
Fabricante de medicamentos: Incyte Corporation (Updated: 2023-03-02)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
JAKAFI® (ruxolitinib) tabletas, para uso oral
Aprobación inicial en EE. UU.: 2011
CAMBIOS RECIENTES IMPORTANTES
INDICACIONES Y USO
Jakafi es un inhibidor de la quinasa indicado para el tratamiento de:
- mielofibrosis de riesgo intermedio o alto, incluyendo mielofibrosis primaria, mielofibrosis posterior a policitemia vera y mielofibrosis posterior a trombocitemia esencial en adultos. (1.1)
- policitemia vera en adultos que han tenido una respuesta inadecuada o son intolerantes a la hidroxiurea. (1.2)
- enfermedad de injerto contra huésped aguda resistente a esteroides en pacientes adultos y pediátricos de 12 años o mayores (1.3)
- enfermedad crónica de injerto contra huésped después del fracaso de una o dos líneas de terapia sistémica en pacientes adultos y pediátricos de 12 años o mayores. (1.4)
DOSIFICACIÓN Y ADMINISTRACIÓN
Las dosis deben individualizarse en función de la seguridad y la eficacia. Las dosis iniciales por indicación se indican a continuación.
Mielofibrosis (2.2)
- La dosis inicial de Jakafi se basa en el recuento de plaquetas basal del paciente:
• Mayor que 200 x 109/L: 20 mg administrados por vía oral dos veces al día
• 100 x 109/L a 200 x 109/L: 15 mg administrados por vía oral dos veces al día
• 50 x 109/L a menos de 100 x 109/L: 5 mg administrados por vía oral dos veces al día - Controle los hemogramas completos cada 2 a 4 semanas hasta que las dosis se estabilicen, y luego según esté clínicamente indicado. Modifique o interrumpa la dosificación para la trombocitopenia.
Policitemia Vera (2.3)
- La dosis inicial de Jakafi es de 10 mg administrados por vía oral dos veces al día.
Enfermedad Aguda de Injerto Contra Huésped (2.4)
-
La dosis inicial de Jakafi es de 5 mg administrados por vía oral dos veces al día.
Enfermedad Crónica de Injerto Contra Huésped (2.5)
- La dosis inicial de Jakafi es de 10 mg administrados por vía oral dos veces al día.
FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
Comprimidos: 5 mg, 10 mg, 15 mg, 20 mg y 25 mg. (3)
CONTRAINDICACIONES
Ninguna. (4)
ADVERTENCIAS Y PRECAUCIONES
- Trombocitopenia, Anemia y Neutropenia: Controlar mediante reducción de la dosis, interrupción o transfusión. (5.1)
- Riesgo de infección: Evaluar a los pacientes para detectar signos y síntomas de infección e iniciar el tratamiento adecuado con prontitud. Las infecciones graves deben haberse resuelto antes de iniciar el tratamiento con Jakafi. (5.2)
- Exacerbación de los síntomas tras la interrupción o suspensión: Controlar con cuidados de apoyo y considerar la posibilidad de reanudar el tratamiento con Jakafi. (5.3)
- Riesgo de cáncer de piel no melanoma: Realizar exámenes periódicos de la piel. (5.4)
- Elevaciones de lípidos: Evaluar los niveles de lípidos de 8 a 12 semanas después del inicio del tratamiento y tratar según sea necesario. (5.5)
- Eventos cardiovasculares adversos mayores (MACE): Monitorizar el desarrollo de MACE. (5.6)
- Trombosis: Evaluar y tratar los síntomas de trombosis con prontitud. (5.7)
- Tumores malignos secundarios: Monitorizar el desarrollo de tumores malignos secundarios, especialmente en pacientes fumadores actuales o anteriores. (5.8)
REACCIONES ADVERSAS
- En la mielofibrosis y la policitemia vera, las reacciones adversas hematológicas más comunes (incidencia > 20%) son trombocitopenia y anemia. Las reacciones adversas no hematológicas más comunes (incidencia ≥ 15%) son hematomas, mareos, dolor de cabeza y diarrea. (6.1)
- En la enfermedad aguda de injerto contra huésped, las reacciones adversas hematológicas más comunes (incidencia > 50%) son anemia, trombocitopenia y neutropenia. Las reacciones adversas no hematológicas más comunes (incidencia > 50%) son infecciones (patógeno no especificado) y edema. (6.1)
- En la enfermedad crónica de injerto contra huésped, las reacciones adversas hematológicas más comunes (incidencia > 35%) son anemia y trombocitopenia. Las reacciones adversas no hematológicas más comunes (incidencia ≥ 20%) son infecciones (patógeno no especificado) e infecciones virales. (6.1)
Para reportar REACCIONES ADVERSAS SOSPECHOSAS, contacte a Incyte Corporation al 1-855-463-3463 o a la FDA al 1-800-FDA-1088 o www.fda.gov/medwatch
INTERACCIONES CON MEDICAMENTOS
- Fluconazole: Evitar el uso concomitante con dosis de fluconazol mayores a 200 mg. Reducir la dosis de Jakafi con dosis de fluconazol menores o iguales a 200 mg. (2.6, 7)
- Inhibidores potentes de CYP3A4: Reducir, interrumpir o suspender las dosis de Jakafi según lo recomendado, excepto en pacientes con enfermedad de injerto contra huésped aguda o crónica. (2.6, 7)
USO EN POBLACIONES ESPECÍFICAS
Ver 17 para INFORMACIÓN DE ASESORAMIENTO AL PACIENTE y etiqueta del paciente aprobada por la FDA.
Revisado: 3/2023
Tabla de contenido
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDO*
1. INDICACIONES Y USO
1.1 Mielofibrosis
1.2 Policitemia Vera
1.3 Enfermedad injerto contra huésped aguda
1.4 Enfermedad injerto contra huésped crónica
2. POSOLOGÍA Y ADMINISTRACIÓN
2.1 Monitorización para evaluar la seguridad
2.2 Posología recomendada para Mielofibrosis
2.3 Posología recomendada para Policitemia Vera
2.4 Posología recomendada para Enfermedad injerto contra huésped aguda
2.5 Posología recomendada para Enfermedad injerto contra huésped crónica
2.6 Modificaciones de la dosis para el uso concomitante con inhibidores potentes del CYP3A4 o fluconazol
2.7 Modificaciones de la dosis para insuficiencia renal o hepática
2.8 Método de administración
3. FORMAS Y CONCENTRACIONES FARMACÉUTICAS
4. CONTRAINDICACIONES
5. ADVERTENCIAS Y PRECAUCIONES
5.1 Trombocitopenia, anemia y neutropenia
5.2 Riesgo de infección
5.3 Exacerbación de los síntomas tras la interrupción o suspensión del tratamiento con Jakafi
5.4 Cáncer de piel no melanoma (CPNM)
5.5
Elevación de lípidos
5.6 Eventos cardiovasculares adversos mayores (ECAM)
5.7 Trombosis
5.8 Neoplasias secundarias
6. REACCIONES ADVERSAS
6.1 Experiencia en ensayos clínicos
6.2 Experiencia postcomercialización
7. INTERACCIONES MEDICAMENTOSAS
7.1 Efecto de otros medicamentos sobre Jakafi
8. USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.4 Uso pediátrico
8.5 Uso geriátrico
8.6 Insuficiencia renal
8.7 Insuficiencia hepática
10. SOBREDOSIFICACIÓN
11. DESCRIPCIÓN
12. FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
12.2 Farmacodinamia
12.3 Farmacocinética
13. TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, mutagénesis, deterioro de la fertilidad
14. ESTUDIOS CLÍNICOS
14.1 Mielofibrosis
14.2 Policitemia Vera
14.3
Enfermedad injerto contra huésped aguda
14.4 Enfermedad injerto contra huésped crónica
16. PRESENTACIÓN/CONSERVACIÓN Y MANIPULACIÓN
17. INFORMACIÓN PARA EL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información completa de prescripción no figuran en la lista.
1. INDICACIONES Y USO
1.1 Mielofibrosis
Jakafi está indicado para el tratamiento de la mielofibrosis (MF) de riesgo intermedio o alto, incluyendo MF primaria, MF post-policitemia vera y MF post-trombocitemia esencial en adultos.
1.2 Policitemia Vera
Jakafi está indicado para el tratamiento de la policitemia vera (PV) en adultos que han tenido una respuesta inadecuada a la hidroxiurea o son intolerantes a ella.
1.3 Enfermedad Injerto contra Huésped Aguda
Jakafi está indicado para el tratamiento de la enfermedad injerto contra huésped aguda (EICH) refractaria a esteroides en pacientes adultos y pediátricos de 12 años o más.
1.4 Enfermedad Injerto contra Huésped Crónica
Jakafi está indicado para el tratamiento de la enfermedad injerto contra huésped crónica (EICH crónica) después del fracaso de una o dos líneas de terapia sistémica en pacientes adultos y pediátricos de 12 años o más.
2. DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Monitorización para evaluar la seguridad
Antes del tratamiento con Jakafi:
- Realizar un hemograma completo [ver Advertencias y precauciones (5.1)].
- Preguntar sobre infecciones previas, incluyendo tuberculosis, herpes simple, herpes zóster y hepatitis B [ver Advertencias y precauciones (5.2)].
Durante el tratamiento con Jakafi:
- Realizar un hemograma completo cada 2 a 4 semanas hasta que las dosis se estabilicen, y luego según esté clínicamente indicado [ver Advertencias y precauciones (5.1)].
- Evaluar los parámetros lipídicos aproximadamente 8-12 semanas después del inicio del tratamiento con Jakafi [ver Advertencias y precauciones (5.5)].
2.2 Dosis recomendada para la mielofibrosis
La dosis inicial recomendada de Jakafi se basa en el recuento de plaquetas (Tabla 1). Las dosis pueden ajustarse según la seguridad y la eficacia.
| Recuento de plaquetas | Dosis inicial |
| Mayor de 200 x 109/L | 20 mg por vía oral dos veces al día |
| 100 x 109/L a 200 x 109/L | 15 mg por vía oral dos veces al día |
| 50 x 109/L a menos de 100 x 109/L | 5 mg por vía oral dos veces al día |
Pautas de modificación de la dosis para la toxicidad hematológica para pacientes con mielofibrosis que inician el tratamiento con un recuento de plaquetas de 100 x 109/L o superior
Interrupción del tratamiento y reinicio de la dosificación
Interrumpir el tratamiento para recuentos de plaquetas inferiores a 50 x 109/L o recuento absoluto de neutrófilos (RAN) inferior a 0,5 x 109/L.
Después de la recuperación de los recuentos de plaquetas por encima de 50 x 109/L y el RAN por encima de 0,75 x 109/L, la dosificación puede reiniciarse. La Tabla 2 ilustra la dosis máxima permitida que puede utilizarse para reiniciar Jakafi después de una interrupción previa.
|
|
| Recuento de plaquetas actual | Dosis máxima al reiniciar el tratamiento con Jakafi* |
| Mayor o igual a 125 x 109/L | 20 mg dos veces al día |
| 100 a menos de 125 x 109/L | 15 mg dos veces al día |
| 75 a menos de 100 x 109/L | 10 mg dos veces al día durante al menos 2 semanas; si es estable, puede aumentarse a 15 mg dos veces al día |
| 50 a menos de 75 x 109/L | 5 mg dos veces al día durante al menos 2 semanas; si es estable, puede aumentarse a 10 mg dos veces al día |
| Menos de 50 x 109/L | Continuar con la suspensión |
Tras la interrupción del tratamiento por ANC inferior a 0,5 x 109/L, después de que el ANC se recupere a 0,75 x 109/L o más, reinicie la dosificación con la dosis más alta entre 5 mg una vez al día o 5 mg dos veces al día, inferior a la dosis más alta de la semana anterior a la interrupción del tratamiento.
Reducciones de la dosis
Se deben considerar reducciones de la dosis si el recuento de plaquetas disminuye como se describe en la Tabla 3, con el objetivo de evitar interrupciones del tratamiento por trombocitopenia.
| Dosis en el momento de la disminución de plaquetas | |||||
| Recuento de plaquetas | 25 mg dos veces al día |
20 mg dos veces al día |
15 mg dos veces al día |
10 mg dos veces al día |
5 mg dos veces al día |
| Nueva Dosis |
Nueva Dosis |
Nueva Dosis |
Nueva Dosis |
Nueva Dosis |
|
| 100 a menos de 125 x 109/L |
20 mg dos veces al día |
15 mg dos veces al día |
Sin cambio |
Sin cambio |
Sin cambio |
| 75 a menos de 100 x 109/L |
10 mg dos veces al día |
10 mg dos veces al día |
10 mg dos veces al día |
Sin cambio |
Sin cambio |
| 50 a menos de 75 x 109/L |
5 mg dos veces al día |
5 mg dos veces al día |
5 mg dos veces al día |
5 mg dos veces al día |
Sin cambio |
| Menos de 50 x 109/L | Suspender | Suspender | Suspender | Suspender | Suspender |
Modificación de la dosis según la respuesta insuficiente para pacientes con mielofibrosis que comienzan el tratamiento con un recuento plaquetario de 100 x 109/L o superior
Si la respuesta es insuficiente y los recuentos de plaquetas y neutrófilos son adecuados, las dosis pueden aumentarse en incrementos de 5 mg dos veces al día hasta un máximo de 25 mg dos veces al día. Las dosis no deben aumentarse durante las primeras 4 semanas de tratamiento y no con más frecuencia que cada 2 semanas.
Considere aumentos de dosis en pacientes que cumplen todas las siguientes condiciones:
- Imposibilidad de lograr una reducción desde el valor basal previo al tratamiento en la longitud del bazo palpable del 50% o una reducción del 35% en el volumen del bazo medido mediante tomografía computarizada (TC) o resonancia magnética (RM);
- Recuento de plaquetas superior a 125 x 109/L a las 4 semanas y recuento de plaquetas nunca inferior a 100 x 109/L;
- Niveles de ANC superiores a 0,75 x 109/L.
Según datos clínicos limitados, el mantenimiento a largo plazo a una dosis de 5 mg dos veces al día no ha mostrado respuestas y el uso continuado a esta dosis debe limitarse a pacientes en los que los beneficios superen los riesgos potenciales. Suspenda Jakafi si no hay reducción del tamaño del bazo o mejoría de los síntomas después de 6 meses de tratamiento.
Modificaciones de la dosis para la toxicidad hematológica para pacientes con mielofibrosis que comienzan el tratamiento con recuentos plaquetarios de 50 x 109/L a menos de 100 x 109/L
Esta sección solo se aplica a pacientes con recuentos de plaquetas de 50 x 109/L a menos de 100 x 109/L antes de cualquier tratamiento con Jakafi. Consulte las modificaciones de la dosis en la Sección 2.2 (Pautas de modificación de la dosis para la toxicidad hematológica para pacientes con mielofibrosis que comienzan el tratamiento con un recuento plaquetario de 100 x 109/L o superior) para la toxicidad hematológica en pacientes cuyos recuentos de plaquetas fueron de 100 x 109/L o más antes de comenzar el tratamiento con Jakafi.
Interrupción del tratamiento y reinicio de la dosificación
Interrompa el tratamiento para recuentos de plaquetas inferiores a 25 x 109/L o ANC inferiores a 0,5 x 109/L.
Después de la recuperación de los recuentos de plaquetas por encima de 35 x 109/L y ANC por encima de 0,75 x 109/L, la dosificación puede reiniciarse. Reinicie la dosificación en la dosis más alta de 5 mg una vez al día o 5 mg dos veces al día por debajo de la dosis más alta en la semana anterior a la disminución del recuento de plaquetas por debajo de 25 x 109/L o ANC por debajo de 0,5 x 109/L que provocó la interrupción de la dosis.
Reducciones de la dosis
Reduzca la dosis de Jakafi para recuentos de plaquetas inferiores a 35 x 109/L como se describe en la Tabla 4.
| Recuento de plaquetas | Recomendaciones de dosificación |
| Menos de 25 x 109/L |
|
|
25 x 109/L a menos de 35 x 109/L Y la disminución del recuento de plaquetas es |
|
|
25 x 109/L a menos de 35 x 109/L Y la disminución del recuento de plaquetas es |
|
Modificaciones de la dosis según la respuesta insuficiente para pacientes con mielofibrosis y recuento plaquetario inicial de 50 x 109/L a menos de 100 x 109/L
No aumente las dosis durante las primeras 4 semanas de tratamiento, y no aumente la dosis con más frecuencia que cada 2 semanas.
Si la respuesta es insuficiente, según se define en la Sección 2.2 (véase Modificación de la dosis según la respuesta insuficiente con mielofibrosis que inicia el tratamiento con un recuento plaquetario de 100 x 109/L o superior), las dosis pueden aumentarse en incrementos de 5 mg diarios hasta un máximo de 10 mg dos veces al día si:
- el recuento plaquetario se ha mantenido en al menos 40 x 109/L, y
- el recuento plaquetario no ha disminuido en más del 20% en las 4 semanas anteriores, y
- el ANC es superior a 1 x 109/L, y
- la dosis no se ha reducido ni interrumpido por un evento adverso o toxicidad hematológica en las 4 semanas anteriores.
La continuación del tratamiento durante más de 6 meses debe limitarse a los pacientes en los que los beneficios superen los riesgos potenciales. Suspenda Jakafi si no hay reducción del tamaño del bazo o mejora de los síntomas después de 6 meses de tratamiento.
Modificación de la dosis para hemorragia
Interrompa el tratamiento en caso de hemorragia que requiera intervención, independientemente del recuento plaquetario actual. Una vez que el evento hemorrágico se haya resuelto, considere reanudar el tratamiento con la dosis anterior si se ha controlado la causa subyacente de la hemorragia. Si el evento hemorrágico se ha resuelto pero la causa subyacente persiste, considere reanudar el tratamiento con Jakafi a una dosis menor.
2.3 Dosis recomendada para la policitemia vera
La dosis inicial recomendada de Jakafi es de 10 mg dos veces al día. Las dosis pueden ajustarse según la seguridad y la eficacia.
Pautas de modificación de la dosis para pacientes con policitemia vera
Reducciones de la dosis
Se deben considerar reducciones de la dosis para las disminuciones del recuento de hemoglobina y plaquetas, como se describe en la Tabla 5.
| Recuento de hemoglobina y/o plaquetas | Recomendaciones de dosificación |
| Hemoglobina mayor o igual a 12 g/dL Y recuento plaquetario mayor o igual a 100 x 109/L |
|
| Hemoglobina de 10 a menos de 12 g/dL Y recuento plaquetario de 75 a menos de 100 x 109/L |
|
| Hemoglobina de 8 a menos de 10 g/dL O recuento plaquetario de 50 a menos de 75 x 109/L |
|
| Hemoglobina menor de 8 g/dL O recuento plaquetario menor de 50 x 109/L |
|
Interrupción del tratamiento y reinicio de la dosificación
Interrumpir el tratamiento si la hemoglobina es inferior a 8 g/dL, el recuento de plaquetas es inferior a 50 x 109/L o el ANC es inferior a 1,0 x 109/L.
Después de la recuperación del o los parámetros hematológicos a niveles aceptables, se puede reiniciar la dosificación.
La Tabla 6 ilustra la dosis que se puede utilizar para reiniciar Jakafi después de una interrupción previa.
Tabla 6: Vera Policitemia: Reinicio de las dosis de Jakafi después de la interrupción de seguridad para los parámetros hematológicos
Utilice la categoría más grave de la anomalía de hemoglobina, recuento de plaquetas o ANC del paciente para determinar la dosis máxima de reinicio correspondiente.
|
|
| Hemoglobina, Recuento de plaquetas o ANC | Dosis máxima de reinicio |
| Hemoglobina inferior a 8 g/dL O recuento de plaquetas inferior a 50 x 109/L O ANC inferior a 1 x 109/L |
Mantener la suspensión |
| Hemoglobina de 8 a menos de 10 g/dL O recuento de plaquetas de 50 a menos de 75 x 109/L O ANC de 1 a menos de 1,5 x 109/L |
5 mg dos veces al día* o no más de 5 mg dos veces al día menos que la dosis que provocó la interrupción de la dosis |
| Hemoglobina de 10 a menos de 12 g/dL O recuento de plaquetas de 75 a menos de 100 x 109/L O ANC de 1,5 a menos de 2 x 109/L |
10 mg dos veces al día* o no más de 5 mg dos veces al día menos que la dosis que provocó la interrupción de la dosis |
| Hemoglobina mayor o igual a 12 g/dL O recuento de plaquetas mayor o igual a 100 x 109/L O ANC mayor o igual a 2 x 109/L |
15 mg dos veces al día* o no más de 5 mg dos veces al día menos que la dosis que provocó la interrupción de la dosis |
Los pacientes que hayan requerido la interrupción de la dosis mientras recibían una dosis de 5 mg dos veces al día, pueden reiniciar con una dosis de 5 mg dos veces al día o 5 mg una vez al día, pero no más, una vez que la hemoglobina sea mayor o igual a 10 g/dL, el recuento de plaquetas sea mayor o igual a 75 x 109/L, y el ANC sea mayor o igual a 1.5 x 109/L.
Manejo de la dosis después de reiniciar el tratamiento
Después de reiniciar Jakafi tras la interrupción del tratamiento, las dosis pueden ajustarse, pero la dosis diaria máxima total no debe superar los 5 mg menos que la dosis que provocó la interrupción de la dosis. Una excepción a esto es la interrupción de la dosis tras la anemia asociada a flebotomía, en cuyo caso la dosis diaria máxima total permitida después de reiniciar Jakafi no estaría limitada.
Modificaciones de la dosis basadas en una respuesta insuficiente para pacientes con policitemia vera
Si la respuesta es insuficiente y los recuentos de plaquetas, hemoglobina y neutrófilos son adecuados, las dosis pueden aumentarse en incrementos de 5 mg dos veces al día hasta un máximo de 25 mg dos veces al día. Las dosis no deben aumentarse durante las primeras 4 semanas de tratamiento y no con más frecuencia que cada dos semanas.
Considere aumentos de dosis en pacientes que cumplen todas las siguientes condiciones:
- Eficacia inadecuada demostrada por una o más de las siguientes:
- Necesidad continua de flebotomía
- GB mayor que el límite superior del rango normal
- Recuento de plaquetas mayor que el límite superior del rango normal
- Bazo palpable que se reduce en menos del 25% con respecto al valor basal
- Recuento de plaquetas mayor o igual a 140 x 109/L
- Hemoglobina mayor o igual a 12 g/dL
- ANC mayor o igual a 1.5 x 109/L
2.4 Dosis recomendada para la enfermedad aguda del injerto contra el huésped
La dosis inicial recomendada de Jakafi es de 5 mg administrados por vía oral dos veces al día. Considere aumentar la dosis a 10 mg dos veces al día después de al menos 3 días de tratamiento si el recuento de ANC y plaquetas no disminuye en un 50% o más en relación con el primer día de dosificación con Jakafi.
Considere la reducción gradual de Jakafi después de 6 meses de tratamiento en pacientes con respuesta que hayan interrumpido las dosis terapéuticas de corticosteroides. Reduzca gradualmente Jakafi en un nivel de dosis aproximadamente cada 8 semanas (10 mg dos veces al día a 5 mg dos veces al día a 5 mg una vez al día). Si los signos o síntomas de la enfermedad injerto contra huésped aguda reaparecen durante o después de la reducción gradual de Jakafi, considere el retratamiento.
Pautas de modificación de la dosis para pacientes con enfermedad aguda del injerto contra el huésped
Controle los hemogramas completos (CBC), incluido el recuento de plaquetas y el ANC, y la bilirrubina antes de iniciar el tratamiento, cada 2 a 4 semanas hasta que las dosis se estabilicen, y luego según lo indicado clínicamente.
Modifique la dosis de Jakafi para las reacciones adversas como se describe en la Tabla 7. Para las reducciones de dosis, los pacientes que actualmente reciben Jakafi 10 mg dos veces al día pueden reducir su dosis a 5 mg dos veces al día; los pacientes que reciben 5 mg dos veces al día pueden reducir su dosis a 5 mg una vez al día. Los pacientes que no pueden tolerar Jakafi a una dosis de 5 mg una vez al día deben interrumpir el tratamiento hasta que sus parámetros clínicos y/o de laboratorio se recuperen.
| Parámetro de laboratorio | Recomendaciones de dosificación |
| Trombocitopenia clínicamente significativa después de medidas de apoyo |
Reducir la dosis en 1 nivel de dosis. |
| ANC menor que 1 x 109/L considerado relacionado con Jakafi |
Suspender Jakafi hasta por 14 días; reanudar en 1 nivel de dosis inferior tras la recuperación. |
| Elevación de la bilirrubina total, sin EIGH hepática |
3.0−5.0 x ULN: Continuar con Jakafi en 1 nivel de dosis inferior hasta la recuperación. > 5.0−10.0 x ULN: Suspender Jakafi hasta por 14 días hasta Bilirrubina total > 10.0 x ULN: Suspender Jakafi hasta por |
| Elevación de la bilirrubina total, EIGH hepática |
> 3.0 × ULN: Continuar con Jakafi en 1 nivel de dosis inferior hasta la recuperación. |
2.5 Dosis recomendada para la enfermedad crónica del injerto contra el huésped
La dosis inicial recomendada de Jakafi es de 10 mg administrada por vía oral dos veces al día.
Considere la posibilidad de reducir la dosis de Jakafi después de 6 meses de tratamiento en pacientes con respuesta que hayan suspendido las dosis terapéuticas de corticosteroides. Reduzca la dosis de Jakafi en un nivel de dosis aproximadamente cada 8 semanas (10 mg dos veces al día a 5 mg dos veces al día a 5 mg una vez al día). Si los signos o síntomas de la EICH reaparecen durante o después de la reducción gradual de Jakafi, considere la posibilidad de volver a administrar el tratamiento.
Pautas de modificación de la dosis para pacientes con enfermedad crónica del injerto contra el huésped
Controle los hemogramas completos (CBC), incluido el recuento de plaquetas y el ANC, y la bilirrubina antes de iniciar el tratamiento, cada 2 a 4 semanas hasta que las dosis se estabilicen, y luego según lo indicado clínicamente.
Modifique la dosis de Jakafi para las reacciones adversas como se describe en la Tabla 8. Para las reducciones de dosis, los pacientes que actualmente reciben Jakafi 10 mg dos veces al día pueden reducir su dosis a 5 mg dos veces al día; los pacientes que reciben 5 mg dos veces al día pueden reducir su dosis a 5 mg una vez al día. Los pacientes que no toleran Jakafi a una dosis de 5 mg una vez al día deben interrumpir el tratamiento hasta que sus parámetros clínicos y/o de laboratorio se recuperen.
| Parámetro | Recomendaciones de dosificación |
| Recuento de plaquetas inferior a 20 × 109/L | Reducir Jakafi en 1 nivel de dosis. Si se resuelve en 7 días, la dosificación puede volver al nivel de dosis inicial. Si no se resuelve en 7 días, mantener en 1 nivel de dosis inferior. |
|
ANC inferior a 0,75 × 109/L considerado relacionado con Jakafi |
Reducir Jakafi en 1 nivel de dosis; reanudar en el nivel de dosis inicial tras la recuperación. |
|
ANC inferior a 0,5 × 109/L considerado relacionado con Jakafi |
Suspender Jakafi hasta por 14 días; reanudar en 1 nivel de dosis inferior tras la recuperación. Puede reanudarse el nivel de dosis inicial cuando el ANC sea superior a 1,0 × 109/L. |
|
Bilirrubina total: 3,0-5,0 × ULN |
Continuar con Jakafi en 1 nivel de dosis inferior hasta la recuperación. Si se resuelve en 14 días, aumentar en un nivel de dosis. Si no se resuelve en 14 días, mantener el nivel de dosis disminuido. |
|
Bilirrubina total: > 5,0-10,0 × ULN |
Suspender Jakafi hasta por 14 días hasta que se resuelva; reanudar con la dosis actual tras la recuperación. Si no se resuelve en 14 días, reanudar en 1 nivel de dosis inferior tras la recuperación. |
| Bilirrubina total: > 10,0 × ULN |
Suspender Jakafi hasta por 14 días hasta que se resuelva; reanudar en 1 nivel de dosis inferior tras la recuperación. Si no se resuelve en 14 días, suspender. |
| Otras reacciones adversas: Grado 3 | Continuar con Jakafi en 1 nivel de dosis inferior hasta la recuperación. |
| Otras reacciones adversas: Grado 4 | Suspender Jakafi. |
2.6 Modificaciones de la dosis para el uso concomitante con inhibidores potentes del CYP3A4 o fluconazol
Modifique la dosis de Jakafi cuando se coadministre con inhibidores potentes del CYP3A4 o dosis de menos de o igual a 200 mg de fluconazol [ver Interacciones medicamentosas (7)], de acuerdo con la Tabla 9. Evite el uso concomitante de Jakafi con dosis de fluconazol superiores a 200 mg diarios.
| Para pacientes coadministrados con inhibidores potentes del CYP3A4 o dosis de menos de o igual a 200 mg de fluconazol | Modificación de la dosis de Jakafi recomendada |
| Dosis inicial para pacientes con MF con un recuento plaquetario: | |
|
10 mg dos veces al día |
|
5 mg una vez al día |
| Dosis inicial para pacientes con PV: | 5 mg dos veces al día |
| Si está en dosis estable para pacientes con MF o PV: | |
|
Disminuya la dosis en un 50% (redondee hacia arriba a la concentración de comprimido disponible más cercana) |
|
5 mg una vez al día |
|
Evite el tratamiento con inhibidores potentes del CYP3A4 o fluconazol o interrumpa el tratamiento con Jakafi durante la duración del uso de inhibidores potentes del CYP3A4 o fluconazol |
| Dosis inicialpara pacientes con aGVHD o cGVHD: | |
| Dosis de fluconazol de menos de o igual a 200 mg |
5 mg una vez al día para pacientes con aGVHD; |
| Otros inhibidores del CYP3A4 | Controle los recuentos sanguíneos con más frecuencia para detectar toxicidad y modifique la dosis de Jakafi para las reacciones adversas si se producen [ver Dosis y administración (2.4, 2.5)]. |
2.7 Modificaciones de la dosis en caso de insuficiencia renal o hepática
Insuficiencia renal moderada a grave o enfermedad renal en etapa terminal con diálisis
Modifique la dosis de Jakafi para pacientes con insuficiencia renal moderada (CLcr 30 a 59 mL/min) a grave (CLcr 15 a 29 mL/min) o enfermedad renal en etapa terminal (ERET) con diálisis según la Tabla 10. Evite el uso de Jakafi en pacientes con ERET (CLcr inferior a 15 mL/min) que no requieren diálisis [véase Uso en poblaciones específicas (8.6)].
| ERET = enfermedad renal en etapa terminal, y CLcr = aclaramiento de creatinina | ||
| Estado de insuficiencia renal | Recuento de plaquetas | Dosis inicial recomendada |
| Pacientes con MF |
||
|
Moderada o grave |
Mayor de 150 x 109/L | Sin ajuste de dosis |
| 100 a 150 x 109/L | 10 mg dos veces al día | |
| 50 a menos de 100 x 109/L | 5 mg al día | |
| Menos de 50 x 109/L |
Evitar el uso [véase Uso en poblaciones específicas |
|
|
ERET con diálisis |
100 a 200 x 109/L | 15 mg una vez después de la sesión de diálisis |
|
Mayor de 200 x 109/L |
20 mg una vez después de la sesión de diálisis | |
| Pacientes con PV | ||
| Moderada o grave | Cualquiera | 5 mg dos veces al día |
| ERET con diálisis | Cualquiera | 10 mg una vez después de la sesión de diálisis |
| Pacientes con aGVHD | ||
| Moderada o grave | Cualquiera | 5 mg una vez al día |
| ERET con diálisis | Cualquiera | 5 mg una vez después de la sesión de diálisis |
| Pacientes con cGVHD |
||
| Moderada o grave | Cualquiera | 5 mg dos veces al día |
| ERET con diálisis | Cualquiera | 10 mg una vez después de la sesión de diálisis |
Insuficiencia hepática
Modifique la dosis de Jakafi para pacientes con insuficiencia hepática según la Tabla 11.
| Estado de insuficiencia hepática | Recuento de plaquetas | Dosis inicial recomendada |
|
Pacientes con MF |
Mayor de 150 x 109/L | Sin ajuste de dosis |
| 100 x 109/L a 150 x 109/L | 10 mg dos veces al día | |
| 50 a menos de 100 x 109/L | 5 mg al día | |
| Menos de 50 x 109/L | Evitar el uso [ver Uso en poblaciones específicas (8.7)] | |
| Pacientes con PV Leve, moderada o grave (Child-Pugh clase A, B, C) |
Cualquiera | 5 mg dos veces al día |
| Pacientes con aGVHD | ||
|
Leve, moderada o grave según los criterios del NCI sin GVHD hepática |
Cualquiera | Sin ajuste de dosis |
|
Estadio 1, 2 o 3 de aGVHD hepática |
Cualquiera | Sin ajuste de dosis |
|
Estadio 4 de aGVHD hepática |
Cualquiera | 5 mg una vez al día |
| Pacientes con cGVHD |
||
|
Leve, moderada o grave según los criterios del NCI sin GVHD hepática |
Cualquiera | Sin ajuste de dosis |
|
Puntuación 1 o 2 de cGVHD hepática |
Cualquiera | Sin ajuste de dosis |
|
Puntuación 3 de cGVHD hepática |
Cualquiera | Controlar los recuentos sanguíneos con más frecuencia para detectar toxicidad y modificar la dosis de Jakafi si se producen reacciones adversas [ver Dosis y administración (2.4, 2.5)]. |
2.8 Método de administración
Jakafi se administra por vía oral y puede administrarse con o sin alimentos.
Si se olvida una dosis, el paciente no debe tomar una dosis adicional, sino que debe tomar la siguiente dosis prescrita habitual.
Cuando se interrumpe el tratamiento con Jakafi por razones distintas a la trombocitopenia, puede considerarse una reducción gradual de la dosis de Jakafi, por ejemplo, 5 mg dos veces al día cada semana.
Para los pacientes que no pueden ingerir comprimidos, Jakafi puede administrarse a través de una sonda nasogástrica (8 French o superior) de la siguiente manera:
- Suspender un comprimido en aproximadamente 40 ml de agua removiendo durante aproximadamente 10 minutos.
- En las 6 horas siguientes a la dispersión del comprimido, la suspensión puede administrarse a través de una sonda nasogástrica utilizando una jeringa adecuada.
La sonda debe enjuagarse con aproximadamente 75 ml de agua. No se ha evaluado el efecto de las preparaciones de alimentación por sonda en la exposición a Jakafi durante la administración a través de una sonda nasogástrica.
3. FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
Comprimidos de 5 mg – redondos y blancos con “INCY” en un lado y “5” en el otro.
Comprimidos de 10 mg – redondos y blancos con “INCY” en un lado y “10” en el otro.
Comprimidos de 15 mg – ovalados y blancos con “INCY” en un lado y “15” en el otro.
Comprimidos de 20 mg – con forma de cápsula y blancos con “INCY” en un lado y “20” en el otro.
Comprimidos de 25 mg – ovalados y blancos con “INCY” en un lado y “25” en el otro.
4. CONTRAINDICACIONES
Ninguna.
5. ADVERTENCIAS Y PRECAUCIONES
5.1 Trombocitopenia, Anemia y Neutropenia
El tratamiento con Jakafi puede causar trombocitopenia, anemia y neutropenia [véase Reacciones adversas (6.1)].
Controle la trombocitopenia reduciendo la dosis o interrumpiendo temporalmente Jakafi. Pueden ser necesarias transfusiones de plaquetas [véase Posología y administración (2)].
Los pacientes que desarrollan anemia pueden requerir transfusiones de sangre y/o modificaciones de la dosis de Jakafi.
La neutropenia grave (ANC inferior a 0,5 × 109/L) fue generalmente reversible al suspender Jakafi hasta la recuperación.
Realice un hemograma completo (CBC) antes del tratamiento y controle los CBC cada 2 a 4 semanas hasta que las dosis se estabilicen, y luego según lo clínicamente indicado [véase Posología y administración (2)].
5.2 Riesgo de infección
Se han producido infecciones bacterianas, micobacterianas, fúngicas y víricas graves [véase Reacciones adversas (6.1)]. Retrase el inicio del tratamiento con Jakafi hasta que las infecciones graves activas se hayan resuelto. Observe a los pacientes que reciben Jakafi para detectar signos y síntomas de infección y trátelos con prontitud. Utilice la vigilancia activa y los antibióticos profilácticos de acuerdo con las guías clínicas.
Tuberculosis
Se ha notificado infección por tuberculosis en pacientes que reciben Jakafi. Observe a los pacientes que reciben Jakafi para detectar signos y síntomas de tuberculosis activa y trátelos con prontitud.
Antes de iniciar Jakafi, se debe evaluar a los pacientes para detectar factores de riesgo de tuberculosis, y aquellos con mayor riesgo deben someterse a pruebas de infección latente. Los factores de riesgo incluyen, entre otros, la residencia anterior o los viajes a países con una alta prevalencia de tuberculosis, el contacto cercano con una persona con tuberculosis activa y un historial de tuberculosis activa o latente donde no se puede confirmar un tratamiento adecuado.
Para los pacientes con evidencia de tuberculosis activa o latente, consulte a un médico con experiencia en el tratamiento de la tuberculosis antes de comenzar con Jakafi. La decisión de continuar con Jakafi durante el tratamiento de la tuberculosis activa debe basarse en la determinación general de riesgo-beneficio.
Leucoencefalopatía multifocal progresiva
Se ha producido leucoencefalopatía multifocal progresiva (LMP) con el tratamiento con Jakafi. Si se sospecha LMP, suspenda Jakafi y evalúe.
Herpes Zoster y Herpes Simplex
Se ha notificado infección por herpes zóster en pacientes que reciben Jakafi [véase Reacciones adversas (6.1)]. Avise a los pacientes sobre los signos y síntomas tempranos del herpes zóster y que busquen tratamiento lo antes posible si lo sospechan.
Se ha notificado reactivación y/o diseminación del virus del herpes simple en pacientes que reciben Jakafi [véase Reacciones adversas (6.2)]. Controle a los pacientes para detectar el desarrollo de infecciones por herpes simple. Si un paciente desarrolla evidencia de diseminación del herpes simple, considere interrumpir el tratamiento con Jakafi; los pacientes deben ser tratados y controlados rápidamente de acuerdo con las guías clínicas.
Hepatitis B
Se ha notificado un aumento de la carga viral de la hepatitis B (título de ADN del VHB), con o sin elevaciones asociadas de alanino aminotransferasa y aspartato aminotransferasa, en pacientes con infecciones crónicas por VHB que toman Jakafi. Se desconoce el efecto de Jakafi sobre la replicación viral en pacientes con infección crónica por VHB. Los pacientes con infección crónica por VHB deben ser tratados y controlados de acuerdo con las guías clínicas.
5.3 Exacerbación de los síntomas tras la interrupción o suspensión del tratamiento con Jakafi
Tras la suspensión de Jakafi, los síntomas de las neoplasias mieloproliferativas pueden volver a los niveles previos al tratamiento en un período de aproximadamente una semana. Algunos pacientes con MF han experimentado uno o más de los siguientes eventos adversos después de suspender Jakafi: fiebre, dificultad respiratoria, hipotensión, CID o insuficiencia multiorgánica. Si se produce uno o más de estos después de suspender o mientras se reduce la dosis de Jakafi, evalúe y trate cualquier enfermedad intercurrente y considere reiniciar o aumentar la dosis de Jakafi. Indique a los pacientes que no interrumpan ni suspendan el tratamiento con Jakafi sin consultar a su médico. Al suspender o interrumpir el tratamiento con Jakafi por razones distintas a la trombocitopenia o la neutropenia [véase Posología y administración (2.8)], considere reducir la dosis de Jakafi gradualmente en lugar de suspenderla abruptamente.
5.4 Cáncer de piel no melanoma (CPNM)
Se han producido cánceres de piel no melanoma, incluidos el carcinoma basocelular, el carcinoma espinocelular y el carcinoma de células de Merkel, en pacientes tratados con Jakafi. Realice exámenes periódicos de la piel.
5.5 Elevación de Lípidos
El tratamiento con Jakafi se ha asociado con aumentos en los parámetros lipídicos, incluyendo el colesterol total, el colesterol de lipoproteínas de baja densidad (LDL) y los triglicéridos [ver Reacciones Adversas (6.1)]. No se ha determinado el efecto de estas elevaciones de los parámetros lipídicos en la morbilidad y mortalidad cardiovascular en pacientes tratados con Jakafi. Evalúe los parámetros lipídicos aproximadamente 8-12 semanas después del inicio del tratamiento con Jakafi. Monitoree y trate de acuerdo con las guías clínicas para el manejo de la hiperlipidemia.
5.6 Eventos Cardiovasculares Adversos Mayores (ECAM)
Otro inhibidor de JAK ha aumentado el riesgo de ECAM, incluyendo muerte cardiovascular, infarto de miocardio y accidente cerebrovascular (en comparación con aquellos tratados con bloqueadores del TNF) en pacientes con artritis reumatoide, una afección para la cual Jakafi no está indicado.
Considere los beneficios y riesgos para el paciente individual antes de iniciar o continuar el tratamiento con Jakafi, particularmente en pacientes que son fumadores actuales o pasados y pacientes con otros factores de riesgo cardiovascular. Se debe informar a los pacientes sobre los síntomas de eventos cardiovasculares graves y los pasos a seguir si ocurren.
5.7 Trombosis
Otro inhibidor de JAK ha aumentado el riesgo de trombosis, incluyendo trombosis venosa profunda (TVP), embolia pulmonar (EP) y trombosis arterial (en comparación con aquellos tratados con bloqueadores del TNF) en pacientes con artritis reumatoide, una afección para la cual Jakafi no está indicado. En pacientes con MF y PV tratados con Jakafi en ensayos clínicos, las tasas de eventos tromboembólicos fueron similares en pacientes tratados con Jakafi y en el grupo control.
Los pacientes con síntomas de trombosis deben ser evaluados y tratados de inmediato de manera apropiada.
5.8 Neoplasias Secundarias
Otro inhibidor de JAK ha aumentado el riesgo de linfoma y otras neoplasias malignas excluyendo el CNM (en comparación con aquellos tratados con bloqueadores del TNF) en pacientes con artritis reumatoide, una afección para la cual Jakafi no está indicado. Los pacientes que son fumadores actuales o pasados tienen un riesgo adicional aumentado.
Considere los beneficios y riesgos para el paciente individual antes de iniciar o continuar el tratamiento con Jakafi, particularmente en pacientes con una neoplasia maligna secundaria conocida (que no sea un CNM tratado con éxito), pacientes que desarrollan una neoplasia maligna y pacientes que son fumadores actuales o pasados.
6. Reacciones adversas
Las siguientes reacciones adversas clínicamente significativas se describen con mayor detalle en otras secciones del etiquetado:
- Trombocitopenia, anemia y neutropenia [ver Advertencias y precauciones (5.1)]
- Riesgo de infección [ver Advertencias y precauciones (5.2)]
- Exacerbación de los síntomas tras la interrupción o suspensión del tratamiento con Jakafi [ver Advertencias y precauciones (5.3)]
- Cáncer de piel no melanoma [ver Advertencias y precauciones (5.4)]
- Elevaciones de lípidos [ver Advertencias y precauciones (5.5)]
- Eventos cardiovasculares adversos mayores (MACE) [ver Advertencias y precauciones (5.6)]
- Trombosis [ver Advertencias y precauciones (5.7)]
- Tumores malignos secundarios [ver Advertencias y precauciones (5.8)]
6.1 Experiencia en ensayos clínicos
Dado que los ensayos clínicos se realizan bajo condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un medicamento no pueden compararse directamente con las tasas de los ensayos clínicos de otro medicamento y es posible que no reflejen las tasas observadas en la práctica.
Mielofibrosis
La seguridad de Jakafi se evaluó en 617 pacientes en seis estudios clínicos con una duración media de seguimiento de 10,9 meses, incluidos 301 pacientes con MF en dos estudios de fase 3.
En estos dos estudios de fase 3, los pacientes tuvieron una duración media de exposición a Jakafi de 9,5 meses (intervalo de 0,5 a 17 meses), con un 89 % de los pacientes tratados durante más de 6 meses y un 25 % tratados durante más de 12 meses. Ciento once (111) pacientes comenzaron el tratamiento con 15 mg dos veces al día y 190 pacientes comenzaron con 20 mg dos veces al día. En los pacientes que comenzaron el tratamiento con 15 mg dos veces al día (recuento de plaquetas previo al tratamiento de 100 a 200 x 109/L) y 20 mg dos veces al día (recuento de plaquetas previo al tratamiento superior a 200 x 109/L), el 65 % y el 25 % de los pacientes, respectivamente, requirieron una reducción de la dosis por debajo de la dosis inicial en las primeras 8 semanas de terapia.
En un estudio doble ciego, aleatorizado y controlado con placebo de Jakafi, entre los 155 pacientes tratados con Jakafi, las reacciones adversas más frecuentes fueron trombocitopenia y anemia [ver Tabla 13]. La trombocitopenia, la anemia y la neutropenia son efectos relacionados con la dosis. Las tres reacciones adversas no hematológicas más frecuentes fueron hematomas, mareos y dolor de cabeza [ver Tabla 12].
Se observó la interrupción del tratamiento debido a eventos adversos, independientemente de la causalidad, en el 11 % de los pacientes tratados con Jakafi y en el 11 % de los pacientes tratados con placebo.
La Tabla 12 presenta las reacciones adversas no hematológicas más comunes que ocurrieron en pacientes que recibieron Jakafi en el estudio doble ciego, controlado con placebo durante el tratamiento aleatorizado.
|
||||||
| Jakafi (N=155) |
Placebo (N=151) |
|||||
| Reacciones adversas | Todos los grados* (%) |
Grado 3 (%) |
Grado 4 (%) |
Todos los grados (%) |
Grado 3 (%) |
Grado 4 (%) |
| Moretones† | 23 | < 1 | 0 | 15 | 0 | 0 |
| Mareo‡ | 18 | < 1 | 0 | 7 | 0 | 0 |
| Dolor de cabeza | 15 | 0 | 0 | 5 | 0 | 0 |
| Infecciones del tracto urinario§ | 9 | 0 | 0 | 5 | < 1 | < 1 |
| Aumento de peso¶ | 7 | < 1 | 0 | 1 | < 1 | 0 |
| Flatulencia | 5 | 0 | 0 | < 1 | 0 | 0 |
| Herpes Zoster# | 2 | 0 | 0 | < 1 | 0 | 0 |
Descripción de las Reacciones Adversas Seleccionadas
Anemia
En los dos estudios clínicos de Fase 3, la mediana del tiempo hasta la aparición de la primera anemia CTCAE de Grado 2 o superior fue de aproximadamente 6 semanas. Un paciente (< 1%) interrumpió el tratamiento debido a la anemia. En pacientes que recibieron Jakafi, las disminuciones medias de hemoglobina alcanzaron un nadir de aproximadamente 1.5 a 2.0 g/dL por debajo del valor inicial después de 8 a 12 semanas de terapia y luego se recuperaron gradualmente para alcanzar un nuevo estado estacionario que fue aproximadamente 1.0 g/dL por debajo del valor inicial. Este patrón se observó en pacientes independientemente de si habían recibido transfusiones durante la terapia.
En el estudio aleatorizado, controlado con placebo, el 60% de los pacientes tratados con Jakafi y el 38% de los pacientes que recibieron placebo recibieron transfusiones de glóbulos rojos durante el tratamiento aleatorizado. Entre los pacientes transfundidos, la mediana del número de unidades transfundidas por mes fue de 1.2 en pacientes tratados con Jakafi y de 1.7 en pacientes tratados con placebo.
Trombocitopenia
En los dos estudios clínicos de Fase 3, en pacientes que desarrollaron trombocitopenia de Grado 3 o 4, la mediana del tiempo hasta la aparición fue de aproximadamente 8 semanas. La trombocitopenia fue generalmente reversible con la reducción de la dosis o la interrupción de la dosis. La mediana del tiempo hasta la recuperación del recuento de plaquetas por encima de 50 x 109/L fue de 14 días. Se administraron transfusiones de plaquetas al 5% de los pacientes que recibieron Jakafi y al 4% de los pacientes que recibieron regímenes de control. La interrupción del tratamiento debido a la trombocitopenia ocurrió en < 1% de los pacientes que recibieron Jakafi y en < 1% de los pacientes que recibieron regímenes de control. Los pacientes con un recuento de plaquetas de 100 x 109/L a 200 x 109/L antes de comenzar con Jakafi tuvieron una mayor frecuencia de trombocitopenia de Grado 3 o 4 en comparación con los pacientes con un recuento de plaquetas superior a 200 x 109/L (17% versus 7%).
Neutropenia
En los dos estudios clínicos de Fase 3, el 1% de los pacientes redujeron o suspendieron Jakafi debido a la neutropenia.
La Tabla 13 proporciona la frecuencia y la gravedad de las anomalías hematológicas clínicas informadas para los pacientes que recibieron tratamiento con Jakafi o placebo en el estudio controlado con placebo.
| Jakafi (N=155) |
Placebo (N=151) |
|||||
|
Parámetro de Laboratorio |
Todos los Grados† (%) |
Grado 3 (%) |
Grado 4 (%) |
Todos los Grados (%) |
Grado 3 (%) |
Grado 4 (%) |
| Trombocitopenia | 70 | 9 | 4 | 31 | 1 | 0 |
| Anemia | 96 | 34 | 11 | 87 | 16 | 3 |
| Neutropenia | 19 | 5 | 2 | 4 | < 1 | 1 |
Datos Adicionales del Estudio Controlado con Placebo
- El 25% de los pacientes tratados con Jakafi y el 7% de los pacientes tratados con placebo desarrollaron anomalías de Grado 1 nuevas o que empeoraron en la alanina transaminasa (ALT). La incidencia de elevaciones mayores o iguales al Grado 2 fue del 2% para Jakafi con un 1% de Grado 3 y ninguna elevación de ALT de Grado 4.
- El 17% de los pacientes tratados con Jakafi y el 6% de los pacientes tratados con placebo desarrollaron anomalías de Grado 1 nuevas o que empeoraron en la aspartato transaminasa (AST). La incidencia de elevaciones de AST de Grado 2 fue < 1% para Jakafi sin elevaciones de AST de Grado 3 o 4.
- El 17% de los pacientes tratados con Jakafi y < 1% de los pacientes tratados con placebo desarrollaron elevaciones de colesterol de Grado 1 nuevas o que empeoraron. La incidencia de elevaciones de colesterol de Grado 2 fue < 1% para Jakafi sin elevaciones de colesterol de Grado 3 o 4.
Policitemia Vera
En un estudio aleatorizado, abierto y con control activo, 110 pacientes con PV resistente o intolerante a la hidroxiurea recibieron Jakafi y 111 pacientes recibieron la mejor terapia disponible [ver Estudios Clínicos (14.2)]. La reacción adversa más frecuente fue la anemia. Se observó la interrupción del tratamiento por eventos adversos, independientemente de la causalidad, en el 4% de los pacientes tratados con Jakafi. La Tabla 14 presenta las reacciones adversas no hematológicas más frecuentes que ocurrieron hasta la Semana 32.
|
||||
| Jakafi (N=110) |
Mejor Terapia Disponible (N=111) |
|||
|
Reacciones Adversas |
Todos los Grados* (%) |
Grado 3-4 (%) |
Todos los Grados (%) |
Grado 3-4 (%) |
| Diarrea | 15 | 0 | 7 | < 1 |
| Mareos† | 15 | 0 | 13 | 0 |
| Disnea‡ | 13 | 3 | 4 | 0 |
| Espasmos Musculares | 12 | < 1 | 5 | 0 |
| Estreñimiento | 8 | 0 | 3 | 0 |
| Herpes Zóster§ | 6 | < 1 | 0 | 0 |
| Náuseas | 6 | 0 | 4 | 0 |
| Aumento de Peso¶ | 6 | 0 | < 1 | 0 |
| Infecciones del tracto urinario# | 6 | 0 | 3 | 0 |
| Hipertensión | 5 | < 1 | 3 | < 1 |
Las anomalías clínicamente relevantes de laboratorio se muestran en la Tabla 15.
| Jakafi (N=110) |
Mejor Terapia Disponible (N=111) |
|||||
| Parámetro de Laboratorio | Todos los Grados† (%) |
Grado 3 (%) |
Grado 4 (%) |
Todos los Grados (%) |
Grado 3 (%) |
Grado 4 (%) |
| Hematología | ||||||
| Anemia | 72 | < 1 | < 1 | 58 | 0 | 0 |
| Trombocitopenia | 27 | 5 | < 1 | 24 | 3 | < 1 |
| Neutropenia | 3 | 0 | < 1 | 10 | < 1 | 0 |
| Química | ||||||
| Hipercolesterolemia | 35 | 0 | 0 | 8 | 0 | 0 |
| ALT elevada | 25 | < 1 | 0 | 16 | 0 | 0 |
| AST elevada | 23 | 0 | 0 | 23 | < 1 | 0 |
| Hipertrigliceridemia | 15 | 0 | 0 | 13 | 0 | 0 |
Enfermedad Aguda de Injerto Contra Huésped
En un estudio abierto de un solo brazo, 71 adultos (de 18 a 73 años) fueron tratados con Jakafi para la EICH aguda que no respondió al tratamiento con esteroides con o sin otros fármacos inmunosupresores [ver Estudios Clínicos (14.3)]. La mediana de la duración del tratamiento con Jakafi fue de 46 días (rango, 4 a 382 días).
No hubo reacciones adversas fatales a Jakafi. Se produjo una reacción adversa que provocó la interrupción del tratamiento en el 31 % de los pacientes. La reacción adversa más común que condujo a la interrupción del tratamiento fue la infección (10 %). La Tabla 16 muestra las reacciones adversas distintas de las anomalías de laboratorio.
| Jakafi (N=71) | ||
| Reacciones Adversas* | Todos los Grados† (%) |
Grado 3-4 (%) |
|
Infecciones (patógeno no especificado) |
55 |
41 |
| Edema | 51 | 13 |
| Hemorragia | 49 | 20 |
| Fatiga | 37 | 14 |
| Infecciones bacterianas | 32 | 28 |
| Disnea | 32 | 7 |
| Infecciones virales | 31 | 14 |
| Trombosis | 25 | 11 |
| Diarrea | 24 | 7 |
| Erupción cutánea | 23 | 3 |
| Dolor de cabeza | 21 | 4 |
| Hipertensión | 20 | 13 |
| Mareos | 16 | 0 |
Las anormalidades de laboratorio seleccionadas durante el tratamiento con Jakafi se muestran en la Tabla 17.
|
||
| Jakafi (N=71) | ||
| Peor grado durante el tratamiento | ||
| Parámetro de Laboratorio | Todos los Grados* (%) |
Grado 3-4 |
| Hematología | ||
| Anemia | 75 | 45 |
| Trombocitopenia | 75 | 61 |
| Neutropenia | 58 | 40 |
| Química | ||
| Elevated ALT | 48 | 8 |
| Elevated AST | 48 | 6 |
| Hipertrigliceridemia | 11 | 1 |
Enfermedad crónica de injerto contra huésped
En un estudio de fase 3, aleatorizado, abierto, multicéntrico, 165 pacientes fueron tratados con Jakafi y 158 pacientes fueron tratados con la mejor terapia disponible para cGVHD que falló el tratamiento con esteroides con o sin otros fármacos inmunosupresores [ver Estudios Clínicos (14.4)]; sesenta y cinco pacientes pasaron de la mejor terapia disponible al tratamiento con Jakafi, para un total de 230 pacientes tratados con Jakafi. La mediana de la duración de la exposición a Jakafi para el estudio fue de 49.7 semanas (rango, 0.7 a 144.9 semanas) en el grupo de Jakafi. Ciento nueve (47%) pacientes estuvieron con Jakafi durante al menos 1 año.
Hubo cinco reacciones adversas fatales a Jakafi, incluyendo 1 por necrólisis epidérmica tóxica y 4 por neutropenia, anemia y/o trombocitopenia. Se produjo una reacción adversa que provocó la interrupción del tratamiento en el 18% de los pacientes tratados con Jakafi. Una reacción adversa que provocó una modificación de la dosis se produjo en el 27%, y una reacción adversa que provocó la interrupción del tratamiento se produjo en el 23%. Las reacciones adversas hematológicas más comunes (incidencia > 35%) son anemia y trombocitopenia. Las reacciones adversas no hematológicas más comunes (incidencia ≥ 20%) son infecciones (patógeno no especificado) e infección viral.
La Tabla 18 presenta las reacciones adversas no relacionadas con el laboratorio más frecuentes que ocurren hasta el Día 1 del Ciclo 7 del tratamiento aleatorizado.
| Reacción adversa* |
Jakafi (N = 165) |
Mejor terapia disponible (N = 158) |
||
|
Todos los grados† |
Grado ≥ 3 |
Todos los grados |
Grado ≥ 3 |
|
| Infecciones e infestaciones |
||||
| Infecciones (patógeno no especificado) | 45 | 15 | 44 | 16 |
| Infecciones virales | 28 | 5 | 23 | 5 |
| Trastornos musculoesqueléticos y del tejido conjuntivo |
||||
| Dolor musculoesquelético | 18 | 1 | 13 | 0 |
| Trastornos generales y afecciones en el lugar de administración | ||||
| Pirexia | 16 | 2 | 9 | 1 |
| Fatiga | 13 | 1 | 10 | 2 |
| Edema | 10 | 1 | 12 | 1 |
| Trastornos vasculares |
||||
| Hipertensión | 16 | 5 | 13 | 7 |
| Hemorragia | 12 | 2 | 15 | 2 |
| Trastornos respiratorios, torácicos y mediastínicos | ||||
| Tos | 13 | 0 | 8 | 0 |
| Disnea | 11 | 1 | 8 | 1 |
| Trastornos gastrointestinales |
||||
| Náuseas | 12 | 0 | 13 | 2 |
| Diarrea | 10 | 1 | 13 | 1 |
Las anomalías clínicamente relevantes de laboratorio se muestran en la Tabla 19.
| Prueba de laboratorio |
Jakafi (N = 165) |
Mejor terapia disponible (N = 158) |
||
|
Todos los grados† |
Grado ≥ 3 |
Todos los grados |
Grado ≥ 3 |
|
| Hematología |
||||
| Anemia | 82 | 13 | 75 | 8 |
| Neutropenia | 27 | 12 | 23 | 9 |
| Trombocitopenia | 58 | 20 | 54 | 17 |
| Química |
||||
| Hipercolesterolemia | 88 | 10 | 85 | 8 |
| AST elevada | 65 | 5 | 54 | 6 |
| ALT elevada | 73 | 11 | 71 | 16 |
| Gamma glutamiltransferasa aumentada | 81 | 42 | 75 | 38 |
| Creatinina aumentada | 47 | 1 | 40 | 2 |
| Lipasa elevada | 38 | 12 | 30 | 9 |
| Amilasa elevada | 35 | 8 | 25 | 4 |
6.2 Experiencia Poscomercialización
Las siguientes reacciones adversas se han identificado durante el uso poscomercialización de Jakafi. Debido a que estas reacciones se reportan voluntariamente de una población de tamaño incierto, no siempre es posible estimar de forma fiable su frecuencia o establecer una relación causal con la exposición al fármaco:
- Infecciones e Infestaciones: Reactivación y/o diseminación del virus del herpes simple
7. INTERACCIONES MEDICAMENTOSAS
7.1 Efecto de otros medicamentos sobre Jakafi
Fluconazol
El uso concomitante de Jakafi con fluconazol aumenta la exposición a ruxolitinib [ver Farmacología Clínica (12.3)], lo que puede aumentar el riesgo de reacciones adversas relacionadas con la exposición. Evite el uso concomitante de Jakafi con dosis de fluconazol superiores a 200 mg diarios. Reduzca la dosis de Jakafi cuando se use concomitantemente con dosis de fluconazol inferiores o iguales a 200 mg [ver Posología y administración (2.6)].
Inhibidores potentes del CYP3A4
El uso concomitante de Jakafi con inhibidores potentes del CYP3A4 aumenta la exposición a ruxolitinib [ver Farmacología Clínica (12.3)], lo que puede aumentar el riesgo de reacciones adversas relacionadas con la exposición. Reduzca la dosis de Jakafi cuando se use concomitantemente con inhibidores potentes del CYP3A4, excepto en pacientes con aGVHD o cGVHD [ver Posología y administración (2.6)].
Inductores potentes del CYP3A4
El uso concomitante de Jakafi con inductores potentes del CYP3A4 puede disminuir la exposición a ruxolitinib [ver Farmacología Clínica (12.3)], lo que puede reducir la eficacia de Jakafi. Controle a los pacientes con frecuencia y ajuste la dosis de Jakafi según la seguridad y la eficacia [ver Farmacología Clínica (12.3)].
8. USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de Riesgo
Cuando se administró ruxolitinib a ratas y conejas embarazadas durante el período de organogénesis, se produjeron resultados de desarrollo adversos a dosis asociadas con toxicidad materna (ver Datos). No hay estudios con el uso de Jakafi en mujeres embarazadas que informen sobre los riesgos asociados con el fármaco.
Se desconoce el riesgo de base de defectos congénitos importantes y aborto espontáneo para las poblaciones indicadas. Los resultados adversos en el embarazo ocurren independientemente de la salud de la madre o del uso de medicamentos. El riesgo de base en la población general de EE. UU. de defectos congénitos importantes es del 2% al 4% y el aborto espontáneo del 15% al 20% de los embarazos clínicamente reconocidos.
Datos
Datos en animales
Se administró ruxolitinib por vía oral a ratas o conejas embarazadas durante el período de organogénesis, a dosis de 15, 30 o 60 mg/kg/día en ratas y 10, 30 o 60 mg/kg/día en conejas. No hubo malformaciones relacionadas con el tratamiento. Se observaron resultados de desarrollo adversos, como disminuciones de aproximadamente el 9% en el peso fetal en ratas a la dosis más alta y tóxica para la madre de 60 mg/kg/día. Esta dosis produce una exposición (AUC) que es aproximadamente 2 veces la exposición clínica a la dosis máxima recomendada de 25 mg dos veces al día. En conejas, se observaron pesos fetales más bajos de aproximadamente el 8% y un aumento de las reabsorciones tardías a la dosis más alta y tóxica para la madre de 60 mg/kg/día. Esta dosis es aproximadamente el 7% de la exposición clínica a la dosis máxima recomendada.
En un estudio de desarrollo pre y postnatal en ratas, se administraron dosis de ruxolitinib a animales preñadas desde la implantación hasta la lactancia a dosis de hasta 30 mg/kg/día. No hubo hallazgos adversos relacionados con el fármaco en las crías para los índices de fertilidad o para los parámetros de supervivencia, crecimiento y desarrollo materno o embriofetal a la dosis más alta evaluada (34% de la exposición clínica a la dosis máxima recomendada de 25 mg dos veces al día).
8.2 Lactancia
Resumen de Riesgo
No hay datos disponibles sobre la presencia de ruxolitinib en la leche materna, los efectos en el niño amamantado o los efectos en la producción de leche. La ruxolitinib y/o sus metabolitos estuvieron presentes en la leche de ratas lactantes (ver Datos). Debido a que muchos fármacos están presentes en la leche materna y debido al potencial de trombocitopenia y anemia que se muestra para Jakafi en estudios humanos, suspenda la lactancia materna durante el tratamiento con Jakafi y durante dos semanas después de la dosis final.
Datos
Datos en animales
Se administró a ratas lactantes una dosis única de ruxolitinib marcado con [14C] (30 mg/kg) en el día postnatal 10, después de lo cual se recogieron muestras de plasma y leche durante un máximo de 24 horas. El AUC para la radiactividad total en la leche fue aproximadamente 13 veces el AUC del plasma materno. El análisis adicional mostró la presencia de ruxolitinib y varios de sus metabolitos en la leche, todos a niveles superiores a los del plasma materno.
8.4 Uso pediátrico
Mielofibrosis
No se ha establecido la seguridad y eficacia de Jakafi para el tratamiento de la mielofibrosis en pacientes pediátricos.
Policitemia Vera
No se ha establecido la seguridad y eficacia de Jakafi para el tratamiento de la policitemia vera en pacientes pediátricos.
Enfermedad injerto contra huésped aguda
La seguridad y eficacia de Jakafi para el tratamiento de la aGVHD refractaria a esteroides se ha establecido para el tratamiento de pacientes pediátricos de 12 años o más. El uso de Jakafi en pacientes pediátricos con aGVHD refractaria a esteroides se basa en evidencia de ensayos adecuados y bien controlados de Jakafi en adultos [ver Estudios clínicos (14.3)] y datos adicionales de farmacocinética y seguridad en pacientes pediátricos. No se ha establecido la seguridad y eficacia de Jakafi para el tratamiento de la aGVHD refractaria a esteroides en pacientes pediátricos menores de 12 años.
Enfermedad injerto contra huésped crónica
La seguridad y eficacia de Jakafi para el tratamiento de la cGVHD después del fracaso de una o dos líneas de terapia sistémica se ha establecido para el tratamiento de pacientes pediátricos de 12 años o más. El uso de Jakafi en pacientes pediátricos con cGVHD después del fracaso de una o dos líneas de terapia sistémica se basa en evidencia de ensayos adecuados y bien controlados de Jakafi en adultos y adolescentes [ver Estudios clínicos (14.4)] y datos adicionales de farmacocinética y seguridad en pacientes pediátricos. No se ha establecido la seguridad y eficacia de Jakafi para el tratamiento de la cGVHD en pacientes pediátricos menores de 12 años.
Otras neoplasias mieloproliferativas, leucemias y tumores sólidos
La seguridad y eficacia de la ruxolitinib se evaluaron pero no se establecieron en un ensayo de un solo brazo (NCT01164163) en pacientes con tumores sólidos, leucemias o neoplasias mieloproliferativas recidivantes o refractarias. Los pacientes incluyeron 18 niños (de 2 a < 12 años) y 14 adolescentes (de 12 a < 17 años). En general, el 19% de los pacientes recibieron más de un ciclo. No se observaron nuevas señales de seguridad en pacientes pediátricos en este ensayo.
La seguridad y eficacia de la ruxolitinib en combinación con quimioterapia para el tratamiento de la leucemia linfoblástica aguda (LLA) de alto riesgo, de novo con reordenamiento de CRLF2 o mutación de la vía JAK similar a la Ph se evaluaron pero no se establecieron en un ensayo de un solo brazo (NCT02723994). Los pacientes incluyeron 2 lactantes (edad < 2 años), 42 niños (edad de 2 a < 12 años) y 62 adolescentes (edad de 12 a < 17 años). No se observaron nuevas señales de seguridad en pacientes pediátricos en este ensayo.
Datos de toxicidad en animales jóvenes
La administración de ruxolitinib a ratas juveniles provocó efectos en el crecimiento y las medidas óseas. Cuando se administró a partir del día postnatal 7 (equivalente a un recién nacido humano) en dosis de 1,5 a 75 mg/kg/día, se observaron fracturas en dosis ≥ 30 mg/kg/día, y efectos en el peso corporal y otras medidas óseas [p. ej., contenido mineral óseo, tomografía computarizada cuantitativa periférica y análisis de rayos X] en dosis ≥ 5 mg/kg/día. Cuando se administró a partir del día postnatal 21 (equivalente a un humano de 2-3 años de edad) en dosis de 5 a 60 mg/kg/día, se observaron efectos en el peso corporal y los huesos en dosis ≥ 15 mg/kg/día, que se consideraron adversos a 60 mg/kg/día. Los machos se vieron más afectados que las hembras en todos los grupos de edad, y los efectos fueron generalmente más graves cuando la administración se inició antes en el período postnatal. Estos hallazgos se observaron en exposiciones que son al menos el 27% de la exposición clínica a la dosis máxima recomendada de 25 mg dos veces al día.
8.5 Uso en pacientes geriátricos
Del número total de pacientes con MF en estudios clínicos con Jakafi, el 52% tenía 65 años o más, mientras que el 15% tenía 75 años o más. No se observaron diferencias generales en la seguridad o eficacia de Jakafi entre estos pacientes y los pacientes más jóvenes.
Los estudios clínicos de Jakafi en pacientes con aGVHD no incluyeron un número suficiente de sujetos de 65 años o más para determinar si responden de manera diferente a los sujetos más jóvenes.
Del número total de pacientes con cGVHD tratados con Jakafi en ensayos clínicos, el 11% tenía 65 años o más. No se observaron diferencias generales en la seguridad o eficacia de Jakafi entre estos pacientes y los pacientes más jóvenes.
8.6 Insuficiencia renal
La exposición total a ruxolitinib y sus metabolitos activos aumentó con la insuficiencia renal moderada (CLcr 30 a 59 mL/min) y grave (CLcr 15 a 29 mL/min), y la ERGE (CLcr inferior a 15 mL/min) en diálisis [ver Farmacología clínica (12.3)]. Modifique la dosis de Jakafi según lo recomendado [ver Dosis y administración (2.7)].
8.7 Insuficiencia hepática
La exposición a ruxolitinib aumentó con la insuficiencia hepática leve (Child-Pugh A), moderada (Child-Pugh B) y grave (Child-Pugh C) [ver Farmacología clínica (12.3)].
Reduzca la dosis de Jakafi según lo recomendado en pacientes con MF o PV con insuficiencia hepática [ver Dosis y administración (2.7)]. Reduzca la dosis de Jakafi según lo recomendado para pacientes con aGVHD hepática en estadio 4.
Controle los recuentos sanguíneos con más frecuencia para detectar toxicidad y modifique la dosis de Jakafi para las reacciones adversas si se producen en pacientes con cGVHD hepática de puntuación 3 [ver Dosis y administración (2.7) y Farmacología clínica (12.3)].
10. SOBREDOSIS
No se conoce antídoto para las sobredosis con Jakafi. Se han administrado dosis únicas de hasta 200 mg con una tolerabilidad aguda aceptable. Las dosis repetidas superiores a las recomendadas se asocian a un aumento de la mielosupresión, incluida la leucopenia, la anemia y la trombocitopenia. Debe administrarse un tratamiento de soporte adecuado.
No se espera que la hemodiálisis mejore la eliminación de Jakafi.
11. DESCRIPCIÓN
El fosfato de ruxolitinib es un inhibidor de la cinasa con el nombre químico (R)-3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-ciclopentilpropanonitrilo fosfato y un peso molecular de 404,36. El fosfato de ruxolitinib tiene la siguiente fórmula estructural:
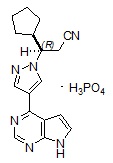
El fosfato de ruxolitinib es un polvo blanco a blanquecino a rosa claro y es soluble en tampones acuosos en un rango de pH de 1 a 8.
Los comprimidos de Jakafi (ruxolitinib) son para administración oral. Cada comprimido contiene 6,6 mg, 13,2 mg, 19,8 mg, 26,4 mg o 33 mg de fosfato de ruxolitinib, equivalente a 5 mg, 10 mg, 15 mg, 20 mg o 25 mg de base libre de ruxolitinib, respectivamente, junto con celulosa microcristalina, lactosa monohidrato, estearato de magnesio, dióxido de silicio coloidal, glicolato de almidón sódico, povidona e hidroxipropilcelulosa.
12. FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
El ruxolitinib, un inhibidor de quinasas, inhibe las quinasas Janus asociadas (JAK) JAK1 y JAK2, que median la señalización de varias citocinas y factores de crecimiento importantes para la hematopoyesis y la función inmunitaria. La señalización JAK implica el reclutamiento de STAT (transductores de señales y activadores de la transcripción) a los receptores de citocinas, la activación y la posterior localización de STAT al núcleo, lo que lleva a la modulación de la expresión génica.
La MF y la PV son neoplasias mieloproliferativas (NMP) que se sabe que están asociadas con una señalización JAK1 y JAK2 desregulada. En un modelo de ratón de NMP positiva para JAK2V617F, la administración oral de ruxolitinib previno la esplenomegalia, disminuyó preferentemente las células mutantes JAK2V617F en el bazo y disminuyó las citocinas inflamatorias circulantes (por ejemplo, TNF-α, IL-6).
Las vías de señalización JAK-STAT desempeñan un papel en la regulación del desarrollo, la proliferación y la activación de varios tipos de células inmunitarias importantes para la patogenia de la EICH. En un modelo de ratón de aEICH, la administración oral de ruxolitinib se asoció con una disminución de la expresión de citocinas inflamatorias en los homogeneizados de colon y una reducción de la infiltración de células inmunitarias en el colon.
12.2 Farmacodinamia
Jakafi inhibe la fosforilación de STAT3 inducida por citocinas en sangre completa de pacientes con MF y PV. La fosforilación de STAT3 alcanzó la inhibición máxima 2 horas después de la administración de Jakafi y volvió a la línea de base en aproximadamente 10 horas en pacientes con MF y PV.
Electrofisiología cardíaca
A una dosis de 1,25 a 10 veces la dosis inicial máxima recomendada, Jakafi no prolonga el intervalo QT en ningún grado clínicamente relevante.
12.3 Farmacocinética
La concentración plasmática máxima (Cmax) y el AUC medias de ruxolitinib aumentaron proporcionalmente en un rango de dosis única de 5 mg a 200 mg (4 veces la dosis diaria total máxima aprobada de 25 mg dos veces al día). La Cmax media de ruxolitinib osciló entre 205 nM y 7100 nM y el AUC osciló entre 862 nM*h y 30700 nM*h en un rango de dosis única de 5 mg a 200 mg.
Absorción
El ruxolitinib alcanza la Cmax entre 1 y 2 horas después de la administración. Se estima que la absorción oral de ruxolitinib es de al menos el 95%.
Efecto de los alimentos
No se observaron cambios clínicamente relevantes en la farmacocinética del ruxolitinib tras la administración de Jakafi con una comida alta en grasas y calorías (aproximadamente 800 a 1000 calorías, de las cuales el 50% procedían de grasas).
Distribución
El volumen de distribución medio del ruxolitinib en estado estacionario es de 72 L (coeficiente de variación [CV] 29%) en pacientes con MF y de 75 L (23%) en pacientes con PV.
La unión a proteínas del ruxolitinib es de aproximadamente el 97%, principalmente a la albúmina.
Eliminación
La semivida de eliminación media del ruxolitinib es de aproximadamente 3 horas y la semivida de eliminación media del ruxolitinib y sus metabolitos es de aproximadamente 5,8 horas en voluntarios sanos.
El aclaramiento de ruxolitinib (%CV) fue de 17,7 L/h en mujeres y de 22,1 L/h en hombres con MF (39%).
El aclaramiento de ruxolitinib (%CV) fue de 12,7 L/h (42%) en pacientes con PV.
El aclaramiento de ruxolitinib (%CV) fue de 11,8 L/h (63%) en pacientes con aEICH.
El aclaramiento de ruxolitinib (%CV) fue de 9,7 L/h (51%) en pacientes con cEICH.
Metabolismo
El ruxolitinib se metaboliza mediante el CYP3A4 y, en menor medida, mediante el CYP2C9.
Excreción
Tras una dosis oral única de ruxolitinib radiomarcado, el 74% de la radiactividad se excretó en la orina y el 22% por las heces. El fármaco inalterado representó menos del 1% de la radiactividad total excretada.
Poblaciones específicas
No se observaron diferencias clínicamente relevantes en la farmacocinética del ruxolitinib en función de la edad (12-73 años), la raza (blanca, asiática), el sexo o el peso (29-139 kg).
Pacientes con insuficiencia renal
El AUC total de ruxolitinib y sus metabolitos activos aumentó en 1,3, 1,5, 1,9 y 1,6 veces en sujetos con insuficiencia renal leve, moderada, grave y con ERGE después de la diálisis, respectivamente, en comparación con los sujetos con función renal normal (CLcr ≥ 90 mL/min). El cambio en el marcador farmacodinámico, la inhibición de pSTAT3, fue consistente con el aumento correspondiente en la exposición a metabolitos con insuficiencia renal. El ruxolitinib no se elimina mediante diálisis; sin embargo, no se puede descartar la eliminación de algunos metabolitos activos mediante diálisis.
Pacientes con insuficiencia hepática
No se observó ningún efecto clínicamente relevante en la farmacocinética del ruxolitinib en función de la insuficiencia hepática leve a grave según los criterios del NCI (bilirrubina total > ULN y cualquier AST) en pacientes con aEICH o cEICH.
El AUC de ruxolitinib aumentó en sujetos con insuficiencia hepática leve (Child-Pugh A) en 1,9 veces, moderada (Child-Pugh B) en 1,3 veces y grave (Child-Pugh C) en 1,7 veces en comparación con la de los sujetos con función hepática normal.
El cambio en el marcador farmacodinámico, la inhibición de pSTAT3, fue consistente con el aumento correspondiente en la exposición al ruxolitinib, excepto en la cohorte con insuficiencia hepática grave, donde la actividad farmacodinámica fue más prolongada en algunos sujetos de lo esperado en función de las concentraciones plasmáticas de ruxolitinib.
Pacientes con afectación hepática en la enfermedad de injerto contra huésped
No se observó ningún efecto clínicamente relevante en la farmacocinética del ruxolitinib en función de la aEICH hepática de estadio 1, 2 o 3, o de la cEICH hepática de puntuación 1 o 2.
Se observó un aclaramiento aparente menor de ruxolitinib en pacientes con aEICH hepática de estadio 4 en comparación con pacientes sin aEICH hepática.
Se desconoce el efecto de la cEICH hepática de puntuación 3 en la farmacocinética del ruxolitinib.
Estudios de interacción medicamentosa
Estudios clínicos y enfoques basados en modelos
Fluconazol: Fluconazol 100 a 400 mg una vez al día (un inhibidor moderado de CYP3A4 y CYP2C9) aumenta el AUC en estado estacionario de ruxolitinib en aproximadamente un 100% a 300% [véase Dosis y administración (2.6) e Interacciones medicamentosas (7)].
Inhibidores potentes de CYP3A4: Ketoconazol (inhibidor potente de CYP3A4) aumentó la Cmax de ruxolitinib en un 33% y el AUC en un 91% y prolongó la semivida de ruxolitinib de 3,7 horas a 6 horas [véase Dosis y administración (2.6) e Interacciones medicamentosas (7)].
Inhibidores moderados de CYP3A4: Eritromicina (inhibidor moderado de CYP3A4) aumentó la Cmax de ruxolitinib en un 8% y el AUC en un 27% [véase Interacciones medicamentosas (7)].
Inductores potentes de CYP3A4: Rifampicina (inductor potente de CYP3A4) disminuyó la Cmax de ruxolitinib en un 32% y el AUC en un 61%. La exposición relativa a los metabolitos activos de ruxolitinib aumentó aproximadamente un 100% [véase Interacciones medicamentosas (7)].
Estudios In Vitro
Enzimas del citocromo P450 (CYP): El ruxolitinib y su metabolito M18 no inhibieron CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 o CYP3A4. El ruxolitinib no indujo CYP1A2, CYP2B6 o CYP3A4 a concentraciones clínicamente relevantes.
Sistemas de transporte: El ruxolitinib y su metabolito M18 no inhibieron P-gp, BCRP, OATP1B1, OATP1B3, OCT1, OCT2, OAT1 u OAT3 a concentraciones clínicamente relevantes. El ruxolitinib no fue un sustrato de P-gp.
13. TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
El ruxolitinib no fue carcinogénico en el modelo de ratón transgénico Tg.rasH2 de 6 meses ni en un estudio de carcinogenicidad de 2 años en ratas.
El ruxolitinib no fue mutagénico en un ensayo de mutagenicidad bacteriana (prueba de Ames) ni clastogénico en un ensayo de aberración cromosómica in vitro (linfocitos de sangre periférica humana cultivados) o in vivo en un ensayo de micronúcleos de médula ósea de rata.
En un estudio de fertilidad, se administró ruxolitinib a ratas macho antes y durante el apareamiento y a ratas hembra antes del apareamiento y hasta el día de la implantación (día 7 de gestación). El ruxolitinib no tuvo ningún efecto sobre la fertilidad ni la función reproductiva en ratas macho o hembra a dosis de 10, 30 o 60 mg/kg/día. Sin embargo, en ratas hembras, dosis iguales o superiores a 30 mg/kg/día provocaron un aumento de la pérdida postimplantacional. La exposición (AUC) a la dosis de 30 mg/kg/día es aproximadamente el 34% de la exposición clínica a la dosis máxima recomendada de 25 mg dos veces al día.
14. ESTUDIOS CLÍNICOS
14.1 Mielofibrosis
Se realizaron dos estudios aleatorizados de Fase 3 (Estudios 1 y 2) en pacientes con MF (MF primaria, MF post-policitemia vera o MF post-trombocitemia esencial). En ambos estudios, los pacientes presentaban esplenomegalia palpable de al menos 5 cm por debajo del margen costal y una categoría de riesgo intermedia 2 (2 factores pronósticos) o de alto riesgo (3 o más factores pronósticos) según los Criterios de Consenso del Grupo de Trabajo Internacional (IWG).
La dosis inicial de Jakafi se basó en el recuento de plaquetas. Los pacientes con un recuento de plaquetas entre 100 y 200 x 109/L comenzaron con Jakafi 15 mg dos veces al día y los pacientes con un recuento de plaquetas superior a 200 x 109/L comenzaron con Jakafi 20 mg dos veces al día. Las dosis se individualizaron posteriormente en función de la tolerabilidad y la eficacia, con dosis máximas de 20 mg dos veces al día para pacientes con recuentos de plaquetas entre 100 y ≤ 125 x 109/L, de 10 mg dos veces al día para pacientes con recuentos de plaquetas entre 75 y ≤ 100 x 109/L, y de 5 mg dos veces al día para pacientes con recuentos de plaquetas entre 50 y ≤ 75 x 109/L.
Estudio 1
El Estudio 1 (NCT00952289) fue un estudio doble ciego, aleatorizado y controlado con placebo en 309 pacientes que eran refractarios o no eran candidatos a la terapia disponible. La mediana de edad fue de 68 años (rango de 40 a 91 años), con el 61% de los pacientes mayores de 65 años y el 54% eran hombres. El cincuenta por ciento (50%) de los pacientes tenían MF primaria, el 31% tenían MF post-policitemia vera y el 18% tenían MF post-trombocitemia esencial. El veintiún por ciento (21%) de los pacientes recibieron transfusiones de glóbulos rojos en las 8 semanas previas a la inscripción en el estudio. La mediana del recuento de hemoglobina fue de 10,5 g/dL y la mediana del recuento de plaquetas fue de 251 x 109/L. Los pacientes tenían una longitud esplénica palpable mediana de 16 cm por debajo del margen costal, con el 81% presentando una longitud esplénica de 10 cm o más por debajo del margen costal. Los pacientes tenían un volumen esplénico mediano, medido mediante resonancia magnética (RM) o tomografía computarizada (TC), de 2595 cm3 (rango de 478 cm3 a 8881 cm3). (El límite superior de la normalidad es de aproximadamente 300 cm3).
Los pacientes recibieron dosis de Jakafi o placebo coincidente. El criterio principal de valoración de la eficacia fue la proporción de pacientes que lograron una reducción ≥ 35% con respecto al valor basal en el volumen del bazo en la semana 24, medido mediante RM o TC.
Los criterios de valoración secundarios incluyeron la duración de una reducción ≥ 35% en el volumen del bazo y la proporción de pacientes con una reducción ≥ 50% en la puntuación total de los síntomas desde el inicio hasta la semana 24, medida mediante el diario del formulario modificado de evaluación de los síntomas de la mielofibrosis (MFSAF) v2.0.
Estudio 2
El Estudio 2 (NCT00934544) fue un estudio abierto y aleatorizado en 219 pacientes. Los pacientes fueron aleatorizados en una proporción de 2:1 a Jakafi frente al mejor tratamiento disponible. El mejor tratamiento disponible fue seleccionado por el investigador caso por caso. En el brazo del mejor tratamiento disponible, los medicamentos recibidos por más del 10% de los pacientes fueron hidroxiurea (47%) y glucocorticoides (16%). La mediana de edad fue de 66 años (rango de 35 a 85 años), con el 52% de los pacientes mayores de 65 años y el 57% eran hombres. El cincuenta y tres por ciento (53%) de los pacientes tenían MF primaria, el 31% tenían MF post-policitemia vera y el 16% tenían MF post-trombocitemia esencial. El veintiún por ciento (21%) de los pacientes recibieron transfusiones de glóbulos rojos en las 8 semanas previas a la inscripción en el estudio. La mediana del recuento de hemoglobina fue de 10,4 g/dL y la mediana del recuento de plaquetas fue de 236 x 109/L. Los pacientes tenían una longitud esplénica palpable mediana de 15 cm por debajo del margen costal, con el 70% presentando una longitud esplénica de 10 cm o más por debajo del margen costal. Los pacientes tenían un volumen esplénico mediano, medido mediante RM o TC, de 2381 cm3 (rango de 451 cm3 a 7765 cm3).
El criterio principal de valoración de la eficacia fue la proporción de pacientes que lograron una reducción ≥ 35% con respecto al valor basal en el volumen del bazo en la semana 48, medido mediante RM o TC.
Un criterio de valoración secundario en el Estudio 2 fue la proporción de pacientes que lograron una reducción ≥ 35% del volumen del bazo, medido mediante RM o TC, desde el inicio hasta la semana 24.
Resultados de eficacia de los estudios 1 y 2
Los análisis de eficacia del criterio principal de valoración en los Estudios 1 y 2 se presentan en la Tabla 20 a continuación. Una proporción significativamente mayor de pacientes del grupo Jakafi logró una reducción ≥ 35% en el volumen del bazo desde el inicio en ambos estudios en comparación con el placebo en el Estudio 1 y el mejor tratamiento disponible en el Estudio 2. Una proporción similar de pacientes del grupo Jakafi logró una reducción ≥ 50% en la longitud esplénica palpable.
| Estudio 1 | Estudio 2 | |||
| Jakafi (N=155) |
Placebo (N=154) |
Jakafi (N=146) |
Mejor tratamiento disponible (N=73) |
|
| Puntos temporales | Semana 24 | Semana 48 | ||
| Número (%) de pacientes con reducción del volumen del bazo en un 35% o más |
65 (42) | 1 (< 1) | 41 (29) | 0 |
| Valor de P | < 0.0001 | < 0.0001 | ||
La Figura 1 muestra el cambio porcentual respecto al valor inicial en el volumen del bazo para cada paciente en la semana 24 (Jakafi N=139, placebo N=106) o la última evaluación anterior a la semana 24 para los pacientes que no completaron las 24 semanas de tratamiento aleatorizado (Jakafi N=16, placebo N=47). No se incluye un (1) paciente (placebo) con un volumen del bazo basal faltante.
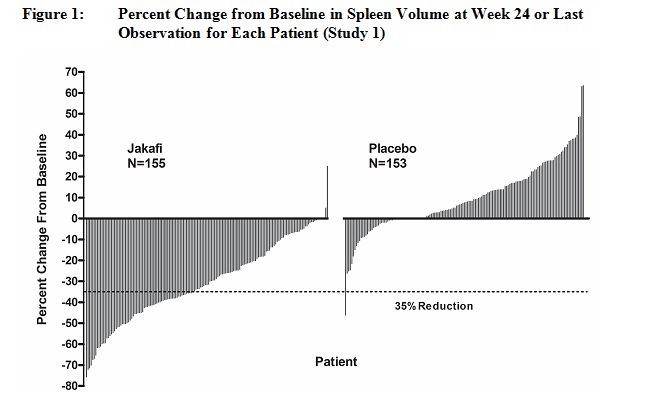
En el Estudio 1, los síntomas de MF fueron un criterio de valoración secundario y se midieron utilizando el formulario diario modificado de evaluación de los síntomas de la mielofibrosis (MFSAF) v2.0. El MFSAF modificado es un diario diario que capta los síntomas principales de la MF (malestar abdominal, dolor debajo de las costillas izquierdas, sudores nocturnos, picazón, dolor óseo/muscular y saciedad precoz). Las puntuaciones de los síntomas oscilaron entre 0 y 10, donde 0 representa síntomas “ausentes” y 10 representa síntomas “peores imaginables”. Estas puntuaciones se sumaron para crear la puntuación total diaria, que tiene un máximo de 60.
La Tabla 21 presenta evaluaciones de la puntuación total de los síntomas desde el inicio hasta la semana 24 en el Estudio 1, incluida la proporción de pacientes con al menos una reducción del 50% (es decir, mejoría de los síntomas). Al inicio, la puntuación total media de los síntomas fue de 18,0 en el grupo de Jakafi y de 16,5 en el grupo de placebo. Una mayor proporción de pacientes en el grupo de Jakafi tuvo una reducción del 50% o mayor en la puntuación total de los síntomas que en el grupo de placebo, con un tiempo medio hasta la respuesta de menos de 4 semanas.
| Jakafi (N=148) |
Placebo (N=152) |
|
| Número (%) de pacientes con una reducción del 50% o mayor en la puntuación total de los síntomas en la semana 24 |
68 (46) | 8 (5) |
| Valor de P | < 0.0001 | |
La Figura 2 muestra el cambio porcentual respecto al valor inicial en la puntuación total de síntomas para cada paciente en la semana 24 (Jakafi N=129, placebo N=103) o la última evaluación en el tratamiento aleatorizado antes de la semana 24 para los pacientes que no completaron 24 semanas de tratamiento aleatorizado (Jakafi N=16, placebo N=42). Los resultados se excluyen para 5 pacientes con una puntuación total de síntomas inicial de cero, 8 pacientes con datos iniciales faltantes y 6 pacientes con datos post-iniciales insuficientes.
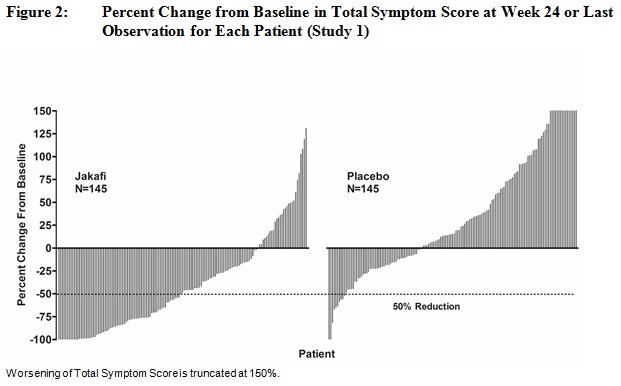
La Figura 3 muestra la proporción de pacientes con al menos un 50% de mejora en cada uno de los síntomas individuales que componen la puntuación total de síntomas, indicando que los 6 síntomas contribuyeron a la mayor tasa de respuesta de la puntuación total de síntomas en el grupo tratado con Jakafi.
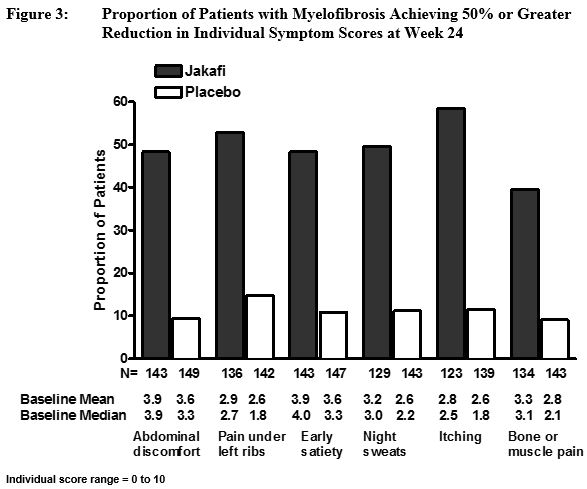
Un análisis exploratorio de los pacientes que recibieron Jakafi también mostró una mejora en los síntomas relacionados con la fatiga (es decir, cansancio, agotamiento, cansancio mental y falta de energía) y los impactos asociados en las actividades diarias (es decir, limitaciones de la actividad relacionadas con el trabajo, el autocuidado y el ejercicio) según lo medido por la puntuación total del formulario corto de 7 ítems de PROMIS® Fatigue en la semana 24. Los pacientes que lograron una reducción de 4.5 puntos o más desde el inicio hasta la semana 24 en la puntuación total de fatiga de PROMIS® se consideraron que habían logrado una respuesta a la fatiga. La respuesta a la fatiga se informó en el 35% de los pacientes del grupo Jakafi frente al 14% de los pacientes del grupo placebo.
La supervivencia general fue un criterio de valoración secundario tanto en el Estudio 1 como en el Estudio 2. Los pacientes de los grupos de control fueron elegibles para el cambio en ambos estudios, y los tiempos medios para el cambio fueron de 9 meses en el Estudio 1 y 17 meses en el Estudio 2.
Las Figuras 4 y 5 muestran las curvas de Kaplan-Meier de la supervivencia general en los análisis planificados prospectivamente después de que todos los pacientes que permanecieron en el estudio hubieran completado 144 semanas en el estudio.
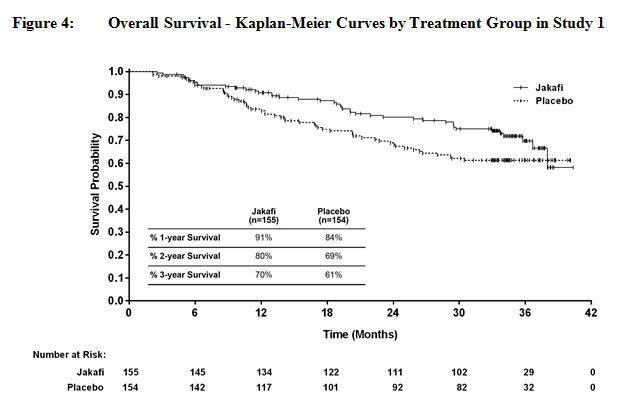
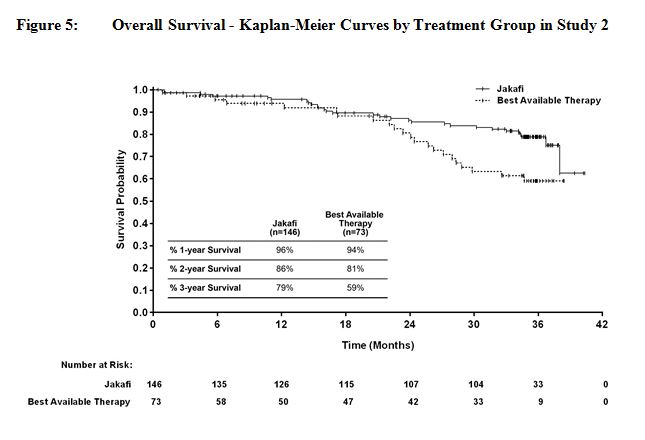
14.2 Policitemia Vera
El Estudio 3 (NCT01243944) fue un estudio de fase 3 aleatorizado, abierto y controlado con activo realizado en 222 pacientes con PV. Los pacientes habían sido diagnosticados con PV durante al menos 24 semanas, habían tenido una respuesta inadecuada a la hidroxiurea o eran intolerantes a ella, requerían flebotomía y presentaban esplenomegalia. Todos los pacientes debieron demostrar un control del hematocrito entre 40-45% antes de la aleatorización. La edad osciló entre 33 y 90 años, con un 30% de pacientes mayores de 65 años y un 66% eran hombres. Los pacientes tenían un volumen esplénico mediano, medido por RM o TC, de 1272 cm3 (rango 254 cm3 a 5147 cm3) y una longitud esplénica palpable mediana por debajo del margen costal de 7 cm.
Los pacientes fueron aleatorizados a Jakafi o al mejor tratamiento disponible. La dosis inicial de Jakafi fue de 10 mg dos veces al día. Las dosis se individualizaron luego en función de la tolerabilidad y la eficacia, con una dosis máxima de 25 mg dos veces al día. En la semana 32, 98 pacientes aún estaban con Jakafi, con un 8% recibiendo más de 20 mg dos veces al día, un 15% recibiendo 20 mg dos veces al día, un 33% recibiendo 15 mg dos veces al día, un 34% recibiendo 10 mg dos veces al día y un 10% recibiendo menos de 10 mg dos veces al día. El mejor tratamiento disponible (BAT) fue seleccionado por el investigador caso por caso e incluyó hidroxiurea (60%), interferón/interferón pegilado (12%), anagrelida (7%), pipobroman (2%), lenalidomida/talidomida (5%) y observación (15%).
El criterio de valoración principal fue la proporción de sujetos que lograron una respuesta en la semana 32, con la respuesta definida como haber logrado tanto el control del hematocrito (la ausencia de elegibilidad para flebotomía a partir de la visita de la semana 8 y continuando hasta la semana 32) como la reducción del volumen del bazo (una reducción mayor o igual al 35% respecto al valor inicial en el volumen del bazo en la semana 32). La elegibilidad para flebotomía se definió como un hematocrito confirmado mayor del 45% que es al menos 3 puntos porcentuales mayor que el hematocrito obtenido al inicio o un hematocrito confirmado mayor del 48%, el que sea menor. Los criterios de valoración secundarios incluyeron la proporción de todos los sujetos aleatorizados que lograron el criterio de valoración principal y que mantuvieron su respuesta 48 semanas después de la aleatorización, y la proporción de sujetos que lograron la remisión hematológica completa en la semana 32, con la remisión hematológica completa definida como el logro del control del hematocrito, un recuento de plaquetas menor o igual a 400 x 109/L y un recuento de leucocitos menor o igual a 10 x 109/L.
Los resultados de los criterios de valoración principales y secundarios se presentan en la Tabla 22. Una proporción significativamente mayor de pacientes en el brazo de Jakafi logró una respuesta para el criterio de valoración principal en comparación con el mejor tratamiento disponible en la semana 32 y mantuvo su respuesta 48 semanas después de la aleatorización. Una proporción significativamente mayor de pacientes en el brazo de Jakafi en comparación con el mejor tratamiento disponible también logró la remisión hematológica completa en la semana 32.
| Respuesta principal definida como haber logrado tanto la ausencia de elegibilidad para flebotomía a partir de la visita de la semana 8 y continuando hasta la semana 32 como una reducción mayor o igual al 35% respecto al valor inicial en el volumen del bazo en la semana 32. | ||
| Jakafi (N=110) |
Mejor tratamiento disponible (N=112) |
|
| Número (%) de pacientes que lograron una respuesta primaria en la semana 32 | 25 (23%) | 1 (< 1%) |
| IC del 95% de la tasa de respuesta (%) | (15%, 32%) | (0%, 5%) |
| Valor de P | < 0.0001 | |
| Número (%) de pacientes que lograron una respuesta primaria duradera en la semana 48 | 22 (20%) | 1 (< 1%) |
| IC del 95% de la tasa de respuesta (%) | (13%, 29%) | (0%, 5%) |
| Valor de P | < 0.0001 | |
| Número (%) de pacientes que lograron una remisión hematológica completa en la semana 32 | 26 (24%) | 9 (8%) |
| IC del 95% de la tasa de respuesta (%) | (16%, 33%) | (4%, 15%) |
| Valor de P | 0.0016 | |
Se realizaron análisis adicionales para el Estudio 3 para evaluar la durabilidad de la respuesta en la semana 80 solo en el brazo de Jakafi. En este brazo, 91 (83%) pacientes aún estaban en tratamiento en el momento del corte de datos de la semana 80. De los 25 pacientes que lograron una respuesta primaria en la semana 32, 19 (76% de los que respondieron) mantuvieron su respuesta hasta la semana 80, y de los 26 pacientes que lograron una remisión hematológica completa en la semana 32, 15 (58% de los que respondieron) mantuvieron su respuesta hasta la semana 80.
En una evaluación de los componentes individuales que conforman el criterio de valoración principal, hubo 66 (60%) pacientes con control del hematocrito en el brazo de Jakafi frente a 21 (19%) pacientes con el mejor tratamiento disponible en la semana 32; 51 (77% de los pacientes que respondieron al hematocrito) en el brazo de Jakafi mantuvieron el control del hematocrito hasta la semana 80. Hubo 44 (40%) pacientes con una reducción del volumen del bazo desde el inicio mayor o igual al 35% en el brazo de Jakafi frente a 1 (< 1%) paciente con el mejor tratamiento disponible en la semana 32; 43 (98% de los pacientes que respondieron a la reducción del volumen del bazo) en el brazo de Jakafi mantuvieron la reducción del volumen del bazo hasta la semana 80.
14.3
Enfermedad injerto contra huésped aguda
El Estudio 4 (NCT02953678) fue un estudio abierto, de un solo brazo y multicéntrico de Jakafi para el tratamiento de pacientes con aGVHD refractaria a esteroides de grados 2 a 4 (criterios del Consorcio Internacional de aGVHD Aguda de Mount Sinai (MAGIC)) que se produce después del trasplante alogénico de células madre hematopoyéticas. Jakafi se administró a 5 mg dos veces al día, y la dosis podría aumentarse a 10 mg dos veces al día después de 3 días en ausencia de toxicidad.
Hubo 49 pacientes con aGVHD refractaria a los esteroides solos. Estos pacientes tenían una edad media de 57 años (rango, 18-72 años), el 47% eran hombres, el 92% eran caucásicos y el 14% eran hispanos. Al inicio del estudio, la aGVHD fue de grado 2 en el 27%, de grado 3 en el 55% y de grado 4 en el 18%; el 84% tenía aGVHD visceral; la puntuación media del biomarcador MAGIC fue de 0,47 (rango, 0,10-0,92); y el nivel medio de ST2 fue de 334 mcg/L (rango, 55-1286 mcg/L). La duración media de la exposición previa a corticosteroides al inicio del estudio fue de 15 días (rango: 3-106 días).
La eficacia de Jakafi se basó en la tasa de respuesta global (TRG) del día 28 (respuesta completa, respuesta parcial muy buena o respuesta parcial según los criterios del Centro de Investigación Internacional de Trasplantes de Médula Ósea y Sangre (CIBMTR)) y la duración de la respuesta. Los resultados de la TRG se presentan en la Tabla 23; la TRG del día 28 fue del 100% para la aGVHD de grado 2, del 40,7% para la aGVHD de grado 3 y del 44,4% para la aGVHD de grado 4.
La duración media de la respuesta, calculada desde la respuesta del día 28 hasta la progresión, la nueva terapia de rescate para la aGVHD o la muerte por cualquier causa (siendo la progresión la definida como el empeoramiento en una etapa en cualquier órgano sin mejoría en otros órganos en comparación con la evaluación de la respuesta anterior) fue de 16 días (IC del 95% 9, 83). Además, para los que respondieron el día 28, el tiempo medio desde la respuesta del día 28 hasta la muerte o la necesidad de una nueva terapia para la aGVHD (terapia de rescate adicional o aumento de esteroides) fue de 173 días (IC del 95% 66, NE).
| Refractaria a esteroides solos (n=49) |
|
| Respuesta global (%) (IC del 95%) | 28 (57.1%) (42.2, 71.2) |
| Respuesta completa | 15 (30.6%) |
| Respuesta parcial muy buena | 2 (4.1%) |
| Respuesta parcial | 11 (22.4%) |
14.4 Enfermedad crónica injerto contra huésped
El estudio 5 (REACH-3; NCT03112603) fue un estudio multicéntrico, abierto, aleatorizado de Jakafi en comparación con el mejor tratamiento disponible (BAT) para el tratamiento de la enfermedad injerto contra huésped crónica (cGVHD) refractaria a corticosteroides después del trasplante de células madre alogénicas. Los pacientes elegibles tenían ≥ 12 años de edad con cGVHD moderada o grave según lo definido por los Criterios de Consenso del NIH que requieren terapia adicional después del fracaso de la terapia con corticosteroides y no más de un tratamiento de rescate adicional. Se excluyó a los pacientes si tenían ANC < 1 Gi/L y recuento de plaquetas < 25 Gi/L, aclaramiento de creatinina estimado < 30 ml/min, inicio progresivo de cGVHD, saturación de oxígeno < 90%, bilirrubina total > 2 mg/dL o diarrea debido a GVHD.
Un total de 329 pacientes fueron aleatorizados 1:1 para recibir Jakafi 10 mg dos veces al día (n=165) o BAT (n=164). El BAT fue seleccionado por el investigador antes de la aleatorización e incluyó los siguientes tratamientos: fotoferesis extracorpórea (ECP), metotrexato (MTX) en dosis bajas, micofenolato de mofetilo (MMF), inhibidores de mTOR (everolimus o sirolimus), infliximab, rituximab, pentostatina, imatinib o ibrutinib. La aleatorización se estratificó por la gravedad de la cGVHD (moderada versus grave). El día 1 del ciclo 7 y posteriores, los pacientes aleatorizados a BAT podrían cambiar a Jakafi si tenían progresión de la enfermedad, respuesta mixta, respuesta sin cambios, brote de cGVHD o toxicidad al BAT. Todos los pacientes también recibieron atención de apoyo estándar, incluidos medicamentos antiinfecciosos. Se permitió continuar la profilaxis de GVHD y los medicamentos para el tratamiento de cGVHD iniciados antes de la aleatorización, incluidos los corticosteroides sistémicos, los inhibidores de la calcineurina y la terapia con corticosteroides tópicos o inhalados, según las pautas institucionales. La Tabla 24 muestra las características demográficas y basales de la enfermedad de la población aleatorizada.
|
||
|
Jakafi |
Mejor tratamiento disponible |
|
| Edad mediana, años (rango) | 49 (13, 73) | 50 (12, 76) |
|
Edad de 12 a < 18 años, n (%) |
4 (2) | 8 (5) |
|
Edad > 65 años, n (%) |
18 (11) | 22 (13) |
| Masculino, n (%) | 109 (66) | 92 (56) |
| Raza, n (%) | ||
|
Blanca |
116 (70) | 132 (81) |
|
Negra |
2 (1) | 0 |
|
Asiática |
33 (20) | 21 (13) |
|
Indígena americana o nativa de Alaska |
2 (1) | 0 |
|
Otra |
9 (6) | 4 (2) |
|
Desconocida |
3 (2) | 7 (4) |
| Tiempo mediano (rango) (días) desde el diagnóstico de cGVHD hasta la aleatorización | 174 (7-2017) | 150 (10-1947) |
| Tratamiento previo | ||
|
Sin tratamiento previo para cGVHD |
2 (1) | 1 (1) |
|
Fracaso de los esteroides de primera línea solos |
115 (70) | 125 (76) |
|
Fracaso de la combinación de primera línea que incluye esteroides |
42 (25) | 30 (18) |
|
Fracaso de dos líneas de terapia |
6 (4) | 8 (5) |
| ≥ 4 órganos afectados, n (%) | 67 (41) | 63 (38) |
| cGVHD grave, n (%) | 86 (52) | 79 (48) |
| Mediana (rango) de la puntuación total de síntomas de cGVHD | 19 (0-80) | 18 (1-54) |
| Mediana (rango) de la dosis de corticosteroides al inicio (mg/kg de PE)* | 0.29 (0.01-1.81) | 0.26 (0.06-1.21) |
La eficacia de Jakafi se basó en la tasa de respuesta general (TRG) hasta el día 1 del ciclo 7, donde la respuesta general incluyó la respuesta completa o la respuesta parcial según los Criterios de Respuesta NIH de 2014 y la durabilidad de la respuesta. Los resultados de la TRG se presentan en la Tabla 25; la diferencia en la TRG entre los brazos de Jakafi y BAT fue del 13% (IC del 95% 3%, 23%). El tiempo medio hasta la primera respuesta en los respondedores fue de 3 semanas (rango, 2 a 24) para el brazo de Jakafi y de 4 semanas (rango, 2 a 25) para el brazo de BAT. La duración mediana de la respuesta, calculada desde la primera respuesta hasta la progresión, la muerte o las nuevas terapias sistémicas para la cGVHD, fue de 4,2 meses (IC del 95% 3,2, 6,7) para el brazo de Jakafi y de 2,1 meses (IC del 95% 1,6, 3,2) para el brazo de BAT; y el tiempo medio desde la primera respuesta hasta la muerte o las nuevas terapias sistémicas para la cGVHD fue de 25 meses (IC del 95% 16,8, NE) para el brazo de Jakafi y de 5,6 meses (IC del 95% 4,1, 7,8) para el brazo de BAT.
|
||
| Jakafi (N=165) |
Mejor terapia disponible (N=164) |
|
| Respuesta general (%) (IC del 95%)* |
116 (70%) (63%, 77%) |
94 (57%) (49%, 65%) |
| Respuesta completa (%) | 14 (8%) | 8 (5%) |
| Respuesta parcial (%) | 102 (62%) | 86 (52%) |
Los resultados de la TRG se apoyaron en análisis exploratorios de la gravedad de los síntomas notificados por los pacientes, que mostraron al menos una disminución de 7 puntos en la puntuación total de síntomas de cGVHD en cualquier momento hasta el día 1 del ciclo 7 en 66 (40%; IC del 95% 32, 48) pacientes en el brazo de Jakafi y en 47 (29%; IC del 95% 22, 36) pacientes en el brazo de BAT.
16. CÓMO SE SUMINISTRA/ALMACENAMIENTO Y MANIPULACIÓN
Las tabletas de Jakafi (ruxolitinib) están disponibles de la siguiente manera:
| Consérvese a temperatura ambiente de 20°C a 25°C (68°F a 77°F); se permiten excursiones entre 15°C y 30°C (59°F y 86°F) [véase Temperatura ambiente controlada USP]. | |||
| Número NDC | Concentración | Descripción | Tabletas por Frasco |
| 50881-005-60 | 5 mg | Tableta redonda con “INCY” en un lado y “5” en el otro | 60 |
| 50881-010-60 | 10 mg | Tableta redonda con “INCY” en un lado y “10” en el otro | 60 |
| 50881-015-60 | 15 mg | Tableta ovalada con “INCY” en un lado y “15” en el otro | 60 |
| 50881-020-60 | 20 mg | Tableta con forma de cápsula con “INCY” en un lado y “20” en el otro | 60 |
| 50881-025-60 | 25 mg | Tableta ovalada con “INCY” en un lado y “25” en el otro | 60 |
17. INFORMACIÓN DE ASESORAMIENTO AL PACIENTE
Aconseje al paciente que lea el etiquetado del paciente aprobado por la FDA (Información para el paciente).
Trombocitopenia, Anemia y Neutropenia
Informe a los pacientes que Jakafi se asocia con trombocitopenia, anemia y neutropenia, y de la necesidad de controlar los hemogramas completos antes y durante el tratamiento. Aconseje a los pacientes que observen e informen cualquier sangrado [ver Advertencias y precauciones (5.1)].
Infecciones
Informe a los pacientes sobre los signos y síntomas de infección y que informen cualquier signo o síntoma de inmediato.
Informe a los pacientes sobre los signos y síntomas tempranos de herpes zóster y leucoencefalopatía multifocal progresiva, y aconseje a los pacientes que busquen el consejo de un médico si se observan dichos síntomas [ver Advertencias y precauciones (5.2)].
Exacerbación de los síntomas después de la interrupción o suspensión del tratamiento con Jakafi
Informe a los pacientes que después de la suspensión del tratamiento, se espera que regresen los signos y síntomas de las neoplasias mieloproliferativas. Indique a los pacientes que no interrumpan ni suspendan el tratamiento con Jakafi sin consultar a su médico [ver Advertencias y precauciones (5.3)].
Cáncer de piel no melanoma
Informe a los pacientes que Jakafi puede aumentar su riesgo de ciertos cánceres de piel no melanoma. Aconseje a los pacientes que informen a su proveedor de atención médica si alguna vez han tenido algún tipo de cáncer de piel o si observan alguna lesión cutánea nueva o cambiante [ver Advertencias y precauciones (5.4)].
Elevaciones de lípidos
Informe a los pacientes que Jakafi puede aumentar el colesterol en sangre y la necesidad de controlar los niveles de colesterol en sangre [ver Advertencias y precauciones (5.5)].
Eventos cardiovasculares adversos mayores (MACE)
Avise a los pacientes que se han notificado eventos de eventos cardiovasculares adversos mayores (MACE), incluido infarto de miocardio, accidente cerebrovascular y muerte cardiovascular, en estudios clínicos con otro inhibidor de JAK utilizado para tratar la artritis reumatoide, una afección para la que no está indicado Jakafi. Aconseje a los pacientes, especialmente a los fumadores actuales o pasados o a los pacientes con otros factores de riesgo cardiovascular, que estén atentos al desarrollo de signos y síntomas de eventos cardiovasculares [ver Advertencias y precauciones (5.6)].
Trombosis
Avise a los pacientes que se han notificado eventos de trombosis venosa profunda (TVP) y embolia pulmonar (EP) en estudios clínicos con otro inhibidor de JAK utilizado para tratar la artritis reumatoide, una afección para la que no está indicado Jakafi. Aconseje a los pacientes que informen a su proveedor de atención médica si desarrollan algún signo o síntoma de TVP o EP [ver Advertencias y precauciones (5.7)].
Neoplasias secundarias
Avise a los pacientes, especialmente a los fumadores actuales o pasados y a los pacientes con una neoplasia secundaria conocida (que no sea un carcinoma espinocelular no melanoma tratado con éxito), que se han notificado linfoma y otras neoplasias (excluyendo el carcinoma espinocelular no melanoma) en estudios clínicos con otro inhibidor de JAK utilizado para tratar la artritis reumatoide, una afección para la que no está indicado Jakafi [ver Advertencias y precauciones (5.8)].
Interacciones medicamentosas
Aconseje a los pacientes que informen a sus proveedores de atención médica de todos los medicamentos que están tomando, incluidos los medicamentos de venta libre, los productos herbales y los suplementos dietéticos [ver Interacciones medicamentosas (7.1) y Farmacología clínica (12.3)].
Diálisis
Informe a los pacientes en diálisis que su dosis no debe tomarse antes de la diálisis, sino solo después de la diálisis [ver Posología y administración (2.7)].
Lactancia
Informe a las mujeres que no deben amamantar durante el tratamiento con Jakafi y durante dos semanas después de la dosis final [ver Uso en poblaciones específicas (8.2)].
Cumplimiento
Aconseje a los pacientes que continúen tomando Jakafi todos los días durante el tiempo que su médico se lo indique y que este es un tratamiento a largo plazo. Los pacientes no deben cambiar la dosis ni dejar de tomar Jakafi sin consultar primero a su médico. Los pacientes deben tener en cuenta que después de la suspensión del tratamiento, se espera que regresen los signos y síntomas de las neoplasias mieloproliferativas.
Fabricado para:
Incyte Corporation
1801 Augustine Cut-off
Wilmington, DE 19803
Jakafi es una marca registrada de Incyte. Todos los derechos reservados.
Números de patente de EE. UU. 7598257; 8415362; 8722693; 8822481; 8829013; 9079912; 9814722; 10016429
© 2011-2023 Incyte Corporation. Todos los derechos reservados.
PROSPECTO PARA EL PACIENTE
|
Información para el paciente |
|
|
¿Qué es Jakafi? Jakafi es un medicamento recetado que se usa para tratar:
No se sabe si Jakafi es seguro o eficaz en niños para el tratamiento de la mielofibrosis o la policitemia vera. |
|
|
Antes de tomar Jakafi, informe a su proveedor de atención médica sobre sus afecciones médicas, incluso si:
Informe a su proveedor de atención médica sobre todos los medicamentos que toma, incluidos los medicamentos recetados y de venta libre, las vitaminas y los suplementos herbales. Tomar Jakafi con ciertos otros medicamentos puede afectar la forma en que funciona Jakafi. Conozca los medicamentos que toma. Lleve una lista de ellos para mostrársela a su proveedor de atención médica y farmacéutico cuando reciba un medicamento nuevo. |
|
¿Cómo debo tomar Jakafi?
|
|
|
¿Cuáles son los posibles efectos secundarios de Jakafi? Jakafi puede causar efectos secundarios graves, que incluyen: Recuentos bajos de células sanguíneas. Jakafi puede causar recuentos bajos de plaquetas (trombocitopenia), recuentos bajos de glóbulos rojos (anemia) y recuentos bajos de glóbulos blancos (neutropenia). Si presenta sangrado, deje de tomar Jakafi y llame a su proveedor de atención médica. Su proveedor de atención médica le hará un análisis de sangre para verificar sus recuentos de células sanguíneas antes de comenzar a tomar Jakafi y regularmente durante su tratamiento con Jakafi. Informe a su proveedor de atención médica de inmediato si desarrolla o empeora alguno de estos síntomas: |
|
|
|
|
Infección. Puede correr el riesgo de desarrollar una infección grave durante el tratamiento con Jakafi. Informe a su proveedor de atención médica si desarrolla alguno de los siguientes síntomas de infección: |
|
|
|
|
Cáncer. Algunas personas han presentado ciertos tipos de cánceres de piel no melanoma durante el tratamiento con Jakafi. Su proveedor de atención médica revisará regularmente su piel durante el tratamiento con Jakafi. Informe a su proveedor de atención médica si desarrolla alguna lesión cutánea nueva o cambiante durante el tratamiento con Jakafi.
Aumento del colesterol. Puede experimentar cambios en sus niveles de colesterol en sangre durante el tratamiento con Jakafi. Su proveedor de atención médica le realizará análisis de sangre para controlar sus niveles de colesterol aproximadamente cada 8 a 12 semanas después de comenzar a tomar Jakafi, y según sea necesario.
Mayor riesgo de eventos cardiovasculares importantes como ataque cardíaco, accidente cerebrovascular o muerte en personas que tienen factores de riesgo cardiovascular y que son fumadoras actuales o pasadas mientras usan otro inhibidor de JAK para tratar la artritis reumatoide.
Busque ayuda de emergencia de inmediato si tiene algún síntoma de ataque cardíaco o accidente cerebrovascular mientras toma Jakafi, incluyendo:
Mayor riesgo de coágulos sanguíneos. Se han producido coágulos sanguíneos en las venas de las piernas (trombosis venosa profunda, TVP) o los pulmones (embolia pulmonar, EP) en personas que toman otro inhibidor de JAK para la artritis reumatoide y pueden ser potencialmente mortales.
Los efectos secundarios más comunes de Jakafi en adultos con ciertos tipos de MF y PV incluyen: |
|
|
|
|
Los efectos secundarios más comunes de Jakafi en personas con aGVHD incluyen: |
|
|
|
| Los efectos secundarios más comunes de Jakafi en personas con cGVHD incluyen: |
|
|
|
| Estos no son todos los posibles efectos secundarios de Jakafi. Llame a su médico para obtener asesoramiento médico sobre los efectos secundarios. Puede informar los efectos secundarios a la FDA al 1-800-FDA-1088. También puede informar los efectos secundarios a Incyte Corporation al 1-855-463-3463. |
|
|
¿Cómo debo guardar Jakafi?
Mantenga Jakafi y todos los medicamentos fuera del alcance de los niños. |
|
| Información general sobre el uso seguro y eficaz de Jakafi.
Los medicamentos a veces se recetan para fines distintos de los que figuran en la información para el paciente. No use Jakafi para una afección para la que no esté recetado. No le dé Jakafi a otras personas, incluso si tienen los mismos síntomas que usted. Podría causarles daño. Puede pedirle a su farmacéutico o proveedor de atención médica información escrita para profesionales de la salud. |
|
|
¿Cuáles son los ingredientes de Jakafi? Ingredientes inactivos: celulosa microcristalina, monohidrato de lactosa, estearato de magnesio, dióxido de silicio coloidal, glicolato de almidón sódico, povidona y hidroxipropilcelulosa Fabricado para: Incyte Corporation, 1801 Augustine Cut-off, Wilmington, DE 19803 Jakafi es una marca registrada de Incyte. Todos los derechos reservados. © 2011-2021 Incyte Corporation. Todos los derechos reservados. |
|
Esta información para el paciente ha sido aprobada por la Administración de Alimentos y Medicamentos de EE. UU. Revisado: septiembre de 2021
Etiqueta del frasco de comprimidos de 5 mg
Rx solamente
NDC 50881-005-60
Jakafi® (Ruxolitinib) Tabletas
5 mg
60 tabletas
Cada tableta contiene 6.6 mg de fosfato de ruxolitinib equivalente a 5 mg de base libre de ruxolitinib.

Etiqueta del frasco de tabletas de 10 mg
Rx solamente
NDC 50881-010-60
Jakafi® (Ruxolitinib) Tabletas
10 mg
60 tabletas
Cada tableta contiene 13.2 mg de fosfato de ruxolitinib equivalente a 10 mg de base libre de ruxolitinib.

Etiqueta del frasco de comprimidos de 15 mg
Rx solamente
NDC 50881-015-60
Jakafi® (Ruxolitinib) Tabletas
15 mg
60 tabletas
Cada tableta contiene 19.8 mg de fosfato de ruxolitinib equivalente a 15 mg de base libre de ruxolitinib.

Etiqueta del frasco de comprimidos de 20 mg
Rx solamente
NDC 50881-020-60
Jakafi® (Ruxolitinib) Tabletas
20 mg
60 tabletas
Cada tableta contiene 26.4 mg de fosfato de ruxolitinib equivalente a 20 mg de base libre de ruxolitinib.

Etiqueta del frasco de comprimidos de 25 mg
Rx solamente
NDC 50881-025-60
Jakafi® (Ruxolitinib) Tabletas
25 mg
60 tabletas
Cada tableta contiene 33 mg de fosfato de ruxolitinib equivalente a 25 mg de ruxolitinib base libre.
