Fabricante de medicamentos: Bayer HealthCare LLC (Updated: 2022-12-01)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
KOVALTRY [Factor VIII Antihemofílico (Recombinante)]
Polvo liofilizado para solución para inyección intravenosa – Reconstitución con adaptador de vial
Aprobación inicial en EE. UU.: 2016
INDICACIONES Y USO
KOVALTRY®, Factor VIII Antihemofílico (Recombinante), es un concentrado de Factor VIII de longitud completa derivado de una secuencia de ADN humano recombinante, indicado para su uso en adultos y niños con hemofilia A (deficiencia congénita de Factor VIII) para:
• Tratamiento a demanda y control de episodios hemorrágicos
• Manejo perioperatorio de la hemorragia
• Profilaxis de rutina para reducir la frecuencia de episodios hemorrágicos
KOVALTRY no está indicado para el tratamiento de la enfermedad de von Willebrand (1).
DOSIFICACIÓN Y ADMINISTRACIÓN
Para uso intravenoso después de la reconstitución únicamente.
Control de episodios hemorrágicos y manejo perioperatorio (2.1)
- •
- Dosis requerida (UI) = peso corporal (kg) x aumento deseado de Factor VIII (% de lo normal o UI/dL) x recíproco de la recuperación esperada/observada (por ejemplo, 0,5 para una recuperación de 2 UI/dL por UI/kg).
- •
- Incremento estimado de Factor VIII (UI/dL o % de lo normal) = [Dosis total (UI)/peso corporal (kg)] x 2 (UI/dL por UI/kg).
Profilaxis de rutina (2.1)
- •
- Adultos y adolescentes: 20-40 UI/kg 2 o 3 veces por semana.
- •
- Niños ≤12 años de edad: 25-50 UI/kg 2 veces por semana, 3 veces por semana o cada dos días.
FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
- KOVALTRY está disponible como polvo liofilizado en viales de dosis única que contienen nominalmente 250, 500, 1000, 2000 o 3000 UI. Cada vial de KOVALTRY contiene la cantidad etiquetada de Factor VIII recombinante en UI (3).
CONTRAINDICACIONES
No lo use en pacientes que tengan antecedentes de reacciones de hipersensibilidad a la sustancia activa, proteína de ratón o hámster, u otros componentes del producto (4).
ADVERTENCIAS Y PRECAUCIONES
- •
- Las reacciones de hipersensibilidad, incluida la anafilaxia, son posibles. Si se presentan síntomas, suspenda el tratamiento con KOVALTRY y administre el tratamiento adecuado (5.1).
- •
- Se ha producido el desarrollo de anticuerpos neutralizantes del Factor VIII. Realice un ensayo que mida la concentración de inhibidores del Factor VIII si no se alcanzan los niveles esperados de actividad del Factor VIII en plasma, o si la hemorragia no se controla como se esperaba con la dosis administrada (5.2, 5.5).
REACCIONES ADVERSAS
Las reacciones adversas más frecuentes informadas en los ensayos clínicos (≥5%) fueron inhibidores en pacientes previamente no tratados (PUP)/pacientes mínimamente tratados (MTP), pirexia, cefalea y erupción cutánea (6).
Para informar sobre REACCIONES ADVERSAS SOSPECHOSAS, comuníquese con Bayer HealthCare al 1-888-842-2937 o con la FDA al 1-800-FDA-1088 o www.fda.gov/medwatch.
USO EN POBLACIONES ESPECÍFICAS
- Uso pediátrico: Debido a una mayor depuración (ajustada al peso corporal) en niños ≤12 años de edad, es posible que se necesiten dosis más altas o más frecuentes (8.4).
Consulte el 17 para obtener INFORMACIÓN PARA EL PACIENTE y el etiquetado del paciente aprobado por la FDA.
Revisado: 12/2022
Tabla de Contenido
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDO*
1 INDICACIONES Y USO
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Dosis
2.2 Preparación y Reconstitución
2.3 Administración
3 FORMAS Y FUERZAS DE DOSIFICACIÓN
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Reacciones de Hipersensibilidad
5.2 Anticuerpos Neutralizantes
5.3 Factores de Riesgo Cardiovascular
5.4 Infecciones Relacionadas con el Catéter
5.5 Monitoreo de Pruebas de Laboratorio
6 REACCIONES ADVERSAS
6.1 Experiencia en Ensayos Clínicos
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.4 Uso Pediátrico
8.5 Uso Geriátrico
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de Acción
12.2 Farmacodinamia
12.3 Farmacocinética
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
14 ESTUDIOS CLÍNICOS
16 CÓMO SE SUMINISTRA/ALMACENAMIENTO Y MANEJO
17 INFORMACIÓN PARA EL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información completa de prescripción no están enumeradas.
1 INDICACIONES Y USO
- KOVALTRY, Factor VIII (Recombinant), es un concentrado de Factor VIII de longitud completa, derivado de una secuencia de ADN humano recombinante, indicado para su uso en adultos y niños con hemofilia A (deficiencia congénita de Factor VIII) para:
- •
- Tratamiento a demanda y control de episodios hemorrágicos
- •
- Manejo perioperatorio de la hemorragia
- •
- Profilaxis de rutina para reducir la frecuencia de episodios hemorrágicos
KOVALTRY no está indicado para el tratamiento de la enfermedad de von Willebrand.
2 DOSIS Y ADMINISTRACIÓN
Solo para uso intravenoso después de la reconstitución.
2.1 Dosis
- •
- La dosis y la duración del tratamiento dependen de la gravedad de la deficiencia de Factor VIII, la ubicación y la extensión de la hemorragia, y el estado clínico del paciente. El control cuidadoso de la terapia de reemplazo es especialmente importante en casos de cirugía mayor o episodios de sangrado potencialmente mortales.
- •
- Cada etiqueta de vial de KOVALTRY indica la potencia del Factor VIII en unidades internacionales (UI). Una UI se define según el estándar internacional (IS) actual de la OMS (Organización Mundial de la Salud) para el concentrado de Factor VIII.
- •
- La asignación de potencia para KOVALTRY se determina mediante un ensayo de sustrato cromogénico. Un estudio de campo en el que participaron 41 laboratorios clínicos de todo el mundo midió las recuperaciones de KOVALTRY añadido al plasma hemofílico. Los resultados del estudio de campo indicaron que la actividad del Factor VIII de KOVALTRY puede medirse con precisión en plasma utilizando un ensayo de coagulación de una etapa o un ensayo de sustrato cromogénico de acuerdo con los métodos de rutina del laboratorio de análisis.
- •
- La dosis requerida para un nivel deseado de Factor VIII expresado como UI/dL (o % de lo normal) puede estimarse utilizando la siguiente fórmula:
-
Dosis requerida (UI) = peso corporal (kg) x aumento deseado de Factor VIII (% de lo normal o UI/dL)
x recíproco de la recuperación esperada/observada (p. ej., 0,5 para una recuperación de 2 UI/dL por UI/kg) - El aumento máximo esperado in vivo del nivel de Factor VIII expresado como UI/dL (o % de lo normal) puede estimarse utilizando la siguiente fórmula:
-
Incremento estimado de Factor VIII (UI/dL o % de lo normal) = [Dosis total (UI)/peso corporal (kg)]
x 2 (UI/dL por UI/kg) - Ejemplos (asumiendo que el Factor VIII basal del paciente es <1%):
- •
- Se requiere un pico del 50% [50 UI/dL] en un niño de 20 kg. En esta situación, la dosis requerida de KOVALTRY sería 20 kg x 50 UI/dL x 0,5 (para una recuperación de 2 UI/dL por UI/kg) = 500 UI
- •
- Se espera que una dosis de 2000 UI de KOVALTRY administrada a un paciente de 50 kg dé como resultado un aumento del Factor VIII después de la infusión de 2000 UI / 50 kg (peso corporal) x 2 UI/dL por UI/kg = 80 UI/dL (80% de lo normal)
- •
- Ajuste la dosis a la respuesta clínica del paciente. Los pacientes pueden variar en sus respuestas farmacocinéticas (p. ej., semivida, recuperación incremental) y clínicas a KOVALTRY.
- •
- Tratamiento a demanda y control de episodios de sangrado
En la Tabla 1 se proporciona una guía para la dosificación de KOVALTRY para el tratamiento a demanda y el control de los episodios de sangrado. El objetivo del tratamiento es mantener un nivel de actividad del Factor VIII en plasma igual o superior a los niveles plasmáticos (en % de lo normal o en UI/dL) descritos en la Tabla 1.
|
Grado de sangrado |
Nivel de Factor VIII requerido |
Frecuencia de las dosis (horas) |
Duración del tratamiento (días) |
|
Leve (Hemartrosis temprana, sangrado leve muscular, oral) |
20–40 |
Repetir cada |
Al menos 1 día, hasta que el episodio de sangrado, según lo indique el dolor, se haya resuelto o se haya logrado la curación |
|
Moderado (Hemartrosis más extensa, sangrado muscular o hematoma) |
30–60 |
Repetir cada |
De 3 a 4 días o más hasta que el dolor y la discapacidad aguda se hayan resuelto |
|
Grave (hemorragias intracraneales, intraabdominales o intratorácicas, hemorragia gastrointestinal, hemorragia del sistema nervioso central, hemorragia en los espacios retrofaríngeos o retroperitoneales, o vaina del iliopsoas, hemorragia potencialmente mortal o de extremidades) |
60–100 |
Repetir cada |
Hasta que se resuelva el sangrado |
Manejo perioperatorio de la hemorragia
En la Tabla 2 se proporciona una guía para la dosificación de KOVALTRY durante la cirugía (manejo perioperatorio). El objetivo del tratamiento es mantener un nivel de actividad del factor VIII en plasma igual o superior al nivel plasmático (en % del normal o en UI/dl) descrito en la Tabla 2. Durante una cirugía mayor, se recomienda encarecidamente la monitorización con pruebas de laboratorio adecuadas, incluidos ensayos seriados de la actividad del factor VIII [véanse Advertencias y precauciones (5.5)].
|
Tipo de cirugía |
Nivel de factor VIII requerido |
Frecuencia de las dosis (horas) |
Duración del tratamiento (días) |
|
Menor (como la extracción de un diente) |
30–60 |
Repetir cada 24 horas |
Al menos 1 día hasta que se logre la cicatrización |
|
Mayor (como cirugía intracraneal, intraabdominal, intratorácica o de reemplazo articular) |
80–100 |
Repetir cada |
Hasta que se complete la cicatrización adecuada de la herida, luego continuar el tratamiento durante al menos otros 7 días para mantener la actividad del factor VIII de |
Profilaxis de rutina
- •
- Individualice la dosis del paciente según la respuesta clínica.
- •
- Adultos y adolescentes: de 20 a 40 UI de KOVALTRY por kg de peso corporal dos o tres veces por semana.
- •
- Niños ≤12 años: de 25 a 50 UI de KOVALTRY por kg de peso corporal dos veces por semana, tres veces por semana o en días alternos según las necesidades individuales [consulte Uso en poblaciones específicas (8.4)].
2.2 Preparación y reconstitución
- •
- Reconstituya y administre KOVALTRY con los componentes proporcionados con cada envase. Si algún componente del envase está abierto o dañado, no lo utilice.
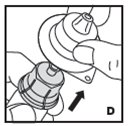
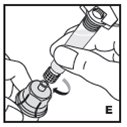
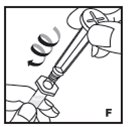
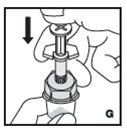
Los procedimientos siguientes se proporcionan como pautas generales para la reconstitución de KOVALTRY utilizando el adaptador de vial estéril con un filtro de 15 micrómetros y una jeringa de diluyente precargada, que juntos sirven como un sistema alternativo de reconstitución sin aguja.
Prueba de usabilidad del adaptador de vial
Se realizaron pruebas de usabilidad con 60 usuarios, incluidos 15 pacientes pediátricos con hemofilia A (entre 10 y 17 años de edad), 15 pacientes adultos con hemofilia A (≥18 años de edad), 15 cuidadores y 15 profesionales sanitarios. Para simular la vida real, los pacientes pediátricos y adultos y los cuidadores recibieron una formación mínima, que incluía que los participantes realizaran una reconstitución supervisada y posteriormente una única reconstitución sin ayuda. Los profesionales sanitarios no recibieron formación en este estudio y pudieron aprender el procedimiento a partir de las Instrucciones de uso proporcionadas. Todos los participantes pudieron utilizar con éxito y seguridad el dispositivo adaptador de vial para la reconstitución.
Reconstitución
- •
- Trabaje sobre una superficie limpia y lávese bien las manos con agua tibia y jabón antes de realizar los procedimientos.
- •
- Reconstituya KOVALTRY con los componentes proporcionados con cada envase. Si algún componente del envase está abierto o dañado, no lo utilice.
- •
- Filtre el producto reconstituido para eliminar posibles partículas en la solución. El filtrado se realiza utilizando el adaptador de vial.
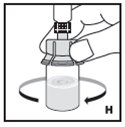
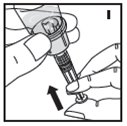
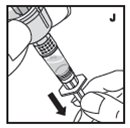
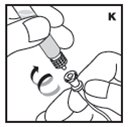
Combinación
Si la dosis requiere más de un vial, reconstituir cada vial como se describe anteriormente con la jeringa diluyente provista. Use una jeringa de plástico más grande (no provista) para combinar el contenido de los viales en la jeringa.
2.3 Administración
Sólo para uso intravenoso.
- •
- Inspeccione visualmente el KOVALTRY reconstituido en busca de materia particulada y decoloración antes de la administración. No lo use si nota cualquier materia particulada o decoloración.
- •
- Administre el KOVALTRY reconstituido tan pronto como sea posible. Si no, almacénelo a temperatura ambiente durante no más de 3 horas.
- •
- Infunda el KOVALTRY por vía intravenosa durante un período de 1 a 15 minutos. Ajuste la velocidad de administración a la respuesta de cada paciente individual.
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
KOVALTRY está disponible como polvo liofilizado en viales de vidrio de dosis única que contienen nominalmente 250, 500, 1000, 2000 o 3000 UI de Factor VIII recombinante por vial.
Cada vial de KOVALTRY está etiquetado con la potencia real del Factor VIII expresada en UI determinada mediante un ensayo de sustrato cromogénico. Esta asignación de potencia emplea un estándar de concentrado de Factor VIII que se refiere al Estándar Internacional actual de la OMS para el concentrado de Factor VIII, y se evalúa mediante una metodología adecuada para garantizar la precisión de los resultados.
4 CONTRAINDICACIONES
KOVALTRY está contraindicado en pacientes con antecedentes de reacciones de hipersensibilidad a la sustancia activa, a cualquiera de los excipientes o a proteínas de ratón o hámster [ver Descripción (11)].
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Reacciones de Hipersensibilidad
Las reacciones de hipersensibilidad, incluida la anafilaxia, son posibles con KOVALTRY. Los signos tempranos de reacciones de hipersensibilidad, que pueden progresar a anafilaxia, pueden incluir opresión en el pecho o la garganta, mareos, hipotensión leve y náuseas. Suspenda KOVALTRY si se presentan síntomas y busque atención médica de emergencia inmediata.
KOVALTRY puede contener trazas de proteínas de ratón y hámster [ver Descripción (11)]. Los pacientes tratados con este producto pueden desarrollar hipersensibilidad a estas proteínas de mamíferos no humanos.
5.2 Anticuerpos Neutralizantes
Se ha producido la formación de anticuerpos neutralizantes (inhibidores) después de la administración de KOVALTRY. Los pacientes previamente no tratados (PUP) tienen el mayor riesgo de desarrollar inhibidores con todos los productos del Factor VIII [ver Reacciones adversas (6.1)]. Monitoree cuidadosamente a los pacientes para detectar el desarrollo de inhibidores del Factor VIII, utilizando observaciones clínicas y pruebas de laboratorio apropiadas. Si no se alcanzan los niveles esperados de actividad del Factor VIII en plasma o si el sangrado no se controla como se esperaba con la dosis administrada, sospeche la presencia de un inhibidor (anticuerpo neutralizante) [ver Advertencias y precauciones (5.5)].
5.3 Factores de Riesgo Cardiovascular
Los pacientes hemofílicos con factores de riesgo cardiovascular o enfermedades pueden tener el mismo riesgo de desarrollar eventos cardiovasculares que los pacientes no hemofílicos cuando la coagulación se ha normalizado mediante el tratamiento con Factor VIII.
5.4 Infecciones Relacionadas con el Catéter
Se pueden observar infecciones relacionadas con el catéter cuando KOVALTRY se administra a través de dispositivos de acceso venoso central (CVAD). Estas infecciones no se han asociado con el producto en sí.
5.5 Monitoreo de Pruebas de Laboratorio
- •
- Monitoree los niveles de actividad del Factor VIII en plasma utilizando una prueba validada para confirmar que se han logrado y mantenido los niveles adecuados de Factor VIII [ver Dosificación y administración (2.1)].
- •
- Monitoree el desarrollo de inhibidores del Factor VIII. Realice un ensayo de inhibición de Bethesda si no se alcanzan los niveles esperados de Factor VIII en plasma o si el sangrado no se controla con la dosis esperada de KOVALTRY. Utilice unidades de Bethesda (BU) para informar los títulos de inhibidores.
6 REACCIONES ADVERSAS
Las reacciones adversas más frecuentes en los ensayos clínicos (≥5%) fueron inhibidores en pacientes previamente no tratados (PUP)/pacientes mínimamente tratados (MTP), pirexia, cefalea y erupción cutánea (ver Tabla 3).
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se llevan a cabo en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no se pueden comparar directamente con las tasas en los ensayos clínicos de otro fármaco y pueden no reflejar las tasas observadas en la práctica clínica.
El perfil de seguridad de KOVALTRY se evaluó en 236 (193 previamente tratados (PTPs), 43 PUP/MTP) pacientes, incluidos 94 pacientes pediátricos <12 años de edad. El análisis de seguridad se realizó utilizando una base de datos agrupada de tres estudios clínicos multicéntricos, prospectivos y de etiqueta abierta. El tiempo medio en el ensayo clínico para la población de seguridad agrupada fue de 558 días con una mediana de 183 días de exposición (ED). La mayoría de los pacientes (N=201) acumularon ≥ 100 ED.
El número total de días de exposición a KOVALTRY para todos los tratamientos, incluida la gestión perioperatoria, fue de 65029 ED. Los sujetos que recibieron KOVALTRY para la gestión perioperatoria (N=5) con un período de tratamiento de 2 a 3 semanas y aquellos que recibieron dosis únicas de KOVALTRY para estudios de PK (N=6) fueron excluidos del análisis de seguridad agrupado. La Tabla 3 enumera las reacciones adversas notificadas durante los estudios clínicos. Para los PTP, la frecuencia, el tipo y la gravedad de las reacciones adversas en los niños son similares a las de los adultos.
| Clase de órgano del sistema primario MedDRA Término preferido |
Frecuencia de las reacciones adversas a los medicamentos N (%) |
|---|---|
|
Trastornos de la sangre y del sistema linfático |
|
|
Linfadenopatía |
2 (0.8%) |
|
Trastornos cardíacos |
|
|
Palpitaciones Taquicardia sinusal |
2 (0.8%) 2 (0.8%) |
|
Trastornos gastrointestinales |
|
|
Dolor abdominal Malestar abdominal Dispepsia |
9 (3.8%) 3 (1.3%) 4 (1.7%) |
|
Trastornos generales y condiciones del lugar de administración |
|
|
Pirexia Dolor en el pecho Reacciones en el lugar de la inyección* |
22 (9.3%) 2 (0.8%) 6 (2.5%) |
|
Trastornos del sistema inmunitario |
|
|
Hipersensibilidad |
1 (0.4%) |
|
Trastornos del sistema nervioso |
|
|
Mareos Disgeusia Cefalea |
3 (1.3%) 1 (0.4%) 20 (8.5%) |
|
Trastornos psiquiátricos |
|
|
Insomnio |
5 (2.1%) |
|
Trastornos de la piel y del tejido subcutáneo |
|
|
Dermatitis alérgica Prurito Erupción cutánea† Urticaria |
2 (0.8%) 6 (2.5%) 13 (5.5%) 3 (1.3%) |
|
Trastornos vasculares |
|
|
Rubor |
1 (0.4%) |
En PTP (N=193), los inhibidores del Factor VIII ocurrieron en 1 paciente con una frecuencia del 0.5% (1/193) y en PUP/MTP (N=43) con una frecuencia del 54.8% (23/42); el análisis de inhibidores se basó en 42 de 43 PUP/MTP, ya que un paciente no fue evaluable para el análisis de inhibidores y por lo tanto fue excluido.
Inmunogenicidad
Todos los sujetos de los ensayos clínicos fueron monitoreados para detectar anticuerpos neutralizantes (inhibidores) al Factor VIII mediante el ensayo de Bethesda modificado utilizando muestras de sangre obtenidas antes de la primera infusión de KOVALTRY, a intervalos definidos durante los estudios y en la visita de finalización.
Los ensayos clínicos con KOVALTRY evaluaron un total de 204 pacientes pediátricos y adultos tratados diagnosticados con hemofilia A grave (Factor VIII <1%) con exposición previa a concentrados de Factor VIII ≥50 EDs, ningún PTP desarrolló anticuerpos neutralizantes al Factor VIII. Un caso de inhibidor de bajo título transitorio (0.6 BU/mL (título máximo: 1.0 BU/mL)) ocurrió en un PTP de 13 años después de 549 EDs concurrentes con una infección aguda y anticuerpos anticardiolipina IgG positivos. La recuperación del Factor VIII fue normal (2.7 UI/dL por UI/kg), la tasa anualizada de sangrado (ABR) fue cero y no se requirió ningún cambio en la terapia.
En la fase principal del ensayo clínico en PUP/MTP, se detectaron inhibidores del Factor VIII en 23 de 42 pacientes (54.8%, IC del 95%: 39 a 70%) con una mediana (rango) de 9 (4-42) EDs en el momento de la primera prueba de inhibidor positiva. De estos, 6 (14.3%) pacientes tuvieron inhibidores de bajo título (≤ 5.0 BU) que continuaron con el tratamiento y tuvieron resultados de inhibidores negativos al final del estudio. En 17 (40.5%) pacientes que desarrollaron inhibidores de alto título (> 5.0 BU), se identificaron mutaciones de alto riesgo para el desarrollo de inhibidores en 12 (85.7%) de 14 pacientes con datos disponibles de mutación de FVIII.
La detección de la formación de anticuerpos depende de la sensibilidad y especificidad del ensayo. Además, la incidencia observada de positividad de anticuerpos (incluidos los anticuerpos neutralizantes) en un ensayo puede verse influenciada por varios factores, incluida la metodología del ensayo, el manejo de las muestras, el momento de la recolección de las muestras, los medicamentos concomitantes y la enfermedad subyacente. Por estas razones, puede ser engañoso comparar la incidencia de anticuerpos a KOVALTRY con la incidencia de anticuerpos a otros productos.
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de Riesgos
No hay datos sobre el uso de KOVALTRY en mujeres embarazadas para informar sobre el riesgo asociado al fármaco. No se han realizado estudios de reproducción en animales utilizando KOVALTRY. Se desconoce si KOVALTRY puede causar daño fetal cuando se administra a una mujer embarazada o puede afectar la capacidad de reproducción. En la población general de los EE. UU., el riesgo de fondo estimado de defectos de nacimiento mayores y aborto espontáneo en embarazos clínicamente reconocidos es del 2-4% y del 15-20%, respectivamente.
8.2 Lactancia
Resumen de Riesgos
No hay información sobre la presencia de KOVALTRY en la leche materna, los efectos en el lactante amamantado o los efectos sobre la producción de leche. Los beneficios para el desarrollo y la salud de la lactancia materna deben considerarse junto con la necesidad clínica de la madre de KOVALTRY y cualquier posible efecto adverso en el lactante amamantado por KOVALTRY o por la condición materna subyacente.
8.4 Uso Pediátrico
Se han realizado estudios de seguridad y eficacia con KOVALTRY en 51 PTP pediátricos ≤12 años de edad y 43 PUP/MTP pediátricos <6 años de edad [ver Estudios Clínicos (14)].
El aclaramiento del Factor VIII ajustado al peso corporal en niños ≤12 años de edad es mayor que en adultos y adolescentes. Considere dosis más altas o más frecuentes en niños para tener en cuenta esta diferencia en el aclaramiento [ver Farmacología Clínica (12.3)].
8.5 Uso Geriátrico
Los estudios clínicos con KOVALTRY no incluyeron pacientes de 65 años o más para determinar si responden de manera diferente a los pacientes más jóvenes. Sin embargo, la experiencia clínica con otros productos de Factor VIII no ha identificado diferencias entre los pacientes ancianos y los pacientes más jóvenes. Al igual que con cualquier paciente que recibe Factor VIII recombinante, la selección de la dosis para un paciente anciano debe ser individualizada.
11 DESCRIPCIÓN
KOVALTRY, Factor Antihemofílico (Recombinante), es un polvo estéril, no pirógeno, de blanco a ligeramente amarillo para reconstitución contenido en un vial de dosis única. El producto final no contiene ningún conservante. El producto reconstituido está indicado para administración intravenosa. El producto está disponible en potencias nominales de 250 UI, 500 UI, 1000 UI, 2000 UI o 3000 UI; sin embargo, para cada concentración de dosis, la potencia real de Factor VIII ensayada se imprime directamente en la etiqueta de cada vial. El sistema de cierre del contenedor consiste en un vial de vidrio Tipo I de 10 mL sellado con un tapón gris de bromobutilo y un sello de crimpado de aluminio con tapa abatible de plástico más adaptador de vial. El adaptador de vial fue diseñado para conectarse con la agua estéril para inyección (sWFI), jeringa de diluyente prellenada. KOVALTRY está formulado con los siguientes excipientes: 2.2% glicina, 1% sacarosa, 30 mM cloruro de sodio, 2.5 mM cloruro de calcio, 20 mM histidina y 80 ppm polisorbato 80. El pH del producto reconstituido es de 6.6 a 7.0. La administración intravenosa de sacarosa contenida en KOVALTRY no afectará el nivel de glucosa en sangre.
La sustancia activa en KOVALTRY es la glicoproteína Factor VIII recombinante de longitud completa no modificada que comprende la secuencia de aminoácidos derivada de humanos. Las modificaciones postraduccionales son similares a las del Factor VIII endógeno, incluidos los sitios de glicosilación y la sulfatación de los sitios de tirosina. Los controles de fabricación y calidad aseguran que tanto el contenido de galactosa-alfa-1,3-galactosa (alfa-Gal) como el ácido N-glicolneuramínico (NGNA) estén por debajo del límite de detección del 1% establecido para cada método analítico.
KOVALTRY se produce mediante una línea celular de riñón de hámster bebé (BHK) genéticamente modificada en la que se introdujo el gen del Factor VIII humano junto con el gen de la proteína de choque térmico 70 (HSP 70) humana. HSP 70 es una proteína intracelular que mejora el plegamiento adecuado de la proteína Factor VIII. Si bien KOVALTRY y Kogenate FS tienen la misma estructura proteica, las materias primas de origen humano y animal no se agregan a los procesos de cultivo celular, purificación o formulación de KOVALTRY. En el proceso de fabricación de KOVALTRY, el Factor VIII recombinante se secreta en el medio de cultivo celular y se purifica de las impurezas relacionadas con el proceso y el producto mediante una serie de pasos de cromatografía y filtración. El proceso de producción incorpora dos pasos dedicados de eliminación viral: (1) un paso de tratamiento con detergente para la inactivación y (2) un paso de filtración de 20 nanómetros para la eliminación de virus y posibles agregados de proteínas.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
KOVALTRY reemplaza temporalmente el Factor VIII de coagulación faltante que se necesita para una hemostasia eficaz.
12.2 Farmacodinamia
El tiempo de coagulación del plasma, medido por el tiempo de tromboplastina parcial activada (aPTT), se prolonga en pacientes con hemofilia A. El tratamiento con KOVALTRY normaliza el aPTT.
12.3 Farmacocinética
La farmacocinética (PK) de KOVALTRY se investigó en PTP (de 0 a 61 años de edad) con hemofilia A grave después de la administración de 50 UI/kg de KOVALTRY. Los parámetros PK de KOVALTRY se presentan en la Tabla 4 (ensayo de coagulación de una etapa) y la Tabla 5 (ensayo de sustrato cromogénico). La PK de KOVALTRY fue similar entre la dosificación única y la repetida (en 19 sujetos después de 6 a 12 meses de profilaxis).
|
Parámetro [unidad] |
12 a 17 años (N=5) |
≥18 años |
|
AUC [UI*h/dL] |
1013.9 ± 286.8 |
1601.3 ± 520.0 |
|
Cmax [IU/dL] |
91.7 ± 28.7 |
99.7 ± 14.9 |
|
t½ [h] |
11.7 ± 1.11 |
14.3 ± 3.7 |
|
MRTIV [h] |
16.1 ± 0.8 |
19.8 ± 5.7 |
|
Vss [dL/kg] |
0.85 ± 0.24 |
0.63 ± 0.11 |
|
CL [dL/h/kg] |
0.053 ± 0.017 |
0.035 ± 0.012 |
AUC: área bajo la curva
Cmax: concentración máxima del fármaco en plasma después de una dosis única
t½ : vida media terminal
MRTIV: tiempo medio de residencia después de una administración IV
Vss: volumen de distribución aparente en estado estacionario
CL: aclaramiento
Los parámetros farmacocinéticos de KOVALTRY para sujetos de todos los grupos de edad se muestran en la Tabla 5. En general, los niños <12 años de edad demostraron concentraciones plasmáticas más bajas en comparación con los niños PTP ≥12 años de edad.
|
Parámetro [unidad] |
0 a <2 años (N=4) |
2 a <6 años (N=6) |
6 a <12 años |
12 a 17 años |
≥18 años |
|
AUC [UI*h/dL] |
1232.5 ± 581.3 |
1484.8 ± 411.3a |
1214.5 ± 395.1 |
1572.0 ± 448.0 |
2103.4 ± 702.8 |
|
Cmax [UI/dL] |
96.1 ± 20.4 |
83.3 ± 28.7 |
81.6± 17.8 |
132.5 ± 46.3 |
133.1 ± 20.4 |
|
t½ [h] |
9.6 ± 3.1 |
12.2 ± 3.1a |
12.0 ± 2.1 |
14.4 ± 5.5 |
14.2 ± 3.5 |
|
MRTIV [h] |
14.1 ± 4.7 |
17.9 ± 4.1a |
17.8 ± 2.9 |
19.8 ± 5.8 |
19.9 ± 4.9 |
|
Vss [dL/kg] |
0.63 ± 0.18 |
0.60 ± 0.15a |
0.79 ± 0.23 |
0.71 ± 0.39 |
0.50 ± 0.11 |
|
CL [dL/h/kg] |
0.050 ± 0.024 |
0.034 ± 0.011a |
0.045 ± 0.016 |
0.034 ± 0.010 |
0.027 ± 0.010 |
a n=5
b Un sujeto considerado un valor atípico de PK fue excluido
El análisis de recuperación incremental después de 6 meses de tratamiento profiláctico arrojó resultados comparables con la recuperación incremental después de la primera dosis (consulte la Tabla 6).
|
0 a <6 años |
6 a 12 años |
≥12 años |
|
|
Resultados del ensayo de sustrato cromogénicoa Mediana (Q1; Q3) (UI/dL por UI/kg) |
1.6 (1.3; 1.9) |
1.7 (1.4; 2.0) |
2.3 (1.8; 2.6) |
|
Resultados del ensayo de una etapaa Mediana (Q1; Q3) (UI/dL por UI/kg) |
– |
– |
2.2 (1.8; 2.4) |
aInicio del ensayo
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
No se han realizado estudios a largo plazo en animales para evaluar el potencial carcinogénico de KOVALTRY u otros estudios para determinar los efectos de KOVALTRY sobre la fertilidad. KOVALTRY fue negativo en la prueba de genotoxicidad modificada in-vitro (Ensayo de Mutación y Aberración Cromosómica de Mamíferos con Células de Linfoma de Ratón). Se espera que KOVALTRY no tenga potencial mutagénico.
14 ESTUDIOS CLÍNICOS
La seguridad y eficacia de KOVALTRY para el tratamiento a demanda y el control de episodios hemorrágicos, el manejo perioperatorio de la hemorragia y la profilaxis de rutina en sujetos con hemofilia A grave (<1% de Factor VIII) se evaluó en tres estudios clínicos internacionales (incluidos los EE. UU.). Los sujetos inmunocompetentes con hemofilia A grave (actividad del Factor VIII ≤1%) y sin antecedentes de inhibidores del Factor VIII fueron elegibles para los ensayos.
Ensayo 1: un estudio multicéntrico, abierto, cruzado, no controlado, en adolescentes y adultos (edad ≥12 años a <65 años) PTP (≥150 ED) evaluó la farmacocinética, eficacia y seguridad de la profilaxis de rutina y el manejo perioperatorio de la hemorragia de KOVALTRY (ver Tabla 7). La variable principal de eficacia fue ABR. El régimen profiláctico fue de 20 a 50 UI/kg dos o tres veces por semana, en el que la frecuencia de dosificación fue asignada por el investigador en función de las necesidades individuales del sujeto.
Ensayo 2: un estudio multicéntrico, abierto, cruzado, no controlado, aleatorizado en adolescentes y adultos (edad ≥12 años a <65 años) PTP (≥150 ED) evaluó la superioridad de la profilaxis sobre el tratamiento a demanda con KOVALTRY durante un período de tratamiento de un año (ver Tabla 7). La variable principal de eficacia fue ABR. El régimen profiláctico fue de 20 a 30 UI/kg dos veces por semana o de 30 a 40 UI/kg tres veces por semana y el grupo de tratamiento fue asignado por aleatorización.
Ensayo 3: un estudio multicéntrico, abierto, no controlado que evaluó la farmacocinética, eficacia y seguridad de la profilaxis de rutina y el manejo perioperatorio de la hemorragia de KOVALTRY (ver Tabla 8) en pacientes previamente tratados (PTP; ≥50 ED) ≤12 años de edad (Parte A) y pacientes previamente no tratados (PUP) y pacientes mínimamente tratados (MTP; ≤3 ED) <6 años de edad (Parte B). La variable principal de eficacia fue el número anualizado de hemorragias totales durante la profilaxis de rutina que ocurrieron dentro de las 48 horas posteriores a la infusión de profilaxis previa. También se analizó el ABR durante la profilaxis, independientemente del momento de la infusión. En la Parte A, el régimen profiláctico fue de 25 a 50 UI/kg con frecuencias de 2 veces por semana, 3 veces por semana o cada dos días y podría adaptarse a las necesidades individuales del sujeto por el investigador.
En todos los estudios, los tratamientos de hemorragias de avance y el manejo perioperatorio quedaron a discreción del investigador según el estándar de atención.
Un total de 204 sujetos previamente tratados (153 sujetos ≥12 años de edad y 51 sujetos <12 años de edad) y 43 PUP/MTP <6 años de edad fueron tratados en los ensayos clínicos. Entre los PTP, 174 sujetos fueron tratados durante al menos 12 meses y 78 de estos sujetos fueron tratados durante 24 meses. Entre los PUP/MTP, 34 sujetos fueron tratados durante al menos 12 meses y 20 de estos sujetos fueron tratados durante 24 meses.
| Ensayo 1 (N=62) |
Ensayo 2 (N=80) |
|
|---|---|---|
|
Edad: media ± DE |
31.5 ± 12.7 años |
29.6 ± 11.0 años |
|
Tratamiento previo: % |
Profilaxis: 80.6% |
A demanda: 100% |
|
Número de articulaciones diana en la línea de base: |
1.4 ± 1.3 |
3.0 ± 2.1 |
|
Historia de hemorragia articular |
8.0 ± 11.9 |
32.1 ± 23.8 |
| Ensayo 3 | ||
|---|---|---|
| PTP de 0 a <6 años (N=25) |
PTP de 6 a 12 años (N=26) |
|
|
Edad: media ± DE (rango) |
3.8 ± 1.3 años (1-5) |
8.8 ± 1.8 años (6-11) |
|
Tratamiento previo: % |
Profilaxis: 92.0% |
Profilaxis: 65.4% |
|
Número de articulaciones diana en la línea de base: media ± DE |
0.2 ± 0.4 |
0.7 ± 1.1 |
Tratamiento a demanda y control de episodios de sangrado
Adolescentes y adultos
Se trataron un total de 1892 episodios de sangrado en 110 sujetos con KOVALTRY en el ensayo 1 y el ensayo 2 (ver Tabla 9). La mayoría de los episodios de sangrado fueron espontáneos, localizados en las articulaciones y de gravedad leve a moderada.
En el ensayo 1 y el ensayo 2, las respuestas al tratamiento en un total de 1859 sangrados tratados fueron evaluadas por los sujetos en comparación con su experiencia previa de tratamiento.
|
Características de los episodios de sangrado |
Ensayo 1 |
Ensayo 2 |
||
|
Profilaxis |
Profilaxis |
Profilaxis |
A demanda |
|
|
Número total de sangrados |
241 |
154 |
293 |
1204 |
|
Espontáneos: n/total (%) |
153/241 (63.5%) |
79/150* (52.7%) |
209/283* (73.9%) |
943/1202* (78.5%) |
|
Trauma: n/total (%) |
79/241 (32.8%) |
70/150* (46.7%) |
74/283* (26.1%) |
258/1202* (21.5%) |
|
Sangrados en articulaciones: n/total (%) |
191/241 (79.3%) |
120/154 (77.9%) |
255/293 (87.0%) |
924/1197* (77.2%) |
|
Leve/moderado: n/total (%) |
215/241 (89.2%) |
130/153* (84.9%) |
260/293 (88.8%) |
1092/1196* (91.3%) |
|
% de sangrados tratados con ≤2 infusiones |
87.0% |
96.2% |
95.3% |
|
|
Respuesta al tratamiento de sangrados evaluada como “Excelente” o “Buena”: n/total† (%) |
190/235 (80.9%) |
107/149 (71.8%) |
172/279 (61.6%) |
834/1196 (69.7%) |
|
Dosis mediana por infusión (rango) |
31.6 IU/kg |
29.4 IU/kg |
22.0 IU/kg |
|
Niños de 12 años de edad o menos
Se trataron un total de 97 episodios hemorrágicos en 28 sujetos pediátricos previamente tratados y 105 episodios hemorrágicos en 37 PUP/MTP con KOVALTRY. La mayoría (96,9% en PTP y 97,1% en PUP/MTP) de las hemorragias fueron de leve a moderada en gravedad para ambos grupos. Cincuenta y nueve (72,8%) y 62 (59,0%) hemorragias estuvieron relacionadas con traumatismos para los sujetos previamente tratados y los PUP/MTP, respectivamente. Durante el período de tratamiento de 6 meses, la dosis mediana de KOVALTRY para el tratamiento de hemorragias de ruptura en los sujetos previamente tratados fue de 36,94 UI/kg por infusión (rango 20,8–71,6 UI/kg).
La evaluación de la respuesta al tratamiento de las hemorragias fue la siguiente:
Excelente: Alivio del dolor abrupto y/o mejora en los signos de hemorragia sin administración de infusión adicional; Bueno: Alivio del dolor definido y/o mejora en los signos de hemorragia, pero posiblemente requiriendo más de una infusión para la resolución completa; Moderado: Mejora probable o leve en los signos de hemorragia con al menos una infusión adicional para la resolución completa; Pobre: Ninguna mejora en absoluto entre las infusiones o la condición empeora.
La eficacia hemostática en el tratamiento a demanda de las hemorragias se evaluó como “buena” o “excelente” en el 90,1% de los casos (97,8% en el grupo de edad más joven y 81,0% en el grupo de edad más viejo). La mayoría de las hemorragias (89,7%) se trataron con éxito con ≤2 infusiones. La respuesta al tratamiento fue similar para los niños de 0 a <6 años en comparación con los de 6 a 12 años de edad (ver Tabla 10).
| Características de los episodios hemorrágicos | Ensayo 3 | ||
|---|---|---|---|
| PTP de 0 a <6 años (N=25) |
PTP de 6 a 12 años (N=26) |
PTP de 0 a 12 años (N=51) |
|
|
Número total de hemorragias |
52 |
45 |
97 |
|
Espontáneas: n/total (%) |
8/44* (18,2%) |
12/37* (32,4%) |
20/81* (24,7%) |
|
Traumatismos: n/total (%) |
36/44* (81,8%) |
23/37* (62,2%) |
59/81* (72,8%) |
|
Hemorragias articulares: n/total (%) |
10/52 (19,2%) |
22/45 (48,9%) |
32/97 (33,0%) |
|
Leve/moderada: n/total (%) |
50/52 (96,2%) |
44/45 (97,8%) |
94/97 (96,9%) |
|
% de hemorragias tratadas con ≤2 infusiones |
92,4% |
86,7% |
89,7% |
|
Respuesta al tratamiento de las hemorragias evaluada como “Excelente” o “Buena”: n/total† (%) |
43/44 (97,8%) |
30/37 (81,0%) |
73/81 (90,1%) |
|
Dosis mediana por infusión (rango) |
38,7 UI/kg |
32,4 UI/kg |
36,9 UI/kg |
Manejo Perioperatorio
Se realizaron un total de 14 cirugías mayores y 46 menores en 44 sujetos previamente tratados (43 adultos y adolescentes y 1 niño menor de 12 años) con hemofilia A grave. Siete de las 14 cirugías mayores en PTP fueron procedimientos ortopédicos, incluida la artroplastia. Aproximadamente el 51% de las cirugías menores en PTP fueron extracciones dentales. Todos los sujetos recibieron KOVALTRY como infusiones en bolo. En los sujetos adolescentes y adultos, las dosis iniciales de KOVALTRY administradas oscilaron entre 3000 y 5000 UI. La dosis total mediana el día de la cirugía fue de 107,5 UI/kg (rango 60-207 UI/kg). En un solo sujeto previamente tratado menor de 12 años que se sometió a una cirugía mayor, la dosis inicial total de KOVALTRY administrada fue de 2500 UI (108,7 UI/kg).
La pérdida de sangre, durante y después de la cirugía, estuvo dentro de los rangos esperados. El control hemostático fue evaluado por los cirujanos como “bueno” (sangrado perioperatorio leve pero no clínicamente significativo en comparación con las expectativas para el paciente no hemofílico; tratamiento similar al paciente no hemofílico) o “excelente” (pérdida de sangre perioperatoria similar a la del paciente no hemofílico).
Profilaxis de rutina
Adolescentes y adultos
Un total de 140 sujetos fueron tratados con KOVALTRY durante al menos 12 meses con una mediana (rango) de 157 ED (25-178) en el ensayo 1, [305 ED (25-355) inclusive de la fase de extensión], y 153 ED (103-187) en el ensayo 2 (ver Tabla 11). En ambos estudios, los sujetos en la población de intención de tratar (ITT) recibieron del 95% al 100% del número prescrito de infusiones de profilaxis.
| Ensayo 1 (N=62)* |
Ensayo 2 (N=59) |
|
|---|---|---|
|
||
|
Dosis nominal mediana de profilaxis/infusión (rango) Todos Profilaxis 2 veces por semana Profilaxis 3 veces por semana |
31,2 UI/kg (21-43 UI/kg) 35,0 UI/kg (21-42 UI/kg) 31,1 UI/kg (24-43 UI/kg) |
31,7 UI/kg (21-42 UI/kg) 30,4 UI/kg (21-34 UI/kg) 37,4 UI/kg (30-42 UI/kg) |
|
Duración del tratamiento |
1 año estudio principal |
1 año |
|
Ensayo 1: 2 veces por semana (n=18); 3 veces por semana (n=44) Ensayo 2: 2 veces por semana (n=28); 3 veces por semana (n=31) |
||
La media y la mediana de ABR para la población ITT en el ensayo 1 fue de 3,8 ± 5,2 y 1 sangrado/año, respectivamente. En el ensayo 2, la comparación de las tasas de sangrado entre los sujetos que recibieron terapia a demanda versus profilaxis en un ANOVA demostró una diferencia estadísticamente significativa (p<0,0001) en la mediana de ABR en los sujetos que recibieron terapia a demanda (60 sangrados por año) en comparación con los sujetos que recibieron profilaxis (2 sangrados por año). En el ensayo 2, la media de ABR en los sujetos que recibieron terapia a demanda fue de 57,7 ± 24,6 frente a 4,9 ± 6,8 en los sujetos que recibieron profilaxis.
|
||||
|
Ensayo 1 |
Ensayo 2 |
|||
|
2 veces por semana |
3 veces por semana |
2 veces por semana |
3 veces por semana |
|
|
ABR Mediana (IQR*Q1; Q3) |
||||
|
1.0 (0.0; 8.0) |
2.0 (0.5; 5.0) |
||
|
0.5 (0.0; 2.0) |
1.0 (0.0; 3.9) |
2.0 (0.0; 6.5) |
0.0 (0.0; 3.0) |
|
0.5 (0.0; 7.0) |
1.8 (0.0; 3.0) |
2.5 (0.0; 7.5) |
1.0 (0.0; 4.0) |
|
Sujetos con cero episodios de sangrado‡ % (n) |
37.5% (6/16§) |
62.5% (10/16§) |
28.6% (8/28¶) |
25.8% (8/31¶) |
La ABR para los sujetos (n=21) que recibieron terapia a demanda en el Ensayo 2 [mediana (IQR Q1; Q3)] para todos los sangrados: 60 (41.7; 76.3); sangrados espontáneos: 42.1 (24.3; 61.3); sangrados articulares: 38.8 (24.3; 60.0).
Niños de 12 años de edad o menores
En la Parte A, un total de 51 PTPs fueron tratados con KOVALTRY durante al menos 6 meses con una mediana (rango) de 73 EDs (37-103) (ver Tabla 13). Los sujetos recibieron >95% del número prescrito de infusiones de profilaxis.
| Ensayo 3 | ||
|---|---|---|
| PTPs de 0 a <6 años (N=25) |
PTPs de 6 a 12 años (N=26) |
|
|
||
|
Régimen de tratamiento* durante el estudio (6 meses) n (%) 2 veces por semana 3 veces por semana o cada dos días |
9 (36%) 16 (64%) |
13 (50%) 13 (50%) |
|
Dosis nominal de profilaxis por infusión, mediana (rango) |
36.4 IU/kg (21-58 IU/kg) |
31.8 IU/kg (22-50 IU/kg) |
En niños de 12 años de edad o menores (n=51), la mediana (IQR Q1; Q3) ABR dentro de las 48 horas posteriores a la infusión profiláctica fue de 0 (0; 4) para todos los sangrados, y 0 (0; 0) para los sangrados espontáneos y articulares. La mediana (IQR Q1; Q3) ABR durante el tratamiento profiláctico independientemente del momento de la infusión fue de 1.9 (0; 6) para todos los sangrados, 0 (0; 0) para los sangrados espontáneos y 0 (0; 2) para los sangrados articulares. La ABR media dentro de las 48 horas posteriores a la infusión profiláctica fue de 2.04 ± 2.91. La ABR media en cualquier momento durante el régimen de profilaxis fue de 3.75 ± 4.98.
En ambos grupos de edad (0 a <6 años y 6 a 12 años), la ABR para los sangrados espontáneos y articulares dentro de las 48 horas posteriores al tratamiento profiláctico [ABR mediana (IQR Q1; Q3)] fue de 0 (0; 0). La mediana (IQR Q1; Q3) del número anualizado de sangrados espontáneos durante el tratamiento profiláctico independientemente del momento de la infusión fue de 0 (0; 0). La mediana (IQR Q1; Q3) del número anualizado de sangrados articulares durante el tratamiento profiláctico independientemente del momento de la infusión fue de 0 (0; 1.9) en el grupo de edad de 0 a <6 años y 0 (0; 2.1) en el grupo de edad de 6 a 12 años (ver Tabla 14).
La mayoría (32/53) de los sangrados que ocurrieron dentro de las 48 horas posteriores a una infusión profiláctica previa estuvieron relacionados con traumatismos. Veintitrés (45.1%) sujetos no reportaron sangrados durante el período de profilaxis de seis meses.
| Ensayo 3 | ||||
|---|---|---|---|---|
| PTPs de 0 a <6 años (N=25) |
PTPs de 6 a 12 años (N=26) |
|||
|
Dentro de las 48 hrs posteriores al tratamiento profiláctico |
Durante el tratamiento profiláctico* |
Dentro de las 48 hrs posteriores al tratamiento profiláctico |
Durante el tratamiento profiláctico* |
|
|
Todos los sangrados ABR Mediana (IQR†Q1; Q3) |
1.9 (0.0; 4.0) |
2.0 (0.0; 6.0) |
0.0 (0.0; 2.0) |
0.9 (0.0; 5.8) |
|
Número de sujetos con cero episodios de sangrado (%) |
10 (40%) |
13 (50%) |
||
16 FORMA DE SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
Presentación
KOVALTRY está disponible como polvo liofilizado en viales de vidrio de dosis única, un vial por caja. Se suministra con un adaptador de vial estéril con filtro de 15 micrómetros y una jeringa de barril de vidrio con diluyente prellenado, que juntos sirven como un sistema de reconstitución sin aguja. La jeringa de diluyente prellenada contiene Agua estéril para inyección, USP. También se proporciona un equipo de administración en el paquete. Tamaños disponibles:
|
Concentración nominal (UI) |
Diluyente (mL) |
Número de NDC del kit |
Código de color |
|
250 |
2.5 |
0026-3821-25 |
Azul |
|
500 |
2.5 |
0026-3822-25 |
Verde |
|
1000 |
2.5 |
0026-3824-25 |
Rojo |
|
2000 |
5.0 |
0026-3826-50 |
Amarillo |
|
3000 |
5.0 |
0026-3828-50 |
Gris |
La actividad real del Factor VIII en UI se indica en la etiqueta de cada vial de KOVALTRY.
El vial del producto y la jeringa de diluyente no están hechos con látex de caucho natural.
Almacenamiento y manipulación
Producto envasado para la venta
- •
- Almacene KOVALTRY a +2°C a +8°C (36°F a 46°F) durante un máximo de 30 meses a partir de la fecha de fabricación. No congelar. Dentro de este período, KOVALTRY puede almacenarse durante un período único de hasta 12 meses a temperaturas de hasta +25°C o 77°F.
- •
- Registre la fecha de inicio del almacenamiento a temperatura ambiente en la caja del producto sin abrir. Una vez almacenado a temperatura ambiente, no devuelva el producto al refrigerador. La vida útil expira después del almacenamiento a temperatura ambiente durante 12 meses, o después de la fecha de caducidad en el vial del producto, lo que ocurra primero.
- •
- No utilice KOVALTRY después de la fecha de caducidad indicada en el vial.
- •
- Proteja KOVALTRY de la exposición extrema a la luz y almacene el vial con el polvo liofilizado en la caja antes de su uso.
Producto después de la reconstitución
- •
- Administre KOVALTRY reconstituido lo antes posible. De lo contrario, almacene a temperatura ambiente durante no más de 3 horas.
- •
- No utilice KOVALTRY si la solución reconstituida está turbia o tiene partículas.
- •
- Utilice el equipo de administración proporcionado.
17 INFORMACIÓN PARA EL PACIENTE
- •
- Aconseje al paciente que lea el etiquetado del paciente aprobado por la FDA (Información del paciente e Instrucciones de uso).
- •
- Las reacciones de hipersensibilidad son posibles con KOVALTRY [ver Advertencias y precauciones (5.1)]. Advierta a los pacientes sobre los signos tempranos de reacciones de hipersensibilidad (incluida la opresión en el pecho o la garganta, mareos, hipotensión leve y náuseas durante la infusión) que pueden progresar a anafilaxia. Aconseje a los pacientes que interrumpan el uso del producto si se presentan estos síntomas y busquen atención médica de emergencia inmediata con medidas de reanimación, como la administración de epinefrina y oxígeno.
- •
- La formación de inhibidores puede ocurrir en cualquier momento en el tratamiento de un paciente con hemofilia A [ver Advertencias y precauciones (5.2)]. Aconseje a los pacientes que se pongan en contacto con su médico o centro de tratamiento para recibir tratamiento y/o evaluación adicionales, si experimentan una falta de respuesta clínica a la terapia de reemplazo del factor VIII, ya que esto puede ser una manifestación de un inhibidor.
INSERTO PARA EL PACIENTE
Etiquetado del paciente aprobado por la FDA
Información para el paciente
KOVALTRY (KOH-vahl-tree)
Factor antihemofílico (recombinante)
Este folleto resume información importante sobre KOVALTRY con adaptador de vial. Léalo detenidamente antes de usar este medicamento. Esta información no reemplaza la conversación con su proveedor de atención médica y no incluye toda la información importante sobre KOVALTRY. Si tiene alguna pregunta después de leer esto, consulte a su proveedor de atención médica.
No intente autoinfundirse a menos que su proveedor de atención médica o centro de hemofilia le haya enseñado cómo hacerlo.
¿Qué es KOVALTRY?
KOVALTRY es un medicamento que se usa para reemplazar el factor de coagulación (Factor VIII o factor antihemofílico) que falta en las personas con hemofilia A (también llamada hemofilia “clásica”). La hemofilia A es un trastorno hemorrágico hereditario que evita que la sangre coagule normalmente.
KOVALTRY se usa para tratar y controlar el sangrado en adultos y niños con hemofilia A. Su proveedor de atención médica puede administrarle KOVALTRY cuando se someta a una cirugía. KOVALTRY puede reducir la cantidad de episodios de sangrado en adultos y niños con hemofilia A cuando se usa regularmente (profilaxis).
KOVALTRY no se usa para tratar la enfermedad de von Willebrand.
¿Quién no debe usar KOVALTRY?
No debe usar KOVALTRY si usted
- •
- es alérgico a los roedores (como ratones y hámsters).
- •
- es alérgico a cualquiera de los ingredientes de KOVALTRY.
¿Qué debo decirle a mi proveedor de atención médica antes de usar KOVALTRY?
- •
- Informe a su proveedor de atención médica sobre todas sus afecciones médicas.
- •
- Informe a su proveedor de atención médica y farmacéutico sobre todos los medicamentos que toma, incluidos todos los medicamentos recetados y de venta libre, como medicamentos de venta libre, suplementos o remedios herbales.
- •
- Informe a su proveedor de atención médica si le han dicho que tiene una enfermedad cardíaca o si está en riesgo de tener una enfermedad cardíaca.
- •
- Informe a su proveedor de atención médica si le han dicho que tiene inhibidores del Factor VIII (porque KOVALTRY puede no funcionar para usted).
¿Cuáles son los posibles efectos secundarios de KOVALTRY?
Los efectos secundarios comunes de KOVALTRY son fiebre, dolor de cabeza y erupción cutánea, además de inhibidores en pacientes que no fueron tratados previamente o que fueron tratados mínimamente con productos del Factor VIII.
Su cuerpo puede producir anticuerpos, llamados “inhibidores” contra KOVALTRY, que pueden impedir que KOVALTRY funcione correctamente. Si su sangrado no se controla adecuadamente, podría deberse al desarrollo de inhibidores del Factor VIII. Consulte con su proveedor de atención médica para asegurarse de que lo monitoreen cuidadosamente con análisis de sangre para detectar el desarrollo de inhibidores del Factor VIII.
Pueden ocurrir reacciones alérgicas con KOVALTRY. Llame a su proveedor de atención médica de inmediato y suspenda el tratamiento si experimenta opresión en el pecho o la garganta, mareos, disminución de la presión arterial y náuseas.
Estos no son todos los posibles efectos secundarios de KOVALTRY. Puede pedirle a su proveedor de atención médica información que esté escrita para profesionales de la salud.
Informe a su proveedor de atención médica sobre cualquier efecto secundario que le moleste o que no desaparezca.
¿Cuáles son las concentraciones de dosis de KOVALTRY?
KOVALTRY con 2.5 mL o 5 mL de Agua estéril para inyección (SWFI) viene en cinco concentraciones de dosis diferentes etiquetadas como Unidades Internacionales (UI): 250 UI, 500 UI, 1000 UI, 2000 UI y 3000 UI. Las cinco concentraciones diferentes están codificadas por colores de la siguiente manera:
|
Azul |
250 UI con 2.5 mL SWFI |
|
Verde |
500 UI con 2.5 mL SWFI |
|
Rojo |
1000 UI con 2.5 mL SWFI |
|
Amarillo |
2000 UI con 5 mL SWFI |
|
Gris |
3000 UI con 5 mL SWFI |
¿Cómo guardo KOVALTRY?
No congele KOVALTRY.
Almacene KOVALTRY a +2°C a +8°C (36°F a 46°F) por hasta 30 meses a partir de la fecha de fabricación. Dentro de este período, KOVALTRY puede almacenarse durante un período de hasta 12 meses a temperaturas de hasta +25°C o 77°F.
Anote la fecha de inicio del almacenamiento a temperatura ambiente claramente en la caja del producto sin abrir. Una vez almacenado a temperatura ambiente, no vuelva a colocar el producto en el refrigerador. El producto caduca después del almacenamiento a temperatura ambiente durante 12 meses, o después de la fecha de caducidad en el vial del producto, lo que ocurra primero. Almacene los viales en su caja original y protéjalos de la exposición extrema a la luz.
Administre KOVALTRY reconstituido lo antes posible. De lo contrario, almacene a temperatura ambiente durante no más de 3 horas.
Deseche cualquier KOVALTRY no utilizado después de la fecha de caducidad.
No use KOVALTRY reconstituido si no está claro.
¿Qué más debo saber sobre KOVALTRY y la hemofilia A?
Encontrar venas para inyecciones puede ser difícil en niños pequeños. Cuando se requieren inyecciones frecuentes, su proveedor de atención médica puede proponer que se coloque un dispositivo quirúrgicamente debajo de la piel para facilitar el acceso al torrente sanguíneo. Estos dispositivos pueden provocar infecciones.
Los medicamentos a veces se recetan para fines distintos de los que se enumeran aquí. No use KOVALTRY para una afección para la que no esté recetado. No comparta KOVALTRY con otras personas, incluso si tienen los mismos síntomas que usted.
Este folleto resume la información más importante sobre KOVALTRY. Si desea obtener más información, hable con su proveedor de atención médica. Puede pedirle a su proveedor de atención médica o farmacéutico información sobre KOVALTRY que esté escrita para profesionales de la salud.
Instrucciones de uso
KOVALTRY (KOH-vahl-tree)
Factor antihemofílico (recombinante)
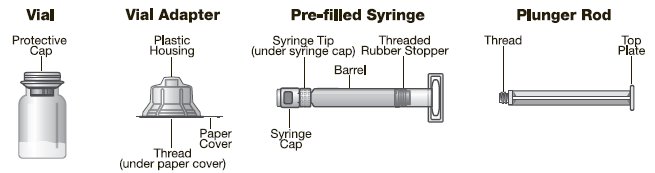
No intente autoinfundirse a menos que su proveedor de atención médica o centro de hemofilia le haya enseñado cómo hacerlo.
Siempre debe seguir las instrucciones específicas que le dé su proveedor de atención médica. Los pasos que se enumeran a continuación son pautas generales para usar KOVALTRY. Si no está seguro de los procedimientos, llame a su proveedor de atención médica antes de usar.
Llame a su proveedor de atención médica de inmediato si el sangrado no se controla después de usar KOVALTRY.
Su proveedor de atención médica le recetará la dosis que debe tomar.
Su proveedor de atención médica puede necesitar tomarle análisis de sangre de vez en cuando.
Hable con su proveedor de atención médica antes de viajar. Debe planificar llevar suficiente KOVALTRY para su tratamiento durante este tiempo.
Vea las instrucciones paso a paso a continuación para reconstituir KOVALTRY con el adaptador de vial. Siga el folleto de instrucciones de infusión específico incluido con el juego de infusión proporcionado.
Maneje KOVALTRY con cuidado. Deseche todos los materiales, incluido cualquier producto KOVALTRY reconstituido sobrante, en un contenedor adecuado.
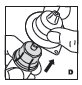
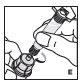
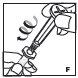
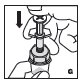
Reconstitución
Siempre trabaje en una superficie limpia y lávese las manos antes de realizar el siguiente procedimiento. Use solo los componentes para la reconstitución y administración que se proporcionan con cada paquete de KOVALTRY. Si un paquete está abierto o dañado, no use este componente. Si estos componentes no se pueden usar, comuníquese con su proveedor de atención médica.
Prepare una superficie plana limpia y reúna todos los materiales necesarios para la infusión.
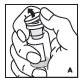
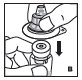
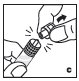
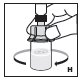
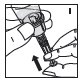
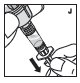
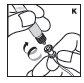
Agrupación
Si la dosis requiere más de un vial, reconstituya cada vial como se describió anteriormente con la jeringa de diluyente proporcionada. Para combinar el contenido de los viales, use una jeringa de plástico más grande (no proporcionada) para agrupar la solución en la jeringa y administrar como de costumbre.
Velocidad de administración
La dosis completa de KOVALTRY generalmente se puede infundir en 1 a 15 minutos. Su proveedor de atención médica determinará la velocidad de administración que sea mejor para usted.
Recursos de Bayer disponibles para el paciente:
Para reportar reacciones adversas, comuníquese con Bayer Medical Communications 1-888-84-BAYER (1-888-842-2937)
Para recibir más información sobre el producto, comuníquese con el Servicio al Cliente de KOVALTRY 1-888-606-3780
Línea de ayuda de reembolso de Bayer 1-800-288-8374
Para obtener más información, visite www.KOVALTRY-us.com
Bayer HealthCare LLC
Whippany, NJ 07981 USA
Licencia de EE. UU. No. 8
Esta información para el paciente e instrucciones de uso han sido aprobadas por la Administración de Alimentos y Medicamentos de los EE. UU. Revisado: 10/2021
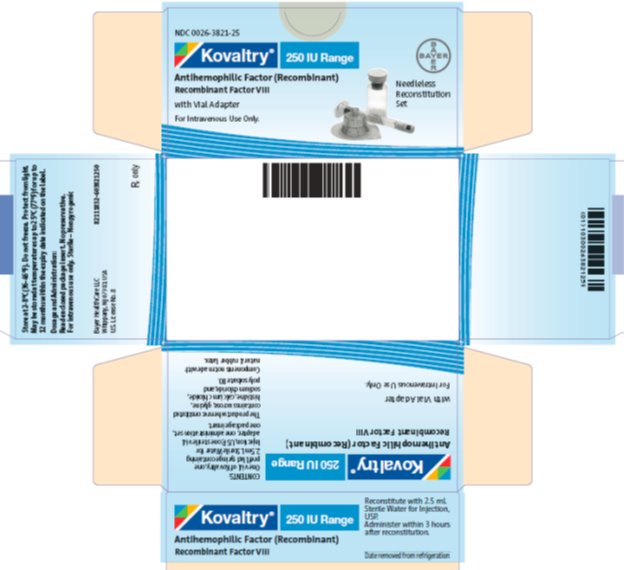
PANEL DE VISUALIZACIÓN PRINCIPAL
NDC 0026-3821-25
Kovaltry 250 IU Rango
Antihemophilic Factor (Recombinant)
Factor VIII Recombinante
con adaptador de vial
Solo para uso intravenoso
Set de reconstitución sin aguja
PANEL DE VISUALIZACIÓN PRINCIPAL
NDC 0026-3822-25
Kovaltry 500 IU Range
Antihemophilic Factor (Recombinant)
Recombinant Factor VIII
with Vial Adapter
For Intravenous Use Only
Needleless Reconstitution Set

PANEL DE VISUALIZACIÓN PRINCIPAL
NDC 0026-3824-25
Kovaltry 1000 IU Range
Antihemophilic Factor (Recombinant)
Recombinant Factor VIII
with Vial Adapter
For Intravenous Use Only
Needleless Reconstitution Set

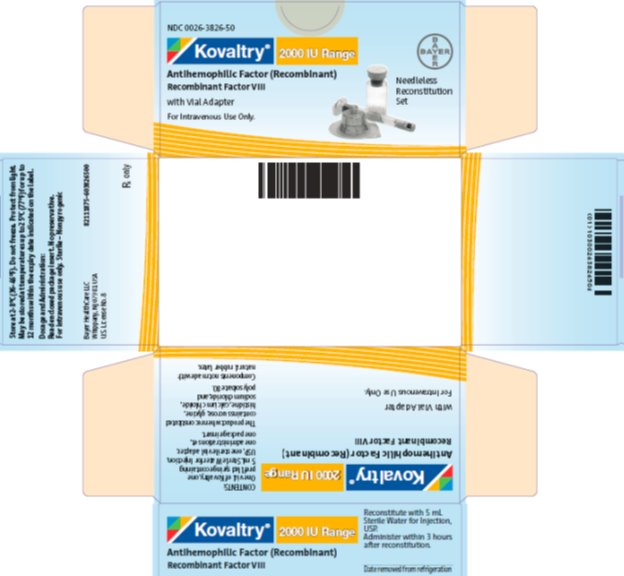
PANEL DE VISUALIZACIÓN PRINCIPAL
NDC 0026-3826-50
Kovaltry 2000 IU Range
Antihemophilic Factor (Recombinant)
Recombinant Factor VIII
with Vial Adapter
For Intravenous Use Only
Needleless Reconstitution Set
PANEL DE VISUALIZACIÓN PRINCIPAL
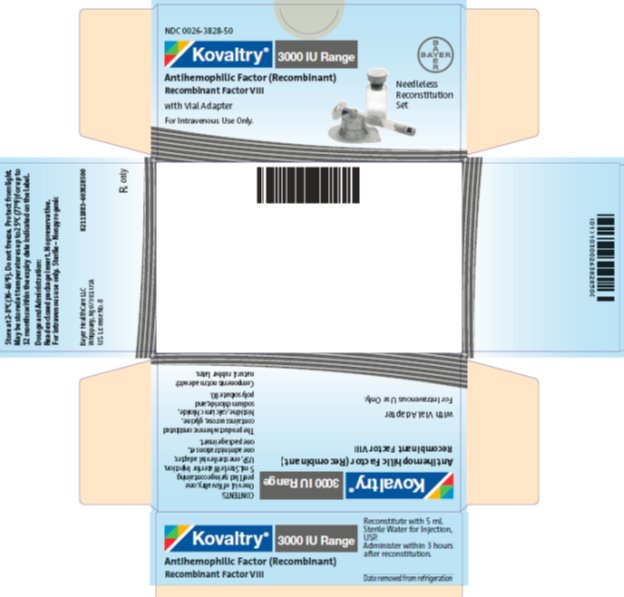
NDC 0026-3828-50
Kovaltry 3000 IU Range
Antihemophilic Factor (Recombinant)
Recombinant Factor VIII
with Vial Adapter
For Intravenous Use Only
Needleless Reconstitution Set