ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
Estos aspectos destacados no incluyen toda la información necesaria para usar LATUDA de forma segura y eficaz. Consulte la información completa de prescripción de LATUDA.
LATUDA (clorhidrato de lurasidona) comprimidos, para administración oral Aprobación inicial en EE. UU.: 2010
ADVERTENCIA: AUMENTO DE LA MORTALIDAD EN PACIENTES ANCIANOS CON PSICOSIS ASOCIADA A DEMENTIA; Y PENSAMIENTOS Y COMPORTAMIENTOS SUICIDAS
Consulte la información completa de la advertencia en recuadro.
Los pacientes ancianos con psicosis relacionada con la demencia tratados con fármacos antipsicóticos presentan un mayor riesgo de muerte. LATUDA no está aprobado para el tratamiento de pacientes con psicosis relacionada con la demencia (5.1).
Los antidepresivos aumentaron el riesgo de pensamientos y comportamientos suicidas en pacientes pediátricos y adultos jóvenes. Supervisar estrechamente la aparición de empeoramiento clínico y la aparición de pensamientos y comportamientos suicidas (5.2).
LATUDA es un antipsicótico atípico indicado para el tratamiento de:
Esquizofrenia en adultos y adolescentes (13 a 17 años) (1, 14.1)
Episodio depresivo asociado con el trastorno bipolar I (depresión bipolar) en adultos y pacientes pediátricos (10 a 17 años) como monoterapia (1, 14.2)
Episodio depresivo asociado con el trastorno bipolar I (depresión bipolar) en adultos como terapia complementaria con litio o valproato (1, 14.2)
POSOLOGÍA Y ADMINISTRACIÓN
LATUDA debe tomarse con alimentos (al menos 350 calorías). La administración con alimentos aumenta sustancialmente la absorción de LATUDA (2.3, 12.3).
Depresión bipolar – pacientes pediátricos (10 a 17 años) (2.2)
20 mg al día
20 mg a 80 mg al día
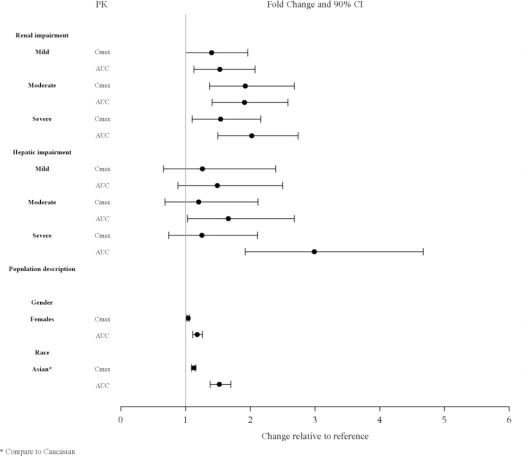
Insuficiencia renal moderada y grave: La dosis inicial recomendada es de 20 mg por día, y la dosis máxima recomendada es de 80 mg por día (2.4, 8.6).
Insuficiencia hepática moderada y grave: La dosis inicial recomendada es de 20 mg por día. La dosis máxima recomendada es de 80 mg por día en insuficiencia hepática moderada y de 40 mg por día en insuficiencia hepática grave (2.5, 8.7).
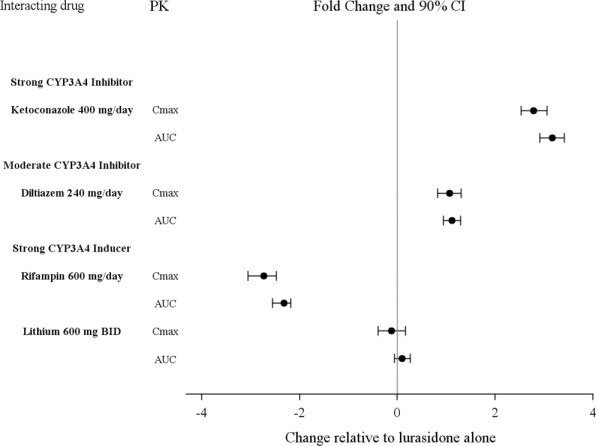
Uso concomitante de un inhibidor moderado del CYP3A4 (p. ej., diltiazem):
La dosis de LATUDA debe reducirse a la mitad del nivel de dosis original. La dosis inicial recomendada es de 20 mg por día. La dosis máxima recomendada es de 80 mg por día (2.6, 7.1).
Uso concomitante de un inductor moderado del CYP3A4: Puede ser necesario aumentar la dosis de LATUDA (2.6, 7.1).
Hipersensibilidad conocida a LATUDA o a cualquiera de los componentes de la formulación (4).
Uso concomitante con un inhibidor potente del CYP3A4 (p. ej., ketoconazol) (2.6, 4, 7.1).
Uso concomitante con un inductor potente del CYP3A4 (p. ej., rifampicina) (2.6, 4, 7.1).
ADVERTENCIAS Y PRECAUCIONES
Reacciones adversas cerebrovasculares en pacientes de edad avanzada con psicosis relacionada con la demencia: Mayor incidencia de eventos adversos cerebrovasculares (p. ej., accidente cerebrovascular, accidente isquémico transitorio) (5.3).
Síndrome neuroléptico maligno: Manejar con la interrupción inmediata y una estrecha vigilancia (5.4).
Discinesia tardía: Interrumpir si es clínicamente apropiado (5.5).
Cambios metabólicos: Controlar la hiperglucemia/diabetes mellitus, la dislipidemia y el aumento de peso (5.6).
Hiperprolactinemia: Pueden producirse elevaciones de prolactina (5.7).
Leucopenia, neutropenia y agranulocitosis: Realizar hemogramas completos (CBC) en pacientes con un recuento bajo de leucocitos (WBC) preexistente o antecedentes de leucopenia o neutropenia. Considere la posibilidad de interrumpir LATUDA si se produce una disminución clínicamente significativa de los leucocitos en ausencia de otros factores causales (5.8).
Hipotensión ortostática y síncope: Controlar la frecuencia cardíaca y la presión arterial y advertir a los pacientes con enfermedad cardiovascular o cerebrovascular conocida y riesgo de deshidratación o síncope (5.9).
REACCIONES ADVERSAS
Las reacciones adversas observadas comúnmente (incidencia ≥ 5% y al menos el doble de la tasa para el placebo) fueron (6.1):
Pacientes adultos con esquizofrenia: somnolencia, acatisia, síntomas extrapiramidales y náuseas
Pacientes adolescentes (13-17 años) con esquizofrenia: somnolencia, náuseas, acatisia, SEP (no acatisia), rinitis (solo 80 mg) y vómitos
Pacientes adultos con depresión bipolar: acatisia, síntomas extrapiramidales y somnolencia
Pacientes pediátricos (10-17 años) con depresión bipolar: náuseas, aumento de peso e insomnio.
Para notificar SOSPECHAS DE REACCIONES ADVERSAS, comuníquese con Sumitomo Pharma America, Inc. al 1-877-737-7226 o con la FDA al 1-800-FDA-1088 o www.fda.gov/medwatch.
USO EN POBLACIONES ESPECÍFICAS
Embarazo: Puede causar síntomas extrapiramidales y/o de abstinencia en neonatos con exposición en el tercer trimestre (8.1).
Ver 17 para INFORMACIÓN AL PACIENTE y Guía de Medicamentos.
Las secciones o subsecciones omitidas de la información completa de prescripción no se enumeran.
ADVERTENCIA RECUADRO
ADVERTENCIA: AUMENTO DE LA MORTALIDAD EN PACIENTES ANCIANOS CON PSICOSIS ASOCIADA A DEMENTIA; Y PENSAMIENTOS Y COMPORTAMIENTOS SUICIDAS
Aumento de la mortalidad en pacientes ancianos con psicosis asociada a demencia
Los pacientes ancianos con psicosis asociada a demencia tratados con fármacos antipsicóticos presentan un mayor riesgo de muerte. LATUDA no está aprobado para el tratamiento de pacientes con psicosis asociada a demencia [ver Advertencias y precauciones (5.1)].
Pensamientos y comportamientos suicidas
Los antidepresivos aumentaron el riesgo de pensamientos y comportamientos suicidas en niños y adultos jóvenes en estudios a corto plazo. Monitorice estrechamente a todos los pacientes tratados con antidepresivos para detectar cualquier empeoramiento clínico y la aparición de pensamientos y comportamientos suicidas [ver Advertencias y precauciones (5.2)].
1 INDICACIONES Y USO
LATUDA está indicado para:
Tratamiento de pacientes adultos y adolescentes (de 13 a 17 años) con esquizofrenia [ver Estudios Clínicos (14.1)].
Monoterapia para el tratamiento de pacientes adultos y pediátricos (de 10 a 17 años) con episodio depresivo mayor asociado con trastorno bipolar I (depresión bipolar) [ver Estudios Clínicos (14.2)].
Tratamiento adyuvante con litio o valproato en pacientes adultos con episodio depresivo mayor asociado con trastorno bipolar I (depresión bipolar) [ver Estudios Clínicos (14.2)].
2 DOSIS Y ADMINISTRACIÓN
2.1 Esquizofrenia
Adultos
La dosis inicial recomendada de LATUDA es de 40 mg una vez al día. No se requiere titulación de la dosis inicial. Se ha demostrado que LATUDA es eficaz en un rango de dosis de 40 mg por día a 160 mg por día [ver Estudios Clínicos (14.1)]. La dosis máxima recomendada es de 160 mg por día.
Adolescentes (13 – 17 años)
La dosis inicial recomendada de LATUDA es de 40 mg una vez al día. No se requiere titulación de la dosis inicial. Se ha demostrado que LATUDA es eficaz en un rango de dosis de 40 mg por día a 80 mg por día [ver Estudios Clínicos (14.1)]. La dosis máxima recomendada es de 80 mg por día.
2.2 Episodios depresivos asociados con el trastorno bipolar I
Adultos
La dosis inicial recomendada de LATUDA es de 20 mg una vez al día como monoterapia o como terapia adjunta con litio o valproato. No se requiere titulación de la dosis inicial. Se ha demostrado que LATUDA es eficaz en un rango de dosis de 20 mg por día a 120 mg por día como monoterapia o como terapia adjunta con litio o valproato [ver Estudios Clínicos (14.2)]. La dosis máxima recomendada, como monoterapia o como terapia adjunta con litio o valproato, es de 120 mg por día. En el estudio de monoterapia, el rango de dosis más alto (80 mg a 120 mg por día) no proporcionó una eficacia adicional, en promedio, en comparación con el rango de dosis más bajo (20 a 60 mg por día) [ver Estudios Clínicos (14.2)].
Pacientes pediátricos (10 – 17 años)
La dosis inicial recomendada de LATUDA es de 20 mg una vez al día como monoterapia. No se requiere titulación de la dosis inicial. La dosis se puede aumentar después de una semana según la respuesta clínica. Se ha demostrado que LATUDA es eficaz en un rango de dosis de 20 mg por día a 80 mg por día como monoterapia. Al final del estudio clínico, la mayoría de los pacientes (67%) recibieron 20 mg o 40 mg una vez al día [ver Estudios Clínicos (14.2)]. La dosis máxima recomendada es de 80 mg por día.
No se ha establecido la eficacia de LATUDA en el tratamiento de la manía asociada con el trastorno bipolar.
2.3 Información de administración
LATUDA debe tomarse con alimentos (al menos 350 calorías). La administración con alimentos aumenta sustancialmente la absorción de LATUDA. La administración con alimentos aumenta el AUC aproximadamente 2 veces y aumenta la Cmax aproximadamente 3 veces. En los estudios clínicos, LATUDA se administró con alimentos [ver Farmacología Clínica (12.3)].
La eficacia de LATUDA para uso a largo plazo, es decir, durante más de 6 semanas, no se ha establecido en estudios controlados. Por lo tanto, el médico que opte por usar LATUDA durante períodos prolongados debe reevaluar periódicamente la utilidad a largo plazo del medicamento para el paciente individual [ver Dosis y Administración (2.1 y 2.2)].
2.4 Modificaciones de la dosis para insuficiencia renal
Se recomienda el ajuste de la dosis en pacientes con insuficiencia renal moderada (aclaramiento de creatinina: 30 a <50 mL/min) y grave (aclaramiento de creatinina <30 mL/min). La dosis inicial recomendada es de 20 mg por día. La dosis en estos pacientes no debe exceder los 80 mg por día [ver Uso en poblaciones específicas (8.6)].
2.5 Modificaciones de la dosis para insuficiencia hepática
Se recomienda el ajuste de la dosis en pacientes con insuficiencia hepática moderada (puntuación de Child-Pugh = 7 a 9) y grave (puntuación de Child-Pugh = 10 a 15). La dosis inicial recomendada es de 20 mg por día. La dosis en pacientes con insuficiencia hepática moderada no debe exceder los 80 mg por día y la dosis en pacientes con insuficiencia hepática grave no debe exceder los 40 mg/día [ver Uso en poblaciones específicas (8.7)].
2.6 Modificaciones de la dosis debido a interacciones medicamentosas de inhibidores e inductores del CYP3A4
Uso concomitante con inhibidores del CYP3A4
LATUDA no debe usarse concomitantemente con un inhibidor potente del CYP3A4 (p. ej., ketoconazol, claritromicina, ritonavir, voriconazol, mibefradil, etc.) [ver Contraindicaciones (4)].
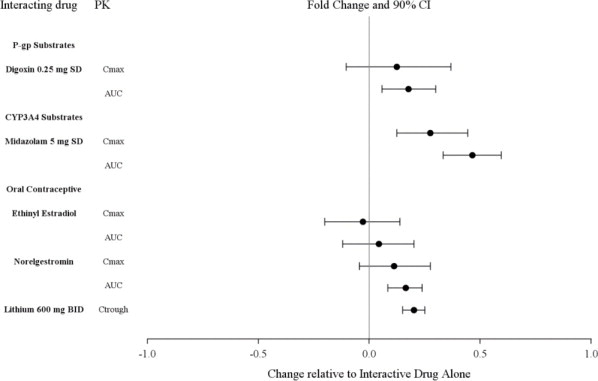
Si se prescribe LATUDA y se agrega un inhibidor moderado del CYP3A4 (p. ej., diltiazem, atazanavir, eritromicina, fluconazol, verapamilo, etc.) a la terapia, la dosis de LATUDA debe reducirse a la mitad del nivel de dosis original. De manera similar, si se prescribe un inhibidor moderado del CYP3A4 y se agrega LATUDA a la terapia, la dosis inicial recomendada de LATUDA es de 20 mg por día, y la dosis máxima recomendada de LATUDA es de 80 mg por día [ver Contraindicaciones (4), Interacciones medicamentosas (7.1)].
Se debe evitar el pomelo y el zumo de pomelo en pacientes que toman LATUDA, ya que estos pueden inhibir el CYP3A4 y alterar las concentraciones de LATUDA [ver Interacciones medicamentosas (7.1)].
Uso concomitante con inductores del CYP3A4
LATUDA no debe usarse concomitantemente con un inductor potente del CYP3A4 (p. ej., rifampicina, avasimibe, hierba de San Juan, fenitoína, carbamazepina, etc.) [ver Contraindicaciones (4); Interacciones medicamentosas (7.1)]. Si LATUDA se usa concomitantemente con un inductor moderado del CYP3A4, puede ser necesario aumentar la dosis de LATUDA después del tratamiento crónico (7 días o más) con el inductor del CYP3A4.
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
Los comprimidos de LATUDA están disponibles en las siguientes formas y colores (Tabla 1) con respectiva impresión en una sola cara.
Tabla 1: Presentaciones de comprimidos de LATUDA
Concentración del comprimido
Color/Forma del comprimido
Marcas del comprimido
20 mg
blanco a blanquecino redondo
L20
40 mg
blanco a blanquecino redondo
L40
60 mg
oblongo blanco a blanquecino
L60
80 mg
ovalado verde pálido
L80
120 mg
ovalado blanco a blanquecino
L120
4 CONTRAINDICACIONES
Hipersensibilidad conocida a hidrocloruro de lurasidona o a cualquier componente de la formulación. Se ha observado angioedema con lurasidona [ver Reacciones adversas (6.1)].
Inductores potentes del CYP3A4 (p. ej., rifampicina, avasimibe, hierba de San Juan, fenitoína, carbamazepina, etc.) [ver Interacciones medicamentosas (7.1)].
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Aumento de la Mortalidad en Pacientes Ancianos con Psicosis Relacionada con la Demencia
Los pacientes ancianos con psicosis relacionada con la demencia tratados con fármacos antipsicóticos presentan un mayor riesgo de muerte. Los análisis de 17 ensayos controlados con placebo (duración modal de 10 semanas), en gran parte en pacientes que tomaban fármacos antipsicóticos atípicos, revelaron un riesgo de muerte en los pacientes tratados con fármacos de entre 1,6 y 1,7 veces el riesgo de muerte en los pacientes tratados con placebo. En el transcurso de un ensayo controlado típico de 10 semanas, la tasa de mortalidad en los pacientes tratados con fármacos fue de aproximadamente el 4,5 %, en comparación con una tasa de aproximadamente el 2,6 % en el grupo placebo. Aunque las causas de muerte fueron variadas, la mayoría de las muertes parecieron ser de naturaleza cardiovascular (p. ej., insuficiencia cardíaca, muerte súbita) o infecciosa (p. ej., neumonía). LATUDA no está aprobado para el tratamiento de pacientes con psicosis relacionada con la demencia [véase Advertencia en recuadro, Advertencias y precauciones (5.3)].
5.2 Pensamientos y Comportamientos Suicidas en Pacientes Pediátricos y Adultos Jóvenes
En análisis agrupados de ensayos controlados con placebo de fármacos antidepresivos (ISRS y otras clases de antidepresivos) que incluyeron aproximadamente 77 000 pacientes adultos y más de 4400 pacientes pediátricos, la incidencia de pensamientos y comportamientos suicidas en pacientes pediátricos y adultos jóvenes fue mayor en los pacientes tratados con antidepresivos que en los pacientes tratados con placebo. Las diferencias entre el fármaco y el placebo en el número de casos de pensamientos y comportamientos suicidas por cada 1000 pacientes tratados se proporcionan en la Tabla 2.
No se produjeron suicidios en ninguno de los estudios pediátricos. Hubo suicidios en los estudios de adultos, pero el número no fue suficiente para llegar a ninguna conclusión sobre el efecto de los fármacos antidepresivos en el suicidio.
Tabla 2: Diferencias de Riesgo del Número de Casos de Pensamientos o Comportamientos Suicidas en los Ensayos Agrupados Controlados con Placebo de Antidepresivos en Pacientes Pediátricos y Adultos
Rango de Edad
Diferencia entre Fármaco y Placebo en el Número de Pacientes con Pensamientos o Comportamientos Suicidas por cada 1000 Pacientes Tratados
Aumentos en Comparación con Placebo
<18
14 pacientes adicionales
18-24
5 pacientes adicionales
Disminuciones en Comparación con Placebo
25-64
1 paciente menos
≥65
6 pacientes menos
Se desconoce si el riesgo de pensamientos y comportamientos suicidas en pacientes pediátricos y adultos jóvenes se extiende a un uso a más largo plazo, es decir, más allá de los cuatro meses. Sin embargo, hay evidencia sustancial de estudios de mantenimiento controlados con placebo en adultos con TDM que los antidepresivos retrasan la recurrencia de la depresión.
Monitoree a todos los pacientes tratados con antidepresivos para detectar el empeoramiento clínico y la aparición de pensamientos y comportamientos suicidas, especialmente durante los primeros meses de la terapia farmacológica y en momentos de cambios de dosis. Oriente a los familiares o cuidadores de los pacientes para que controlen los cambios de comportamiento y alerten al proveedor de atención médica. Considere cambiar el régimen terapéutico, incluso posiblemente suspender LATUDA, en pacientes cuya depresión empeora persistentemente o que experimentan pensamientos o comportamientos suicidas emergentes.
5.3 Reacciones adversas cerebrovasculares, incluido el accidente cerebrovascular en pacientes ancianos con psicosis relacionada con la demencia
En ensayos controlados con placebo con risperidona, aripiprazol y olanzapina en sujetos ancianos con demencia, hubo una mayor incidencia de reacciones adversas cerebrovasculares (accidentes cerebrovasculares y ataques isquémicos transitorios), incluidas las muertes, en comparación con los sujetos tratados con placebo. LATUDA no está aprobado para el tratamiento de pacientes con psicosis relacionada con la demencia [ver Advertencia en recuadro, Advertencias y precauciones (5.1)].
5.4 Síndrome neuroléptico maligno
Se ha informado un complejo de síntomas potencialmente mortal, a veces denominado síndrome neuroléptico maligno (SNM), en asociación con la administración de fármacos antipsicóticos, incluido LATUDA. Las manifestaciones clínicas del SNM son hipertermia, rigidez muscular, alteración del estado mental y evidencia de inestabilidad autonómica. Los signos adicionales pueden incluir creatina fosfocinasa elevada, mioglobinuria (rabdomiólisis) e insuficiencia renal aguda.
Si se sospecha SNM, suspenda inmediatamente LATUDA y proporcione tratamiento sintomático intensivo y monitoreo.
5.5 Discinesia tardía
La discinesia tardía es un síndrome que consiste en movimientos discinéticos involuntarios, potencialmente irreversibles, que pueden desarrollarse en pacientes tratados con fármacos antipsicóticos. Aunque la prevalencia del síndrome parece ser más alta entre las personas mayores, especialmente las mujeres mayores, es imposible confiar en las estimaciones de prevalencia para predecir, al inicio del tratamiento antipsicótico, qué pacientes probablemente desarrollarán el síndrome. Se desconoce si los productos farmacológicos antipsicóticos difieren en su potencial para causar discinesia tardía.
Se cree que el riesgo de desarrollar discinesia tardía y la probabilidad de que se vuelva irreversible aumentan a medida que aumentan la duración del tratamiento y la dosis acumulativa total de fármacos antipsicóticos administrados al paciente. Sin embargo, el síndrome puede desarrollarse, aunque con mucha menos frecuencia, después de períodos de tratamiento relativamente breves a dosis bajas o incluso puede surgir después de la interrupción del tratamiento.
El síndrome puede remitir, parcial o completamente, si se retira el tratamiento antipsicótico. Sin embargo, el tratamiento antipsicótico en sí mismo puede suprimir (o suprimir parcialmente) los signos y síntomas del síndrome y, por lo tanto, posiblemente enmascarar el proceso subyacente. Se desconoce el efecto que la supresión sintomática tiene sobre el curso a largo plazo del síndrome.
Dadas estas consideraciones, LATUDA debe prescribirse de la manera más probable para minimizar la aparición de discinesia tardía. El tratamiento antipsicótico crónico generalmente debe reservarse para pacientes que padecen una enfermedad crónica que (1) se sabe que responde a los fármacos antipsicóticos y (2) para quienes no hay tratamientos alternativos, igualmente efectivos pero potencialmente menos dañinos, disponibles o apropiados. En pacientes que requieren tratamiento crónico, se debe buscar la dosis más pequeña y la duración más corta del tratamiento que produzcan una respuesta clínica satisfactoria. La necesidad de tratamiento continuo debe reevaluarse periódicamente.
Si aparecen signos y síntomas de discinesia tardía en un paciente con LATUDA, se debe considerar la interrupción del fármaco. Sin embargo, algunos pacientes pueden requerir tratamiento con LATUDA a pesar de la presencia del síndrome.
5.6 Cambios metabólicos
Los fármacos antipsicóticos atípicos se han asociado con cambios metabólicos que pueden aumentar el riesgo cardiovascular/cerebrovascular. Estos cambios metabólicos incluyen hiperglucemia, dislipidemia y aumento de peso corporal. Si bien se ha demostrado que todos los fármacos de la clase producen algunos cambios metabólicos, cada fármaco tiene su propio perfil de riesgo específico.
Hiperglucemia y diabetes mellitus
Se ha informado hiperglucemia, en algunos casos extrema y asociada con cetoacidosis o coma hiperosmolar o muerte, en pacientes tratados con antipsicóticos atípicos. La evaluación de la relación entre el uso de antipsicóticos atípicos y las anomalías de la glucosa se complica por la posibilidad de un mayor riesgo de fondo de diabetes mellitus en pacientes con esquizofrenia y la creciente incidencia de diabetes mellitus en la población general. Dados estos factores de confusión, la relación entre el uso de antipsicóticos atípicos y los eventos adversos relacionados con la hiperglucemia no se comprende completamente. Sin embargo, los estudios epidemiológicos sugieren un mayor riesgo de eventos adversos relacionados con la hiperglucemia en pacientes tratados con antipsicóticos atípicos.
Los pacientes con un diagnóstico establecido de diabetes mellitus a quienes se les inicia tratamiento con antipsicóticos atípicos deben ser controlados regularmente para detectar un empeoramiento del control de la glucosa. Los pacientes con factores de riesgo de diabetes mellitus (p. ej., obesidad, antecedentes familiares de diabetes) que comienzan el tratamiento con antipsicóticos atípicos deben someterse a una prueba de glucosa en sangre en ayunas al comienzo del tratamiento y periódicamente durante el mismo. Cualquier paciente tratado con antipsicóticos atípicos debe ser controlado para detectar síntomas de hiperglucemia, incluyendo polidipsia, poliuria, polifagia y debilidad. Los pacientes que desarrollen síntomas de hiperglucemia durante el tratamiento con antipsicóticos atípicos deben someterse a una prueba de glucosa en sangre en ayunas. En algunos casos, la hiperglucemia se resolvió cuando se suspendió el antipsicótico atípico; sin embargo, algunos pacientes requirieron la continuación del tratamiento antidiabético a pesar de la suspensión del fármaco sospechoso.
Esquizofrenia
Adultos
Los datos agrupados de estudios de esquizofrenia a corto plazo controlados con placebo se presentan en la Tabla 3.
Tabla 3: Cambio en la glucosa en ayunas en estudios de esquizofrenia en adultos
LATUDA
Placebo
20 mg/día
40 mg/día
80 mg/día
120 mg/día
160 mg/día
Cambio medio desde el inicio (mg/dL)
n=680
n=71
n=478
n=508
n=283
n=113
Glucosa sérica
-0.0
-0.6
+2.6
-0.4
+2.5
+2.5
Proporción de pacientes con cambios a ≥ 126 mg/dL
Glucosa sérica (≥ 126 mg/dL)
8.3% (52/628)
11.7% (7/60)
12.7% ( 57/449)
6.8% (32/472)
10.0% (26/260)
5.6% (6/108)
En los estudios de esquizofrenia no controlados a largo plazo (principalmente estudios de extensión de etiqueta abierta), LATUDA se asoció con un cambio medio en la glucosa de +1,8 mg/dL en la semana 24 (n=355), +0,8 mg/dL en la semana 36 (n=299) y +2,3 mg/dL en la semana 52 (n=307).
Adolescentes
En estudios de adolescentes y adultos con esquizofrenia, los cambios en la glucosa en ayunas fueron similares. En el estudio a corto plazo, controlado con placebo, en adolescentes, los valores medios de glucosa sérica en ayunas fueron de -1,3 mg/dL para placebo (n=95), +0,1 mg/dL para 40 mg/día (n=90) y +1,8 mg/dL para 80 mg/día (n=92).
Depresión bipolar
Adultos
Monoterapia
Los datos del estudio de monoterapia para la depresión bipolar a corto plazo, de dosis flexible y controlado con placebo en adultos se presentan en la Tabla 4.
Tabla 4: Cambio en la glucosa en ayunas en el estudio de monoterapia para la depresión bipolar en adultos
LATUDA
Placebo
20 a 60 mg/día
80 a 120 mg/día
Cambio medio desde el inicio (mg/dL)
Los pacientes fueron aleatorizados a LATUDA con dosis flexible de 20 a 60 mg/día, LATUDA de 80 a 120 mg/día o placebo.
n=148
n=140
n=143
Glucosa sérica
+1,8
-0,8
+1,8
Proporción de pacientes con cambios a ≥ 126 mg/dL
Glucosa sérica (≥ 126 mg/dL)
4,3% (6/141)
2,2% (3/138)
6,4% (9/141)
En el estudio no controlado, abierto y a largo plazo de la depresión bipolar, los pacientes que recibieron LATUDA como monoterapia en el estudio a corto plazo y continuaron en el estudio a largo plazo tuvieron un cambio medio en la glucosa de +1,2 mg/dL en la semana 24 (n=129).
Terapia adjunta con litio o valproato
Los datos de los estudios de terapia adjunta para la depresión bipolar a corto plazo, de dosis flexible y controlados con placebo en adultos se presentan en la Tabla 5.
Tabla 5: Cambio en la glucosa en ayunas en los estudios de terapia adjunta para la depresión bipolar en adultos
LATUDA
Placebo
20 a 120 mg/día
Cambio medio desde el inicio (mg/dL)
Los pacientes fueron aleatorizados a LATUDA con dosis flexible de 20 a 120 mg/día o placebo como terapia adjunta con litio o valproato.
n=302
n=319
Glucosa sérica
-0,9
+1,2
Proporción de pacientes con cambios a ≥ 126 mg/dL
Glucosa sérica (≥ 126 mg/dL)
1,0% (3/290)
1,3% (4/316)
En el estudio de depresión bipolar a largo plazo, abierto y sin control, los pacientes que recibieron LATUDA como terapia complementaria con litio o valproato en el estudio a corto plazo y continuaron en el estudio a largo plazo, tuvieron un cambio medio en la glucosa de +1,7 mg/dL en la semana 24 (n=88).
Pacientes pediátricos (10 a 17 años)
En estudios de pacientes pediátricos de 10 a 17 años y adultos con depresión bipolar, los cambios en la glucosa en ayunas fueron similares. En el estudio de 6 semanas, controlado con placebo, de pacientes pediátricos con depresión bipolar, el cambio medio en la glucosa en ayunas fue de +1,6 mg/dL para LATUDA 20 a 80 mg/día (n=145) y -0,5 mg/dL para placebo (n=145).
Pacientes pediátricos (6 a 17 años)
En un estudio abierto de 104 semanas en pacientes pediátricos con esquizofrenia, depresión bipolar o trastorno autista, el 7% de los pacientes con una glucosa en ayunas basal normal experimentaron un cambio a alta al final del tratamiento mientras tomaban lurasidona.
Dislipidemia
Se han observado alteraciones indeseables en los lípidos en pacientes tratados con antipsicóticos atípicos.
Esquizofrenia
Adultos
Los datos agrupados de los estudios de esquizofrenia a corto plazo controlados con placebo se presentan en la Tabla 6.
Tabla 6: Cambio en los lípidos en ayunas en estudios de esquizofrenia en adultos
LATUDA
Placebo
20 mg/día
40 mg/día
80 mg/día
120 mg/día
160 mg/día
Cambio medio desde la línea base (mg/dL)
n=660
n=71
n=466
n=499
n=268
n=115
Colesterol total
-5,8
-12,3
-5,7
-6,2
-3,8
-6,9
Triglicéridos
-13,4
-29,1
-5,1
-13,0
-3,1
-10,6
Proporción de pacientes con cambios
Colesterol total (≥ 240 mg/dL)
5,3% (30/571)
13,8% (8/58)
6,2% (25/402)
5,3% (23/434)
3,8% (9/238)
4,0% (4/101)
Triglicéridos (≥ 200 mg/dL)
10,1% (53/526)
14,3% (7/49)
10,8% (41/379)
6,3% (25/400)
10,5% (22/209)
7,0% (7/100)
En los estudios de esquizofrenia no controlados a largo plazo (principalmente estudios de extensión de etiqueta abierta), LATUDA se asoció con un cambio medio en el colesterol total y los triglicéridos de -3,8 (n=356) y -15,1 (n=357) mg/dL en la semana 24, -3,1 (n=303) y -4,8 (n=303) mg/dL en la semana 36 y -2,5 (n=307) y -6,9 (n=307) mg/dL en la semana 52, respectivamente.
Adolescentes
En el estudio a corto plazo, controlado con placebo, en adolescentes, los valores medios del colesterol sérico en ayunas fueron -9,6 mg/dL para placebo (n=95), -4,4 mg/dL para 40 mg/día (n=89) y +1,6 mg/dL para 80 mg/día (n=92), y los valores medios de triglicéridos séricos en ayunas fueron +0,1 mg/dL para placebo (n=95), -0,6 mg/dL para 40 mg/día (n=89) y +8,5 mg/dL para 80 mg/día (n=92).
Depresión Bipolar
Adultos
Monoterapia
Los datos del estudio de depresión bipolar en adultos a corto plazo, con dosis flexible, controlado con placebo y monoterapia se presentan en Tabla 7.
Tabla 7: Cambio en los lípidos en ayunas en el estudio de depresión bipolar en adultos con monoterapia
LATUDA
Placebo
20 a 60 mg/día
80 a 120 mg/día
Cambio medio desde el inicio (mg/dL)
Los pacientes fueron aleatorizados a LATUDA con dosis flexible de 20 a 60 mg/día, LATUDA de 80 a 120 mg/día o placebo
n=147
n=140
n=144
Colesterol total
-3,2
+1,2
-4,6
Triglicéridos
+6,0
+5,6
+0,4
Proporción de pacientes con cambios
Colesterol total (≥ 240 mg/dL)
4,2% (5/118)
4,4% (5/113)
4,4% (5/114)
Triglicéridos (≥ 200 mg/dL)
4,8% (6/126)
10,1% (12/119)
9,8% (12/122)
En el estudio de depresión bipolar a largo plazo, abierto y sin control, los pacientes que recibieron LATUDA como monoterapia a corto plazo y continuaron en el estudio a largo plazo tuvieron un cambio medio en el colesterol total y los triglicéridos de -0.5 mg/dL (n=130) y -1.0 mg/dL (n=130) en la semana 24, respectivamente.
Terapia adjunta con litio o valproato
Los datos de los estudios de depresión bipolar de terapia adjunta a corto plazo, dosis flexible, controlados con placebo en adultos se presentan en Tabla 8.
Tabla 8: Cambio en los lípidos en ayunas en los estudios de depresión bipolar con terapia adjunta en adultos
LATUDA
Placebo
20 to 120 mg/day
Cambio medio desde el inicio (mg/dL)
Los pacientes fueron aleatorizados a LATUDA con dosis flexible de 20 a 120 mg/día o placebo como terapia adjunta con litio o valproato.
n=303
n=321
Colesterol total
-2.9
-3.1
Triglicéridos
-4.6
+4.6
Proporción de pacientes con cambios
Colesterol total (≥ 240 mg/dL)
5.7% (15/263)
5.4% (15/276)
Triglicéridos (≥ 200 mg/dL)
8.6% (21/243)
10.8% (28/260)
En el estudio de depresión bipolar a largo plazo, abierto y sin control, los pacientes que recibieron LATUDA como terapia complementaria con litio o valproato en el estudio a corto plazo y continuaron en el estudio a largo plazo, tuvieron un cambio medio en el colesterol total y los triglicéridos de -0.9 (n=88) y +5.3 (n=88) mg/dL en la semana 24, respectivamente.
Pacientes pediátricos (10 a 17 años)
En el estudio de depresión bipolar de 6 semanas, controlado con placebo, con pacientes pediátricos de 10 a 17 años, el cambio medio en el colesterol en ayunas fue de -6.3 mg/dL para LATUDA 20 a 80 mg/día (n=144) y -1.4 mg/dL para placebo (n=145), y el cambio medio en los triglicéridos en ayunas fue de -7.6 mg/dL para LATUDA 20 a 80 mg/día (n=144) y +5.9 mg/dL para placebo (n=145).
Pacientes pediátricos (6 a 17 años)
En un estudio abierto de 104 semanas de pacientes pediátricos con esquizofrenia, depresión bipolar o trastorno autista, se informaron cambios en el colesterol en ayunas basal de normal a alto al final del estudio en el 12% (colesterol total), 3% (colesterol LDL), y cambios en la línea de base de normal a bajo en el 27% (colesterol HDL) de los pacientes que tomaban lurasidona. De los pacientes con triglicéridos en ayunas basales normales, el 12% experimentó cambios a altos.
Aumento de peso
Se ha observado aumento de peso con el uso de antipsicóticos atípicos. Se recomienda la monitorización clínica del peso.
Esquizofrenia
Adultos
Los datos agrupados de los estudios de esquizofrenia a corto plazo controlados con placebo se presentan en Tabla 9. El aumento de peso medio fue de +0.43 kg para los pacientes tratados con LATUDA en comparación con -0.02 kg para los pacientes tratados con placebo. El cambio de peso desde la línea de base para olanzapina fue de +4.15 kg y para quetiapina de liberación prolongada fue de +2.09 kg en los Estudios 3 y 5 [ver Estudios clínicos (14.1)], respectivamente. La proporción de pacientes con un aumento ≥7% en el peso corporal (al final del estudio) fue del 4.8% para los pacientes tratados con LATUDA y del 3.3% para los pacientes tratados con placebo.
Tabla 9: Cambio medio de peso (kg) desde la línea de base en estudios de esquizofrenia en adultos
LATUDA
Placebo (n=696)
20 mg/día (n=71)
40 mg/día (n=484)
80 mg/día (n=526)
120 mg/día (n=291)
160 mg/día (n=114)
Todos los pacientes
-0.02
-0.15
+0.22
+0.54
+0.68
+0.60
En los estudios de esquizofrenia a largo plazo no controlados (principalmente estudios de extensión abiertos), LATUDA se asoció con un cambio medio de peso de -0.69 kg en la semana 24 (n=755), -0.59 kg en la semana 36 (n=443) y -0.73 kg en la semana 52 (n=377).
Adolescentes
Los datos del estudio de esquizofrenia en adolescentes a corto plazo controlado con placebo se presentan en Tabla 10. El cambio medio en el aumento de peso fue de +0.5 kg para los pacientes tratados con LATUDA en comparación con +0.2 kg para los pacientes tratados con placebo. La proporción de pacientes con un aumento ≥7% en el peso corporal (al final del estudio) fue del 3.3% para los pacientes tratados con LATUDA y del 4.5% para los pacientes tratados con placebo.
Tabla 10: Cambio medio de peso (kg) desde la línea de base en el estudio de esquizofrenia en adolescentes
Placebo (n=111)
LATUDA
40 mg/día (n=109)
80 mg/día (n=104)
Todos los pacientes
+0.2
+0.3
+0.7
Depresión Bipolar
Adultos
Monoterapia
Los datos del estudio de monoterapia para la depresión bipolar en adultos, a corto plazo, con dosis flexibles y controlado con placebo, se presentan en la Tabla 11. El cambio medio en el aumento de peso fue de +0,29 kg para los pacientes tratados con LATUDA en comparación con -0,04 kg para los pacientes tratados con placebo. La proporción de pacientes con un aumento de peso ≥7% (al final del estudio) fue del 2,4% para los pacientes tratados con LATUDA y del 0,7% para los pacientes tratados con placebo.
Tabla 11: Cambio medio de peso (kg) desde el inicio en el estudio de monoterapia para la depresión bipolar en adultos
LATUDA
Placebo (n=151)
20 a 60 mg/día (n=143)
80 a 120 mg/día (n=147)
Los pacientes fueron aleatorizados a LATUDA con dosis flexibles de 20 a 60 mg/día, LATUDA de 80 a 120 mg/día o placebo.
Todos los pacientes
-0,04
+0,56
+0,02
En el estudio de depresión bipolar a largo plazo, no controlado, abierto, los pacientes que recibieron LATUDA como monoterapia a corto plazo y continuaron en el estudio a largo plazo tuvieron un cambio medio de peso de -0,02 kg en la semana 24 (n=130).
Terapia adjunta con litio o valproato
Los datos de los estudios de terapia adjunta para la depresión bipolar en adultos, a corto plazo, con dosis flexibles y controlados con placebo, se presentan en la Tabla 12. El cambio medio en el aumento de peso fue de +0,11 kg para los pacientes tratados con LATUDA en comparación con +0,16 kg para los pacientes tratados con placebo. La proporción de pacientes con un aumento de peso ≥7% (al final del estudio) fue del 3,1% para los pacientes tratados con LATUDA y del 0,3% para los pacientes tratados con placebo.
Tabla 12: Cambio medio de peso (kg) desde el inicio en los estudios de terapia adjunta para la depresión bipolar en adultos
LATUDA
Placebo (n=307)
20 a 120 mg/día (n=327)
Los pacientes fueron aleatorizados a LATUDA con dosis flexibles de 20 a 120 mg/día o placebo como terapia adjunta con litio o valproato.
Todos los pacientes
+0,16
+0,11
En el estudio de depresión bipolar a largo plazo, no controlado, abierto, los pacientes tratados con LATUDA como terapia adjunta con litio o valproato a corto plazo y que continuaron en el estudio a largo plazo tuvieron un cambio medio de peso de +1,28 kg en la semana 24 (n=86).
Pacientes pediátricos (10 a 17 años)
Los datos del estudio de depresión bipolar de 6 semanas, controlado con placebo, en pacientes de 10 a 17 años se presentan en la Tabla 13. El cambio medio en el aumento de peso fue de +0,7 kg para los pacientes tratados con LATUDA en comparación con +0,5 kg para los pacientes tratados con placebo. La proporción de pacientes con un aumento de peso ≥7% (al final del estudio) fue del 4,0% para los pacientes tratados con LATUDA y del 5,3% para los pacientes tratados con placebo.
Tabla 13: Cambio medio de peso (kg) desde el inicio en el estudio de depresión bipolar en pacientes pediátricos (10 a 17 años)
LATUDA
Placebo (n=170)
20 a 80 mg/día (n=175)
Todos los pacientes
+0,5
+0,7
Pacientes pediátricos (6 a 17 años)
En un estudio abierto a largo plazo en el que participaron pacientes pediátricos con esquizofrenia, depresión bipolar o trastorno autista de tres ensayos controlados con placebo a corto plazo, el 54% (378/701) recibió lurasidona durante 104 semanas. El aumento medio de peso desde el inicio del estudio abierto hasta la semana 104 fue de 5,85 kg. Para ajustar el crecimiento normal, se derivaron puntuaciones z (medidas en desviaciones estándar [DE]), que se normalizan para el crecimiento natural de niños y adolescentes mediante comparaciones con los estándares de la población ajustados por edad y sexo. Un cambio en la puntuación z <0,5 DE se considera no clínicamente significativo. En este ensayo, el cambio medio en la puntuación z desde el inicio del estudio abierto hasta la semana 104 fue de -0,06 DE para el peso corporal y de -0,13 DE para el índice de masa corporal (IMC), lo que indica una desviación mínima de la curva normal para el aumento de peso.
5.7 Hiperprolactinemia
Al igual que con otros fármacos que antagonizan los receptores D2 de dopamina, LATUDA eleva los niveles de prolactina.
La hiperprolactinemia puede suprimir la GnRH hipotalámica, lo que resulta en una reducción de la secreción de gonadotropina pituitaria. Esto, a su vez, puede inhibir la función reproductiva al deteriorar la esteroidogénesis gonadal tanto en pacientes mujeres como hombres. Se han notificado galactorrea, amenorrea, ginecomastia e impotencia con compuestos que elevan la prolactina. La hiperprolactinemia prolongada, cuando se asocia con hipogonadismo, puede provocar una disminución de la densidad ósea tanto en pacientes mujeres como hombres [véase Reacciones adversas (6)].
Los experimentos de cultivo de tejidos indican que aproximadamente un tercio de los cánceres de mama humanos dependen de la prolactina in vitro, un factor de importancia potencial si se considera la prescripción de estos fármacos en una paciente con cáncer de mama previamente detectado. Como es común con los compuestos que aumentan la liberación de prolactina, se observó un aumento en la neoplasia de la glándula mamaria en un estudio de carcinogenicidad realizado con lurasidona en ratas y ratones [véase Toxicología no clínica (13)]. Los estudios epidemiológicos publicados han mostrado resultados inconsistentes al explorar la posible asociación entre la hiperprolactinemia y el cáncer de mama.
Esquizofrenia
Adultos
En estudios de esquizofrenia a corto plazo controlados con placebo, el cambio mediano desde el inicio hasta el punto final en los niveles de prolactina para los pacientes tratados con LATUDA fue de +0,4 ng/mL y de -1,9 ng/mL en los pacientes tratados con placebo. El cambio mediano desde el inicio hasta el punto final para los hombres fue de +0,5 ng/mL y para las mujeres de -0,2 ng/mL. Los cambios medianos de prolactina por dosis se muestran en la Tabla 14.
Tabla 14: Cambio mediano en la prolactina (ng/mL) desde el inicio en estudios de esquizofrenia en adultos
LATUDA
Placebo
20 mg/día
40 mg/día
80 mg/día
120 mg/día
160 mg/día
Todos los pacientes
-1,9 (n=672)
-1,1 (n=70)
-1,4 (n=476)
-0,2 (n=495)
+3,3 (n=284)
+3,3 (n=115)
Mujeres
-5,1 (n=200)
-0,7 (n=19)
-4,0 (n=149)
-0,2 (n=150)
+6,7 (n=70)
+7,1 (n=36)
Hombres
-1,3 (n=472)
-1,2 (n=51)
-0,7 (n=327)
-0,2 (n=345)
+3,1 (n=214)
+2,4 (n=79)
La proporción de pacientes con elevaciones de prolactina ≥5 veces el límite superior normal (LSN) fue del 2,8% para los pacientes tratados con LATUDA y del 1,0% para los pacientes tratados con placebo. La proporción de pacientes mujeres con elevaciones de prolactina ≥5 veces el LSN fue del 5,7% para las pacientes tratadas con LATUDA y del 2,0% para las pacientes mujeres tratadas con placebo. La proporción de pacientes varones con elevaciones de prolactina ≥5 veces el LSN fue del 1,6% y del 0,6% para los pacientes varones tratados con placebo.
En los estudios no controlados a largo plazo de esquizofrenia (principalmente estudios de extensión abiertos), LATUDA se asoció con un cambio mediano en la prolactina de -0,9 ng/mL en la semana 24 (n=357), -5,3 ng/mL en la semana 36 (n=190) y -2,2 ng/mL en la semana 52 (n=307).
Adolescentes
En el estudio de esquizofrenia en adolescentes a corto plazo y controlado con placebo, el cambio mediano desde el inicio hasta el final en los niveles de prolactina para los pacientes tratados con LATUDA fue de +1,1 ng/mL y de +0,1 ng/mL para los pacientes tratados con placebo. Para los pacientes tratados con LATUDA, el cambio mediano desde el inicio hasta el final para los varones fue de +1,0 ng/mL y para las mujeres de +2,6 ng/mL. Los cambios medianos de prolactina por dosis se muestran en Tabla 15.
Tabla 15: Cambio mediano en la prolactina (ng/mL) desde el inicio en el estudio de esquizofrenia en adolescentes
Placebo
LATUDA
40 mg/día
LATUDA
80 mg/día
Todos los pacientes
+0,10 (n=103)
+0,75 (n=102)
+1,20 (n=99)
Mujeres
+0,70 (n=39)
+0,60 (n=42)
+4,40 (n=33)
Varones
0,00 (n=64)
+0,75 (n=60)
+1,00 (n=66)
La proporción de pacientes con elevaciones de prolactina ≥5 veces el LSN fue del 0,5% para los pacientes tratados con LATUDA y del 1,0% para los pacientes tratados con placebo. La proporción de pacientes mujeres con elevaciones de prolactina ≥5 veces el LSN fue del 1,3% para las pacientes tratadas con LATUDA y del 0% para las pacientes mujeres tratadas con placebo. La proporción de pacientes varones con elevaciones de prolactina ≥5 veces el LSN fue del 0% para los pacientes tratados con LATUDA y del 1,6% para los pacientes varones tratados con placebo.
Depresión bipolar
Adultos
Monoterapia
El cambio mediano desde el inicio hasta el final en los niveles de prolactina, en el estudio de depresión bipolar en adultos a corto plazo, con dosis flexibles y controlado con placebo, fue de +1,7 ng/mL y +3,5 ng/mL con LATUDA 20 a 60 mg/día y 80 a 120 mg/día, respectivamente, en comparación con +0,3 ng/mL con los pacientes tratados con placebo. El cambio mediano desde el inicio hasta el final para los varones fue de +1,5 ng/mL y para las mujeres de +3,1 ng/mL. Los cambios medianos de prolactina por rango de dosis se muestran en Tabla 16.
Tabla 16: Cambio mediano en la prolactina (ng/mL) desde el inicio en el estudio de depresión bipolar en adultos con monoterapia
LATUDA
Placebo
20 a 60 mg/día
80 a 120 mg/día
Los pacientes fueron aleatorizados a LATUDA con dosis flexibles de 20 a 60 mg/día, LATUDA de 80 a 120 mg/día o placebo
Todos los pacientes
+0,3 (n=147)
+1,7 (n=140)
+3,5 (n=144)
Mujeres
0,0 (n=82)
+1,8 (n=78)
+5,3 (n=88)
Varones
+0,4 (n=65)
+1,2 (n=62)
+1,9 (n=56)
La proporción de pacientes con elevaciones de prolactina ≥5 veces el límite superior normal (LSN) fue del 0,4% para los pacientes tratados con LATUDA y del 0,0% para los pacientes tratados con placebo. La proporción de pacientes mujeres con elevaciones de prolactina ≥5 veces el LSN fue del 0,6% para las pacientes tratadas con LATUDA y del 0% para las pacientes tratadas con placebo. La proporción de pacientes varones con elevaciones de prolactina ≥5 veces el LSN fue del 0% y del 0% para los pacientes varones tratados con placebo.
En el estudio de depresión bipolar a largo plazo, no controlado, abierto, los pacientes que fueron tratados con LATUDA como monoterapia a corto plazo y continuaron en el estudio a largo plazo, tuvieron un cambio mediano en la prolactina de -1,15 ng/mL en la semana 24 (n=130).
Terapia adjunta con litio o valproato
El cambio mediano desde el inicio hasta el final en los niveles de prolactina, en los estudios de terapia adjunta para la depresión bipolar a corto plazo, de dosis flexible y controlados con placebo en adultos, fue de +2,8 ng/mL con LATUDA 20 a 120 mg/día en comparación con 0,0 ng/mL con pacientes tratados con placebo. El cambio mediano desde el inicio hasta el final para los varones fue de +2,4 ng/mL y para las mujeres de +3,2 ng/mL. Los cambios medianos de prolactina en todo el rango de dosis se muestran en Tabla 17.
Tabla 17: Cambio mediano en la prolactina (ng/mL) desde el inicio en los estudios de terapia adjunta para la depresión bipolar en adultos
LATUDA
Placebo
20 a 120 mg/día
Los pacientes fueron aleatorizados a LATUDA con dosis flexible de 20 a 120 mg/día o placebo como terapia adjunta con litio o valproato.
Todos los pacientes
0,0 (n=301)
+2,8 (n=321)
Mujeres
+0,4 (n=156)
+3,2 (n=162)
Varones
-0,1 (n=145)
+2,4 (n=159)
La proporción de pacientes con elevaciones de prolactina ≥5 veces el límite superior normal (LSN) fue del 0,0% para los pacientes tratados con LATUDA y del 0,0% para los pacientes tratados con placebo. La proporción de pacientes mujeres con elevaciones de prolactina ≥5 veces el LSN fue del 0% para las pacientes tratadas con LATUDA y del 0% para las pacientes tratadas con placebo. La proporción de pacientes varones con elevaciones de prolactina ≥5 veces el LSN fue del 0% y del 0% para los pacientes varones tratados con placebo.
En el estudio de depresión bipolar a largo plazo, no controlado, abierto, los pacientes que fueron tratados con LATUDA, como terapia adjunta con litio o valproato, a corto plazo y continuaron en el estudio a largo plazo, tuvieron un cambio mediano en la prolactina de -2,9 ng/mL en la semana 24 (n=88).
Pacientes pediátricos (10 a 17 años)
En el estudio de depresión bipolar de 6 semanas, controlado con placebo, con pacientes pediátricos de 10 a 17 años, el cambio mediano desde el inicio hasta el final en los niveles de prolactina para los pacientes tratados con LATUDA fue de +1,10 ng/mL y de +0,50 ng/mL para los pacientes tratados con placebo. Para los pacientes tratados con LATUDA, el cambio mediano desde el inicio hasta el final para los varones fue de +0,85 ng/mL y para las mujeres de +2,50 ng/mL. Los cambios medianos de prolactina se muestran en Tabla 18.
Tabla 18: Cambio mediano en la prolactina (ng/mL) desde el inicio en el estudio de depresión bipolar en pacientes pediátricos (10 a 17 años)
LATUDA
Placebo
20 a 80 mg/día
Todos los pacientes
+0,50 (n=157)
+1,10 (n=165)
Mujeres
+0,55 (n=78)
+2,50 (n=83)
Varones
+0,50 (n=79)
+0,85 (n=82)
La proporción de pacientes con elevaciones de prolactina ≥5x ULN fue del 0% para los pacientes tratados con LATUDA y del 0,6% para los pacientes tratados con placebo. La proporción de pacientes mujeres con elevaciones de prolactina ≥5x ULN fue del 0% para las pacientes tratadas con LATUDA y del 1,3% para las pacientes mujeres tratadas con placebo. Ningún paciente varón en los grupos de tratamiento con placebo o LATUDA presentó elevaciones de prolactina ≥5x ULN.
Pacientes pediátricos (6 a 17 años)
En un estudio abierto de 104 semanas de pacientes pediátricos con esquizofrenia, depresión bipolar o trastorno autista, los cambios medianos desde el inicio hasta el final en los niveles de prolactina sérica fueron de -0,20 ng/mL (todos los pacientes), -0,30 ng/mL (mujeres) y -0,05 ng/mL (varones). Las proporciones de pacientes con un nivel de prolactina marcadamente alto (≥5 veces el límite superior de la normalidad) en cualquier momento durante el tratamiento abierto fueron del 2% (todos los pacientes), del 3% (mujeres) y del 1% (varones).
Los eventos adversos entre las mujeres en este ensayo que son potencialmente relacionados con la prolactina incluyen galactorrea (0,6%). Entre los pacientes varones en este estudio, se informó disminución de la libido en un paciente (0,2%) y no hubo informes de impotencia, ginecomastia o galactorrea.
5.8 Leucopenia, neutropenia y agranulocitosis
Se ha notificado leucopenia/neutropenia durante el tratamiento con agentes antipsicóticos. Se ha notificado agranulocitosis (incluidos casos mortales) con otros agentes de la clase.
Los posibles factores de riesgo de leucopenia/neutropenia incluyen un recuento bajo preexistente de leucocitos (WBC) y antecedentes de leucopenia/neutropenia inducida por fármacos. Los pacientes con un recuento bajo preexistente de leucocitos o antecedentes de leucopenia/neutropenia inducida por fármacos deben controlar su hemograma completo (CBC) con frecuencia durante los primeros meses de terapia y se debe interrumpir LATUDA a la primera señal de disminución de los leucocitos, en ausencia de otros factores causales.
Los pacientes con neutropenia deben ser cuidadosamente monitoreados para detectar fiebre u otros síntomas o signos de infección y tratados rápidamente si ocurren dichos síntomas o signos. Los pacientes con neutropenia grave (recuento absoluto de neutrófilos < 1000/mm3) deben interrumpir LATUDA y controlar sus leucocitos hasta la recuperación.
5.9 Hipotensión ortostática y síncope
LATUDA puede causar hipotensión ortostática y síncope, quizás debido a su antagonismo del receptor α1-adrenérgico. Las reacciones adversas asociadas pueden incluir mareos, aturdimiento, taquicardia y bradicardia. En general, estos riesgos son mayores al comienzo del tratamiento y durante el aumento de la dosis. Los pacientes con mayor riesgo de estas reacciones adversas o con mayor riesgo de desarrollar complicaciones por hipotensión incluyen aquellos con deshidratación, hipovolemia, tratamiento con medicamentos antihipertensivos, antecedentes de enfermedad cardiovascular (p. ej., insuficiencia cardíaca, infarto de miocardio, isquemia o anomalías de conducción), antecedentes de enfermedad cerebrovascular, así como pacientes que no han recibido tratamiento con antipsicóticos. En tales pacientes, considere usar una dosis inicial más baja y una titulación más lenta, y controle los signos vitales ortostáticos.
La hipotensión ortostática, evaluada mediante la medición de los signos vitales, se definió mediante los siguientes cambios en los signos vitales: disminución de ≥ 20 mm Hg en la presión arterial sistólica y aumento de ≥10 lpm en el pulso de la posición sentada a la de pie o de la posición supina a la de pie.
Esquizofrenia
Adultos
La incidencia de hipotensión ortostática y síncope notificada como eventos adversos en estudios de esquizofrenia a corto plazo controlados con placebo fue (incidencia de LATUDA, incidencia de placebo): hipotensión ortostática [0,3% (5/1508), 0,1% (1/708)] y síncope [0,1% (2/1508), 0% (0/708)].
En estudios clínicos de esquizofrenia a corto plazo, la hipotensión ortostática, evaluada mediante signos vitales, se produjo con una frecuencia del 0,8% con LATUDA 40 mg, del 2,1% con LATUDA 80 mg, del 1,7% con LATUDA 120 mg y del 0,8% con LATUDA 160 mg en comparación con el 0,7% con placebo.
Adolescentes
La incidencia de hipotensión ortostática notificada como eventos adversos en el estudio de esquizofrenia en adolescentes a corto plazo controlado con placebo fue del 0,5% (1/214) en los pacientes tratados con LATUDA y del 0% (0/112) en los pacientes tratados con placebo. No se notificó ningún evento de síncope.
La hipotensión ortostática, evaluada mediante signos vitales, se produjo con una frecuencia del 0% con LATUDA 40 mg y del 2,9% con LATUDA 80 mg, en comparación con el 1,8% con placebo.
Depresión bipolar
Adultos
Monoterapia
En el estudio de depresión bipolar de monoterapia a corto plazo, de dosis flexible y controlado con placebo en adultos, no hubo eventos adversos notificados de hipotensión ortostática y síncope.
La hipotensión ortostática, evaluada mediante signos vitales, se produjo con una frecuencia del 0,6% con LATUDA 20 a 60 mg y del 0,6% con LATUDA 80 a 120 mg en comparación con el 0% con placebo.
Terapia adjunta con litio o valproato
En los estudios de terapia de depresión bipolar de terapia adjunta a corto plazo, de dosis flexible y controlada con placebo en adultos, no hubo eventos adversos notificados de hipotensión ortostática y síncope. La hipotensión ortostática, evaluada mediante signos vitales, se produjo con una frecuencia del 1,1% con LATUDA 20 a 120 mg en comparación con el 0,9% con placebo.
Pacientes pediátricos (10 a 17 años)
En el estudio de 6 semanas, controlado con placebo, de depresión bipolar en pacientes pediátricos de 10 a 17 años, no se reportaron eventos adversos de hipotensión ortostática o síncope.
La hipotensión ortostática, evaluada mediante signos vitales, se produjo con una frecuencia del 1,1% con LATUDA de 20 a 80 mg/día, en comparación con el 0,6% con placebo.
5.10 Caídas
LATUDA puede causar somnolencia, hipotensión postural, inestabilidad motora y sensorial, lo que puede llevar a caídas y, en consecuencia, fracturas u otras lesiones. Para pacientes con enfermedades, afecciones o medicamentos que puedan exacerbar estos efectos, realice evaluaciones completas del riesgo de caídas al iniciar el tratamiento antipsicótico y de forma recurrente para pacientes en tratamiento antipsicótico a largo plazo.
5.11 Convulsiones
Al igual que con otros fármacos antipsicóticos, LATUDA debe usarse con precaución en pacientes con antecedentes de convulsiones o con afecciones que disminuyan el umbral convulsivo, por ejemplo, la demencia de Alzheimer. Las condiciones que disminuyen el umbral convulsivo pueden ser más prevalentes en pacientes de 65 años o más.
Esquizofrenia
En estudios de corto plazo, controlados con placebo, de esquizofrenia en adultos, se produjeron convulsiones en el 0,1% (2/1508) de los pacientes tratados con LATUDA en comparación con el 0,1% (1/708) de los pacientes tratados con placebo.
Depresión bipolar
Monoterapia
En los estudios de 6 semanas, de dosis flexible, controlados con placebo, de monoterapia de depresión bipolar en adultos y pediátricos, ningún paciente experimentó convulsiones.
Terapia adyuvante con litio o valproato
En los estudios de corto plazo, de dosis flexible, controlados con placebo, de terapia adyuvante de depresión bipolar en adultos, ningún paciente experimentó convulsiones.
5.12 Potencial de deterioro cognitivo y motor
LATUDA, al igual que otros antipsicóticos, tiene el potencial de afectar el juicio, el pensamiento o las habilidades motoras. Advierta a los pacientes sobre el manejo de maquinaria peligrosa, incluidos vehículos de motor, hasta que estén razonablemente seguros de que el tratamiento con LATUDA no les afecta de manera adversa.
En estudios clínicos con LATUDA, la somnolencia incluyó: hipersomnia, hipersomnolencia, sedación y somnolencia.
Esquizofrenia
Adultos
En estudios de corto plazo, controlados con placebo, de esquizofrenia en adultos, la somnolencia fue reportada por el 17,0% (256/1508) de los pacientes tratados con LATUDA (15,5% LATUDA 20 mg, 15,6% LATUDA 40 mg, 15,2% LATUDA 80 mg, 26,5% LATUDA 120 mg y 8,3% LATUDA 160 mg/día) en comparación con el 7,1% (50/708) de los pacientes tratados con placebo.
Adolescentes
En el estudio de corto plazo, controlado con placebo, de esquizofrenia en adolescentes, la somnolencia fue reportada por el 14,5% (31/214) de los pacientes tratados con LATUDA (15,5% LATUDA 40 mg y 13,5% LATUDA 80 mg/día) en comparación con el 7,1% (8/112) de los pacientes tratados con placebo.
Depresión bipolar
Adultos
Monoterapia
En el estudio de corto plazo, de dosis flexible, controlado con placebo, de monoterapia de depresión bipolar en adultos, la somnolencia fue reportada por el 7,3% (12/164) y el 13,8% (23/167) con LATUDA de 20 a 60 mg y de 80 a 120 mg, respectivamente, en comparación con el 6,5% (11/168) de los pacientes tratados con placebo.
Terapia adyuvante con litio o valproato
En los estudios de corto plazo, de dosis flexible, controlados con placebo, de terapia adyuvante de depresión bipolar en adultos, la somnolencia fue reportada por el 11,4% (41/360) de los pacientes tratados con LATUDA de 20 – 120 mg en comparación con el 5,1% (17/334) de los pacientes tratados con placebo.
Pacientes pediátricos (de 10 a 17 años)
En el estudio de 6 semanas, controlado con placebo, de depresión bipolar en pacientes pediátricos de 10 a 17 años, la somnolencia fue reportada por el 11,4% (20/175) de los pacientes tratados con LATUDA de 20 a 80 mg/día en comparación con el 5,8% (10/172) de los pacientes tratados con placebo.
5.13 Desregulación de la temperatura corporal
La interrupción de la capacidad del cuerpo para reducir la temperatura corporal central se ha atribuido a los agentes antipsicóticos. Se recomienda una atención adecuada al prescribir LATUDA a pacientes que estarán expuestos a condiciones que pueden contribuir a un aumento de la temperatura corporal central, por ejemplo, hacer ejercicio vigoroso, exponerse a un calor extremo, recibir medicamentos concomitantes con actividad anticolinérgica o estar sujetos a deshidratación.
5.14 Activación de manía/hipomanía
El tratamiento antidepresivo puede aumentar el riesgo de desarrollar un episodio maníaco o hipomaníaco, particularmente en pacientes con trastorno bipolar. Monitoree a los pacientes para la aparición de dichos episodios.
En los estudios de monoterapia y terapia adjunta (con litio o valproato) en depresión bipolar en adultos, menos del 1% de los sujetos en los grupos de LATUDA y placebo desarrollaron episodios maníacos o hipomaníacos.
5.15 Disfagia
La dismotilidad esofágica y la aspiración se han asociado con el uso de fármacos antipsicóticos. La neumonía por aspiración es una causa frecuente de morbilidad y mortalidad en pacientes ancianos, en particular aquellos con demencia de Alzheimer avanzada. LATUDA y otros fármacos antipsicóticos deben usarse con precaución en pacientes con riesgo de neumonía por aspiración.
5.16 Reacciones adversas neurológicas en pacientes con enfermedad de Parkinson o demencia con cuerpos de Lewy
Se ha informado que los pacientes con enfermedad de Parkinson o demencia con cuerpos de Lewy tienen una mayor sensibilidad a la medicación antipsicótica. Las manifestaciones de esta mayor sensibilidad incluyen confusión, obnubilación, inestabilidad postural con caídas frecuentes, síntomas extrapiramidales y características clínicas compatibles con el síndrome neuroléptico maligno.
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas se discuten con más detalle en otras secciones del etiquetado:
Mayor mortalidad en pacientes ancianos con psicosis relacionada con la demencia [see Boxed Warning and Warnings and Precautions (5.1)]
Pensamientos y comportamientos suicidas [see Boxed Warning and Warnings and Precautions (5.2)]
Reacciones adversas cerebrovasculares, incluido accidente cerebrovascular, en pacientes ancianos con psicosis relacionada con la demencia [see Warnings and Precautions (5.3)]
Síndrome neuroléptico maligno [see Warnings and Precautions (5.4)]
Discinesia tardía [see Warnings and Precautions (5.5)]
Cambios metabólicos [see Warnings and Precautions (5.6)]
Hiperprolactinemia [see Warnings and Precautions (5.7)]
Leucopenia, neutropenia y agranulocitosis [see Warnings and Precautions (5.8)]
Hipotensión ortostática y síncope [see Warnings and Precautions (5.9)]
Reacciones adversas neurológicas en pacientes con enfermedad de Parkinson o demencia con cuerpos de Lewy [see Warnings and Precautions (5.16)]
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se realizan bajo condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un medicamento no se pueden comparar directamente con las tasas en los ensayos clínicos de otro medicamento y es posible que no reflejen las tasas observadas en la práctica clínica.
Adultos
La información a continuación se deriva de una base de datos integrada de estudios clínicos para LATUDA que consta de 3799 pacientes adultos expuestos a una o más dosis de LATUDA para el tratamiento de la esquizofrenia y la depresión bipolar en estudios controlados con placebo. Esta experiencia corresponde a una experiencia total de 1250.9 pacientes-año. Un total de 1106 pacientes tratados con LATUDA tuvieron al menos 24 semanas y 371 pacientes tratados con LATUDA tuvieron al menos 52 semanas de exposición.
Los eventos adversos durante la exposición al tratamiento del estudio se obtuvieron mediante consultas generales y experiencias adversas informadas voluntariamente, así como los resultados de exámenes físicos, signos vitales, ECG, pesos e investigaciones de laboratorio. Las experiencias adversas fueron registradas por investigadores clínicos utilizando su propia terminología. Para proporcionar una estimación significativa de la proporción de individuos que experimentan eventos adversos, los eventos se agruparon en categorías estandarizadas utilizando la terminología MedDRA.
Esquizofrenia
Los siguientes hallazgos se basan en los estudios precomercialización controlados con placebo a corto plazo para la esquizofrenia en adultos en los que se administró LATUDA en dosis diarias que oscilaron entre 20 y 160 mg (n=1508).
Reacciones adversas comúnmente observadas: Las reacciones adversas más comunes (incidencia ≥ 5% y al menos el doble de la tasa de placebo) en pacientes tratados con LATUDA fueron somnolencia, acatisia, síntomas extrapiramidales y náuseas.
Reacciones adversas asociadas con la interrupción del tratamiento: Un total de 9.5% (143/1508) de los pacientes tratados con LATUDA y 9.3% (66/708) de los pacientes tratados con placebo interrumpieron el tratamiento debido a reacciones adversas. No hubo reacciones adversas asociadas con la interrupción en sujetos tratados con LATUDA que fueran al menos del 2% y al menos el doble de la tasa de placebo.
Reacciones adversas que ocurren con una incidencia del 2% o más en pacientes tratados con LATUDA: Las reacciones adversas asociadas con el uso de LATUDA (incidencia del 2% o mayor, redondeada al porcentaje más cercano e incidencia de LATUDA mayor que placebo) que ocurrieron durante el tratamiento agudo (hasta 6 semanas en pacientes con esquizofrenia) se muestran en Tabla 19.
Tabla 19: Reacciones adversas en el 2% o más de los pacientes tratados con LATUDA y que ocurrieron con mayor incidencia que en los pacientes tratados con placebo en estudios de esquizofrenia a corto plazo en adultos
Nota: Cifras redondeadas al entero más cercano
* Somnolencia incluye términos de eventos adversos: hipersomnia, hipersomnolencia, sedación y somnolencia
** Los síntomas extrapiramidales incluyen términos de eventos adversos: bradicinesia, rigidez en rueda dentada, babeo, distonía, trastorno extrapiramidal, hipocinesia, rigidez muscular, crisis oculogira, distonía oromandibular, parkinsonismo, retraso psicomotor, espasmo lingual, tortícolis, temblor y trismus
Porcentaje de Pacientes que Reportaron la Reacción
LATUDA
Sistema Corporal o
Clase de Órgano
Placebo
(N=708)
(%)
20
mg/día
(N=71)
(%)
40
mg/día
(N=487)
(%)
80
mg/día
(N=538)
(%)
120
mg/día
(N=291)
(%)
160
mg/día
(N=121)
(%)
Todos
LATUDA
(N=1508)
(%)
Trastornos Gastrointestinales
Náuseas
5
11
10
9
13
7
10
Vómitos
6
7
6
9
9
7
8
Dispepsia
5
11
6
5
8
6
6
Hipersecreción Salivar
<1
1
1
2
4
2
2
Trastornos Musculoesqueléticos y del Tejido Conectivo
Dolor de Espalda
2
0
4
3
4
0
3
Trastornos del Sistema Nervioso
Somnolencia*
7
15
16
15
26
8
17
Akathisia
3
6
11
12
22
7
13
Trastorno extrapiramidal**
6
6
11
12
22
13
14
Mareo
2
6
4
4
5
6
4
Trastornos psiquiátricos
Insomnio
8
8
10
11
9
7
10
Agitación
4
10
7
3
6
5
5
Ansiedad
4
3
6
4
7
3
5
Inquietud
1
1
3
1
3
2
2
Reacciones adversas relacionadas con la dosis en los estudios de esquizofrenia
La acatisia y los síntomas extrapiramidales estuvieron relacionados con la dosis. La frecuencia de acatisia aumentó con la dosis hasta 120 mg/día (5.6% para LATUDA 20 mg, 10.7% para LATUDA 40 mg, 12.3% para LATUDA 80 mg y 22.0% para LATUDA 120 mg). Se reportó acatisia en el 7.4% (9/121) de los pacientes que recibieron 160 mg/día. La acatisia ocurrió en el 3.0% de los sujetos que recibieron placebo. La frecuencia de síntomas extrapiramidales aumentó con la dosis hasta 120 mg/día (5.6% para LATUDA 20 mg, 11.5% para LATUDA 40 mg, 11.9% para LATUDA 80 mg y 22.0% para LATUDA 120 mg).
Depresión bipolar (monoterapia)
Los siguientes hallazgos se basan en el estudio precomercialización, controlado con placebo, a corto plazo en adultos para la depresión bipolar en el que se administró LATUDA en dosis diarias que oscilaron entre 20 y 120 mg (n=331).
Reacciones adversas comúnmente observadas: Las reacciones adversas más comunes (incidencia ≥5%, en cualquier grupo de dosis y al menos el doble de la tasa de placebo) en pacientes tratados con LATUDA fueron acatisia, síntomas extrapiramidales, somnolencia, náuseas, vómitos, diarrea y ansiedad.
Reacciones adversas asociadas con la interrupción del tratamiento: Un total del 6.0% (20/331) de los pacientes tratados con LATUDA y el 5.4% (9/168) de los pacientes tratados con placebo interrumpieron el tratamiento debido a reacciones adversas. No hubo reacciones adversas asociadas con la interrupción en sujetos tratados con LATUDA que fueran al menos del 2% y al menos el doble de la tasa de placebo.
Reacciones adversas que ocurren con una incidencia del 2% o más en pacientes tratados con LATUDA: Las reacciones adversas asociadas con el uso de LATUDA (incidencia del 2% o mayor, redondeada al porcentaje más cercano e incidencia de LATUDA mayor que placebo) que ocurrieron durante el tratamiento agudo (hasta 6 semanas en pacientes con depresión bipolar) se muestran en la Tabla 20.
Tabla 20: Reacciones adversas en el 2% o más de los pacientes tratados con LATUDA y que ocurrieron con mayor incidencia que en los pacientes tratados con placebo en el estudio de depresión bipolar de monoterapia a corto plazo en adultos
Porcentaje de pacientes que reportan reacción
Sistema corporal o clase de órgano
Término derivado del diccionario
Placebo
(N=168)
(%)
LATUDA
20-60 mg/día
(N=164)
(%)
LATUDA
80-120 mg/día
(N=167)
(%)
Todo LATUDA
(N=331)
(%)
Nota: Cifras redondeadas al entero más cercano
*Síntomas extrapiramidales incluyen términos de eventos adversos: bradicinesia, rigidez en rueda dentada, babeo, distonía, trastorno extrapiramidal, reflejo glabelar anormal, hipocinesia, rigidez muscular, crisis oculógira, distonía oromandibular, parkinsonismo, retardo psicomotor, espasmo de la lengua, tortícolis, temblor y trismo.
** Somnolencia incluye términos de eventos adversos: hipersomnia, hipersomnolencia, sedación y somnolencia
Trastornos gastrointestinales
Náuseas
8
10
17
14
Boca seca
4
6
4
5
Vómitos
2
2
6
4
Diarrea
2
5
3
4
Infecciones e infestaciones
Nasofaringitis
1
4
4
4
Influenza
1
<1
2
2
Infección del tracto urinario
<1
2
1
2
Trastornos musculoesqueléticos y del tejido conjuntivo
Trastornos
Dolor de espalda
<1
3
<1
2
Trastornos del sistema nervioso
Síntomas extrapiramidales*
2
5
9
7
Acatisia
2
8
11
9
Somnolencia**
7
7
14
11
Trastornos psiquiátricos
Ansiedad
1
4
5
4
Reacciones adversas relacionadas con la dosis en el estudio de monoterapia:
En el estudio a corto plazo en adultos controlado con placebo (que incluyó rangos de dosis de LATUDA más bajas y más altas) [véase Estudios clínicos (14.2)] las reacciones adversas que ocurrieron con una incidencia superior al 5% en los pacientes tratados con LATUDA en cualquier grupo de dosis y mayor que el placebo en ambos grupos fueron náuseas (10,4%, 17,4%), somnolencia (7,3%, 13,8%), acatisia (7,9%, 10,8%) y síntomas extrapiramidales (4,9%, 9,0%) para LATUDA 20 a 60 mg/día y LATUDA 80 a 120 mg/día, respectivamente.
Depresión bipolar
Terapia adjunta con litio o valproato
Los siguientes hallazgos se basan en dos estudios precomercialización a corto plazo en adultos controlados con placebo para la depresión bipolar en los que se administró LATUDA en dosis diarias que oscilaban entre 20 y 120 mg como terapia adjunta con litio o valproato (n=360).
Reacciones adversas comúnmente observadas: Las reacciones adversas más comunes (incidencia ≥5% y al menos el doble de la tasa de placebo) en los sujetos tratados con LATUDA fueron acatisia y somnolencia.
Reacciones adversas asociadas con la interrupción del tratamiento: Un total del 5,8% (21/360) de los pacientes tratados con LATUDA y el 4,8% (16/334) de los pacientes tratados con placebo interrumpieron el tratamiento debido a reacciones adversas. No hubo reacciones adversas asociadas con la interrupción en los sujetos tratados con LATUDA que fueran al menos del 2% y al menos el doble de la tasa de placebo.
Reacciones adversas que ocurrieron con una incidencia del 2% o más en pacientes tratados con LATUDA: Las reacciones adversas asociadas con el uso de LATUDA (incidencia del 2% o mayor, redondeada al porcentaje más cercano e incidencia de LATUDA mayor que la del placebo) que ocurrieron durante la terapia aguda (hasta 6 semanas en pacientes con depresión bipolar) se muestran en la Tabla 21.
Tabla 21: Reacciones adversas en el 2% o más de los pacientes tratados con LATUDA y que ocurrieron con una incidencia mayor que en los pacientes tratados con placebo en los estudios de terapia adjunta a corto plazo en adultos con depresión bipolar
Porcentaje de pacientes que informaron la reacción
Sistema orgánico o clase de órgano
Término derivado del diccionario
Placebo
(N=334)
(%)
LATUDA 20 a 120 mg/día (N=360)
(%)
Nota: Las cifras se redondean al entero más cercano
** La somnolencia incluye términos de eventos adversos: hipersomnia, hipersomnolencia, sedación y somnolencia
Trastornos gastrointestinales
Náuseas
10
14
Vómitos
1
4
Trastornos generales
Fatiga
1
3
Infecciones e infestaciones
Nasofaringitis
2
4
Exploraciones complementarias
Aumento de peso
<1
3
Trastornos del metabolismo y de la nutrición
Aumento del apetito
1
3
Trastornos del sistema nervioso
Síntomas extrapiramidales*
9
14
Somnolencia**
5
11
Acatisia
5
11
Trastornos psiquiátricos
Inquietud
<1
4
Adolescentes
Esquizofrenia
Los siguientes hallazgos se basan en el estudio adolescente a corto plazo controlado con placebo para la esquizofrenia en el que se administró LATUDA en dosis diarias que oscilan entre 40 (N=110) y 80 mg (N=104).
Reacciones adversas observadas comúnmente: Las reacciones adversas más comunes (incidencia ≥5% y al menos el doble de la tasa de placebo) en pacientes adolescentes (13 a 17 años) tratados con LATUDA fueron somnolencia, náuseas, acatisia, síntomas extrapiramidales (no acatisia, solo 40 mg), vómitos y rinorrea/rinitis (solo 80 mg).
Reacciones adversas asociadas con la interrupción del tratamiento: La incidencia de interrupción debido a reacciones adversas entre los pacientes adolescentes (13 a 17 años) tratados con LATUDA y placebo fue del 4% y el 8%, respectivamente.
Reacciones adversas que ocurren con una incidencia del 2% o más en pacientes tratados con LATUDA: Las reacciones adversas asociadas con el uso de LATUDA (incidencia del 2% o mayor, redondeada al porcentaje más cercano e incidencia de LATUDA mayor que placebo) que ocurrieron durante la terapia aguda (hasta 6 semanas en pacientes adolescentes con esquizofrenia) se muestran en la Tabla 22.
Tabla 22: Reacciones adversas en 2% o más de los pacientes tratados con LATUDA y que ocurrieron con mayor incidencia que en los pacientes tratados con placebo en el estudio de esquizofrenia a corto plazo en adolescentes
Porcentaje de pacientes que reportaron la reacción
Sistema corporal o clase de órgano
Término derivado del diccionario
Placebo
(N=112)
LATUDA
40 mg/día
(N=110)
LATUDA
80 mg/día
(N=104)
Todos los LATUDA
(N=214)
Nota: Las cifras se redondean al entero más cercano
* La somnolencia incluye términos de eventos adversos: hipersomnia, sedación y somnolencia
** La infección viral incluye términos de eventos adversos: nasofaringitis, influenza, infección viral, infección del tracto respiratorio superior
*** La rinitis incluye términos de eventos adversos: rinitis, rinitis alérgica, rinorrea y congestión nasal
Trastornos gastrointestinales
Náuseas
3
13
14
14
Vómitos
2
8
6
8
Diarrea
1
3
5
4
Boca seca
0
2
3
2
Infecciones e infestaciones
Infección viral**
6
11
10
10
Rinitis***
2
<1
8
4
Dolor orofaríngeo
0
<1
3
2
Taquicardia
0
0
3
1
Trastornos del sistema nervioso
Somnolencia*
7
15
13
15
Acatisia
2
9
9
9
Mareo
1
5
5
5
Pacientes pediátricos (10 a 17 años)
Depresión bipolar
Los siguientes hallazgos se basan en el estudio de 6 semanas, controlado con placebo, para la depresión bipolar en pacientes pediátricos de 10 a 17 años en el que se administró LATUDA en dosis diarias que oscilan entre 20 y 80 mg (N=175).
Reacciones adversas comúnmente observadas: Las reacciones adversas más comunes (incidencia ≥5%, y al menos el doble de la tasa de placebo) en pacientes pediátricos (10 a 17 años) tratados con LATUDA fueron náuseas, aumento de peso e insomnio.
Reacciones adversas asociadas con la interrupción del tratamiento: La incidencia de interrupción debido a reacciones adversas entre los pacientes pediátricos de 10 a 17 años tratados con LATUDA y placebo fue del 2% y el 2%, respectivamente.
Reacciones adversas que ocurren con una incidencia del 2% o más en pacientes tratados con LATUDA: Las reacciones adversas asociadas con el uso de LATUDA (incidencia del 2% o mayor, redondeada al porcentaje más cercano e incidencia de LATUDA mayor que el placebo) que ocurrieron durante la terapia aguda (hasta 6 semanas en pacientes pediátricos con depresión bipolar) se muestran en la Tabla 23.
Tabla 23: Reacciones adversas en 2% o más de los pacientes tratados con LATUDA y que ocurrieron con mayor incidencia que en los pacientes tratados con placebo en el estudio de depresión bipolar de 6 semanas en pacientes pediátricos (10 a 17 años)
Porcentaje de pacientes que reportaron la reacción
Sistema orgánico o clase de órgano
Término derivado del diccionario
Placebo
(N=172)
LATUDA 20 a 80 mg/día (N=175)
Nota: Las cifras se redondean al entero más cercano
*Somnolencia incluye términos de eventos adversos: hipersomnia, hipersomnolencia, sedación y somnolencia
**EPS incluye términos de eventos adversos: acatisia, rigidez en rueda dentada, discinesia, distonía, hipercinesia, rigidez articular, rigidez muscular, espasmos musculares, rigidez musculoesquelética, crisis oculogírica, parkinsonismo, discinesia tardía y temblor
Trastornos gastrointestinales
Náuseas
6
16
Vómitos
4
6
Dolor abdominal superior
2
3
Diarrea
2
3
Dolor abdominal
1
3
Trastornos generales y afecciones en el sitio de administración
Fatiga
2
3
Investigaciones
Aumento de peso
2
7
Trastornos del metabolismo y la nutrición
Disminución del apetito
2
4
Trastornos del sistema nervioso
Somnolencia*
6
11
Síntomas extrapiramidales**
5
6
Mareos
5
6
Trastornos psiquiátricos
Insomnio
2
5
Sueños anormales
2
2
Trastornos respiratorios, torácicos y mediastínicos
Dolor orofaríngeo
2
2
Síntomas extrapiramidales
Esquizofrenia
Adultos
En los estudios de esquizofrenia controlados con placebo a corto plazo, para los pacientes tratados con LATUDA, la incidencia de eventos reportados relacionados con síntomas extrapiramidales (SEP), excluyendo acatisia e inquietud, fue del 13.5% y del 5.8% para los pacientes tratados con placebo. La incidencia de acatisia para los pacientes tratados con LATUDA fue del 12.9% y del 3.0% para los pacientes tratados con placebo. La incidencia de SEP por dosis se proporciona en Tabla 24.
Tabla 24: Incidencia de SEP en comparación con placebo en estudios de esquizofrenia en adultos
LATUDA
Término de evento adverso
Placebo
(N=708)
(%)
20 mg/día
(N=71)
(%)
40 mg/día
(N=487)
(%)
80 mg/día
(N=538)
(%)
120 mg/día
(N=291)
(%)
160 mg/día
(N=121)
(%)
Todos los eventos SEP
9
10
21
23
39
20
Todos los eventos SEP, excluyendo acatisia/inquietud
6
6
11
12
22
13
Nota: Cifras redondeadas al entero más cercano
* Distonía incluye los términos de eventos adversos: distonía, crisis oculógira, distonía oromandibular, espasmo de lengua, tortícolis y trismo.
** Parkinsonismo incluye los términos de eventos adversos: bradicinesia, rigidez en rueda dentada, babeo, trastorno extrapiramidal, hipocinesia, rigidez muscular, parkinsonismo, retraso psicomotor y temblor
Acatisia
3
6
11
12
22
7
Distonía*
<1
0
4
5
7
2
Parkinsonismo**
5
6
9
8
17
11
Inquietud
1
1
3
1
3
2
Adolescentes
En el estudio a corto plazo, controlado con placebo, de esquizofrenia en adolescentes, la incidencia de EPS, excluyendo eventos relacionados con acatisia, para pacientes tratados con LATUDA fue mayor en los grupos de tratamiento de 40 mg (10%) y 80 mg (7,7%) vs. placebo (3,6%); y la incidencia de eventos relacionados con acatisia para pacientes tratados con LATUDA fue del 8,9% vs. 1,8% para pacientes tratados con placebo. La incidencia de EPS por dosis se proporciona en Tabla 25.
Tabla 25: Incidencia de EPS en comparación con placebo en el estudio de esquizofrenia en adolescentes
LATUDA
Término del evento adverso
Placebo
(N=112)
(%)
40 mg/día
(N=110)
(%)
80 mg/día
(N=104)
(%)
Todos los eventos de EPS
5
14
14
Todos los eventos de EPS, excluyendo acatisia/inquietud
4
7
7
Nota: Las cifras se redondean al entero más cercano
* La distonía incluye términos de eventos adversos: distonía, trismus, crisis oculógiras, distonía oromandibular, espasmo de la lengua y tortícolis
** El parkinsonismo incluye términos de eventos adversos: bradicinesia, babeo, trastorno extrapiramidal, reflejo glabellar anormal, hipocinesia, parkinsonismo y retraso psicomotor
Acatisia
2
9
9
Parkinsonismo**
<1
4
0
Discinesia
<1
<1
1
Distonía*
0
<1
1
Depresión Bipolar
Adultos
Monoterapia
En el estudio de depresión bipolar de monoterapia a corto plazo, controlado con placebo, en adultos, para los pacientes tratados con LATUDA, la incidencia de eventos relacionados con EPS, excluyendo acatisia e inquietud, fue del 6,9% y del 2,4% para los pacientes tratados con placebo. La incidencia de acatisia para los pacientes tratados con LATUDA fue del 9,4% y del 2,4% para los pacientes tratados con placebo. La incidencia de EPS por grupos de dosis se proporciona en Tabla 26.
Tabla 26: Incidencia de EPS en comparación con placebo en el estudio de depresión bipolar en monoterapia en adultos
LATUDA
Término del evento adverso
Placebo
(N=168)
(%)
20 a 60 mg/día
(N=164)
(%)
80 a 120 mg/día
(N=167)
(%)
Todos los eventos de EPS
5
12
20
Todos los eventos de EPS, excluyendo acatisia/inquietud
2
5
9
Nota: Las cifras se redondean al entero más cercano
* La distonía incluye términos de eventos adversos: distonía, crisis oculogírica, distonía oromandibular, espasmo de la lengua, tortícolis y trismus
** El parkinsonismo incluye términos de eventos adversos: bradicinesia, rigidez en rueda dentada, babeo, trastorno extrapiramidal, reflejo glabellar anormal, hipocinesia, rigidez muscular, parkinsonismo, retraso psicomotor y temblor
Acatisia
2
8
11
Distonía*
0
0
2
Parkinsonismo**
2
5
8
Inquietud
<1
0
3
Terapia adjunta con litio o valproato
En los estudios de terapia adjunta a corto plazo, controlados con placebo, en adultos con depresión bipolar, para los pacientes tratados con LATUDA, la incidencia de EPS, excluyendo acatisia e inquietud, fue del 13,9% y del 8,7% para el placebo. La incidencia de acatisia para los pacientes tratados con LATUDA fue del 10,8% y del 4,8% para los pacientes tratados con placebo. La incidencia de EPS se proporciona en Tabla 27.
Tabla 27: Incidencia de EPS en comparación con placebo en los estudios de terapia adjunta para la depresión bipolar en adultos
Término del evento adverso
Placebo
(N=334)
(%)
LATUDA 20 a 120 mg/día (N=360)
(%)
Todos los eventos de EPS
13
24
Todos los eventos de EPS, excluyendo acatisia/inquietud
9
14
Nota: Las cifras se redondean al entero más cercano
* La distonía incluye los términos de eventos adversos: distonía, crisis oculógiras, distonía oromandibular, espasmo de la lengua, tortícolis y trismus
** El parkinsonismo incluye los términos de eventos adversos: bradicinesia, rigidez en rueda dentada, babeo, trastorno extrapiramidal, reflejo glabellar anormal, hipocinesia, rigidez muscular, parkinsonismo, retraso psicomotor y temblor
Acatisia
5
11
Distonía*
<1
1
Parkinsonismo**
8
13
Inquietud
<1
4
En los estudios a corto plazo, controlados con placebo, sobre esquizofrenia y depresión bipolar, los datos se recogieron objetivamente en la Escala de Calificación de Simpson Angus (SAS) para los síntomas extrapiramidales (SEP), la Escala de Acatisia de Barnes (BAS) para la acatisia y la Escala de Movimientos Involuntarios Anormales (AIMS) para las discinesias.
Pacientes pediátricos (10 a 17 años)
En el estudio de 6 semanas, controlado con placebo, sobre depresión bipolar en pacientes pediátricos de 10 a 17 años, la incidencia de SEP, excluyendo los eventos relacionados con acatisia, para los pacientes tratados con LATUDA fue similar en el grupo de tratamiento con LATUDA 20 a 80 mg/día (3,4%) frente a placebo (3,5%); y la incidencia de eventos relacionados con acatisia para los pacientes tratados con LATUDA fue del 2,9% frente al 3,5% para los pacientes tratados con placebo. La incidencia de SEP por dosis se proporciona en Tabla 28.
Tabla 28: Incidencia de SEP en comparación con placebo en el estudio de depresión bipolar en pacientes pediátricos (10 a 17 años)
Término del evento adverso
Placebo
(N=172)
(%)
LATUDA
20 a 80 mg/día
(N=175)
(%)
Todos los eventos SEP*
5
6
Todos los eventos SEP, excluyendo acatisia/inquietud
4
3
Nota: Las cifras se redondean al entero más próximo
* Los SEP incluyen los términos de eventos adversos: acatisia, rigidez en rueda dentada, discinesia, distonía, hipercinesia, rigidez articular, rigidez muscular, espasmos musculares, rigidez musculoesquelética, crisis oculogírica, parkinsonismo, discinesia tardía y temblor
** El parkinsonismo incluye los términos de eventos adversos: bradicinesia, babeo, trastorno extrapiramidal, reflejo glabellar anormal, hipocinesia, parkinsonismo y retraso psicomotor
***La distonía incluye los términos de eventos adversos: distonía, crisis oculogírica, distonía oromandibular, espasmo lingual, tortícolis y trismus
Acatisia
4
3
Parkinsonismo**
<1
<1
Distonía***
1
<1
Hipersecreción salival
<1
<1
Hiperactividad psicomotora
0
<1
Discinesia tardía
<1
0
Esquizofrenia
Adultos
El cambio medio desde la línea de base para los pacientes tratados con LATUDA para la SAS, BAS y AIMS fue comparable a los pacientes tratados con placebo, con la excepción de la puntuación global de la escala de acatisia de Barnes (LATUDA, 0.1; placebo, 0.0). El porcentaje de pacientes que pasaron de normal a anormal fue mayor en los pacientes tratados con LATUDA y placebo para la BAS (LATUDA, 14.4%; placebo, 7.1%), la SAS (LATUDA, 5.0%; placebo, 2.3%) y la AIMS (LATUDA, 7.4%; placebo, 5.8%).
Adolescentes
El cambio medio desde la línea de base para los pacientes tratados con LATUDA con esquizofrenia adolescente para la SAS, BAS y AIMS fue comparable a los pacientes tratados con placebo. El porcentaje de pacientes que pasaron de normal a anormal fue mayor en los pacientes tratados con LATUDA y placebo para la BAS (LATUDA, 7.0%; placebo, 1.8%), la SAS (LATUDA, 8.3%; placebo, 2.7%) y la AIMS (LATUDA, 2.8%; placebo, 0.9%).
Depresión Bipolar
Adultos
Monoterapia
El cambio medio desde la línea de base para los pacientes adultos tratados con LATUDA para la SAS, BAS y AIMS fue comparable a los pacientes tratados con placebo. El porcentaje de pacientes que pasaron de normal a anormal fue mayor en los pacientes tratados con LATUDA y placebo para la BAS (LATUDA, 8.4%; placebo, 5.6%), la SAS (LATUDA, 3.7%; placebo, 1.9%) y la AIMS (LATUDA, 3.4%; placebo, 1.2%).
Terapia adjunta con Litio o Valproato
El cambio medio desde la línea de base para los pacientes adultos tratados con LATUDA para la SAS, BAS y AIMS fue comparable a los pacientes tratados con placebo. El porcentaje de pacientes que pasaron de normal a anormal fue mayor en los pacientes tratados con LATUDA y placebo para la BAS (LATUDA, 8.7%; placebo, 2.1%), la SAS (LATUDA, 2.8%; placebo, 2.1%) y la AIMS (LATUDA, 2.8%; placebo, 0.6%).
Pacientes pediátricos (10 a 17 años)
El cambio medio desde la línea de base para los pacientes pediátricos tratados con LATUDA de 10 a 17 años con depresión bipolar para la SAS, BAS y AIMS fue comparable a los pacientes tratados con placebo. El porcentaje de pacientes que pasaron de normal a anormal fue mayor en los pacientes tratados con LATUDA y placebo para la BAS (LATUDA, 4.6%; placebo, 2.4%), la SAS (LATUDA, 0.6%; placebo, 0%) y fue el mismo para la AIMS (LATUDA, 0%; placebo, 0%).
Distonía
Efecto de clase: Los síntomas de distonía, contracciones musculares anormales prolongadas, pueden ocurrir en individuos susceptibles durante los primeros días de tratamiento. Los síntomas distónicos incluyen: espasmo de los músculos del cuello, a veces progresando a tensión de la garganta, dificultad para tragar, dificultad para respirar y/o protrusión de la lengua. Si bien estos síntomas pueden ocurrir a dosis bajas, ocurren con mayor frecuencia y con mayor gravedad con fármacos antipsicóticos de primera generación de alta potencia y a dosis más altas. Se observa un mayor riesgo de distonía aguda en hombres y grupos de edad más jóvenes.
Esquizofrenia
Adultos
En los estudios clínicos de esquizofrenia a corto plazo controlados con placebo, la distonía se produjo en el 4.2% de los sujetos tratados con LATUDA (0.0% LATUDA 20 mg, 3.5% LATUDA 40 mg, 4.5% LATUDA 80 mg, 6.5% LATUDA 120 mg y 2.5% LATUDA 160 mg) en comparación con el 0.8% de los sujetos que recibieron placebo. Siete sujetos (0.5%, 7/1508) abandonaron los ensayos clínicos debido a eventos distónicos: cuatro estaban recibiendo LATUDA 80 mg/día y tres estaban recibiendo LATUDA 120 mg/día.
Adolescentes
En el estudio de esquizofrenia adolescente a corto plazo, controlado con placebo, la distonía se produjo en el 1% de los pacientes tratados con LATUDA (1% LATUDA 40 mg y 1% LATUDA 80 mg) en comparación con el 0% de los pacientes que recibieron placebo. Ningún paciente abandonó el estudio clínico debido a eventos distónicos.
Depresión Bipolar
Adultos
Monoterapia
En el estudio de depresión bipolar de monoterapia a corto plazo, de dosis flexible, controlado con placebo en adultos, la distonía se produjo en el 0.9% de los sujetos tratados con LATUDA (0.0% y 1.8% para LATUDA 20 a 60 mg/día y LATUDA 80 a 120 mg/día, respectivamente) en comparación con el 0.0% de los sujetos que recibieron placebo. Ningún sujeto abandonó el estudio clínico debido a eventos distónicos.
Terapia adjunta con Litio o Valproato
En los estudios de depresión bipolar de terapia adjunta a corto plazo, de dosis flexible, controlados con placebo en adultos, la distonía se produjo en el 1.1% de los sujetos tratados con LATUDA (20 a 120 mg) en comparación con el 0.6% de los sujetos que recibieron placebo. Ningún sujeto abandonó el estudio clínico debido a eventos distónicos.
Pacientes pediátricos (10 a 17 años)
En el estudio de depresión bipolar de 6 semanas, controlado con placebo, en pacientes pediátricos de 10 a 17 años, la distonía se produjo en el 0,6% de los pacientes tratados con LATUDA en comparación con el 1,2% de los pacientes que recibieron placebo. Ningún paciente abandonó el estudio clínico debido a eventos distónicos.
Otras reacciones adversas observadas durante la evaluación precomercialización de LATUDA
A continuación, se muestra una lista de reacciones adversas notificadas por pacientes adultos tratados con LATUDA a dosis múltiples de ≥ 20 mg una vez al día dentro de la base de datos precomercialización de 2905 pacientes con esquizofrenia. Las reacciones enumeradas son aquellas que podrían tener importancia clínica, así como las reacciones que están plausiblemente relacionadas con el fármaco por motivos farmacológicos u otros. No se incluyen las reacciones enumeradas en Tabla 19 o aquellas que aparecen en otra parte de la etiqueta de LATUDA.
Las reacciones se categorizan además por clase de órgano y se enumeran en orden de frecuencia decreciente según las siguientes definiciones: aquellas que ocurren en al menos 1/100 pacientes (frecuentes) (solo aquellas que no están ya enumeradas en los resultados tabulados de los estudios controlados con placebo aparecen en esta lista); aquellas que ocurren en 1/100 a 1/1000 pacientes (infrecuentes); y aquellas que ocurren en menos de 1/1000 pacientes (raras).
Trastornos de la sangre y del sistema linfático: Infrecuentes: anemia
Trastornos cardíacos: Frecuentes: taquicardia; Infrecuentes: bloqueo AV de primer grado, angina de pecho, bradicardia
Trastornos del oído y del laberinto: Infrecuentes: vértigo
Trastornos generales y condiciones del lugar de administración: Raras: muerte súbita
Pruebas: Frecuentes: aumento de la CPK
Trastornos del metabolismo y del sistema nutricional: Frecuentes: disminución del apetito
Trastornos musculoesqueléticos y del tejido conjuntivo: Raras: rabdomiólisis
Trastornos del sistema nervioso: Infrecuentes: accidente cerebrovascular, disartria
Trastornos psiquiátricos: Infrecuentes: sueños anormales, ataque de pánico, trastorno del sueño
Trastornos renales y urinarios: Infrecuentes: disuria; Raras: insuficiencia renal
Trastornos del aparato reproductor y de la mama: Infrecuentes: amenorrea, dismenorrea; Raras: aumento de mamas, dolor de mamas, galactorrea, disfunción eréctil, priapismo
Trastornos de la piel y del tejido subcutáneo: Frecuentes: erupción, prurito; Raras: angioedema
Trastornos vasculares: Frecuentes: hipertensión
Cambios en los análisis de laboratorio clínico
Esquizofrenia
Adultos
Creatinina sérica: En ensayos a corto plazo controlados con placebo, el cambio medio desde el inicio en la creatinina sérica fue de +0,05 mg/dL para los pacientes tratados con LATUDA en comparación con +0,02 mg/dL para los pacientes tratados con placebo. Se produjo un cambio de creatinina de normal a alta en el 3,0% (43/1453) de los pacientes tratados con LATUDA y en el 1,6% (11/681) con placebo. El umbral para el valor alto de creatinina varió de > 0,79 a > 1,3 mg/dL según la definición del laboratorio centralizado para cada estudio (Tabla 29).
Tabla 29: Cambios en la creatinina sérica de normal al inicio a alta al final del estudio en estudios de esquizofrenia en adultos
Parámetro de laboratorio
Placebo
(N=708)
LATUDA 20 mg/día
(N=71)
LATUDA 40 mg/día
(N=487)
LATUDA 80 mg/día
(N=538)
LATUDA 120 mg/día
(N=291)
LATUDA 160 mg/día
(N=121)
Creatinina sérica elevada
2%
1%
2%
2%
5%
7%
Adolescentes
Creatinina sérica: En el estudio de esquizofrenia en adolescentes a corto plazo, controlado con placebo, el cambio medio desde el valor basal en la creatinina sérica fue de –0,009 mg/dL para los pacientes tratados con LATUDA en comparación con +0,017 mg/dL para los pacientes tratados con placebo. Se produjo un cambio en la creatinina de normal a alta (según la definición del laboratorio centralizado) en el 7,2% (14/194) de los pacientes tratados con LATUDA y en el 2,9% (3/103) con placebo (Tabla 30).
Tabla 30: Cambios en la creatinina sérica de normal al inicio del estudio a alta al final del estudio en el estudio de esquizofrenia en adolescentes
Parámetro de laboratorio
Placebo
(N=103)
LATUDA 40 mg/día
(N=97)
LATUDA 80 mg/día
(N=97)
Creatinina sérica elevada
2,9%
7,2%
7,2%
Depresión bipolar
Adultos
Monoterapia
Creatinina sérica: En el estudio de depresión bipolar en adultos a corto plazo, de dosis flexible, controlado con placebo y en monoterapia, el cambio medio desde el valor basal en la creatinina sérica fue de +0,01 mg/dL para los pacientes tratados con LATUDA en comparación con -0,02 mg/dL para los pacientes tratados con placebo. Se produjo un cambio en la creatinina de normal a alta en el 2,8% (9/322) de los pacientes tratados con LATUDA y en el 0,6% (1/162) con placebo (Tabla 31).
Tabla 31: Cambios en la creatinina sérica de normal al inicio del estudio a alta al final del estudio en el estudio de depresión bipolar en adultos en monoterapia
Parámetro de laboratorio
Placebo
(N=168)
LATUDA 20 a 60 mg/día
(N=164)
LATUDA 80 a 120 mg/día
(N=167)
Creatinina sérica elevada
<1%
2%
4%
Terapia Adyuvante con Litio o Valproato
Creatinina Sérica: En estudios precomercialización a corto plazo, controlados con placebo, en adultos para la depresión bipolar adyuvante, el cambio medio desde el inicio en la creatinina sérica fue de +0.04 mg/dL para los pacientes tratados con LATUDA en comparación con -0.01 mg/dL para los pacientes tratados con placebo. Un cambio de creatinina de normal a alta se produjo en el 4.3% (15/360) de los pacientes tratados con LATUDA y en el 1.6% (5/334) con placebo (Tabla 32).
Tabla 32: Cambios en la Creatinina Sérica de Normal al Inicio a Alta al Final del Estudio en los Estudios de Depresión Bipolar de Terapia Adyuvante en Adultos
Parámetro de Laboratorio
Placebo
(N=334)
LATUDA
20 a 120 mg/día (N=360)
Creatinina Sérica Elevada
2%
4%
Pacientes Pediátricos (10 a 17 años)
Creatinina Sérica: En el estudio de depresión bipolar de 6 semanas, controlado con placebo, en pacientes pediátricos de 10 a 17 años, el cambio medio desde el inicio en la creatinina sérica fue de +0.021 mg/dL para los pacientes tratados con LATUDA en comparación con +0.009 mg/dL para los pacientes tratados con placebo. Un cambio de creatinina de normal a alta (basado en la definición de laboratorio centralizada) se produjo en el 6.7% (11/163) de los pacientes tratados con LATUDA y en el 4.5% (7/155) con placebo (Tabla 33).
Tabla 33: Cambios en la Creatinina Sérica de Normal al Inicio a Alta al Final del Estudio en el Estudio de Depresión Bipolar en Pacientes Pediátricos (10 a 17 años)
Parámetro de Laboratorio
Placebo
(N=155)
LATUDA 20 a 80 mg/día
(N=163)
Creatinina Sérica Elevada
4.5%
6.7%
Pacientes Pediátricos (6 a 17 años)
En un estudio abierto de 104 semanas en pacientes pediátricos con esquizofrenia, depresión bipolar o trastorno autista, el cambio medio desde el inicio hasta la semana 104 en la creatinina sérica fue de +0.07 mg/dL. En pacientes con una creatinina sérica normal al inicio, el 6% experimentó un cambio a alta al final del estudio.
6.2 Experiencia Postcomercialización
Las siguientes reacciones adversas se han identificado durante el uso posterior a la aprobación de LATUDA. Debido a que estas reacciones se notifican voluntariamente a partir de una población de tamaño incierto, no siempre es posible estimar de forma fiable su frecuencia o establecer una relación causal con la exposición al fármaco.
Reacciones de Hipersensibilidad: Urticaria, hinchazón de garganta, hinchazón de lengua, disnea y erupción cutánea.
Trastornos del Metabolismo y la Nutrición: Hiponatremia
7 INTERACCIONES MEDICAMENTOSAS
7.1 Medicamentos con Interacciones Clínicamente Importantes con LATUDA
Tabla 34: Interacciones Medicamentosas Clínicamente Importantes con LATUDA
Inhibidores Potentes del CYP3A4
Impacto Clínico:
El uso concomitante de LATUDA con inhibidores potentes del CYP3A4 aumentó la exposición a lurasidona en comparación con el uso de LATUDA solo [ver Farmacología Clínica (12.3)].
Intervención:
LATUDA no debe usarse concomitantemente con inhibidores potentes del CYP3A4 [ver Contraindicaciones (4)].
El uso concomitante de LATUDA con inhibidores moderados del CYP3A4 aumentó la exposición a lurasidona en comparación con el uso de LATUDA solo [ver Farmacología Clínica (12.3)].
Intervención:
La dosis de LATUDA debe reducirse a la mitad del nivel original cuando se usa concomitantemente con inhibidores moderados del CYP3A4 [ver Dosis y Administración (2.6)].
El uso concomitante de LATUDA con inductores potentes del CYP3A4 disminuyó la exposición a lurasidona en comparación con el uso de LATUDA solo [ver Farmacología Clínica (12.3)].
Intervención:
LATUDA no debe usarse concomitantemente con inductores potentes del CYP3A4 [ver Contraindicaciones (4)].
Ejemplos:
Rifampicina, avasimibe, hierba de San Juan, fenitoína, carbamazepina
Inductores Moderados del CYP3A4
Impacto Clínico:
El uso concomitante de LATUDA con inductores moderados del CYP3A4 disminuyó la exposición a lurasidona en comparación con el uso de LATUDA solo [ver Farmacología Clínica (12.3)].
Intervención:
La dosis de LATUDA debe aumentarse cuando se usa concomitantemente con inductores moderados del CYP3A4 [ver Dosis y Administración (2.6)].
7.2 Medicamentos sin Interacciones Clínicamente Importantes con LATUDA
Según estudios farmacocinéticos, no se requiere ajuste de dosis de LATUDA cuando se administra concomitantemente con litio, valproato o sustratos de P-gp o CYP3A4 [ver Farmacología Clínica (12.3)].
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Registro de exposición durante el embarazo
Existe un registro de exposición durante el embarazo que monitoriza los resultados del embarazo en mujeres expuestas a LATUDA durante la gestación. Para más información, contacte con el Registro Nacional de Embarazos para Antipsicóticos Atípicos al 1-866-961-2388 o visite http://womensmentalhealth.org/clinical-and-research-programs/pregnancyregistry/.
Resumen de riesgo
Los recién nacidos expuestos a fármacos antipsicóticos durante el tercer trimestre del embarazo presentan riesgo de síntomas extrapiramidales y/o de abstinencia después del parto [ver Consideraciones clínicas]. No hay estudios sobre el uso de LATUDA en mujeres embarazadas. Los datos limitados disponibles no son suficientes para determinar un riesgo asociado al fármaco de defectos congénitos o aborto espontáneo. En estudios de reproducción animal, no se observaron efectos teratogénicos en ratas y conejas embarazadas a las que se administró lurasidona durante el período de organogénesis a dosis aproximadamente 1,5 y 6 veces, respectivamente, la dosis máxima recomendada en humanos (MRHD) de 160 mg/día, basada en mg/m2 de superficie corporal [ver Datos].
Se desconoce el riesgo de base estimado de defectos congénitos importantes y aborto espontáneo para la(s) población(es) indicada(s). Todos los embarazos tienen un riesgo de base de defectos congénitos, pérdida u otros resultados adversos. En la población general de EE. UU., el riesgo de base estimado de defectos congénitos importantes y aborto espontáneo en embarazos clínicamente reconocidos es del 2-4 % y del 15-20 %, respectivamente.
Consideraciones clínicas
Reacciones adversas fetales/neonatales
Se han notificado síntomas extrapiramidales y/o de abstinencia, incluyendo agitación, hipertonía, hipotonía, temblor, somnolencia, dificultad respiratoria y trastornos de la alimentación en recién nacidos expuestos a fármacos antipsicóticos durante el tercer trimestre del embarazo. Estos síntomas han variado en gravedad. Algunos recién nacidos se recuperaron en horas o días sin tratamiento específico; otros requirieron hospitalización prolongada. Se debe monitorizar a los recién nacidos para detectar síntomas extrapiramidales y/o de abstinencia y controlar los síntomas adecuadamente.
Datos
Datos en animales
Las ratas embarazadas fueron tratadas con lurasidona oral a dosis de 3, 10 y 25 mg/kg/día durante el período de organogénesis. Estas dosis son 0,2, 0,6 y 1,5 veces la MRHD de 160 mg/día basada en mg/m2 de superficie corporal. No se observaron efectos teratogénicos ni embriofetales hasta 1,5 veces la MRHD de 160 mg/día, basada en mg/m2.
Las conejas embarazadas fueron tratadas con lurasidona oral a dosis de 2, 10 y 50 mg/kg/día durante el período de organogénesis. Estas dosis son 0,2, 1,2 y 6 veces la MRHD de 160 mg/día basada en mg/m2. No se observaron efectos teratogénicos ni embriofetales hasta 6 veces la MRHD de 160 mg/día basada en mg/m2.
Las ratas embarazadas fueron tratadas con lurasidona oral a dosis de 0,4, 2 y 10 mg/kg/día durante los períodos de organogénesis y lactancia. Estas dosis son 0,02, 0,1 y 0,6 veces la MRHD de 160 mg/día basada en mg/m2. No se observaron efectos en el desarrollo pre y postnatal hasta 0,6 veces la MRHD de 160 mg/día, basada en mg/m2.
8.2 Lactancia
Resumen de riesgo
No se han realizado estudios de lactancia para evaluar la presencia de lurasidona en la leche materna, los efectos en el lactante o los efectos sobre la producción de leche. La lurasidona está presente en la leche de rata. Se deben considerar los beneficios para el desarrollo y la salud de la lactancia materna junto con la necesidad clínica de la madre de LATUDA y cualquier posible efecto adverso en el lactante amamantado por LATUDA o por la enfermedad materna subyacente.
8.4 Uso pediátrico
Esquizofrenia
La seguridad y eficacia de LATUDA 40 mg/día y 80 mg/día para el tratamiento de la esquizofrenia en adolescentes (13 a 17 años) se estableció en un estudio clínico de 6 semanas, controlado con placebo, en 326 pacientes adolescentes [ver Posología y administración (2.1), Reacciones adversas (6.1), y Estudios clínicos (14.1)].
No se ha establecido la seguridad y eficacia de LATUDA en pacientes pediátricos menores de 13 años con esquizofrenia.
Depresión bipolar
La seguridad y eficacia de LATUDA 20 a 80 mg/día para el tratamiento de la depresión bipolar en pacientes pediátricos (10 a 17 años) se estableció en un estudio clínico de 6 semanas, controlado con placebo, en 347 pacientes pediátricos [ver Posología y administración (2.2), Reacciones adversas (6.1), y Estudios clínicos (14.2)].
No se ha establecido la seguridad y eficacia de LATUDA en pacientes pediátricos menores de 10 años con depresión bipolar.
Irritabilidad asociada al trastorno del espectro autista
La efectividad de LATUDA en pacientes pediátricos para el tratamiento de la irritabilidad asociada con el trastorno autista no se ha establecido.
No se demostró eficacia en un estudio de 6 semanas que evaluó LATUDA 20 mg/día y 60 mg/día para el tratamiento de pacientes pediátricos de 6 a 17 años de edad con irritabilidad asociada con el trastorno autista diagnosticado por el Manual Diagnóstico y Estadístico de los Trastornos Mentales, 4ª Ed., Revisión del texto [DSM-IV-TR] criterios. El objetivo principal del estudio, medido por la mejora desde el inicio en la subescala de irritabilidad de la Lista de verificación de comportamiento aberrante (ABC) en el punto final (semana 6), no se cumplió. Un total de 149 pacientes fueron aleatorizados a LATUDA o placebo. Los vómitos ocurrieron a una tasa más alta que la reportada en otros estudios de LATUDA (4/49 u 8% para 20 mg, 14/51 o 27% para 60 mg y 2/49 o 4% para placebo), particularmente en niños de 6 a 12 años (13 de 18 pacientes con LATUDA con vómitos).
En un estudio abierto a largo plazo que incluyó a pacientes pediátricos (de 6 a 17 años) con esquizofrenia, depresión bipolar o trastorno autista de tres ensayos a corto plazo controlados con placebo, el 54 % (378/701) recibió lurasidona durante 104 semanas. Hubo un evento adverso en este ensayo que se consideró posiblemente relacionado con el fármaco y no se ha informado en adultos que reciben lurasidona: un varón de 10 años experimentó una erección prolongada y dolorosa, compatible con priapismo, que condujo a la interrupción del tratamiento.
En este ensayo, el aumento medio de la altura desde el inicio abierto hasta la semana 104 fue de 4,94 cm. Para ajustar el crecimiento normal, se derivaron puntuaciones z (medidas en desviaciones estándar [SD]), que normalizan el crecimiento natural de niños y adolescentes mediante comparaciones con estándares de población emparejados por edad y sexo. Un cambio en la puntuación z <0,5 SD se considera no clínicamente significativo. En este ensayo, el cambio medio en la puntuación z de altura desde el inicio abierto hasta la semana 104 fue de +0,05 SD, lo que indica una desviación mínima de la curva de crecimiento normal.
Estudios en animales juveniles
Se observaron efectos adversos sobre el crecimiento, el desarrollo físico y neuroconductual a dosis tan bajas como 0,2 veces la MRHD en función de mg/m2. La lurasidona se administró por vía oral a ratas desde los días posnatales 21 hasta 91 (este período corresponde a la infancia, la adolescencia y la edad adulta temprana en humanos) en dosis de 3, 30 y 150 (machos) o 300 (hembras) mg/kg/día, que son de 0,2 a 10 veces (machos) y 20 veces (hembras) la dosis máxima recomendada para humanos adultos (MRHD) de 160 mg/día en función de mg/m2. Los efectos adversos incluyeron disminuciones dependientes de la dosis en la longitud femoral, el contenido mineral óseo, el peso corporal y cerebral a 2 veces la MRHD en ambos sexos, y hiperactividad motora a 0,2 y 2 veces la MRHD en ambos sexos en función de mg/m2. En las hembras, hubo un retraso en el logro de la madurez sexual a 2 veces la MRHD, asociado con una disminución del estradiol sérico. La mortalidad ocurrió en ambos sexos durante el período temprano posterior al destete y algunas de las crías machos murieron después de solo 4 tratamientos a dosis tan bajas como 2 veces la MRHD en función de mg/m2. Los hallazgos histopatológicos incluyeron aumento de coloide en la tiroides e inflamación de la próstata en machos a 10 veces la MRHD en función de mg/m2 e hiperplasia de las glándulas mamarias, aumento de la mucificación vaginal y aumento de folículos atrésicos ováricos a dosis tan bajas como 0,2 veces la MRHD en función de mg/m2. Algunos de estos hallazgos se atribuyeron a la prolactina sérica transitoriamente elevada que se observó en ambos sexos en todas las dosis. Sin embargo, no hubo cambios en ningún nivel de dosis en los parámetros reproductivos (fertilidad, índices de concepción, espermatogénesis, ciclo estral, duración de la gestación, parto, número de crías nacidas). La dosis sin efecto para los cambios neuroconductuales en los machos es 0,2 veces la MRHD en función de mg/m2 y no se pudo determinar en las hembras. La dosis sin efecto para el crecimiento y el desarrollo físico en ambos sexos es 0,2 veces la MRHD en función de mg/m2.
8.5 Uso geriátrico
Los estudios clínicos con LATUDA no incluyeron un número suficiente de pacientes de 65 años o más para determinar si responden de manera diferente a los pacientes más jóvenes. En pacientes ancianos con psicosis (65 a 85), las concentraciones de LATUDA (20 mg/día) fueron similares a las de los sujetos jóvenes. Se desconoce si es necesario un ajuste de dosis solo por la edad.
Los pacientes ancianos con psicosis relacionada con la demencia tratados con LATUDA tienen un mayor riesgo de muerte en comparación con el placebo. LATUDA no está aprobado para el tratamiento de pacientes con psicosis relacionada con la demencia [ver Recuadro de advertencia, Advertencias y precauciones (5.1, 5.3)].
8.6 Insuficiencia renal
Reduzca la dosis máxima recomendada en pacientes con insuficiencia renal moderada o grave (CLcr<50 mL/minuto). Los pacientes con insuficiencia renal (CLcr<50 mL/minuto) tuvieron una mayor exposición a lurasidona que los pacientes con función renal normal [ver Farmacología clínica (12.3)]. Una mayor exposición puede aumentar el riesgo de reacciones adversas asociadas con LATUDA [ver Dosis y administración (2.4)].
8.7 Insuficiencia hepática
Reduzca la dosis máxima recomendada en pacientes con insuficiencia hepática moderada a grave (puntuación Child-Pugh ≥7). Los pacientes con insuficiencia hepática moderada a grave (puntuación Child-Pugh ≥7) generalmente tuvieron una mayor exposición a lurasidona que los pacientes con función hepática normal [ver Farmacología clínica (12.3)]. Una mayor exposición puede aumentar el riesgo de reacciones adversas asociadas con LATUDA [ver Dosis y administración (2.5)].
8.8 Otras poblaciones específicas
No se requiere ajuste de dosis para LATUDA en función del sexo, la raza o el estado de fumador del paciente [ver Farmacología clínica (12.3)].
9 ABUSO DE DROGAS Y DEPENDENCIA
9.1 Sustancia controlada
LATUDA no es una sustancia controlada.
9.2 Abuso
LATUDA no se ha estudiado sistemáticamente en humanos por su potencial de abuso o dependencia física o su capacidad para inducir tolerancia. Si bien los estudios clínicos con LATUDA no revelaron ninguna tendencia al comportamiento de búsqueda de drogas, estas observaciones no fueron sistemáticas y no es posible predecir el grado en que un fármaco activo del SNC será mal utilizado, desviado y/o abusado una vez que se comercialice. Los pacientes deben ser evaluados cuidadosamente para detectar antecedentes de abuso de drogas, y dichos pacientes deben ser observados cuidadosamente para detectar signos de mal uso o abuso de LATUDA (por ejemplo, desarrollo de tolerancia, comportamiento de búsqueda de drogas, aumentos en la dosis).
10 SOBREDOSIS
10.1 Experiencia en humanos
En estudios clínicos previos a la comercialización, se identificó una sobredosis accidental o intencional de LATUDA en un paciente que ingirió aproximadamente 560 mg de LATUDA. Este paciente se recuperó sin secuelas. Este paciente reanudó el tratamiento con LATUDA durante dos meses adicionales.
10.2 Manejo de la sobredosis
No se conocen antídotos específicos para LATUDA. En el manejo de la sobredosis, proporcione atención de apoyo, incluida la supervisión y monitorización médica estrecha, y considere la posibilidad de la participación de múltiples fármacos. Si se produce una sobredosis, consulte a un Centro de Control de Envenenamiento Certificado (1-800-222-1222 o www.poison.org).
La monitorización cardiovascular debe comenzar inmediatamente, incluyendo la monitorización electrocardiográfica continua para detectar posibles arritmias. Si se administra terapia antiarrítmica, la disopiramida, la procainamida y la quinidina conllevan un riesgo teórico de efectos aditivos de prolongación del intervalo QT cuando se administran en pacientes con una sobredosis aguda de LATUDA. De manera similar, las propiedades alfa-bloqueantes del bretilio podrían ser aditivas a las de LATUDA, lo que resulta en una hipotensión problemática.
La hipotensión y el colapso circulatorio deben tratarse con medidas apropiadas. No se deben usar epinefrina y dopamina, u otros simpaticomiméticos con actividad beta-agonista, ya que la estimulación beta puede empeorar la hipotensión en el contexto del bloqueo alfa inducido por LATUDA. En caso de síntomas extrapiramidales graves, se debe administrar medicación anticolinérgica.
Se debe considerar el lavado gástrico (después de la intubación si el paciente está inconsciente) y la administración de carbón activado junto con un laxante.
La posibilidad de obnubilación, convulsiones o reacción distónica de la cabeza y el cuello después de una sobredosis puede crear un riesgo de aspiración con la inducción de emesis.
11 DESCRIPCIÓN
LATUDA es un antipsicótico atípico que pertenece a la clase química de los derivados de benzisotiazol.
Su nombre químico es (3aR,4S,7R,7aS)-2-{(1R,2R)-2-[4-(1,2-benzisotiazol-3-il)piperazin-1-ilmetil] ciclohexilmetil}hexahidro-4,7-metano-2H-isoindol-1,3-diona hidrocloruro. Su fórmula molecular es C28H36N4O2S•HCl y su peso molecular es 529,14.
La estructura química es:
El hidrocloruro de lurasidona es un polvo blanco o blanquecino. Es muy poco soluble en agua, prácticamente insoluble o insoluble en HCl 0,1 N, ligeramente soluble en etanol, escasamente soluble en metanol, prácticamente insoluble o insoluble en tolueno y muy poco soluble en acetona.
Los comprimidos de LATUDA son únicamente para administración oral. Cada comprimido contiene 20 mg, 40 mg, 60 mg, 80 mg o 120 mg de hidrocloruro de lurasidona.
Los excipientes inactivos son manitol, almidón pregelatinizado, croscarmelosa sódica, hipromelosa, estearato de magnesio, Opadry® y cera de carnauba. Además, el comprimido de 80 mg contiene óxido férrico amarillo y FD&C Blue No. 2 Aluminum Lake.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
El mecanismo de acción de la lurasidona en el tratamiento de la esquizofrenia y la depresión bipolar no está claro. Sin embargo, su eficacia en la esquizofrenia y la depresión bipolar podría estar mediada a través de una combinación de antagonismo de los receptores centrales de dopamina D2 y serotonina tipo 2 (5HT2A).
12.2 Farmacodinámica
La lurasidona es un antagonista con alta afinidad de unión a los receptores de dopamina D2 (Ki de 1 nM) y a la serotonina 5-HT2A (Ki de 0,5 nM) y 5-HT7 (Ki de 0,5 nM). También se une con afinidad moderada a los receptores adrenérgicos α2C humanos (Ki de 11 nM), es un agonista parcial en los receptores de serotonina 5-HT1A (Ki de 6,4 nM), y es un antagonista en los receptores adrenérgicos α2A (Ki de 41 nM). La lurasidona exhibe poca o ninguna afinidad por los receptores de histamina H1 y muscarínicos M1 (IC50 > 1000 nM).
Cambios en el ECG
Los efectos de LATUDA en el intervalo QTc se evaluaron en un estudio QT completo, aleatorizado, doble ciego, de dosis múltiples y paralelo dedicado en 43 pacientes con esquizofrenia o trastorno esquizoafectivo, que fueron tratados con dosis de LATUDA de 120 mg diarios, 600 mg diarios y completaron el estudio. El aumento máximo medio (límite superior unilateral, IC del 95%) en los intervalos QTc ajustados a la línea de base según el método de corrección individual (QTcI) fue de 7,5 (11,7) ms y 4,6 (9,5) ms, para los grupos de dosis de 120 mg y 600 mg respectivamente, observado de 2 a 4 horas después de la administración. En este estudio, no hubo una relación aparente dosis (exposición)-respuesta.
En estudios a corto plazo, controlados con placebo, en esquizofrenia y depresión bipolar, no se informaron prolongaciones del QT posteriores a la línea de base que superaran los 500 mseg en pacientes tratados con LATUDA o placebo.
12.3 Farmacocinética
Adultos
La actividad de LATUDA se debe principalmente al fármaco original. La farmacocinética de LATUDA es proporcional a la dosis dentro de un rango de dosis diaria total de 20 mg a 160 mg. Las concentraciones en estado estacionario de LATUDA se alcanzan dentro de los 7 días de iniciar el tratamiento con LATUDA.
Después de la administración de 40 mg de LATUDA, la vida media de eliminación media (%CV) fue de 18 (7) horas.
Absorción y distribución: LATUDA se absorbe y alcanza concentraciones séricas máximas en aproximadamente 1-3 horas. Se estima que se absorbe entre el 9 y el 19% de una dosis administrada. Después de la administración de 40 mg de LATUDA, el volumen de distribución aparente medio (%CV) fue de 6173 (17,2) L. LATUDA se une en gran medida (~99%) a las proteínas séricas.
En un estudio de efecto de los alimentos, la Cmax y el AUC medias de LATUDA fueron aproximadamente 3 y 2 veces mayores, respectivamente, cuando se administraron con alimentos en comparación con los niveles observados en ayunas. La exposición a LATUDA no se vio afectada al aumentar el tamaño de la comida de 350 a 1000 calorías y fue independiente del contenido de grasa de la comida [ver Posología y administración (2.3)].
En estudios clínicos que establecieron la seguridad y eficacia de LATUDA, se indicó a los pacientes que tomaran su dosis diaria con alimentos [ver Posología y administración (2.3)].
Metabolismo y eliminación: LATUDA se metaboliza principalmente a través del CYP3A4. Las principales vías de biotransformación son la N-desalquilación oxidativa, la hidroxilación del anillo norbornano y la S-oxidación. LATUDA se metaboliza en dos metabolitos activos (ID-14283 e ID-14326) y dos metabolitos principales no activos (ID-20219 e ID-20220). Según los estudios in vitro, LATUDA no es un sustrato de las enzimas CYP1A1, CYP1A2, CYP2A6, CYP4A11, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 o CYP2E1. Debido a que LATUDA no es un sustrato del CYP1A2, no se espera que fumar tenga un efecto en la farmacocinética de LATUDA.
Proteínas transportadoras: Los estudios in vitro sugieren que LATUDA no es un sustrato de OATP1B1 u OATP1B3, sin embargo, probablemente sea un sustrato de P-gp y BCRP. Los estudios in vitro indican que no se espera que LATUDA inhiba los transportadores OATP1B1, OATP1B3, OCT1, OCT2, OAT1, OAT3, MATE1, MATE2-K y BSEP a concentraciones clínicamente relevantes. LATUDA no es un inhibidor clínicamente significativo de P-gp. Sin embargo, puede inhibir BCRP.
La excreción total de radiactividad en orina y heces combinadas fue de aproximadamente el 89%, con aproximadamente el 80% recuperado en heces y el 9% recuperado en orina, después de una dosis única de LATUDA marcada con [14C].
Después de la administración de 40 mg de LATUDA, el aclaramiento aparente medio (%CV) fue de 3902 (18,0) mL/min.
Estudios de interacción farmacológica
Los efectos de otros fármacos en la exposición a la lurasidona se resumen en la Figura 1. Un análisis de PK de población concluyó que la administración conjunta de litio 300-2400 mg/día o valproato 300-2000 mg/día con lurasidona durante un máximo de 6 semanas tiene un efecto mínimo en la exposición a la lurasidona.
Y los efectos de LATUDA en las exposiciones a otros fármacos se resumen en la Figura 2. Un análisis de PK de población concluyó que la administración conjunta de lurasidona tiene un efecto mínimo en la exposición al litio y al valproato cuando se administra conjuntamente con litio 300-2400 mg/día o valproato 300-2000 mg/día.
Figura 1: Impacto de otros fármacos en la farmacocinética de LATUDA
Figura 2: Impacto de LATUDA en otros fármacos
Estudios en poblaciones específicas
El efecto de los factores intrínsecos del paciente en la farmacocinética de LATUDA se presenta en la Figura 3.
Pacientes pediátricos
La exposición a LATUDA (es decir, Cmax y AUC en estado estacionario) en niños y adolescentes (de 10 a 17 años de edad) fue generalmente similar a la de los adultos en el rango de dosis de 40 a 160 mg, sin ajustar según el peso corporal.
Figura 3: Impacto de otros factores del paciente en la farmacocinética de LATUDA
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogenicidad, Mutagénesis, Deterioro de la Fertilidad
Carcinogenicidad: La lurasidona aumentó la incidencia de tumores malignos de la glándula mamaria y adenomas de la glándula pituitaria en ratones hembra a los que se administraron dosis orales de 30, 100, 300 o 650 mg/kg/día. La dosis más baja produjo niveles plasmáticos (AUC) aproximadamente iguales a los de los humanos que recibieron la dosis máxima recomendada para humanos (MRHD) de 160 mg/día. No se observaron aumentos en los tumores en ratones machos hasta la dosis más alta probada, que produjo niveles plasmáticos (AUC) 14 veces superiores a los de los humanos que recibieron la MRHD.
La lurasidona aumentó la incidencia de carcinomas de la glándula mamaria en ratas hembra a las que se administraron dosis orales de 12 y 36 mg/kg/día: la dosis más baja; 3 mg/kg/día es la dosis sin efecto que produjo niveles plasmáticos (AUC) 0,4 veces superiores a los de los humanos que recibieron la MRHD. No se observaron aumentos en los tumores en ratas macho hasta la dosis más alta probada, que produjo niveles plasmáticos (AUC) 6 veces superiores a los de los humanos que recibieron la MRHD.
Se han observado cambios proliferativos y/o neoplásicos en las glándulas mamarias y pituitarias de roedores después de la administración crónica de fármacos antipsicóticos y se consideran mediados por la prolactina [véase Advertencias y precauciones (5.7)].
Mutagénesis: La lurasidona no causó mutaciones ni aberraciones cromosómicas cuando se probó in vitro y en una batería de pruebas in vivo. La lurasidona fue negativa en la prueba de mutación genética de Ames, en las células de pulmón de hámster chino (CHL) y en la prueba de micronúcleos de médula ósea de ratón in vivo hasta 2000 mg/kg, que es 61 veces la MRHD de 160 mg/día basada en mg/m2 de superficie corporal.
Deterioro de la fertilidad: Se observaron irregularidades en el ciclo estral en ratas a las que se administró lurasidona por vía oral a dosis de 1,5, 15 y 150 mg/kg/día durante 15 días consecutivos antes del apareamiento, durante el período de apareamiento y hasta el día 7 de la gestación. No se observó ningún efecto a la dosis más baja de 0,1 mg/kg, que es aproximadamente 0,006 veces la MRHD de 160 mg/día basada en mg/m2. La fertilidad se redujo solo a la dosis más alta, lo que fue reversible después de un período de 14 días sin fármaco. La dosis sin efecto para la reducción de la fertilidad fue aproximadamente igual a la MRHD basada en mg/m2.
La lurasidona no tuvo ningún efecto sobre la fertilidad en ratas macho tratadas por vía oral durante 64 días consecutivos antes del apareamiento y durante el período de apareamiento a dosis de hasta 9 veces la MRHD basada en mg/m2.
14 ESTUDIOS CLÍNICOS
14.1 Esquizofrenia
Adultos
La eficacia de LATUDA para el tratamiento de la esquizofrenia se estableció en cinco estudios a corto plazo (6 semanas), controlados con placebo, en pacientes adultos (edad media de 38,4 años, rango 18-72) que cumplían los criterios del DSM-IV para la esquizofrenia. Se incluyó un brazo de control activo (olanzapina o quetiapina de liberación prolongada) en dos estudios para evaluar la sensibilidad del ensayo.
Se utilizaron varios instrumentos para evaluar los signos y síntomas psiquiátricos en estos estudios:
Positive and Negative Syndrome Scale (PANSS), es un inventario de múltiples ítems de psicopatología general utilizado para evaluar los efectos del tratamiento farmacológico en la esquizofrenia. Las puntuaciones totales de la PANSS pueden oscilar entre 30 y 210.
Brief Psychiatric Rating Scale derived (BPRSd), derivada de la PANSS, es un inventario de múltiples ítems que se centra principalmente en los síntomas positivos de la esquizofrenia, mientras que la PANSS incluye una gama más amplia de síntomas positivos, negativos y otros de la esquizofrenia. La BPRSd consta de 18 ítems calificados en una escala de 1 (no presente) a 7 (grave). Las puntuaciones de la BPRSd pueden oscilar entre 18 y 126.
La escala de gravedad de la impresión clínica global (CGI-S) es una escala calificada por el clínico que mide el estado de la enfermedad actual del sujeto en una escala de 1 a 7 puntos.
El punto final asociado con cada instrumento es el cambio desde el inicio en la puntuación total hasta el final de la semana 6. Estos cambios se comparan luego con los cambios del placebo para el fármaco y los grupos de control.
Los resultados de los estudios son los siguientes:
Estudio 1: En un ensayo controlado con placebo de 6 semanas (N=145) que incluyó dos dosis fijas de LATUDA (40 o 120 mg/día), ambas dosis de LATUDA en el punto final fueron superiores al placebo en la puntuación total de la BPRSd y la CGI-S.
Estudio 2: En un ensayo controlado con placebo de 6 semanas (N=180) que incluyó una dosis fija de LATUDA (80 mg/día), LATUDA en el punto final fue superior al placebo en la puntuación total de la BPRSd y la CGI-S.
Estudio 3: En un ensayo controlado con placebo y activo (N=473) que incluyó dos dosis fijas de LATUDA (40 o 120 mg/día) y un control activo (olanzapina), ambas dosis de LATUDA y el control activo en el punto final fueron superiores al placebo en la puntuación total de la PANSS y la CGI-S.
Estudio 4: En un ensayo controlado con placebo de 6 semanas (N=489) que incluyó tres dosis fijas de LATUDA (40, 80 o 120 mg/día), solo la dosis de 80 mg/día de LATUDA en el punto final fue superior al placebo en la puntuación total de la PANSS y la CGI-S.
Estudio 5: En un ensayo controlado con placebo y activo (N=482) que incluyó dos dosis fijas de LATUDA (80 o 160 mg/día) y un control activo (quetiapina de liberación prolongada), ambas dosis de LATUDA y el control activo en el punto final fueron superiores al placebo en la puntuación total de la PANSS y la CGI-S.
Por lo tanto, se ha establecido la eficacia de LATUDA a dosis de 40, 80, 120 y 160 mg/día (Tabla 35).
Tabla 35: Resultados principales de eficacia para estudios en pacientes adultos con esquizofrenia (puntuaciones BPRSd o PANSS)
DE: desviación estándar; EE: error estándar; Media LS: media de mínimos cuadrados; IC: intervalo de confianza, no ajustado para comparaciones múltiples.
a Diferencia (fármaco menos placebo) en el cambio de la media de mínimos cuadrados desde el inicio.
b Incluido para la sensibilidad del ensayo.
* Dosis estadísticamente significativamente superiores al placebo.
Estudio
Grupo de tratamiento
Medida principal de eficacia: BPRSd
Puntuación basal media (DE)
Cambio medio LS desde el inicio (EE)
Diferencia sustraída del placeboa (IC del 95%)
1
LATUDA (40 mg/día)*
54.2 (8.8)
-9.4 (1.6)
-5.6 (-9.8, -1.4)
LATUDA (120 mg/día)*
52.7 (7.6)
-11.0 (1.6)
-6.7 (-11.0, -2.5)
Placebo
54.7 (8.1)
-3.8 (1.6)
—
2
LATUDA (80 mg/día)*
55.1 (6.0)
-8.9 (1.3)
-4.7 (-8.3, -1.1)
Placebo
56.1 (6.8)
-4.2 (1.4)
—
Medida principal de eficacia: PANSS
3
LATUDA (40 mg/día)*
96.6 (10.7)
-25.7 (2.0)
-9.7 (-15.3, -4.1)
LATUDA (120 mg/día)*
97.9 (11.3)
-23.6 (2.1)
-7.5 (-13.4, -1.7)
Olanzapina (15 mg/día)*b
96.3 (12.2)
-28.7 (1.9)
-12.6 (-18.2, -7.9)
Placebo
95.8 (10.8)
-16.0 (2.1)
—
4
LATUDA (40 mg/día)
96.5 (11.5)
-19.2 (1.7)
-2.1 (-7.0, 2.8)
LATUDA (80 mg/día)*
96.0 (10.8)
-23.4 (1.8)
-6.4 (-11.3, -1.5)
LATUDA (120 mg/día)
96.0 (9.7)
-20.5 (1.8)
-3.5 (-8.4, 1.4)
Placebo
96.8 (11.1)
-17.0 (1.8)
—
5
LATUDA (80 mg/día)*
97.7 (9.7)
-22.2 (1.8)
-11.9 (-16.9, -6.9)
LATUDA (160 mg/día)*
97.5 (11.8)
-26.5 (1.8)
-16.2 (-21.2, -11.2)
Quetiapina de liberación prolongada (600 mg/día)*b
97.7 (10.2)
-27.8 (1.8)
-17.5 (-22.5, -12.4)
Placebo
96.6 (10.2)
-10.3 (1.8)
—
El examen de los subgrupos de población según la edad (había pocos pacientes mayores de 65 años), el sexo y la raza no reveló ninguna evidencia clara de una respuesta diferencial.
Adolescentes (13-17 años)
La eficacia de LATUDA se estableció en un estudio multicéntrico, aleatorizado, doble ciego, controlado con placebo de 6 semanas de duración en adolescentes (de 13 a 17 años) que cumplían los criterios del DSM-IV-TR para la esquizofrenia (N=326). Los pacientes fueron aleatorizados a una de dos dosis fijas de LATUDA (40 u 80 mg/día) o placebo.
El instrumento de evaluación principal utilizado para evaluar los signos y síntomas psiquiátricos fue el PANSS. El instrumento secundario clave fue el CGI-S.
Para ambos grupos de dosis, LATUDA fue superior al placebo en la reducción de las puntuaciones PANSS y CGI-S en la semana 6. En promedio, la dosis de 80 mg/día no proporcionó un beneficio adicional en comparación con la dosis de 40 mg/día.
Los resultados principales de eficacia se proporcionan en Tabla 36.
Tabla 36: Resultados principales de eficacia (puntuación total PANSS) para el estudio de esquizofrenia en adolescentes
Grupo de tratamiento
Medida principal de eficacia: PANSS
Puntuación basal media (DE)
Cambio medio de mínimos cuadrados desde la línea de base (EE)
Diferencia sustraída del placeboa (IC del 95%)
DE: desviación estándar; EE: error estándar; Media de mínimos cuadrados: media de mínimos cuadrados; IC: intervalo de confianza, no ajustado para comparaciones múltiples.
a Diferencia (fármaco menos placebo) en el cambio medio de mínimos cuadrados desde la línea de base.
* Dosis estadísticamente superiores al placebo.
LATUDA (40 mg/día)*
94.5 (10.97)
-18.6 (1.59)
-8.0 (-12.4, -3.7)
LATUDA (80 mg/día)*
94.0 (11.12)
-18.3 (1.60)
-7.7 (-12.1, -3.4)
Placebo
92.8 (11.08)
-10.5 (1.59)
—
14.2 Episodios depresivos asociados con el trastorno bipolar I
Adultos
Monoterapia
La eficacia de LATUDA, como monoterapia, se estableció en un estudio multicéntrico, aleatorizado, doble ciego, controlado con placebo de 6 semanas de duración en pacientes adultos (edad media de 41,5 años, rango de 18 a 74) que cumplían los criterios del DSM-IV-TR para episodios depresivos mayores asociados con el trastorno bipolar I, con o sin ciclos rápidos, y sin características psicóticas (N=485). Los pacientes fueron aleatorizados a uno de dos rangos de dosis flexibles de LATUDA (20 a 60 mg/día, u 80 a 120 mg/día) o placebo.
El instrumento de evaluación principal utilizado para evaluar los síntomas depresivos en este estudio fue la Escala de Calificación de Depresión de Montgomery-Asberg (MADRS), una escala de 10 ítems calificada por el clínico con puntuaciones totales que van de 0 (sin características depresivas) a 60 (puntuación máxima). El criterio de valoración principal fue el cambio desde la línea de base en la puntuación MADRS en la semana 6. El instrumento secundario clave fue la escala de Impresión Clínica Global-Bipolar-Gravedad de la Enfermedad (CGI-BP-S), una escala calificada por el clínico que mide el estado actual de la enfermedad del sujeto en una escala de 7 puntos, donde una puntuación más alta se asocia con una mayor gravedad de la enfermedad.
Para ambos grupos de dosis, LATUDA fue superior al placebo en la reducción de las puntuaciones MADRS y CGI-BP-S en la semana 6. Los resultados principales de eficacia se proporcionan en la Tabla 37. El rango de dosis alta (80 a 120 mg por día) no proporcionó una eficacia adicional en promedio, en comparación con el rango de dosis baja (20 a 60 mg por día).
Terapia adjunta con litio o valproato
La eficacia de LATUDA, como terapia adjunta con litio o valproato, se estableció en un estudio multicéntrico, aleatorizado, doble ciego, controlado con placebo de 6 semanas de duración en pacientes adultos (edad media de 41,7 años, rango de 18 a 72) que cumplían los criterios del DSM-IV-TR para episodios depresivos mayores asociados con el trastorno bipolar I, con o sin ciclos rápidos, y sin características psicóticas (N=340). Los pacientes que permanecieron sintomáticos después del tratamiento con litio o valproato fueron aleatorizados a LATUDA con dosis flexibles de 20 a 120 mg/día o placebo.
El instrumento de evaluación principal utilizado para evaluar los síntomas depresivos en este estudio fue la MADRS. El criterio de valoración principal fue el cambio desde la línea de base en la puntuación MADRS en la semana 6. El instrumento secundario clave fue la escala CGI-BP-S.
LATUDA fue superior al placebo en la reducción de las puntuaciones MADRS y CGI-BP-S en la semana 6, como terapia adjunta con litio o valproato (Tabla 37).
Tabla 37: Resultados principales de eficacia para estudios en adultos con episodios depresivos asociados con el trastorno bipolar I (puntuaciones MADRS)
DE: desviación estándar; EE: error estándar; Media LS: media de mínimos cuadrados; IC: intervalo de confianza, no ajustado para comparaciones múltiples.
a Diferencia (fármaco menos placebo) en el cambio de la media de mínimos cuadrados desde la línea de base. * Grupo de tratamiento estadísticamente superior al placebo.
Estudio
Grupo de tratamiento
Medida principal de eficacia: MADRS
Puntuación media basal (DE)
Cambio de la media LS desde la línea de base (EE)
Diferencia sustraída del placeboa (IC del 95%)
Estudio de monoterapia
LATUDA (20-60 mg/día)*
30.3 (5.0)
-15.4 (0.8)
-4.6 (-6.9, -2.3)
LATUDA (80-120 mg/día)*
30.6 (4.9)
-15.4 (0.8)
-4.6 (-6.9, -2.3)
Placebo
30.5 (5.0)
-10.7 (0.8)
—
Estudio de terapia adjunta
LATUDA (20-120 mg/día)* + litio o valproato
30.6 (5.3)
-17.1 (0.9)
-3.6 (-6.0, -1.1)
Placebo + litio o valproato
30.8 (4.8)
-13.5 (0.9)
—
Pacientes pediátricos (10 a 17 años)
La eficacia de LATUDA se estableció en un estudio multicéntrico, aleatorizado, doble ciego, controlado con placebo de 6 semanas de duración en pacientes pediátricos (10 a 17 años) que cumplían los criterios del DSM-5 para un episodio depresivo mayor asociado con trastorno bipolar I, con o sin ciclos rápidos, y sin características psicóticas (N=343). Los pacientes fueron aleatorizados a LATUDA con dosis flexible de 20 a 80 mg/día o placebo. Al final del estudio clínico, la mayoría de los pacientes (67%) recibieron 20 mg/día o 40 mg/día.
La escala de evaluación principal utilizada para evaluar los síntomas depresivos en este estudio fue la puntuación total de la Children’s Depression Rating Scale, Revised (CDRS-R). La CDRS-R es una escala de 17 ítems calificada por el clínico con puntuaciones totales que van de 17 a 113. El criterio de valoración principal fue el cambio desde la línea de base en la puntuación CDRS-R en la semana 6. El criterio de valoración secundario principal fue el cambio desde la línea de base en la puntuación de depresión CGI-BP-S.
LATUDA fue superior al placebo en la reducción de la puntuación total de CDRS-R y la puntuación de depresión CGI-BP-S en la semana 6. Los resultados principales de eficacia se proporcionan en la Tabla 38.
Tabla 38: Resultados principales de eficacia para el estudio en episodios depresivos asociados con trastorno bipolar I (puntuación total CDRS-R) en pacientes pediátricos (10 a 17 años)
Grupo de tratamiento
Medida principal de eficacia: CDRS-R
Puntuación media basal (DE)
Cambio medio LS desde la línea de base (EE)
Diferencia sustraída del placeboa (IC del 95%)
DE: desviación estándar; EE: error estándar; Media LS: media de mínimos cuadrados; IC: intervalo de confianza, no ajustado para comparaciones múltiples.
a Diferencia (fármaco menos placebo) en el cambio medio de mínimos cuadrados desde la línea de base.
* Grupo de tratamiento estadísticamente superior al placebo.
LATUDA (20 a 80 mg/día)*
59.2 (8.24)
-21.0 (1.06)
-5.7 (-8.4, -3.0)
Placebo
58.6 (8.26)
-15.3 (1.08)
—
16 SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
Los comprimidos de LATUDA son de color blanco a blanquecino, redondos (20 mg o 40 mg), blanco a blanquecino, oblongos (60 mg), verde pálido, ovalados (80 mg) o blanco a blanquecino, ovalados (120 mg) e identificados con un grabado en relieve unilateral específico de la concentración, “L20” (20 mg), “L40” (40 mg), “L80” (80 mg) o “L120” (120 mg). Los comprimidos se suministran en las siguientes concentraciones y configuraciones de envases (Tabla 39).
Tabla 39: Configuración del envase para los comprimidos de LATUDA
Concentración del comprimido
Configuración del envase
Código NDC
20 mg
Frascos de 30
63402-302-30
Frascos de 90
63402-302-90
Frascos de 500
63402-302-50
Caja de 100 (Unidad de dosis hospitalaria) 10 blíster, 10 comprimidos cada uno
63402-302-10 Cartón 63402-302-01 Blíster
40 mg
Frascos de 30
63402-304-30
Frascos de 90
63402-304-90
Frascos de 500
63402-304-50
Caja de 100 (Unidad de dosis hospitalaria) 10 blíster, 10 comprimidos cada uno
63402-304-10 Cartón 63402-304-01 Blíster
60 mg
Frascos de 30
63402-306-30
Frascos de 90
63402-306-90
Frascos de 500
63402-306-50
Caja de 100 (Unidad de dosis hospitalaria) 10 blíster, 10 comprimidos cada uno
63402-306-10 Cartón 63402-306-01 Blíster
80 mg
Frascos de 30
63402-308-30
Frascos de 90
63402-308-90
Frascos de 500
63402-308-50
Caja de 100 (Unidad de dosis hospitalaria) 10 blíster, 10 comprimidos cada uno
63402-308-10 Cartón 63402-308-01 Blíster
120 mg
Frascos de 30
63402-312-30
Frascos de 90
63402-312-90
Frascos de 500
63402-312-50
Caja de 100 (Unidad de dosis hospitalaria) 10 blíster, 10 comprimidos cada uno
63402-312-10 Carton 63402-312-01 Blíster
Almacenamiento
Conserve los comprimidos de LATUDA a 25°C (77°F); se permiten excursiones a 15° – 30°C (59° – 86°F) [Véase Temperatura ambiente controlada USP].
17 INFORMACIÓN DE ASESORAMIENTO AL PACIENTE
Aconseje al paciente que lea el etiquetado del paciente aprobado por la FDA (Guía del Medicamento).
Pensamientos y Conducta Suicidas
Aconseje a los pacientes y cuidadores que observen la aparición de suicidabilidad, especialmente al principio del tratamiento y cuando se ajuste la dosis hacia arriba o hacia abajo, e indíqueles que informen de dichos síntomas al proveedor de atención médica [ver Advertencia en recuadro, Advertencias y precauciones (5.2)].
Síndrome Neuroléptico Maligno
Informe a los pacientes sobre una reacción adversa potencialmente mortal denominada Síndrome Neuroléptico Maligno (SNM). Aconseje a los pacientes, familiares o cuidadores que se pongan en contacto con el proveedor de atención médica o que acudan a la sala de urgencias si experimentan signos y síntomas de SNM [ver Advertencias y precauciones (5.4)].
Discinesia Tardía
Informe a los pacientes sobre los signos y síntomas de la discinesia tardía y que se pongan en contacto con su proveedor de atención médica si se producen estos movimientos anormales [ver Advertencias y precauciones (5.5)].
Cambios Metabólicos
Eduque a los pacientes sobre el riesgo de cambios metabólicos, cómo reconocer los síntomas de hiperglucemia y diabetes mellitus, y la necesidad de una monitorización específica, incluyendo glucosa en sangre, lípidos y peso [ver Advertencias y precauciones (5.6)].
Hiperprolactinemia
Informe a los pacientes sobre los signos y síntomas de hiperprolactinemia que pueden estar asociados con el uso crónico de LATUDA. Aconseje a los pacientes que busquen atención médica si experimentan alguno de los siguientes síntomas: amenorrea o galactorrea en mujeres, disfunción eréctil o ginecomastia en hombres [ver Advertencias y precauciones (5.7)].
Leucopenia/Neutropenia
Avise a los pacientes con un recuento bajo de leucocitos preexistente o un historial de leucopenia/neutropenia inducida por fármacos que deben controlar su hemograma completo mientras toman LATUDA [ver Advertencias y precauciones (5.8)].
Hipotensión Ortostática
Eduque a los pacientes sobre el riesgo de hipotensión ortostática, particularmente en el momento de iniciar el tratamiento, reiniciar el tratamiento o aumentar la dosis [ver Advertencias y precauciones (5.9)].
Interferencia con el Rendimiento Cognitivo y Motor
Advierta a los pacientes sobre la realización de actividades que requieren alerta mental, como el manejo de maquinaria peligrosa o la conducción de un vehículo de motor, hasta que estén razonablemente seguros de que la terapia con LATUDA no les afecta adversamente [ver Advertencias y precauciones (5.12)].
Exposición al Calor y Deshidratación
Eduque a los pacientes sobre el cuidado adecuado para evitar el sobrecalentamiento y la deshidratación [ver Advertencias y precauciones (5.13)].
Activación de la Manía o Hipomanía
Avise a los pacientes y a sus cuidadores que observen los signos de activación de la manía/hipomanía [ver Advertencias y precauciones (5.14)].
Medicamentos Concomitantes
Aconseje a los pacientes que informen a sus médicos si están tomando o planean tomar cualquier medicamento recetado o de venta libre, ya que existe la posibilidad de interacciones medicamentosas [ver Interacciones medicamentosas (7)].
Embarazo
Avise a las pacientes que LATUDA puede causar síntomas extrapiramidales y/o de abstinencia en un recién nacido. Aconseje a las pacientes que notifiquen a su proveedor de atención médica si tienen un embarazo conocido o sospechoso [ver Uso en poblaciones específicas (8.1)]. Avise a las pacientes que existe un registro de exposición al embarazo que monitoriza los resultados del embarazo en mujeres expuestas a LATUDA durante el embarazo [ver Uso en poblaciones específicas (8.1)].
Sumitomo Pharma Fabricado para: Sumitomo Pharma America, Inc. Marlborough, MA 01752 USA
Para Servicio al Cliente, llame al 1-888-394-7377. Para Información Médica, llame al 1-800-739-0565. Para reportar sospechas de reacciones adversas, llame al 1-877-737-7226.
Esta Guía del Medicamento ha sido aprobada por la Administración de Alimentos y Medicamentos de los EE. UU.
Revisado: 1/2025
GUÍA DEL MEDICAMENTO
LATUDA (luh-TOO-duh)
(lurasidone hydrochloride)
tabletas
¿Cuál es la información más importante que debo saber sobre LATUDA?
LATUDA puede causar efectos secundarios graves, que incluyen:
Mayor riesgo de muerte en personas mayores con psicosis relacionada con la demencia. Los medicamentos como LATUDA pueden aumentar el riesgo de muerte en personas mayores que han perdido el contacto con la realidad (psicosis) debido a confusión y pérdida de memoria (demencia). LATUDA no está aprobado para el tratamiento de personas con psicosis relacionada con la demencia.
Mayor riesgo de pensamientos o acciones suicidas en niños y adultos jóvenes. Los medicamentos antidepresivos pueden aumentar los pensamientos o acciones suicidas en algunos niños y adultos jóvenes durante los primeros meses de tratamiento y cuando se cambia la dosis.
La depresión y otras enfermedades mentales graves son las causas más importantes de pensamientos y acciones suicidas. Algunas personas pueden tener un riesgo particularmente alto de tener pensamientos o acciones suicidas. Estos incluyen personas que tienen (o tienen antecedentes familiares de) depresión, trastorno bipolar (también llamado enfermedad maníaco-depresiva) o antecedentes de pensamientos o acciones suicidas.
¿Cómo puedo estar atento e intentar prevenir pensamientos y acciones suicidas en mí mismo o en un miembro de mi familia?
Preste mucha atención a cualquier cambio, especialmente a los cambios repentinos en el estado de ánimo, los comportamientos, los pensamientos o los sentimientos. Esto es muy importante cuando se comienza a tomar un medicamento antidepresivo o cuando se cambia la dosis.
Llame al profesional de la salud de inmediato para informar cambios nuevos o repentinos en el estado de ánimo, el comportamiento, los pensamientos o los sentimientos.
Asista a todas las visitas de seguimiento con el profesional de la salud según lo programado. Llame al profesional de la salud entre visitas según sea necesario, especialmente si tiene inquietudes sobre los síntomas. Llame a un profesional de la salud de inmediato si usted o un miembro de su familia tiene alguno de los siguientes síntomas, especialmente si son nuevos, empeoran o le preocupan:
pensamientos sobre el suicidio o la muerte
depresión nueva o peor
sentirse muy agitado o inquieto
dificultad para dormir (insomnio)
actuar agresivamente, estar enojado o violento
un aumento extremo en la actividad y el habla (manía)
intentos de suicidio
ansiedad nueva o peor
ataques de pánico
irritabilidad nueva o peor
actuar por impulsos peligrosos
otros cambios inusuales en el comportamiento o el estado de ánimo
¿Qué es LATUDA?
LATUDA es un medicamento recetado que se usa:
Para tratar a personas de 13 años o mayores con esquizofrenia.
Solo para tratar a personas de 10 años o mayores con episodios depresivos que ocurren con el trastorno bipolar I (depresión bipolar).
Con el medicamento litio o valproato para tratar adultos con episodios depresivos que ocurren con el trastorno bipolar I (depresión bipolar).
Se desconoce si LATUDA es seguro y efectivo en niños:
menores de 13 años con esquizofrenia.
menores de 10 años con depresión bipolar.
para el tratamiento de la irritabilidad asociada con el trastorno autista.
No tome LATUDA si usted:
es alérgico al clorhidrato de lurasidona o a cualquiera de los ingredientes de LATUDA. Consulte la lista completa de ingredientes de LATUDA al final de esta Guía del Medicamento.
está tomando otros medicamentos llamados inhibidores o inductores de CYP3A4, incluidos ketoconazol, claritromicina, ritonavir, voriconazol, mibefradil, rifampicina, avasimiba, hierba de San Juan, fenitoína o carbamazepina. Pregúntele a su proveedor de atención médica si no está seguro de si está tomando alguno de estos medicamentos.
Antes de tomar LATUDA, informe a su proveedor de atención médica sobre todas sus afecciones médicas, incluso si usted:
tiene o ha tenido problemas cardíacos o un derrame cerebral
tiene o ha tenido presión arterial baja o alta
tiene o ha tenido diabetes o niveles altos de azúcar en la sangre, o tiene antecedentes familiares de diabetes o niveles altos de azúcar en la sangre.
tiene o ha tenido niveles altos de colesterol total o triglicéridos
tiene o ha tenido niveles altos de prolactina
tiene o ha tenido un recuento bajo de glóbulos blancos
tiene o ha tenido convulsiones
tiene o ha tenido problemas renales o hepáticos
está embarazada o planea quedar embarazada. Se desconoce si LATUDA dañará a su bebé nonato. Hable con su proveedor de atención médica sobre el riesgo para su bebé nonato si toma LATUDA durante el embarazo.
Informe a su proveedor de atención médica si queda embarazada o cree que está embarazada durante el tratamiento con LATUDA.
Si queda embarazada durante el tratamiento con LATUDA, hable con su proveedor de atención médica sobre cómo registrarse en el Registro Nacional de Embarazos para Antipsicóticos Atípicos. Puede registrarse llamando al 1-866-961-2388 o visitando http://womensmentalhealth.org/clinical-and-researchprograms/pregnancyregistry/.
está amamantando o planea amamantar. Se desconoce si LATUDA pasa a la leche materna. Hable con su proveedor de atención médica sobre la mejor manera de alimentar a su bebé durante el tratamiento con LATUDA.
Informe a su proveedor de atención médica sobre todos los medicamentos que toma, incluidos los medicamentos recetados y de venta libre, las vitaminas y los suplementos herbales. LATUDA y otros medicamentos pueden afectarse entre sí y causar posibles efectos secundarios graves. LATUDA puede afectar la forma en que funcionan otros medicamentos, y otros medicamentos pueden afectar la forma en que funciona LATUDA. Su proveedor de atención médica puede decirle si es seguro tomar LATUDA con sus otros medicamentos. No comience ni deje de tomar ningún otro medicamento durante el tratamiento con LATUDA sin antes hablar con su proveedor de atención médica. Conozca los medicamentos que toma. Mantenga una lista de sus medicamentos para mostrársela a su proveedor de atención médica y farmacéutico cuando reciba un nuevo medicamento.
¿Cómo debo tomar LATUDA?
Tome LATUDA exactamente como se lo indique su proveedor de atención médica. No cambie la dosis ni deje de tomar LATUDA sin antes hablar con su proveedor de atención médica.
Tome LATUDA por vía oral, con alimentos (al menos 350 calorías).
Si toma demasiado LATUDA, llame a su proveedor de atención médica o al centro de control de intoxicaciones o vaya a la sala de emergencias del hospital más cercano de inmediato.
¿Qué debo evitar mientras tomo LATUDA?
No conduzca, opere maquinaria pesada ni realice otras actividades peligrosas hasta que sepa cómo le afecta LATUDA. LATUDA puede causarle somnolencia.
Evite comer toronja o beber jugo de toronja durante el tratamiento con LATUDA. La toronja y el jugo de toronja pueden afectar la cantidad de LATUDA en su sangre.
No se caliente demasiado ni se deshidrate durante el tratamiento con LATUDA.
No haga demasiado ejercicio.
Cuando haga calor, permanezca adentro en un lugar fresco si es posible.
Manténgase alejado del sol.
No use demasiada ropa ni ropa pesada.
Beba mucha agua.
¿Cuáles son los posibles efectos secundarios de LATUDA?
LATUDA puede causar efectos secundarios graves, que incluyen:
Accidente cerebrovascular (problemas cerebrovasculares) en personas mayores con psicosis relacionada con la demencia que puede provocar la muerte.
Síndrome neuroléptico maligno (SNM), una afección grave que puede provocar la muerte. Llame a su proveedor de atención médica o vaya a la sala de emergencias del hospital más cercano de inmediato si tiene algunos o todos los siguientes signos y síntomas de SNM:
fiebre alta
confusión
cambios en su respiración, frecuencia cardíaca y presión arterial
rigidez muscular
aumento de la sudoración
Movimientos corporales incontrolables (discinesia tardía). LATUDA puede causar movimientos que usted no puede controlar en su cara, lengua u otras partes del cuerpo. La discinesia tardía puede no desaparecer, incluso si deja de tomar LATUDA. La discinesia tardía también puede comenzar después de que deje de tomar LATUDA.
Problemas con su metabolismo, tales como:
nivel alto de azúcar en la sangre (hiperglucemia) y diabetes. Aumentos en el azúcar en la sangre pueden ocurrir en algunas personas que toman LATUDA. Un nivel extremadamente alto de azúcar en la sangre puede provocar coma o la muerte. Si tiene diabetes o factores de riesgo de diabetes (como sobrepeso o antecedentes familiares de diabetes), su proveedor de atención médica debe controlar su nivel de azúcar en la sangre antes de comenzar y durante el tratamiento con LATUDA. Llame a su proveedor de atención médica si tiene alguno de estos síntomas de nivel alto de azúcar en la sangre durante el tratamiento con LATUDA:
mucha sed
mucha hambre
náuseas
necesidad de orinar con más frecuencia de lo habitual
debilidad o cansancio
confusión o aliento con olor a fruta
aumento de los niveles de grasa (colesterol y triglicéridos) en la sangre.
aumento de peso. Usted y su proveedor de atención médica deben controlar su peso regularmente durante el tratamiento con LATUDA.
Aumento de los niveles de prolactina en la sangre (hiperprolactinemia). Su proveedor de atención médica puede realizar análisis de sangre para controlar sus niveles de prolactina durante el tratamiento con LATUDA. Informe a su proveedor de atención médica si tiene alguno de los siguientes signos y síntomas de hiperprolactinemia: Mujeres:
ausencia de su ciclo menstrual
secreción de leche materna cuando no está amamantando
Hombres:
problemas para conseguir o mantener una erección (disfunción eréctil)
agrandamiento de los senos (ginecomastia)
Recuento bajo de glóbulos blancos. Su proveedor de atención médica puede realizar análisis de sangre durante los primeros meses de tratamiento con LATUDA.
Disminución de la presión arterial (hipotensión ortostática). Puede sentirse mareado o débil al levantarse demasiado rápido de una posición sentada o acostada.
Caídas. LATUDA puede causarle sueño o mareos, puede causar una disminución de su presión arterial al cambiar de posición (hipotensión ortostática) y puede ralentizar su pensamiento y sus habilidades motoras, lo que puede provocar caídas que pueden causar fracturas u otras lesiones.
Convulsiones.Problemas para controlar la temperatura corporal, por lo que se siente demasiado caliente. Consulte “¿¿Qué debo evitar mientras tomo LATUDA?” Manía o hipomanía (episodios maníacos) en personas con antecedentes de trastorno bipolar. Los síntomas pueden incluir:
gran aumento de energía
pensamientos acelerados
ideas inusualmente grandiosas
hablar más o más rápido de lo habitual
problemas graves para dormir
comportamiento imprudente
felicidad o irritabilidad excesiva
Dificultad para tragar Los efectos secundarios más comunes de LATUDA incluyen:
Adultos con esquizofrenia:
somnolencia o adormecimiento
inquietud y sensación de que necesita moverse (acatisia)
dificultad para moverse, movimientos lentos, rigidez muscular o temblor
náuseas
Niños de 13 a 17 años con esquizofrenia:
somnolencia o adormecimiento
náuseas
inquietud y sensación de que necesita moverse (acatisia)
dificultad para moverse, movimientos lentos, rigidez muscular o temblor
secreción nasal
vómitos
Adultos con depresión bipolar:
inquietud y sensación de que necesita moverse (acatisia)
dificultad para moverse, movimientos lentos, rigidez muscular o temblor
somnolencia o adormecimiento
Niños de 10 a 17 años con depresión bipolar:
náuseas
aumento de peso
problemas para dormir (insomnio)
Estos no son todos los posibles efectos secundarios de LATUDA. Llame a su médico para obtener asesoramiento médico sobre los efectos secundarios. Puede informar los efectos secundarios a la FDA al 1-800-FDA-1088.
¿Cómo debo almacenar LATUDA?
Guarde las tabletas de LATUDA a temperatura ambiente entre 20 °C y 25 °C (68 °F y 77 °F).
Mantenga LATUDA y todos los medicamentos fuera del alcance de los niños.
Información general sobre el uso seguro y eficaz de LATUDA.
Los medicamentos a veces se recetan para fines distintos de los que se enumeran en una Guía del medicamento. No use LATUDA para una afección para la que no fue recetado. No le dé LATUDA a otras personas, incluso si tienen los mismos síntomas que usted. Puede perjudicarlos. Puede pedirle a su farmacéutico o proveedor de atención médica información sobre LATUDA que esté escrita para profesionales de la salud.
ETIQUETA DEL ENVASE – PANEL PRINCIPAL DE PRESENTACIÓN – 20 mg, Etiqueta de 30 comprimidos
NDC 63402-302-30
30 Comprimidos
Latuda®
(lurasidone HCl) comprimidos
20 mg
ATENCIÓN AL DISPENSADOR: Cada vez que se dispense Latuda entregue al paciente la Guía de Medicamentos adjunta, también disponible en www.latuda.com o 1-888-394-7377.
Rx solamente
Sunovion
PANEL DE VISUALIZACIÓN PRINCIPAL
ETIQUETA DEL ENVASE – PANEL PRINCIPAL DE PRESENTACIÓN – 40 mg, Etiqueta de 30 comprimidos
NDC 63402-304-30
30 Comprimidos
Latuda®
(lurasidone HCl) comprimidos
40 mg
ATENCIÓN AL DISPENSADOR: Cada vez que se dispense Latuda entregue al paciente la Guía de Medicamentos adjunta, también disponible en www.latuda.com o 1-888-394-7377.
Rx solamente
Sunovion
PANEL DE VISUALIZACIÓN PRINCIPAL
ETIQUETA DEL ENVASE – PANEL DE VISUALIZACIÓN PRINCIPAL – 60 mg, etiqueta de 30 tabletas
NDC 63402-306-30
30 Tabletas
Latuda®
(lurasidone HCl) tabletas
60 mg
ATENCIÓN DISPENSADOR: Cada vez que se dispense Latuda entregue al paciente la Guía de Medicamentos adjunta, también disponible en www.latuda.com o 1-888-394-7377.
Rx Only
Sunovion
PANEL DE VISUALIZACIÓN PRINCIPAL
ETIQUETA DEL ENVASE – PANEL DE VISUALIZACIÓN PRINCIPAL – 60 mg, etiqueta de 90 comprimidos
NDC 63402-306-90
90 Tabletas/Comprimidos
Latuda®
(lurasidone HCl) tabletas/comprimidos
60 mg
ATENCIÓN DISPENSADOR: Cada vez que se dispense Latuda, entregue al paciente la Guía del Medicamento adjunta, también disponible en www.latuda.com o al 1-888-394-7377.
Rx Only
Sunovion
PANEL DE VISUALIZACIÓN PRINCIPAL
ETIQUETA DEL ENVASE – PANEL PRINCIPAL DE PRESENTACIÓN – 60 mg, envase de 500 comprimidos
NDC 63402-306-50
500 Comprimidos
Latuda®
(lurasidone HCl) comprimidos
60 mg
ATENCIÓN AL DISPENSADOR: Cada vez que se dispense Latuda, entregue al paciente la Guía de Medicamentos adjunta, también disponible en www.latuda.com o al 1-888-394-7377.
Receta médica solamente
Sunovion
PANEL DE VISUALIZACIÓN PRINCIPAL
ETIQUETA DEL ENVASE – PANEL PRINCIPAL DE PRESENTACIÓN – Blíster de 60 mg
NDC 63402-306-07
7 Comprimidos
MUESTRA PROFESIONAL
NO APTO PARA VENTA O REEMBOLSO
Latuda®
(lurasidone HCl) comprimidos
60 mg por comprimido
Rx solamente
ATENCIÓN AL DISPENSADOR: Entregue la Guía del medicamento adjunta a cada paciente.
Sunovion
PANEL DE VISUALIZACIÓN PRINCIPAL
ETIQUETA DEL PAQUETE – PANEL DE VISUALIZACIÓN PRINCIPAL – Caja de 60 mg
NDC 63402-306-70
28 Tabletas (4 Blísters, 7 Tabletas Cada Uno)
MUESTRA PROFESIONAL
NO PARA LA VENTA O REEMBOLSO
Latuda®
(lurasidone HCl) tabletas
60 mg por tableta
Rx only
ATENCIÓN DISPENSADOR: Entregue la Guía de Medicamentos adjunta a cada paciente.
Sunovion
PANEL DE VISUALIZACIÓN PRINCIPAL
ETIQUETA DEL ENVASE – PANEL PRINCIPAL DE PRESENTACIÓN – 60 mg Blíster
NDC 63402-306-01
10 Tabletas
Latuda®
(lurasidone HCl) tablets
60 mg
EXP: LOT:
Sunovion Pharmaceuticals Inc.
PANEL DE VISUALIZACIÓN PRINCIPAL
ETIQUETA DEL ENVASE – PANEL PRINCIPAL DE PRESENTACIÓN – 60 mg, Cartón HUD
NDC 63402-306-10
100 Tabletas (10 Cartones, 10 Tabletas Cada Uno)
Latuda®
(lurasidone HCl) tabletas
60 mg por tableta
ATENCIÓN AL DISPENSADOR: Cada vez que se dispense Latuda, entregue al paciente la Guía de Medicamentos adjunta, también disponible en www.latuda.com o al 1-888-394-7377.
Rx solamente
Sunovion
PANEL DE VISUALIZACIÓN PRINCIPAL
ETIQUETA DEL ENVASE – PANEL PRINCIPAL DE PRESENTACIÓN – 80 mg, Etiqueta de 30 comprimidos
NDC 63402-308-30
30 Comprimidos
Latuda®
(lurasidone HCl) comprimidos
80 mg
ATENCIÓN AL DISPENSADOR: Cada vez que se dispense Latuda entregue al paciente la Guía de Medicamentos adjunta, también disponible en www.latuda.com o 1-888-394-7377.
Rx solamente
Sunovion
PANEL DE VISUALIZACIÓN PRINCIPAL
ETIQUETA DEL ENVASE – PANEL PRINCIPAL DE PRESENTACIÓN – 120 mg, Etiqueta de 30 comprimidos
NDC 63402-312-30
30 Comprimidos
Latuda®
(lurasidone HCl) comprimidos
120 mg
ATENCIÓN AL DISPENSADOR: Cada vez que se dispense Latuda entregue al paciente la Guía de Medicamentos adjunta, también disponible en www.latuda.com o 1-888-394-7377.
Fabricante de medicamentos: Boehringer Ingelheim Pharmaceuticals, Inc. (Updated: 2024-10-15) Tags: RANKL ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓNEstos aspectos destacados no incluyen toda la información necesaria para usar JARDIANCE de forma segura y eficaz. Consulte la información completa de prescripción de JARDIANCE. JARDIANCE® (comprimidos de empagliflozina), para administración…
Fabricante de medicamentos: sanofi-aventis U.S. LLC (Updated: 2024-09-27) Tags: Proton pump ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓNEstos aspectos destacados no incluyen toda la información necesaria para usar DUPIXENT de forma segura y eficaz. Consulte la información completa de prescripción de DUPIXENT. DUPIXENT® (dupilumab) inyección, para uso subcutáneoAprobación…
Fabricante de medicamentos: Takeda Pharmaceuticals America, Inc. (Updated: 2023-07-25) Tags: IL6 ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓNEstos aspectos destacados no incluyen toda la información necesaria para usar DEXILANT de forma segura y eficaz. Consulte la información completa de prescripción de DEXILANT. DEXILANT (dexlansoprazol) cápsulas de liberación retardada,…