Fabricante de medicamentos: Takeda Pharmaceuticals America, Inc. (Updated: 2024-09-09)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
NINLARO® (ixazomib) cápsulas, para administración oral
Aprobación inicial en EE. UU.: 2015
CAMBIOS IMPORTANTES RECIENTES
| Advertencias y precauciones, reacciones cutáneas (5.5) | 3/2024 |
INDICACIONES Y USO
NINLARO es un inhibidor del proteasoma indicado en combinación con lenalidomida y dexametasona para el tratamiento de pacientes con mieloma múltiple que han recibido al menos una terapia previa. (1)
Limitaciones de uso: NINLARO no se recomienda para su uso en el contexto de mantenimiento ni en el mieloma múltiple recién diagnosticado en combinación con lenalidomida y dexametasona fuera de ensayos clínicos controlados. (1)
DOSIFICACIÓN Y ADMINISTRACIÓN
FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
Cápsulas: 4 mg, 3 mg y 2.3 mg (3)
CONTRAINDICACIONES
Ninguna. (4)
ADVERTENCIAS Y PRECAUCIONES
- Trombocitopenia: Controle los recuentos de plaquetas al menos una vez al mes durante el tratamiento y ajuste la dosis, según sea necesario. (2.2, 5.1)
- Toxicidades gastrointestinales: Ajuste la dosis para la diarrea grave, el estreñimiento, las náuseas y los vómitos, según sea necesario. (2.2, 5.2)
- Neuropatía periférica: Controle a los pacientes para detectar síntomas de neuropatía periférica y ajuste la dosis, según sea necesario. (2.2, 5.3)
- Edema periférico: Controle la retención de líquidos. Investigue las causas subyacentes, cuando corresponda. Ajuste la dosis, según sea necesario. (2.2, 5.4)
- Reacciones cutáneas: Controle a los pacientes para detectar erupciones cutáneas y ajuste la dosis, según sea necesario. (2.2, 5.5)
- Microangiopatía trombótica: Controle los signos y síntomas. Suspenda NINLARO si se sospecha. (5.6)
- Hepatotoxicidad: Controle las enzimas hepáticas durante el tratamiento. (5.7)
- Toxicidad embriofetal: NINLARO puede causar daño fetal. Informe a las pacientes sobre el riesgo potencial para el feto y sobre el uso de anticonceptivos no hormonales eficaces. (5.8, 8.1, 8.3)
- Mayor mortalidad en pacientes tratados con NINLARO en el contexto de mantenimiento: No se recomienda el tratamiento de pacientes con NINLARO para el mieloma múltiple en el contexto de mantenimiento fuera de ensayos controlados. (5.9)
REACCIONES ADVERSAS
Las reacciones adversas más comunes (≥20%) son trombocitopenia, neutropenia, diarrea, estreñimiento, neuropatía periférica, náuseas, edema periférico, erupción cutánea, vómitos y bronquitis. (6.1)
Para informar REACCIONES ADVERSAS SOSPECHOSAS, comuníquese con Takeda Pharmaceuticals al 1-844-617-6468 o con la FDA al 1-800-FDA-1088 o www.fda.gov/medwatch.
INTERACCIONES CON OTROS MEDICAMENTOS
USO EN POBLACIONES ESPECÍFICAS
- Insuficiencia hepática: Reduzca la dosis inicial de NINLARO a 3 mg en pacientes con insuficiencia hepática moderada o grave. (2.3, 8.6)
- Insuficiencia renal: Reduzca la dosis inicial de NINLARO a 3 mg en pacientes con insuficiencia renal grave o enfermedad renal en etapa terminal que requieren diálisis. (2.4, 8.7)
- Lactancia: Aconseje no amamantar. (8.2)
Consulte 17 para obtener INFORMACIÓN DE ASESORAMIENTO AL PACIENTE y el etiquetado del paciente aprobado por la FDA.
Revisado: 7/2024
Tabla de Contenido
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDO*
1 INDICACIONES Y USO
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Pautas de dosificación y administración
2.2 Pautas de modificación de la dosis
2.3 Dosificación en pacientes con insuficiencia hepática
2.4 Dosificación en pacientes con insuficiencia renal
3 FORMAS Y FUERZAS DE DOSIFICACIÓN
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Trombocitopenia
5.2 Toxicidades gastrointestinales
5.3 Neuropatía periférica
5.4 Edema periférico
5.5 Reacciones cutáneas
5.6 Microangiopatía trombótica
5.7 Hepatotoxicidad
5.8 Toxicidad embrio-fetal
5.9 Aumento de la mortalidad en pacientes tratados con NINLARO en el entorno de mantenimiento
6 REACCIONES ADVERSAS
6.1 Experiencia en ensayos clínicos
6.2 Experiencia postcomercialización
7 INTERACCIONES MEDICAMENTOSAS
7.1 Inductores fuertes del CYP3A
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.3 Mujeres y hombres en edad fértil
8.4 Uso pediátrico
8.5 Uso geriátrico
8.6 Insuficiencia hepática
8.7 Insuficiencia renal
10 SOBREDOSIS
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
12.2 Farmacodinamia
12.3 Farmacocinética
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, mutagénesis, deterioro de la fertilidad
14 ESTUDIOS CLÍNICOS
14.1 Mieloma múltiple en pacientes que han recibido al menos una terapia previa
14.2 Aumento de la mortalidad en pacientes tratados con NINLARO en el entorno de mantenimiento
14.3 Falta de eficacia en pacientes con mieloma múltiple de novo
15 REFERENCIAS
16 CÓMO SE SUMINISTRA/ALMACENAMIENTO Y MANEJO
17 INFORMACIÓN PARA EL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información completa de prescripción no están enumeradas.
1 INDICACIONES Y USO
NINLARO está indicado en combinación con lenalidomida y dexametasona para el tratamiento de pacientes con mieloma múltiple que han recibido al menos una terapia previa.
Limitaciones de uso: NINLARO no se recomienda para su uso en el contexto de mantenimiento ni en el mieloma múltiple recién diagnosticado en combinación con lenalidomida y dexametasona fuera de ensayos clínicos controlados [ver Advertencias y precauciones (5.9) y Estudios clínicos (14.2, 14.3)].
2 DOSIS Y ADMINISTRACIÓN
2.1 Directrices de dosificación y administración
NINLARO en combinación con lenalidomida y dexametasona
La dosis inicial recomendada de NINLARO es de 4 mg administrada por vía oral una vez a la semana los días 1, 8 y 15 de un ciclo de tratamiento de 28 días.
La dosis inicial recomendada de lenalidomida es de 25 mg administrada diariamente los días 1 a 21 de un ciclo de tratamiento de 28 días.
La dosis inicial recomendada de dexametasona es de 40 mg administrada los días 1, 8, 15 y 22 de un ciclo de tratamiento de 28 días.
| Ciclo de 28 días (un ciclo de 4 semanas) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Semana 1 | Semana 2 | Semana 3 | Semana 4 | |||||
| Día 1 | Días 2-7 | Día 8 | Días 9-14 | Día 15 | Días 16-21 | Día 22 | Días 23-28 | |
| NINLARO | ✔ | ✔ | ✔ | |||||
| Lenalidomida | ✔ | ✔ Diariamente | ✔ | ✔ Diariamente | ✔ | ✔ Diariamente | ||
| Dexametasona | ✔ | ✔ | ✔ | ✔ | ||||
2.1 Posología y administración
Para obtener información adicional sobre lenalidomida y dexametasona, consulte su información de prescripción.
NINLARO debe tomarse una vez por semana el mismo día y aproximadamente a la misma hora durante las tres primeras semanas de un ciclo de cuatro semanas. Se debe hablar con los pacientes que comienzan el tratamiento sobre la importancia de seguir cuidadosamente todas las instrucciones de dosificación. Indique a los pacientes que tomen la dosis recomendada según las indicaciones, ya que la sobredosis ha provocado muertes [ver Sobredosis (10)].
NINLARO debe tomarse al menos una hora antes o al menos dos horas después de las comidas [ver Farmacología clínica (12.3)]. La cápsula entera debe tragarse con agua. La cápsula no debe triturarse, masticarse ni abrirse [ver Presentación/Conservación y manipulación (16)].
Si se retrasa o se olvida una dosis de NINLARO, la dosis debe tomarse solo si la siguiente dosis programada está a ≥72 horas de distancia. No se debe tomar una dosis olvidada dentro de las 72 horas de la siguiente dosis programada. No se debe tomar una dosis doble para compensar la dosis olvidada.
Si se produce vómito después de tomar una dosis, el paciente no debe repetir la dosis. El paciente debe reanudar la dosificación en el momento de la siguiente dosis programada.
Antes de iniciar un nuevo ciclo de terapia:
- El recuento absoluto de neutrófilos debe ser de al menos 1.000/mm3
- El recuento de plaquetas debe ser de al menos 75.000/mm3
- Las toxicidades no hematológicas, a discreción del profesional sanitario, generalmente deben recuperarse hasta el estado basal del paciente o hasta el grado 1 o inferior
El tratamiento debe continuarse hasta la progresión de la enfermedad o la toxicidad inaceptable.
Medicamentos concomitantes
Considere la profilaxis antiviral en pacientes tratados con NINLARO para disminuir el riesgo de reactivación del herpes zóster [ver Reacciones adversas (6.1)].
2.2 Pautas de modificación de la dosis
Los pasos de reducción de la dosis de NINLARO se presentan en la Tabla 2 y las pautas de modificación de la dosis se proporcionan en la Tabla 3.
|
|||
| Dosis inicial recomendada* | Primera reducción a | Segunda reducción a | Interrumpir |
| 4 mg | 3 mg | 2.3 mg | |
Se recomienda un enfoque de modificación de dosis alterna para NINLARO y lenalidomida para trombocitopenia, neutropenia y erupción cutánea, como se describe en la Tabla 3. Consulte la información de prescripción de lenalidomida si es necesario reducir la dosis de lenalidomida.
| Toxicidades hematológicas | Acciones recomendadas |
| Trombocitopenia (Recuento de plaquetas) | |
| Recuento de plaquetas inferior a 30 000/mm3 |
|
| Neutropenia (Recuento absoluto de neutrófilos) | |
| Recuento absoluto de neutrófilos inferior a 500/mm3 |
|
| Toxicidades no hematológicas | Acciones recomendadas |
| Erupción cutánea | |
| Grado† 2 o 3 |
|
| Grado 4 | Suspenda el régimen de tratamiento. |
| Neuropatía periférica | |
| Neuropatía periférica de Grado 1 con dolor o Neuropatía periférica de Grado 2 |
|
| Neuropatía periférica de Grado 2 con dolor o Neuropatía periférica de Grado 3 |
|
| Neuropatía periférica de Grado 4 | Suspenda el régimen de tratamiento. |
| Otras toxicidades no hematológicas | |
| Otras toxicidades no hematológicas de Grado 3 o 4 |
|
2.3 Dosificación en pacientes con insuficiencia hepática
Reduzca la dosis inicial de NINLARO a 3 mg en pacientes con insuficiencia hepática moderada (bilirrubina total mayor que 1,5-3 × LSN) o grave (bilirrubina total mayor que 3 × LSN) [ver Uso en poblaciones específicas (8.6) y Farmacología clínica (12.3)].
2.4 Dosificación en pacientes con insuficiencia renal
Reduzca la dosis inicial de NINLARO a 3 mg en pacientes con insuficiencia renal grave (aclaramiento de creatinina menor que 30 mL/min) o enfermedad renal en etapa terminal (ESRD) que requiere diálisis. NINLARO no es dializable y, por lo tanto, se puede administrar sin tener en cuenta el momento de la diálisis [ver Uso en poblaciones específicas (8.7) y Farmacología clínica (12.3)].
Consulte la información de prescripción de lenalidomida para obtener recomendaciones de dosificación en pacientes con insuficiencia renal.
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
NINLARO está disponible en las siguientes cápsulas:
- 4 mg de ixazomib: Cápsula de gelatina naranja claro con “Takeda” impreso en la tapa y “4 mg” en el cuerpo en tinta negra.
- 3 mg de ixazomib: Cápsula de gelatina gris claro con “Takeda” impreso en la tapa y “3 mg” en el cuerpo en tinta negra.
- 2.3 mg de ixazomib: Cápsula de gelatina rosa claro con “Takeda” impreso en la tapa y “2.3 mg” en el cuerpo en tinta negra.
4 CONTRAINDICACIONES
Ninguna.
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Trombocitopenia
Se ha informado trombocitopenia con NINLARO, con nadirs de plaquetas que ocurren típicamente entre los días 14-21 de cada ciclo de 28 días y recuperación a la línea de base para el inicio del siguiente ciclo [ver Reacciones adversas (6.1)]. Se informó trombocitopenia de grado 3 en el 17% de los pacientes en el régimen de NINLARO y trombocitopenia de grado 4 se informó en el 13% en el régimen de NINLARO. La tasa de transfusiones de plaquetas fue del 10% en el régimen de NINLARO y del 7% en el régimen de placebo.
Monitorear los recuentos de plaquetas al menos mensualmente durante el tratamiento con NINLARO. Considere un monitoreo más frecuente durante los primeros tres ciclos. Manejar la trombocitopenia con modificaciones de dosis [ver Dosificación y administración (2.2)] y transfusiones de plaquetas según las pautas médicas estándar.
5.2 Toxicidades gastrointestinales
Se ha informado diarrea, estreñimiento, náuseas y vómitos con NINLARO, ocasionalmente requiriendo el uso de medicamentos antidiarreicos y antieméticos, y atención de apoyo. La diarrea se informó en el 52% de los pacientes en el régimen de NINLARO y el 43% en el régimen de placebo, el estreñimiento en el 35% y el 28%, respectivamente, las náuseas en el 32% y el 23%, respectivamente, y los vómitos en el 26% y el 13%, respectivamente. La diarrea resultó en la interrupción de uno o más de los tres medicamentos en el 3% de los pacientes en el régimen de NINLARO y el 2% de los pacientes en el régimen de placebo [ver Reacciones adversas (6.1)]. Ajuste la dosis para síntomas de grado 3 o 4 [ver Dosificación y administración (2.2)].
5.3 Neuropatía periférica
La mayoría de las reacciones adversas de neuropatía periférica fueron de grado 1 (18% en el régimen de NINLARO y 16% en el régimen de placebo) y de grado 2 (11% en el régimen de NINLARO y 6% en el régimen de placebo) [ver Reacciones adversas (6.1)]. Se informaron reacciones adversas de grado 3 de neuropatía periférica en el 2% en ambos regímenes.
La reacción más comúnmente informada fue la neuropatía sensorial periférica (24% y 17% en el régimen de NINLARO y placebo, respectivamente). La neuropatía motora periférica no se informó comúnmente en ninguno de los regímenes (<1%). La neuropatía periférica resultó en la interrupción de uno o más de los tres medicamentos en el 4% de los pacientes en el régimen de NINLARO y <1% de los pacientes en el régimen de placebo. Los pacientes deben ser monitoreados para detectar síntomas de neuropatía. Los pacientes que experimenten neuropatía periférica nueva o que empeore pueden requerir una modificación de la dosis [ver Dosificación y administración (2.2)].
5.4 Edema periférico
Se informó edema periférico en el 27% y el 21% de los pacientes en los regímenes de NINLARO y placebo, respectivamente. La mayoría de las reacciones adversas de edema periférico fueron de grado 1 (17% en el régimen de NINLARO y 14% en el régimen de placebo) y de grado 2 (7% en el régimen de NINLARO y 6% en el régimen de placebo).
Se informó edema periférico de grado 3 en el 2% y el 1% de los pacientes en los regímenes de NINLARO y placebo, respectivamente [ver Reacciones adversas (6.1)]. El edema periférico resultó en la interrupción de uno o más de los tres medicamentos en <1% de los pacientes en ambos regímenes. Evaluar las causas subyacentes y brindar atención de apoyo, según sea necesario. Ajuste la dosis de dexametasona según su información de prescripción o NINLARO para síntomas de grado 3 o 4 [ver Dosificación y administración (2.2)].
5.5 Reacciones cutáneas
Se informó erupción cutánea en el 27% de los pacientes en el régimen de NINLARO y el 16% de los pacientes en el régimen de placebo. La mayoría de las reacciones adversas de erupción cutánea fueron de grado 1 (15% en el régimen de NINLARO y 9% en el régimen de placebo) o de grado 2 (9% en el régimen de NINLARO y 4% en el régimen de placebo) [ver Reacciones adversas (6.1)]. Se informó erupción cutánea de grado 3 en el 3% de los pacientes en el régimen de NINLARO y el 2% de los pacientes en el régimen de placebo. Se informaron reacciones adversas graves de erupción cutánea en <1% de los pacientes en el régimen de NINLARO. El tipo más común de erupción cutánea informado en ambos regímenes incluyó erupción maculo-papular y macular. La erupción cutánea resultó en la interrupción de uno o más de los tres medicamentos en <1% de los pacientes en ambos regímenes. Manejar la erupción cutánea con atención de apoyo o con modificación de la dosis si es de grado 2 o superior [ver Dosificación y administración (2.2)].
Se ha informado síndrome de Stevens-Johnson y necrólisis epidérmica tóxica, incluidos casos fatales, con NINLARO [ver Reacciones adversas (6.1, 6.2)]. Si se produce síndrome de Stevens-Johnson o necrólisis epidérmica tóxica, suspenda NINLARO y maneje como se indique clínicamente.
5.6 Microangiopatía trombótica
Se han informado casos, a veces fatales, de microangiopatía trombótica, incluida la púrpura trombocitopénica trombótica/síndrome urémico hemolítico (TTP/HUS), en pacientes que recibieron NINLARO [ver Reacciones adversas (6.1)]. Monitorear los signos y síntomas de TTP/HUS. Si se sospecha el diagnóstico, suspenda NINLARO y evalúe. Si se excluye el diagnóstico de TTP/HUS, considere reiniciar NINLARO. La seguridad de reiniciar el tratamiento con NINLARO en pacientes que previamente experimentaron TTP/HUS no se conoce.
5.7 Hepatotoxicidad
Se ha notificado lesión hepática inducida por fármacos, lesión hepatocelular, esteatosis hepática, hepatitis colestásica y hepatotoxicidad en <1% de los pacientes tratados con NINLARO [ver Reacciones adversas (6.1)]. Se ha notificado hepatotoxicidad (10% en el régimen de NINLARO y 9% en el régimen de placebo). Controle las enzimas hepáticas regularmente y ajuste la dosis para los síntomas de Grado 3 o 4 [ver Dosificación y administración (2.2)].
5.8 Toxicidad embrio-fetal
NINLARO puede causar daño fetal cuando se administra a una mujer embarazada en base al mecanismo de acción y los hallazgos en estudios en animales. Ixazomib causó toxicidad embrio-fetal en ratas y conejos embarazadas a dosis que resultaron en exposiciones que fueron ligeramente superiores a las observadas en pacientes que recibieron la dosis recomendada. Advierta a las mujeres embarazadas del riesgo potencial para el feto. Aconseje a las mujeres en edad fértil que utilicen una contracepción no hormonal eficaz durante el tratamiento con NINLARO y durante 90 días después de la última dosis. Aconseje a los hombres con parejas femeninas en edad fértil que utilicen una contracepción eficaz durante el tratamiento con NINLARO y durante 90 días después de la última dosis [ver Interacciones medicamentosas (7.1) y Uso en poblaciones específicas (8.1, 8.3)].
5.9 Aumento de la mortalidad en pacientes tratados con NINLARO en el entorno de mantenimiento
En dos ensayos clínicos prospectivos aleatorizados en mieloma múltiple en el entorno de mantenimiento, el tratamiento con NINLARO resultó en un aumento de las muertes. No se recomienda el tratamiento de pacientes con NINLARO para el mieloma múltiple en el entorno de mantenimiento fuera de los ensayos controlados [ver Estudios clínicos (14.2)].
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas se describen en detalle en otras secciones de la información de prescripción:
- Trombocitopenia [ver Advertencias y precauciones (5.1)]
- Toxicidades gastrointestinales [ver Advertencias y precauciones (5.2)]
- Neuropatía periférica [ver Advertencias y precauciones (5.3)]
- Edema periférico [ver Advertencias y precauciones (5.4)]
- Reacciones cutáneas [ver Advertencias y precauciones (5.5)]
- Microangiopatía trombótica [ver Advertencias y precauciones (5.6)]
- Hepatotoxicidad [ver Advertencias y precauciones (5.7)]
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se llevan a cabo en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no se pueden comparar directamente con las tasas en los ensayos clínicos de otro fármaco y pueden no reflejar las tasas observadas en la práctica.
La población de seguridad del estudio clínico aleatorizado, doble ciego, controlado con placebo incluyó 720 pacientes con mieloma múltiple recidivante y/o refractario, que recibieron NINLARO en combinación con lenalidomida y dexametasona (régimen NINLARO; N=361) o placebo en combinación con lenalidomida y dexametasona (régimen placebo; N=359).
Las reacciones adversas más frecuentes (≥20% con una diferencia de ≥5% en comparación con placebo) en el régimen NINLARO fueron trombocitopenia, neutropenia, diarrea, estreñimiento, neuropatía periférica, náuseas, edema periférico, erupción cutánea, vómitos y bronquitis. Las reacciones adversas graves notificadas en ≥2% de los pacientes en el régimen NINLARO incluyeron diarrea (3%), trombocitopenia (2%) y bronquitis (2%). Uno o más de los tres fármacos se suspendieron permanentemente en el 4% de los pacientes que notificaron neuropatía periférica, el 3% de los pacientes que notificaron diarrea y el 2% de los pacientes que notificaron trombocitopenia. La suspensión permanente de NINLARO debido a una reacción adversa se produjo en el 10% de los pacientes.
La Tabla 4 resume las reacciones adversas no hematológicas que se producen en al menos el 5% de los pacientes con una diferencia de al menos el 5% entre el régimen NINLARO y el régimen placebo.
| Clase de órgano del sistema / Término preferido |
NINLARO + Lenalidomida y Dexametasona N=361 |
Placebo + Lenalidomida y Dexametasona N=359 |
||||
|---|---|---|---|---|---|---|
| % | % | |||||
| Todos los grados | Grado 3 | Grado 4 | Todos los grados | Grado 3 | Grado 4 | |
| Nota: Las reacciones adversas incluidas como términos preferidos se basan en la versión 23.0 de MedDRA. | ||||||
| Trastornos gastrointestinales | ||||||
| Diarrea | 52 | 10 | 0 | 43 | 3 | 0 |
| Estreñimiento | 35 | <1 | 0 | 28 | <1 | 0 |
| Náuseas | 32 | 2 | 0 | 23 | 0 | 0 |
| Vómitos | 26 | 1 | 0 | 13 | <1 | 0 |
| Trastornos del sistema nervioso | ||||||
| Neuropatías periféricas* | 32 | 2 | 0 | 24 | 2 | 0 |
| Trastornos musculoesqueléticos y del tejido conjuntivo | ||||||
| Dolor de espalda† | 27 | <1 | 0 | 24 | 3 | 0 |
| Infecciones e infestaciones | ||||||
| Infección de las vías respiratorias altas† | 27 | 1 | 0 | 23 | 1 | 0 |
| Bronquitis | 22 | 2 | 0 | 17 | 2 | <1 |
| Trastornos de la piel y del tejido subcutáneo | ||||||
| Erupción* | 27 | 3 | 0 | 16 | 2 | 0 |
| Trastornos generales y condiciones del lugar de administración | ||||||
| Edema periférico | 27 | 2 | 0 | 21 | 1 | 0 |
La tabla 5 representa información agrupada de datos de eventos adversos y de laboratorio.
| NINLARO + Lenalidomida y Dexametasona N=361 |
Placebo + Lenalidomida y Dexametasona N=359 |
|||
|---|---|---|---|---|
| % | % | |||
| Cualquier Grado | Grado 3-4 | Cualquier Grado | Grado 3-4 | |
| Trombocitopenia | 85 | 30 | 67 | 14 |
| Neutropenia | 74 | 34 | 70 | 37 |
Herpes Zoster
Se informó herpes zóster en el 6% de los pacientes en el régimen de NINLARO y en el 3% de los pacientes en el régimen de placebo. La profilaxis antiviral se permitió a discreción del profesional sanitario. Los pacientes tratados en el régimen de NINLARO que recibieron profilaxis antiviral tuvieron una incidencia menor (1%) de infección por herpes zóster en comparación con los pacientes que no recibieron profilaxis (10%).
Trastornos oculares
Se informaron trastornos oculares con muchos términos preferidos diferentes, pero en conjunto, la frecuencia fue del 38% en los pacientes en el régimen de NINLARO. Las reacciones adversas más comunes de los ojos fueron cataratas (15%), conjuntivitis (9%), visión borrosa (7%) y ojo seco (6%).
Otra experiencia en ensayos clínicos
Las siguientes reacciones adversas graves se han informado cada una con una frecuencia de <1% en pacientes tratados con NINLARO: dermatosis neutrófila febril aguda (síndrome de Sweet), síndrome de Stevens-Johnson, mielitis transversa, síndrome de encefalopatía reversible posterior, síndrome de lisis tumoral y púrpura trombocitopénica trombótica.
6.2 Experiencia postcomercialización
Las siguientes reacciones adversas se han identificado durante el uso posterior a la aprobación de NINLARO. Debido a que estas reacciones se notifican voluntariamente de una población de tamaño incierto, no siempre es posible estimar de forma fiable su frecuencia o establecer una relación causal con la exposición al fármaco.
Trastornos del sistema inmunitario: Angioedema
Trastornos de la piel y del tejido subcutáneo: Necrólisis epidérmica tóxica
7 INTERACCIONES MEDICAMENTOSAS
7.1 Inductores potentes del CYP3A
Evite la administración concomitante de NINLARO con inductores potentes del CYP3A (como rifampicina, fenitoína, carbamazepina y hierba de San Juan) [consulte Farmacología clínica (12.3)].
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de Riesgos
Basado en su mecanismo de acción [ver Farmacología Clínica (12.1)] y datos de estudios de reproducción en animales, NINLARO puede causar daño fetal cuando se administra a una mujer embarazada. No hay datos disponibles sobre el uso de NINLARO en mujeres embarazadas para evaluar el riesgo asociado al fármaco. Ixazomib causó toxicidad embrio-fetal en ratas y conejas embarazadas a dosis que resultaron en exposiciones ligeramente superiores a las observadas en pacientes que recibieron la dosis recomendada (ver Datos). Advierta a las mujeres embarazadas sobre el riesgo potencial para el feto.
En la población general de los EE. UU., el riesgo de fondo estimado de defectos de nacimiento mayores y aborto espontáneo en embarazos clínicamente reconocidos es del 2-4% y del 15-20%, respectivamente.
Datos
Datos en Animales
En un estudio de desarrollo embrio-fetal en conejas embarazadas, hubo aumentos en las variaciones/anomalías esqueléticas fetales (vértebras caudales fusionadas, número de vértebras lumbares y costillas supernumerarias completas) a dosis que también fueron tóxicas para la madre (≥0.3 mg/kg). Las exposiciones en el conejo a 0.3 mg/kg fueron 1.9 veces las exposiciones promedio en el tiempo clínico a la dosis recomendada de 4 mg. En un estudio de desarrollo embrio-fetal de búsqueda de rango de dosis en ratas, a dosis que fueron tóxicas para la madre, hubo disminuciones en los pesos fetales, una tendencia hacia la disminución de la viabilidad fetal y un aumento de las pérdidas postimplantación a 0.6 mg/kg. Las exposiciones en ratas a la dosis de 0.6 mg/kg fueron 2.5 veces las exposiciones promedio en el tiempo clínico a la dosis recomendada de 4 mg.
8.2 Lactancia
Resumen de Riesgos
No hay datos sobre la presencia de ixazomib o sus metabolitos en la leche materna, los efectos del fármaco en el lactante amamantado o los efectos del fármaco en la producción de leche. Debido al potencial de reacciones adversas graves de NINLARO en un lactante amamantado, se debe aconsejar a las mujeres que no amamanten durante el tratamiento con NINLARO y durante 90 días después de la última dosis.
8.3 Mujeres y Hombres en Edad Reproductiva
NINLARO puede causar daño fetal cuando se administra a mujeres embarazadas [ver Uso en Poblaciones Específicas (8.1)].
Prueba de Embarazo
Verifique el estado de embarazo en mujeres en edad reproductiva antes de iniciar el tratamiento con NINLARO.
Anticoncepción
Mujeres
Se debe aconsejar a las mujeres en edad reproductiva que utilicen métodos anticonceptivos no hormonales efectivos durante el tratamiento con NINLARO y durante 90 días después de la última dosis. Se sabe que la dexametasona es un inductor débil a moderado de CYP3A4, así como de otras enzimas y transportadores. Debido a que NINLARO se administra con dexametasona, es necesario considerar el riesgo de reducción de la eficacia de los anticonceptivos [ver Interacciones Medicamentosas (7.1)].
8.4 Uso Pediátrico
No se ha establecido la seguridad y eficacia de NINLARO en pacientes pediátricos.
8.5 Uso Geriátrico
Del total de sujetos en los estudios clínicos de NINLARO, el 55% tenía 65 años o más, mientras que el 17% tenía 75 años o más. No se observaron diferencias generales en la seguridad o eficacia entre estos sujetos y los sujetos más jóvenes, y otras experiencias clínicas reportadas no han identificado diferencias en las respuestas entre los pacientes ancianos y los pacientes más jóvenes, pero no se puede descartar una mayor sensibilidad de algunos individuos mayores.
8.6 Insuficiencia Hepática
En pacientes con insuficiencia hepática moderada o grave, el AUC promedio aumentó un 20% en comparación con los pacientes con función hepática normal. Reduzca la dosis inicial de NINLARO en pacientes con insuficiencia hepática moderada o grave [ver Dosificación y Administración (2.3), Farmacología Clínica (12.3)].
8.7 Insuficiencia Renal
En pacientes con insuficiencia renal grave o ESRD que requieren diálisis, el AUC promedio aumentó un 39% en comparación con los pacientes con función renal normal. Reduzca la dosis inicial de NINLARO en pacientes con insuficiencia renal grave o ESRD que requieren diálisis. NINLARO no es dializable y, por lo tanto, se puede administrar sin tener en cuenta el momento de la diálisis [ver Dosificación y Administración (2.4), Farmacología Clínica (12.3)].
10 SOBREDOSIS
Se ha notificado sobredosis, incluida la sobredosis mortal, en pacientes que toman NINLARO. Las manifestaciones de sobredosis incluyen reacciones adversas notificadas a la dosis recomendada [ver Dosificación y administración (2.1), Reacciones adversas (6.1)]. Las reacciones adversas graves notificadas con sobredosis incluyen náuseas graves, vómitos, diarrea, neumonía por aspiración, insuficiencia multiorgánica y muerte.
En caso de sobredosis, controlar las reacciones adversas y proporcionar atención de apoyo adecuada. NINLARO no es dializable.
11 DESCRIPCIÓN
Ixazomib es un inhibidor del proteasoma. Ixazomib citrato, un profármaco, se hidroliza rápidamente en condiciones fisiológicas a su forma biológicamente activa, ixazomib. El nombre químico del citrato de ixazomib es ácido 1,3,2-dioxaborolano-4,4-diacético, 2 – [(1 R ) -1 – [[2 – [(2,5-diclorobenzoyl)amino]acetil]amino] -3-metilbutil] -5-oxo y la fórmula estructural es:
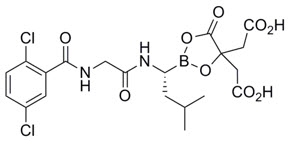
La fórmula molecular del citrato de ixazomib es C 20H 23BCl 2N 2O 9 y su peso molecular es 517,12. El citrato de ixazomib tiene un centro quirál y es el estereoisómero R. La solubilidad del citrato de ixazomib en 0,1N HCl (pH 1,2) a 37 °C es 0,61 mg/mL (reportado como ixazomib). La solubilidad aumenta a medida que aumenta el pH.
NINLARO (ixazomib) cápsulas para uso oral contienen 4, 3 o 2,3 mg de ixazomib equivalente a 5,7, 4,3 o 3,3 mg de citrato de ixazomib, respectivamente. Los ingredientes inactivos incluyen celulosa microcristalina, estearato de magnesio y talco. Las cápsulas contienen gelatina y dióxido de titanio. La cápsula de 4 mg contiene óxido de hierro rojo y amarillo, la cápsula de 3 mg contiene óxido de hierro negro y la cápsula de 2,3 mg contiene óxido de hierro rojo. La tinta de impresión contiene goma laca, propilenglicol, hidróxido de potasio y óxido de hierro negro.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
La ixazomib es un inhibidor reversible del proteasoma. La ixazomib se une preferentemente e inhibe la actividad quimotripsina-like de la subunidad beta 5 del proteasoma 20S.
La ixazomib indujo apoptosis en líneas celulares de mieloma múltiple in vitro. La ixazomib demostró citotoxicidad in vitro contra células de mieloma de pacientes que habían recaído después de múltiples terapias previas, incluyendo bortezomib, lenalidomida y dexametasona. La combinación de ixazomib y lenalidomida demostró efectos citotóxicos sinérgicos en líneas celulares de mieloma múltiple. In vivo, la ixazomib demostró actividad antitumoral en un modelo de xenoinjerto tumoral de mieloma múltiple en ratones.
12.3 Farmacocinética
Absorción
Después de la administración oral, el tiempo medio para alcanzar las concentraciones plasmáticas máximas de ixazomib fue una hora. La biodisponibilidad oral absoluta media fue del 58%, basada en el análisis PK poblacional. El AUC de la ixazomib aumenta de manera proporcional a la dosis en un rango de dosis de 0.2 a 10.6 mg.
Un estudio del efecto de la comida realizado en pacientes con una sola dosis de 4 mg de ixazomib mostró que una comida alta en grasas disminuyó el AUC de la ixazomib en un 28% y el Cmax en un 69% [véase Dosis y administración (2.1)].
Distribución
La ixazomib se une al 99% a las proteínas plasmáticas y se distribuye en los glóbulos rojos con una relación sangre-plasma de 10. El volumen de distribución en estado estacionario es de 543 L.
Eliminación
Basado en un análisis PK poblacional, el aclaramiento sistémico fue de aproximadamente 1.9 L/hora con una variabilidad interindividual del 44%. La vida media terminal (t1/2) de la ixazomib fue de 9.5 días. Después de la administración oral semanal, se determinó que la razón de acumulación era de 2 veces.
Metabolismo
Después de la administración oral de una dosis radiomarcada, la ixazomib representó el 70% del material relacionado con el fármaco en el plasma. Se espera que el metabolismo por múltiples enzimas CYP y proteínas no CYP sea el mecanismo de aclaramiento principal de la ixazomib. En concentraciones clínicamente relevantes de ixazomib, los estudios in vitro utilizando isozimas del citocromo P450 expresadas en cDNA humano mostraron que ninguna isoforma CYP específica contribuye predominantemente al metabolismo de la ixazomib. A concentraciones superiores a las clínicas, la ixazomib fue metabolizada por múltiples isoformas CYP con contribuciones relativas estimadas de 3A4 (42%), 1A2 (26%), 2B6 (16%), 2C8 (6%), 2D6 (5%), 2C19 (5%) y 2C9 (<1%).
Poblaciones específicas
No hubo un efecto clínicamente significativo de la edad (rango 23-91 años), sexo, área de superficie corporal (rango 1.2-2.7 m2) o raza en el aclaramiento de la ixazomib basado en el análisis PK poblacional.
Pacientes con insuficiencia hepática
La PK de la ixazomib fue similar en pacientes con función hepática normal y en pacientes con insuficiencia hepática leve (bilirrubina total ≤ ULN y AST >ULN o bilirrubina total >1-1.5 × ULN y cualquier AST) basado en el análisis PK poblacional.
La PK de la ixazomib se caracterizó en pacientes con función hepática normal a 4 mg (N=12), insuficiencia hepática moderada a 2.3 mg (bilirrubina total >1.5-3 × ULN, N=13) o insuficiencia hepática severa a 1.5 mg (bilirrubina total >3 × ULN, N=18). El AUC medio normalizado por dosis fue un 20% más alto en pacientes con insuficiencia hepática moderada o severa en comparación con pacientes con función hepática normal [véase Dosis y administración (2.3)].
Pacientes con insuficiencia renal
La PK de la ixazomib fue similar en pacientes con función renal normal y en pacientes con insuficiencia renal leve o moderada (aclaramiento de creatinina ≥30 mL/min) basado en el análisis PK poblacional.
La PK de la ixazomib se caracterizó en una dosis de 3 mg en pacientes con función renal normal (aclaramiento de creatinina ≥90 mL/min, N=18), insuficiencia renal severa (aclaramiento de creatinina <30 mL/min, N=14) o ESRD que requiere diálisis (N=6). El AUC medio fue un 39% más alto en pacientes con insuficiencia renal severa o ESRD que requiere diálisis en comparación con pacientes con función renal normal. Las concentraciones de ixazomib antes y después del dializador medidas durante la sesión de hemodiálisis fueron similares, lo que sugiere que la ixazomib no es dializable [véase Dosis y administración (2.4)].
Estudios de interacción farmacológica
Efecto de otros fármacos en NINLARO
Inductores fuertes de CYP3A
Inductores fuertes de CYP3A
La coadministración de NINLARO con rifampicina disminuyó la Cmax de ixazomib en un 54% y el AUC en un 74% [ver Interacciones medicamentosas (7.1)].
Efecto de NINLARO sobre otros medicamentos
Ixazomib no es un inhibidor reversible ni dependiente del tiempo de las CYPs 1A2, 2B6, 2C8, 2C9, 2C19, 2D6 o 3A4/5. Ixazomib no indujo la actividad de CYP1A2, CYP2B6 y CYP3A4/5 o los niveles correspondientes de proteína inmunorreactiva. No se espera que NINLARO produzca interacciones medicamentosas a través de la inhibición o inducción de CYP.
Interacciones basadas en transportadores
Ixazomib es un sustrato de baja afinidad de P-gp. Ixazomib no es un sustrato de BCRP, MRP2 o OATPs hepáticos. Ixazomib no es un inhibidor de P-gp, BCRP, MRP2, OATP1B1, OATP1B3, OCT2, OAT1, OAT3, MATE1 o MATE2-K. No se espera que NINLARO cause interacciones medicamentosas mediadas por transportadores.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
Ixazomib no fue mutagénico en una prueba de mutación inversa bacteriana (prueba de Ames). Ixazomib se consideró positivo en una prueba de clastogenicidad in vitro en linfocitos de sangre periférica humana. Sin embargo, in vivo, ixazomib no fue clastogénico en una prueba de micronúcleos de médula ósea en ratones y fue negativo en una prueba de cometa in vivo en ratones, según se evaluó en el estómago y el hígado. No se han realizado estudios de carcinogenicidad con ixazomib.
Los estudios de toxicidad del desarrollo en ratas y conejos no mostraron toxicidad embrio-fetal directa por debajo de las dosis maternas tóxicas de ixazomib. No se realizaron estudios de fertilidad y desarrollo embrionario temprano y toxicología pre y posnatal con ixazomib, pero se realizó una evaluación de los tejidos reproductivos en los estudios de toxicidad general. No hubo efectos debido al tratamiento con ixazomib en los órganos reproductivos masculinos o femeninos en estudios de hasta 6 meses de duración en ratas y hasta 9 meses de duración en perros.
14 ESTUDIOS CLÍNICOS
14.1 Mieloma múltiple en pacientes que han recibido al menos un tratamiento previo
La eficacia y seguridad de NINLARO en combinación con lenalidomida y dexametasona se evaluó en un estudio multicéntrico, aleatorizado, doble ciego, controlado con placebo en pacientes con mieloma múltiple recurrente y/o refractario que habían recibido al menos una línea previa de tratamiento. Los pacientes refractarios a lenalidomida o inhibidores de la proteasoma fueron excluidos del estudio.
Un total de 722 pacientes fueron aleatorizados en una proporción de 1:1 para recibir ya sea la combinación de NINLARO, lenalidomida y dexametasona (N = 360; régimen de NINLARO) o la combinación de placebo, lenalidomida y dexametasona (N = 362; régimen de placebo) hasta la progresión de la enfermedad o toxicidad inaceptable. La aleatorización se estratificó según el número de líneas de tratamiento previo (1 frente a 2 o 3), el sistema de estadificación internacional del mieloma (ISS) (etapa I o II frente a III) y el tratamiento previo con un inhibidor de la proteasoma (expuesto o no expuesto). El veintitrés por ciento (N = 166) de los pacientes tenían enfermedad de cadena ligera y el 12% (N = 87) de los pacientes tenían enfermedad medible solo por cadenas ligeras libres.
Se recomendó la profilaxis antitrombótica para todos los pacientes en ambos grupos de tratamiento de acuerdo con la información de prescripción de lenalidomida. Los antieméticos se utilizaron en el 19% de los pacientes en el régimen de NINLARO y en el 12% de los pacientes en el régimen de placebo; los antivirales en el 64% y 60%, respectivamente, y los antihistamínicos en el 27% y 19%, respectivamente. Estos medicamentos se administraron a los pacientes a discreción del proveedor de atención médica como profilaxis y/o tratamiento de síntomas.
Los pacientes recibieron NINLARO 4 mg o placebo en los días 1, 8 y 15 más lenalidomida (25 mg) en los días 1 al 21 y dexametasona (40 mg) en los días 1, 8, 15 y 22 de un ciclo de 28 días. Los pacientes con insuficiencia renal recibieron una dosis inicial de lenalidomida de acuerdo con su información de prescripción. El tratamiento continuó hasta la progresión de la enfermedad o toxicidades inaceptables.
La Tabla 6 resume las características basales de los pacientes y la enfermedad en el estudio. Las características demográficas y de la enfermedad basales fueron equilibradas y comparables entre los regímenes de estudio.
| NINLARO + Lenalidomida y Dexametasona (N = 360) |
Placebo + Lenalidomida y Dexametasona (N = 362) |
|
|---|---|---|
|
||
| Características de los pacientes | ||
| Edad media en años (rango) | 66 (38, 91) | 66 (30, 89) |
| Género (%) Hombre/ Mujer | 58/42 | 56/44 |
| Grupo de edad (% [<65/ ≥65 años]) | 41/59 | 43/57 |
| Raza n (%) | ||
| Blanco | 310 (86) | 301 (83) |
| Negro | 7 (2) | 6 (2) |
| Asiático | 30 (8) | 34 (9) |
| Otro o no especificado | 13 (4) | 21 (6) |
| Estado de rendimiento ECOG, n (%) | ||
| 0 o 1 | 336 (93) | 334 (92) |
| 2 | 18 (5) | 24 (7) |
| Faltante | 6 (2) | 4 (1) |
| Aclaramiento de creatinina, n (%) | ||
| <30 mL/min | 5 (1) | 5 (1) |
| 30-59 mL/min | 74 (21) | 95 (26) |
| ≥60 mL/min | 281 (78) | 261 (72) |
| Características de la enfermedad | ||
| Etapa del ISS del mieloma, n (%) | ||
| Etapa I o II | 315 (87) | 320 (88) |
| Etapa III | 45 (13) | 42 (12) |
| Tratamientos previos n (%) | ||
| Mediana (rango) | 1 (1, 3) | 1 (1, 3) |
| 1 | 224 (62) | 217 (60) |
| 2 or 3 | 136 (38) | 145 (40) |
| Status at Baseline n (%) | ||
| Relapsed | 276 (77) | 280 (77) |
| Refractory* | 42 (12) | 40 (11) |
| Relapsed and Refractory | 41 (11) | 42 (12) |
| Type of Prior Therapy n (%) | ||
| Bortezomib containing | 248 (69) | 250 (69) |
| Carfilzomib containing | 1 (<1) | 4 (1) |
| Thalidomide containing | 157 (44) | 170 (47) |
| Lenalidomide containing | 44 (12) | 44 (12) |
| Melphalan containing | 293 (81) | 291 (80) |
| Stem cell transplantation | 212 (59) | 199 (55) |
| High risk (deletion (del) 17, t(4:14) and/or t(14:16) | 75 (21) | 62 (17) |
| deletion del (17) | 36 (10) | 33 (9) |
La eficacia de NINLARO se evaluó mediante la supervivencia sin progresión (SSP) según los Criterios de Respuesta Uniformes del Consenso del Grupo de Trabajo Internacional sobre Mieloma (IMWG) de 2011, según la evaluación de un comité de revisión independiente ciego (CRI) basada en los resultados del laboratorio central. La respuesta se evaluó cada cuatro semanas hasta la progresión de la enfermedad.
La aprobación de NINLARO se basó en una mejora estadísticamente significativa en la SSP del régimen de NINLARO en comparación con el régimen de placebo. Los resultados de la SSP se resumen en la Tabla 7 y se muestran en la Figura 1.
| NINLARO + Lenalidomida y Dexametasona (N = 360) |
Placebo + Lenalidomida y Dexametasona (N = 362) |
|
|---|---|---|
| NE: No evaluable. | ||
| Supervivencia sin progresión | ||
| Eventos de SSP, n (%) | 129 (36) | 157 (43) |
| Mediana (meses) (IC del 95%) |
20.6 (17.0, NE) |
14.7 (12.9, 17.6) |
| Hazard Ratio* (IC del 95%) |
0.74 (0.59, 0.94) |
|
| Valor p† | 0.012 | |
| Tasa de respuesta | ||
| Tasa de respuesta global, n (%) | 282 (78) | 259 (72) |
| Respuesta completa | 42 (12) | 24 (7) |
| Respuesta parcial muy buena | 131 (36) | 117 (32) |
| Respuesta parcial | 109 (30) | 118 (33) |
La mediana del tiempo hasta la respuesta fue de 1,1 meses en el régimen de NINLARO y de 1,9 meses en el régimen de placebo. La mediana de la duración de la respuesta fue de 20,5 meses en el régimen de NINLARO y de 15 meses en el régimen de placebo para los respondedores en la población evaluable de la respuesta.
Figura 1: Gráfico de Kaplan-Meier de la supervivencia libre de progresión
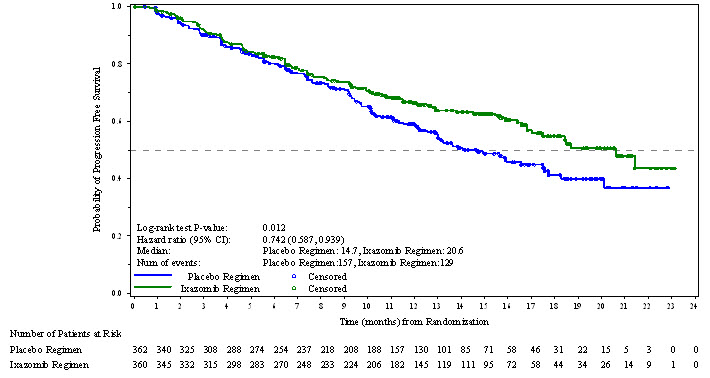
Se realizó un análisis de SLP no inferencial con un seguimiento mediano de 23 meses con 372 eventos de SLP. La razón de riesgo de SLP fue de 0,82 (intervalo de confianza del 95% [0,67, 1,0]) para el régimen de NINLARO frente al régimen de placebo, y la SLP mediana estimada fue de 20 meses en el régimen de NINLARO y de 15,9 meses en el régimen de placebo.
En el análisis final para la SO con una duración mediana de seguimiento de aproximadamente 85 meses, la SO mediana en la población ITT fue de 53,6 meses para los pacientes en el régimen de NINLARO y de 51,6 meses para los pacientes en el régimen de placebo (HR = 0,94 [IC del 95%: 0,78, 1,13]).
14.2 Aumento de la mortalidad en pacientes tratados con NINLARO en el contexto de mantenimiento
En C16019 (NCT02181413), los pacientes con mieloma múltiple de novo que se sometieron a trasplante autólogo de células madre, continuaron con la terapia de mantenimiento durante 24 meses. Hubo un 27% (105/395) de muertes en el brazo de NINLARO en comparación con un 26% (69/261) en el brazo de placebo. La razón de riesgo para la supervivencia general fue de 1,008 (IC del 95%: 0,744 – 1,367).
En C16021 (NCT02312258), los pacientes con mieloma múltiple de novo, no tratados con trasplante de células madre que lograron una respuesta parcial o mejor, continuaron con la terapia de mantenimiento durante 24 meses. Hubo un 30% (127/425) de muertes en el brazo de NINLARO en comparación con un 27% (76/281) en el brazo de placebo. La razón de riesgo para la supervivencia general fue de 1,136 (IC del 95%: 0,853 – 1,514).
No se recomienda el uso de NINLARO en el contexto de mantenimiento para el mieloma múltiple fuera de los ensayos clínicos controlados [ver Indicaciones y uso (1) y Advertencias y precauciones (5.9)].
14.3 Falta de eficacia en pacientes con mieloma múltiple de novo
La falta de eficacia en pacientes con mieloma múltiple de novo se determinó en un ensayo clínico aleatorizado prospectivo.
En C16014 (NCT01850524), en pacientes con mieloma múltiple de novo, el estudio no cumplió con el criterio de valoración primario preespecificado para la SLP. Hubo 136 (39%) muertes en el brazo de NINLARO, lenalidomida y dexametasona en comparación con 148 (42%) en el brazo de lenalidomida y dexametasona. La razón de riesgo para la supervivencia general fue de 0,998 (IC del 95%: 0,79 – 1,261).
No se recomienda el uso de NINLARO en combinación con lenalidomida y dexametasona en el mieloma múltiple de novo fuera de los ensayos clínicos controlados [ver Indicaciones y uso (1)].
15 REFERENCIAS
- OSHA Hazardous Drugs. OSHA. http://www.osha.gov/SLTC/hazardousdrugs/index.html
16 FORMA DE SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
Presentación
NINLARO se suministra de la siguiente manera:
| Concentración por cápsula | Descripción de la cápsula | Caja exterior | Blister de 3 unidades | Blister de 1 unidad | NDC |
|---|---|---|---|---|---|
| 4 mg | Naranja claro, tamaño 3, con la impresión “Takeda” en la tapa y “4 mg” en el cuerpo en tinta negra. | Tres cápsulas de 4 mg en una caja | Cada blister contiene tres cápsulas de 4 mg | Cada blister contiene una cápsula de 4 mg | Caja exterior NDC 63020-400-02 Blister de 3 unidades NDC 63020-400-03 Blister de 1 unidad NDC 63020-400-01 |
| 3 mg | Gris claro, tamaño 4, con la impresión “Takeda” en la tapa y “3 mg” en el cuerpo en tinta negra. | Tres cápsulas de 3 mg en una caja | Cada blister contiene tres cápsulas de 3 mg | Cada blister contiene una cápsula de 3 mg | Caja exterior NDC 63020-390-02 Blister de 3 unidades NDC 63020-390-03 Blister de 1 unidad NDC 63020-390-01 |
| 2.3 mg | Rosa claro, tamaño 4, con la impresión “Takeda” en la tapa y “2.3 mg” en el cuerpo en tinta negra. | Tres cápsulas de 2.3 mg en una caja | Cada blister contiene tres cápsulas de 2.3 mg | Cada blister contiene una cápsula de 2.3 mg | Caja exterior NDC 63020-230-02 Blister de 3 unidades NDC 63020-230-03 Blister de 1 unidad NDC 63020-230-01 |
Las cápsulas se envasan individualmente en un blister de PVC-Aluminio/Aluminio.
Almacenamiento
Almacenar NINLARO a temperatura ambiente. No almacenar por encima de 30°C (86°F). No congelar.
Almacenar las cápsulas en su envase original hasta inmediatamente antes de su uso.
Manipulación y eliminación
NINLARO es un fármaco peligroso. Siga los procedimientos especiales de manipulación y eliminación aplicables1.
No abra ni triture las cápsulas. Evite el contacto directo con el contenido de la cápsula. En caso de rotura de la cápsula, evite el contacto directo del contenido de la cápsula con la piel o los ojos. Si se produce contacto con la piel, lávese bien con agua y jabón. Si se produce contacto con los ojos, enjuague bien con agua.
Cualquier medicamento no utilizado o material de desecho debe eliminarse de acuerdo con los requisitos locales.
17 INFORMACIÓN PARA EL PACIENTE
Aconseje al paciente que lea el etiquetado del paciente aprobado por la FDA (Información para el paciente).
Instrucciones de dosificación
- Indique a los pacientes que tomen NINLARO exactamente como se lo recetaron.
- Aconseje a los pacientes que tomen NINLARO una vez a la semana el mismo día y aproximadamente a la misma hora durante las primeras tres semanas de un ciclo de cuatro semanas. Se debe discutir con los pacientes que comienzan el tratamiento la importancia de seguir cuidadosamente todas las instrucciones de dosificación. Aconseje a los pacientes que tomen la dosis recomendada según las indicaciones, ya que la sobredosis ha provocado muertes [ver Sobredosis (10)].
- Aconseje a los pacientes que tomen NINLARO al menos una hora antes o al menos dos horas después de las comidas.
- Aconseje a los pacientes que NINLARO y dexametasona no deben tomarse al mismo tiempo, ya que la dexametasona debe tomarse con alimentos y NINLARO no debe tomarse con alimentos.
- Aconseje a los pacientes que traguen la cápsula entera con agua. La cápsula no debe triturarse, masticarse ni abrirse.
- Aconseje a los pacientes que eviten el contacto directo con el contenido de la cápsula. En caso de rotura de la cápsula, evite el contacto directo del contenido de la cápsula con la piel o los ojos. Si se produce contacto con la piel, lávese bien con agua y jabón. Si se produce contacto con los ojos, enjuague bien con agua.
- Si un paciente olvida una dosis, aconseje que tome la dosis olvidada siempre que la próxima dosis programada esté a ≥72 horas de distancia. Aconseje a los pacientes que no tomen una dosis olvidada si está dentro de las 72 horas de su próxima dosis programada.
- Si un paciente vomita después de tomar una dosis, aconseje que no repita la dosis, sino que reanude la dosificación en el momento de la próxima dosis programada.
- Aconseje a los pacientes que guarden las cápsulas en el envase original y que no las saquen del envase hasta justo antes de tomar NINLARO.
[ver Dosificación y administración (2.1)]
Trombocitopenia
Aconseje a los pacientes que pueden experimentar recuentos bajos de plaquetas (trombocitopenia). Los signos de trombocitopenia pueden incluir sangrado y hematomas fáciles. [ver Advertencias y precauciones (5.1)].
Toxicidades gastrointestinales
Aconseje a los pacientes que pueden experimentar diarrea, estreñimiento, náuseas y vómitos y que se pongan en contacto con sus proveedores de atención médica si estas reacciones adversas persisten. [ver Advertencias y precauciones (5.2)].
Neuropatía periférica
Aconseje a los pacientes que se pongan en contacto con sus proveedores de atención médica si experimentan síntomas nuevos o que empeoran de neuropatía periférica, como hormigueo, entumecimiento, dolor, sensación de ardor en los pies o las manos, o debilidad en los brazos o las piernas. [ver Advertencias y precauciones (5.3)].
Edema periférico
Aconseje a los pacientes que se pongan en contacto con sus proveedores de atención médica si experimentan hinchazón inusual de las extremidades o aumento de peso debido a la hinchazón [ver Advertencias y precauciones (5.4)].
Reacciones cutáneas
Aconseje a los pacientes que se pongan en contacto con sus proveedores de atención médica de inmediato si experimentan erupción cutánea nueva o que empeora [ver Advertencias y precauciones (5.5)].
Microangiopatía trombótica
Aconseje a los pacientes que busquen atención médica inmediata si se presenta algún signo o síntoma de microangiopatía trombótica [ver Advertencias y precauciones (5.6)].
Hepatotoxicidad
Aconseje a los pacientes que se pongan en contacto con sus proveedores de atención médica si experimentan ictericia o dolor abdominal en el cuadrante superior derecho [ver Advertencias y precauciones (5.7)].
Otras reacciones adversas
Aconseje a los pacientes que se pongan en contacto con sus proveedores de atención médica si experimentan signos y síntomas de dermatosis neutrófila febril aguda (síndrome de Sweet), síndrome de Stevens-Johnson, mielitis transversa, síndrome de encefalopatía posterior reversible, síndrome de lisis tumoral, herpes zóster, cataratas, ojos secos, visión borrosa, conjuntivitis y púrpura trombocitopénica trombótica [ver Reacciones adversas (6.1)].
Toxicidad embrio-fetal
Aconseje a las mujeres embarazadas y a las mujeres en edad fértil sobre el riesgo potencial para el feto. Aconseje a las mujeres en edad fértil que informen a su proveedor de atención médica sobre un embarazo conocido o sospechado [ver Advertencias y precauciones (5.8) y Uso en poblaciones específicas (8.1)].
Aconseje a las mujeres en edad fértil que usen métodos anticonceptivos eficaces durante el tratamiento con NINLARO y durante 90 días después de la última dosis. Aconseje a las mujeres que usan anticonceptivos hormonales que también usen un método anticonceptivo de barrera [ver Uso en poblaciones específicas (8.1)].
Aconseje a los hombres con parejas femeninas en edad fértil que usen métodos anticonceptivos eficaces durante el tratamiento con NINLARO y durante 90 días después de la última dosis [ver Uso en poblaciones específicas (8.1)].
Lactancia
Aconseje a las mujeres que no amamanten durante el tratamiento con NINLARO y durante 90 días después de la última dosis [ver Uso en poblaciones específicas (8.2)].
Medicamentos concomitantes
Aconseje a los pacientes que hablen con sus proveedores de atención médica sobre cualquier otro medicamento que estén tomando actualmente y antes de comenzar cualquier medicamento nuevo.
SECCIÓN NO CLASIFICADA DE SPL
Distribuido por:
Takeda Pharmaceuticals America, Inc.
Cambridge, MA 02142
NINLARO es una marca registrada de Millennium Pharmaceuticals, Inc.
©2024 Takeda Pharmaceuticals U.S.A., Inc. Todos los derechos reservados.
Para obtener más información, también puede visitar www.NINLARO.com o llamar al 1-844-617-6468.
IXB349 R10
INSERTO PARA EL PACIENTE
| Esta información para el paciente ha sido aprobada por la Administración de Alimentos y Medicamentos de los Estados Unidos. | Revisado: Julio/2024 | |
| INFORMACIÓN PARA EL PACIENTE NINLARO® (nin-LAR-oh) (ixazomib) cápsulas |
||
| NINLARO se usa con otros dos medicamentos recetados llamados REVLIMID® (lenalidomida) y dexametasona. Lea la Guía de medicamentos que viene con REVLIMID® (lenalidomida). Puede pedirle información sobre la dexametasona a su proveedor de atención médica o farmacéutico. | ||
| ¿Qué es NINLARO? NINLARO es un medicamento recetado que se usa para tratar el mieloma múltiple en combinación con los medicamentos REVLIMID® (lenalidomida) y dexametasona, en personas que han recibido al menos un tratamiento previo para su mieloma múltiple. NINLARO no debe usarse para tratar a personas:
No se sabe si NINLARO es seguro y eficaz en niños. |
||
Antes de tomar NINLARO, informe a su proveedor de atención médica sobre todas sus afecciones médicas, incluso si:
Informe a su proveedor de atención médica sobre todos los medicamentos que toma, incluidos los medicamentos recetados y de venta libre, las vitaminas y los suplementos herbales. Hable con su proveedor de atención médica antes de comenzar cualquier medicamento nuevo durante el tratamiento con NINLARO. |
||
¿Cómo debo tomar NINLARO?
|
||
| ¿Cuáles son los posibles efectos secundarios de NINLARO? NINLARO puede causar efectos secundarios graves, incluyendo:
|
||
|
|
|
|
||
|
|
|
Otros efectos secundarios comunes de NINLARO incluyen: |
||
|
|
|
Dígale a su proveedor de atención médica si presenta signos o síntomas nuevos o empeoramiento de los siguientes durante el tratamiento con NINLARO:
Estos no son todos los posibles efectos secundarios de NINLARO. |
||
¿Cómo debo almacenar NINLARO?
Mantenga NINLARO y todos los medicamentos fuera del alcance de los niños. |
||
| Información general sobre el uso seguro y eficaz de NINLARO. Los medicamentos a veces se prescriben para fines distintos a los enumerados en un prospecto de información para el paciente. No use NINLARO para una condición para la que no se ha prescrito. No le dé NINLARO a otras personas, incluso si tienen los mismos síntomas que usted. Puede dañarles. Puede pedir a su farmacéutico o proveedor de atención médica información sobre NINLARO que esté escrita para profesionales de la salud. |
||
| ¿Cuáles son los ingredientes de NINLARO? Ingrediente activo: ixazomib Ingredientes inactivos: celulosa microcristalina, estearato de magnesio y talco Cápsulas: gelatina y dióxido de titanio. La cápsula de 4 mg contiene óxido de hierro rojo y amarillo. La cápsula de 3 mg contiene óxido de hierro negro. La cápsula de 2,3 mg contiene óxido de hierro rojo. La tinta de impresión contiene laca, propilenglicol, hidróxido de potasio y óxido de hierro negro. |
||
|
Distribuido por: Takeda Pharmaceuticals America, Inc. Cambridge, MA 02142 NINLARO es una marca registrada de Millennium Pharmaceuticals, Inc. ©2024 Takeda Pharmaceuticals U.S.A., Inc. Todos los derechos reservados. Para más información, también puede visitar www.NINLARO.com o llamar al 1-844-617-6468. IXB349 R10 |
||
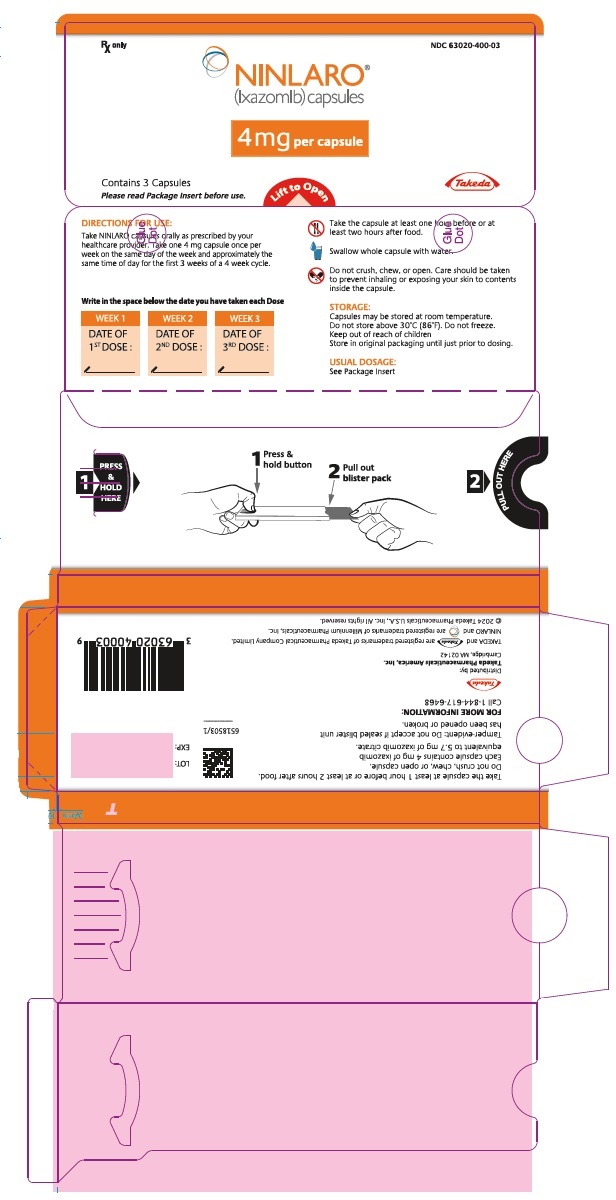
PANEL PRINCIPAL DE VISUALIZACIÓN – Cápsula de 4 mg, blíster
Rx only NDC 63020-400-03
NINLARO®
(ixazomib) capsules
4 mg por cápsula
Contiene 3 Cápsulas
Lea el Folleto de Información para el Paciente antes de usar.
Levante para abrir
Takeda
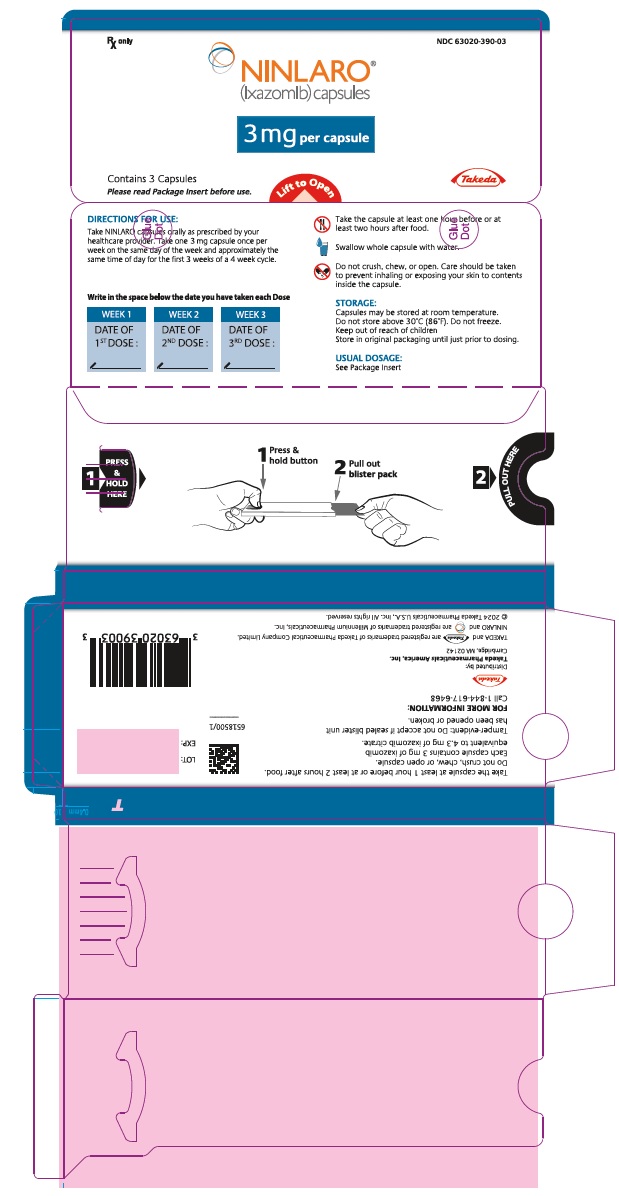
PANEL PRINCIPAL DE VISUALIZACIÓN – Cápsula de 3 mg, blíster
Rx only NDC 63020-390-03
NINLARO®
(ixazomib) capsules
3 mg per capsule
Contiene 3 Cápsulas
Lea el Folleto de Información para el Paciente antes de usar.
Levante para abrir
Takeda
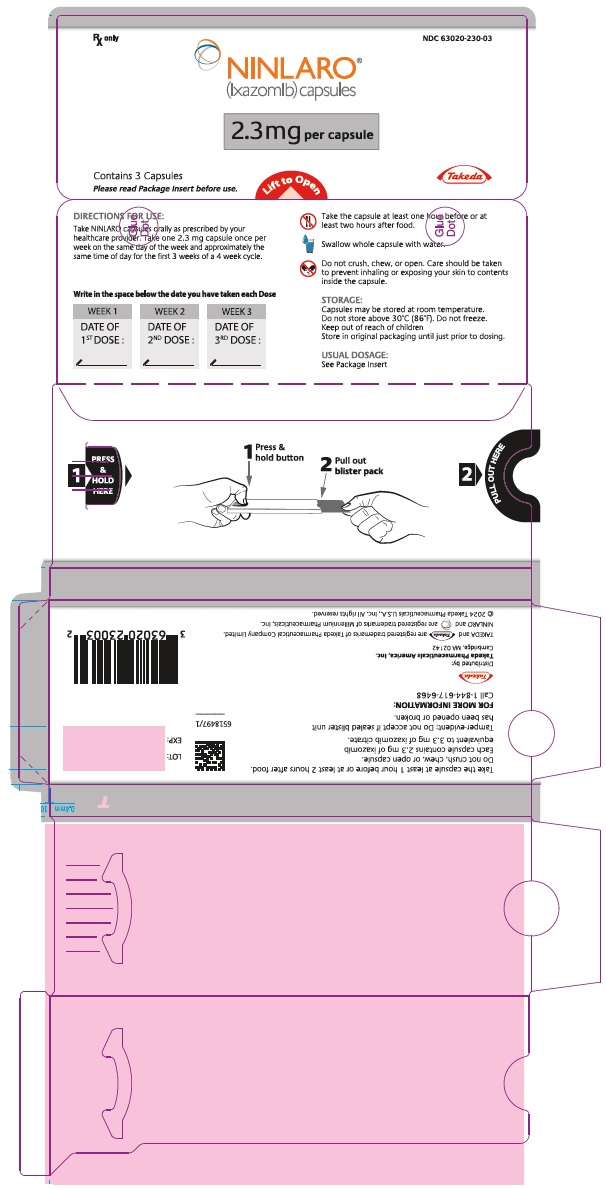
PANEL PRINCIPAL DE EXHIBICIÓN – Envase blíster de cápsulas de 2,3 mg
Rx only NDC 63020-230-03
NINLARO®
(ixazomib) capsules
2.3 mg por cápsula
Contiene 3 Cápsulas
Lea el Folleto de Información para el Paciente antes de usar.
Levante para abrir
Takeda