el tratamiento con OFEV. En los ensayos clínicos y en el período posterior a la comercialización, se notificaron casos no graves y graves de DILI. Se han notificado casos de lesión hepática grave con desenlace mortal en el período posterior a la comercialización. La mayoría de los eventos hepáticos ocurren dentro de los primeros tres meses de tratamiento. En los ensayos clínicos, la administración de OFEV se asoció con elevaciones de las enzimas hepáticas (ALT, AST, ALKP, GGT) y bilirrubina. Los aumentos de las enzimas hepáticas y la bilirrubina fueron reversibles con la modificación o interrupción de la dosis en la mayoría de los casos. En los estudios de IPF (Estudio 1, Estudio 2 y Estudio 3), la mayoría (94%) de los pacientes con elevaciones de ALT y/o AST tuvieron elevaciones menores de 5 veces el LSN y la mayoría (95%) de los pacientes con elevaciones de bilirrubina tuvieron elevaciones menores de 2 veces el LSN. En el estudio de EPIc con fenotipo progresivo (Estudio 5), la mayoría (95%) de los pacientes con elevaciones de ALT y/o AST tuvieron elevaciones menores de 5 veces el LSN y la mayoría (94%) de los pacientes con elevaciones de bilirrubina tuvieron elevaciones menores de 2 veces el LSN. En el estudio de EPIc-esclerodermia sistémica (Estudio 4), se observó una ALT y/o AST máxima mayor o igual a 3 veces el LSN en el 4.9% de los pacientes en el grupo de OFEV y en el 0.7% de los pacientes en el grupo placebo [ver Uso en poblaciones específicas (8.6) y Farmacología clínica (12.3)]. Los pacientes con bajo peso corporal (menos de 65 kg), asiáticos y mujeres pueden tener un mayor riesgo de elevaciones en las enzimas hepáticas. La exposición a nintedanib aumentó con la edad del paciente, lo que también puede resultar en un mayor riesgo de aumento de las enzimas hepáticas [ver Farmacología clínica (12.3)].
Realice pruebas de función hepática (ALT, AST y bilirrubina) antes de iniciar el tratamiento con OFEV, a intervalos regulares durante los primeros tres meses de tratamiento y periódicamente a partir de entonces o según esté clínicamente indicado. Mida las pruebas hepáticas con prontitud en pacientes que reporten síntomas que puedan indicar lesión hepática, incluyendo fatiga, anorexia, molestias en la parte superior derecha del abdomen, orina oscura o ictericia. Pueden ser necesarias modificaciones de la dosis o interrupciones para las elevaciones de las enzimas hepáticas [ver Posología y administración (2.1, 2.4)].
5.3 Trastornos gastrointestinales
Diarrea
En los ensayos clínicos, la diarrea fue el evento gastrointestinal más frecuente reportado. En la mayoría de los pacientes, el evento fue de intensidad leve a moderada y ocurrió dentro de los primeros 3 meses de tratamiento. En los estudios de IPF (Estudio 1, Estudio 2 y Estudio 3), se reportó diarrea en el 62% versus 18% de los pacientes tratados con OFEV y placebo, respectivamente [ver Reacciones adversas (6.1)]. La diarrea condujo a una reducción permanente de la dosis en el 11% de los pacientes tratados con OFEV en comparación con el 0 de los pacientes tratados con placebo. La diarrea condujo a la interrupción de OFEV en el 5% de los pacientes en comparación con menos del 1% de los pacientes tratados con placebo. En el estudio de EPIc con fenotipo progresivo (Estudio 5), se reportó diarrea en el 67% versus 24% de los pacientes tratados con OFEV y placebo, respectivamente [ver Reacciones adversas (6.1)]. La diarrea condujo a una reducción permanente de la dosis en el 16% de los pacientes tratados con OFEV en comparación con menos del 1% de los pacientes tratados con placebo. La diarrea condujo a la interrupción de OFEV en el 6% de los pacientes en comparación con menos del 1% de los pacientes tratados con placebo. En el estudio de EPIc-esclerodermia sistémica (Estudio 4), se reportó diarrea en el 76% versus 32% de los pacientes tratados con OFEV y placebo, respectivamente [ver Reacciones adversas (6.1)]. La diarrea condujo a una reducción permanente de la dosis en el 22% de los pacientes tratados con OFEV en comparación con el 1% de los pacientes tratados con placebo. La diarrea condujo a la interrupción de OFEV en el 7% de los pacientes en comparación con el 0.3% de los pacientes tratados con placebo.
Las modificaciones de la dosis o las interrupciones del tratamiento pueden ser necesarias en pacientes con reacciones adversas de diarrea. Trate la diarrea a las primeras señales con una hidratación adecuada y medicamentos antidiarreicos (p. ej., loperamida), y considere la reducción de la dosis o la interrupción del tratamiento si la diarrea continúa [ver Posología y administración (2.4)]. El tratamiento con OFEV puede reanudarse con la dosis completa (150 mg dos veces al día) o con la dosis reducida (100 mg dos veces al día), que posteriormente puede aumentarse a la dosis completa. Si la diarrea grave persiste a pesar del tratamiento sintomático, suspenda el tratamiento con OFEV.
Náuseas y vómitos
En los estudios de FPI (Estudio 1, Estudio 2 y Estudio 3), se reportaron náuseas en el 24% versus 7% y vómitos en el 12% versus 3% de los pacientes tratados con OFEV y placebo, respectivamente. En el estudio de EPIc con fenotipo progresivo (Estudio 5), se reportaron náuseas en el 29% versus 9% y vómitos en el 18% versus 5% de los pacientes tratados con OFEV y placebo, respectivamente. En el estudio de EPI-esclerodermia (Estudio 4), se reportaron náuseas en el 32% versus 14% y vómitos en el 25% versus 10% de los pacientes tratados con OFEV y placebo, respectivamente [ver Reacciones Adversas (6.1)]. En la mayoría de los pacientes, estos eventos fueron de intensidad leve a moderada. En los estudios de FPI (Estudio 1, Estudio 2 y Estudio 3), las náuseas llevaron a la interrupción de OFEV en el 2% de los pacientes y los vómitos llevaron a la interrupción de OFEV en el 1% de los pacientes. En el estudio de EPIc con fenotipo progresivo (Estudio 5), las náuseas llevaron a la interrupción de OFEV en menos del 1% de los pacientes y los vómitos llevaron a la interrupción de OFEV en el 1% de los pacientes. En el estudio de EPI-esclerodermia (Estudio 4), las náuseas llevaron a la interrupción de OFEV en el 2% de los pacientes y los vómitos llevaron a la interrupción de OFEV en el 1% de los pacientes.
Para las náuseas o vómitos que persisten a pesar del cuidado de apoyo apropiado, incluyendo la terapia antiemética, puede ser necesaria la reducción de la dosis o la interrupción del tratamiento [ver Dosis y Administración (2.4)]. El tratamiento con OFEV puede reanudarse con la dosis completa (150 mg dos veces al día), o con la dosis reducida (100 mg dos veces al día), que posteriormente puede aumentarse a la dosis completa. Si las náuseas o vómitos graves no se resuelven, suspenda el tratamiento con OFEV.
5.4 Toxicidad Embrio-Fetal
Basado en los hallazgos de estudios en animales y su mecanismo de acción, OFEV puede causar daño fetal cuando se administra a una mujer embarazada. Nintedanib causó muertes embrio-fetales y anomalías estructurales en ratas y conejos cuando se administró durante la organogénesis a menos de (ratas) y aproximadamente 5 veces (conejos) la dosis máxima recomendada en humanos (MRHD) en adultos. Advierta a las mujeres embarazadas sobre el riesgo potencial para el feto. Aconseje a las mujeres en edad fértil que eviten quedar embarazadas mientras reciben tratamiento con OFEV y que utilicen métodos anticonceptivos altamente efectivos al inicio, durante el tratamiento y al menos 3 meses después de la última dosis de OFEV. Nintedanib no cambia la exposición a los anticonceptivos orales que contienen etinilestradiol y levonorgestrel en pacientes con EPI-esclerodermia. Sin embargo, la eficacia de los anticonceptivos hormonales orales puede verse comprometida por vómitos y/o diarrea u otras afecciones en las que la absorción del fármaco puede verse reducida. Aconseje a las mujeres que toman anticonceptivos hormonales orales que experimenten estas afecciones que utilicen métodos anticonceptivos alternativos altamente efectivos. Verifique el estado de embarazo antes del tratamiento con OFEV y durante el tratamiento según corresponda [ver Uso en Poblaciones Específicas (8.1, 8.3) y Farmacología Clínica (12.1, 12.3)].
5.5 Eventos Tromboembólicos Arteriales
Se han reportado eventos tromboembólicos arteriales en pacientes que toman OFEV. En los estudios de FPI (Estudio 1, Estudio 2 y Estudio 3), se reportaron eventos tromboembólicos arteriales en el 2.5% de los pacientes tratados con OFEV y en menos del 1% de los pacientes tratados con placebo. El infarto de miocardio fue la reacción adversa más común entre los eventos tromboembólicos arteriales, ocurriendo en el 1.5% de los pacientes tratados con OFEV en comparación con menos del 1% de los pacientes tratados con placebo. En el estudio de EPIc con fenotipo progresivo (Estudio 5), se reportaron eventos tromboembólicos arteriales en menos del 1% de los pacientes en ambos brazos de tratamiento. Se observó infarto de miocardio en menos del 1% de los pacientes en ambos brazos de tratamiento. En el estudio de EPI-esclerodermia (Estudio 4), se reportaron eventos tromboembólicos arteriales en el 0.7% de los pacientes en ambos brazos de tratamiento. Hubo 0 casos de infarto de miocardio en pacientes tratados con OFEV en comparación con el 0.7% de los pacientes tratados con placebo.
Tenga precaución al tratar a pacientes con mayor riesgo cardiovascular, incluyendo enfermedad arterial coronaria conocida. Considere la interrupción del tratamiento en pacientes que desarrollen signos o síntomas de isquemia miocárdica aguda.
5.6 Riesgo de Sangrado
Basado en el mecanismo de acción (inhibición de VEGFR), OFEV puede aumentar el riesgo de sangrado. En los estudios de FPI (Estudio 1, Estudio 2 y Estudio 3), se reportaron eventos de sangrado en el 10% de los pacientes tratados con OFEV y en el 7% de los pacientes tratados con placebo. En el estudio de EPIc con fenotipo progresivo (Estudio 5), se reportaron eventos de sangrado en el 11% de los pacientes tratados con OFEV y en el 13% de los pacientes tratados con placebo. En el estudio de EPI-esclerodermia (Estudio 4), se reportaron eventos de sangrado en el 11% de los pacientes tratados con OFEV y en el 8% de los pacientes tratados con placebo. En ensayos clínicos, la epistaxis fue el evento de sangrado más frecuente reportado.
En el período posterior a la comercialización se han observado eventos de sangrado no graves y graves, algunos de los cuales fueron fatales.
Use OFEV en pacientes con riesgo conocido de sangrado solo si el beneficio anticipado supera el riesgo potencial.
5.7 Perforación Gastrointestinal
Basado en el mecanismo de acción, OFEV puede aumentar el riesgo de perforación gastrointestinal. En los estudios de FPI (Estudio 1, Estudio 2 y Estudio 3), se reportó perforación gastrointestinal en menos del 1% de los pacientes tratados con OFEV, en comparación con 0 casos en los pacientes tratados con placebo. En el estudio de EPIc con fenotipo progresivo (Estudio 5), no se reportó perforación gastrointestinal en ningún paciente en ningún brazo de tratamiento. En el estudio de EPI-esclerodermia (Estudio 4), no se reportaron casos de perforación gastrointestinal en pacientes tratados con OFEV o en pacientes tratados con placebo.
En el período posterior a la comercialización, se han reportado casos de perforaciones gastrointestinales, algunos de los cuales fueron fatales.
Tenga precaución al tratar a pacientes que se han sometido recientemente a cirugía abdominal, antecedentes de enfermedad diverticular o que reciben corticosteroides o AINE concomitantes. Suspenda el tratamiento con OFEV en pacientes que desarrollen perforación gastrointestinal. Solo use OFEV en pacientes con riesgo conocido de perforación gastrointestinal si el beneficio anticipado supera el riesgo potencial.
5.8 Proteinuria en Rango Nefrótico
Se han reportado casos de proteinuria en rango nefrótico en el período posterior a la comercialización. Los hallazgos histológicos, cuando estaban disponibles, fueron consistentes con microangiopatía glomerular con o sin trombos renales. Se ha observado una mejoría en la proteinuria después de la interrupción de OFEV; sin embargo, en algunos casos, la proteinuria residual persistió. Considere la interrupción del tratamiento en pacientes que desarrollan proteinuria nueva o que empeora.
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas clínicamente significativas se analizan con mayor detalle en otras secciones de la etiqueta:
Debido a que los ensayos clínicos se llevan a cabo en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no se pueden comparar directamente con las tasas en los ensayos clínicos de otro fármaco y es posible que no reflejen las tasas observadas en la práctica.
La seguridad de OFEV se evaluó en más de 1000 pacientes con FPI, 332 pacientes con ILD fibrosante crónica con un fenotipo progresivo y más de 280 pacientes con ILD-ESc. Más de 200 pacientes con FPI estuvieron expuestos a OFEV durante más de 2 años en ensayos clínicos.
Fibrosis pulmonar idiopática
OFEV se estudió en tres ensayos aleatorizados, doble ciego, controlados con placebo de 52 semanas. En los ensayos de fase 2 (Estudio 1) y fase 3 (Estudio 2 y Estudio 3), 723 pacientes con FPI recibieron OFEV 150 mg dos veces al día y 508 pacientes recibieron placebo. La duración media de la exposición fue de 10 meses para los pacientes tratados con OFEV y 11 meses para los pacientes tratados con placebo. Los sujetos tenían edades comprendidas entre los 42 y los 89 años (edad media de 67 años). La mayoría de los pacientes eran hombres (79%) y caucásicos (60%).
Las reacciones adversas graves más frecuentes notificadas en pacientes tratados con OFEV, más que con placebo, fueron bronquitis (1,2% frente a 0,8%) e infarto de miocardio (1,5% frente a 0,4%). Los eventos adversos más comunes que llevaron a la muerte en pacientes tratados con OFEV, más que con placebo, fueron neumonía (0,7% frente a 0,6%), neoplasia pulmonar maligna (0,3% frente a 0%) e infarto de miocardio (0,3% frente a 0,2%). En la categoría predefinida de eventos cardiovasculares adversos mayores (MACE) que incluyen IM, se notificaron eventos fatales en el 0,6% de los pacientes tratados con OFEV y el 1,8% de los pacientes tratados con placebo.
Se notificaron reacciones adversas que llevaron a reducciones permanentes de la dosis en el 16% de los pacientes tratados con OFEV y el 1% de los pacientes tratados con placebo. La reacción adversa más frecuente que llevó a una reducción permanente de la dosis en los pacientes tratados con OFEV fue la diarrea (11%).
Se notificaron reacciones adversas que llevaron a la interrupción del tratamiento en el 21% de los pacientes tratados con OFEV y el 15% de los pacientes tratados con placebo. Las reacciones adversas más frecuentes que llevaron a la interrupción del tratamiento en los pacientes tratados con OFEV fueron la diarrea (5%), las náuseas (2%) y la disminución del apetito (2%).
Las reacciones adversas más comunes con una incidencia mayor o igual al 5% y más frecuentes en el grupo de tratamiento con OFEV que con placebo se enumeran en la Tabla 1.
Tabla 1 Reacciones adversas que se producen en ≥5% de los pacientes tratados con OFEV con fibrosis pulmonar idiopática y con mayor frecuencia que el placebo en el Estudio 1, el Estudio 2 y el Estudio 3
Reacción adversa
OFEV, 150 mg n=723
Placebo n=508
a Incluye dolor abdominal, dolor abdominal superior, dolor abdominal inferior, dolor gastrointestinal y sensibilidad abdominal. b Incluye aumento de la gamma-glutamiltransferasa, aumento de las enzimas hepáticas, aumento de la alanina aminotransferasa, aumento de la aspartato aminotransferasa, función hepática anormal, prueba de función hepática anormal, aumento de las transaminasas, aumento de la fosfatasa alcalina en sangre, alanina aminotransferasa anormal, aspartato aminotransferasa anormal y gamma-glutamiltransferasa anormal. c Incluye hipertensión, aumento de la presión arterial, crisis hipertensiva y cardiomiopatía hipertensiva.
Trastornos gastrointestinales
Diarrea
62%
18%
Náuseas
24%
7%
Dolor abdominala
15%
6%
Vómitos
12%
3%
Trastornos hepatobiliares
Elevación de las enzimas hepáticasb
14%
3%
Trastornos del metabolismo y la nutrición
Disminución del apetito
11%
5%
Trastornos del sistema nervioso
Dolor de cabeza
8%
5%
Investigaciones
Pérdida de peso
10%
3%
Trastornos vasculares
Hipertensiónc
5%
4%
Además, se notificó hipotiroidismo en pacientes tratados con OFEV, más que con placebo (1.1% frente a 0.6%). También se notificó alopecia en más pacientes tratados con OFEV que con placebo (0.8% frente a 0.4%).
Combinación con Pirfenidona
Se investigó el tratamiento concomitante con nintedanib y pirfenidona en un ensayo exploratorio abierto, aleatorizado (1:1) de nintedanib 150 mg dos veces al día con pirfenidona adicional (titulada a 801 mg tres veces al día) en comparación con nintedanib 150 mg dos veces al día solo en 105 pacientes aleatorizados durante 12 semanas. El punto final primario fue el porcentaje de pacientes con eventos adversos gastrointestinales desde el inicio hasta la Semana 12. Los eventos adversos gastrointestinales estaban en línea con el perfil de seguridad establecido de cada componente y se experimentaron en 37 (70%) pacientes tratados con pirfenidona agregada a nintedanib frente a 27 (53%) pacientes tratados solo con nintedanib.
Diarrea, náuseas, vómitos y dolor abdominal (incluye dolor abdominal superior, molestia abdominal y dolor abdominal) fueron los eventos adversos más frecuentes notificados en 20 (38%) frente a 16 (31%), en 22 (42%) frente a 6 (12%), en 15 (28%) frente a 6 (12%) y en 15 (28%) frente a 7 (14%) pacientes tratados con pirfenidona agregada a nintedanib frente a nintedanib solo, respectivamente. Más sujetos notificaron elevaciones de AST o ALT (mayores o iguales a 3 veces el límite superior de lo normal) cuando se usó pirfenidona en combinación con nintedanib (n = 3 (6%)) en comparación con nintedanib solo (n = 0) [véase Advertencias y Precauciones (5.2, 5.3)].
Enfermedades pulmonares intersticiales fibrosantes crónicas con fenotipo progresivo
OFEV se estudió en un ensayo clínico de fase 3, doble ciego, controlado con placebo (Estudio 5) en el que 663 pacientes con ILDs fibrosantes crónicas con fenotipo progresivo fueron aleatorizados para recibir OFEV 150 mg dos veces al día (n = 332) o placebo (n = 331) durante al menos 52 semanas. A las 52 semanas, la duración mediana de exposición fue de 12 meses para los pacientes en ambos brazos de tratamiento. Los sujetos tenían edades que oscilaban entre los 27 y los 87 años (edad mediana de 67 años). La mayoría de los pacientes eran caucásicos (74%) o asiáticos (25%). La mayoría de los pacientes eran hombres (54%).
El evento adverso grave más frecuente notificado en pacientes tratados con OFEV, más que con placebo, fue la neumonía (4% frente a 3%). Se notificaron eventos adversos que condujeron a la muerte en el 3% de los pacientes tratados con OFEV y en el 5% de los pacientes tratados con placebo. No se identificó un patrón en los eventos adversos que condujeron a la muerte.
Se notificaron reacciones adversas que llevaron a reducciones de dosis permanentes en el 33% de los pacientes tratados con OFEV y en el 4% de los pacientes tratados con placebo. La reacción adversa más frecuente que condujo a una reducción de dosis permanente en los pacientes tratados con OFEV fue la diarrea (16%).
Se notificaron reacciones adversas que llevaron a la discontinuación en el 20% de los pacientes tratados con OFEV y en el 10% de los pacientes tratados con placebo. La reacción adversa más frecuente que condujo a la discontinuación en los pacientes tratados con OFEV fue la diarrea (6%).
El perfil de seguridad en pacientes con ILDs fibrosantes crónicas con fenotipo progresivo tratados con OFEV fue consistente con el observado en pacientes con FPI. Además, los siguientes eventos adversos se notificaron en OFEV más que en placebo en ILD fibrosante crónica progresiva: nasofaringitis (13% frente a 12%), infección del tracto respiratorio superior (7% frente a 6%), infección del tracto urinario (6% frente a 4%), fatiga (10% frente a 6%) y dolor de espalda (6% frente a 5%).
Enfermedad pulmonar intersticial asociada a esclerosis sistémica
OFEV se estudió en un ensayo clínico de fase 3, aleatorizado, doble ciego, controlado con placebo (Estudio 4) en el que 576 pacientes con SSc-ILD recibieron OFEV 150 mg dos veces al día (n = 288) o placebo (n = 288). Los pacientes debían recibir tratamiento durante al menos 52 semanas; los pacientes individuales fueron tratados hasta 100 semanas. La duración mediana de exposición fue de 15 meses para los pacientes tratados con OFEV y de 16 meses para los pacientes tratados con placebo. Los sujetos tenían edades que oscilaban entre los 20 y los 79 años (edad mediana de 55 años). La mayoría de los pacientes eran mujeres (75%). Los pacientes eran principalmente caucásicos (67%), asiáticos (25%) o negros (6%). En el punto de partida, el 49% de los pacientes estaban en tratamiento estable con micofenolato.
Los eventos adversos graves más frecuentes notificados en pacientes tratados con OFEV, más que con placebo, fueron la enfermedad pulmonar intersticial (2.4% nintedanib frente a 1.7% placebo) y la neumonía (2.8% nintedanib frente a 0.3% placebo). Dentro de las 52 semanas, 5 pacientes tratados con OFEV (1.7%) y 4 pacientes tratados con placebo (1.4%) murieron. No hubo un patrón entre los eventos adversos que condujeron a la muerte en ninguno de los brazos de tratamiento.
Se notificaron reacciones adversas que llevaron a reducciones de dosis permanentes en el 34% de los pacientes tratados con OFEV y en el 4% de los pacientes tratados con placebo. La reacción adversa más frecuente que condujo a una reducción de dosis permanente en los pacientes tratados con OFEV fue la diarrea (22%).
Se notificaron reacciones adversas que llevaron a la discontinuación en el 16% de los pacientes tratados con OFEV y en el 9% de los pacientes tratados con placebo. Las reacciones adversas más frecuentes que condujeron a la discontinuación en los pacientes tratados con OFEV fueron la diarrea (7%), náuseas (2%), vómitos (1%), dolor abdominal (1%) y enfermedad pulmonar intersticial (1%).
El perfil de seguridad en pacientes tratados con OFEV con o sin micofenolato en el punto de partida fue comparable.
Las reacciones adversas más comunes con una incidencia de mayor o igual a 5% en pacientes tratados con OFEV y más comúnmente que en placebo se enumeran en Tabla 2.
Tabla 2 Reacciones Adversas que Ocurren en ≥5% de los Pacientes Tratados con OFEV con Enfermedad Pulmonar Intersticial Asociada a Esclerosis Sistémica y Más Comúnmente que con Placebo en el Estudio 4
Reacción Adversa
OFEV, 150 mg n = 288
Placebo n = 288
a Incluye dolor abdominal, dolor abdominal superior, dolor abdominal inferior y dolor esofágico.
c Incluye hipertensión, presión arterial aumentada y crisis hipertensiva.
Diarrea
76%
32%
Náuseas
32%
14%
Vómitos
25%
10%
Úlcera cutánea
18%
17%
Dolor abdominala
18%
11%
Elevación de las enzimas hepáticasb
13%
3%
Pérdida de peso
12%
4%
Fatiga
11%
7%
Disminución del apetito
9%
4%
Dolor de cabeza
9%
8%
Fiebre
6%
5%
Dolor de espalda
6%
4%
Mareos
6%
4%
Hipertensiónc
5%
2%
Además, se informó alopecia en pacientes tratados con OFEV, más que con placebo (1,4% frente a 1,0%).
6.2 Experiencia postcomercialización
Las siguientes reacciones adversas se han identificado durante el uso posterior a la aprobación de OFEV. Debido a que estas reacciones se informan voluntariamente de una población de tamaño incierto, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal con la exposición al fármaco.
Trastornos de la sangre y del sistema linfático: Trombocitopenia Trastornos gastrointestinales: Pancreatitis Trastornos hepatobiliares: Lesión hepática inducida por fármacos Trastornos del sistema nervioso: Síndrome de encefalopatía posterior reversible Trastornos renales y urinarios: Proteinuria Trastornos de la piel y del tejido subcutáneo: Prurito, erupción cutánea Trastornos vasculares: Eventos hemorrágicos no graves y graves, algunos de los cuales fueron fatales
7 INTERACCIONES MEDICAMENTOSAS
7.1 Inhibidores e inductores de la P-glicoproteína (P-gp) y CYP3A4
Nintedanib es un sustrato de la P-gp y, en menor medida, del CYP3A4 [see Clinical Pharmacology (12.3)]. La administración conjunta con dosis orales de ketoconazol, un inhibidor de la P-gp y del CYP3A4, aumentó la exposición a nintedanib en un 60%. El uso concomitante de inhibidores de la P-gp y del CYP3A4 (p. ej., eritromicina) con OFEV puede aumentar la exposición a nintedanib [see Clinical Pharmacology (12.3)]. En tales casos, los pacientes deben ser monitoreados de cerca para determinar la tolerabilidad de OFEV. El manejo de las reacciones adversas puede requerir la interrupción, la reducción de la dosis o la suspensión del tratamiento con OFEV [see Dosage and Administration (2.4)].
La administración conjunta con dosis orales de rifampicina, un inductor de la P-gp y del CYP3A4, disminuyó la exposición a nintedanib en un 50%. Debe evitarse el uso concomitante de inductores de la P-gp y del CYP3A4 (p. ej., carbamazepina, fenitoína y hierba de San Juan) con OFEV, ya que estos medicamentos pueden disminuir la exposición a nintedanib [see Clinical Pharmacology (12.3)].
7.2 Anticoagulantes
Nintedanib es un inhibidor del VEGFR y puede aumentar el riesgo de sangrado. Monitoree de cerca a los pacientes que reciben terapia anticoagulante completa para detectar sangrado y ajuste el tratamiento anticoagulante según sea necesario [see Warnings and Precautions (5.6)].
7.3 Pirfenidona
En un estudio de dosis múltiples realizado para evaluar los efectos farmacocinéticos del tratamiento concomitante con nintedanib y pirfenidona, la administración conjunta de nintedanib con pirfenidona no alteró la exposición de ninguno de los agentes [see Clinical Pharmacology (12.3)]. Por lo tanto, no es necesario ajustar la dosis durante la administración concomitante de nintedanib con pirfenidona.
7.4 Bosentan
La administración conjunta de nintedanib con bosentan no alteró la farmacocinética de nintedanib [see Clinical Pharmacology (12.3)].
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de Riesgos
Basado en los hallazgos de estudios en animales y su mecanismo de acción [ver Farmacología Clínica (12.1)], OFEV puede causar daño fetal cuando se administra a una mujer embarazada. No hay datos sobre el uso de OFEV durante el embarazo. En estudios en animales de ratas y conejos embarazadas tratados durante la organogénesis, nintedanib causó muertes embrio-fetales y anomalías estructurales a menos de (ratas) y aproximadamente 5 veces (conejos) la dosis humana máxima recomendada [ver Datos]. Advierta a las mujeres embarazadas sobre el riesgo potencial para un feto.
El riesgo de fondo estimado de defectos de nacimiento mayores y aborto espontáneo para la población indicada es desconocido. En la población general de los EE. UU., el riesgo de fondo estimado de defectos de nacimiento mayores es del 2% al 4% y el aborto espontáneo en embarazos clínicamente reconocidos es del 15% al 20%.
Datos
Datos en Animales
En estudios de toxicidad reproductiva en animales, nintedanib causó muertes embrio-fetales y anomalías estructurales en ratas y conejos a menos de y aproximadamente 5 veces la dosis humana máxima recomendada (MRHD) en adultos (sobre una base de AUC plasmática a dosis orales maternas de 2.5 y 15 mg/kg/día en ratas y conejos, respectivamente). Las malformaciones incluyeron anomalías en los sistemas vascular, urogenital y esquelético. Las anomalías vasculares incluyeron vasos sanguíneos principales faltantes o adicionales. Las anomalías esqueléticas incluyeron anomalías en las vértebras torácicas, lumbares y caudales (por ejemplo, hemivértebra, faltantes o osificadas asimétricamente), costillas (bífidas o fusionadas) y esternón (fusionadas, divididas o osificadas unilateralmente). En algunos fetos, faltaban órganos en el sistema urogenital. En conejos, se observó un cambio significativo en la proporción de sexos en los fetos (proporción hembra:macho de aproximadamente 71%:29%) a aproximadamente 15 veces la MRHD en adultos (sobre una base de AUC a una dosis oral materna de 60 mg/kg/día). Nintedanib disminuyó la viabilidad postnatal de las crías de rata durante los primeros 4 días postnatales cuando las madres estuvieron expuestas a menos de la MRHD (sobre una base de AUC a una dosis oral materna de 10 mg/kg/día).
8.2 Lactancia
Resumen de Riesgos
No hay información sobre la presencia de nintedanib en la leche materna, los efectos en el lactante o los efectos en la producción de leche. Nintedanib y/o sus metabolitos están presentes en la leche de ratas lactantes [ver Datos]. Debido al potencial de reacciones adversas graves en los lactantes de OFEV, se debe aconsejar a las mujeres que la lactancia no se recomienda durante el tratamiento con OFEV.
Datos
La leche y el plasma de ratas lactantes tienen concentraciones similares de nintedanib y sus metabolitos.
8.3 Mujeres y Hombres en Potencial Reproductivo
Basado en los hallazgos de estudios en animales y su mecanismo de acción, OFEV puede causar daño fetal cuando se administra a una mujer embarazada y puede reducir la fertilidad en mujeres en potencial reproductivo [ver Uso en Poblaciones Específicas (8.1), Farmacología Clínica (12.1), 12.3), y Toxicología No Clínica (13.1)]. Aconseje a los pacientes sobre la prevención y planificación del embarazo.
OFEV puede causar daño fetal cuando se administra a una mujer embarazada. Aconseje a las mujeres en potencial reproductivo que eviten quedar embarazadas mientras reciben tratamiento con OFEV. Aconseje a las mujeres en potencial reproductivo que usen métodos anticonceptivos altamente efectivos al inicio, durante el tratamiento y durante al menos 3 meses después de tomar la última dosis de OFEV. Nintedanib no cambia la exposición al anticonceptivo oral que contiene etinilestradiol y levonorgestrel en pacientes con SSc-ILD. Sin embargo, la eficacia de los anticonceptivos hormonales orales puede verse comprometida por vómitos y/o diarrea u otras afecciones donde la absorción del medicamento puede reducirse. Aconseje a las mujeres que toman anticonceptivos hormonales orales que experimentan estas afecciones que usen métodos anticonceptivos alternativos altamente efectivos.
Infertilidad
Basado en datos de animales, OFEV puede reducir la fertilidad en mujeres en potencial reproductivo [ver Toxicología No Clínica (13.1)].
8.4 Uso Pediátrico
La seguridad y eficacia de OFEV no se han establecido en pacientes pediátricos para el tratamiento de enfermedades pulmonares intersticiales fibrosantes. La eficacia no se demostró en un estudio aleatorizado, doble ciego, controlado con placebo realizado en 26 pacientes pediátricos tratados con OFEV de 6 a 17 años con enfermedades pulmonares intersticiales fibrosantes, que fueron tratados con OFEV en función del peso.
Datos de Toxicidad en Animales
En estudios de toxicología de dosis repetidas, animales jóvenes (ratones, ratas y monos) tratados con nintedanib mostraron cambios en los huesos y los dientes de rápido crecimiento. Los cambios óseos incluyen el engrosamiento de la placa de crecimiento en todas las especies. Estos cambios fueron totalmente o al menos parcialmente reversibles en ratas y monos; la reversibilidad en ratones no se ha estudiado.
Los cambios en los dientes incluyen incisivos rotos y decoloración en roedores. Estos cambios fueron irreversibles después de la interrupción del tratamiento con nintedanib.
8.5 Uso en Geriatría
Del número total de sujetos en los estudios clínicos de fase 2 y 3 de OFEV en EPI (Estudio 1, Estudio 2 y Estudio 3), el 61% tenía 65 años o más, mientras que el 16% tenía 75 años o más. En el estudio clínico de EILD fibrosante crónica con fenotipo progresivo (Estudio 5), el 61% tenía 65 años o más, mientras que el 19% tenía 75 años o más. En SSc-ILD (Estudio 4), el 21,4% tenía 65 años o más, mientras que el 1,9% tenía 75 años o más. En los estudios de fase 3, no se observaron diferencias generales en la eficacia entre los sujetos que tenían 65 años o más y los sujetos más jóvenes; no se observaron diferencias generales en la seguridad entre los sujetos que tenían 65 años o más o 75 años o más y los sujetos más jóvenes, pero no se puede descartar una mayor sensibilidad de algunos individuos mayores.
8.6 Insuficiencia Hepática
Nintedanib se elimina principalmente a través de la excreción biliar/fecal (más del 90%). En un estudio de PK realizado en pacientes con insuficiencia hepática (Child Pugh A, Child Pugh B), la exposición a nintedanib aumentó [ver Farmacología Clínica (12.3)]. En pacientes con insuficiencia hepática leve (Child Pugh A), la dosis recomendada de OFEV es de 100 mg dos veces al día [ver Dosificación y Administración (2.3)]. Supervisar las reacciones adversas y considerar la interrupción del tratamiento o la suspensión para el manejo de las reacciones adversas en estos pacientes [ver Dosificación y Administración (2.4)]. No se recomienda el tratamiento de pacientes con insuficiencia hepática moderada (Child Pugh B) y grave (Child Pugh C) con OFEV [ver Advertencias y Precauciones (5.1)].
8.7 Insuficiencia Renal
Según un estudio de dosis única, menos del 1% de la dosis total de nintedanib se excreta a través del riñón [ver Farmacología Clínica (12.3)]. No se requiere ajuste de la dosis inicial en pacientes con insuficiencia renal leve a moderada. La seguridad, eficacia y farmacocinética de nintedanib no se han estudiado en pacientes con insuficiencia renal grave (menos de 30 mL/min CrCl) y enfermedad renal terminal.
8.8 Fumadores
El tabaquismo se asoció con una disminución de la exposición a OFEV [ver Farmacología Clínica (12.3)], lo que puede alterar el perfil de eficacia de OFEV. Anime a los pacientes a dejar de fumar antes del tratamiento con OFEV y a evitar fumar cuando usen OFEV.
10 SOBREDOSIS
En los ensayos de IPF, un paciente se expuso inadvertidamente a una dosis de 600 mg diarios durante un total de 21 días. Se produjo un evento adverso no grave (nasofaringitis) que se resolvió durante el período de dosificación incorrecta, sin aparición de otros eventos reportados. También se informó sobredosis en dos pacientes en estudios de oncología que estuvieron expuestos a un máximo de 600 mg dos veces al día durante un máximo de 8 días. Los eventos adversos reportados fueron consistentes con el perfil de seguridad existente de OFEV. Ambos pacientes se recuperaron. En caso de sobredosis, interrumpa el tratamiento e inicie medidas generales de apoyo según corresponda.
11 DESCRIPCIÓN
Las cápsulas de OFEV contienen nintedanib, un inhibidor de la cinasa [ver Mecanismo de acción (12.1)]. Nintedanib se presenta como la sal de etanosulfonato (esilato), con el nombre químico ácido 1H-indol-6-carboxílico, 2,3-dihidro-3-[[[4-[metil[(4-metil-1-piperazinil)acetil]amino]fenil]amino]fenilmetileno]-2-oxo-, éster metílico, (3Z)-, etanosulfonato (1:1).
Su fórmula estructural es:
El esilato de nintedanib es un polvo amarillo brillante con una fórmula empírica de C31H33N5O4∙C2H6O3S y un peso molecular de 649,76 g/mol.
Las cápsulas de OFEV para administración oral están disponibles en 2 dosis que contienen 100 mg o 150 mg de nintedanib (equivalente a 120,40 mg o 180,60 mg de etanosulfonato de nintedanib, respectivamente). Los ingredientes inactivos de OFEV son los siguientes: Material de relleno: triglicéridos, grasa dura, lecitina. Cápsula: gelatina, glicerol, dióxido de titanio, óxido férrico rojo, óxido férrico amarillo, tinta negra.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
Nintedanib es una molécula pequeña que inhibe múltiples receptores de tirosina quinasa (RTK) y tirosina quinasas no receptoras (nRTK). Nintedanib inhibe los siguientes RTK: receptor del factor de crecimiento derivado de plaquetas (PDGFR) α y β, receptor del factor de crecimiento de fibroblastos (FGFR) 1-3, receptor del factor de crecimiento endotelial vascular (VEGFR) 1-3, receptor del factor estimulante de colonias 1 (CSF1R) y tirosina quinasa similar a Fms-3 (FLT-3). Estas quinasas, excepto FLT-3, se han visto implicadas en la patogenia de las enfermedades pulmonares intersticiales (ILD). Nintedanib se une de forma competitiva al bolsillo de unión del trifosfato de adenosina (ATP) de estas quinasas y bloquea las cascadas de señalización intracelular, que se ha demostrado que están implicadas en la patogenia de la remodelación del tejido fibrótico en la ILD. Nintedanib también inhibe las siguientes nRTK: quinasas Lck, Lyn y Src. Se desconoce la contribución de la inhibición de FLT-3 y nRTK a la eficacia de nintedanib en la ILD.
12.2 Farmacodinamia
Electrofisiología cardíaca
En un estudio en pacientes con cáncer de células renales, se registraron las mediciones de QT/QTc y mostraron que una dosis oral única de 200 mg de nintedanib, así como dosis orales múltiples de 200 mg de nintedanib administradas dos veces al día durante 15 días, no prolongaron el intervalo QTcF.
12.3 Farmacocinética
Las propiedades farmacocinéticas de nintedanib fueron similares en voluntarios sanos, pacientes con IPF, pacientes con ILD fibrosantes crónicas con un fenotipo progresivo, pacientes con SSc-ILD y pacientes con cáncer. La farmacocinética de nintedanib es lineal. La proporcionalidad de la dosis se demostró mediante un aumento de la exposición a nintedanib con el aumento de las dosis (rango de dosis de 50 a 450 mg una vez al día y de 150 a 300 mg dos veces al día). La acumulación tras la administración múltiple en pacientes con IPF fue de 1,76 veces para el AUC. Las concentraciones plasmáticas en estado estacionario se alcanzaron en una semana de dosificación. Las concentraciones mínimas de nintedanib se mantuvieron estables durante más de un año. La variabilidad interindividual en la farmacocinética de nintedanib fue de moderada a alta (coeficiente de variación de los parámetros farmacocinéticos estándar en el rango del 30% al 70%), la variabilidad intraindividual de baja a moderada (coeficientes de variación por debajo del 40%).
Absorción
Nintedanib alcanzó concentraciones plasmáticas máximas aproximadamente de 2 a 4 horas después de la administración oral como cápsula de gelatina blanda en condiciones de alimentación. La biodisponibilidad absoluta de una dosis de 100 mg fue del 4,7% (IC del 90%: 3,62 a 6,08) en voluntarios sanos. La absorción y la biodisponibilidad se reducen por los efectos del transportador y el metabolismo de primer paso sustancial.
Después de la ingesta de alimentos, la exposición a nintedanib aumentó aproximadamente un 20% en comparación con la administración en ayunas (IC del 90%: 95,3% a 152,5%) y la absorción se retrasó (tmax mediana en ayunas: 2,00 horas; alimentado: 3,98 horas), independientemente del tipo de alimento.
Distribución
Nintedanib sigue una cinética de disposición bifásica. Después de la infusión intravenosa, se observó un alto volumen de distribución que fue mayor que el volumen corporal total (Vss: 1050 L).
La unión a proteínas in vitro de nintedanib en plasma humano fue alta, con una fracción unida del 97,8%. Se considera que la albúmina sérica es la principal proteína de unión. Nintedanib se distribuye preferentemente en plasma con una relación sangre/plasma de 0,87.
Eliminación
La semivida efectiva de nintedanib en pacientes con IPF fue de 9,5 horas (gCV 31,9%). El aclaramiento plasmático total después de la infusión intravenosa fue alto (CL: 1390 mL/min; gCV 28,8%). La excreción urinaria de fármaco inalterado en las 48 horas fue de aproximadamente el 0,05% de la dosis después de la administración oral y de aproximadamente el 1,4% de la dosis después de la administración intravenosa; el aclaramiento renal fue de 20 mL/min.
Metabolismo
La reacción metabólica predominante para nintedanib es la escisión hidrolítica por esterasas, lo que da como resultado la fracción de ácido libre BIBF 1202. BIBF 1202 se glucuronida posteriormente por las enzimas UGT, a saber, UGT 1A1, UGT 1A7, UGT 1A8 y UGT 1A10 a glucurónido de BIBF 1202. Sólo una parte menor de la biotransformación de nintedanib consistió en vías CYP, siendo CYP3A4 la principal enzima implicada. El principal metabolito dependiente de CYP no pudo detectarse en plasma en el estudio de absorción, distribución, metabolismo y eliminación en humanos. In vitro, el metabolismo dependiente de CYP representó aproximadamente el 5% en comparación con aproximadamente el 25% de la escisión de ésteres.
Excreción
La principal vía de eliminación de la radiactividad relacionada con el fármaco después de la administración oral de [14C] nintedanib fue a través de la excreción fecal/biliar (93,4% de la dosis), y la mayoría de OFEV se excretó como BIBF 1202. La contribución de la excreción renal al aclaramiento total fue baja (0,65% de la dosis). La recuperación general se consideró completa (por encima del 90%) en los 4 días posteriores a la dosificación.
Poblaciones específicas
Edad, peso corporal y sexo
Basándose en el análisis farmacocinético poblacional, la edad y el peso corporal se correlacionaron con la exposición a nintedanib. Sin embargo, los efectos sobre la exposición no son suficientes para justificar un ajuste de la dosis. No hubo influencia del sexo en la exposición a nintedanib.
Pacientes con insuficiencia renal
Basado en un análisis de PK poblacional de datos de 933 pacientes con IPF, la exposición a nintedanib no se vio influenciada por el deterioro renal leve (CrCl: 60 a 90 mL/min; n=399) o moderado (CrCl: 30 a 60 mL/min; n=116). Los datos en insuficiencia renal grave (CrCl por debajo de 30 mL/min) fueron limitados.
Pacientes con deterioro hepático
Un estudio dedicado de farmacocinética de fase I de dosis única de OFEV comparó 8 sujetos con deterioro hepático leve (Child Pugh A) y 8 sujetos con deterioro hepático moderado (Child Pugh B) con 17 sujetos con función hepática normal. En sujetos con deterioro hepático leve, la exposición media a nintedanib fue 2,4 veces mayor según Cmax (IC del 90%: 1,6 a 3,6) y 2,2 veces mayor según AUC0-inf (IC del 90%: 1,4 a 3,5). En sujetos con deterioro hepático moderado, la exposición fue 6,9 veces mayor según Cmax (IC del 90%: 4,4 a 11,0) y 7,6 veces mayor según AUC0-inf (IC del 90%: 5,1 a 11,3). Los sujetos con deterioro hepático grave (Child Pugh C) no se han estudiado.
Fumadores
En el análisis de PK poblacional, la exposición a nintedanib fue un 21% menor en los fumadores actuales en comparación con los exfumadores y los que nunca fumaron. El efecto no es suficiente para justificar un ajuste de dosis.
Estudios de interacción medicamentosa
Potencial de nintedanib para afectar otros medicamentos
El efecto de la coadministración de nintedanib en el AUC y Cmax de pirfenidona se evaluó en un estudio de dosis múltiples. Nintedanib no tuvo efecto en la exposición a pirfenidona.
Quince pacientes mujeres con SSc-ILD recibieron una dosis única de una combinación de 30 mcg de etinilestradiol y 150 mcg de levonorgestrel antes y después de la dosificación dos veces al día de 150 mg de nintedanib durante al menos 10 días. La coadministración de nintedanib no cambió la exposición a etinilestradiol y levonorgestrel [ver Advertencias y precauciones (5.4) y Uso en poblaciones específicas (8.3)].
En estudios in vitro, se demostró que nintedanib no es un inhibidor de OATP-1B1, OATP-1B3, OATP-2B1, OCT-2 o MRP-2. Los estudios in vitro también mostraron que nintedanib tiene un potencial inhibitorio débil sobre OCT-1, BCRP y P-gp; estos hallazgos se consideran de baja relevancia clínica. Nintedanib y sus metabolitos, BIBF 1202 y BIBF 1202 glucurónido, no inhibieron ni indujeron enzimas CYP in vitro.
Potencial de otros medicamentos para afectar nintedanib
Nintedanib es un sustrato de P-gp y, en menor medida, CYP3A4. La coadministración con el inhibidor de P-gp y CYP3A4, ketoconazol, aumentó la exposición a nintedanib 1,61 veces según el AUC y 1,83 veces según el Cmax en un estudio dedicado de interacción fármaco-fármaco. En un estudio de interacción fármaco-fármaco con el inductor de P-gp y CYP3A4, rifampicina, la exposición a nintedanib disminuyó al 50,3% según el AUC y al 60,3% según el Cmax tras la coadministración con rifampicina en comparación con la administración de nintedanib solo.
El efecto de la coadministración de pirfenidona en el AUC y Cmax de nintedanib se evaluó en un estudio de interacción fármaco-fármaco de dosis múltiples. Pirfenidona no tuvo efecto en la exposición a nintedanib. El tratamiento concomitante con nintedanib y pirfenidona también se investigó en un ensayo separado, que fue un ensayo exploratorio abierto, aleatorizado (1:1) de nintedanib 150 mg dos veces al día con pirfenidona adicional (titulado a 801 mg tres veces al día) en comparación con nintedanib 150 mg dos veces al día solo en 105 pacientes aleatorizados durante 12 semanas. Se observaron concentraciones plasmáticas valle de nintedanib similares al comparar pacientes que recibieron nintedanib solo con pacientes que recibieron nintedanib con pirfenidona adicional.
Los voluntarios sanos recibieron una dosis única de 150 mg de nintedanib antes y después de la dosificación múltiple de 125 mg de bosentan dos veces al día en estado estacionario. La coadministración de nintedanib con bosentan no alteró la farmacocinética de nintedanib.
Nintedanib muestra un perfil de solubilidad dependiente del pH con mayor solubilidad a un pH ácido inferior a 3. Sin embargo, en los ensayos clínicos, la coadministración con inhibidores de la bomba de protones o antagonistas de la histamina H2 no influyó en la exposición (concentraciones valle) de nintedanib.
En estudios in vitro, se demostró que nintedanib no es un sustrato de OATP-1B1, OATP-1B3, OATP-2B1, OCT-2, MRP-2 o BCRP. Los estudios in vitro también mostraron que nintedanib era un sustrato de OCT-1; estos hallazgos se consideran de baja relevancia clínica.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
Los estudios de carcinogenicidad oral de dos años de nintedanib en ratas y ratones no han revelado ninguna evidencia de potencial carcinogénico. Nintedanib se administró hasta 10 y 30 mg/kg/día en ratas y ratones, respectivamente. Estas dosis fueron menores y aproximadamente 4 veces la MRHD sobre una base de AUC de fármaco plasmático.
Nintedanib fue negativo para genotoxicidad en la prueba de mutación inversa bacteriana in vitro, la prueba de mutación directa de células de linfoma de ratón y la prueba de micronúcleos de rata in vivo.
En ratas, nintedanib redujo la fertilidad femenina a niveles de exposición aproximadamente 3 veces la MRHD (sobre una base de AUC a una dosis oral de 100 mg/kg/día). Los efectos incluyeron aumentos en la resorción y la pérdida postimplantación, y una disminución en el índice de gestación. Se observaron cambios en el número y tamaño de los cuerpos lúteos en los ovarios en estudios de toxicidad crónica en ratas y ratones. Se observó un aumento en el número de hembras con solo resorciones a exposiciones aproximadamente iguales a la MRHD (sobre una base de AUC a una dosis oral de 20 mg/kg/día). Nintedanib no tuvo efectos sobre la fertilidad masculina en ratas a niveles de exposición aproximadamente 3 veces la MRHD (sobre una base de AUC a una dosis oral de 100 mg/kg/día).
14 ESTUDIOS CLÍNICOS
14.1 Fibrosis Pulmonar Idiopática
La eficacia clínica de OFEV se ha estudiado en 1231 pacientes con FPI en un estudio de fase 2 (Estudio 1 [NCT00514683]) y dos estudios de fase 3 (Estudio 2 [NCT01335464] y Estudio 3 [NCT01335477]). Estos fueron estudios aleatorizados, doble ciego, controlados con placebo que compararon el tratamiento con OFEV 150 mg dos veces al día con placebo durante 52 semanas.
El Estudio 2 y el Estudio 3 fueron idénticos en diseño. El Estudio 1 fue muy similar en diseño. Los pacientes fueron aleatorizados en una proporción de 3:2 (1:1 para el Estudio 1) a OFEV 150 mg o placebo dos veces al día durante 52 semanas. El Estudio 1 también incluyó otros brazos de tratamiento (50 mg diarios, 50 mg dos veces al día y 100 mg dos veces al día) que no se discuten más. El criterio de valoración principal fue la tasa anual de disminución de la Capacidad Vital Forzada (CVF). El tiempo hasta la primera exacerbación aguda de FPI fue un criterio de valoración secundario clave en el Estudio 2 y el Estudio 3 y un criterio de valoración secundario en el Estudio 1. El cambio desde el inicio en el porcentaje de CVF predicho y la supervivencia fueron criterios de valoración secundarios adicionales en todos los estudios.
Los pacientes debían tener un diagnóstico de FPI (criterios ATS/ERS/JRS/ALAT) durante menos de 5 años. Los diagnósticos fueron adjudicados centralmente en función de la confirmación radiológica y, si corresponde, histopatológica. Los pacientes debían tener 40 años o más con una CVF mayor o igual al 50% de la predicha y una capacidad de difusión de monóxido de carbono (DLCO, corregida para la hemoglobina) del 30% al 79% de la predicha. Los pacientes con obstrucción de las vías respiratorias relevantes (es decir, FEV1/CVF pre-broncodilatador menor que 0,7) o, en opinión del investigador, con probabilidad de recibir un trasplante de pulmón durante los estudios fueron excluidos (estar en lista de espera para un trasplante de pulmón fue aceptable para la inclusión). Los pacientes con más de 1,5 veces el LSN de ALT, AST o bilirrubina, los pacientes con un riesgo o predisposición conocida a sangrado, los pacientes que recibían una dosis completa de tratamiento anticoagulante y los pacientes con antecedentes recientes de infarto de miocardio o accidente cerebrovascular fueron excluidos de los estudios. Los pacientes también fueron excluidos si recibieron otra terapia de investigación, azatioprina, ciclofosfamida o ciclosporina A dentro de las 8 semanas previas a la entrada en este ensayo, o n-acetilcisteína y prednisona (más de 15 mg/día o equivalente) dentro de las 2 semanas previas. La mayoría de los pacientes eran caucásicos (60%) o asiáticos (30%) y hombres (79%). Los pacientes tenían una edad media de 67 años y un porcentaje de CVF predicho medio del 80%.
Tasa anual de disminución de la CVF
Se demostró una reducción estadísticamente significativa en la tasa anual de disminución de la CVF (en mL) en los pacientes que recibieron OFEV en comparación con los pacientes que recibieron placebo en función del modelo de regresión de coeficientes aleatorios, ajustado por sexo, altura y edad. El efecto del tratamiento sobre la CVF fue consistente en los 3 estudios. Consulte Tabla 3 para obtener los resultados de cada estudio.
Tabla 3 Tasa anual de disminución de la CVF (mL) en el Estudio 1, el Estudio 2 y el Estudio 3a
Estudio 1
Estudio 2
Estudio 3
OFEV 150 mg dos veces al día
Placebo
OFEV 150 mg dos veces al día
Placebo
OFEV 150 mg dos veces al día
Placebo
aConjunto aleatorizado en el Estudio 1; conjunto tratado en el Estudio 2 y el Estudio 3 bEstimado en función de un modelo de regresión de coeficientes aleatorios
Número de pacientes analizados
84
83
309
204
329
219
Tasaa de disminución durante 52 semanas
-60
-191
-115
-240
-114
-207
Comparación con placebo
Diferenciab
131
125
94
IC del 95%
(27, 235)
(78, 173)
(45, 143)
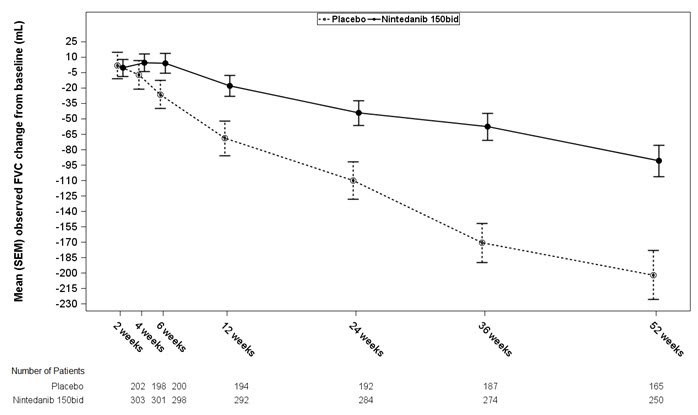
Figura 1 muestra el cambio desde la línea de base con el tiempo en ambos grupos de tratamiento para el Estudio 2. Cuando el cambio medio observado en FVC desde la línea de base se trazó con el tiempo, las curvas divergieron en todos los puntos de tiempo hasta la semana 52. Se observaron gráficos similares para el Estudio 1 y el Estudio 3.
Figura 1 Cambio medio (SEM) observado en FVC desde la línea de base (mL) con el tiempo en el Estudio 2
bid = dos veces al día
Cambio desde la línea de base en el porcentaje de capacidad vital forzada predicha
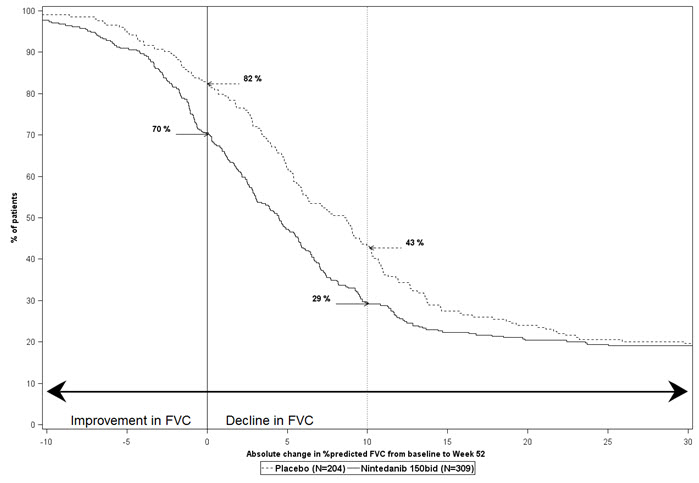
Figura 2 presenta la distribución acumulativa para todos los puntos de corte para el cambio desde la línea de base en el porcentaje de FVC predicha en la semana 52 para el Estudio 2. Para todas las disminuciones categóricas en la función pulmonar, la proporción de pacientes que disminuyeron fue menor en OFEV que en placebo. El Estudio 3 mostró resultados similares.
Los datos faltantes para el cambio desde la línea de base en la semana 52 en el porcentaje de FVC predicha (debido a la muerte, la pérdida de seguimiento o la censura antes de las 52 semanas) se imputaron utilizando la peor disminución desde la línea de base en la semana 52 observada entre todos los pacientes con datos disponibles, independientemente del tratamiento. bid = dos veces al día
Figura 2 Distribución acumulativa de pacientes por cambio en el porcentaje de FVC predicha desde la línea de base hasta la semana 52 (Estudio 2).* Las líneas verticales indican una disminución ≥0% o una disminución ≥10%.
Tiempo hasta la primera exacerbación aguda de la IPF
La exacerbación aguda de la IPF se definió como un empeoramiento o desarrollo inexplicable de disnea dentro de los 30 días, nuevos infiltrados pulmonares difusos en la radiografía de tórax y/o nuevas anomalías parenquimatosas en la TC de alta resolución sin neumotórax o derrame pleural, y exclusión de causas alternativas. La exacerbación aguda de la IPF se adjudicó en el Estudio 2 y el Estudio 3. En el Estudio 1 (informado por el investigador) y el Estudio 3 (adjudicado), el riesgo de primera exacerbación aguda de la IPF durante 52 semanas se redujo significativamente en los pacientes que recibieron OFEV en comparación con el placebo (razón de riesgo [HR]: 0,16, IC del 95%: 0,04, 0,71) y (HR: 0,20, IC del 95%: 0,07, 0,56), respectivamente. En el Estudio 2 (adjudicado), no hubo diferencia entre los grupos de tratamiento (HR: 0,55, IC del 95%: 0,20, 1,54).
Supervivencia
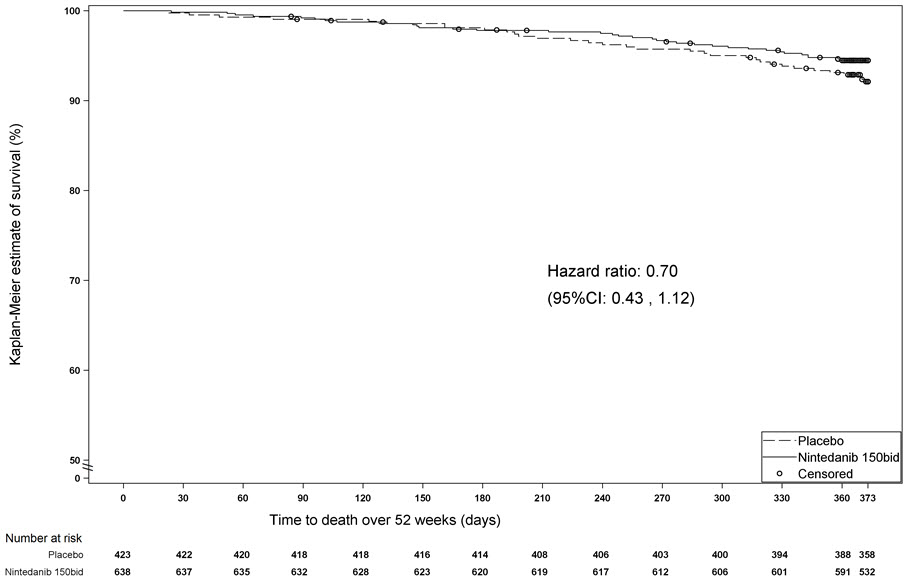
La supervivencia se evaluó para OFEV en comparación con el placebo en el Estudio 2 y el Estudio 3 como un análisis exploratorio para apoyar el criterio de valoración principal (FVC). La mortalidad por todas las causas se evaluó durante la duración del estudio y el período de seguimiento disponible, independientemente de la causa de la muerte y de si los pacientes continuaron el tratamiento. La mortalidad por todas las causas no mostró una diferencia estadísticamente significativa (ver Figura 3).
Figura 3 Estimaciones de Kaplan-Meier de la mortalidad por todas las causas en el estado vital – Fin del estudio: Estudio 2 y Estudio 3
bid = dos veces al día
14.2 Enfermedades pulmonares intersticiales fibrosantes crónicas con un fenotipo progresivo
La eficacia clínica de OFEV se ha estudiado en pacientes con EPI fibrosantes crónicas con un fenotipo progresivo en un ensayo de fase 3 aleatorizado, doble ciego, controlado con placebo (Estudio 5 [NCT02999178]). Un total de 663 pacientes fueron aleatorizados en una proporción de 1:1 para recibir OFEV 150 mg dos veces al día o placebo coincidente durante al menos 52 semanas. La aleatorización se estratificó en función del patrón fibrótico de la tomografía computarizada de alta resolución (TCAR) evaluado por lectores centrales: 412 pacientes con patrón fibrótico de TCAR similar al UIP y 251 pacientes con otros patrones fibróticos de TCAR fueron aleatorizados. Hubo 2 poblaciones coprimarios definidas para los análisis en este ensayo: todos los pacientes (la población general) y los pacientes con TCAR con patrón fibrótico de TCAR similar al UIP.
El criterio de valoración principal fue la tasa anual de disminución de la FVC (en mL) durante 52 semanas. Otros criterios de valoración incluyeron el tiempo hasta la primera exacerbación aguda de la EPI y el tiempo hasta la muerte.
Los pacientes con un diagnóstico clínico de EPI fibrosante crónica fueron seleccionados si tenían fibrosis relevante (más del 10% de características fibróticas) en la TCAR y presentaban signos clínicos de progresión (definidos como una disminución de la FVC ≥10%, una disminución de la FVC ≥5% y <10% con empeoramiento de los síntomas o la imagen, o empeoramiento de los síntomas y empeoramiento de la imagen, todo en los 24 meses anteriores a la selección). Los pacientes debían tener una FVC mayor o igual al 45% de la predicha y una DLCO del 30% al menos del 80% de la predicha. Los pacientes debían haber progresado a pesar de la gestión considerada apropiada en la práctica clínica por los investigadores para la EPI relevante del paciente.
Los pacientes con IPF, obstrucción de las vías respiratorias relevante (es decir, FEV1/FVC prebroncodilatador menor de 0,7) o hipertensión pulmonar significativa fueron excluidos del ensayo. Los pacientes con más de 1,5 veces el LSN de ALT, AST o bilirrubina, los pacientes con un riesgo o predisposición conocida a la hemorragia, los pacientes que recibían una dosis completa de tratamiento anticoagulante y los pacientes con antecedentes recientes de infarto de miocardio o accidente cerebrovascular fueron excluidos. Los pacientes también fueron excluidos si recibieron otra terapia de investigación, azatioprina, ciclosporina, micofenolato mofetil, tacrolimus, corticosteroides orales superiores a 20 mg/día o la combinación de corticosteroides orales + azatioprina + N-acetilcisteína dentro de las 4 semanas de la aleatorización, ciclofosfamida dentro de las 8 semanas anteriores a la aleatorización, rituximab dentro de los 6 meses o tratamiento previo con nintedanib o pirfenidona.
La mayoría de los pacientes eran caucásicos (74%) o asiáticos (25%). Los pacientes eran en su mayoría hombres (54%) y tenían una edad media de 66 años y un porcentaje medio de FVC predicho del 69%, y el 49% nunca fueron fumadores. Los diagnósticos clínicos subyacentes de EPI en los grupos representados en el ensayo fueron neumonitis por hipersensibilidad (26%), EPI autoinmunes (26%), neumonía intersticial inespecífica idiopática (19%), neumonía intersticial idiopática no clasificable (17%) y otras EPI (12%).
Tasa anual de disminución de la FVC
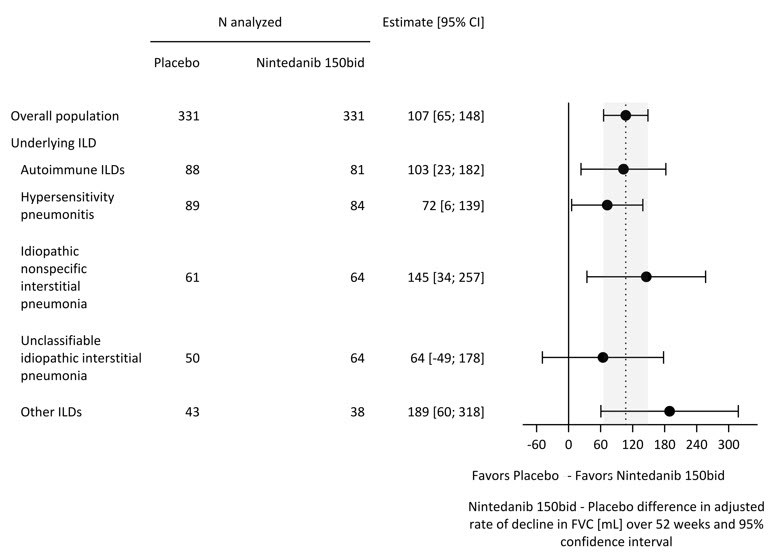
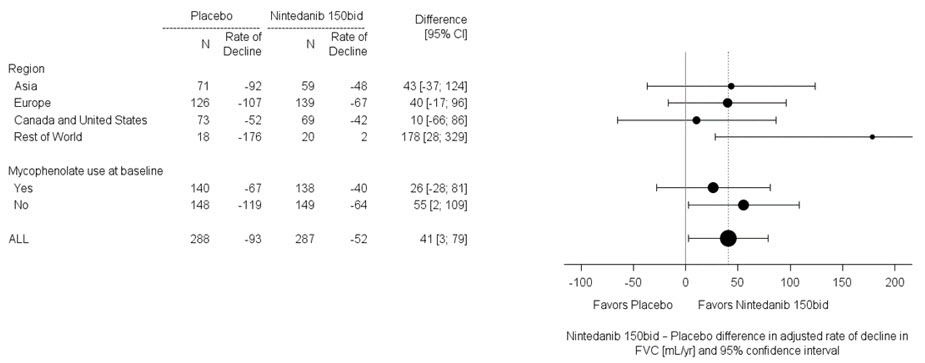
Hubo una reducción estadísticamente significativa en la tasa anual de disminución de la FVC (en mL) durante 52 semanas en los pacientes que recibieron OFEV en comparación con los pacientes que recibieron placebo. La tasa anual de disminución de la FVC (en mL) durante 52 semanas se redujo significativamente en 107 mL en los pacientes que recibieron OFEV en comparación con los pacientes que recibieron placebo. Los resultados en las subpoblaciones de pacientes con TCAR con patrón fibrótico similar al UIP y pacientes con otros patrones fibróticos (Otro TCAR) se incluyen con la población general en Tabla 4.
Tabla 4 Tasa anual de disminución de la FVC (mL) en el Estudio 5
General
Subpoblación similar al UIP
Subpoblación de otro TCAR
OFEV
Placebo
OFEV
Placebo
OFEV
Placebo
aBasado en un modelo de regresión de coeficientes aleatorios con efectos categóricos fijos del tratamiento, patrón de HRCT, efectos continuos fijos del tiempo, FVC basal (mL), e incluyendo las interacciones tratamiento por tiempo y basal por tiempo
Número de pacientes analizados
331
331
206
206
125
125
Tasa anual ajustada de disminución durante 52 semanas
-81
-188
-83
-211
-79
-154
Diferencia de comparación vs placeboa
107
128
75*
95% IC
(65, 148)
(71, 186)
(16, 135)*
*La comparación basada en la subpoblación de Otro HRCT no se incluyó en el procedimiento de pruebas múltiples. Los valores que se muestran aquí son para fines descriptivos.
Se realizó un análisis exploratorio posterior por diagnóstico de EILD y se muestra en Figura 4. La respuesta al tratamiento en todos los diagnósticos de EILD fue consistente para la CVF.
Figura 4 Tasa anual de disminución de la CVF (mL) durante 52 semanas según el diagnóstico subyacente de EILD en el Estudio 5*
EILD = enfermedad pulmonar intersticial; EILD autoinmunes: incluye EILD asociada a artritis reumatoide, enfermedad del tejido conectivo mixto, EILD asociada a esclerosis sistémica y otros términos; Otros EILD: incluye EILD fibroantes que no se clasifican bajo EILD autoinmunes, neumonitis de hipersensibilidad, neumonía intersticial no específica idiopática o neumonía intersticial idiopática no clasificable. Las tres EILD más comunes en esta categoría son EILD relacionada con la exposición, sarcoidosis y fibroelastosis pleuroparenquimatosa. *Estos resultados son de un análisis exploratorio posterior. Los valores mostrados aquí son para fines descriptivos.
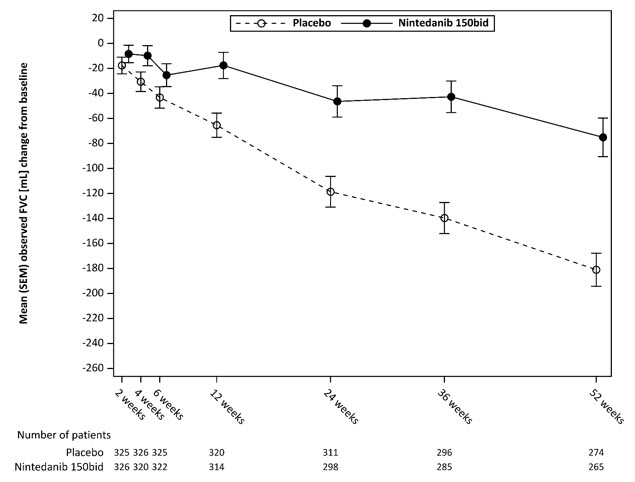
Figura 5 muestra el cambio en la CVF desde el punto de referencia a lo largo del tiempo en los grupos de tratamiento. Cuando se trazó el cambio medio observado de la CVF desde el punto de referencia a lo largo del tiempo, las curvas se divergieron en todos los puntos temporales hasta la Semana 52.
Figura 5 Cambio medio (SEM) observado de la CVF desde el punto de referencia (mL) durante 52 semanas en el Estudio 5
bid = dos veces al día
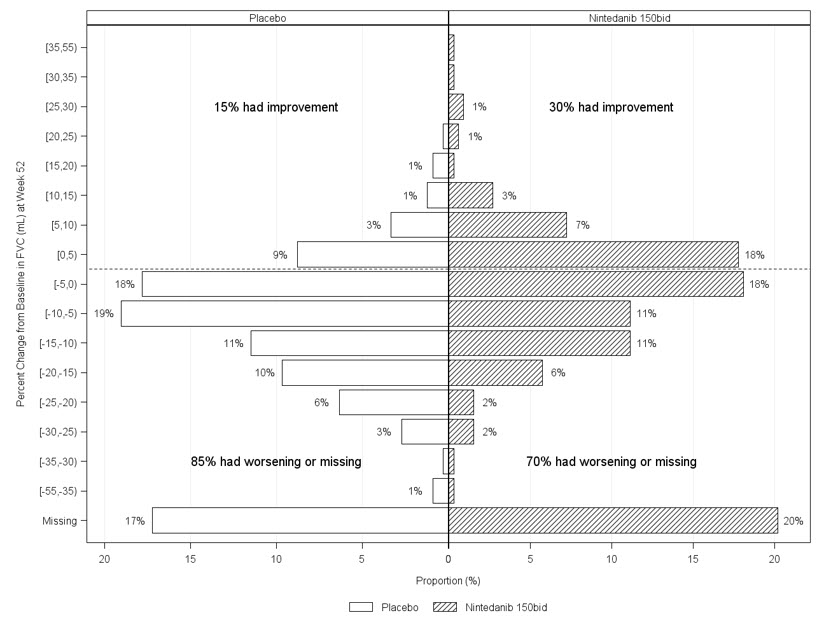
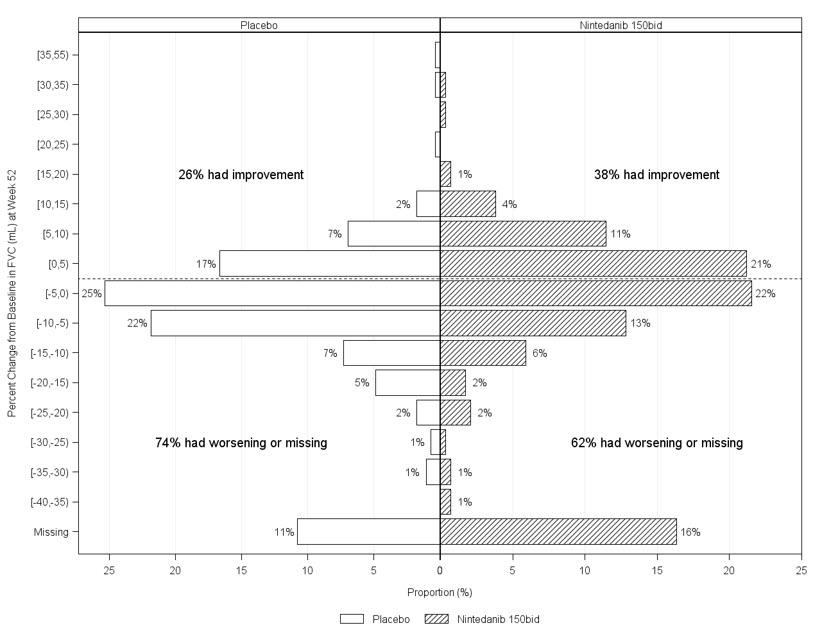
Porcentaje de cambio desde el punto de referencia en la Capacidad Vital Forzada
Figura 6 presenta el porcentaje de cambio desde el punto de referencia en la CVF en mL en la Semana 52 para el Estudio 5. Para la mayoría de los pacientes, la disminución de la función pulmonar fue menor con OFEV que con el placebo.
Figura 6 Histograma del porcentaje de cambio en la CVF (mL) desde el punto de referencia hasta la Semana 52 según el tratamiento y los incrementos o decrementos porcentuales de 5 (Estudio 5)a
a Los pacientes clasificados como que tienen datos de CVF faltantes en la Semana 52 son aquellos sin evaluación de CVF entre el Día 310 y el Día 373. bid = dos veces al día
Tiempo hasta la primera exacerbación aguda de EILD
La exacerbación aguda de EILD se definió como un empeoramiento o desarrollo inexplicable de disnea en un plazo de 30 días, nuevos infiltrados pulmonares difusos en la radiografía de tórax y/o nuevas anomalías parenquimatosas en la TCAR de alta resolución sin neumotórax o derrame pleural, y la exclusión de causas alternativas. Las exacerbaciones agudas de EILD no fueron arbitradas.
El riesgo de la primera exacerbación aguda de EILD no mostró una diferencia estadísticamente significativa entre el grupo OFEV en comparación con el placebo (período de tratamiento de 52 semanas: HR 0,72, (95% CI: 0,38, 1,37); todo el ensayo: HR 0,63 (95% CI: 0,37, 1,07)).
Supervivencia
Se evaluó la supervivencia de OFEV en comparación con el placebo en el Estudio 5 para apoyar el punto final primario (CVF). La mortalidad por todas las causas se evaluó durante la duración del estudio y el período de seguimiento disponible, independientemente de la causa de la muerte y si los pacientes continuaron el tratamiento. La mortalidad por todas las causas no mostró una diferencia estadísticamente significativa (período de tratamiento de 52 semanas: HR 0,94 (95% CI: 0,47, 1,86); todo el ensayo: HR 0,78 (95% CI: 0,50, 1,21)).
14.3 Enfermedad pulmonar intersticial asociada a esclerosis sistémica
La eficacia clínica de nintedanib se ha estudiado en pacientes con SSc-ILD en un ensayo aleatorizado, doble ciego, controlado con placebo de fase 3 (Estudio 4 [NCT02597933]). Un total de 580 pacientes fueron aleatorizados en una proporción de 1:1 para recibir ya sea OFEV 150 mg dos veces al día o un placebo idéntico durante al menos 52 semanas, de los cuales 576 pacientes fueron tratados. La aleatorización se estratificó por el estado del anticuerpo antitopoisomerasa (ATA). Los pacientes individuales permanecieron en el tratamiento de ensayo ciego durante hasta 100 semanas. El punto final primario fue la tasa anual de disminución de la CVF durante 52 semanas. El cambio absoluto desde el punto de referencia en la puntuación de la piel modificada de Rodnan (mRSS) en la Semana 52 fue un punto final secundario clave. La mortalidad durante todo el ensayo fue un punto final secundario adicional.
Los pacientes fueron diagnosticados con SSc-ILD según los criterios de clasificación de la American College of Rheumatology / European League Against Rheumatism de 2013 para la SSc con el inicio de la enfermedad (primer síntoma no-Raynaud) de menos de 7 años y más de o igual a 10% de fibrosis en un escáner de tomografía computarizada de alta resolución torácica (TCAR) realizado en los últimos 12 meses. Los pacientes debían tener una CVF mayor o igual al 40% del previsto y un DLCO del 30 al 89% del previsto. Los pacientes con obstrucción de las vías respiratorias relevante (es decir, FEV1/CVF prebroncodilatador menor de 0,7) o trasplante de células madre hematopoyéticas previo o planeado fueron excluidos del ensayo. Los pacientes con más de 1,5 veces el límite superior de la normalidad de ALT, AST o bilirrubina, los pacientes con un riesgo o predisposición conocida a la hemorragia, los pacientes que recibían una dosis completa de tratamiento anticoagulante y los pacientes con un historial reciente de infarto de miocardio o accidente cerebrovascular fueron excluidos del estudio. Los pacientes fueron excluidos si tenían hipertensión pulmonar significativa, más de tres úlceras digitales en las yemas de los dedos, un historial de necrosis digital severa que requiriera hospitalización o un historial de crisis renal esclerodérmica. Los pacientes también fueron excluidos si recibieron otra terapia investigacional, azatioprina en las 8 semanas anteriores a la aleatorización, ciclofosfamida o ciclosporina A en los 6 meses anteriores a la aleatorización o tratamiento previo con nintedanib o pirfenidona.
La mayoría de los pacientes eran mujeres (75%). Los pacientes eran en su mayoría caucásicos (67%), asiáticos (25%) o negros (6%). La edad media era de 54 años. En general, el 52% de los pacientes tenían esclerosis sistémica cutánea difusa (SSc) y el 48% tenían SSc cutánea limitada. El tiempo medio desde el primer inicio de un síntoma no-Raynaud fue de 3,49 años. En el punto de referencia, el 49% de los pacientes estaban en tratamiento estable con micofenolato.
Tasa anual de disminución de la CVF
La tasa anual de disminución de la FVC (en mL) durante 52 semanas se redujo significativamente en 41 mL en los pacientes que recibieron OFEV en comparación con los pacientes que recibieron placebo, lo que corresponde a un efecto de tratamiento relativo del 44%. Consulte Tabla 5.
Tabla 5 Tasa anual de disminución de la FVC (mL) en el estudio 4
OFEV 150 mg dos veces al día
Placebo
aBasado en un modelo de regresión de coeficientes aleatorios, ajustado por sexo, altura, edad, estado de ATA, FVC al inicio, FVC al inicio por tiempo
Número de pacientes analizados
287
288
Tasa ajustada de disminución durante 52 semanas
-52
-93
Comparación vs placebo
Diferenciaa
41
IC del 95%
(3, 79)
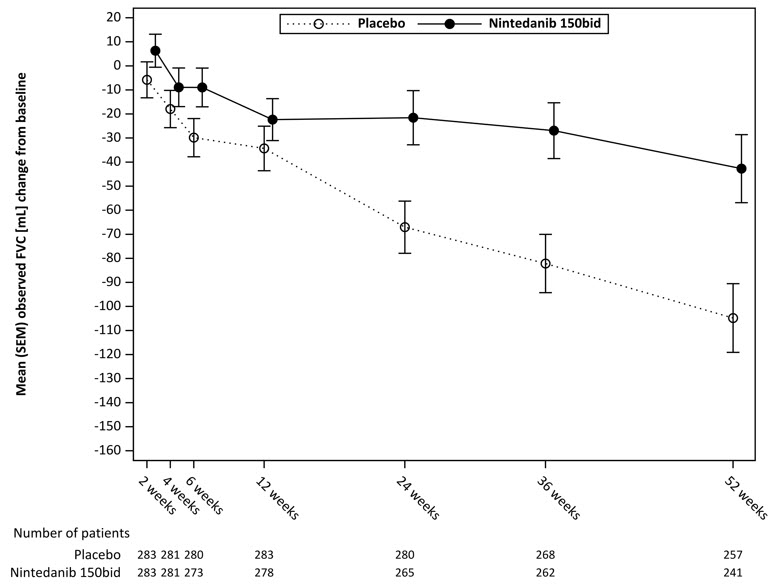
Figura 7 muestra el cambio desde el inicio con el tiempo en ambos grupos de tratamiento. Cuando el cambio medio observado de la FVC desde el inicio se trazó con el tiempo, las curvas divergieron en todos los puntos de tiempo hasta la semana 52. La separación de los valores medios se observa después de 12 semanas de tratamiento.
Figura 7 Cambio medio (SEM) observado de la FVC desde el inicio (mL) con el tiempo en el estudio 4
bid = dos veces al día
En dos análisis de eficacia de subgrupos preespecificados, se examinó la diferencia media de tratamiento en la disminución de la FVC a las 52 semanas en los pacientes por región y uso de micofenolato (Figura 8).
Figura 8 Análisis de subgrupos de la diferencia media de tratamiento en la disminución de la FVC (mL) a la semana 52 por región y uso de micofenolato (estudio 4)
Porcentaje de cambio desde el inicio en la capacidad vital forzada
Figura 9 presenta el porcentaje de cambio desde el inicio en la FVC en mL a la semana 52 para el estudio 4. Para la mayoría de los pacientes, la disminución de la función pulmonar fue menor con OFEV que con placebo.
a Los pacientes clasificados como que tienen datos de FVC faltantes a la semana 52 son aquellos que no tienen una evaluación de FVC entre el día 310 y el día 373. bid = dos veces al día
Figura 9 Histograma del porcentaje de cambio en la FVC (mL) desde el inicio hasta la semana 52 según el tratamiento y los incrementos o decrementos porcentuales de 5 (estudio 4)a
Puntuación modificada de la piel de Rodnan
No se observó ningún beneficio en la mRSS en los pacientes que recibieron OFEV. El cambio medio absoluto ajustado desde el inicio en la mRSS a la semana 52 fue comparable entre el grupo OFEV (-2,17 (IC del 95%: -2,69, -1,65)) y el grupo placebo (-1,96 (IC del 95%: -2,48, -1,45)). La diferencia media ajustada entre los grupos de tratamiento fue -0,21 (IC del 95%: -0,94, 0,53).
Supervivencia
No se observó ninguna diferencia en la supervivencia en un análisis exploratorio de la mortalidad durante todo el ensayo (OFEV: n=10 (3,5%) vs. placebo: n=9 (3,1%)). El análisis del tiempo hasta la muerte durante todo el ensayo dio como resultado una HR de 1,16 (IC del 95%: 0,47, 2,84).
16 FORMA DE SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
150 mg: Cápsulas blandas, de color marrón, opacas, oblongas, impresas en negro con el símbolo de la compañía Boehringer Ingelheim y “150”. Se envasan en frascos de HDPE con tapa resistente a los niños y están disponibles de la siguiente manera:
Frascos de 60
NDC: 0597-0145-60
100 mg: Cápsulas blandas, de color melocotón, opacas, oblongas, impresas en negro con el símbolo de la compañía Boehringer Ingelheim y “100”. Se envasan en frascos de HDPE con tapa resistente a los niños y están disponibles de la siguiente manera:
Frascos de 60
NDC: 0597-0143-60
Almacenamiento
Almacenar a 20 °C a 25 °C (68 °F a 77 °F); se permiten fluctuaciones de 15 °C a 30 °C (59 °F a 86 °F) [ver Temperatura Controlada de la Sala de la USP]. Proteger de la exposición a la humedad alta y evitar el calor excesivo. Si se reempaqueta, utilizar un recipiente hermético de la USP.
17 INFORMACIÓN PARA EL PACIENTE
Aconseje al paciente que lea la etiqueta del paciente aprobada por la FDA (Información para el paciente).
Elevación de las enzimas hepáticas y lesión hepática inducida por fármacos
Avise a los pacientes que deberán someterse a pruebas de función hepática periódicamente. Avise a los pacientes que informen inmediatamente cualquier síntoma de un problema hepático (por ejemplo, la piel o la parte blanca de los ojos se vuelven amarillas, la orina se vuelve oscura o marrón (color té), dolor en el lado derecho del estómago, sangrado o moretones más fácilmente de lo normal, letargo, pérdida de apetito) [ver Advertencias y precauciones (5.2)].
Trastornos gastrointestinales
Informe a los pacientes que los trastornos gastrointestinales como la diarrea, las náuseas y los vómitos fueron los eventos gastrointestinales más comúnmente reportados que ocurrieron en pacientes que recibieron OFEV. Avise a los pacientes que su proveedor de atención médica puede recomendar hidratación, medicamentos antidiarreicos (por ejemplo, loperamida) o medicamentos antieméticos para tratar estos efectos secundarios. Pueden ser necesarias reducciones o interrupciones temporales de la dosis. Indique a los pacientes que se pongan en contacto con su proveedor de atención médica a la primera señal de diarrea o por cualquier diarrea grave o persistente, náuseas o vómitos [ver Advertencias y precauciones (5.3) y Reacciones adversas (6.1)].
Toxicidad embrio-fetal
Oriente a los pacientes sobre la prevención y la planificación del embarazo. Avise a las mujeres en edad fértil del riesgo potencial para un feto y que eviten quedar embarazadas mientras reciben tratamiento con OFEV. Avise a las mujeres en edad fértil que utilicen métodos anticonceptivos altamente efectivos al inicio, durante el tratamiento y durante al menos 3 meses después de tomar la última dosis de OFEV. Avise a las mujeres que toman anticonceptivos hormonales orales que experimentan vómitos y/o diarrea u otras afecciones donde la absorción del medicamento puede reducirse que se pongan en contacto con su médico para discutir métodos anticonceptivos alternativos altamente efectivos. Avise a las pacientes que notifiquen a su médico si quedan embarazadas o sospechan que están embarazadas durante la terapia con OFEV [ver Advertencias y precauciones (5.4) y Uso en poblaciones específicas (8.1, 8.3)].
Eventos tromboembólicos arteriales
Avise a los pacientes sobre los signos y síntomas de la isquemia miocárdica aguda y otros eventos tromboembólicos arteriales y la urgencia de buscar atención médica inmediata para estas afecciones [ver Advertencias y precauciones (5.5)].
Riesgo de sangrado
Se han reportado eventos de sangrado. Avise a los pacientes que reporten cualquier sangrado inusual [ver Advertencias y precauciones (5.6)].
Perforación gastrointestinal
Se han reportado eventos graves de perforación gastrointestinal. Avise a los pacientes que reporten signos y síntomas de perforación gastrointestinal [ver Advertencias y precauciones (5.7)].
Proteinuria de rango nefrótico
Se ha reportado proteinuria de rango nefrótico. Avise a los pacientes que reporten signos y síntomas de proteinuria (por ejemplo, retención de líquidos, orina espumosa) [ver Advertencias y precauciones (5.8)].
Anime a los pacientes a dejar de fumar antes del tratamiento con OFEV y a evitar fumar cuando usen OFEV [ver Farmacología clínica (12.3)].
Administración
Indique a los pacientes que tomen OFEV con alimentos, que traguen las cápsulas de OFEV enteras con líquido y que no mastiquen las cápsulas debido al sabor amargo. Avise a los pacientes o cuidadores que no abran ni trituren las cápsulas de OFEV y que se laven las manos inmediatamente y a fondo si entran en contacto con el contenido de la cápsula. Avise a los pacientes que no compensen una dosis olvidada [ver Dosificación y administración (2.2)].
SECCIÓN NO CLASIFICADA DE SPL
Distribuido por: Boehringer Ingelheim Pharmaceuticals, Inc. Ridgefield, CT 06877 USA
Con licencia de: Boehringer Ingelheim International GmbH
OFEV es una marca registrada de y utilizada bajo licencia de Boehringer Ingelheim International GmbH.
Esta información para el paciente ha sido aprobada por la Administración de Alimentos y Medicamentos de los Estados Unidos.
Revisado: Octubre 2024
Información para el paciente OFEV® (OH-fev) (cápsulas de nintedanib)
¿Cuál es la información más importante que debo saber sobre OFEV?
OFEV puede causar defectos de nacimiento o la muerte de un bebé por nacer. Las mujeres no deben quedar embarazadas mientras toman OFEV. Las mujeres que pueden quedar embarazadas deben hacerse una prueba de embarazo antes de comenzar el tratamiento con OFEV.
Las mujeres que pueden quedar embarazadas deben usar un método anticonceptivo altamente efectivo al inicio del tratamiento, durante el tratamiento y durante al menos 3 meses después del tratamiento. Hable con su médico sobre qué método anticonceptivo es adecuado para usted durante este tiempo.
Las píldoras anticonceptivas pueden no funcionar tan bien en mujeres que tienen vómitos, diarrea u otros problemas que reducen la absorción del medicamento. Si tiene alguno de estos problemas, hable con su médico sobre qué método anticonceptivo altamente efectivo es adecuado para usted.
Si queda embarazada o cree que está embarazada mientras toma OFEV, informe a su médico de inmediato.
¿Qué es OFEV?
OFEV es un medicamento recetado que se usa:
para tratar a adultos con una enfermedad pulmonar llamada fibrosis pulmonar idiopática (FPI).
para tratar a adultos con una enfermedad pulmonar intersticial crónica (de larga duración) en la que la fibrosis pulmonar continúa empeorando (progresando).
para ralentizar la tasa de disminución de la función pulmonar en adultos con enfermedad pulmonar intersticial asociada a esclerodermia (ILD-EE) (también conocida como ILD asociada a esclerodermia).
No se sabe si OFEV es seguro y eficaz en niños.
¿Qué debo decirle a mi médico antes de tomar OFEV? Antes de tomar OFEV, informe a su médico sobre todas sus afecciones médicas, incluso si:
tiene problemas hepáticos.
tiene problemas cardíacos.
tiene antecedentes de coágulos de sangre.
tiene un problema de sangrado o antecedentes familiares de un problema de sangrado.
ha tenido una cirugía reciente en el área del estómago (abdominal).
está amamantando o planea amamantar. No se sabe si OFEV pasa a la leche materna. Usted no debe amamantar mientras toma OFEV.
Informe a su médico sobre todos los medicamentos que toma, incluidos los medicamentos recetados y de venta libre, las vitaminas y los suplementos herbales como la hierba de San Juan. Mantenga una lista de los medicamentos que toma y muéstresela a su médico y farmacéutico cuando obtenga un nuevo medicamento.
¿Cómo debo tomar OFEV?
Tome OFEV exactamente como se lo indique su médico.
Su médico le dirá cuánta OFEV debe tomar y cuándo debe tomarla.
Tome OFEV con alimentos. Trague las cápsulas de OFEV enteras con un líquido.
No mastique, triture ni abra las cápsulas de OFEV. Si usted o su cuidador entran en contacto accidentalmente con el contenido de la cápsula, lávese bien las manos de inmediato.
Si olvida una dosis de OFEV, tome la siguiente dosis a la hora habitual. No tome la dosis olvidada.
No tome más de 300 mg de OFEV en 1 día.
Si toma demasiado OFEV, llame a su médico o vaya a la sala de emergencias del hospital más cercano de inmediato.
Su médico debe realizar ciertos análisis de sangre antes de que comience a tomar OFEV.
¿Cuáles son los posibles efectos secundarios de OFEV? OFEV puede causar efectos secundarios graves, que incluyen:
problemas hepáticos. Llame a su médico de inmediato si tiene síntomas inexplicables como coloración amarillenta de la piel o la parte blanca de los ojos (ictericia), orina oscura o marrón (color té), dolor en la parte superior derecha del área del estómago (abdomen), sangrado o moretones más fácilmente de lo normal, sensación de cansancio o pérdida de apetito. Su médico le hará análisis de sangre para comprobar el funcionamiento de su hígado antes de comenzar y durante su tratamiento con OFEV.
diarrea, náuseas y vómitos. Mientras esté tomando OFEV, su médico puede recomendarle que beba líquidos o tome medicamentos para tratar estos efectos secundarios. Informe a su médico si tiene diarrea, náuseas o vómitos, o si estos síntomas no desaparecen o empeoran. Informe a su médico si está tomando laxantes de venta libre, ablandadores de heces y otros medicamentos o suplementos dietéticos que pueden causar diarrea.
ataque cardíaco. Informe a su médico de inmediato si tiene síntomas de un problema cardíaco. Estos síntomas pueden incluir dolor o presión en el pecho, dolor en los brazos, la espalda, el cuello o la mandíbula, o dificultad para respirar.
derrame cerebral. Informe a su médico de inmediato si tiene síntomas de un derrame cerebral. Estos síntomas pueden incluir entumecimiento o debilidad en un lado del cuerpo, dificultad para hablar, dolor de cabeza o mareos.
problemas de sangrado. OFEV puede aumentar sus posibilidades de tener problemas de sangrado. Informe a su médico si tiene sangrado inusual, moretones o heridas que no cicatrizan. Informe a su médico si está tomando un anticoagulante, incluidos los anticoagulantes recetados y la aspirina de venta libre.
desgarro en el estómago o la pared intestinal (perforación). OFEV puede aumentar sus posibilidades de tener un desgarro en el estómago o la pared intestinal. Informe a su médico si tiene dolor o hinchazón en el área del estómago.
aumento de proteína en la orina (proteinuria). OFEV puede aumentar sus posibilidades de tener proteína en la orina. Informe a su médico si tiene algún signo o síntoma de proteína en la orina, como orina espumosa, hinchazón, incluso en las manos, los brazos, las piernas o los pies, o aumento repentino de peso.
Los efectos secundarios más comunes de OFEV son diarrea, náuseas, dolor de estómago, vómitos, problemas hepáticos, disminución del apetito, dolor de cabeza, pérdida de peso e hipertensión arterial. Estos no son todos los posibles efectos secundarios de OFEV. Para obtener más información, consulte a su médico o farmacéutico. Llame a su médico para obtener asesoramiento médico sobre los efectos secundarios. Puede informar los efectos secundarios a la FDA al 1-800-FDA-1088.
¿Cómo debo almacenar OFEV?
Almacene OFEV a temperatura ambiente entre 68°F y 77°F (20°C y 25°C).
Mantenga OFEV seco y protegido del calor intenso.
Mantenga OFEV y todos los medicamentos fuera del alcance de los niños.
Información general sobre el uso seguro y eficaz de OFEV.
Los medicamentos a veces se recetan para fines distintos de los que se enumeran en un folleto de información para el paciente. No use OFEV para una afección para la que no fue recetado. No le dé OFEV a otras personas, incluso si tienen los mismos síntomas que usted. Puede hacerles daño. Este folleto de información para el paciente resume la información más importante sobre OFEV. Si desea obtener más información, hable con su médico. Puede pedirle a su farmacéutico o médico información sobre OFEV que esté escrita para profesionales de la salud. Para obtener más información sobre OFEV, incluida la información de prescripción actual y el apoyo al paciente, visite www.ofev.com/support o llame a Boehringer Ingelheim Pharmaceuticals, Inc. al 1-800-542-6257, o escanee el código para ir a www.ofev.com/support.
Fabricante de medicamentos: Dr. Reddys Laboratories Inc. (Updated: 2021-12-20) Tags: PAHFenilcetonuria ASPECTOS DESTACADOS DE INFORMACIÓN DE PRESCRIPCIÓN Estos aspectos destacados no incluyen toda la información necesaria para usar SAPROPTERIN DIHYDROCHLORIDE TABLETS y SAPROPTERIN DIHYDROCHLORIDE POWDER FOR ORAL SOLUTION de manera segura y efectiva. Ver la información completa de…
Fabricante de medicamentos: Novo Nordisk (Updated: 2020-07-10) Tags: F10 ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓNEstos aspectos destacados no incluyen toda la información necesaria para usar NOVOSEVEN RT de forma segura y eficaz. Consulte la información completa de prescripción para NOVOSEVEN RT. NOVOSEVEN RT (Factor VIIa de coagulación,…
Fabricante de medicamentos: Puma Biotechnology, Inc. (Updated: 2024-03-25) Tags: PTHR ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓNEstas aspectos destacados no incluyen toda la información necesaria para usar NERLYNX de forma segura y eficaz. Consulte la información de prescripción completa para NERLYNX. NERLYNX ®(neratinib) comprimidos, para uso oral Aprobación…