
Fabricante de medicamentos: AstraZeneca Pharmaceuticals LP (Updated: 2024-02-08)
PUNTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
LOKELMA® (cicloslicato de circonio sódico) para suspensión oral
Aprobación inicial en EE.UU.: 2018
INDICACIONES Y USO
DOSIFICACIÓN Y ADMINISTRACIÓN
- •
- La dosis inicial recomendada es de 10 g administrados tres veces al día durante un máximo de 48 horas. (2.1)
- •
- Para el tratamiento de mantenimiento, la dosis recomendada es de 10 g una vez al día. Ajuste la dosis en intervalos de una semana según sea necesario (por 5 g diarios) para obtener el rango objetivo deseado de potasio sérico. (2.1)
- Pacientes en hemodiálisis crónica
- •
- La dosis inicial recomendada es de 5 g una vez al día en los días sin diálisis. (2.2)
- Consulte la información de prescripción completa para obtener instrucciones adicionales sobre la dosificación, así como instrucciones de reconstitución y administración para la suspensión oral.
FORMAS FARMACÉUTICAS Y CONCENTRACIONES
CONTRAINDICACIONES
Ninguna. (4)
ADVERTENCIAS Y PRECAUCIONES
- •
- Eventos adversos gastrointestinales en pacientes con trastornos de la motilidad. (5.1)
- •
- Edema. (5.2)
- •
- Hipopotasemia en pacientes en hemodiálisis. (5.3)
- •
- LOKELMA tiene propiedades radiopacas y, por lo tanto, puede presentar la apariencia típica de un agente de imagen durante los procedimientos de rayos X abdominales. (5.4)
REACCIONES ADVERSAS
Las reacciones adversas más comunes con LOKELMA: edema leve a moderado. (6.1)
Para informar SOSPECHAS DE REACCIONES ADVERSAS, comuníquese con AstraZeneca al 1-800-236-9933 o con la FDA al 1-800-FDA-1088 o www.fda.gov/medwatch.
INTERACCIONES FARMACOLÓGICAS
Consulte la sección 17 para INFORMACIÓN DE ASESORAMIENTO PARA EL PACIENTE.
Revisado: 2/2024
Tabla de contenido
INFORMACIÓN DE PRESCRIPCIÓN COMPLETA: CONTENIDO*
1 INDICACIONES Y USO
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Dosificación recomendada
2.2 Ajuste de la dosificación para pacientes en hemodiálisis crónica
2.3 Reconstitución y administración
3 FORMAS FARMACÉUTICAS Y CONCENTRACIONES
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Eventos adversos gastrointestinales en pacientes con trastornos de la motilidad
5.2 Edema
5.3 Hipopotasemia en pacientes en hemodiálisis
5.4 Pruebas diagnósticas
6 REACCIONES ADVERSAS
6.1 Experiencia en estudios clínicos
7 INTERACCIONES FARMACOLÓGICAS
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.4 Uso pediátrico
8.5 Uso geriátrico
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
12.2 Farmacodinámica
12.3 Farmacocinética
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, mutagénesis, alteración de la fertilidad
14 ESTUDIOS CLÍNICOS
14.1 Estudio 1
14.2 Estudio 2
14.3 Estudio de extensión de once meses
14.4 Estudio 3
14.5 Estudio 4
16 PRESENTACIÓN/ALMACENAMIENTO Y MANIPULACIÓN
17 INFORMACIÓN PARA EL PACIENTE
- *
- No se enumeran las secciones o subsecciones omitidas de la información de prescripción completa.
1 INDICACIONES Y USO
LOKELMA está indicado para el tratamiento de la hiperpotasemia en adultos.
Limitación de uso
LOKELMA no debe utilizarse como tratamiento de emergencia para la hiperpotasemia potencialmente mortal debido a su inicio de acción retardado [ver Farmacología clínica (12.2) y Estudios clínicos (14)].
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Dosificación recomendada
Para el tratamiento inicial de la hiperpotasemia, la dosis recomendada de LOKELMA es de 10 g administrada tres veces al día durante un máximo de 48 horas. Administre LOKELMA por vía oral como una suspensión en agua [ver Dosificación y administración (2.3)].
Para el tratamiento continuado, la dosis recomendada es de 10 g una vez al día. Monitoree el potasio sérico y ajuste la dosis de LOKELMA según el nivel de potasio sérico y el rango objetivo deseado. Durante el tratamiento de mantenimiento, aumente la dosis basándose en el nivel de potasio sérico en intervalos de 1 semana o más y en incrementos de 5 g. Disminuya la dosis de LOKELMA o suspéndala si el potasio sérico se encuentra por debajo del rango objetivo deseado. El rango de dosis de mantenimiento recomendado es de 5 g cada dos días a 15 g diarios.
2.2 Ajuste de dosis para pacientes en hemodiálisis crónica
Para pacientes en hemodiálisis crónica, administre LOKELMA solamente en los días sin diálisis.
La dosis inicial recomendada es de 5 g una vez al día en los días sin diálisis. Considere una dosis inicial de 10 g una vez al día en los días sin diálisis en pacientes con potasio sérico superior a 6,5 mEq/L. Monitoree el potasio sérico y ajuste la dosis de LOKELMA según el valor de potasio sérico pre-diálisis después del intervalo interdiálisis prolongado y el rango objetivo deseado.
Durante el inicio y después de un ajuste de dosis, evalúe el potasio sérico después de una semana. El rango de dosis de mantenimiento recomendado es de 5 g a 15 g una vez al día, en los días sin diálisis.
Suspenda o disminuya la dosis de LOKELMA si:
- •
- el potasio sérico cae por debajo del rango objetivo deseado según el valor pre-diálisis después del intervalo interdiálisis prolongado, o;
- •
- el paciente desarrolla hipopotasemia clínicamente significativa
2.3 Reconstitución y administración
En general, otros medicamentos orales deben administrarse al menos 2 horas antes o 2 horas después de LOKELMA [ver Interacciones farmacológicas (7)].
Indique a los pacientes que vacíen el contenido completo del/los paquete(s) en un vaso con aproximadamente 3 cucharadas de agua o más si lo desean. Revuelva bien y beba de inmediato. Si queda polvo en el vaso, agregue agua, revuelva y beba de inmediato. Repita hasta que no quede polvo para asegurarse de tomar toda la dosis.
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
Para suspensión oral: 5 g o 10 g de polvo blanco a gris en un paquete recubierto con papel de aluminio.
4 CONTRAINDICACIONES
Ninguna.
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Eventos adversos gastrointestinales en pacientes con trastornos de la motilidad
Evite el uso de LOKELMA en pacientes con estreñimiento severo, obstrucción intestinal o impactación, incluyendo trastornos de motilidad intestinal anormales postoperatorios, porque LOKELMA no se ha estudiado en pacientes con estas afecciones y puede ser ineficaz y empeorar las afecciones gastrointestinales.
5.2 Edema
Cada dosis de 5 g de LOKELMA contiene aproximadamente 400 mg de sodio, pero la extensión de la absorción por el paciente es desconocida. En los ensayos clínicos de LOKELMA en pacientes que no estaban en diálisis, se observó edema y generalmente fue de leve a moderado en severidad y fue más común en pacientes tratados con 15 g una vez al día. Monitoree los signos de edema, particularmente en pacientes que deban restringir su ingesta de sodio o que sean propensos a la sobrecarga de líquidos (p. ej., insuficiencia cardíaca o enfermedad renal). Aconseje a los pacientes que ajusten la ingesta de sodio en la dieta, si corresponde. Aumente la dosis de diuréticos según sea necesario [ver Reacciones adversas (6)].
En un ensayo clínico de LOKELMA en pacientes en hemodiálisis crónica en los que la mayoría de los pacientes fueron tratados con dosis de 5 a 10 g una vez al día en los días sin diálisis, no hubo diferencia en el cambio promedio desde el inicio en la ganancia de peso interdialítica (una medida de retención de líquidos) entre los grupos de LOKELMA y placebo.
5.3 Hipopotasemia en pacientes en hemodiálisis
Los pacientes en hemodiálisis pueden ser propensos a enfermedades agudas que pueden aumentar el riesgo de hipopotasemia con LOKELMA (p. ej., enfermedades asociadas con disminución de la ingesta oral, diarrea). Considere ajustar la dosis de Lokelma según los niveles de potasio en estos entornos.
5.4 Pruebas diagnósticas
LOKELMA tiene propiedades radio-opacas y, por lo tanto, puede dar la apariencia típica de un agente de imagen durante los procedimientos de rayos X abdominales.
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas se discuten con más detalle en otras partes de la etiqueta:
- •
- Edema [ver Advertencias y precauciones (5.2)].
6.1 Experiencia en estudios clínicos
Debido a que los estudios clínicos se llevan a cabo en condiciones muy variadas, las tasas de reacciones adversas observadas en los estudios clínicos de un medicamento no pueden compararse directamente con las tasas de los estudios clínicos de otro medicamento y pueden no reflejar las tasas observadas en la práctica.
La exposición total a LOKELMA en los estudios clínicos de seguridad y eficacia de pacientes no en diálisis con hiperpotasemia fue de 1.760 pacientes, con 652 pacientes expuestos a LOKELMA durante al menos 6 meses y 507 pacientes expuestos durante al menos un año.
La población (n=1.009) en los ensayos controlados con placebo incluyó pacientes de 22 a 96 años, mujeres (n=454), caucásicos (n=859) y negros (n=130). Los pacientes tenían hiperpotasemia asociada con enfermedades concomitantes como enfermedad renal crónica, insuficiencia cardíaca y diabetes mellitus.
En los ensayos controlados con placebo en los que los pacientes que no estaban en diálisis fueron tratados con dosis diarias únicas de LOKELMA por hasta 28 días, se reportó edema en el 4.4% de los pacientes que recibieron 5 g, el 5.9% de los pacientes que recibieron 10 g y el 16.1% de los pacientes que recibieron 15 g de LOKELMA en comparación con el 2.4% de los pacientes que recibieron placebo. En ensayos no controlados a más largo plazo en los que la mayoría de los pacientes recibieron dosis <15 g una vez al día, se reportaron reacciones adversas de edema (edema, edema generalizado y edema periférico) en el 8% al 11% de los pacientes.
En un análisis agrupado de estudios clínicos realizados en países con una población predominantemente asiática, el estreñimiento ocurrió en pacientes que recibieron LOKELMA con una incidencia estimada del 9% y 5% para las dosis de 10 g y 5 g, respectivamente. El estreñimiento se resolvió con el ajuste de la dosis o la interrupción del tratamiento. No se reportaron casos de estreñimiento en pacientes que recibieron placebo.
Anomalías de laboratorio
En los ensayos clínicos en pacientes que no estaban en diálisis, el 4.1% de los pacientes tratados con LOKELMA desarrollaron hipopotasemia con un valor de potasio sérico inferior a 3.5 mEq/L, que se resolvió con la reducción de la dosis o la interrupción de LOKELMA. En un ensayo clínico de LOKELMA en pacientes en hemodiálisis crónica, el 5% de los pacientes desarrollaron hipopotasemia previa a la diálisis (potasio sérico <3.5 mEq/L) tanto en los grupos de LOKELMA como de placebo; el 3% y el 1% de los pacientes desarrollaron un potasio sérico < 3.0 mEq/L en los grupos de LOKELMA y placebo, respectivamente.
7 INTERACCIONES FARMACOLÓGICAS
LOKELMA puede aumentar transitoriamente el pH gástrico. Como resultado, LOKELMA puede cambiar la absorción de medicamentos coadministrados que presentan solubilidad dependiente del pH, lo que potencialmente podría conducir a una eficacia o seguridad alterada de estos medicamentos cuando se toman cerca del momento en que se administra LOKELMA [ver Dosificación y administración (2.3) y Farmacología clínica (12.3)]. En general, otros medicamentos orales deben administrarse al menos 2 horas antes o 2 horas después de LOKELMA. No se espera que LOKELMA impacte la exposición sistémica de medicamentos que no presentan solubilidad dependiente del pH y, por lo tanto, no se necesita espaciamiento si se ha determinado que el medicamento concomitante no presenta solubilidad dependiente del pH.
8 USO EN POBLACIONES ESPECÍFICAS
8.5 Uso geriátrico
Del número total de sujetos en los estudios clínicos de LOKELMA, el 58% tenía 65 años o más, mientras que el 25% tenía 75 años o más. No se observaron diferencias generales en seguridad o eficacia entre estos pacientes y pacientes más jóvenes.
11 DESCRIPCIÓN
LOKELMA es un polvo para suspensión oral. El ingrediente activo en LOKELMA es el ciclosilicato de circonio sódico, un fijador de potasio. El ciclosilicato de circonio sódico es un silicato de circonio no absorbido que intercambia preferentemente potasio por hidrógeno y sodio. LOKELMA es un polvo inodoro, insoluble de color blanco a gris para suspensión oral. Tiene un tamaño medio de partícula de 20 µm y no incluye más del 3% de partículas con un diámetro inferior a 3 µm. Cada 5 g de ciclosilicato de circonio sódico contienen 400 mg de sodio.
La fórmula química del ciclosilicato de circonio sódico es Na~1.5H~0.5ZrSi3O9•2–3H2O.
Figura 1: Estructura cristalina del ciclosilicato de circonio sódico
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de Acción
LOKELMA (silicato cíclico de circonio sódico) es un silicato de circonio no absorbido que captura preferencialmente el potasio a cambio de hidrógeno y sodio. In vitro, LOKELMA tiene una alta afinidad por los iones potasio, incluso en presencia de otros cationes como calcio y magnesio. LOKELMA aumenta la excreción fecal de potasio mediante la unión del potasio en la luz del tracto gastrointestinal. La unión del potasio reduce la concentración de potasio libre en la luz gastrointestinal, lo que disminuye los niveles séricos de potasio.
12.2 Farmacodinámica
En un estudio en adultos sanos, LOKELMA administrado como 5 g o 10 g una vez al día durante cuatro días causó un aumento dependiente de la dosis en la excreción fecal de potasio. También se observaron disminuciones correspondientes dependientes de la dosis en la excreción urinaria de potasio y los niveles séricos de potasio.
En pacientes con hiperpotasemia tratados con LOKELMA 10 g tres veces al día durante hasta 48 horas, se observaron reducciones en el potasio sérico una hora después del inicio de la terapia; las concentraciones de potasio sérico continuaron disminuyendo durante el período de tratamiento de 48 horas [ver Estudios Clínicos (14.2)]. En pacientes que no continuaron con LOKELMA, los niveles de potasio aumentaron. Los pacientes con niveles más altos de potasio sérico inicial o que recibieron una dosis más alta presentaron mayores reducciones en el potasio sérico.
LOKELMA causa un pequeño aumento dependiente de la dosis en las concentraciones séricas de bicarbonato (1.1 mmol/L a 5 g una vez al día, 2.3 mmol/L a 10 g una vez al día y 2.6 mmol/L a 15 g una vez al día en comparación con un aumento medio de 0.6 mmol/L en pacientes tratados con placebo). El significado clínico de este hallazgo no está claro.
12.3 Farmacocinética
LOKELMA es un compuesto inorgánico insoluble que no está sujeto a metabolismo enzimático. En un estudio clínico en pacientes con hiperpotasemia en el que se midieron las concentraciones de circonio en la orina y la sangre, las concentraciones de circonio fueron similares en pacientes tratados y no tratados (es decir, indetectables o alrededor del límite inferior de cuantificación del ensayo). Un estudio de balance de masa in vivo en ratas mostró que LOKELMA se recuperó en las heces sin evidencia de absorción sistémica.
Interacciones Farmacológicas
Treinta y seis (36) medicamentos se probaron in vitro para determinar posibles interacciones con LOKELMA. Dieciséis (16) medicamentos evaluados no mostraron interacción in vitro con LOKELMA (alopurinol, apixaban, aspirina, captopril, ciclosporina, digoxina, etinilestradiol, lisinopril, magnesio, metformina, fenitoína, prednisona, propranolol, quinapril, espironolactona y ticagrelor).
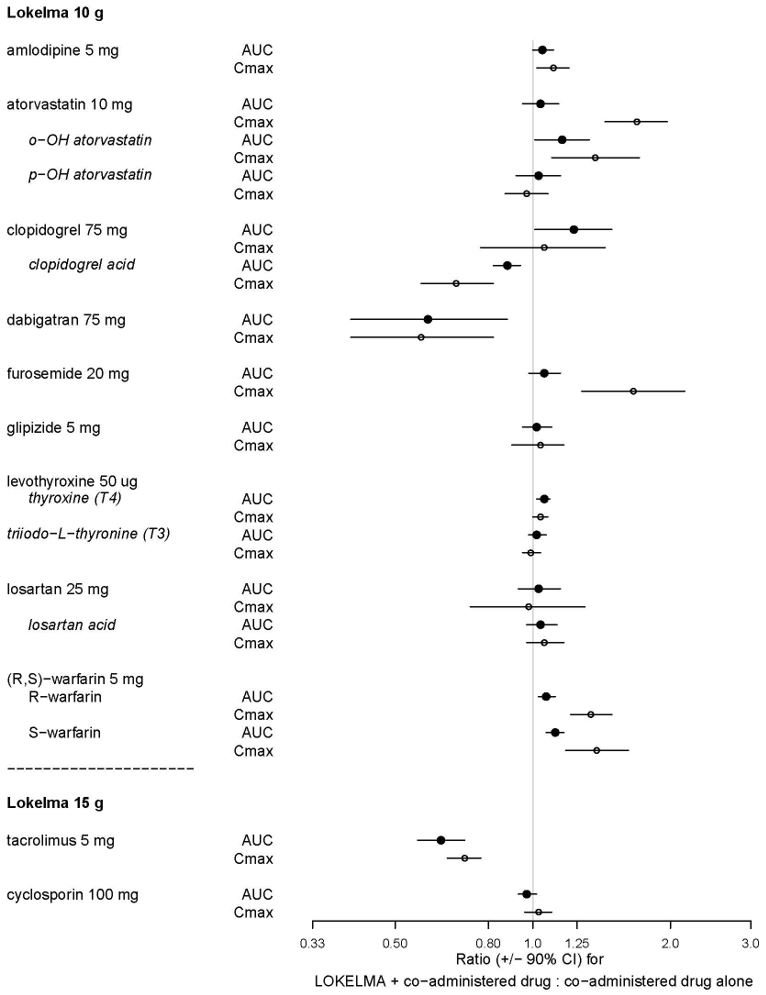
Nueve (9) de los 20 medicamentos que mostraron una interacción in vitro se probaron posteriormente in vivo con LOKELMA 10 g en voluntarios sanos. Losartán, glipizida y levotiroxina no mostraron cambios en la exposición cuando se administraron junto con LOKELMA. Sin embargo, hubo un aumento en la exposición sistémica a ácidos débiles como furosemida y atorvastatina, y una disminución en la exposición sistémica a bases débiles como dabigatrán cuando se administraron junto con LOKELMA, como se muestra en la Figura 2. Estos cambios son consistentes con la hipótesis de que LOKELMA, al elevar el pH gástrico, afecta la exposición sistémica de los medicamentos co-administrados cuya solubilidad depende del pH [ver Interacciones Farmacológicas (7)].
En otro estudio de interacción farmacológica en voluntarios sanos, la co-administración de LOKELMA 15 g disminuyó las exposiciones sistémicas de tacrolimus (Figura 2), probablemente debido a la acción de LOKELMA para elevar el pH gástrico. En el mismo estudio, la co-administración de LOKELMA y ciclosporina no mostró una interacción clínicamente significativa.
Figura 2: Efectos de LOKELMA 10 g o 15 g sobre las Exposiciones Farmacocinéticas de Otros Medicamentos Administrados por Vía Oral
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
Las siguientes pruebas de potencial mutagénico del ciclosilicato de zirconio sódico fueron negativas: (1) la prueba de Ames (S. typhimurium y E. coli); (2) ensayo de aberración cromosómica en células de ovario de hámster chino (CHO); y (3) ensayo de micronúcleos in vivo en ratas. Dado que el ciclosilicato de zirconio no es genotóxico, no se absorbe desde el tracto gastrointestinal y no causó alteraciones gastrointestinales locales en un estudio de toxicidad crónica en perros, no se consideró necesario realizar estudios de carcinogenicidad en animales para evaluar el potencial tumorigénico del ciclosilicato de zirconio sódico.
La fertilidad en ratas machos y hembras se ha evaluado con dosis de hasta una dosis equivalente humana (HED) de 58 g por día (la dosis máxima factible) sin efectos adversos.
14 ESTUDIOS CLÍNICOS
14.1 Estudio 1
La efectividad de LOKELMA en disminuir el potasio sérico se demostró en un ensayo clínico de dos partes, doble ciego, aleatorizado, controlado con placebo (NCT01737697) en pacientes con hiperpotasemia (5 a 6.5 mEq/L, potasio promedio 5.3 mEq/L), Estudio 1.
En la primera fase del ensayo (la fase aguda), 753 pacientes fueron aleatorizados para recibir una de cuatro dosis de LOKELMA (1.25, 2.5, 5 o 10 g) o placebo, administrados tres veces al día durante las primeras 48 horas con las comidas.
La edad media de los pacientes fue de 66 años, el 59% de los pacientes eran hombres y el 86% eran caucásicos. Aproximadamente el 60% de los pacientes tenían enfermedad renal crónica, el 10% tenía insuficiencia cardíaca, el 62% tenía diabetes mellitus y el 67% tomaban inhibidores del sistema renina-angiotensina-aldosterona (RAAS) al inicio del estudio.
El criterio principal de valoración en la fase aguda fue la diferencia en la tasa de cambio exponencial en los niveles de potasio sérico durante las primeras 48 horas de tratamiento con el medicamento del estudio, comparando pacientes tratados con placebo y pacientes tratados con LOKELMA. El estudio cumplió su criterio principal de valoración demostrando una mayor reducción en los niveles de potasio sérico para los grupos de dosis de 2.5, 5 y 10 g (tres veces al día) en comparación con el grupo placebo (p<0.001). Como se muestra en la Tabla 1 para el criterio secundario de cambio de potasio desde el inicio, LOKELMA mostró reducciones dependientes de la dosis en el potasio sérico a 2.5, 5 y 10 g. En pacientes a los que se administró 10 g TID, la reducción media de potasio sérico fue de -0.7 mEq/L a las 48 horas. Los pacientes con niveles más altos de potasio al inicio tuvieron una respuesta mayor a LOKELMA. LOKELMA fue eficaz para disminuir los niveles de potasio en pacientes con enfermedad renal crónica, insuficiencia cardíaca, diabetes mellitus y en aquellos que tomaban terapia con inhibidores de RAAS.
[La Tabla 1 se mantiene sin traducir]
Los pacientes que lograron un nivel de potasio entre 3.5 y 5 mEq/L después de recibir LOKELMA durante la fase aguda fueron reasignados aleatoriamente a recibir placebo una vez al día o 1.25, 2.5, 5 o 10 g de LOKELMA una vez al día durante 12 días junto con el desayuno.
El criterio principal de valoración en la fase de mantenimiento fue la diferencia en la tasa de cambio exponencial en los niveles de potasio sérico durante el intervalo de tratamiento de 12 días, comparando pacientes que recibían LOKELMA y pacientes que recibían placebo. El estudio cumplió el criterio principal de eficacia en las dosis de 5 y 10 g en comparación con sus respectivos grupos placebo (p<0.01 y p<0.001).
14.2 Estudio 2
La eficacia de LOKELMA también se demostró en un ensayo de dos partes con una fase aguda de etiqueta abierta y una fase de retirada aleatoria, doble ciego, controlada con placebo de un mes de duración (Estudio 2; NCT02088073).
En la fase aguda de etiqueta abierta del Estudio 2, 258 pacientes con hiperpotasemia (línea base promedio 5.6 mEq/L, rango 5.1 a 7.4 mEq/L) recibieron 10 g de LOKELMA administrados tres veces al día con las comidas durante 48 horas. Como se muestra en la Figura 3, izquierda, los niveles promedio de potasio sérico disminuyeron de 5.6 a 4.5 mEq/L durante el tratamiento con LOKELMA en la fase aguda.
Después de la fase aguda del estudio, hubo una fase de retirada aleatorizada, doble ciego, donde los pacientes que lograron niveles de potasio entre 3.5 y 5 mEq/L fueron aleatorizados a una de tres dosis de LOKELMA administradas una vez al día durante 28 días, o placebo justo antes del desayuno. Del 92% de los pacientes inscritos en la fase aguda, lograron un nivel de potasio dentro de este rango y fueron inscritos en la segunda fase del ensayo.
El criterio principal de valoración en la fase de retirada aleatorizada fue el valor medio de potasio sérico durante el período del Día 8 al Día 29, comparando pacientes tratados con LOKELMA y pacientes tratados con placebo. Las tres dosis (5, 10 y 15 g) de LOKELMA una vez al día mantuvieron los niveles medios de potasio más bajos que el placebo (el potasio sérico medio fue 4.8, 4.5 y 4.4 mEq/L para los grupos de dosis de 5, 10 y 15 g, respectivamente, vs. 5.1 mEq/L en el grupo placebo, p≤0.001 para todas las dosis, Figura 3, derecha). Una mayor proporción de pacientes tuvo niveles medios de potasio sérico en el rango normal (3.5 a 5 mEq/L) mientras tomaban LOKELMA que mientras tomaban placebo (80%, 90% y 94% en las dosis de 5, 10 y 15 g, respectivamente, vs. 46% en placebo).
Figura 3: Estudio 2 – Niveles medios de potasio sérico en las fases aguda y de retirada aleatorizada
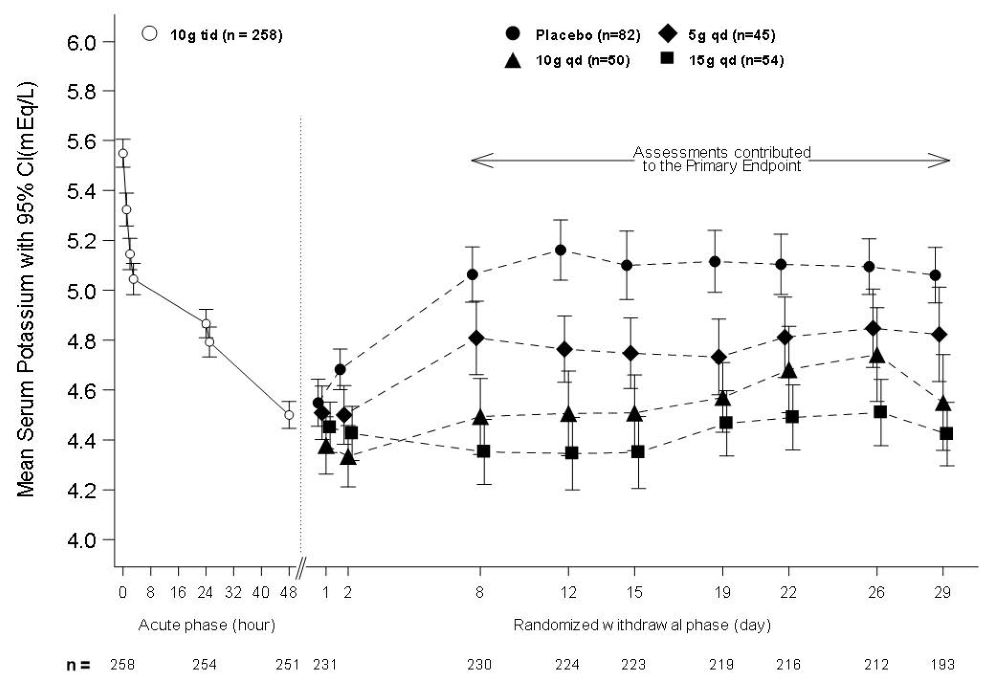
La población de intención de tratar incluye sujetos con al menos una medición válida de potasio sérico en o después del Día 8
14.3 Estudio de extensión de once meses
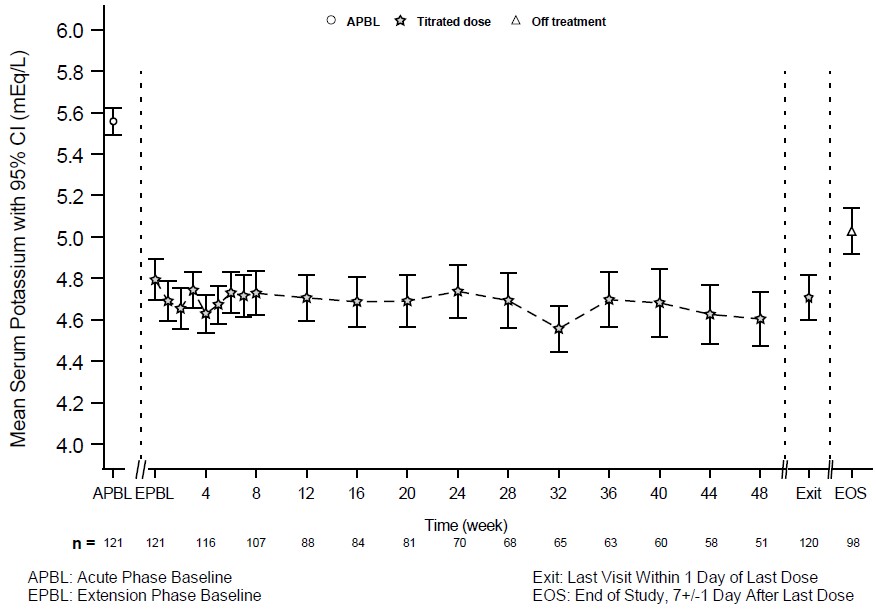
Los pacientes que completaron la fase de retiro aleatorizada de 28 días tuvieron la opción de continuar el tratamiento con LOKELMA, tomado justo antes del desayuno, en una fase de extensión abierta durante hasta 11 meses (n=123; NCT02107092). La Figura 4 muestra que el efecto del tratamiento sobre el potasio sérico se mantuvo durante la terapia continua.
Figura 4: Fase de extensión abierta de 11 meses del Estudio 2 – Potasio sérico promedio (mEq/L)
14.4 Estudio 3
LOKELMA fue evaluado en un estudio abierto de 12 meses en 751 pacientes hipercalémicos (NCT02163499). El nivel basal promedio de potasio en este estudio fue de 5.6 mEq/L. Después de la fase aguda de tratamiento con LOKELMA 10 g tres veces al día, los pacientes que lograron normokalemia (3.5-5.0 mEq/L) dentro de las 72 horas (n=746; 99%) ingresaron a la fase de mantenimiento. Para el tratamiento de mantenimiento, la dosificación inicial de LOKELMA fue de 5 g una vez al día y se ajustó a un mínimo de 5 g cada dos días hasta un máximo de 15 g una vez al día, según el nivel de potasio sérico. El efecto del tratamiento sobre el potasio sérico se mantuvo durante la terapia continua.
14.5 Estudio 4
La efectividad de LOKELMA para disminuir el potasio sérico se estudió en un ensayo doble ciego, controlado con placebo, de 196 pacientes en hemodiálisis crónica (edad promedio 58 años, rango 20 a 86 años) con hipercalemia pre-diálisis persistente (potasio basal promedio 5.8 mEq/L) que fueron aleatorizados para recibir LOKELMA 5 g o placebo una vez al día en los días sin diálisis (NCT03303521). Durante el período de ajuste de dosis (4 semanas iniciales), la dosis se ajustó semanalmente en incrementos de 5 g hasta 15 g una vez al día según la medición de potasio sérico pre-diálisis después del intervalo interdiálisis largo para lograr un nivel de potasio sérico pre-diálisis entre 4.0-5.0 mEq/L. La dosis alcanzada al final del período de ajuste de dosis se mantuvo durante las 4 semanas posteriores del período de evaluación.
El criterio principal en el ensayo fue la proporción de respondedores, definidos como pacientes que mantuvieron un potasio sérico pre-diálisis entre 4.0 y 5.0 mEq/L en al menos 3 de 4 tratamientos de diálisis después del intervalo interdiálisis largo y que no recibieron terapia de rescate durante el período de evaluación. Una mayor proporción de pacientes fueron respondedores en el brazo de LOKELMA en comparación con el placebo (41% vs 1%, respectivamente; p<0.001). El efecto del tratamiento sobre los niveles promedio de potasio sérico pre-diálisis se mantuvo durante el tratamiento continuo. Los niveles promedio de potasio sérico pre-diálisis durante el estudio se presentan en la Figura 5.
Figura 5: Niveles promedio de potasio sérico pre-diálisis a lo largo del tiempo en pacientes en hemodiálisis crónica
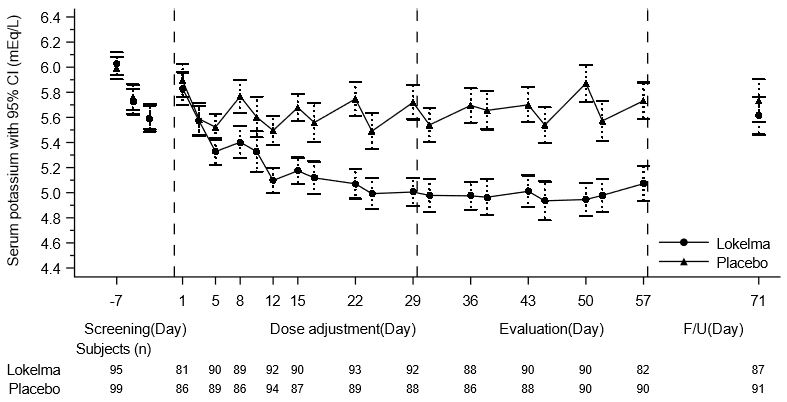
F/U – período de seguimiento
Las barras de error mostradas corresponden a intervalos de confianza del 95%.
n = Número de pacientes con mediciones de potasio no faltantes en una visita en particular.
16 PRESENTACIÓN/ALMACENAMIENTO Y MANIPULACIÓN
LOKELMA (ciclosilicato de circonio sódico) para suspensión oral se suministra como un polvo blanco a gris en paquetes forrados con lámina de aluminio de la siguiente manera:
|
LOKELMA (gramos) |
Paquete individual |
Caja de 11 paquetes |
Caja de 30 paquetes |
|
5 |
NDC 0310-1105-01 |
NDC 0310-1105-39 |
NDC 0310-1105-30 |
|
10 |
NDC 0310-1110-01 |
NDC 0310-1110-39 |
NDC 0310-1110-30 |
Almacenamiento y manipulación
Almacenar LOKELMA a 15°C-30°C (59°F-86°F).
17 INFORMACIÓN PARA ACONSEJAR AL PACIENTE
Dosificación
Instruir al paciente sobre cómo reconstituir LOKELMA para su administración. Informar al paciente que es necesario beber la dosis completa [ver Dosificación y Administración (2.3)].
Instruir a los pacientes en diálisis que experimenten una enfermedad aguda (p. ej., disminución de la ingesta oral de alimentos o líquidos, diarrea) a contactar con el proveedor de atención médica. La dosis de Lokelma puede necesitar ajustarse [ver Advertencias y Precauciones (5.3)].
Pruebas diagnósticas
Aconsejar a los pacientes que notifiquen a su médico antes de una radiografía abdominal [ver Advertencias y Precauciones (5.4)].
Interacciones con medicamentos
Aconsejar a los pacientes que toman otros medicamentos orales separar la dosis de LOKELMA por al menos 2 horas (antes o después) [ver Interacciones con Medicamentos (7)].
Dieta
Aconsejar a los pacientes que ajusten la ingesta de sodio en la dieta, si corresponde [ver Advertencias y Precauciones (5.2)].
Patente de EE.UU. N.º: 6332985, 8808750, 8877255, 8802152, 9592253
© AstraZeneca 2024
Fabricado por: AstraZeneca Pharmaceuticals LP, Wilmington, DE 19850
PAQUETE/PANEL DE EXHIBICIÓN PRINCIPAL – 5 g
NDC 0310-1105-30 Contiene 30 paquetes
LOKELMA®
(ciclosilicato de zirconio y sodio)
para suspensión oral
5 g por paquete
Sólo con receta
AstraZeneca

PAQUETE/ETIQUETA PRINCIPAL DE EXHIBICIÓN – 10 g
NDC 0310-1110-30 Contiene 30 paquetes
LOKELMA®
(ciclosilicato de zirconio sódico)
para suspensión oral
10 g por paquete
Sólo con receta
AstraZeneca
