Fabricante de medicamentos: Merck Sharp & Dohme LLC (Updated: 2024-12-11)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
KEYTRUDA® (pembrolizumab) inyección, para administración intravenosa
Aprobación inicial en EE. UU.: 2014
CAMBIOS IMPORTANTES RECIENTES
INDICACIONES Y USO
KEYTRUDA es un anticuerpo bloqueante del receptor 1 de muerte programada (PD-1) indicado para:
Melanoma
- el tratamiento de pacientes con melanoma irresecable o metastásico. (1.1)
- el tratamiento adyuvante de pacientes adultos y pediátricos (12 años o más) con melanoma en estadio IIB, IIC o III después de la resección completa. (1.1)
Cáncer de pulmón de células no pequeñas (CPNP)
- en combinación con pemetrexed y quimioterapia con platino, como tratamiento de primera línea para pacientes con CPNP no escamoso metastásico, sin aberraciones genómicas tumorales EGFR o ALK. (1.2)
- en combinación con carboplatino y paclitaxel o paclitaxel unido a proteínas, como tratamiento de primera línea para pacientes con CPNP escamoso metastásico. (1.2)
- como agente único para el tratamiento de primera línea de pacientes con CPNP que expresan PD-L1 [Puntuación de proporción tumoral (TPS) ≥1%] según lo determine una prueba aprobada por la FDA, sin aberraciones genómicas tumorales EGFR o ALK, y que son:
- como agente único para el tratamiento de pacientes con CPNP metastásico cuyos tumores expresan PD-L1 (TPS ≥1%) según lo determine una prueba aprobada por la FDA, con progresión de la enfermedad durante o después de la quimioterapia con platino. Los pacientes con aberraciones genómicas tumorales EGFR o ALK deben presentar progresión de la enfermedad con terapia aprobada por la FDA para estas aberraciones antes de recibir KEYTRUDA. (1.2, 2.1)
- para el tratamiento de pacientes con CPNP resecable (tumores ≥4 cm o ganglios positivos) en combinación con quimioterapia con platino como tratamiento neoadyuvante, y luego continuado como agente único como tratamiento adyuvante después de la cirugía. (1.2)
- como agente único, para el tratamiento adyuvante después de la resección y la quimioterapia a base de platino para pacientes adultos con CPNP en estadio IB (T2a ≥4 cm), II o IIIA. (1.2)
Mesotelioma pleural maligno (MPM)
- en combinación con pemetrexed y quimioterapia con platino, como tratamiento de primera línea para pacientes adultos con MPM avanzado o metastásico irresecable. (1.3)
Cáncer de células escamosas de cabeza y cuello (HNSCC)
- en combinación con platino y FU para el tratamiento de primera línea de pacientes con HNSCC metastásico o con HNSCC recurrente irresecable. (1.4)
- como agente único para el tratamiento de primera línea de pacientes con HNSCC metastásico o con HNSCC recurrente irresecable cuyos tumores expresan PD-L1 [Puntuación positiva combinada (CPS) ≥1] según lo determine una prueba aprobada por la FDA. (1.4, 2.1)
- como agente único para el tratamiento de pacientes con HNSCC recurrente o metastásico con progresión de la enfermedad durante o después de la quimioterapia con platino. (1.4)
Linfoma de Hodgkin clásico (cHL)
- para el tratamiento de pacientes adultos con cHL recidivante o refractario. (1.5)
- para el tratamiento de pacientes pediátricos con cHL refractario, o cHL que ha recidivado después de 2 o más líneas de terapia. (1.5)
Linfoma difuso de células B grandes del mediastino primario (PMBCL)
- para el tratamiento de pacientes adultos y pediátricos con PMBCL refractario, o que han recidivado después de 2 o más líneas de terapia previas. (1.6)
- Limitaciones de uso: KEYTRUDA no se recomienda para el tratamiento de pacientes con PMBCL que requieren terapia citoreductora urgente.
Cáncer urotelial
- en combinación con enfortumab vedotina, para el tratamiento de pacientes adultos con cáncer urotelial localmente avanzado o metastásico. (1.7)
- como agente único para el tratamiento de pacientes con carcinoma urotelial localmente avanzado o metastásico que:
- no son elegibles para ninguna quimioterapia con platino, o
- que presentan progresión de la enfermedad durante o después de la quimioterapia con platino o dentro de los 12 meses del tratamiento neoadyuvante o adyuvante con quimioterapia con platino. (1.7)
- como agente único para el tratamiento de pacientes con cáncer de vejiga no invasivo de alto riesgo, no responsivo al bacilo de Calmette-Guérin (BCG), con carcinoma in situ (CIS) con o sin tumores papilares que no son elegibles o han optado por no someterse a cistectomía. (1.7)
Cáncer con alta inestabilidad de microsatélites o deficiencia de reparación de desajuste
- para el tratamiento de pacientes adultos y pediátricos con tumores sólidos irresecables o metastásicos con alta inestabilidad de microsatélites (MSI-H) o deficiencia de reparación de desajuste (dMMR), según lo determine una prueba aprobada por la FDA, que han progresado después del tratamiento previo y que no tienen opciones de tratamiento alternativas satisfactorias. (1.8, 2.1)
Cáncer colorrectal (CCR) con alta inestabilidad de microsatélites o deficiencia de reparación de desajuste
- para el tratamiento de pacientes con cáncer colorrectal (CCR) MSI-H o dMMR irresecable o metastásico, según lo determine una prueba aprobada por la FDA. (1.9, 2.1)
Cáncer gástrico
- en combinación con trastuzumab, quimioterapia con fluoropirimidina y platino, para el tratamiento de primera línea de adultos con adenocarcinoma gástrico o de la unión gastroesofágica (UGE) HER2 positivo localmente avanzado irresecable o metastásico cuyos tumores expresan PD-L1 (CPS ≥1) según lo determinado por una prueba aprobada por la FDA.1 (1.10)
- en combinación con quimioterapia con fluoropirimidina y platino, para el tratamiento de primera línea de adultos con adenocarcinoma gástrico o de la unión gastroesofágica (UGE) HER2 negativo localmente avanzado irresecable o metastásico. (1.10)
Cáncer de Esófago
- para el tratamiento de pacientes con carcinoma esofágico o de la unión gastroesofágica (UGE) (tumores con epicentro de 1 a 5 centímetros por encima de la UGE) localmente avanzado o metastásico que no es susceptible de resección quirúrgica o quimiorradiación definitiva, ya sea:
- en combinación con quimioterapia a base de platino y fluoropirimidina, o
- como agente único después de una o más líneas previas de terapia sistémica para pacientes con tumores de histología de células escamosas que expresan PD-L1 (CPS ≥10) según lo determinado por una prueba aprobada por la FDA. (1.11, 2.1)
Cáncer de Cérvix
- en combinación con quimioradioterapia, para el tratamiento de pacientes con cáncer de cérvix en estadio III-IVA según la FIGO 2014. (1.12)
- en combinación con quimioterapia, con o sin bevacizumab, para el tratamiento de pacientes con cáncer de cérvix persistente, recurrente o metastásico cuyos tumores expresan PD-L1 (CPS ≥1) según lo determinado por una prueba aprobada por la FDA. (1.12, 2.1)
- como agente único para el tratamiento de pacientes con cáncer de cérvix recurrente o metastásico con progresión de la enfermedad durante o después de la quimioterapia cuyos tumores expresan PD-L1 (CPS ≥1) según lo determinado por una prueba aprobada por la FDA. (1.12, 2.1)
Carcinoma Hepatocelular (CHC)
- para el tratamiento de pacientes con CHC secundario a hepatitis B que han recibido terapia sistémica previa que no sea un régimen que contenga PD-1/PD-L1. (1.13)
Cáncer de Vías Biliares (CVB)
- en combinación con gemcitabina y cisplatino, para el tratamiento de pacientes con cáncer de vías biliares localmente avanzado irresecable o metastásico. (1.14)
Carcinoma de Células de Merkel (CCM)
- para el tratamiento de pacientes adultos y pediátricos con carcinoma de células de Merkel recurrente localmente avanzado o metastásico. (1.15)
Carcinoma de Células Renales (CCR)
- en combinación con axitinib, para el tratamiento de primera línea de pacientes adultos con CCR avanzado. (1.16)
- en combinación con lenvatinib, para el tratamiento de primera línea de pacientes adultos con CCR avanzado. (1.16)
- para el tratamiento adyuvante de pacientes con CCR con riesgo intermedio-alto o alto de recurrencia después de la nefrectomía, o después de la nefrectomía y la resección de lesiones metastásicas. (1.16)
Carcinoma de Endometrio
- en combinación con carboplatino y paclitaxel, seguido de KEYTRUDA como agente único, para el tratamiento de pacientes adultos con carcinoma de endometrio primario avanzado o recurrente. (1.17)
- en combinación con lenvatinib, para el tratamiento de pacientes adultos con carcinoma de endometrio avanzado con reparación de desajuste de coincidencia (pMMR) según lo determinado por una prueba aprobada por la FDA o no MSI-H, que presentan progresión de la enfermedad después de la terapia sistémica previa en cualquier contexto y que no son candidatos a cirugía o radiación curativa. (1.17, 2.1)
- como agente único, para el tratamiento de pacientes adultos con carcinoma de endometrio avanzado que es MSI-H o dMMR, según lo determinado por una prueba aprobada por la FDA, que presentan progresión de la enfermedad después de la terapia sistémica previa en cualquier contexto y que no son candidatos a cirugía o radiación curativa. (1.17, 2.1)
Cáncer con Alta Carga Mutacional Tumoral (TMB-A)
- para el tratamiento de pacientes adultos y pediátricos con tumores sólidos irresecables o metastásicos con alta carga mutacional tumoral (TMB-A) [≥10 mutaciones/megabase (mut/Mb)], según lo determinado por una prueba aprobada por la FDA, que han progresado después del tratamiento previo y que no tienen opciones de tratamiento alternativas satisfactorias.1 (1.18, 2.1)
- Limitaciones de Uso: No se ha establecido la seguridad y eficacia de KEYTRUDA en pacientes pediátricos con cánceres del sistema nervioso central con TMB-A.
Carcinoma de Células Escamosas Cutáneo (CCEC)
- para el tratamiento de pacientes con CCEC recurrente o metastásico o CCEC localmente avanzado que no es curable mediante cirugía o radiación. (1.19)
Cáncer de Mama Triple Negativo (CMTN)
- para el tratamiento de pacientes con CMTN de estadio temprano de alto riesgo en combinación con quimioterapia como tratamiento neoadyuvante, y luego continuado como agente único como tratamiento adyuvante después de la cirugía. (1.20)
- en combinación con quimioterapia, para el tratamiento de pacientes con CMTN localmente recurrente irresecable o metastásico cuyos tumores expresan PD-L1 (CPS ≥10) según lo determinado por una prueba aprobada por la FDA. (1.20, 2.1)
Linfoma de Hodgkin Clásico Adulto y Linfoma de Células B Grandes Mediastínico Primario Adulto: Régimen de Dosificación Adicional de 400 mg Cada 6 Semanas
- para su uso a una dosis recomendada adicional de 400 mg cada 6 semanas para el linfoma de Hodgkin clásico y el linfoma de células B grandes mediastínico primario en adultos.2 (1.21, 2.2)
1 Esta indicación está aprobada bajo aprobación acelerada basada en la tasa de respuesta tumoral y la durabilidad de la respuesta. La aprobación continua para esta indicación puede estar supeditada a la verificación y descripción del beneficio clínico en los ensayos confirmatorios.
2 Esta indicación está aprobada bajo aprobación acelerada basada en datos farmacocinéticos, la relación entre la exposición y la eficacia, y la relación entre la exposición y la seguridad. La aprobación continua para esta dosificación puede estar supeditada a la verificación y descripción del beneficio clínico en los ensayos confirmatorios.
POSOLOGÍA Y ADMINISTRACIÓN
- Melanoma: 200 mg cada 3 semanas o 400 mg cada 6 semanas; 2 mg/kg (hasta 200 mg) cada 3 semanas para pediatría. (2.2)
- Cáncer de pulmón de células no pequeñas (CPNM): 200 mg cada 3 semanas o 400 mg cada 6 semanas. (2.2)
- Mesotelioma pleural maligno (MPM): 200 mg cada 3 semanas o 400 mg cada 6 semanas. (2.2)
- Cáncer de cabeza y cuello (HNSCC): 200 mg cada 3 semanas o 400 mg cada 6 semanas. (2.2)
- Linfoma de Hodgkin clásico (cHL) o linfoma de células B grandes mediastínico primario (PMBCL): 200 mg cada 3 semanas o 400 mg cada 6 semanas para adultos; 2 mg/kg (hasta 200 mg) cada 3 semanas para pediatría. (2.2)
- Cáncer urotelial: 200 mg cada 3 semanas o 400 mg cada 6 semanas. (2.2)
- Cáncer con alta inestabilidad de microsatélites (MSI-H) o deficiencia de reparación de errores de apareamiento (dMMR): 200 mg cada 3 semanas o 400 mg cada 6 semanas para adultos; 2 mg/kg (hasta 200 mg) cada 3 semanas para pediatría. (2.2)
- Cáncer colorrectal (CRC) MSI-H o dMMR: 200 mg cada 3 semanas o 400 mg cada 6 semanas. (2.2)
- Cáncer gástrico: 200 mg cada 3 semanas o 400 mg cada 6 semanas. (2.2)
- Cáncer esofágico: 200 mg cada 3 semanas o 400 mg cada 6 semanas. (2.2)
- Cáncer cervical: 200 mg cada 3 semanas o 400 mg cada 6 semanas. (2.2)
- Carcinoma hepatocelular (HCC): 200 mg cada 3 semanas o 400 mg cada 6 semanas. (2.2)
- Cáncer de vías biliares (BTC): 200 mg cada 3 semanas o 400 mg cada 6 semanas. (2.2)
- Carcinoma de células de Merkel (MCC): 200 mg cada 3 semanas o 400 mg cada 6 semanas para adultos; 2 mg/kg (hasta 200 mg) cada 3 semanas para pediatría. (2.2)
- Carcinoma de células renales (RCC): 200 mg cada 3 semanas o 400 mg cada 6 semanas como agente único en el contexto adyuvante, o en el contexto avanzado con:
- axitinib 5 mg por vía oral dos veces al día o
- lenvatinib 20 mg por vía oral una vez al día. (2.2)
- Carcinoma endometrial: 200 mg cada 3 semanas o 400 mg cada 6 semanas
- en combinación con carboplatino y paclitaxel independientemente del estado de MMR o MSI, o
- en combinación con lenvatinib 20 mg por vía oral una vez al día para tumores pMMR o no MSI-H, o
- como agente único para tumores MSI-H o dMMR. (2.2)
- Cáncer con alta carga mutacional tumoral (TMB-H): 200 mg cada 3 semanas o 400 mg cada 6 semanas para adultos; 2 mg/kg (hasta 200 mg) cada 3 semanas para pediatría. (2.2)
- Carcinoma espinocelular de células cutáneas (cSCC): 200 mg cada 3 semanas o 400 mg cada 6 semanas. (2.2)
- Cáncer de mama triple negativo (TNBC): 200 mg cada 3 semanas o 400 mg cada 6 semanas. (2.2)
- Administrar KEYTRUDA como infusión intravenosa durante 30 minutos después de la dilución. (2.4)
- Consulte la información completa de prescripción para las modificaciones de la dosificación en caso de reacciones adversas e instrucciones de preparación y administración. (2.3, 2.4)
FORMAS Y CONCENTRACIONES FARMACÉUTICAS
- Inyección: 100 mg/4 mL (25 mg/mL) solución en un vial de dosis única (3)
CONTRAINDICACIONES
Ninguna. (4)
ADVERTENCIAS Y PRECAUCIONES
- Reacciones adversas inmunomediadas (5.1)
- Pueden producirse reacciones adversas inmunomediadas, que pueden ser graves o mortales, en cualquier sistema orgánico o tejido, incluyendo las siguientes: neumonitis inmunomediada, colitis inmunomediada, hepatitis inmunomediada, endocrinopatías inmunomediadas, nefritis inmunomediada con disfunción renal, reacciones adversas dermatológicas inmunomediadas y rechazo de trasplante de órgano sólido.
- Monitorizar para la identificación y el manejo tempranos. Evaluar las enzimas hepáticas, la creatinina y la función tiroidea al inicio y periódicamente durante el tratamiento.
- Suspender o interrumpir permanentemente en función de la gravedad y el tipo de reacción.
- Reacciones relacionadas con la infusión: Interrumpir, disminuir la velocidad de infusión o interrumpir permanentemente KEYTRUDA en función de la gravedad de la reacción. (5.2)
- Complicaciones del trasplante de células hematopoyéticas alogénico (TCHA): Pueden producirse complicaciones mortales y otras complicaciones graves en pacientes que reciben un TCHA antes o después de ser tratados con un anticuerpo bloqueante de PD-1/PD-L1. (5.3)
- No se recomienda el tratamiento de pacientes con mieloma múltiple con un anticuerpo bloqueante de PD-1 o PD-L1 en combinación con un análogo de talidomida más dexametasona fuera de ensayos clínicos controlados. (5.4)
- Toxicidad embriofetal: Puede causar daño fetal. Aconsejar a las mujeres en edad fértil sobre el riesgo potencial para el feto y el uso de un método anticonceptivo eficaz. (5.5, 8.1, 8.3)
REACCIONES ADVERSAS
Las reacciones adversas más frecuentes (notificadas en ≥20% de los pacientes) fueron:
- KEYTRUDA como agente único: fatiga, dolor musculoesquelético, erupción cutánea, diarrea, pirexia, tos, disminución del apetito, prurito, disnea, estreñimiento, dolor, dolor abdominal, náuseas e hipotiroidismo. (6.1)
- KEYTRUDA en combinación con quimioterapia o quimioradioterapia: fatiga/astenia, náuseas, estreñimiento, diarrea, disminución del apetito, erupción cutánea, vómitos, tos, disnea, pirexia, alopecia, neuropatía periférica, inflamación de la mucosa, estomatitis, cefalea, pérdida de peso, dolor abdominal, artralgia, mialgia, insomnio, eritrodisestesia palmar-plantar, infección del tracto urinario e hipotiroidismo. (6.1)
- KEYTRUDA en combinación con quimioterapia y bevacizumab: neuropatía periférica, alopecia, anemia, fatiga/astenia, náuseas, neutropenia, diarrea, hipertensión, trombocitopenia, estreñimiento, artralgia, vómitos, infección del tracto urinario, erupción cutánea, leucopenia, hipotiroidismo y disminución del apetito. (6.1)
- KEYTRUDA en combinación con axitinib: diarrea, fatiga/astenia, hipertensión, hepatotoxicidad, hipotiroidismo, disminución del apetito, eritrodisestesia palmar-plantar, náuseas, estomatitis/inflamación de la mucosa, disfonía, erupción cutánea, tos y estreñimiento. (6.1)
- KEYTRUDA en combinación con lenvatinib: hipotiroidismo, hipertensión, fatiga, diarrea, trastornos musculoesqueléticos, náuseas, disminución del apetito, vómitos, estomatitis, pérdida de peso, dolor abdominal, infección del tracto urinario, proteinuria, estreñimiento, cefalea, eventos hemorrágicos, eritrodisestesia palmar-plantar, disfonía, erupción cutánea, hepatotoxicidad e insuficiencia renal aguda. (6.1)
- KEYTRUDA en combinación con enfortumab vedotina: erupción cutánea, neuropatía periférica, fatiga, prurito, diarrea, alopecia, pérdida de peso, disminución del apetito, ojo seco, náuseas, estreñimiento, disgeusia e infección del tracto urinario. (6.1)
Para notificar REACCIONES ADVERSAS SOSPECHOSAS, comuníquese con Merck Sharp & Dohme LLC al 1-877-888-4231 o con la FDA al 1-800-FDA-1088 o www.fda.gov/medwatch.
Ver 17 para INFORMACIÓN AL PACIENTE y Guía de medicamentos.
Revisado: 12/2024
Tabla de contenido
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDO*
1 INDICACIONES Y USO
1.1 Melanoma
1.2 Cáncer de pulmón de células no pequeñas
1.3 Mesotelioma pleural maligno
1.4 Cáncer de células escamosas de cabeza y cuello
1.5 Linfoma de Hodgkin clásico
1.6 Linfoma difuso de células B grandes del mediastino primario
1.7 Cáncer urotelial
1.8 Cáncer con alta inestabilidad de microsatélites o deficiencia de reparación de desajuste
1.9 Cáncer colorrectal con alta inestabilidad de microsatélites o deficiencia de reparación de desajuste
1.10 Cáncer gástrico
1.11 Cáncer esofágico
1.12 Cáncer cervical
1.13 Carcinoma hepatocelular
1.14 Cáncer de las vías biliares
1.15 Carcinoma de células de Merkel
1.16 Carcinoma de células renales
1.17 Carcinoma endometrial
1.18 Cáncer con alta carga mutacional tumoral
1.19 Carcinoma de células escamosas cutáneo
1.20 Cáncer de mama triple negativo
1.21 Linfoma de Hodgkin clásico en adultos y linfoma difuso de células B grandes del mediastino primario en adultos: Régimen de dosificación adicional de 400 mg cada 6 semanas
2 POSOLOGÍA Y ADMINISTRACIÓN
2.1 Selección del paciente
2.2 Dosis recomendada
2.3 Modificaciones de la dosis
2.4 Preparación y administración
3 FORMAS Y CONCENTRACIONES FARMACÉUTICAS
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Reacciones adversas inmunomediadas graves y mortales
5.2 Reacciones relacionadas con la infusión
5.3 Complicaciones del TCH alogénico
5.4 Aumento de la mortalidad en pacientes con mieloma múltiple cuando se añade KEYTRUDA a un análogo de la talidomida y dexametasona
5.5 Toxicidad embriofetal
6 REACCIONES ADVERSAS
6.1 Experiencia en ensayos clínicos
6.2 Experiencia postcomercialización
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.3 Mujeres y hombres en edad fértil
8.4 Uso pediátrico
8.5 Uso geriátrico
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
12.2 Farmacodinámica
12.3 Farmacocinética
12.6 Inmunogenicidad
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, mutagénesis, deterioro de la fertilidad
13.2 Toxicología y/o farmacología animal
14 ESTUDIOS CLÍNICOS
14.1 Melanoma
14.2 Cáncer de pulmón de células no pequeñas
14.3 Mesotelioma pleural maligno
14.4 Cáncer de células escamosas de cabeza y cuello
14.5 Linfoma de Hodgkin clásico
14.6 Linfoma difuso de células B grandes del mediastino primario
14.7 Cáncer urotelial
14.8 Cáncer con alta inestabilidad de microsatélites o deficiencia de reparación de desajuste
14.9 Cáncer colorrectal con alta inestabilidad de microsatélites o deficiencia de reparación de desajuste
14.10 Cáncer gástrico
14.11 Cáncer esofágico
14.12 Cáncer cervical
14.13 Carcinoma hepatocelular
14.14 Cáncer de las vías biliares
14.15 Carcinoma de células de Merkel
14.16 Carcinoma de células renales
14.17 Carcinoma endometrial
14.18 Cáncer con alta carga mutacional tumoral
14.19 Carcinoma de células escamosas cutáneo
14.20 Cáncer de mama triple negativo
14.21 Linfoma de Hodgkin clásico en adultos y linfoma difuso de células B grandes del mediastino primario en adultos: Régimen de dosificación adicional de 400 mg cada 6 semanas
16 PRESENTACIÓN/CONSERVACIÓN Y MANIPULACIÓN
17 INFORMACIÓN PARA EL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información completa de prescripción no figuran en la lista.
1 INDICACIONES Y USO
1.1 Melanoma
KEYTRUDA® está indicado para el tratamiento de pacientes con melanoma irresecable o metastásico.
KEYTRUDA está indicado para el tratamiento adyuvante de pacientes adultos y pediátricos (12 años o mayores) con melanoma en estadio IIB, IIC o III después de la resección completa.
1.2 Cáncer de pulmón de células no pequeñas
KEYTRUDA, en combinación con pemetrexed y quimioterapia con platino, está indicado para el tratamiento de primera línea de pacientes con cáncer de pulmón de células no pequeñas (CPNP) no escamoso metastásico, sin aberraciones genómicas tumorales EGFR o ALK.
KEYTRUDA, en combinación con carboplatino y paclitaxel o paclitaxel unido a proteínas, está indicado para el tratamiento de primera línea de pacientes con CPNP escamoso metastásico.
KEYTRUDA, como agente único, está indicado para el tratamiento de primera línea de pacientes con CPNP que expresan PD-L1 [Puntuación de proporción tumoral (TPS) ≥1%] según lo determine una prueba aprobada por la FDA [ver Posología y administración (2.1)], sin aberraciones genómicas tumorales EGFR o ALK, y es:
- Estadio III donde los pacientes no son candidatos a resección quirúrgica o quimiorradiación definitiva, o
- metastásico.
KEYTRUDA, como agente único, está indicado para el tratamiento de pacientes con CPNP metastásico cuyos tumores expresan PD-L1 (TPS ≥1%) según lo determine una prueba aprobada por la FDA [ver Posología y administración (2.1)], con progresión de la enfermedad durante o después de la quimioterapia con platino. Los pacientes con aberraciones genómicas tumorales EGFR o ALK deben presentar progresión de la enfermedad en la terapia aprobada por la FDA para estas aberraciones antes de recibir KEYTRUDA.
KEYTRUDA está indicado para el tratamiento de pacientes con CPNP resecable (tumores ≥4 cm o ganglios positivos) en combinación con quimioterapia con platino como tratamiento neoadyuvante, y luego continuado como agente único como tratamiento adyuvante después de la cirugía.
KEYTRUDA, como agente único, está indicado como tratamiento adyuvante después de la resección y la quimioterapia a base de platino para pacientes adultos con CPNP en estadio IB (T2a ≥4 cm), II o IIIA.
1.3 Mesotelioma pleural maligno
KEYTRUDA, en combinación con pemetrexed y quimioterapia con platino, está indicado para el tratamiento de primera línea de pacientes adultos con mesotelioma pleural maligno (MPM) avanzado o metastásico irresecable.
1.4 Cáncer de células escamosas de cabeza y cuello
KEYTRUDA, en combinación con platino y fluorouracilo (FU), está indicado para el tratamiento de primera línea de pacientes con carcinoma de células escamosas de cabeza y cuello (CCECC) metastásico o irresecable recurrente.
KEYTRUDA, como agente único, está indicado para el tratamiento de primera línea de pacientes con CCECC metastásico o irresecable recurrente cuyos tumores expresan PD-L1 [Puntuación positiva combinada (CPS) ≥1] según lo determine una prueba aprobada por la FDA [ver Posología y administración (2.1)].
KEYTRUDA, como agente único, está indicado para el tratamiento de pacientes con CCECC recurrente o metastásico con progresión de la enfermedad durante o después de la quimioterapia con platino.
1.5 Linfoma de Hodgkin clásico
KEYTRUDA está indicado para el tratamiento de pacientes adultos con linfoma de Hodgkin clásico (LCH) recidivante o refractario.
KEYTRUDA está indicado para el tratamiento de pacientes pediátricos con LCH refractario, o LCH que ha recidivado después de 2 o más líneas de terapia.
1.6 Linfoma difuso de células B grandes del mediastino primario
KEYTRUDA está indicado para el tratamiento de pacientes adultos y pediátricos con linfoma difuso de células B grandes del mediastino primario (LDCBGMP) refractario, o que han recidivado después de 2 o más líneas de terapia previas.
Limitaciones de uso: No se recomienda KEYTRUDA para el tratamiento de pacientes con LDCBGMP que requieren terapia citoreductora urgente.
1.7 Cáncer urotelial
KEYTRUDA, en combinación con enfortumab vedotina, está indicado para el tratamiento de pacientes adultos con cáncer urotelial localmente avanzado o metastásico.
KEYTRUDA, como agente único, está indicado para el tratamiento de pacientes con carcinoma urotelial localmente avanzado o metastásico:
- que no son elegibles para ninguna quimioterapia con platino, o
- que presentan progresión de la enfermedad durante o después de la quimioterapia con platino o dentro de los 12 meses del tratamiento neoadyuvante o adyuvante con quimioterapia con platino.
KEYTRUDA, como agente único, está indicado para el tratamiento de pacientes con cáncer de vejiga no invasivo de alto riesgo (NMIBC) no responsivo al bacilo de Calmette-Guérin (BCG) con carcinoma in situ (CIS) con o sin tumores papilares que no son elegibles o han optado por no someterse a cistectomía.
1.8 Cáncer con alta inestabilidad de microsatélites o deficiencia de reparación de desajuste
KEYTRUDA está indicado para el tratamiento de pacientes adultos y pediátricos con tumores sólidos irresecables o metastásicos con alta inestabilidad de microsatélites (MSI-H) o deficiencia de reparación de desajuste (dMMR), según lo determine una prueba aprobada por la FDA, que han progresado después del tratamiento previo y que no tienen opciones de tratamiento alternativas satisfactorias [ver Posología y administración (2.1)].
1.9 Cáncer colorrectal con inestabilidad de microsatélites alta o deficiencia de reparación de desajuste
KEYTRUDA está indicado para el tratamiento de pacientes con cáncer colorrectal (CCR) MSI-H o dMMR irresecable o metastásico, según lo determine una prueba aprobada por la FDA [ver Posología y administración (2.1)].
1.10 Cáncer gástrico
KEYTRUDA, en combinación con trastuzumab, quimioterapia con fluoropirimidina y platino, está indicado para el tratamiento de primera línea de adultos con adenocarcinoma gástrico o de la unión gastroesofágica (UGE) HER2-positivo localmente avanzado irresecable o metastásico cuyos tumores expresan PD-L1 (CPS ≥1) según lo determine una prueba aprobada por la FDA [ver Posología y administración (2.1)].
Esta indicación está aprobada bajo aprobación acelerada basada en la tasa de respuesta tumoral y la durabilidad de la respuesta [ver Estudios clínicos (14.10)]. La aprobación continua de esta indicación puede estar supeditada a la verificación y descripción del beneficio clínico en los ensayos confirmatorios.
KEYTRUDA, en combinación con quimioterapia con fluoropirimidina y platino, está indicado para el tratamiento de primera línea de adultos con adenocarcinoma gástrico o de la unión gastroesofágica (UGE) HER2-negativo localmente avanzado irresecable o metastásico.
1.11 Cáncer esofágico
KEYTRUDA está indicado para el tratamiento de pacientes con carcinoma esofágico o de la unión gastroesofágica (UGE) (tumores con epicentro de 1 a 5 centímetros por encima de la UGE) localmente avanzado o metastásico que no es susceptible de resección quirúrgica o quimiorradiación definitiva:
- en combinación con quimioterapia a base de platino y fluoropirimidina, o
- como agente único después de una o más líneas previas de terapia sistémica para pacientes con tumores de histología de células escamosas que expresan PD-L1 (CPS ≥10) según lo determine una prueba aprobada por la FDA [ver Posología y administración (2.1)].
1.12 Cáncer cervical
KEYTRUDA, en combinación con quimioradiación (CRT), está indicado para el tratamiento de pacientes con cáncer cervical en estadio FIGO 2014 III-IVA.
KEYTRUDA, en combinación con quimioterapia, con o sin bevacizumab, está indicado para el tratamiento de pacientes con cáncer cervical persistente, recurrente o metastásico cuyos tumores expresan PD-L1 (CPS ≥1) según lo determine una prueba aprobada por la FDA [ver Posología y administración (2.1)].
KEYTRUDA, como agente único, está indicado para el tratamiento de pacientes con cáncer cervical recurrente o metastásico con progresión de la enfermedad después de la quimioterapia cuyos tumores expresan PD-L1 (CPS ≥1) según lo determine una prueba aprobada por la FDA [ver Posología y administración (2.1)].
1.13 Carcinoma hepatocelular
KEYTRUDA está indicado para el tratamiento de pacientes con carcinoma hepatocelular (CHC) secundario a hepatitis B que han recibido terapia sistémica previa que no sea un régimen que contenga PD-1/PD-L1.
1.14 Cáncer de vías biliares
KEYTRUDA, en combinación con gemcitabina y cisplatino, está indicado para el tratamiento de pacientes con cáncer de vías biliares (CVB) localmente avanzado irresecable o metastásico.
1.15 Carcinoma de células de Merkel
KEYTRUDA está indicado para el tratamiento de pacientes adultos y pediátricos con carcinoma de células de Merkel (CCM) recurrente localmente avanzado o metastásico.
1.16 Carcinoma de células renales
KEYTRUDA, en combinación con axitinib, está indicado para el tratamiento de primera línea de pacientes adultos con carcinoma de células renales (CCR) avanzado.
KEYTRUDA, en combinación con lenvatinib, está indicado para el tratamiento de primera línea de pacientes adultos con CCR avanzado.
KEYTRUDA está indicado para el tratamiento adyuvante de pacientes con CCR con riesgo intermedio-alto o alto de recurrencia después de la nefrectomía, o después de la nefrectomía y la resección de lesiones metastásicas [ver Estudios clínicos (14.16)].
1.17 Carcinoma endometrial
KEYTRUDA, en combinación con carboplatino y paclitaxel, seguido de KEYTRUDA como agente único, está indicado para el tratamiento de pacientes adultos con carcinoma endometrial primario avanzado o recurrente.
KEYTRUDA, en combinación con lenvatinib, está indicado para el tratamiento de pacientes adultos con carcinoma endometrial avanzado que es competente para la reparación de desajuste (pMMR) según lo determine una prueba aprobada por la FDA o no MSI-H, que presentan progresión de la enfermedad después de la terapia sistémica previa en cualquier contexto y no son candidatos a cirugía o radiación curativa [ver Posología y administración (2.1)].
KEYTRUDA, como agente único, está indicado para el tratamiento de pacientes adultos con carcinoma endometrial avanzado que es MSI-H o dMMR, según lo determine una prueba aprobada por la FDA, que presentan progresión de la enfermedad después de la terapia sistémica previa en cualquier contexto y no son candidatos a cirugía o radiación curativa [ver Posología y administración (2.1)].
1.18 Cáncer con alta carga mutacional tumoral
KEYTRUDA está indicado para el tratamiento de pacientes adultos y pediátricos con tumores sólidos irresecables o metastásicos con alta carga mutacional tumoral (TMB-H) [≥10 mutaciones/megabase (mut/Mb)], según lo determine una prueba aprobada por la FDA [véase Posología y administración (2.1)], que han progresado después del tratamiento previo y que no tienen opciones de tratamiento alternativas satisfactorias.
Esta indicación está aprobada bajo aprobación acelerada basada en la tasa de respuesta tumoral y la durabilidad de la respuesta [véase Estudios clínicos (14.18)]. La aprobación continua para esta indicación puede estar supeditada a la verificación y descripción del beneficio clínico en los ensayos confirmatorios.
Limitaciones de uso: No se ha establecido la seguridad y eficacia de KEYTRUDA en pacientes pediátricos con cánceres del sistema nervioso central con TMB-H.
1.19 Carcinoma epidermoide cutáneo
KEYTRUDA está indicado para el tratamiento de pacientes con carcinoma epidermoide cutáneo (CEC) recurrente o metastásico, o CEC localmente avanzado que no es curable mediante cirugía o radiación.
1.20 Cáncer de mama triple negativo
KEYTRUDA está indicado para el tratamiento de pacientes con cáncer de mama triple negativo (TNBC) de alto riesgo en estadio temprano en combinación con quimioterapia como tratamiento neoadyuvante, y luego continuado como agente único como tratamiento adyuvante después de la cirugía.
KEYTRUDA, en combinación con quimioterapia, está indicado para el tratamiento de pacientes con TNBC localmente recurrente irresecable o metastásico cuyos tumores expresan PD-L1 (CPS ≥10) según lo determine una prueba aprobada por la FDA [véase Posología y administración (2.1)].
1.21 Linfoma de Hodgkin clásico en adultos y linfoma difuso de células B grandes del mediastino anterior en adultos: Régimen de dosificación adicional de 400 mg cada 6 semanas
KEYTRUDA está indicado para su uso con una dosis recomendada adicional de 400 mg cada 6 semanas para el linfoma de Hodgkin clásico y el linfoma difuso de células B grandes del mediastino anterior en adultos [véase Indicaciones y uso (1.5, 1.6), Posología y administración (2.2)]. Esta indicación está aprobada bajo aprobación acelerada basada en datos farmacocinéticos, la relación entre la exposición y la eficacia, y la relación entre la exposición y la seguridad [véase Farmacología clínica (12.2), Estudios clínicos (14.21)]. La aprobación continua para esta dosis puede estar supeditada a la verificación y descripción del beneficio clínico en los ensayos confirmatorios.
2 DOSIS Y ADMINISTRACIÓN
2.1 Selección de pacientes
La información sobre las pruebas aprobadas por la FDA para la selección de pacientes está disponible en:
http://www.fda.gov/CompanionDiagnostics.
Selección de pacientes para tratamiento con un solo agente
Seleccione a los pacientes para el tratamiento con KEYTRUDA como agente único según la presencia de expresión positiva de PD-L1 en:
- NSCLC en estadio III que no son candidatos para resección quirúrgica o quimiorradiación definitiva [ver Estudios clínicos (14.2)].
- NSCLC metastásico [ver Estudios clínicos (14.2)].
- tratamiento de primera línea de HNSCC metastásico o irresecable, recurrente [ver Estudios clínicos (14.4)].
- cáncer de esófago recurrente localmente avanzado o metastásico previamente tratado [ver Estudios clínicos (14.11)].
- cáncer de cuello uterino recurrente o metastásico con progresión de la enfermedad durante o después de la quimioterapia [ver Estudios clínicos (14.12)].
Para las indicaciones MSI-H/dMMR, seleccione pacientes para el tratamiento con KEYTRUDA como agente único según el estado MSI-H/dMMR en muestras tumorales [ver Estudios clínicos (14.8, 14.9)].
Para la indicación TMB-H, seleccione pacientes para el tratamiento con KEYTRUDA como agente único según el estado TMB-H en muestras tumorales [ver Estudios clínicos (14.18)].
Debido a que las mutaciones dMMR subclonales y la inestabilidad de microsatélites pueden surgir en gliomas de alto grado durante la terapia con temozolomida, se recomienda realizar pruebas de TMB-H, MSI-H y dMMR en las muestras tumorales primarias obtenidas antes del inicio de la quimioterapia con temozolomida en pacientes con gliomas de alto grado.
Información adicional sobre la selección de pacientes para MSI-H o dMMR en pacientes con tumores sólidos que no son CRC
Debido a la discrepancia entre las pruebas locales y las pruebas aprobadas por la FDA, se recomienda la confirmación del estado MSI-H o dMMR mediante una prueba aprobada por la FDA en pacientes con tumores sólidos MSI-H o dMMR, si es factible. Si no se pueden realizar pruebas confirmatorias de MSI-H/dMMR, la presencia de TMB ≥10 mut/Mb, según lo determine una prueba aprobada por la FDA, puede usarse para seleccionar pacientes para el tratamiento [ver Estudios clínicos (14.8)].
Selección de pacientes para terapia combinada
Para el uso de KEYTRUDA en combinación con quimioterapia y trastuzumab, seleccione pacientes según la presencia de expresión positiva de PD-L1 (CPS ≥1) en adenocarcinoma gástrico o de la unión gastroesofágica (GEJ) HER2-positivo localmente avanzado irresecable o metastásico [ver Estudios clínicos (14.10)].
Para el uso de KEYTRUDA en combinación con quimioterapia, con o sin bevacizumab, seleccione pacientes según la presencia de expresión positiva de PD-L1 en cáncer de cuello uterino persistente, recurrente o metastásico [ver Estudios clínicos (14.12)].
Para la indicación de carcinoma endometrial avanzado pMMR/no MSI-H, seleccione pacientes para el tratamiento con KEYTRUDA en combinación con lenvatinib según el estado MSI o MMR en muestras tumorales [ver Estudios clínicos (14.17)].
Para el uso de KEYTRUDA en combinación con quimioterapia, seleccione pacientes según la presencia de expresión positiva de PD-L1 en TNBC localmente recurrente irresecable o metastásico [ver Estudios clínicos (14.20)].
Información adicional sobre la selección de pacientes
- Actualmente no hay disponible una prueba aprobada por la FDA para la detección de no MSI-H para la selección de pacientes con carcinoma endometrial no MSI-H para el tratamiento con KEYTRUDA en combinación con lenvatinib [ver Estudios clínicos (14.17)].
2.2 Dosis recomendada
| Indicación | Dosis recomendada de KEYTRUDA |
Duración/Tiempo de tratamiento |
|---|---|---|
|
||
| Monoterapia | ||
| Pacientes adultos con melanoma irresectable o metastásico |
200 mg cada 3 semanas* o 400 mg cada 6 semanas* |
Hasta la progresión de la enfermedad o toxicidad inaceptable |
| Tratamiento adyuvante de pacientes adultos con melanoma, NSCLC o RCC |
200 mg cada 3 semanas* o 400 mg cada 6 semanas* |
Hasta la recurrencia de la enfermedad, toxicidad inaceptable o hasta 12 meses |
| Pacientes adultos con NSCLC, HNSCC, cHL, PMBCL, Carcinoma Urotelial localmente avanzado o metastásico, Cáncer MSI-H o dMMR, MSI-H o dMMR CRC, Carcinoma Endometrial MSI-H o dMMR, Cáncer de Esófago, Cáncer de Cuello Uterino, HCC, MCC, Cáncer TMB-H o cSCC |
200 mg cada 3 semanas* o 400 mg cada 6 semanas* |
Hasta la progresión de la enfermedad, toxicidad inaceptable o hasta 24 meses |
| Pacientes adultos con NMIBC de alto riesgo que no responde al BCG |
200 mg cada 3 semanas* o 400 mg cada 6 semanas* |
Hasta NMIBC de alto riesgo persistente o recurrente, progresión de la enfermedad, toxicidad inaceptable o hasta 24 meses |
| Pacientes pediátricos con cHL, PMBCL, Cáncer MSI-H o dMMR, MCC o Cáncer TMB-H |
2 mg/kg cada 3 semanas (hasta un máximo de 200 mg)* |
Hasta la progresión de la enfermedad, toxicidad inaceptable o hasta 24 meses |
| Pacientes pediátricos (12 años y mayores) para el tratamiento adyuvante del melanoma |
2 mg/kg cada 3 semanas (hasta un máximo de 200 mg)* |
Hasta la recurrencia de la enfermedad, toxicidad inaceptable o hasta 12 meses |
| Terapia de combinación† | ||
| Pacientes adultos con NSCLC resecable | 200 mg cada 3 semanas* o 400 mg cada 6 semanas* Administrar KEYTRUDA antes de la quimioterapia cuando se administren el mismo día. |
Tratamiento neoadyuvante en combinación con quimioterapia durante 12 semanas o hasta la progresión de la enfermedad que imposibilite la cirugía definitiva o toxicidad inaceptable, seguido de tratamiento adyuvante con KEYTRUDA como agente único después de la cirugía durante 39 semanas o hasta la recurrencia de la enfermedad o toxicidad inaceptable |
| Pacientes adultos con NSCLC, MPM, HNSCC, Cáncer Gástrico HER2-negativo, Cáncer de Esófago o BTC |
200 mg cada 3 semanas* o 400 mg cada 6 semanas* Administrar KEYTRUDA antes de la quimioterapia cuando se administren el mismo día. |
Hasta la progresión de la enfermedad, toxicidad inaceptable o hasta 24 meses |
| Pacientes adultos con cáncer urotelial localmente avanzado o metastásico |
200 mg cada 3 semanas* o 400 mg cada 6 semanas* Administrar KEYTRUDA después de enfortumab vedotin cuando se administren el mismo día. |
Hasta la progresión de la enfermedad, toxicidad inaceptable o hasta 24 meses |
| Pacientes adultos con Cáncer Gástrico HER2-positivo |
200 mg cada 3 semanas* o 400 mg cada 6 semanas* Administrar KEYTRUDA antes de trastuzumab y quimioterapia cuando se administren el mismo día. |
Hasta la progresión de la enfermedad, toxicidad inaceptable o hasta 24 meses |
| Pacientes adultos con Cáncer de Cuello Uterino |
200 mg cada 3 semanas* o 400 mg cada 6 semanas* Administrar KEYTRUDA antes de la quimiorradioterapia o antes de la quimioterapia con o sin bevacizumab cuando se administren el mismo día. |
Hasta la progresión de la enfermedad, toxicidad inaceptable o, para KEYTRUDA, hasta 24 meses |
| Pacientes adultos con RCC | 200 mg cada 3 semanas* o 400 mg cada 6 semanas* Administrar KEYTRUDA en combinación con axitinib 5 mg por vía oral dos veces al día‡ o Administrar KEYTRUDA en combinación con lenvatinib 20 mg por vía oral una vez al día. |
Hasta la progresión de la enfermedad, toxicidad inaceptable o, para KEYTRUDA, hasta 24 meses |
| Pacientes adultos con carcinoma endometrial |
200 mg cada 3 semanas* o 400 mg cada 6 semanas* Administrar KEYTRUDA antes de carboplatino y paclitaxel cuando se administren el mismo día. o Administrar KEYTRUDA en combinación con lenvatinib 20 mg por vía oral una vez al día. |
Hasta la progresión de la enfermedad, toxicidad inaceptable o, para KEYTRUDA, hasta 24 meses |
| Pacientes adultos con TNBC de estadio temprano de alto riesgo |
200 mg cada 3 semanas* o 400 mg cada 6 semanas* Administrar KEYTRUDA antes de la quimioterapia cuando se administren el mismo día. |
Tratamiento neoadyuvante en combinación con quimioterapia durante 24 semanas (8 dosis de 200 mg cada 3 semanas o 4 dosis de 400 mg cada 6 semanas) o hasta la progresión de la enfermedad o toxicidad inaceptable, seguido de tratamiento adyuvante con KEYTRUDA como agente único durante un máximo de 27 semanas (9 dosis de 200 mg cada 3 semanas o 5 dosis de 400 mg cada 6 semanas) o hasta la recurrencia de la enfermedad o toxicidad inaceptable§ |
| Pacientes adultos con TNBC localmente recurrente irresecable o metastásico |
200 mg cada 3 semanas* o 400 mg cada 6 semanas* Administrar KEYTRUDA antes de la quimioterapia cuando se administre el mismo día. |
Hasta la progresión de la enfermedad, toxicidad inaceptable o hasta 24 meses |
2.3 Modificaciones de la dosis
No se recomienda la reducción de la dosis de KEYTRUDA. En general, suspenda KEYTRUDA para las reacciones adversas inmunomediadas graves (Grado 3). Suspenda permanentemente KEYTRUDA para las reacciones adversas inmunomediadas potencialmente mortales (Grado 4), las reacciones inmunomediadas graves recurrentes (Grado 3) que requieren tratamiento inmunosupresor sistémico o la incapacidad de reducir la dosis de corticosteroides a 10 mg o menos de prednisona o equivalente por día dentro de las 12 semanas posteriores al inicio de los esteroides.
Las modificaciones de la dosis de KEYTRUDA para las reacciones adversas que requieren un manejo diferente de estas pautas generales se resumen en la Tabla 2.
| Reacción adversa | Severidad* | Modificación de la dosis |
|---|---|---|
| ALT = alanina aminotransferasa, AST = aspartato aminotransferasa, DRESS = erupción cutánea por fármacos con eosinofilia y síntomas sistémicos, SJS = síndrome de Stevens Johnson, TEN = necrólisis epidérmica tóxica, ULN = límite superior normal | ||
|
||
| Reacciones adversas inmunomediadas [ver Advertencias y precauciones (5.1)] | ||
| Neumonitis | Grado 2 | Suspender† |
| Grado 3 o 4 | Suspender permanentemente | |
| Colitis | Grado 2 o 3 | Suspender† |
| Grado 4 | Suspender permanentemente | |
|
Hepatitis sin afectación tumoral |
AST o ALT aumenta a más de 3 y hasta 8 veces ULN o La bilirrubina total aumenta a más de 1.5 y hasta 3 veces ULN |
Suspender† |
| Para elevaciones de enzimas hepáticas en pacientes tratados con terapia de combinación con axitinib, ver Tabla 3. |
AST o ALT aumenta a más de 8 veces ULN o La bilirrubina total aumenta a más de 3 veces ULN |
Suspender permanentemente |
| Hepatitis con afectación tumoral del hígado‡ |
AST o ALT basal es más de 1 y hasta 3 veces ULN y aumenta a más de 5 y hasta 10 veces ULN o AST o ALT basal es más de 3 y hasta 5 veces ULN y aumenta a más de 8 y hasta 10 veces ULN |
Suspender† |
| ALT o AST aumenta a más de 10 veces ULN o La bilirrubina total aumenta a más de 3 veces ULN |
Suspender permanentemente | |
| Endocrinopatías | Grado 3 o 4 | Suspender hasta que esté clínicamente estable o suspender permanentemente dependiendo de la gravedad |
| Nefritis con disfunción renal | Aumento de creatinina en sangre de grado 2 o 3 | Suspender† |
| Aumento de creatinina en sangre de grado 4 | Suspender permanentemente | |
| Afecciones dermatológicas exfoliativas | Sospecha de SJS, TEN o DRESS | Suspender† |
| SJS, TEN o DRESS confirmados | Suspender permanentemente | |
| Miocarditis | Grado 2, 3 o 4 | Suspender permanentemente |
| Toxicidades neurológicas | Grado 2 | Suspender† |
| Grado 3 o 4 | Suspender permanentemente | |
| Hematologic toxicity in patients with cHL or PMBCL |
Grade 4 | Suspender hasta la resolución a Grados 0 o 1 |
| Otras Reacciones Adversas | ||
| Reacciones relacionadas con la infusión [see Warnings and Precautions (5.2)] |
Grade 1 or 2 | Interrumpir o reducir la velocidad de infusión |
| Grade 3 or 4 | Interrumpir permanentemente | |
La siguiente tabla representa las modificaciones de dosis que son diferentes de las descritas anteriormente para KEYTRUDA o en la Información de Prescripción Completa para el medicamento administrado en combinación.
| Tratamiento | Reacción Adversa | Severidad | Modificación de la Dosis |
|---|---|---|---|
| ALT = alanina aminotransferasa, AST = aspartato aminotransferasa, ULN = límite superior normal | |||
|
|||
| KEYTRUDA en combinación con axitinib |
Elevaciones de enzimas hepáticas* | Los aumentos de ALT o AST son al menos 3 veces pero menos de 10 veces el ULN sin bilirrubina total concurrente al menos 2 veces el ULN | Suspender KEYTRUDA y axitinib hasta la resolución a Grados 0 o 1† |
| ALT o AST aumenta a más de 3 veces ULN con bilirrubina total concurrente al menos 2 veces ULN o ALT o AST ≥10 veces ULN |
Interrumpir permanentemente KEYTRUDA y axitinib |
||
Modificaciones de Dosis Recomendadas para Reacciones Adversas para KEYTRUDA en Combinación con Lenvatinib
Cuando se administra KEYTRUDA en combinación con lenvatinib, modifique la dosis de uno o ambos medicamentos. Suspenda o interrumpa KEYTRUDA como se muestra en la Tabla 2. Consulte la información de prescripción de lenvatinib para obtener información adicional sobre la modificación de la dosis.
2.4 Preparación y Administración
Preparación para la infusión intravenosa
- Inspeccione visualmente la solución en busca de partículas y decoloración. La solución es de transparente a ligeramente opalescente, de incolora a ligeramente amarilla. Deseche el vial si se observan partículas visibles.
- Diluya la inyección de KEYTRUDA (solución) antes de la administración intravenosa.
- Extraiga el volumen requerido del(los) vial(es) de KEYTRUDA y transfiéralo a una bolsa intravenosa (IV) que contenga inyección de cloruro de sodio al 0,9 %, USP o inyección de dextrosa al 5 %, USP. Mezcle la solución diluida invirtiéndola suavemente. No agitar. La concentración final de la solución diluida debe estar entre 1 mg/mL y 10 mg/mL.
- Deseche cualquier porción no utilizada que quede en el vial.
Almacenamiento de la solución diluida
El producto no contiene conservantes.
Almacene la solución diluida del vial de KEYTRUDA 100 mg/4 mL de la siguiente manera:
- A temperatura ambiente durante no más de 6 horas desde el momento de la dilución. Esto incluye el almacenamiento a temperatura ambiente de la solución diluida y la duración de la infusión.
- En refrigeración a 2°C a 8°C (36°F a 46°F) durante no más de 96 horas desde el momento de la dilución. Si se refrigera, deje que la solución diluida alcance la temperatura ambiente antes de la administración. No agitar.
Deseche después de 6 horas a temperatura ambiente o después de 96 horas en refrigeración.
No congelar.
Administración
- Administre la solución diluida por vía intravenosa durante 30 minutos a través de una vía intravenosa que contenga un filtro estéril, no pirogénico, de baja unión a proteínas de 0,2 micras a 5 micras en línea o adicional.
- No coadministre otros medicamentos a través de la misma vía de infusión.
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
- Inyección: 100 mg/4 mL (25 mg/mL) solución transparente a ligeramente opalescente, incolora a ligeramente amarilla en un vial de dosis única
4 CONTRAINDICACIONES
Ninguna.
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Reacciones adversas inmunitarias graves y fatales mediadas por el sistema inmune
KEYTRUDA es un anticuerpo monoclonal que pertenece a una clase de fármacos que se unen ya sea al receptor de muerte programada 1 (PD-1) o al ligando 1 de PD (PD-L1), bloqueando la vía PD-1/PD-L1, eliminando así la inhibición de la respuesta inmune, potencialmente rompiendo la tolerancia periférica e induciendo reacciones adversas inmunitarias mediadas. Las importantes reacciones adversas inmunitarias mediadas que se enumeran en ADVERTENCIAS Y PRECAUCIONES pueden no incluir todas las posibles reacciones adversas inmunitarias graves y fatales mediadas.
Las reacciones adversas inmunitarias mediadas, que pueden ser graves o fatales, pueden ocurrir en cualquier sistema orgánico o tejido y pueden afectar más de un sistema corporal simultáneamente. Las reacciones adversas inmunitarias mediadas pueden ocurrir en cualquier momento después de iniciar el tratamiento con un anticuerpo que bloquea PD-1/PD-L1. Si bien las reacciones adversas inmunitarias mediadas generalmente se manifiestan durante el tratamiento con anticuerpos que bloquean PD-1/PD-L1, también pueden manifestarse después de la suspensión de los anticuerpos que bloquean PD-1/PD-L1.
La identificación temprana y el manejo de las reacciones adversas inmunitarias mediadas son esenciales para garantizar el uso seguro de los anticuerpos que bloquean PD-1/PD-L1. Monitorice a los pacientes de cerca para detectar síntomas y signos que puedan ser manifestaciones clínicas de reacciones adversas inmunitarias mediadas subyacentes. Evalúe las enzimas hepáticas, la creatinina y la función tiroidea al inicio y periódicamente durante el tratamiento. Para pacientes con TNBC tratados con KEYTRUDA en el entorno neoadyuvante, controle el cortisol sanguíneo al inicio, antes de la cirugía y según lo indique la clínica. En casos de sospecha de reacciones adversas inmunitarias mediadas, inicie las pruebas apropiadas para excluir etiologías alternativas, incluyendo infecciones. Inicie el manejo médico de inmediato, incluyendo la consulta especializada según corresponda.
Suspenda o descontinúe definitivamente KEYTRUDA dependiendo de la gravedad [véase Dosificación y Administración (2.3) ] . En general, si KEYTRUDA requiere interrupción o descontinuación, administre terapia con corticosteroides sistémicos (1 a 2 mg/kg/día de prednisona o equivalente) hasta que mejore a Grado 1 o menos. Una vez que mejore a Grado 1 o menos, inicie la disminución gradual de los corticosteroides y continúe disminuyéndolos durante al menos 1 mes. Considere la administración de otros inmunosupresores sistémicos en pacientes cuyas reacciones adversas inmunitarias mediadas no se controlan con terapia con corticosteroides.
Se discuten a continuación las pautas de manejo de toxicidad para las reacciones adversas que no necesariamente requieren esteroides sistémicos (por ejemplo, endocrinopatías y reacciones dermatológicas).
Neumonitis inmunomediada
KEYTRUDA puede causar neumonitis inmunomediada. La incidencia de neumonitis es mayor en pacientes que han recibido radioterapia torácica previa. La neumonitis inmunomediada se produjo en el 3,4% (94/2799) de los pacientes que recibieron KEYTRUDA, incluidas reacciones adversas fatales (0,1%), Grado 4 (0,3%), Grado 3 (0,9%) y Grado 2 (1,3%). Se requirieron corticosteroides sistémicos en el 67% (63/94) de los pacientes con neumonitis. La neumonitis llevó a la descontinuación permanente de KEYTRUDA en el 1,3% (36) de los pacientes y a la retención de KEYTRUDA en el 0,9% (26) de los pacientes. Todos los pacientes que fueron retenidos reiniciaron KEYTRUDA después de la mejora de los síntomas; de estos, el 23% tuvo recurrencia de neumonitis. La neumonitis se resolvió en el 59% de los 94 pacientes.
En estudios clínicos que incluyeron a 389 pacientes adultos con LCH que recibieron KEYTRUDA como agente único, la neumonitis se produjo en 31 (8%) pacientes, incluidas neumonitis de Grados 3-4 en el 2,3% de los pacientes. Los pacientes recibieron corticosteroides en dosis altas durante una mediana de 10 días (rango: 2 días a 53 meses). Las tasas de neumonitis fueron similares en pacientes con y sin radioterapia torácica previa. La neumonitis llevó a la descontinuación de KEYTRUDA en 21 (5,4%) pacientes. De los pacientes que desarrollaron neumonitis, el 42% interrumpió KEYTRUDA, el 68% descontinuó KEYTRUDA y el 77% tuvo resolución.
En un estudio clínico que incluyó a 580 pacientes adultos con NSCLC resecado (KEYNOTE-091) que recibieron KEYTRUDA como agente único para el tratamiento adyuvante, la neumonitis se produjo en 41 (7%) pacientes, incluidas reacciones adversas fatales (0,2%), Grado 4 (0,3%) y Grado 3 (1%). Los pacientes recibieron corticosteroides en dosis altas durante una mediana de 10 días (rango: 1 día a 2,3 meses). La neumonitis llevó a la descontinuación de KEYTRUDA en 26 (4,5%) de los pacientes. De los pacientes que desarrollaron neumonitis, el 54% interrumpió KEYTRUDA, el 63% descontinuó KEYTRUDA y el 71% tuvo resolución.
Colitis inmunomediada
KEYTRUDA puede causar colitis inmunomediada, que puede presentarse con diarrea. Se ha informado de infección/reactivación por citomegalovirus (CMV) en pacientes con colitis inmunomediada refractaria a los corticosteroides. En casos de colitis refractaria a los corticosteroides, considere repetir las pruebas infecciosas para excluir etiologías alternativas. La colitis inmunomediada se produjo en el 1,7% (48/2799) de los pacientes que recibieron KEYTRUDA, incluidas reacciones adversas de Grado 4 (<0,1%), Grado 3 (1,1%) y Grado 2 (0,4%). Se requirieron corticosteroides sistémicos en el 69% (33/48) de los pacientes con colitis. Se requirió terapia inmunosupresora adicional en el 4,2% de los pacientes. La colitis llevó a la descontinuación permanente de KEYTRUDA en el 0,5% (15) de los pacientes y a la retención de KEYTRUDA en el 0,5% (13) de los pacientes. Todos los pacientes que fueron retenidos reiniciaron KEYTRUDA después de la mejora de los síntomas; de estos, el 23% tuvo recurrencia de colitis. La colitis se resolvió en el 85% de los 48 pacientes.
Hepatotoxicidad e hepatitis inmunomediada
KEYTRUDA como agente único
KEYTRUDA puede causar hepatitis inmunomediada. La hepatitis inmunomediada se produjo en el 0,7% (19/2799) de los pacientes que recibieron KEYTRUDA, incluidas reacciones adversas de Grado 4 (<0,1%), Grado 3 (0,4%) y Grado 2 (0,1%). Se requirieron corticosteroides sistémicos en el 68% (13/19) de los pacientes con hepatitis. El once por ciento de estos pacientes requirió terapia inmunosupresora adicional. La hepatitis llevó a la descontinuación permanente de KEYTRUDA en el 0,2% (6) de los pacientes y a la retención de KEYTRUDA en el 0,3% (9) de los pacientes. Todos los pacientes que fueron retenidos reiniciaron KEYTRUDA después de la mejora de los síntomas; de estos, ninguno tuvo recurrencia de hepatitis. La hepatitis se resolvió en el 79% de los 19 pacientes.
KEYTRUDA con axitinib
KEYTRUDA en combinación con axitinib puede causar toxicidad hepática con frecuencias mayores a las esperadas de elevaciones de ALT y AST de Grados 3 y 4 en comparación con KEYTRUDA solo. Monitorear las enzimas hepáticas antes del inicio y periódicamente durante el tratamiento. Considere un monitoreo más frecuente de las enzimas hepáticas en comparación con cuando los medicamentos se administran como agentes únicos. Para las enzimas hepáticas elevadas, interrumpa KEYTRUDA y axitinib, y considere administrar corticosteroides según sea necesario [ver Dosis y Administración (2.3)].
Con la combinación de KEYTRUDA y axitinib, se observaron ALT aumentada de Grados 3 y 4 (20%) y AST aumentada (13%). El cincuenta y nueve por ciento de los pacientes con ALT aumentada recibieron corticosteroides sistémicos. En pacientes con ALT ≥3 veces el LSN (Grados 2-4, n=116), la ALT se resolvió a Grados 0-1 en el 94%. Entre los 92 pacientes que fueron retados con KEYTRUDA (n=3) o axitinib (n=34) administrados como agente único o con ambos (n=55), se observó la recurrencia de ALT ≥3 veces el LSN en 1 paciente que recibió KEYTRUDA, 16 pacientes que recibieron axitinib y 24 pacientes que recibieron KEYTRUDA y axitinib. Todos los pacientes con una recurrencia de ALT ≥3 LSN se recuperaron posteriormente del evento.
Endocrinopatías Mediadas por el Sistema Inmunitario
Insuficiencia Suprarrenal
KEYTRUDA puede causar insuficiencia suprarrenal primaria o secundaria. Para la insuficiencia suprarrenal de Grado 2 o superior, inicie el tratamiento sintomático, incluyendo la terapia de reemplazo hormonal según esté clínicamente indicado. Suspender KEYTRUDA dependiendo de la gravedad [ver Dosis y Administración (2.3)].
La insuficiencia suprarrenal se produjo en el 0,8% (22/2799) de los pacientes que recibieron KEYTRUDA, incluyendo reacciones adversas de Grado 4 (<0,1%), Grado 3 (0,3%) y Grado 2 (0,3%). Se requirieron corticosteroides sistémicos en el 77% (17/22) de los pacientes con insuficiencia suprarrenal; de estos, la mayoría permaneció con corticosteroides sistémicos. La insuficiencia suprarrenal provocó la interrupción permanente de KEYTRUDA en <0,1% (1) de los pacientes y la suspensión de KEYTRUDA en el 0,3% (8) de los pacientes. Todos los pacientes a quienes se les suspendió el tratamiento reiniciaron KEYTRUDA después de la mejoría de los síntomas.
Hipofisitis
KEYTRUDA puede causar hipofisitis mediada por el sistema inmunitario. La hipofisitis puede presentarse con síntomas agudos asociados con el efecto de masa, como dolor de cabeza, fotofobia o defectos del campo visual. La hipofisitis puede causar hipopituitarismo. Iniciar la terapia de reemplazo hormonal según esté indicado. Suspender o interrumpir permanentemente KEYTRUDA dependiendo de la gravedad [ver Dosis y Administración (2.3)].
La hipofisitis se produjo en el 0,6% (17/2799) de los pacientes que recibieron KEYTRUDA, incluyendo reacciones adversas de Grado 4 (<0,1%), Grado 3 (0,3%) y Grado 2 (0,2%). Se requirieron corticosteroides sistémicos en el 94% (16/17) de los pacientes con hipofisitis; de estos, la mayoría permaneció con corticosteroides sistémicos. La hipofisitis provocó la interrupción permanente de KEYTRUDA en el 0,1% (4) de los pacientes y la suspensión de KEYTRUDA en el 0,3% (7) de los pacientes. Todos los pacientes a quienes se les suspendió el tratamiento reiniciaron KEYTRUDA después de la mejoría de los síntomas.
Trastornos Tiroideos
KEYTRUDA puede causar trastornos tiroideos mediados por el sistema inmunitario. La tiroiditis puede presentarse con o sin endocrinopatía. El hipotiroidismo puede seguir a la hipertiroidismo. Iniciar la terapia de reemplazo hormonal para el hipotiroidismo o instituir el manejo médico de la hipertiroidismo según esté clínicamente indicado. Suspender o interrumpir permanentemente KEYTRUDA dependiendo de la gravedad [ver Dosis y Administración (2.3)].
La tiroiditis se produjo en el 0,6% (16/2799) de los pacientes que recibieron KEYTRUDA, incluyendo Grado 2 (0,3%). Ningún paciente interrumpió KEYTRUDA debido a la tiroiditis. KEYTRUDA se suspendió en <0,1% (1) de los pacientes.
La hipertiroidismo se produjo en el 3,4% (96/2799) de los pacientes que recibieron KEYTRUDA, incluyendo Grado 3 (0,1%) y Grado 2 (0,8%). La hipertiroidismo provocó la interrupción permanente de KEYTRUDA en <0,1% (2) de los pacientes y la suspensión de KEYTRUDA en el 0,3% (7) de los pacientes. Todos los pacientes a quienes se les suspendió el tratamiento reiniciaron KEYTRUDA después de la mejoría de los síntomas.
La incidencia de hipertiroidismo nuevo o que empeoró fue mayor en 580 pacientes con CPN resecado, que se produjo en el 11% de los pacientes que recibieron KEYTRUDA como agente único como tratamiento adyuvante (KEYNOTE-091), incluyendo hipertiroidismo de Grado 3 (0,2%).
El hipotiroidismo se produjo en el 8% (237/2799) de los pacientes que recibieron KEYTRUDA, incluyendo Grado 3 (0,1%) y Grado 2 (6,2%). El hipotiroidismo provocó la interrupción permanente de KEYTRUDA en <0,1% (1) de los pacientes y la suspensión de KEYTRUDA en el 0,5% (14) de los pacientes. Todos los pacientes a quienes se les suspendió el tratamiento reiniciaron KEYTRUDA después de la mejoría de los síntomas. La mayoría de los pacientes con hipotiroidismo requirieron terapia de reemplazo de hormona tiroidea a largo plazo.
La incidencia de hipotiroidismo nuevo o que empeoró fue mayor en 1185 pacientes con CCH, que se produjo en el 16% de los pacientes que recibieron KEYTRUDA como agente único o en combinación con platino y FU, incluyendo hipotiroidismo de Grado 3 (0,3%). La incidencia de hipotiroidismo nuevo o que empeoró fue mayor en 389 pacientes con LNH (17%) que recibieron KEYTRUDA como agente único, incluyendo hipotiroidismo de Grado 1 (6,2%) y Grado 2 (10,8%).
La incidencia de hipotiroidismo nuevo o que empeoró fue mayor en 580 pacientes con CPN resecado, que se produjo en el 22% de los pacientes que recibieron KEYTRUDA como agente único como tratamiento adyuvante (KEYNOTE-091), incluyendo hipotiroidismo de Grado 3 (0,3%).
Diabetes Mellitus Tipo 1, que puede presentarse con Cetoacidosis Diabética
Monitorear a los pacientes para detectar hiperglucemia u otros signos y síntomas de diabetes. Iniciar el tratamiento con insulina según esté clínicamente indicado. Suspender KEYTRUDA dependiendo de la gravedad [ver Dosis y Administración (2.3)].
La diabetes mellitus tipo 1 se produjo en el 0,2% (6/2799) de los pacientes que recibieron KEYTRUDA. La diabetes mellitus tipo 1 provocó la interrupción permanente en <0,1% (1) de los pacientes y la suspensión de KEYTRUDA en <0,1% (1) de los pacientes. Todos los pacientes a quienes se les suspendió el tratamiento reiniciaron KEYTRUDA después de la mejoría de los síntomas. Todos los pacientes con diabetes mellitus tipo 1 requirieron terapia con insulina a largo plazo.
Nefritis Mediada por el Sistema Inmunitario con Disfunción Renal
KEYTRUDA puede causar nefritis mediada por el sistema inmunitario. La nefritis mediada por el sistema inmunitario se produjo en el 0,3 % (9/2799) de los pacientes que recibieron KEYTRUDA, incluidas reacciones adversas de Grado 4 (<0,1 %), Grado 3 (0,1 %) y Grado 2 (0,1 %). Se requirieron corticosteroides sistémicos en el 89 % (8/9) de los pacientes con nefritis. La nefritis provocó la interrupción permanente de KEYTRUDA en el 0,1 % (3) de los pacientes y la suspensión de KEYTRUDA en el 0,1 % (3) de los pacientes. Todos los pacientes a quienes se les suspendió el tratamiento reiniciaron KEYTRUDA después de la mejoría de los síntomas; de estos, ninguno tuvo una recurrencia de nefritis. La nefritis se resolvió en el 56 % de los 9 pacientes.
Reacciones adversas dermatológicas mediadas por el sistema inmunitario
KEYTRUDA puede causar erupción o dermatitis mediada por el sistema inmunitario. Se ha producido dermatitis exfoliativa, incluido el síndrome de Stevens-Johnson, DRESS y la necrolisis epidérmica tóxica (NET), con anticuerpos bloqueantes de PD-1/PD-L1. Los emolientes tópicos y/o los corticosteroides tópicos pueden ser adecuados para tratar las erupciones no exfoliativas leves o moderadas. Suspender o interrumpir permanentemente KEYTRUDA según la gravedad [ver Posología y administración (2.3)].
Las reacciones adversas dermatológicas mediadas por el sistema inmunitario se produjeron en el 1,4 % (38/2799) de los pacientes que recibieron KEYTRUDA, incluidas reacciones adversas de Grado 3 (1 %) y Grado 2 (0,1 %). Se requirieron corticosteroides sistémicos en el 40 % (15/38) de los pacientes con reacciones adversas dermatológicas mediadas por el sistema inmunitario. Las reacciones adversas dermatológicas mediadas por el sistema inmunitario provocaron la interrupción permanente de KEYTRUDA en el 0,1 % (2) de los pacientes y la suspensión de KEYTRUDA en el 0,6 % (16) de los pacientes. Todos los pacientes a quienes se les suspendió el tratamiento reiniciaron KEYTRUDA después de la mejoría de los síntomas; de estos, el 6 % tuvo una recurrencia de reacciones adversas dermatológicas mediadas por el sistema inmunitario. Las reacciones adversas dermatológicas mediadas por el sistema inmunitario se resolvieron en el 79 % de los 38 pacientes.
Otras reacciones adversas mediadas por el sistema inmunitario
Las siguientes reacciones adversas mediadas por el sistema inmunitario clínicamente significativas se produjeron con una incidencia de <1 % (a menos que se indique lo contrario) en pacientes que recibieron KEYTRUDA o se informaron con el uso de otros anticuerpos bloqueantes de PD-1/PD-L1. Se han notificado casos graves o mortales para algunas de estas reacciones adversas.
Cardiovasculares: Miocarditis, pericarditis, vasculitis
Sistema nervioso: Meningitis, encefalitis, mielitis y desmielinización, síndrome miasténico/miastenia gravis (incluida la exacerbación), síndrome de Guillain-Barré, paresia nerviosa, neuropatía autoinmune
Oculares: Puede producirse uveítis, iritis y otras toxicidades inflamatorias oculares. Algunos casos pueden estar asociados con desprendimiento de retina. Pueden producirse diversos grados de discapacidad visual, incluida la ceguera. Si se produce uveítis en combinación con otras reacciones adversas mediadas por el sistema inmunitario, considere un síndrome similar al de Vogt-Koyanagi-Harada, ya que esto puede requerir tratamiento con esteroides sistémicos para reducir el riesgo de pérdida permanente de la visión.
Gastrointestinales: Pancreatitis, que incluye aumentos en los niveles séricos de amilasa y lipasa, gastritis, duodenitis
Musculoesqueléticas y del tejido conjuntivo: Miositis/polimiositis, rabdomiolisis (y secuelas asociadas, incluida la insuficiencia renal), artritis (1,5 %), polimialgia reumática
Endocrinas: Hipoparatiroidismo
Hematologías/Inmunológicas: Anemia hemolítica, anemia aplásica, linfistiocitosis hemofagocítica, síndrome de respuesta inflamatoria sistémica, linfadenitis necrotizante histiocítica (linfadenitis de Kikuchi), sarcoidosis, púrpura trombocitopénica inmunitaria, rechazo de trasplante de órgano sólido, rechazo de otro trasplante (incluido el injerto corneal)
5.2 Reacciones relacionadas con la infusión
KEYTRUDA puede causar reacciones relacionadas con la infusión graves o potencialmente mortales, incluidas hipersensibilidad y anafilaxia, que se han notificado en el 0,2 % de 2799 pacientes que recibieron KEYTRUDA. Controle a los pacientes para detectar signos y síntomas de reacciones relacionadas con la infusión, como escalofríos, temblores, sibilancias, prurito, rubor, erupción, hipotensión, hipoxemia y fiebre. Interrumpa o disminuya la velocidad de infusión para las reacciones relacionadas con la infusión leves (Grado 1) o moderadas (Grado 2). Para las reacciones relacionadas con la infusión graves (Grado 3) o potencialmente mortales (Grado 4), detenga la infusión e interrumpa permanentemente KEYTRUDA [ver Posología y administración (2.3)].
5.3 Complicaciones del TCHC alogénico
Pueden producirse complicaciones mortales y otras complicaciones graves en pacientes que reciben un trasplante de células madre hematopoyéticas alogénico (TCHC) antes o después de ser tratados con un anticuerpo bloqueante de PD-1/PD-L1. Las complicaciones relacionadas con el trasplante incluyen enfermedad de injerto contra huésped hiperaguda (EICH), EICH aguda, EICH crónica, enfermedad veno-oclusiva hepática (EVO) después del acondicionamiento de intensidad reducida y síndrome febril que requiere esteroides (sin una causa infecciosa identificada). Estas complicaciones pueden producirse a pesar de la terapia intermedia entre el bloqueo de PD-1/PD-L1 y el TCHC alogénico.
5.4 Aumento de la mortalidad en pacientes con mieloma múltiple cuando se añade KEYTRUDA a un análogo de la talidomida y dexametasona
En dos ensayos aleatorizados en pacientes con mieloma múltiple, la adición de KEYTRUDA a un análogo de la talidomida más dexametasona, un uso para el cual no está indicado ningún anticuerpo bloqueante de PD-1 o PD-L1, provocó un aumento de la mortalidad. No se recomienda el tratamiento de pacientes con mieloma múltiple con un anticuerpo bloqueante de PD-1 o PD-L1 en combinación con un análogo de la talidomida más dexametasona fuera de los ensayos controlados.
5.5 Toxicidad embriofetal
Basándose en su mecanismo de acción, KEYTRUDA puede causar daño fetal cuando se administra a una mujer embarazada. Los modelos animales vinculan la vía de señalización PD-1/PD-L1 con el mantenimiento del embarazo a través de la inducción de la tolerancia inmunitaria materna al tejido fetal. Advierta a las mujeres del riesgo potencial para un feto. Aconseje a las mujeres en edad fértil que utilicen métodos anticonceptivos efectivos durante el tratamiento con KEYTRUDA y durante 4 meses después de la última dosis [ver Uso en Poblaciones Específicas (8.1, 8.3)].
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas clínicamente significativas se describen en otras partes del etiquetado.
- Reacciones adversas inmunomediadas graves y mortales [ver Advertencias y precauciones (5.1)].
- Reacciones relacionadas con la infusión [ver Advertencias y precauciones (5.2)].
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se realizan en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no se pueden comparar directamente con las tasas en los ensayos clínicos de otro fármaco y pueden no reflejar las tasas observadas en la práctica.
Los datos descritos en las ADVERTENCIAS Y PRECAUCIONES reflejan la exposición a KEYTRUDA como agente único en 2799 pacientes en tres ensayos aleatorizados, abiertos y controlados con activo (KEYNOTE-002, KEYNOTE-006 y KEYNOTE-010), que incluyeron 912 pacientes con melanoma y 682 pacientes con CPNM, y un ensayo de un solo brazo (KEYNOTE-001), que incluyó 655 pacientes con melanoma y 550 pacientes con CPNM. Además de los 2799 pacientes, ciertas subsecciones en las ADVERTENCIAS Y PRECAUCIONES describen reacciones adversas observadas con la exposición a KEYTRUDA como agente único en un ensayo aleatorizado, controlado con placebo (KEYNOTE-091), que incluyó 580 pacientes con CPNM resecada, un ensayo no aleatorizado, abierto, multicóhorte (KEYNOTE-012), un ensayo no aleatorizado, abierto, de una sola cohorte (KEYNOTE-055) y dos ensayos aleatorizados, abiertos y controlados con activo (brazos de agente único KEYNOTE-040 y KEYNOTE-048), que incluyeron 909 pacientes con CNHCC; en dos ensayos no aleatorizados, abiertos (KEYNOTE-013 y KEYNOTE-087) y un ensayo aleatorizado, abierto y controlado con activo (KEYNOTE-204), que incluyó 389 pacientes con LNH; en un ensayo aleatorizado, abierto y controlado con activo (brazo de combinación KEYNOTE-048), que incluyó 276 pacientes con CNHCC; en combinación con axitinib en un ensayo aleatorizado y controlado con activo (KEYNOTE-426), que incluyó 429 pacientes con CCR; y en el uso posterior a la comercialización. En todos los ensayos, KEYTRUDA se administró en dosis de 2 mg/kg por vía intravenosa cada 3 semanas, 10 mg/kg por vía intravenosa cada 2 semanas, 10 mg/kg por vía intravenosa cada 3 semanas o 200 mg por vía intravenosa cada 3 semanas. Entre los 2799 pacientes, el 41% estuvo expuesto durante 6 meses o más y el 21% estuvo expuesto durante 12 meses o más.
Melanoma
Melanoma sin tratamiento previo con ipilimumab
La seguridad de KEYTRUDA para el tratamiento de pacientes con melanoma irresecable o metastásico que no habían recibido ipilimumab previamente y que habían recibido no más de una terapia sistémica previa se investigó en KEYNOTE-006. KEYNOTE-006 fue un ensayo multicéntrico, abierto y controlado con activo en el que los pacientes fueron aleatorizados (1:1:1) y recibieron KEYTRUDA 10 mg/kg cada 2 semanas (n=278) o KEYTRUDA 10 mg/kg cada 3 semanas (n=277) hasta la progresión de la enfermedad o toxicidad inaceptable o ipilimumab 3 mg/kg cada 3 semanas durante 4 dosis a menos que se interrumpiera antes por progresión de la enfermedad o toxicidad inaceptable (n=256) [ver Estudios clínicos (14.1)]. Los pacientes con enfermedad autoinmune, una condición médica que requirió corticosteroides sistémicos u otra medicación inmunosupresora; antecedentes de enfermedad pulmonar intersticial; o infección activa que requiriera terapia, incluyendo VIH o hepatitis B o C, fueron inelegibles.
La duración media de la exposición fue de 5,6 meses (rango: 1 día a 11,0 meses) para KEYTRUDA y similar en ambos brazos de tratamiento. Cincuenta y un y 46% de los pacientes recibieron KEYTRUDA 10 mg/kg cada 2 o 3 semanas, respectivamente, durante ≥6 meses. Ningún paciente en ninguno de los brazos recibió tratamiento durante más de un año.
Las características de la población del estudio fueron: edad media de 62 años (rango: 18 a 89); 60% hombres; 98% blancos; 32% tenían un valor elevado de lactato deshidrogenasa (LDH) al inicio del estudio; 65% tenían enfermedad en estadio M1c; 9% con antecedentes de metástasis cerebral; y aproximadamente el 36% habían sido tratados previamente con terapia sistémica que incluía un inhibidor de BRAF (15%), quimioterapia (13%) e inmunoterapia (6%).
En KEYNOTE-006, el perfil de reacciones adversas fue similar para el programa cada 2 semanas y cada 3 semanas, por lo que los resultados de seguridad resumidos se proporcionan en un análisis agrupado (n=555) de ambos brazos de KEYTRUDA. Las reacciones adversas que llevaron a la interrupción permanente de KEYTRUDA ocurrieron en el 9% de los pacientes. Las reacciones adversas que llevaron a la interrupción de KEYTRUDA en más de un paciente fueron colitis (1,4%), hepatitis autoinmune (0,7%), reacción alérgica (0,4%), polineuropatía (0,4%) e insuficiencia cardíaca (0,4%). Las reacciones adversas que llevaron a la interrupción de KEYTRUDA ocurrieron en el 21% de los pacientes; la más común (≥1%) fue diarrea (2,5%). Las tablas 4 y 5 resumen las reacciones adversas y las anormalidades de laboratorio seleccionadas, respectivamente, en pacientes con KEYTRUDA en KEYNOTE-006.
| Reacción adversa | KEYTRUDA 10 mg/kg cada 2 o 3 semanas |
Ipilimumab | ||||
|---|---|---|---|---|---|---|
| n=555 | n=256 | |||||
| Todos los grados† (%) |
Grados 3-4 (%) |
Todos los grados (%) |
Grados 3-4 (%) |
|||
|
||||
| General | ||||
| Fatiga | 28 | 0.9 | 28 | 3.1 |
| Piel y tejido subcutáneo | ||||
| Erupción‡ | 24 | 0.2 | 23 | 1.2 |
| Vitiligo§ | 13 | 0 | 2 | 0 |
| Sistema musculoesquelético y tejido conectivo | ||||
| Artralgia | 18 | 0.4 | 10 | 1.2 |
| Dolor de espalda | 12 | 0.9 | 7 | 0.8 |
| Respiratorio, torácico y mediastínico | ||||
| Tos | 17 | 0 | 7 | 0.4 |
| Disnea | 11 | 0.9 | 7 | 0.8 |
| Metabolismo y nutrición | ||||
| Disminución del apetito | 16 | 0.5 | 14 | 0.8 |
| Sistema nervioso | ||||
| Cefalea | 14 | 0.2 | 14 | 0.8 |
Otras reacciones adversas clínicamente importantes que ocurrieron en ≥10% de los pacientes que recibieron KEYTRUDA fueron diarrea (26%), náuseas (21%) y prurito (17%).
| Prueba de laboratorio† | KEYTRUDA 10 mg/kg cada 2 o 3 semanas |
Ipilimumab | ||
|---|---|---|---|---|
| Todos los grados‡ % |
Grados 3-4 % |
Todos los grados % |
Grados 3-4 % |
|
|
||||
| Química | ||||
| Hiperglucemia | 45 | 4.2 | 45 | 3.8 |
| Hipertrigliceridemia | 43 | 2.6 | 31 | 1.1 |
| Hiponatremia | 28 | 4.6 | 26 | 7 |
| Aumento de AST | 27 | 2.6 | 25 | 2.5 |
| Hipercolesterolemia | 20 | 1.2 | 13 | 0 |
| Hematología | ||||
| Anemia | 35 | 3.8 | 33 | 4.0 |
| Linfopenia | 33 | 7 | 25 | 6 |
Otras anormalidades de laboratorio que ocurrieron en ≥20% de los pacientes que recibieron KEYTRUDA fueron aumento de hipoalbuminemia (27% de todos los grados; 2.4% grados 3-4), aumento de ALT (23% de todos los grados; 3.1% grados 3-4) y aumento de fosfatasa alcalina (21% de todos los grados, 2% grados 3-4).
Melanoma refractario a ipilimumab
La seguridad de KEYTRUDA en pacientes con melanoma irresecable o metastásico con progresión de la enfermedad después de ipilimumab y, si la mutación BRAF V600 es positiva, un inhibidor de BRAF, se investigó en KEYNOTE-002. KEYNOTE-002 fue un ensayo multicéntrico, parcialmente ciego (dosis de KEYTRUDA), aleatorizado (1:1:1), controlado con activo en el que 528 pacientes recibieron KEYTRUDA 2 mg/kg (n=178) o 10 mg/kg (n=179) cada 3 semanas o la quimioterapia a elección del investigador (n=171), que consistió en dacarbazina (26%), temozolomida (25%), paclitaxel y carboplatino (25%), paclitaxel (16%) o carboplatino (8%) [ver Estudios clínicos (14.1)]. Los pacientes con enfermedad autoinmune, toxicidad relacionada con el sistema inmunitario grave relacionada con ipilimumab, definida como cualquier toxicidad de grado 4 o toxicidad de grado 3 que requiera tratamiento con corticosteroides (más de 10 mg/día de prednisona o dosis equivalente) durante más de 12 semanas; afecciones médicas que requirieron corticosteroides sistémicos u otros medicamentos inmunosupresores; antecedentes de enfermedad pulmonar intersticial; o una infección activa que requiera terapia, incluidos VIH o hepatitis B o C, no fueron elegibles.
La duración media de la exposición a KEYTRUDA 2 mg/kg cada 3 semanas fue de 3,7 meses (rango: 1 día a 16,6 meses) y a KEYTRUDA 10 mg/kg cada 3 semanas fue de 4,8 meses (rango: 1 día a 16,8 meses). En el brazo de KEYTRUDA 2 mg/kg, el 36% de los pacientes estuvieron expuestos a KEYTRUDA durante ≥6 meses y el 4% estuvieron expuestos durante ≥12 meses. En el brazo de KEYTRUDA 10 mg/kg, el 41% de los pacientes estuvieron expuestos a KEYTRUDA durante ≥6 meses y el 6% de los pacientes estuvieron expuestos a KEYTRUDA durante ≥12 meses.
Las características de la población del estudio fueron: edad media de 62 años (rango: 15 a 89); 61% hombres; 98% blancos; 41% tenían un valor de LDH elevado al inicio del estudio; 83% tenían enfermedad en estadio M1c; 73% recibieron dos o más terapias previas para enfermedad avanzada o metastásica (100% recibieron ipilimumab y 25% un inhibidor de BRAF); y 15% con antecedentes de metástasis cerebral.
En KEYNOTE-002, el perfil de reacciones adversas fue similar para la dosis de 2 mg/kg y la dosis de 10 mg/kg, por lo que los resultados de seguridad resumidos se proporcionan en un análisis agrupado (n=357) de ambos brazos de KEYTRUDA. Las reacciones adversas que provocaron la interrupción permanente ocurrieron en el 12% de los pacientes que recibieron KEYTRUDA; las más comunes (≥1%) fueron el deterioro general de la salud física (1%), astenia (1%), disnea (1%), neumonitis (1%) y edema generalizado (1%). Las reacciones adversas que llevaron a la interrupción de KEYTRUDA ocurrieron en el 14% de los pacientes; las más comunes (≥1%) fueron disnea (1%), diarrea (1%) y erupción maculopapular (1%). Los cuadros 6 y 7 resumen las reacciones adversas y las anormalidades de laboratorio, respectivamente, en pacientes con KEYTRUDA en KEYNOTE-002.
| Reacción adversa | KEYTRUDA 2 mg/kg o 10 mg/kg cada 3 semanas |
Quimioterapia† | ||
|---|---|---|---|---|
| n=357 | n=171 | |||
| Todos los grados‡ (%) |
Grados 3-4 (%) |
Todos los grados (%) |
Grados 3-4 (%) |
|
|
||||
| Piel y tejido subcutáneo | ||||
| Prurito | 28 | 0 | 8 | 0 |
| Erupción§ | 24 | 0.6 | 8 | 0 |
| Gastrointestinal | ||||
| Estreñimiento | 22 | 0.3 | 20 | 2.3 |
| Diarrea | 20 | 0.8 | 20 | 2.3 |
| Dolor abdominal | 13 | 1.7 | 8 | 1.2 |
| Respiratorio, torácico y mediastínico | ||||
| Tos | 18 | 0 | 16 | 0 |
| General | ||||
| Pirexia | 14 | 0.3 | 9 | 0.6 |
| Astenia | 10 | 2.0 | 9 | 1.8 |
| Musculoesquelético y del tejido conjuntivo | ||||
| Artralgia | 14 | 0.6 | 10 | 1.2 |
Otras reacciones adversas clínicamente importantes que se produjeron en pacientes que recibieron KEYTRUDA fueron fatiga (43 %), náuseas (22 %), disminución del apetito (20 %), vómitos (13 %) y neuropatía periférica (1,7 %).
| Prueba de laboratorio† | KEYTRUDA 2 mg/kg o 10 mg/kg cada 3 semanas |
Quimioterapia | ||
|---|---|---|---|---|
| Todos los grados‡ % |
Grados 3-4 % |
Todos los grados % |
Grados 3-4 % |
|
|
||||
| Química | ||||
| Hiperglucemia | 49 | 6 | 44 | 6 |
| Hipoalbuminemia | 37 | 1.9 | 33 | 0.6 |
| Hiponatremia | 37 | 7 | 24 | 3.8 |
| Hipertrigliceridemia | 33 | 0 | 32 | 0.9 |
| Aumento de la fosfatasa alcalina | 26 | 3.1 | 18 | 1.9 |
| Aumento de la AST | 24 | 2.2 | 16 | 0.6 |
| Disminución del bicarbonato | 22 | 0.4 | 13 | 0 |
| Hipocalcemia | 21 | 0.3 | 18 | 1.9 |
| Aumento de la ALT | 21 | 1.8 | 16 | 0.6 |
Otras anormalidades de laboratorio que ocurrieron en ≥20% de los pacientes que recibieron KEYTRUDA fueron anemia (44% de todos los grados; 10% de grados 3-4) y linfopenia (40% de todos los grados; 9% de grados 3-4).
Tratamiento adyuvante del melanoma resecado en estadio IIB o IIC
Entre los 969 pacientes con melanoma en estadio IIB o IIC inscritos en KEYNOTE-716 [ver Estudios clínicos (14.1)] tratados con KEYTRUDA, la duración media de la exposición a KEYTRUDA fue de 9,9 meses (rango: 0 a 15,4 meses). Los pacientes con enfermedad autoinmune o una afección médica que requiriera inmunosupresión o melanoma mucoso u ocular fueron inelegibles. Las reacciones adversas que ocurrieron en pacientes con melanoma en estadio IIB o IIC fueron similares a las que ocurrieron en 1011 pacientes con melanoma en estadio III de KEYNOTE-054 o los 2799 pacientes con melanoma o CNCP tratados con KEYTRUDA como agente único.
Tratamiento adyuvante del melanoma resecado en estadio III
La seguridad de KEYTRUDA como agente único se investigó en KEYNOTE-054, un ensayo aleatorizado (1:1) doble ciego en el que 1019 pacientes con melanoma en estadio IIIA (>1 mm de metástasis en los ganglios linfáticos), IIIB o IIIC completamente resecado recibieron 200 mg de KEYTRUDA por infusión intravenosa cada 3 semanas (n=509) o placebo (n=502) durante un máximo de un año [ver Estudios clínicos (14.1)]. Los pacientes con enfermedad autoinmune activa o una afección médica que requiriera inmunosupresión o melanoma mucoso u ocular fueron inelegibles. El setenta y seis por ciento de los pacientes recibieron KEYTRUDA durante 6 meses o más.
Las características de la población del estudio fueron: edad media de 54 años (rango: 19 a 88), 25% de edad 65 o más; 62% hombres; y 94% ECOG PS de 0 y 6% ECOG PS de 1. El dieciséis por ciento tenía estadio IIIA, el 46% tenía estadio IIIB, el 18% tenía estadio IIIC (1-3 ganglios linfáticos positivos) y el 20% tenía estadio IIIC (≥4 ganglios linfáticos positivos).
Dos pacientes tratados con KEYTRUDA murieron por causas distintas a la progresión de la enfermedad; las causas de muerte fueron reacción medicamentosa con eosinofilia y síntomas sistémicos y miositis autoinmune con insuficiencia respiratoria. Se produjeron reacciones adversas graves en el 25% de los pacientes que recibieron KEYTRUDA. Las reacciones adversas que llevaron a la interrupción permanente ocurrieron en el 14% de los pacientes que recibieron KEYTRUDA; las más comunes (≥1%) fueron neumonitis (1,4%), colitis (1,2%) y diarrea (1%). Las reacciones adversas que llevaron a la interrupción de KEYTRUDA ocurrieron en el 19% de los pacientes; las más comunes (≥1%) fueron diarrea (2,4%), neumonitis (2%), aumento de ALT (1,4%), artralgia (1,4%), aumento de AST (1,4%), disnea (1%) y fatiga (1%). Los cuadros 8 y 9 resumen las reacciones adversas y las anormalidades de laboratorio, respectivamente, en pacientes con KEYTRUDA en KEYNOTE-054.
| Reacción adversa | KEYTRUDA 200 mg cada 3 semanas n=509 |
Placebo
n=502 |
||
|---|---|---|---|---|
| Todos los grados† (%) |
Grados 3-4 (%) |
Todos los grados (%) |
Grados 3-4 (%) |
|
| Gastrointestinales | ||||
| Diarrea | 28 | 1.2 | 26 | 1.2 |
| Náuseas | 17 | 0.2 | 15 | 0 |
| Piel y tejido subcutáneo | ||||
| Prurito | 19 | 0 | 12 | 0 |
| Erupción | 13 | 0.2 | 9 | 0 |
| Musculoesquelético y tejido conectivo | ||||
| Artralgia | 16 | 1.2 | 14 | 0 |
| Endocrino | ||||
| Hipotiroidismo | 15 | 0 | 2.8 | 0 |
| Hipertiroidismo | 10 | 0.2 | 1.2 | 0 |
| Respiratorio, torácico y mediastínico | ||||
| Tos | 14 | 0 | 11 | 0 |
| General | ||||
| Astenia | 11 | 0.2 | 8 | 0 |
| Enfermedad tipo influenza | 11 | 0 | 8 | 0 |
| Investigaciones | ||||
| Pérdida de peso | 11 | 0 | 8 | 0 |
| Prueba de laboratorio† | KEYTRUDA 200 mg cada 3 semanas |
Placebo | ||
|---|---|---|---|---|
| Todos los grados‡ % |
Grados 3-4 % |
Todos los grados % |
Grados 3-4 % |
|
|
||||
| Química | ||||
| ALT aumentada | 25 | 2.4 | 15 | 0.2 |
| AST aumentada | 22 | 1.8 | 14 | 0.4 |
| Hematología | ||||
| Linfopenia | 22 | 1 | 15 | 1.2 |
NSCLC
Tratamiento de primera línea del NSCLC no escamoso metastásico con quimioterapia a base de pemetrexed y platino
La seguridad de KEYTRUDA en combinación con pemetrexed y la elección del investigador de platino (carboplatino o cisplatino) se investigó en KEYNOTE-189, un ensayo multicéntrico, doble ciego, aleatorizado (2:1), controlado con activo en pacientes con NSCLC no escamoso metastásico previamente no tratado sin aberraciones genómicas tumorales EGFR o ALK [ver Estudios Clínicos (14.2)]. Un total de 607 pacientes recibieron KEYTRUDA 200 mg, pemetrexed y platino cada 3 semanas durante 4 ciclos, seguidos de KEYTRUDA y pemetrexed (n=405) o placebo, pemetrexed y platino cada 3 semanas durante 4 ciclos, seguidos de placebo y pemetrexed (n=202). Los pacientes con enfermedad autoinmune que requirieron terapia sistémica dentro de los 2 años previos al tratamiento; una condición médica que requirió inmunosupresión; o que habían recibido más de 30 Gy de radiación torácica en las 26 semanas anteriores fueron inelegibles.
La duración mediana de la exposición a KEYTRUDA 200 mg cada 3 semanas fue de 7,2 meses (rango: 1 día a 20,1 meses). El sesenta por ciento de los pacientes en el brazo de KEYTRUDA estuvieron expuestos a KEYTRUDA durante ≥6 meses. El setenta y dos por ciento de los pacientes recibieron carboplatino.
Las características de la población del estudio fueron: edad mediana de 64 años (rango: 34 a 84), 49% de edad 65 o mayor; 59% hombres; 94% blancos y 3% asiáticos; y 18% con antecedentes de metástasis cerebrales al inicio del estudio.
KEYTRUDA se suspendió por reacciones adversas en el 20% de los pacientes. Las reacciones adversas más comunes que provocaron la suspensión permanente de KEYTRUDA fueron neumonitis (3%) e insuficiencia renal aguda (2%). Las reacciones adversas que llevaron a la interrupción de KEYTRUDA se produjeron en el 53% de los pacientes; las reacciones adversas o anomalías de laboratorio más comunes que llevaron a la interrupción de KEYTRUDA (≥2%) fueron neutropenia (13%), astenia/fatiga (7%), anemia (7%), trombocitopenia (5%), diarrea (4%), neumonía (4%), aumento de creatinina en sangre (3%), disnea (2%), neutropenia febril (2%), infección del tracto respiratorio superior (2%), aumento de ALT (2%) y pirexia (2%). Las tablas 10 y 11 resumen las reacciones adversas y las anomalías de laboratorio, respectivamente, en pacientes con KEYTRUDA en KEYNOTE-189.
| Reacción adversa | KEYTRUDA 200 mg cada 3 semanas Pemetrexed Quimioterapia con platino n=405 |
Placebo
Pemetrexed |
||
|---|---|---|---|---|
| Todos los grados* (%) |
Grados 3-4 (%) |
Todos los grados (%) |
Grados 3-4 (%) |
|
| Gastrointestinal | ||||
| Náuseas | 56 | 3.5 | 52 | 3.5 |
| Estreñimiento | 35 | 1.0 | 32 | 0.5 |
| Diarrea | 31 | 5 | 21 | 3.0 |
| Vómitos | 24 | 3.7 | 23 | 3.0 |
| General | ||||
| Fatiga† | 56 | 12 | 58 | 6 |
| Pirexia | 20 | 0.2 | 15 | 0 |
| Metabolismo y Nutrición | ||||
| Disminución del apetito | 28 | 1.5 | 30 | 0.5 |
| Piel y Tejido Subcutáneo | ||||
| Erupción‡ | 25 | 2.0 | 17 | 2.5 |
| Respiratorio, Torácico y Mediastinico | ||||
| Tos | 21 | 0 | 28 | 0 |
| Disnea | 21 | 3.7 | 26 | 5 |
| Prueba de laboratorio* | KEYTRUDA 200 mg cada 3 semanas Pemetrexed Quimioterapia con platino |
Placebo
Pemetrexed |
||
|---|---|---|---|---|
| Todos los grados† % |
Grados 3-4 % |
Todos los grados % |
Grados 3-4 % |
|
|
||||
| Hematología | ||||
| Anemia | 85 | 17 | 81 | 18 |
| Linfopenia | 65 | 22 | 64 | 25 |
| Neutropenia | 50 | 21 | 41 | 19 |
| Trombocitopenia | 30 | 12 | 29 | 8 |
| Química | ||||
| Hiperglucemia | 63 | 9 | 60 | 7 |
| Aumento de ALT | 47 | 3.8 | 42 | 2.6 |
| Aumento de AST | 47 | 2.8 | 40 | 1.0 |
| Hipoalbuminemia | 39 | 2.8 | 39 | 1.1 |
| Aumento de creatinina | 37 | 4.2 | 25 | 1.0 |
| Hiponatremia | 32 | 7 | 23 | 6 |
| Hipofosfatemia | 30 | 10 | 28 | 14 |
| Aumento de fosfatasa alcalina | 26 | 1.8 | 29 | 2.1 |
| Hipocalcemia | 24 | 2.8 | 17 | 0.5 |
| Hiperkalemia | 24 | 2.8 | 19 | 3.1 |
| Hipokalemia | 21 | 5 | 20 | 5 |
Tratamiento de primera línea del CPNM escamoso metastásico con carboplatino y paclitaxel o quimioterapia con paclitaxel unido a proteínas
La seguridad de KEYTRUDA en combinación con carboplatino y la elección del investigador de paclitaxel o paclitaxel unido a proteínas se investigó en KEYNOTE-407, un ensayo multicéntrico, doble ciego, aleatorizado (1:1), controlado con placebo en 558 pacientes con CPNM escamoso metastásico previamente no tratado [véase Estudios clínicos (14.2)]. Hay datos de seguridad disponibles para los primeros 203 pacientes que recibieron KEYTRUDA y quimioterapia (n=101) o placebo y quimioterapia (n=102). Los pacientes con enfermedad autoinmune que requirieron terapia sistémica dentro de los 2 años anteriores al tratamiento; una condición médica que requirió inmunosupresión; o que habían recibido más de 30 Gy de radiación torácica en las 26 semanas anteriores fueron inelegibles.
La duración media de la exposición a KEYTRUDA fue de 7 meses (rango: 1 día a 12 meses). El sesenta y un por ciento de los pacientes del brazo de KEYTRUDA estuvieron expuestos a KEYTRUDA durante ≥6 meses. Un total de 139 de 203 pacientes (68%) recibieron paclitaxel y 64 pacientes (32%) recibieron paclitaxel unido a proteínas en combinación con carboplatino.
Las características de la población del estudio fueron: edad media de 65 años (rango: 40 a 83), 52% de edad igual o superior a 65 años; 78% hombres; 83% blancos; y 9% con antecedentes de metástasis cerebrales.
KEYTRUDA se suspendió por reacciones adversas en el 15% de los pacientes, sin que ningún tipo de reacción adversa representara la mayoría. Las reacciones adversas que llevaron a la interrupción de KEYTRUDA se produjeron en el 43% de los pacientes; las más comunes (≥2%) fueron trombocitopenia (20%), neutropenia (11%), anemia (6%), astenia (2%) y diarrea (2%). Las reacciones adversas graves más frecuentes (≥2%) fueron neutropenia febril (6%), neumonía (6%) e infección del tracto urinario (3%).
Las reacciones adversas observadas en KEYNOTE-407 fueron similares a las observadas en KEYNOTE-189, con la excepción de que se observaron mayores incidencias de alopecia (47% vs. 36%) y neuropatía periférica (31% vs. 25%) en el brazo de KEYTRUDA y quimioterapia en comparación con el brazo de placebo y quimioterapia en KEYNOTE-407.
CPNM previamente no tratado
La seguridad de KEYTRUDA se investigó en KEYNOTE-042, un ensayo multicéntrico, abierto, aleatorizado (1:1), controlado con activo en 1251 pacientes con CPNM en estadio III que expresaban PD-L1 y que no eran candidatos a resección quirúrgica o quimiorradiación definitiva o CPNM metastásico [véase Estudios clínicos (14.2)]. Los pacientes recibieron KEYTRUDA 200 mg cada 3 semanas (n=636) o la quimioterapia elegida por el investigador (n=615), que consistió en pemetrexed y carboplatino seguidos de pemetrexed opcional (n=312) o paclitaxel y carboplatino seguidos de pemetrexed opcional (n=303) cada 3 semanas. Los pacientes con aberraciones genómicas tumorales EGFR o ALK; enfermedad autoinmune que requirió terapia sistémica dentro de los 2 años anteriores al tratamiento; una condición médica que requirió inmunosupresión; o que habían recibido más de 30 Gy de radiación torácica en las 26 semanas anteriores fueron inelegibles.
La duración media de la exposición a KEYTRUDA fue de 5,6 meses (rango: 1 día a 27,3 meses). El cuarenta y ocho por ciento de los pacientes del brazo de KEYTRUDA estuvieron expuestos a KEYTRUDA 200 mg durante ≥6 meses.
Las características de la población del estudio fueron: edad media de 63 años (rango: 25 a 90), 45% de edad igual o superior a 65 años; 71% hombres; y 64% blancos, 30% asiáticos y 2% negros. El diecinueve por ciento eran hispanos o latinos. El ochenta y siete por ciento tenía enfermedad metastásica (estadio IV), el 13% tenía enfermedad en estadio III (2% estadio IIIA y 11% estadio IIIB), y el 5% tenía metástasis cerebrales tratadas al inicio del estudio.
KEYTRUDA se suspendió por reacciones adversas en el 19% de los pacientes. Las reacciones adversas más comunes que provocaron la suspensión permanente de KEYTRUDA fueron neumonitis (3,0%), muerte por causa desconocida (1,6%) y neumonía (1,4%). Las reacciones adversas que llevaron a la interrupción de KEYTRUDA se produjeron en el 33% de los pacientes; las reacciones adversas o anomalías de laboratorio más comunes que llevaron a la interrupción de KEYTRUDA (≥2%) fueron neumonitis (3,1%), neumonía (3,0%), hipotiroidismo (2,2%) y aumento de ALT (2,0%). Las reacciones adversas graves más frecuentes (≥2%) fueron neumonía (7%), neumonitis (3,9%), embolia pulmonar (2,4%) y derrame pleural (2,2%).
Los cuadros 12 y 13 resumen las reacciones adversas y las anomalías de laboratorio, respectivamente, en los pacientes tratados con KEYTRUDA en KEYNOTE-042.
| Reacción adversa | KEYTRUDA 200 mg cada 3 semanas n=636 |
Quimioterapia
n=615 |
||
|---|---|---|---|---|
| Todos los grados* (%) |
Grados 3-5 (%) |
Todos los grados (%) |
Grados 3-5 (%) |
|
| General | ||||
| Fatiga† | 25 | 3.1 | 33 | 3.9 |
| Pirexia | 10 | 0.3 | 8 | 0 |
| Metabolismo y Nutrición | ||||
| Disminución del apetito | 17 | 1.7 | 21 | 1.5 |
| Respiratorio, Torácico y Mediastínico | ||||
| Disnea | 17 | 2.0 | 11 | 0.8 |
| Tos | 16 | 0.2 | 11 | 0.3 |
| Piel y Tejido Subcutáneo | ||||
| Erupción‡ | 15 | 1.3 | 8 | 0.2 |
| Gastrointestinal | ||||
| Estreñimiento | 12 | 0 | 21 | 0.2 |
| Diarrea | 12 | 0.8 | 12 | 0.5 |
| Náuseas | 12 | 0.5 | 32 | 1.1 |
| Endocrino | ||||
| Hipotiroidismo | 12 | 0.2 | 1.5 | 0 |
| Infecciones | ||||
| Neumonía | 12 | 7 | 9 | 6 |
| Pruebas | ||||
| Pérdida de peso | 10 | 0.9 | 7 | 0.2 |
| Prueba de laboratorio* | KEYTRUDA 200 mg cada 3 semanas |
Quimioterapia | ||
|---|---|---|---|---|
| Todos los grados† % |
Grados 3-4 % |
Todos los grados % |
Grados 3-4 % |
|
|
||||
| Química | ||||
| Hiperglucemia | 52 | 4.7 | 51 | 5 |
| Aumento de ALT | 33 | 4.8 | 34 | 2.9 |
| Hipoalbuminemia | 33 | 2.2 | 29 | 1.0 |
| Aumento de AST | 31 | 3.6 | 32 | 1.7 |
| Hiponatremia | 31 | 9 | 32 | 8 |
| Aumento de fosfatasa alcalina | 29 | 2.3 | 29 | 0.3 |
| Hipocalcemia | 25 | 2.5 | 19 | 0.7 |
| Hiperkalemia | 23 | 3.0 | 20 | 2.2 |
| Aumento de la INR de protrombina | 21 | 2.0 | 15 | 2.9 |
| Hipofosfatemia | 20 | 4.7 | 17 | 4.3 |
| Hematología | ||||
| Anemia | 43 | 4.4 | 79 | 19 |
| Linfopenia | 30 | 7 | 42 | 13 |
Cáncer de pulmón de células no pequeñas (CPNC) previamente tratado
La seguridad de KEYTRUDA se investigó en KEYNOTE-010, un ensayo multicéntrico, abierto, aleatorizado (1:1:1) y controlado con activo, en pacientes con CPNC avanzado que habían presentado progresión de la enfermedad tras el tratamiento con quimioterapia a base de platino y, si eran positivos para aberraciones genéticas de EGFR o ALK, terapia apropiada para estas aberraciones [véase Estudios clínicos (14.2)]. Un total de 991 pacientes recibieron KEYTRUDA 2 mg/kg (n=339) o 10 mg/kg (n=343) cada 3 semanas o docetaxel (n=309) a 75 mg/m2 cada 3 semanas. Los pacientes con enfermedad autoinmune, afecciones médicas que requerían corticosteroides sistémicos u otros medicamentos inmunosupresores, o que habían recibido más de 30 Gy de radiación torácica en las 26 semanas anteriores fueron inelegibles.
La duración media de la exposición a KEYTRUDA 2 mg/kg cada 3 semanas fue de 3,5 meses (rango: 1 día a 22,4 meses) y a KEYTRUDA 10 mg/kg cada 3 semanas fue de 3,5 meses (rango: 1 día a 20,8 meses). Los datos que se describen a continuación reflejan la exposición a KEYTRUDA 2 mg/kg en el 31% de los pacientes expuestos a KEYTRUDA durante ≥6 meses. En el brazo de KEYTRUDA 10 mg/kg, el 34% de los pacientes estuvieron expuestos a KEYTRUDA durante ≥6 meses.
Las características de la población del estudio fueron: edad media de 63 años (rango: 20 a 88), 42% de edad igual o superior a 65 años; 61% hombres; 72% blancos y 21% asiáticos; y 8% con enfermedad localizada avanzada, 91% con enfermedad metastásica y 15% con antecedentes de metástasis cerebrales. El veintinueve por ciento recibió dos o más tratamientos sistémicos previos para la enfermedad avanzada o metastásica.
En KEYNOTE-010, el perfil de reacciones adversas fue similar para la dosis de 2 mg/kg y 10 mg/kg, por lo que los resultados de seguridad resumidos se proporcionan en un análisis agrupado (n=682). El tratamiento se interrumpió por reacciones adversas en el 8% de los pacientes que recibieron KEYTRUDA. El evento adverso más común que provocó la interrupción permanente de KEYTRUDA fue la neumonitis (1,8%). Las reacciones adversas que llevaron a la interrupción de KEYTRUDA se produjeron en el 23% de los pacientes; las más comunes (≥1%) fueron diarrea (1%), fatiga (1,3%), neumonía (1%), elevación de las enzimas hepáticas (1,2%), disminución del apetito (1,3%) y neumonitis (1%). Las tablas 14 y 15 resumen las reacciones adversas y las anormalidades de laboratorio, respectivamente, en pacientes con KEYTRUDA en KEYNOTE-010.
| Reacción adversa | KEYTRUDA 2 o 10 mg/kg cada 3 semanas n=682 |
Docetaxel 75 mg/m2 cada 3 semanas n=309 |
||
|---|---|---|---|---|
| Todos los grados† (%) |
Grados 3-4 (%) |
Todos los grados† (%) |
Grados 3-4 (%) |
|
| Metabolismo y nutrición | ||||
| Disminución del apetito | 25 | 1.5 | 23 | 2.6 |
| Respiratorio, torácico y mediastínico | ||||
| Disnea | 23 | 3.7 | 20 | 2.6 |
| Tos | 19 | 0.6 | 14 | 0 |
| Gastrointestinal | ||||
| Náuseas | 20 | 1.3 | 18 | 0.6 |
| Estreñimiento | 15 | 0.6 | 12 | 0.6 |
| Vómitos | 13 | 0.9 | 10 | 0.6 |
| Piel y tejido subcutáneo | ||||
| Erupción cutánea‡ | 17 | 0.4 | 8 | 0 |
| Prurito | 11 | 0 | 3 | 0.3 |
| Musculoesquelético y tejido conjuntivo | ||||
| Artralgia | 11 | 1.0 | 9 | 0.3 |
| Dolor de espalda | 11 | 1.5 | 8 | 0.3 |
Otras reacciones adversas clínicamente importantes que ocurrieron en pacientes que recibieron KEYTRUDA fueron fatiga (25%), diarrea (14%), astenia (11%) y pirexia (11%).
| Prueba de laboratorio† | KEYTRUDA 2 o 10 mg/kg cada 3 semanas |
Docetaxel 75 mg/m2 cada 3 semanas |
||
|---|---|---|---|---|
| Todos los grados‡ % |
Grados 3-4 % |
Todos los grados‡ % |
Grados 3-4 % |
|
|
||||
| Química | ||||
| Hiponatremia | 32 | 8 | 27 | 2.9 |
| Aumento de la fosfatasa alcalina | 28 | 3.0 | 16 | 0.7 |
| Aumento de AST | 26 | 1.6 | 12 | 0.7 |
| Aumento de ALT | 22 | 2.7 | 9 | 0.4 |
| Hipocalcemia | 20 | 0.9 | 20 | 1.8 |
Otras anormalidades de laboratorio que ocurrieron en ≥20% de los pacientes que recibieron KEYTRUDA fueron hiperglucemia (44% de todos los grados; 4.1% de grados 3-4), anemia (37% de todos los grados; 3.8% de grados 3-4), hipertrigliceridemia (36% de todos los grados; 1.8% de grados 3-4), linfopenia (32% de todos los grados; 9% de grados 3-4), hipoalbuminemia (34% de todos los grados; 1.6% de grados 3-4) e hipercolesterolemia (20% de todos los grados; 0.7% de grados 3-4).
Tratamiento neoadyuvante y adyuvante del CPNM resecable
La seguridad de KEYTRUDA en combinación con quimioterapia neoadyuvante que contiene platino seguida de cirugía y tratamiento adyuvante continuo con KEYTRUDA como agente único después de la cirugía se investigó en KEYNOTE-671, un ensayo multicéntrico, aleatorizado (1:1), doble ciego, controlado con placebo en pacientes con CPNM en estadio II, IIIA o IIIB (N2) previamente no tratado y resecable según la 8.ª edición de AJCC [ver Estudios clínicos (14.2)]. Los pacientes con enfermedad autoinmune activa que requirió terapia sistémica dentro de los 2 años del tratamiento o una condición médica que requirió inmunosupresión fueron inelegibles.
La duración media de la exposición a KEYTRUDA 200 mg cada 3 semanas fue de 10,9 meses (rango: 1 día a 18,6 meses). Las características de la población del estudio fueron: edad media de 64 años (rango: 26 a 83), 45% de edad 65 o más, 7% de edad 75 o más; 71% hombres; 61% blancos, 31% asiáticos, 2% negros, 4% raza no informada; 9% hispanos o latinos.
Las reacciones adversas que ocurrieron en pacientes con CPNM resecable que recibieron KEYTRUDA en combinación con quimioterapia que contiene platino, administrada como tratamiento neoadyuvante y continuada como tratamiento adyuvante en monoterapia, fueron generalmente similares a las que ocurrieron en pacientes en otros ensayos clínicos en diferentes tipos de tumores que recibieron KEYTRUDA en combinación con quimioterapia.
Fase neoadyuvante de KEYNOTE-671
Un total de 396 pacientes recibieron al menos 1 dosis de KEYTRUDA en combinación con quimioterapia que contiene platino como tratamiento neoadyuvante y 399 pacientes recibieron al menos 1 dosis de placebo en combinación con quimioterapia que contiene platino como tratamiento neoadyuvante.
Las reacciones adversas graves ocurrieron en el 34% de los pacientes que recibieron KEYTRUDA en combinación con quimioterapia que contiene platino como tratamiento neoadyuvante; las reacciones adversas graves más frecuentes (≥2%) fueron neumonía (4,8%), tromboembolismo venoso (3,3%) y anemia (2%). Las reacciones adversas fatales ocurrieron en el 1,3% de los pacientes, incluida la muerte por causa desconocida (0,8%), sepsis (0,3%) y enfermedad pulmonar inmunomediada (0,3%).
La interrupción permanente de cualquier medicamento del estudio debido a una reacción adversa ocurrió en el 18% de los pacientes que recibieron KEYTRUDA en combinación con quimioterapia que contiene platino como tratamiento neoadyuvante; las reacciones adversas más frecuentes (≥1%) que llevaron a la interrupción permanente de cualquier medicamento del estudio fueron lesión renal aguda (1,8%), enfermedad pulmonar intersticial (1,8%), anemia (1,5%), neutropenia (1,5%) y neumonía (1,3%).
De los 396 pacientes tratados con KEYTRUDA y los 399 pacientes tratados con placebo que recibieron tratamiento neoadyuvante, el 6% (n=25) y el 4,3% (n=17), respectivamente, no recibieron cirugía debido a reacciones adversas. La reacción adversa más frecuente (≥1%) que provocó la cancelación de la cirugía en el brazo de KEYTRUDA fue la enfermedad pulmonar intersticial (1%).
De los 325 pacientes tratados con KEYTRUDA que recibieron cirugía, el 3,1% (n=10) experimentaron un retraso en la cirugía (cirugía a más de 8 semanas del último tratamiento neoadyuvante si el paciente recibió menos de 4 ciclos de terapia neoadyuvante o más de 20 semanas después de la primera dosis de tratamiento neoadyuvante si el paciente recibió 4 ciclos de terapia neoadyuvante) debido a reacciones adversas. De los 317 pacientes tratados con placebo que recibieron cirugía, el 2,5% (n=8) experimentaron un retraso en la cirugía debido a reacciones adversas.
De los 325 pacientes tratados con KEYTRUDA que recibieron cirugía, el 7% (n=22) no recibieron tratamiento adyuvante debido a reacciones adversas. De los 317 pacientes tratados con placebo que recibieron cirugía, el 3,2% (n=10) no recibieron tratamiento adyuvante debido a reacciones adversas.
Fase adyuvante de KEYNOTE-671
Un total de 290 pacientes en el brazo de KEYTRUDA y 267 pacientes en el brazo de placebo recibieron al menos 1 dosis de tratamiento adyuvante.
De los pacientes que recibieron KEYTRUDA como agente único como tratamiento adyuvante, el 14% experimentó reacciones adversas graves; la reacción adversa grave más frecuente fue la neumonía (3,4%). Se produjo una reacción adversa fatal de hemorragia pulmonar.
La interrupción permanente de KEYTRUDA adyuvante debido a una reacción adversa ocurrió en el 12% de los pacientes; las reacciones adversas más frecuentes (≥1%) que llevaron a la interrupción permanente de KEYTRUDA adyuvante fueron diarrea (1,7%), enfermedad pulmonar intersticial (1,4%), aumento de AST (1%) y dolor musculoesquelético (1%).
Tratamiento adyuvante del CPNM resecado
La seguridad de KEYTRUDA como agente único se investigó en KEYNOTE-091, un ensayo multicéntrico, aleatorizado (1:1), triple ciego, controlado con placebo en pacientes con CPNM en estadio IB (T2a ≥4 cm), II o IIIA completamente resecado; la quimioterapia adyuvante hasta 4 ciclos fue opcional [ver Estudios clínicos (14.2)]. Un total de 1161 pacientes recibieron KEYTRUDA 200 mg (n=580) o placebo (n=581) cada 3 semanas. Los pacientes fueron inelegibles si tenían una enfermedad autoinmune activa, estaban tomando agentes inmunosupresores crónicos o tenían antecedentes de enfermedad pulmonar intersticial o neumonitis.
La duración media de la exposición a KEYTRUDA fue de 11,7 meses (rango: 1 día a 18,9 meses). El sesenta y ocho por ciento de los pacientes en el brazo de KEYTRUDA estuvieron expuestos a KEYTRUDA durante ≥6 meses.
Las reacciones adversas observadas en KEYNOTE-091 fueron generalmente similares a las que ocurrieron en otros pacientes con CPNM que recibieron KEYTRUDA como agente único, con la excepción del hipotiroidismo (22%), el hipertiroidismo (11%) y la neumonitis (7%). Ocurrieron dos reacciones adversas fatales de miocarditis.
Mesotelioma pleural maligno (MPM)
Tratamiento de primera línea del MPM avanzado o metastásico irresecable con pemetrexed y quimioterapia con platino
La seguridad de KEYTRUDA en combinación con pemetrexed y quimioterapia con platino (carboplatino o cisplatino) se investigó en KEYNOTE-483, un ensayo multicéntrico, abierto, aleatorizado (1:1), controlado con activo en pacientes con MPM avanzado o metastásico irresecable previamente no tratado [ver Estudios Clínicos (14.3)]. Un total de 473 pacientes recibieron KEYTRUDA 200 mg, pemetrexed y platino cada 3 semanas hasta por 6 ciclos, seguidos de KEYTRUDA (n=241), o pemetrexed y quimioterapia con platino cada 3 semanas hasta por 6 ciclos (n=232). Los pacientes con enfermedad autoinmune que requirieron terapia sistémica dentro de los 3 años del tratamiento o una condición médica que requirió inmunosupresión fueron inelegibles.
La duración mediana de la exposición a KEYTRUDA 200 mg cada 3 semanas fue de 6,9 meses (rango: 1 día a 25,2 meses). El sesenta y un por ciento de los pacientes en el brazo de KEYTRUDA estuvieron expuestos a KEYTRUDA durante ≥6 meses.
Las reacciones adversas que ocurrieron en pacientes con MPM fueron generalmente similares a las de otros pacientes que recibieron KEYTRUDA en combinación con pemetrexed y quimioterapia con platino.
HNSCC
Tratamiento de primera línea de HNSCC metastásico o irresecable, recurrente
La seguridad de KEYTRUDA, como agente único y en combinación con platino (cisplatino o carboplatino) y quimioterapia con FU, se investigó en KEYNOTE-048, un ensayo multicéntrico, abierto, aleatorizado (1:1:1), controlado con activo en pacientes con HNSCC recurrente o metastásico previamente no tratado [ver Estudios Clínicos (14.4)]. Los pacientes con enfermedad autoinmune que requirieron terapia sistémica dentro de los 2 años del tratamiento o una condición médica que requirió inmunosupresión fueron inelegibles. Un total de 576 pacientes recibieron KEYTRUDA 200 mg cada 3 semanas, ya sea como agente único (n=300) o en combinación con platino y FU (n=276) cada 3 semanas durante 6 ciclos seguidos de KEYTRUDA, en comparación con 287 pacientes que recibieron cetuximab semanalmente en combinación con platino y FU cada 3 semanas durante 6 ciclos seguidos de cetuximab.
La duración mediana de la exposición a KEYTRUDA fue de 3,5 meses (rango: 1 día a 24,2 meses) en el brazo de agente único de KEYTRUDA y de 5,8 meses (rango: 3 días a 24,2 meses) en el brazo de combinación. El diecisiete por ciento de los pacientes en el brazo de agente único de KEYTRUDA y el 18% de los pacientes en el brazo de combinación estuvieron expuestos a KEYTRUDA durante ≥12 meses. El cincuenta y siete por ciento de los pacientes que recibieron KEYTRUDA en combinación con quimioterapia comenzaron el tratamiento con carboplatino.
KEYTRUDA se suspendió por reacciones adversas en el 12% de los pacientes en el brazo de agente único de KEYTRUDA. Las reacciones adversas más comunes que provocaron la suspensión permanente de KEYTRUDA fueron sepsis (1,7%) y neumonía (1,3%). Las reacciones adversas que llevaron a la interrupción de KEYTRUDA ocurrieron en el 31% de los pacientes; las reacciones adversas más comunes que llevaron a la interrupción de KEYTRUDA (≥2%) fueron neumonía (2,3%), neumonitis (2,3%) e hiponatremia (2%).
KEYTRUDA se suspendió por reacciones adversas en el 16% de los pacientes en el brazo de combinación. Las reacciones adversas más comunes que provocaron la suspensión permanente de KEYTRUDA fueron neumonía (2,5%), neumonitis (1,8%) y shock séptico (1,4%). Las reacciones adversas que llevaron a la interrupción de KEYTRUDA ocurrieron en el 45% de los pacientes; las reacciones adversas más comunes que llevaron a la interrupción de KEYTRUDA (≥2%) fueron neutropenia (14%), trombocitopenia (10%), anemia (6%), neumonía (4,7%) y neutropenia febril (2,9%).
Las tablas 16 y 17 resumen las reacciones adversas y las anormalidades de laboratorio, respectivamente, en pacientes con KEYTRUDA en KEYNOTE-048.
| KEYTRUDA 200 mg cada 3 semanas |
KEYTRUDA 200 mg cada 3 semanas Platino FU |
Cetuximab Platino FU |
||||
|---|---|---|---|---|---|---|
| Reacción adversa | n=300 | n=276 | n=287 | |||
| Todos los grados* (%) |
Grados 3-4 (%) |
Todos los grados* (%) |
Grados 3-4 (%) |
Todos los grados* (%) |
Grados 3-4 (%) |
|
|
||||||
| General | ||||||
| Fatiga† | 33 | 4 | 49 | 11 | 48 | 8 |
| Pirexia | 13 | 0.7 | 16 | 0.7 | 12 | 0 |
| Inflamación de la mucosa | 4.3 | 1.3 | 31 | 10 | 28 | 5 |
| Gastrointestinal | ||||||
| Estreñimiento | 20 | 0.3 | 37 | 0 | 33 | 1.4 |
| Náuseas | 17 | 0 | 51 | 6 | 51 | 6 |
| Diarrea‡ | 16 | 0.7 | 29 | 3.3 | 35 | 3.1 |
| Vómitos | 11 | 0.3 | 32 | 3.6 | 28 | 2.8 |
| Disfagia | 8 | 2.3 | 12 | 2.9 | 10 | 2.1 |
| Estomatitis | 3 | 0 | 26 | 8 | 28 | 3.5 |
| Piel | ||||||
| Erupción§ | 20 | 2.3 | 17 | 0.7 | 70 | 8 |
| Prurito | 11 | 0 | 8 | 0 | 10 | 0.3 |
| Respiratorio, torácico y mediastínico | ||||||
| Tos¶ | 18 | 0.3 | 22 | 0 | 15 | 0 |
| Disnea# | 14 | 2.0 | 10 | 1.8 | 8 | 1.0 |
| Endocrino | ||||||
| Hipotiroidismo | 18 | 0 | 15 | 0 | 6 | 0 |
| Metabolismo y Nutrición | ||||||
| Disminución del apetito | 15 | 1.0 | 29 | 4.7 | 30 | 3.5 |
| Pérdida de peso | 15 | 2 | 16 | 2.9 | 21 | 1.4 |
| Infecciones | ||||||
| NeumoníaÞ | 12 | 7 | 19 | 11 | 13 | 6 |
| Sistema Nervioso | ||||||
| Dolor de cabeza | 12 | 0.3 | 11 | 0.7 | 8 | 0.3 |
| Mareo | 5 | 0.3 | 10 | 0.4 | 13 | 0.3 |
| Neuropatía sensitiva periféricaß | 1 | 0 | 14 | 1.1 | 7 | 1 |
| Musculoesquelético | ||||||
| Mialgiaà | 12 | 1.0 | 13 | 0.4 | 11 | 0.3 |
| Dolor de cuello | 6 | 0.7 | 10 | 1.1 | 7 | 0.7 |
| Psiquiátrico | ||||||
| Insomnio | 7 | 0.7 | 10 | 0 | 8 | 0 |
| KEYTRUDA 200 mg cada 3 semanas |
KEYTRUDA 200 mg cada 3 semanas Platino FU |
Cetuximab Platino FU |
||||
|---|---|---|---|---|---|---|
| Prueba de laboratorio* | Todos los grados† (%) |
Grados 3-4 (%) |
Todos los grados† (%) |
Grados 3-4 (%) |
Todos los grados† (%) |
Grados 3-4 (%) |
|
||||||
| Hematología | ||||||
| Linfopenia | 54 | 25 | 70 | 35 | 75 | 46 |
| Anemia | 52 | 7 | 89 | 29 | 79 | 20 |
| Trombocitopenia | 12 | 3.8 | 73 | 18 | 76 | 18 |
| Neutropenia | 8 | 1.4 | 68 | 37 | 73 | 43 |
| Química | ||||||
| Hiperglucemia | 47 | 3.8 | 54 | 6 | 65 | 4.7 |
| Hiponatremia | 46 | 18 | 55 | 20 | 59 | 20 |
| Hipoalbuminemia | 44 | 3.5 | 46 | 3.9 | 49 | 1.1 |
| AST aumentada | 28 | 3.1 | 25 | 1.9 | 37 | 3.6 |
| ALT aumentada | 25 | 2.1 | 22 | 1.5 | 38 | 1.8 |
| Fosfatasa alcalina aumentada | 25 | 2.1 | 26 | 1.1 | 33 | 1.1 |
| Hipercalcemia | 22 | 4.5 | 16 | 4.2 | 13 | 2.5 |
| Hipocalcemia | 22 | 1.0 | 32 | 3.8 | 58 | 6 |
| Hiperkalemia | 21 | 2.8 | 28 | 4.2 | 29 | 4.6 |
| Hipofosfatemia | 20 | 5 | 34 | 12 | 49 | 20 |
| Hipokalemia | 19 | 5 | 33 | 12 | 47 | 15 |
| Aumento de creatinina | 17 | 1.0 | 36 | 2.3 | 27 | 2.1 |
| Hipomagnesemia | 15 | 0.4 | 40 | 1.7 | 76 | 9 |
HNSCC recurrente o metastásico previamente tratado
Entre los 192 pacientes con HNSCC inscritos en KEYNOTE-012 [véase Estudios clínicos (14.4)], la duración media de la exposición a KEYTRUDA fue de 3,3 meses (rango: 1 día a 27,9 meses). Los pacientes con enfermedad autoinmune o una afección médica que requiriera inmunosupresión no fueron elegibles para KEYNOTE-012.
Las características de la población del estudio fueron: edad media de 60 años (rango: 20 a 84), 35% de edad igual o superior a 65 años; 83% hombres; y 77% blancos, 15% asiáticos y 5% negros. El sesenta y un por ciento de los pacientes habían recibido dos o más líneas de terapia en el contexto recurrente o metastásico, y el 95% habían recibido radioterapia previa. El estado de rendimiento ECOG basal fue 0 (30%) o 1 (70%) y el 86% tenía enfermedad M1.
KEYTRUDA se suspendió debido a reacciones adversas en el 17% de los pacientes. Se produjeron reacciones adversas graves en el 45% de los pacientes que recibieron KEYTRUDA. Las reacciones adversas graves más frecuentes notificadas en al menos el 2% de los pacientes fueron neumonía, disnea, estado confusional, vómitos, derrame pleural e insuficiencia respiratoria. La incidencia de reacciones adversas, incluidas las reacciones adversas graves, fue similar entre los regímenes de dosificación (10 mg/kg cada 2 semanas o 200 mg cada 3 semanas); por lo tanto, los resultados de seguridad resumidos se proporcionan en un análisis agrupado. Las reacciones adversas más comunes (que ocurrieron en ≥20% de los pacientes) fueron fatiga, disminución del apetito y disnea. Las reacciones adversas que ocurrieron en pacientes con HNSCC fueron generalmente similares a las que ocurrieron en 2799 pacientes con melanoma o CNCP tratados con KEYTRUDA como agente único, con la excepción de un aumento en la incidencia de edema facial (10% de todos los grados; 2,1% de grados 3-4) e hipotiroidismo nuevo o empeorado [véase Advertencias y precauciones (5.1)].
Linfoma de Hodgkin (LH) recidivante o refractario
KEYNOTE-204
La seguridad de KEYTRUDA se evaluó en KEYNOTE-204 [véase Estudios clínicos (14.5)]. Los adultos con LH recidivante o refractario recibieron KEYTRUDA 200 mg por vía intravenosa cada 3 semanas (n=148) o brentuximab vedotina (BV) 1,8 mg/kg por vía intravenosa cada 3 semanas (n=152). El ensayo requirió un recuento absoluto de neutrófilos (RAN) ≥1000/µL, recuento de plaquetas ≥75.000/µL, transaminasas hepáticas ≤2,5 veces el límite superior de la normalidad (LSN), bilirrubina ≤1,5 veces el LSN y estado de rendimiento ECOG de 0 o 1. El ensayo excluyó a los pacientes con neumonitis no infecciosa activa, neumonitis previa que requiriera corticosteroides, enfermedad autoinmune activa, una afección médica que requiriera inmunosupresión o un trasplante de células madre hematopoyéticas (TCMH) alogénico en los últimos 5 años. La duración media de la exposición a KEYTRUDA fue de 10 meses (rango: 1 día a 2,2 años), con un 68% recibiendo al menos 6 meses de tratamiento y un 48% recibiendo al menos 1 año de tratamiento.
Se produjeron reacciones adversas graves en el 30% de los pacientes que recibieron KEYTRUDA. Las reacciones adversas graves en ≥1% incluyeron neumonitis, neumonía, pirexia, miocarditis, lesión renal aguda, neutropenia febril y sepsis. Tres pacientes (2%) murieron por causas distintas a la progresión de la enfermedad: dos por complicaciones después de un TCMH alogénico y uno por causa desconocida.
La interrupción permanente de KEYTRUDA debido a una reacción adversa se produjo en el 14% de los pacientes; el 7% de los pacientes interrumpieron el tratamiento debido a neumonitis. La interrupción de la dosis de KEYTRUDA debido a una reacción adversa se produjo en el 30% de los pacientes. Las reacciones adversas que requirieron interrupción de la dosis en ≥3% de los pacientes fueron infección del tracto respiratorio superior, neumonitis, aumento de las transaminasas y neumonía.
El treinta y ocho por ciento de los pacientes tuvieron una reacción adversa que requirió terapia con corticosteroides sistémicos.
La Tabla 18 resume las reacciones adversas en KEYNOTE-204.
| Reacción adversa | KEYTRUDA 200 mg cada 3 semanas N=148 |
Brentuximab Vedotina 1,8 mg/kg cada 3 semanas N=152 |
||
|---|---|---|---|---|
| Todos los grados* (%) |
Grados 3-4 (%) |
Todos los grados* (%) |
Grados 3-4† (%) |
|
|
||||
| Infecciones | ||||
| Infección del tracto respiratorio superior‡ | 41 | 1.4 | 24 | 0 |
| Infección del tracto urinario | 11 | 0 | 3 | 0.7 |
| Musculoesquelético y del tejido conjuntivo | ||||
| Dolor musculoesquelético§ | 32 | 0 | 29 | 1.3 |
| Gastrointestinales | ||||
| Diarrea¶ | 22 | 2.7 | 17 | 1.3 |
| Náuseas | 14 | 0 | 24 | 0.7 |
| Vómitos | 14 | 1.4 | 20 | 0 |
| Dolor abdominal# | 11 | 0.7 | 13 | 1.3 |
| Generales | ||||
| Pirexia | 20 | 0.7 | 13 | 0.7 |
| FatigaÞ | 20 | 0 | 22 | 0.7 |
| Piel y tejido subcutáneo | ||||
| Erupciónß | 20 | 0 | 19 | 0.7 |
| Prurito | 18 | 0 | 12 | 0 |
| Respiratorio, torácico y mediastínico | ||||
| Tosà | 20 | 0.7 | 14 | 0.7 |
| Neumonitisè | 11 | 5 | 3 | 1.3 |
| Disneað | 11 | 0.7 | 7 | 0.7 |
| Endocrino | ||||
| Hipotiroidismo | 19 | 0 | 3 | 0 |
| Sistema Nervioso | ||||
| Neuropatía periféricaø | 11 | 0.7 | 43 | 7 |
| Cefaleaý | 11 | 0 | 11 | 0 |
Las reacciones adversas clínicamente relevantes en <10% de los pacientes que recibieron KEYTRUDA incluyeron infección por virus del herpes (9%), neumonía (8%), dolor orofaríngeo (8%), hipertiroidismo (5%), hipersensibilidad (4.1%), reacciones a la infusión (3.4%), alteración del estado mental (2.7%), y en 1.4% cada una, uveítis, miocarditis, tiroiditis, neutropenia febril, sepsis y exacerbación tumoral.
La Tabla 19 resume las anormalidades de laboratorio en KEYNOTE-204.
| Anormalidad de laboratorio* | KEYTRUDA 200 mg cada 3 semanas |
Brentuximab Vedotin 1.8 mg/kg cada 3 semanas |
||
|---|---|---|---|---|
| Todos los grados† (%) |
Grados 3-4 (%) |
Todos los grados† (%) |
Grados 3-4 (%) |
|
|
||||
| Química | ||||
| Hiperglucemia | 45 | 4.1 | 36 | 2.0 |
| Aumento de AST | 38 | 4.7 | 38 | 2.0 |
| Aumento de ALT | 31 | 5 | 43 | 2.6 |
| Hipofosfatemia | 31 | 4.9 | 17 | 2.8 |
| Aumento de creatinina | 26 | 3.4 | 13 | 2.6 |
| Hipomagnesemia | 23 | 0 | 13 | 0 |
| Hiponatremia | 24 | 4.1 | 20 | 3.3 |
| Hipocalcemia | 21 | 2.0 | 15 | 0 |
| Aumento de fosfatasa alcalina | 19 | 2.1 | 21 | 2.0 |
| Hipoalbuminemia | 16 | 0.7 | 18 | 0.7 |
| Hiperbilirrubinemia | 15 | 1.4 | 8 | 0.7 |
| Hematología | ||||
| Linfopenia | 34 | 8 | 32 | 13 |
| Trombocitopenia | 32 | 9 | 24 | 4.0 |
| Neutropenia | 27 | 8 | 42 | 16 |
| Anemia | 22 | 4.1 | 32 | 7 |
KEYNOTE-087
Entre los 210 pacientes con linfoma de Hodgkin clásico (LCH) que recibieron KEYTRUDA en KEYNOTE-087 [véase Estudios Clínicos (14.5)], la duración media de la exposición a KEYTRUDA fue de 8,4 meses (rango: 1 día a 15,2 meses). Se produjeron reacciones adversas graves en el 16 % de los pacientes que recibieron KEYTRUDA. Las reacciones adversas graves que se produjeron en ≥1 % de los pacientes incluyeron neumonía, neumonitis, pirexia, disnea, enfermedad de injerto contra huésped (EICH) y herpes zóster. Dos pacientes murieron por causas distintas a la progresión de la enfermedad; uno por EICH después de un trasplante de células madre hematopoyéticas (TCMH) alogénico posterior y otro por shock séptico.
La interrupción permanente de KEYTRUDA debido a una reacción adversa se produjo en el 5 % de los pacientes y la interrupción de la dosis debido a una reacción adversa se produjo en el 26 %. El quince por ciento de los pacientes presentaron una reacción adversa que requirió tratamiento con corticosteroides sistémicos. Los cuadros 20 y 21 resumen las reacciones adversas y las alteraciones de laboratorio, respectivamente, en KEYNOTE-087.
| Reacción adversa | KEYTRUDA 200 mg cada 3 semanas N=210 |
|
|---|---|---|
| Todos los grados* (%) |
Grado 3 (%) |
|
|
||
| General | ||
| Fatiga† | 26 | 1.0 |
| Pirexia | 24 | 1.0 |
| Respiratorio, torácico y mediastínico | ||
| Tos‡ | 24 | 0.5 |
| Disnea§ | 11 | 1.0 |
| Musculoesquelético y del tejido conjuntivo | ||
| Dolor musculoesquelético¶ | 21 | 1.0 |
| Artralgia | 10 | 0.5 |
| Gastrointestinal | ||
| Diarrea# | 20 | 1.4 |
| Vómitos | 15 | 0 |
| Náuseas | 13 | 0 |
| Piel y tejido subcutáneo | ||
| Erupción cutánea Þ | 20 | 0.5 |
| Prurito | 11 | 0 |
| Endocrino | ||
| Hipotiroidismo | 14 | 0.5 |
| Infecciones | ||
| Infección del tracto respiratorio superior | 13 | 0 |
| Sistema Nervioso | ||
| Dolor de cabeza | 11 | 0.5 |
| Neuropatía periféricaß | 10 | 0 |
Las reacciones adversas clínicamente relevantes en <10% de los pacientes que recibieron KEYTRUDA incluyeron reacciones a la infusión (9%), hipertiroidismo (3%), neumonitis (3%), uveítis y miositis (1% cada una), y mielitis y miocarditis (0.5% cada una).
| Anormalidad de laboratorio* | KEYTRUDA 200 mg cada 3 semanas |
|
|---|---|---|
| Todos los grados† (%) |
Grados 3-4 (%) |
|
| Química | ||
| Hipertransaminasemia‡ | 35 | 2.4 |
| Aumento de la fosfatasa alcalina | 17 | 0 |
| Aumento de creatinina | 15 | 0.5 |
| Hematología | ||
| Anemia | 30 | 6 |
| Trombocitopenia | 27 | 4.3 |
| Neutropenia | 25 | 7 |
La hiperbilirrubinemia se produjo en menos del 15 % de los pacientes en KEYNOTE-087 (10 % de todos los grados, 2,4 % de grado 3-4).
PMBCL
Entre los 53 pacientes con PMBCL que recibieron KEYTRUDA en KEYNOTE-170 [véase Estudios clínicos (14.6)], la duración media de la exposición a KEYTRUDA fue de 3,5 meses (rango: 1 día a 22,8 meses). Se produjeron reacciones adversas graves en el 26 % de los pacientes. Las reacciones adversas graves que se produjeron en >2 % de los pacientes incluyeron arritmia (4 %), taponamiento cardíaco (2 %), infarto de miocardio (2 %), derrame pericárdico (2 %) y pericarditis (2 %). Seis (11 %) pacientes murieron en los 30 días posteriores al inicio del tratamiento. La interrupción permanente de KEYTRUDA debido a una reacción adversa se produjo en el 8 % de los pacientes y la interrupción de la dosis debido a una reacción adversa se produjo en el 15 %. El veinticinco por ciento de los pacientes presentaron una reacción adversa que requirió tratamiento con corticosteroides sistémicos. Los cuadros 22 y 23 resumen las reacciones adversas y las anomalías de laboratorio, respectivamente, en KEYNOTE-170.
| Reacción adversa | KEYTRUDA 200 mg cada 3 semanas N=53 |
|
|---|---|---|
| Todos los grados* (%) |
Grados 3-4 (%) |
|
|
||
| Musculoesquelético y del tejido conjuntivo | ||
| Dolor musculoesquelético† | 30 | 0 |
| Infecciones | ||
| Infección de las vías respiratorias superiores‡ | 28 | 0 |
| General | ||
| Pirexia | 28 | 0 |
| Fatiga§ | 23 | 2 |
| Respiratorio, torácico y mediastínico | ||
| Tos¶ | 26 | 2 |
| Disnea | 21 | 11 |
| Gastrointestinal | ||
| Diarrea# | 13 | 2 |
| Dolor abdominal Þ | 13 | 0 |
| Náuseas | 11 | 0 |
| Cardíaco | ||
| Arritmia ß | 11 | 4 |
| Sistema nervioso | ||
| Cefalea | 11 | 0 |
Las reacciones adversas clínicamente relevantes en <10% de los pacientes que recibieron KEYTRUDA incluyeron hipotiroidismo (8%), hipertiroidismo y pericarditis (4% cada una), y tiroiditis, derrame pericárdico, neumonitis, artritis e insuficiencia renal aguda (2% cada una).
| Anormalidad de laboratorio* | KEYTRUDA 200 mg cada 3 semanas |
|
|---|---|---|
| Todos los grados† (%) |
Grados 3-4 (%) |
|
| Hematología | ||
| Anemia | 23 | 0 |
| Leucopenia | 47 | 12 |
| Linfopenia | 27 | 10 |
| Neutropenia | 39 | 15 |
| Química | ||
| Hiperglucemia | 33 | 2.2 |
| Hipofosfatemia | 24 | 11 |
| Hipertransaminasemia‡ | 24 | 4.4 |
| Hipoglucemia | 20 | 0 |
| Creatinina aumentada | 16 | 0 |
Cáncer urotelial
Pacientes con cáncer urotelial en combinación con enfortumab vedotina
La seguridad de KEYTRUDA en combinación con enfortumab vedotina se investigó en KEYNOTE-A39 en pacientes con cáncer urotelial localmente avanzado o metastásico [véase Estudios clínicos (14.7)]. Un total de 440 pacientes recibieron KEYTRUDA 200 mg el día 1 y enfortumab vedotina 1,25 mg/kg los días 1 y 8 de cada ciclo de 21 días en comparación con 433 pacientes que recibieron gemcitabina los días 1 y 8 y la elección del investigador de cisplatino o carboplatino el día 1 de cada ciclo de 21 días. Entre los pacientes que recibieron KEYTRUDA y enfortumab vedotina, la duración media de la exposición a KEYTRUDA fue de 8,5 meses (rango: 9 días a 28,5 meses).
Se produjeron reacciones adversas mortales en el 3,9% de los pacientes tratados con KEYTRUDA en combinación con enfortumab vedotina, incluyendo insuficiencia respiratoria aguda (0,7%), neumonía (0,5%) y neumonitis/ILD (0,2%).
Se produjeron reacciones adversas graves en el 50% de los pacientes que recibieron KEYTRUDA en combinación con enfortumab vedotina. Las reacciones adversas graves en ≥2% de los pacientes que recibieron KEYTRUDA en combinación con enfortumab vedotina fueron erupción cutánea (6%), lesión renal aguda (5%), neumonitis/ILD (4,5%), infección del tracto urinario (3,6%), diarrea (3,2%), neumonía (2,3%), pirexia (2%) e hiperglucemia (2%).
La interrupción permanente de KEYTRUDA se produjo en el 27% de los pacientes. Las reacciones adversas más comunes (≥2%) que provocaron la interrupción permanente de KEYTRUDA fueron neumonitis/ILD (4,8%) y erupción cutánea (3,4%).
Se produjeron interrupciones de la dosis de KEYTRUDA en el 61% de los pacientes. Las reacciones adversas más comunes (≥2%) que provocaron la interrupción de KEYTRUDA fueron erupción cutánea (17%), neuropatía periférica (7%), COVID-19 (5%), diarrea (4,3%), neumonitis/ILD (3,6%), neutropenia (3,4%), fatiga (3%), aumento de la alanina aminotransferasa (2,7%), hiperglucemia (2,5%), neumonía (2%) y prurito (2%).
Los cuadros 24 y 25 resumen las reacciones adversas y las alteraciones de laboratorio, respectivamente, en pacientes con KEYTRUDA en combinación con enfortumab vedotina en KEYNOTE-A39.
| Reacción adversa | KEYTRUDA en combinación con Enfortumab vedotina n=440 |
Quimioterapia
n=433 |
||
|---|---|---|---|---|
| Todos los grados* % |
Grados 3-4 % |
Todos los grados* % |
Grados 3-4 % |
|
| Trastornos de la piel y del tejido subcutáneo | ||||
| Erupción cutánea† | 68 | 15 | 15 | 0 |
| Prurito | 41 | 1.1 | 7 | 0 |
| Alopecia | 35 | 0.5 | 8 | 0.2 |
| Trastornos generales y afecciones en el lugar de administración | ||||
| Fatiga† | 51 | 6 | 57 | 7 |
| Trastornos del sistema nervioso | ||||
| Neuropatía periférica† | 67 | 8 | 14 | 0 |
| Disgeusia | 21 | 0 | 9 | 0 |
| Trastornos del metabolismo y la nutrición | ||||
| Disminución del apetito | 33 | 1.8 | 26 | 1.8 |
| Trastornos gastrointestinales | ||||
| Diarrea | 38 | 4.5 | 16 | 1.4 |
| Náuseas | 26 | 1.6 | 41 | 2.8 |
| Constipación | 26 | 0 | 34 | 0.7 |
| Exploraciones complementarias | ||||
| Pérdida de peso | 33 | 3.6 | 9 | 0.2 |
| Trastornos oculares | ||||
| Sequedad ocular† | 24 | 0 | 2.1 | 0 |
| Infecciones e infestaciones | ||||
| Infección del tracto urinario | 21 | 5 | 19 | 8 |
Las reacciones adversas clínicamente relevantes (<20%) incluyen pirexia (18%), piel seca (17%), vómitos (12%), neumonitis/ILD (10%), hipotiroidismo (10%), visión borrosa (6%), extravasación en el sitio de infusión (2%) y miositis (0.5%).
| Prueba de laboratorio* | KEYTRUDA 200 mg cada 3 semanas y Enfortumab Vedotina |
Quimioterapia | ||
|---|---|---|---|---|
| Todos los grados† % |
Grados 3-4 % |
Todos los grados† % |
Grados 3-4 % |
|
| Química | ||||
| Aumento de la aspartato aminotransferasa | 75 | 4.6 | 39 | 3.3 |
| Aumento de creatinina | 71 | 3.2 | 68 | 2.6 |
| Hiperglucemia | 66 | 14 | 54 | 4.7 |
| Aumento de la alanina aminotransferasa | 59 | 5 | 49 | 3.3 |
| Hiponatremia | 46 | 13 | 47 | 13 |
| Hipofosfatemia | 44 | 9 | 36 | 9 |
| Hipoalbuminemia | 39 | 1.8 | 35 | 0.5 |
| Hipokalemia | 26 | 5 | 16 | 3.1 |
| Hiperkalemia | 24 | 1.4 | 36 | 4.0 |
| Hipercalcemia | 21 | 1.2 | 14 | 0.2 |
| Hematología | ||||
| Linfopenia | 58 | 15 | 59 | 17 |
| Anemia | 53 | 7 | 89 | 33 |
| Neutropenia | 30 | 9 | 80 | 50 |
Pacientes con cáncer urotelial no elegibles para cisplatino en combinación con enfortumab vedotina
La seguridad de KEYTRUDA en combinación con enfortumab vedotina se investigó en KEYNOTE-869 en pacientes con cáncer urotelial localmente avanzado o metastásico que no son elegibles para quimioterapia a base de cisplatino [ver Estudios Clínicos (14.7)]. Un total de 121 pacientes recibieron KEYTRUDA 200 mg el día 1 y enfortumab vedotina 1,25 mg/kg los días 1 y 8 de cada ciclo de 21 días. La duración media de la exposición a KEYTRUDA fue de 6,9 meses (rango de 1 día a 29,6 meses).
Se produjeron reacciones adversas mortales en el 5% de los pacientes tratados con KEYTRUDA en combinación con enfortumab vedotina, incluyendo sepsis (1,6%), dermatitis bullosa (0,8%), miastenia gravis (0,8%) y neumonitis (0,8%).
Se produjeron reacciones adversas graves en el 50% de los pacientes que recibieron KEYTRUDA y enfortumab vedotina. Las reacciones adversas graves en ≥2% de los pacientes que recibieron KEYTRUDA en combinación con enfortumab vedotina fueron lesión renal aguda (7%), infección del tracto urinario (7%), urosepsis (5%), hematuria (3,3%), neumonía (3,3%), neumonitis (3,3%), sepsis (3,3%), anemia (2,5%), diarrea (2,5%), hipotensión (2,5%), miastenia gravis (2,5%), miositis (2,5%) y retención urinaria (2,5%).
La interrupción permanente de KEYTRUDA se produjo en el 32% de los pacientes. Las reacciones adversas más comunes (≥2%) que provocaron la interrupción permanente de KEYTRUDA fueron neumonitis (5%), neuropatía periférica (5%), erupción cutánea (3,3%) y miastenia gravis (2,5%).
Se produjeron interrupciones de la dosis de KEYTRUDA en el 69% de los pacientes. Las reacciones adversas más comunes (≥2%) que provocaron la interrupción de KEYTRUDA fueron neuropatía periférica (22%), erupción cutánea (17%), neutropenia (7%), fatiga (6%), diarrea (5%), aumento de lipasa (5%), lesión renal aguda (3,3%), aumento de ALT (2,5%) y COVID-19 (2,5%).
Los cuadros 26 y 27 resumen las reacciones adversas y las alteraciones de laboratorio, respectivamente, en pacientes con KEYTRUDA en combinación con enfortumab vedotina en KEYNOTE-869.
| Reacción adversa | KEYTRUDA en combinación con Enfortumab Vedotina n=121 |
|
|---|---|---|
| Todos los grados* % |
Grado 3-4 % |
|
|
||
| Trastornos de la piel y del tejido subcutáneo | ||
| Erupción cutánea† | 71 | 21 |
| Alopecia | 52 | 0 |
| Prurito | 40 | 3.3 |
| Sequedad de la piel | 21 | 0.8 |
| Trastornos del sistema nervioso | ||
| Neuropatía periférica‡ | 65 | 3.3 |
| Disgeusia | 35 | 0 |
| Mareo | 23 | 0 |
| Trastornos generales y condiciones en el lugar de administración | ||
| Fatiga | 60 | 11 |
| Edema periférico | 26 | 0 |
| Investigaciones | ||
| Pérdida de peso | 48 | 5 |
| Trastornos gastrointestinales | ||
| Diarrea | 45 | 7 |
| Náuseas | 36 | 0.8 |
| Estreñimiento | 27 | 0 |
| Trastornos del metabolismo y la nutrición | ||
| Disminución del apetito | 38 | 0.8 |
| Infecciones e infestaciones | ||
| Infección del tracto urinario | 30 | 12 |
| Trastornos oculares | ||
| Sequedad ocular | 25 | 0 |
| Trastornos musculoesqueléticos y del tejido conjuntivo | ||
| Artralgia | 23 | 1.7 |
Las reacciones adversas clínicamente relevantes (<20%) incluyen vómitos (19.8%), fiebre (18%), hipotiroidismo (11%), neumonitis/ILD (10%), miositis (3.3%), miastenia gravis (2.5%) y extravasación en el sitio de infusión (0.8%).
| Prueba de laboratorio* | KEYTRUDA 200 mg cada 3 semanas y Enfortumab Vedotina |
|
|---|---|---|
| Todos los grados† % |
Grados 3-4 % |
|
| Química | ||
| Hiperglucemia | 74 | 13 |
| Aumento de la aspartato aminotransferasa | 73 | 9 |
| Aumento de creatinina | 69 | 3.3 |
| Hiponatremia | 60 | 19 |
| Aumento de la alanina aminotransferasa | 60 | 7 |
| Aumento de lipasa | 59 | 32 |
| Hipoalbuminemia | 59 | 4.2 |
| Hipofosfatemia | 51 | 15 |
| Hipokalemia | 35 | 8 |
| Aumento de potasio | 27 | 1.7 |
| Aumento de calcio | 27 | 4.2 |
| Hematología | ||
| Anemia | 69 | 15 |
| Linfopenia | 64 | 17 |
| Neutropenia | 32 | 12 |
Pacientes con Carcinoma Urotelial no Elegibles para Platino
La seguridad de KEYTRUDA se investigó en KEYNOTE-052, un ensayo de un solo brazo en el que se inscribió a 370 pacientes con carcinoma urotelial localmente avanzado o metastásico que tenían una o más comorbilidades. Los pacientes con enfermedades autoinmunitarias o afecciones médicas que requerían corticosteroides sistémicos u otros medicamentos inmunosupresores no fueron elegibles [ver Estudios Clínicos (14.7)]. Los pacientes recibieron KEYTRUDA 200 mg cada 3 semanas hasta que se produjo una toxicidad inaceptable o bien progresión de la enfermedad radiográfica o clínica.
La duración media de la exposición a KEYTRUDA fue de 2,8 meses (rango: 1 día a 15,8 meses).
KEYTRUDA se suspendió debido a reacciones adversas en el 11% de los pacientes. Dieciocho pacientes (5%) murieron por causas distintas a la progresión de la enfermedad. Cinco pacientes (1,4%) tratados con KEYTRUDA experimentaron sepsis que provocó la muerte, y tres pacientes (0,8%) experimentaron neumonía que provocó la muerte. Las reacciones adversas que llevaron a la interrupción de KEYTRUDA se produjeron en el 22% de los pacientes; las más comunes (≥1%) fueron aumento de las enzimas hepáticas, diarrea, infección del tracto urinario, lesión renal aguda, fatiga, dolor articular y neumonía. Se produjeron reacciones adversas graves en el 42% de los pacientes. Las reacciones adversas graves más frecuentes (≥2%) fueron infección del tracto urinario, hematuria, lesión renal aguda, neumonía y urosepsis.
Las reacciones adversas relacionadas con el sistema inmunitario que requirieron glucocorticoides sistémicos se produjeron en el 8% de los pacientes, el uso de suplementos hormonales debido a una reacción adversa relacionada con el sistema inmunitario se produjo en el 8% de los pacientes, y el 5% de los pacientes requirieron al menos una dosis de esteroides ≥40 mg de equivalente a prednisona oral.
La Tabla 28 resume las reacciones adversas en pacientes tratados con KEYTRUDA en KEYNOTE-052.
| Reacción adversa | KEYTRUDA 200 mg cada 3 semanas N=370 |
|||||
|---|---|---|---|---|---|---|
| Todos los grados* (%) |
Grados 3–4 (%) |
|||||
|
||||||
| General | ||||||
| Fatiga† | 38 | 6 | ||||
| Pirexia | 11 | 0.5 | ||||
| Pérdida de peso | 10 | 0 | ||||
| Musculoesquelético y del tejido conjuntivo | ||||||
| Dolor musculoesquelético‡ | 24 | 4.9 | ||||
| Artralgia | 10 | 1.1 | ||||
| Metabolismo y nutrición | ||||||
| Disminución del apetito | 22 | 1.6 | ||||
| Hiponatremia | 10 | 4.1 | ||||
| Gastrointestinal | ||||||
| Estreñimiento | 21 | 1.1 | ||||
| Diarrea§ | 20 | 2.4 | ||||
| Náuseas | 18 | 1.1 | ||||
| Dolor abdominal¶ | 18 | 2.7 |
| Elevación de las transaminasas hepáticas# | 13 | 3.5 |
| Vómitos | 12 | 0 |
| Piel y tejido subcutáneo | ||
| ErupciónÞ | 21 | 0.5 |
| Prurito | 19 | 0.3 |
| Edema periféricoß | 14 | 1.1 |
| Infecciones | ||
| Infección del tracto urinario | 19 | 9 |
| Sangre y sistema linfático | ||
| Anemia | 17 | 7 |
| Respiratorio, torácico y mediastínico | ||
| Tos | 14 | 0 |
| Disnea | 11 | 0.5 |
| Renal y urinario | ||
| Aumento de creatinina en sangre | 11 | 1.1 |
| Hematuria | 13 | 3.0 |
Carcinoma urotelial previamente tratado
La seguridad de KEYTRUDA para el tratamiento de pacientes con carcinoma urotelial localmente avanzado o metastásico con progresión de la enfermedad después de la quimioterapia que contiene platino se investigó en KEYNOTE-045. KEYNOTE-045 fue un ensayo multicéntrico, abierto, aleatorizado (1:1), controlado con activo en el que 266 pacientes recibieron KEYTRUDA 200 mg cada 3 semanas o la quimioterapia a elección del investigador (n=255), que consistió en paclitaxel (n=84), docetaxel (n=84) o vinflunina (n=87) [ver Estudios Clínicos (14.7)]. Los pacientes con enfermedad autoinmune o una condición médica que requiriera corticosteroides sistémicos u otros medicamentos inmunosupresores fueron inelegibles.
La duración media de la exposición fue de 3,5 meses (rango: 1 día a 20 meses) en pacientes que recibieron KEYTRUDA y de 1,5 meses (rango: 1 día a 14 meses) en pacientes que recibieron quimioterapia.
KEYTRUDA se suspendió debido a reacciones adversas en el 8% de los pacientes. La reacción adversa más común que provocó la suspensión permanente de KEYTRUDA fue la neumonitis (1,9%). Las reacciones adversas que llevaron a la interrupción de KEYTRUDA se produjeron en el 20% de los pacientes; las más comunes (≥1%) fueron la infección del tracto urinario (1,5%), la diarrea (1,5%) y la colitis (1,1%). Se produjeron reacciones adversas graves en el 39% de los pacientes tratados con KEYTRUDA. Las reacciones adversas graves más frecuentes (≥2%) en los pacientes tratados con KEYTRUDA fueron infección del tracto urinario, neumonía, anemia y neumonitis. Los cuadros 29 y 30 resumen las reacciones adversas y las anormalidades de laboratorio, respectivamente, en pacientes con KEYTRUDA en KEYNOTE-045.
| Reacción adversa | KEYTRUDA 200 mg cada 3 semanas |
Quimioterapia* | ||
|---|---|---|---|---|
| n=266 | n=255 | |||
| Todos los grados† (%) |
Grados 3-4 (%) |
Todos los grados† (%) |
Grados 3-4 (%) |
|
|
||||
| General | ||||
| Fatiga‡ | 38 | 4.5 | 56 | 11 |
| Pirexia | 14 | 0.8 | 13 | 1.2 |
| Musculoesquelético y tejido conjuntivo | ||||
| Dolor musculoesquelético§ | 32 | 3.0 | 27 | 2.0 |
| Piel y tejido subcutáneo | ||||
| Prurito | 23 | 0 | 6 | 0.4 |
| Erupción¶ | 20 | 0.4 | 13 | 0.4 |
| Gastrointestinal | ||||
| Náuseas | 21 | 1.1 | 29 | 1.6 |
| Constipación | 19 | 1.1 | 32 | 3.1 |
| Diarrea# | 18 | 2.3 | 19 | 1.6 |
| Vómitos | 15 | 0.4 | 13 | 0.4 |
| Dolor abdominal | 13 | 1.1 | 13 | 2.7 |
| Infecciones | ||||
| Infección del tracto urinario | 15 | 4.9 | 14 | 4.3 |
| Metabolismo y Nutrición | ||||
| Disminución del apetito | 21 | 3.8 | 21 | 1.2 |
| Respiratorio, Torácico y Mediastínico | ||||
| TosÞ | 15 | 0.4 | 9 | 0 |
| Disneaß | 14 | 1.9 | 12 | 1.2 |
| Renal y Urinario | ||||
| Hematuria à | 12 | 2.3 | 8 | 1.6 |
| Prueba de laboratorio* | KEYTRUDA 200 mg cada 3 semanas |
Quimioterapia | ||
|---|---|---|---|---|
| Todos los grados† % |
Grados 3-4 % |
Todos los grados† % |
Grados 3-4 % |
|
|
||||
| Química | ||||
| Hiperglucemia | 52 | 8 | 60 | 7 |
| Anemia | 52 | 13 | 68 | 18 |
| Linfopenia | 45 | 15 | 55 | 26 |
| Hipoalbuminemia | 43 | 1.7 | 50 | 3.8 |
| Hiponatremia | 37 | 9 | 47 | 13 |
| Aumento de la fosfatasa alcalina | 37 | 7 | 33 | 4.9 |
| Aumento de creatinina | 35 | 4.4 | 28 | 2.9 |
| Hipofosfatemia | 29 | 8 | 34 | 14 |
| Aumento de AST | 28 | 4.1 | 20 | 2.5 |
| Hiperkalemia | 28 | 0.8 | 27 | 6 |
| Hipocalcemia | 26 | 1.6 | 34 | 2.1 |
NMIBC de alto riesgo no responsiva a BCG
La seguridad de KEYTRUDA se investigó en KEYNOTE-057, un ensayo multicéntrico, abierto y de un solo brazo que incluyó a 148 pacientes con cáncer de vejiga no invasivo de alto riesgo (NMIBC), 96 de los cuales tenían carcinoma in situ (CIS) no responsivo a BCG con o sin tumores papilares. Los pacientes recibieron KEYTRUDA 200 mg cada 3 semanas hasta que se produjo una toxicidad inaceptable, NMIBC de alto riesgo persistente o recurrente o enfermedad progresiva, o hasta 24 meses de terapia sin progresión de la enfermedad.
La duración media de la exposición a KEYTRUDA fue de 4,3 meses (rango: 1 día a 25,6 meses).
KEYTRUDA se suspendió debido a reacciones adversas en el 11% de los pacientes. La reacción adversa más común (>1%) que provocó la suspensión permanente de KEYTRUDA fue la neumonitis (1,4%). Las reacciones adversas que llevaron a la interrupción de KEYTRUDA se produjeron en el 22% de los pacientes; las más comunes (≥2%) fueron diarrea (4%) e infección del tracto urinario (2%). Se produjeron reacciones adversas graves en el 28% de los pacientes tratados con KEYTRUDA. Las reacciones adversas graves más frecuentes (≥2%) en los pacientes tratados con KEYTRUDA fueron neumonía (3%), isquemia cardíaca (2%), colitis (2%), embolia pulmonar (2%), sepsis (2%) e infección del tracto urinario (2%). Los cuadros 31 y 32 resumen las reacciones adversas y las anormalidades de laboratorio, respectivamente, en los pacientes tratados con KEYTRUDA en KEYNOTE-057.
| Reacción adversa | KEYTRUDA 200 mg cada 3 semanas N=148 |
|
|---|---|---|
| Todos los grados* (%) |
Grados 3–4 (%) |
|
|
||
| General | ||
| Fatiga† | 29 | 0.7 |
| Edema periférico‡ | 11 | 0 |
| Gastrointestinal | ||
| Diarrea§ | 24 | 2.0 |
| Náuseas | 13 | 0 |
| Estreñimiento | 12 | 0 |
| Piel y tejido subcutáneo | ||
| Erupción¶ | 24 | 0.7 |
| Prurito | 19 | 0.7 |
| Musculoesquelético y tejido conjuntivo | ||
| Dolor musculoesquelético# | 19 | 0 |
| Artralgia | 14 | 1.4 |
| Renal y urinario | ||
| Hematuria | 19 | 1.4 |
| Respiratorio, torácico y mediastínico | ||
| TosÞ | 19 | 0 |
| Infecciones | ||
| Infección del tracto urinario | 12 | 2.0 |
| Nasofaringitis | 10 | 0 |
| Endocrino | ||
| Hipotiroidismo | 11 | 0 |
| Prueba de laboratorio* | KEYTRUDA 200 mg cada 3 semanas |
|
|---|---|---|
| Todos los grados† (%) |
Grados 3-4 (%) |
|
| Química | ||
| Hiperglucemia | 59 | 7 |
| Aumento de ALT | 25 | 2.7 |
| Hiponatremia | 24 | 7 |
| Hipofosfatemia | 24 | 6 |
| Hipoalbuminemia | 24 | 1.4 |
| Hiperkalemia | 23 | 1.4 |
| Hipocalcemia | 22 | 0.7 |
| Aumento de AST | 20 | 2.7 |
| Aumento de creatinina | 20 | 0.7 |
| Hematología | ||
| Anemia | 35 | 1.4 |
| Linfopenia | 29 | 1.6 |
Cáncer con alta inestabilidad de microsatélites o deficiencia de reparación de desajuste
La seguridad de KEYTRUDA se investigó en 504 pacientes con cáncer MSI-H o dMMR inscritos en KEYNOTE-158, KEYNOTE-164 y KEYNOTE-051 [véase Estudios clínicos (14.8)]. La duración mediana de la exposición a KEYTRUDA fue de 6,2 meses (rango: 1 día a 53,5 meses). Las reacciones adversas que se produjeron en pacientes con cáncer MSI-H o dMMR fueron similares a las que se produjeron en pacientes con otros tumores sólidos que recibieron KEYTRUDA como agente único.
Cáncer colorrectal con alta inestabilidad de microsatélites o deficiencia de reparación de desajuste
Entre los 153 pacientes con CRC MSI-H o dMMR inscritos en KEYNOTE-177 [véase Estudios clínicos (14.9)] tratados con KEYTRUDA, la duración mediana de la exposición a KEYTRUDA fue de 11,1 meses (rango: 1 día a 30,6 meses). Los pacientes con enfermedad autoinmune o una afección médica que requiriera inmunosupresión no fueron elegibles. Las reacciones adversas que se produjeron en pacientes con CRC MSI-H o dMMR fueron similares a las que se produjeron en 2799 pacientes con melanoma o CPNM tratados con KEYTRUDA como agente único.
Cáncer gástrico
Tratamiento de primera línea del adenocarcinoma gástrico o de la unión gastroesofágica HER2-positivo localmente avanzado irresecable o metastásico
La seguridad de KEYTRUDA se evaluó en 433 pacientes con cáncer gástrico o de la unión gastroesofágica HER2-positivo inscritos en KEYNOTE-811, que incluyó a 217 pacientes tratados con KEYTRUDA 200 mg, trastuzumab y CAPOX (n=189) o FP (n=28) cada 3 semanas, en comparación con 216 pacientes tratados con placebo, trastuzumab y CAPOX (n=187) o FP (n=29) cada 3 semanas [véase Estudios clínicos (14.10)].
La duración mediana de la exposición a KEYTRUDA fue de 5,8 meses (rango: 1 día a 17,7 meses).
Las características de la población del estudio fueron: edad mediana de 63 años (rango: 19 a 84), 43% de edad igual o superior a 65 años; 81% hombres; 58% blancos, 35% asiáticos y 0,9% negros; 44% ECOG PS de 0 y 56% ECOG PS de 1.
KEYTRUDA y el placebo se interrumpieron debido a reacciones adversas en el 6% de los pacientes de cada brazo. La reacción adversa más común que provocó la interrupción permanente de KEYTRUDA fue la neumonitis (1,4%). Las reacciones adversas que provocaron la interrupción de KEYTRUDA se produjeron en el 58% de los pacientes; las reacciones adversas o anomalías de laboratorio más comunes que provocaron la interrupción de KEYTRUDA (≥2%) fueron neutropenia (18%), trombocitopenia (12%), diarrea (6%), anemia (3,7%), hipopotasemia (3,7%), fatiga/astenia (3,2%), disminución del apetito (3,2%), aumento de AST (2,8%), aumento de bilirrubina en sangre (2,8%), neumonía (2,8%), aumento de ALT (2,3%) y vómitos (2,3%).
En el brazo de KEYTRUDA frente al placebo, hubo una diferencia de ≥5% de incidencia entre los pacientes tratados con KEYTRUDA frente al tratamiento estándar para la diarrea (53% frente a 44%) y las náuseas (49% frente a 44%). No hubo diferencias clínicamente significativas en la incidencia de toxicidad de grado 3-4 entre los brazos.
Hubo una diferencia de ≥5% de incidencia entre los pacientes tratados con KEYTRUDA frente al tratamiento estándar para el aumento de creatinina (20% frente a 10%). No hubo diferencias clínicamente significativas en la incidencia de toxicidad de grado 3-4 entre los brazos.
Tratamiento de primera línea del adenocarcinoma gástrico o de la unión gastroesofágica HER2-negativo localmente avanzado irresecable o metastásico
La seguridad de KEYTRUDA se evaluó en 1572 pacientes con cáncer gástrico o de la unión gastroesofágica HER2-negativo inscritos en KEYNOTE-859, que incluyó a 785 pacientes tratados con KEYTRUDA 200 mg y FP (n=106) o CAPOX (n=674) cada 3 semanas, en comparación con 787 pacientes que recibieron placebo y FP (n=107) o CAPOX (n=679) cada 3 semanas [véase Estudios clínicos (14.10)].
La duración mediana de la exposición a KEYTRUDA fue de 6,2 meses (rango: 1 día a 33,7 meses).
Se produjeron reacciones adversas graves en el 45% de los pacientes que recibieron KEYTRUDA. Las reacciones adversas graves en >2% de los pacientes incluyeron neumonía (4,1%), diarrea (3,9%), hemorragia (3,9%) y vómitos (2,4%). Se produjeron reacciones adversas mortales en el 8% de los pacientes que recibieron KEYTRUDA, incluida la infección (2,3%) y el tromboembolismo (1,3%).
La interrupción permanente de KEYTRUDA debido a reacciones adversas se produjo en el 15% de los pacientes. Las reacciones adversas que provocaron la interrupción permanente de KEYTRUDA en ≥1% fueron infecciones (1,8%) y diarrea (1,0%).
Las interrupciones de la dosis de KEYTRUDA debido a una reacción adversa se produjeron en el 65% de los pacientes. Las reacciones adversas o anomalías de laboratorio que provocaron la interrupción de KEYTRUDA (≥2%) fueron neutropenia (21%), trombocitopenia (13%), diarrea (5,5%), fatiga (4,8%), infección (4,8%), anemia (4,5%), aumento de AST (4,3%), aumento de ALT (3,8%), aumento de bilirrubina en sangre (3,3%), disminución del recuento de leucocitos (2,2%), náuseas (2%), síndrome de eritrodisestesia palmar-plantar (2%) y vómitos (2%).
Los cuadros 33 y 34 resumen las reacciones adversas y las anomalías de laboratorio, respectivamente, en los pacientes tratados con KEYTRUDA en KEYNOTE-859.
| Reacción adversa | KEYTRUDA 200 mg cada 3 semanas y FP o CAPOX n=785 |
Placebo
y FP o CAPOX |
||
|---|---|---|---|---|
| Todos los grados* (%) |
Grados 3-4 (%) |
Todos los grados* (%) |
Grados 3-4 (%) |
|
|
||||
| Sistema Nervioso | ||||
| Neuropatía periférica† | 47 | 5 | 48 | 6 |
| Gastrointestinal | ||||
| Náuseas | 46 | 3.7 | 46 | 4.4 |
| Diarrea | 36 | 6 | 32 | 5 |
| Vómitos | 34 | 5 | 27 | 5 |
| Dolor Abdominal‡ | 26 | 2.8 | 24 | 2.9 |
| Estreñimiento | 22 | 0.5 | 21 | 0.8 |
| General | ||||
| Fatiga§ | 40 | 8 | 39 | 9 |
| Metabolismo y Nutrición | ||||
| Disminución del apetito | 29 | 3.3 | 29 | 2.5 |
| Piel y Tejido Subcutáneo | ||||
| Síndrome de eritrodisestesia palmar-plantar | 25 | 3.1 | 22 | 1.8 |
| Investigaciones | ||||
| Pérdida de peso | 20 | 2.8 | 19 | 2.7 |
| Prueba de laboratorio* | KEYTRUDA 200 mg cada 3 semanas y FP o CAPOX |
Placebo
y FP o CAPOX |
||
|---|---|---|---|---|
| Todos los grados† % |
Grados 3-4 % |
Todos los grados† % |
Grados 3-4 % |
|
|
||||
| Hematología | ||||
| Anemia | 65 | 15 | 69 | 13 |
| Trombocitopenia | 64 | 12 | 62 | 10 |
| Neutropenia | 63 | 25 | 58 | 20 |
| Leucopenia | 59 | 7 | 56 | 6 |
| Linfopenia | 57 | 20 | 51 | 16 |
| Química | ||||
| AST aumentada | 57 | 4.7 | 48 | 3.6 |
| Hipoalbuminemia | 55 | 4.1 | 52 | 2.9 |
| Hiperglucemia | 53 | 6 | 52 | 4.6 |
| Hipocalcemia | 49 | 3.6 | 45 | 3.3 |
| Fosfatasa alcalina aumentada | 48 | 6 | 41 | 5 |
| Hiponatremia | 40 | 13 | 40 | 12 |
| ALT aumentada | 40 | 4.2 | 29 | 2.9 |
| Hipokalemia | 35 | 10 | 27 | 9 |
| Bilirrubina aumentada | 32 | 5 | 30 | 5 |
| Hipofosfatemia | 30 | 10 | 27 | 8 |
| Hipomagnesemia | 29 | 0.3 | 22 | 0.7 |
| Creatinina aumentada | 21 | 3.5 | 18 | 1.7 |
| Hiperkalemia | 20 | 3.7 | 18 | 2.9 |
| INR aumentado | 20 | 1.4 | 22 | 0 |
Cáncer de esófago
Tratamiento de primera línea del cáncer de esófago/unión gastroesofágica localmente avanzado irresecable o metastásico
La seguridad de KEYTRUDA, en combinación con quimioterapia de cisplatino y FU, se investigó en KEYNOTE-590, un ensayo multicéntrico, doble ciego, aleatorizado (1:1), controlado con placebo para el tratamiento de primera línea en pacientes con carcinoma de esófago o unión gastroesofágica metastásico o localmente avanzado (tumores con epicentro de 1 a 5 centímetros por encima de la unión gastroesofágica) que no eran candidatos a resección quirúrgica o quimiorradiación definitiva [ver Estudios clínicos (14.11)]. Un total de 740 pacientes recibieron KEYTRUDA 200 mg (n=370) o placebo (n=370) cada 3 semanas durante un máximo de 35 ciclos, ambos en combinación con hasta 6 ciclos de cisplatino y hasta 35 ciclos de FU.
La duración media de la exposición fue de 5,7 meses (rango: 1 día a 26 meses) en el brazo de combinación de KEYTRUDA y de 5,1 meses (rango: 3 días a 27 meses) en el brazo de quimioterapia.
KEYTRUDA se suspendió por reacciones adversas en el 15% de los pacientes. Las reacciones adversas más comunes que provocaron la suspensión permanente de KEYTRUDA (≥1%) fueron neumonitis (1,6%), lesión renal aguda (1,1%) y neumonía (1,1%). Las reacciones adversas que llevaron a la interrupción de KEYTRUDA se produjeron en el 67% de los pacientes. Las reacciones adversas más comunes que llevaron a la interrupción de KEYTRUDA (≥2%) fueron neutropenia (19%), fatiga/astenia (8%), disminución del recuento de leucocitos (5%), neumonía (5%), disminución del apetito (4,3%), anemia (3,2%), aumento de la creatinina en sangre (3,2%), estomatitis (3,2%), malestar general (3,0%), trombocitopenia (3%), neumonitis (2,7%), diarrea (2,4%), disfagia (2,2%) y náuseas (2,2%).
Los cuadros 35 y 36 resumen las reacciones adversas y las alteraciones de laboratorio, respectivamente, en los pacientes tratados con KEYTRUDA en KEYNOTE-590.
| Reacción adversa | KEYTRUDA 200 mg cada 3 semanas Cisplatino FU n=370 |
Placebo
Cisplatino |
||
|---|---|---|---|---|
| Todos los grados* (%) |
Grados 3-4† (%) |
Todos los grados* (%) |
Grados 3-4† (%) |
|
| Gastrointestinal | ||||
| Náuseas | 67 | 7 | 63 | 7 |
| Estreñimiento | 40 | 0 | 40 | 0 |
| Diarrea | 36 | 4.1 | 33 | 3 |
| Vómitos | 34 | 7 | 32 | 5 |
| Estomatitis | 27 | 6 | 26 | 3.8 |
| General | ||||
| Fatiga‡ | 57 | 12 | 46 | 9 |
| Metabolismo y nutrición | ||||
| Disminución del apetito | 44 | 4.1 | 38 | 5 |
| Investigaciones | ||||
| Pérdida de peso | 24 | 3.0 | 24 | 5 |
| Prueba de laboratorio* | KEYTRUDA 200 mg cada 3 semanas Cisplatino FU |
Quimioterapia (Cisplatino y FU) |
||
|---|---|---|---|---|
| Todos los grados† % |
Grados 3-4 % |
Todos los grados† % |
Grados 3-4 % |
|
|
||||
| Hematología | ||||
| Anemia | 84 | 21 | 87 | 25 |
| Neutropenia | 77 | 44 | 73 | 41 |
| Leucopenia | 73 | 21 | 73 | 17 |
| Linfopenia | 57 | 23 | 53 | 18 |
| Trombocitopenia | 43 | 5 | 46 | 8 |
| Química | ||||
| Hiperglucemia | 56 | 7 | 55 | 6 |
| Hiponatremia | 53 | 19 | 53 | 19 |
| Hipoalbuminemia | 53 | 2.8 | 52 | 2.3 |
| Creatinina aumentada | 45 | 2.5 | 42 | 2.5 |
| Hipocalcemia | 44 | 3.9 | 37 | 2 |
| Hipofosfatemia | 37 | 9 | 31 | 10 |
| Hipokalemia | 30 | 12 | 34 | 15 |
| Fosfatasa alcalina aumentada | 29 | 1.9 | 29 | 1.7 |
| Hiperkalemia | 28 | 3.6 | 28 | 2.5 |
| AST aumentada | 25 | 4.4 | 22 | 2.8 |
| ALT aumentada | 23 | 3.6 | 18 | 1.7 |
Cáncer esofágico recurrente localmente avanzado o metastásico previamente tratado
Entre los 314 pacientes con cáncer de esófago inscritos en KEYNOTE-181 [véase Estudios clínicos (14.11)] tratados con KEYTRUDA, la duración media de la exposición a KEYTRUDA fue de 2,1 meses (rango: 1 día a 24,4 meses). Los pacientes con enfermedad autoinmune o una afección médica que requiriera inmunosupresión no fueron elegibles. Las reacciones adversas que se produjeron en pacientes con cáncer de esófago fueron similares a las que se produjeron en 2799 pacientes con melanoma o CNCP tratados con KEYTRUDA como agente único.
Cáncer de cuello uterino
Cáncer de cuello uterino FIGO 2014 Estadio III-IVA con quimioradioterapia
La seguridad de KEYTRUDA en combinación con CRT (cisplatino más radioterapia de haz externo [EBRT] seguida de braquiterapia [BT]) se investigó en KEYNOTE-A18, un ensayo multicéntrico, doble ciego, aleatorizado (1:1), controlado con placebo que incluyó a 594 pacientes con cáncer de cuello uterino FIGO 2014 Estadio III-IVA [véase Estudios clínicos (14.12)]. Doscientos noventa y dos pacientes recibieron KEYTRUDA en combinación con quimioradioterapia y 302 pacientes recibieron placebo en combinación con quimioradioterapia.
La duración media de la exposición a KEYTRUDA fue de 12,1 meses (rango: 1 día a 27 meses).
Se produjeron reacciones adversas mortales en el 1,4% de los pacientes que recibieron KEYTRUDA en combinación con quimioradioterapia, incluido 1 caso cada uno (0,3%) de perforación intestinal grande, urosepsis, sepsis y hemorragia vaginal.
Se produjeron reacciones adversas graves en el 30% de los pacientes que recibieron KEYTRUDA en combinación con quimioradioterapia. Las reacciones adversas graves que se produjeron en ≥1% de los pacientes incluyeron infección del tracto urinario (2,7%), urosepsis (1,4%) y sepsis (1%).
KEYTRUDA se suspendió por reacciones adversas en el 7% de los pacientes. La reacción adversa más común (≥1%) que provocó la suspensión permanente fue la diarrea (1%).
Las reacciones adversas que llevaron a la interrupción de KEYTRUDA se produjeron en el 43% de los pacientes; las reacciones adversas más comunes que llevaron a la interrupción de KEYTRUDA (≥2%) fueron anemia (8%), COVID-19 (6%), prueba positiva de SARS-CoV-2 (3,1%), disminución del recuento de neutrófilos (2,7%), diarrea (2,7%), infección del tracto urinario (2,7%) y aumento de ALT (2,4%).
Las tablas 37 y 38 resumen las reacciones adversas y las anormalidades de laboratorio, respectivamente, en pacientes con KEYTRUDA en KEYNOTE-A18.
| Reacción adversa | KEYTRUDA 200 mg cada 3 semanas y 400 mg cada 6 semanas con quimioradioterapia n=292 |
Placebo con quimioradioterapia n=302 |
||||
|---|---|---|---|---|---|---|
| Todos los grados* (%) |
Grados 3-4 (%) |
Todos los grados* (%) |
Grados 3-4 (%) |
|||
|
||||||
| Gastrointestinal | ||||||
| Náuseas | 56 | 0 | 61 | 2.3 | ||
| Diarrea | 50 | 3.8 | 50 | 4.3 | ||
| Vómitos | 33 | 1 | 34 | 1.7 | ||
| Estreñimiento | 18 | 0 | 18 | 0.7 | ||
| Dolor abdominal | 12 | 0.7 | 12 | 1.7 | ||
| Infecciones | ||||||
| Infección del tracto urinario† | 32 | 4.1 | 31 | 4.6 | ||
| General | ||||||
| Fatiga‡ | 26 | 1 | 27 | 1.3 |
| Pirexia | 12 | 0.3 | 13 | 0 |
| Endocrino | ||||
| Hipotiroidismo§ | 20 | 0.7 | 5 | 0 |
| Hipertiroidismo | 11 | 0.3 | 2.6 | 0 |
| Metabolismo y Nutrición | ||||
| Disminución del apetito | 17 | 0.7 | 17 | 0.3 |
| Investigaciones | ||||
| Pérdida de peso | 17 | 1.4 | 18 | 1 |
| Renal y Urinario | ||||
| Disuria | 11 | 0.3 | 12 | 0 |
| Trastornos de la piel y del tejido subcutáneo | ||||
| Erupción¶ | 11 | 0.7 | 7 | 0.3 |
| Sistema Reproductor | ||||
| Dolor pélvico | 10 | 1 | 13 | 1.3 |
| Prueba de laboratorio* | KEYTRUDA 200 mg cada 3 semanas y 400 mg cada 6 semanas con quimioradioterapia |
Placebo
con quimioradioterapia |
||
|---|---|---|---|---|
| Todos los grados† (%) |
Grados 3-4 (%) |
Todos los grados† (%) |
Grados 3-4 (%) |
|
|
||||
| Hematología | ||||
| Linfopenia | 99 | 96 | 99 | 92 |
| Leucopenia | 96 | 46 | 94 | 49 |
| Anemia | 88 | 31 | 81 | 25 |
| Neutropenia | 75 | 32 | 74 | 33 |
| Trombocitopenia | 65 | 8 | 61 | 6 |
| Química | ||||
| Hipomagnesemia | 59 | 4.2 | 63 | 3.4 |
| Hiponatremia | 54 | 3.8 | 47 | 4 |
| AST aumentada | 45 | 1 | 39 | 1.7 |
| ALT aumentada | 44 | 2.1 | 44 | 1 |
| Hipocalcemia | 43 | 4.8 | 40 | 4.3 |
| Hipokalemia | 42 | 14 | 38 | 10 |
| Creatinina aumentada | 41 | 6 | 43 | 6 |
| Hipoalbuminemia | 37 | 0.7 | 35 | 1.7 |
| Fosfatasa alcalina aumentada | 34 | 0.3 | 33 | 0.3 |
Cáncer de cuello uterino persistente, recurrente o metastásico
La seguridad de KEYTRUDA en combinación con paclitaxel y cisplatino o paclitaxel y carboplatino, con o sin bevacizumab, se investigó en KEYNOTE-826, un ensayo multicéntrico, doble ciego, aleatorizado (1:1), controlado con placebo en pacientes con cáncer de cuello uterino persistente, recurrente o metastásico de primera línea que no habían sido tratados con quimioterapia, excepto cuando se usó concurrentemente como agente radiosensibilizador [véase Estudios clínicos (14.12)]. Un total de 616 pacientes, independientemente de la expresión de PD-L1 tumoral, recibieron KEYTRUDA 200 mg y quimioterapia con o sin bevacizumab (n=307) cada 3 semanas o placebo y quimioterapia con o sin bevacizumab (n=309) cada 3 semanas.
La duración mediana de la exposición a KEYTRUDA fue de 9,9 meses (rango: 1 día a 26 meses).
Se produjeron reacciones adversas fatales en el 4,6% de los pacientes que recibieron KEYTRUDA en combinación con quimioterapia con o sin bevacizumab, incluidos 3 casos de hemorragia, 2 casos de sepsis, 2 casos por causas desconocidas y 1 caso de infarto agudo de miocardio, encefalitis autoinmune, paro cardíaco, accidente cerebrovascular, fractura de fémur con embolia pulmonar perioperatoria, perforación intestinal e infección pélvica.
Se produjeron reacciones adversas graves en el 50% de los pacientes que recibieron KEYTRUDA en combinación con quimioterapia con o sin bevacizumab. Las reacciones adversas graves en ≥3% de los pacientes incluyeron neutropenia febril (6,8%), infección del tracto urinario (5,2%), anemia (4,6%), lesión renal aguda (3,3%) y sepsis (3,3%).
KEYTRUDA se suspendió por reacciones adversas en el 15% de los pacientes. La reacción adversa más común que provocó la suspensión permanente de KEYTRUDA (≥1%) fue la colitis (1%).
Las reacciones adversas que llevaron a la interrupción de KEYTRUDA se produjeron en el 66% de los pacientes; las reacciones adversas o anomalías de laboratorio más comunes que llevaron a la interrupción de KEYTRUDA (≥2%) fueron trombocitopenia (15%), neutropenia (14%), anemia (11%), aumento de ALT (6%), leucopenia (5%), fatiga/astenia (4,2%), infección del tracto urinario (3,6%), aumento de AST (3,3%), pirexia (3,3%), diarrea (2,6%), lesión renal aguda (2,6%), aumento de creatinina en sangre (2,6%), colitis (2,3%), disminución del apetito (2%) y tos (2%).
Para los pacientes tratados con KEYTRUDA, quimioterapia y bevacizumab (n=196), las reacciones adversas más comunes (≥20%) fueron neuropatía periférica (62%), alopecia (58%), anemia (55%), fatiga/astenia (53%), náuseas (41%), neutropenia (41%), diarrea (39%), hipertensión (35%), trombocitopenia (35%), estreñimiento (31%), artralgia (31%), vómitos (30%), infección del tracto urinario (27%), erupción cutánea (26%), leucopenia (24%), hipotiroidismo (22%) y disminución del apetito (21%).
Las tablas 39 y 40 resumen las reacciones adversas y las anomalías de laboratorio, respectivamente, en pacientes con KEYTRUDA en KEYNOTE-826.
| Reacción adversa | KEYTRUDA 200 mg cada 3 semanas y quimioterapia* con o sin bevacizumab n=307 |
Placebo
y quimioterapia* con o sin bevacizumab |
||
|---|---|---|---|---|
| Todos los grados† (%) |
Grados 3-4 (%) |
Todos los grados† (%) |
Grados 3-4 (%) |
|
|
||||
| Sistema nervioso | ||||
| Neuropatía periférica‡ | 58 | 4.2 | 57 | 6 |
| Piel y tejido subcutáneo | ||||
| Alopecia | 56 | 0 | 58 | 0 |
| Erupción cutánea§ | 22 | 3.6 | 15 | 0.3 |
| General | ||||
| Fatiga¶ | 47 | 7 | 46 | 6 |
| Gastrointestinal | ||||
| Nausea | 40 | 2 | 44 | 1.6 |
| Diarrea | 36 | 2 | 30 | 2.6 |
| Estreñimiento | 28 | 0.3 | 33 | 1 |
| Vómitos | 26 | 2.6 | 27 | 1.9 |
| Musculoesquelético y del tejido conjuntivo | ||||
| Artralgia | 27 | 0.7 | 26 | 1.3 |
| Vascular | ||||
| Hipertensión | 24 | 9 | 23 | 11 |
| Infecciones | ||||
| Infección del tracto urinario | 24 | 9 | 26 | 8 |
| Prueba de laboratorio* | KEYTRUDA 200 mg cada 3 semanas y quimioterapia† con o sin bevacizumab n=307 |
Placebo
y quimioterapia† con o sin bevacizumab |
||
|---|---|---|---|---|
| Todos los grados‡ (%) |
Grados 3-4 (%) |
Todos los grados‡ (%) |
Grados 3-4 (%) |
|
|
||||
| Hematología | ||||
| Anemia | 80 | 35 | 77 | 33 |
| Leucopenia | 76 | 27 | 69 | 19 |
| Neutropenia | 73 | 43 | 62 | 32 |
| Linfopenia | 64 | 35 | 59 | 35 |
| Trombocitopenia | 57 | 19 | 53 | 15 |
| Química | ||||
| Hiperglucemia | 51 | 4.7 | 46 | 2.3 |
| Hipoalbuminemia | 46 | 1.4 | 37 | 5 |
| Hiponatremia | 39 | 14 | 38 | 11 |
| ALT aumentada | 40 | 7 | 38 | 6 |
| AST aumentada | 40 | 6 | 36 | 3.0 |
| Fosfatasa alcalina aumentada | 38 | 3.4 | 40 | 2.3 |
| Hipocalcemia | 37 | 4.1 | 31 | 5 |
| Creatinina aumentada | 34 | 5 | 32 | 6 |
| Hipokalemia | 29 | 7 | 26 | 7 |
| Hiperkalemia | 23 | 3.7 | 27 | 4.7 |
| Hipercalcemia | 21 | 1.0 | 20 | 1.3 |
Cáncer cervical recurrente o metastásico previamente tratado
Entre los 98 pacientes con cáncer cervical inscritos en la Cohorte E de KEYNOTE-158 [véase Estudios Clínicos (14.12)], la duración media de la exposición a KEYTRUDA fue de 2,9 meses (rango: 1 día a 22,1 meses). Los pacientes con enfermedad autoinmune o una condición médica que requiriera inmunosupresión fueron excluidos.
KEYTRUDA se suspendió debido a reacciones adversas en el 8% de los pacientes. Se produjeron reacciones adversas graves en el 39% de los pacientes que recibieron KEYTRUDA. Las reacciones adversas graves más frecuentes fueron anemia (7%), fístula (4,1%), hemorragia (4,1%) e infecciones [excepto ITU] (4,1%). Los cuadros 41 y 42 resumen las reacciones adversas y las anormalidades de laboratorio, respectivamente, en pacientes con KEYTRUDA en KEYNOTE-158.
| Reacción adversa | KEYTRUDA 200 mg cada 3 semanas N=98 |
|
|---|---|---|
| Todos los grados* (%) |
Grados 3–4 (%) |
|
|
||
| General | ||
| Fatiga† | 43 | 5 |
| Dolor‡ | 22 | 2.0 |
| Pirexia | 19 | 1.0 |
| Edema periférico§ | 15 | 2.0 |
| Musculoesquelético y del tejido conjuntivo | ||
| Dolor musculoesquelético¶ | 27 | 5 |
| Gastrointestinal | ||
| Diarrea# | 23 | 2.0 |
| Dolor abdominalÞ | 22 | 3.1 |
| Náuseas | 19 | 0 |
| Vómitos | 19 | 1.0 |
| Estreñimiento | 14 | 0 |
| Metabolismo y nutrición | ||
| Disminución del apetito | 21 | 0 |
| Vascular | ||
| Hemorragiaß | 19 | 5 |
| Infecciones | ||
| ITUà | 18 | 6 |
| Infección (excepto ITU)è | 16 | 4.1 |
| Piel y tejido subcutáneo | ||
| Erupciónð | 17 | 2.0 |
| Endocrino | ||
| Hipotiroidismo | 11 | 0 |
| Sistema nervioso | ||
| Cefalea | 11 | 2.0 |
| Respiratorio, torácico y mediastínico | ||
| Disnea | 10 | 1.0 |
| Prueba de laboratorio* | KEYTRUDA 200 mg cada 3 semanas |
|
|---|---|---|
| Todos los grados† (%) |
Grados 3-4 (%) |
|
| Hematología | ||
| Anemia | 54 | 24 |
| Linfopenia | 45 | 9 |
| Química | ||
| Hipoalbuminemia | 44 | 5 |
| Fosfatasa alcalina aumentada | 40 | 1.3 |
| Hiponatremia | 38 | 13 |
| Hiperglucemia | 38 | 1.3 |
| AST aumentada | 34 | 3.9 |
| Creatinina aumentada | 32 | 5 |
| Hipocalcemia | 27 | 0 |
| ALT aumentada | 21 | 3.9 |
| Hipokalemia | 20 | 6 |
Otras anormalidades de laboratorio que ocurrieron en ≥10% de los pacientes que recibieron KEYTRUDA fueron hipofosfatemia (19% de todos los grados; 6% de grados 3-4), aumento del INR (17% de todos los grados; 0% de grados 3-4), hipercalcemia (14% de todos los grados; 2.6% de grados 3-4), disminución del recuento plaquetario (14% de todos los grados; 1.3% de grados 3-4), prolongación del tiempo de tromboplastina parcial activada (10% de todos los grados; 0% de grados 3-4), hipoglucemia (13% de todos los grados; 1.3% de grados 3-4), disminución de leucocitos (13% de todos los grados; 2.6% de grados 3-4) e hiperkalemia (13% de todos los grados; 1.3% de grados 3-4).
HCC
HCC previamente tratado
La seguridad de KEYTRUDA se investigó en KEYNOTE-394, un ensayo multicéntrico, doble ciego, aleatorizado y controlado con placebo que incluyó pacientes con HCC previamente tratado. Los pacientes fueron aleatorizados (2:1) y recibieron KEYTRUDA 200 mg (n=299) o placebo (n=153) por vía intravenosa cada 3 semanas hasta por 35 ciclos [ver Estudios clínicos (14.13)].
La duración media de la exposición fue de 3,3 meses (rango: 1 día a 27,3 meses) en el brazo de KEYTRUDA y de 2,2 meses (rango: 1 día a 15,5 meses) en el brazo de placebo. Se interrumpió el tratamiento con KEYTRUDA debido a reacciones adversas en el 13% de los pacientes. La reacción adversa más frecuente que provocó la interrupción permanente de KEYTRUDA fue la ascitis (2,3%). Las reacciones adversas que provocaron la interrupción de KEYTRUDA se produjeron en el 26% de los pacientes; las reacciones adversas o anomalías de laboratorio más frecuentes que provocaron la interrupción de KEYTRUDA (≥2%) fueron el aumento de la bilirrubina en sangre (9%), el aumento de la AST (5%) y el aumento de la ALT (2%).
Los cuadros 43 y 44 resumen las reacciones adversas y las anomalías de laboratorio, respectivamente, en pacientes tratados con KEYTRUDA en KEYNOTE-394.
| Reacción adversa | KEYTRUDA 200 mg cada 3 semanas n=299 |
Placebo
n=153 |
||
|---|---|---|---|---|
| Todos los grados* (%) |
Grados 3-5 (%) |
Todos los grados* (%) |
Grados 3-5 (%) |
|
| General | ||||
| Pirexia | 18 | 0.7 | 14 | 0 |
| Piel y tejido subcutáneo | ||||
| Erupción† | 18 | 0.7 | 7 | 0 |
| Prurito | 12 | 0 | 4 | 0 |
| Gastrointestinal | ||||
| Diarrea | 16 | 1.7 | 9 | 0 |
| Metabolismo y nutrición | ||||
| Disminución del apetito | 15 | 0.3 | 9 | 0 |
| Infecciones | ||||
| Infección de las vías respiratorias superiores | 11 | 1.0 | 7 | 0.7 |
| Respiratorio, torácico y mediastínico | ||||
| Tos | 11 | 0 | 9 | 0 |
| Endocrino | ||||
| Hipotiroidismo | 10 | 0 | 7 | 0 |
| Prueba de laboratorio* | KEYTRUDA | Placebo | ||
|---|---|---|---|---|
| Todos los grados† % |
Grados 3-4 % |
Todos los grados† % |
Grados 3-4 % |
|
|
||||
| Química | ||||
| AST aumentado | 54 | 14 | 44 | 12 |
| Bilirrubina aumentada | 47 | 11 | 36 | 7 |
| ALT aumentada | 47 | 7 | 32 | 4.6 |
| Gamma-glutamil transferasa (GGT) aumentada | 40 | 20 | 39 | 15 |
| Hipoalbuminemia | 40 | 0.7 | 20 | 0.7 |
| Fosfatasa alcalina aumentada | 39 | 4.1 | 34 | 4 |
| Hiperglucemia | 36 | 3.3 | 26 | 1.4 |
| Hiponatremia | 36 | 11 | 28 | 5 |
| Hipofosfatemia | 30 | 6 | 17 | 4 |
| Hipocalcemia | 24 | 1.4 | 15 | 0.7 |
| Hematología | ||||
| Linfopenia | 44 | 11 | 34 | 4.6 |
| Anemia | 36 | 7 | 30 | 3.3 |
| Disminución de plaquetas | 32 | 4.7 | 29 | 2 |
| Leucopenia | 30 | 1.3 | 21 | 0.7 |
| Neutropenia | 25 | 4.4 | 21 | 2 |
BTC
La seguridad de KEYTRUDA en combinación con gemcitabina y cisplatino se investigó en KEYNOTE-966, un ensayo multicéntrico, doble ciego, aleatorizado y controlado con placebo en pacientes con BTC localmente avanzado irresecable o metastásico que no habían recibido terapia sistémica previa en el contexto de enfermedad avanzada [véase Estudios Clínicos (14.14)]. Un total de 1063 pacientes recibieron KEYTRUDA 200 mg más quimioterapia con gemcitabina y cisplatino (n=529) o placebo más quimioterapia con gemcitabina y cisplatino (n=534) cada 3 semanas.
La duración media de la exposición a KEYTRUDA fue de 6 meses (rango: 1 día a 28 meses).
KEYTRUDA se suspendió por reacciones adversas en el 15% de los pacientes. La reacción adversa más común que provocó la suspensión permanente de KEYTRUDA (≥1%) fue la neumonitis (1,3%).
Las reacciones adversas que llevaron a la interrupción de KEYTRUDA se produjeron en el 55% de los pacientes. Las reacciones adversas o anomalías de laboratorio más comunes que llevaron a la interrupción de KEYTRUDA (≥2%) fueron la disminución del recuento de neutrófilos (18%), la disminución del recuento de plaquetas (10%), la anemia (6%), la disminución del recuento de leucocitos (4%), la pirexia (3,8%), la fatiga (3,0%), la colangitis (2,8%), el aumento de ALT (2,6%), el aumento de AST (2,5%) y la obstrucción biliar (2,3%).
En los brazos de KEYTRUDA más quimioterapia versus placebo más quimioterapia, hubo una diferencia de ≥5% en la incidencia de reacciones adversas entre los pacientes tratados con KEYTRUDA versus placebo para la pirexia (26% vs 20%), la erupción cutánea (21% vs 13%), el prurito (15% vs 10%) y el hipotiroidismo (9% vs. 2,6%). No hubo diferencias clínicamente significativas en la incidencia de toxicidad de Grado 3-4 entre los brazos.
Hubo una diferencia de ≥5% en la incidencia de anomalías de laboratorio entre los pacientes tratados con KEYTRUDA más quimioterapia versus placebo más quimioterapia para la disminución de linfocitos (69% vs 61%). No hubo diferencias clínicamente significativas en la incidencia de toxicidad de Grado 3-4 entre los brazos.
MCC
Entre los 105 pacientes con MCC inscritos en KEYNOTE-017 y KEYNOTE-913 [véase Estudios Clínicos (14.15)], la duración media de la exposición a KEYTRUDA fue de 6,3 meses (rango de 1 día a 28 meses). Los pacientes con enfermedad autoinmune o una condición médica que requiriera inmunosupresión fueron inelegibles. Las reacciones adversas que ocurrieron en pacientes con MCC fueron similares a las que ocurrieron en 2799 pacientes con melanoma o CNCP tratados con KEYTRUDA como agente único. Las anomalías de laboratorio (Grados 3-4) que ocurrieron con una mayor incidencia incluyeron un aumento de lipasa (17%).
RCC
En combinación con axitinib en el tratamiento de primera línea del RCC avanzado (KEYNOTE-426)
La seguridad de KEYTRUDA en combinación con axitinib se investigó en KEYNOTE-426 [véase Estudios Clínicos (14.16)]. Los pacientes con afecciones médicas que requerían corticosteroides sistémicos u otros medicamentos inmunosupresores o que tenían antecedentes de enfermedad autoinmune grave que no fuera diabetes tipo 1, vitiligo, síndrome de Sjögren e hipotiroidismo estable con terapia de reemplazo hormonal fueron inelegibles. Los pacientes recibieron KEYTRUDA 200 mg por vía intravenosa cada 3 semanas y axitinib 5 mg por vía oral dos veces al día, o sunitinib 50 mg una vez al día durante 4 semanas y luego sin tratamiento durante 2 semanas. La duración media de la exposición a la terapia combinada de KEYTRUDA y axitinib fue de 10,4 meses (rango: 1 día a 21,2 meses).
Las características de la población del estudio fueron: edad media de 62 años (rango: 30 a 89), 40% de edad 65 o mayor; 71% hombres; 80% blancos; y 80% Estado de Rendimiento de Karnofsky (KPS) de 90-100 y 20% KPS de 70-80.
Se produjeron reacciones adversas fatales en el 3,3% de los pacientes que recibieron KEYTRUDA en combinación con axitinib. Estas incluyeron 3 casos de paro cardíaco, 2 casos de embolia pulmonar y 1 caso de cada uno de insuficiencia cardíaca, muerte por causa desconocida, miastenia gravis, miocarditis, gangrena de Fournier, mieloma de células plasmáticas, derrame pleural, neumonitis e insuficiencia respiratoria.
Se produjeron reacciones adversas graves en el 40% de los pacientes que recibieron KEYTRUDA en combinación con axitinib. Las reacciones adversas graves en ≥1% de los pacientes que recibieron KEYTRUDA en combinación con axitinib incluyeron hepatotoxicidad (7%), diarrea (4,2%), lesión renal aguda (2,3%), deshidratación (1%) y neumonitis (1%).
La suspensión permanente debido a una reacción adversa de KEYTRUDA o axitinib se produjo en el 31% de los pacientes; 13% solo KEYTRUDA, 13% solo axitinib y 8% ambos fármacos. La reacción adversa más común (>1%) que provocó la suspensión permanente de KEYTRUDA, axitinib o la combinación fue la hepatotoxicidad (13%), la diarrea/colitis (1,9%), la lesión renal aguda (1,6%) y el accidente cerebrovascular (1,2%).
Las interrupciones o reducciones de la dosis debido a una reacción adversa, excluyendo las interrupciones temporales de las infusiones de KEYTRUDA debido a reacciones relacionadas con la infusión, se produjeron en el 76% de los pacientes que recibieron KEYTRUDA en combinación con axitinib. Esto incluye la interrupción de KEYTRUDA en el 50% de los pacientes. Axitinib se interrumpió en el 64% de los pacientes y la dosis se redujo en el 22% de los pacientes. Las reacciones adversas más comunes (>10%) que provocaron la interrupción de KEYTRUDA fueron la hepatotoxicidad (14%) y la diarrea (11%), y las reacciones adversas más comunes (>10%) que provocaron la interrupción o reducción de axitinib fueron la hepatotoxicidad (21%), la diarrea (19%) y la hipertensión (18%).
Las reacciones adversas más comunes (≥20%) en pacientes que recibieron KEYTRUDA y axitinib fueron diarrea, fatiga/astenia, hipertensión, hipotiroidismo, disminución del apetito, hepatotoxicidad, eritrodisestesia palmar-plantar, náuseas, estomatitis/inflamación de la mucosa, disfonía, erupción cutánea, tos y estreñimiento.
El veintisiete por ciento (27%) de los pacientes tratados con KEYTRUDA en combinación con axitinib recibieron una dosis oral de prednisona equivalente a ≥40 mg diarios para una reacción adversa mediada por el sistema inmunitario.
Los cuadros 45 y 46 resumen las reacciones adversas y las anomalías de laboratorio, respectivamente, que ocurrieron en al menos el 20% de los pacientes tratados con KEYTRUDA y axitinib en KEYNOTE-426.
| Reacción adversa | KEYTRUDA 200 mg cada 3 semanas y Axitinib n=429 |
Sunitinib n=425 |
||
|---|---|---|---|---|
| Todos los grados* (%) |
Grados 3-4 (%) |
Todos los grados (%) |
Grados 3-4 (%) |
|
|
||||
| Gastrointestinal | ||||
| Diarrea† | 56 | 11 | 45 | 5 |
| Náuseas | 28 | 0.9 | 32 | 0.9 |
| Estreñimiento | 21 | 0 | 15 | 0.2 |
| General | ||||
| Fatiga/Astenia | 52 | 5 | 51 | 10 |
| Vascular | ||||
| Hipertensión‡ | 48 | 24 | 48 | 20 |
| Hepatobiliar | ||||
| Hepatotoxicidad§ | 39 | 20 | 25 | 4.9 |
| Endocrino | ||||
| Hipotiroidismo | 35 | 0.2 | 32 | 0.2 |
| Metabolismo y Nutrición | ||||
| Disminución del apetito | 30 | 2.8 | 29 | 0.7 |
| Piel y Tejido Subcutáneo | ||||
| Síndrome eritrodisestésico palmar-plantar | 28 | 5 | 40 | 3.8 |
| Estomatitis/Inflamación de la mucosa | 27 | 1.6 | 41 | 4 |
| Erupción cutánea¶ | 25 | 1.4 | 21 | 0.7 |
| Respiratorio, Torácico y Mediastínico | ||||
| Disfonía | 25 | 0.2 | 3.3 | 0 |
| Tos | 21 | 0.2 | 14 | 0.5 |
| Prueba de laboratorio* | KEYTRUDA 200 mg cada 3 semanas y Axitinib |
Sunitinib | ||
|---|---|---|---|---|
| Todos los grados† % |
Grados 3-4 % |
Todos los grados % |
Grados 3-4 % |
|
|
||||
| Química | ||||
| Hiperglucemia | 62 | 9 | 54 | 3.2 |
| ALT aumentada | 60 | 20 | 44 | 5 |
| AST aumentada | 57 | 13 | 56 | 5 |
| Creatinina aumentada | 43 | 4.3 | 40 | 2.4 |
| Hiponatremia | 35 | 8 | 29 | 8 |
| Hiperkalemia | 34 | 6 | 22 | 1.7 |
| Hipoalbuminemia | 32 | 0.5 | 34 | 1.7 |
| Hipercalcemia | 27 | 0.7 | 15 | 1.9 |
| Hipofosfatemia | 26 | 6 | 49 | 17 |
| Fosfatasa alcalina aumentada | 26 | 1.7 | 30 | 2.7 |
| Hipocalcemia‡ | 22 | 0.2 | 29 | 0.7 |
| Bilirrubina sanguínea aumentada | 22 | 2.1 | 21 | 1.9 |
| Tiempo de tromboplastina parcial activado prolongado§ | 22 | 1.2 | 14 | 0 |
| Hematología | ||||
| Linfopenia | 33 | 11 | 47 | 9 |
| Anemia | 29 | 2.1 | 65 | 8 |
| Trombocitopenia | 27 | 1.4 | 78 | 14 |
En combinación con lenvatinib en el tratamiento de primera línea de RCC avanzado (KEYNOTE-581)
La seguridad de KEYTRUDA se evaluó en KEYNOTE-581 [véase Estudios clínicos (14.16)]. Los pacientes recibieron KEYTRUDA 200 mg por vía intravenosa cada 3 semanas en combinación con lenvatinib 20 mg por vía oral una vez al día (n=352), o lenvatinib 18 mg por vía oral una vez al día en combinación con everolimus 5 mg por vía oral una vez al día (n=355), o sunitinib 50 mg por vía oral una vez al día durante 4 semanas y luego sin tratamiento durante 2 semanas (n=340). La duración media de la exposición a la terapia combinada de KEYTRUDA y lenvatinib fue de 17 meses (rango: 0,1 a 39).
Se produjeron reacciones adversas mortales en el 4,3% de los pacientes tratados con KEYTRUDA en combinación con lenvatinib, incluyendo paro cardiorrespiratorio (0,9%), sepsis (0,9%) y un caso (0,3%) de cada uno de arritmia, hepatitis autoinmune, disnea, crisis hipertensiva, aumento de creatinina en sangre, síndrome de disfunción multiorgánica, síndrome miasténico, miocarditis, nefritis, neumonitis, aneurisma roto y hemorragia subaracnoidea.
Se produjeron reacciones adversas graves en el 51% de los pacientes que recibieron KEYTRUDA y lenvatinib. Las reacciones adversas graves en ≥2% de los pacientes fueron eventos hemorrágicos (5%), diarrea (4%), hipertensión (3%), infarto de miocardio (3%), neumonitis (3%), vómitos (3%), lesión renal aguda (2%), insuficiencia suprarrenal (2%), disnea (2%) y neumonía (2%).
La interrupción permanente de KEYTRUDA, lenvatinib o ambos debido a una reacción adversa se produjo en el 37% de los pacientes que recibieron KEYTRUDA en combinación con lenvatinib; 29% solo KEYTRUDA, 26% solo lenvatinib y 13% ambos. Las reacciones adversas más comunes (≥2%) que provocaron la interrupción permanente de KEYTRUDA, lenvatinib o la combinación fueron neumonitis (3%), infarto de miocardio (3%), hepatotoxicidad (3%), lesión renal aguda (3%), erupción cutánea (3%) y diarrea (2%).
Las interrupciones de la dosis de KEYTRUDA, lenvatinib o ambos debido a una reacción adversa se produjeron en el 78% de los pacientes que recibieron KEYTRUDA en combinación con lenvatinib. KEYTRUDA se interrumpió en el 55% de los pacientes y ambos fármacos se interrumpieron en el 39% de los pacientes. Las reacciones adversas más comunes (≥3%) que provocaron la interrupción de KEYTRUDA fueron diarrea (10%), hepatotoxicidad (8%), fatiga (7%), aumento de lipasa (5%), aumento de amilasa (4%), dolor musculoesquelético (3%), hipertensión (3%), erupción cutánea (3%), lesión renal aguda (3%) y disminución del apetito (3%).
El quince por ciento (15%) de los pacientes tratados con KEYTRUDA en combinación con lenvatinib recibieron un equivalente oral de prednisona ≥40 mg diarios para una reacción adversa inmunomediada.
Los cuadros 47 y 48 resumen las reacciones adversas y las alteraciones de laboratorio, respectivamente, que se produjeron en ≥20% de los pacientes tratados con KEYTRUDA y lenvatinib en KEYNOTE-581.
| Reacción adversa | KEYTRUDA 200 mg cada 3 semanas con lenvatinib N=352 |
Sunitinib 50 mg N=340 |
||
|---|---|---|---|---|
| Todos los grados (%) |
Grados 3-4 (%) |
Todos los grados (%) |
Grados 3-4 (%) |
|
|
||||
| General | ||||
| Fatiga* | 63 | 9 | 56 | 8 |
| Gastrointestinal | ||||
| Diarrea† | 62 | 10 | 50 | 6 |
| Estomatitis‡ | 43 | 2 | 43 | 2 |
| Náuseas | 36 | 3 | 33 | 1 |
| Dolor abdominal§ | 27 | 2 | 18 | 1 |
| Vómitos | 26 | 3 | 20 | 1 |
| Estreñimiento | 25 | 1 | 19 | 0 |
| Musculoesquelético y del tejido conjuntivo | ||||
| Trastornos musculoesqueléticos¶ | 58 | 4 | 41 | 3 |
| Endocrino | ||||
| Hipotiroidismo# | 57 | 1 | 32 | 0 |
| Vascular | ||||
| HipertensiónÞ | 56 | 29 | 43 | 20 |
| Eventos hemorrágicosß | 27 | 5 | 26 | 4 |
| Metabolismo | ||||
| Disminución del apetitoà | 41 | 4 | 31 | 1 |
| Piel y tejido subcutáneo | ||||
| Erupciónè | 37 | 5 | 17 | 1 |
| Síndrome eritrodisestésico palmar-plantarð | 29 | 4 | 38 | 4 |
| Pruebas | ||||
| Pérdida de peso | 30 | 8 | 9 | 0.3 |
| Respiratorio, torácico y mediastínico | ||||
| Disfonía | 30 | 0 | 4 | 0 |
| Renal y urinario | ||||
| Proteinuriaø | 30 | 8 | 13 | 3 |
| Lesión renal agudaý | 21 | 5 | 16 | 2 |
| Hepatobiliar | ||||
| Hepatotoxicidad£ | 25 | 9 | 21 | 5 |
| Sistema nervioso | ||||
| Cefalea | 23 | 1 | 16 | 1 |
Las reacciones adversas clínicamente relevantes (<20%) que se produjeron en pacientes que recibieron KEYTRUDA con lenvatinib fueron infarto de miocardio (3%) y angina de pecho (1%).
| Prueba de laboratorio* | KEYTRUDA 200 mg cada 3 semanas con lenvatinib |
Sunitinib 50 mg | ||
|---|---|---|---|---|
| Todos los grados %† |
Grado 3-4 %† |
Todos los grados %† |
Grado 3-4 %† |
|
|
||||
| Química | ||||
| Hipertrigliceridemia | 80 | 15 | 71 | 15 |
| Hipercolesterolemia | 64 | 5 | 43 | 1 |
| Lipasa aumentada | 61 | 34 | 59 | 28 |
| Creatinina aumentada | 61 | 5 | 61 | 2 |
| Amilasa aumentada | 59 | 17 | 41 | 9 |
| AST aumentada | 58 | 7 | 57 | 3 |
| Hiperglucemia | 55 | 7 | 48 | 3 |
| ALT aumentada | 52 | 7 | 49 | 4 |
| Hiperkalemia | 44 | 9 | 28 | 6 |
| Hipoglucemia | 44 | 2 | 27 | 1 |
| Hiponatremia | 41 | 12 | 28 | 9 |
| Albúmina disminuida | 34 | 0.3 | 22 | 0 |
| Fosfatasa alcalina aumentada | 32 | 4 | 32 | 1 |
| Hipocalcemia | 30 | 2 | 22 | 1 |
| Hipofosfatemia | 29 | 7 | 50 | 8 |
| Hipomagnesemia | 25 | 2 | 15 | 3 |
| Creatina fosfocinasa aumentada | 24 | 6 | 36 | 5 |
| Hipermagnesemia | 23 | 2 | 22 | 3 |
| Hipercalcemia | 21 | 1 | 11 | 1 |
| Hematología | ||||
| Linfopenia | 54 | 9 | 66 | 15 |
| Trombocitopenia | 39 | 2 | 73 | 13 |
| Anemia | 38 | 3 | 66 | 8 |
| Leucopenia | 34 | 1 | 77 | 8 |
| Neutropenia | 31 | 4 | 72 | 16 |
Se observó un aumento de ALT o AST de grado 3 y 4 en el 9 % de los pacientes. Se notificó un aumento de ALT o AST de grado ≥2 en 64 (18 %) pacientes, de los cuales 20 (31 %) recibieron ≥40 mg diarios de prednisona oral equivalente. Se observó la recurrencia de un aumento de ALT o AST de grado ≥2 en el re-desafío en 10 pacientes que recibieron KEYTRUDA y lenvatinib (n=38) y no se observó en el re-desafío con KEYTRUDA solo (n=3).
Tratamiento adyuvante del CCR
La seguridad de KEYTRUDA como agente único se investigó en KEYNOTE-564, un ensayo aleatorizado (1:1) doble ciego controlado con placebo en el que 984 pacientes sometidos a nefrectomía por CCR recibieron 200 mg de KEYTRUDA por infusión intravenosa cada 3 semanas (n=488) o placebo (n=496) durante un máximo de un año [véase Estudios clínicos (14.16)]. La duración media de la exposición a KEYTRUDA fue de 11,1 meses (rango: 1 día a 14,3 meses). Los pacientes con enfermedad autoinmune activa o una afección médica que requiriera inmunosupresión no fueron elegibles.
Se produjeron reacciones adversas graves en el 20 % de estos pacientes que recibieron KEYTRUDA. Las reacciones adversas graves (≥1 %) fueron lesión renal aguda, insuficiencia suprarrenal, neumonía, colitis y cetoacidosis diabética (1 % cada una). Se produjeron reacciones adversas mortales en el 0,2 % de los tratados con KEYTRUDA, incluido un caso de neumonía.
La interrupción de KEYTRUDA debido a una reacción adversa se produjo en el 21 % de los pacientes; las más frecuentes (≥1 %) fueron aumento de ALT (1,6 %), colitis (1 %) e insuficiencia suprarrenal (1 %).
Las interrupciones de la dosis de KEYTRUDA debido a una reacción adversa se produjeron en el 26 % de los pacientes; las más frecuentes (≥1 %) fueron aumento de AST (2,3 %), artralgia (1,6 %), hipotiroidismo (1,6 %), diarrea (1,4 %), aumento de ALT (1,4 %), fatiga (1,4 %), erupción cutánea, disminución del apetito y vómitos (1 % cada uno). Los cuadros 49 y 50 resumen las reacciones adversas y las alteraciones de laboratorio, respectivamente, en pacientes con KEYTRUDA en KEYNOTE-564.
| Reacción adversa | KEYTRUDA 200 mg cada 3 semanas n=488 |
Placebo
n=496 |
||
|---|---|---|---|---|
| Todos los grados† (%) |
Grados 3-4 (%) |
Todos los grados (%) |
Grados 3-4 (%) |
|
|
||||
| Sistema musculoesquelético y tejido conjuntivo | ||||
| Dolor musculoesquelético‡ | 41 | 1.2 | 36 | 0.6 |
| General | ||||
| Fatiga§ | 40 | 1.2 | 31 | 0.2 |
| Piel y tejido subcutáneo | ||||
| Erupción cutánea¶ | 30 | 1.4 | 15 | 0.4 |
| Prurito | 23 | 0.2 | 13 | 0 |
| Gastrointestinal | ||||
| Diarrea# | 27 | 2.7 | 23 | 0.2 |
| Náuseas | 16 | 0.4 | 10 | 0 |
| Dolor abdominalÞ | 11 | 0.4 | 13 | 0.2 |
| Endocrino | ||||
| Hipotiroidismo | 21 | 0.2 | 3.6 | 0 |
| Hipertiroidismo | 12 | 0.2 | 0.2 | 0 |
| Respiratorio, torácico y mediastínico | ||||
| Tos ß | 17 | 0 | 12 | 0 |
| Sistema nervioso | ||||
| Cefaleaà | 15 | 0.2 | 13 | 0 |
| Hepatobiliar | ||||
| Hepatotoxicidadè | 14 | 3.7 | 7 | 0.6 |
| Renal y urinario | ||||
| Lesión renal agudað | 13 | 1.2 | 10 | 0.2 |
| Prueba de laboratorio† | KEYTRUDA 200 mg cada 3 semanas |
Placebo | ||
|---|---|---|---|---|
| Todos los grados‡ % |
Grados 3-4 % |
Todos los grados % |
Grados 3-4 % |
|
|
||||
| Química | ||||
| Hiperglucemia | 48 | 8 | 45 | 4.5 |
| Aumento de creatinina | 39 | 1.1 | 28 | 0.2 |
| Aumento de INR | 29 | 1.0 | 20 | 0.9 |
| Hiponatremia | 21 | 3.3 | 13 | 1.9 |
| Aumento de ALT | 20 | 3.6 | 11 | 0.2 |
| Hematología | ||||
| Anemia | 28 | 0.5 | 20 | 0.4 |
Carcinoma de endometrio
Carcinoma de endometrio primario avanzado o recurrente
La seguridad de KEYTRUDA en combinación con quimioterapia (paclitaxel y carboplatino) se investigó en KEYNOTE-868, un ensayo aleatorizado (1:1), multicéntrico, doble ciego, controlado con placebo que incluyó pacientes con carcinoma de endometrio avanzado o recurrente [véase Estudios clínicos (14.17)]. Un total de 759 pacientes recibieron KEYTRUDA 200 mg cada 3 semanas y quimioterapia durante 6 ciclos seguidos de KEYTRUDA 400 mg cada 6 semanas hasta por 14 ciclos (n=382) o placebo y quimioterapia durante 6 ciclos seguidos de placebo hasta por 14 ciclos (n=377). La duración media de la exposición a KEYTRUDA fue de 5,6 meses (rango: 1 día a 24,0 meses).
Se produjeron reacciones adversas graves en el 35 % de los pacientes que recibieron KEYTRUDA en combinación con quimioterapia, en comparación con el 19 % de los pacientes que recibieron placebo en combinación con quimioterapia.
Se produjeron reacciones adversas mortales en el 1,6 % de los pacientes que recibieron KEYTRUDA en combinación con quimioterapia, incluida la COVID-19 (0,5 %) y el paro cardíaco (0,3 %).
KEYTRUDA se suspendió por una reacción adversa en el 14 % de los pacientes. Se requirió una reducción de la dosis de quimioterapia en el 29 % de los pacientes que recibieron KEYTRUDA en combinación con quimioterapia, en comparación con el 23 % de los pacientes que recibieron placebo en combinación con quimioterapia. No hubo diferencias clínicamente significativas en las suspensiones o interrupciones de la quimioterapia entre los brazos.
Las reacciones adversas que se produjeron en los pacientes tratados con KEYTRUDA y quimioterapia fueron generalmente similares a las observadas con KEYTRUDA solo o quimioterapia sola, con la excepción de la erupción cutánea (33 % de todos los grados; 2,9 % de grados 3-4).
En combinación con lenvatinib para el tratamiento del carcinoma de endometrio avanzado que es pMMR o no MSI-H.
La seguridad de KEYTRUDA en combinación con lenvatinib se investigó en KEYNOTE-775, un ensayo multicéntrico, abierto, aleatorizado (1:1), controlado con activo en pacientes con carcinoma de endometrio avanzado tratados previamente con al menos un régimen de quimioterapia a base de platino en cualquier contexto, incluidos los contextos neoadyuvante y adyuvante [véase Estudios clínicos (14.17)]. Los pacientes con carcinoma de endometrio pMMR o no MSI-H recibieron KEYTRUDA 200 mg cada 3 semanas en combinación con lenvatinib 20 mg por vía oral una vez al día (n=342) o recibieron doxorrubicina o paclitaxel (n=325).
Para los pacientes con estado tumoral pMMR o no MSI-H, la duración media del tratamiento del estudio fue de 7,2 meses (rango de 1 día a 26,8 meses) y la duración media de la exposición a KEYTRUDA fue de 6,8 meses (rango: 1 día a 25,8 meses).
Las reacciones adversas mortales entre estos pacientes se produjeron en el 4,7 % de los tratados con KEYTRUDA y lenvatinib, incluidos 2 casos de neumonía y 1 caso de lo siguiente: lesión renal aguda, infarto agudo de miocardio, colitis, disminución del apetito, perforación intestinal, hemorragia gastrointestinal inferior, obstrucción gastrointestinal maligna, síndrome de disfunción multiorgánica, síndrome mielodisplásico, embolia pulmonar y disfunción ventricular derecha.
Se produjeron reacciones adversas graves en el 50 % de estos pacientes que recibieron KEYTRUDA y lenvatinib. Las reacciones adversas graves (≥3 %) fueron hipertensión (4,4 %) e infecciones del tracto urinario (3,2 %).
La suspensión de KEYTRUDA debido a una reacción adversa se produjo en el 15 % de estos pacientes. La reacción adversa más frecuente que provocó la suspensión de KEYTRUDA (≥1 %) fue el aumento de ALT (1,2 %).
Las interrupciones de la dosis de KEYTRUDA debido a una reacción adversa se produjeron en el 48 % de estos pacientes. Las reacciones adversas más frecuentes que provocaron la interrupción de KEYTRUDA (≥3 %) fueron diarrea (8 %), aumento de ALT (4,4 %), aumento de AST (3,8 %) e hipertensión (3,5 %).
Los cuadros 51 y 52 resumen las reacciones adversas y las anomalías de laboratorio, respectivamente, en pacientes con KEYTRUDA en combinación con lenvatinib en KEYNOTE-775.
| Carcinoma de endometrio (pMMR o no MSI-H) | ||||||
|---|---|---|---|---|---|---|
| Reacción adversa | KEYTRUDA 200 mg cada 3 semanas y lenvatinib n=342 |
Doxorrubicina o Paclitaxel n=325 |
||||
| Todos los grados* (%) |
Grados 3-4 (%) |
Todos los grados* (%) |
Grados 3-4 (%) |
|||
|
||||||
| Endocrino | ||||||
| Hipotiroidismo† | 67 | 0.9 | 0.9 | 0 | ||
| Vascular | ||||||
| Hipertensión‡ | 67 | 39 | 6 | 2.5 | ||
| Eventos hemorrágicos§ | 25 | 2.6 | 15 | 0.9 | ||
| General | ||||||
| Fatiga¶ | 58 | 11 | 54 | 6 | ||
| Gastrointestinal | ||||||
| Diarrea# | 55 | 8 | 20 | 2.8 | ||
| Náuseas | 49 | 2.9 | 47 | 1.5 | ||
| Vómitos | 37 | 2.3 | 21 | 2.2 | ||
| EstomatitisÞ | 35 | 2.6 | 26 | 1.2 | ||
| Dolor abdominalß | 34 | 2.6 | 21 | 1.2 | ||
| Estreñimiento | 27 | 0 | 25 | 0.6 | ||
| Musculoesquelético y del tejido conectivo | ||||||
| Trastornos musculoesqueléticosà | 53 | 5 | 27 | 0.6 |
| Metabolismo | ||||
| Disminución del apetitoè | 44 | 7 | 21 | 0 |
| Exploraciones complementarias | ||||
| Pérdida de peso | 34 | 10 | 6 | 0.3 |
| Renal y urinario | ||||
| Proteinuriað | 29 | 6 | 3.4 | 0.3 |
| Infecciones | ||||
| Infección del tracto urinarioø | 31 | 5 | 13 | 1.2 |
| Sistema nervioso | ||||
| Cefalea | 26 | 0.6 | 9 | 0.3 |
| Respiratorio, torácico y mediastínico | ||||
| Disfonía | 22 | 0 | 0.6 | 0 |
| Piel y tejido subcutáneo | ||||
| Eritrodisestesia palmar-plantarý | 23 | 2.9 | 0.9 | 0 |
| Erupción£ | 20 | 2.3 | 4.9 | 0 |
| Carcinoma endometrial (pMMR o no MSI-H) | ||||
|---|---|---|---|---|
| Prueba de laboratorio† | KEYTRUDA 200 mg cada 3 semanas y Lenvatinib |
Doxorrubicina o Paclitaxel |
||
| Todos los grados‡ % |
Grados 3-4 % |
Todos los grados‡ % |
Grados 3-4 % |
|
|
||||
| Química | ||||
| Hipertrigliceridemia | 70 | 6 | 45 | 1.7 |
| Hipoalbuminemia | 60 | 2.7 | 42 | 1.6 |
| Aumento de la aspartato aminotransferasa | 58 | 9 | 23 | 1.6 |
| Hiperglucemia | 58 | 8 | 45 | 4.4 |
| Hipomagnesemia | 46 | 0 | 27 | 1.3 |
| Aumento de la alanina aminotransferasa | 55 | 9 | 21 | 1.2 |
| Hipercolesterolemia | 53 | 3.2 | 23 | 0.7 |
| Hiponatremia | 46 | 15 | 28 | 7 |
| Aumento de la fosfatasa alcalina | 43 | 4.7 | 18 | 0.9 |
| Hipocalcemia | 40 | 4.7 | 21 | 1.9 |
| Aumento de lipasa | 36 | 14 | 13 | 3.9 |
| Aumento de creatinina | 35 | 4.7 | 18 | 1.9 |
| Hipokalemia | 34 | 10 | 24 | 5 |
| Hipofosfatemia | 26 | 8 | 17 | 3.2 |
| Aumento de amilasa | 25 | 7 | 8 | 1 |
| Hiperkalemia | 23 | 2.4 | 12 | 1.2 |
| Aumento de creatina cinasa | 19 | 3.7 | 7 | 0 |
| Increased bilirubin | 18 | 3.6 | 6 | 1.6 |
| Hematología | ||||
| Linfopenia | 51 | 18 | 66 | 23 |
| Trombocitopenia | 50 | 8 | 30 | 4.7 |
| Anemia | 49 | 8 | 84 | 14 |
| Leucopenia | 43 | 3.5 | 83 | 43 |
| Neutropenia | 34 | 8 | 80 | 60 |
Como agente único para el tratamiento del carcinoma endometrial MSI-H o dMMR avanzado
Entre los 90 pacientes con carcinoma endometrial MSI-H o dMMR inscritos en KEYNOTE-158 [véase Estudios clínicos (14.17)] tratados con KEYTRUDA como agente único, la duración media de la exposición a KEYTRUDA fue de 8,3 meses (rango: 1 día a 26,9 meses). Las reacciones adversas que se produjeron en pacientes con carcinoma endometrial fueron similares a las que se produjeron en 2799 pacientes con melanoma o CNCP tratados con KEYTRUDA como agente único.
Cáncer TMB-H
La seguridad de KEYTRUDA se investigó en 105 pacientes con cáncer TMB-H inscritos en KEYNOTE-158 [véase Estudios clínicos (14.18)]. La duración media de la exposición a KEYTRUDA fue de 4,9 meses (rango: 0,03 a 35,2 meses). Las reacciones adversas que se produjeron en pacientes con cáncer TMB-H fueron similares a las que se produjeron en pacientes con otros tumores sólidos que recibieron KEYTRUDA como agente único.
cSCC
Entre los 159 pacientes con cSCC avanzado (enfermedad recurrente o metastásica o localmente avanzada) inscritos en KEYNOTE-629 [véase Estudios clínicos (14.19)], la duración media de la exposición a KEYTRUDA fue de 6,9 meses (rango: 1 día a 28,9 meses). Los pacientes con enfermedad autoinmune o una afección médica que requiriera corticosteroides sistémicos u otros medicamentos inmunosupresores no fueron elegibles. Las reacciones adversas que se produjeron en pacientes con cSCC recurrente o metastásico o cSCC localmente avanzado fueron similares a las que se produjeron en 2799 pacientes con melanoma o CNCP tratados con KEYTRUDA como agente único. Las anomalías de laboratorio (grados 3-4) que se produjeron con una incidencia mayor incluyeron linfopenia (10%) y disminución del sodio (10%).
TNBC
Tratamiento neoadyuvante y adyuvante del TNBC de estadio temprano de alto riesgo
La seguridad de KEYTRUDA en combinación con quimioterapia neoadyuvante (carboplatino y paclitaxel seguidos de doxorrubicina o epirrubicina y ciclofosfamida) seguida de cirugía y tratamiento adyuvante continuado con KEYTRUDA como agente único se investigó en KEYNOTE-522, un ensayo aleatorizado (2:1), multicéntrico, doble ciego, controlado con placebo en pacientes con TNBC de estadio temprano de alto riesgo, recién diagnosticado y previamente no tratado.
Un total de 778 pacientes del brazo de KEYTRUDA recibieron al menos 1 dosis de KEYTRUDA en combinación con quimioterapia neoadyuvante seguida de KEYTRUDA como tratamiento adyuvante después de la cirugía, en comparación con 389 pacientes que recibieron al menos 1 dosis de placebo en combinación con quimioterapia neoadyuvante seguida de placebo como tratamiento adyuvante después de la cirugía [véase Estudios clínicos (14.20)].
La duración media de la exposición a KEYTRUDA 200 mg cada 3 semanas fue de 13,3 meses (rango: 1 día a 21,9 meses).
Se produjeron reacciones adversas mortales en el 0,9% de los pacientes que recibieron KEYTRUDA, incluyendo 1 caso de crisis suprarrenal, encefalitis autoinmune, hepatitis, neumonía, neumonitis, embolia pulmonar y sepsis en asociación con síndrome de disfunción multiorgánica e infarto de miocardio.
Se produjeron reacciones adversas graves en el 44% de los pacientes que recibieron KEYTRUDA. Las reacciones adversas graves en ≥2% de los pacientes que recibieron KEYTRUDA incluyeron neutropenia febril (15%), pirexia (3,7%), anemia (2,6%) y neutropenia (2,2%).
KEYTRUDA se suspendió por reacciones adversas en el 20% de los pacientes. Las reacciones adversas más comunes (≥1%) que provocaron la suspensión permanente de KEYTRUDA fueron aumento de ALT (2,7%), aumento de AST (1,5%) y erupción cutánea (1%). Las reacciones adversas que llevaron a la interrupción de KEYTRUDA se produjeron en el 57% de los pacientes. Las reacciones adversas más comunes que llevaron a la interrupción de KEYTRUDA (≥2%) fueron neutropenia (26%), trombocitopenia (6%), aumento de ALT (6%), aumento de AST (3,7%), anemia (3,5%), erupción cutánea (3,2%), neutropenia febril (2,8%), leucopenia (2,8%), infección del tracto respiratorio superior (2,6%), pirexia (2,2%) y fatiga (2,1%).
Los cuadros 53 y 54 resumen las reacciones adversas y las anomalías de laboratorio, respectivamente, en los pacientes tratados con KEYTRUDA en KEYNOTE-522.
| Reacción adversa | KEYTRUDA 200 mg cada 3 semanas con quimioterapia*/KEYTRUDA n=778 |
Placebo con quimioterapia*/Placebo n=389 |
||
|---|---|---|---|---|
| Todos los grados† (%) |
Grados 3-4 (%) |
Todos los grados† (%) |
Grados 3-4 (%) |
|
|
||||
| General | ||||
| Fatiga‡ | 70 | 8 | 66 | 3.9 |
| Pirexia | 28 | 1.3 | 19 | 0.3 |
| Gastrointestinal | ||||
| Náuseas | 67 | 3.7 | 66 | 1.8 |
| Estreñimiento | 42 | 0 | 39 | 0.3 |
| Diarrea | 41 | 3.2 | 34 | 1.8 |
| Estomatitis§ | 34 | 2.7 | 29 | 1 |
| Vómitos | 31 | 2.7 | 28 | 1.5 |
| Dolor abdominal¶ | 24 | 0.5 | 23 | 0.8 |
| Piel y tejido subcutáneo | ||||
| Alopecia | 61 | 0 | 58 | 0 |
| Erupción# | 52 | 5 | 41 | 0.5 |
| Sistema nervioso | ||||
| Neuropatía periféricaÞ | 41 | 3.3 | 42 | 2.3 |
| Cefalea | 30 | 0.5 | 29 | 1 |
| Sistema musculoesquelético y tejido conjuntivo | ||||
| Artralgia | 29 | 0.5 | 31 | 0.3 |
| Mialgia | 20 | 0.5 | 19 | 0 |
| Respiratorio, torácico y mediastínico | ||||
| Tosß | 26 | 0.1 | 24 | 0 |
| Metabolismo y nutrición | ||||
| Disminución del apetito | 23 | 0.9 | 17 | 0.3 |
| Psiquiátricos | ||||
| Insomnio | 21 | 0.5 | 19 | 0 |
| Prueba de laboratorio* | KEYTRUDA 200 mg cada 3 semanas con quimioterapia†/KEYTRUDA |
Placebo con quimioterapia†/Placebo |
||
|---|---|---|---|---|
| Todos los grados‡ % |
Grados 3-4 % |
Todos los grados‡ % |
Grados 3-4 % |
|
|
||||
| Hematología | ||||
| Anemia | 97 | 22 | 96 | 19 |
| Leucopenia | 93 | 41 | 91 | 32 |
| Neutropenia | 88 | 62 | 89 | 62 |
| Linfopenia | 79 | 28 | 74 | 22 |
| Trombocitopenia | 57 | 10 | 56 | 8 |
| Química | ||||
| ALT elevada | 70 | 9 | 67 | 3.9 |
| AST elevada | 65 | 6 | 56 | 1.5 |
| Hiperglucemia | 63 | 4.3 | 61 | 2.8 |
| Fosfatasa alcalina elevada | 37 | 1 | 35 | 0.5 |
| Hiponatremia | 35 | 9 | 25 | 4.6 |
| Hipoalbuminemia | 34 | 1.0 | 30 | 1.3 |
| Hipocalcemia | 31 | 2.2 | 28 | 3.1 |
| Hipokalemia | 31 | 6 | 22 | 2.8 |
| Hipofosfatemia | 20 | 6 | 15 | 4.2 |
Recidiva local unresecable o metastásica de TNBC
La seguridad de KEYTRUDA en combinación con paclitaxel, paclitaxel unido a proteínas o gemcitabina y carboplatino se investigó en KEYNOTE-355, un ensayo multicéntrico, doble ciego, aleatorizado (2:1), controlado con placebo en pacientes con recidiva local unresecable o TNBC metastásica que no habían sido tratados previamente con quimioterapia en el contexto metastásico [véase Estudios clínicos (14.20)]. Un total de 596 pacientes (incluidos 34 pacientes de un período de seguridad inicial) recibieron KEYTRUDA 200 mg cada 3 semanas en combinación con paclitaxel, paclitaxel unido a proteínas o gemcitabina y carboplatino.
La duración media de la exposición a KEYTRUDA fue de 5,7 meses (rango: 1 día a 33,0 meses).
Se produjeron reacciones adversas mortales en el 2,5% de los pacientes que recibieron KEYTRUDA en combinación con quimioterapia, incluyendo parada cardiorrespiratoria (0,7%) y shock séptico (0,3%).
Se produjeron reacciones adversas graves en el 30% de los pacientes que recibieron KEYTRUDA en combinación con paclitaxel, paclitaxel unido a proteínas o gemcitabina y carboplatino. Las reacciones adversas graves en ≥2% de los pacientes fueron neumonía (2,9%), anemia (2,2%) y trombocitopenia (2%).
KEYTRUDA se suspendió por reacciones adversas en el 11% de los pacientes. Las reacciones adversas más comunes que provocaron la suspensión permanente de KEYTRUDA (≥1%) fueron aumento de ALT (2,2%), aumento de AST (1,5%) y neumonitis (1,2%). Las reacciones adversas que llevaron a la interrupción de KEYTRUDA se produjeron en el 50% de los pacientes. Las reacciones adversas más comunes que llevaron a la interrupción de KEYTRUDA (≥2%) fueron neutropenia (22%), trombocitopenia (14%), anemia (7%), aumento de ALT (6%), leucopenia (5%), aumento de AST (5%), disminución del recuento de leucocitos (3,9%) y diarrea (2%).
Los cuadros 55 y 56 resumen las reacciones adversas y las alteraciones de laboratorio en los pacientes tratados con KEYTRUDA en KEYNOTE-355.
| Reacción adversa | KEYTRUDA 200 mg cada 3 semanas con quimioterapia n=596 |
Placebo cada 3 semanas con quimioterapia n=281 |
||
|---|---|---|---|---|
| Todos los grados* (%) |
Grados 3-4 (%) |
Todos los grados* (%) |
Grados 3-4 (%) |
|
|
||||
| General | ||||
| Fatiga† | 48 | 5 | 49 | 4.3 |
| Gastrointestinal | ||||
| Náuseas | 44 | 1.7 | 47 | 1.8 |
| Diarrea | 28 | 1.8 | 23 | 1.8 |
| Estreñimiento | 28 | 0.5 | 27 | 0.4 |
| Vómitos | 26 | 2.7 | 22 | 3.2 |
| Piel y tejido subcutáneo | ||||
| Alopecia | 34 | 0.8 | 35 | 1.1 |
| Erupción‡ | 26 | 2 | 16 | 0 |
| Respiratorio, torácico y mediastínico | ||||
| Tos§ | 23 | 0 | 20 | 0.4 |
| Metabolismo y nutrición | ||||
| Disminución del apetito | 21 | 0.8 | 14 | 0.4 |
| Sistema Nervioso | ||||
| Dolor de cabeza¶ | 20 | 0.7 | 23 | 0.7 |
| Prueba de laboratorio* | KEYTRUDA 200 mg cada 3 semanas con quimioterapia |
Placebo cada 3 semanas con quimioterapia |
||
|---|---|---|---|---|
| Todos los grados† % |
Grados 3-4 % |
Todos los grados† % |
Grados 3-4 % |
|
|
||||
| Hematología | ||||
| Anemia | 90 | 20 | 85 | 19 |
| Leucopenia | 85 | 39 | 86 | 39 |
| Neutropenia | 78 | 50 | 79 | 53 |
| Linfopenia | 73 | 28 | 71 | 19 |
| Trombocitopenia | 54 | 19 | 53 | 21 |
| Química | ||||
| ALT elevada | 60 | 11 | 58 | 8 |
| AST elevada | 57 | 9 | 55 | 6 |
| Hiperglucemia | 52 | 4.4 | 51 | 2.2 |
| Hipoalbuminemia | 36 | 2.0 | 32 | 2.2 |
| Fosfatasa alcalina elevada | 35 | 3.9 | 39 | 2.2 |
| Hipocalcemia | 29 | 3.3 | 27 | 1.8 |
| Hiponatremia | 28 | 5 | 26 | 6 |
| Hipofosfatemia | 21 | 7 | 18 | 4.8 |
| Hipokalemia | 20 | 4.4 | 18 | 4.0 |
6.2 Experiencia postcomercialización
Las siguientes reacciones adversas se han identificado durante el uso posterior a la aprobación de KEYTRUDA. Debido a que estas reacciones se notifican voluntariamente a partir de una población de tamaño incierto, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal con la exposición al fármaco.
Gastrointestinal: Insuficiencia pancreática exocrina
Hepatobiliar: colangitis esclerosante
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de Riesgo
Basándose en su mecanismo de acción, KEYTRUDA puede causar daño fetal cuando se administra a una mujer embarazada. No hay datos humanos disponibles que informen sobre el riesgo de toxicidad embriofetal. En modelos animales, la vía de señalización PD-1/PD-L1 es importante para el mantenimiento del embarazo a través de la inducción de la tolerancia inmunitaria materna al tejido fetal (ver Datos). Se sabe que la IgG4 humana (inmunoglobulinas) atraviesa la placenta; por lo tanto, pembrolizumab tiene el potencial de transmitirse de la madre al feto en desarrollo. Advierta a las mujeres embarazadas sobre el riesgo potencial para un feto.
En la población general de EE. UU., el riesgo estimado de fondo de defectos congénitos importantes y aborto espontáneo en embarazos clínicamente reconocidos es del 2-4 % y del 15-20 %, respectivamente.
Datos
Datos en animales
No se han realizado estudios de reproducción en animales con KEYTRUDA para evaluar su efecto en la reproducción y el desarrollo fetal. Una evaluación basada en la literatura de los efectos de la vía PD-1 en la reproducción demostró que una función central de la vía PD-1/PD-L1 es preservar el embarazo manteniendo la tolerancia inmunitaria materna al feto. Se ha demostrado en modelos murinos de embarazo que el bloqueo de la señalización de PD-L1 interrumpe la tolerancia al feto y provoca un aumento de la pérdida fetal; por lo tanto, los riesgos potenciales de administrar KEYTRUDA durante el embarazo incluyen un aumento de las tasas de aborto o muerte fetal. Según lo informado en la literatura, no hubo malformaciones relacionadas con el bloqueo de la señalización de PD-1 en la descendencia de estos animales; sin embargo, se produjeron trastornos mediados por el sistema inmunitario en ratones knockout de PD-1. Basándose en su mecanismo de acción, la exposición fetal a pembrolizumab puede aumentar el riesgo de desarrollar trastornos mediados por el sistema inmunitario o de alterar la respuesta inmunitaria normal.
8.2 Lactancia
Resumen de Riesgo
No hay datos sobre la presencia de pembrolizumab en la leche animal o humana ni sobre sus efectos en el niño amamantado o en la producción de leche. Se sabe que la IgG materna está presente en la leche humana. Se desconocen los efectos de la exposición gastrointestinal local y la exposición sistémica limitada en el niño amamantado a KEYTRUDA. Debido al potencial de reacciones adversas graves en los niños amamantados, se recomienda a las mujeres que no amamanten durante el tratamiento con KEYTRUDA y durante 4 meses después de la última dosis.
8.3 Mujeres y hombres en edad fértil
Prueba de embarazo
Verifique el estado del embarazo en mujeres en edad fértil antes de iniciar el tratamiento con KEYTRUDA [ver Uso en poblaciones específicas (8.1)].
Anticoncepción
KEYTRUDA puede causar daño fetal cuando se administra a una mujer embarazada [ver Advertencias y precauciones (5.5), Uso en poblaciones específicas (8.1)]. Aconseje a las mujeres en edad fértil que utilicen un método anticonceptivo eficaz durante el tratamiento con KEYTRUDA y durante 4 meses después de la última dosis.
8.4 Uso pediátrico
La seguridad y eficacia de KEYTRUDA como agente único se han establecido en pacientes pediátricos con melanoma, linfoma de Hodgkin clásico (cHL), linfoma de células B grandes del manto (PMBCL), carcinoma de células de Merkel (MCC), cáncer con alta inestabilidad de microsatélites (MSI-H) o deficiencia de reparación de errores de apareamiento (dMMR), y cáncer con alta carga mutacional tumoral (TMB-H). El uso de KEYTRUDA en pacientes pediátricos para estas indicaciones está respaldado por evidencia de estudios adecuados y bien controlados en adultos con datos adicionales de farmacocinética y seguridad en pacientes pediátricos [ver Reacciones adversas (6.1), Farmacología clínica (12.3), Estudios clínicos (14.1, 14.5, 14.6, 14.8, 14.15, 14.18)].
En KEYNOTE-051, 173 pacientes pediátricos (65 pacientes pediátricos de 6 meses a menos de 12 años y 108 pacientes pediátricos de 12 a 17 años) con melanoma avanzado, linfoma o tumores sólidos positivos para PD-L1 o MSI-H recibieron KEYTRUDA 2 mg/kg cada 3 semanas. La duración media de la exposición fue de 2,1 meses (rango: 1 día a 25 meses). Las reacciones adversas que ocurrieron a una tasa ≥10 % mayor en pacientes pediátricos en comparación con los adultos incluyeron pirexia (33 %), vómitos (29 %), cefalea (25 %), dolor abdominal (23 %), disminución del recuento de linfocitos (13 %) y disminución del recuento de leucocitos (11 %). Las anormalidades de laboratorio que ocurrieron a una tasa ≥10 % mayor en pacientes pediátricos en comparación con los adultos fueron leucopenia (30 %), neutropenia (28 %), trombocitopenia (22 %) y anemia de grado 3 (17 %).
La seguridad y eficacia de KEYTRUDA en pacientes pediátricos no se han establecido en las otras indicaciones aprobadas [ver Indicaciones y uso (1)].
8.5 Uso en geriatría
De 3781 pacientes con melanoma, cáncer de pulmón no microcítico (CPNM), cáncer de cabeza y cuello (CCHC) o carcinoma urotelial que fueron tratados con KEYTRUDA en estudios clínicos, el 48 % tenía 65 años o más y el 17 % tenía 75 años o más. No se observaron diferencias generales en la seguridad o la eficacia entre los pacientes de edad avanzada y los pacientes más jóvenes.
De 389 pacientes adultos con LNH de células grandes que recibieron KEYTRUDA en estudios clínicos, 46 (12%) tenían 65 años o más. Los pacientes de 65 años o más tuvieron una mayor incidencia de reacciones adversas graves (50%) que los pacientes menores de 65 años (24%). Los estudios clínicos de KEYTRUDA en LNH no incluyeron un número suficiente de pacientes de 65 años o más para determinar si la eficacia difiere de la de los pacientes más jóvenes.
De 506 pacientes adultos con CPNM de estadio IB (T2a ≥4 cm), II o IIIA después de la resección completa y quimioterapia a base de platino que recibieron KEYTRUDA en KEYNOTE-091, 242 (48%) tenían 65 años o más. No se observaron diferencias generales en la seguridad o la eficacia entre los pacientes de edad avanzada y los pacientes más jóvenes.
De 596 pacientes adultos con CNTB que recibieron KEYTRUDA en combinación con paclitaxel, paclitaxel unido a proteínas o gemcitabina y carboplatino en KEYNOTE-355, 137 (23%) tenían 65 años o más. No se observaron diferencias generales en la seguridad o la eficacia entre los pacientes de edad avanzada y los pacientes más jóvenes.
De 406 pacientes adultos con carcinoma endometrial que recibieron KEYTRUDA en combinación con lenvatinib en KEYNOTE-775, 201 (50%) tenían 65 años o más. No se observaron diferencias generales en la seguridad o la eficacia entre los pacientes de edad avanzada y los pacientes más jóvenes.
De los 564 pacientes con cáncer urotelial localmente avanzado o metastásico tratados con KEYTRUDA en combinación con enfortumab vedotina, el 44% (n=247) tenían entre 65 y 74 años y el 26% (n=144) tenían 75 años o más. No se observaron diferencias generales en la seguridad o la eficacia entre los pacientes de 65 años o más y los pacientes más jóvenes. Los pacientes de 75 años o más tratados con KEYTRUDA en combinación con enfortumab vedotina experimentaron una mayor incidencia de reacciones adversas fatales que los pacientes más jóvenes. La incidencia de reacciones adversas fatales fue del 4% en pacientes menores de 75 años y del 7% en pacientes de 75 años o más.
De los 432 pacientes aleatorizados a KEYTRUDA en combinación con axitinib en el ensayo KEYNOTE-426, el 40% tenía 65 años o más. No se informó ninguna diferencia general en la seguridad o la eficacia entre los pacientes de ≥65 años de edad y los más jóvenes.
De 292 pacientes adultos con cáncer de cuello uterino en estadio III-IVA según la FIGO 2014 que recibieron KEYTRUDA en combinación con CRT en KEYNOTE-A18, 42 (14%) tenían 65 años o más. No se observaron diferencias generales en la seguridad o la eficacia entre los pacientes de edad avanzada y los pacientes más jóvenes.
11 DESCRIPCIÓN
Pembrolizumab es un anticuerpo bloqueante del receptor 1 de muerte programada (PD-1). Pembrolizumab es un anticuerpo monoclonal IgG4 kappa humanizado con un peso molecular aproximado de 149 kDa. Pembrolizumab se produce en células de ovario de hámster chino (CHO) recombinantes.
KEYTRUDA (pembrolizumab) inyección es una solución estéril, sin conservantes, de transparente a ligeramente opalescente, de incolora a ligeramente amarilla para uso intravenoso. Cada vial contiene 100 mg de pembrolizumab en 4 mL de solución. Cada 1 mL de solución contiene 25 mg de pembrolizumab y está formulado en: L-histidina (1.55 mg), polisorbato 80 (0.2 mg), sacarosa (70 mg) y agua para inyección, USP.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
La unión de los ligandos de PD-1, PD-L1 y PD-L2, al receptor PD-1 que se encuentra en los linfocitos T, inhibe la proliferación de linfocitos T y la producción de citocinas. La regulación al alza de los ligandos PD-1 se produce en algunos tumores y la señalización a través de esta vía puede contribuir a la inhibición de la vigilancia inmunitaria activa de los linfocitos T en los tumores. Pembrolizumab es un anticuerpo monoclonal que se une al receptor PD-1 y bloquea su interacción con PD-L1 y PD-L2, liberando la inhibición mediada por la vía PD-1 de la respuesta inmunitaria, incluida la respuesta inmunitaria antitumoral. En modelos de tumores de ratón singenéticos, el bloqueo de la actividad de PD-1 provocó una disminución del crecimiento tumoral.
En modelos de tumores de ratón singenéticos, el tratamiento combinado con un anticuerpo bloqueante de PD-1 y el inhibidor de la cinasa lenvatinib disminuyó los macrófagos asociados a tumores, aumentó los linfocitos T citotóxicos activados y redujo el crecimiento tumoral en comparación con cualquiera de los tratamientos por separado.
12.2 Farmacodinamia
No existen relaciones exposición-respuesta clínicamente significativas para la eficacia o la seguridad a dosis de pembrolizumab de 200 mg o 2 mg/kg cada 3 semanas, independientemente del tipo de cáncer. No existen relaciones exposición-respuesta clínicamente significativas para la eficacia o la seguridad a dosis de pembrolizumab de 200 mg o 2 mg/kg cada 3 semanas y 400 mg cada 6 semanas en pacientes con tumores sólidos, según los datos observados en pacientes adultos con melanoma. Las relaciones exposición-respuesta para la eficacia o la seguridad a dosis de pembrolizumab de 400 mg cada 6 semanas en pacientes con linfoma de Hodgkin clásico o linfoma de células B grandes mediastínico no se han caracterizado completamente.
12.3 Farmacocinética
La farmacocinética (FC) de pembrolizumab se caracterizó utilizando un análisis de FC poblacional con datos de concentración recogidos de 2993 pacientes con diversos tipos de cáncer que recibieron dosis de pembrolizumab de 1 a 10 mg/kg cada 2 semanas, de 2 a 10 mg/kg cada 3 semanas o 200 mg cada 3 semanas.
Las concentraciones en estado estacionario de pembrolizumab se alcanzaron a las 16 semanas de administración repetida con un régimen de cada 3 semanas y la acumulación sistémica fue de 2,1 veces. La concentración máxima (Cmax), la concentración mínima (Cmin) y el área bajo la curva de concentración plasmática frente al tiempo en estado estacionario (AUCss) de pembrolizumab aumentaron proporcionalmente a la dosis en el intervalo de dosis de 2 a 10 mg/kg cada 3 semanas.
Distribución
El valor medio geométrico (CV%) para el volumen de distribución en estado estacionario es de 6,0 L (20%).
Eliminación
El aclaramiento de pembrolizumab (CV%) es aproximadamente un 23% menor [media geométrica, 195 mL/día (40%)] en estado estacionario que después de la primera dosis [252 mL/día (37%)]; esta disminución del aclaramiento con el tiempo no se considera clínicamente importante. La semivida terminal (t1/2) es de 22 días (32%).
Poblaciones específicas
Los siguientes factores no tuvieron ningún efecto clínicamente importante en el aclaramiento de pembrolizumab: edad (intervalo: 15 a 94 años), sexo, raza (89% blancos), insuficiencia renal (eGFR ≥15 mL/min/1,73 m2), insuficiencia hepática leve a moderada (bilirrubina total ≤3 veces el LSN y cualquier AST), o carga tumoral. Se desconoce el impacto de la insuficiencia hepática grave (bilirrubina total >3 veces el LSN y cualquier AST) en la farmacocinética de pembrolizumab.
12.6 Inmunogenicidad
La incidencia observada de anticuerpos anti-fármaco (AAF) depende en gran medida de la sensibilidad y la especificidad del ensayo. Las diferencias en los métodos de ensayo impiden comparaciones significativas de la incidencia de AAF en los estudios descritos en esta sección con la incidencia de AAF en otros estudios, incluidos los de KEYTRUDA o de otros productos de pembrolizumab.
Los niveles mínimos de pembrolizumab interfieren con los resultados del ensayo electroquimioluminiscente (ECL); por lo tanto, se realizó un análisis de subgrupos en los pacientes tratados con KEYTRUDA con una concentración de pembrolizumab por debajo del nivel de tolerancia del fármaco del ensayo de AAF.
En estudios clínicos en pacientes tratados con KEYTRUDA a una dosis de 2 mg/kg cada 3 semanas, 200 mg cada 3 semanas o 10 mg/kg cada 2 o 3 semanas, 27 (2,1%) de 1289 pacientes evaluables dieron positivo para anticuerpos anti-pembrolizumab emergentes durante el tratamiento, de los cuales 6 (0,5%) pacientes tenían anticuerpos neutralizantes contra pembrolizumab. No se identificaron efectos clínicamente significativos de los AAF en la farmacocinética de pembrolizumab ni en el riesgo de reacciones de infusión. Debido a la baja incidencia de AAF, se desconoce el efecto de estos AAF en la eficacia de KEYTRUDA.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogenicidad, Mutagénesis, Deterioro de la Fertilidad
No se han realizado estudios para evaluar el potencial carcinogénico o genotóxico del pembrolizumab.
No se han realizado estudios de fertilidad con pembrolizumab. En estudios de toxicología de dosis repetidas de 1 y 6 meses en monos, no hubo efectos notables en los órganos reproductivos masculinos y femeninos; sin embargo, la mayoría de los animales en estos estudios no eran sexualmente maduros.
13.2 Toxicología y/o Farmacología Animal
En modelos animales, la inhibición de la señalización PD-1/PD-L1 aumentó la gravedad de algunas infecciones y mejoró las respuestas inflamatorias. Los ratones knockout para PD-1 infectados con Mycobacterium tuberculosis muestran una disminución notable de la supervivencia en comparación con los controles de tipo salvaje, lo que se correlacionó con un aumento de la proliferación bacteriana y las respuestas inflamatorias en estos animales. También se demostró que el bloqueo de PD-1 utilizando un anticuerpo anti-PD-1 de primates exacerbaba la infección por M. tuberculosis en macacos rhesus. Los ratones knockout para PD-1 y PD-L1 y los ratones que recibieron anticuerpos bloqueantes de PD-L1 también mostraron una disminución de la supervivencia después de la infección con el virus de la coriomeningitis linfocítica. La administración de pembrolizumab en chimpancés con infección crónica por hepatitis B natural provocó que dos de cuatro animales presentaran niveles significativamente aumentados de ALT, AST y GGT en suero, que persistieron durante al menos 1 mes después de la interrupción del pembrolizumab.
14 ESTUDIOS CLÍNICOS
14.1 Melanoma
Melanoma sin tratamiento previo con ipilimumab
La eficacia de KEYTRUDA se investigó en KEYNOTE-006 (NCT01866319), un ensayo aleatorizado (1:1:1), abierto, multicéntrico y controlado con activo en 834 pacientes. Los pacientes fueron aleatorizados para recibir KEYTRUDA a una dosis de 10 mg/kg por vía intravenosa cada 2 semanas o 10 mg/kg por vía intravenosa cada 3 semanas hasta la progresión de la enfermedad o toxicidad inaceptable o ipilimumab 3 mg/kg por vía intravenosa cada 3 semanas durante 4 dosis a menos que se interrumpiera antes por progresión de la enfermedad o toxicidad inaceptable. Los pacientes con progresión de la enfermedad podrían recibir dosis adicionales de tratamiento a menos que la progresión de la enfermedad fuera sintomática, rápidamente progresiva, requiriera intervención urgente, ocurriera con una disminución en el estado funcional o se confirmara en 4 a 6 semanas con imágenes repetidas. La aleatorización se estratificó por línea de terapia (0 vs. 1), PS ECOG (0 vs. 1) y expresión de PD-L1 (≥1% de las células tumorales [positivas] vs. <1% de las células tumorales [negativas]) según un ensayo de uso exclusivamente para investigación (IUO). Los criterios de elegibilidad clave fueron melanoma irresecable o metastásico; sin ipilimumab previo; y no más de un tratamiento sistémico previo para el melanoma metastásico. No se requirió que los pacientes con melanoma con mutación BRAF V600E positiva hubieran recibido terapia previa con inhibidores de BRAF. Los pacientes con enfermedad autoinmune; una condición médica que requiriera inmunosupresión; hipersensibilidad grave previa a otros anticuerpos monoclonales; e infección por VIH, hepatitis B o hepatitis C, no fueron elegibles. La evaluación del estado tumoral se realizó a las 12 semanas, luego cada 6 semanas hasta la semana 48, seguida de cada 12 semanas a partir de entonces. Las principales medidas de resultado de eficacia fueron la supervivencia general (SG) y la supervivencia libre de progresión (SLP; según la evaluación de la revisión central independiente ciega [BICR] utilizando los Criterios de Evaluación de la Respuesta en Tumores Sólidos [RECIST v1.1, modificados para seguir un máximo de 10 lesiones diana y un máximo de 5 lesiones diana por órgano]). Las medidas de resultado de eficacia adicionales fueron la tasa de respuesta objetiva (TRO) y la duración de la respuesta (DOR).
Las características de la población del estudio fueron: edad media de 62 años (rango: 18 a 89); 60% hombres; 98% blancos; 66% no tuvieron terapia sistémica previa para la enfermedad metastásica; 69% PS ECOG de 0; 80% tuvieron melanoma PD-L1 positivo, 18% tuvieron melanoma PD-L1 negativo y 2% tuvieron estado PD-L1 desconocido usando el ensayo IUO; 65% tuvieron enfermedad en estadio M1c; 68% con LDH normal; 36% con melanoma con mutación BRAF positiva reportada; y 9% con antecedentes de metástasis cerebrales. Entre los pacientes con melanoma con mutación BRAF positiva, 139 (46%) habían sido tratados previamente con un inhibidor de BRAF.
El estudio demostró mejoras estadísticamente significativas en la SG y la SLP para los pacientes aleatorizados a KEYTRUDA en comparación con ipilimumab. Entre los 91 pacientes aleatorizados a KEYTRUDA 10 mg/kg cada 3 semanas con una respuesta objetiva, las duraciones de la respuesta oscilaron entre 1.4+ y 8.1+ meses. Entre los 94 pacientes aleatorizados a KEYTRUDA 10 mg/kg cada 2 semanas con una respuesta objetiva, las duraciones de la respuesta oscilaron entre 1.4+ y 8.2 meses. Los resultados de eficacia se resumen en la Tabla 57 y la Figura 1.
| Variable principal | KEYTRUDA 10 mg/kg cada 3 semanas n=277 |
KEYTRUDA 10 mg/kg cada 2 semanas n=279 |
Ipilimumab 3 mg/kg cada 3 semanas n=278 |
|---|---|---|---|
|
|||
| SG | |||
| Defunciones (%) | 92 (33%) | 85 (30%) | 112 (40%) |
| Hazard ratio* (IC 95%) | 0.69 (0.52, 0.90) | 0.63 (0.47, 0.83) | — |
| p-Valor (log-rank estratificado) | 0.004 | <0.001 | — |
| SLP según BICR | |||
| Eventos (%) | 157 (57%) | 157 (56%) | 188 (68%) |
| Mediana en meses (IC 95%) | 4.1 (2.9, 6.9) | 5.5 (3.4, 6.9) | 2.8 (2.8, 2.9) |
| Hazard ratio* (IC 95%) | 0.58 (0.47, 0.72) | 0.58 (0.46, 0.72) | — |
| p-Valor (log-rank estratificado) | <0.001 | <0.001 | — |
| Mejor respuesta objetiva según BICR | |||
| ORR (IC 95%) | 33% (27, 39) | 34% (28, 40) | 12% (8, 16) |
| Tasa de respuesta completa | 6% | 5% | 1% |
| Tasa de respuesta parcial | 27% | 29% | 10% |
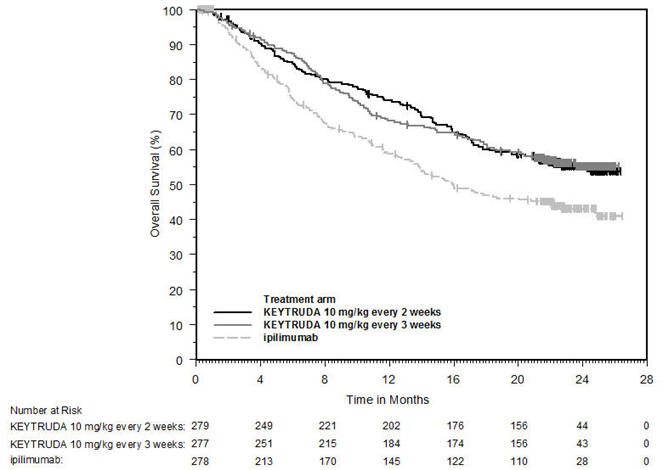
Melanoma refractario a Ipilimumab
La eficacia de KEYTRUDA se investigó en KEYNOTE-002 (NCT01704287), un ensayo multicéntrico, aleatorizado (1:1:1), controlado con activo en 540 pacientes aleatorizados para recibir una de dos dosis de KEYTRUDA de forma ciega o quimioterapia a elección del investigador. Los brazos de tratamiento consistieron en KEYTRUDA 2 mg/kg o 10 mg/kg por vía intravenosa cada 3 semanas o la quimioterapia a elección del investigador de cualquiera de los siguientes regímenes: dacarbazina 1000 mg/m2 por vía intravenosa cada 3 semanas (26%), temozolomida 200 mg/m2 por vía oral una vez al día durante 5 días cada 28 días (25%), carboplatino AUC 6 mg/mL/min por vía intravenosa más paclitaxel 225 mg/m2 por vía intravenosa cada 3 semanas durante cuatro ciclos, luego carboplatino AUC de 5 mg/mL/min más paclitaxel 175 mg/m2 cada 3 semanas (25%), paclitaxel 175 mg/m2 por vía intravenosa cada 3 semanas (16%) o carboplatino AUC 5 o 6 mg/mL/min por vía intravenosa cada 3 semanas (8%). La aleatorización se estratificó por PS ECOG (0 vs. 1), niveles de LDH (normal vs. elevado [≥110% ULN]) y estado de mutación BRAF V600 (tipo salvaje [WT] o V600E). El ensayo incluyó pacientes con melanoma irresecable o metastásico con progresión de la enfermedad; refractario a dos o más dosis de ipilimumab (3 mg/kg o superior) y, si es positivo para la mutación BRAF V600, un inhibidor de BRAF o MEK; y progresión de la enfermedad dentro de las 24 semanas posteriores a la última dosis de ipilimumab. El ensayo excluyó a pacientes con melanoma uveal y metástasis cerebral activa. Los pacientes recibieron KEYTRUDA hasta que se produjo una toxicidad inaceptable; progresión de la enfermedad que fue sintomática, rápidamente progresiva, requirió intervención urgente, ocurrió con una disminución en el estado funcional o se confirmó en 4 a 6 semanas con imágenes repetidas; retiro del consentimiento; o decisión del médico de suspender el tratamiento para el paciente. La evaluación del estado tumoral se realizó a las 12 semanas después de la aleatorización, luego cada 6 semanas hasta la semana 48, seguida de cada 12 semanas a partir de entonces. A los pacientes en quimioterapia que experimentaron progresión de la enfermedad se les ofreció KEYTRUDA. Los principales resultados de eficacia fueron la PFS evaluada por BICR según RECIST v1.1, modificada para seguir un máximo de 10 lesiones diana y un máximo de 5 lesiones diana por órgano, y la OS. Las medidas adicionales de resultado de eficacia fueron la ORR confirmada evaluada por BICR según RECIST v1.1, modificada para seguir un máximo de 10 lesiones diana y un máximo de 5 lesiones diana por órgano, y la DoR.
Las características de la población del estudio fueron: edad media de 62 años (rango: 15 a 89), 43% de edad 65 o más; 61% hombres; 98% blancos; y 55% PS ECOG de 0 y 45% PS ECOG de 1. El veintitrés por ciento de los pacientes fueron positivos para la mutación BRAF V600, el 40% tuvo LDH elevada al inicio, el 82% tuvo enfermedad M1c y el 73% tuvo dos o más terapias previas para enfermedad avanzada o metastásica.
El estudio demostró una mejora estadísticamente significativa en la PFS para los pacientes aleatorizados a KEYTRUDA en comparación con el brazo de control. No hubo una diferencia estadísticamente significativa entre KEYTRUDA 2 mg/kg y la quimioterapia o entre KEYTRUDA 10 mg/kg y la quimioterapia en el análisis de OS en el que el 55% de los pacientes que habían sido aleatorizados para recibir quimioterapia habían pasado a recibir KEYTRUDA. Entre los 38 pacientes aleatorizados a KEYTRUDA 2 mg/kg con una respuesta objetiva, las duraciones de la respuesta oscilaron entre 1.3+ y 11.5+ meses. Entre los 46 pacientes aleatorizados a KEYTRUDA 10 mg/kg con una respuesta objetiva, las duraciones de la respuesta oscilaron entre 1.1+ y 11.1+ meses. Los resultados de eficacia se resumen en la Tabla 58 y la Figura 2.
| Variable principal | KEYTRUDA 2 mg/kg cada 3 semanas |
KEYTRUDA 10 mg/kg cada 3 semanas |
Quimioterapia |
|---|---|---|---|
| n=180 | n=181 | n=179 | |
|
|||
| PFS | |||
| Número de eventos, n (%) | 129 (72%) | 126 (70%) | 155 (87%) |
| Progresión, n (%) | 105 (58%) | 107 (59%) | 134 (75%) |
| Muerte, n (%) | 24 (13%) | 19 (10%) | 21 (12%) |
| Mediana en meses (IC del 95%) | 2.9 (2.8, 3.8) | 2.9 (2.8, 4.7) | 2.7 (2.5, 2.8) |
| p-Valor (log-rank estratificado) | <0.001 | <0.001 | — |
| Hazard ratio* (IC del 95%) | 0.57 (0.45, 0.73) | 0.50 (0.39, 0.64) | — |
| OS† | |||
| Defunciones (%) | 123 (68%) | 117 (65%) | 128 (72%) |
| Hazard ratio* (IC 95%) | 0.86 (0.67, 1.10) | 0.74 (0.57, 0.96) | — |
| Valor p (log-rank estratificado) | 0.117 | 0.011‡ | — |
| Mediana en meses (IC 95%) | 13.4 (11.0, 16.4) | 14.7 (11.3, 19.5) | 11.0 (8.9, 13.8) |
| Tasa de respuesta objetiva | |||
| TRO (IC 95%) | 21% (15, 28) | 25% (19, 32) | 4% (2, 9) |
| Tasa de respuesta completa | 2% | 3% | 0% |
| Tasa de respuesta parcial | 19% | 23% | 4% |
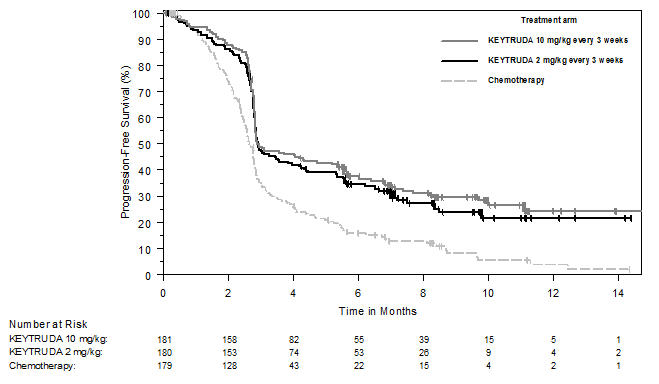 |
Tratamiento adyuvante del melanoma resecado en estadio IIB o IIC
La eficacia de KEYTRUDA se investigó en KEYNOTE-716 (NCT03553836), un ensayo multicéntrico, aleatorizado (1:1), doble ciego, controlado con placebo en pacientes con melanoma en estadio IIB o IIC completamente resecado. Los pacientes fueron aleatorizados a KEYTRUDA 200 mg o la dosis pediátrica (≥12 años) de KEYTRUDA 2 mg/kg por vía intravenosa (hasta un máximo de 200 mg) cada tres semanas o placebo durante un máximo de un año hasta la recurrencia de la enfermedad o toxicidad inaceptable. La aleatorización se estratificó por el estadio T de la 8ª edición de AJCC (>2.0-4.0 mm con ulceración vs. >4.0 mm sin ulceración vs. >4.0 mm con ulceración). Los pacientes no deben haber recibido tratamiento previo para el melanoma más allá de la resección quirúrgica completa de su melanoma antes de entrar en el estudio. La principal medida de resultado de eficacia fue la supervivencia sin recurrencia (SSR) evaluada por el investigador (definida como el tiempo transcurrido entre la fecha de aleatorización y la fecha de la primera recurrencia [ganglios linfáticos locales, en tránsito o regionales o recurrencia a distancia] o la muerte, lo que ocurra primero). Los nuevos melanomas primarios se excluyeron de la definición de SSR. La supervivencia sin metástasis a distancia (SMD), definida como la propagación del tumor a órganos distantes o ganglios linfáticos distantes, fue una medida de resultado de eficacia adicional. Los pacientes se sometieron a pruebas de imagen cada seis meses durante un año a partir de la aleatorización, cada 6 meses de los años 2 a 4, y luego una vez en el año 5 a partir de la aleatorización o hasta la recurrencia, lo que ocurra primero.
Las características de la población del estudio fueron: mediana de edad de 61 años (rango: 16 a 87), 39% de edad 65 o superior; 60% hombres; 98% blancos; y 93% ECOG PS de 0 y 7% ECOG PS de 1. Sesenta y cuatro por ciento tenían estadio IIB y 35% tenían estadio IIC.
El ensayo demostró una mejora estadísticamente significativa en la SSR y la SMD para los pacientes aleatorizados al brazo de KEYTRUDA en comparación con el placebo. Los resultados de eficacia se resumen en la Tabla 59 y la Figura 3.
| Variable principal | KEYTRUDA 200 mg cada 3 semanas n=487 |
Placebo n=489 |
|---|---|---|
| NR = no alcanzado | ||
|
||
| SSR | ||
| Número (%) de pacientes con evento | 54 (11%) | 82 (17%) |
| Mediana en meses (IC del 95%) | NR (22.6, NR) | NR (NR, NR) |
| Razón de riesgo*,† (IC del 95%) | 0.65 (0.46, 0.92) | |
| Valor p† | 0.0132‡ | |
| SMD | ||
| Número (%) de pacientes con evento | 63 (13%) | 95 (19%) |
| Mediana en meses (IC del 95%) | NR (NR, NR) | NR (NR, NR) |
| Razón de riesgo*,† (IC del 95%) | 0.64 (0.47, 0.88) | |
| Valor p† | 0.0058§ | |
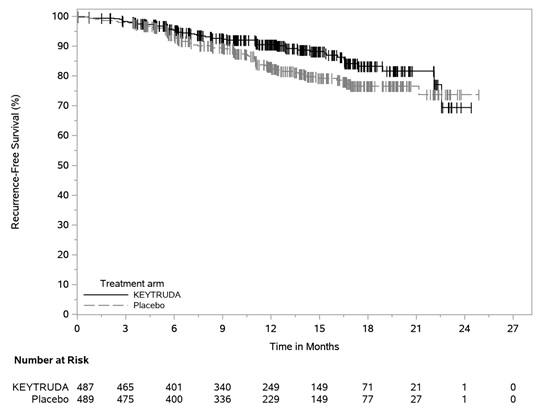 |
Tratamiento adyuvante del melanoma resecado en estadio III
La eficacia de KEYTRUDA se investigó en KEYNOTE-054 (NCT02362594), un ensayo multicéntrico, aleatorizado (1:1), doble ciego, controlado con placebo en pacientes con melanoma completamente resecado en estadio IIIA (>1 mm de metástasis en ganglios linfáticos), IIIB o IIIC. Los pacientes fueron aleatorizados a KEYTRUDA 200 mg por vía intravenosa cada tres semanas o placebo durante un máximo de un año hasta la recurrencia de la enfermedad o la toxicidad inaceptable. La aleatorización se estratificó por estadio de la 7.ª edición del American Joint Committee on Cancer (AJCC) (IIIA frente a IIIB frente a IIIC 1-3 ganglios linfáticos positivos frente a IIIC ≥4 ganglios linfáticos positivos) y región geográfica (América del Norte, países europeos, Australia y otros países según se designó). Los pacientes debieron someterse a disección de ganglios linfáticos y, si estaba indicado, a radioterapia dentro de las 13 semanas anteriores al inicio del tratamiento. La principal medida de resultado de eficacia fue la supervivencia sin recurrencia (SSR) evaluada por el investigador en toda la población y en la población con tumores PD-L1 positivos, donde la SSR se definió como el tiempo transcurrido entre la fecha de aleatorización y la fecha de la primera recurrencia (metástasis local, regional o a distancia) o la muerte, lo que ocurra primero. Los nuevos melanomas primarios se excluyeron de la definición de SSR. La SMFS en toda la población y en la población con tumores PD-L1 positivos fueron medidas adicionales de resultado de eficacia. La SMFS se definió como una diseminación del tumor a órganos distantes o ganglios linfáticos distantes. Los pacientes se sometieron a pruebas de imagen cada 12 semanas después de la primera dosis de KEYTRUDA durante los dos primeros años, luego cada 6 meses del año 3 al 5, y luego anualmente.
Las características de la población del estudio fueron: mediana de edad de 54 años (rango: 19 a 88), 25 % de edad 65 o más; 62 % hombres; y 94 % ECOG PS de 0 y 6 % ECOG PS de 1. Dieciséis por ciento tenía estadio IIIA, 46 % tenía estadio IIIB, 18 % tenía estadio IIIC (1-3 ganglios linfáticos positivos) y 20 % tenía estadio IIIC (≥4 ganglios linfáticos positivos); 50 % eran positivos para la mutación BRAF V600 y 44 % eran de tipo salvaje BRAF; y 84 % tenían melanoma PD-L1 positivo con TPS ≥1 % según un ensayo IUO.
El ensayo demostró una mejora estadísticamente significativa en la SSR y la SMFS para los pacientes aleatorizados al brazo KEYTRUDA en comparación con el placebo. Los resultados de eficacia se resumen en la Tabla 60 y la Figura 4.
| Variable principal | KEYTRUDA 200 mg cada 3 semanas n=514 |
Placebo n=505 |
|---|---|---|
| NR = no alcanzado | ||
|
||
| SSR | ||
| Número (%) de pacientes con evento | 135 (26%) | 216 (43%) |
| Mediana en meses (IC del 95%) | NR | 20,4 (16,2; NR) |
| Razón de riesgos*,† (IC del 95%) | 0,57 (0,46; 0,70) | |
| Valor p† (log-rank) | <0,001‡ | |
| SMFS | ||
| Número (%) de pacientes con evento | 173 (34%) | 245 (49%) |
| Mediana en meses (IC del 95%) | NR (49,6; NR) | 40,0 (27,7; NR) |
| Razón de riesgos*,† (IC del 95%) | 0,60 (0,49; 0,73) | |
| Valor p† (log-rank) | <0,0001§ | |
Para los pacientes con tumores PD-L1 positivos, la RR de SSR fue de 0,54 (IC del 95%: 0,42; 0,69); p<0,0001. Para los pacientes con tumores PD-L1 positivos, la RR de SMFS fue de 0,61 (IC del 95%: 0,49; 0,76); p<0,0001. El beneficio de la SSR y la SMFS para KEYTRUDA en comparación con el placebo se observó independientemente de la expresión de PD-L1 tumoral.
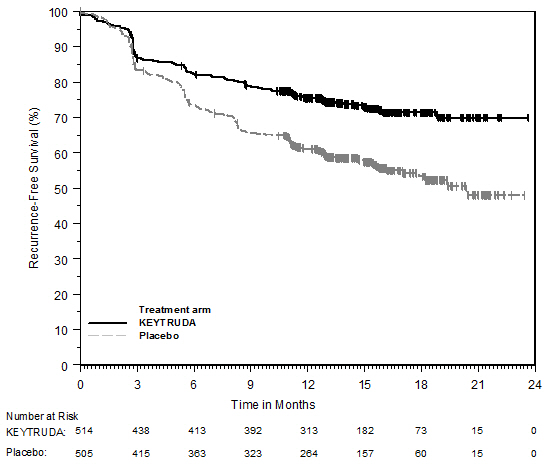 |
14.2 Cáncer de pulmón de células no pequeñas
Tratamiento de primera línea del cáncer de pulmón de células no pequeñas (CPNC) no escamoso metastásico con quimioterapia a base de pemetrexed y platino
La eficacia de KEYTRUDA en combinación con quimioterapia a base de pemetrexed y platino se investigó en KEYNOTE-189 (NCT02578680), un ensayo aleatorizado, multicéntrico, doble ciego y controlado con activo realizado en 616 pacientes con CPNC no escamoso metastásico, independientemente del estado de expresión tumoral de PD-L1, que no habían recibido previamente terapia sistémica para la enfermedad metastásica y en quienes no existían aberraciones genómicas tumorales de EGFR o ALK. Los pacientes con enfermedad autoinmune que requirieron terapia sistémica dentro de los 2 años previos al tratamiento; una condición médica que requirió inmunosupresión; o que habían recibido más de 30 Gy de radiación torácica en las 26 semanas anteriores fueron inelegibles. La aleatorización se estratificó por estado de tabaquismo (nunca fumador frente a exfumador/fumador), elección de platino (cisplatino frente a carboplatino) y estado de PD-L1 tumoral (TPS <1% [negativo] frente a TPS ≥1%). Los pacientes fueron aleatorizados (2:1) a uno de los siguientes brazos de tratamiento:
- KEYTRUDA 200 mg, pemetrexed 500 mg/m2, y la elección del investigador de cisplatino 75 mg/m2 o carboplatino AUC 5 mg/mL/min por vía intravenosa el día 1 de cada ciclo de 21 días durante 4 ciclos, seguidos de KEYTRUDA 200 mg y pemetrexed 500 mg/m2 por vía intravenosa cada 3 semanas. KEYTRUDA se administró antes de la quimioterapia el día 1.
- Placebo, pemetrexed 500 mg/m2, y la elección del investigador de cisplatino 75 mg/m2 o carboplatino AUC 5 mg/mL/min por vía intravenosa el día 1 de cada ciclo de 21 días durante 4 ciclos, seguidos de placebo y pemetrexed 500 mg/m2 por vía intravenosa cada 3 semanas.
El tratamiento con KEYTRUDA continuó hasta la progresión de la enfermedad según la definición de RECIST v1.1 (modificada para seguir un máximo de 10 lesiones diana y un máximo de 5 lesiones diana por órgano) determinada por el investigador, toxicidad inaceptable o un máximo de 24 meses. Se permitió la administración de KEYTRUDA más allá de la progresión de la enfermedad según la definición de RECIST si el paciente era clínicamente estable y el investigador consideraba que estaba obteniendo un beneficio clínico. A los pacientes aleatorizados a placebo y quimioterapia se les ofreció KEYTRUDA como agente único en el momento de la progresión de la enfermedad. La evaluación del estado tumoral se realizó en la semana 6, la semana 12 y luego cada 9 semanas a partir de entonces. Las principales medidas de resultado de eficacia fueron la supervivencia general (SG) y la supervivencia libre de progresión (SLP) evaluadas por el comité de revisión independiente de datos (BICR) según RECIST v1.1, modificada para seguir un máximo de 10 lesiones diana y un máximo de 5 lesiones diana por órgano. Las medidas de resultado de eficacia adicionales fueron la tasa de respuesta objetiva (TRO) y la duración de la respuesta (DR), evaluadas por el BICR según RECIST v1.1, modificada para seguir un máximo de 10 lesiones diana y un máximo de 5 lesiones diana por órgano.
Las características de la población del estudio fueron: edad media de 64 años (rango: 34 a 84), 49% de edad igual o superior a 65 años; 59% hombres; 94% blancos y 3% asiáticos; 56% con estado de rendimiento ECOG de 1; y 18% con antecedentes de metástasis cerebrales. El 31% tenía una expresión tumoral de PD-L1 TPS <1% [negativa]. El 72% recibió carboplatino y el 12% nunca fueron fumadores. Un total de 85 pacientes en el brazo de placebo y quimioterapia recibieron un anticuerpo monoclonal anti-PD-1/PD-L1 en el momento de la progresión de la enfermedad.
El ensayo demostró una mejora estadísticamente significativa en la SG y la SLP para los pacientes aleatorizados a KEYTRUDA en combinación con quimioterapia a base de pemetrexed y platino en comparación con placebo, pemetrexed y quimioterapia a base de platino. La Tabla 61 y la Figura 5 resumen los resultados de eficacia de KEYNOTE-189.
| Variable principal | KEYTRUDA 200 mg cada 3 semanas Pemetrexed Quimioterapia con platino n=410 |
Placebo Pemetrexed Quimioterapia con platino n=206 |
|---|---|---|
| NR = no alcanzado | ||
|
||
| SG | ||
| Número (%) de pacientes con evento | 127 (31%) | 108 (52%) |
| Mediana en meses (IC del 95%) | NR (NR, NR) |
11.3 (8.7, 15.1) |
| Razón de riesgo* (IC del 95%) | 0.49 (0.38, 0.64) | |
| Valor p† | <0.0001 | |
| SLP | ||
| Número de pacientes con evento (%) | 245 (60%) | 166 (81%) |
| Mediana en meses (IC del 95%) | 8.8 (7.6, 9.2) | 4.9 (4.7, 5.5) |
| Hazard ratio* (IC 95%) | 0.52 (0.43, 0.64) | |
| Valor de p† | <0.0001 | |
| Tasa de respuesta objetiva | ||
| ORR‡ (IC 95%) | 48% (43, 53) | 19% (14, 25) |
| Respuesta completa | 0.5% | 0.5% |
| Respuesta parcial | 47% | 18% |
| Valor de p§ | <0.0001 | |
| Duración de la respuesta | ||
| Mediana en meses (rango) | 11.2 (1.1+, 18.0+) | 7.8 (2.1+, 16.4+) |
En el análisis de supervivencia global (OS) final especificado en el protocolo, la mediana en el brazo de KEYTRUDA en combinación con pemetrexed y quimioterapia con platino fue de 22,0 meses (IC 95%: 19,5, 24,5) en comparación con 10,6 meses (IC 95%: 8,7, 13,6) en el brazo de placebo con pemetrexed y quimioterapia con platino, con una HR de 0,56 (IC 95%: 0,46, 0,69).
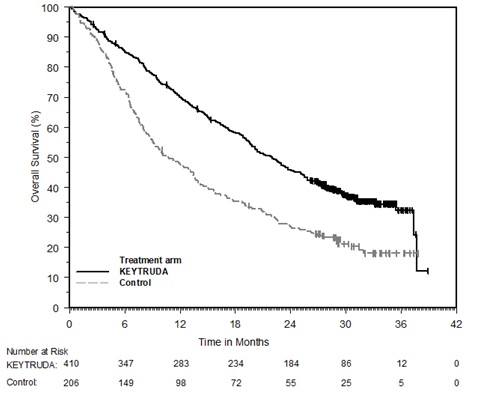
Tratamiento de primera línea del NSCC epidermoide metastásico con carboplatino y quimioterapia con paclitaxel o paclitaxel unido a proteínas
La eficacia de KEYTRUDA en combinación con carboplatino y la elección del investigador de paclitaxel o paclitaxel unido a proteínas se investigó en KEYNOTE-407 (NCT02775435), un ensayo aleatorizado, multicéntrico, doble ciego y controlado con placebo realizado en 559 pacientes con NSCC epidermoide metastásico, independientemente del estado de expresión tumoral de PD-L1, que no habían recibido previamente terapia sistémica para la enfermedad metastásica. Los pacientes con enfermedad autoinmune que requirieron terapia sistémica dentro de los 2 años del tratamiento; una condición médica que requirió inmunosupresión; o que habían recibido más de 30 Gy de radiación torácica en las 26 semanas anteriores fueron inelegibles. La aleatorización se estratificó por el estado de PD-L1 tumoral (TPS <1% [negativo] vs. TPS ≥1%), la elección de paclitaxel o paclitaxel unido a proteínas y la región geográfica (Asia oriental vs. no Asia oriental). Los pacientes fueron aleatorizados (1:1) a uno de los siguientes brazos de tratamiento; todos los medicamentos del estudio se administraron mediante infusión intravenosa:
- KEYTRUDA 200 mg y carboplatino AUC 6 mg/mL/min el día 1 de cada ciclo de 21 días durante 4 ciclos, y paclitaxel 200 mg/m2 el día 1 de cada ciclo de 21 días durante 4 ciclos o paclitaxel unido a proteínas 100 mg/m2 los días 1, 8 y 15 de cada ciclo de 21 días durante 4 ciclos, seguido de KEYTRUDA 200 mg cada 3 semanas. KEYTRUDA se administró antes de la quimioterapia el día 1.
- Placebo y carboplatino AUC 6 mg/mL/min el día 1 de cada ciclo de 21 días durante 4 ciclos y paclitaxel 200 mg/m2 el día 1 de cada ciclo de 21 días durante 4 ciclos o paclitaxel unido a proteínas 100 mg/m2 los días 1, 8 y 15 de cada ciclo de 21 días durante 4 ciclos, seguido de placebo cada 3 semanas.
El tratamiento con KEYTRUDA y quimioterapia o placebo y quimioterapia continuó hasta la progresión de la enfermedad definida por RECIST v1.1 (modificada para seguir un máximo de 10 lesiones diana y un máximo de 5 lesiones diana por órgano), según lo determinado por BICR, toxicidad inaceptable o un máximo de 24 meses. Se permitió la administración de KEYTRUDA más allá de la progresión de la enfermedad definida por RECIST si el paciente era clínicamente estable y obtenía un beneficio clínico según lo determinado por el investigador. A los pacientes aleatorizados al brazo de placebo y quimioterapia se les ofreció KEYTRUDA como agente único en el momento de la progresión de la enfermedad. La evaluación del estado tumoral se realizó cada 6 semanas hasta la semana 18, cada 9 semanas hasta la semana 45 y cada 12 semanas a partir de entonces. Las principales medidas de resultado de eficacia fueron la PFS y la ORR, según lo evaluado por BICR utilizando RECIST v1.1, modificada para seguir un máximo de 10 lesiones diana y un máximo de 5 lesiones diana por órgano, y la OS. Una medida de resultado de eficacia adicional fue la DoR, según lo evaluado por BICR de acuerdo con RECIST v1.1, modificada para seguir un máximo de 10 lesiones diana y un máximo de 5 lesiones diana por órgano.
Las características de la población del estudio fueron: edad media de 65 años (rango: 29 a 88), 55% de edad 65 o más; 81% hombres; 77% blancos; 71% ECOG PS de 1; y 8% con antecedentes de metástasis cerebrales. El 35% tenía expresión tumoral de PD-L1 TPS <1%; el 19% pertenecía a la región del este de Asia; y el 60% recibió paclitaxel.
El ensayo demostró una mejora estadísticamente significativa en la OS, la PFS y la ORR en los pacientes aleatorizados a KEYTRUDA en combinación con carboplatino y quimioterapia con paclitaxel o paclitaxel unido a proteínas en comparación con los pacientes aleatorizados a placebo con carboplatino y quimioterapia con paclitaxel o paclitaxel unido a proteínas. La Tabla 62 y la Figura 6 resumen los resultados de eficacia de KEYNOTE-407.
| Variable principal | KEYTRUDA 200 mg cada 3 semanas Carboplatino Paclitaxel/Paclitaxel unido a proteínas n=278 |
Placebo Carboplatino Paclitaxel/Paclitaxel unido a proteínas n=281 |
|---|---|---|
| NE = no estimable | ||
| OS | ||
| Número de eventos (%) | 85 (31%) | 120 (43%) |
| Mediana en meses (IC del 95%) | 15.9 (13.2, NE) | 11.3 (9.5, 14.8) |
| Razón de riesgo* (IC del 95%) | 0.64 (0.49, 0.85) | |
| Valor p† | 0.0017 | |
| PFS | ||
| Número de eventos (%) | 152 (55%) | 197 (70%) |
| Mediana en meses (IC del 95%) | 6.4 (6.2, 8.3) | 4.8 (4.2, 5.7) |
| Hazard ratio* (IC 95%) | 0.56 (0.45, 0.70) | |
| Valor de p† | <0.0001 | |
| n=101 | n=103 | |
| Tasa de respuesta objetiva‡ | ||
| ORR (IC 95%) | 58% (48, 68) | 35% (26, 45) |
| Diferencia (IC 95%) | 23.6% (9.9, 36.4) | |
| Valor de p§ | 0.0008 | |
| Duración de la respuesta‡ | ||
| Duración mediana de la respuesta en meses (rango) | 7.2 (2.4, 12.4+) | 4.9 (2.0, 12.4+) |
En el análisis de supervivencia global (OS) final especificado en el protocolo, la mediana en el brazo de KEYTRUDA en combinación con carboplatino y ya sea paclitaxel o quimioterapia con paclitaxel unido a proteínas fue de 17,1 meses (IC 95%: 14,4, 19,9) en comparación con 11,6 meses (IC 95%: 10,1, 13,7) en el brazo de placebo con carboplatino y ya sea paclitaxel o quimioterapia con paclitaxel unido a proteínas, con una HR de 0,71 (IC 95%: 0,58, 0,88).
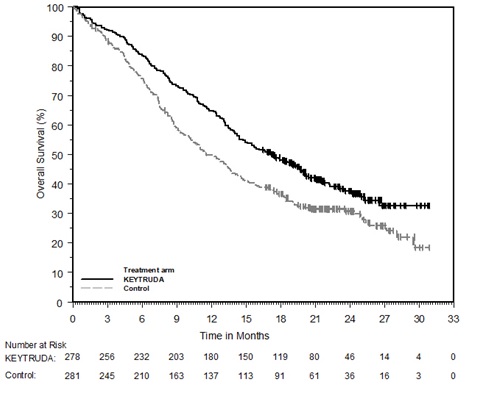
Tratamiento de primera línea del NSCLC metastásico como agente único
KEYNOTE-042
La eficacia de KEYTRUDA se investigó en KEYNOTE-042 (NCT02220894), un ensayo aleatorizado, multicéntrico, abierto y controlado con activo realizado en 1274 pacientes con NSCLC en estadio III que no eran candidatos a resección quirúrgica o quimiorradiación definitiva, o pacientes con NSCLC metastásico. Solo eran elegibles los pacientes cuyos tumores expresaban PD-L1 (TPS ≥1%) mediante un ensayo de inmunohistoquímica utilizando el kit pharmDx PD-L1 IHC 22C3 y que no habían recibido tratamiento sistémico previo para el NSCLC metastásico. Los pacientes con aberraciones genómicas tumorales EGFR o ALK; enfermedad autoinmune que requiriera terapia sistémica dentro de los 2 años previos al tratamiento; una afección médica que requiriera inmunosupresión; o que hubieran recibido más de 30 Gy de radiación en la región torácica en las 26 semanas previas al inicio del estudio no eran elegibles. La aleatorización se estratificó por ECOG PS (0 frente a 1), histología (escamosa frente a no escamosa), región geográfica (Asia oriental frente a no Asia oriental) y expresión de PD-L1 (TPS ≥50% frente a TPS 1 a 49%). Los pacientes se asignaron aleatoriamente (1:1) para recibir KEYTRUDA 200 mg por vía intravenosa cada 3 semanas o la quimioterapia con platino a elección del investigador, según los siguientes regímenes:
- Pemetrexed 500 mg/m2 cada 3 semanas y carboplatino AUC 5 a 6 mg/mL/min cada 3 semanas el día 1 durante un máximo de 6 ciclos, seguidos de pemetrexed 500 mg/m2 opcional cada 3 semanas para pacientes con histologías no escamosas;
- Paclitaxel 200 mg/m2 cada 3 semanas y carboplatino AUC 5 a 6 mg/mL/min cada 3 semanas el día 1 durante un máximo de 6 ciclos, seguidos de pemetrexed 500 mg/m2 opcional cada 3 semanas para pacientes con histologías no escamosas.
El tratamiento con KEYTRUDA continuó hasta la progresión de la enfermedad según la definición de RECIST v1.1 (modificada para seguir un máximo de 10 lesiones diana y un máximo de 5 lesiones diana por órgano), toxicidad inaceptable o un máximo de 24 meses. Se permitió la administración de KEYTRUDA más allá de la progresión de la enfermedad según la definición de RECIST si el paciente era clínicamente estable y obtenía un beneficio clínico según lo determinado por el investigador. El tratamiento con KEYTRUDA podría reiniciarse en el momento de la progresión posterior de la enfermedad y administrarse durante un máximo de 12 meses. La evaluación del estado tumoral se realizó cada 9 semanas. La principal medida de resultado de eficacia fue la OS en el subgrupo de pacientes con NSCLC TPS ≥50%, el subgrupo de pacientes con NSCLC TPS ≥20% y la población general con NSCLC TPS ≥1%. Las medidas de resultado de eficacia adicionales fueron la PFS y la ORR en el subgrupo de pacientes con NSCLC TPS ≥50%, el subgrupo de pacientes con NSCLC TPS ≥20% y la población general con NSCLC TPS ≥1%, según la evaluación del BICR de acuerdo con RECIST v1.1, modificada para seguir un máximo de 10 lesiones diana y un máximo de 5 lesiones diana por órgano.
Las características de la población del estudio fueron: mediana de edad de 63 años (rango: 25 a 90), 45% de edad igual o superior a 65 años; 71% hombres; y 64% blancos, 30% asiáticos y 2% negros. Diecinueve por ciento eran hispanos o latinos. Sesenta y nueve por ciento tenían ECOG PS de 1; 39% con histología escamosa y 61% con histología no escamosa; 87% tenían enfermedad M1 y 13% tenían estadio IIIA (2%) o estadio IIIB (11%) y no eran candidatos a resección quirúrgica o quimiorradiación definitiva según la evaluación del investigador; y 5% con metástasis cerebrales tratadas al inicio del estudio. Cuarenta y siete por ciento de los pacientes tenían NSCLC TPS ≥50% y 53% tenían NSCLC TPS 1 a 49%.
El ensayo demostró una mejora estadísticamente significativa en la OS para los pacientes (PD-L1 TPS ≥50%, TPS ≥20%, TPS ≥1%) asignados aleatoriamente a KEYTRUDA en comparación con la quimioterapia. La Tabla 63 y la Figura 7 resumen los resultados de eficacia en el subgrupo de pacientes con TPS ≥50% y en todos los pacientes aleatorizados con TPS ≥1%.
| TPS ≥1% | TPS ≥50% | |||
|---|---|---|---|---|
| Variable principal | KEYTRUDA 200 mg cada 3 semanas |
Quimioterapia | KEYTRUDA 200 mg cada 3 semanas |
Quimioterapia |
| n=637 | n=637 | n=299 | n=300 | |
|
||||
| OS | ||||
| Número de eventos (%) | 371 (58%) | 438 (69%) | 157 (53%) | 199 (66%) |
| Mediana en meses (IC del 95%) | 16.7 (13.9, 19.7) | 12.1 (11.3, 13.3) | 20.0 (15.4, 24.9) | 12.2 (10.4, 14.2) |
| Hazard ratio* (IC 95%) | 0.81 (0.71, 0.93) | 0.69 (0.56, 0.85) | ||
| Valor de p† | 0.0036 | 0.0006 | ||
| SPT | ||||
| Número de eventos (%) | 507 (80%) | 506 (79%) | 221 (74%) | 233 (78%) |
| Mediana en meses (IC 95%) | 5.4 (4.3, 6.2) | 6.5 (6.3, 7.0) | 6.9 (5.9, 9.0) | 6.4 (6.1, 6.9) |
| Hazard ratio*, ‡ (IC 95%) | 1.07 (0.94, 1.21) |
0.82 (0.68, 0.99) |
||
| Valor de p† | –‡ | NS§ | ||
| Tasa de respuesta objetiva | ||||
| TRO‡ (IC 95%) | 27% (24, 31) | 27% (23, 30) | 39% (33.9, 45.3) | 32% (26.8, 37.6) |
| Tasa de respuesta completa | 0.5% | 0.5% | 0.7% | 0.3% |
| Tasa de respuesta parcial | 27% | 26% | 39% | 32% |
| Duración de la respuesta | ||||
| % con duración ≥12 meses¶ | 47% | 16% | 42% | 17% |
| % con duración ≥18 meses¶ | 26% | 6% | 25% | 5% |
Los resultados de todas las medidas de resultado de eficacia en el subgrupo de pacientes con CPNM PD-L1 ≥20% de CPNM fueron intermedios entre los resultados de aquellos con CPNM PD-L1 ≥1% y aquellos con CPNM PD-L1 ≥50%. En un análisis de subgrupo exploratorio preespecificado para pacientes con CPNM TPS 1-49%, la mediana de la OS fue de 13,4 meses (IC del 95%: 10,7, 18,2) para el grupo de pembrolizumab y de 12,1 meses (IC del 95%: 11,0, 14,0) en el grupo de quimioterapia, con una HR de 0,92 (IC del 95%: 0,77, 1,11).
| Figura 7: Curva de Kaplan-Meier para la supervivencia general en todos los pacientes aleatorizados en KEYNOTE-042 (TPS ≥1%) |
|
|
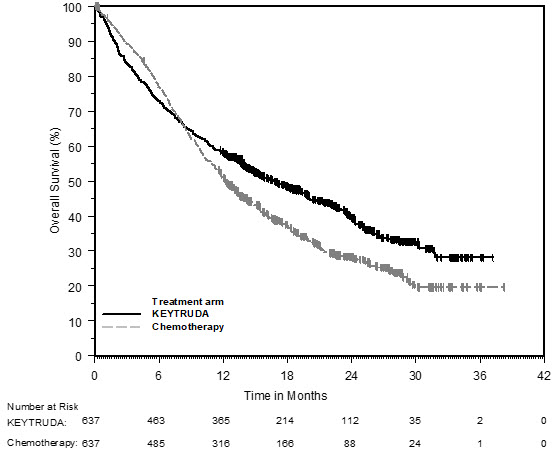
KEYNOTE-024
La eficacia de KEYTRUDA también se investigó en KEYNOTE-024 (NCT02142738), un ensayo aleatorizado, multicéntrico, abierto y controlado con activo en 305 pacientes previamente no tratados con CPNM metastásico. El diseño del estudio fue similar al de KEYNOTE-042, excepto que solo eran elegibles los pacientes cuyos tumores tenían una alta expresión de PD-L1 (TPS del 50% o superior) mediante un ensayo de inmunohistoquímica utilizando el kit pharmDx PD-L1 IHC 22C3. Los pacientes fueron aleatorizados (1:1) para recibir KEYTRUDA 200 mg por vía intravenosa cada 3 semanas o la quimioterapia con platino a elección del investigador, según los siguientes regímenes:
- Pemetrexed 500 mg/m2 cada 3 semanas y carboplatino AUC 5 a 6 mg/mL/min cada 3 semanas el día 1 durante 4 a 6 ciclos, seguidos de pemetrexed 500 mg/m2 opcional cada 3 semanas para pacientes con histologías no escamosas;
- Pemetrexed 500 mg/m2 cada 3 semanas y cisplatino 75 mg/m2 cada 3 semanas el día 1 durante 4 a 6 ciclos, seguidos de pemetrexed 500 mg/m2 opcional cada 3 semanas para pacientes con histologías no escamosas;
- Gemcitabina 1250 mg/m2 los días 1 y 8 y cisplatino 75 mg/m2 cada 3 semanas el día 1 durante 4 a 6 ciclos;
- Gemcitabina 1250 mg/m2 los días 1 y 8 y carboplatino AUC 5 a 6 mg/mL/min cada 3 semanas el día 1 durante 4 a 6 ciclos;
- Paclitaxel 200 mg/m2 cada 3 semanas y carboplatino AUC 5 a 6 mg/mL/min cada 3 semanas el día 1 durante 4 a 6 ciclos, seguidos de mantenimiento opcional con pemetrexed (para histologías no escamosas).
A los pacientes aleatorizados a quimioterapia se les ofreció KEYTRUDA en el momento de la progresión de la enfermedad.
La principal medida de resultado de eficacia fue la PFS, según la evaluación del BICR de acuerdo con RECIST v1.1, modificada para seguir un máximo de 10 lesiones diana y un máximo de 5 lesiones diana por órgano. Las medidas de resultado de eficacia adicionales fueron la OS y la ORR, según la evaluación del BICR de acuerdo con RECIST v1.1, modificada para seguir un máximo de 10 lesiones diana y un máximo de 5 lesiones diana por órgano.
Las características de la población del estudio fueron: mediana de edad de 65 años (rango: 33 a 90), 54% de edad igual o superior a 65 años; 61% hombres; 82% blancos y 15% asiáticos; 65% con ECOG PS de 1; 18% con histología escamosa y 82% con histología no escamosa y 9% con antecedentes de metástasis cerebrales. Un total de 66 pacientes del grupo de quimioterapia recibieron KEYTRUDA en el momento de la progresión de la enfermedad.
El ensayo demostró una mejora estadísticamente significativa tanto en la PFS como en la OS para los pacientes aleatorizados a KEYTRUDA en comparación con la quimioterapia. La Tabla 64 y la Figura 8 resumen los resultados de eficacia de KEYNOTE-024.
| Variable principal | KEYTRUDA 200 mg cada 3 semanas |
Quimioterapia |
|---|---|---|
| n=154 | n=151 | |
| NR = no alcanzado | ||
|
||
| PFS | ||
| Número (%) de pacientes con evento | 73 (47%) | 116 (77%) |
| Mediana en meses (IC del 95%) | 10.3 (6.7, NR) | 6.0 (4.2, 6.2) |
| Razón de riesgo* (IC del 95%) | 0.50 (0.37, 0.68) | |
| Valor p (log-rank estratificado) | <0.001 | |
| OS | ||
| Número (%) de pacientes con evento | 44 (29%) | 64 (42%) |
| Mediana en meses (IC del 95%)† | 30.0 (18.3, NR) |
14.2 (9.8, 19.0) |
| Razón de riesgo* (IC del 95%) | 0.60 (0.41, 0.89) | |
| Valor p (log-rank estratificado) | 0.005‡ | |
| Tasa de respuesta objetiva | ||
| ORR (IC del 95%) | 45% (37, 53) | 28% (21, 36) |
| Tasa de respuesta completa | 4% | 1% |
| Tasa de respuesta parcial | 41% | 27% |
| Valor p (Miettinen-Nurminen) | 0.001 | |
| Duración mediana de la respuesta en meses (rango) | NR (1.9+, 14.5+) |
6.3 (2.1+, 12.6+) |
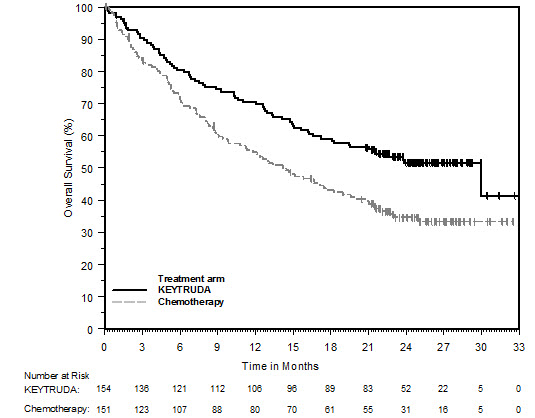
Cáncer de pulmón de células no pequeñas (CPNC) previamente tratado
La eficacia de KEYTRUDA se investigó en KEYNOTE-010 (NCT01905657), un ensayo aleatorizado, multicéntrico, abierto y controlado con activo realizado en 1033 pacientes con CPNC metastásico que había progresado después de la quimioterapia con platino y, si era apropiado, terapia dirigida para aberraciones tumorales genómicas EGFR o ALK. Los pacientes elegibles tenían una expresión de PD-L1 TPS del 1 % o superior mediante un ensayo de inmunohistoquímica utilizando el kit pharmDx PD-L1 IHC 22C3. Los pacientes con enfermedad autoinmune; una condición médica que requería inmunosupresión; o que habían recibido más de 30 Gy de radiación torácica en las 26 semanas anteriores fueron inelegibles. La aleatorización se estratificó por la expresión tumoral de PD-L1 (expresión de PD-L1 TPS ≥50 % frente a expresión de PD-L1 TPS = 1-49 %), PS ECOG (0 frente a 1) y región geográfica (Asia oriental frente a no Asia oriental). Los pacientes se asignaron aleatoriamente (1:1:1) para recibir KEYTRUDA 2 mg/kg por vía intravenosa cada 3 semanas, KEYTRUDA 10 mg/kg por vía intravenosa cada 3 semanas o docetaxel por vía intravenosa 75 mg/m2 cada 3 semanas hasta que se produjera una toxicidad inaceptable o progresión de la enfermedad. Los pacientes aleatorizados a KEYTRUDA pudieron continuar hasta la progresión de la enfermedad que fuera sintomática, rápidamente progresiva, requiriera intervención urgente, ocurriera con una disminución en el estado funcional o confirmación de la progresión en 4 a 6 semanas con imágenes repetidas o hasta por 24 meses sin progresión de la enfermedad. La evaluación del estado tumoral se realizó cada 9 semanas. Las principales medidas de resultado de eficacia fueron la OS y la PFS, según la evaluación de BICR de acuerdo con RECIST v1.1, modificada para seguir un máximo de 10 lesiones diana y un máximo de 5 lesiones diana por órgano, en el subgrupo de pacientes con TPS ≥50 % y en la población general con TPS ≥1 %. Las medidas de resultado de eficacia adicionales fueron la ORR y la DoR en el subgrupo de pacientes con TPS ≥50 % y en la población general con TPS ≥1 %.
Las características de la población del estudio fueron: mediana de edad de 63 años (rango: 20 a 88), 42 % de edad 65 o superior; 61 % hombres; 72 % blancos y 21 % asiáticos; 66 % PS ECOG de 1; 43 % con alta expresión tumoral de PD-L1; 21 % con histología escamosa, 70 % con histología no escamosa y 8 % con histología mixta, otra o desconocida; 91 % enfermedad metastásica (M1); 15 % con antecedentes de metástasis cerebrales; y 8 % y 1 % con aberraciones genómicas EGFR y ALK, respectivamente. Todos los pacientes habían recibido previamente terapia con un régimen de doblete de platino, el 29 % recibió dos o más terapias previas para su enfermedad metastásica.
Las tablas 65 y 66 y la figura 9 resumen los resultados de eficacia en el subgrupo con población TPS ≥50 % y en todos los pacientes, respectivamente.
| Variable principal | KEYTRUDA 2 mg/kg cada 3 semanas n=139 |
KEYTRUDA 10 mg/kg cada 3 semanas n=151 |
Docetaxel 75 mg/m2 cada 3 semanas n=152 |
|---|---|---|---|
| NR = no alcanzado | |||
| OS | |||
| Defunciones (%) | 58 (42%) | 60 (40%) | 86 (57%) |
| Mediana en meses (IC del 95%) | 14.9 (10.4, NR) | 17.3 (11.8, NR) | 8.2 (6.4, 10.7) |
| Razón de riesgos* (IC del 95%) | 0.54 (0.38, 0.77) | 0.50 (0.36, 0.70) | — |
| Valor p (log-rank estratificado) | <0.001 | <0.001 | — |
| PFS | |||
| Eventos (%) | 89 (64%) | 97 (64%) | 118 (78%) |
| Mediana en meses (IC del 95%) | 5.2 (4.0, 6.5) | 5.2 (4.1, 8.1) | 4.1 (3.6, 4.3) |
| Razón de riesgos* (IC del 95%) | 0.58 (0.43, 0.77) | 0.59 (0.45, 0.78) | — |
| Valor p (log-rank estratificado) | <0.001 | <0.001 | — |
| Tasa de respuesta objetiva | |||
| ORR† (IC 95%) | 30% (23, 39) | 29% (22, 37) | 8% (4, 13) |
| Valor p (Miettinen-Nurminen) | <0.001 | <0.001 | — |
| Duración mediana de la respuesta en meses (rango) | NR (0.7+, 16.8+) |
NR (2.1+, 17.8+) |
8.1 (2.1+, 8.8+) |
| Variable principal | KEYTRUDA 2 mg/kg cada 3 semanas n=344 |
KEYTRUDA 10 mg/kg cada 3 semanas n=346 |
Docetaxel 75 mg/m2 cada 3 semanas n=343 |
|---|---|---|---|
| NR = no alcanzado | |||
| OS | |||
| Defunciones (%) | 172 (50%) | 156 (45%) | 193 (56%) |
| Mediana en meses (IC 95%) | 10.4 (9.4, 11.9) | 12.7 (10.0, 17.3) | 8.5 (7.5, 9.8) |
| Hazard ratio* (IC 95%) | 0.71 (0.58, 0.88) | 0.61 (0.49, 0.75) | — |
| Valor p (log-rank estratificado) | <0.001 | <0.001 | — |
| PFS | |||
| Eventos (%) | 266 (77%) | 255 (74%) | 257 (75%) |
| Mediana en meses (IC 95%) | 3.9 (3.1, 4.1) | 4.0 (2.6, 4.3) | 4.0 (3.1, 4.2) |
| Hazard ratio* (IC 95%) | 0.88 (0.73, 1.04) | 0.79 (0.66, 0.94) | — |
| Valor p (log-rank estratificado) | 0.068 | 0.005 | — |
| Tasa de respuesta objetiva | |||
| ORR† (IC 95%) | 18% (14, 23) | 19% (15, 23) | 9% (7, 13) |
| Valor p (Miettinen-Nurminen) | <0.001 | <0.001 | — |
| Duración mediana de la respuesta en meses (rango) | NR (0.7+, 20.1+) |
NR (2.1+, 17.8+) |
6.2 (1.4+, 8.8+) |
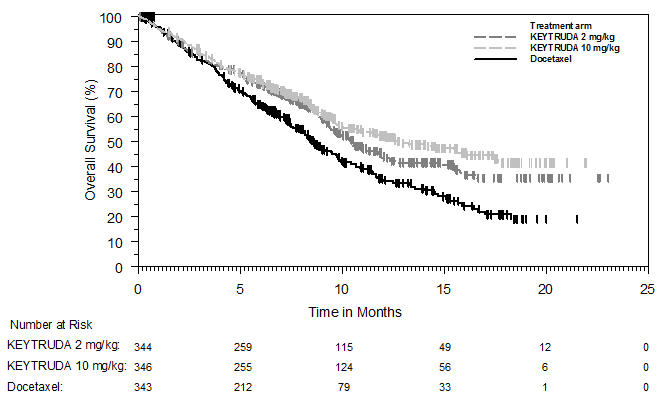 |
Tratamiento neoadyuvante y adyuvante del CPNM resecable
La eficacia de KEYTRUDA en combinación con quimioterapia neoadyuvante seguida de cirugía y tratamiento adyuvante continuado con KEYTRUDA como agente único se investigó en KEYNOTE-671 (NCT03425643), un ensayo multicéntrico, aleatorizado, doble ciego y controlado con placebo realizado en 797 pacientes con CPNM en estadio II, IIIA o IIIB (N2) previamente no tratado y resecable según la 8.ª edición de AJCC. Los pacientes se incluyeron independientemente de la expresión de PD-L1 tumoral. Los pacientes con enfermedad autoinmune activa que requiriera terapia sistémica en los 2 años anteriores al tratamiento, una afección médica que requiriera inmunosupresión o antecedentes de enfermedad pulmonar intersticial o neumonitis que requiriera corticosteroides no fueron elegibles. La aleatorización se estratificó por estadio (II frente a III), expresión de PD-L1 tumoral (TPS ≥50% o <50%), histología (escamosa frente a no escamosa) y región geográfica (Asia oriental frente a no Asia oriental).
Los pacientes se aleatorizaron (1:1) a uno de los siguientes brazos de tratamiento:
- Brazo de tratamiento A: KEYTRUDA 200 mg en el Día 1 en combinación con cisplatino 75 mg/m2 y pemetrexed 500 mg/m2 en el Día 1 o gemcitabina 1000 mg/m2 en los Días 1 y 8 de cada ciclo de 21 días durante un máximo de 4 ciclos. En un plazo de 4 a 12 semanas después de la cirugía, se administraron 200 mg de KEYTRUDA cada 3 semanas durante un máximo de 13 ciclos.
- Brazo de tratamiento B: placebo neoadyuvante en el Día 1 en combinación con cisplatino 75 mg/m2 y pemetrexed 500 mg/m2 en el Día 1 o gemcitabina 1000 mg/m2 en los Días 1 y 8 de cada ciclo de 21 días durante un máximo de 4 ciclos. En un plazo de 4 a 12 semanas después de la cirugía, se administró placebo cada 3 semanas durante un máximo de 13 ciclos.
Todos los medicamentos del estudio se administraron mediante infusión intravenosa. El tratamiento con KEYTRUDA o placebo continuó hasta la finalización del tratamiento (17 ciclos), la progresión de la enfermedad que impidió la cirugía definitiva, la recurrencia de la enfermedad en la fase adyuvante, la progresión de la enfermedad para aquellos que no se sometieron a cirugía o tuvieron una resección incompleta y entraron en la fase adyuvante, o una toxicidad inaceptable. La evaluación del estado tumoral se realizó al inicio del estudio, en la semana 7 y en la semana 13 en la fase neoadyuvante y en las 4 semanas previas al inicio de la fase adyuvante. Tras el inicio de la fase adyuvante, la evaluación del estado tumoral se realizó cada 16 semanas hasta el final del año 3, y luego cada 6 meses a partir de entonces.
El ensayo no fue diseñado para aislar el efecto de KEYTRUDA en cada fase (neoadyuvante o adyuvante) del tratamiento.
Las principales medidas de resultado de eficacia fueron la supervivencia general (SG) y la supervivencia sin eventos (SSE) evaluada por el investigador. Las medidas de resultado de eficacia adicionales fueron la tasa de respuesta patológica completa (RPC) y la tasa de respuesta patológica mayor (RPM) evaluadas mediante una revisión patológica independiente ciega.
Las características de la población del estudio fueron: mediana de edad de 64 años (rango: 26 a 83); 45% de edad igual o superior a 65 años y 7% de edad igual o superior a 75 años; 71% hombres; 61% blancos, 31% asiáticos, 2% negros, 4% raza no declarada; 9% hispanos o latinos; 63% ECOG PS de 0 y 37% ECOG PS de 1. El treinta por ciento tenía enfermedad en estadio II y el 70% tenía enfermedad en estadio III; el 33% tenía TPS ≥50% y el 67% tenía TPS <50%; el 43% tenía tumores con histología escamosa y el 57% tenía tumores con histología no escamosa; el 31% pertenecía a la región del este de Asia.
El ochenta y un por ciento de los pacientes del brazo de KEYTRUDA en combinación con quimioterapia con platino recibieron cirugía definitiva en comparación con el 76% de los pacientes del brazo de placebo en combinación con quimioterapia con platino.
El ensayo demostró mejoras estadísticamente significativas en la SG y la SSE para los pacientes aleatorizados a KEYTRUDA en combinación con quimioterapia con platino seguida de KEYTRUDA como agente único en comparación con los pacientes aleatorizados a placebo en combinación con quimioterapia con platino seguida de placebo solo.
La Tabla 67 y la Figura 10 resumen los resultados de eficacia de KEYNOTE-671.
| Variable principal | KEYTRUDA 200 mg cada 3 semanas con quimioterapia/KEYTRUDA n=397 |
Placebo con quimioterapia/Placebo n=400 |
|---|---|---|
| NR = no alcanzado | ||
|
||
| SG | ||
| Número de pacientes con evento (%) | 110 (28%) | 144 (36%) |
| Mediana en meses* (IC del 95%) | NR (NR, NR) | 52.4 (45.7, NR) |
| Hazard ratio† (IC del 95%) | 0.72 (0.56, 0.93) | |
| Valor p‡,§ | 0.0103 | |
| EFS | ||
| Número de pacientes con evento (%) | 139 (35%) | 205 (51%) |
| Mediana en meses* (IC del 95%) | NR (34.1, NR) | 17.0 (14.3, 22.0) |
| Hazard ratio† (IC del 95%) | 0.58 (0.46, 0.72) | |
| Valor p‡,¶ | <0.0001 | |
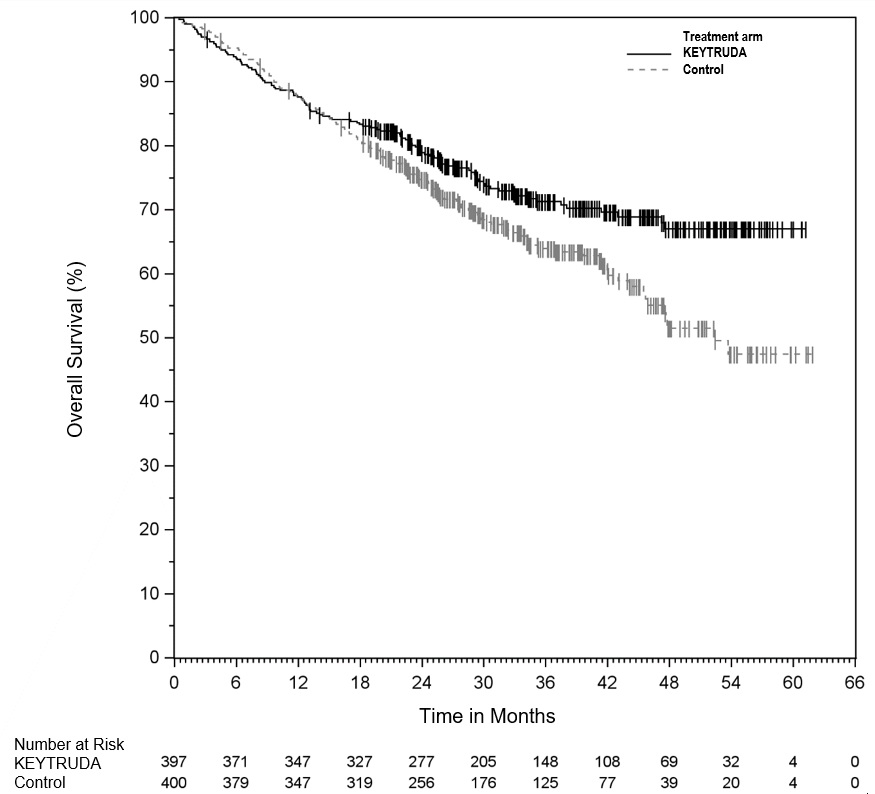 |
El ensayo demostró una diferencia estadísticamente significativa en la tasa de pCR (18,1% vs. 4,0%; p<0,0001) y la tasa de mPR (30,2% vs. 11,0%; p<0,0001).
Tratamiento adyuvante de CPNM resecado
La eficacia de KEYTRUDA se investigó en KEYNOTE-091 (NCT02504372), un ensayo multicéntrico, aleatorizado, triple ciego, controlado con placebo realizado en 1177 pacientes con CPNM en estadio IB (T2a ≥4 cm), II o IIIA completamente resecado según la 7.ª edición de AJCC. Los pacientes no habían recibido radioterapia o quimioterapia neoadyuvantes. La quimioterapia adyuvante de hasta 4 ciclos era opcional. Los pacientes fueron inelegibles si tenían una enfermedad autoinmune activa, estaban tomando agentes inmunosupresores crónicos o tenían antecedentes de enfermedad pulmonar intersticial o neumonitis. La aleatorización se estratificó por estadio (IB vs. II vs. IIIA), recepción de quimioterapia adyuvante (sí vs. no), estado de PD-L1 (TPS <1% [negativo] vs. TPS 1-49% vs. TPS ≥50%) y región geográfica (Europa occidental vs. Europa oriental vs. Asia vs. Resto del mundo). Los pacientes se asignaron aleatoriamente (1:1) para recibir KEYTRUDA 200 mg o placebo por vía intravenosa cada 3 semanas.
El tratamiento continuó hasta la recurrencia de la enfermedad definida por RECIST v1.1 según lo determinado por el investigador, toxicidad inaceptable o hasta un año. Las evaluaciones tumorales se realizaron cada 12 semanas durante el primer año, luego cada 6 meses durante los años 2 a 3, y luego anualmente hasta el año 5. Después del año 5, se realizaron imágenes según el estándar de atención local. La principal medida de resultado de eficacia fue la supervivencia sin enfermedad (DFS) evaluada por el investigador. Una medida de resultado de eficacia adicional fue la SO.
De los 1177 pacientes aleatorizados, 1010 (86%) recibieron quimioterapia basada en platino adyuvante después de la resección. Entre estos 1010 pacientes, la mediana de edad fue de 64 años (rango: 35 a 84), 49% de edad 65 o más; 68% hombres; 77% blancos, 18% asiáticos; 86% fumadores actuales o anteriores; y 39% con ECOG PS de 1. El once por ciento tenía enfermedad en estadio IB, el 57% en estadio II y el 31% en estadio IIIA. El treinta y nueve por ciento tenía PD-L1 TPS <1% [negativo], el 33% tenía TPS 1-49% y el 28% tenía TPS ≥50%. El cincuenta y dos por ciento procedía de Europa occidental, el 20% de Europa oriental, el 17% de Asia y el 11% del resto del mundo.
El ensayo cumplió su objetivo principal, demostrando una mejora estadísticamente significativa en la DFS en la población general para los pacientes aleatorizados al brazo de KEYTRUDA en comparación con los pacientes aleatorizados al brazo de placebo. En un análisis de subgrupo exploratorio de los 167 pacientes (14%) que no recibieron quimioterapia adyuvante, el HR de DFS fue de 1,25 (IC del 95%: 0,76, 2,05). Los resultados de la SO no estaban maduros con solo el 42% de los eventos de SO preespecificados en la población general.
La Tabla 68 y la Figura 11 resumen los resultados de eficacia de KEYNOTE-091 en pacientes que recibieron quimioterapia adyuvante.
| Variable principal | KEYTRUDA 200 mg cada 3 semanas n=506 |
Placebo
n=504 |
|---|---|---|
| NR = no alcanzado | ||
|
||
| DFS | ||
| Número (%) de pacientes con evento | 177 (35%) | 231 (46%) |
| Mediana en meses (IC del 95%) | 58.7 (39.2, NR) |
34.9 (28.6, NR) |
| Hazard ratio* (IC del 95%) | 0.73 (0.60, 0.89) | |
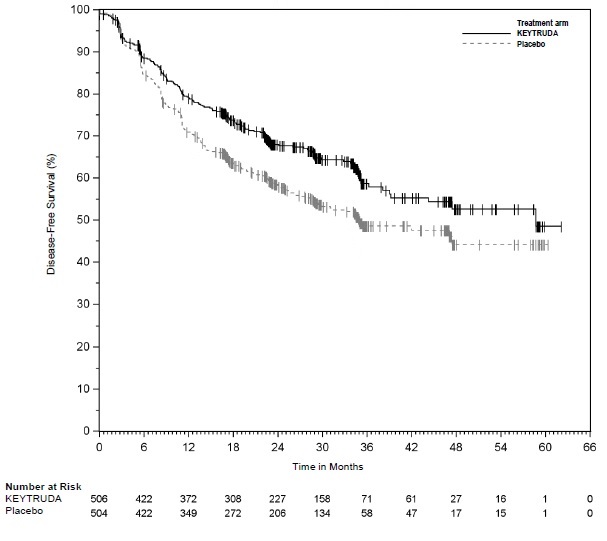 |
14.3 Mesotelioma pleural maligno
Tratamiento de primera línea del mesotelioma pleural maligno (MPM) avanzado o metastásico irresecable con quimioterapia con pemetrexed y platino
La eficacia de KEYTRUDA en combinación con quimioterapia con pemetrexed y platino se investigó en KEYNOTE-483 (NCT02784171), un ensayo multicéntrico, aleatorizado, abierto y controlado activo en el que se inscribieron 440 pacientes con MPM avanzado o metastásico irresecable y sin terapia sistémica previa para la enfermedad avanzada/metastásica. Los pacientes se inscribieron independientemente de la expresión de PD-L1 tumoral. Los pacientes con enfermedad autoinmune que requirieron terapia sistémica dentro de los 3 años del tratamiento o una afección médica que requiriera inmunosupresión no fueron elegibles. La aleatorización se estratificó por subtipo histológico (epitelioide frente a no epitelioide). Los pacientes se aleatorizaron (1:1) a uno de los siguientes brazos de tratamiento; todos los medicamentos del estudio se administraron mediante infusión intravenosa:
- KEYTRUDA 200 mg con pemetrexed 500 mg/m2 y cisplatino 75 mg/m2 o carboplatino AUC 5-6 mg/mL/min el día 1 de cada ciclo de 21 días durante un máximo de 6 ciclos, seguido de KEYTRUDA 200 mg cada 3 semanas. KEYTRUDA se administró antes de la quimioterapia el día 1.
- Pemetrexed 500 mg/m2 y cisplatino 75 mg/m2 o carboplatino AUC 5-6 mg/mL/min el día 1 de cada ciclo de 21 días durante un máximo de 6 ciclos.
El tratamiento con KEYTRUDA continuó hasta la progresión de la enfermedad según lo determinado por el investigador de acuerdo con mRECIST 1.1 modificado para mesotelioma (mRECIST), toxicidad inaceptable o un máximo de 24 meses. La evaluación del estado tumoral se realizó cada 6 semanas durante 18 semanas, seguida de cada 12 semanas a partir de entonces. La principal medida de resultado de eficacia fue la SO. Las medidas de resultado de eficacia adicionales fueron la SLP, la TEO y la DuR, según lo evaluado por BICR de acuerdo con mRECIST.
Las características de la población del estudio fueron: edad media de 70 años (77% de edad 65 o más); 76% hombres; 79% blancos, 21% raza no informada o desconocida; 2% hispanos o latinos; y 53% estado de rendimiento ECOG de 1. El setenta y ocho por ciento tenía histología epitelioide y el 22% tenía histología no epitelioide; el 60% tenía tumores con CPS de PD-L1 ≥1 y el 30% tenía tumores con CPS de PD-L1 <1.
El ensayo demostró una mejora estadísticamente significativa en la SO, la SLP y la TEO en pacientes aleatorizados a KEYTRUDA en combinación con quimioterapia en comparación con los pacientes aleatorizados a quimioterapia sola. La Tabla 69 y la Figura 12 resumen los resultados de eficacia de KEYNOTE-483.
| Variable principal | KEYTRUDA 200 mg cada 3 semanas Quimioterapia con pemetrexed y platino |
Quimioterapia con pemetrexed y platino |
|---|---|---|
| (n=222) | (n=218) | |
|
||
| SO | ||
| Número (%) de pacientes con evento | 167 (75%) | 175 (80%) |
| Mediana en meses (IC del 95%) | 17.3 (14.4, 21.3) | 16.1 (13.1, 18.2) |
| Razón de riesgo* (IC del 95%) | 0.79 (0.64, 0.98) | |
| Valor p† | 0.0162 | |
| SLP | ||
| Número (%) de pacientes con evento | 190 (86%) | 166 (76%) |
| Mediana en meses (IC del 95%) | 7.1 (6.9, 8.1) | 7.1 (6.8, 7.7) |
| Razón de riesgo* (IC del 95%) | 0.80 (0.65, 0.99) | |
| Valor p† | 0.0194 | |
| Tasa de respuesta objetiva | ||
| TEO % (IC del 95%) | 52% (45.5, 59.0) | 29% (23.0, 35.4) |
| Respuestas completas | 1 (0.5%) | 0 (0%) |
| Respuestas parciales | 115 (52%) | 63 (29%) |
| Valor p‡ | <0.00001 | |
| Duración de la respuesta§ | ||
| Mediana en meses (IC del 95%) | 6.9 (5.8, 8.3) | 6.8 (5.5, 8.5) |
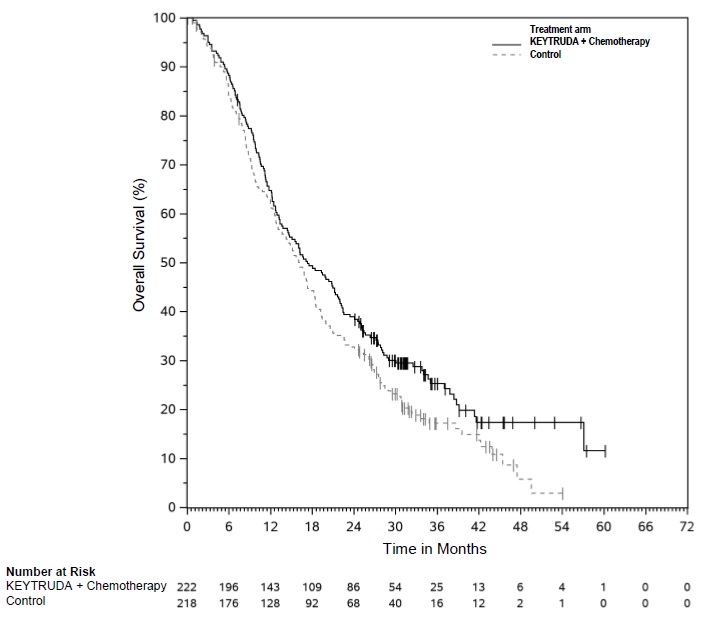 |
En un análisis exploratorio preespecificado basado en la histología, en el subgrupo de pacientes con histología epitelioide (n=345), la razón de riesgo (HR) para la SO fue de 0,89 (IC del 95%: 0,70, 1,13), con una SO mediana de 19,8 meses en KEYTRUDA en combinación con quimioterapia y 18,2 meses en quimioterapia sola. En el subgrupo de pacientes con histología no epitelioide (n=95), la HR para la SO fue de 0,57 (IC del 95%: 0,36, 0,89), con una SO mediana de 12,3 meses en KEYTRUDA en combinación con quimioterapia y 8,2 meses en quimioterapia sola.
14.4 Cáncer de células escamosas de cabeza y cuello
Tratamiento de primera línea del CECC metastásico o irresecable, recurrente
La eficacia de KEYTRUDA se investigó en KEYNOTE-048 (NCT02358031), un ensayo aleatorizado, multicéntrico, abierto y controlado activo realizado en 882 pacientes con CECC metastásico que no habían recibido previamente terapia sistémica para la enfermedad metastásica o con enfermedad recurrente que se consideraba incurable mediante terapias locales. Los pacientes con enfermedad autoinmune activa que requiriera terapia sistémica dentro de los dos años posteriores al tratamiento o una afección médica que requiriera inmunosupresión no fueron elegibles. La aleatorización se estratificó por la expresión de PD-L1 tumoral (TPS ≥50% o <50%) según el kit pharmDx 22C3 de IHC PD-L1, el estado del VPH según la IHC p16 (positiva o negativa) y el ECOG PS (0 vs. 1). Los pacientes se aleatorizaron 1:1:1 a uno de los siguientes brazos de tratamiento:
- KEYTRUDA 200 mg por vía intravenosa cada 3 semanas
- KEYTRUDA 200 mg por vía intravenosa cada 3 semanas, carboplatino AUC 5 mg/mL/min por vía intravenosa cada 3 semanas o cisplatino 100 mg/m2 por vía intravenosa cada 3 semanas, y FU 1000 mg/m2/día como infusión intravenosa continua durante 96 horas cada 3 semanas (máximo de 6 ciclos de platino y FU)
- Cetuximab 400 mg/m2 por vía intravenosa como dosis inicial y luego 250 mg/m2 por vía intravenosa una vez por semana, carboplatino AUC 5 mg/mL/min por vía intravenosa cada 3 semanas o cisplatino 100 mg/m2 por vía intravenosa cada 3 semanas, y FU 1000 mg/m2/día como infusión intravenosa continua durante 96 horas cada 3 semanas (máximo de 6 ciclos de platino y FU)
El tratamiento con KEYTRUDA continuó hasta la progresión de la enfermedad definida por RECIST v1.1 según lo determinado por el investigador, toxicidad inaceptable o un máximo de 24 meses. Se permitió la administración de KEYTRUDA más allá de la progresión de la enfermedad definida por RECIST si el paciente era clínicamente estable y se consideraba que estaba obteniendo un beneficio clínico por parte del investigador. La evaluación del estado tumoral se realizó en la semana 9 y luego cada 6 semanas durante el primer año, seguida de cada 9 semanas hasta los 24 meses. Se realizó una reclasificación retrospectiva del estado de PD-L1 tumoral de los pacientes según CPS utilizando el kit pharmDx 22C3 de IHC PD-L1 utilizando las muestras tumorales utilizadas para la aleatorización.
Las principales medidas de resultado de eficacia fueron la SO y la PFS evaluadas por BICR según RECIST v1.1 (modificada para seguir un máximo de 10 lesiones diana y un máximo de 5 lesiones diana por órgano) probadas secuencialmente en el subgrupo de pacientes con CPS ≥20, el subgrupo de pacientes con CPS ≥1 y la población general.
Las características de la población del estudio fueron: edad media de 61 años (rango: 20 a 94), 36% de edad 65 o mayor; 83% hombres; 73% blancos, 20% asiáticos y 2,4% negros; 61% tenían ECOG PS de 1; y el 79% eran fumadores anteriores/actuales. El 22% de los tumores de los pacientes eran positivos para el VPH, el 23% tenían PD-L1 TPS ≥50% y el 95% tenían enfermedad en estadio IV (estadio IVA 19%, estadio IVB 6% y estadio IVC 70%). El 85% de los tumores de los pacientes tenían una expresión de PD-L1 de CPS ≥1 y el 43% tenían CPS ≥20.
El ensayo demostró una mejora estadísticamente significativa en la SO para los pacientes aleatorizados a KEYTRUDA en combinación con quimioterapia en comparación con aquellos aleatorizados a cetuximab en combinación con quimioterapia en un análisis interino preespecificado en la población general. La Tabla 70 y la Figura 13 resumen los resultados de eficacia de KEYTRUDA en combinación con quimioterapia.
| Variable principal | KEYTRUDA 200 mg cada 3 semanas Platino FU |
Cetuximab Platino FU |
|---|---|---|
| n=281 | n=278 | |
| SO | ||
| Número (%) de pacientes con evento | 197 (70%) | 223 (80%) |
| Mediana en meses (IC del 95%) | 13,0 (10,9, 14,7) | 10,7 (9,3, 11,7) |
| Razón de riesgo† (IC del 95%) | 0,77 (0,63, 0,93) | |
| Valor p‡ | 0,0067 | |
| PFS | ||
| Número de pacientes con evento (%) | 244 (87%) | 253 (91%) |
| Mediana en meses (IC del 95%) | 4.9 (4.7, 6.0) | 5.1 (4.9, 6.0) |
| Hazard ratio† (IC del 95%) | 0.92 (0.77, 1.10) | |
| Valor p‡ | 0.3394 | |
| Tasa de respuesta objetiva | ||
| ORR§ (IC del 95%) | 36% (30.0, 41.5) | 36% (30.7, 42.3) |
| Tasa de respuesta completa | 6% | 3% |
| Tasa de respuesta parcial | 30% | 33% |
| Duración de la respuesta | ||
| Mediana en meses (rango) | 6.7 (1.6+, 30.4+) | 4.3 (1.2+, 27.9+) |
En el análisis final de supervivencia global (SG) preespecificado para la población ITT, el hazard ratio fue de 0.72 (IC del 95%: 0.60, 0.87). Además, KEYNOTE-048 demostró una mejora estadísticamente significativa en la SG para los subgrupos de pacientes con CPS de PD-L1 ≥1 (HR=0.65, IC del 95%: 0.53, 0.80) y CPS ≥20 (HR=0.60, IC del 95%: 0.45, 0.82).
|
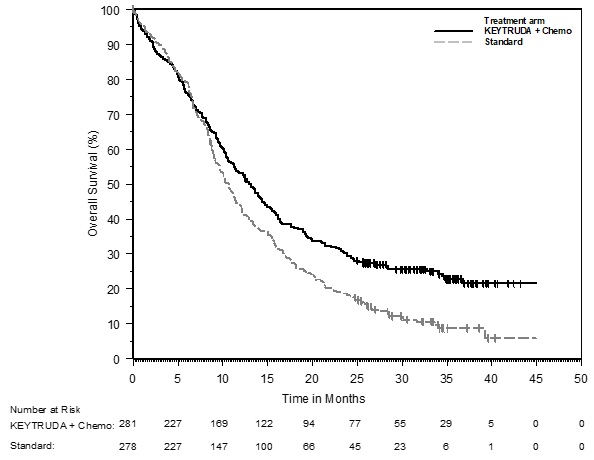 |
El ensayo también demostró una mejora estadísticamente significativa en la SO para el subgrupo de pacientes con CPS de PD-L1 ≥1 asignados al azar a KEYTRUDA como agente único en comparación con aquellos asignados al azar a cetuximab en combinación con quimioterapia en un análisis intermedio preespecificado. En el momento de los análisis intermedios y finales, no hubo una diferencia significativa en la SO entre el brazo de agente único KEYTRUDA y el brazo de control para la población general.
La Tabla 71 resume los resultados de eficacia de KEYTRUDA como agente único en los subgrupos de pacientes con CPH ≥1 CCHC y CPH ≥20 CCHC. La Figura 14 resume los resultados de SO en el subgrupo de pacientes con CPH ≥1 CCHC.
| Variable principal | CPS ≥1 | CPS ≥20 | ||
|---|---|---|---|---|
| KEYTRUDA 200 mg cada 3 semanas |
Cetuximab Platino FU |
KEYTRUDA 200 mg cada 3 semanas |
Cetuximab Platino FU |
|
| n=257 | n=255 | n=133 | n=122 | |
| SO | ||||
| Número de eventos (%) | 177 (69%) | 206 (81%) | 82 (62%) | 95 (78%) |
| Mediana en meses (IC del 95%) | 12.3 (10.8, 14.9) | 10.3 (9.0, 11.5) | 14.9 (11.6, 21.5) | 10.7 (8.8, 12.8) |
| Razón de riesgo† (IC del 95%) | 0.78 (0.64, 0.96) | 0.61 (0.45, 0.83) | ||
| Valor p‡ | 0.0171 | 0.0015 | ||
| SPT | ||||
| Número de eventos (%) | 225 (88%) | 231 (91%) | 113 (85%) | 111 (91%) |
| Mediana en meses (IC del 95%) | 3.2 (2.2, 3.4) | 5.0 (4.8, 5.8) | 3.4 (3.2, 3.8) | 5.0 (4.8, 6.2) |
| Razón de riesgo† (IC del 95%) | 1.15 (0.95, 1.38) | 0.97 (0.74, 1.27) | ||
| Tasa de respuesta objetiva | ||||
| TRO§ (IC del 95%) | 19% (14.5, 24.4) | 35% (29.1, 41.1) | 23% (16.4, 31.4) | 36% (27.6, 45.3) |
| Tasa de respuesta completa | 5% | 3% | 8% | 3% |
| Tasa de respuesta parcial | 14% | 32% | 16% | 33% |
| Duración de la respuesta | ||||
| Mediana en meses (rango) | 20.9 (1.5+, 34.8+) | 4.5 (1.2+, 28.6+) | 20.9 (2.7, 34.8+) | 4.2 (1.2+, 22.3+) |
En el análisis de supervivencia general (OS) final preespecificado que comparó KEYTRUDA como agente único con cetuximab en combinación con quimioterapia, la razón de riesgo para el subgrupo de pacientes con CPS ≥1 fue de 0,74 (IC del 95%: 0,61; 0,90) y la razón de riesgo para el subgrupo de pacientes con CPS ≥20 fue de 0,58 (IC del 95%: 0,44; 0,78).
En un análisis de subgrupos exploratorio para pacientes con CPS 1-19 CCHNC en el momento del análisis de OS final preespecificado, la mediana de OS fue de 10,8 meses (IC del 95%: 9,0; 12,6) para KEYTRUDA como agente único y de 10,1 meses (IC del 95%: 8,7; 12,1) para cetuximab en combinación con quimioterapia, con una HR de 0,86 (IC del 95%: 0,66; 1,12).
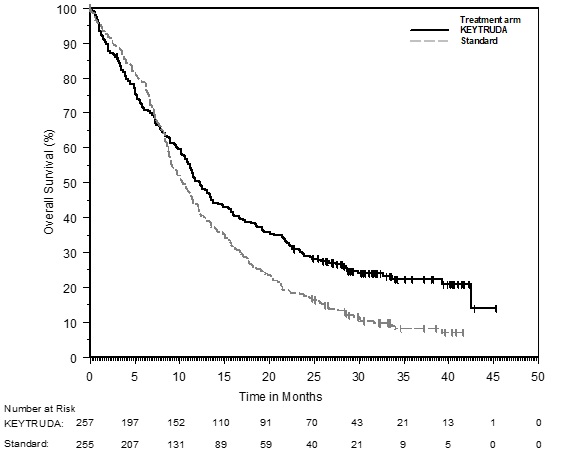
Carcinoma de células escamosas de cabeza y cuello (HNSCC) recurrente o metastásico previamente tratado
La eficacia de KEYTRUDA se investigó en KEYNOTE-012 (NCT01848834), un estudio multicéntrico, no aleatorizado, abierto y multicóhorte que incluyó a 174 pacientes con HNSCC recurrente o metastásico que habían presentado progresión de la enfermedad durante o después de la quimioterapia con platino administrada para HNSCC recurrente o metastásico o después de la quimioterapia con platino administrada como parte de la terapia de inducción, concomitante o adyuvante. Los pacientes con enfermedad autoinmune activa, una afección médica que requiriera inmunosupresión, evidencia de enfermedad pulmonar intersticial o ECOG PS ≥2 fueron inelegibles.
Los pacientes recibieron KEYTRUDA 10 mg/kg cada 2 semanas (n=53) o 200 mg cada 3 semanas (n=121) hasta que se produjo una toxicidad inaceptable o una progresión de la enfermedad que fuera sintomática, rápidamente progresiva, requiriera intervención urgente, ocurriera con una disminución en el estado funcional o se confirmara al menos 4 semanas después con imágenes repetidas. Los pacientes sin progresión de la enfermedad fueron tratados hasta por 24 meses. El tratamiento con pembrolizumab podría reiniciarse para la progresión posterior de la enfermedad y administrarse hasta por 1 año adicional. La evaluación del estado tumoral se realizó cada 8 semanas. Las principales medidas de resultado de eficacia fueron la TEO según RECIST v1.1, modificada para seguir un máximo de 10 lesiones diana y un máximo de 5 lesiones diana por órgano, según la evaluación de BICR, y la DR.
Las características de la población del estudio fueron una mediana de edad de 60 años, 32% de edad 65 o mayor; 82% hombres; 75% blancos, 16% asiáticos y 6% negros; 87% tenían enfermedad M1; 33% tenían tumores positivos para VPH; 63% habían recibido cetuximab previamente; 29% tenían un ECOG PS de 0 y 71% tenían un ECOG PS de 1; y la mediana del número de líneas de tratamiento previas administradas para el tratamiento de HNSCC fue de 2.
La TEO fue del 16% (IC del 95%: 11, 22) con una tasa de respuesta completa del 5%. El tiempo medio de seguimiento fue de 8,9 meses. Entre los 28 pacientes que respondieron, la mediana de la DR no se había alcanzado (rango: 2,4+ a 27,7+ meses), con 23 pacientes que tuvieron respuestas de 6 meses o más. La TEO y la DR fueron similares independientemente del régimen de dosificación (10 mg/kg cada 2 semanas o 200 mg cada 3 semanas) o del estado del VPH.
14.5 Linfoma de Hodgkin clásico
KEYNOTE-204
La eficacia de KEYTRUDA se investigó en KEYNOTE-204 (NCT02684292), un ensayo controlado activo, abierto y aleatorizado realizado en 304 pacientes con Linfoma de Hodgkin (LNH) recidivante o refractario. El ensayo incluyó a adultos con enfermedad recidivante o refractaria después de al menos un régimen de quimioterapia multiagente. Los pacientes fueron aleatorizados (1:1) para recibir:
- KEYTRUDA 200 mg por vía intravenosa cada 3 semanas o
- Brentuximab vedotina (BV) 1,8 mg/kg por vía intravenosa cada 3 semanas
El tratamiento continuó hasta que se produjo una toxicidad inaceptable, progresión de la enfermedad o un máximo de 35 ciclos (hasta aproximadamente 2 años). La evaluación de la enfermedad se realizó cada 12 semanas. La aleatorización se estratificó por trasplante autólogo de células madre hematopoyéticas (TCMH) previo (sí frente a no) y estado de la enfermedad después de la terapia de primera línea (refractario primario frente a recidiva <12 meses después de la finalización frente a recidiva ≥12 meses después de la finalización). La principal medida de eficacia fue la SLP según la evaluación de BICR utilizando los criterios del Grupo de Trabajo Internacional revisados de 2007.
Las características de la población del estudio fueron: mediana de edad de 35 años (rango: 18 a 84); 57% hombres; 77% blancos, 9% asiáticos, 3,9% negros. La mediana del número de tratamientos previos fue de 2 (rango: 1 a 10) en el brazo de KEYTRUDA y de 3 (rango: 1 a 11) en el brazo de BV, con un 18% en ambos brazos que tenían 1 línea previa. El 42% de los pacientes fueron refractarios a la última terapia previa, el 29% tuvieron enfermedad refractaria primaria, el 37% tuvieron un TCMH autólogo previo, el 5% habían recibido BV previamente y el 39% habían recibido radioterapia previa.
La eficacia se resume en la Tabla 72 y la Figura 15.
| Variable principal | KEYTRUDA 200 mg cada 3 semanas n=151 |
Brentuximab Vedotina 1,8 mg/kg cada 3 semanas n=153 |
|---|---|---|
| + Denota un valor censurado. | ||
| SLP | ||
| Número de pacientes con evento (%) | 81 (54%) | 88 (58%) |
| Mediana en meses (IC del 95%)* | 13,2 (10,9; 19,4) | 8,3 (5,7; 8,8) |
| Razón de riesgos† (IC del 95%) | 0,65 (0,48; 0,88) | |
| Valor p‡ | 0,0027 | |
| Tasa de respuesta objetiva | ||
| ORR§ (IC 95%) | 66% (57, 73) | 54% (46, 62) |
| Respuesta completa | 25% | 24% |
| Respuesta parcial | 41% | 30% |
| Duración de la respuesta | ||
| Mediana en meses (intervalo)* | 20.7 (0.0+, 33.2+) | 13.8 (0.0+, 33.9+) |
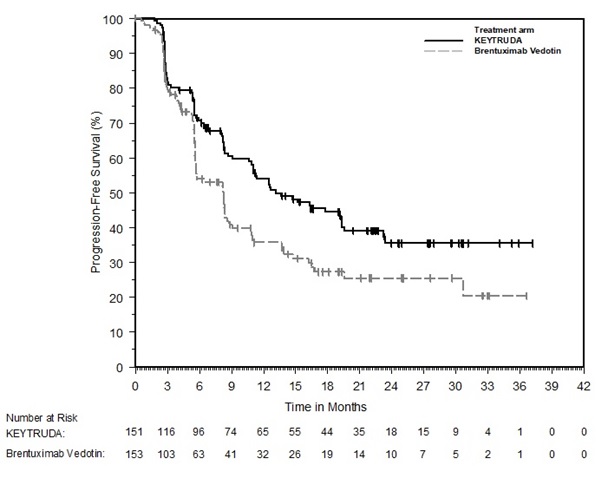 |
KEYNOTE-087
La eficacia de KEYTRUDA se investigó en KEYNOTE-087 (NCT02453594), un ensayo multicéntrico, no aleatorizado, abierto en 210 pacientes con LNH de células grandes recidivante o refractaria. Los pacientes con neumonitis activa no infecciosa, un TCHC alogénico en los últimos 5 años (o >5 años pero con síntomas de EIGH), enfermedad autoinmunitaria activa, una afección médica que requiriera inmunosupresión o una infección activa que requiriera terapia sistémica no fueron elegibles para el ensayo. Los pacientes recibieron KEYTRUDA 200 mg por vía intravenosa cada 3 semanas hasta que se produjo una toxicidad inaceptable o una progresión de la enfermedad documentada, o hasta por 24 meses en pacientes que no progresaron. La evaluación de la enfermedad se realizó cada 12 semanas. Las principales medidas de resultado de eficacia (TMO, tasa de respuesta completa y DuR) fueron evaluadas por BICR de acuerdo con los criterios del Grupo de Trabajo Internacional (IWG) revisados en 2007.
Las características de la población del estudio fueron: mediana de edad de 35 años (intervalo: 18 a 76), 9% de edad igual o superior a 65 años; 54% hombres; 88% blancos; y 49% ECOG PS de 0 y 51% ECOG PS de 1. La mediana del número de líneas de terapia previas administradas para el tratamiento de la LNH de células grandes fue de 4 (intervalo: 1 a 12). El 58% fueron refractarios a la última terapia previa, incluyendo el 35% con enfermedad refractaria primaria y el 14% cuya enfermedad fue quimiorrefractaria a todos los regímenes previos. El 61% de los pacientes se habían sometido a un TCHC autólogo previo, el 83% habían recibido brentuximab vedotina previo y el 36% de los pacientes habían recibido radioterapia previa.
Los resultados de eficacia para KEYNOTE-087 se resumen en la Tabla 73.
| Variable principal | KEYTRUDA 200 mg cada 3 semanas n=210* |
|---|---|
| Tasa de respuesta objetiva | |
| TMO (IC 95%) | 69% (62, 75) |
| Tasa de respuesta completa | 22% |
| Tasa de respuesta parcial | 47% |
| Duración de la respuesta | |
| Mediana en meses (intervalo) | 11.1 (0.0+, 11.1)† |
14.6 Linfoma de células B grandes mediastínico primario
La eficacia de KEYTRUDA se investigó en KEYNOTE-170 (NCT02576990), un ensayo multicéntrico, abierto, de un solo brazo en 53 pacientes con LCBGM de recaída o refractario. Los pacientes no fueron elegibles si tenían neumonitis no infecciosa activa, TCH alologénica en los últimos 5 años (o >5 años pero con síntomas de EIH), enfermedad autoinmune activa, una condición médica que requiriera inmunosupresión o una infección activa que requiriera terapia sistémica. Los pacientes fueron tratados con KEYTRUDA 200 mg por vía intravenosa cada 3 semanas hasta que se produjo una toxicidad inaceptable o progresión de la enfermedad documentada, o hasta por 24 meses para los pacientes que no progresaron. Las evaluaciones de la enfermedad se realizaron cada 12 semanas y fueron evaluadas por BICR de acuerdo con los criterios IWG revisados de 2007. Las medidas de resultado de eficacia fueron la TEO y la DuR.
Las características de la población del estudio fueron: mediana de edad de 33 años (rango: 20 a 61 años); 43% hombres; 92% blancos; y 43% ECOG PS de 0 y 57% ECOG PS de 1. La mediana del número de líneas de terapia previas administradas para el tratamiento del LCBGM fue de 3 (rango 2 a 8). El 36% tenía enfermedad refractaria primaria, el 49% tenía enfermedad recidivante refractaria a la última terapia previa, y el 15% tenía una recaída no tratada. El 26% de los pacientes se habían sometido a un TCH autólogo previo, y el 32% de los pacientes habían recibido radioterapia previa. Todos los pacientes habían recibido rituximab como parte de una línea de terapia previa.
Para los 24 respondedores, la mediana del tiempo hasta la primera respuesta objetiva (respuesta completa o parcial) fue de 2,8 meses (rango 2,1 a 8,5 meses). Los resultados de eficacia para KEYNOTE-170 se resumen en la Tabla 74.
| Variable principal | KEYTRUDA 200 mg cada 3 semanas n=53* |
|---|---|
| NR = no alcanzado | |
| Tasa de respuesta objetiva | |
| TRO (IC del 95%) | 45% (32, 60) |
| Tasa de respuesta completa | 11% |
| Tasa de respuesta parcial | 34% |
| Duración de la respuesta | |
| Mediana en meses (rango) | NR (1,1+, 19,2+)† |
14.7 Cáncer urotelial
En combinación con enfortumab vedotina para el tratamiento de pacientes con cáncer urotelial
La eficacia de KEYTRUDA en combinación con enfortumab vedotina se evaluó en KEYNOTE-A39 (NCT04223856), un ensayo abierto, aleatorizado y multicéntrico en el que se inscribieron 886 pacientes con cáncer urotelial localmente avanzado o metastásico que no habían recibido terapia sistémica previa para la enfermedad localmente avanzada o metastásica. Se excluyeron los pacientes con metástasis cerebrales activas, neuropatía sensitiva o motora en curso de grado ≥2 o diabetes no controlada definida como hemoglobina A1C (HbA1c) ≥8% o HbA1c ≥7% con síntomas de diabetes asociados.
Los pacientes fueron aleatorizados 1:1 para recibir:
- KEYTRUDA 200 mg durante 30 minutos en el Día 1 y enfortumab vedotina 1,25 mg/kg en los Días 1 y 8 de cada ciclo de 21 días. KEYTRUDA se administró aproximadamente 30 minutos después de enfortumab vedotina. El tratamiento continuó hasta la progresión de la enfermedad o la toxicidad inaceptable. En ausencia de progresión de la enfermedad o toxicidad inaceptable, KEYTRUDA continuó hasta por 2 años.
- Gemcitabina 1000 mg/m2 en los Días 1 y 8 de un ciclo de 21 días con cisplatino 70 mg/m2 o carboplatino (AUC = 4,5 o 5) en el Día 1 de un ciclo de 21 días. El tratamiento continuó hasta la progresión de la enfermedad o la toxicidad inaceptable hasta por 6 ciclos.
La aleatorización se estratificó por la elegibilidad para cisplatino, la expresión de PD-L1 y la presencia de metástasis hepáticas.
La mediana de edad fue de 69 años (rango: 22 a 91); el 77% eran hombres; el 67% eran blancos, el 22% eran asiáticos, el 1% eran negros o afroamericanos, y el 10% eran desconocidos u otros; el 12% eran hispanos o latinos. Los pacientes tenían un estado de rendimiento ECOG basal de 0 (49%), 1 (47%) o 2 (3%). El cuarenta y siete por ciento de los pacientes tenían una HbA1c basal documentada de <5,7%. Al inicio del estudio, el 95% de los pacientes tenían cáncer urotelial metastásico, incluyendo el 72% con metástasis viscerales y el 22% con metástasis hepáticas, y el 5% tenían cáncer urotelial localmente avanzado. El ochenta y cinco por ciento de los pacientes tenían histología de carcinoma urotelial (CU), incluyendo el 6% con CU con diferenciación escamosa mixta y el 2% con CU con otras variantes histológicas mixtas. El cuarenta y seis por ciento de los pacientes se consideraron no elegibles para cisplatino y el 54% se consideraron elegibles para cisplatino en el momento de la aleatorización.
Las principales medidas de resultado de eficacia fueron la SO y la SPP, evaluadas por BICR según RECIST v1.1. Las medidas de resultado de eficacia adicionales incluyeron la TRR, evaluada por BICR.
El ensayo demostró mejoras estadísticamente significativas en la SO, la SPP y la TRR para los pacientes aleatorizados a KEYTRUDA en combinación con enfortumab vedotina en comparación con la quimioterapia a base de platino. Los resultados de eficacia fueron consistentes en todos los subgrupos de pacientes estratificados.
La Tabla 75 y las Figuras 16 y 17 resumen los resultados de eficacia de KEYNOTE-A39.
| Variable principal | KEYTRUDA 200 mg cada 3 semanas en combinación con Enfortumab Vedotina n=442 |
Cisplatino o carboplatino con gemcitabina n=444 |
|---|---|---|
| NR = no alcanzado | ||
|
||
| SO | ||
| Número (%) de pacientes con evento | 133 (30%) | 226 (51%) |
| Mediana en meses (IC del 95%) | 31,5 (25,4, NR) | 16,1 (13,9, 18,3) |
| Razón de riesgos* (IC del 95%) | 0,47 (0,38, 0,58) | |
| Valor p† | <0,0001 | |
| SPP | ||
| Número (%) de pacientes con evento | 223 (50%) | 307 (69%) |
| Mediana en meses (IC del 95%) | 12,5 (10,4, 16,6) | 6,3 (6,2, 6,5) |
| Razón de riesgos* (IC del 95%) | 0,45 (0,38, 0,54) | |
| Valor p† | <0,0001 | |
| Tasa de respuesta objetiva confirmada‡ | ||
| ORR§ % (IC 95%) | 68% (63, 72) | 44% (40, 49) |
| Valor p¶ | <0.0001 | |
| Respuesta completa | 29% | 12% |
| Respuesta parcial | 39% | 32% |
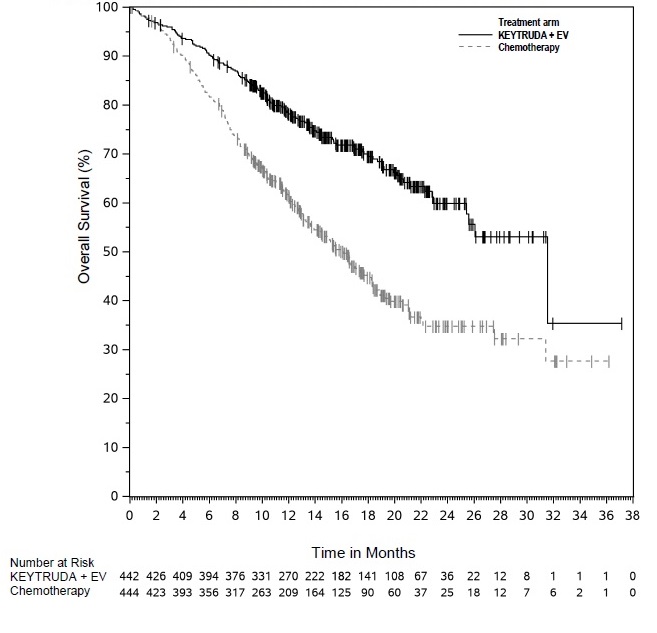 |
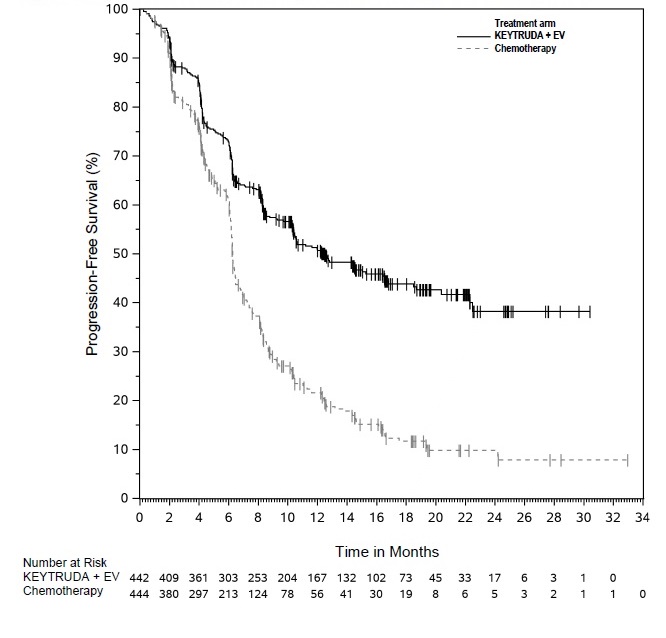 |
En combinación con enfortumab vedotina para el tratamiento de pacientes con cáncer urotelial no elegibles para cisplatino
La eficacia de KEYTRUDA en combinación con enfortumab vedotina se evaluó en KEYNOTE-869 (NCT03288545), un estudio abierto, multicóhorte (cohorte de escalada de dosis, Cohorte A, Cohorte K) en pacientes con cáncer urotelial localmente avanzado o metastásico que no eran elegibles para quimioterapia con cisplatino y no habían recibido terapia sistémica previa para la enfermedad localmente avanzada o metastásica. Los pacientes con metástasis cerebrales activas, neuropatía sensitiva o motora en curso de grado ≥2 o diabetes no controlada definida como hemoglobina A1C (HbA1c) ≥8% o HbA1c ≥7% con síntomas de diabetes asociados fueron excluidos de la participación en el estudio.
Los pacientes en la cohorte de escalada de dosis (n=5), la Cohorte A (n=40) y la Cohorte K (n=76) recibieron enfortumab vedotina 1,25 mg/kg como infusión IV durante 30 minutos en los Días 1 y 8 de un ciclo de 21 días, seguido de KEYTRUDA 200 mg como infusión IV en el Día 1 de un ciclo de 21 días aproximadamente 30 minutos después de enfortumab vedotina. Los pacientes fueron tratados hasta la progresión de la enfermedad o la toxicidad inaceptable.
Un total de 121 pacientes recibieron KEYTRUDA en combinación con enfortumab vedotina. La mediana de edad fue de 71 años (rango: 51 a 91); el 74% eran hombres; el 85% eran blancos, el 5% eran negros, el 4% eran asiáticos y el 6% eran de otra raza, desconocida o no informada. El diez por ciento de los pacientes eran hispanos o latinos. El cuarenta y cinco por ciento de los pacientes tenían un estado de rendimiento ECOG de 1 y el 15% tenían un estado de rendimiento ECOG de 2. El cuarenta y siete por ciento de los pacientes tenían una HbA1c basal documentada de <5,7%. Las razones para la no elegibilidad para cisplatino incluyeron: 60% con aclaramiento de creatinina basal de 30-59 mL/min, 10% con estado de rendimiento ECOG de 2, 13% con pérdida auditiva de grado 2 o superior, y 16% con más de un criterio de no elegibilidad para cisplatino.
Al inicio del estudio, el 97,5% de los pacientes tenían cáncer urotelial metastásico y el 2,5% de los pacientes tenían cáncer urotelial localmente avanzado. El treinta y siete por ciento de los pacientes tenían enfermedad de las vías urinarias altas. El ochenta y cuatro por ciento de los pacientes tenían metástasis visceral al inicio del estudio, incluyendo el 22% con metástasis hepáticas. El treinta y nueve por ciento de los pacientes tenían histología de TCC; el 13% tenían TCC con diferenciación escamosa, y el 48% tenían TCC con otras variantes histológicas.
Las principales medidas de resultado de eficacia fueron la ORR y la DoR, evaluadas por BICR según RECIST v1.1.
El tiempo medio de seguimiento para la cohorte de escalada de dosis + Cohorte A fue de 44,7 meses (rango 0,7 a 52,4) y para la Cohorte K fue de 14,8 meses (rango: 0,6 a 26,2).
Los resultados de eficacia se presentan en la Tabla 76 a continuación.
| Variable principal | KEYTRUDA en combinación con Enfortumab Vedotina n=121 |
|---|---|
| ORR confirmada (IC 95%) | 68% (58,7, 76,0) |
| Tasa de respuesta completa | 12% |
| Tasa de respuesta parcial | 55% |
La duración mediana de la respuesta para la cohorte de escalada de dosis + Cohorte A fue de 22,1 meses (rango: 1,0+ a 46,3+) y para la Cohorte K no se alcanzó (rango: 1,2 a 24,1+).
Pacientes con carcinoma urotelial no elegibles para platino
La eficacia de KEYTRUDA se investigó en KEYNOTE-052 (NCT02335424), un ensayo multicéntrico, abierto y de un solo brazo en 370 pacientes con carcinoma urotelial localmente avanzado o metastásico que tenían una o más comorbilidades, incluidos pacientes que no eran elegibles para ningún quimioterapia que contuviera platino. El ensayo excluyó a pacientes con enfermedad autoinmune o una condición médica que requiriera inmunosupresión. Los pacientes recibieron KEYTRUDA 200 mg cada 3 semanas hasta que se produjo una toxicidad inaceptable o una progresión de la enfermedad. Los pacientes con progresión inicial de la enfermedad radiográfica podrían recibir dosis adicionales de tratamiento durante la confirmación de la progresión a menos que la progresión de la enfermedad fuera sintomática, fuera rápidamente progresiva, requiriera intervención urgente o ocurriera con una disminución en el estado funcional. Los pacientes sin progresión de la enfermedad podrían tratarse hasta por 24 meses. Las evaluaciones de la respuesta tumoral se realizaron a las 9 semanas después de la primera dosis, luego cada 6 semanas durante el primer año y luego cada 12 semanas a partir de entonces. Las principales medidas de resultado de eficacia fueron la TEO y la DuR según lo evaluado por BICR de acuerdo con RECIST v1.1, modificado para seguir un máximo de 10 lesiones diana y un máximo de 5 lesiones diana por órgano.
Las características de la población del estudio fueron: edad media de 74 años; 77% hombres; y 89% blancos. El ochenta y siete por ciento tenía enfermedad M1 y el 13% tenía enfermedad M0. El ochenta y un por ciento tenía un tumor primario en el tracto inferior, y el 19% de los pacientes tenían un tumor primario en el tracto superior. El ochenta y cinco por ciento de los pacientes tenían metástasis viscerales, incluido el 21% con metástasis hepáticas. El cincuenta por ciento de los pacientes tenían un aclaramiento de creatinina basal de <60 mL/min, el 32% tenía un EPCOG de 2, el 9% tenía un EPCOG de 2 y un aclaramiento de creatinina basal de <60 mL/min, y el 9% tenía una o más de insuficiencia cardíaca de Clase III, neuropatía periférica de grado 2 o superior y pérdida auditiva de grado 2 o superior. El noventa por ciento de los pacientes fueron ingenuos al tratamiento, y el 10% recibió quimioterapia basada en platino adyuvante o neoadyuvante previa.
El tiempo medio de seguimiento para los 370 pacientes tratados con KEYTRUDA fue de 11,4 meses (rango 0,1 a 63,8 meses). Los resultados de eficacia se resumen en la Tabla 77.
| Variable principal | KEYTRUDA 200 mg cada 3 semanas |
|---|---|
| Todos los sujetos n=370 |
|
| + Denota respuesta en curso | |
| Tasa de respuesta objetiva | |
| TRO (IC del 95%) | 29% (24, 34) |
| Tasa de respuesta completa | 10% |
| Tasa de respuesta parcial | 20% |
| Duración de la respuesta | |
| Mediana en meses (rango) | 33.4 (1.4+, 60.7+) |
Pacientes con Carcinoma Urotelial Previamente No Tratado Elegibles para Platino
La eficacia de KEYTRUDA para el tratamiento de primera línea de pacientes con carcinoma urotelial localmente avanzado o metastásico elegibles para platino se investigó en KEYNOTE-361 (NCT02853305), un estudio multicéntrico, aleatorizado, abierto y controlado con activo en 1010 pacientes previamente no tratados. No se ha establecido la seguridad y eficacia de KEYTRUDA en combinación con quimioterapia a base de platino para pacientes previamente no tratados con carcinoma urotelial localmente avanzado o metastásico.
El estudio comparó KEYTRUDA con o sin quimioterapia a base de platino (es decir, cisplatino o carboplatino con gemcitabina) con quimioterapia a base de platino sola. Entre los pacientes que recibieron KEYTRUDA más quimioterapia a base de platino, el 44% recibió cisplatino y el 56% recibió carboplatino.
El estudio no cumplió con sus principales medidas de resultado de eficacia de mejora de la PFS o la OS en el brazo de KEYTRUDA más quimioterapia en comparación con el brazo de quimioterapia sola. No se pudieron probar formalmente los puntos finales de eficacia adicionales, incluida la mejora de la OS en el brazo de monoterapia con KEYTRUDA.
Carcinoma Urotelial Previamente Tratado
La eficacia de KEYTRUDA se investigó en KEYNOTE-045 (NCT02256436), un ensayo multicéntrico, aleatorizado (1:1), controlado con activo en 542 pacientes con carcinoma urotelial localmente avanzado o metastásico con progresión de la enfermedad en o después de la quimioterapia con platino. El ensayo excluyó a pacientes con enfermedad autoinmune o una condición médica que requiriera inmunosupresión.
Los pacientes fueron aleatorizados para recibir KEYTRUDA 200 mg cada 3 semanas (n=270) o la quimioterapia a elección del investigador de cualquiera de los siguientes regímenes, todos administrados por vía intravenosa cada 3 semanas (n=272): paclitaxel 175 mg/m2 (n=90), docetaxel 75 mg/m2 (n=92), o vinflunina 320 mg/m2 (n=90). El tratamiento continuó hasta que se produjo una toxicidad inaceptable o una progresión de la enfermedad. Los pacientes con progresión inicial de la enfermedad radiográfica podrían recibir dosis adicionales de tratamiento durante la confirmación de la progresión, a menos que la progresión de la enfermedad fuera sintomática, rápidamente progresiva, requiriera intervención urgente o ocurriera con una disminución en el estado funcional. Los pacientes sin progresión de la enfermedad podrían ser tratados hasta por 24 meses. La evaluación del estado del tumor se realizó a las 9 semanas después de la aleatorización, luego cada 6 semanas durante el primer año, seguido de cada 12 semanas a partir de entonces. Los principales resultados de eficacia fueron la OS y la PFS según la evaluación de BICR según RECIST v1.1, modificada para seguir un máximo de 10 lesiones diana y un máximo de 5 lesiones diana por órgano. Las medidas de resultado de eficacia adicionales fueron la ORR según la evaluación de BICR según RECIST v1.1, modificada para seguir un máximo de 10 lesiones diana y un máximo de 5 lesiones diana por órgano, y la DoR.
Las características de la población del estudio fueron: edad media de 66 años (rango: 26 a 88), 58% de edad 65 o mayor; 74% hombres; 72% blancos y 23% asiáticos; 42% ECOG PS de 0 y 56% ECOG PS de 1; y 96% de enfermedad M1 y 4% de enfermedad M0. El ochenta y siete por ciento de los pacientes tenían metástasis viscerales, incluyendo el 34% con metástasis hepáticas. El ochenta y seis por ciento tenía un tumor primario en el tracto inferior y el 14% tenía un tumor primario en el tracto superior. El quince por ciento de los pacientes tuvieron progresión de la enfermedad después de la quimioterapia neoadyuvante o adyuvante con platino previa. El veintiún por ciento había recibido 2 o más regímenes sistémicos previos en el contexto metastásico. El setenta y seis por ciento de los pacientes recibieron cisplatino previo, el 23% recibió carboplatino previo y el 1% fue tratado con otros regímenes a base de platino.
El estudio demostró mejoras estadísticamente significativas en la OS y la ORR para los pacientes aleatorizados a KEYTRUDA en comparación con la quimioterapia. No hubo una diferencia estadísticamente significativa entre KEYTRUDA y la quimioterapia con respecto a la PFS. El tiempo medio de seguimiento para este ensayo fue de 9,0 meses (rango: 0,2 a 20,8 meses). La Tabla 78 y la Figura 18 resumen los resultados de eficacia de KEYNOTE-045.
| KEYTRUDA 200 mg cada 3 semanas |
Quimioterapia | |
|---|---|---|
| n=270 | n=272 | |
| + Denota respuesta en curso NR = no alcanzado |
||
|
||
| OS | ||
| Defunciones (%) | 155 (57%) | 179 (66%) |
| Mediana en meses (IC del 95%) | 10.3 (8.0, 11.8) | 7.4 (6.1, 8.3) |
| Hazard ratio* (IC del 95%) | 0.73 (0.59, 0.91) | |
| p-Valor (log-rank estratificado) | 0.004 | |
| PFS por BICR | ||
| Eventos (%) | 218 (81%) | 219 (81%) |
| Mediana en meses (IC del 95%) | 2.1 (2.0, 2.2) | 3.3 (2.3, 3.5) |
| Hazard ratio* (IC 95%) | 0.98 (0.81, 1.19) | |
| Valor de p (log-rank estratificado) | 0.833 | |
| Tasa de respuesta objetiva | ||
| TRO (IC 95%) | 21% (16, 27) | 11% (8, 16) |
| Tasa de respuesta completa | 7% | 3% |
| Tasa de respuesta parcial | 14% | 8% |
| Valor de p (Miettinen-Nurminen) | 0.002 | |
| Duración mediana de la respuesta en meses (intervalo) | NR (1.6+, 15.6+) |
4.3 (1.4+, 15.4+) |
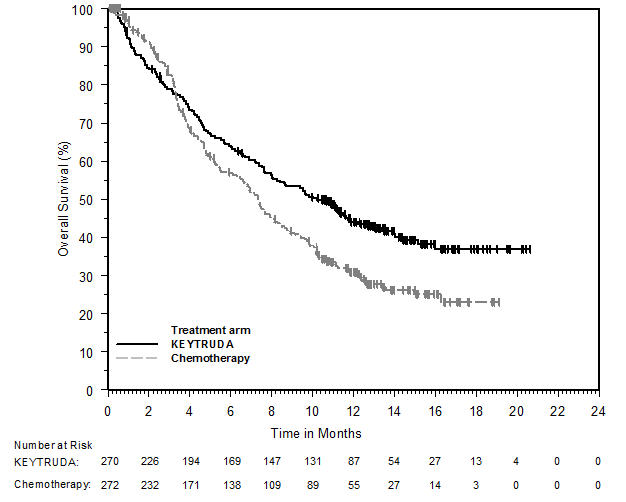 |
Cáncer de vejiga no invasivo de alto riesgo sin respuesta a BCG
La eficacia de KEYTRUDA se investigó en KEYNOTE-057 (NCT02625961), un ensayo multicéntrico, abierto y de un solo brazo en 96 pacientes con cáncer de vejiga no invasivo de alto riesgo (NMIBC) sin respuesta al bacilo de Calmette-Guérin (BCG) con carcinoma in situ (CIS) con o sin tumores papilares que no son elegibles o que han optado por no someterse a una cistectomía. El NMIBC de alto riesgo sin respuesta a BCG se definió como enfermedad persistente a pesar de una terapia adecuada con BCG, recurrencia de la enfermedad después de un estado inicial sin tumor tras una terapia adecuada con BCG, o enfermedad T1 después de un único ciclo de inducción con BCG. La terapia adecuada con BCG se definió como la administración de al menos cinco de seis dosis de un ciclo de inducción inicial más cualquiera de las siguientes: al menos dos de tres dosis de terapia de mantenimiento o al menos dos de seis dosis de un segundo ciclo de inducción. Antes del tratamiento, todos los pacientes se habían sometido a una resección transuretral del tumor vesical (TURBT) para extirpar toda la enfermedad resecable (componentes Ta y T1). Se permitió el CIS residual (componentes Tis) no susceptible de resección completa. El ensayo excluyó a pacientes con carcinoma urotelial metastásico o localmente avanzado invasivo muscular (es decir, T2, T3, T4), carcinoma de células transicionales no invasivo muscular extravesical concomitante (es decir, uretra, uréter o pelvis renal), o enfermedad autoinmune o una afección médica que requiriera inmunosupresión.
Los pacientes recibieron KEYTRUDA 200 mg cada 3 semanas hasta que se produjo una toxicidad inaceptable, un NMIBC de alto riesgo persistente o recurrente, o una enfermedad progresiva. La evaluación del estado del tumor se realizó cada 12 semanas durante dos años y luego cada 24 semanas durante tres años, y los pacientes sin progresión de la enfermedad pudieron recibir tratamiento hasta por 24 meses. Las principales medidas de resultado de eficacia fueron la respuesta completa (definida por resultados negativos para cistoscopia [con TURBT/biopsias según corresponda], citología de orina e imágenes de urografía computarizada [TCU]) y la duración de la respuesta.
Las características de la población del estudio fueron: mediana de edad de 73 años (intervalo: 44 a 92); 44% de edad ≥75; 84% hombres; 67% blancos; y 73% y 27% con un estado de rendimiento ECOG de 0 o 1, respectivamente. El patrón tumoral al inicio del estudio fue CIS con T1 (13%), CIS con TA de alto grado (25%) y CIS (63%). El estado de la enfermedad NMIBC de alto riesgo basal fue 27% persistente y 73% recurrente. La mediana del número de instilaciones previas de BCG fue de 12.
El tiempo medio de seguimiento fue de 28,0 meses (intervalo: 4,6 a 40,5 meses). Los resultados de eficacia se resumen en la Tabla 79.
14.8 Cáncer con alta inestabilidad de microsatélites o deficiencia de reparación de desajuste
La eficacia de KEYTRUDA se investigó en 504 pacientes con cánceres MSI-H o dMMR inscritos en tres ensayos multicéntricos, no aleatorizados, abiertos y multicóhorte: KEYNOTE-164 (NCT02460198), KEYNOTE-158 (NCT02628067) y KEYNOTE-051 (NCT02332668). Todos los ensayos excluyeron a pacientes con enfermedad autoinmune o una condición médica que requiriera inmunosupresión. Independientemente de la histología, el estado tumoral MSI o MMR se determinó mediante reacción en cadena de la polimerasa (PCR; local o central) o inmunohistoquímica (IHC; local o central), respectivamente.
- KEYNOTE-164 inscribió a 124 pacientes con cáncer colorrectal (CCR) MSI-H o dMMR avanzado que progresó después del tratamiento con fluoropirimidina y oxaliplatino o irinotecán +/- terapia basada en mAb anti-VEGF/EGFR.
- KEYNOTE-158 inscribió a 373 pacientes con cánceres no colorrectales (no CCR) MSI-H o dMMR avanzados que habían presentado progresión de la enfermedad después de la terapia previa. Los pacientes fueron inscritos prospectivamente con tumores MSI-H/dMMR (Cohorte K) o identificados retrospectivamente en una de las 10 cohortes de tumores sólidos (Cohortes A-J).
- KEYNOTE-051 inscribió a 7 pacientes pediátricos con cánceres MSI-H o dMMR.
Los pacientes adultos recibieron KEYTRUDA 200 mg cada 3 semanas (los pacientes pediátricos recibieron 2 mg/kg cada 3 semanas) hasta que se produjo una toxicidad inaceptable, progresión de la enfermedad o un máximo de 24 meses. En KEYNOTE-164 y KEYNOTE-158, la evaluación del estado tumoral se realizó cada 9 semanas durante el primer año, y luego cada 12 semanas a partir de entonces. En KEYNOTE-051, la evaluación del estado tumoral se realizó cada 8 semanas durante 24 semanas, y luego cada 12 semanas a partir de entonces. Las principales medidas de resultado de eficacia fueron la TEO y la DuR, según la evaluación del BICR de acuerdo con RECIST v1.1 (modificado para seguir un máximo de 10 lesiones diana y un máximo de 5 lesiones diana por órgano en KEYNOTE-158) y según la evaluación del investigador de acuerdo con RECIST v1.1 en KEYNOTE-051.
En KEYNOTE-164 y KEYNOTE-158, las características de la población del estudio fueron una mediana de edad de 60 años, 36% de edad igual o superior a 65 años; 44% hombres; 78% blancos, 14% asiáticos, 4% amerindios o nativos de Alaska y 3% negros; y 45% ECOG PS de 0 y 55% ECOG PS de 1. El noventa y dos por ciento de los pacientes tenían enfermedad metastásica y el 4% tenían enfermedad localmente avanzada e irresecable. El treinta y siete por ciento de los pacientes recibieron una línea de terapia previa y el 61% recibieron dos o más líneas de terapia previas.
En KEYNOTE-051, las características de la población del estudio fueron una mediana de edad de 11 años (rango: 3 a 16); 71% mujeres; 86% blancos y 14% asiáticos; y el 57% tenía una puntuación de Lansky/Karnofsky de 100. El setenta y un por ciento de los pacientes tenían enfermedad en estadio IV y el 14% tenían enfermedad en estadio III. El cincuenta y siete por ciento de los pacientes recibieron una línea de terapia previa y el 29% recibieron dos líneas de terapia previas.
Se observaron resultados discordantes entre las pruebas locales MSI-H o dMMR y las pruebas centrales entre los pacientes inscritos en la Cohorte K de KEYNOTE-158. Entre las 104 muestras de tumor que fueron MSI-H o dMMR mediante pruebas locales y también se analizaron utilizando la prueba FoundationOne®CDx (F1CDx), 59 (56,7%) fueron MSI-H y 45 (43,3%) no fueron MSI-H. Entre las 169 muestras de tumor que fueron MSI-H o dMMR mediante pruebas locales y también se analizaron utilizando el panel VENTANA MMR RxDx, 105 (62,1%) fueron dMMR y 64 (37,9%) fueron pMMR.
Los resultados de eficacia se resumen en las tablas 80 y 81.
| Variable principal | KEYTRUDA n=504* |
|---|---|
| + Denota respuesta en curso | |
| Tasa de respuesta objetiva | |
| TRO (IC del 95%)† | 33,3% (29,2, 37,6) |
| Tasa de respuesta completa | 10,3% |
| Tasa de respuesta parcial | 23,0% |
| Duración de la respuesta | n=168 |
| Mediana en meses (rango) | 63,2 (1,9+, 63,9+) |
| % con duración ≥12 meses | 77% |
| % con duración ≥36 meses | 39% |
| Tasa de respuesta objetiva | Duración del rango de respuesta |
|||
|---|---|---|---|---|
| N | n (%) | IC del 95% | (meses) | |
| + Denota respuesta en curso | ||||
|
||||
| CRC | 124 | 42 (34%) | (26%, 43%) | (4.4, 58.5+) |
| No CRC* | 380 | 126 (33%) | (28%, 38%) | (1.9+, 63.9+) |
| Cáncer endometrial | 94 | 47 (50%) | (40%, 61%) | (2.9, 63.2) |
| Cáncer gástrico o de la unión gastroesofágica | 51 | 20 (39%) | (26%, 54%) | (1.9+, 63.0+) |
| Cáncer de intestino delgado | 27 | 16 (59%) | (39%, 78%) | (3.7+, 57.3+) |
| Cáncer cerebral | 27† | 1 (4%)‡ | (0%, 19%) | 18.9 |
| Cáncer de ovario | 25 | 8 (32%) | (15%, 54%) | (4.2, 56.6+) |
| Cáncer biliar | 22 | 9 (41%) | (21%, 64%) | (6.2, 49.0+) |
| Cáncer de páncreas | 22 | 4 (18%) | (5%, 40%) | (8.1, 24.3+) |
| Sarcoma | 14 | 3 (21%) | (5%, 51%) | (35.4+, 57.2+) |
| Cáncer de mama | 13 | 1 (8%) | (0%,36%) | 24.3+ |
| Otros§ | 13 | 4 (31%) | (9%, 61%) | (6.2+, 32.3+) |
| Cáncer cervical | 11 | 1 (9%) | (0%, 41%) | 63.9+ |
| Cáncer neuroendocrino | 11 | 1 (9%) | (0%, 41%) | 13.3 |
| Cáncer de próstata | 8 | 1 (13%) | (0%, 53%) | 24.5+ |
| Cáncer adrenocortical | 7 | 1 (14%) | (0%, 58%) | 4.2 |
| Mesotelioma | 7 | 0 (0%) | (0%, 41%) | |
| Cáncer de tiroides | 7 | 1 (14%) | (0%, 58%) | 8.2 |
| Cáncer de pulmón de células pequeñas | 6 | 2 (33%) | (4%, 78%) | (20.0, 47.5) |
| Cáncer de vejiga | 6 | 3 (50%) | (12%, 88%) | (35.6+, 57.5+) |
| Cáncer de saliva | 5 | 2 (40%) | (5%, 85%) | (42.6+, 57.8+) |
| Cáncer de células renales | 4 | 1 (25%) | (0%, 81%) | 22.0 |
Análisis exploratorio por TMB
En un análisis exploratorio realizado en 138 pacientes (Cohorte K de KEYNOTE-158) a quienes se les realizó una prueba retrospectiva de carga mutacional tumoral (TMB) utilizando una prueba aprobada por la FDA, 45 (33%) tenían tumores con una puntuación de TMB de <10 mut/Mb; la ORR en estos 45 pacientes fue del 6,7% (IC del 95%: 1,4, 18,3). Entre los 45 pacientes con una puntuación de TMB de <10 mut/Mb, 39 no eran MSI-H/dMMR cuando se les realizó una prueba aprobada por la FDA.
14.9 Cáncer colorrectal con alta inestabilidad de microsatélites o deficiencia de reparación de desajuste
La eficacia de KEYTRUDA se investigó en KEYNOTE-177 (NCT02563002), un ensayo multicéntrico, aleatorizado, abierto y controlado con activo en el que se inscribieron 307 pacientes con CRC MSI-H o dMMR metastásico o irresecable previamente no tratado. El estado tumoral de MSI o MMR se determinó localmente mediante reacción en cadena de la polimerasa (PCR) o inmunohistoquímica (IHC), respectivamente. Los pacientes con enfermedad autoinmune o una afección médica que requiriera inmunosupresión no fueron elegibles.
Los pacientes se asignaron aleatoriamente (1:1) para recibir KEYTRUDA 200 mg por vía intravenosa cada 3 semanas o la elección del investigador de los siguientes regímenes de quimioterapia administrados por vía intravenosa cada 2 semanas:
- mFOLFOX6 (oxaliplatino, leucovorina y FU) o mFOLFOX6 en combinación con bevacizumab o cetuximab: Oxaliplatino 85 mg/m2, leucovorina 400 mg/m2 (o levoleucovorina 200 mg/m2), y FU 400 mg/m2 en bolo el día 1, luego FU 2400 mg/m2 durante 46-48 horas. Bevacizumab 5 mg/kg el día 1 o cetuximab 400 mg/m2 en la primera infusión, luego 250 mg/m2 semanalmente.
- FOLFIRI (irinotecán, leucovorina y FU) o FOLFIRI en combinación con bevacizumab o cetuximab: Irinotecán 180 mg/m2, leucovorina 400 mg/m2 (o levoleucovorina 200 mg/m2), y FU 400 mg/m2 en bolo el día 1, luego FU 2400 mg/m2 durante 46-48 horas. Bevacizumab 5 mg/kg el día 1 o cetuximab 400 mg/m2 en la primera infusión, luego 250 mg/m2 semanalmente.
El tratamiento con KEYTRUDA o quimioterapia continuó hasta la progresión de la enfermedad definida por RECIST v1.1 según lo determinado por el investigador o toxicidad inaceptable. Los pacientes tratados con KEYTRUDA sin progresión de la enfermedad podrían recibir tratamiento hasta por 24 meses. La evaluación del estado tumoral se realizó cada 9 semanas. A los pacientes asignados aleatoriamente a quimioterapia se les ofreció KEYTRUDA en el momento de la progresión de la enfermedad. Las principales medidas de resultado de eficacia fueron la PFS (según lo evaluado por BICR de acuerdo con RECIST v1.1, modificado para seguir un máximo de 10 lesiones diana y un máximo de 5 lesiones diana por órgano) y la OS. Las medidas de resultado de eficacia adicionales fueron la ORR y la DoR.
Se inscribieron y asignaron aleatoriamente un total de 307 pacientes a KEYTRUDA (n=153) o quimioterapia (n=154). Las características basales de estos 307 pacientes fueron: edad media de 63 años (rango: 24 a 93), 47% de edad 65 o mayor; 50% hombres; 75% blancos y 16% asiáticos; 52% tenían una puntuación ECOG PS de 0 y 48% tenían una puntuación ECOG PS de 1; y el 27% recibió quimioterapia adyuvante o neoadyuvante previa. De los 154 pacientes asignados aleatoriamente para recibir quimioterapia, 143 recibieron quimioterapia según el protocolo. De los 143 pacientes, el 56% recibió mFOLFOX6, el 44% recibió FOLFIRI, el 70% recibió bevacizumab más mFOLFOX6 o FOLFIRI, y el 11% recibió cetuximab más mFOLFOX6 o FOLFIRI.
El ensayo demostró una mejora estadísticamente significativa en la PFS para los pacientes asignados aleatoriamente a KEYTRUDA en comparación con la quimioterapia. No hubo una diferencia estadísticamente significativa entre KEYTRUDA y la quimioterapia en el análisis final de OS. El sesenta por ciento de los pacientes que habían sido asignados aleatoriamente para recibir quimioterapia habían pasado a recibir terapias anti-PD-1/PD-L1 posteriores, incluida KEYTRUDA. El tiempo medio de seguimiento en el análisis final fue de 38,1 meses (rango: 0,2 a 58,7 meses). La Tabla 82 y la Figura 19 resumen las principales medidas de eficacia para KEYNOTE-177.
| Variable principal | KEYTRUDA 200 mg cada 3 semanas n=153 |
Quimioterapia n=154 |
|---|---|---|
| + Denota respuesta en curso NR = no alcanzado |
||
|
||
| PFS | ||
| Número (%) de pacientes con evento | 82 (54%) | 113 (73%) |
| Mediana en meses (IC del 95%) | 16,5 (5,4, 32,4) | 8,2 (6,1, 10,2) |
| Razón de riesgo* (IC del 95%) | 0,60 (0,45, 0,80) | |
| Valor p† | 0.0004 | |
| SP‡ | ||
| Número (%) de pacientes con evento | 62 (41%) | 78 (51%) |
| Mediana en meses (IC del 95%) | NR (49.2, NR) | 36.7 (27.6, NR) |
| Razón de riesgo* (IC del 95%) | 0.74 (0.53, 1.03) | |
| Valor p§ | 0.0718 | |
| Tasa de respuesta objetiva¶ | ||
| TRO (IC del 95%) | 44% (35.8, 52.0) | 33% (25.8, 41.1) |
| Tasa de respuesta completa | 11% | 4% |
| Tasa de respuesta parcial | 33% | 29% |
| Duración de la respuesta¶,# | ||
| Mediana en meses (rango) | NR (2.3+, 41.4+) | 10.6 (2.8, 37.5+) |
| % con duración ≥12 mesesÞ | 75% | 37% |
| % con duración ≥24 mesesÞ | 43% | 18% |
|
|
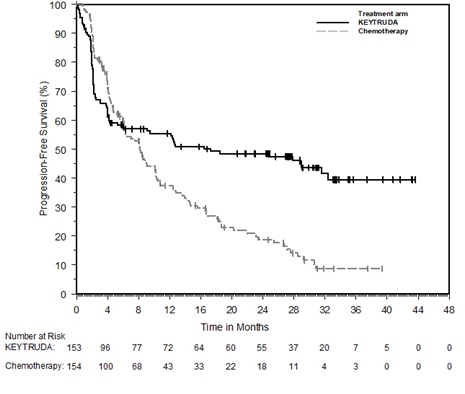
14.10 Cáncer Gástrico
Tratamiento de primera línea del adenocarcinoma gástrico o de la unión gastroesofágica HER2-positivo localmente avanzado irresecable o metastásico
La eficacia de KEYTRUDA en combinación con trastuzumab más quimioterapia con fluoropirimidina y platino se investigó en KEYNOTE-811 (NCT03615326), un ensayo multicéntrico, aleatorizado, doble ciego y controlado con placebo en el que se inscribieron 698 pacientes con adenocarcinoma gástrico o de la unión gastroesofágica (UGE) HER2-positivo avanzado que no habían recibido previamente terapia sistémica para la enfermedad metastásica. El estado de PD-L1 se determinó utilizando el kit PD-L1 IHC 22C3 pharmDx™. Los pacientes con una enfermedad autoinmune que requiriera terapia sistémica dentro de los 2 años del tratamiento o una condición médica que requiriera inmunosupresión no fueron elegibles. La aleatorización se estratificó por la expresión de PD-L1 (CPS ≥1 o CPS <1), el régimen de quimioterapia (5-FU más cisplatino [FP] o capecitabina más oxaliplatino [CAPOX]) y la región geográfica (Europa/Israel/América del Norte/Australia, Asia o Resto del Mundo). Los pacientes se asignaron aleatoriamente (1:1) a uno de los siguientes brazos de tratamiento:
- KEYTRUDA 200 mg, trastuzumab 8 mg/kg en la primera infusión y 6 mg/kg en ciclos posteriores, seguido de la quimioterapia combinada a elección del investigador de cisplatino 80 mg/m2 durante un máximo de 6 ciclos y 5-FU 800 mg/m2/día durante 5 días (FP) u oxaliplatino 130 mg/m2 hasta 6-8 ciclos y capecitabina 1000 mg/m2 bid durante 14 días (CAPOX). KEYTRUDA se administró antes del trastuzumab y la quimioterapia el día 1 de cada ciclo.
- Placebo, trastuzumab 8 mg/kg en la primera infusión y 6 mg/kg en ciclos posteriores, seguido de la quimioterapia combinada a elección del investigador de cisplatino 80 mg/m2 durante un máximo de 6 ciclos y 5-FU 800 mg/m2/día durante 5 días (FP) u oxaliplatino 130 mg/m2 hasta 6-8 ciclos y capecitabina 1000 mg/m2 bid durante 14 días (CAPOX).
Todos los medicamentos del estudio, excepto la capecitabina oral, se administraron como infusión intravenosa cada 3 semanas. El tratamiento con KEYTRUDA continuó hasta la progresión de la enfermedad definida por RECIST v1.1 según lo determinado por BICR, toxicidad inaceptable o un máximo de 24 meses. En un análisis de eficacia interino, las principales medidas de resultado evaluadas fueron la ORR y la DoR por BICR utilizando RECIST v1.1, modificado para seguir un máximo de 10 lesiones diana y un máximo de 5 lesiones diana por órgano.
En el momento del análisis interino, se evaluaron la ORR y la DoR en los primeros 264 pacientes aleatorizados. Entre los 264 pacientes, las características de la población fueron: edad media de 62 años (rango: 19 a 84), 41% de edad 65 o más; 82% hombres; 63% blancos, 31% asiáticos y 0,8% negros; 47% ECOG PS de 0 y 53% ECOG PS de 1. El noventa y siete por ciento de los pacientes tenían enfermedad metastásica (Estadio IV) y el 3% tenían enfermedad localmente avanzada irresecable. El noventa y uno por ciento (n=240) tenían tumores que no eran MSI-H, el 1% (n=2) tenían tumores que eran MSI-H, y en el 8% (n=22) el estado no se conocía. El ochenta y siete por ciento de los pacientes recibieron CAPOX.
Se demostró una mejora estadísticamente significativa en la ORR en los pacientes aleatorizados a KEYTRUDA en combinación con trastuzumab y quimioterapia en comparación con el placebo en combinación con trastuzumab y quimioterapia. Los resultados de eficacia se resumen en la Tabla 83.
| Variable principal | KEYTRUDA 200 mg cada 3 semanas Trastuzumab Quimioterapia con fluoropirimidina y platino n=133 |
Placebo
Trastuzumab |
|---|---|---|
| + Denota respuesta en curso | ||
| Tasa de respuesta objetiva | ||
| ORR* (IC del 95%) | 74% (66, 82) | 52% (43, 61) |
| Tasa de respuesta completa | 11% | 3.1% |
| Tasa de respuesta parcial | 63% | 49% |
| Valor p† | <0.0001 | |
| Duración de la respuesta | n=99 | n=68 |
| Mediana en meses (rango) | 10.6 (1.1+, 16.5+) | 9.5 (1.4+, 15.4+) |
| % con duración ≥6 meses‡ | 65% | 53% |
En un análisis de subgrupos preespecificado de la TMR basado en el estado de PD-L1, la TMR en pacientes con enfermedad PD-L1 positiva (CPS ≥1) fue del 76% (IC del 95%: 67, 83) en el brazo de pembrolizumab (n=117) frente al 51% (IC del 95%: 41, 60) en el brazo de control (n=112). En pacientes con tumores con CPS de PD-L1 <1, la TMR fue del 63% (IC del 95%: 35, 85) en el brazo de pembrolizumab (n=16) frente al 58% (IC del 95%: 34, 80) en el brazo de control (n=19).
En un análisis provisional posterior de subgrupos preespecificados basados en el estado de PD-L1 en la población total del estudio (n=698), la HR para la SPP y la SO en pacientes con CPS de PD-L1 <1 (N=104) fue de 1,03 (IC del 95%: 0,65, 1,64) y 1,41 (IC del 95%: 0,90, 2,20), respectivamente.
Tratamiento de primera línea del adenocarcinoma gástrico o de la unión gastroesofágica HER2 negativo localmente irresecable o metastásico
La eficacia de KEYTRUDA en combinación con quimioterapia con fluoropirimidina y platino se investigó en KEYNOTE-859 (NCT03675737), un ensayo multicéntrico, aleatorizado, doble ciego y controlado con placebo en el que se inscribieron 1579 pacientes con adenocarcinoma gástrico o de la unión gastroesofágica (UGE) avanzado HER2 negativo que no habían recibido previamente terapia sistémica para la enfermedad metastásica. Los pacientes con una enfermedad autoinmune que requiriera terapia sistémica en los 2 años anteriores al tratamiento o una afección médica que requiriera inmunosupresión no eran elegibles. La aleatorización se estratificó por la expresión de PD-L1 (CPS ≥1 o CPS <1), el régimen de quimioterapia (FP o CAPOX) y la región geográfica (Europa/Israel/América del Norte/Australia, Asia o Resto del Mundo). Los pacientes se aleatorizaron (1:1) a uno de los siguientes brazos de tratamiento; el tratamiento se administró antes de la quimioterapia el día 1 de cada ciclo:
- KEYTRUDA 200 mg, quimioterapia de combinación a elección del investigador de cisplatino 80 mg/m2 y 5-FU 800 mg/m2/día durante 5 días (FP) u oxaliplatino 130 mg/m2 y capecitabina 1000 mg/m2 bid durante 14 días (CAPOX).
- Placebo, quimioterapia de combinación a elección del investigador de cisplatino 80 mg/m2 y 5-FU 800 mg/m2/día durante 5 días (FP) u oxaliplatino 130 mg/m2 y capecitabina 1000 mg/m2 bid durante 14 días (CAPOX).
Todos los medicamentos del estudio, excepto la capecitabina oral, se administraron como infusión intravenosa cada 3 semanas. Los agentes de platino se podían administrar durante 6 o más ciclos según las directrices locales. El tratamiento con KEYTRUDA continuó hasta la progresión de la enfermedad definida por RECIST v1.1, según lo determinado por BICR, toxicidad inaceptable o un máximo de 24 meses. La principal medida de resultado de eficacia fue la SO. Las medidas de resultado de eficacia secundarias adicionales incluyeron la SPP, la TMR y la DoR, según la evaluación de BICR utilizando RECIST v1.1, modificada para seguir un máximo de 10 lesiones diana y un máximo de 5 lesiones diana por órgano.
Las características de la población fueron: mediana de edad de 62 años (rango: 21 a 86), 39% de edad igual o superior a 65 años; 68% hombres y 32% mujeres; 55% blancos, 34% asiáticos, 4,6% múltiples, 4,2% amerindios o nativos de Alaska, 1,3% negros y 0,2% nativos de Hawái u otros isleños del Pacífico; 76% no hispanos o latinos y 21% hispanos o latinos; 37% ECOG PS de 0 y 63% ECOG PS de 1. El noventa y siete por ciento de los pacientes tenían enfermedad metastásica (estadio IV) y el 3% tenían enfermedad localmente avanzada irresecable. El setenta y ocho por ciento tenían tumores que expresaban PD-L1 con una CPS ≥1 y el 5% (n=74) tenían tumores MSI-H. El ochenta y seis por ciento de los pacientes recibieron CAPOX.
Se demostró una mejora estadísticamente significativa en la SO, la SPP y la TMR en los pacientes aleatorizados a KEYTRUDA en combinación con quimioterapia en comparación con placebo en combinación con quimioterapia en el momento de un análisis provisional preespecificado de la SO. Los resultados de eficacia se resumen en la Tabla 84 y en las Figuras 20 y 21.
| Variable principal | KEYTRUDA 200 mg cada 3 semanas y FP o CAPOX n=790 |
Placebo
y |
KEYTRUDA 200 mg cada 3 semanas y FP o CAPOX n=618 |
Placebo
y |
KEYTRUDA 200 mg cada 3 semanas y FP o CAPOX n=279 |
Placebo
y |
|---|---|---|---|---|---|---|
| Todos los pacientes | CPS≥1 | CPS≥10 | ||||
| + Denota respuesta en curso | ||||||
|
||||||
| SO | ||||||
| Número (%) de pacientes con evento | 603 (76) | 666 (84) | 464 (75) | 526 (85) | 188 (67) | 226 (83) |
| Mediana en meses (IC del 95%) | 12.9 (11.9, 14.0) | 11.5 (10.6, 12.1) | 13.0 (11.6, 14.2) | 11.4 (10.5, 12.0) | 15.7 (13.8, 19.3) | 11.8 (10.3, 12.7) |
| Hazard ratio† (IC del 95%) | 0.78 (0.70, 0.87) | 0.74 (0.65, 0.84) | 0.65 (0.53, 0.79) | |||
| Valor p (log-rank estratificado)‡ | <0.0001 | <0.0001 | <0.0001 | |||
| SPV | ||||||
| Número (%) de pacientes con evento | 572 (72) | 608 (77) | 443 (72%) | 483 (78%) | 190 (68) | 210 (77) |
| Mediana en meses (IC del 95%) | 6.9 (6.3, 7.2) | 5.6 (5.5, 5.7) | 6.9 (6.0, 7.2) | 5.6 (5.4, 5.7) | 8.1 (6.8, 8.5) | 5.6 (5.4, 6.7) |
| Hazard ratio† (IC del 95%) | 0.76 (0.67, 0.85) | 0.72 (0.63, 0.82) | 0.62 (0.51, 0.76) | |||
| Valor p (log-rank estratificado)‡ | <0.0001 | <0.0001 | <0.0001 | |||
| Tasa de respuesta objetiva | ||||||
| TRO§ (IC del 95%) | 51% (48, 55) | 42% (38, 45) | 52% (48, 56) | 43% (39, 47) | 61% (55, 66) | 43% (37, 49) |
| Tasa de respuesta completa | 9% | 6% | 10% | 6% | 13% | 5% |
| Tasa de respuesta parcial | 42% | 36% | 42% | 37% | 48% | 38% |
| Valor p¶ | <0.0001 | 0.0004 | <0.0001 | |||
| Duración de la respuesta | n=405 | n=331 | n=322 | n=263 | n=169 | n=117 |
| Mediana en meses# (IC del 95%) | 8.0 (7.0, 9.7) | 5.7 (5.5, 6.9) | 8.3 (7.0, 10.9) | 5.6 (5.4, 6.9) | 10.9 (8.0, 13.8) | 5.8 (5.3, 7.0) |
| Rango en meses | 1.2+, 41.5+ | 1.3+, 34.7+ | 1.2+, 41.5+ | 1.3+, 34.2+ | 1.2+, 41.5+ | 1.4+, 31.2+ |
|
|
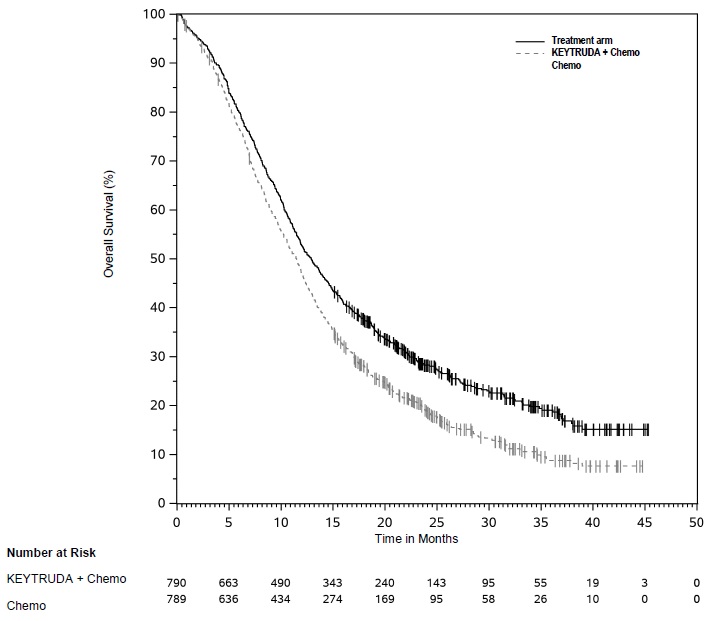
|
|
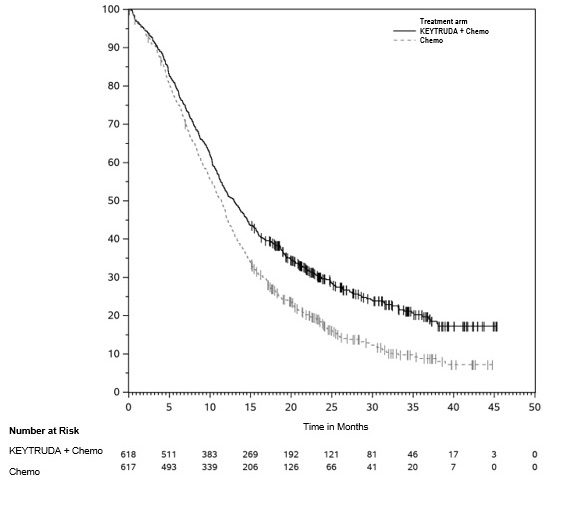
En un análisis de subgrupos exploratorio en pacientes con PD-L1 CPS<1 (n=344) en el momento del análisis intermedio preespecificado de OS, la OS mediana fue de 12,7 meses (IC del 95%: 11,4, 15,0) para el brazo KEYTRUDA y de 12,2 meses (IC del 95%: 9,5, 14,0) para el brazo placebo, con una HR de 0,92 (IC del 95%: 0,73, 1,17).
14.11 Cáncer de esófago
Tratamiento de primera línea del cáncer de esófago/unión gastroesofágica localmente avanzado irresecable o metastásico
KEYNOTE-590
La eficacia de KEYTRUDA se investigó en KEYNOTE-590 (NCT03189719), un ensayo multicéntrico, aleatorizado y controlado con placebo en el que se inscribieron 749 pacientes con carcinoma de esófago o unión gastroesofágica (tumores con epicentro de 1 a 5 centímetros por encima de la unión gastroesofágica) metastásico o localmente avanzado que no eran candidatos a resección quirúrgica o quimiorradiación definitiva. El estado de PD-L1 se determinó centralmente en muestras de tumor en todos los pacientes utilizando el kit pharmDx PD-L1 IHC 22C3. Los pacientes con enfermedad autoinmune activa, una afección médica que requiriera inmunosupresión o que hubieran recibido terapia sistémica previa en el contexto localmente avanzado o metastásico no fueron elegibles. La aleatorización se estratificó por histología tumoral (carcinoma de células escamosas frente a adenocarcinoma), región geográfica (Asia frente a ex-Asia) y estado de rendimiento ECOG (0 frente a 1).
Los pacientes se asignaron aleatoriamente (1:1) a uno de los siguientes brazos de tratamiento; todos los medicamentos del estudio se administraron mediante infusión intravenosa:
- KEYTRUDA 200 mg el día 1 de cada ciclo de tres semanas en combinación con cisplatino 80 mg/m2 IV el día 1 de cada ciclo de tres semanas durante un máximo de seis ciclos y FU 800 mg/m2 IV por día del día 1 al día 5 de cada ciclo de tres semanas, o según el estándar local para la administración de FU, durante un máximo de 24 meses.
- Placebo el día 1 de cada ciclo de tres semanas en combinación con cisplatino 80 mg/m2 IV el día 1 de cada ciclo de tres semanas durante un máximo de seis ciclos y FU 800 mg/m2 IV por día del día 1 al día 5 de cada ciclo de tres semanas, o según el estándar local para la administración de FU, durante un máximo de 24 meses.
El tratamiento con KEYTRUDA o quimioterapia continuó hasta que se produjo una toxicidad inaceptable o progresión de la enfermedad. Los pacientes pudieron recibir tratamiento con KEYTRUDA durante un máximo de 24 meses en ausencia de progresión de la enfermedad. Las principales medidas de resultado de eficacia fueron la OS y la PFS, evaluadas por el investigador según RECIST v1.1 (modificada para seguir un máximo de 10 lesiones diana y un máximo de 5 lesiones diana por órgano). Las medidas de resultado de eficacia adicionales fueron la ORR y la DoR, según RECIST v1.1 modificada, evaluadas por el investigador.
Las características de la población del estudio fueron: edad media de 63 años (rango: 27 a 94), 43% de edad igual o superior a 65 años; 83% hombres; 37% blancos, 53% asiáticos y 1% negros; 40% tenían un PS ECOG de 0 y 60% tenían un PS ECOG de 1. El noventa y un por ciento tenía enfermedad M1 y el 9% tenía enfermedad M0. El setenta y tres por ciento tenía una histología tumoral de carcinoma de células escamosas y el 27% tenía adenocarcinoma.
El ensayo demostró una mejora estadísticamente significativa en la OS y la PFS para los pacientes asignados aleatoriamente a KEYTRUDA en combinación con quimioterapia, en comparación con la quimioterapia.
La Tabla 85 y la Figura 22 resumen los resultados de eficacia de KEYNOTE-590 en todos los pacientes.
| Variable principal | KEYTRUDA 200 mg cada 3 semanas Cisplatino FU n=373 |
Placebo Cisplatino FU n=376 |
|---|---|---|
| OS | ||
| Número (%) de eventos | 262 (70) | 309 (82) |
| Mediana en meses (IC del 95%) |
12.4 (10.5, 14.0) |
9.8 (8.8, 10.8) |
| Razón de riesgos* (IC del 95%) | 0.73 (0.62, 0.86) | |
| Valor p† | <0.0001 | |
| PFS | ||
| Número de eventos (%) | 297 (80) | 333 (89) |
| Mediana en meses (IC del 95%) |
6.3 (6.2, 6.9) |
5.8 (5.0, 6.0) |
| Razón de riesgos* (IC del 95%) | 0.65 (0.55, 0.76) | |
| Valor p† | <0.0001 | |
| Tasa de respuesta objetiva | ||
| ORR, %‡ (IC 95%) |
45 (40, 50) |
29 (25, 34) |
| Número (%) de respuestas completas | 24 (6) | 9 (2.4) |
| Número (%) de respuestas parciales | 144 (39) | 101 (27) |
| Valor p§ | <0.0001 | |
| Duración de la respuesta | ||
| Mediana en meses (rango) |
8.3 (1.2+, 31.0+) |
6.0 (1.5+, 25.0+) |
|
|
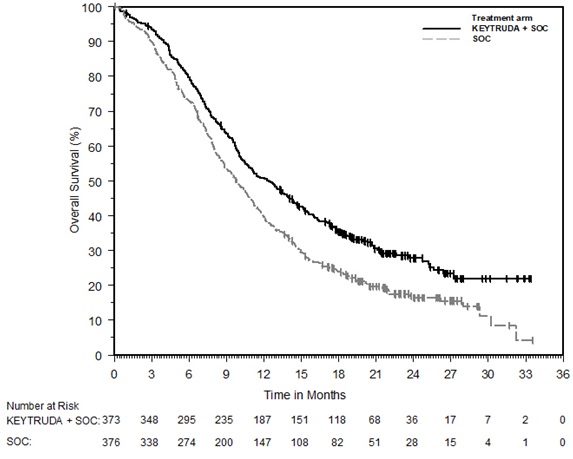
En una prueba formal preespecificada de SO en pacientes con CPS de PD-L1 ≥ 10 (n=383), la mediana fue de 13,5 meses (IC del 95%: 11,1; 15,6) para el brazo KEYTRUDA y de 9,4 meses (IC del 95%: 8,0; 10,7) para el brazo placebo, con una HR de 0,62 (IC del 95%: 0,49; 0,78; p-valor < 0,0001). En un análisis exploratorio, en pacientes con CPS de PD-L1 < 10 (n=347), la SO mediana fue de 10,5 meses (IC del 95%: 9,7; 13,5) para el brazo KEYTRUDA y de 10,6 meses (IC del 95%: 8,8; 12,0) para el brazo placebo, con una HR de 0,86 (IC del 95%: 0,68; 1,10).
Cáncer esofágico metastásico o localmente avanzado recidivante previamente tratado
KEYNOTE-181
La eficacia de KEYTRUDA se investigó en KEYNOTE-181 (NCT02564263), un ensayo multicéntrico, aleatorizado, abierto y controlado con activo en el que se inscribieron 628 pacientes con cáncer esofágico metastásico o localmente avanzado recidivante que progresaron después de una o más líneas de tratamiento sistémico previo para la enfermedad avanzada. Los pacientes con cáncer esofágico positivo para HER2/neu debían haber recibido tratamiento con terapia dirigida a HER2/neu aprobada. Todos los pacientes debían tener muestras de tumor para la prueba de PD-L1 en un laboratorio central; el estado de PD-L1 se determinó utilizando el kit pharmDx PD-L1 IHC 22C3. Los pacientes con antecedentes de neumonitis no infecciosa que requiriera esteroides o neumonitis actual, enfermedad autoinmune activa o una afección médica que requiriera inmunosupresión no eran elegibles.
Los pacientes se asignaron aleatoriamente (1:1) para recibir KEYTRUDA 200 mg cada 3 semanas o el tratamiento a elección del investigador de cualquiera de los siguientes regímenes de quimioterapia, todos administrados por vía intravenosa: paclitaxel 80-100 mg/m2 en los días 1, 8 y 15 de cada ciclo de 4 semanas, docetaxel 75 mg/m2 cada 3 semanas o irinotecán 180 mg/m2 cada 2 semanas. La aleatorización se estratificó por histología tumoral (carcinoma de células escamosas esofágico [ESCC] frente a adenocarcinoma esofágico [EAC]/EAC tipo I de Siewert de la unión gastroesofágica [UGE]) y región geográfica (Asia frente a ex-Asia). El tratamiento con KEYTRUDA o quimioterapia continuó hasta que se produjo una toxicidad inaceptable o la progresión de la enfermedad. Se permitió que los pacientes aleatorizados a KEYTRUDA continuaran más allá de la primera progresión de la enfermedad definida por RECIST v1.1 (modificada para seguir un máximo de 10 lesiones diana y un máximo de 5 lesiones diana por órgano) si estaban clínicamente estables hasta que la primera evidencia radiográfica de progresión de la enfermedad se confirmara al menos 4 semanas después con imágenes repetidas. Los pacientes tratados con KEYTRUDA sin progresión de la enfermedad podían recibir tratamiento hasta por 24 meses. La evaluación del estado del tumor se realizó cada 9 semanas. La principal medida de resultado de eficacia fue la SO evaluada en las siguientes poblaciones coprimas: pacientes con ESCC, pacientes con tumores que expresan PD-L1 CPS ≥10 y todos los pacientes aleatorizados. Las medidas de resultado de eficacia adicionales fueron la PFS, la ORR y la DoR, según RECIST v1.1, modificada para seguir un máximo de 10 lesiones diana y un máximo de 5 lesiones diana por órgano, según la evaluación de BICR.
Se inscribieron y asignaron aleatoriamente un total de 628 pacientes a KEYTRUDA (n=314) o al tratamiento a elección del investigador (n=314). De estos 628 pacientes, 167 (27%) tenían ESCC que expresaba PD-L1 con una CPS ≥10. De estos 167 pacientes, 85 pacientes fueron asignados aleatoriamente a KEYTRUDA y 82 pacientes al tratamiento a elección del investigador [paclitaxel (n=50), docetaxel (n=19) o irinotecán (n=13)]. Las características basales de estos 167 pacientes fueron: edad media de 65 años (rango: 33 a 80), 51% de edad igual o superior a 65 años; 84% hombres; 32% blancos y 68% asiáticos; 38% tenían una ECOG PS de 0 y 62% tenían una ECOG PS de 1. El noventa por ciento tenía enfermedad M1 y el 10% tenía enfermedad M0. Antes de la inscripción, el 99% de los pacientes habían recibido tratamiento a base de platino y el 84% también habían recibido tratamiento con una fluoropirimidina. El treinta y tres por ciento de los pacientes recibieron tratamiento previo con un taxano.
La razón de riesgo de SO observada fue de 0,77 (IC del 95%: 0,63; 0,96) en pacientes con ESCC, de 0,70 (IC del 95%: 0,52; 0,94) en pacientes con tumores que expresan PD-L1 CPS ≥10 y de 0,89 (IC del 95%: 0,75; 1,05) en todos los pacientes aleatorizados. Tras un examen más detenido en pacientes cuyos tumores de ESCC expresaban PD-L1 (CPS ≥10), se observó una mejora en la SO entre los pacientes aleatorizados a KEYTRUDA en comparación con la quimioterapia. La tabla 86 y la figura 23 resumen las principales medidas de eficacia de KEYNOTE-181 para pacientes con ESCC CPS ≥10.
| Variable principal | KEYTRUDA 200 mg cada 3 semanas n=85 |
Quimioterapia n=82 |
|---|---|---|
|
||
| SO | ||
| Número (%) de pacientes con evento | 68 (80%) | 72 (88%) |
| Mediana en meses (IC del 95%) | 10,3 (7,0; 13,5) | 6,7 (4,8; 8,6) |
| Razón de riesgos* (IC del 95%) | 0,64 (0,46; 0,90) | |
| PFS | ||
| Número (%) de pacientes con evento | 76 (89%) | 76 (93%) |
| Mediana en meses (IC del 95%) | 3,2 (2,1; 4,4) | 2,3 (2,1; 3,4) |
| Razón de riesgos* (IC del 95%) | 0,66 (0,48; 0,92) | |
| Tasa de respuesta objetiva | ||
| TRO (IC del 95%) | 22 (14, 33) | 7 (3, 15) |
| Número (%) de respuestas completas | 4 (5) | 1 (1) |
| Número (%) de respuestas parciales | 15 (18) | 5 (6) |
| Duración mediana de la respuesta en meses (intervalo) | 9.3 (2.1+, 18.8+) | 7.7 (4.3, 16.8+) |
|
|
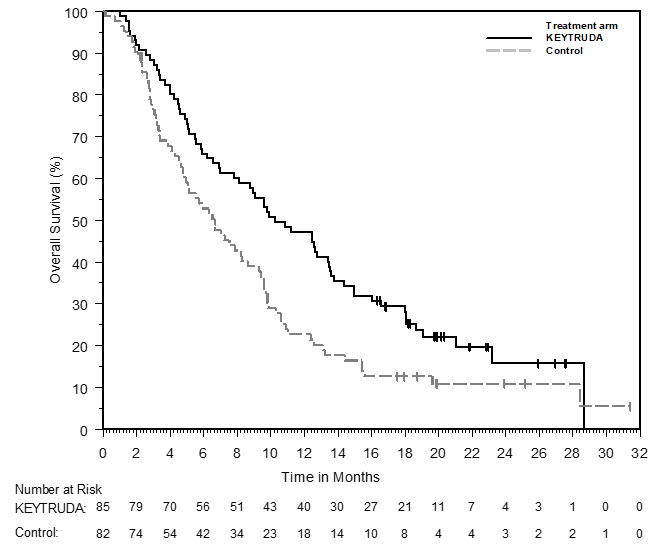
KEYNOTE-180
La eficacia de KEYTRUDA se investigó en KEYNOTE-180 (NCT02559687), un ensayo multicéntrico, no aleatorizado, abierto en el que se inscribieron 121 pacientes con cáncer de esófago localmente avanzado o metastásico que progresaron después de al menos 2 tratamientos sistémicos previos para la enfermedad avanzada. Con la excepción del número de líneas de tratamiento previas, los criterios de elegibilidad fueron similares y el régimen de dosificación idéntico a KEYNOTE-181.
Las principales medidas de resultado de eficacia fueron la TMR y la DR según RECIST v1.1, modificada para seguir un máximo de 10 lesiones diana y un máximo de 5 lesiones diana por órgano, según la evaluación de BICR.
De los 121 pacientes inscritos, el 29% (n=35) tenían CECC que expresaba PD-L1 CPS ≥10. Las características basales de estos 35 pacientes fueron: mediana de edad de 65 años (rango: 47 a 81), 51% de edad 65 o superior; 71% hombres; 26% blancos y 69% asiáticos; 40% tenían una puntuación ECOG PS de 0 y 60% tenían una puntuación ECOG PS de 1. El cien por cien tenían enfermedad M1.
La TMR en los 35 pacientes con CECC que expresaban PD-L1 fue del 20% (IC del 95%: 8, 37). Entre los 7 pacientes que respondieron, la DR osciló entre 4,2 y 25,1+ meses, con 5 pacientes (71%) que tuvieron respuestas de 6 meses o más y 3 pacientes (57%) que tuvieron respuestas de 12 meses o más.
14.12 Cáncer de cuello uterino
Cáncer de cuello uterino en estadio FIGO 2014 III-IVA con quimioradioterapia
La eficacia de KEYTRUDA en combinación con CRT (cisplatino y radioterapia de haz externo [EBRT] seguida de braquiterapia [BT]) se investigó en KEYNOTE-A18 (NCT04221945), un ensayo multicéntrico, aleatorizado, doble ciego, controlado con placebo en el que se inscribieron 1060 pacientes con cáncer de cuello uterino que no habían recibido previamente ninguna cirugía definitiva, radiación o terapia sistémica para el cáncer de cuello uterino. Hubo 596 pacientes con estadio FIGO 2014 III-IVA (afectación tumoral de la vagina inferior con o sin extensión a la pared pélvica o hidronefrosis/riñón no funcionante o que se ha extendido a órganos pélvicos adyacentes) con enfermedad con o sin ganglios positivos, y 462 pacientes con estadio FIGO 2014 IB2-IIB (lesiones tumorales >4 cm o lesiones clínicamente visibles que se han extendido más allá del útero pero que no se han extendido a la pared pélvica o al tercio inferior de la vagina) con enfermedad con ganglios positivos; dos pacientes tenían enfermedad en estadio FIGO 2014 IVB. La aleatorización se estratificó por el tipo previsto de EBRT (radioterapia modulada por intensidad [IMRT] o terapia de arco modulada volumétrica [VMAT] frente a no IMRT y no VMAT), estadio en la evaluación del cáncer de cuello uterino (estadio FIGO 2014 IB2-IIB frente a estadio FIGO 2014 III-IVA) y dosis total de radioterapia prevista (dosis de EBRT + braquiterapia de <70 Gy frente a ≥70 Gy según la dosis equivalente [EQD2]).
Los pacientes fueron aleatorizados (1:1) a uno de dos brazos de tratamiento:
- KEYTRUDA 200 mg IV cada 3 semanas (5 ciclos) de forma concomitante con cisplatino 40 mg/m2 IV semanal (5 ciclos, se podría administrar una sexta infusión opcional según la práctica local), y radioterapia (EBRT seguida de BT), seguida de KEYTRUDA 400 mg IV cada 6 semanas (15 ciclos)
- Placebo IV cada 3 semanas (5 ciclos) de forma concomitante con cisplatino 40 mg/m2 IV semanal (5 ciclos, se podría administrar una sexta infusión opcional según la práctica local), y radioterapia (EBRT seguida de BT), seguida de placebo IV cada 6 semanas (15 ciclos)
El tratamiento continuó hasta la progresión de la enfermedad definida por RECIST v1.1 según lo determinado por el investigador o toxicidad inaceptable.
La evaluación del estado del tumor se realizó cada 12 semanas a partir de la finalización de la CRT durante los dos primeros años, seguida de cada 24 semanas en el año 3 y luego anualmente. Las principales medidas de resultado de eficacia fueron la SLP según la evaluación del investigador de acuerdo con RECIST v1.1, modificada para seguir un máximo de 10 lesiones diana y un máximo de 5 lesiones diana por órgano, o confirmación histopatológica, y la SO.
Entre los 596 pacientes con enfermedad en estadio FIGO 2014 III-IVA, las características basales fueron: mediana de edad de 52 años (rango: 22 a 87), 17% de edad 65 o superior; 36% blancos, 34% asiáticos, 1% negros; 38% hispanos o latinos; 68% ECOG PS 0 y 32% ECOG PS 1; 93% con CPS ≥1; 70% tenían ganglios linfáticos pélvicos y/o para-aórticos positivos y 30% no tenían ganglios linfáticos pélvicos ni para-aórticos positivos; 83% tenían carcinoma de células escamosas y 17% tenían histología no escamosa. Con respecto a la radiación, el 85% de los pacientes recibieron EBRT IMRT o VMAT, y la dosis mediana de EQD2 fue de 87 Gy (rango: 7 a 114).
El ensayo demostró una mejora estadísticamente significativa en la SLP en la población general. En un análisis de subgrupo exploratorio para los 462 pacientes (44%) con enfermedad en estadio FIGO 2014 IB2-IIB, la estimación de la HR de la SLP fue de 0,91 (IC del 95%: 0,63, 1,31), lo que indica que la mejora de la SLP en la población general se atribuyó principalmente a los resultados observados en el subgrupo de pacientes con enfermedad en estadio FIGO 2014 III-IVA. Los datos de SO no estaban maduros en el momento del análisis de la SLP, con un 10% de muertes en la población general.
Los resultados de eficacia en el análisis de subgrupo exploratorio de 596 pacientes con enfermedad en estadio FIGO 2014 III-IVA se resumen en la Tabla 87 y la Figura 24.
| Variable principal | KEYTRUDA 200 mg cada 3 semanas y 400 mg cada 6 semanas con CRT n=293 |
Placebo
con CRT |
|---|---|---|
| CRT = Quimioradioterapia NR = no alcanzado |
||
|
||
| SLP según el investigador | ||
| Número de pacientes con evento (%) | 61 (21%) | 94 (31%) |
| Mediana en meses (IC del 95%) | NR (NR, NR) | NR (18.8, NR) |
| Tasa de supervivencia libre de progresión a 12 meses (IC del 95%) | 81% (75, 85) | 70% (64, 76) |
| Hazard ratio* (IC del 95%) | 0.59 (0.43, 0.82) | |
|
|
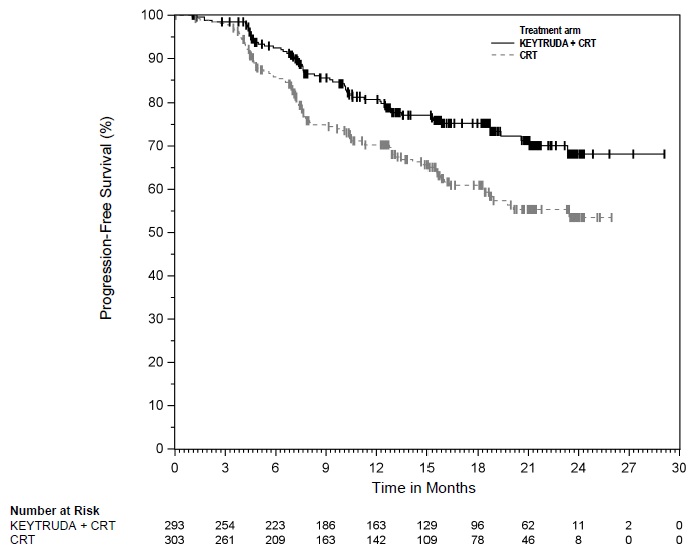
Cáncer de cuello uterino persistente, recurrente o metastásico
La eficacia de KEYTRUDA en combinación con paclitaxel y cisplatino o paclitaxel y carboplatino, con o sin bevacizumab, se investigó en KEYNOTE-826 (NCT03635567), un ensayo multicéntrico, aleatorizado, doble ciego, controlado con placebo en el que se inscribieron 617 pacientes con cáncer de cuello uterino persistente, recurrente o metastásico de primera línea que no habían sido tratados con quimioterapia, excepto cuando se usó concurrentemente como agente radiosensibilizador. Los pacientes fueron inscritos independientemente del estado de expresión de PD-L1 tumoral. Los pacientes con enfermedad autoinmune que requirieron terapia sistémica dentro de los 2 años del tratamiento o una condición médica que requiriera inmunosupresión fueron inelegibles. La aleatorización se estratificó por el estado metastásico en el diagnóstico inicial, la decisión del investigador de usar bevacizumab y el estado de PD-L1 (CPS <1 vs. CPS 1 a <10 vs. CPS ≥10). Los pacientes fueron aleatorizados (1:1) a uno de los dos grupos de tratamiento:
- Grupo de tratamiento 1: KEYTRUDA 200 mg más quimioterapia con o sin bevacizumab
- Grupo de tratamiento 2: Placebo más quimioterapia con o sin bevacizumab
El investigador seleccionó uno de los siguientes cuatro regímenes de tratamiento antes de la aleatorización:
- Paclitaxel 175 mg/m2 + cisplatino 50 mg/m2
- Paclitaxel 175 mg/m2 + cisplatino 50 mg/m2 + bevacizumab 15 mg/kg
- Paclitaxel 175 mg/m2 + carboplatino AUC 5 mg/mL/min
- Paclitaxel 175 mg/m2 + carboplatino AUC 5 mg/mL/min + bevacizumab 15 mg/kg
Todos los medicamentos del estudio se administraron como infusión intravenosa. Todos los tratamientos del estudio se administraron el día 1 de cada ciclo de tratamiento de 3 semanas. La cisplatina podría administrarse el día 2 de cada ciclo de tratamiento de 3 semanas. El tratamiento con KEYTRUDA continuó hasta la progresión de la enfermedad definida por RECIST v1.1, toxicidad inaceptable o un máximo de 24 meses. Se permitió la administración de KEYTRUDA más allá de la progresión de la enfermedad definida por RECIST si el paciente era clínicamente estable y se consideraba que estaba obteniendo un beneficio clínico por parte del investigador. La evaluación del estado tumoral se realizó cada 9 semanas durante el primer año, seguida de cada 12 semanas a partir de entonces. Las principales medidas de resultado de eficacia fueron la SO y la SPP, según la evaluación del investigador de acuerdo con RECIST v1.1, modificada para seguir un máximo de 10 lesiones diana y un máximo de 5 lesiones diana por órgano. Las medidas de resultado de eficacia adicionales fueron la TTR y la DR, según RECIST v1.1, según la evaluación del investigador.
De los 617 pacientes inscritos, 548 pacientes (89%) tenían tumores que expresaban PD-L1 con una CPS ≥1. Entre estos 548 pacientes inscritos con tumores que expresaban PD-L1, 273 pacientes fueron aleatorizados a KEYTRUDA en combinación con quimioterapia con o sin bevacizumab, y 275 pacientes fueron aleatorizados a placebo en combinación con quimioterapia con o sin bevacizumab. El sesenta y tres por ciento de los 548 pacientes recibieron bevacizumab como parte del tratamiento del estudio. Las características basales de los 548 pacientes fueron: edad media de 51 años (rango: 22 a 82), 16% de edad 65 o mayor; 59% blancos, 18% asiáticos, 6% amerindios o nativos de Alaska, y 1% negros; 37% hispanos o latinos; 56% estado de rendimiento ECOG 0 y 43% estado de rendimiento ECOG 1. El setenta y cinco por ciento tenía carcinoma de células escamosas, 21% adenocarcinoma y 5% histología adenoescamosa, y el 32% de los pacientes tenían enfermedad metastásica en el diagnóstico. Al inicio del estudio, el 21% de los pacientes tenían solo enfermedad metastásica y el 79% tenían enfermedad persistente o recurrente con o sin metástasis a distancia, de los cuales el 39% había recibido quimiorradiación previa solamente y el 17% había recibido quimiorradiación previa más cirugía.
Se demostró una mejora estadísticamente significativa en la SO y la SPP en los pacientes aleatorizados para recibir KEYTRUDA en comparación con los pacientes aleatorizados para recibir placebo. Se realizó un análisis actualizado de la SO en el momento del análisis final cuando se observaron 354 muertes en la población de CPS ≥1. La Tabla 88 y la Figura 25 resumen las principales medidas de eficacia para KEYNOTE-826 para pacientes con tumores que expresan PD-L1 (CPS ≥1).
| Variable principal | KEYTRUDA 200 mg cada 3 semanas y quimioterapia* con o sin bevacizumab n=273 |
Placebo
y quimioterapia* con o sin bevacizumab |
|---|---|---|
| + Denota respuesta en curso NR = no alcanzado |
||
|
||
| SO | ||
| Número de pacientes con evento (%) | 118 (43.2) | 154 (56.0) |
| Mediana en meses (IC del 95%) | NR (19.8, NR) | 16.3 (14.5, 19.4) |
| Hazard ratio† (IC 95%) | 0.64 (0.50, 0.81) | |
| p-Value‡ | 0.0001 | |
| Supervivencia global actualizada | ||
| Número de pacientes con evento (%) | 153 (56.0%) | 201 (73.1%) |
| Mediana en meses (IC 95%) | 28.6 (22.1, 38.0) | 16.5 (14.5, 20.0) |
| Hazard ratio† (IC 95%) | 0.60 (0.49, 0.74) | |
| SPV | ||
| Número de pacientes con evento (%) | 157 (57.5) | 198 (72.0) |
| Mediana en meses (IC 95%) | 10.4 (9.7, 12.3) | 8.2 (6.3, 8.5) |
| Hazard ratio† (IC 95%) | 0.62 (0.50, 0.77) | |
| p-Value§ | < 0.0001 | |
| Tasa de respuesta objetiva | ||
| ORR¶ (IC 95%) | 68% (62, 74) | 50% (44, 56) |
| Tasa de respuesta completa | 23% | 13% |
| Tasa de respuesta parcial | 45% | 37% |
| Duración de la respuesta | ||
| Mediana en meses (rango) | 18.0 (1.3+, 24.2+) | 10.4 (1.5+, 22.0+) |
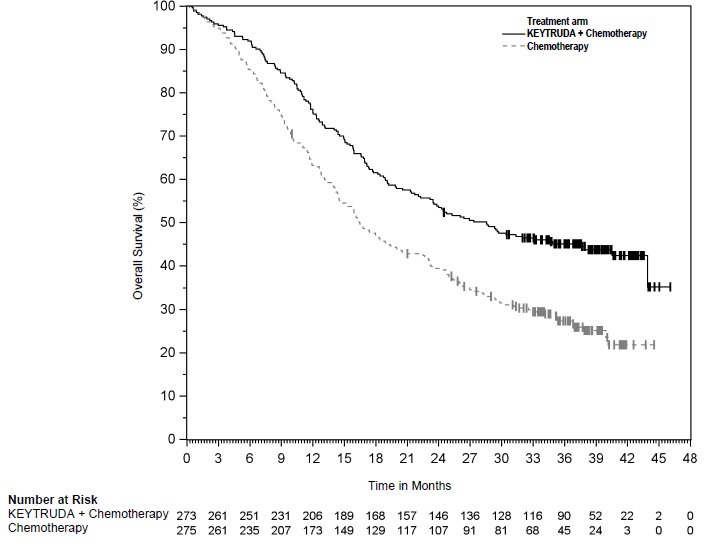
Cáncer cervical recidivante o metastásico previamente tratado
La eficacia de KEYTRUDA se investigó en 98 pacientes con cáncer cervical recidivante o metastásico inscritos en una única cohorte (Cohorte E) en KEYNOTE-158 (NCT02628067), un ensayo multicéntrico, no aleatorizado, abierto y multicóhorte. El ensayo excluyó a pacientes con enfermedad autoinmune o una afección médica que requiriera inmunosupresión. Los pacientes recibieron KEYTRUDA 200 mg por vía intravenosa cada 3 semanas hasta que se produjo una toxicidad inaceptable o una progresión de la enfermedad documentada. Los pacientes con progresión inicial de la enfermedad radiográfica podrían recibir dosis adicionales de tratamiento durante la confirmación de la progresión, a menos que la progresión de la enfermedad fuera sintomática, rápidamente progresiva, requiriera intervención urgente o se produjera con una disminución del estado funcional. Los pacientes sin progresión de la enfermedad podrían recibir tratamiento hasta por 24 meses. La evaluación del estado tumoral se realizó cada 9 semanas durante los primeros 12 meses y cada 12 semanas a partir de entonces. Las principales medidas de resultado de eficacia fueron la ORR según RECIST v1.1, modificada para seguir un máximo de 10 lesiones diana y un máximo de 5 lesiones diana por órgano, según la evaluación de BICR, y la DoR.
De los 98 pacientes de la Cohorte E, 77 (79%) tenían tumores que expresaban PD-L1 con una CPS ≥ 1 y recibieron al menos una línea de quimioterapia en el contexto metastásico. El estado de PD-L1 se determinó utilizando el kit pharmDx IHC 22C3. Las características basales de estos 77 pacientes fueron: mediana de edad de 45 años (rango: 27 a 75); 81% blancos, 14% asiáticos y 3% negros; 32% ECOG PS de 0 y 68% ECOG PS de 1; 92% tenían carcinoma de células escamosas, 6% adenocarcinoma y 1% histología adenoescamosa; 95% tenían enfermedad M1 y 5% tenían enfermedad recidivante; y 35% tenían una y 65% tenían dos o más líneas de terapia previas en el contexto recidivante o metastásico.
No se observaron respuestas en pacientes cuyos tumores no tenían expresión de PD-L1 (CPS <1). Los resultados de eficacia se resumen en la Tabla 89 para pacientes con expresión de PD-L1 (CPS ≥1).
| Variable principal | KEYTRUDA 200 mg cada 3 semanas n=77* |
|---|---|
| + Denota respuesta en curso NR = no alcanzado |
|
| Tasa de respuesta objetiva | |
| ORR (IC del 95%) | 14,3% (7,4, 24,1) |
| Tasa de respuesta completa | 2,6% |
| Tasa de respuesta parcial | 11,7% |
| Duración de la respuesta | |
| Mediana en meses (rango) | NR (4,1, 18,6+)† |
| % con duración ≥6 meses | 91% |
14.13 Carcinoma Hepatocelular
HCC previamente tratado
La eficacia de KEYTRUDA se investigó en KEYNOTE-394 (NCT03062358), un ensayo multicéntrico, aleatorizado, controlado con placebo y doble ciego realizado en Asia en pacientes con HCC en estadio B o C de la Clasificación Clínica del Cáncer Hepático de Barcelona (BCLC), que habían sido tratados previamente con sorafenib o quimioterapia a base de oxaliplatino y que no eran aptos para o eran refractarios a la terapia locorregional. Los pacientes también debían tener una función hepática Child-Pugh A.
Los pacientes con hepatitis B tenían la enfermedad controlada con tratamiento (carga viral del VHB <2000 UI/mL o <104 copias/mL). Los pacientes con una enfermedad autoinmune que requiriera terapia sistémica dentro de los 2 años del tratamiento o una condición médica que requiriera inmunosupresión no eran elegibles. Los pacientes con encefalopatía hepática, invasión venosa portal de rama principal, ascitis clínicamente aparente o hemorragia varicosa esofágica o gástrica en los últimos 6 meses tampoco eran elegibles.
La aleatorización se estratificó por tratamiento previo: sorafenib frente a quimioterapia a base de oxaliplatino, invasión macrovascular y etiología (VHB activo frente a otros (VHC activo, no infectados)). Los pacientes se aleatorizaron (2:1) para recibir pembrolizumab 200 mg por vía intravenosa cada 3 semanas o placebo.
El tratamiento con KEYTRUDA continuó hasta la progresión de la enfermedad definida por RECIST v1.1 según lo determinado por BICR, toxicidad inaceptable o un máximo de 24 meses. La evaluación del estado tumoral se realizó cada 6 semanas. La principal medida de resultado de eficacia fue la SO. Las medidas de resultado de eficacia adicionales fueron la SPP, la TEO y la DuR, según lo evaluado por BICR utilizando RECIST v1.1, modificado para seguir un máximo de 10 lesiones diana y un máximo de 5 lesiones diana por órgano.
El estudio incluyó a 453 pacientes, y 360 (79%) tenían hepatitis B activa. Las características de la población en pacientes con hepatitis B activa fueron: mediana de edad de 52 años (rango: 23 a 82), 16% de edad igual o superior a 65 años; 86% hombres; 100% asiáticos; 42% ECOG PS de 0 y 58% ECOG PS de 1; 90% recibieron sorafenib previo y 10% recibieron quimioterapia previa a base de oxaliplatino. Las características de los pacientes también incluyeron enfermedad extrahepática (77%), invasión macrovascular (10%), estadio C de BCLC (93%) y B (7%), y AFP basal ≥200 ng/mL (57%).
KEYNOTE-394 demostró una mejora en la SO en pacientes con HCC secundario a hepatitis B aleatorizados a KEYTRUDA en comparación con placebo. Los resultados de eficacia se resumen en la Tabla 90 y la Figura 26.
| Variable principal | KEYTRUDA 200 mg cada 3 semanas n=236 |
Placebo
n=124 |
|---|---|---|
| + Denota respuesta en curso | ||
| SO* | ||
| Número (%) de pacientes con eventos | 172 (73) | 105 (85) |
| Mediana en meses (IC del 95%) | 13.9 (12.5, 17.9) | 13.0 (10.1, 15.6) |
| Razón de riesgo† (IC del 95%) | 0.78 (0.61, 0.99) | |
| SPP‡ | ||
| Número (%) de pacientes con eventos | 189 (80) | 108 (87) |
| Mediana en meses (IC del 95%) | 2 (1.4, 2.7) | 2.3 (1.4, 2.8) |
| Razón de riesgo† (IC del 95%) | 0.78 (0.61, 1.00) | |
| Tasa de respuesta objetiva‡ | ||
| TER§ (IC del 95%) | 11% (7, 16) | 1.6% (0.2, 5.7) |
| Número (%) de respuestas completas | 2 (0.9%) | 1 (0.8%) |
| Número (%) de respuestas parciales | 24 (10%) | 1 (0.8%) |
| Duración de la respuesta* | n=28 | n=2 |
| Mediana en meses¶ (intervalo) | 23.9 (2.6+, 44.4+) | 5.6 (3.0+, 5.6) |
| Figura 26: Curva de Kaplan-Meier para la supervivencia general en KEYNOTE-394 |
|
|
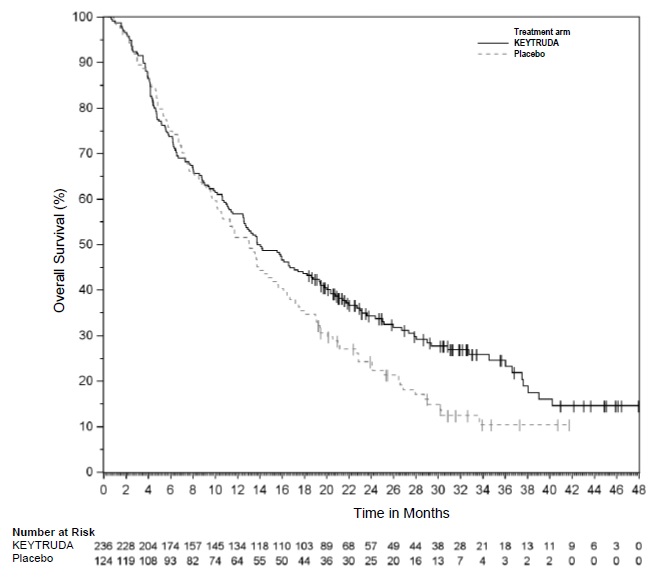
14.14 Cáncer de las vías biliares
La eficacia de KEYTRUDA en combinación con quimioterapia de gemcitabina y cisplatino se investigó en KEYNOTE-966 (NCT04003636), un ensayo multicéntrico, aleatorizado, doble ciego y controlado con placebo en el que se inscribieron 1069 pacientes con BTC localmente avanzado irresecable o metastásico, que no habían recibido terapia sistémica previa en el contexto de enfermedad avanzada. Los pacientes con enfermedad autoinmune que requirieron terapia sistémica dentro de los 2 años del tratamiento o una condición médica que requiriera inmunosupresión fueron inelegibles. La aleatorización se estratificó por región (Asia frente a no Asia), localmente avanzado frente a metastásico y sitio de origen (vesícula biliar, colangiocarcinoma intrahepático o extrahepático).
Los pacientes fueron aleatorizados (1:1) a KEYTRUDA 200 mg en el Día 1 más gemcitabina 1000 mg/m2 y cisplatino 25 mg/m2 en el Día 1 y el Día 8 cada 3 semanas, o placebo en el Día 1 más gemcitabina 1000 mg/m2 y cisplatino 25 mg/m2 en el Día 1 y el Día 8 cada 3 semanas. Los medicamentos del estudio se administraron mediante infusión intravenosa. El tratamiento continuó hasta que se produjo una toxicidad inaceptable o progresión de la enfermedad. Para pembrolizumab, el tratamiento continuó durante un máximo de 35 ciclos, o aproximadamente 24 meses. Para gemcitabina, el tratamiento podría continuar más allá de los 8 ciclos, mientras que para cisplatino, el tratamiento podría administrarse durante un máximo de 8 ciclos.
La administración de KEYTRUDA con quimioterapia se permitió más allá de la progresión de la enfermedad definida por RECIST si el paciente era clínicamente estable y el investigador consideraba que estaba obteniendo un beneficio clínico. La evaluación del estado tumoral se realizó al inicio y luego cada 6 semanas hasta las 54 semanas, seguidas de cada 12 semanas a partir de entonces.
Las características de la población del estudio fueron una edad media de 64 años (rango: 23 a 85), 47% de edad 65 o mayor; 52% hombres; 49% blancos, 46% asiáticos, 1.3% negros o afroamericanos; 10% hispanos o latinos; 46% ECOG PS de 0 y 54% ECOG PS de 1; 31% de los pacientes tenían antecedentes de infección por hepatitis B y 3% tenían antecedentes de infección por hepatitis C.
La principal medida de resultado de eficacia fue la OS. Las medidas de resultado de eficacia adicionales fueron PFS, ORR y DoR según lo evaluado por BICR de acuerdo con RECIST v1.1, modificado para seguir un máximo de 10 lesiones diana y un máximo de 5 lesiones diana por órgano.
La Tabla 91 y la Figura 27 resumen los resultados de eficacia de KEYNOTE-966.
| Variable principal | KEYTRUDA 200 mg cada 3 semanas con gemcitabina/cisplatino n=533 |
Placebo con gemcitabina/cisplatino n=536 |
|---|---|---|
| NS = no significativo | ||
|
||
| OS* | ||
| Número de pacientes con evento (%) | 414 (78%) | 443 (83%) |
| Mediana en meses (IC del 95%) | 12.7 (11.5, 13.6) | 10.9 (9.9, 11.6) |
| Razón de riesgo† (IC del 95%) | 0.83 (0.72, 0.95) | |
| Valor p‡ | 0.0034 | |
| PFS§ | ||
| Número (%) de pacientes con evento | 361 (68%) | 391 (73%) |
| Mediana en meses (IC del 95%) | 6.5 (5.7, 6.9) | 5.6 (5.1, 6.6) |
| Razón de riesgo† (IC del 95%) | 0.86 (0.75, 1.00) | |
| Valor p‡ | NS | |
| Tasa de respuesta objetiva§ | ||
| ORR¶ (IC del 95%) | 29% (25, 33) | 29% (25, 33) |
| Número (%) de respuestas completas | 11 (2.1%) | 7 (1.3%) |
| Número (%) de respuestas parciales | 142 (27%) | 146 (27%) |
| Valor p # | NS | |
| Duración de la respuesta* | n=156 | n=152 |
| Mediana en meses Þ (IC del 95%) | 8.3 (6.9, 10.2) | 6.8 (5.7, 7.1) |
| Figura 27: Curva de Kaplan-Meier para la supervivencia general en KEYNOTE-966 |
|
|
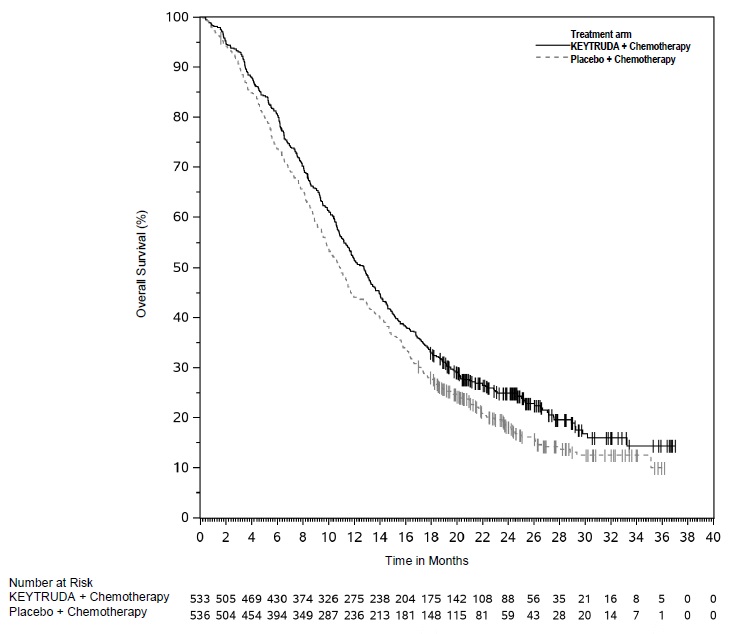
14.15 Carcinoma de células de Merkel
La eficacia de KEYTRUDA se investigó en KEYNOTE-017 (NCT02267603) y KEYNOTE-913 (NCT03783078), dos ensayos multicéntricos, no aleatorizados y abiertos en los que se inscribieron 105 pacientes con MCC metastásico o localmente avanzado recurrente que no habían recibido terapia sistémica previa para su enfermedad avanzada. Los pacientes con enfermedad autoinmune activa o una afección médica que requiriera inmunosupresión no fueron elegibles.
Los pacientes recibieron KEYTRUDA 2 mg/kg (KEYNOTE-017) o 200 mg (KEYNOTE-913) cada 3 semanas hasta que se produjo una toxicidad inaceptable o progresión de la enfermedad que fuera sintomática, rápidamente progresiva, requiriera intervención urgente, ocurriera con una disminución en el estado funcional o se confirmara al menos 4 semanas después con imágenes repetidas. Los pacientes sin progresión de la enfermedad fueron tratados hasta por 24 meses.
Las principales medidas de resultado de eficacia fueron la TEO y la DuR, según la evaluación de BICR según RECIST v1.1.
Entre los 105 pacientes inscritos, la mediana de edad fue de 73 años (rango: 38 a 91), el 79% tenía 65 años o más; el 62% eran hombres; el 80% eran blancos, la raza del 19% era desconocida o faltaba, y el 1% eran asiáticos; el 53% tenía un estado funcional ECOG de 0 y el 47% tenía un estado funcional ECOG de 1. El trece por ciento tenía enfermedad en estadio IIIB y el 84% tenía enfermedad en estadio IV. El setenta y seis por ciento de los pacientes habían sido sometidos a cirugía previa y el 51% habían recibido radioterapia previa.
Los resultados de eficacia se resumen en la Tabla 92.
| Variable principal | KEYNOTE-017 KEYTRUDA 2 mg/kg cada 3 semanas n=50 |
KEYNOTE-913 KEYTRUDA 200 mg o 2 mg/kg cada 3 semanas n=55 |
|---|---|---|
| + Denota respuesta en curso | ||
| NR = no alcanzado | ||
| Tasa de respuesta objetiva | ||
| TRO (IC del 95%) | 56% (41, 70) | 49% (35, 63) |
| Respuestas completas, n (%) | 12 (24%) | 9 (16%) |
| Respuestas parciales, n (%) | 16 (32%) | 18 (33%) |
| Duración de la respuesta | n=28 | n=27 |
| DoR mediana en meses (rango) | NR (5.9, 34.5+) | NR (4.8, 25.4+) |
| Pacientes con duración ≥6 meses, n (%) | 27 (96%) | 25 (93%) |
| Pacientes con duración ≥12 meses, n (%) | 15 (54%) | 19 (70%) |
14.16 Carcinoma de células renales
Tratamiento de primera línea con axitinib
KEYNOTE-426
La eficacia de KEYTRUDA en combinación con axitinib se investigó en KEYNOTE-426 (NCT02853331), un ensayo aleatorizado, multicéntrico y abierto realizado en 861 pacientes que no habían recibido terapia sistémica para CCR avanzada. Los pacientes fueron incluidos independientemente del estado de expresión tumoral de PD-L1. Los pacientes con enfermedad autoinmune activa que requirieran inmunosupresión sistémica en los últimos 2 años fueron inelegibles. La aleatorización se estratificó por las categorías de riesgo del Consorcio Internacional de la Base de Datos de RCC metastásico (IMDC) (favorable versus intermedio versus pobre) y la región geográfica (América del Norte versus Europa Occidental versus “Resto del mundo”).
Los pacientes fueron aleatorizados (1:1) a uno de los siguientes brazos de tratamiento:
- KEYTRUDA 200 mg por vía intravenosa cada 3 semanas hasta 24 meses en combinación con axitinib 5 mg por vía oral, dos veces al día. Los pacientes que toleraron axitinib 5 mg dos veces al día durante 2 ciclos consecutivos (6 semanas) pudieron aumentar a 7 mg y posteriormente a 10 mg dos veces al día. Axitinib podría interrumpirse o reducirse a 3 mg dos veces al día y posteriormente a 2 mg dos veces al día para controlar la toxicidad.
- Sunitinib 50 mg por vía oral, una vez al día durante 4 semanas y luego sin tratamiento durante 2 semanas.
El tratamiento con KEYTRUDA y axitinib continuó hasta la progresión de la enfermedad definida por RECIST v1.1 o una toxicidad inaceptable. La administración de KEYTRUDA y axitinib se permitió más allá de la progresión de la enfermedad definida por RECIST si el paciente era clínicamente estable y se consideraba que estaba obteniendo un beneficio clínico por parte del investigador. La evaluación del estado tumoral se realizó al inicio, después de la aleatorización en la semana 12, luego cada 6 semanas hasta la semana 54, y luego cada 12 semanas a partir de entonces.
Las características de la población del estudio fueron: edad media de 62 años (rango: 26 a 90), 38% de edad 65 o más; 73% hombres; 79% blancos y 16% asiáticos; 20% y 80% de los pacientes tenían una KPS basal de 70 a 80 y 90 a 100, respectivamente; y la distribución de los pacientes por categorías de riesgo de IMDC fue del 31% favorable, 56% intermedio y 13% pobre.
Las principales medidas de resultado de eficacia fueron la SO y la SPP, evaluadas por BICR según RECIST v1.1, modificadas para seguir un máximo de 10 lesiones diana y un máximo de 5 lesiones diana por órgano. Las medidas de resultado de eficacia adicionales incluyeron la TTR, evaluada por BICR. Se demostró una mejora estadísticamente significativa en la SO en el primer análisis interino preespecificado en pacientes aleatorizados a KEYTRUDA en combinación con axitinib en comparación con sunitinib. El ensayo también demostró mejoras estadísticamente significativas en la SPP y la TTR. Se realizó un análisis actualizado de la SO cuando se observaron 418 muertes según el número de muertes planificado para el análisis final preespecificado. La Tabla 93 y la Figura 28 resumen los resultados de eficacia de KEYNOTE-426.
| Variable principal | KEYTRUDA 200 mg cada 3 semanas y Axitinib |
Sunitinib |
|---|---|---|
| n=432 | n=429 | |
| NR = no alcanzado | ||
|
||
| SO | ||
| Número de pacientes con evento (%) | 59 (14%) | 97 (23%) |
| Mediana en meses (IC del 95%) | NR (NR, NR) | NR (NR, NR) |
| Razón de riesgo* (IC del 95%) | 0,53 (0,38, 0,74) | |
| Valor p† | <0,0001‡ | |
| SO actualizada | ||
| Número de pacientes con evento (%) | 193 (45%) | 225 (52%) |
| Mediana en meses (IC del 95%) | 45,7 (43,6, NR) | 40,1 (34,3, 44,2) |
| Razón de riesgo* (IC del 95%) | 0,73 (0,60, 0,88) | |
| SPP | ||
| Número de pacientes con evento (%) | 183 (42%) | 213 (50%) |
| Mediana en meses (IC del 95%) | 15.1 (12.6, 17.7) | 11.0 (8.7, 12.5) |
| Hazard ratio* (IC del 95%) | 0.69 (0.56, 0.84) | |
| Valor p† | 0.0001§ | |
| Tasa de respuesta objetiva | ||
| ORR¶ (IC del 95%) | 59% (54, 64) | 36% (31, 40) |
| Tasa de respuesta completa | 6% | 2% |
| Tasa de respuesta parcial | 53% | 34% |
| Valor p# | <0.0001 | |
| Figura 28: Curva de Kaplan-Meier para la supervivencia general actualizada en KEYNOTE-426 |
|
|
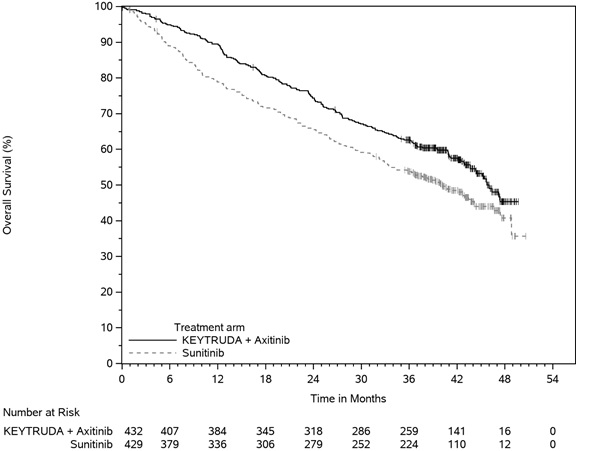
En un análisis exploratorio, el análisis actualizado de la supervivencia general (SG) en pacientes con riesgo IMDC favorable, intermedio, intermedio/pobre y pobre demostró una HR de 1,17 (IC del 95%: 0,76, 1,80), 0,67 (IC del 95%: 0,52, 0,86), 0,64 (IC del 95%: 0,52, 0,80) y 0,51 (IC del 95%: 0,32, 0,81), respectivamente.
Tratamiento de primera línea con lenvatinib
KEYNOTE-581
La eficacia de KEYTRUDA en combinación con lenvatinib se investigó en KEYNOTE-581 (NCT02811861), un ensayo multicéntrico, abierto y aleatorizado realizado en 1069 pacientes con CCR avanzado en el contexto de primera línea. Se inscribieron pacientes independientemente del estado de expresión tumoral de PD-L1. Los pacientes con enfermedad autoinmune activa o una afección médica que requiriera inmunosupresión no fueron elegibles. La aleatorización se estratificó por región geográfica (América del Norte frente a Europa Occidental frente a “Resto del mundo”) y grupos pronósticos del Memorial Sloan Kettering Cancer Center (MSKCC) (riesgo favorable frente a intermedio frente a pobre).
Los pacientes fueron aleatorizados (1:1:1) a uno de los siguientes brazos de tratamiento:
- KEYTRUDA 200 mg por vía intravenosa cada 3 semanas hasta 24 meses en combinación con lenvatinib 20 mg por vía oral una vez al día.
- Lenvatinib 18 mg por vía oral una vez al día en combinación con everolimus 5 mg por vía oral una vez al día.
- Sunitinib 50 mg por vía oral una vez al día durante 4 semanas y luego sin tratamiento durante 2 semanas.
El tratamiento continuó hasta que se produjo una toxicidad inaceptable o progresión de la enfermedad. Se permitió la administración de KEYTRUDA con lenvatinib más allá de la progresión de la enfermedad definida por RECIST si el paciente estaba clínicamente estable y el investigador consideraba que estaba obteniendo un beneficio clínico. KEYTRUDA se continuó durante un máximo de 24 meses; sin embargo, el tratamiento con lenvatinib podría continuarse más allá de los 24 meses. La evaluación del estado tumoral se realizó al inicio y luego cada 8 semanas.
Las características de la población del estudio fueron: mediana de edad de 62 años (rango: 29 a 88 años), 42% de edad igual o superior a 65 años; 75% hombres; 74% blancos, 21% asiáticos, 1% negros y 2% de otras razas; 18% y 82% de los pacientes tenían una KPS basal de 70 a 80 y de 90 a 100, respectivamente; la distribución de los pacientes por categorías de riesgo del MSKCC fue del 27% favorable, 64% intermedio y 9% pobre. Los sitios comunes de metástasis en los pacientes fueron el pulmón (68%), los ganglios linfáticos (45%) y los huesos (25%).
Las principales medidas de resultado de eficacia fueron la supervivencia libre de progresión (SLP), evaluada mediante revisión radiológica independiente (RRI) según RECIST v1.1, y la supervivencia general (SG). Las medidas de resultado de eficacia adicionales incluyeron la tasa de respuesta objetiva (TRO) confirmada evaluada mediante RRI. KEYTRUDA en combinación con lenvatinib demostró mejoras estadísticamente significativas en la SLP, la SG y la TRO en comparación con sunitinib. Se realizó un análisis actualizado de la SG cuando se observaron 304 muertes según el número de muertes previsto para el análisis final preespecificado. La Tabla 94 y las Figuras 29 y 30 resumen los resultados de eficacia de KEYNOTE-581.
| Variable principal | KEYTRUDA 200 mg cada 3 semanas y Lenvatinib |
Sunitinib |
|---|---|---|
| n=355 | n=357 | |
| Las evaluaciones tumorales se basaron en RECIST 1.1; solo se incluyen las respuestas confirmadas para la TRO. Fecha límite de los datos = 28 de agosto de 2020, Fecha límite de la SG actualizada = 31 de julio de 2022 IC = intervalo de confianza; NR= No alcanzado |
||
| Supervivencia libre de progresión (SLP) | ||
| Número de eventos, n (%) | 160 (45%) | 205 (57%) |
| Enfermedad progresiva | 145 (41%) | 196 (55%) |
| Muerte | 15 (4%) | 9 (3%) |
| SLP mediana en meses (IC del 95%) | 23,9 (20,8, 27,7) | 9,2 (6,0, 11,0) |
| Cociente de riesgos instantáneos* (IC del 95%) | 0,39 (0,32, 0,49) | |
| Valor p† | <0,0001 | |
| Supervivencia general (SG) | ||
| Número de muertes, n (%) | 80 (23%) | 101 (28%) |
| SG mediana en meses (IC del 95%) | NR (33,6, NR) | NR (NR, NR) |
| Cociente de riesgos instantáneos* (IC del 95%) | 0,66 (0,49, 0,88) | |
| Valor p† | 0,0049 | |
| SG actualizada | ||
| Número de muertes, n (%) | 149 (42%) | 159 (45%) |
| Supervivencia mediana en meses (IC del 95%) | 53.7 (48.7, NR) | 54.3 (40.9, NR) |
| Hazard ratio* (IC del 95%) | 0.79 (0.63, 0.99) | |
| Tasa de respuesta objetiva (confirmada) | ||
| ORR, n (%) | 252 (71%) | 129 (36%) |
| (IC del 95%) | (66, 76) | (31, 41) |
| Tasa de respuesta completa | 16% | 4% |
| Tasa de respuesta parcial | 55% | 32% |
| Valor p‡ | <0.0001 | |
| Figura 29: Curva de Kaplan-Meier para la SPP en KEYNOTE-581 |
|
|
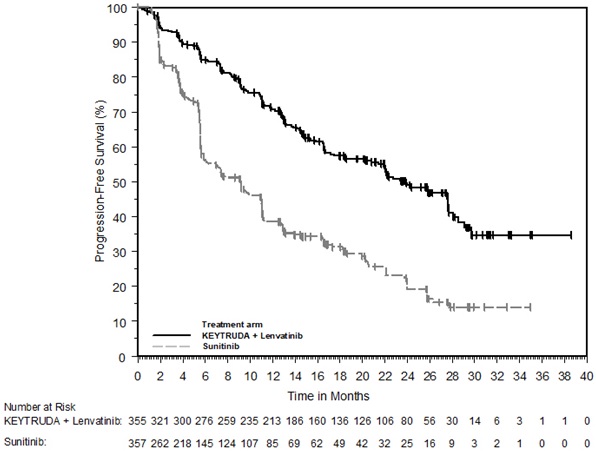
| Figura 30: Curva de Kaplan-Meier para la supervivencia general actualizada en KEYNOTE-581 |
|
|
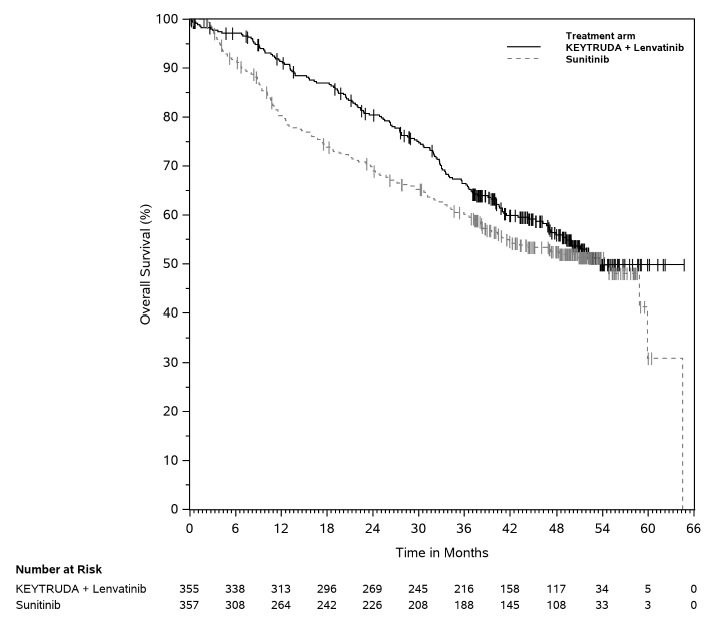
KEYNOTE-B61
La eficacia de KEYTRUDA en combinación con lenvatinib se investigó en KEYNOTE-B61 (NCT04704219), un ensayo multicéntrico de un solo brazo que incluyó a 160 pacientes con RCC no de células claras avanzada o metastásica en el contexto de primera línea. Los pacientes con enfermedad autoinmune activa o una afección médica que requiriera inmunosupresión no fueron elegibles.
Los pacientes recibieron KEYTRUDA 400 mg cada 6 semanas en combinación con lenvatinib 20 mg por vía oral una vez al día. KEYTRUDA se continuó durante un máximo de 24 meses; sin embargo, lenvatinib podría continuarse más allá de los 24 meses. El tratamiento continuó hasta que se produjo una toxicidad inaceptable o progresión de la enfermedad. La administración de KEYTRUDA con lenvatinib se permitió más allá de la progresión de la enfermedad definida por RECIST si el investigador consideraba que el paciente estaba obteniendo un beneficio clínico.
Entre los 158 pacientes tratados, las características basales fueron: mediana de edad de 60 años (rango: 24 a 87 años); 71% hombres; 86% blancos, 8% asiáticos y 3% negros; <1% hispanos o latinos; 22% y 78% de los pacientes tenían una puntuación KPS basal de 70 a 80 y de 90 a 100, respectivamente; los subtipos histológicos fueron 59% papilar, 18% cromófobo, 4% translocación, <1% medular, 13% no clasificado y 6% otros; la distribución de los pacientes según las categorías de riesgo IMDC fue 35% favorable, 54% intermedia y 10% desfavorable. Los sitios comunes de metástasis en los pacientes fueron ganglios linfáticos (65%), pulmón (35%), hueso (30%) e hígado (21%).
La principal medida de resultado de eficacia fue la ORR según la evaluación del BICR utilizando RECIST 1.1. Las medidas de resultado de eficacia adicionales incluyeron la DOR según la evaluación del BICR utilizando RECIST 1.1. Los resultados de eficacia se resumen en la Tabla 95.
| Variable principal | KEYTRUDA 400 mg cada 6 semanas y Lenvatinib n=158 |
|---|---|
| IC = intervalo de confianza + Denota respuesta en curso |
|
|
|
| Tasa de respuesta objetiva (confirmada) | |
| ORR (IC del 95%) | 51% (43, 59) |
| Respuesta completa | 8% |
| Respuesta parcial | 42% |
| Duración de la respuesta* | |
| Mediana en meses (rango) | 19.5 (1.5+, 23.5+) |
Tratamiento adyuvante del CCR (KEYNOTE‑564)
La eficacia de KEYTRUDA se investigó como terapia adyuvante para el CCR en KEYNOTE-564 (NCT03142334), un ensayo multicéntrico, aleatorizado (1:1), doble ciego, controlado con placebo en 994 pacientes con riesgo intermedio-alto o alto de recurrencia de CCR, o M1 sin evidencia de enfermedad (NED). La categoría de riesgo intermedio-alto incluyó: pT2 con características de grado 4 o sarcomatoides; pT3, cualquier grado sin afectación ganglionar (N0) o metástasis a distancia (M0). La categoría de alto riesgo incluyó: pT4, cualquier grado N0 y M0; cualquier pT, cualquier grado con afectación ganglionar y M0. La categoría M1 NED incluyó pacientes con enfermedad metastásica que se habían sometido a una resección completa de las lesiones primarias y metastásicas. Los pacientes debieron someterse a una nefrectomía parcial nefropreservadora o radical completa (y resección completa de la(s) lesión(es) metastásica(s) sólida(s), aislada(s), de tejido blando en participantes con M1 NED) con márgenes quirúrgicos negativos ≥4 semanas antes del momento de la selección. Los pacientes fueron excluidos del ensayo si habían recibido terapia sistémica previa para CCR avanzado. Los pacientes con enfermedad autoinmune activa o una condición médica que requiriera inmunosupresión también fueron inelegibles. Los pacientes fueron aleatorizados a KEYTRUDA 200 mg administrado por vía intravenosa cada 3 semanas o placebo durante hasta 1 año hasta la recurrencia de la enfermedad o toxicidad inaceptable. La aleatorización se estratificó por estado de metástasis (M0, M1 NED); el grupo M0 se estratificó además por ECOG PS (0,1) y región geográfica (EE. UU., fuera de EE. UU.).
Las características de la población del estudio fueron: edad media de 60 años (rango: 25 a 84), 33% de edad 65 o mayor; 71% hombres; 75% blancos, 14% asiáticos, 9% desconocidos, 1% negros o afroamericanos, 1% amerindios o nativos de Alaska, 1% multirraciales; 13% hispanos o latinos, 78% no hispanos o latinos, 8% desconocidos; y 85% ECOG PS de 0 y 15% ECOG PS de 1. El noventa y cuatro por ciento de los pacientes inscritos tenían enfermedad N0; el 11% tenían características sarcomatoides; el 86% tenían riesgo intermedio-alto; el 8% tenían riesgo alto; y el 6% eran M1 NED. El noventa y dos por ciento de los pacientes se sometieron a una nefrectomía radical, y el 8% a una nefrectomía parcial.
La principal medida de resultado de eficacia fue la supervivencia sin enfermedad (DFS) evaluada por el investigador, definida como el tiempo hasta la recurrencia, metástasis o muerte. Una medida de resultado adicional fue la SO. Se demostraron mejoras estadísticamente significativas en la DFS y la SO en los análisis intermedios preespecificados en pacientes aleatorizados al brazo KEYTRUDA en comparación con el placebo. Los resultados de eficacia se resumen en la Tabla 96 y las Figuras 31 y 32.
| Variable principal | KEYTRUDA 200 mg cada 3 semanas n=496 |
Placebo
n=498 |
|---|---|---|
| NR = no alcanzado | ||
| DFS | ||
| Número (%) de pacientes con evento | 109 (22%) | 151 (30%) |
| Mediana en meses (IC del 95%) | NR | NR |
| Razón de riesgo* (IC del 95%) | 0.68 (0.53, 0.87) | |
| Valor p† | 0.0010‡ | |
| Tasa de DFS a 24 meses (IC del 95%) | 77% (73, 81) | 68% (64, 72) |
| SO | ||
| Número (%) de pacientes con evento | 55 (11%) | 86 (17%) |
| Mediana en meses (IC del 95%) | NR (NR, NR) | NR (NR, NR) |
| Razón de riesgo* (IC del 95%) | 0.62 (0.44, 0.87) | |
| Valor p† | 0.0024§ | |
| Tasa de SO a 48 meses (IC del 95%) | 91% (88, 93) | 86% (83, 89) |
| Figura 31: Curva de Kaplan-Meier para la supervivencia sin enfermedad en KEYNOTE-564 |
|
|
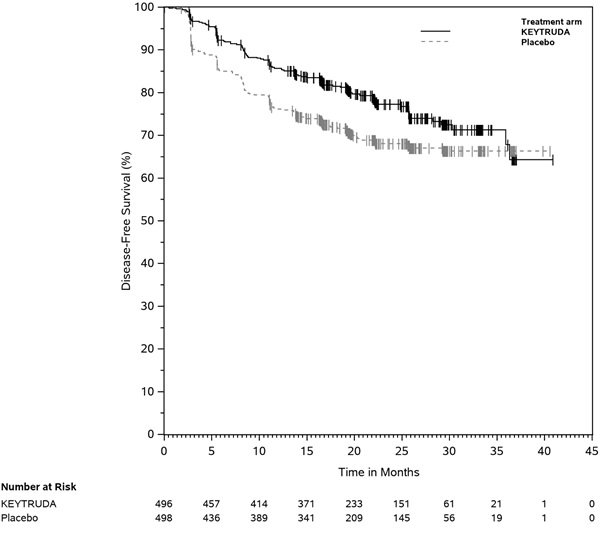
| Figura 32: Curva de Kaplan-Meier para la Supervivencia Global en KEYNOTE-564 |
|
|
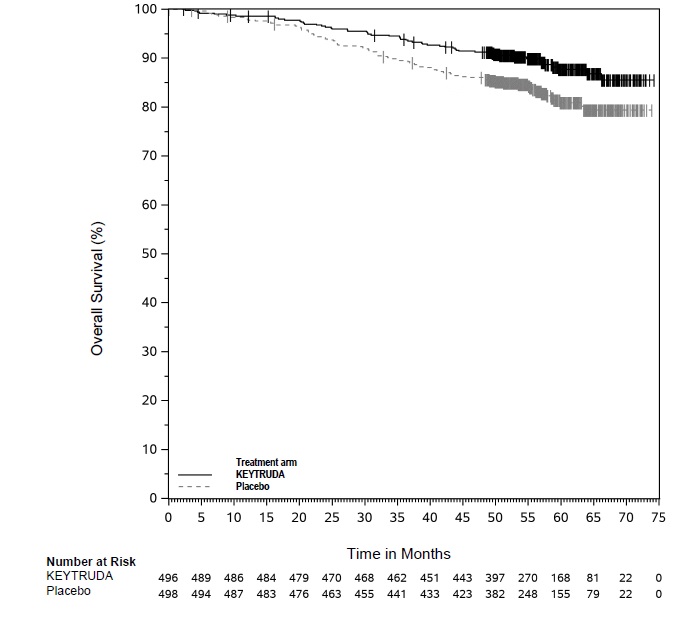
14.17 Carcinoma de endometrio
En combinación con paclitaxel y carboplatino para el tratamiento del carcinoma de endometrio primario avanzado o recurrente
La eficacia de KEYTRUDA en combinación con paclitaxel y carboplatino se investigó en KEYNOTE-868/NRG-GY018 (NCT03914612), un ensayo multicéntrico, aleatorizado, doble ciego, controlado con placebo en 810 pacientes con carcinoma de endometrio avanzado o recurrente. El diseño del estudio incluyó dos cohortes separadas según el estado de MMR; 222 (27%) pacientes estaban en la cohorte dMMR, 588 (73%) pacientes estaban en la cohorte pMMR. El ensayo incluyó cáncer de endometrio en estadio III medible, estadio IVA medible, estadio IVB o recurrente (con o sin enfermedad medible). Los pacientes que no habían recibido terapia sistémica previa o que habían recibido quimioterapia previa en el contexto adyuvante fueron elegibles. Los pacientes que habían recibido quimioterapia adyuvante previa solo fueron elegibles si su intervalo libre de quimioterapia fue de al menos 12 meses. Los pacientes con sarcoma de endometrio, incluido el carcinosarcoma, o pacientes con enfermedad autoinmune activa o una condición médica que requiriera inmunosupresión no fueron elegibles. La aleatorización se estratificó según el estado de MMR, el estado funcional ECOG (0 o 1 vs. 2) y la quimioterapia adyuvante previa.
Los pacientes fueron aleatorizados (1:1) a uno de los siguientes brazos de tratamiento:
- KEYTRUDA 200 mg cada 3 semanas, paclitaxel 175 mg/m2 y carboplatino AUC 5 mg/mL/min durante 6 ciclos, seguido de KEYTRUDA 400 mg cada 6 semanas hasta por 14 ciclos.
- Placebo cada 3 semanas, paclitaxel 175 mg/m2 y carboplatino AUC 5 mg/mL/min durante 6 ciclos, seguido de placebo cada 6 semanas hasta por 14 ciclos.
Todos los medicamentos del estudio se administraron como infusión intravenosa el día 1 de cada ciclo de tratamiento. El tratamiento continuó hasta la progresión de la enfermedad, toxicidad inaceptable o un máximo de 20 ciclos (hasta aproximadamente 24 meses). Los pacientes con enfermedad medible que tuvieron enfermedad estable o respuesta parcial definida por RECIST al completar el ciclo 6 pudieron continuar recibiendo paclitaxel y carboplatino con KEYTRUDA o placebo hasta por 10 ciclos según lo determine el investigador. La evaluación del estado tumoral se realizó cada 9 semanas durante los primeros 9 meses y luego cada 12 semanas a partir de entonces. La principal medida de resultado de eficacia fue la PFS según la evaluación del investigador de acuerdo con RECIST 1.1. Una medida de resultado de eficacia adicional fue la OS.
Las características de la población dMMR fueron: edad media de 66 años (rango: 37 a 86), 55% de edad 65 o mayor; 79% blancos, 9% negros y 3% asiáticos; 5% hispanos o latinos; 64% con estado funcional ECOG de 0, 33% con estado funcional ECOG de 1 y 3% con estado funcional ECOG de 2; 61% tuvieron enfermedad recurrente y 39% tuvieron enfermedad primaria o persistente; 5% recibieron quimioterapia adyuvante previa y 43% recibieron radioterapia previa. Los subtipos histológicos fueron carcinoma endometrioide (81%), adenocarcinoma NOS (11%), carcinoma seroso (2%) y otros (6%).
Las características de la población pMMR fueron: edad media de 66 años (rango: 29 a 94), 54% de edad 65 o mayor; 72% blancos, 16% negros y 5% asiáticos; 6% hispanos o latinos; 67% con estado funcional ECOG de 0, 30% con estado funcional ECOG de 1 y 3% con estado funcional ECOG de 2; 56% tuvieron enfermedad recurrente y 44% tuvieron enfermedad primaria o persistente; 26% recibieron quimioterapia adyuvante previa y 41% recibieron radioterapia previa. Los subtipos histológicos fueron carcinoma endometrioide (52%), carcinoma seroso (26%), adenocarcinoma NOS (10%), carcinoma de células claras (7%) y otros (5%).
El ensayo demostró mejoras estadísticamente significativas en la PFS para los pacientes aleatorizados a KEYTRUDA en combinación con paclitaxel y carboplatino en comparación con placebo en combinación con paclitaxel y carboplatino tanto en las poblaciones dMMR como pMMR. La Tabla 97 y las Figuras 33 y 34 resumen los resultados de eficacia de KEYNOTE-868 por estado de MMR. En el momento del análisis de PFS, los datos de OS no estaban maduros con un 12% de muertes en la población dMMR y un 17% de muertes en la población pMMR.
| Variable principal | Población dMMR | Población pMMR | ||
|---|---|---|---|---|
| KEYTRUDA con paclitaxel y carboplatino n=110 |
Placebo con paclitaxel y carboplatino n=112 |
KEYTRUDA con paclitaxel y carboplatino n=294 |
Placebo con paclitaxel y carboplatino n=294 |
|
| NR = no alcanzado | ||||
| PFS* | ||||
| Número (%) de pacientes con evento | 26 (24%) | 57 (51%) | 91 (31%) | 124 (42%) |
| Mediana en meses (IC del 95%) | NR (30.7, NR) | 6.5 (6.4, 8.7) | 11.1 (8.7, 13.5) | 8.5 (7.2, 8.8) |
| Hazard ratio† (IC 95%) | 0.30 (0.19, 0.48) | 0.60 (0.46, 0.78) | ||
| Valor de p‡ | <0.0001 | <0.0001 | ||
| Figura 33: Curva de Kaplan-Meier para la Supervivencia Libre de Progresión en KEYNOTE-868 (Población dMMR) |
|
|
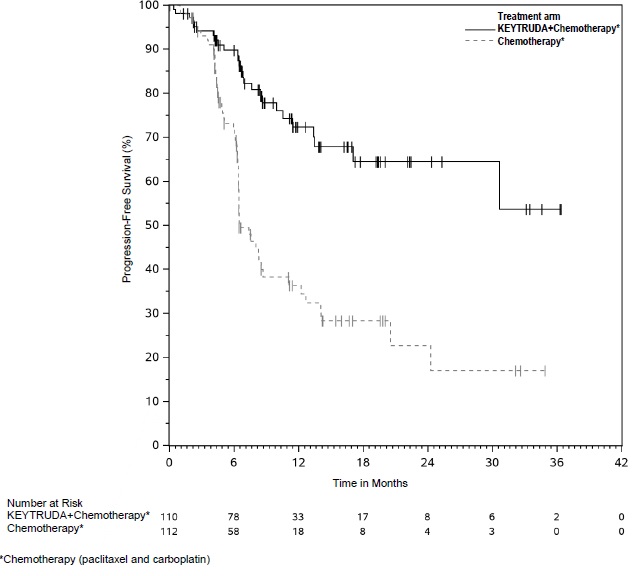
| Figura 34: Curva de Kaplan-Meier para la Supervivencia Libre de Progresión en KEYNOTE-868 (Población pMMR) |
|
|
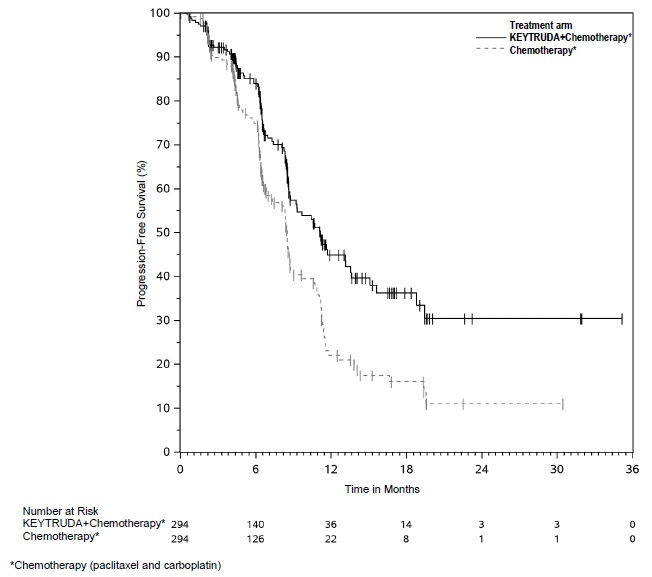
En combinación con lenvatinib para el tratamiento del carcinoma endometrial avanzado que es pMMR o no MSI-H
La eficacia de KEYTRUDA en combinación con lenvatinib se investigó en KEYNOTE-775 (NCT03517449), un ensayo multicéntrico, abierto, aleatorizado y controlado con activo en el que se inscribieron 827 pacientes con carcinoma endometrial avanzado que habían recibido previamente al menos un régimen de quimioterapia a base de platino en cualquier contexto, incluidos los contextos neoadyuvante y adyuvante. Los pacientes con sarcoma endometrial, incluido el carcinosarcoma, o los pacientes que tenían una enfermedad autoinmune activa o una afección médica que requería inmunosupresión no eran elegibles. Los pacientes con carcinoma endometrial que eran pMMR (utilizando la prueba VENTANA MMR RxDx Panel) o no MSI-H se estratificaron por estado de rendimiento ECOG, región geográfica e historial de radiación pélvica. Los pacientes se asignaron aleatoriamente (1:1) a uno de los siguientes brazos de tratamiento:
- KEYTRUDA 200 mg por vía intravenosa cada 3 semanas en combinación con lenvatinib 20 mg por vía oral una vez al día.
- A elección del investigador, que consiste en doxorrubicina 60 mg/m2 cada 3 semanas o paclitaxel 80 mg/m2 administrado semanalmente, 3 semanas de tratamiento/1 semana de descanso.
El tratamiento con KEYTRUDA y lenvatinib continuó hasta la progresión de la enfermedad definida por RECIST v1.1, verificada por BICR, toxicidad inaceptable o, para KEYTRUDA, un máximo de 24 meses. Se permitió el tratamiento más allá de la progresión de la enfermedad definida por RECIST v1.1 si el investigador tratante consideraba que el paciente se estaba beneficiando clínicamente y el tratamiento era tolerado. La evaluación del estado tumoral se realizó cada 8 semanas. Las principales medidas de resultado de eficacia fueron la OS y la PFS, evaluadas por BICR según RECIST v1.1, modificadas para seguir un máximo de 10 lesiones diana y un máximo de 5 lesiones diana por órgano. Las medidas de resultado de eficacia adicionales incluyeron la ORR y la DoR, evaluadas por BICR.
Entre los 697 pacientes con pMMR, 346 pacientes se asignaron aleatoriamente a KEYTRUDA en combinación con lenvatinib, y 351 pacientes se asignaron aleatoriamente a la elección del investigador de doxorrubicina (n=254) o paclitaxel (n=97). Las características de la población pMMR fueron: edad media de 65 años (rango: 30 a 86), 52% de edad igual o superior a 65 años; 62% blancos, 22% asiáticos y 3% negros; 60% ECOG PS de 0 y 40% ECOG PS de 1. Los subtipos histológicos fueron carcinoma endometrioide (55%), seroso (30%), carcinoma de células claras (7%), mixto (4%) y otros (3%). Los 697 pacientes recibieron terapia sistémica previa para el carcinoma endometrial: el 67% recibió una, el 30% recibió dos y el 3% recibió tres o más terapias sistémicas previas. El 37% de los pacientes recibieron solo terapia neoadyuvante o adyuvante previa.
Los resultados de eficacia para los pacientes pMMR o no MSI-H se resumen en la Tabla 98 y las Figuras 35 y 36.
| Carcinoma endometrial (pMMR o no MSI-H) | ||
|---|---|---|
| Variable principal | KEYTRUDA 200 mg cada 3 semanas y Lenvatinib n=346 |
Doxorrubicina o Paclitaxel
n=351 |
|
||
| OS | ||
| Número (%) de pacientes con evento | 165 (48%) | 203 (58%) |
| Mediana en meses (IC del 95%) | 17.4 (14.2, 19.9) | 12.0 (10.8, 13.3) |
| Razón de riesgo* (IC del 95%) | 0.68 (0.56, 0.84) | |
| Valor p† | 0.0001 | |
| PFS | ||
| Número (%) de pacientes con evento | 247 (71%) | 238 (68%) |
| Mediana en meses (IC del 95%) | 6.6 (5.6, 7.4) | 3.8 (3.6, 5.0) |
| Razón de riesgo* (IC del 95%) | 0.60 (0.50, 0.72) | |
| Valor p† | <0.0001 | |
| Tasa de respuesta objetiva | ||
| ORR‡ (IC del 95%) | 30% (26, 36) | 15% (12, 19) |
| Tasa de respuesta completa | 5% | 3% |
| Tasa de respuesta parcial | 25% | 13% |
| Valor p§ | <0.0001 | |
| Duración de la respuesta | n=105 | n=53 |
| Mediana en meses (rango) | 9.2 (1.6+, 23.7+) | 5.7 (0.0+, 24.2+) |
| Figura 35: Curva de Kaplan-Meier para la supervivencia general en KEYNOTE-775 (pMMR o no MSI-H) |
|
|
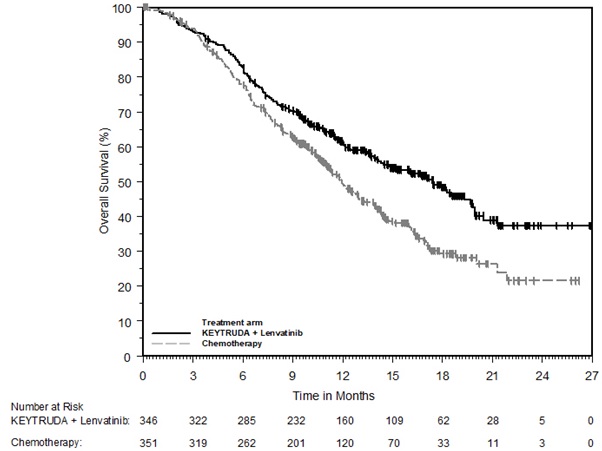
| Figura 36: Curva de Kaplan-Meier para la supervivencia libre de progresión en KEYNOTE-775 (pMMR o no MSI-H) |
|
|
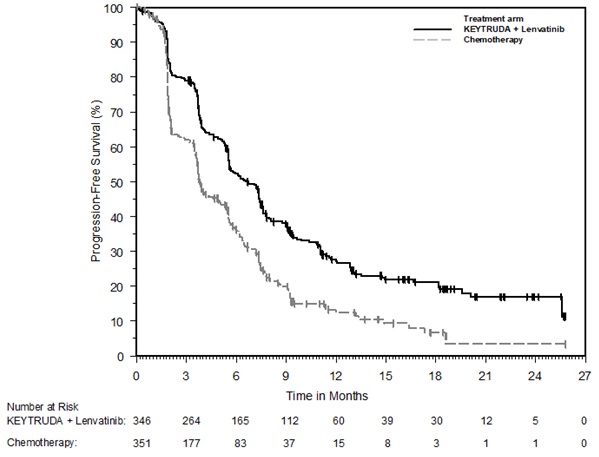
Como agente único para el tratamiento del carcinoma endometrial MSI-H o dMMR avanzado
La eficacia de KEYTRUDA se investigó en KEYNOTE-158 (NCT02628067), un ensayo multicéntrico, no aleatorizado, abierto y multicóhorte. El ensayo incluyó a 90 pacientes con carcinoma endometrial MSI-H o dMMR irresecable o metastásico en las cohortes D y K que recibieron al menos una dosis de KEYTRUDA. El estado tumoral MSI o MMR se determinó mediante reacción en cadena de la polimerasa (PCR) o inmunohistoquímica (IHC), respectivamente. Los pacientes con enfermedad autoinmune o una afección médica que requiriera inmunosupresión no fueron elegibles. Los pacientes recibieron KEYTRUDA 200 mg por vía intravenosa cada 3 semanas hasta que se produjo una toxicidad inaceptable o una progresión de la enfermedad documentada. Los pacientes tratados con KEYTRUDA sin progresión de la enfermedad pudieron recibir tratamiento hasta por 24 meses. La evaluación del estado tumoral se realizó cada 9 semanas durante los primeros 12 meses y cada 12 semanas a partir de entonces. Las principales medidas de resultado de eficacia fueron la TMR y la DuR, según la evaluación del BICR de acuerdo con RECIST v1.1, modificada para seguir un máximo de 10 lesiones diana y un máximo de 5 lesiones diana por órgano.
Entre los 90 pacientes evaluados, las características basales fueron: mediana de edad de 64 años (rango: 42 a 86); 83% blancos, 8% asiáticos y 3% negros; 12% hispanos o latinos; 39% ECOG PS de 0 y 61% ECOG PS de 1; 96% tenían enfermedad M1 y 4% tenían enfermedad M0 al inicio del estudio; y el 51% tenían una y el 48% tenían dos o más líneas de terapia previas. Nueve pacientes recibieron solo terapia adyuvante y un paciente recibió solo terapia neoadyuvante y adyuvante antes de participar en el estudio.
Los resultados de eficacia se resumen en la Tabla 99.
| Variable principal | KEYTRUDA n=90* |
|---|---|
| + Denota respuesta en curso NR = no alcanzado |
|
|
|
| Tasa de respuesta objetiva | |
| TMR (IC del 95%) | 46% (35, 56) |
| Tasa de respuesta completa | 12% |
| Tasa de respuesta parcial | 33% |
| Duración de la respuesta | n=41 |
| Mediana en meses (rango) | NR (2.9, 55.7+) |
| % con duración ≥12 meses | 68% |
| % con duración ≥24 meses | 44% |
14.18 Cáncer con alta carga mutacional tumoral
La eficacia de KEYTRUDA se investigó en un análisis retrospectivo planificado prospectivamente de 10 cohortes (A a J) de pacientes con varios tumores sólidos metastásicos o irresecables previamente tratados con alta carga mutacional tumoral (TMB-H) que fueron incluidos en un ensayo multicéntrico, no aleatorizado, abierto, KEYNOTE-158 (NCT02628067). El ensayo excluyó a pacientes que habían recibido previamente un anticuerpo monoclonal anti-PD-1 u otro anticuerpo monoclonal inmunomodulador, o que tenían una enfermedad autoinmune o una condición médica que requería inmunosupresión. Los pacientes recibieron KEYTRUDA 200 mg por vía intravenosa cada 3 semanas hasta que se produjo una toxicidad inaceptable o una progresión de la enfermedad documentada. La evaluación del estado del tumor se realizó cada 9 semanas durante los primeros 12 meses y cada 12 semanas a partir de entonces.
El plan de análisis estadístico preespecificó ≥10 y ≥13 mutaciones por megabase utilizando el ensayo FoundationOne CDx como puntos de corte para evaluar la TMB. La prueba de TMB se realizó de forma ciega con respecto a los resultados clínicos. Las principales medidas de resultado de eficacia fueron la ORR y la DoR en pacientes que recibieron al menos una dosis de KEYTRUDA, según la evaluación del BICR de acuerdo con RECIST v1.1, modificado para seguir un máximo de 10 lesiones diana y un máximo de 5 lesiones diana por órgano.
En KEYNOTE-158, se incluyeron 1050 pacientes en la población de análisis de eficacia. La TMB se analizó en el subgrupo de 790 pacientes con tejido suficiente para la prueba según los requisitos de prueba especificados en el protocolo. De los 790 pacientes, 102 (13%) tenían tumores identificados como TMB-H, definidos como TMB ≥10 mutaciones por megabase. Entre los 102 pacientes con tumores sólidos avanzados TMB-H, las características de la población del estudio fueron: edad media de 61 años (rango: 27 a 80), 34% de edad ≥65 años; 34% hombres; 81% blancos; y 41% ECOG PS de 0 y 58% ECOG PS de 1. El cincuenta y seis por ciento de los pacientes habían recibido al menos dos líneas de terapia previas.
Los resultados de eficacia se resumen en las Tablas 100 y 101.
|
Variable principal |
KEYTRUDA 200 mg cada 3 semanas |
|
|---|---|---|
| TMB ≥10 mut/Mb n=102* |
TMB ≥13 mut/Mb n=70 |
|
| + Indica respuesta en curso NR = no alcanzado |
||
| Tasa de respuesta objetiva | ||
| ORR (IC del 95%) | 29% (21, 39) | 37% (26, 50) |
| Tasa de respuesta completa | 4% | 3% |
| Tasa de respuesta parcial | 25% | 34% |
| Duración de la respuesta | n=30 | n=26 |
| Mediana en meses (rango)† | NR (2.2+, 34.8+) | NR (2.2+, 34.8+) |
| % con duración ≥12 meses | 57% | 58% |
| % con duración ≥24 meses | 50% | 50% |
| Tasa de respuesta objetiva | Duración del rango de respuesta | |||
|---|---|---|---|---|
| N | n (%) | IC del 95% | (meses) | |
| CR = respuesta completa PR = respuesta parcial SD = enfermedad estable PD = enfermedad progresiva |
||||
|
||||
| General* | 102 | 30 (29%) | (21%, 39%) | (2.2+, 34.8+) |
| Cáncer de pulmón de células pequeñas | 34 | 10 (29%) | (15%, 47%) | (4.1, 32.5+) |
| Cáncer de cuello uterino | 16 | 5 (31%) | (11%, 59%) | (3.7+, 34.8+) |
| Cáncer de endometrio | 15 | 7 (47%) | (21%, 73%) | (8.4+, 33.9+) |
| Cáncer anal | 14 | 1 (7%) | (0.2%, 34%) | 18.8+ |
| Cáncer de vulva | 12 | 2 (17%) | (2%, 48%) | (8.8, 11.0) |
| Cáncer neuroendocrino | 5 | 2 (40%) | (5%, 85%) | (2.2+, 32.6+) |
| Cáncer salival | 3 | PR, SD, PD | 31.3+ | |
| Cáncer de tiroides | 2 | CR, CR | (8.2, 33.2+) | |
| Cáncer de mesotelioma | 1 | PD | ||
En un análisis exploratorio en 32 pacientes inscritos en KEYNOTE-158 cuyo cáncer tenía TMB ≥10 mut/Mb y <13 mut/Mb, la ORR fue del 13% (IC del 95%: 4%, 29%), incluidas dos respuestas completas y dos respuestas parciales.
14.19 Carcinoma de células escamosas cutáneo
La eficacia de KEYTRUDA se investigó en pacientes con cSCC recurrente o metastásico o cSCC localmente avanzado inscritos en KEYNOTE-629 (NCT03284424), un ensayo multicéntrico, multicóhorte, no aleatorizado y abierto. El ensayo excluyó a pacientes con enfermedad autoinmune o una condición médica que requiriera inmunosupresión.
Los pacientes recibieron KEYTRUDA 200 mg por vía intravenosa cada 3 semanas hasta la progresión documentada de la enfermedad, toxicidad inaceptable o un máximo de 24 meses. Los pacientes con progresión inicial de la enfermedad radiográfica podrían recibir dosis adicionales de KEYTRUDA durante la confirmación de la progresión a menos que la progresión de la enfermedad fuera sintomática, rápidamente progresiva, requiriera intervención urgente o ocurriera con una disminución en el estado funcional.
La evaluación del estado del tumor se realizó cada 6 semanas durante el primer año y cada 9 semanas durante el segundo año. Las principales medidas de resultado de eficacia fueron la ORR y la DoR según la evaluación del BICR de acuerdo con RECIST v1.1, modificado para seguir un máximo de 10 lesiones diana y un máximo de 5 lesiones diana por órgano.
Entre los 105 pacientes con cSCC recurrente o metastásico tratados, las características de la población del estudio fueron: edad media de 72 años (rango: 29 a 95), 71% de edad 65 o superior; 76% hombres; 70% blancos, 25% raza desconocida; 34% ECOG PS de 0 y 66% ECOG PS de 1. El cuarenta y cinco por ciento de los pacientes tenían cSCC solo recurrente localmente, el 24% tenían cSCC solo metastásico y el 31% tenían cSCC tanto recurrente localmente como metastásico. El ochenta y siete por ciento recibió una o más líneas de terapia previas; el 73% recibió radioterapia previa.
Entre los 54 pacientes con cSCC localmente avanzado tratados, las características de la población del estudio fueron: edad media de 76 años (rango: 35 a 95), 80% de edad 65 o superior; 72% hombres; 83% blancos, 13% raza desconocida; 41% ECOG PS de 0 y 59% ECOG PS de 1. El veintidós por ciento recibió una o más líneas de terapia previas; el 63% recibió radioterapia previa.
Los resultados de eficacia se resumen en la Tabla 102.
| Variable principal | KEYTRUDA cSCC recurrente o metastásico n=105 |
KEYTRUDA cSCC localmente avanzado n=54 |
|---|---|---|
| + Denota respuesta en curso | ||
|
||
| Tasa de respuesta objetiva | ||
| ORR (IC del 95%) | 35% (26, 45) | 52% (38, 66) |
| Tasa de respuesta completa | 12% | 22% |
| Tasa de respuesta parcial | 23% | 30% |
| Duración de la respuesta* | n=37 | n=28 |
| Mediana en meses (rango) | NR (2,7, 64,2+) | 47,2 (1,0+, 49,9+) |
| % con duración ≥6 meses | 76% | 89% |
| % con duración ≥12 meses | 68% | 75% |
14.20 Cáncer de mama triple negativo
Tratamiento neoadyuvante y adyuvante de TNBC de estadio temprano de alto riesgo
La eficacia de KEYTRUDA en combinación con quimioterapia neoadyuvante seguida de cirugía y tratamiento adyuvante continuado con KEYTRUDA como agente único se investigó en KEYNOTE-522 (NCT03036488), un ensayo aleatorizado (2:1), multicéntrico, doble ciego, controlado con placebo realizado en 1174 pacientes con TNBC de estadio temprano de alto riesgo, recién diagnosticado y previamente no tratado (tamaño tumoral >1 cm pero ≤2 cm de diámetro con afectación ganglionar o tamaño tumoral >2 cm de diámetro independientemente de la afectación ganglionar). Se incluyeron pacientes independientemente de la expresión de PD-L1 tumoral. Los pacientes con enfermedad autoinmune activa que requiriera terapia sistémica en los dos años anteriores al tratamiento o una afección médica que requiriera inmunosupresión fueron inelegibles. La aleatorización se estratificó por estado ganglionar (positivo frente a negativo), tamaño tumoral (T1/T2 frente a T3/T4) y elección de carboplatino (dosis cada 3 semanas frente a semanal).
Los pacientes fueron aleatorizados (2:1) a uno de los dos brazos de tratamiento siguientes; todos los medicamentos del estudio se administraron por vía intravenosa:
-
Brazo 1:
- Cuatro ciclos de KEYTRUDA preoperatorio 200 mg cada 3 semanas el día 1 de los ciclos 1-4 del régimen de tratamiento en combinación con:
- Carboplatino
- AUC 5 mg/mL/min cada 3 semanas el día 1 de los ciclos 1-4 del régimen de tratamiento
-o- - AUC 1,5 mg/mL/min cada semana los días 1, 8 y 15 de los ciclos 1-4 del régimen de tratamiento
-y-
- AUC 5 mg/mL/min cada 3 semanas el día 1 de los ciclos 1-4 del régimen de tratamiento
- Paclitaxel 80 mg/m2 cada semana los días 1, 8 y 15 de los ciclos 1-4 del régimen de tratamiento
- Carboplatino
- Seguido de cuatro ciclos adicionales de KEYTRUDA preoperatorio 200 mg cada 3 semanas el día 1 de los ciclos 5-8 del régimen de tratamiento en combinación con:
- Doxorrubicina 60 mg/m2 -o- epirrubicina 90 mg/m2 cada 3 semanas el día 1 de los ciclos 5-8 del régimen de tratamiento -y-
- Ciclofosfamida 600 mg/m2 cada 3 semanas el día 1 de los ciclos 5-8 del régimen de tratamiento
- Tras la cirugía, se administraron nueve ciclos de KEYTRUDA 200 mg cada 3 semanas.
- Cuatro ciclos de KEYTRUDA preoperatorio 200 mg cada 3 semanas el día 1 de los ciclos 1-4 del régimen de tratamiento en combinación con:
-
Brazo 2:
- Cuatro ciclos de placebo preoperatorio cada 3 semanas el día 1 de los ciclos 1-4 del régimen de tratamiento en combinación con:
- Carboplatino
- AUC 5 mg/mL/min cada 3 semanas el día 1 de los ciclos 1-4 del régimen de tratamiento
-o- - AUC 1,5 mg/mL/min cada semana los días 1, 8 y 15 de los ciclos 1-4 del régimen de tratamiento
-y-
- AUC 5 mg/mL/min cada 3 semanas el día 1 de los ciclos 1-4 del régimen de tratamiento
- Paclitaxel 80 mg/m2 cada semana los días 1, 8 y 15 de los ciclos 1-4 del régimen de tratamiento
- Carboplatino
- Seguido de cuatro ciclos de placebo preoperatorio cada 3 semanas el día 1 de los ciclos 5-8 del régimen de tratamiento en combinación con:
- Doxorrubicina 60 mg/m2 -o- epirrubicina 90 mg/m2 cada 3 semanas el día 1 de los ciclos 5-8 del régimen de tratamiento -y-
- Ciclofosfamida 600 mg/m2 cada 3 semanas el día 1 de los ciclos 5-8 del régimen de tratamiento
- Tras la cirugía, se administraron nueve ciclos de placebo cada 3 semanas.
- Cuatro ciclos de placebo preoperatorio cada 3 semanas el día 1 de los ciclos 1-4 del régimen de tratamiento en combinación con:
Las principales variables de eficacia fueron la tasa de RCP y la LFS. La RCP se definió como la ausencia de cáncer invasivo en la mama y los ganglios linfáticos (ypT0/Tis ypN0) y fue evaluada por el patólogo local ciego en el momento de la cirugía definitiva. La LFS se definió como el tiempo desde la aleatorización hasta la primera aparición de cualquiera de los siguientes eventos: progresión de la enfermedad que impide la cirugía definitiva, recurrencia local o a distancia, segunda neoplasia maligna o muerte por cualquier causa. Una variable de eficacia adicional fue la supervivencia general (SG).
Las características de la población del estudio fueron: mediana de edad de 49 años (rango: 22 a 80), 11% de edad ≥65 años; 99,9% mujeres; 64% blancas, 20% asiáticas, 4,5% negras y 1,8% amerindias o nativas de Alaska; 87% ECOG PS de 0 y 13% ECOG PS de 1; 56% fueron premenopáusicas y 44% posmenopáusicas; 7% fueron Tumor primario 1 (T1), 68% T2, 19% T3 y 7% T4; 49% fueron afectación ganglionar 0 (N0), 40% N1, 11% N2 y 0,2% N3; 75% de los pacientes fueron en general Estadio II y 25% Estadio III.
La Tabla 103 y la Figura 37 resumen los resultados de eficacia de KEYNOTE-522. En el análisis intermedio IA4 preespecificado del protocolo de SG, los datos de SG no estaban maduros con el 45% de los eventos requeridos para el análisis final.
| Variable principal | KEYTRUDA 200 mg cada 3 semanas con quimioterapia/KEYTRUDA n=784 |
Placebo con quimioterapia/Placebo n=390 |
|---|---|---|
|
||
| pCR (ypT0/Tis ypN0)* | ||
| Número de pacientes con pCR | 494 | 217 |
| Tasa de pCR (%), (IC del 95%) | 63.0 (59.5, 66.4) | 55.6 (50.6, 60.6) |
| Estimación de la diferencia de tratamiento (%) (IC del 95%)†,‡ | 7.5 (1.6, 13.4) | |
| EFS | ||
| Número de pacientes con evento (%) | 123 (16%) | 93 (24%) |
| Hazard ratio (IC del 95%)§ | 0.63 (0.48, 0.82) | |
| Valor p¶,# | 0.00031 | |
| Figura 37: Curva de Kaplan-Meier para la supervivencia libre de eventos en KEYNOTE-522 |
|
|
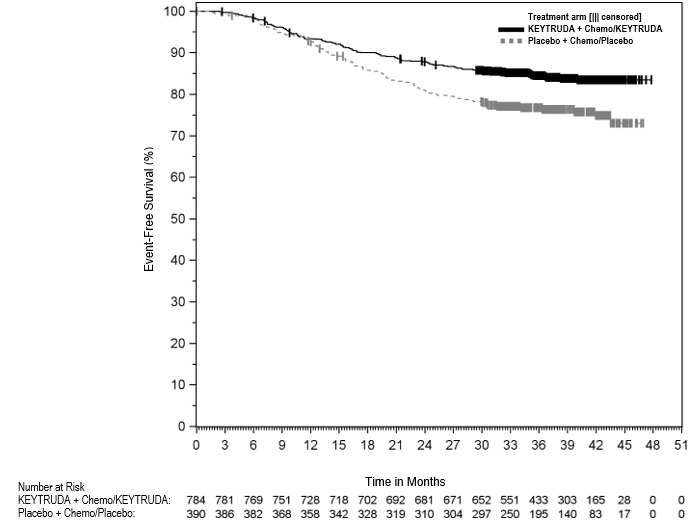
Recidiva local irresecable o metastásica de TNBC
La eficacia de KEYTRUDA en combinación con paclitaxel, paclitaxel unido a proteínas o gemcitabina y carboplatino se investigó en KEYNOTE-355 (NCT02819518), un ensayo multicéntrico, doble ciego, aleatorizado y controlado con placebo realizado en 847 pacientes con recidiva local irresecable o TNBC metastásica, independientemente de la expresión de PD-L1 tumoral, que no habían sido tratados previamente con quimioterapia en el contexto metastásico. Los pacientes con enfermedad autoinmune activa que requiriera terapia sistémica dentro de los 2 años del tratamiento o una condición médica que requiriera inmunosupresión fueron inelegibles. La aleatorización se estratificó por tratamiento de quimioterapia (paclitaxel o paclitaxel unido a proteínas frente a gemcitabina y carboplatino), expresión de PD-L1 tumoral (CPS ≥1 frente a CPS <1) según el kit pharmDx PD-L1 IHC 22C3, y tratamiento previo con la misma clase de quimioterapia en el contexto neoadyuvante (sí frente a no).
Los pacientes fueron aleatorizados (2:1) a uno de los siguientes brazos de tratamiento; todos los medicamentos del estudio se administraron mediante infusión intravenosa:
- KEYTRUDA 200 mg el día 1 cada 3 semanas en combinación con paclitaxel unido a proteínas 100 mg/m2 los días 1, 8 y 15 cada 28 días, paclitaxel 90 mg/m2 los días 1, 8 y 15 cada 28 días, o gemcitabina 1000 mg/m2 y carboplatino AUC 2 mg/mL/min los días 1 y 8 cada 21 días.
- Placebo el día 1 cada 3 semanas en combinación con paclitaxel unido a proteínas 100 mg/m2 los días 1, 8 y 15 cada 28 días, paclitaxel 90 mg/m2 los días 1, 8 y 15 cada 28 días, o gemcitabina 1000 mg/m2 y carboplatino AUC 2 mg/mL/min los días 1 y 8 cada 21 días.
La evaluación del estado tumoral se realizó en las semanas 8, 16 y 24, luego cada 9 semanas durante el primer año y cada 12 semanas a partir de entonces. Las principales medidas de resultado de eficacia fueron la OS y la PFS, evaluadas por BICR según RECIST v1.1, modificadas para seguir un máximo de 10 lesiones diana y un máximo de 5 lesiones diana por órgano, probadas en el subgrupo de pacientes con CPS ≥10. Las medidas de resultado de eficacia adicionales fueron la ORR y la DoR, evaluadas por BICR.
Las características de la población del estudio para los pacientes fueron: edad media de 53 años (rango: 22 a 85), 21% de edad 65 o superior; 100% mujeres; 68% blancas, 21% asiáticas y 4% negras; 60% ECOG PS de 0 y 40% ECOG PS de 1; y el 68% tenían estado posmenopáusico. El setenta y cinco por ciento de los pacientes tenían una expresión de PD-L1 tumoral CPS ≥1 y el 38% tenían una expresión de PD-L1 tumoral CPS ≥10.
La Tabla 104 y las Figuras 38 y 39 resumen los resultados de eficacia de KEYNOTE-355.
| Variable principal | KEYTRUDA 200 mg cada 3 semanas con quimioterapia n=220 |
Placebo cada 3 semanas con quimioterapia n=103 |
|---|---|---|
|
||
| OS* | ||
| Número de pacientes con evento (%) | 155 (70%) | 84 (82%) |
| Mediana en meses (IC del 95%) | 23 (19.0, 26.3) | 16.1 (12.6, 18.8) |
| Razón de riesgo† (IC del 95%) | 0.73 (0.55, 0.95) | |
| Valor p‡ | 0.0093 | |
| PFS§ | ||
| Número de pacientes con evento (%) |
136 (62%) | 79 (77%) |
| Mediana en meses (IC del 95%) | 9.7 (7.6, 11.3) | 5.6 (5.3, 7.5) |
| Razón de riesgo† (IC del 95%) | 0.65 (0.49, 0.86) | |
| Valor p¶ | 0.0012 | |
| Tasa de respuesta objetiva (confirmada)* | ||
| ORR (IC del 95%) | 53% (46, 59) | 41% (31, 51) |
| Tasa de respuesta completa | 17% | 14% |
| Tasa de respuesta parcial | 35% | 27% |
| Duración de la respuesta* | n=116 | n=42 |
| Mediana en meses (IC del 95%) | 12.8 (9.9, 25.9) | 7.3 (5.5, 15.4) |
| Figura 38: Curva de Kaplan-Meier para la supervivencia general en KEYNOTE-355 (CPS ≥10) |
|
|
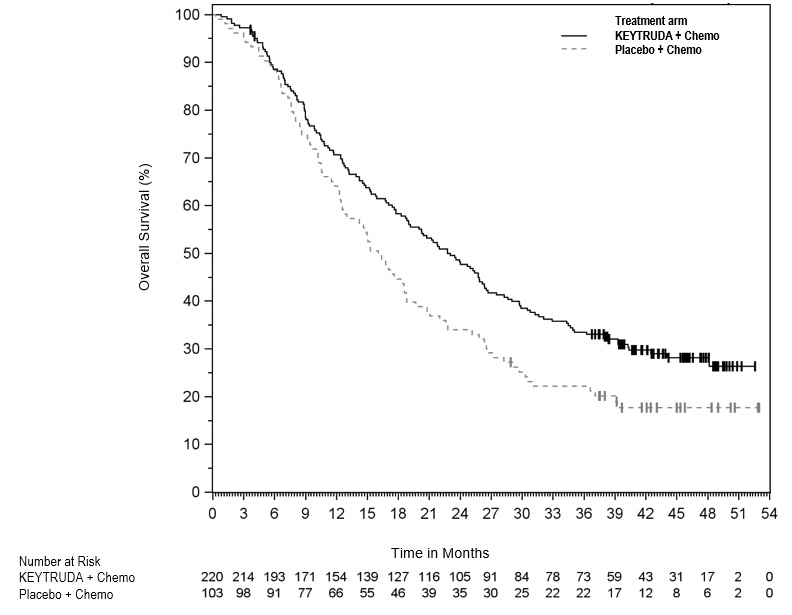
| Figura 39: Curva de Kaplan-Meier para la supervivencia libre de progresión en KEYNOTE-355 (CPS ≥10) |
|
|
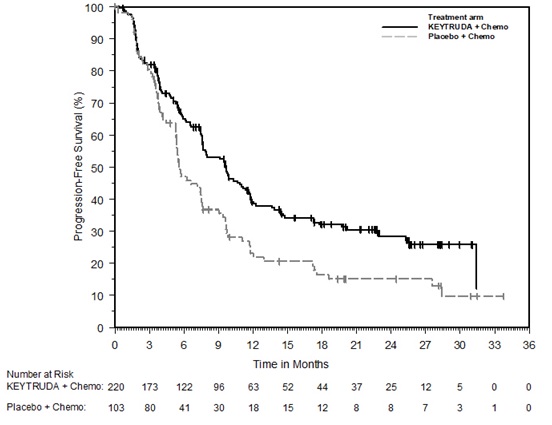
14.21 Linfoma de Hodgkin clásico en adultos y linfoma difuso de células B grandes del mediastino anterior en adultos: Régimen de dosificación adicional de 400 mg cada 6 semanas
La eficacia y seguridad de KEYTRUDA utilizando una dosis de 400 mg cada 6 semanas para las indicaciones de linfoma de Hodgkin clásico y linfoma difuso de células B grandes del mediastino anterior en adultos se basó principalmente en las relaciones de eficacia y seguridad de la dosis/exposición y los datos farmacocinéticos observados en pacientes con melanoma [véase Farmacología clínica (12.2)].
16 SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
KEYTRUDA inyección (solución transparente a ligeramente opalescente, incolora a ligeramente amarilla):
Caja conteniendo un vial de dosis única de 100 mg/4 mL (25 mg/mL) (NDC 0006-3026-02)
Caja conteniendo dos viales de dosis única de 100 mg/4 mL (25 mg/mL) (NDC 0006-3026-04)
Almacenar los viales bajo refrigeración a 2°C a 8°C (36°F a 46°F) en su caja original para protegerlos de la luz. No congelar. No agitar.
17 INFORMACIÓN DE ASESORAMIENTO AL PACIENTE
Aconseje al paciente que lea el etiquetado del paciente aprobado por la FDA (Guía de medicamentos).
Reacciones adversas inmunomediadas
- Informe a los pacientes sobre el riesgo de reacciones adversas inmunomediadas que pueden ser graves o mortales, pueden ocurrir después de la interrupción del tratamiento y pueden requerir tratamiento con corticosteroides e interrupción o suspensión de KEYTRUDA. Estas reacciones pueden incluir:
- Neumonitis: Aconseje a los pacientes que se pongan en contacto con su proveedor de atención médica inmediatamente si presentan tos nueva o que empeora, dolor en el pecho o dificultad para respirar [ver Advertencias y precauciones (5.1)].
- Colitis: Aconseje a los pacientes que se pongan en contacto con su proveedor de atención médica inmediatamente si presentan diarrea o dolor abdominal intenso [ver Advertencias y precauciones (5.1)].
- Hepatitis: Aconseje a los pacientes que se pongan en contacto con su proveedor de atención médica inmediatamente si presentan ictericia, náuseas o vómitos intensos, o facilidad para la aparición de hematomas o sangrado [ver Advertencias y precauciones (5.1)].
- Endocrinopatías: Aconseje a los pacientes que se pongan en contacto con su proveedor de atención médica inmediatamente si presentan signos o síntomas de insuficiencia suprarrenal, hipofisitis, hipotiroidismo, hipertiroidismo o diabetes mellitus tipo 1 [ver Advertencias y precauciones (5.1)].
- Nefritis: Aconseje a los pacientes que se pongan en contacto con su proveedor de atención médica inmediatamente si presentan signos o síntomas de nefritis [ver Advertencias y precauciones (5.1)].
- Reacciones cutáneas graves: Aconseje a los pacientes que se pongan en contacto con su proveedor de atención médica inmediatamente si presentan cualquier signo o síntoma de reacciones cutáneas graves, SJS o TEN [ver Advertencias y precauciones (5.1)].
- Otras reacciones adversas inmunomediadas:
- Aconseje a los pacientes que pueden producirse reacciones adversas inmunomediadas y que pueden afectar a cualquier sistema orgánico, y que deben ponerse en contacto con su proveedor de atención médica inmediatamente si presentan cualquier signo o síntoma nuevo o que empeora [ver Advertencias y precauciones (5.1)].
- Aconseje a los pacientes sobre el riesgo de rechazo de trasplante de órgano sólido y otros rechazos de trasplante (incluido el injerto de córnea). Aconseje a los pacientes que se pongan en contacto con su proveedor de atención médica inmediatamente si presentan signos o síntomas de rechazo de trasplante de órgano y otros rechazos de trasplante (incluido el injerto de córnea) [ver Advertencias y precauciones (5.1)].
Reacciones relacionadas con la infusión
- Aconseje a los pacientes que se pongan en contacto con su proveedor de atención médica inmediatamente si presentan signos o síntomas de reacciones relacionadas con la infusión [ver Advertencias y precauciones (5.2)].
Complicaciones del TCHC alogénico
- Aconseje a los pacientes sobre el riesgo de complicaciones posteriores al trasplante de células madre hematopoyéticas alogénicas [ver Advertencias y precauciones (5.3)].
Toxicidad embriofetal
- Aconseje a las mujeres en edad fértil sobre el riesgo potencial para el feto y que informen a su proveedor de atención médica sobre un embarazo conocido o sospechoso [ver Advertencias y precauciones (5.5), Uso en poblaciones específicas (8.1, 8.3)].
- Aconseje a las mujeres en edad fértil que utilicen un método anticonceptivo eficaz durante el tratamiento con KEYTRUDA y durante 4 meses después de la última dosis [ver Advertencias y precauciones (5.5), Uso en poblaciones específicas (8.1, 8.3)].
Lactancia
- Aconseje a las mujeres que no deben amamantar durante el tratamiento con KEYTRUDA y durante 4 meses después de la última dosis [ver Uso en poblaciones específicas (8.2)].
Pruebas de laboratorio
- Aconseje a los pacientes sobre la importancia de cumplir con las citas programadas para análisis de sangre u otras pruebas de laboratorio [ver Advertencias y precauciones (5.1)].
SECCIÓN NO CLASIFICADA DE SPL
Fabricado por: Merck Sharp & Dohme LLC
Rahway, NJ 07065, EE. UU.
Licencia de EE. UU. n.° 0002
Para obtener información sobre patentes: www.msd.com/research/patent
Las marcas comerciales que se muestran aquí son propiedad de sus respectivas compañías.
Copyright © 2014-2024 Merck & Co., Inc., Rahway, NJ, EE. UU. y sus filiales.
Todos los derechos reservados.
uspi-mk3475-iv-2412r082
Guía de medicación
| Esta Guía del Medicamento ha sido aprobada por la Administración de Alimentos y Medicamentos de los EE. UU. | Revised: September 2024 | ||
| GUÍA DEL MEDICAMENTO KEYTRUDA® (key-true-duh) (pembrolizumab) injection |
|||
| ¿Cuál es la información más importante que debo saber sobre KEYTRUDA? | |||
| KEYTRUDA es un medicamento que puede tratar ciertos tipos de cáncer al trabajar con su sistema inmunitario. KEYTRUDA puede hacer que su sistema inmunitario ataque los órganos y tejidos normales en cualquier área de su cuerpo y puede afectar la forma en que funcionan. Estos problemas a veces pueden volverse graves o potencialmente mortales y pueden causar la muerte. Puede tener más de uno de estos problemas al mismo tiempo. Estos problemas pueden ocurrir en cualquier momento durante el tratamiento o incluso después de que haya finalizado su tratamiento. | |||
| Llame o consulte a su proveedor de atención médica de inmediato si desarrolla cualquier signo o síntoma nuevo o que empeora, incluyendo: | |||
| Problemas pulmonares | |||
|
|
|
|
| Problemas intestinales | |||
|
|||
| Problemas hepáticos | |||
|
|
||
| Problemas de las glándulas hormonales | |||
|
|
||
| Problemas renales | |||
|
|
||
| Problemas de la piel | |||
|
|
||
| También pueden ocurrir problemas en otros órganos y tejidos. Estos no son todos los signos y síntomas de problemas del sistema inmunitario que pueden ocurrir con KEYTRUDA. Llame o consulte a su proveedor de atención médica de inmediato si presenta cualquier signo o síntoma nuevo o que empeora, que puede incluir: | |||
|
|||
| Reacciones a la infusión que a veces pueden ser graves o potencialmente mortales. Los signos y síntomas de las reacciones a la infusión pueden incluir: | |||
|
|
||
| Rechazo de un órgano o tejido trasplantado. Su proveedor de atención médica debe informarle qué signos y síntomas debe reportar y monitorearlo según el tipo de trasplante de órgano o tejido que haya recibido. | |||
| Complicaciones, incluida la enfermedad de injerto contra huésped (EICH), en personas que han recibido un trasplante de médula ósea (células madre) que utiliza células madre de donantes (alogénicas). Estas complicaciones pueden ser graves y pueden provocar la muerte. Estas complicaciones pueden ocurrir si se sometió a un trasplante antes o después de recibir tratamiento con KEYTRUDA. Su proveedor de atención médica lo controlará para detectar estas complicaciones. | |||
|
Buscar atención médica de inmediato puede ayudar a evitar que estos problemas se agraven. Su proveedor de atención médica lo revisará para detectar estos problemas durante el tratamiento con KEYTRUDA. Su proveedor de atención médica puede tratarlo con corticosteroides o medicamentos de reemplazo hormonal. Es posible que su proveedor de atención médica también deba retrasar o suspender por completo el tratamiento con KEYTRUDA si tiene efectos secundarios graves. |
|||
| ¿Qué es KEYTRUDA? | |||
| KEYTRUDA es un medicamento recetado que se usa para tratar: | |||
|
|||
Antes de recibir KEYTRUDA, informe a su profesional de la salud sobre todas sus afecciones médicas, incluso si:
|
|||
| Informe a su profesional de la salud sobre todos los medicamentos que toma, incluyendo medicamentos recetados y de venta libre, vitaminas y suplementos herbales. | |||
| ¿Cómo recibiré KEYTRUDA? | |||
|
|||
| ¿Cuáles son los posibles efectos secundarios de KEYTRUDA? | |||
| KEYTRUDA puede causar efectos secundarios graves. Consulte “¿Cuál es la información más importante que debo saber sobre KEYTRUDA?”. | |||
| Los efectos secundarios comunes de KEYTRUDA cuando se usa solo incluyen: sensación de cansancio, dolor, incluido dolor muscular, sarpullido, diarrea, fiebre, tos, disminución del apetito, picazón, dificultad para respirar, estreñimiento, dolor en los huesos o articulaciones y en el área del estómago (abdominal), náuseas y niveles bajos de hormona tiroidea. | |||
| Los efectos secundarios de KEYTRUDA cuando se usa solo que son más comunes en niños que en adultos incluyen: fiebre, vómitos, dolor de cabeza, dolor en el área del estómago (abdominal) y niveles bajos de glóbulos blancos. | |||
| Los efectos secundarios comunes de KEYTRUDA cuando se administra con cierta quimioterapia o quimioterapia con medicamentos de radioterapia incluyen: sensación de cansancio o debilidad, náuseas, estreñimiento, diarrea, disminución del apetito, sarpullido, vómitos, tos, dificultad para respirar, fiebre, pérdida de cabello, inflamación de los nervios que puede causar dolor, debilidad y parálisis en brazos y piernas, hinchazón del revestimiento de la boca, nariz, ojos, garganta, intestinos o vagina, llagas en la boca, dolor de cabeza, pérdida de peso, dolor en el área del estómago (abdominal), dolor muscular y articular, dificultad para dormir, sangrado, ampollas o sarpullido en las palmas de las manos y plantas de los pies, infección del tracto urinario y niveles bajos de hormona tiroidea. | |||
| Los efectos secundarios comunes de KEYTRUDA cuando se administra con quimioterapia y bevacizumab incluyen: hormigueo o entumecimiento de brazos o piernas, pérdida de cabello, recuento bajo de glóbulos rojos, sensación de cansancio o debilidad, náuseas, recuento bajo de glóbulos blancos, diarrea, presión arterial alta, recuento bajo de plaquetas, estreñimiento, dolores articulares, vómitos, infección del tracto urinario, sarpullido, niveles bajos de hormona tiroidea y disminución del apetito. | |||
| Los efectos secundarios comunes de KEYTRUDA cuando se administra con axitinib incluyen: diarrea, sensación de cansancio o debilidad, presión arterial alta, problemas hepáticos, niveles bajos de hormona tiroidea, disminución del apetito, ampollas o sarpullido en las palmas de las manos y plantas de los pies, náuseas, llagas en la boca o hinchazón del revestimiento de la boca, nariz, ojos, garganta, intestinos o vagina, ronquera, sarpullido, tos y estreñimiento. | |||
| Los efectos secundarios comunes de KEYTRUDA cuando se administra con lenvatinib incluyen: niveles bajos de hormona tiroidea, presión arterial alta, sensación de cansancio, diarrea, dolor muscular y articular, náuseas, disminución del apetito, vómitos, llagas en la boca, pérdida de peso, dolor en el área del estómago (abdominal), infección del tracto urinario, proteínas en la orina, estreñimiento, dolor de cabeza, sangrado, ampollas o sarpullido en las palmas de las manos y plantas de los pies, ronquera, sarpullido, problemas hepáticos y problemas renales. | |||
| Los efectos secundarios comunes de KEYTRUDA cuando se administra con enfortumab vedotin incluyen: sarpullido, hormigueo o entumecimiento de brazos o piernas, sensación de cansancio, picazón, diarrea, pérdida de cabello, pérdida de peso, disminución del apetito, sequedad ocular, náuseas, estreñimiento, cambios en el sentido del gusto e infección del tracto urinario. | |||
| Estos no son todos los posibles efectos secundarios de KEYTRUDA. | |||
| Llame a su médico para obtener asesoramiento médico sobre los efectos secundarios. Puede informar los efectos secundarios a la FDA al 1-800-FDA-1088. | |||
| Información general sobre el uso seguro y eficaz de KEYTRUDA | |||
| A veces, los medicamentos se recetan para fines distintos a los que se enumeran en una Guía del Medicamento. Puede preguntarle a su farmacéutico o proveedor de atención médica para obtener información sobre KEYTRUDA que esté escrita para profesionales de la salud. | |||
| ¿Cuáles son los ingredientes de KEYTRUDA? | |||
| Ingrediente activo: pembrolizumab | |||
| Ingredientes inactivos: Inyección KEYTRUDA: L-histidina, polisorbato 80, sacarosa y agua para inyección. | |||
| Fabricado por: Merck Sharp & Dohme LLC Rahway, NJ 07065, USA |
U.S. License No. 0002 Para obtener información sobre patentes: www.msd.com/research/patent Copyright © 2014-2024 Merck & Co., Inc., Rahway, NJ, USA, and its affiliates. All rights reserved. usmg-mk3475-iv-2409r066 Para obtener más información, visite www.keytruda.com. |
||
PANEL PRINCIPAL DE EXHIBICIÓN – Envase de vial de 50 mg
NDC 0006-3029-02
Keytruda®
(pembrolizumab)
para inyección
50 mg / vial
Sólo para infusión intravenosa
Entregue la Guía de Medicamentos adjunta a cada paciente.
El polvo liofilizado estéril debe reconstituirse con agua esterilizada para
inyección, USP. La solución reconstituida requiere una dilución adicional antes
de la administración.
Rx only
Vial de dosis única. Deseche la porción no utilizada.

PRINCIPAL PANEL DE PRESENTACIÓN – Vial de 100 mg/4 mL en caja
NDC 0006-3026-02
Keytruda®
(pembrolizumab)
Inyección
100 mg/4 mL
(25 mg/mL)
Sólo para infusión intravenosa
Entregue la Guía de Medicamentos adjunta a cada paciente.
Requiere dilución antes de la administración.
Rx only
Vial de dosis única. Deseche la porción no utilizada.