
Fabricante de medicamentos: Puma Biotechnology, Inc. (Updated: 2024-03-25)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
NERLYNX
®(neratinib) comprimidos, para uso oral
Aprobación inicial en EE. UU.: 2017
CAMBIOS IMPORTANTES RECIENTES
INDICACIONES Y USO
NERLYNX es un inhibidor de la cinasa indicado:
- Como agente único, para el tratamiento adyuvante extendido de pacientes adultos con cáncer de mama HER2 positivo en estadio temprano, después de la terapia adyuvante basada en trastuzumab. (
1.1)
- En combinación con capecitabina, para el tratamiento de pacientes adultos con cáncer de mama HER2 positivo avanzado o metastásico que han recibido dos o más regímenes previos basados en anti-HER2 en el entorno metastásico. (
1.2)
DOSIFICACIÓN Y ADMINISTRACIÓN
- Premedicación para la diarrea: cuando no se utiliza el aumento gradual de la dosis, inicie la loperamida con la primera dosis de NERLYNX y continúe durante los primeros 56 días de tratamiento. Después del día 56, use loperamida para mantener de 1 a 2 evacuaciones intestinales por día. (
2.1,
2.2)
- Tratamiento adyuvante extendido del cáncer de mama en estadio temprano: 240 mg (6 tabletas) administrados por vía oral una vez al día, con alimentos, de forma continua hasta la recurrencia de la enfermedad hasta por un año. (
2.2)
- Cáncer de mama avanzado o metastásico: 240 mg (6 tabletas) administrados por vía oral una vez al día con alimentos los días 1 a 21 de un ciclo de 21 días más capecitabina (750 mg/m
2administrados por vía oral dos veces al día) en los días 1 a 14 de un ciclo de 21 días hasta la progresión de la enfermedad o toxicidades inaceptables. (
2.2)
- Aumento gradual de la dosis: también se puede iniciar un aumento gradual de la dosis de NERLYNX durante dos semanas. (
2.2)
- Se recomiendan interrupciones de la dosis y/o reducciones de la dosis según la seguridad y la tolerabilidad individuales. (
2.3)
- Insuficiencia hepática: Reducir la dosis inicial a 80 mg en pacientes con insuficiencia hepática grave. (
2.4)
FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
Tabletas: 40 mg. (
3)
CONTRAINDICACIONES
Ninguna. (
4)
ADVERTENCIAS Y PRECAUCIONES
- Diarrea: controle la diarrea mediante el aumento gradual de la dosis de NERLYNX o la profilaxis con loperamida (
2.1,
2.2). Si se produce diarrea a pesar de la profilaxis recomendada, trátela con antidiarreicos, líquidos y electrolitos adicionales según esté clínicamente indicado. Suspenda NERLYNX en pacientes que experimenten diarrea grave y/o persistente. Suspenda permanentemente NERLYNX en pacientes que experimenten diarrea de Grado 4 o diarrea de Grado ≥2 que ocurra después de la reducción máxima de la dosis. (
2.3,
5.1)
- Hepatotoxicidad: controle las pruebas de función hepática mensualmente durante los primeros 3 meses de tratamiento, luego cada 3 meses mientras esté en tratamiento y según esté clínicamente indicado. Suspenda NERLYNX en pacientes que experimenten anomalías hepáticas de Grado 3 y suspenda permanentemente NERLYNX en pacientes que experimenten anomalías hepáticas de Grado 4. (
2.3,
5.2)
- Toxicidad embriofetal: NERLYNX puede causar daño fetal. Aconseje a las pacientes sobre el riesgo potencial para el feto y que utilicen métodos anticonceptivos eficaces. (
5.3,
8.1,
8.3)
REACCIONES ADVERSAS
Las reacciones adversas más comunes (reportadas en ≥5% de los pacientes) fueron:
- NERLYNX como agente único: diarrea, náuseas, dolor abdominal, fatiga, vómitos, sarpullido, estomatitis, disminución del apetito, espasmos musculares, dispepsia, aumento de AST o ALT, trastorno de las uñas, piel seca, distensión abdominal, epistaxis, disminución de peso e infección del tracto urinario. (
6)
- NERLYNX en combinación con capecitabina: diarrea, náuseas, vómitos, disminución del apetito, estreñimiento, fatiga/astenia, disminución de peso, mareos, dolor de espalda, artralgia, infección del tracto urinario, infección del tracto respiratorio superior, distensión abdominal, insuficiencia renal y espasmos musculares. (
6)
Para informar REACCIONES ADVERSAS SOSPECHOSAS, comuníquese con Puma Biotechnology, Inc. al 1-844-NERLYNX (1-844-637-5969) o con la FDA al 1-800-FDA-1088 owww.fda.gov/medwatch.
INTERACCIONES CON LA MEDICINA
- Agentes reductores del ácido gástrico: evite el uso concomitante con inhibidores de la bomba de protones. Separe NERLYNX por lo menos 2 horas antes o 10 horas después de H
2-antagonistas de receptores. O separe NERLYNX por lo menos 3 horas después de los antiácidos. (
2.5,
7.1)
- Inhibidores potentes de CYP3A4: evite el uso concomitante. (
7.1)
- Inhibidores duales de P-gp y CYP3A4 moderados: evite el uso concomitante. (
7.1)
- Inductores potentes o moderados de CYP3A4: evite el uso concomitante. (
7.1)
- Ciertos sustratos de P-gp: controle las reacciones adversas de los sustratos de P-gp para los cuales un cambio mínimo en la concentración puede provocar reacciones adversas graves cuando se usan concomitantemente con NERLYNX. (
7.2)
Consulte 17 para obtener INFORMACIÓN DE ASESORAMIENTO AL PACIENTE y el etiquetado del paciente aprobado por la FDA.
Revisado: 3/2022
Tabla de Contenido
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDO*
1 INDICACIONES Y USO
1.1 Tratamiento Adyuvante Extendido del Cáncer de Mama en Estadio Temprano
1.2 Cáncer de Mama Avanzado o Metastásico
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Premedicación para la Diarrea
2.2 Dosis y Programa Recomendados
2.3 Modificaciones de la Dosis para Reacciones Adversas
2.4 Modificaciones de la Dosis para Insuficiencia Hepática
2.5 Modificaciones de la Dosis para Agentes Reductores del Ácido Gástrico
3 FORMAS Y FUERZAS DE DOSIFICACIÓN
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Diarrea
5.2 Hepatotoxicidad
5.3 Toxicidad Embriofetal
6 REACCIONES ADVERSAS
6.1 Experiencia en Ensayos Clínicos
7 INTERACCIONES MEDICAMENTOSAS
7.1 Efecto de Otros Medicamentos en NERLYNX
7.2 Efecto de NERLYNX en Otros Medicamentos
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.3 Mujeres y Hombres en Potencial Reproductivo
8.4 Uso Pediátrico
8.5 Uso Geriátrico
8.6 Insuficiencia Hepática
10 SOBREDOSIS
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de Acción
12.2 Farmacodinamia
12.3 Farmacocinética
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
14 ESTUDIOS CLÍNICOS
14.1 Tratamiento Adyuvante Extendido del Cáncer de Mama en Estadio Temprano
14.2 Cáncer de Mama Avanzado o Metastásico
16 FORMA DE SUMINISTRO/ALMACENAMIENTO Y MANEJO
17 INFORMACIÓN PARA EL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información completa de prescripción no están enumeradas.
1 INDICACIONES Y USO
1.1 Tratamiento adyuvante extendido del cáncer de mama en estadio temprano
NERLYNX como agente único está indicado para el tratamiento adyuvante extendido de pacientes adultas con cáncer de mama positivo para el receptor 2 del factor de crecimiento epidérmico humano (HER2) en estadio temprano, después de la terapia adyuvante basada en trastuzumab
[ver Estudios clínicos (
14.1)]
.
1.2 Cáncer de mama avanzado o metastásico
NERLYNX en combinación con capecitabina está indicado para el tratamiento de pacientes adultas con cáncer de mama HER2 positivo avanzado o metastásico que han recibido dos o más regímenes previos basados en anti-HER2 en el entorno metastásico
[ver Estudios clínicos (
14.2)]
.
2 DOSIS Y ADMINISTRACIÓN
2.1 Medicación previa para la diarrea
Cuando no se utiliza el aumento gradual de la dosis
[ver Posología y administración (
2.2)],
administre profilaxis antidiarreica durante los primeros 56 días de tratamiento e inicie con la primera dosis de NERLYNX
[ver Advertencias y precauciones (5.1) y Reacciones adversas (
6.1)]
.
Indique a los pacientes que tomen loperamida como se indica en la
Tabla 1. Ajuste la dosis de loperamida a 1 o 2 defecaciones al día.
| Tiempo con NERLYNX | Dosis y frecuencia de loperamida |
|---|---|
| Semanas 1 a 2 (días 1 a 14) | 4 mg tres veces al día |
| Semanas 3 a 8 (días 15 a 56) | 4 mg dos veces al día |
| Semanas 9 a la suspensión de NERLYNX | 4 mg según sea necesario, sin exceder los 16 mg al día; ajuste la dosis para lograr 1 o 2 defecaciones al día |
Si se produce diarrea a pesar de la profilaxis, trátela con antidiarreicos, líquidos y electrolitos adicionales según esté clínicamente indicado. También pueden ser necesarias interrupciones y reducciones de la dosis de NERLYNX para controlar la diarrea
[ver Posología y administración (
2.3)]
.
2.2 Dosis y esquema recomendados
Tratamiento adyuvante extendido del cáncer de mama en estadio temprano
La dosis recomendada de NERLYNX es de 240 mg (seis comprimidos) administrados por vía oral una vez al día, con alimentos, de forma continua hasta la recurrencia de la enfermedad o durante un máximo de un año.
Cáncer de mama avanzado o metastásico
La dosis recomendada de NERLYNX es de 240 mg (seis comprimidos) administrados por vía oral una vez al día con alimentos los días 1 a 21 de un ciclo de 21 días más capecitabina (750 mg/m
2administrados por vía oral dos veces al día) los días 1 a 14 de un ciclo de 21 días hasta la progresión de la enfermedad o toxicidades inaceptables.
Aumento gradual de la dosis
Se puede considerar un aumento gradual de la dosis de NERLYNX durante dos semanas en lugar de comenzar con la dosis diaria de 240 mg para pacientes con cáncer de mama en estadio temprano y cáncer de mama metastásico, como se describe en la
Tabla 2[ver Advertencias y precauciones (5.1) y Reacciones adversas (
6.1)]
.
| Tiempo con NERLYNX | Dosis de NERLYNX |
|---|---|
| Semana 1 (días 1 a 7) | 120 mg al día (tres comprimidos de 40 mg) |
| Semana 2 (días 8 a 14) | 160 mg al día (cuatro comprimidos de 40 mg) |
| Semana 3 y siguientes | 240 mg al día (seis comprimidos de 40 mg, dosis recomendada) |
Si se produce diarrea, trátela con medicamentos antidiarreicos, líquidos y electrolitos según esté clínicamente indicado. También pueden ser necesarias las interrupciones de la dosis y las reducciones de la dosis de NERLYNX para controlar la diarrea
[ver Posología y administración (
2.3)]
.
Instrucciones de administración
Indique a los pacientes que tomen NERLYNX aproximadamente a la misma hora todos los días. Los comprimidos de NERLYNX deben tragarse enteros (los comprimidos no deben masticarse, triturarse ni partirse antes de tragarlos).
Si un paciente olvida una dosis, no la reponga e indíquele que vuelva a tomar NERLYNX con la siguiente dosis diaria programada.
2.3 Modificaciones de la dosis por reacciones adversas
Se recomienda la modificación de la dosis de NERLYNX en función de la seguridad y la tolerabilidad individuales. El manejo de algunas reacciones adversas puede requerir la interrupción de la dosis y/o la reducción de la dosis, como se muestra en
Suspenda NERLYNX en pacientes con reacciones adversas que no se recuperen a Grado 0-1 o al valor inicial, con toxicidades que provoquen un retraso en el tratamiento > 3 semanas o si no pueden tolerar 120 mg diarios. Otras situaciones clínicas pueden dar lugar a ajustes de la dosis según esté clínicamente indicado (por ejemplo, toxicidades intolerables, reacciones adversas persistentes de Grado 2, etc.).
Cuando NERLYNX se utiliza en combinación con capecitabina, consulte la información de prescripción de capecitabina para conocer las modificaciones de la dosis de capecitabina.
| Nivel de dosis | Dosis de NERLYNX |
|---|---|
| Dosis inicial recomendada | 240 mg diarios (seis comprimidos de 40 mg) |
| Primera reducción de la dosis | 200 mg diarios (cinco comprimidos de 40 mg) |
| Segunda reducción de la dosis | 160 mg diarios (cuatro comprimidos de 40 mg) |
| Tercera reducción de la dosis | 120 mg diarios (tres comprimidos de 40 mg) |
| Reacción adversa | Severidad† | Medidas/Modificación de la dosis |
|---|---|---|
|
ALT=Alanina aminotransferasa; AST=Aspartato aminotransferasa; ULN=Límite superior normal |
||
|
† Según CTCAE v4.0 |
||
|
* Las características complicadas incluyen deshidratación, fiebre, hipotensión, insuficiencia renal o neutropenia de Grado 3 o 4. |
||
|
‡ A pesar de recibir tratamiento médico óptimo |
||
| Diarrea [ver Advertencias y precauciones ( 5.1)]
|
|
|
|
|
|
|
|
|
|
|
|
| Hepatotoxicidad [ver Advertencias y precauciones ( 5.2)]
|
|
|
|
|
|
| Otros [ver Reacciones adversas ( 6.1)]
|
|
|
|
|
|
| Nivel de dosis | Dosis de NERLYNX |
|---|---|
| Dosis inicial recomendada | 240 mg al día (seis comprimidos de 40 mg) |
| Primera reducción de la dosis | 160 mg al día (cuatro comprimidos de 40 mg) |
| Segunda reducción de la dosis | 120 mg al día (tres comprimidos de 40 mg) |
|
ALT=Alanina Aminotransferasa; AST=Aspartato Aminotransferasa; ULN=Límite superior normal |
||
|
† Según CTCAE v4.0 |
||
|
aDado que la capecitabina se proporciona en comprimidos de 150 mg o 500 mg, se recomienda que la(s) reducción(es) de la dosis de capecitabina se redondeen a la baja al múltiplo de 500 mg o 150 mg más cercano para la dosis dos veces al día. Si la superficie corporal del paciente es >2,0, se puede utilizar el estándar de atención del centro de estudio para la dosificación de capecitabina en mg/m 2. |
||
| Reacción adversa | Gravedad† | Acción/Modificación de la dosis |
| Diarrea [ver Advertencias y precauciones ( 5.1)]
|
|
|
|
|
|
| Hepatotoxicidad [ver Advertencias y precauciones ( 5.2)]
|
|
|
|
|
|
| Otros [ver Reacciones adversas ( 6.1)]
|
|
|
|
|
|
2.4 Modificaciones de la dosis en pacientes con insuficiencia hepática
Reduzca la dosis inicial de NERLYNX a 80 mg en pacientes con insuficiencia hepática grave (Child Pugh C). No se recomiendan modificaciones de la dosis en pacientes con insuficiencia hepática leve a moderada (Child Pugh A o B)
[consulte Uso en poblaciones específicas (
8.6) y Farmacología clínica (
12.3)]
.
2.5 Modificaciones de la dosis para agentes reductores del ácido gástrico
Inhibidores de la bomba de protones (IBP):Evite el uso concomitante con NERLYNX
[consulte Interacciones medicamentosas (
7.1)]
.
H2-antagonistas de los receptores:Tome NERLYNX al menos 2 horas antes de la siguiente dosis del antagonista de los receptores H
2 o 10 horas después del antagonista de los receptores H
2
[consulte Interacciones medicamentosas (
7.1)]
.
Antiácidos:Separe la dosificación de NERLYNX por 3 horas después de los antiácidos
[consulte Interacciones medicamentosas (
7.1)]
.
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
Tabletas: 40 mg de neratinib (equivalente a 48.31 mg de maleato de neratinib).
Recubiertas con película, de color rojo, de forma ovalada y con la inscripción ‘W104’ en una cara y lisa en la otra.
4 CONTRAINDICACIONES
Ninguna.
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Diarrea
Se produjo diarrea grave y secuelas, como deshidratación, hipotensión e insuficiencia renal durante el tratamiento con NERLYNX. La diarrea se informó en el 95% de los pacientes tratados con NERLYNX en ExteNET, un ensayo aleatorizado controlado con placebo en el entorno adyuvante extendido que no requirió profilaxis antidiarreica. En el brazo de NERLYNX, la diarrea de grado 3 se produjo en el 40% y la diarrea de grado 4 se produjo en el 0,1% de los pacientes. La mayoría de los pacientes (93%) tuvieron diarrea en el primer mes de tratamiento, el tiempo medio hasta la aparición de la diarrea de grado ≥3 fue de 8 días (rango, 1–350) y la duración media acumulada de la diarrea de grado ≥3 fue de 5 días (rango, 1–139)
[ver Reacciones adversas (
6.1)]
.
Se informó diarrea en el 83% de los pacientes tratados con NERLYNX más capecitabina en NALA, un ensayo aleatorizado controlado con placebo en el entorno de cáncer de mama metastásico que requirió profilaxis antidiarreica en el primer ciclo de 21 días. La mayoría de los pacientes (70%) tuvieron diarrea en los primeros 21 días de tratamiento, el tiempo medio hasta la aparición de la diarrea de grado ≥3 fue de 11 días (rango, 2–728) y la duración media acumulada de la diarrea de grado ≥3 fue de 3 días (rango, 1–21). En el brazo de NERLYNX más capecitabina, la diarrea de grado 3 se produjo en el 24% de los pacientes
[ver Reacciones adversas (
6.1)]
.
Se ha demostrado que la profilaxis antidiarreica reduce la incidencia y la gravedad de la diarrea. Indique a los pacientes que inicien la profilaxis antidiarreica con loperamida junto con la primera dosis de NERLYNX y que la continúen durante los primeros 56 días de tratamiento; después del día 56, ajuste la dosis para lograr de 1 a 2 deposiciones al día y no exceda los 16 mg de loperamida al día
[ver Posología y administración (2.1)]. Considere agregar otros agentes a la loperamida según lo indicado clínicamente
[ver Reacciones adversas (
6.1)]
.
Alternativamente, también se puede considerar un enfoque de escalada de dosis de NERLYNX de 2 semanas antes de iniciar el régimen de tratamiento recomendado con NERLYNX para el manejo de la diarrea
[ver Posología y administración (
2.2)].
Para los pacientes que utilizaron la escalada de dosis de NERLYNX, el tiempo medio hasta la aparición de la diarrea de grado ≥3 fue de 45 días (rango, 15–132) y la duración media acumulada de la diarrea de grado ≥3 fue de 2,5 días (rango, 1–6). La diarrea de grado 3 se produjo en el 13% de los pacientes que utilizaron la escalada de dosis de NERLYNX
[ver Reacciones adversas (
6.1)].
Controle a los pacientes para detectar diarrea y trate con antidiarreicos adicionales según sea necesario. Cuando se produzca diarrea grave con deshidratación, administre líquidos y electrolitos según sea necesario, interrumpa NERLYNX y reduzca las dosis posteriores
[ver Posología y administración (2.3)]. Realice cultivos de heces según lo indicado clínicamente para excluir causas infecciosas de diarrea de grado 3 o 4 o diarrea de cualquier grado con características complicadas (deshidratación, fiebre, neutropenia).
5.2 Hepatotoxicidad
NERLYNX se ha asociado con hepatotoxicidad caracterizada por un aumento de las enzimas hepáticas. En ExteNET, el 10% de los pacientes experimentaron un aumento de la alanina aminotransferasa (ALT) ≥2× ULN, el 5% de los pacientes experimentaron un aumento de la aspartato aminotransferasa (AST) ≥2× ULN y el 1,7% de los pacientes experimentaron un aumento de la AST o la ALT >5× ULN (≥grado 3). La hepatotoxicidad o los aumentos de las transaminasas hepáticas llevaron a la interrupción del fármaco en el 1,7% de los pacientes tratados con NERLYNX.
En el estudio NALA, en los pacientes tratados con NERLYNX y capecitabina, el 7% experimentó un aumento de la ALT o la AST >3× ULN, el 2% experimentó un aumento de la ALT o la AST >5× ULN, el 7% experimentó un aumento de la bilirrubina >1,5× ULN y el 1,3% experimentó un aumento de la bilirrubina >3× ULN. La hepatotoxicidad o los aumentos de las transaminasas hepáticas llevaron a la interrupción del fármaco en el 0,3% de los pacientes tratados con NERLYNX y capecitabina.
La bilirrubina total, la AST, la ALT y la fosfatasa alcalina deben medirse antes de iniciar el tratamiento con NERLYNX mensualmente durante los primeros 3 meses de tratamiento, luego cada 3 meses mientras se esté en tratamiento y según lo indicado clínicamente. Estas pruebas también deben realizarse en pacientes que experimenten diarrea de grado 3 o cualquier signo o síntoma de hepatotoxicidad, como empeoramiento de la fatiga, náuseas, vómitos, dolor en el cuadrante superior derecho, fiebre, erupción cutánea o eosinofilia
[ver Posología y administración (
2.3) y Reacciones adversas (
6.1)]
.
5.3 Toxicidad embrio-fetal
Con base en los hallazgos de estudios en animales y su mecanismo de acción, NERLYNX puede causar daño fetal cuando se administra a una mujer embarazada. En estudios de reproducción en animales, la administración de neratinib a conejas embarazadas durante la organogénesis provocó abortos, muerte embrio-fetal y anomalías fetales en conejas a AUC maternas aproximadamente 0,2 veces el AUC en pacientes que recibieron la dosis recomendada. Advierta a las mujeres embarazadas sobre el riesgo potencial para un feto. Avise a las mujeres en edad fértil que usen métodos anticonceptivos eficaces durante el tratamiento y durante al menos 1 mes después de la última dosis.
[ver Uso en poblaciones específicas (
8.1,
8.3) y Farmacología clínica (
12.1)]
.
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas clínicamente significativas se describen en otras secciones del prospecto:
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se realizan en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un medicamento no pueden compararse directamente con las tasas en los ensayos clínicos de otro medicamento y pueden no reflejar las tasas observadas en la práctica.
Tratamiento adyuvante extendido del cáncer de mama en estadio temprano
ExteNET
Los datos que se describen a continuación reflejan los datos de seguridad de NERLYNX como agente único en ExteNET, un estudio multicéntrico, aleatorizado, doble ciego y controlado con placebo de NERLYNX en los 2 años posteriores a la finalización del tratamiento adyuvante con terapia basada en trastuzumab en mujeres con cáncer de mama HER2 positivo en estadio temprano. Las pacientes que recibieron NERLYNX en este ensayo no estaban obligadas a recibir ninguna profilaxis con agentes antidiarreicos para prevenir la diarrea relacionada con NERLYNX. Las pacientes fueron tratadas con 240 mg de NERLYNX administrados por vía oral una vez al día con alimentos, de forma continua hasta la recurrencia de la enfermedad o durante un máximo de un año. La mediana de duración del tratamiento fue de 11,6 meses en el grupo de NERLYNX y de 11,8 meses en el grupo de placebo. La mediana de edad fue de 52 años (el 60% tenía ≥50 años, el 12% tenía ≥65 años); el 81% eran caucásicas, el 3% negras o afroamericanas, el 14% asiáticas y el 3% de otras razas. Un total de 1408 pacientes fueron tratadas con NERLYNX.
La reducción de la dosis de NERLYNX debido a una reacción adversa de cualquier grado se produjo en el 31% de las pacientes que recibieron NERLYNX en comparación con el 2,6% de las pacientes que recibieron placebo. Se notificó la interrupción permanente debido a cualquier reacción adversa en el 28% de las pacientes tratadas con NERLYNX. La reacción adversa más común que condujo a la interrupción fue la diarrea, que representó el 17% de las pacientes tratadas con NERLYNX.
Las reacciones adversas más comunes (≥5%) fueron diarrea, náuseas, dolor abdominal, fatiga, vómitos, erupción cutánea, estomatitis, disminución del apetito, espasmos musculares, dispepsia, aumento de AST o ALT, trastorno de las uñas, sequedad de la piel, distensión abdominal, epistaxis, disminución de peso e infección del tracto urinario. Las reacciones adversas de grado 3 o 4 notificadas con mayor frecuencia fueron diarrea, vómitos, náuseas y dolor abdominal.
Las reacciones adversas graves en el grupo de NERLYNX incluyeron diarrea (1,6%), vómitos (0,9%), deshidratación (0,6%), celulitis (0,4%), insuficiencia renal (0,4%), erisipela (0,4%), aumento de ALT (0,3%), aumento de AST (0,3%), náuseas (0,3%), fatiga (0,2%) y dolor abdominal (0,2%).
La Tabla 7 resume las reacciones adversas en ExteNET.
|
* Incluye dolor abdominal, dolor abdominal superior y dolor abdominal inferior |
||||||
|
† Incluye estomatitis, estomatitis aftosa, ulceración bucal, ampollas en la mucosa oral, inflamación de la mucosa, dolor orofaríngeo, dolor oral, glosodinia, glositis y queilitis |
||||||
|
‡ Incluye erupción cutánea, erupción cutánea eritematosa, erupción cutánea folicular, erupción cutánea generalizada, erupción cutánea pruriginosa, erupción cutánea pustulosa, erupción cutánea maculopapular, erupción cutánea papular, dermatitis, dermatitis acneiforme y erupción cutánea tóxica |
||||||
|
§ Incluye trastorno de las uñas, paroniquia, onicoclasis, decoloración de las uñas, toxicidad de las uñas, crecimiento anormal de las uñas y distrofia de las uñas |
||||||
| Sistema de clasificación de órganos (Término preferido) |
NERLYNX n=1408 |
Placebo n=1408 |
||||
| Todos los grados (%) |
Grado 3 (%) | Grado 4 (%) | Todos los grados (%) | Grado 3 (%) | Grado 4 (%) | |
| Trastornos gastrointestinales | ||||||
| Diarrhea | 95 | 40 | 0.1 | 35 | 2 | 0 |
| Nausea | 43 | 2 | 0 | 22 | 0.1 | 0 |
| Abdominal pain
* |
36 | 2 | 0 | 15 | 0.4 | 0 |
| Vómitos | 26 | 3 | 0 | 8 | 0.4 | 0 |
| Estomatitis
† |
14 | 0.6 | 0 | 6 | 0.1 | 0 |
| Dispepsia | 10 | 0.4 | 0 | 4 | 0 | 0 |
| Distensión abdominal | 5 | 0.3 | 0 | 3 | 0 | 0 |
| Sequedad de boca | 3 | 0.1 | 0 | 2 | 0 | 0 |
| Trastornos generales y afecciones en el lugar de administración | ||||||
| Fatiga | 27 | 2 | 0 | 20 | 0.4 | 0 |
| Trastornos hepatobiliares | ||||||
| Alanina aminotransferasa elevada | 9 | 1 | 0.2 | 3 | 0.2 | 0 |
| Aspartato aminotransferasa elevada | 7 | 0.5 | 0.2 | 3 | 0.3 | 0 |
| Infecciones e infestaciones | ||||||
| Infección del tracto urinario | 5 | 0.1 | 0 | 2 | 0 | 0 |
| Investigaciones | ||||||
| Pérdida de peso | 5 | 0.1 | 0 | 0.5 | 0 | 0 |
| Trastornos del metabolismo y de la nutrición | ||||||
| Disminución del apetito | 12 | 0.2 | 0 | 3 | 0 | 0 |
| Deshidratación | 4 | 0.9 | 0.1 | 0.4 | 0.1 | 0 |
| Trastornos musculoesqueléticos y del tejido conjuntivo | ||||||
| Espasmos musculares | 11 | 0.1 | 0 | 3 | 0.1 | 0 |
| Trastornos respiratorios, torácicos y mediastínicos | ||||||
| Epistaxis | 5 | 0 | 0 | 1 | 0.1 | 0 |
| Trastornos de la piel y del tejido subcutáneo | ||||||
| Erupción cutánea
‡ |
18 | 0.6 | 0 | 9 | 0 | 0 |
| Piel seca | 6 | 0 | 0 | 2 | 0 | 0 |
| Trastorno de las uñas
§ |
8 | 0.3 | 0 | 2 | 0 | 0 |
| Fisuras cutáneas | 2 | 0.1 | 0 | 0.1 | 0 | 0 |
Cáncer de mama avanzado o metastásico
NALA
Los datos que se describen a continuación reflejan los datos de seguridad de NERLYNX más capecitabina en NALA, un estudio aleatorizado, multicéntrico, multinacional, abierto, con control activo de cáncer de mama metastásico HER2 positivo en pacientes, con o sin metástasis cerebrales, que han recibido dos o más regímenes previos basados en HER2 en el contexto metastásico.
Las pacientes fueron tratadas con 240 mg de NERLYNX por vía oral una vez al día los días 1 a 21 de un ciclo de 21 días en combinación con capecitabina (750 mg/m
2administrados por vía oral dos veces al día) los días 1 a 14 de un ciclo de 21 días, o 1250 mg de lapatinib por vía oral una vez al día los días 1 a 21 de un ciclo de 21 días en combinación con capecitabina (1000 mg/m
2administrados por vía oral dos veces al día) los días 1 a 14 de un ciclo de 21 días hasta la progresión de la enfermedad. La mediana de la duración del tratamiento fue de 5.7 meses en el grupo de NERLYNX más capecitabina y de 4.4 meses en el grupo de lapatinib más capecitabina.
La reducción de la dosis de NERLYNX debido a una reacción adversa de cualquier grado se produjo en el 10 % de las pacientes que recibieron NERLYNX más capecitabina. Se notificó la interrupción permanente debido a cualquier reacción adversa en el 14 % de las pacientes tratadas con NERLYNX más capecitabina. Las reacciones adversas más comunes que provocaron la interrupción del tratamiento fueron vómitos (3.6 %), diarrea (2.6 %), náuseas (2.6 %) y síndrome de eritrodisestesia palmo-plantar (2.3 %) de las pacientes tratadas con NERLYNX más capecitabina.
Las reacciones adversas más comunes de cualquier grado (≥5 %) en el grupo de NERLYNX más capecitabina fueron diarrea, náuseas, vómitos, disminución del apetito, estreñimiento, fatiga/astenia, pérdida de peso, mareos, dolor de espalda, artralgia, infección del tracto urinario, infección del tracto respiratorio superior, distensión abdominal, insuficiencia renal y espasmos musculares. Las reacciones adversas de grado 3 o 4 notificadas con mayor frecuencia fueron diarrea, náuseas, vómitos, fatiga y disminución del apetito.
Las reacciones adversas graves ≥2 % en el grupo de NERLYNX más capecitabina incluyeron diarrea (7 %), vómitos (3 %), náuseas (2.3 %) y lesión renal aguda (2.3 %).
La Tabla 8resume las reacciones adversas en NALA.
|
* La insuficiencia renal incluye lesión renal aguda, aumento de la creatinina en sangre, insuficiencia renal y deterioro de la función renal. |
||||||
| Sistema de clasificación de órganos (Término preferido) |
NERLYNX + Capecitabina n=303 |
Lapatinib + Capecitabina n=311 |
||||
| Todos los grados (%) |
Grado 3 (%) | Grado 4 (%) | Todos los grados (%) | Grado 3 (%) | Grado 4 (%) | |
| Trastornos gastrointestinales | ||||||
| Diarrea | 83 | 25 | 0 | 66 | 13 | 0 |
| Náuseas | 53 | 4.3 | 0 | 42 | 2.9 | 0 |
| Vómitos | 46 | 4 | 0 | 31 | 1.9 | 0 |
| Estreñimiento | 31 | 1 | 0 | 13 | 0 | 0 |
| Distensión abdominal | 8 | 0.3 | 0 | 3.2 | 0.6 | 0 |
| Trastornos generales y alteraciones en el lugar de administración | ||||||
| Fatiga/astenia | 45 | 6 | 0 | 40 | 4.5 | 0 |
| Malestar | 4.3 | 0 | 0 | 2.3 | 0.3 | 0 |
| Enfermedad tipo influenza | 4 | 0 | 0 | 1.3 | 0 | 0 |
| Infecciones e infestaciones | ||||||
| Infección del tracto urinario | 9 | 0.7 | 0 | 4.2 | 0.6 | 0 |
| Infección de las vías respiratorias superiores | 8 | 0.3 | 0 | 4.5 | 0.3 | 0 |
| Investigaciones | ||||||
| Pérdida de peso | 20 | 0.3 | 0 | 13 | 0.6 | 0 |
| Trastornos del metabolismo y la nutrición | ||||||
| Disminución del apetito | 35 | 2.6 | 0 | 22 | 2.3 | 0 |
| Trastornos musculoesqueléticos y del tejido conjuntivo | ||||||
| Dorsalgia | 10 | 0.3 | 0 | 8 | 0.3 | 0 |
| Artralgia | 10 | 0 | 0 | 6 | 1 | 0 |
| Espasmos musculares | 5 | 0 | 0 | 1.9 | 0 | 0 |
| Trastorno del sistema nervioso | ||||||
| Mareo | 14 | 0.3 | 0 | 10 | 0.6 | 0 |
| Trastornos renales y urinarios | ||||||
| Insuficiencia renal* | 7 | 2 | 0.3 | 1 | 0 | 0.3 |
| Disuria | 4.6 | 0 | 0 | 1.9 | 0 | 0 |
Manejo de la diarrea
CONTROL
El estudio CONTROL (NCT02400476) fue un ensayo multicéntrico, abierto y de múltiples cohortes que evaluó a pacientes con cáncer de mama HER2 positivo en estadio temprano tratadas con NERLYNX 240 mg al día durante un máximo de un año que recibieron profilaxis con loperamida y tratamiento antidiarreico adicional según fuera necesario o escalado de la dosis de NERLYNX con loperamida según fuera necesario. Todas las pacientes de la cohorte de profilaxis recibieron una dosis de carga de loperamida de 4 mg, seguida de 4 mg tres veces al día desde los días 1 a 14, seguida de 4 mg dos veces al día en los días 15 a 56, seguida de loperamida según fuera necesario durante 1 año de tratamiento con NERLYNX
[ver Posología y administración (
2.1)].
Todas las pacientes de la cohorte de escalado de dosis recibieron 120 mg de NERLYNX durante la semana 1, seguidos de 160 mg de NERLYNX durante la semana 2, seguidos de 240 mg de NERLYNX durante la semana 3 y posteriormente
[ver Posología y administración (
2.2)]
.
La Tabla 9 resume las reacciones adversas de diarrea para NERLYNX con profilaxis con loperamida y escalado de la dosis de NERLYNX.
| Profilaxis con loperamida n=109 |
Escalado de la dosis de NERLYNX n=60 |
|
| Duración del tratamiento, meses | ||
| Mediana | 11.8 | 12.0 |
| Rango | 0.1, 12.8 | 0.2, 12.4 |
| Intensidad de la dosis, mg por día | ||
| Mediana | 234 | 230 |
| Rango | 46, 240 | 32, 236 |
| Incidencia de diarrea, % | ||
| Cualquier grado | 78 | 98 |
| Grado 2 | 25 | 45 |
| Grado 3 | 32 | 13 |
| Medidas tomadas, % | ||
| Interrupción debido a la diarrea | 18 | 3.3 |
7 INTERACCIONES MEDICAMENTOSAS
7.1 Efecto de otros medicamentos en NERLYNX
Tabla 10 incluye interacciones medicamentosas que afectan la farmacocinética de neratinib.
|
AUC=Área bajo la curva; C max=Concentración máxima |
|
| Agentes reductores del ácido gástrico | |
| Impacto clínico | El uso concomitante de NERLYNX con un inhibidor de la bomba de protones (PPI), antagonista del receptor H
2, o antiácido puede disminuir el AUC de neratinib [ver Farmacología clínica ( 12.3)], lo que puede reducir la actividad de NERLYNX. |
| Prevención o manejo [ver Dosis y administración ( 2.5)]
|
Evite el uso concomitante de PPI. |
| Separe la administración de NERLYNX al menos 2 horas antes o 10 horas después de la dosis del antagonista del receptor H
2. |
|
| Separe la administración de NERLYNX por al menos 3 horas después de los antiácidos. | |
| Inhibidores fuertes de CYP3A4 | |
| Impacto clínico | El uso concomitante de NERLYNX con un inhibidor fuerte de CYP3A4 aumentó la C
maxy el AUC de neratinib [ver Farmacología clínica ( 12.3)] , lo que puede aumentar el riesgo de toxicidad de NERLYNX. |
| Prevención o manejo | Evite el uso concomitante de NERLYNX con inhibidores fuertes de CYP3A4. |
| Inhibidores duales de P-gp y CYP3A4 moderados | |
| Impacto clínico | El uso concomitante de NERLYNX con un inhibidor dual de P-gp y CYP3A4 moderado puede aumentar la C
maxy el AUC de neratinib [ver Farmacología clínica ( 12.3)] , lo que puede aumentar el riesgo de toxicidad de NERLYNX. |
| Prevención o manejo | Evite el uso concomitante de NERLYNX con inhibidores duales de P-gp y CYP3A4 moderados. |
| Inductores fuertes o moderados de CYP3A4 | |
| Impacto clínico | El uso concomitante de NERLYNX con un inductor fuerte de CYP3A4 redujo la C
maxy el AUC de neratinib [ver Farmacología clínica ( 12.3)] , lo que puede reducir la actividad de NERLYNX. |
| Prevención o manejo | Evite el uso concomitante de NERLYNX con inductores fuertes o moderados de CYP3A4. |
7.2 Efecto de NERLYNX en otros medicamentos
Ciertos sustratos de la glicoproteína P (P-gp)
El uso concomitante de NERLYNX aumentó las concentraciones de un sustrato de P-gp
[ver Farmacología clínica (
12.3)]
, lo que puede aumentar el riesgo de reacciones adversas de estos sustratos. Monitoree las reacciones adversas de ciertos sustratos de P-gp para los cuales los cambios mínimos de concentración pueden conducir a reacciones adversas graves.
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de Riesgos
Basado en los hallazgos de estudios en animales y el mecanismo de acción, NERLYNX puede causar daño fetal cuando se administra a una mujer embarazada
[ver Farmacología Clínica (
12.1)]
.
No hay datos disponibles en mujeres embarazadas para informar el riesgo asociado al fármaco. En estudios de reproducción en animales, la administración de neratinib a conejas embarazadas durante la organogénesis resultó en abortos, muerte embrio-fetal y anomalías fetales en conejas a exposiciones maternas (AUC) aproximadamente 0.2 veces las exposiciones en pacientes a la dosis recomendada (
ver
Datos). Advierta a las mujeres embarazadas sobre el riesgo potencial para un feto.
El riesgo de fondo de defectos de nacimiento mayores y aborto espontáneo para la población indicada es desconocido. Sin embargo, el riesgo de fondo de defectos de nacimiento mayores es del 2% al 4% y de aborto espontáneo del 15% al 20% de los embarazos clínicamente reconocidos en la población general de los EE. UU.
Datos
Datos en Animales
En un estudio de fertilidad y desarrollo embrionario temprano en ratas hembras, neratinib se administró por vía oral durante 15 días antes del apareamiento hasta el día 7 del embarazo, lo que no causó toxicidad embrionaria a dosis de hasta 12 mg/kg/día en presencia de toxicidad materna. Una dosis de 12 mg/kg/día en ratas es aproximadamente 0.5 veces la dosis máxima recomendada de 240 mg/día en pacientes en una base de mg/m
2.
En un estudio de desarrollo embrio-fetal en ratas, los animales preñados recibieron dosis orales de neratinib de hasta 15 mg/kg/día durante el período de organogénesis. No se observaron efectos sobre el desarrollo embrio-fetal o la supervivencia. La toxicidad materna fue evidente a 15 mg/kg/día (aproximadamente 0.6 veces el AUC en pacientes que reciben la dosis máxima recomendada de 240 mg/día).
En un estudio de desarrollo embrio-fetal en conejas, los animales preñados recibieron dosis orales de neratinib de hasta 9 mg/kg/día durante el período de organogénesis. La administración de neratinib a dosis ≥6 mg/kg/día resultó en toxicidad materna, abortos y muerte embrio-fetal (aumento de las resorciones). La administración de neratinib resultó en un aumento de la incidencia de anomalías fetales externas brutas (cabeza abovedada), de tejidos blandos (dilatación de los ventrículos cerebrales y defecto septal ventricular) y esqueléticas (fontanelas anteriores deformadas y fontanelas anteriores y/o posteriores agrandadas) a ≥3 mg/kg/día. El AUC
(0-t)a 6 mg/kg/día y 9 mg/kg/día en conejas fueron aproximadamente 0.5 y 0.8 veces, respectivamente, los AUC en pacientes que reciben la dosis máxima recomendada de 240 mg/día.
En un estudio de desarrollo peri y postnatal en ratas, la administración oral de neratinib desde el día 7 de gestación hasta el día 20 de lactancia resultó en toxicidad materna a ≥10 mg/kg/día (aproximadamente 0.4 veces la dosis máxima recomendada de 240 mg/día en pacientes en una base de mg/m
2) incluyendo disminución del peso corporal, aumento de peso corporal y consumo de alimentos. Se observaron efectos sobre la memoria a largo plazo en la descendencia masculina a dosis maternas ≥5 mg/kg/día (aproximadamente 0.2 veces la dosis máxima recomendada de 240 mg/día en pacientes en una base de mg/m
2).
8.2 Lactancia
Resumen de Riesgos
No hay datos disponibles sobre la presencia de neratinib o sus metabolitos en la leche materna o sus efectos en el lactante o en la producción de leche. Debido al potencial de reacciones adversas graves en los lactantes amamantados por NERLYNX, se debe aconsejar a las mujeres lactantes que no amamanten mientras toman NERLYNX y durante al menos 1 mes después de la última dosis.
8.3 Mujeres y Hombres con Potencial Reproductivo
Embarazo
Basado en estudios en animales, NERLYNX puede causar daño fetal cuando se administra a una mujer embarazada
[ver Uso en Poblaciones Específicas (
8.1)]
. Las mujeres con potencial reproductivo deben hacerse una prueba de embarazo antes de comenzar el tratamiento con NERLYNX.
Anticoncepción
Mujeres
Basado en estudios en animales, NERLYNX puede causar daño fetal cuando se administra a una mujer embarazada
[ver Uso en Poblaciones Específicas (
8.1)]
. Se debe aconsejar a las mujeres con potencial reproductivo que utilicen métodos anticonceptivos efectivos durante el tratamiento con NERLYNX y durante al menos 1 mes después de la última dosis.
Hombres
Basado en los hallazgos de estudios de reproducción en animales, se debe aconsejar a los pacientes masculinos con parejas femeninas con potencial reproductivo que utilicen métodos anticonceptivos efectivos durante el tratamiento y durante 3 meses después de la última dosis de NERLYNX
[ver Uso en Poblaciones Específicas (
8.1)]
.
8.4 Uso Pediátrico
No se ha establecido la seguridad y eficacia de NERLYNX en pacientes pediátricos.
8.5 Uso en poblaciones específicas
En el ensayo ExteNET, en el brazo de NERLYNX; 1236 pacientes tenían <65 años, 172 pacientes tenían ≥65 años, de los cuales 25 pacientes tenían 75 años o más. Hubo una mayor frecuencia de interrupciones del tratamiento debido a reacciones adversas en el grupo de edad ≥65 años que en el grupo de edad <65 años; en el brazo de NERLYNX, los porcentajes fueron 45% en comparación con 25%, respectivamente, y en el brazo de placebo 6% y 5%, respectivamente. La incidencia de reacciones adversas graves en el brazo de NERLYNX frente al brazo de placebo fue del 7% frente al 6% (<65 años) y del 10% frente al 8% (≥65 años). Las reacciones adversas graves más frecuentemente reportadas en el grupo de ≥65 años fueron vómitos (2.3%), diarrea (1.7%), insuficiencia renal (1.7%) y deshidratación (1.2%).
En el ensayo NALA, en el brazo de NERLYNX más capecitabina; 242 pacientes tenían <65 años, 61 pacientes tenían ≥65 años, de los cuales 12 pacientes tenían 75 años o más. La incidencia de reacciones adversas graves en el brazo de NERLYNX más capecitabina en el grupo de edad ≥65 años fue del 36% y en el grupo de edad <65 años fue del 34%. Las reacciones adversas graves más frecuentemente reportadas en el grupo de ≥65 años fueron diarrea (16%), lesión renal aguda (8%) y deshidratación (7%). No se observaron diferencias generales en la eficacia entre los pacientes ≥65 años y los pacientes <65 años.
8.6 Insuficiencia hepática
No se recomiendan modificaciones de la dosis para pacientes con insuficiencia hepática leve a moderada (Child Pugh A o B).
El aclaramiento de neratinib se reduce, y C
maxy AUC aumentan en pacientes con insuficiencia hepática preexistente grave (Child Pugh C). Reduzca la dosis de NERLYNX para pacientes con insuficiencia hepática grave
[ver Dosificación y administración (
2.4) y Farmacología clínica (
12.3)]
.
10 SOBREDOSIS
No existe un antídoto específico, y se desconoce el beneficio de la hemodiálisis en el tratamiento de la sobredosis de NERLYNX. En caso de sobredosis, se debe suspender la administración y tomar medidas generales de apoyo.
En el entorno de ensayos clínicos, un número limitado de pacientes reportó sobredosis. Las reacciones adversas experimentadas por estos pacientes fueron diarrea, náuseas, vómitos y deshidratación. La frecuencia y gravedad de los trastornos gastrointestinales (diarrea, dolor abdominal, náuseas y vómitos) parecen estar relacionados con la dosis.
11 DESCRIPCIÓN
NERLYNX (neratinib) es un medicamento en tabletas de liberación inmediata recubiertas con película para administración oral que contiene 40 mg de neratinib, equivalente a 48.31 mg de neratinib maleato. Neratinib pertenece a la clase de inhibidores de quinolidina 4-anilino de la quinasa de proteínas. La fórmula molecular del neratinib maleato es C
30H
29ClN
6O
3•C
4H
4O
4 y su peso molecular es de 673.11 Daltons. El nombre químico es (E)-N-{4-[3-cloro-4-(piridin-2-il metoxi)anilino]-3-ciano-7-etoxiquinolin-6-il}-4-(dimetilamino)but-2-enamida maleato, y su fórmula estructural es:
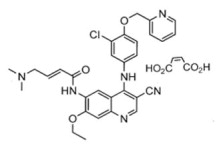
El neratinib maleato es un polvo de color blanco a amarillo con pK
as de 7.65 y 4.66. La solubilidad del neratinib maleato aumenta drásticamente a medida que el neratinib se protona a un pH ácido. El neratinib maleato es ligeramente soluble a pH 1.2 (32.90 mg/mL) e insoluble a aproximadamente pH 5.0 y superior (0.08 mg/mL o menos).
Ingredientes inactivos: Núcleo de la tableta: dióxido de silicio coloidal, manitol, celulosa microcristalina, crospovidona, povidona, estearato de magnesio y agua purificada. Recubrimiento: recubrimiento de película roja: alcohol polivinílico, dióxido de titanio, polietilenglicol, talco y óxido de hierro rojo.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
Neratinib es un inhibidor de la cinasa intracelular que se une irreversiblemente al receptor del factor de crecimiento epidérmico (EGFR), HER2 y HER4.
In vitro, neratinib reduce la autofosforilación de EGFR y HER2, las vías de señalización descendentes de MAPK y AKT, y mostró actividad antitumoral en líneas celulares de carcinoma que expresan EGFR y/o HER2. Los metabolitos humanos de neratinib M3, M6, M7 y M11 inhibieron la actividad de EGFR, HER2 y HER4
in vitro. In vivo, la administración oral de neratinib inhibió el crecimiento tumoral en modelos de xenotrasplantes de ratón con líneas celulares tumorales que expresan HER2 y EGFR.
12.2 Farmacodinamia
Se desconocen las relaciones exposición-respuesta de neratinib y la evolución temporal de la respuesta farmacodinámica.
Electrofisiología cardíaca
El efecto de NERLYNX en el intervalo QTc se evaluó en un estudio aleatorizado, controlado con placebo y positivo, doble ciego, de dosis única, cruzado en 60 sujetos sanos. A las exposiciones terapéuticas del 140% de NERLYNX, no hubo ningún efecto clínicamente relevante en el intervalo QTc.
12.3 Farmacocinética
El AUC de neratinib aumenta de forma menos que proporcional a la dosis en un rango de dosis diario de 40 a 400 mg (0,17 a 1,7 veces la dosis máxima recomendada aprobada).
Absorción
Las concentraciones máximas de neratinib y los principales metabolitos activos M3, M6 y M7 se alcanzan en un rango de 2 a 8 horas después de la administración oral.
Efecto de los alimentos
Una comida rica en grasas (aproximadamente 55% de grasas, 31% de carbohidratos y 14% de proteínas) aumentó la C
maxy el AUC
infde neratinib en un 70% (IC del 90%: 1,1–2,7) y un 120% (IC del 90%: 1,4–3,5), respectivamente, en sujetos sanos en comparación con las condiciones de ayuno. Un desayuno estándar (aproximadamente 50% de carbohidratos, 35% de grasas y 15% de proteínas) aumentó la C
maxy el AUC
infen un 20% (IC del 90%: 0,97–1,42) y un 10% (IC del 90%: 1,02–1,24), respectivamente, en sujetos sanos
[ver Posología y forma de administración (
2.2)]
.
Distribución
El volumen de distribución aparente medio (%CV) en estado estacionario (V
ss/F) fue de 6433 (19%) L en los pacientes. La unión a proteínas in vitro de neratinib fue superior al 99%, principalmente a la albúmina sérica y a la alfa-1 glicoproteína ácida, y fue independiente de la concentración.
Eliminación
La semivida plasmática media (%CV) de neratinib, M3, M6 y M7 fue de 14,6 (38%), 21,6 (77%), 13,8 (50%) y 10,4 (33%) horas, respectivamente, en sujetos sanos. La semivida de eliminación media de neratinib osciló entre 7 y 17 horas después de una dosis oral única en pacientes. La CL/F media (%CV) después de la primera dosis y en estado estacionario (día 21) fue de 216 (34%) y 281 (40%) L/hora, respectivamente, en los pacientes.
Metabolismo
Neratinib se metaboliza principalmente en el hígado por CYP3A4 y en menor medida por la monooxigenasa que contiene flavina (FMO).
Neratinib representa el componente más prominente en plasma. Las exposiciones sistémicas (AUC) de los metabolitos activos M3, M6, M7 y M11 fueron del 15%, 33%, 22% y 4% de la exposición sistémica a neratinib, respectivamente, en estado estacionario en sujetos sanos.
Excreción
Después de la administración oral de neratinib radiomarcado de 200 mg (0,83 veces la dosis máxima recomendada aprobada), la excreción fecal representó aproximadamente el 97% y la excreción urinaria representó el 1,1% de la dosis total. El 61% de la radiactividad excretada se recuperó en 96 horas y el 98% se recuperó después de 10 días.
Poblaciones específicas
La edad, el sexo, la raza y la función renal no tienen un efecto clínicamente significativo en la farmacocinética de neratinib.
Pacientes con insuficiencia hepática
Las exposiciones a neratinib en pacientes con insuficiencia hepática leve (Child Pugh A) y moderada (Child Pugh B) fueron similares a las de los sujetos sanos con función hepática normal. La C
maxy el AUC de neratinib aumentaron un 173% y un 181%, respectivamente, en pacientes con insuficiencia hepática grave (Child Pugh C) en comparación con los sujetos con función hepática normal
[ver Posología y forma de administración (
2.4) y Uso en poblaciones específicas (
8.6)]
.
Estudios de interacción medicamentosa
Agentes reductores del ácido gástrico:El uso concomitante con lansoprazol (inhibidor de la bomba de protones) disminuyó la C
maxde neratinib en un 71% y el AUC en un 65%. Cuando NERLYNX se administró 2 horas después de ranitidina (antagonista del receptor H
2), la C
maxde neratinib se redujo en un 57% y el AUC en un 48%. Cuando NERLYNX se administró 2 horas antes de ranitidina, la C
maxde neratinib se redujo en un 44% y el AUC en un 32%
[ver Posología y forma de administración (
2.5) e Interacciones medicamentosas (
7.1)]
.
Inhibidores fuertes de CYP3A4:El uso concomitante de ketoconazol (inhibidor fuerte de CYP3A4 e inhibidor de P-gp) aumentó la C
maxde neratinib en un 221% y el AUC en un 381%
[ver Interacciones medicamentosas (
7.1)]
.
Inhibidores duales de P-gp y CYP3A4 moderados:Verapamilo (inhibidor dual moderado de CYP3A4 y P-gp) aumentó la C
maxy el AUC de neratinib en un 203% y un 299%, respectivamente
[ver Interacciones medicamentosas (
7.1)]
.
Inhibidores moderados de CYP3A4:Fluconazol (inhibidor moderado de CYP3A4) aumentó la C
maxy el AUC de neratinib en un 30% y un 68%, respectivamente.
Inductores fuertes y moderados de CYP3A4:El uso concomitante de rifampicina (inductor fuerte de CYP3A4) disminuyó la C
maxde neratinib en un 76% y el AUC en un 87%. El AUC de los metabolitos activos M6 y M7 también se redujo en un 37-49% en comparación con NERLYNX administrado solo. Efavirenz (inductor moderado de CYP3A4) disminuyó la C
maxde neratinib en un 36% y el AUC en un 52%
[ver Interacciones medicamentosas (
7.1)]
.
Efecto de NERLYNX en los transportadores de P-gp:El uso concomitante de NERLYNX aumentó la C
maxmedia de digoxina (sustrato de P-gp) en un 54% y el AUC en un 32%
[ver Interacciones medicamentosas (
7.2)]
.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
Se llevó a cabo un estudio de carcinogenicidad de dos años en ratas a dosis orales de neratinib de 1, 3 y 10 mg/kg/día. Neratinib no fue carcinogénico en ratas macho y hembra a niveles de exposición >25 veces el AUC en pacientes que recibieron la dosis máxima recomendada de 240 mg/día. Neratinib no fue carcinogénico en un estudio de 26 semanas en ratones transgénicos Tg.rasH2 cuando se administró diariamente por sonda gástrica a dosis de hasta 50 mg/kg/día en machos y 125 mg/kg/día en hembras.
Neratinib no fue mutagénico en una prueba de mutación inversa bacteriana
in vitro(AMES) o clastogénico en una prueba de aberración cromosómica de linfocitos humanos
in vitroo en una prueba de micronúcleos de médula ósea de rata
in vivo.
En un estudio de fertilidad en ratas, la administración de neratinib hasta 12 mg/kg/día (aproximadamente 0,5 veces la dosis máxima recomendada de 240 mg/día en pacientes en una base de mg/m
2) no causó efectos en el apareamiento o la capacidad de los animales para quedar embarazadas. En estudios de toxicidad de dosis repetidas en perros con administración oral de neratinib diariamente durante un máximo de 39 semanas, se observó hipoplasia tubular de los testículos a ≥0,5 mg/kg/día. Este hallazgo se observó a AUC que eran aproximadamente 0,4 veces el AUC en pacientes a la dosis máxima recomendada de 240 mg.
14 ESTUDIOS CLÍNICOS
14.1 Tratamiento Adyuvante Extendido del Cáncer de Mama en Estadio Temprano
La seguridad y eficacia de NERLYNX se investigaron en el ensayo ExteNET (NCT00878709), un estudio multicéntrico, aleatorizado, doble ciego, controlado con placebo de NERLYNX después del tratamiento adyuvante con una terapia basada en trastuzumab en mujeres con cáncer de mama HER2 positivo.
Se aleatorizó a un total de 2840 pacientes con cáncer de mama HER2 positivo en estadio temprano (estadios 1 a 3c) dentro de los dos años posteriores a la finalización del tratamiento con trastuzumab adyuvante para recibir NERLYNX (n = 1420) o placebo (n = 1420). La aleatorización se estratificó por los siguientes factores: estado del receptor hormonal, estado ganglionar (0, 1–3, frente a 4 o más ganglios positivos) y si el trastuzumab se administró de forma secuencial o concurrente con la quimioterapia. NERLYNX 240 mg o placebo se administró por vía oral una vez al día durante un año. La principal medida de resultado de eficacia fue la supervivencia libre de enfermedad invasiva (iDFS), definida como el tiempo transcurrido entre la fecha de aleatorización y la primera aparición de una recurrencia invasiva (cáncer de mama local/regional, ipsilateral o contralateral), recurrencia a distancia o muerte por cualquier causa, con 2 años y 28 días de seguimiento.
Las características demográficas de los pacientes y las características tumorales se equilibraron generalmente entre los brazos de tratamiento. Los pacientes tenían una edad media de 52 años (rango de 23 a 83) y el 12% de los pacientes tenían 65 años o más. La mayoría de los pacientes eran blancos (81%) y la mayoría de los pacientes (99,7%) tenían un estado de rendimiento ECOG de 0 o 1. El 57% (57%) de los pacientes tenían enfermedad con receptor hormonal positivo (definida como ER positivo y/o PR positivo), el 24% eran negativos para ganglios, el 47% tenían de uno a tres ganglios positivos y el 30% tenían cuatro o más ganglios positivos. El 10% (10%) de los pacientes tenían enfermedad en estadio I, el 41% tenían enfermedad en estadio II y el 31% tenían enfermedad en estadio III. La mayoría de los pacientes (81%) se inscribieron dentro de un año de la finalización del tratamiento con trastuzumab. El tiempo medio transcurrido desde el último tratamiento adyuvante con trastuzumab hasta la aleatorización fue de 4,4 meses en el brazo de NERLYNX frente a 4,6 meses en el brazo de placebo. La duración media del tratamiento fue de 11,6 meses en el brazo de NERLYNX frente a 11,8 meses en el brazo de placebo.
Los resultados de eficacia del ensayo ExteNET se resumen en
|
IC= Intervalo de Confianza; HR= Razón de Riesgo; iDFS= Supervivencia Libre de Enfermedad Invasiva; ITT= Intención de Tratar |
|||||
|
* Estimación de Kaplan-Meier |
|||||
|
† Estratificado por trastuzumab previo (concurrente frente a secuencial), estado ganglionar (0–3 ganglios positivos frente a ≥4 ganglios positivos) y estado ER/PR (positivo frente a negativo) |
|||||
|
‡ Prueba de rango logarítmico estratificada |
|||||
| Número de Eventos/Total N (%) |
iDFS a los 24 meses,* % (IC del 95%) |
HR estratificado† (IC del 95%) |
Valor p‡ | ||
| NERLYNX | Placebo | NERLYNX | Placebo | ||
| 67/1420 (4.7) |
106/1420 (7.5) |
94.2 (92.6, 95.4) |
91.9 (90.2, 93.2) |
0.66 (0.49, 0.90) |
0.008 |
Figura 1: iDFS en el ensayo ExteNET – Población ITT
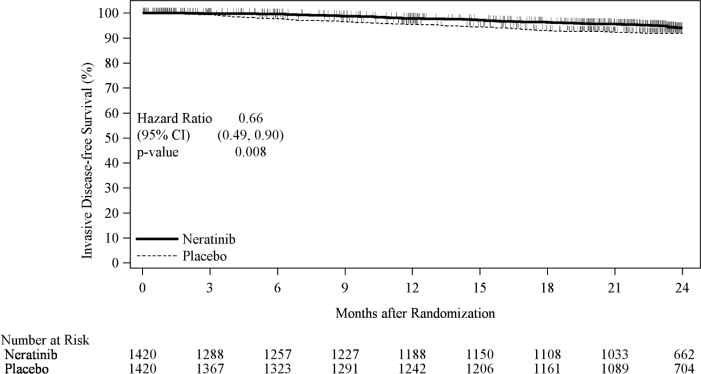
CI=Intervalo de confianza; iDFS=Supervivencia libre de enfermedad invasiva; ITT=Intención de tratar
|
CI=Intervalo de confianza; HR=Razón de riesgo |
|||||
|
* Análisis exploratorios sin ajustar las comparaciones múltiples |
|||||
|
† Estimación de Kaplan-Meier |
|||||
| Población | Número de eventos/Total N (%) |
iDFS a los 24 meses,†% (95% CI) |
HR no estratificado (95% CI) | ||
| NERLYNX | Placebo | NERLYNX | Placebo | ||
| Estado del receptor hormonal | |||||
| Positivo | 29/816 (3.6) |
63/815 (7.7) |
95.6 (93.8, 96.9) |
91.5 (89.2, 93.3) |
0.49 (0.31, 0.75) |
| Negativo | 38/604 (6.3) |
43/605 (7.1) |
92.2 (89.4, 94.3) |
92.4 (89.8, 94.3) |
0.93 (0.60, 1.43) |
| Estado ganglionar | |||||
| Negativo | 7/335 (2.1) |
11/336 (3.3) |
97.2 (94.1, 98.7) |
96.5 (93.7, 98.0) |
0.72 (0.26, 1.83) |
| 1–3 Ganglios positivos | 31/664 (4.7) |
47/664 (7.1) |
94.4 (92.2, 96.1) |
92.4 (90.0, 94.2) |
0.68 (0.43, 1.07) |
| ≥4 Ganglios positivos | 29/421 (6.9) |
48/420 (11.4) |
91.4 (87.9, 94.0) |
87.3 (83.4, 90.2) |
0.62 (0.39, 0.97) |
| Trastuzumab previo | |||||
| Concurrente | 49/884 (5.5) |
66/886 (7.4) |
93.2 (91.0, 94.8) |
92.0 (89.9, 93.7) |
0.80 (0.55, 1.16) |
| Secuencial | 18/536 (3.4) |
40/534 (7.5) |
95.8 (93.4, 97.3) |
91.6 (88.7, 93.8) |
0.46 (0.26, 0.78) |
| Finalización del trastuzumab previo | |||||
| ≤1 año | 58/1152 (5.0) |
95/1145 (8.3) |
93.8 (92.0, 95.2) |
90.9 (89.0, 92.5) |
0.63 (0.45, 0.88) |
| 1–2 años | 9/262 (3.4) |
11/270 (4.1) |
95.8 (92.0, 97.8) |
95.7 (92.3, 97.6) |
0.92 (0.37, 2.22) |
Aproximadamente el 75% de los pacientes fueron reconsentidos para un seguimiento extendido más allá de los 24 meses. Las observaciones con datos faltantes se censuraron en la última fecha de evaluación. Este análisis exploratorio sugiere que los resultados de iDFS a los 5 años son consistentes con los resultados de iDFS a los 2 años observados en ExteNET. Después de un seguimiento mediano de 8 años, no hubo una diferencia estadísticamente significativa en la OS entre el brazo de NERLYNX y el brazo de placebo [HR 0.95 (IC del 95%: 0.75, 1.21)]. La estimación de OS a los 5 años fue del 94.1% (IC del 95%, 92.7%, 95.3%) en el brazo de NERLYNX y del 93.3% (IC del 95%, 91.8%, 94.5%) en el brazo de placebo.
14.2 Cáncer de mama avanzado o metastásico
La seguridad y eficacia de NERLYNX en combinación con capecitabina se estudió en NALA (NCT01808573), un ensayo clínico aleatorizado, multicéntrico, abierto en pacientes (n=621) con cáncer de mama HER2 positivo metastásico que habían recibido 2 o más regímenes previos basados en anti-HER2 en el entorno metastásico. La expresión de HER2 se basó en tejido de archivo probado en un laboratorio central antes de la inscripción. La positividad de HER2 se definió como una puntuación de inmunohistoquímica (IHC) de 3+ o IHC 2+ con hibridación in situ (ISH) confirmatoria positiva. El cincuenta y nueve por ciento de estos pacientes fueron receptores hormonales positivos (HR+) y el 41% fueron receptores hormonales negativos (HR-); el 69% había recibido dos regímenes previos basados en anti-HER2, el 31% había recibido tres o más regímenes previos basados en anti-HER2, el 81% tenía enfermedad visceral y el 19% tenía enfermedad no visceral únicamente. Los pacientes con metástasis cerebrales asintomáticas o estables se incluyeron en el ensayo NALA (16%).
Los pacientes fueron aleatorizados (1:1) para recibir NERLYNX 240 mg por vía oral una vez al día en los días 1–21 en combinación con capecitabina 750 mg/m
2administrado por vía oral dos veces al día en los días 1–14 de cada ciclo de 21 días (n=307) o lapatinib 1250 mg por vía oral una vez al día en los días 1–21 en combinación con capecitabina 1000 mg/m
2administrado por vía oral dos veces al día en los días 1–14 de cada ciclo de 21 días (n=314). Los pacientes fueron tratados hasta la progresión de la enfermedad o la toxicidad inaceptable.
Los resultados de eficacia del ensayo NALA se resumen en
Figura 2, y
|
HR=Hazard Ratio |
||
|
* La razón de riesgo se presenta como NERLYNX más Capecitabina (N+C) vs Lapatinib más Capecitabina (L+C). |
||
|
† Prueba de rango logarítmico estratificada |
||
|
‡ El número total de pacientes que permanecieron en el estudio a los 24 meses es 11; con 9 pacientes en N+C y 2 pacientes en L+C. |
||
|
§ ORR confirmada en pacientes con enfermedad medible en la selección (256 en el brazo N+C y 270 en el brazo L+C) |
||
| NERLYNX + Capecitabina (n=307) |
Lapatinib + Capecitabina (n=314) |
|
| Supervivencia libre de progresión (PFS) | ||
| Número de eventos (%) | 210 (68.4) | 223 (71.0) |
| PFS mediana, meses (IC del 95%) | 5.6 (4.9, 6.9) | 5.5 (4.3, 5.6) |
| HR (IC del 95%)
* |
0.76 (0.63,0.93) | |
| Valor p
† |
0.0059 | |
| Tasas de PFS a los 12 meses, % (IC del 95%) | 29 (23, 35) | 15 (10, 20) |
| Tasas de PFS a los 24 meses, % (IC del 95%)
‡ |
12 (7, 18) | 3 (1, 8) |
| Supervivencia general (OS) | ||
| Número de eventos (%) | 192 (62.5) | 218 (69.4) |
| OS mediana, meses (IC del 95%) | 21.0 (17.7, 23.8) | 18.7 (15.5, 21.2) |
| HR (IC del 95%)
* |
0.88 (0.72, 1.07) | |
| Valor p
† |
0.2086 | |
| Tasa de respuesta objetiva (ORR)§ | ||
| ORR, % (IC del 95%) | 32.8 (27.1, 38.9) | 26.7 (21.5, 32.4) |
| Duración de la respuesta (DOR) | ||
| DOR mediana, meses (IC del 95%) | 8.5 (5.6, 11.2) | 5.6 (4.2, 6.4) |
Figura 2. Supervivencia libre de progresión (Evaluación central – Población ITT)
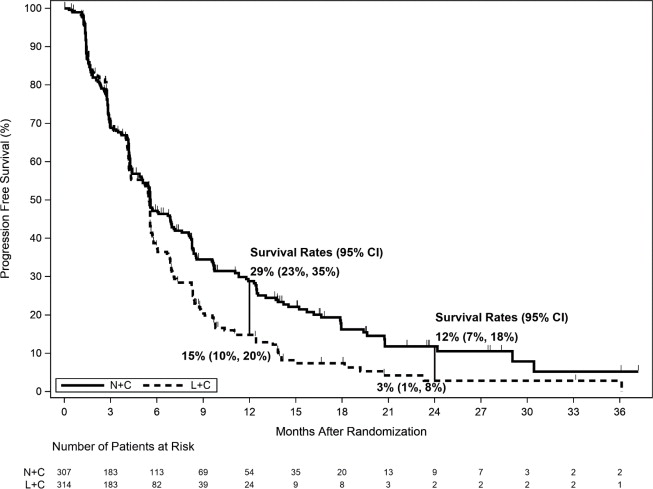
IC=Intervalo de confianza; ITT=Intención de tratar; L+C=Lapatinib más Capecitabina; N+C=NERLYNX más Capecitabina
Figura 3. Supervivencia general (Población ITT)
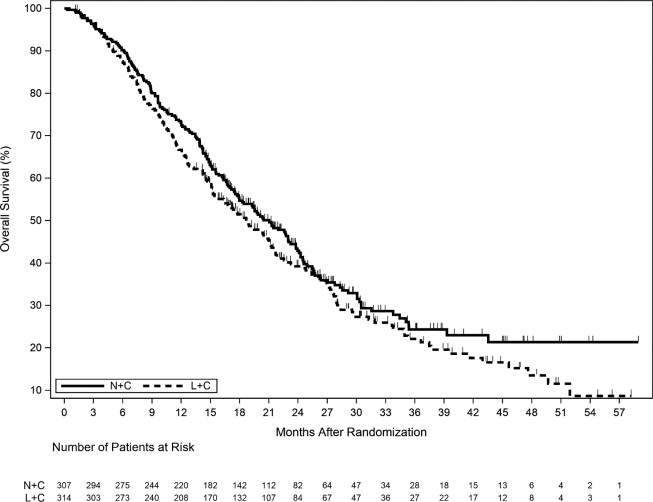
ITT=Intención de tratar; L+C=Lapatinib más Capecitabina; N+C=NERLYNX más Capecitabina
|
IC=Intervalo de confianza; SLP=Supervivencia libre de progresión |
||||
|
α Análisis exploratorio |
||||
| Población | Número de eventos/Total N (%) | Tasas de SLP (%) a los 12 meses (IC del 95%) | ||
| NERLYNX + Capecitabina |
Lapatinib + Capecitabina |
NERLYNX + Capecitabina |
Lapatinib + Capecitabina |
|
| Ubicación de la enfermedad | ||||
| Visceral | 181/247 (73.3) | 185/253 (73.1) | 23 (17, 30) | 14 (10, 20) |
| No visceral | 29/60 (48.3) | 38/61 (62.3) | 53 (38, 66) | 18 (7, 32) |
| Estado del receptor hormonal | ||||
| Positivo | 128/181 (70.7) | 115/186 (61.8) | 27 (19, 34) | 23 (15, 31) |
| Negativo | 82/126 (65.1) | 108/128 (84.4) | 32 (23, 41) | 5 (2, 11) |
| Regímenes previos de HER2 | ||||
| 2 Regímenes | 148/215 (68.8) | 151/215 (70.2) | 26 (20, 33) | 13 (8, 19) |
| ≥3 Regímenes | 62/92 (67.4) | 72/99 (72.7) | 34 (24, 45) | 19 (11, 29) |
16 FORMA DE SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
Las tabletas recubiertas con película NERLYNX de 40 mg son de color rojo, de forma ovalada y con la marca ‘W104’ en un lado y lisas en el otro.
NERLYNX está disponible en:
- Frascos de 180 tabletas: NDC 70437-240-18
- Frascos de 133 tabletas: NDC 70437-240-33
- Frascos de 126 tabletas: NDC 70437-240-26
Almacenar a temperatura ambiente controlada, de 20°C a 25°C (68°F a 77°F); se permiten excursiones de 15°C a 30°C (59°F a 86°F)
[ver USP Temperatura ambiente controlada].
17 INFORMACIÓN PARA EL PACIENTE
Aconseje al paciente que lea la etiqueta del paciente aprobada por la FDA (
Información para el paciente).
Diarrea
- Informe a los pacientes que NERLYNX se ha asociado con diarrea, que puede ser grave en algunos casos.
- Cuando no se utiliza la escalada de dosis, indique a los pacientes que inicien la profilaxis antidiarreica con la primera dosis de NERLYNX.
- Cuando se utiliza la escalada de dosis, indique a los pacientes que inicien 2 semanas de NERLYNX a dosis más bajas antes de recibir la dosis completa recomendada de NERLYNX.
- Indique a los pacientes que mantengan de 1 a 2 deposiciones al día y cómo utilizar los regímenes de tratamiento antidiarreico.
- Aconseje a los pacientes que informen a su médico inmediatamente si se produce diarrea grave (≥Grado 3) o diarrea asociada con debilidad, mareos o fiebre durante el tratamiento con NERLYNX
[ver Dosificación y administración (
2.1,
2.2) y Advertencias y precauciones (
5.1)]
.
Hepatotoxicidad
- Informe a los pacientes que NERLYNX se ha asociado con hepatotoxicidad, que puede ser grave en algunos casos.
- Informe a los pacientes que deben informar a su médico inmediatamente sobre los signos y síntomas de disfunción hepática
[ver Advertencias y precauciones (
5.2)]
.
Toxicidad embrio-fetal
- Aconseje a las mujeres que informen a su médico si están embarazadas o quedan embarazadas. Informe a las pacientes del riesgo para el feto y la posible pérdida del embarazo
[ver Uso en poblaciones específicas (
8.1)]
.
- Aconseje a las mujeres en edad fértil que utilicen métodos anticonceptivos eficaces durante el tratamiento y durante 1 mes después de recibir la última dosis de NERLYNX
[ver Advertencias y precauciones (
5.3) y Uso en poblaciones específicas (
8.1,
8.3)]
.
- Aconseje a las mujeres lactantes que no den el pecho durante el tratamiento con NERLYNX y durante al menos 1 mes después de la última dosis
[ver Uso en poblaciones específicas (
8.2)]
.
Interacciones medicamentosas
- NERLYNX puede interactuar con muchos medicamentos; por lo tanto, aconseje a los pacientes que informen a su médico sobre el uso de cualquier otro medicamento recetado o de venta libre o productos herbales
[ver Dosificación y administración (
2.5) y Farmacología clínica (
12.3)]
.
- NERLYNX puede interactuar con los agentes reductores del ácido gástrico. Aconseje a los pacientes que eviten el uso concomitante de inhibidores de la bomba de protones. Cuando los pacientes requieran agentes reductores del ácido gástrico, utilice un antagonista del receptor H
2 o un antiácido. Aconseje a los pacientes que separen la dosificación de NERLYNX por 3 horas después de la medicina antiácida, y que tomen NERLYNX al menos 2 horas antes o 10 horas después de un antagonista del receptor H
2
[ver Dosificación y administración (
2.5) e Interacciones medicamentosas (
7.1)]
.
- NERLYNX puede interactuar con el pomelo. Aconseje a los pacientes que eviten tomar NERLYNX con productos de pomelo
[ver Interacciones medicamentosas (
7.1)]
.
Dosificación y administración
- Para los pacientes que se someten a tratamiento adyuvante prolongado para el cáncer de mama en estadio temprano, indique a los pacientes que tomen NERLYNX con alimentos aproximadamente a la misma hora todos los días de forma consecutiva hasta la recurrencia de la enfermedad o durante un máximo de un año.
- Para los pacientes que se someten a tratamiento para el cáncer de mama metastásico, indique a los pacientes que tomen NERLYNX con alimentos los días 1 a 21 de un ciclo de 21 días, con capecitabina los días 1 a 14 de un ciclo de 21 días hasta la progresión de la enfermedad o toxicidades inaceptables.
- Si un paciente olvida una dosis, indique al paciente que no reemplace la dosis olvidada y que reanude NERLYNX con la siguiente dosis diaria programada
[ver Dosificación y administración (
2.2)]
.
Fabricado para Puma Biotechnology, Inc.
10880 Wilshire Blvd., Suite 2150
Los Ángeles, CA 90024-4106
©2022, Puma Biotechnology, Inc. Todos los derechos reservados.
INSERTO PARA EL PACIENTE
|
Esta información para el paciente ha sido aprobada por la Administración de Alimentos y Medicamentos de los EE. UU. |
Revisión: 03/2022 |
|
| INFORMACIÓN PARA EL PACIENTE NERLYNX®(ner linkss) (neratinib) tabletas |
||
|
¿Cuál es la información más importante que debo saber sobre NERLYNX?
|
||
| Su proveedor de atención médica puede cambiar su dosis de NERLYNX, suspender temporalmente o detener completamente NERLYNX si es necesario para controlar su diarrea. Consulte ” ¿Cuáles son los posibles efectos secundarios de NERLYNX?” para más información sobre los efectos secundarios. |
||
¿Qué es NERLYNX?
|
||
| No se sabe si NERLYNX es seguro y eficaz en niños. | ||
Antes de tomar NERLYNX, informe a su proveedor de atención médica sobre todas sus condiciones médicas, incluso si:
|
||
| Informe a su proveedor de atención médica sobre todos los medicamentos que toma,incluidos los medicamentos recetados y de venta libre, las vitaminas y los suplementos herbales. Especialmente informe a su proveedor de atención médica sitoma medicamentos utilizados para disminuir el ácido estomacal, llamados inhibidores de la bomba de protones o IBP. Debe evitar tomar estos medicamentos durante el tratamiento con NERLYNX. |
||
¿Cómo debo tomar NERLYNX?
|
||
| ¿Qué debo evitar mientras tomo NERLYNX? Debe evitar comer productos que contengan toronja (pomelo) durante el tratamiento con NERLYNX. |
||
|
¿Cuáles son los posibles efectos secundarios de NERLYNX? NERLYNX puede causar efectos secundarios graves, que incluyen: ¿Cuál es la información más importante que debo saber sobre NERLYNX?”
|
||
|
|
|
| Los efectos secundarios más comunes de NERLYNX cuando se usa solo incluyen: | ||
|
|
|
| Los efectos secundarios más comunes de NERLYNX cuando se usa con capecitabina incluyen: | ||
|
|
|
| Estos no son todos los posibles efectos secundarios de NERLYNX. Para obtener más información, consulte a su proveedor de atención médica. Informe a su proveedor de atención médica si tiene algún efecto secundario que le moleste o que no desaparezca. Llame a su médico para obtener asesoramiento médico sobre los efectos secundarios. Puede informar los efectos secundarios a la FDA al 1-800-FDA-1088. |
||
¿Cómo debo almacenar NERLYNX?
|
||
| Mantenga NERLYNX y todos los medicamentos fuera del alcance de los niños. | ||
| Información general sobre el uso seguro y eficaz de NERLYNX. Los medicamentos a veces se recetan para fines distintos a los que se enumeran en un folleto de información para el paciente. No use NERLYNX para una afección para la que no fue recetado. No le dé NERLYNX a otras personas, incluso si tienen los mismos síntomas que usted. Puede dañarlos. Puede pedirle a su farmacéutico o proveedor de atención médica información sobre NERLYNX que esté escrita para profesionales de la salud. |
||
| ¿Cuáles son los ingredientes de NERLYNX? Ingrediente activo: neratinib Ingredientes inactivos: Núcleo del comprimido: dióxido de silicio coloidal, manitol, celulosa microcristalina, crospovidona, povidona, estearato de magnesio y agua purificada. Recubrimiento: recubrimiento pelicular rojo: alcohol polivinílico, dióxido de titanio, polietilenglicol, talco y óxido de hierro rojo. Fabricado para: Puma Biotechnology, Inc. 10880 Wilshire Blvd., Suite 2150 Los Angeles, CA 90024-4106 ©2022, Puma Biotechnology, Inc. Reservados todos los derechos. Para obtener más información, visite www.NERLYNX.com o llame al 1-844-637-5969. |
||
PANEL DE VISUALIZACIÓN PRINCIPAL
Panel de visualización principal – Etiqueta de la caja de 180 tabletas de Nerlynx
NDC 70437-240-18
nerlynx™
(neratinib) tabletas
40 mg
Cada tableta recubierta contiene 40 mg de neratinib
equivalente a 48.31 mg de maleato de neratinib.
180 tabletas solo con receta médica

PANEL DE VISUALIZACIÓN PRINCIPAL
Panel de exhibición principal – Etiqueta de botella de 180 tabletas de Nerlynx
NDC 70437-240-18
nerlynx™
(neratinib) tabletas
40 mg
Solo con receta médica 180 tabletas

PANEL DE VISUALIZACIÓN PRINCIPAL
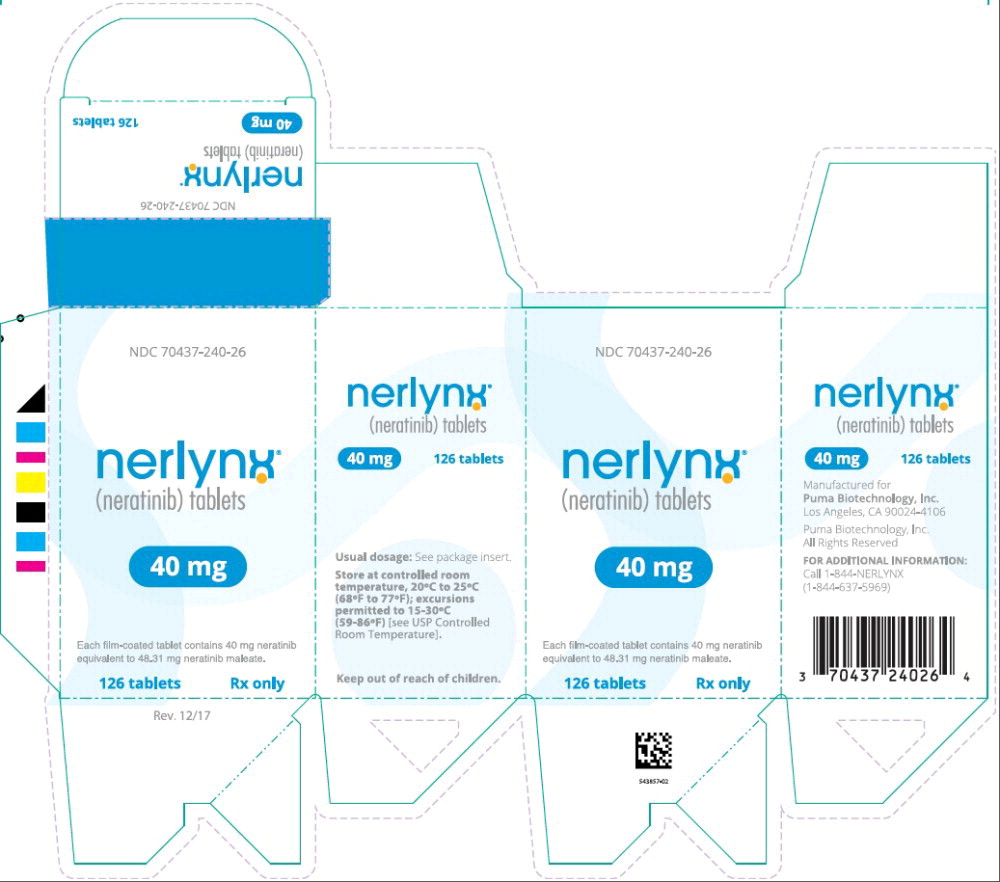
Panel de visualización principal – Etiqueta de la caja de 126 tabletas de Nerlynx
NDC 70437-240-26
nerlynx™
(neratinib) tabletas
40 mg
Cada tableta recubierta contiene 40 mg de neratinib
equivalente a 48.31 mg de neratinib maleato.
126 tabletas Solo con receta médica
PANEL DE VISUALIZACIÓN PRINCIPAL
Panel de exhibición principal – Etiqueta del frasco de 126 tabletas de Nerlynx
NDC 70437-240-26
nerlynx™
(neratinib) tabletas
40 mg
Solo con receta médica 126 tabletas

PANEL DE VISUALIZACIÓN PRINCIPAL
Panel de exhibición principal – Etiqueta de la caja de 133 tabletas de Nerlynx
NDC 70437-240-33
nerlynx™
(neratinib) tabletas
40 mg
Cada tableta recubierta contiene 40 mg de neratinib
equivalente a 48.31 mg de maleato de neratinib.
133 tabletas Solo con receta médica

PANEL DE VISUALIZACIÓN PRINCIPAL
Panel de visualización principal – Etiqueta del frasco de 133 tabletas de Nerlynx
NDC 70437-240-33
nerlynx™
(neratinib) tabletas
40 mg
Solo con receta médica 133 tabletas
