
Fabricante de medicamentos: Novartis Pharmaceuticals Corporation (Updated: 2024-10-01)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
MEKINIST® (trametinib) tabletas, para uso oral
MEKINIST® (trametinib) para solución oral
Aprobación inicial en EE. UU.: 2013
INDICACIONES Y USO
MEKINIST es un inhibidor de la cinasa indicado como agente único para el tratamiento de pacientes con melanoma irresecable o metastásico con mutaciones BRAF V600E o V600K que no han recibido tratamiento con inhibidores de BRAF, detectadas mediante una prueba aprobada por la FDA. (1.1, 2.1)
MEKINIST está indicado, en combinación con dabrafenib, para:
- el tratamiento de pacientes con melanoma irresecable o metastásico con mutaciones BRAF V600E o V600K, detectadas mediante una prueba aprobada por la FDA. (1.1, 2.1)
- el tratamiento adyuvante de pacientes con melanoma con mutaciones BRAF V600E o V600K, detectadas mediante una prueba aprobada por la FDA, e invasión de ganglio(s) linfático(s), después de la resección completa. (1.2, 2.1)
- el tratamiento de pacientes con cáncer de pulmón de células no pequeñas (CPCNP) metastásico con mutación BRAF V600E, detectada mediante una prueba aprobada por la FDA. (1.3, 2.1)
- el tratamiento de pacientes con cáncer de tiroides anaplásico (CTA) localmente avanzado o metastásico con mutación BRAF V600E y sin opciones de tratamiento locoregional satisfactorias. (1.4, 2.1)
- el tratamiento de pacientes adultos y pediátricos de 1 año de edad o mayores con tumores sólidos irresecables o metastásicos con mutación BRAF V600E que han progresado después del tratamiento previo y no tienen opciones de tratamiento alternativas satisfactorias. Esta indicación está aprobada bajo aprobación acelerada en función de la tasa de respuesta global y la duración de la respuesta. La aprobación continua para esta indicación puede estar supeditada a la verificación y descripción del beneficio clínico en un ensayo(s) confirmatorio(s). (1.5, 2.1)
- el tratamiento de pacientes pediátricos de 1 año de edad o mayores con glioma de bajo grado (GBG) con una mutación BRAF V600E que requieren terapia sistémica. (1.6, 2.1)
Limitaciones de uso: MEKINIST no está indicado para el tratamiento de pacientes con cáncer colorrectal debido a la resistencia intrínseca conocida a la inhibición de BRAF. (1.7, 12.1)
DOSIFICACIÓN Y ADMINISTRACIÓN
- La dosis recomendada de MEKINIST en pacientes adultos es de 2 mg por vía oral una vez al día. La dosis recomendada de MEKINIST en pacientes pediátricos se basa en el peso corporal. Tome MEKINIST al menos 1 hora antes o al menos 2 horas después de una comida. (2)
FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
CONTRAINDICACIONES
Ninguna. (4)
ADVERTENCIAS Y PRECAUCIONES
- Nuevas neoplasias malignas primarias, cutáneas y no cutáneas: Pueden ocurrir cuando MEKINIST se usa con dabrafenib. Monitorear a los pacientes para detectar nuevas neoplasias malignas antes o durante la terapia, y después de la interrupción del tratamiento. (5.1)
- Hemorragia: Pueden ocurrir eventos hemorrágicos importantes. Monitorear los signos y síntomas de sangrado. (5.2)
- Colitis y perforación gastrointestinal: La colitis y la perforación gastrointestinal pueden ocurrir en pacientes que reciben MEKINIST. (5.3)
- Eventos tromboembólicos venosos: La trombosis venosa profunda (TVP) y la embolia pulmonar (EP) pueden ocurrir en pacientes que reciben MEKINIST. (5.4, 2.4)
- Cardiomiopatía: Evaluar la fracción de eyección del ventrículo izquierdo (FEVI) antes del tratamiento, después de un mes de tratamiento, y luego cada 2 a 3 meses a partir de entonces. (5.5, 2.4)
- Toxicidades oculares: Realizar una evaluación oftalmológica para cualquier trastorno visual. Para la oclusión de la vena retiniana (OVR), suspender permanentemente MEKINIST. (5.6, 2.4)
- Enfermedad pulmonar intersticial (EPI)/Neumonitis: Suspender MEKINIST para síntomas pulmonares inexplicables nuevos o progresivos. Suspender permanentemente MEKINIST para EPI o neumonitis relacionada con el tratamiento. (5.7, 2.4)
- Reacciones febriles graves: Pueden ocurrir cuando MEKINIST se usa con dabrafenib. (5.8, 2.4)
- Toxicidades cutáneas graves: Monitorear las toxicidades cutáneas y las infecciones secundarias. Suspender permanentemente MEKINIST para una erupción de Grado 2 intolerable o para una erupción de Grado 3 o 4 que no mejore dentro de las 3 semanas a pesar de la interrupción de MEKINIST. Suspender permanentemente para reacciones adversas cutáneas graves (SCAR). (5.9, 2.4)
- Hiperglucemia: Monitorear los niveles de glucosa en suero en pacientes con diabetes preexistente o hiperglucemia. (5.10)
- Linfohistiocitosis hemofagocítica (HLH): Interrumpir el tratamiento para HLH sospechosa. Suspender el tratamiento si se confirma la HLH. (5.12)
- Toxicidad embrio-fetal: Puede causar daño fetal. Advertir a las mujeres en edad fértil sobre el riesgo potencial para un feto y el uso de métodos anticonceptivos efectivos. (5.13, 8.1, 8.3)
REACCIONES ADVERSAS
Las reacciones adversas más comunes (≥ 20%) para MEKINIST como agente único incluyen erupción cutánea, diarrea y linfedema. (6.1)
Las reacciones adversas más comunes (≥ 20%) para MEKINIST en combinación con dabrafenib incluyen:
- Melanoma irresecable o metastásico: pirexia, náuseas, erupción cutánea, escalofríos, diarrea, vómitos, hipertensión y edema periférico. (6.1)
- Tratamiento adyuvante del melanoma: pirexia, fatiga, náuseas, dolor de cabeza, erupción cutánea, escalofríos, diarrea, vómitos, artralgia y mialgia. (6.1)
- CNPC: pirexia, fatiga, náuseas, vómitos, diarrea, piel seca, disminución del apetito, edema, erupción cutánea, escalofríos, hemorragia, tos y disnea. (6.1)
- Pacientes adultos con tumores sólidos: pirexia, fatiga, náuseas, erupción cutánea, escalofríos, dolor de cabeza, hemorragia, tos, vómitos, estreñimiento, diarrea, mialgia, artralgia y edema. (6.1)
- Pacientes pediátricos con tumores sólidos: pirexia, erupción cutánea, vómitos, fatiga, piel seca, tos, diarrea, dermatitis acneiforme, dolor de cabeza, dolor abdominal, náuseas, hemorragia, estreñimiento y paroniquia. (6.1)
- Pacientes pediátricos con LGG: pirexia, erupción cutánea, dolor de cabeza, vómitos, dolor musculoesquelético, fatiga, diarrea, piel seca, náuseas, hemorragia, dolor abdominal y dermatitis acneiforme. (6.1)
Para reportar REACCIONES ADVERSAS SOSPECHOSAS, comuníquese con Novartis Pharmaceuticals Corporation al 1-888-669-6682 o con la FDA al 1-800-FDA-1088 o www.fda.gov/medwatch.
USO EN POBLACIONES ESPECÍFICAS
Ver 17 para INFORMACIÓN PARA EL PACIENTE y etiquetado del paciente aprobado por la FDA.
Revisado: 10/2024
Tabla de Contenido
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDO*
1
INDICACIONES Y USO
1.1
Melanoma Inoperable o Metastásico Positivo a la Mutación BRAF V600E o V600K
1.2
Tratamiento Adyuvante del Melanoma Positivo a la Mutación BRAF V600E o V600K
1.3
Cáncer de Pulmón de Células No Pequeñas Metastásico Positivo a la Mutación BRAF V600E
1.4
Cáncer Tiroideo Anaplásico Localmente Avanzado o Metastásico Positivo a la Mutación BRAF V600E
1.5
Tumores Sólidos Inoperables o Metastásicos Positivos a la Mutación BRAF V600E
1.6
Glioma de Bajo Grado Positivo a la Mutación BRAF V600E
1.7
Limitaciones de Uso
2
DOSIFICACIÓN Y ADMINISTRACIÓN
2.1
Selección del Paciente
2.2
Dosis Recomendada
2.3
Administración
2.4
Modificaciones de la Dosis para Reacciones Adversas
3
FORMAS Y FUERZAS DE DOSIFICACIÓN
4
CONTRAINDICACIONES
5
ADVERTENCIAS Y PRECAUCIONES
5.1
Nuevas Neoplasias Primarias
5.2
Hemorragia
5.3
Colitis y Perforación Gastrointestinal
5.4
Eventos Tromboembólicos Venosos
5.5
Cardiomiopatía
5.6
Toxicidades Oculares
5.7
Enfermedad Pulmonar Intersticial/Neumonitis
5.8
Reacciones Febriles Graves
5.9
Toxicidades Cutáneas Graves
5.10
Hiperglucemia
5.11
Riesgos Asociados con el Tratamiento Combinado
5.12
Linfohistiocitosis Hemofagocítica
5.13
Toxicidad Embriofetal
6
REACCIONES ADVERSAS
6.1
Experiencia en Ensayos Clínicos
6.2
Experiencia Postcomercialización
7
INTERACCIONES MEDICAMENTOSAS
8
USO EN POBLACIONES ESPECÍFICAS
8.1
Embarazo
8.2
Lactancia
8.3
Mujeres y Hombres en Edad Reproductiva
8.4
Uso Pediátrico
8.5
Uso Geriátrico
8.6
Insuficiencia Hepática
10
SOBREDOSIS
11
DESCRIPCIÓN
12
FARMACOLOGÍA CLÍNICA
12.1
Mecanismo de Acción
12.2
Farmacodinamia
12.3
Farmacocinética
13
TOXICOLOGÍA NO CLÍNICA
13.1
Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
14
ESTUDIOS CLÍNICOS
14.1
Melanoma Inoperable o Metastásico Positivo a la Mutación BRAF V600E o V600K
14.2
Tratamiento Adyuvante del Melanoma Positivo a la Mutación BRAF V600E o V600K
14.3
Cáncer de Pulmón de Células No Pequeñas Metastásico Positivo a la Mutación BRAF V600E
14.4
Cáncer Tiroideo Anaplásico Localmente Avanzado o Metastásico Positivo a la Mutación BRAF V600E
14.5
Falta de Actividad Clínica en Melanoma Metastásico Después de la Terapia con Inhibidores de BRAF
14.6
Tumores Sólidos Inoperables o Metastásicos Positivos a la Mutación BRAF V600E
14.7
Glioma de Bajo Grado Positivo a la Mutación BRAF V600E
16
FORMA DE SUMINISTRO/ALMACENAMIENTO Y MANEJO
17
INFORMACIÓN PARA EL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información completa de prescripción no están enumeradas.
1 INDICACIONES Y USO
1.1
Melanoma irresecable o metastásico con mutación BRAF V600E o V600K positiva
MEKINIST® está indicado, como agente único en pacientes que no han recibido tratamiento con inhibidores de BRAF o en combinación con dabrafenib, para el tratamiento de pacientes con melanoma irresecable o metastásico con mutaciones BRAF V600E o V600K, detectadas mediante una prueba aprobada por la FDA [ver Dosificación y administración (2.1)].
1.2
Tratamiento adyuvante del melanoma con mutación BRAF V600E o V600K positiva
MEKINIST está indicado, en combinación con dabrafenib, para el tratamiento adyuvante de pacientes con melanoma con mutaciones BRAF V600E o V600K, detectadas mediante una prueba aprobada por la FDA, e invasión de ganglio(s) linfático(s), tras la resección completa [ver Dosificación y administración (2.1)].
1.3
Cáncer de pulmón de células no pequeñas (CPNP) metastásico con mutación BRAF V600E positiva
MEKINIST está indicado, en combinación con dabrafenib, para el tratamiento de pacientes con cáncer de pulmón de células no pequeñas (CPNP) metastásico con mutación BRAF V600E, detectada mediante una prueba aprobada por la FDA [ver Dosificación y administración (2.1)].
1.4
Cáncer de tiroides anaplásico localmente avanzado o metastásico con mutación BRAF V600E positiva
MEKINIST está indicado, en combinación con dabrafenib, para el tratamiento de pacientes con cáncer de tiroides anaplásico (CTA) localmente avanzado o metastásico con mutación BRAF V600E y sin opciones de tratamiento locoregional satisfactorias [ver Dosificación y administración (2.1)].
1.5
Tumores sólidos irresecables o metastásicos con mutación BRAF V600E positiva
MEKINIST está indicado, en combinación con dabrafenib, para el tratamiento de pacientes adultos y pediátricos de 1 año de edad o mayores con tumores sólidos irresecables o metastásicos con mutación BRAF V600E que han progresado tras el tratamiento previo y no tienen opciones de tratamiento alternativas satisfactorias [ver Dosificación y administración (2.1)]. Esta indicación está aprobada bajo aprobación acelerada en base a la tasa de respuesta global y la duración de la respuesta [ver Estudios clínicos (14.6)]. La aprobación continua para esta indicación puede estar supeditada a la verificación y descripción del beneficio clínico en un ensayo(s) confirmatorio(s).
1.6
Glioma de bajo grado con mutación BRAF V600E positiva
MEKINIST está indicado, en combinación con dabrafenib, para el tratamiento de pacientes pediátricos de 1 año de edad o mayores con glioma de bajo grado (GBG) con una mutación BRAF V600E que requieren terapia sistémica [ver Dosificación y administración (2.1)].
1.7
Limitaciones de uso
MEKINIST no está indicado para el tratamiento de pacientes con cáncer colorrectal debido a la conocida resistencia intrínseca a la inhibición de BRAF [ver Indicaciones y uso (1.5), Farmacología clínica (12.1)].
2 DOSIS Y ADMINISTRACIÓN
2.1
Selección del Paciente
Melanoma
- Confirme la presencia de la mutación BRAF V600E o V600K en las muestras tumorales antes de iniciar el tratamiento con MEKINIST como agente único o en combinación con dabrafenib [ver Estudios Clínicos (14.1, 14.2)].
- La información sobre las pruebas aprobadas por la FDA para la detección de mutaciones BRAF V600 en melanoma está disponible en: http://www.fda.gov/CompanionDiagnostics.
CNPC
- Confirme la presencia de la mutación BRAF V600E en las muestras tumorales antes de iniciar el tratamiento con MEKINIST y dabrafenib [ver Estudios Clínicos (14.3)].
- La información sobre las pruebas aprobadas por la FDA para la detección de mutaciones BRAF V600E en CNPC está disponible en: http://www.fda.gov/CompanionDiagnostics.
ATC
- Confirme la presencia de la mutación BRAF V600E en las muestras tumorales antes de iniciar el tratamiento con MEKINIST y dabrafenib [ver Estudios Clínicos (14.4)]. Actualmente no hay una prueba aprobada por la FDA para la detección de la mutación BRAF V600E en ATC.
Tumores Sólidos
- Confirme la presencia de la mutación BRAF V600E en las muestras tumorales antes de iniciar el tratamiento con MEKINIST y dabrafenib [ver Estudios Clínicos (14.6)]. Actualmente no hay una prueba aprobada por la FDA para la detección de la mutación BRAF V600E en tumores sólidos distintos del melanoma y el CNPC.
Glioma de Bajo Grado
- Confirme la presencia de la mutación BRAF V600E en las muestras tumorales antes de iniciar el tratamiento con MEKINIST y dabrafenib [ver Estudios Clínicos (14.7)]. Actualmente no hay una prueba aprobada por la FDA para la detección de la mutación BRAF V600E en LGG.
2.2
Dosis Recomendada
Tabletas de MEKINIST
Pacientes Adultos
La dosis recomendada de tabletas de MEKINIST en pacientes adultos es de 2 mg por vía oral una vez al día [ver Dosis y Administración (2.3)].
Pacientes Pediátricos
La dosis recomendada de tabletas de MEKINIST en pacientes pediátricos que pesan al menos 26 kg se basa en el peso corporal (Tabla 1) [ver Dosis y Administración (2.3)]. No se ha establecido una dosis recomendada de tabletas de MEKINIST en pacientes que pesan menos de 26 kg.
| Peso Corporal | Dosis Recomendada |
| 26 a 37 kg | 1 mg por vía oral una vez al día |
| 38 a 50 kg | 1.5 mg por vía oral una vez al día |
| 51 kg o más | 2 mg por vía oral una vez al día |
MEKINIST para Solución Oral
La dosis recomendada de MEKINIST para solución oral se basa en el peso corporal (Tabla 2) [ver Dosis y Administración (2.3)].
| Peso Corporal | Dosis Recomendada Volumen Total de Solución Oral Una Vez al Día (Contenido de Trametinib) |
| 8 kg | 0.3 mg (6 mL) |
| 9 kg | 0.35 mg (7 mL) |
| 10 kg | 0.35 mg (7 mL) |
| 11 kg | 0.4 mg (8 mL) |
| 12 a 13 kg | 0.45 mg (9 mL) |
| 14 a 17 kg | 0.55 mg (11 mL) |
| 18 a 21 kg | 0.7 mg (14 mL) |
| 22 a 25 kg | 0.85 mg (17 mL) |
| 26 a 29 kg | 0.9 mg (18 mL) |
| 30 a 33 kg | 1 mg (20 mL) |
| 34 a 37 kg | 1.15 mg (23 mL) |
| 38 a 41 kg | 1.25 mg (25 mL) |
| 42 a 45 kg | 1.4 mg (28 mL) |
| 46 a 50 kg | 1.6 mg (32 mL) |
| ≥ 51 kg | 2 mg (40 mL) |
- La duración recomendada del tratamiento para pacientes con melanoma o tumores sólidos irresecables o metastásicos, NSCLC metastásico o cáncer de tiroides anaplásico localmente avanzado o metastásico es hasta la progresión de la enfermedad o toxicidad inaceptable.
- La duración recomendada del tratamiento en el contexto del melanoma adyuvante es hasta la recurrencia de la enfermedad o toxicidad inaceptable por hasta 1 año.
- La duración recomendada del tratamiento para pacientes pediátricos con LGG es hasta la progresión de la enfermedad o hasta toxicidad inaceptable.
Consulte la información de prescripción de dabrafenib para obtener información sobre la dosificación recomendada de dabrafenib.
2.3
Administración
- Tome MEKINIST a la misma hora todos los días, aproximadamente 24 horas de diferencia.
- Tome MEKINIST al menos 1 hora antes o 2 horas después de una comida [ver Farmacología clínica (12.3)].
- No tome una dosis olvidada de MEKINIST dentro de las 12 horas de la siguiente dosis de MEKINIST.
- Si se produce vómito después de la administración de MEKINIST, no tome una dosis adicional. Tome la siguiente dosis a la hora programada.
Tabletas de MEKINIST
- No triture ni rompa las tabletas de MEKINIST.
MEKINIST para solución oral
- MEKINIST para solución oral está destinado a ser administrado por un cuidador. Antes de usar la solución oral, asegúrese de que los cuidadores reciban capacitación sobre la dosificación y administración adecuadas de MEKINIST para solución oral.
Preparación y administración
- Para preparar MEKINIST para solución oral, golpee la botella hasta que el polvo fluya libremente. Agregue 90 mL de agua destilada o purificada al polvo en la botella e invierta o agite suavemente la botella con la tapa puesta hasta por 5 minutos hasta que el polvo se disuelva completamente, obteniendo una solución clara. Separe el adaptador de la botella de la jeringa oral. Inserte el adaptador de la botella en el cuello de la botella después de reconstituir la solución. Escriba la fecha de descarte. Una vez reconstituido, MEKINIST para solución oral se puede usar durante 35 días.
- La concentración final de la solución es de 0.05 mg/mL.
- Administre MEKINIST para solución oral desde una jeringa oral o una sonda de alimentación (calibre 4 francés o más grande).
- Después de la reconstitución, almacene en la botella original por debajo de 25 °C (77 °F) y no congele.
2.4
Modificaciones de la dosis para reacciones adversas
Las reducciones de dosis para reacciones adversas asociadas con MEKINIST se presentan en las Tablas 3 y 4.
| Dosis recomendada | 1 mg por vía oral una vez al día | 1.5 mg por vía oral una vez al día | 2 mg por vía oral una vez al día |
| Primera reducción de dosis | 0.5 mg por vía oral una vez al día | 1 mg por vía oral una vez al día | 1.5 mg por vía oral una vez al día |
| Segunda reducción de dosis | N/A | 0.5 mg por vía oral una vez al día | 1 mg por vía oral una vez al día |
| Modificación posterior | Suspenda permanentemente las tabletas de MEKINIST si no puede tolerar un máximo de dos reducciones de dosis. | ||
| Peso corporal (Dosis recomendada una vez al día) |
Primera reducción de dosis (Administrar una vez al día) |
Segunda reducción de dosis (Administrar una vez al día) |
| 8 kg [0.3 mg (6 mL)] |
0.25 mg (5 mL) | 0.15 mg (3 mL) |
| 9 kg [0.35 mg (7 mL)] |
0.25 mg (5 mL) | 0.2 mg (4 mL) |
| 10 kg [0.35 mg (7 mL)] |
0.25 mg (5 mL) | 0.2 mg (4 mL) |
| 11 kg [0.4 mg (8 mL)] |
0.3 mg (6 mL) | 0.2 mg (4 mL) |
| 12 to 13 kg [0.45 mg (9 mL)] |
0.35 mg (7 mL) | 0.25 mg (5 mL) |
| 14 to 17 kg [0.55 mg (11 mL)] |
0.4 mg (8 mL) | 0.3 mg (6 mL) |
| 18 to 21 kg [0.7 mg (14 mL)] |
0.55 mg (11 mL) | 0.35 mg (7 mL) |
| 22 to 25 kg [0.85 mg (17 mL)] |
0.65 mg (13 mL) | 0.45 mg (9 mL) |
| 26 to 29 kg [0.9 mg (18 mL)] |
0.7 mg (14 mL) | 0.45 mg (9 mL) |
| 30 to 33 kg [1 mg (20 mL)] |
0.75 mg (15 mL) | 0.5 mg (10 mL) |
| 34 to 37 kg [1.15 mg (23 mL)] |
0.85 mg (17 mL) | 0.6 mg (12 mL) |
| 38 to 41 kg [1.25 mg (25 mL)] |
0.95 mg (19 mL) | 0.65 mg (13 mL) |
| 42 to 45 kg [1.4 mg (28 mL)] |
1.05 mg (21 mL) | 0.7 mg (14 mL) |
| 46 to 50 kg [1.6 mg (32 mL)] |
1.2 mg (24 mL) | 0.8 mg (16 mL) |
| ≥ 51 kg [2 mg (40 mL)] |
1.5 mg (30 mL) | 1 mg (20 mL) |
| Suspenda permanentemente MEKINIST para solución oral si no puede tolerar un máximo de dos reducciones de dosis. | ||
Las modificaciones de la dosis para las reacciones adversas asociadas con MEKINIST se presentan en la Tabla 5.
| aCriterios de terminología común del Instituto Nacional del Cáncer para eventos adversos (NCI CTCAE) versión 4.0. bConsulte las Tablas 3 y 4 para las reducciones de dosis recomendadas de MEKINIST. cNo se recomiendan modificaciones de la dosis para MEKINIST cuando se administra con dabrafenib para las siguientes reacciones adversas de dabrafenib: neoplasias no cutáneas y uveítis. No se requiere modificación de la dosis de MEKINIST para nuevas neoplasias cutáneas primarias. |
|
| Gravedad de la reacción adversaa | Modificación de la dosis para MEKINISTb |
| Hemorragia [ver Advertencias y precauciones (5.2)] | |
|
Suspenda MEKINIST.
|
|
Suspenda MEKINIST de forma permanente. |
| Eventos tromboembólicos venosos [ver Advertencias y precauciones (5.4)] | |
|
Suspenda MEKINIST hasta por 3 semanas.
|
|
Suspenda MEKINIST de forma permanente. |
| Cardiomiopatía [ver Advertencias y precauciones (5.5)] | |
|
Suspenda MEKINIST hasta por 4 semanas.
|
|
Suspenda MEKINIST de forma permanente. |
| Toxicidades oculares [ver Advertencias y precauciones (5.6)] | |
|
Suspenda MEKINIST hasta por 3 semanas.
|
|
Suspenda MEKINIST de forma permanente. |
| Pulmonar [ver Advertencias y precauciones (5.7)] | |
|
Suspenda MEKINIST de forma permanente. |
| Reacciones febriles [ver Advertencias y precauciones (5.8)] | |
|
Suspenda MEKINIST hasta que la fiebre desaparezca, luego reanude MEKINIST a la misma dosis o a una dosis más baja. |
|
O
|
| Toxicidades cutáneas [ver Advertencias y precauciones (5.9)] | |
|
Suspenda MEKINIST hasta por 3 semanas.
|
|
Suspenda MEKINIST de forma permanente. |
| Otras reacciones adversasc | |
|
Suspenda MEKINIST.
|
|
O
|
- Recurrent Grade 4
Consulte la información de prescripción de dabrafenib para las modificaciones de dosis para las reacciones adversas asociadas con dabrafenib.
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
Tabletas MEKINIST:
- Tabletas de 0.5 mg: Tabletas de color amarillo, ovaladas modificadas, biconvexas, recubiertas con película con ‘GS’ grabado en relieve en una cara y ‘TFC’ en la cara opuesta.
- Tabletas de 0.5 mg: Tabletas de color amarillo, ovaloides, biconvexas, recubiertas con película sin ranura con bordes biselados y con el logotipo de Novartis grabado en relieve en un lado y ‘TT’ en el otro lado.
- Tabletas de 2 mg: Tabletas de color rosa, redondas, biconvexas, recubiertas con película con ‘GS’ grabado en relieve en una cara y ‘HMJ’ en la cara opuesta.
- Tabletas de 2 mg: Tabletas de color rosa, redondas, biconvexas, recubiertas con película sin ranura con bordes biselados y con el logotipo de Novartis grabado en relieve en un lado y ‘LL’ en el otro lado.
MEKINIST para solución oral:
- Polvo de color blanco a casi blanco que contiene 4.7 mg de trametinib por frasco. Cada mL de solución de trametinib reconstituida con sabor a fresa contiene 0.05 mg de trametinib.
4 CONTRAINDICACIONES
Ninguna.
5 ADVERTENCIAS Y PRECAUCIONES
5.1
Nuevas neoplasias primarias
Neoplasias cutáneas
MEKINIST administrado con dabrafenib (adultos): En la población de seguridad agrupada [ver Reacciones adversas (6.1)], los carcinomas de células escamosas cutáneos (cuSCC) y los queratoacantomas ocurrieron en el 2% de los pacientes. El carcinoma de células basales y el melanoma primario nuevo ocurrieron en el 3% y < 1% de los pacientes, respectivamente.
MEKINIST administrado con dabrafenib (pediátrico): En la población de seguridad agrupada, el melanoma primario nuevo ocurrió en < 1% de los pacientes.
Realice evaluaciones dermatológicas antes de iniciar MEKINIST cuando se use con dabrafenib, cada 2 meses mientras esté en terapia y hasta por 6 meses después de la interrupción de la combinación.
Neoplasias no cutáneas
Con base en su mecanismo de acción, dabrafenib puede promover el crecimiento y desarrollo de neoplasias con activación de RAS a través de mutación u otros mecanismos; consulte la información de prescripción para dabrafenib.
En la población de seguridad agrupada de MEKINIST administrado con dabrafenib, las neoplasias no cutáneas ocurrieron en el 1% de los pacientes.
Monitoree de cerca a los pacientes que reciben MEKINIST y dabrafenib para detectar signos o síntomas de neoplasias no cutáneas. No se requiere modificación de la dosis para MEKINIST en pacientes que desarrollan neoplasias no cutáneas.
5.2
Hemorragia
Las hemorragias, incluida la hemorragia mayor definida como sangrado sintomático en un área o órgano crítico, pueden ocurrir con MEKINIST. Se han reportado casos fatales.
MEKINIST administrado con dabrafenib (adultos): En la población de seguridad agrupada [ver Reacciones adversas (6.1)], los eventos hemorrágicos ocurrieron en el 17% de los pacientes; la hemorragia gastrointestinal ocurrió en el 3% de los pacientes; la hemorragia intracraneal ocurrió en el 0.6% de los pacientes; la hemorragia fatal ocurrió en el 0.5% de los pacientes. Los eventos fatales fueron hemorragia cerebral y hemorragia del tronco encefálico.
MEKINIST administrado con dabrafenib (pediátrico): En la población de seguridad agrupada, los eventos hemorrágicos ocurrieron en el 25% de los pacientes; el tipo más común de sangrado fue la epistaxis (16%). Los eventos graves de sangrado ocurrieron en el 3.6% de los pacientes e incluyeron hemorragia gastrointestinal (1.2%), hemorragia cerebral (0.6%) hemorragia uterina (0.6%), hemorragia postprocedimiento (0.6%) y epistaxis (0.6%).
Suspenda permanentemente MEKINIST para todos los eventos hemorrágicos de Grado 4 y para cualquier evento hemorrágico de Grado 3 que no mejore. Suspenda MEKINIST para los eventos hemorrágicos de Grado 3; si mejora, reanude MEKINIST en el siguiente nivel de dosis más bajo.
5.3
Colitis y perforación gastrointestinal
Se han reportado colitis y perforación gastrointestinal, incluidos los resultados fatales, en pacientes que toman:
MEKINIST en monoterapia y administrado con dabrafenib (adultos): En la población de seguridad agrupada [ver Reacciones adversas (6.1)], la colitis ocurrió en < 1% de los pacientes y la perforación gastrointestinal ocurrió en < 1% de los pacientes.
MEKINIST administrado con dabrafenib (pediátrico): En la población de seguridad agrupada, los eventos de colitis ocurrieron en <1% de los pacientes.
Monitoree de cerca a los pacientes para detectar colitis y perforaciones gastrointestinales.
5.4
Eventos tromboembólicos venosos
MEKINIST administrado con dabrafenib (adultos): En la población de seguridad agrupada [ver Reacciones adversas (6.1)], la trombosis venosa profunda (TVP) y la embolia pulmonar (EP) ocurrieron en el 2% de los pacientes.
MEKINIST administrado con dabrafenib (pediátrico): En la población de seguridad agrupada, los eventos de embolia ocurrieron en < 1% de los pacientes.
Aconseje a los pacientes que busquen atención médica de inmediato si desarrollan síntomas de TVP o EP, como dificultad para respirar, dolor en el pecho o hinchazón en los brazos o piernas. Suspenda permanentemente MEKINIST para la EP que amenaza la vida. Suspenda MEKINIST para la TVP y la EP no complicadas hasta por 3 semanas; si mejora, MEKINIST puede reanudarse a un nivel de dosis más bajo [ver Dosificación y administración (2.4)].
5.5
Cardiomiopatía
La cardiomiopatía, incluida la insuficiencia cardíaca, puede ocurrir con MEKINIST.
MEKINIST administrado con dabrafenib (adultos): En la población de seguridad agrupada [ver Reacciones adversas (6.1)], la cardiomiopatía, definida como una disminución en la fracción de eyección del ventrículo izquierdo (FEVI) ≥ 10% con respecto al valor inicial y por debajo del límite inferior institucional de lo normal (LLN), ocurrió en el 6% de los pacientes. El desarrollo de cardiomiopatía resultó en la interrupción de la dosis o la interrupción de MEKINIST en el 3% y < 1% de los pacientes, respectivamente. La cardiomiopatía se resolvió en 45 de 50 pacientes que recibieron MEKINIST administrado con dabrafenib.
MEKINIST administrado con dabrafenib (pediátrico): En la población de seguridad agrupada, la cardiomiopatía, definida como una disminución en la FEVI ≥ 10% con respecto al valor inicial y por debajo del LLN institucional, ocurrió en el 9% de los pacientes.
Evalúe la FEVI mediante ecocardiograma o exploración de adquisición multigated (MUGA) antes de iniciar MEKINIST como agente único o con dabrafenib, un mes después del inicio y luego a intervalos de 2 a 3 meses mientras esté en tratamiento. Para una disminución absoluta asintomática en la FEVI del 10% o más con respecto al valor inicial que está por debajo del LLN, suspenda MEKINIST hasta por 4 semanas. Si mejora a un valor normal de FEVI, reanude MEKINIST a una dosis más baja. Si no hay mejora a un valor normal de FEVI dentro de las 4 semanas, suspenda permanentemente MEKINIST. Para la cardiomiopatía sintomática o una disminución absoluta en la FEVI de más del 20% con respecto al valor inicial que está por debajo del LLN, suspenda permanentemente MEKINIST [ver Dosificación y administración (2.4)].
5.6
Toxicidades Oculares
Oclusión de la Vena Retiniana
En la población de seguridad agrupada [ver Reacciones Adversas (6.1)] de MEKINIST en monoterapia, la incidencia de oclusión de la vena retiniana (OVR) fue del 0,6%. En la población de seguridad agrupada [ver Reacciones Adversas (6.1)] de MEKINIST administrado con dabrafenib, no hubo casos de OVR. La OVR puede provocar edema macular, disminución de la función visual, neovascularización y glaucoma.
Realice una evaluación oftalmológica urgentemente (dentro de las 24 horas) en caso de pérdida de visión o cualquier otro trastorno visual notificado por el paciente. Suspenda permanentemente MEKINIST en pacientes con OVR documentada [ver Posología y Administración (2.4)].
Desprendimiento del Epitelio Pigmentario Retiniano
El desprendimiento del epitelio pigmentario retiniano (DEPR) puede ocurrir con MEKINIST. Los desprendimientos de retina pueden ser bilaterales y multifocales, ocurriendo en la región macular central de la retina o en otras partes de la retina. En los ensayos de melanoma y CNPC, no se realizó un seguimiento rutinario de los pacientes para detectar DEPR asintomático; por lo tanto, se desconoce la verdadera incidencia de este hallazgo.
MEKINIST administrado con dabrafenib (Pediátrico): En la población de seguridad agrupada, los eventos de DEPR ocurrieron en < 1% de los pacientes.
Realice una evaluación oftalmológica periódicamente y en cualquier momento que un paciente reporte trastornos visuales. Suspenda MEKINIST si se diagnostica DEPR. Si se documenta la resolución del DEPR en una evaluación oftalmológica de repetición dentro de las 3 semanas, reanude MEKINIST a la misma dosis o a una dosis reducida. Si no hay mejoría después de 3 semanas, reanude MEKINIST a una dosis reducida o suspenda permanentemente MEKINIST [ver Posología y Administración (2.4)].
5.7
Enfermedad Pulmonar Intersticial/Neumonitis
En la población de seguridad agrupada [ver Reacciones Adversas (6.1)] de MEKINIST en monoterapia, la enfermedad pulmonar intersticial o neumonitis ocurrió en el 2% de los pacientes. En la población de seguridad agrupada [ver Reacciones Adversas (6.1)] de MEKINIST administrado con dabrafenib, la EPI o neumonitis ocurrió en el 1% de los pacientes.
Suspenda MEKINIST en pacientes que presenten síntomas o hallazgos pulmonares nuevos o progresivos, incluyendo tos, disnea, hipoxia, derrame pleural o infiltrados, en espera de investigaciones clínicas. Suspenda permanentemente MEKINIST en pacientes diagnosticados con EPI o neumonitis relacionada con el tratamiento [ver Posología y Administración (2.4)].
5.8
Reacciones Febriles Graves
Las reacciones febriles graves y la fiebre de cualquier gravedad acompañada de hipotensión, escalofríos o temblores, deshidratación o insuficiencia renal, pueden ocurrir cuando MEKINIST se administra con dabrafenib.
MEKINIST administrado con dabrafenib (Adulto): En la población de seguridad agrupada [ver Reacciones Adversas (6.1)], la fiebre ocurrió en el 58% de los pacientes. Las reacciones febriles graves y la fiebre de cualquier gravedad complicada por hipotensión, escalofríos o temblores, deshidratación o insuficiencia renal ocurrieron en el 5% de los pacientes. La fiebre se complicó con hipotensión en el 4%, deshidratación en el 3%, síncope en el 2%, insuficiencia renal en el 1% y escalofríos/temblores graves en < 1% de los pacientes.
MEKINIST administrado con dabrafenib (Pediátrico): En la población de seguridad agrupada [ver Reacciones Adversas (6.1)], la pirexia ocurrió en el 66% de los pacientes.
Suspenda MEKINIST cuando se use como monoterapia, y tanto MEKINIST como dabrafenib cuando se usen en combinación, si la temperatura del paciente es ≥ 100,4°F. En caso de recurrencia, el tratamiento también puede interrumpirse al primer síntoma de pirexia [ver Reacciones Adversas (6.1)]. La fiebre puede complicarse con hipotensión, escalofríos o temblores, deshidratación o insuficiencia renal. Evalúe los signos y síntomas de infección y controle la creatinina sérica y otras pruebas de función renal durante y después de la pirexia grave. Si es apropiado, MEKINIST, o tanto MEKINIST como dabrafenib cuando se usen en combinación, pueden reiniciarse si el paciente se ha recuperado de la reacción febril durante al menos 24 horas, ya sea a la misma dosis o a una dosis más baja [ver Posología y Administración (2.4)]. Administre antipiréticos como profilaxis secundaria al reanudar MEKINIST si el paciente tuvo un episodio previo de reacción febril grave o fiebre asociada con complicaciones. Administre corticosteroides (por ejemplo, prednisona 10 mg diarios) durante al menos 5 días para la segunda o subsiguiente pirexia si la temperatura no vuelve a la línea de base dentro de los 3 días del inicio de la pirexia, o para la pirexia asociada con complicaciones, como deshidratación, hipotensión, insuficiencia renal o escalofríos/temblores graves, y no hay evidencia de infección activa.
5.9
Toxicidades Cutáneas Graves
Se han notificado reacciones adversas cutáneas graves (RACS), incluyendo síndrome de Stevens-Johnson (SSJ) y reacción medicamentosa con eosinofilia y síntomas sistémicos (DRESS), que pueden ser potencialmente mortales o fatales, durante el tratamiento con MEKINIST administrado con dabrafenib [ver Reacciones Adversas (6.2)].
MEKINIST administrado con dabrafenib (Adulto): En la población de seguridad agrupada [ver Reacciones Adversas (6.1)], otras toxicidades cutáneas graves ocurrieron en < 1% de los pacientes.
MEKINIST administrado con dabrafenib (Pediátrico): En la población de seguridad agrupada, los eventos adversos graves de trastornos de la piel y el tejido subcutáneo ocurrieron en el 1,8% de los pacientes.
Controle las reacciones cutáneas graves nuevas o que empeoren. Suspenda permanentemente MEKINIST para las RACS [ver Posología y Administración (2.4)]. Para otras toxicidades cutáneas, suspenda MEKINIST para la toxicidad cutánea intolerable o grave. Reanude MEKINIST a una dosis más baja en pacientes con mejoría o recuperación de la toxicidad cutánea dentro de las 3 semanas. Suspenda permanentemente MEKINIST si la toxicidad cutánea no ha mejorado en 3 semanas [ver Posología y Administración (2.4)].
5.10
Hiperglucemia
MEKINIST administrado con dabrafenib (Adultos): En la población de seguridad combinada [ver Reacciones adversas (6.1)], el 15% de los pacientes con antecedentes de diabetes que recibieron MEKINIST con dabrafenib requirieron una terapia hipoglucémica más intensiva. La hiperglucemia de Grado 3 y Grado 4 se produjo en el 2% de los pacientes.
MEKINIST administrado con dabrafenib (Pediátrico): En la población de seguridad combinada, los eventos de hiperglucemia de Grado 3 y Grado 4 se produjeron en < 1% de los pacientes.
Controle los niveles de glucosa en suero al inicio y según sea clínicamente apropiado cuando MEKINIST se administre con dabrafenib en pacientes con diabetes preexistente o hiperglucemia. Inicie u optimice los medicamentos antihiperglucémicos según sea clínicamente indicado.
5.11
Riesgos asociados con el tratamiento combinado
MEKINIST está indicado para su uso en combinación con dabrafenib. Revise la información de prescripción para dabrafenib para obtener información sobre los riesgos graves de dabrafenib antes de iniciar MEKINIST con dabrafenib.
5.12
Linfohistiocitosis hemofagocítica
Se ha observado linfohistiocitosis hemofagocítica (HLH) en el entorno post-comercialización cuando MEKINIST se administró con dabrafenib. Si se sospecha HLH, interrumpa el tratamiento. Si se confirma HLH, suspenda el tratamiento e inicie la gestión adecuada de HLH.
5.13
Toxicidad embrio-fetal
Con base en los hallazgos de estudios en animales y su mecanismo de acción, MEKINIST puede causar daño fetal cuando se administra a una mujer embarazada. Trametinib fue embriotóxico y abortivo en conejos a dosis mayores o iguales a las que resultaron en exposiciones aproximadamente 0.3 veces la exposición humana a la dosis clínica adulta recomendada. Advierta a las mujeres embarazadas sobre el riesgo potencial para un feto. Avise a las pacientes de potencial reproductivo que usen métodos anticonceptivos efectivos durante el tratamiento con MEKINIST y durante 4 meses después del tratamiento [ver Uso en poblaciones específicas (8.1, 8.3)].
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas clínicamente significativas se describen en otras partes de la etiqueta:
- Nuevas neoplasias malignas primarias [ver Advertencias y precauciones (5.1)]
- Hemorragia [ver Advertencias y precauciones (5.2)]
- Colitis y perforación gastrointestinal [ver Advertencias y precauciones (5.3)]
- Eventos tromboembólicos venosos [ver Advertencias y precauciones (5.4)]
- Cardiomiopatía [ver Advertencias y precauciones (5.5)]
- Toxicidades oculares [ver Advertencias y precauciones (5.6)]
- Enfermedad pulmonar intersticial/neumonitis [ver Advertencias y precauciones (5.7)]
- Reacciones febriles graves [ver Advertencias y precauciones (5.8)]
- Toxicidades cutáneas graves [ver Advertencias y precauciones (5.9)]
- Hiperglucemia [ver Advertencias y precauciones (5.10)]
- Linfohistiocitosis hemofagocítica [ver Advertencias y precauciones (5.12)]
Hay reacciones adversas adicionales asociadas con dabrafenib. Consulte la información de prescripción de dabrafenib para obtener información adicional.
6.1
Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se llevan a cabo en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no se pueden comparar directamente con las tasas en los ensayos clínicos de otro fármaco y es posible que no reflejen las tasas observadas en la práctica.
Grupos de seguridad para adultos
La población de seguridad agrupada descrita en las ADVERTENCIAS Y PRECAUCIONES refleja la exposición a MEKINIST 2 mg por vía oral, una vez al día como agente único en 329 pacientes con varios tumores sólidos inscritos en METRIC, MEK113583 y MEK111054. Entre estos 329 pacientes que recibieron MEKINIST como agente único, el 33% estuvo expuesto durante 6 meses o más y el 9% estuvo expuesto durante más de un año.
La población de seguridad agrupada descrita en las ADVERTENCIAS Y PRECAUCIONES refleja la exposición a MEKINIST 2 mg por vía oral, una vez al día administrado en combinación con dabrafenib 150 mg por vía oral, dos veces al día, en 1087 pacientes inscritos en COMBI-d, COMBI-v, COMBI-AD y BRF113928 con melanoma irresecable o metastásico, melanoma adyuvante o NSCLC. Entre estos 1087 pacientes que recibieron MEKINIST administrado con dabrafenib, el 70% estuvo expuesto durante 6 meses o más y el 21% estuvo expuesto durante más de un año.
Grupo de seguridad pediátrica
La población de seguridad pediátrica agrupada descrita en las ADVERTENCIAS Y PRECAUCIONES refleja la exposición a MEKINIST por vía oral, una vez al día, basado en el peso, administrado en combinación con dabrafenib en 166 pacientes pediátricos en dos ensayos: un estudio multicéntrico, abierto, de múltiples cohortes en pacientes pediátricos con glioma positivo para la mutación BRAF V600E que requiere terapia sistémica (Estudio G2201; n = 123) y un estudio multicéntrico, abierto, de múltiples cohortes en pacientes pediátricos con tumores sólidos refractarios o recurrentes con activación de la vía MAPK (Estudio X2101; n = 43) [ver Estudios clínicos (14.6, 14.7)]. Entre 166 pacientes que recibieron MEKINIST administrado con dabrafenib, el 85% estuvo expuesto durante 6 meses y el 69% estuvo expuesto durante más de un año. Las reacciones adversas más comunes (> 20%) fueron pirexia (66%), erupción cutánea (54%), cefalea (40%), vómitos (38%), dolor musculoesquelético (36%), fatiga (31%), piel seca (31%), diarrea (30%), náuseas (26%), epistaxis y otros eventos hemorrágicos (25%), dolor abdominal (24%) y dermatitis acneiforme (23%). Las anomalías de laboratorio de grado 3 o 4 más comunes (> 2%) fueron disminución del recuento de neutrófilos (20%), aumento de la alanina aminotransferasa (3.1%) y aumento de la aspartato aminotransferasa (3.1%).
Melanoma irresecable o metastásico positivo para la mutación BRAF V600E o V600K
MEKINIST como agente único
La seguridad de MEKINIST se evaluó en el estudio METRIC, un ensayo aleatorizado, abierto, de pacientes con melanoma irresecable o metastásico positivo para la mutación BRAF V600E o V600K que recibieron MEKINIST (N = 211) 2 mg por vía oral una vez al día o quimioterapia (N = 99) (ya sea dacarbazina 1000 mg/m2 cada 3 semanas o paclitaxel 175 mg/m2 cada 3 semanas) [ver Estudios clínicos (14.1)]. Los pacientes con LVEF anormal, antecedentes de síndrome coronario agudo dentro de los 6 meses o evidencia actual de insuficiencia cardíaca congestiva de clase II o superior (Asociación Americana del Corazón) fueron excluidos. La duración media del tratamiento con MEKINIST fue de 4.3 meses.
En este estudio, el 9% de los pacientes que recibieron MEKINIST experimentaron reacciones adversas que dieron lugar a la interrupción permanente de la medicación del ensayo. Las reacciones adversas más frecuentes que dieron lugar a la interrupción permanente de MEKINIST fueron la disminución de la LVEF, la neumonitis, la insuficiencia renal, la diarrea y la erupción cutánea. Las reacciones adversas llevaron a reducciones de dosis en el 27% de los pacientes tratados con MEKINIST. La erupción cutánea y la disminución de la LVEF fueron las razones más frecuentes citadas para las reducciones de dosis de MEKINIST. La Tabla 6 y la Tabla 7 presentan las reacciones adversas y las anomalías de laboratorio, respectivamente, de MEKINIST como agente único en el estudio METRIC.
| aNCI CTCAE versión 4.0. bReacciones adversas de grado 4 limitadas a erupción cutánea (n = 1) en el brazo de trametinib y diarrea (n = 1) en el brazo de quimioterapia. cIncluye estomatitis, estomatitis aftosa, ulceración bucal e inflamación de la mucosa. dIncluye dolor abdominal, dolor abdominal inferior, dolor abdominal superior y sensibilidad abdominal. eIncluye linfedema, edema y edema periférico. fIncluye epistaxis, hemorragia gingival, hematochezia, hemorragia rectal, melena, hemorragia vaginal, hemorragia hemorroidal, hematuria y hemorragia conjuntival. |
||||
| Reacciones adversas | MEKINIST | Quimioterapia | ||
| N = 211 | N = 99 | |||
| Todos Gradosa (%) |
Grados 3 y 4b (%) |
Todos Gradosa (%) |
Grados 3 y 4b (%) |
|
| Piel y tejido subcutáneo | ||||
| Erupción cutánea | 57 | 8 | 10 | 0 |
| Dermatitis acneiforme | 19 | < 1 | 1 | 0 |
| Piel seca | 11 | 0 | 0 | 0 |
| Prurito | 10 | 2 | 1 | 0 |
| Paroniquia | 10 | 0 | 1 | 0 |
| Gastrointestinal | ||||
| Diarrea | 43 | 0 | 16 | 2 |
| Estomatitisc | 15 | 2 | 2 | 0 |
| Dolor abdominald | 13 | 1 | 5 | 1 |
| Vascular | ||||
| Linfedemae | 32 | 1 | 4 | 0 |
| Hipertensión | 15 | 12 | 7 | 3 |
| Hemorragiaf | 13 | < 1 | 0 | 0 |
| Abreviaturas: ALT, alanina aminotransferasa; AST, aspartato aminotransferasa. aSolo se informaron reacciones adversas de grado 3 en cualquiera de los brazos de tratamiento. |
||||
| Anormalidad de laboratorio | MEKINIST | Quimioterapia | ||
| N = 211 | N = 99 | |||
| Todos Grados (%) |
Grados 3 y 4 (%) |
Todos Grados (%) |
Grados 3 y 4 (%) |
|
| AST aumentado | 60 | 2 | 16 | 1 |
| Hipoalbuminemia | 42 | 2 | 23 | 1 |
| ALT aumentado | 39 | 3 | 20 | 3 |
| Anemia | 38 | 2 | 26 | 3 |
| Fosfatasa alcalina aumentada | 24 | 2 | 18 | 3 |
Otras reacciones adversas clínicamente importantes para MEKINIST en un grupo de estudios clínicos de monoterapia con MEKINIST observadas en menos del 10% de los pacientes que recibieron MEKINIST fueron:
Cardíacas: Bradicardia, bloqueo auriculoventricular, bloqueo de rama
Gastrointestinales: Boca seca
Infecciones e Infestaciones: Foliculitis, erupción pustular, celulitis
Musculoesqueléticas y del tejido conectivo: Rabdomiólisis
Sistema nervioso: Mareos, disgeusia, neuropatía periférica
Oculares: Visión borrosa, ojo seco
MEKINIST con Dabrafenib
La seguridad de MEKINIST cuando se administra con dabrafenib se evaluó en 559 pacientes con melanoma previamente no tratado, irresecable o metastásico, con mutación BRAF V600 positiva que recibieron MEKINIST en dos ensayos, el estudio COMBI-d (n = 209), un ensayo multicéntrico, doble ciego, aleatorizado (1:1), controlado con activo y el estudio COMBI-v (n = 350), un ensayo multicéntrico, abierto, aleatorizado (1:1), controlado con activo. En ambos ensayos, los pacientes recibieron MEKINIST 2 mg por vía oral una vez al día y dabrafenib 150 mg por vía oral dos veces al día hasta la progresión de la enfermedad o la toxicidad inaceptable. Ambos ensayos excluyeron a pacientes con LVEF anormal, antecedentes de síndrome coronario agudo en los últimos 6 meses, antecedentes de insuficiencia cardíaca congestiva de clase II o superior (Asociación Americana del Corazón), antecedentes de RVO o RPED, intervalo QTcB ≥ 480 mseg, hipertensión no controlada, arritmias no controladas, metástasis cerebrales activas o antecedentes conocidos de deficiencia de glucosa-6-fosfato deshidrogenasa [ver Estudios clínicos (14.1)].
Entre estos 559 pacientes, 197 (35%) estuvieron expuestos a MEKINIST durante > 6 meses a 12 meses, mientras que 185 (33%) estuvieron expuestos a MEKINIST durante > 1 año. La edad media fue de 55 años (rango: 18 a 91), el 57% eran hombres y el 98% eran blancos, el 72% tenía un estado de rendimiento ECOG basal de 0 y el 28% tenía un estado de rendimiento ECOG de 1, el 64% tenía enfermedad M1c, el 35% tenía deshidrogenasa láctica (LDH) elevada al inicio y el 0,5% tenía antecedentes de metástasis cerebrales.
Las reacciones adversas más comunes (≥ 20%) para MEKINIST en pacientes que recibieron MEKINIST más dabrafenib en los estudios COMBI-d y COMBI-v fueron: pirexia, náuseas, erupción cutánea, escalofríos, diarrea, vómitos, hipertensión y edema periférico.
La demografía y las características tumorales basales de los pacientes inscritos en el estudio COMBI-d se resumen en Estudios clínicos [ver Estudios clínicos (14.1)]. Los pacientes que recibieron MEKINIST más dabrafenib tuvieron una duración media de exposición de 11 meses (rango: 3 días a 30 meses) a MEKINIST. Entre los 209 pacientes que recibieron MEKINIST más dabrafenib, el 26% estuvo expuesto a MEKINIST durante > 6 meses a 12 meses, mientras que el 46% estuvo expuesto a MEKINIST durante > 1 año.
En el estudio COMBI-d, las reacciones adversas que llevaron a la interrupción de MEKINIST ocurrieron en el 11% de los pacientes que recibieron MEKINIST más dabrafenib; las más frecuentes fueron pirexia (1,4%) y disminución de la fracción de eyección (1,4%). Las reacciones adversas que llevaron a reducciones de dosis de MEKINIST ocurrieron en el 18% de los pacientes que recibieron MEKINIST más dabrafenib; las más frecuentes fueron pirexia (2,9%), neutropenia (1,9%), disminución de la fracción de eyección (1,9%) y erupción cutánea (1,9%). Las reacciones adversas que llevaron a interrupciones de la dosis de MEKINIST ocurrieron en el 46% de los pacientes que recibieron MEKINIST más dabrafenib; las más frecuentes fueron pirexia (18%), escalofríos (7%), vómitos (6%) y disminución de la fracción de eyección (4,8%).
La Tabla 8 y la Tabla 9 presentan reacciones adversas y anormalidades de laboratorio seleccionadas, respectivamente, de MEKINIST observadas en el estudio COMBI-d.
|
* ≥ 5% para todos los grados o ≥ 2% para la incidencia de grados 3–4 en pacientes que recibieron MEKINIST con dabrafenib en comparación con pacientes que recibieron dabrafenib como agente único. aNCI CTCAE versión 4.0. bIncluye edema periférico, edema, linfedema, edema localizado y edema generalizado. cIncluye dolor abdominal, dolor abdominal superior, dolor abdominal inferior y molestias abdominales. dIncluye erupción cutánea, erupción cutánea generalizada, erupción cutánea pruriginosa, erupción cutánea eritematosa, erupción cutánea papular, erupción cutánea vesicular, erupción cutánea macular, erupción cutánea maculo-papular y erupción cutánea folicular. eLos eventos más comunes (≥ 1%) incluyen epistaxis, hematochezia, disminución de la hemoglobina, púrpura y hemorragia rectal. Los eventos de grado 4 se limitaron a hematoma hepático y hemorragia de úlcera duodenal (cada uno n = 1 en el brazo de combinación agrupada). |
||||||
| Reacciones adversas | MEKINIST agrupado más Dabrafenib N = 559 |
Estudio COMBI-d | ||||
| MEKINIST más Dabrafenib N = 209 |
Dabrafenib N = 211 |
|||||
| Todas Grados (%) |
Grados 3 y 4 (%) |
Todas Grados (%) |
Grados 3 y 4 (%) |
Todas Grados (%) |
Grados 3 y 4 (%) |
|
| General | ||||||
|
Fiebre |
54 | 5 | 57 | 7 | 33 | 1.9 |
|
Escalofríos |
31 | 0.5 | 31 | 0 | 17 | 0.5 |
|
Edema periféricob |
21 | 0.7 | 25 | 1.4 | 11 | 0.5 |
| Gastrointestinal | ||||||
|
Náuseas |
35 | 0.4 | 34 | 0.5 | 27 | 1.4 |
|
Diarrea |
31 | 1.3 | 30 | 1.4 | 16 | 0.9 |
|
Vómitos |
27 | 1.1 | 25 | 1.0 | 14 | 0.5 |
|
Dolor abdominalc |
18 | 0.9 | 26 | 1.0 | 14 | 2.4 |
| Piel y tejido subcutáneo | ||||||
|
Erupciónd |
32 | 1.1 | 42 | 0 | 27 | 1.4 |
| Vascular | ||||||
|
Hipertensión |
26 | 11 | 25 | 6 | 16 | 6 |
|
Hemorragiae |
18 | 2.0 | 19 | 1.9 | 15 | 1.9 |
| Sistema nervioso | ||||||
|
Mareos |
11 | 0.2 | 14 | 0 | 7 | 0 |
Otras reacciones adversas clínicamente importantes para MEKINIST en los estudios COMBI-d y COMBI-v (N = 559) observadas en menos del 10% de los pacientes que recibieron MEKINIST en combinación con dabrafenib fueron:
Cardíaco: Bradicardia, bloqueo auriculoventricular, bloqueo de rama
Sistema inmunitario: Sarcoidosis
Musculoesquelético y tejido conectivo: Rabdomiólisis
Sistema nervioso: Neuropatía periférica
Piel y tejido subcutáneo: Fotosensibilidad
| Abreviaturas: ALT, alanina aminotransferasa; AST, aspartato aminotransferasa. * ≥ 5% para todos los grados o ≥ 2% para la incidencia de grados 3–4 en pacientes que recibieron MEKINIST con dabrafenib en comparación con pacientes que recibieron dabrafenib como agente único. aPara estas pruebas de laboratorio, el denominador es 556. bPara estas pruebas de laboratorio, el denominador es 208 para el brazo de combinación, 207-209 para el brazo de dabrafenib. cReacciones adversas de grado 4 limitadas a linfopenia e hiperglucemia (cada una n = 4), aumento de ALT y aumento de AST (cada una n = 3), neutropenia (n = 2) e hiponatremia (n = 1) en el brazo de combinación agrupado; neutropenia, linfopenia, aumento de ALT, aumento de AST e hiperglucemia (cada una n = 1) en el brazo de combinación del estudio COMBI-d; neutropenia, trombocitopenia, aumento de ALT y aumento de AST (cada una n = 1) en el brazo de dabrafenib. |
||||||
| Anormalidad de laboratorio | MEKINIST más Dabrafenib agrupado N = 559a |
Estudio COMBI-d | ||||
| MEKINIST más Dabrafenib N = 209b |
Dabrafenib N = 211b |
|||||
| Todos Grados (%) |
Grados 3 y 4c (%) |
Todos Grados (%) |
Grados 3 y 4c (%) |
Todos Grados (%) |
Grados 3 y 4c (%) |
|
| Química | ||||||
|
Hiperglucemia |
60 | 4.7 | 65 | 6 | 57 | 4.3 |
|
Hipoalbuminemia |
48 | 1.1 | 53 | 1.4 | 27 | 0 |
|
Hiponatremia |
25 | 8 | 24 | 6 | 14 | 2.9 |
| Hepático | ||||||
|
Aumento de AST |
59 | 4.1 | 60 | 4.3 | 21 | 1.0 |
| Aumento de la fosfatasa alcalina en sangre | 49 | 2.7 | 50 | 1.0 | 25 | 0.5 |
|
Aumento de ALT |
48 | 4.5 | 44 | 3.8 | 28 | 1.0 |
| Hematología | ||||||
|
Neutropenia |
46 | 7 | 50 | 6 | 16 | 1.9 |
|
Anemia |
43 | 2.3 | 43 | 2.4 | 38 | 4.3 |
|
Lymphopenia |
32 | 8 | 38 | 9 | 28 | 7 |
|
Thrombocytopenia |
21 | 0.7 | 19 | 0.5 | 10 | 0.5 |
Tratamiento Adyuvante del Melanoma Positivo para la Mutación BRAF V600E o V600K
La seguridad de MEKINIST cuando se administra con dabrafenib se evaluó en 435 pacientes con melanoma en estadio III con mutaciones BRAF V600E o V600K después de la resección completa que recibieron al menos una dosis de terapia de estudio en el estudio COMBI-AD [ver Estudios Clínicos (14.2)]. Los pacientes recibieron MEKINIST 2 mg por vía oral una vez al día y dabrafenib 150 mg por vía oral dos veces al día durante 12 meses. El ensayo excluyó a pacientes con LVEF anormal; antecedentes de síndromes coronarios agudos, angioplastia coronaria o colocación de stent dentro de los 6 meses; insuficiencia cardíaca congestiva de clase II o superior (Asociación Americana del Corazón); intervalo QTc ≥ 480 mseg; hipertensión refractaria al tratamiento; arritmias no controladas; o antecedentes de RVO.
Los pacientes que recibieron MEKINIST en combinación con dabrafenib tuvieron una duración media de exposición de 11 meses (rango: 0 a 12) a MEKINIST. Entre los 435 pacientes que recibieron MEKINIST en combinación con dabrafenib, el 72% estuvo expuesto a MEKINIST durante > 6 meses. La edad media de los pacientes que recibieron MEKINIST en combinación con dabrafenib fue de 50 años (rango: 18 a 89), el 56% eran hombres, el 99% eran blancos, el 92% tenía un estado de rendimiento ECOG basal de 0 y el 8% tenía un estado de rendimiento ECOG basal de 1.
Las reacciones adversas más comunes (≥ 20%) en pacientes que recibieron MEKINIST en combinación con dabrafenib fueron: pirexia, fatiga, náuseas, dolor de cabeza, erupción cutánea, escalofríos, diarrea, vómitos, artralgia y mialgia.
Las reacciones adversas que provocaron la interrupción y las interrupciones de la dosis de MEKINIST ocurrieron en el 24% y el 54% de los pacientes, respectivamente; las más frecuentes para cada una fueron pirexia y escalofríos. Las reacciones adversas que llevaron a reducciones de dosis de MEKINIST ocurrieron en el 23% de los pacientes; las más frecuentes fueron pirexia y disminución de la fracción de eyección.
La Tabla 10 resume las reacciones adversas que ocurrieron en al menos el 20% de los pacientes que recibieron MEKINIST en combinación con dabrafenib.
| aNCI CTCAE versión 4.0. bIncluye pirexia e hiperpirexia. cIncluye fatiga, astenia y malestar. dIncluye dolor de cabeza y cefalea tensional. eIncluye erupción cutánea, erupción maculo-papular, erupción macular, erupción generalizada, erupción eritematosa, erupción papular, erupción pruriginosa, erupción nodular, erupción vesicular y erupción pustular. fIncluye mialgia, dolor musculoesquelético y dolor musculoesquelético en el pecho. |
||||
| Reacciones Adversas | MEKINIST más Dabrafenib N = 435 |
Placebo N = 432 |
||
| Todas Grados (%) |
Grados 3 y 4 (%) |
Todas Grados (%) |
Grados 3 y 4 (%) |
|
| General | ||||
| Pirexiab | 63 | 5 | 11 | < 1 |
| Fatigac | 59 | 5 | 37 | < 1 |
| Escalofríos | 37 | 1 | 4 | 0 |
| Gastrointestinal | ||||
| Náuseas | 40 | < 1 | 20 | 0 |
| Diarrea | 33 | < 1 | 15 | < 1 |
| Vómitos | 28 | < 1 | 10 | 0 |
| Sistema nervioso | ||||
| Dolor de cabezad | 39 | 1 | 24 | 0 |
| Piel y tejido subcutáneo | ||||
| Erupción cutáneae | 37 | < 1 | 16 | < 1 |
| Musculoesquelético y tejido conectivo | ||||
| Artralgia | 28 | < 1 | 14 | 0 |
| Mialgiaf | 20 | < 1 | 14 | 0 |
Otras reacciones adversas clínicamente importantes para MEKINIST en el estudio COMBI-AD observadas en menos del 20% de los pacientes que recibieron MEKINIST en combinación con dabrafenib fueron: visión borrosa (6%), disminución de la fracción de eyección (5%), neuropatía periférica (2.5%), rabdomiólisis (< 1%), bloqueo auriculoventricular (< 1%) y sarcoidosis (< 1%).
Las anormalidades de laboratorio se resumen en la Tabla 11.
| Abreviaturas: ALT, alanina aminotransferasa; AST, aspartato aminotransferasa. aLa incidencia se basa en el número de pacientes que tuvieron tanto una medición de laboratorio inicial como al menos una medición de laboratorio durante el estudio: MEKINIST más dabrafenib (rango: 429 a 431) y brazo placebo (rango: 426 a 428). |
||||
| Anormalidad de laboratorio | MEKINIST más Dabrafeniba N = 435 |
Placeboa N = 432 |
||
| Todos Grados (%) |
Grados 3 y 4 (%) |
Todos Grados (%) |
Grados 3 y 4 (%) |
|
| Química | ||||
| Hiperglucemia | 63 | 3 | 47 | 2 |
| Hipofosfatemia | 42 | 7 | 10 | < 1 |
| Hipoalbuminemia | 25 | < 1 | < 1 | 0 |
| Hepático | ||||
| AST aumentado | 57 | 6 | 11 | < 1 |
| ALT aumentado | 48 | 5 | 18 | < 1 |
| Fosfatasa alcalina sanguínea aumentada | 38 | 1 | 6 | < 1 |
| Hematología | ||||
| Neutropenia | 47 | 6 | 12 | < 1 |
| Linfopenia | 26 | 5 | 6 | < 1 |
| Anemia | 25 | < 1 | 6 | < 1 |
Estudio COMBI-APlus (Estudio de manejo de pirexia)
COMBI-APlus evaluó el impacto de los resultados relacionados con la pirexia de un algoritmo de manejo de pirexia revisado en pacientes que recibieron dabrafenib administrado con trametinib en el tratamiento adyuvante del melanoma positivo para la mutación BRAF V600 después de la resección completa. El algoritmo de manejo de pirexia interrumpió tanto dabrafenib como trametinib cuando la temperatura del paciente es ≥ 100.4°F.
La pirexia de grado 3-4 ocurrió en el 4.3% de los pacientes, las hospitalizaciones debido a pirexia ocurrieron en el 5.1% de los pacientes, la pirexia con complicaciones (deshidratación, hipotensión, disfunción renal, síncope, escalofríos severos) ocurrió en el 2.2% de los pacientes, y la interrupción del tratamiento debido a pirexia ocurrió en el 2.5% de los pacientes.
Cáncer de pulmón de células no pequeñas metastásico, positivo para la mutación BRAF V600E
La seguridad de MEKINIST cuando se administra con dabrafenib se evaluó en 93 pacientes con NSCLC metastásico positivo para la mutación BRAF V600E previamente no tratado (n = 36) y previamente tratado (n = 57) en un ensayo multicéntrico, multicóhorte, no aleatorizado, de etiqueta abierta (Estudio BRF113928). Los pacientes recibieron MEKINIST 2 mg por vía oral una vez al día y dabrafenib 150 mg por vía oral dos veces al día hasta la progresión de la enfermedad o la toxicidad inaceptable. El ensayo excluyó a los pacientes con LVEF anormal, antecedentes de síndrome coronario agudo dentro de los 6 meses, antecedentes de insuficiencia cardíaca congestiva de clase II o superior (Asociación Americana del Corazón), intervalo QTc ≥ 480 mseg, hipertensión refractaria al tratamiento, arritmias no controladas, metástasis cerebrales activas, antecedentes de ILD o neumonitis, o antecedentes o RVO actual [ver Estudios clínicos (14.3)].
Entre estos 93 pacientes, 53 (57%) estuvieron expuestos a MEKINIST y dabrafenib durante > 6 meses y 27 (29%) estuvieron expuestos a MEKINIST y dabrafenib durante ≥ 1 año. La edad media fue de 65 años (rango: 41 a 91), el 46% eran hombres, el 85% eran blancos; el 32% tenía un estado de rendimiento ECOG basal de 0 y el 61% tenía un estado de rendimiento ECOG de 1; el 98% tenía histología no escamosa; y el 12% eran fumadores actuales, el 60% eran exfumadores y el 28% nunca había fumado.
Las reacciones adversas más comunes (≥ 20%) en estos 93 pacientes fueron: pirexia, fatiga, náuseas, vómitos, diarrea, piel seca, disminución del apetito, edema, erupción cutánea, escalofríos, hemorragia, tos y disnea.
Las reacciones adversas que llevaron a la interrupción de MEKINIST ocurrieron en el 19% de los pacientes; las más frecuentes fueron pirexia (2.2%), disminución de la fracción de eyección (2.2%) y dificultad respiratoria (2.2%). Las reacciones adversas que llevaron a reducciones de dosis de MEKINIST ocurrieron en el 30% de los pacientes; las más frecuentes fueron pirexia (5%), náuseas (4.3%), vómitos (4.3%), diarrea (3.2%) y neutropenia (3.2%). Las reacciones adversas que llevaron a interrupciones de dosis de MEKINIST ocurrieron en el 57% de los pacientes; las más frecuentes fueron pirexia (16%), vómitos (10%), neutropenia (8%), náuseas (5%) y disminución de la fracción de eyección (5%).
La Tabla 12 y la Tabla 13 presentan las reacciones adversas y las anormalidades de laboratorio, respectivamente, de MEKINIST en combinación con dabrafenib en el Estudio BRF113928.
| aNCI CTCAE versión 4.0. bIncluye fatiga, malestar y astenia. cIncluye edema periférico, edema y edema generalizado. dIncluye erupción cutánea, erupción cutánea generalizada, erupción cutánea papular, erupción cutánea macular, erupción cutánea maculo-papular y erupción cutánea pustular. eIncluye hemoptisis, hematoma, epistaxis, púrpura, hematuria, hemorragia subaracnoidea, hemorragia gástrica, hemorragia de la vejiga urinaria, contusión, hematochezia, hemorragia en el sitio de inyección, hemorragia pulmonar y hemorragia retroperitoneal. |
||
| Reacciones adversas | MEKINIST más Dabrafenib N = 93 |
|
| Todos Grados (%) |
Grados 3 y 4 (%) |
|
| General | ||
| Pirexia | 55 | 5 |
| Fatigab | 51 | 5 |
| Edemac | 28 | 0 |
| Escalofríos | 23 | 1.1 |
| Gastrointestinal | ||
| Náuseas | 45 | 0 |
| Vómitos | 33 | 3.2 |
| Diarrea | 32 | 2.2 |
| Disminución del apetito | 29 | 0 |
| Piel y tejido subcutáneo | ||
| Piel seca | 31 | 1.1 |
| Erupción cutánead | 28 | 3.2 |
| Vascular | ||
| Hemorragiae | 23 | 3.2 |
| Sistema respiratorio | ||
| Tos | 22 | 0 |
| Disnea | 20 | 5 |
Otras reacciones adversas clínicamente importantes para MEKINIST en el Estudio BRF113928 observadas en menos del 20% de los pacientes que recibieron MEKINIST administrado con dabrafenib fueron:
Cardíaco: Bloqueo auriculoventricular
Sistema nervioso: Neuropatía periférica
| Abreviaturas: ALT, alanina aminotransferasa; AST, aspartato aminotransferasa. aPara estas pruebas de laboratorio, el denominador es 90. bPara estas pruebas de laboratorio, el denominador es 91. |
||
| Anormalidad de laboratorio | MEKINIST más Dabrafenib N = 93 |
|
| Todos Grados (%) |
Grados 3 y 4 (%) |
|
| Químicaa | ||
| Hiperglucemia | 71 | 9 |
| Hiponatremia | 57 | 17 |
| Hipofosfatemia | 36 | 7 |
| Creatinina aumentada | 21 | 1.1 |
| Hepáticoa | ||
| Fosfatasa alcalina sanguínea aumentada | 64 | 0 |
| AST aumentada | 61 | 4.4 |
| ALT aumentada | 32 | 6 |
| Hematologíab | ||
| Leucopenia | 48 | 8 |
| Anemia | 46 | 10 |
| Neutropenia | 44 | 8 |
| Linfopenia | 42 | 14 |
Tumores avanzados positivos para la mutación BRAF V600E
Estudio BRF117019
La seguridad de MEKINIST cuando se administra con dabrafenib se evaluó en un estudio multicóhorte, multicéntrico, no aleatorizado y abierto en pacientes adultos con cánceres con la mutación BRAF V600E (Estudio BRF117019). Se inscribieron un total de 206 pacientes en el ensayo, 36 de los cuales se inscribieron en la cohorte ATC, 105 se inscribieron en cohortes específicas de tumores sólidos y 65 en otras malignidades [ver Estudios clínicos (14.4, 14.6)]. Los pacientes recibieron MEKINIST 2 mg por vía oral una vez al día y dabrafenib 150 mg por vía oral dos veces al día hasta la progresión de la enfermedad o la toxicidad inaceptable.
Entre estos 206 pacientes, 101 (49%) estuvieron expuestos a MEKINIST durante ≥ 1 año y 103 (50%) estuvieron expuestos a dabrafenib durante ≥ 1 año. La edad media fue de 60 años (rango: 18 a 89); el 56% eran hombres; el 79% eran blancos; y el 34% tenía un estado de rendimiento ECOG de 0 y el 60% tenía un estado de rendimiento ECOG de 1.
Las reacciones adversas graves ocurrieron en el 45% de los pacientes que recibieron MEKINIST en combinación con dabrafenib. Las reacciones adversas graves en > 5% de los pacientes incluyeron pirexia (11%) y neumonía (6%). Las reacciones adversas fatales ocurrieron en el 3.9% de los pacientes que recibieron MEKINIST en combinación con dabrafenib. Las reacciones adversas fatales que ocurrieron en > 1% de los pacientes incluyeron sepsis (1.9%).
La interrupción permanente del tratamiento debido a una reacción adversa ocurrió en el 13% de los pacientes. Las reacciones adversas que dieron lugar a la interrupción permanente del tratamiento en > 1% de los pacientes incluyeron náuseas (1.5%).
Las interrupciones de la dosis debido a una reacción adversa ocurrieron en el 55% de los pacientes. Las reacciones adversas que requirieron interrupción de la dosis en > 5% de los pacientes incluyeron pirexia (22%), escalofríos (9%), fatiga (6%), neutropenia (6%) y náuseas (5%).
Las reducciones de dosis debido a una reacción adversa ocurrieron en el 44% de los pacientes. Las reacciones adversas que requirieron reducciones de dosis en > 5% de los pacientes incluyeron pirexia (18%), escalofríos (8%) y fatiga (6%).
Las reacciones adversas más comunes (≥ 20%), incluidas las anormalidades de laboratorio, se enumeran en la Tabla 14 y la Tabla 15.
La Tabla 14 resume las reacciones adversas en el Estudio BRF117019.
| aNCI CTCAE versión 4.0. bIncluye fatiga, astenia y malestar. cIncluye edema periférico e hinchazón periférica. dIncluye erupción cutánea, erupción maculopapular, erupción eritematosa, erupción pustular y erupción papular. eIncluye epistaxis, hematuria, contusión, hematoma, hemoptisis, hemorragia conjuntival, hematochezia, hemorragia rectal, hemorragia hemorroidal, melena, púrpura, contusión ocular, hemorragia ocular, hemorragia gástrica, sangrado gingival, hematemesis, hemorragia intracraneal, accidente cerebrovascular hemorrágico, hemotórax, mayor tendencia a los hematomas, hemorragia del intestino grueso, hemorragia bucal, petequias, hemorragia faríngea, tiempo de protrombina prolongado, hematoma pulmonar, hemorragia retiniana, hemorragia vaginal y hemorragia vítrea. fIncluye tos y tos productiva. gIncluye mialgia, dolor torácico musculoesquelético y dolor musculoesquelético. |
||
| Reacciones adversas | MEKINIST más dabrafeniba (N = 206) |
|
| Todos los grados (%) |
Grado 3 o 4 (%) |
|
| General | ||
| Pirexia | 55 | 4.95 |
| Fatigab | 50 | 5 |
| Escalofríos | 30 | 0.5 |
| Edema periféricoc | 22 | 0 |
| Gastrointestinal | ||
| Náuseas | 40 | 1.5 |
| Estreñimiento | 27 | 0 |
| Vómitos | 27 | 1.5 |
| Diarrea | 26 | 2.93 |
| Piel y tejido subcutáneo | ||
| Erupción cutánead | 40 | 2.4 |
| Sistema nervioso | ||
| Dolor de cabeza | 30 | 1.5 |
| Vascular | ||
| Hemorragiae | 29 | 4.4 |
| Sistema respiratorio | ||
| Tosf | 29 | 0 |
| Musculoesquelético y tejido conectivo | ||
| Mialgiag | 24 | 0.5 |
| Artralgia | 23 | 0.5 |
Las reacciones adversas clínicamente relevantes para MEKINIST en el Estudio BRF117019 observadas en menos del 20% de los pacientes que recibieron MEKINIST en combinación con dabrafenib fueron: neuropatía periférica (9%), disminución de la fracción de eyección (8%), bloqueo auriculoventricular (2.9%), uveítis (1.9%) e hipersensibilidad (1.9%).
La Tabla 15 resume las anormalidades de laboratorio en el Estudio BRF117019.
| Abreviaturas: ALT, alanina aminotransferasa; AST, aspartato aminotransferasa. aEl denominador utilizado para calcular la tasa varió de 199 a 202 en función del número de pacientes con un valor inicial y al menos un valor posterior al tratamiento. |
||
| Anormalidad de laboratorio | MEKINIST más Dabrafeniba | |
| Todos los grados (%) |
Grado 3 o 4 (%) |
|
| Química | ||
| Hiperglucemia | 61 | 8 |
| Disminución de sodio | 35 | 10 |
| Disminución de magnesio | 24 | 0 |
| Aumento de creatinina | 21 | 1.5 |
| Hepático | ||
| Aumento de fosfatasa alcalina | 51 | 5 |
| Aumento de AST | 51 | 4.6 |
| Aumento de ALT | 39 | 3 |
| Hematología | ||
| Disminución de hemoglobina | 44 | 9 |
Tumores sólidos positivos para la mutación BRAF V600E en pacientes pediátricos
Estudio CTMT212X2101 (X2101)
La seguridad de MEKINIST cuando se administra con dabrafenib se evaluó en el Estudio X2101, un estudio multicéntrico, abierto, de múltiples cohortes en pacientes pediátricos (n = 48) con tumores sólidos refractarios o recurrentes activación [ver Estudios clínicos (14.6)]. La duración media de la exposición a MEKINIST en las Partes C (escalada de dosis) y D (expansión de la cohorte) fue de 20,8 y 24,4 meses, respectivamente. La duración media de la exposición a dabrafenib en las Partes C y D fue de 20,8 y 24,9 meses, respectivamente. La edad media de los pacientes pediátricos que recibieron MEKINIST con dabrafenib fue de 9 años (rango: 1 a 17).
Se produjeron reacciones adversas graves en el 46% de los pacientes que recibieron MEKINIST en combinación con dabrafenib. Las reacciones adversas graves en > 5% de los pacientes incluyeron pirexia (25%) y disminución de la fracción de eyección (6%). La interrupción permanente del tratamiento debido a una reacción adversa se produjo en el 21% de los pacientes. Las reacciones adversas que dieron lugar a la interrupción permanente del tratamiento en > 3% de los pacientes incluyeron aumento de ALT (6%), aumento de AST (4,2%) y disminución de la fracción de eyección (4,2%). Las interrupciones de la dosificación debido a una reacción adversa se produjeron en el 73% de los pacientes. Las reacciones adversas que requirieron interrupción de la dosificación en > 5% de los pacientes incluyeron pirexia (56%), vómitos (19%), neutropenia (13%), erupción cutánea (13%), disminución de la fracción de eyección (6%) y uveítis (6%). Las reducciones de dosis debido a una reacción adversa se produjeron en el 25% de los pacientes. Las reacciones adversas que requirieron reducciones de dosis en > 5% de los pacientes incluyeron pirexia (13%).
Las reacciones adversas más comunes (≥ 20%), incluidas las anomalías de laboratorio, se enumeran en la Tabla 16 y la Tabla 17.
La Tabla 16 resume las reacciones adversas en el Estudio X2101.
| aNCI CTCAE versión 4.0. bIncluye fatiga, astenia y malestar. cIncluye erupción cutánea, erupción maculo-papular, erupción eritematosa, erupción papular, erupción pustular y erupción macular. dIncluye dermatitis acneiforme y acné. eIncluye dolor abdominal y dolor abdominal superior. fIncluye epistaxis, hematuria, contusión, hematoma, petequias, hemorragia rectal y recuento de glóbulos rojos disminuido. |
||
| Reacciones adversas | MEKINIST más Dabrafeniba (N = 48) |
|
| Todos los grados (%) |
Grado 3 o 4 (%) |
|
| General | ||
| Pirexia | 75 | 17 |
| Fatigab | 48 | 0 |
| Piel y tejido subcutáneo | ||
| Erupción cutáneac | 73 | 2.1 |
| Piel seca | 48 | 0 |
| Dermatitis acneiformed | 40 | 0 |
| Gastrointestinal | ||
| Vómitos | 52 | 4.2 |
| Diarrea | 42 | 2.1 |
| Dolor abdominale | 33 | 4.2 |
| Náuseas | 33 | 2.1 |
| Estreñimiento | 23 | 0 |
| Sistema respiratorio | ||
| Tos | 44 | 0 |
| Sistema nervioso | ||
| Dolor de cabeza | 35 | 0 |
| Vascular | ||
| Hemorragiaf | 33 | 0 |
| Infecciones e infestaciones | ||
| Paroniquia | 23 | 0 |
Las reacciones adversas clínicamente relevantes para MEKINIST en el Estudio X2101 observadas en menos del 20% de los pacientes (N=48) que recibieron MEKINIST en combinación con dabrafenib fueron: bloqueo auriculoventricular (2,1%).
La Tabla 17 resume las anormalidades de laboratorio en el Estudio X2101.
| Abreviaturas: ALT, alanina aminotransferasa; AST, aspartato aminotransferasa. aEl denominador utilizado para calcular la tasa varió de 39 a 48 en función del número de pacientes con un valor inicial y al menos un valor posterior al tratamiento. |
||
| Anormalidad de laboratorio | MEKINIST más Dabrafeniba | |
| Todos los grados (%) |
Grado 3 o 4 (%) |
|
| Química | ||
| Hiperglucemia | 65 | 2.2 |
| Hipoalbuminemia | 48 | 2.1 |
| Hipocalcemia | 40 | 2.1 |
| Fosfato disminuido | 38 | 0 |
| Magnesio disminuido | 33 | 2.1 |
| Hipernatremia | 27 | 0 |
| Hipokalemia | 21 | 2.1 |
| Hepático | ||
| AST aumentado | 55 | 4.2 |
| ALT aumentado | 40 | 6 |
| Fosfatasa alcalina aumentada | 28 | 6 |
| Bilirrubina total aumentada | 21 | 2.1 |
| Hematología | ||
| Hemoglobina disminuida | 60 | 6 |
| Neutrófilos disminuidos | 49 | 28 |
Glioma de bajo grado positivo para la mutación BRAF V600E en pacientes pediátricos
Estudio CDRB436G2201 (G2201)
La seguridad de MEKINIST en combinación con dabrafenib se evaluó en pacientes pediátricos de 1 a < 18 años de edad en el estudio G2201. Los pacientes con glioma de bajo grado (LGG) que requerían la primera terapia sistémica se aleatorizaron (2:1) a MEKINIST más dabrafenib (n = 73) o carboplatino más vincristina (n = 33). Nueve pacientes cambiaron del brazo de carboplatino más vincristina al brazo de MEKINIST y dabrafenib. Los pacientes pediátricos recibieron MEKINIST basado en el peso por vía oral una vez al día en combinación con dabrafenib hasta la progresión de la enfermedad o toxicidad intolerable. Los pacientes en el brazo de control recibieron carboplatino y vincristina en dosis de 175 mg/m2 y 1.5 mg/m2, respectivamente, en un curso de inducción de 10 semanas seguido de ocho ciclos de mantenimiento de 6 semanas o hasta la progresión de la enfermedad o toxicidad intolerable. Entre los pacientes con glioma de bajo grado que se aleatorizaron a MEKINIST más dabrafenib (n = 73), el 95% estuvieron expuestos durante 6 meses o más y el 71% durante más de un año.
La edad mediana de estos pacientes fue de 10 años (rango: 1 a 17); 60% mujeres; 75% blancos, 7% asiáticos, 2.7% negros o afroamericanos, 4% de otra raza y 11% en los que la raza era desconocida o no se informó.
Se produjeron reacciones adversas graves en el 40% de estos pacientes. Las reacciones adversas graves en > 3% de los pacientes incluyeron fiebre (14%) y vómitos (4%).
Se produjo la suspensión permanente de MEKINIST debido a una reacción adversa en el 4% de los pacientes. Las reacciones adversas que resultaron en la suspensión permanente de MEKINIST incluyeron escalofríos, fatiga, fiebre, aumento de peso y cefalea.
Se produjeron interrupciones de la dosis de MEKINIST debido a una reacción adversa en el 70% de los pacientes. Las reacciones adversas que requirieron una interrupción de la dosis en > 5% de los pacientes incluyeron fiebre (52%).
Se produjeron reducciones de la dosis de MEKINIST debido a una reacción adversa en el 12% de los pacientes. Las reacciones adversas que requirieron reducciones de la dosis en > 2% de los pacientes incluyeron aumento de peso (2.7%).
Las reacciones adversas más comunes (≥ 15%) fueron fiebre (68%), erupción cutánea (51%), cefalea (47%), vómitos (34%), dolor musculoesquelético (34%), fatiga (33%), diarrea (29%), piel seca (26%), náuseas (25%), hemorragia (25%), dolor abdominal (25%), dermatitis acneiforme (22%), mareo (15%), infección de las vías respiratorias superiores (15%) y aumento de peso (15%).
Las anomalías de laboratorio más comunes (≥ 20%) que empeoraron desde la línea de base fueron leucopenia (59%), fosfatasa alcalina aumentada (55%), anemia (46%), neutrófilos disminuidos (44%), AST aumentado (37%), magnesio disminuido (34%), magnesio aumentado (32%), plaquetas disminuidas (30%), ALT aumentado (29%) y linfocitos aumentados (24%).
La Tabla 18 resume las reacciones adversas en el estudio G2201.
| aVersión 4.03 de NCI CTCAE. bIncluye diarrea, colitis, enterocolitis y enteritis. cIncluye dolor abdominal y dolor abdominal superior. dIncluye estomatitis, queilitis, úlceras bucales, úlcera aftosa y glositis. eIncluye fiebre y aumento de la temperatura corporal. fIncluye fatiga y astenia. gIncluye cefalea y migraña con aura. hIncluye mareo y vértigo. iIncluye neuropatía periférica, neuropatía motora periférica, neuropatía sensorio-motora periférica, parestesia, neuralgia, hipoestesia y neuropatía sensitiva periférica. jIncluye epistaxis, hemorragia post-procedimiento, hematuria, hemorragia gastrointestinal superior e hemorragia intracraneal. kIncluye erupción cutánea, erupción macular, erupción maculo-papular, erupción pústular, erupción papular, erupción eritematosa, eczema, eritema multiforme, dermatitis, dermatitis exfoliativa, exfoliación cutánea, síndrome de eritrodisestesia palmo-plantar y dermatitis ampollosa. lIncluye dermatitis acneiforme, acné y acné pústular. mIncluye dolor de espalda, mialgia, dolor en extremidades, artralgia, dolor óseo, dolor torácico no cardíaco, dolor de cuello y rigidez musculoesquelética. |
||||
| Reacciones adversas | MEKINIST más Dabrafenib N = 73 |
Carboplatino más Vincristina N = 33 |
||
| Todos los grados (%) |
Grado ≥ 3 (%) |
Todos los grados (%) |
Grado ≥ 3 (%) |
|
| Gastrointestinal | ||||
| Vómitos | 34 | 1 | 48 | 3 |
| Diarreab | 29 | 0 | 18 | 6 |
| Náuseas | 25 | 0 | 45 | 0 |
| Dolor abdominalc | 25 | 0 | 24 | 0 |
| Estreñimiento | 12 | 0 | 36 | 0 |
| Estomatitisd | 10 | 0 | 18 | 0 |
| General | ||||
| Fiebree | 68 | 8 | 18 | 3 |
| Fatigaf | 33 | 0 | 39 | 0 |
| Sistema nervioso | ||||
| Cefaleag | 47 | 1 | 33 | 3 |
| Mareoh | 15 | 0 | 9 | 3 |
| Neuropatía periféricai | 7 | 0 | 45 | 6 |
| Vascular | ||||
| Hemorragiaj | 25 | 0 | 12 | 0 |
| Piel y tejido subcutáneo | ||||
| Erupciónk | 51 | 2.7 | 18 | 3 |
| Piel seca | 26 | 0 | 3 | 0 |
| Dermatitis acneiformel | 22 | 0 | 0 | 0 |
| Alopecia | 3 | 0 | 24 | 0 |
| Musculoesquelético y tejido conjuntivo | ||||
| Dolor musculoesqueléticom | 34 | 0 | 30 | 0 |
| Dolor en la mandíbula | 1.4 | 0 | 18 | 0 |
| Metabolismo y nutrición | ||||
| Disminución del apetito | 5 | 0 | 24 | 0 |
| Respiratorio, torácico y mediastínico | ||||
| Dolor orofaríngeo | 11 | 0 | 18 | 0 |
| Psiquiátrico | ||||
| Ansiedad | 1.4 | 0 | 15 | 3 |
| Sistema inmunitario | ||||
| Hipersensibilidad | 0 | 0 | 15 | 3 |
| Infecciones e infestaciones | ||||
| Infección de las vías respiratorias altas | 15 | 0 | 6 | 0 |
| Lesión, envenenamiento y complicaciones de procedimientos | ||||
| Reacción relacionada con la infusión | 0 | 0 | 15 | 3 |
| Investigaciones | ||||
| Aumento de peso | 15 | 7 | 0 | 0 |
La Tabla 19 resume las anormalidades de laboratorio en el Estudio G2201.
| Abreviaturas: ALT, alanina aminotransferasa; AST, aspartato aminotransferasa. aEl denominador utilizado para calcular la tasa varió de 70 a 73 en el brazo D + T y de 9 a 33 en el brazo C + V en función del número de pacientes con un valor basal y al menos un valor posterior al tratamiento. |
||||
| Anormalidad de laboratorio | MEKINIST más Dabrafenib N = 73 |
Carboplatino más Vincristina N = 33 |
||
| Todos los grados (%) |
Grado 3 o 4 (%) |
Todos los grados (%) |
Grado 3 o 4 (%) |
|
| Hepático | ||||
| Fosfatasa alcalina aumentada | 55 | 0 | 13 | 0 |
| AST aumentada | 37 | 1.4 | 55 | 0 |
| ALT aumentada | 29 | 3 | 61 | 9 |
| Química | ||||
| Magnesio disminuido | 34 | 4.1 | 76 | 6 |
| Magnesio aumentado | 32 | 0 | 24 | 3 |
| Potasio aumentado | 15 | 4.2 | 21 | 6 |
| Calcio disminuido | 14 | 4.1 | 22 | 9 |
| Potasio disminuido | 8 | 1.4 | 70 | 0 |
| Fosfato disminuido | 7 | 2.7 | 33 | 3 |
| Sodio disminuido | 5 | 1.4 | 27 | 6 |
| Glucosa en ayunas en suero aumentada | 0 | 0 | 44 | 0 |
| Hematología | ||||
| Leucocitos disminuidos | 59 | 0 | 91 | 18 |
| Hemoglobina disminuida | 46 | 0 | 94 | 36 |
| Neutrófilos disminuidos | 44 | 17 | 84 | 75 |
| Plaquetas disminuidas | 30 | 0 | 73 | 18 |
| Linfocitos aumentados | 24 | 0 | 13 | 3.1 |
| Decreased lymphocytes | 16 | 1.4 | 56 | 6 |
6.2
Experiencia postcomercialización
Las siguientes reacciones adversas se han identificado durante el uso posterior a la aprobación de MEKINIST en combinación con dabrafenib. Debido a que estas reacciones se reportan voluntariamente de una población de tamaño incierto, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal con la exposición al fármaco.
Cardíaco: Bloqueo auriculoventricular completo. Esta reacción adversa también se observó con la monoterapia con MEKINIST.
Sistema inmunitario: Linfohistiocitosis hemofagocítica (HLH) [ver Advertencias y precauciones (5.12)]
Piel y tejido subcutáneo: SCAR (incluyendo DRESS y SJS) [ver Advertencias y precauciones (5.9)]
7 INTERACCIONES MEDICAMENTOSAS
MEKINIST está indicado para su uso en combinación con dabrafenib. Consulte la información de prescripción de dabrafenib para obtener información adicional sobre riesgos que se aplica al tratamiento de uso combinado.
8 USO EN POBLACIONES ESPECÍFICAS
8.1
Embarazo
Resumen de riesgos
En función de su mecanismo de acción [ver Farmacología clínica (12.1)] y los hallazgos de estudios de reproducción animal, MEKINIST puede causar daño fetal cuando se administra a una mujer embarazada. No hay datos suficientes sobre mujeres embarazadas expuestas a MEKINIST para evaluar los riesgos. El trametinib fue embriotóxico y abortivo en conejos a dosis mayores o iguales a las que dieron como resultado exposiciones de aproximadamente 0,3 veces la exposición humana a la dosis clínica recomendada en adultos (ver Datos). Se debe advertir a las mujeres embarazadas sobre el riesgo potencial para el feto.
En la población general de EE. UU., el riesgo de base estimado de defectos congénitos importantes y aborto espontáneo en embarazos clínicamente reconocidos es del 2 % al 4 % y del 15 % al 20 %, respectivamente.
Datos
Datos en animales
En estudios de toxicidad reproductiva, la administración de trametinib a ratas durante el período de organogénesis dio como resultado una disminución del peso fetal a dosis mayores o iguales a 0,031 mg/kg/día [aproximadamente 0,3 veces la exposición humana a la dosis recomendada en adultos según el área bajo la curva (AUC)]. En ratas, a una dosis que dio como resultado exposiciones 1,8 veces mayores que la exposición humana a la dosis recomendada en adultos, hubo toxicidad materna y un aumento de la pérdida posterior a la implantación.
En conejas preñadas, la administración de trametinib durante el período de organogénesis dio como resultado una disminución del peso corporal fetal y una mayor incidencia de variaciones en la osificación a dosis mayores o iguales a 0,039 mg/kg/día (aproximadamente 0,08 veces la exposición humana a la dosis recomendada en adultos según el AUC). En conejos a los que se les administró trametinib a 0,15 mg/kg/día (aproximadamente 0,3 veces la exposición humana a la dosis recomendada en adultos según el AUC), hubo un aumento en la pérdida posterior a la implantación, incluida la pérdida total del embarazo, en comparación con los animales de control.
8.2
Lactancia
Resumen de riesgos
No hay datos sobre la presencia de trametinib en la leche materna ni sobre los efectos del trametinib en el lactante o en la producción de leche. Debido a la posibilidad de reacciones adversas graves en lactantes, se debe indicar a las mujeres que no amamanten durante el tratamiento con MEKINIST y durante 4 meses después de la última dosis.
8.3
Mujeres y hombres con potencial reproductivo
Prueba de embarazo
Se debe verificar el estado de embarazo en mujeres con potencial reproductivo antes de iniciar el tratamiento con MEKINIST.
Anticoncepción
Según los datos de estudios en animales y su mecanismo de acción, MEKINIST puede causar daño fetal cuando se administra a mujeres embarazadas [ver Uso en poblaciones específicas (8.1)].
Mujeres
Se debe indicar a las pacientes con potencial reproductivo que utilicen un método anticonceptivo eficaz durante el tratamiento con MEKINIST y durante 4 meses después de la última dosis.
Hombres
Para evitar la posible exposición al fármaco de las parejas embarazadas y de las parejas con potencial reproductivo, se debe indicar a los pacientes varones (incluidos aquellos que se han sometido a una vasectomía) con parejas con potencial reproductivo que utilicen preservativos durante el tratamiento con MEKINIST y durante 4 meses después de la última dosis.
Infertilidad
Mujeres
Se debe advertir a las pacientes con potencial reproductivo que MEKINIST puede afectar a la fertilidad. Se observaron un aumento de los quistes foliculares y una disminución de los cuerpos lúteos en ratas hembra a exposiciones a dosis equivalentes a 0,3 veces la exposición humana a la dosis recomendada en adultos [ver Toxicología no clínica (13.1)].
8.4
Uso pediátrico
Tumores sólidos irresecables o metastásicos con mutación BRAF V600E y LGG
Se ha establecido la seguridad y eficacia de MEKINIST en combinación con dabrafenib en pacientes pediátricos de 1 año de edad en adelante con tumores sólidos irresecables o metastásicos con mutación BRAF V600E que han progresado después de un tratamiento previo y no tienen otras opciones de tratamiento satisfactorias, o con LGG con mutación BRAF V600E que requieren tratamiento sistémico. El uso de MEKINIST en combinación con dabrafenib para estas indicaciones está respaldado por la evidencia de los estudios X2101 y G2201, en los que se reclutó a 171 pacientes (de 1 a < 18 años) con tumores sólidos avanzados con mutación BRAF V600E, de los cuales 4 (2,3 %) pacientes tenían entre 1 y < 2 años, 39 (23 %) pacientes tenían entre 2 y < 6 años, 54 (32 %) pacientes tenían entre 6 y < 12 años y 74 (43 %) pacientes tenían entre 12 y < 18 años [ver Reacciones adversas (6.1), Farmacología clínica (12.3), Estudios clínicos (14.6, 14.7)].
No se ha establecido la seguridad y eficacia de MEKINIST en combinación con dabrafenib para estas indicaciones en pacientes pediátricos menores de 1 año.
No se ha establecido la seguridad y eficacia de MEKINIST en monoterapia en pacientes pediátricos.
Datos de toxicidad en animales jóvenes
En un estudio de toxicidad a dosis repetidas en ratas jóvenes, se observaron una disminución de la longitud de los huesos y distrofia corneal a dosis que dieron como resultado exposiciones tan bajas como 0,3 veces la exposición humana a la dosis recomendada en adultos según el AUC. Además, se observó un retraso en la maduración sexual a dosis que dieron como resultado exposiciones tan bajas como 1,6 veces la exposición humana a la dosis recomendada en adultos según el AUC.
8.5
Uso geriátrico
De los 214 pacientes con melanoma que recibieron MEKINIST en monoterapia en el estudio METRIC, el 27 % tenía 65 años o más y el 4 % tenía más de 75 años [ver Estudios clínicos (14.1)]. Este estudio de MEKINIST en monoterapia en melanoma no incluyó un número suficiente de pacientes geriátricos para determinar si responden de forma diferente a los adultos más jóvenes.
De los 994 pacientes con melanoma que recibieron MEKINIST más dabrafenib en los estudios COMBI-d, COMBI-v y COMBI-AD [ver Estudios clínicos (14.1, 14.2)], el 21 % tenía 65 años o más y el 5 % tenía 75 años o más. No se observaron diferencias generales en la efectividad de MEKINIST más dabrafenib en pacientes geriátricos en comparación con adultos más jóvenes en estos estudios de melanoma. Las incidencias de edema periférico (26 % frente a 12 %) y anorexia (21 % frente a 9 %) aumentaron en pacientes geriátricos en comparación con adultos más jóvenes en estos estudios.
De los 93 pacientes con NSCLC que recibieron MEKINIST en el estudio BRF113928, no hubo suficientes pacientes geriátricos de 65 años o más para determinar si responden de manera diferente a los adultos más jóvenes [ver Estudios clínicos (14.4)].
De los 26 pacientes con ATC que recibieron MEKINIST en el estudio BRF117019, el 77 % tenía 65 años o más y el 31 % tenía 75 años o más [ver Estudios clínicos (14.4)]. Este estudio en ATC no incluyó suficientes adultos más jóvenes para determinar si responden de manera diferente en comparación con los pacientes geriátricos.
8.6
Insuficiencia hepática
No se recomienda ajustar la dosis en pacientes con insuficiencia hepática leve (bilirrubina ≤ límite superior normal (LSN) y aspartato aminotransferasa (AST) > LSN o bilirrubina > 1x a 1.5x LSN y cualquier AST).
No se ha establecido una dosis recomendada de MEKINIST para pacientes con insuficiencia hepática moderada (bilirrubina > 1.5x a 3x LSN y cualquier AST) o grave (bilirrubina > 3x a 10x LSN y cualquier AST). Considere el perfil de riesgo-beneficio de MEKINIST relacionado con la dosificación antes de determinar si administrar MEKINIST a pacientes con insuficiencia hepática moderada o grave.
En pacientes con insuficiencia hepática moderada, 3 pacientes que recibieron una dosis inicial de 1.5 mg por vía oral una vez al día y dos pacientes que recibieron una dosis inicial de 2 mg por vía oral una vez al día no experimentaron toxicidades limitantes de la dosis (DLT) durante el primer ciclo de terapia.
En pacientes con insuficiencia hepática grave, 3 pacientes que recibieron una dosis inicial de 1 mg por vía oral una vez al día no experimentaron DLT durante el primer ciclo; un paciente que recibió una dosis inicial de 1.5 mg por vía oral una vez al día experimentó una DLT (erupción acneiforme de grado 3).
En comparación con los pacientes con función hepática normal, no hubo aumento en la exposición a trametinib en pacientes con insuficiencia hepática moderada o grave [ver Farmacología clínica (12.3)].
10 SOBREDOSIS
Las dosis más altas de MEKINIST evaluadas en ensayos clínicos fueron 4 mg por vía oral una vez al día y 10 mg administrados por vía oral una vez al día durante 2 días consecutivos seguidos de 3 mg una vez al día. En siete pacientes tratados en uno de estos dos esquemas, hubo dos casos de RPED para una incidencia del 28%.
Dado que trametinib se une en gran medida a las proteínas plasmáticas, es probable que la hemodiálisis sea ineficaz en el tratamiento de la sobredosis con MEKINIST.
11 DESCRIPCIÓN
El trametinib dimetilsulfóxido es un inhibidor de la cinasa. El nombre químico es acetamida, N-[3-[3-ciclopropil-5-[(2-fluoro-4- yodofenil)amino]-3,4,6,7-tetrahidro-6,8-dimetil- 2,4,7-trioxopirido[4,3-d]pirimidin-1(2H)-il]fenil]-, compuesto con 1,1’-sulfinilbis[metano] (1:1). Tiene una fórmula molecular C26H23FIN5O4•C2H6OS con una masa molecular de 693,53 g/mol. El trametinib dimetilsulfóxido tiene la siguiente estructura química:

El trametinib dimetilsulfóxido es un polvo blanco o casi blanco. Es prácticamente insoluble en el rango de pH de 2 a 8 en medios acuosos.
MEKINIST (trametinib) comprimidos para administración oral se suministran como comprimidos de 0,5 mg y 2 mg para administración oral. Cada comprimido de 0,5 mg contiene 0,5635 mg de trametinib dimetilsulfóxido equivalente a 0,5 mg de trametinib parental no solvatado. Cada comprimido de 2 mg contiene 2,254 mg de trametinib dimetilsulfóxido equivalente a 2 mg de trametinib parental no solvatado.
Los ingredientes inactivos de los comprimidos de MEKINIST son: Núcleo del comprimido: dióxido de silicio coloidal, croscarmelosa sódica, hipromelosa, estearato de magnesio (de origen vegetal), manitol, celulosa microcristalina y laurilsulfato de sodio. Recubrimiento: hipromelosa, óxido de hierro rojo (comprimidos de 2 mg), óxido de hierro amarillo (comprimidos de 0,5 mg), polietilenglicol, polisorbato 80 (comprimidos de 2 mg) y dióxido de titanio.
MEKINIST (trametinib) para solución oral es un polvo blanco o casi blanco que produce una solución clara e incolora cuando se reconstituye con agua. Cada frasco contiene 4,7 mg de trametinib equivalente a 5,3 mg de trametinib dimetilsulfóxido. Cada mL de solución de trametinib reconstituida contiene 0,05 mg de trametinib parental no solvatado. Los ingredientes inactivos de MEKINIST para solución oral son betadex sulfobutyl éter sódico, ácido cítrico monohidratado, fosfato disódico, metilparabeno, sorbato de potasio, sucralosa y sabor a fresa.
12 FARMACOLOGÍA CLÍNICA
12.1
Mecanismo de acción
Trametinib es un inhibidor reversible de la activación de la cinasa 1 (MEK1) y MEK2 regulada por señales extracelulares activadas por mitógenos y de la actividad de la cinasa MEK1 y MEK2. Las proteínas MEK son reguladores ascendentes de la vía de la cinasa regulada por señales extracelulares (ERK), que promueve la proliferación celular. Las mutaciones BRAF V600E dan como resultado la activación constitutiva de la vía BRAF que incluye MEK1 y MEK2. Trametinib inhibe el crecimiento celular de varios tumores positivos a la mutación BRAF V600 in vitro e in vivo.
Trametinib y dabrafenib se dirigen a dos cinasas diferentes en la vía RAS/RAF/MEK/ERK. El uso de trametinib y dabrafenib en combinación resultó en una mayor inhibición del crecimiento de líneas celulares tumorales positivas a la mutación BRAF V600 in vitro y una inhibición prolongada del crecimiento tumoral en xenotrasplantes tumorales positivos a la mutación BRAF V600 en comparación con cualquiera de los fármacos solos.
En el contexto del cáncer colorrectal con mutación BRAF, la inducción de la reactivación de la vía MAPK mediada por EGFR se ha identificado como un mecanismo de resistencia intrínseca a los inhibidores de BRAF [ver Indicaciones y uso (1.7)].
12.2
Farmacodinamia
La administración de comprimidos de MEKINIST de 1 mg y 2 mg a pacientes con melanoma positivo a la mutación BRAF V600 dio como resultado cambios dependientes de la dosis en los biomarcadores tumorales, incluida la inhibición de ERK fosforilado, la inhibición de Ki67 (un marcador de proliferación celular) y aumentos en p27 (un marcador de apoptosis).
Electrofisiología cardíaca
El potencial de prolongación del intervalo QT corregido por la frecuencia cardíaca (QTc) de trametinib se evaluó en un estudio dedicado en 32 pacientes que recibieron placebo el día 1 y comprimidos de MEKINIST de 2 mg una vez al día del día 2 al 14, seguido de comprimidos de MEKINIST de 3 mg el día 15. No se detectaron cambios grandes en el intervalo QTc medio (es decir, > 20 ms) en el estudio.
Se observó una disminución desde el valor basal en la FC de 9 latidos/min (IC del 90%: -11.4 a -6.1) y un aumento desde el valor basal en el PR de 20 ms (IC del 90%: 13.0 a 27.4) en relación con el placebo a las dos horas posteriores a la dosis en el mismo estudio.
En los ensayos clínicos en pacientes que recibieron MEKINIST con dabrafenib, la prolongación del QTc > 500 ms se produjo en el 0.8% de los pacientes y el QTc aumentó > 60 ms desde el valor basal en el 3.8% de los pacientes.
12.3
Farmacocinética
La farmacocinética de trametinib se caracterizó después de una dosis única y dosis múltiples en pacientes con tumores sólidos y melanoma metastásico positivo a la mutación BRAF V600. Después de la administración de comprimidos de MEKINIST de 0.125 mg (0.0625 veces la dosis recomendada para adultos aprobada) a 4 mg (2 veces la dosis recomendada para adultos aprobada) al día, tanto Cmax como AUC aumentan proporcionalmente con la dosis. La variabilidad interindividual en AUC y Cmax en estado estacionario es del 22% y el 28%, respectivamente.
Absorción
El tiempo medio para alcanzar las concentraciones plasmáticas máximas (Tmax) es de 1.5 horas después de la dosis. La biodisponibilidad absoluta media de los comprimidos de MEKINIST es del 72% y MEKINIST para solución oral es del 81%.
Efecto de los alimentos
Después de la administración de comprimidos de MEKINIST, una comida rica en grasas y calorías (aproximadamente 1000 calorías) disminuyó el AUC de trametinib en un 24%, Cmax en un 70% y retrasó Tmax en aproximadamente 4 horas en comparación con las condiciones en ayunas.
Distribución
Trametinib está unido en un 97.4% a las proteínas plasmáticas humanas. El volumen de distribución aparente (Vc/F) es de 214 L.
Eliminación
La vida media de eliminación estimada es de 3.9 a 4.8 días. El aclaramiento aparente es de 4.9 L/h.
Metabolismo
Trametinib se metaboliza principalmente a través de la desacetilación sola o con mono-oxigenación o en combinación con vías de biotransformación de glucuronidación in vitro. La desacetilación está mediada por carboxilesterasas (es decir, carboxilesterasa 1b/c y 2) y también puede estar mediada por otras enzimas hidrolíticas.
Después de una dosis única de [14C]-trametinib, aproximadamente el 50% de la radiactividad circulante se representa como el compuesto original; sin embargo, ≥ 75% del material relacionado con el fármaco en plasma es el compuesto original según el perfil de metabolitos después de la administración repetida de trametinib.
Excreción
Después de la administración oral de [14C]-trametinib, se recuperó más del 80% de la radiactividad excretada en las heces, mientras que se recuperó menos del 20% de la radiactividad excretada en la orina con menos del 0.1% de la dosis excretada como compuesto original.
Poblaciones específicas
La edad (de 18 a 93 años), el sexo, el peso corporal (de 36 a 170 kg) y la insuficiencia renal (eGFR de 15 a 89 mL/min/1.73 m2) no tienen un efecto clínicamente significativo en la exposición a trametinib. No hay datos suficientes para evaluar las posibles diferencias en la exposición a trametinib por raza o etnia.
Pacientes pediátricos: La farmacocinética de trametinib en glioma y otros tumores sólidos se evaluó en 244 pacientes de 1 a < 18 años después de una dosis única o dosis múltiples. Los parámetros farmacocinéticos en pacientes de 1 a < 18 años están dentro del rango de valores observados previamente en adultos a los que se administró la misma dosis en función del peso. Se encontró que el peso (de 6 a 156 kg) tenía un efecto estadísticamente significativo en el aclaramiento oral de trametinib en esta población.
Pacientes con insuficiencia hepática: La insuficiencia hepática (definida por los niveles de bilirrubina y AST) no tuvo un efecto significativo en la exposición a trametinib o el aclaramiento aparente del fármaco en comparación con los pacientes con función hepática normal.
Estudios de interacción medicamentosa
Efecto de dabrafenib en trametinib: La coadministración de comprimidos de MEKINIST de 2 mg al día con dabrafenib no produjo ningún cambio en el AUC de trametinib.
Efecto de trametinib en los sustratos del CYP: La coadministración de comprimidos de MEKINIST de 2 mg una vez al día con un sustrato sensible del CYP3A4 no tuvo un efecto clínicamente relevante en el AUC y Cmax del sustrato sensible del CYP3A4.
Según los estudios in vitro, trametinib es un inhibidor del CYP2C8, pero no es un inhibidor del CYP1A2, CYP2A6, CYP2B6, CYP2C9, CYP2C19 o CYP2D6 a una concentración sistémica clínicamente relevante.
Efecto de los transportadores en trametinib: Trametinib es un sustrato de la glicoproteína P (P-gp) y BSEP. Es poco probable que la inhibición de la P-gp dé como resultado un aumento clínicamente importante en las concentraciones de trametinib, ya que trametinib presenta una alta permeabilidad pasiva y biodisponibilidad. Trametinib no es un sustrato de BCRP, OATP1B1, OATP1B3, OATP2B1, OCT1, MRP2 o MATE1 in vitro.
Efecto de trametinib en los transportadores: Según los estudios in vitro, trametinib no es un inhibidor de la P-gp, BCRP, OATP1B1, OATP1B3, OAT1, OAT3, OCT2, BSEP, MRP2 o MATE1 a una concentración sistémica clínicamente relevante.
13 TOXICOLOGÍA NO CLÍNICA
13.1
Carcinogenesis, Mutagenesis, Impairment of Fertility
No se han realizado estudios de carcinogenicidad con trametinib. Trametinib no fue genotóxico en estudios que evaluaron mutaciones inversas en bacterias, aberraciones cromosómicas en células de mamíferos y micronúcleos en la médula ósea de ratas.
Trametinib puede afectar la fertilidad en humanos. En ratas hembras a las que se administró trametinib durante un máximo de 13 semanas, se observaron quistes foliculares aumentados y cuerpos lúteos disminuidos a dosis ≥ 0,016 mg/kg/día (aproximadamente 0,3 veces la exposición humana a la dosis recomendada para adultos basada en AUC). En estudios de toxicidad en ratas y perros de hasta 13 semanas de duración, no se observaron efectos del tratamiento en los tejidos reproductivos masculinos [ver Uso en poblaciones específicas (8.3)].
14 ESTUDIOS CLÍNICOS
14.1
Melanoma irresecable o metastásico positivo a la mutación BRAF V600E o V600K
MEKINIST como agente único
La seguridad y eficacia de MEKINIST se evaluaron en un ensayo internacional, multicéntrico, aleatorizado (2:1), abierto, controlado con activo (el estudio METRIC; NCT01245062) en 322 pacientes con melanoma irresecable o metastásico positivo a la mutación BRAF V600E o V600K. En el estudio METRIC, no se permitió que los pacientes tuvieran más de un régimen de quimioterapia previo para la enfermedad avanzada o metastásica; no se permitió el tratamiento previo con un inhibidor de BRAF o un inhibidor de MEK. Los pacientes se asignaron aleatoriamente para recibir MEKINIST 2 mg por vía oral una vez al día (N = 214) o quimioterapia (N = 108) que consistía en dacarbazina 1000 mg/m2 por vía intravenosa cada 3 semanas o paclitaxel 175 mg/m2 por vía intravenosa cada 3 semanas. El tratamiento continuó hasta la progresión de la enfermedad o la toxicidad inaceptable. La aleatorización se estratificó de acuerdo con el uso previo de quimioterapia para la enfermedad avanzada o metastásica (sí versus no) y el nivel de LDH (normal versus mayor que ULN). El tejido tumoral se evaluó para detectar mutaciones de BRAF en un sitio de prueba central utilizando un ensayo de prueba clínica. Las muestras tumorales de 289 pacientes (196 pacientes tratados con MEKINIST y 93 pacientes tratados con quimioterapia) también se analizaron retrospectivamente utilizando una prueba de diagnóstico complementaria aprobada por la FDA, THxID®-BRAF assay. La principal medida de resultado de eficacia fue la supervivencia libre de progresión (SLP).
La edad media de los pacientes aleatorizados fue de 54 años, el 54% eran hombres, más del 99% eran blancos y todos los pacientes tenían un estado de rendimiento ECOG basal de 0 o 1. La mayoría de los pacientes tenían enfermedad metastásica (94%), tenían enfermedad M1c (64%), tenían LDH elevado (36%), no tenían antecedentes de metástasis cerebral (97%) y no recibieron quimioterapia previa para la enfermedad avanzada o metastásica (66%). La distribución de las mutaciones BRAF V600 fue BRAF V600E (87%), V600K (12%) o ambas (menos del 1%). Las duraciones medias de seguimiento antes de iniciar el tratamiento alternativo fueron de 4,9 meses para los pacientes tratados con MEKINIST y de 3,1 meses para los pacientes tratados con quimioterapia. Cincuenta y un (47%) pacientes pasaron del brazo de quimioterapia al de MEKINIST en el momento de la progresión de la enfermedad.
El estudio METRIC demostró un aumento estadísticamente significativo en la SLP en los pacientes tratados con MEKINIST. La Tabla 20 y la Figura 1 resumen los resultados de la SLP.
| Abreviaturas: IC, intervalo de confianza; DoR, duración de la respuesta; HR, razón de riesgo; NR, no alcanzado. aEstimador de Pike. |
||
| Investigador–Assessed Endpoints | MEKINIST N = 214 |
Quimioterapia N = 108 |
| Progression-Free Survival | ||
| Número de eventos (%) | 117 (55%) | 77 (71%) |
| Enfermedad progresiva | 107 (50%) | 70 (65%) |
| Muerte | 10 (5%) | 7 (6%) |
| Media, meses (IC del 95%) | 4,8 (4,3, 4,9) | 1,5 (1,4, 2,7) |
| HRa (IC del 95%) | 0,47 (0,34, 0,65) | |
| P valor (prueba de rango logarítmico) | < 0,0001 | |
| Respuestas tumorales confirmadas | ||
| Tasa de respuesta general (IC del 95%) | 22% (17%, 28%) | 8% (4%, 15%) |
| Respuesta completa, n (%) | 4 (2%) | 0 |
| Respuesta parcial, n (%) | 43 (20%) | 9 (8%) |
| Duración de la respuesta | ||
| DoR media, meses (IC del 95%) | 5,5 (4,1, 5,9) | NR (3,5, NR) |
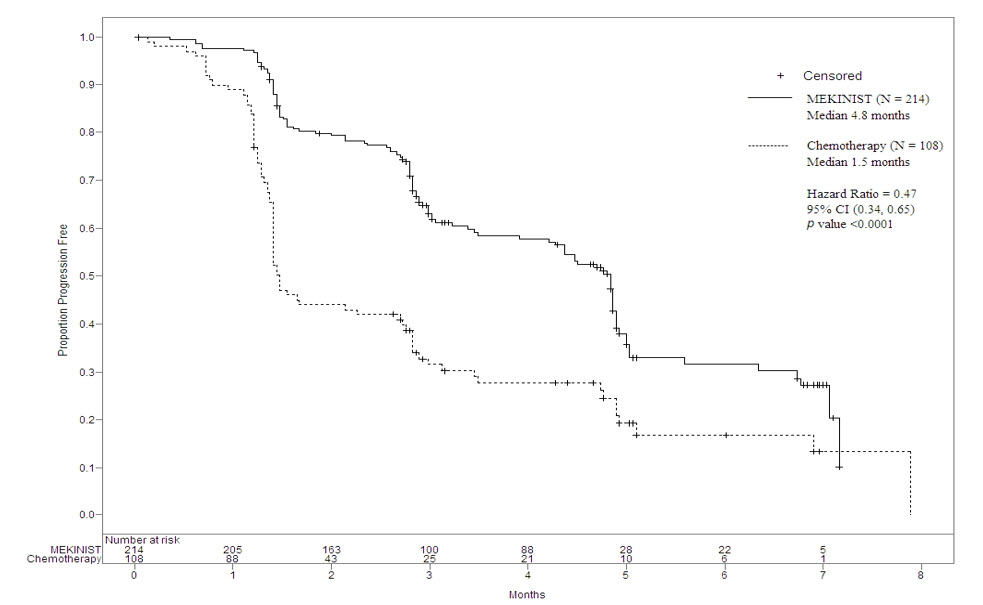
Figura 1. Curvas de Kaplan-Meier de la supervivencia libre de progresión evaluada por el investigador (población ITT) en el estudio METRIC
En los análisis de apoyo basados en la evaluación del comité de revisión radiológica independiente (IRRC), los resultados de la SLP fueron consistentes con los del análisis de eficacia principal.
MEKINIST con Dabrafenib
Estudio COMBI-d
La seguridad y eficacia de MEKINIST administrado con dabrafenib se evaluaron en un ensayo internacional, aleatorizado, doble ciego, controlado con activo (el estudio COMBI-d; NCT01584648). El estudio COMBI-d comparó dabrafenib más MEKINIST con dabrafenib más placebo como tratamiento de primera línea para pacientes con melanoma cutáneo no resecable (Estadio IIIC) o metastásico (Estadio IV) con mutación BRAF V600E o V600K positiva. Los pacientes fueron aleatorizados (1:1) para recibir MEKINIST 2 mg una vez al día más dabrafenib 150 mg dos veces al día o dabrafenib 150 mg dos veces al día más placebo coincidente. La aleatorización se estratificó por nivel de LDH (> ULN vs. ≤ ULN) y subtipo de mutación BRAF (V600E vs. V600K). El principal resultado de eficacia fue la SLP evaluada por el investigador según RECIST v1.1 con medidas de resultado de eficacia adicionales de supervivencia general (SG) y tasa de respuesta general confirmada (TRG).
En el estudio COMBI-d, 423 pacientes fueron aleatorizados a MEKINIST más dabrafenib (n = 211) o dabrafenib más placebo (n = 212). La edad media fue de 56 años (rango: 22 a 89), el 53% eran hombres, > 99% eran blancos, el 72% tenía un estado de rendimiento ECOG de 0, el 4% tenía Estadio IIIC, el 66% tenía enfermedad M1c, el 65% tenía LDH normal y 2 pacientes tenían antecedentes de metástasis cerebrales. Todos los pacientes tenían tumores que contenían mutaciones BRAF V600E o V600K según lo determinado por pruebas centralizadas con la prueba diagnóstica complementaria aprobada por la FDA; el 85% tenía melanoma con mutación BRAF V600E positiva y el 15% tenía melanoma con mutación BRAF V600K positiva.
El estudio COMBI-d demostró mejoras estadísticamente significativas en la SLP y la SG. La Tabla 21 y la Figura 2 resumen los resultados de eficacia.
| Abreviaturas: IC, intervalo de confianza; DoR, duración de la respuesta; HR, razón de riesgos; NR, no alcanzado; TRG, tasa de respuesta general. aLa SLP y la TRG fueron evaluadas por el investigador. bBasado en la prueba de rango logarítmico estratificada. |
||
| Punto final | MEKINIST más Dabrafenib N = 211 |
Placebo más Dabrafenib N = 212 |
| Supervivencia libre de progresióna | ||
| Número de eventos (%) | 102 (48%) | 109 (51%) |
| Mediana, meses (IC del 95%) | 9.3 (7.7, 11.1) | 8.8 (5.9, 10.9) |
| HR (IC del 95%) | 0.75 (0.57, 0.99) | |
| P valorb | 0.035 | |
| Supervivencia general | ||
| Número de muertes (%) | 99 (47%) | 123 (58%) |
| Mediana, meses (IC del 95%) | 25.1 (19.2, NR) | 18.7 (15.2, 23.1) |
| HR (IC del 95%) | 0.71 (0.55, 0.92) | |
| P valorb | 0.01 | |
| Tasa de respuesta generala | ||
| TRG (IC del 95%) | 66% (60%, 73%) | 51% (44%, 58%) |
| P valor | < 0.001 | |
| Respuesta completa | 10% | 8% |
| Respuesta parcial | 56% | 42% |
| DoR mediana, meses (IC del 95%) | 9.2 (7.4, NR) | 10.2 (7.5, NR) |

Figura 2. Curvas de Kaplan-Meier de la supervivencia general en el estudio COMBI-d
Estudio COMBI-MB
La actividad de MEKINIST con dabrafenib para el tratamiento del melanoma positivo para la mutación BRAF V600E o V600K, metastásico al cerebro, se evaluó en un ensayo no aleatorizado, abierto, multicéntrico, multicóhorte (el estudio COMBI-MB; NCT02039947). Los pacientes elegibles debían tener al menos una lesión intracraneal medible y no tener enfermedad leptomeníngea, metástasis cerebral parenquimatosa de más de 4 cm de diámetro, melanoma ocular o melanoma mucoso primario. Los pacientes recibieron MEKINIST 2 mg por vía oral una vez al día y dabrafenib 150 mg por vía oral dos veces al día hasta la progresión de la enfermedad o la toxicidad inaceptable. La principal medida de resultado de eficacia fue la tasa de respuesta intracraneal, definida como el porcentaje de pacientes con una respuesta intracraneal confirmada según RECIST v1.1, modificada para permitir hasta cinco lesiones diana intracraneales de al menos 5 mm de diámetro, según la evaluación de una revisión independiente.
El estudio COMBI-MB reclutó a 121 pacientes con una mutación BRAF V600E (85%) o V600K (15%). La edad media fue de 54 años (rango: 23 a 84), el 58% eran hombres, el 100% eran blancos, el 8% eran de los Estados Unidos, el 65% tenía LDH normal al inicio del estudio y el 97% tenía un estado de rendimiento ECOG de 0 o 1. Las metástasis intracraneales fueron asintomáticas en el 87% y sintomáticas en el 13% de los pacientes, el 22% recibió terapia local previa para las metástasis cerebrales y el 87% también tenía metástasis extracraneales.
La tasa de respuesta intracraneal fue del 50% (IC del 95%: 40, 60), con una tasa de respuesta completa del 4,1% y una tasa de respuesta parcial del 46%. La duración media de la respuesta intracraneal fue de 6,4 meses (rango: 1 a 31). De los pacientes con una respuesta intracraneal, el 9% tuvo una enfermedad estable o progresiva como su mejor respuesta general.
14.2
Tratamiento adyuvante del melanoma positivo para la mutación BRAF V600E o V600K
La seguridad y eficacia de MEKINIST administrado con dabrafenib se evaluaron en un ensayo internacional, multicéntrico, aleatorizado, doble ciego, controlado con placebo (COMBI-AD; NCT01682083) que reclutó a pacientes con melanoma en estadio III con mutaciones BRAF V600E o V600K detectadas mediante el ensayo THxID®-BRAF y afectación patológica de los ganglios linfáticos regionales. La inscripción requirió la resección completa del melanoma con linfadenectomía completa dentro de las 12 semanas previas a la aleatorización. El ensayo excluyó a los pacientes con melanoma mucoso u ocular, metástasis en tránsito irresecables, enfermedad metastásica a distancia o tratamiento sistémico anticancerígeno previo, incluida la radioterapia. Los pacientes fueron aleatorizados (1:1) para recibir MEKINIST 2 mg una vez al día en combinación con dabrafenib 150 mg dos veces al día o dos placebos durante un máximo de 1 año. La aleatorización se estratificó por el estado de la mutación BRAF (V600E o V600K) y el estadio del American Joint Committee on Cancer (AJCC; 7th Edición) (IIIA, IIIB o IIIC). La principal medida de resultado de eficacia fue la supervivencia libre de recaída (SLR), definida como el tiempo desde la aleatorización hasta la recurrencia de la enfermedad (metástasis local, regional o a distancia), nuevo melanoma primario o muerte por cualquier causa, lo que ocurriera primero, según la evaluación del investigador. Los pacientes se sometieron a pruebas de imagen para la recurrencia tumoral cada 3 meses durante los dos primeros años y cada 6 meses a partir de entonces.
En COMBI-AD, se aleatorizaron un total de 870 pacientes: 438 al brazo de MEKINIST en combinación con dabrafenib y 432 al placebo. La edad media fue de 51 años (rango: 18 a 89), el 55% eran hombres, el 99% eran blancos y el 91% tenía un estado de rendimiento ECOG de 0. Las características de la enfermedad fueron: estadio IIIA de AJCC (18%), estadio IIIB de AJCC (41%), estadio IIIC de AJCC (40%), estadio desconocido (1%); mutación BRAF V600E (91%), mutación BRAF V600K (9%); ganglios linfáticos macroscópicos (65%); y ulceración tumoral (41%). La duración media del seguimiento (tiempo desde la aleatorización hasta el último contacto o la muerte) fue de 2,8 años.
COMBI-AD mostró una mejora estadísticamente significativa en la SLR en los pacientes aleatorizados al brazo de MEKINIST en combinación con dabrafenib en comparación con los aleatorizados a placebo. Los resultados de eficacia se presentan en la Tabla 22 y la Figura 3.
| Abreviaturas: HR, razón de riesgos; IC, intervalo de confianza; NE, no estimable. aEstimador de Pike obtenido de la prueba de rango logarítmico estratificada. bPrueba de rango logarítmico estratificada por estadio de la enfermedad (IIIA frente a IIIB frente a IIIC) y tipo de mutación BRAF V600 (V600E frente a V600K). |
||
| Variable | MEKINIST más Dabrafenib N = 438 |
Placebo N = 432 |
| Supervivencia libre de recaída | ||
| Número de eventos (%) | 166 (38) | 248 (57) |
| Mediana, meses (IC del 95%) | NE (44,5, NE) | 16,6 (12,7, 22,1) |
| HR (IC del 95%)a | 0,47 (0,39, 0,58) | |
| P valorb | < 0,0001 | |

Figura 3. Curvas de Kaplan-Meier para la Supervivencia Libre de Recaída en COMBI-AD en el Tratamiento Adyuvante del Melanoma
14.3
Cáncer de Pulmón de Células No Pequeñas Metastásico Positivo para la Mutación BRAF V600E
La seguridad y la eficacia de dabrafenib solo o administrado con MEKINIST se evaluaron en un ensayo clínico multicéntrico, de tres cohortes, no aleatorizado, de estimación de actividad, de etiqueta abierta (Estudio BRF113928; NCT01336634). Los criterios de elegibilidad clave fueron el cáncer de pulmón de células no pequeñas metastásico positivo para la mutación BRAF V600E confirmado localmente, sin exposición previa a un inhibidor de BRAF o MEK, y ausencia de mutación EGFR o reordenamiento ALK (a menos que los pacientes tuvieran progresión en la terapia previa con un inhibidor de la tirosina quinasa). Los pacientes inscritos en las cohortes A y B debían haber recibido al menos un régimen de quimioterapia basado en platino anterior con progresión de la enfermedad demostrada, pero no más de tres regímenes sistémicos previos. Los pacientes de la cohorte C no podían haber recibido terapia sistémica previa para la enfermedad metastásica. Los pacientes de la cohorte A recibieron dabrafenib 150 mg dos veces al día. Los pacientes de las cohortes B y C recibieron MEKINIST 2 mg una vez al día y dabrafenib 150 mg dos veces al día. El principal resultado de eficacia fue la tasa de respuesta objetiva (ORR) según RECIST v1.1 evaluada por un comité de revisión independiente (IRC) y la duración de la respuesta.
Se inscribieron un total de 171 pacientes, que incluyeron 78 pacientes en la cohorte A, 57 pacientes en la cohorte B y 36 pacientes en la cohorte C. Las características de la población fueron: una edad media de 66 años; 48% hombres; 81% blancos, 14% asiáticos, 3% negros y 2% hispanos; 60% exfumadores, 32% no fumadores nunca y 8% fumadores actuales; 27% tenían un estado de rendimiento funcional (PS) de la ECOG de 0, 63% tenían un PS de la ECOG de 1 y 11% tenían un PS de la ECOG de 2; 99% tenían enfermedad metastásica, de los cuales el 6% tenían metástasis cerebral en el momento basal y el 14% tenían metástasis hepática en el momento basal; el 11% había recibido terapia anticancerosa sistémica en el entorno adyuvante, el 58% de los 135 pacientes previamente tratados habían recibido solo una línea de terapia sistémica previa para la enfermedad metastásica; el 98% tenían histología no escamosa.
Los resultados de eficacia se resumen en la Tabla 23.
| Abreviaturas: CI, intervalo de confianza; DoR, duración de la respuesta. aRepresenta los resultados del análisis final (fecha de corte del 24 de febrero de 2021) para las cohortes de respondedores del análisis primario. |
|||
| Tratamiento | Dabrafenib | MEKINIST más Dabrafenib | |
| Población | Previamente Tratado N = 78 |
Previamente Tratado N = 57 |
No Tratado Previamente N = 36 |
| Tasa de Respuesta Objetivaa | |||
| ORR (95% CI) | 27% (18%, 38%) | 61% (48%, 74%) | 61% (44%, 77%) |
| Respuesta completa | 1% | 5% | 8% |
| Respuesta parcial | 26% | 56% | 53% |
| Duración de la Respuestaa | n = 21 | n = 35 | n = 22 |
| Mediana DoR, meses (95% CI) | 18.0 (4.2, 40.1) | 9.0 (5.8, 26.2) | 15.2 (7.8, 23.5) |
En un análisis de subgrupos de pacientes con NSCLC positivo para la mutación BRAF V600E confirmado centralmente de forma retrospectiva con la prueba Oncomine™ Dx Target, los resultados de la ORR fueron similares a los presentados en la Tabla 16.
14.4
Cáncer de tiroides anaplásico positivo para la mutación BRAF V600E localmente avanzado o metastásico
La seguridad y eficacia de MEKINIST administrado con dabrafenib se evaluó en un ensayo de estimación de actividad, multicéntrico, no aleatorizado, de etiqueta abierta, de nueve cohortes (Estudio BRF117019; NCT02034110) en pacientes con cánceres raros con la mutación BRAF V600E, incluido el ATC localmente avanzado, irresecable o metastásico sin opciones estándar de tratamiento locoregional. El ensayo BRF117019 excluyó a los pacientes que no podían tragar o retener la medicación; que recibieron tratamiento previo con inhibidores de BRAF o MEK; con metástasis del SNC sintomáticas o no tratadas; o que tenían obstrucción de las vías respiratorias. Los pacientes recibieron MEKINIST 2 mg una vez al día y dabrafenib 150 mg dos veces al día. La principal medida de resultado de eficacia fue la ORR según RECIST v1.1, evaluada por un comité de revisión independiente (IRC) y la duración de la respuesta (DoR).
Se inscribieron 36 pacientes y fueron evaluables para la respuesta en la cohorte de ATC. La edad media fue de 71 años (rango: 47 a 85); el 44% eran hombres, el 50% blancos, el 44% asiáticos; y el 94% tenía un estado de rendimiento ECOG de 0 o 1. Los tratamientos anticancerosos previos incluyeron cirugía y radioterapia de haz externo (83% cada uno), y terapia sistémica (67%).
Los resultados de eficacia se resumen en la Tabla 24.
| Abreviaturas: ATC, cáncer de tiroides anaplásico; IC, intervalo de confianza; DoR, duración de la respuesta; ORR, tasa de respuesta global; NE, no estimable. | |
| Población de la cohorte de ATC | N = 36 |
| Tasa de respuesta global | |
| ORR (IC del 95%) | 53% (35.5%, 69.6%) |
| Respuesta completa | 6% |
| Respuesta parcial | 47% |
| Duración de la respuesta | n = 19 |
| DoR mediana, meses (IC del 95%) | 13.6 (3.8, NE) |
| % con DoR ≥ 6 meses | 68% |
| % con DoR ≥ 12 meses | 53% |
14.5
Falta de actividad clínica en el melanoma metastásico después del tratamiento con inhibidores de BRAF
La actividad clínica de MEKINIST como agente único se evaluó en un ensayo internacional, unicéntrico, de un solo brazo en 40 pacientes con mutación BRAF V600E o V600K positiva, melanoma irresecable o metastásico que habían recibido tratamiento previo con un inhibidor de BRAF. Todos los pacientes recibieron MEKINIST en una dosis de 2 mg por vía oral una vez al día hasta la progresión de la enfermedad o toxicidad inaceptable.
La edad media fue de 58 años, el 63% eran hombres, todos eran blancos, el 98% tenían un PS ECOG basal de 0 o 1, y la distribución de las mutaciones BRAF V600 fue V600E (83%), V600K (10%) y el resto de los pacientes tenían mutaciones múltiples V600 (5%) o un estado mutacional desconocido (2%). Ningún paciente logró una respuesta parcial o completa confirmada según los investigadores clínicos.
14.6
Tumores sólidos irresecables o metastásicos positivos para la mutación BRAF V600E
La seguridad y eficacia de MEKINIST en combinación con dabrafenib para el tratamiento de tumores sólidos irresecables o metastásicos positivos para la mutación BRAF V600E se evaluaron en los ensayos BRF117019, NCI-MATCH y CTMT212X2101, y se apoyaron en los resultados en COMBI-d, COMBI-v [ver Estudios Clínicos (14.2)] y BRF113928 [ver Estudios Clínicos (14.4)]. En estudios en adultos, los pacientes recibieron MEKINIST 2 mg una vez al día y dabrafenib 150 mg dos veces al día. Las principales medidas de resultado de eficacia fueron la tasa de respuesta objetiva (ORR) según los criterios RECIST v1.1, RANO [HGG] o RANO modificado [LGG] y la duración de la respuesta (DoR).
Estudio BRF117019 y Estudio NCI-MATCH
El estudio BRF117019 (NCT02034110) [ver Estudios Clínicos (14.5)] es un ensayo multicohorte, multicéntrico, no aleatorizado y de etiqueta abierta en pacientes adultos con tumores seleccionados con la mutación BRAF V600E, incluidos glioma de alto grado (HGG) (n = 45), cáncer de vías biliares (BTC) (n = 43), glioma de bajo grado (LGG) (n = 13), adenocarcinoma de intestino delgado (ASI) (n = 3), tumor gastrointestinal estromal (GIST) (n = 1) y cáncer de tiroides anaplásico [ver Estudios Clínicos (14.5)]. Los pacientes se inscribieron en base a las evaluaciones locales del estado de la mutación BRAF V600E; un laboratorio central confirmó la mutación BRAF V600E en 93 de 105 pacientes.
El brazo H (EAY131-H) del estudio NCI-MATCH (NCT02465060) es un estudio de un solo brazo, de etiqueta abierta que inscribió a pacientes con una mutación BRAF V600E. Se excluyeron pacientes con melanoma, cáncer de tiroides o CRC. El estado de la mutación BRAF para la inscripción se determinó por pruebas de laboratorio central o local. El estudio incluyó a pacientes adultos con tumores sólidos, incluidos tumores gastrointestinales (n = 14), tumores pulmonares (n = 7), tumores ginecológicos o peritoneales (n = 6), tumores del SNC (n = 4) y ameloblastoma de la mandíbula (n = 1).
Entre los 131 pacientes inscritos en BRF117019 y NCI-MATCH con los tipos de tumor mostrados en la Tabla 21, las características basales fueron: edad media de 51 años, con un 20% de 65 años o más; 56% mujeres; 85% blancos, 9% asiáticos, 3% negros, 3% otros; y 37% ECOG 0, 56% ECOG 1 y 6% ECOG 2. De los 131 pacientes, el 90% recibió tratamiento sistémico previo.
Los resultados de eficacia en pacientes con tumores sólidos se resumen en la Tabla 25.
| Abreviaturas: PR, respuesta parcial. aExcluye NSCLC (n = 6) y ATC (n = 36) (tipos de tumores previamente aprobados para MEKINIST en combinación con dabrafenib). bMediana DoR 9.8 meses (95% CI: 5.3, 20.4). cMediana DoR 13.6 meses (95% CI: 5.5, 26.7). dDenota una DoR censurada a la derecha. |
||||
| Tipo de tumora | N | Tasa de respuesta objetiva | Duración de la respuesta | |
| % | 95% CI | Rango (meses) | ||
| Cáncer de vías biliaresb | 48 | 46 | (31, 61) | 1.8d, 40d |
| Glioma de alto gradoc | 48 | 33 | (20, 48) | 3.9, 44 |
| Glioblastoma | 32 | 25 | (12, 43) | 3.9, 27 |
| Anaplástico pleomórfico xantoastrocitoma | 6 | 67 | (22, 96) | 6, 43 |
| Anaplástico astrocitoma | 5 | 20 | (0.5, 72) | 15 |
| Astroblastoma | 2 | 100 | (16, 100) | 15, 23d |
| Undifferentiated | 1 | PR | (2.5, 100) | 6 |
| Anaplastic ganglioglioma | 1 | 0 | NA | NA |
| Anaplastic oligodendroglioma | 1 | 0 | NA | NA |
| Low grade glioma | 14 | 50 | (23, 77) | 6, 29d |
| Astrocytoma | 4 | 50 | (7, 93) | 7, 23 |
| Ganglioglioma | 4 | 50 | (7, 93) | 6, 13 |
| Pleomorphic xanthoastrocytoma | 2 | 50 | (1.3, 99) | 6 |
| Pilocytic astrocytoma | 2 | 0 | NA | NA |
| Choroid plexus papilloma | 1 | PR | (2.5, 100) | 29d |
| Gangliocytoma/ganglioglioma | 1 | PR | (2.5, 100) | 18d |
| Low grade serous ovarian carcinoma | 5 | 80 | (28, 100) | 12, 42d |
| Adenocarcinoma small intestine | 4 | 50 | (7, 93) | 7, 8 |
| Adenocarcinoma pancreas | 3 | 0 | NA | NA |
| Mixed ductal/adenoneuroendocrine carcinoma | 2 | 0 | NA | NA |
| Neuroendocrine carcinoma of colon | 2 | 0 | NA | NA |
| Ameloblastoma of mandible | 1 | PR | (2.5, 100) | 30 |
| Combined small cell-squamous carcinoma of lung | 1 | PR | (2.5, 100) | 5 |
| Mucinous-papillary serous adenocarcinoma of peritoneum | 1 | PR | (2.5, 100) | 8 |
| Adenocarcinoma of anus | 1 | 0 | NA | NA |
| Gastrointestinal stromal tumor | 1 | 0 | NA | NA |
Estudio CTMT212X2101 (X2101)
El estudio X2101 (NCT02124772) fue un estudio multicéntrico, de etiqueta abierta, de múltiples cohortes en pacientes pediátricos con tumores sólidos refractarios o recurrentes. La parte C fue una escalada de dosis de MEKINIST en combinación con dabrafenib en pacientes con mutación BRAF V600E. La parte D fue una fase de expansión de la cohorte de MEKINIST en combinación con dabrafenib en pacientes con LGG con mutación BRAF V600E. La medida principal de resultado de eficacia fue la tasa de respuesta objetiva (ORR) evaluada por un comité de revisión independiente según los criterios RANO.
La eficacia de MEKINIST en combinación con dabrafenib se evaluó en 48 pacientes pediátricos, incluidos 34 pacientes con LGG y 2 pacientes con HGG.
Para los pacientes con LGG y HGG mutantes BRAF V600E en las partes C y D, la mediana de edad fue de 10 años (rango: 1 a 17); el 50% eran varones, el 75% blancos, el 8% asiáticos, el 3% negros; y el 58% tenían un estado de rendimiento Karnofsky/Lansky de 100. Los tratamientos anticancerígenos previos incluyeron cirugía (83%), radioterapia externa (2,8%) y terapia sistémica (92%). La ORR fue del 25% (IC 95%: 12%, 42%). De los 9 pacientes que respondieron, la duración de la respuesta (DoR) fue ≥ 6 meses en el 78% de los pacientes y ≥ 24 meses en el 44% de los pacientes.
Estudio CDRB436G2201 (G2201) – Cohorte de glioma de alto grado
El estudio G2201 (NCT02684058) fue un estudio multicéntrico, aleatorizado, de etiqueta abierta, de fase II de dabrafenib y trametinib en pacientes pediátricos naïve a la quimioterapia con glioma de bajo grado (LGG) mutante BRAF V600E y pacientes con HGG mutante BRAF V600E recurrente o progresivo. Los pacientes con HGG se incluyeron en una cohorte de un solo brazo. La medida principal de resultado de eficacia para la cohorte de HGG fue la ORR evaluada por un comité de revisión independiente según los criterios RANO 2010.
La eficacia de MEKINIST en combinación con dabrafenib se evaluó en 41 pacientes pediátricos con HGG recurrente o progresivo.
Para los pacientes con HGG mutante BRAF V600E incluidos en la cohorte de HGG, la mediana de edad fue de 13 años (rango: 2 a 17); el 56% eran mujeres, el 61% blancos, el 27% asiáticos, el 2,4% negros, y el 37% tenían un estado de rendimiento Karnofsky/Lansky de 100. Los tratamientos anticancerígenos previos incluyeron cirugía (98%), radioterapia (90%) y quimioterapia (81%). La ORR fue del 56% (IC 95%: 40, 72). La mediana de DoR no se alcanzó (IC 95%: 9,2, NE). De los 23 pacientes que respondieron en la cohorte de HGG, la DoR fue ≥ 6 meses en el 78% de los pacientes, ≥ 12 meses en el 48% de los pacientes y ≥ 24 meses en el 22% de los pacientes.
14.7
Mutación BRAF V600E positiva en glioma de bajo grado
Estudio CDRB436G2201 (G2201) – Cohorte de glioma de bajo grado
La seguridad y la eficacia de MEKINIST en combinación con dabrafenib para el tratamientoo de glioma de bajo grado (LGG) positivo para la mutación BRAF V600E en pacientes pediátricos de 1 a < 18 años de edad se evaluaron en el ensayo multicéntrico, de etiqueta abierta (Estudio CDRB436G2201; NCT02684058). Los pacientes con LGG (grados 1 y 2 de la OMS) que requerían la primera terapia sistémica se aleatorizaron en una proporción de 2:1 a dabrafenib más trametinib (D + T) o carboplatino más vincristina (C + V).
El estado de mutación BRAF se identificó prospectivamente mediante una evaluación local o una prueba de laboratorio central. Además, se realizó una prueba retrospectiva de muestras tumorales disponibles por el laboratorio central para evaluar el estado de mutación BRAF V600E.
Los pacientes recibieron dosificación basada en la edad y el peso de MEKINIST y dabrafenib hasta la pérdida del beneficio clínico o hasta la toxicidad inaceptable. La carboplatino y la vincristina se dosificaron en función del área de la superficie corporal a dosis de 175 mg/m2 y 1,5 mg/m2 (0,05 mg/kg para pacientes < 12 kg), respectivamente, como un curso de inducción de 10 semanas seguido de ocho ciclos de mantenimiento de 6 semanas.
La medida principal de resultado de eficacia fue la tasa de respuesta global (ORR) por revisión independiente basada en los criterios RANO LGG (2017). Las medidas de resultado de eficacia adicionales fueron la supervivencia libre de progresión y la supervivencia global. El análisis primario se realizó cuando todos los pacientes habían completado al menos 32 semanas de tratamientoo.
En la cohorte de LGG, 110 pacientes se aleatorizaron a D + T (n = 73) o C + V (n = 37). La mediana de edad fue de 9,5 años (rango: 1 a 17); el 60% eran mujeres. El estudio G2201 mostró una mejora estadísticamente significativa en la ORR y la PFS en pacientes con LGG aleatorizados a D + T en comparación con aquellos aleatorizados a C + V. Los resultados de eficacia se muestran en la Tabla 26.
| Abreviaturas: IC, intervalo de confianza; NE, no estimable. aBasado en el intervalo de confianza exacto de Clopper-Pearson bBasado en el método de Kaplan-Meier cBasado en el modelo de riesgos proporcionales |
||||
| MEKINIST más Dabrafenib N = 73 |
Carboplatino más Vincristina N = 37 |
|||
| Tasa de respuesta global | ||||
| ORR% (IC 95%)a | 46,6 (34,8, 58,6) | 10,8 (3,0, 25,4) | ||
| Valor de P | < 0,001 | |||
| Respuesta completa, n (%) | 2 (2,7) | 1 (2,7) | ||
| Respuesta parcial, n (%) | 32 (44) | 3 (8) | ||
| Duración de la respuesta | ||||
| Mediana (IC 95%)b, meses | 23,7 (14,5, NE) | NE (6,6, NE) | ||
| % con DoR observado ≥ 12 meses | 56 | 50 | ||
| % con DoR observado ≥ 24 meses | 15 | 25 | ||
| Supervivencia libre de progresión | ||||
| Mediana (IC del 95%)b, meses | 20.1 (12.8, NE) | 7.4 (3.6, 11.8) | ||
| Razón de riesgo (IC del 95%)c | 0.31 (0.17, 0.55) | |||
| P valor | < 0.001 | |||
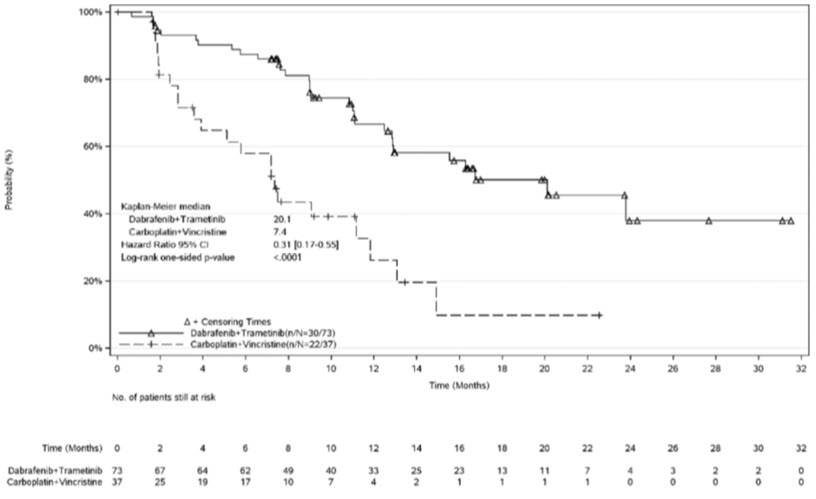
Figura 4. Curvas de Kaplan-Meier para la supervivencia libre de progresión en el estudio G2201 (cohorte LGG)
En el momento del análisis intermedio de la supervivencia general (SG), realizado cuando todos los pacientes habían completado al menos 32 semanas de tratamiento o habían interrumpido antes, hubo una muerte en el brazo C + V. Los resultados de la SG en el análisis intermedio no alcanzaron significación estadística.
16 FORMA DE SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
MEKINIST Tabletas:
Tabletas de 0.5 mg: Tabletas de color amarillo, ovaladas modificadas, biconvexas, recubiertas con película con ‘GS’ grabado en relieve en una cara y ‘TFC’ en la cara opuesta y están disponibles en frascos de 30 (NDC 0078-0666-15).
Tabletas de 0.5 mg: Tabletas de color amarillo, ovaloides, biconvexas, recubiertas con película sin ranurar con bordes biselados y con el logotipo de Novartis grabado en relieve en un lado y ‘TT’ en el otro lado; disponibles en frascos de 30 (NDC 0078-1105-15).
Tabletas de 2 mg: Tabletas de color rosa, redondas, biconvexas, recubiertas con película con ‘GS’ grabado en relieve en una cara y ‘HMJ’ en la cara opuesta y están disponibles en frascos de 30 (NDC 0078-0668-15).
Tabletas de 2 mg: Tabletas de color rosa, redondas, biconvexas, recubiertas con película sin ranurar con bordes biselados y con el logotipo de Novartis grabado en relieve en un lado y ‘LL’ en el otro lado; disponibles en frascos de 30 (NDC 0078-1112-15).
Almacenar a 20°C a 25°C (68°F a 77°F); se permiten excursiones entre 15°C y 30°C (59°F y 86°F) [ver Temperatura ambiente controlada de USP]. Dispensar en el frasco original. No retire el desecante. Proteger de la humedad y la luz. No coloque el medicamento en cajas de pastillas.
MEKINIST para solución oral:
Polvo blanco o casi blanco en frascos de vidrio ámbar, co-envasado con un adaptador de frasco a presión y una jeringa oral. Cada frasco contiene 4.7 mg de trametinib equivalente a 5.3 mg de trametinib dimetilsulfóxido. Cada mL de solución de trametinib reconstituida con sabor a fresa contiene 0.05 mg de trametinib padre no solvatado. (NDC 0078-1161-47).
Almacenar refrigerado a 2°C a 8°C (36°F a 46°F). Almacenar en el envase original para protegerlo de la luz y la humedad.
Después de la reconstitución, almacenar en el frasco original por debajo de 25°C (77°F) y no congelar.
Deseche cualquier solución no utilizada 35 días después de la reconstitución.
17 INFORMACIÓN PARA EL PACIENTE
Aconseje al paciente que lea el etiquetado del paciente aprobado por la FDA (Información para el paciente e Instrucciones de uso).
Nuevas neoplasias malignas cutáneas y no cutáneas primarias
Avise a los pacientes que MEKINIST administrado con dabrafenib puede provocar el desarrollo de nuevas neoplasias malignas cutáneas y no cutáneas primarias. Avise a los pacientes que se pongan en contacto con su profesional sanitario inmediatamente por cualquier lesión nueva, cambios en las lesiones existentes en su piel u otros signos y síntomas de malignidades [ver Advertencias y precauciones (5.1)].
Hemorragia
Avise a los pacientes que MEKINIST administrado con dabrafenib aumenta el riesgo de hemorragia intracraneal y gastrointestinal. Avise a los pacientes que se pongan en contacto con su profesional sanitario para buscar atención médica inmediata por signos o síntomas de sangrado inusual o hemorragia [ver Advertencias y precauciones (5.2)].
Colitis y perforación gastrointestinal
Avise a los pacientes que MEKINIST puede causar colitis y perforación gastrointestinal y que se pongan en contacto con su profesional sanitario por signos o síntomas de colitis o perforación gastrointestinal [ver Advertencias y precauciones (5.3)].
Eventos tromboembólicos venosos
Avise a los pacientes que MEKINIST administrado con dabrafenib aumenta los riesgos de EP y TVP. Avise a los pacientes que busquen atención médica inmediata por la aparición repentina de dificultad para respirar, dolor en las piernas o hinchazón [ver Advertencias y precauciones (5.4)].
Cardiomiopatía
Avise a los pacientes que MEKINIST puede causar cardiomiopatía y que informen inmediatamente a su profesional sanitario de cualquier signo o síntoma de insuficiencia cardíaca [ver Advertencias y precauciones (5.5)].
Toxicidades oculares
Avise a los pacientes que MEKINIST puede causar alteraciones visuales graves que pueden provocar ceguera y que se pongan en contacto con su profesional sanitario si experimentan algún cambio en su visión [ver Advertencias y precauciones (5.6)].
Enfermedad pulmonar intersticial/Neumonitis
Avise a los pacientes que MEKINIST puede causar EPI (o neumonitis). Avise a los pacientes que se pongan en contacto con su profesional sanitario lo antes posible si experimentan signos, como tos o disnea [ver Advertencias y precauciones (5.7)].
Reacciones febriles graves
Avise a los pacientes que MEKINIST administrado con dabrafenib puede causar reacciones febriles graves. Indique a los pacientes que se pongan en contacto con su profesional sanitario si desarrollan fiebre mientras toman MEKINIST con dabrafenib [ver Advertencias y precauciones (5.8)].
Toxicidades cutáneas graves
Avise a los pacientes que MEKINIST puede causar toxicidades cutáneas graves, que pueden requerir hospitalización, y que se pongan en contacto con su profesional sanitario por erupción progresiva o intolerable. Avise a los pacientes que se pongan en contacto con su profesional sanitario inmediatamente si desarrollan signos y síntomas de una reacción cutánea grave [ver Advertencias y precauciones (5.9)].
Hipertensión
Avise a los pacientes que MEKINIST puede causar hipertensión y que deben someterse a la monitorización de la presión arterial y que se pongan en contacto con su profesional sanitario si desarrollan síntomas de hipertensión, como dolor de cabeza intenso, visión borrosa o mareos [ver Reacciones adversas (6.1)].
Diarrea
Avise a los pacientes que MEKINIST a menudo causa diarrea, que puede ser grave en algunos casos. Informe a los pacientes de la necesidad de ponerse en contacto con su profesional sanitario si se produce diarrea grave durante el tratamiento [ver Reacciones adversas (6.1)].
Toxicidad embrio-fetal
- Avise a las mujeres embarazadas y a los hombres con potencial reproductivo del riesgo potencial para un feto [ver Advertencias y precauciones (5.13), Uso en poblaciones específicas (8.1, 8.3)].
- Avise a las mujeres que se pongan en contacto con su profesional sanitario de un embarazo conocido o sospechado.
- Avise a las mujeres con potencial reproductivo que utilicen métodos anticonceptivos eficaces durante el tratamiento con MEKINIST y durante 4 meses después de la última dosis.
- Avise a los pacientes de sexo masculino con parejas femeninas con potencial reproductivo que utilicen condones durante el tratamiento con MEKINIST y durante 4 meses después de la última dosis.
Lactancia
Avise a las mujeres que no deben amamantar durante el tratamiento con MEKINIST y durante 4 meses después de la última dosis [ver Uso en poblaciones específicas (8.2)].
Infertilidad
Avise a las mujeres con potencial reproductivo del riesgo potencial de deterioro de la fertilidad [ver Uso en poblaciones específicas (8.3)].
Administración
Indique a los pacientes que tomen MEKINIST al menos 1 hora antes o al menos 2 horas después de una comida [ver Posología y administración (2.3)].
THxID® BRAF assay is a trademark of bioMérieux.
Oncomine™ Dx Target Test is a trademark of Life Technologies Corporation, a part of Thermo Fisher Scientific Inc.
Distribuido por:
Novartis Pharmaceuticals Corporation
East Hanover, New Jersey 07936
© Novartis
T2024-71
INSERTO PARA EL PACIENTE
| Esta información para el paciente ha sido aprobada por la Administración de Alimentos y Medicamentos de los Estados Unidos. | Revisado: Octubre 2024 |
| Información para el paciente | |
| MEKINIST® (MEK-in-ist) (trametinib) tabletas |
MEKINIST® (MEK-in-ist) (trametinib) para solución oral |
| Información importante: Si su proveedor de atención médica le receta MEKINIST para que lo tome o se lo administren con dabrafenib, también lea la Guía de medicamentos que viene con dabrafenib. | |
| ¿Cuál es la información más importante que debo saber sobre MEKINIST? MEKINIST puede causar efectos secundarios graves, que incluyen: Riesgo de nuevos cánceres de piel. MEKINIST, cuando se usa con dabrafenib, puede causar cánceres de piel, llamados carcinoma de células escamosas cutáneo, queratoacantoma, carcinoma de células basales o melanoma. Hable con su proveedor de atención médica sobre su riesgo de estos cánceres. Revise su piel y dígale a su proveedor de atención médica de inmediato sobre cualquier cambio en la piel, incluido un:
Su proveedor de atención médica debe revisar su piel antes del tratamiento con MEKINIST y dabrafenib, cada 2 meses durante el tratamiento con MEKINIST y dabrafenib, y hasta por 6 meses después de dejar de tomar MEKINIST y dabrafenib para buscar cualquier nuevo cáncer de piel. |
|
| ¿Qué es MEKINIST? MEKINIST es un medicamento recetado que se usa:
MEKINIST no debe usarse para tratar a personas que ya han recibido un inhibidor de BRAF para el tratamiento de su melanoma, y no funcionó o ya no funciona.
MEKINIST no está indicado para el tratamiento de personas con cáncer colorrectal. |
|
Antes de tomar o administrar MEKINIST, informe a su proveedor de atención médica sobre todas sus afecciones médicas, incluso si:
Informe a su proveedor de atención médica sobre todos los medicamentos que toma, incluidos los medicamentos recetados y de venta libre, las vitaminas y los suplementos herbales. |
|
¿Cómo debo tomar o administrar MEKINIST?
Tabletas de MEKINIST:
MEKINIST para solución oral:
|
|
| ¿Cuáles son los posibles efectos secundarios de MEKINIST? MEKINIST puede causar efectos secundarios graves, que incluyen:
|
|
|
○ ampollas o descamación de la piel |
○ ampollas en los labios o alrededor de la boca o los ojos |
Los efectos secundarios más comunes de MEKINIST cuando se toma solo incluyen:
|
|
| Los efectos secundarios más comunes de MEKINIST cuando se toma con dabrafenib en personas con melanoma que se ha diseminado a otras partes del cuerpo o que no se puede extirpar mediante cirugía incluyen: | |
| • fiebre | • diarrea |
| • erupción cutánea | • vómitos |
| • náuseas | • presión arterial alta (hipertensión) |
| • escalofríos | • hinchazón de la cara, los brazos o las piernas |
| Los efectos secundarios más comunes de MEKINIST cuando se toma con dabrafenib para ayudar a prevenir que el melanoma regrese después de que el cáncer se haya extirpado mediante cirugía incluyen: | |
| • fiebre | • escalofríos |
| • cansancio | • diarrea |
| • náuseas | • vómitos |
| • dolor de cabeza | • dolores en las articulaciones |
| • rash | • muscle aches |
| Los efectos secundarios más comunes de MEKINIST cuando se toma con dabrafenib en personas con NSCLC incluyen: | |
| • fever | • rash |
| • tiredness | • swelling of your face, arms, and legs |
| • nausea | • chills |
| • vomiting | • bleeding |
| • diarrhea | • cough |
| • dry skin | • shortness of breath |
| • decreased appetite | |
| Los efectos secundarios más comunes de MEKINIST cuando se toma con dabrafenib en adultos con tumores sólidos que no se pueden extirpar mediante cirugía o que se han diseminado a otras partes del cuerpo incluyen: | |
| • fever | • bleeding |
| • tiredness | • cough |
| • nausea | • vomiting |
| • rash | • constipation |
| • chills | • diarrhea |
| • headache | • muscle and joint aches |
| • swelling of your arms and legs | |
| Los efectos secundarios más comunes de MEKINIST cuando se administra con dabrafenib en niños de 1 año de edad o mayores con tumores sólidos que no se pueden extirpar mediante cirugía o que se han diseminado a otras partes del cuerpo incluyen: | |
| • fever | • acne |
| • rash | • headache |
| • vomiting | • stomach-area (abdominal) pain |
| • tiredness | • nausea |
| • dry skin | • bleeding |
| • cough | • constipation |
| • diarrhea | • skin infection around fingernails or toenails |
| Los efectos secundarios más comunes de MEKINIST cuando se administra con dabrafenib en niños de 1 año de edad o mayores con glioma de bajo grado incluyen: | |
| • fever | • dry skin |
| • rash | • diarrhea |
| • headache | • nausea |
| • vomiting | • bleeding |
| • muscle and bone pain | • stomach-area (abdominal) pain |
| • tiredness | • acne |
| MEKINIST puede causar presión arterial alta nueva o que empeora (hipertensión). Su proveedor de atención médica debe controlar su presión arterial durante el tratamiento con MEKINIST. Llame a su proveedor de atención médica de inmediato si desarrolla presión arterial alta, su presión arterial empeora o tiene dolor de cabeza intenso, mareos, visión borrosa o mareos. MEKINIST puede causar problemas de fertilidad en las mujeres. Esto podría afectar su capacidad para quedar embarazada. Hable con su proveedor de atención médica si esto le preocupa. Estos no son todos los posibles efectos secundarios de MEKINIST. Llame a su proveedor de atención médica para obtener asesoramiento médico sobre los efectos secundarios. Puede informar los efectos secundarios a la FDA al 1-800-FDA-1088. También puede informar los efectos secundarios a Novartis Pharmaceuticals Corporation al 1-888-669-6682. |
|
| ¿Cómo debo almacenar MEKINIST? Tabletas de MEKINIST:
MEKINIST para solución oral:
Mantenga MEKINIST y todos los medicamentos fuera del alcance de los niños. |
|
| Información general sobre el uso seguro y eficaz de MEKINIST Los medicamentos a veces se recetan para fines distintos de los que se enumeran en un folleto de información para el paciente. No use MEKINIST para una condición para la que no fue recetado. No le dé MEKINIST a otras personas, incluso si tienen los mismos síntomas que usted. Puede hacerles daño. Puede pedirle a su proveedor de atención médica o farmacéutico información sobre MEKINIST que esté escrita para profesionales de la salud. |
|
| ¿Cuáles son los ingredientes de MEKINIST? Tabletas MEKINIST: Ingrediente activo: trametinib Ingredientes inactivos: Núcleo de la tableta: dióxido de silicio coloidal, croscarmelosa sódica, hipromelosa, estearato de magnesio (fuente vegetal), manitol, celulosa microcristalina, lauril sulfato de sodio. Recubrimiento de la tableta: hipromelosa, óxido de hierro rojo (tabletas de 2 mg), óxido de hierro amarillo (tabletas de 0,5 mg), polietilenglicol, polisorbato 80 (tabletas de 2 mg), dióxido de titanio. MEKINIST para solución oral: Ingrediente activo: trametinib Ingredientes inactivos: éter sulfobutílico de betadex sódico, ácido cítrico monohidratado, fosfato disódico, metilparabeno, sorbato de potasio, sucralosa y sabor a fresa. Distribuido por: Novartis Pharmaceuticals Corporation, East Hanover, New Jersey 07936 © Novartis Para obtener más información, visite www.us.tafinlarmekinist.com o llame al 1-888-669-6682. |
|
T2024-72
INSTRUCCIONES DE USO
| Estas Instrucciones de uso han sido aprobadas por la Administración de Alimentos y Medicamentos de los Estados Unidos. | Revisado: Julio 2024 |
| INSTRUCCIONES DE USO MEKINIST® (MEK-in-ist) (trametinib) para solución oral |
|
| Estas “Instrucciones de uso” contienen información sobre cómo administrar la solución oral de MEKINIST. | |
| Información importante que necesita saber antes de administrar la solución oral de MEKINIST | |
|
|
| El paquete de MEKINIST debe contener: | |
 |
|
| Partes de la jeringa oral reutilizable: | |
 |
|
| Almacenamiento de la solución oral de MEKINIST | |
|
 |
| Almacenamiento de la jeringa oral | |
|
 |
| Sección A. Medición y administración de una dosis de la solución oral de MEKINIST | |
Para administrar una dosis de la solución oral de MEKINIST, necesitará:
Llame a su proveedor de atención médica o farmacéutico si no tiene uno o más de estos artículos.
|
 |
| Paso 1. Lávese y séquese las manos antes de medir y administrar una dosis de la solución oral de MEKINIST. | |
| Paso 2. Coloque sus suministros sobre una superficie de trabajo limpia y plana. | |
Paso 3. Compruebe si tiene polvo o solución en el frasco ámbar.
|
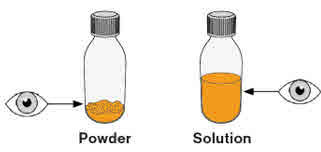 |
Paso 4. Compruebe la fecha de caducidad o “descartar después” de la solución oral de MEKINIST que está escrita a mano o impresa en la etiqueta del frasco ámbar.
|
 |
Paso 5. Agite suavemente el frasco ámbar durante 30 segundos para mezclar la solución oral de MEKINIST.
|
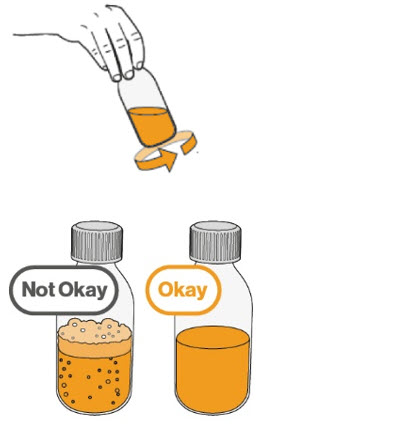 |
| Paso 6. Cuando la espuma haya desaparecido dentro del frasco ámbar, retire la tapa resistente a los niños presionando hacia abajo la tapa y girándola en la dirección de la flecha (en sentido antihorario), como se muestra. Luego coloque el frasco ámbar de nuevo sobre su superficie de trabajo plana. |  |
| Paso 7. Compruebe si hay un adaptador de frasco ya insertado en el cuello del frasco. No retire el adaptador de frasco. Si no está insertado, separe el adaptador de frasco de la jeringa oral e insértelo. Comuníquese con su proveedor de atención médica si no está seguro o el adaptador de frasco falta. |
 |
| Paso 8. A continuación, tome la jeringa oral. Empuje el émbolo hacia arriba en la jeringa oral hasta el máximo para eliminar todo el aire en su interior. |
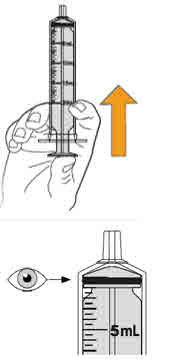 |
| Paso 9. Use una mano para sujetar el frasco ámbar que contiene la solución oral de MEKINIST preparada de forma estable. Con la otra mano, inserte la punta de la jeringa oral en la abertura del adaptador de frasco. Asegúrese de que la jeringa oral esté firmemente conectada. Importante: Debido a la presión del aire, el émbolo puede moverse solo cuando mida su dosis durante el Paso 10. Sujete el extremo del émbolo para evitar que se mueva. |
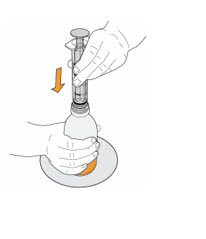 |
| Paso 10. Vire cuidadosamente el frasco ámbar boca abajo y tire del émbolo hacia abajo para extraer la solución oral de MEKINIST en la jeringa oral. Para medir su dosis, mantenga la punta de la jeringa oral hacia arriba. Tira del émbolo hasta que la parte superior del tapón negro se alinee con su dosis prescrita en mL en el cilindro de la jeringa oral. Si aparecen burbujas de aire grandes en la jeringa oral, empuje la solución oral de MEKINIST de vuelta al frasco ámbar y luego tire del émbolo de nuevo para extraer su dosis. |
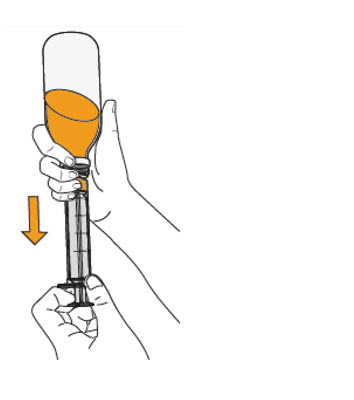 |
| Continúe haciendo esto hasta que no haya burbujas de aire grandes presentes. Las pequeñas burbujas de aire están bien. | 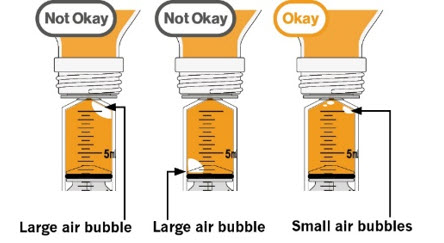 |
| Paso 11. Siga sujetando el émbolo en su lugar y vire cuidadosamente el frasco ámbar derecho. Coloque el frasco ámbar de nuevo en su superficie de trabajo plana. Mientras sigue sujetando el émbolo y el cilindro, retire la jeringa oral del frasco tirando suavemente hacia arriba. |
 |
| Compruebe de nuevo para asegurarse de que la parte superior del tapón negro esté en su dosis prescrita. Si no es así, repita Pasos 9, 10 y 11 de nuevo. Nota: Su dosis puede ser diferente a la dosis que se muestra en esta figura. Si está administrando una dosis de solución oral de MEKINIST por vía oral, pase al Paso 12. Si está administrando una dosis de solución oral de MEKINIST a través de un tubo de alimentación, vaya a la Sección B. |
 |
| Paso 12. Importante: Si le está dando una dosis de solución oral de MEKINIST a un niño, asegúrese de que esté sentado derecho. Coloque la punta de la jeringa oral dentro de la boca. La punta debe tocar el interior de cualquiera de las mejillas. Empuje lentamente el émbolo hasta el final para administrar la dosis completa de solución oral de MEKINIST. Advertencia: Administrar la solución oral de MEKINIST directamente en la garganta o empujar el émbolo demasiado rápido puede causar asfixia. |
 |
| Paso 13. Compruebe que no quede solución oral de MEKINIST en la jeringa oral. Si queda solución oral de MEKINIST en la jeringa oral, adminístrela. Nota: Si su dosis en mL es mayor de lo que puede contener la jeringa oral, repita los pasos 8 a 14 hasta que se administre la dosis total prescrita. |
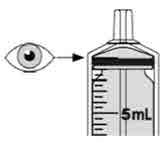 |
| Paso 14. Cuando termine, cierre el frasco ámbar. No retire el adaptador del frasco. Vuelva a colocar la tapa en el frasco ámbar y gírela en la dirección de la flecha (en el sentido de las agujas del reloj), como se muestra, para cerrarla. Asegúrese de que la tapa esté bien sujeta al frasco ámbar. |
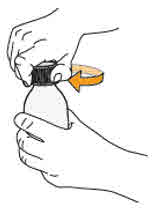 |
| Paso 15. Limpie la jeringa oral reutilizable. Consulte las instrucciones de la sección C “Limpieza de la jeringa oral reutilizable”. |  |
| Sección B. Administración de una dosis de solución oral de MEKINIST a través de una sonda de alimentación | |
| Esta sección es para uso exclusivo si va a administrar una dosis de solución oral de MEKINIST a través de una sonda de alimentación. Antes de administrar una dosis de solución oral de MEKINIST a través de una sonda de alimentación, lea atentamente la siguiente información y luego pase al Paso 1.
|
|
| Paso 1. Enjuague la sonda de alimentación de acuerdo con las instrucciones del fabricante inmediatamente antes de administrar una dosis de solución oral de MEKINIST. | |
| Paso 2. Siga los pasos 1 a 11 de la sección A y luego pase al Paso 3 de esta sección. | |
| Paso 3. Conecte la jeringa oral de 20 mL que contiene la solución oral de MEKINIST a la sonda de alimentación. Es posible que necesite un adaptador ENFIT para conectar la jeringa oral a la sonda de alimentación. | |
| Paso 4. Aplique una presión constante al émbolo para administrar la dosis de solución oral de MEKINIST a través de la sonda de alimentación. | |
| Paso 5. Compruebe que no quede solución oral de MEKINIST en la jeringa oral. Si queda solución oral de MEKINIST en la jeringa oral, repita los pasos 3 a 5. |
|
| Paso 6. Enjuague la sonda de alimentación de nuevo de acuerdo con las instrucciones del fabricante. | |
| Paso 7. Vaya a la sección C para obtener instrucciones sobre “Limpieza de la jeringa oral reutilizable”. | |
| Sección C. Limpieza de la jeringa oral reutilizable | |
| Nota: Limpie la jeringa oral por separado de otros artículos de cocina. | |
| Paso 1. Llene un vaso con agua tibia y jabón. |  |
| Paso 2. Coloque la punta de la jeringa oral en el vaso de agua tibia y jabonosa. Tire hacia arriba y luego empuje hacia abajo el émbolo para introducir y sacar el agua jabonosa de la jeringa oral de 4 a 5 veces. |
 |
| Paso 3. Retire el émbolo del cilindro. |  |
| Paso 4. Enjuague el vaso, el émbolo y el cilindro con agua corriente tibia. |  |
| Paso 5. Deje la jeringa oral y el barril sobre una toalla de papel limpia para que se sequen al aire. Cuando la jeringa oral esté seca, guárdela con el frasco ámbar de solución oral de MEKINIST. Mantenga siempre la jeringa oral fuera del alcance de los niños. Nota: Use una jeringa oral nueva para cada frasco ámbar nuevo de solución oral de MEKINIST. |
 |
| Cómo desechar la solución oral de MEKINIST que está caducada o que ya no se necesita, o las jeringas orales viejas | |
|
|
| Cómo limpiar cualquier derrame de solución oral de MEKINIST | |
| Si accidentalmente derrama alguna solución oral de MEKINIST, limpie el derrame de la siguiente manera: 1. Póngase guantes de plástico. 2. Absorba completamente la solución oral de MEKINIST derramada utilizando un material absorbente, como toallas de papel. 3. Coloque el material absorbente usado en una bolsa de plástico sellable, como una bolsa de almacenamiento de alimentos. 4. Limpie todas las superficies expuestas a la solución oral de MEKINIST con toallitas con alcohol o vierta alcohol para frotar sobre una toalla de papel y luego limpie las superficies expuestas con la toalla de papel. 5. Coloque la bolsa, los guantes y las toallitas con alcohol o la toalla de papel usadas en otra segunda bolsa de plástico y selle. 6. Deseche las bolsas en la basura. 7. Lávese bien las manos con agua y jabón. |
|
| Distribuido por: Novartis Pharmaceuticals Corporation East Hanover, New Jersey 07936 © Novartis |
|
T2024-56
PANEL DE VISUALIZACIÓN PRINCIPAL
NDC 0078-0666-15
Mekinist® (trametinib) Tablets
0.5 mg*
Rx only
30 Tablets
NOVARTIS

PANEL DE VISUALIZACIÓN PRINCIPAL
NDC 0078-0668-15
Mekinist® (trametinib) Tablets
2 mg*
Rx only
30 Tablets
NOVARTIS

PANEL DE VISUALIZACIÓN PRINCIPAL
NDC 0078-1105-15
Mekinist® (trametinib) Tabletas
0.5 mg*
Rx solamente
30 Tabletas
NOVARTIS

PANEL DE VISUALIZACIÓN PRINCIPAL
NDC 0078-1112-15
Mekinist® (trametinib) Tabletas
2 mg*
Rx only
30 Tabletas
NOVARTIS

PANEL DE VISUALIZACIÓN PRINCIPAL
NDC 0078-1161-47
Rx only
Mekinist®
(trametinib)
para Solución Oral
0.05 mg por mL*
90 mL (cuando se reconstituye)
Para el Farmacéutico: Reconstituir antes de
dispensar*. Dispensar con la Información del Paciente y las Instrucciones
de Uso adjuntas.
Para administración por cuidadores únicamente.
NOVARTIS
