Fabricante de medicamentos: Pfizer Laboratories Div Pfizer Inc (Updated: 2025-01-10)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
INLYTA® (axitinib) tabletas, para administración oral
Aprobación inicial en EE. UU.: 2012
INDICACIONES Y USO
INLYTA es un inhibidor de la quinasa indicado:
- •
- en combinación con avelumab, para el tratamiento de primera línea de pacientes con carcinoma de células renales (CCR) avanzado. (1.1)
- •
- en combinación con pembrolizumab, para el tratamiento de primera línea de pacientes con CCR avanzado. (1.1)
- •
- como agente único, para el tratamiento del carcinoma de células renales (CCR) avanzado después del fracaso de una terapia sistémica previa. (1.2)
DOSIFICACIÓN Y ADMINISTRACIÓN
- •
- INLYTA 5 mg por vía oral dos veces al día con avelumab 800 mg cada 2 semanas. (2.1)
- •
- INLYTA 5 mg por vía oral dos veces al día con pembrolizumab 200 mg cada 3 semanas o 400 mg cada 6 semanas. (2.1)
- •
- INLYTA como agente único, la dosis inicial es de 5 mg por vía oral dos veces al día. (2.1)
- •
- Los ajustes de dosis se pueden realizar en función de la seguridad y la tolerabilidad individuales. (2.2)
- •
- Administre la dosis de INLYTA aproximadamente cada 12 horas con o sin alimentos. (2.1)
- •
- INLYTA debe tragarse entero con un vaso de agua. (2.1)
- •
- Consulte la información de prescripción completa para las modificaciones de la dosis por reacciones adversas. (2.2)
- •
- Si se requiere un inhibidor potente de CYP3A4/5, disminuya la dosis de INLYTA aproximadamente a la mitad. (2.2)
- •
- Para pacientes con insuficiencia hepática moderada, disminuya la dosis inicial aproximadamente a la mitad. (2.2)
FORMAS FARMACÉUTICAS Y CONCENTRACIONES
Tabletas de 1 mg y 5 mg (3)
CONTRAINDICACIONES
Ninguna. (4)
ADVERTENCIAS Y PRECAUCIONES
- •
- Hipertensión: Se ha observado hipertensión, incluyendo crisis hipertensiva. La presión arterial debe estar bien controlada antes de iniciar el tratamiento con INLYTA. Controlar la hipertensión y tratar según sea necesario. Suspender y luego reducir la dosis de INLYTA o suspenderla permanentemente según la gravedad de la hipertensión. (5.1)
- •
- Eventos Tromboembólicos Arteriales y Venosos: Se han observado eventos trombóticos arteriales y venosos, que pueden ser fatales. Usar con precaución en pacientes con mayor riesgo de padecer estos eventos. Suspender permanentemente INLYTA si se produce un evento tromboembólico arterial durante el tratamiento. Suspender INLYTA y luego reanudarlo a la misma dosis o suspenderlo permanentemente según la gravedad de la TEV. (5.2, 5.3)
- •
- Hemorragia: Se han notificado eventos hemorrágicos, incluyendo eventos fatales. INLYTA no se ha estudiado en pacientes con evidencia de metástasis cerebral no tratada o hemorragia gastrointestinal activa reciente y no debe utilizarse en estos pacientes. Suspender y luego reducir la dosis de INLYTA o suspenderla según la gravedad y la persistencia de la hemorragia. (5.4)
- •
- Insuficiencia Cardíaca: Se ha observado insuficiencia cardíaca, que puede ser fatal. Controlar los signos o síntomas de insuficiencia cardíaca durante todo el tratamiento con INLYTA. El manejo de la insuficiencia cardíaca puede requerir la reducción de la dosis, la interrupción de la dosis o la suspensión permanente de INLYTA. (5.5)
- •
- Perforación Gastrointestinal y Formación de Fístulas: Se han producido perforaciones gastrointestinales y fístulas, incluyendo la muerte. Usar con precaución en pacientes con riesgo de perforación gastrointestinal o fístula. (5.6)
- •
- Hipotiroidismo: Se ha notificado hipotiroidismo que requiere reemplazo de hormona tiroidea. Controlar la función tiroidea antes de iniciar el tratamiento con INLYTA y periódicamente durante el mismo. (5.7)
- •
- Cicatrización Deficiente de Heridas: Suspender INLYTA durante al menos 2 días antes de una cirugía electiva. No administrar durante al menos 2 semanas después de una cirugía mayor y hasta que la herida haya cicatrizado adecuadamente. Reanudar INLYTA a una dosis reducida o suspenderlo según la gravedad y la persistencia de la cicatrización deficiente de la herida. No se ha establecido la seguridad de la reanudación de INLYTA después de la resolución de las complicaciones de la cicatrización de la herida. (5.8)
- •
- Síndrome de Leucoencefalopatía Posterior Reversible (SLPR): Se ha observado SLPR. Suspender permanentemente INLYTA si se presentan signos o síntomas de SLPR. (5.9)
- •
- Proteinuria: Controlar la proteinuria antes de iniciar el tratamiento con INLYTA y periódicamente durante el mismo. En caso de proteinuria moderada a grave, suspender y luego reducir la dosis de INLYTA. (5.10)
- •
- Hepatotoxicidad: Se ha producido elevación de las enzimas hepáticas durante el tratamiento con INLYTA como agente único. Controlar la ALT, AST y bilirrubina antes de iniciar el tratamiento con INLYTA y periódicamente durante el mismo. Cuando se utiliza en combinación con avelumab o pembrolizumab, pueden producirse mayores frecuencias de elevación de ALT y AST de Grado 3 y 4. Considerar un control más frecuente de las enzimas hepáticas. Suspender INLYTA y avelumab o pembrolizumab, iniciar terapia con corticosteroides según sea necesario y/o suspender permanentemente la combinación en caso de hepatotoxicidad grave o potencialmente mortal. (5.11)
- •
- Uso en Pacientes con Insuficiencia Hepática: Reducir la dosis inicial de INLYTA si se utiliza en pacientes con insuficiencia hepática moderada. INLYTA no se ha estudiado en pacientes con insuficiencia hepática grave. (2.2, 5.12)
- •
- Eventos cardiovasculares adversos mayores (INLYTA en combinación con avelumab): Optimizar el manejo de los factores de riesgo cardiovascular. Suspender permanentemente INLYTA en combinación con avelumab para eventos de Grado 3-4. (5.13)
- •
- Toxicidad Embrio-Fetal: INLYTA puede causar daño fetal. Aconsejar a las pacientes sobre el riesgo potencial para el feto y que utilicen métodos anticonceptivos eficaces. (5.14, 8.1, 8.3)
REACCIONES ADVERSAS
Las reacciones adversas más comunes (≥20%) son:
INLYTA en combinación con avelumab: diarrea, fatiga, hipertensión, dolor musculoesquelético, náuseas, mucositis, eritrodisestesia palmo-plantar, disfonía, disminución del apetito, hipotiroidismo, erupción cutánea, hepatotoxicidad, tos, disnea, dolor abdominal y dolor de cabeza. (6.1)
INLYTA en combinación con pembrolizumab: diarrea, fatiga/astenia, hipertensión, hepatotoxicidad, hipotiroidismo, disminución del apetito, eritrodisestesia palmo-plantar, náuseas, estomatitis/inflamación de la mucosa, disfonía, erupción cutánea, tos y estreñimiento. (6.1)
INLYTA como agente único: diarrea, hipertensión, fatiga, disminución del apetito, náuseas, disfonía, síndrome de eritrodisestesia palmo-plantar (manos-pies), pérdida de peso, vómitos, astenia y estreñimiento. (6.1)
Para reportar REACCIONES ADVERSAS SOSPECHOSAS, contacte a Pfizer Inc. al 1-800-438-1985 o a la FDA al 1-800-FDA-1088 o www.fda.gov/medwatch.
INTERACCIONES CON OTROS MEDICAMENTOS
Consulte la sección 17 para obtener INFORMACIÓN DE ASESORAMIENTO AL PACIENTE y el etiquetado para pacientes aprobado por la FDA.
Revisado: 7/2024
Tabla de contenido
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDO*
1 INDICACIONES Y USO
1.1 Carcinoma de células renales avanzadas de primera línea
1.2 Carcinoma de células renales avanzadas de segunda línea
2 POSOLOGÍA Y ADMINISTRACIÓN
2.1 Posología recomendada
2.2 Pautas de modificación de la dosis
2.3 Modificación de la dosis para interacciones medicamentosas
2.4 Modificación de la dosis para insuficiencia hepática
3 FORMAS Y CONCENTRACIONES FARMACÉUTICAS
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Hipertensión
5.2 Eventos tromboembólicos arteriales
5.3 Eventos tromboembólicos venosos
5.4 Hemorragia
5.5 Insuficiencia cardíaca
5.6 Perforación gastrointestinal y formación de fístulas
5.7 Disfunción tiroidea
5.8 Alteración de la cicatrización de heridas
5.9 Síndrome de leucoencefalopatía posterior reversible
5.10 Proteinuria
5.11 Hepatotoxicidad
5.12 Uso en pacientes con insuficiencia hepática
5.13 Eventos cardiovasculares adversos importantes (MACE)
5.14 Toxicidad embriofetal
6 REACCIONES ADVERSAS
6.1 Experiencia en ensayos clínicos
6.2 Experiencia postcomercialización
7 INTERACCIONES MEDICAMENTOSAS
7.1 Inhibidores de CYP3A4/5
7.2 Inductores de CYP3A4/5
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.3 Mujeres y hombres en edad fértil
8.4 Uso pediátrico
8.5 Uso geriátrico
8.6 Insuficiencia hepática
8.7 Insuficiencia renal
10 SOBREDOSIFICACIÓN
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
12.2 Farmacodinamia
12.3 Farmacocinética
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, mutagénesis, alteración de la fertilidad
14 ESTUDIOS CLÍNICOS
14.1 RCC avanzado de primera línea
14.2 RCC avanzado de segunda línea
16 PRESENTACIÓN/ALMACENAMIENTO Y MANIPULACIÓN
17 INFORMACIÓN PARA EL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información completa de prescripción no se enumeran.
1 INDICACIONES Y USO
1.1 Carcinoma de células renales avanzadas de primera línea
INLYTA en combinación con avelumab está indicado para el tratamiento de primera línea de pacientes con carcinoma de células renales (CCR) avanzado.
INLYTA en combinación con pembrolizumab está indicado para el tratamiento de primera línea de pacientes con CCR avanzado.
1.2 Carcinoma de células renales avanzadas de segunda línea
INLYTA como agente único está indicado para el tratamiento de CCR avanzado después del fracaso de una terapia sistémica previa.
2 DOSIS Y ADMINISTRACIÓN
2.1 Posología recomendada
RCC avanzado de primera línea
INLYTA en combinación con Avelumab
La dosis inicial recomendada de INLYTA es de 5 mg por vía oral dos veces al día (con 12 horas de intervalo) con o sin alimentos, en combinación con avelumab 800 mg administrado como infusión intravenosa durante 60 minutos cada 2 semanas hasta la progresión de la enfermedad o toxicidad inaceptable. Cuando INLYTA se usa en combinación con avelumab, se puede considerar un aumento de la dosis de INLYTA por encima de la dosis inicial de 5 mg a intervalos de dos semanas o más. Consulte la información completa de prescripción para obtener información sobre la dosificación recomendada de avelumab.
INLYTA en combinación con Pembrolizumab
La dosis inicial recomendada de INLYTA es de 5 mg por vía oral dos veces al día (con 12 horas de intervalo) con o sin alimentos, en combinación con pembrolizumab 200 mg cada 3 semanas o 400 mg cada 6 semanas administrado como infusión intravenosa durante 30 minutos hasta la progresión de la enfermedad o toxicidad inaceptable. Cuando INLYTA se usa en combinación con pembrolizumab, se puede considerar un aumento de la dosis de INLYTA por encima de la dosis inicial de 5 mg a intervalos de seis semanas o más. Consulte la información completa de prescripción para obtener información sobre la dosificación recomendada de pembrolizumab.
2.2 Pautas de modificación de la dosis
Se recomienda el aumento o la reducción de la dosis en función de la seguridad y la tolerabilidad individuales.
Los aumentos y reducciones de la dosis de INLYTA recomendados se proporcionan en la Tabla 1.
Durante el curso del tratamiento, los pacientes que toleran INLYTA durante al menos dos semanas consecutivas sin reacciones adversas de grado >2 (según los Criterios comunes de toxicidad para eventos adversos [CTCAE]), son normotensos y no reciben medicación antihipertensiva, pueden tener su dosis aumentada.
| Modificación de la dosis | Régimen de dosificación |
|---|---|
|
Dosis inicial recomendada |
5 mg dos veces al día |
|
Aumento de la dosis |
|
|
Primer aumento de la dosis |
7 mg dos veces al día |
|
Segundo aumento de la dosis |
10 mg dos veces al día |
|
Reducción de la dosis* |
|
|
Primera reducción de la dosis† |
3 mg dos veces al día |
|
Segunda reducción de la dosis |
2 mg dos veces al día |
Las modificaciones posológicas recomendadas para las reacciones adversas de INLYTA se proporcionan en la Tabla 2.
| Reacción adversa | Gravedad | Modificaciones de la dosis para INLYTA |
|---|---|---|
|
Hipertensión [ver Advertencias y precauciones (5.1)] |
PAS >150 mmHg o PAD >100 mmHg a pesar del tratamiento antihipertensivo |
|
|
PAS >160 mmHg o PAD >105 mmHg |
|
|
|
Grado 4 o crisis hipertensiva |
|
|
|
Hemorragia [ver Advertencias y precauciones (5.4)] |
Grado 3 o 4 |
|
|
Insuficiencia cardíaca [ver Advertencias y precauciones (5.5)] |
Cardiomiopatía asintomática (fracción de eyección del ventrículo izquierdo superior al 20% pero inferior al 50% por debajo del valor basal o por debajo del límite inferior de la normalidad si no se obtuvo el valor basal) |
|
|
Insuficiencia cardíaca congestiva clínicamente manifiesta |
|
|
|
Cicatrización deficiente de heridas [ver Advertencias y precauciones (5.8)] |
Cualquier grado |
|
|
Síndrome de leucoencefalopatía posterior reversible [ver Advertencias y precauciones (5.9)] |
Cualquier grado |
|
|
Proteinuria [ver Advertencias y precauciones (5.10)] |
2 o más gramos de proteinuria por 24 horas |
|
|
Otras reacciones adversas |
Grado 3 |
|
|
Grado 4 |
|
La Tabla 3 representa las modificaciones posológicas adicionales recomendadas para las reacciones adversas cuando se administra INLYTA en combinación con avelumab o pembrolizumab.
Consulte la Información completa de prescripción para obtener información posológica adicional sobre avelumab o pembrolizumab, incluidas las modificaciones de la dosis para las reacciones adversas inmunomediadas.
| Tratamiento | Reacción adversa | Gravedad* | Modificaciones de la dosis para INLYTA |
|---|---|---|---|
| ALT = alanina aminotransferasa, AST = aspartato aminotransferasa, ULN = límite superior de la normalidad | |||
|
|||
|
INLYTA en combinación con avelumab O pembrolizumab |
Elevaciones de las enzimas hepáticas† |
ALT o AST al menos 3 veces el ULN pero menos de 10 veces el ULN sin bilirrubina total concurrente al menos 2 veces el ULN |
|
|
Aumentos de ALT o AST a más de 3 veces el ULN con bilirrubina total concurrente al menos 2 veces el ULN o ALT o AST al menos 10 veces el ULN |
|
||
|
Diarrea |
Grado 1-2 |
|
|
|
Grado 3 |
|
||
|
Grado 4 |
|
||
|
INLYTA en combinación con avelumab |
Eventos cardiovasculares adversos importantes (MACE) |
Grado 3 o 4 |
|
2.3 Modificación de la dosis para interacciones medicamentosas
Inhibidores potentes del CYP3A4/5
Debe evitarse el uso concomitante de inhibidores potentes del CYP3A4/5 (p. ej., ketoconazol, itraconazol, claritromicina, atazanavir, indinavir, nefazodona, nelfinavir, ritonavir, saquinavir, telitromicina y voriconazol). Se recomienda la selección de un medicamento concomitante alternativo con un potencial de inhibición del CYP3A4/5 nulo o mínimo. Aunque no se ha estudiado el ajuste de la dosis de INLYTA en pacientes que reciben inhibidores potentes del CYP3A4/5, si se debe administrar conjuntamente un inhibidor potente del CYP3A4/5, se recomienda una reducción de la dosis de INLYTA aproximadamente a la mitad, ya que se predice que esta reducción de la dosis ajustará el área bajo la curva de concentración plasmática frente al tiempo (AUC) de axitinib al rango observado sin inhibidores. Las dosis posteriores se pueden aumentar o disminuir en función de la seguridad y la tolerabilidad individuales. Si se interrumpe la administración conjunta del inhibidor potente, la dosis de INLYTA debe volver (después de 3 a 5 semividas del inhibidor) a la utilizada antes del inicio del inhibidor potente del CYP3A4/5 [véase Interacciones medicamentosas (7.1) y Farmacología clínica (12.3)].
2.4 Modificación de la dosis para insuficiencia hepática
No se requiere ajuste de la dosis inicial cuando se administra INLYTA a pacientes con insuficiencia hepática leve (Child-Pugh clase A). Según los datos farmacocinéticos, la dosis inicial de INLYTA debe reducirse aproximadamente a la mitad en pacientes con insuficiencia hepática moderada basal (Child-Pugh clase B). Las dosis posteriores se pueden aumentar o disminuir en función de la seguridad y la tolerabilidad individuales. INLYTA no se ha estudiado en pacientes con insuficiencia hepática grave (Child-Pugh clase C) [véase Advertencias y precauciones (5.12), Uso en poblaciones específicas (8.6) y Farmacología clínica (12.3)].
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
- •
- Comprimidos de INLYTA de 1 mg: comprimidos ovales, recubiertos con película, de color rojo, con “Pfizer” grabado en una cara y “1 XNB” en la otra.
- •
- Comprimidos de INLYTA de 5 mg: comprimidos triangulares, recubiertos con película, de color rojo, con “Pfizer” grabado en una cara y “5 XNB” en la otra.
4 CONTRAINDICACIONES
Ninguna.
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Hipertensión
En un estudio clínico controlado con INLYTA para el tratamiento de pacientes con CCR, se informó hipertensión en 145/359 pacientes (40%) que recibieron INLYTA y en 103/355 pacientes (29%) que recibieron sorafenib. Se observó hipertensión de grado 3/4 en 56/359 pacientes (16%) que recibieron INLYTA y en 39/355 pacientes (11%) que recibieron sorafenib. Se informó crisis hipertensiva en 2/359 pacientes (<1%) que recibieron INLYTA y en ninguno de los pacientes que recibieron sorafenib. El tiempo medio de inicio de la hipertensión (presión arterial sistólica >150 mmHg o presión arterial diastólica >100 mmHg) se situó dentro del primer mes del inicio del tratamiento con INLYTA y se han observado aumentos de la presión arterial tan pronto como a los 4 días de iniciar el tratamiento con INLYTA. La hipertensión se controló con terapia antihipertensiva estándar. La interrupción del tratamiento con INLYTA debido a la hipertensión se produjo en 1/359 pacientes (<1%) que recibieron INLYTA y en ninguno de los pacientes que recibieron sorafenib [ver Reacciones adversas (6.1)].
Asegúrese de que la presión arterial esté bien controlada antes de iniciar el tratamiento con INLYTA. Controle a los pacientes para detectar hipertensión y trátela según sea necesario con terapia antihipertensiva estándar. Suspenda y luego reduzca la dosis de INLYTA o interrumpa permanentemente el tratamiento en función de la gravedad de la hipertensión [ver Posología y administración (2.2)].
5.2 Eventos tromboembólicos arteriales
En los ensayos clínicos, se han notificado eventos tromboembólicos arteriales, incluidas muertes. En un estudio clínico controlado con INLYTA para el tratamiento de pacientes con CCR, se notificaron eventos tromboembólicos arteriales de grado 3/4 en 4/359 pacientes (1%) que recibieron INLYTA y en 4/355 pacientes (1%) que recibieron sorafenib. Se notificó un accidente cerebrovascular fatal en 1/359 pacientes (<1%) que recibieron INLYTA y en ninguno de los pacientes que recibieron sorafenib [ver Reacciones adversas (6.1)].
INLYTA no se ha estudiado en pacientes que hayan sufrido un evento tromboembólico arterial en los 12 meses anteriores. En los ensayos clínicos con INLYTA, se notificaron eventos tromboembólicos arteriales (incluidos el accidente isquémico transitorio, el accidente cerebrovascular, el infarto de miocardio y la oclusión de la arteria retiniana) en 17/715 pacientes (2%), con dos muertes secundarias a accidente cerebrovascular.
Interrompa permanentemente el tratamiento con INLYTA si se produce un evento tromboembólico arterial durante el tratamiento.
5.3 Eventos tromboembólicos venosos
En los ensayos clínicos, se han notificado eventos tromboembólicos venosos, incluidas muertes. En un estudio clínico controlado con INLYTA para el tratamiento de pacientes con CCR, se notificaron eventos tromboembólicos venosos en 11/359 pacientes (3%) que recibieron INLYTA y en 2/355 pacientes (1%) que recibieron sorafenib. Se notificaron eventos tromboembólicos venosos de grado 3/4 en 9/359 pacientes (3%) que recibieron INLYTA (incluida la embolia pulmonar, la trombosis venosa profunda, la oclusión de la vena retiniana y la trombosis de la vena retiniana) y en 2/355 pacientes (1%) que recibieron sorafenib. Se notificó una embolia pulmonar fatal en 1/359 pacientes (<1%) que recibieron INLYTA y en ninguno de los pacientes que recibieron sorafenib.
INLYTA no se ha estudiado en pacientes que hayan sufrido un evento tromboembólico venoso en los 6 meses anteriores. En los ensayos clínicos con INLYTA, se notificaron eventos tromboembólicos venosos en 22/715 pacientes (3%), con dos muertes secundarias a embolia pulmonar.
Controle los signos y síntomas de TEP y EP. Suspenda INLYTA y luego reanude a la misma dosis o interrumpa permanentemente el tratamiento en función de la gravedad de la TEP.
5.4 Hemorragia
En un estudio clínico controlado con INLYTA para el tratamiento de pacientes con CCR, se notificaron eventos hemorrágicos en 58/359 pacientes (16%) que recibieron INLYTA y en 64/355 pacientes (18%) que recibieron sorafenib. Se notificaron eventos hemorrágicos de grado 3/4 en 5/359 (1%) pacientes que recibieron INLYTA (incluida la hemorragia cerebral, la hematuria, la hemoptisis, la hemorragia gastrointestinal inferior y la melena) y en 11/355 (3%) pacientes que recibieron sorafenib. Se notificó una hemorragia fatal en 1/359 pacientes (<1%) que recibieron INLYTA (hemorragia gástrica) y en 3/355 pacientes (1%) que recibieron sorafenib.
INLYTA no se ha estudiado en pacientes que presenten evidencia de metástasis cerebral no tratada o hemorragia gastrointestinal activa reciente y no debe utilizarse en esos pacientes. Suspenda y luego reduzca la dosis de INLYTA o interrumpa el tratamiento en función de la gravedad y la persistencia de la hemorragia.
5.5 Insuficiencia cardíaca
En un estudio clínico controlado con INLYTA para el tratamiento de pacientes con CCR, se notificó insuficiencia cardíaca en 6/359 pacientes (2%) que recibieron INLYTA y en 3/355 pacientes (1%) que recibieron sorafenib. Se observó insuficiencia cardíaca de grado 3/4 en 2/359 pacientes (1%) que recibieron INLYTA y en 1/355 pacientes (<1%) que recibieron sorafenib. Se notificó insuficiencia cardíaca fatal en 2/359 pacientes (1%) que recibieron INLYTA y en 1/355 pacientes (<1%) que recibieron sorafenib. Controle los signos o síntomas de insuficiencia cardíaca durante todo el tratamiento con INLYTA. El tratamiento de la insuficiencia cardíaca puede requerir una reducción de la dosis, una interrupción de la dosis o la interrupción permanente de INLYTA [ver Posología y administración (2.2)].
5.6 Perforación gastrointestinal y formación de fístulas
En un estudio clínico controlado con INLYTA para el tratamiento de pacientes con CCR, se notificó perforación gastrointestinal en 1/359 pacientes (<1%) que recibieron INLYTA y en ninguno de los pacientes que recibieron sorafenib. En los ensayos clínicos con INLYTA, se notificó perforación gastrointestinal en 5/715 pacientes (1%), incluida una muerte. Además de los casos de perforación gastrointestinal, se notificaron fístulas en 4/715 pacientes (1%).
Controle periódicamente los síntomas de perforación gastrointestinal o fístula durante todo el tratamiento con INLYTA.
5.7 Disfunción Tiroidea
En un estudio clínico controlado con INLYTA para el tratamiento de pacientes con CCR, se notificó hipotiroidismo en 69/359 pacientes (19%) que recibieron INLYTA y en 29/355 pacientes (8%) que recibieron sorafenib. Se notificó hipertiroidismo en 4/359 pacientes (1%) que recibieron INLYTA y en 4/355 pacientes (1%) que recibieron sorafenib. En pacientes que tenían hormona estimulante de la tiroides (TSH) <5 μU/mL antes del tratamiento, se produjeron elevaciones de TSH a ≥10 μU/mL en 79/245 pacientes (32%) que recibieron INLYTA y en 25/232 pacientes (11%) que recibieron sorafenib [ver Reacciones adversas (6.1)].
Controle la función tiroidea antes de iniciar el tratamiento y periódicamente durante el tratamiento con INLYTA. Trate el hipotiroidismo y el hipertiroidismo de acuerdo con la práctica médica estándar para mantener el estado eutiroideo.
5.8 Alteración de la Cicatrización de Heridas
Puede producirse una alteración de la cicatrización de heridas en pacientes que reciben fármacos que inhiben la vía de señalización del factor de crecimiento endotelial vascular (VEGF). Por lo tanto, INLYTA tiene el potencial de afectar negativamente a la cicatrización de heridas.
Suspenda INLYTA durante al menos 2 días antes de una cirugía electiva. No lo administre durante al menos 2 semanas después de una cirugía mayor y hasta que se produzca una cicatrización adecuada de la herida. Reanude la administración de INLYTA a una dosis reducida o interrumpa el tratamiento en función de la gravedad y la persistencia de la alteración de la cicatrización de la herida. No se ha establecido la seguridad de la reanudación de INLYTA después de la resolución de las complicaciones de la cicatrización de heridas [ver Posología y administración (2.2)].
5.9 Síndrome de Leucoencefalopatía Posterior Reversible
En un estudio clínico controlado con INLYTA para el tratamiento de pacientes con CCR, se notificó síndrome de leucoencefalopatía posterior reversible (RPLS) en 1/359 pacientes (<1%) que recibieron INLYTA y en ninguno de los pacientes que recibieron sorafenib [ver Reacciones adversas (6.1)]. Hubo dos informes adicionales de RPLS en otros ensayos clínicos con INLYTA.
El RPLS es un trastorno neurológico que puede presentarse con cefalea, convulsiones, letargo, confusión, ceguera y otras alteraciones visuales y neurológicas. Puede haber hipertensión leve a grave. Es necesaria una resonancia magnética para confirmar el diagnóstico de RPLS. Interrumpa permanentemente el tratamiento con INLYTA en pacientes que desarrollen RPLS. Se desconoce la seguridad de la reiniciación del tratamiento con INLYTA en pacientes que hayan experimentado previamente RPLS [ver Posología y administración (2.2)].
5.10 Proteinuria
En un estudio clínico controlado con INLYTA para el tratamiento de pacientes con CCR, se notificó proteinuria en 39/359 pacientes (11%) que recibieron INLYTA y en 26/355 pacientes (7%) que recibieron sorafenib. Se notificó proteinuria de grado 3 en 11/359 pacientes (3%) que recibieron INLYTA y en 6/355 pacientes (2%) que recibieron sorafenib [ver Reacciones adversas (6.1)].
Se recomienda controlar la proteinuria antes de iniciar el tratamiento y periódicamente durante el tratamiento con INLYTA. En los pacientes que desarrollen proteinuria moderada o grave, suspenda y luego reduzca la dosis de INLYTA [ver Posología y administración (2.2)].
5.11 Hepatotoxicidad
INLYTA como agente único
En un estudio clínico controlado con INLYTA para el tratamiento de pacientes con CCR, se produjeron elevaciones de alanina aminotransferasa (ALT) de todos los grados en el 22% de los pacientes de ambos brazos, con eventos de grado 3/4 en <1% de los pacientes del brazo de INLYTA. Cuando se utiliza como agente único, controle la ALT, la aspartato aminotransferasa (AST) y la bilirrubina antes de iniciar el tratamiento y periódicamente durante el tratamiento con INLYTA.
INLYTA en combinación con avelumab o con pembrolizumab
INLYTA en combinación con avelumab o con pembrolizumab puede causar hepatotoxicidad con frecuencias más altas de lo esperado de elevaciones de ALT y AST de grado 3 y 4. Controle las enzimas hepáticas antes de iniciar el tratamiento y periódicamente durante el mismo. Considere un control más frecuente de las enzimas hepáticas en comparación con cuando los fármacos se administran como agentes únicos. En caso de elevación de las enzimas hepáticas, interrumpa o suspenda permanentemente INLYTA y avelumab o pembrolizumab, y administre corticosteroides según sea necesario [ver Posología y administración (2.3)].
Con la combinación de INLYTA y avelumab, se notificaron ALT aumentada de grados 3 y 4 y AST aumentada en el 9% y el 7% de los pacientes, respectivamente. En pacientes con ALT ≥3 veces el límite superior de la normalidad (LSN) (grados 2–4, n=82), la ALT se resolvió a los grados 0–1 en el 92%. Entre los 73 pacientes a los que se volvió a desafiar con avelumab (n=3) o axitinib (n=25) administrados como agente único o con ambos (n=45), no se observó recurrencia de ALT ≥3 veces el LSN en ningún paciente que recibió avelumab, 6 pacientes que recibieron INLYTA y 15 pacientes que recibieron avelumab e INLYTA. Veintidós (88%) pacientes con una recurrencia de ALT ≥3 LSN posteriormente se recuperaron a grado 0–1 del evento. Se notificó hepatitis inmunomediada en el 7% de los pacientes, incluido el 4,9% con hepatitis inmunomediada de grado 3 o 4. La hepatotoxicidad provocó la interrupción permanente en el 7% y la hepatitis inmunomediada provocó la interrupción permanente de avelumab o INLYTA en el 5% de los pacientes. Treinta y cuatro pacientes fueron tratados con corticosteroides y un paciente fue tratado con un inmunosupresor no esteroideo. La resolución de la hepatitis se produjo en 31 de los 35 pacientes en el momento del corte de datos.
Con la combinación de INLYTA y pembrolizumab, se observaron aumentos de ALT de grados 3 y 4 (20%) y aumentos de AST (13%). El 59% de los pacientes con aumento de ALT recibieron corticosteroides sistémicos. En pacientes con ALT ≥3 veces el LSN (grados 2–4, n=116), la ALT se resolvió a grados 0–1 en el 94%. Entre los 92 pacientes que fueron tratados nuevamente con pembrolizumab (n=3) o INLYTA (n=34) administrados como agente único o con ambos (n=55), se observó una recurrencia de ALT ≥3 veces el LSN en 1 paciente que recibió pembrolizumab, 16 pacientes que recibieron INLYTA y 24 pacientes que recibieron pembrolizumab e INLYTA. Todos los pacientes con una recurrencia de ALT ≥3 LSN se recuperaron posteriormente del evento.
5.12 Uso en pacientes con insuficiencia hepática
La exposición sistémica a axitinib fue mayor en sujetos con insuficiencia hepática moderada (Child-Pugh clase B) en comparación con sujetos con función hepática normal. Se recomienda una reducción de la dosis cuando se administra INLYTA a pacientes con insuficiencia hepática moderada (Child-Pugh clase B). INLYTA no se ha estudiado en pacientes con insuficiencia hepática grave (Child-Pugh clase C) [ver Posología y administración (2.2), Uso en poblaciones específicas (8.6), y Farmacología clínica (12.3)].
5.13 Eventos cardiovasculares adversos mayores (ECAM)
INLYTA en combinación con avelumab puede causar eventos cardiovasculares graves y fatales. Considere las evaluaciones iniciales y periódicas de la fracción de eyección del ventrículo izquierdo. Controle los signos y síntomas de eventos cardiovasculares. Optimice el manejo de los factores de riesgo cardiovascular, como la hipertensión, la diabetes o la dislipidemia. Suspenda permanentemente INLYTA y avelumab para eventos cardiovasculares de grado 3–4.
Los ECAM ocurrieron en el 7% de los pacientes con CCR avanzado tratados con INLYTA en combinación con avelumab en comparación con el 3,4% tratados con sunitinib en un ensayo aleatorizado, JAVELIN Renal 101. Estos eventos incluyeron la muerte por eventos cardíacos (1,4%), infarto de miocardio de grado 3–4 (2,8%) e insuficiencia cardíaca congestiva de grado 3–4 (1,8%). El tiempo medio hasta el inicio de los ECAM fue de 4,2 meses (rango: 2 días a 24,5 meses).
5.14 Toxicidad embriofetal
Basándose en su mecanismo de acción y en los hallazgos de estudios en animales, INLYTA puede causar daño fetal cuando se administra a una mujer embarazada. No hay datos humanos disponibles para informar sobre el riesgo asociado al fármaco. En estudios de toxicidad del desarrollo en ratones, el axitinib fue teratógeno, embriotóxico y fetotóxico a exposiciones maternas que fueron inferiores a las exposiciones humanas a la dosis clínica recomendada. Avise a las mujeres en edad fértil del riesgo potencial para el feto y de que deben usar un método anticonceptivo eficaz durante el tratamiento con INLYTA y durante 1 semana después de la última dosis. Avise a los varones con parejas femeninas en edad fértil que deben usar un método anticonceptivo eficaz durante el tratamiento con INLYTA y durante 1 semana después de la última dosis [ver Uso en poblaciones específicas (8.1, 8.3), Farmacología clínica (12.1)].
Cuando INLYTA se usa en combinación con avelumab o pembrolizumab, consulte la información completa de prescripción de avelumab o pembrolizumab para obtener información sobre el embarazo y la anticoncepción.
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas clínicamente significativas se discuten en otras partes de la etiqueta [ver Advertencias y Precauciones (5)]:
- •
- Hipertensión [ver Advertencias y Precauciones (5.1)]
- •
- Eventos tromboembólicos arteriales [ver Advertencias y Precauciones (5.2)]
- •
- Eventos tromboembólicos venosos [ver Advertencias y Precauciones (5.3)]
- •
- Hemorragia [ver Advertencias y Precauciones (5.4)]
- •
- Insuficiencia cardíaca [ver Advertencias y Precauciones (5.5)]
- •
- Perforación gastrointestinal y formación de fístulas [ver Advertencias y Precauciones (5.6)]
- •
- Disfunción tiroidea [ver Advertencias y Precauciones (5.7)]
- •
- Síndrome de leucoencefalopatía posterior reversible [ver Advertencias y Precauciones (5.9)]
- •
- Proteinuria [ver Advertencias y Precauciones (5.10)]
- •
- Hepatotoxicidad [ver Advertencias y Precauciones (5.11)]
- •
- Insuficiencia hepática [ver Advertencias y Precauciones (5.12)]
6.1 Experiencia en Ensayos Clínicos
Debido a que los ensayos clínicos se llevan a cabo en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no se pueden comparar directamente con las tasas en los ensayos clínicos de otro fármaco y pueden no reflejar las tasas observadas en la práctica clínica.
La seguridad de INLYTA se ha evaluado en combinación con avelumab en JAVELIN Renal 101 y pembrolizumab en KEYNOTE – 426 para el tratamiento de primera línea de pacientes con CCR avanzado [ver Estudios Clínicos (14.1)]. Los datos descritos [ver Reacciones Adversas (6.1)] reflejan la exposición a INLYTA en combinación con avelumab en 434 pacientes y pembrolizumab en 429 pacientes [ver Estudios Clínicos (14.1)].
La seguridad de INLYTA se ha evaluado en 715 pacientes en estudios de monoterapia de segunda línea, que incluyeron a 537 pacientes con CCR avanzado. Los datos descritos [ver Reacciones Adversas (6.1)] reflejan la exposición a INLYTA en 359 pacientes con CCR avanzado que participaron en un estudio clínico aleatorizado frente a sorafenib [ver Estudios Clínicos (14.2)].
CCR Avanzado de Primera Línea
INLYTA en Combinación con Avelumab
La seguridad de INLYTA en combinación con avelumab se evaluó en JAVELIN Renal 101. Se excluyeron los pacientes con enfermedades autoinmunes distintas de la diabetes mellitus tipo I, el vitíligo, la psoriasis o los trastornos tiroideos que no requieran tratamiento inmunosupresor. Los pacientes recibieron INLYTA 5 mg dos veces al día (N = 434) en combinación con avelumab 10 mg / kg cada 2 semanas administrado o sunitinib 50 mg una vez al día durante 4 semanas seguido de 2 semanas de descanso (N = 439).
En el brazo de INLYTA más avelumab, el 70% estuvo expuesto a avelumab durante ≥6 meses y el 29% durante ≥1 año en JAVELIN Renal 101 [ver Estudios Clínicos (14.1)].
La edad mediana de los pacientes tratados con INLYTA en combinación con avelumab fue de 62 años (rango: 29 a 83), el 38% de los pacientes tenían 65 años o más, el 71% eran hombres, el 75% eran blancos y la puntuación de rendimiento del Grupo Oncológico Cooperativo del Este (ECOG) era 0 (64%) o 1 (36%).
Ocurrieron reacciones adversas fatales en el 1,8% de los pacientes que recibieron INLYTA en combinación con avelumab. Estas incluyeron muerte cardíaca súbita (1,2%), accidente cerebrovascular (0,2%), miocarditis (0,2%) y pancreatitis necrotizante (0,2%).
Ocurrieron reacciones adversas graves en el 35% de los pacientes que recibieron INLYTA en combinación con avelumab. Las reacciones adversas graves en ≥1% de los pacientes incluyeron diarrea (2,5%), disnea (1,8%), hepatotoxicidad (1,8%), enfermedad tromboembólica venosa (1,6%), lesión renal aguda (1,4%) y neumonía (1,2%).
La discontinuación permanente debido a una reacción adversa de INLYTA o avelumab se produjo en el 22% de los pacientes: solo avelumab en el 19%, solo INLYTA en el 13% y ambos fármacos en el 8%. Las reacciones adversas más comunes (> 1%) que resultaron en la discontinuación permanente de avelumab o la combinación fueron la hepatotoxicidad (6%) y la reacción relacionada con la infusión (1,8%).
Las interrupciones o reducciones de la dosis debido a una reacción adversa, excluyendo las interrupciones temporales de las infusiones de avelumab debido a reacciones relacionadas con la infusión, ocurrieron en el 76% de los pacientes que recibieron INLYTA en combinación con avelumab. Esto incluye la interrupción de avelumab en el 50% de los pacientes. INLYTA se interrumpió en el 66% y se redujo la dosis en el 19% de los pacientes. La reacción adversa más común (> 10%) que resultó en la interrupción de avelumab fue la diarrea (10%). Las reacciones adversas más comunes que resultaron en la interrupción o reducción de la dosis de INLYTA fueron la diarrea (19%), la hipertensión (18%), la eritrodisestesia palmo – plantar (18%) y la hepatotoxicidad (10%).
Las reacciones adversas más comunes (≥20%) en pacientes que recibieron INLYTA en combinación con avelumab fueron diarrea, fatiga, hipertensión, dolor musculoesquelético, náuseas, mucositis, eritrodisestesia palmo – plantar, disfonía, disminución del apetito, hipotiroidismo, erupción cutánea, hepatotoxicidad, tos, disnea, dolor abdominal y cefalea.
Cuarenta y ocho (11%) de los pacientes tratados con INLYTA en combinación con avelumab recibieron una dosis oral de prednisona equivalente a ≥40 mg diarios por una reacción adversa mediada por el sistema inmune [ver Advertencias y Precauciones (5.12)].
La Tabla 4 resume las reacciones adversas que ocurrieron en ≥20% de los pacientes tratados con INLYTA en combinación con avelumab.
| Reacciones adversas | INLYTA más Avelumab (N=434) | Sunitinib (N=439) | ||
|---|---|---|---|---|
| Todos los grados % |
Grado 3–4 % |
Todos los grados % |
Grado 3–4 % |
|
| La toxicidad se clasificó según los Criterios comunes de terminología para eventos adversos del Instituto Nacional del Cáncer. Versión 4.03 (NCI CTCAE v4). | ||||
|
||||
|
Trastornos gastrointestinales |
||||
|
Diarrea† |
62 |
8 |
48 |
2.7 |
|
Náuseas |
34 |
1.4 |
39 |
1.6 |
|
Mucositis‡ |
34 |
2.8 |
35 |
2.1 |
|
Hepatotoxicidad§ |
24 |
9 |
18 |
3.6 |
|
Dolor abdominal¶ |
22 |
1.4 |
19 |
2.1 |
|
Trastornos generales y afecciones en el sitio de administración |
||||
|
Fatiga# |
53 |
6 |
54 |
6 |
|
Trastornos vasculares |
||||
|
HipertensiónÞ |
50 |
26 |
36 |
17 |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
||||
|
Dolor musculoesqueléticoß |
40 |
3.2 |
33 |
2.7 |
|
Trastornos de la piel y del tejido subcutáneo |
||||
|
Eritrodisestesia palmar-plantar |
33 |
6 |
34 |
4 |
|
Erupciónà |
25 |
0.9 |
16 |
0.5 |
|
Trastornos respiratorios, torácicos y mediastínicos |
||||
|
Disfonía |
31 |
0.5 |
3.2 |
0 |
|
Disneaè |
23 |
3.0 |
16 |
1.8 |
|
Tos |
23 |
0.2 |
19 |
0 |
|
Trastornos del metabolismo y de la nutrición |
||||
|
Disminución del apetito |
26 |
2.1 |
29 |
0.9 |
|
Trastornos endocrinos |
||||
|
Hipotiroidismo |
25 |
0.2 |
14 |
0.2 |
|
Trastornos del sistema nervioso |
||||
|
Cefalea |
21 |
0.2 |
16 |
0.2 |
Otras reacciones adversas clínicamente importantes que ocurrieron en menos del 20% de los pacientes en JAVELIN Renal 101 incluyeron artralgia, disminución de peso y escalofríos.
Los pacientes recibieron premedicación con un antihistamínico y acetaminofeno antes de cada infusión. Las reacciones relacionadas con la infusión ocurrieron en el 12% (Grado 3: 1,6%; ningún Grado 4) de los pacientes tratados con INLYTA en combinación con avelumab.
La Tabla 5 resume las anomalías de laboratorio seleccionadas que ocurrieron en ≥20% de los pacientes tratados con INLYTA en combinación con avelumab.
| Anomalía de laboratorio | INLYTA más Avelumab | Sunitinib† | ||
|---|---|---|---|---|
| Cualquier grado % |
Grado 3–4 % |
Cualquier grado % |
Grado 3–4 % |
|
|
||||
|
Química |
||||
|
Aumento de triglicéridos en sangre |
71 |
13 |
48 |
5 |
|
Aumento de creatinina en sangre |
62 |
2.3 |
68 |
1.4 |
|
Aumento de colesterol en sangre |
57 |
1.9 |
22 |
0.7 |
|
Aumento de alanina aminotransferasa (ALT) |
50 |
9 |
46 |
3.2 |
|
Aumento de aspartato aminotransferasa (AST) |
47 |
7 |
57 |
3.2 |
|
Disminución de sodio en sangre |
38 |
9 |
37 |
10 |
|
Aumento de lipasa |
37 |
14 |
25 |
7 |
|
Aumento de potasio en sangre |
35 |
3.0 |
28 |
3.9 |
|
Aumento de bilirrubina en sangre |
21 |
1.4 |
23 |
1.4 |
|
Hematología |
||||
|
Recuento de plaquetas disminuido |
27 |
0.7 |
80 |
1.5 |
|
Hemoglobina disminuida |
21 |
2.1 |
65 |
8 |
INLYTA en combinación con Pembrolizumab
La seguridad de INLYTA en combinación con pembrolizumab se investigó en KEYNOTE-426 [ver Estudios Clínicos (14.1)]. Los pacientes con afecciones médicas que requerían corticosteroides sistémicos u otros medicamentos inmunosupresores o que tenían antecedentes de enfermedad autoinmune grave que no fuera diabetes tipo 1, vitiligo, síndrome de Sjögren e hipotiroidismo estable con reemplazo hormonal fueron inelegibles. Los pacientes recibieron INLYTA 5 mg por vía oral dos veces al día y pembrolizumab 200 mg por vía intravenosa cada 3 semanas, o sunitinib 50 mg una vez al día durante 4 semanas y luego sin tratamiento durante 2 semanas. La duración media de la exposición a la terapia combinada de INLYTA y pembrolizumab fue de 10,4 meses (rango: 1 día a 21,2 meses).
Las características de la población del estudio fueron: edad media de 62 años (rango: 30 a 89), 40% de edad 65 o más; 71% hombres; 80% blancos; y 80% Estado de Rendimiento de Karnofsky (KPS) de 90–100 y 20% KPS de 70–80.
Se produjeron reacciones adversas fatales en el 3,3% de los pacientes que recibieron INLYTA en combinación con pembrolizumab. Estas incluyeron 3 casos de paro cardíaco, 2 casos de embolia pulmonar y 1 caso de cada uno de insuficiencia cardíaca, muerte por causa desconocida, miastenia gravis, miocarditis, gangrena de Fournier, mieloma de células plasmáticas, derrame pleural, neumonitis e insuficiencia respiratoria.
Se produjeron reacciones adversas graves en el 40% de los pacientes que recibieron INLYTA en combinación con pembrolizumab. Las reacciones adversas graves en ≥1% de los pacientes que recibieron INLYTA en combinación con pembrolizumab incluyeron hepatotoxicidad (7%), diarrea (4,2%), lesión renal aguda (2,3%), deshidratación (1%) y neumonitis (1%).
La interrupción permanente debido a una reacción adversa de INLYTA o pembrolizumab se produjo en el 31% de los pacientes; 13% solo pembrolizumab, 13% solo INLYTA y 8% ambos fármacos. La reacción adversa más común (>1%) que provocó la interrupción permanente de INLYTA, pembrolizumab o la combinación fue hepatotoxicidad (13%), diarrea/colitis (1,9%), lesión renal aguda (1,6%) y accidente cerebrovascular (1,2%).
Las interrupciones o reducciones de la dosis debido a una reacción adversa, excluyendo las interrupciones temporales de las infusiones de pembrolizumab debido a reacciones relacionadas con la infusión, se produjeron en el 76% de los pacientes que recibieron pembrolizumab en combinación con INLYTA. Esto incluye la interrupción de pembrolizumab en el 50% de los pacientes. INLYTA se interrumpió en el 64% de los pacientes y la dosis se redujo en el 22% de los pacientes. Las reacciones adversas más comunes (>10%) que provocaron la interrupción o reducción de INLYTA fueron hepatotoxicidad (21%), diarrea (19%) e hipertensión (18%) y las reacciones adversas más comunes (>10%) que provocaron la interrupción de pembrolizumab fueron hepatotoxicidad (14%) y diarrea (11%).
Las reacciones adversas más comunes (≥20%) en pacientes que recibieron INLYTA y pembrolizumab fueron diarrea, fatiga/astenia, hipertensión, hepatotoxicidad, hipotiroidismo, disminución del apetito, eritrodisestesia palmar-plantar, náuseas, estomatitis/inflamación de la mucosa, disfonía, erupción cutánea, tos y estreñimiento.
El veintisiete por ciento (27%) de los pacientes tratados con INLYTA en combinación con pembrolizumab recibieron una dosis oral de prednisona equivalente a ≥40 mg diarios para una reacción adversa mediada por el sistema inmunitario.
Los cuadros 6 y 7 resumen las reacciones adversas y las anormalidades de laboratorio, respectivamente, que se produjeron en al menos el 20% de los pacientes tratados con INLYTA y pembrolizumab en KEYNOTE-426.
| Reacciones adversas | INLYTA más Pembrolizumab N=429 | Sunitinib N=425 | ||||
|---|---|---|---|---|---|---|
| Todos los grados* % |
Grados 3–4 % |
Todos los grados % |
Grados 3–4 % |
|||
|
||||||
|
Trastornos gastrointestinales |
||||||
|
Diarrea† |
56 |
11 |
45 |
5 |
|
Náuseas |
28 |
0.9 |
32 |
0.9 |
|
Estreñimiento |
21 |
0 |
15 |
0.2 |
|
General |
||||
|
Fatiga/Astenia |
52 |
5 |
51 |
10 |
|
Vascular |
||||
|
Hipertensión‡ |
48 |
24 |
48 |
20 |
|
Hepatobiliar |
||||
|
Hepatotoxicidad§ |
39 |
20 |
25 |
4.9 |
|
Endocrino |
||||
|
Hipotiroidismo |
35 |
0.2 |
32 |
0.2 |
|
Metabolismo y Nutrición |
||||
|
Disminución del apetito |
30 |
2.8 |
29 |
0.7 |
|
Piel y Tejido Subcutáneo |
||||
|
Síndrome de eritrodisestesia palmar-plantar |
28 |
5 |
40 |
3.8 |
|
Estomatitis/Inflamación de la mucosa |
27 |
1.6 |
41 |
4 |
|
Erupción¶ |
25 |
1.4 |
21 |
0.7 |
|
Respiratorio, Torácico y Mediastínico |
||||
|
Disfonía |
25 |
0.2 |
3.3 |
0 |
|
Tos |
21 |
0.2 |
14 |
0.5 |
| Prueba de laboratorio* | INLYTA más Pembrolizumab | Sunitinib | ||||
|---|---|---|---|---|---|---|
| Todos los grados† % |
Grado 3–4 % |
Todos los grados % |
Grado 3–4 % |
|||
|
||||||
|
Química |
||||||
|
Hiperglucemia |
62 |
9 |
54 |
3.2 |
||
|
Aumento de ALT |
60 |
20 |
44 |
5 |
||
|
Aumento de AST |
57 |
13 |
56 |
5 |
||
|
Aumento de creatinina |
43 |
4.3 |
40 |
2.4 |
||
|
Hiponatremia |
35 |
8 |
29 |
8 |
||
|
Hiperkalemia |
34 |
6 |
22 |
1.7 |
||
|
Hipoalbuminemia |
32 |
0.5 |
34 |
1.7 |
||
|
Hipercalcemia |
27 |
0.7 |
15 |
1.9 |
||
|
Hipofosfatemia |
26 |
6 |
49 |
17 |
||
|
Aumento de fosfatasa alcalina |
26 |
1.7 |
30 |
2.7 |
||
|
Hipocalcemia‡ |
22 |
0.2 |
29 |
0.7 |
|
Bilirrubina sanguínea aumentada |
22 |
2.1 |
21 |
1.9 |
|
Tiempo de tromboplastina parcial activado prolongado§ |
22 |
1.2 |
14 |
0 |
|
Hematología |
||||
|
Linfopenia |
33 |
11 |
46 |
8 |
|
Anemia |
29 |
2.1 |
65 |
8 |
|
Trombocitopenia |
27 |
1.4 |
78 |
14 |
RCC avanzado de segunda línea
La duración mediana del tratamiento fue de 6,4 meses (rango 0,03 a 22,0) para los pacientes que recibieron INLYTA y de 5,0 meses (rango 0,03 a 20,1) para los pacientes que recibieron sorafenib. Se produjeron modificaciones de la dosis o retrasos temporales del tratamiento debido a una reacción adversa en 199/359 pacientes (55%) que recibieron INLYTA y en 220/355 pacientes (62%) que recibieron sorafenib. La interrupción permanente debido a una reacción adversa se produjo en 34/359 pacientes (9%) que recibieron INLYTA y en 46/355 pacientes (13%) que recibieron sorafenib.
Las reacciones adversas más frecuentes (≥20%) observadas tras el tratamiento con INLYTA fueron diarrea, hipertensión, fatiga, disminución del apetito, náuseas, disfonía, síndrome de eritrodisestesia palmar-plantar (mano-pie), disminución de peso, vómitos, astenia y estreñimiento. La Tabla 8 presenta las reacciones adversas notificadas en ≥10% de los pacientes que recibieron INLYTA o sorafenib.
| Reacción adversa* | INLYTA | Sorafenib | ||||
|---|---|---|---|---|---|---|
| (N=359) | (N=355) | |||||
| Todos los grados† | Grado 3/4 | Todos los grados† | Grado 3/4 | |||
| % | % | % | % | |||
|
Diarrea |
55 |
11 |
53 |
7 |
||
|
Hipertensión |
40 |
16 |
29 |
11 |
||
|
Fatiga |
39 |
11 |
32 |
5 |
||
|
Disminución del apetito |
34 |
5 |
29 |
4 |
||
|
Náuseas |
32 |
3 |
22 |
1 |
||
|
Disfonía |
31 |
0 |
14 |
0 |
||
|
Síndrome de eritrodisestesia palmar-plantar |
27 |
5 |
51 |
16 |
||
|
Disminución de peso |
25 |
2 |
21 |
1 |
||
|
Vómitos |
24 |
3 |
17 |
1 |
||
|
Astenia |
21 |
5 |
14 |
3 |
||
|
Estreñimiento |
20 |
1 |
20 |
1 |
||
|
Hipotiroidismo |
19 |
<1 |
8 |
0 |
||
|
Tos |
15 |
1 |
17 |
1 |
||
|
Inflamación de la mucosa |
15 |
1 |
12 |
1 |
||
|
Artralgia |
15 |
2 |
11 |
1 |
||
|
Estomatitis |
15 |
1 |
12 |
<1 |
||
|
Disnea |
15 |
3 |
12 |
3 |
||
|
Dolor abdominal |
14 |
2 |
11 |
1 |
||
|
Cefalea |
14 |
1 |
11 |
0 |
||
|
Dolor en extremidades |
13 |
1 |
14 |
1 |
||
|
Erupción |
13 |
<1 |
32 |
4 |
||
|
Proteinuria |
11 |
3 |
7 |
2 |
||
|
Disgeusia |
11 |
0 |
8 |
0 |
|
Piel seca |
10 |
0 |
11 |
0 |
|
Dispepsia |
10 |
0 |
2 |
0 |
|
Prurito |
7 |
0 |
12 |
0 |
|
Alopecia |
4 |
0 |
32 |
0 |
|
Eritema |
2 |
0 |
10 |
<1 |
Las reacciones adversas seleccionadas (de todos los grados) que se notificaron en <10 % de los pacientes tratados con INLYTA incluyeron mareos (9 %), dolor en la parte superior del abdomen (8 %), mialgia (7 %), deshidratación (6 %), epistaxis (6 %), anemia (4 %), hemorroides (4 %), hematuria (3 %), tinnitus (3 %), aumento de lipasa (3 %), glosodia (3 %), embolia pulmonar (2 %), hemorragia rectal (2 %), hemoptisis (2 %), trombosis venosa profunda (1 %), oclusión/trombosis de la vena retiniana (1 %), policitemia (1 %) y accidente isquémico transitorio (1 %).
La Tabla 9 presenta las anomalías de laboratorio más frecuentes notificadas en ≥10 % de los pacientes que recibieron INLYTA o sorafenib.
| Anomalía de laboratorio | N | INLYTA | N | Sorafenib | ||
|---|---|---|---|---|---|---|
| Todos los grados* | Grado 3/4 | Todos los grados* | Grado 3/4 | |||
| % | % | % | % | |||
| ALP: fosfatasa alcalina; ALT: alanina aminotransferasa; AST: aspartato aminotransferasa | ||||||
|
||||||
|
Hematología |
||||||
|
Hemoglobina disminuida |
320 |
35 |
<1 |
316 |
52 |
4 |
|
Linfocitos (absolutos) disminuidos |
317 |
33 |
3 |
309 |
36 |
4 |
|
Plaquetas disminuidas |
312 |
15 |
<1 |
310 |
14 |
0 |
|
Leucocitos disminuidos |
320 |
11 |
0 |
315 |
16 |
<1 |
|
Química |
||||||
|
Creatinina aumentada |
336 |
55 |
0 |
318 |
41 |
<1 |
|
Bicarbonato disminuido |
314 |
44 |
<1 |
291 |
43 |
0 |
|
Hipocalcemia |
336 |
39 |
1 |
319 |
59 |
2 |
|
ALP aumentado |
336 |
30 |
1 |
319 |
34 |
1 |
|
Hiperglucemia |
336 |
28 |
2 |
319 |
23 |
2 |
|
Lipasa aumentada |
338 |
27 |
5 |
319 |
46 |
15 |
|
Amilasa aumentada |
338 |
25 |
2 |
319 |
33 |
2 |
|
ALT aumentado |
331 |
22 |
<1 |
313 |
22 |
2 |
|
AST aumentado |
331 |
20 |
<1 |
311 |
25 |
1 |
|
Hipernatremia |
338 |
17 |
1 |
319 |
13 |
1 |
|
Hipoalbuminemia |
337 |
15 |
<1 |
319 |
18 |
1 |
|
Hiperkalemia |
333 |
15 |
3 |
314 |
10 |
3 |
|
Hipoglucemia |
336 |
11 |
<1 |
319 |
8 |
<1 |
|
Hiponatremia |
338 |
13 |
4 |
319 |
11 |
2 |
|
Hipofosfatemia |
336 |
13 |
2 |
318 |
49 |
16 |
Las anormalidades de laboratorio seleccionadas (todos los grados) que se informaron en <10% de los pacientes tratados con INLYTA incluyeron aumento de hemoglobina (por encima del límite superior de lo normal) (9% para INLYTA versus 1% para sorafenib) e hipercalcemia (6% para INLYTA versus 2% para sorafenib).
6.2 Experiencia postcomercialización
Las siguientes reacciones adversas se han identificado durante el uso posterior a la aprobación de INLYTA. Debido a que estas reacciones se notifican voluntariamente a partir de una población de tamaño incierto, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal con la exposición al fármaco.
Trastornos vasculares: aneurismas, disecciones y roturas arteriales (incluidas las aórticas).
7 INTERACCIONES MEDICAMENTOSAS
7.1 Inhibidores de CYP3A4/5
La administración conjunta de ketoconazol, un inhibidor potente de CYP3A4/5, aumentó la exposición plasmática de axitinib en voluntarios sanos. Debe evitarse la administración conjunta de INLYTA con inhibidores potentes de CYP3A4/5. El pomelo o el zumo de pomelo también pueden aumentar las concentraciones plasmáticas de axitinib y deben evitarse. Se recomienda la selección de medicamentos concomitantes con un potencial de inhibición de CYP3A4/5 nulo o mínimo. Si se debe administrar conjuntamente un inhibidor potente de CYP3A4/5, la dosis de INLYTA debe reducirse [véase Posología y administración (2.2) y Farmacología clínica (12.3)].
7.2 Inductores de CYP3A4/5
La administración conjunta de rifampicina, un inductor potente de CYP3A4/5, redujo la exposición plasmática de axitinib en voluntarios sanos. Debe evitarse la administración conjunta de INLYTA con inductores potentes de CYP3A4/5 (p. ej., rifampicina, dexametasona, fenitoína, carbamazepina, rifabutina, rifapentina, fenobarbital y hierba de San Juan). Se recomienda la selección de medicamentos concomitantes con un potencial de inducción de CYP3A4/5 nulo o mínimo [véase Posología y administración (2.2), Farmacología clínica (12.3)]. Los inductores moderados de CYP3A4/5 (p. ej., bosentán, efavirenz, etravirina, modafinilo y nafcilina) también pueden reducir la exposición plasmática de axitinib y deben evitarse en la medida de lo posible.
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de Riesgo
Basándose en los hallazgos en estudios animales y en su mecanismo de acción, INLYTA puede causar daño fetal cuando se administra a una mujer embarazada. No hay datos humanos disponibles para informar sobre el riesgo asociado al fármaco. En estudios de toxicidad del desarrollo, el axitinib fue teratógeno, embriotóxico y fetotóxico en ratones a exposiciones inferiores a las exposiciones humanas a la dosis inicial recomendada (ver Datos). Avise a las mujeres en edad fértil sobre el riesgo potencial para el feto.
Se desconoce el riesgo de base de defectos congénitos importantes y aborto espontáneo para las poblaciones indicadas. Sin embargo, el riesgo de base en la población general de los Estados Unidos (EE. UU.) de defectos congénitos importantes es del 2% al 4% y de aborto espontáneo del 15% al 20% de los embarazos clínicamente reconocidos.
Cuando INLYTA se usa en combinación con avelumab o pembrolizumab, consulte la información completa de prescripción de avelumab o pembrolizumab para obtener información sobre el embarazo.
Datos en animales
La administración oral de axitinib dos veces al día a ratones hembra antes del apareamiento y durante la primera semana de embarazo provocó un aumento en la pérdida postimplantacional en todas las dosis probadas (≥15 mg/kg/dosis, aproximadamente 10 veces la exposición sistémica (AUC) en pacientes a la dosis inicial recomendada). En un estudio de toxicidad del desarrollo embriofetal, las ratonas embarazadas recibieron dosis orales de 0,15, 0,5 y 1,5 mg/kg/dosis de axitinib dos veces al día durante el período de organogénesis. Las toxicidades embriofetales observadas en ausencia de toxicidad materna incluyeron malformaciones (paladar hendido) a 1,5 mg/kg/dosis (aproximadamente 0,5 veces el AUC en pacientes a la dosis inicial recomendada) y variación en la osificación esquelética a ≥0,5 mg/kg/dosis (aproximadamente 0,15 veces el AUC en pacientes a la dosis inicial recomendada).
8.2 Lactancia
Resumen de Riesgo
No hay datos sobre la presencia de axitinib en la leche materna, ni sobre sus efectos en el niño amamantado o en la producción de leche. Debido al potencial de reacciones adversas graves en un niño amamantado por INLYTA, se debe aconsejar a las mujeres lactantes que no amamanten durante el tratamiento y durante 2 semanas después de la última dosis.
Cuando INLYTA se usa en combinación con avelumab o pembrolizumab, consulte la información completa de prescripción de avelumab o pembrolizumab para obtener información sobre la lactancia.
8.3 Mujeres y hombres en edad fértil
Basándose en los hallazgos en estudios animales, INLYTA puede causar daño fetal cuando se administra a una mujer embarazada [ver Uso en poblaciones específicas (8.1)]. Cuando INLYTA se usa en combinación con avelumab o pembrolizumab, consulte la información completa de prescripción de avelumab o pembrolizumab para obtener información sobre la anticoncepción.
Prueba de embarazo
Verifique el estado del embarazo en las mujeres en edad fértil antes de iniciar el tratamiento con INLYTA.
Anticoncepción
Infertilidad
Mujeres y hombres
Basándose en los hallazgos en animales, INLYTA puede afectar la fertilidad en mujeres y hombres en edad fértil [ver Toxicología no clínica (13.1)].
8.4 Uso pediátrico
No se ha establecido la seguridad y eficacia de INLYTA en pacientes pediátricos.
La seguridad y eficacia de INLYTA se evaluaron, pero no se establecieron, en dos estudios abiertos: un estudio de búsqueda de dosis de INLYTA como agente único en 17 pacientes pediátricos de 5 a <17 años con tumores sólidos recurrentes o refractarios (ADVL1315, NCT02164838) y un estudio aleatorizado de INLYTA como agente único o en combinación en 7 pacientes pediátricos de 7 a <17 años (AREN1721, NCT03595124).
No se observaron nuevas señales de seguridad con INLYTA en pacientes pediátricos en estos estudios.
La exposición en pacientes pediátricos que recibieron INLYTA a la dosis máxima tolerada fue menor que la observada previamente en adultos que recibieron la dosis inicial recomendada aprobada.
Datos de toxicidad en animales jóvenes
Se observaron toxicidades en huesos y dientes en ratones y perros inmaduros a los que se administró axitinib por vía oral dos veces al día durante 1 mes o más. Los efectos en los huesos consistieron en engrosamiento de las placas de crecimiento en ratones y perros a ≥15 mg/kg/dosis (aproximadamente 6 y 15 veces, respectivamente, la exposición sistémica (AUC) en pacientes a la dosis inicial recomendada). Se observaron anomalías en los dientes incisivos en crecimiento (incluidas caries dentales, maloclusiones y dientes rotos o perdidos) en ratones a los que se administró axitinib por vía oral dos veces al día a ≥5 mg/kg/dosis (aproximadamente 1,5 veces el AUC en pacientes a la dosis inicial recomendada). Otras toxicidades de posible interés para pacientes pediátricos no se han evaluado en animales jóvenes.
8.5 Uso en geriatría
En un estudio clínico controlado con INLYTA para el tratamiento de pacientes con CCR, 123/359 pacientes (34%) tratados con INLYTA tenían ≥65 años de edad. Aunque no se puede descartar una mayor sensibilidad en algunos individuos mayores, no se observaron diferencias generales en la seguridad y eficacia de INLYTA entre los pacientes que tenían ≥65 años de edad y los más jóvenes.
De los 434 pacientes aleatorizados a INLYTA 5 mg dos veces al día administrado en combinación con avelumab 10 mg/kg en el ensayo JAVELIN Renal 101, el 38% tenía 65 años o más y el 8% tenía 75 años o más. No se informó ninguna diferencia general en la seguridad o la eficacia entre los pacientes que tenían ≥65 años de edad y los más jóvenes.
De los 432 pacientes aleatorizados a INLYTA 5 mg dos veces al día administrado en combinación con pembrolizumab 200 mg en el ensayo KEYNOTE-426, el 40% tenía 65 años o más. No se informó ninguna diferencia general en la seguridad o la eficacia entre los pacientes que tenían ≥65 años de edad y los más jóvenes.
No se requiere ajuste de la dosis en pacientes de edad avanzada [ver Posología y administración (2.2), Farmacología clínica (12.3)].
8.6 Insuficiencia hepática
En un ensayo específico de insuficiencia hepática, en comparación con los sujetos con función hepática normal, la exposición sistémica después de una dosis única de INLYTA fue similar en los sujetos con insuficiencia hepática leve basal (clase A de Child-Pugh) y mayor en los sujetos con insuficiencia hepática moderada basal (clase B de Child-Pugh).
No se requiere ajuste de la dosis inicial al administrar INLYTA a pacientes con insuficiencia hepática leve (clase A de Child-Pugh). Se recomienda una reducción de la dosis inicial al administrar INLYTA a pacientes con insuficiencia hepática moderada (clase B de Child-Pugh) [ver Posología y administración (2.2), Advertencias y precauciones (5.12), Farmacología clínica (12.3)].
INLYTA no se ha estudiado en sujetos con insuficiencia hepática grave (clase C de Child-Pugh).
8.7 Insuficiencia renal
No se ha realizado ningún ensayo específico de insuficiencia renal para axitinib. Según los análisis farmacocinéticos poblacionales, no se observó ninguna diferencia significativa en el aclaramiento de axitinib en pacientes con insuficiencia renal preexistente leve a grave (15 mL/min ≤ aclaramiento de creatinina [CLcr] <89 mL/min) [ver Farmacología clínica (12.3)]. No es necesario ajustar la dosis inicial para pacientes con insuficiencia renal preexistente leve a grave. Se debe tener precaución en pacientes con enfermedad renal en etapa terminal (CLcr <15 mL/min).
10 SOBREDOSIS
No existe un tratamiento específico para la sobredosis de INLYTA.
En un estudio clínico controlado con INLYTA para el tratamiento de pacientes con CCR, 1 paciente recibió inadvertidamente una dosis de 20 mg dos veces al día durante 4 días y experimentó mareos (Grado 1).
En un estudio clínico de búsqueda de dosis con INLYTA, los sujetos que recibieron dosis iniciales de 10 mg dos veces al día o 20 mg dos veces al día experimentaron reacciones adversas que incluyeron hipertensión, convulsiones asociadas con hipertensión y hemoptisis fatal.
En casos de sospecha de sobredosis, se debe suspender INLYTA e instaurar un tratamiento de soporte.
11 DESCRIPCIÓN
INLYTA (axitinib) es un inhibidor de la quinasa. El axitinib tiene el nombre químico N-metil-2-[3-((E)-2-piridin-2-il-vinil)-1H-indazol-6-ilsulfanyl]-benzamida. La fórmula molecular es C22H18N4OS y el peso molecular es 386,47 Daltons. La estructura química es:
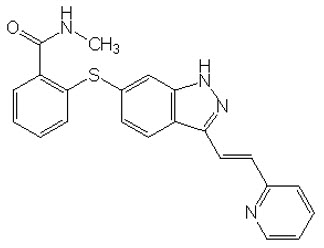
El axitinib es un polvo blanco a amarillo claro con un pKa de 4,8. La solubilidad del axitinib en medios acuosos en el rango de pH 1,1 a pH 7,8 es superior a 0,2 µg/mL. El coeficiente de partición (n-octanol/agua) es 3,5.
INLYTA se suministra como comprimidos recubiertos con película roja que contienen 1 mg o 5 mg de axitinib junto con celulosa microcristalina, lactosa monohidrato, croscarmelosa sódica, estearato de magnesio y Opadry® II red 32K15441 como ingredientes inactivos. El recubrimiento de película Opadry II red 32K15441 contiene lactosa monohidrato, HPMC 2910/Hipromelosa 15cP, dióxido de titanio, triacetina (triacetato de glicerol) y óxido de hierro rojo.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
Se ha demostrado que el axitinib inhibe las tirosina quinasas receptoras, incluidas las del factor de crecimiento endotelial vascular (VEGFR)-1, VEGFR-2 y VEGFR-3 a concentraciones plasmáticas terapéuticas. Estos receptores están implicados en la angiogénesis patológica, el crecimiento tumoral y la progresión del cáncer. La proliferación y supervivencia de las células endoteliales mediadas por VEGF fueron inhibidas por el axitinib in vitro y en modelos de ratón. Se demostró que el axitinib inhibe el crecimiento tumoral y la fosforilación de VEGFR-2 en modelos de ratón con xenotrasplantes tumorales.
12.2 Farmacodinamia
El efecto de una dosis oral única de INLYTA (5 mg) en ausencia y presencia de 400 mg de ketoconazol sobre el intervalo QTc se evaluó en un estudio cruzado de dos vías, aleatorizado y simple ciego, en 35 sujetos sanos. No se detectaron cambios importantes en el intervalo QTc medio (es decir, >20 ms) con respecto al placebo hasta 3 horas después de la administración de la dosis. Sin embargo, no se pueden descartar pequeños aumentos en el intervalo QTc medio (es decir, <10 ms).
12.3 Farmacocinética
El análisis farmacocinético poblacional agrupó datos de 17 ensayos en sujetos sanos y pacientes con cáncer. Un modelo de disposición de dos compartimentos con absorción de primer orden y tiempo de retraso describe adecuadamente el perfil concentración-tiempo del axitinib.
Absorción y distribución
Después de la administración de una dosis oral única de 5 mg, la Tmax mediana osciló entre 2,5 y 4,1 horas. Según la semivida plasmática, se espera que el estado estacionario se alcance en 2 o 3 días de dosificación. La dosificación de axitinib a 5 mg dos veces al día produjo una acumulación de aproximadamente 1,4 veces en comparación con la administración de una dosis única. En estado estacionario, el axitinib exhibe una farmacocinética aproximadamente lineal dentro del rango de dosis de 1 mg a 20 mg. La biodisponibilidad absoluta media del axitinib después de una dosis oral de 5 mg es del 58 %.
En comparación con el ayuno nocturno, la administración de INLYTA con una comida con moderado contenido de grasa produjo un 10 % menos de AUC y una comida alta en grasa y calorías produjo un 19 % más de AUC. INLYTA se puede administrar con o sin alimentos [ver Posología y administración (2.1)].
El axitinib se une en gran medida (>99 %) a las proteínas plasmáticas humanas con unión preferencial a la albúmina y unión moderada a la glicoproteína ácida α1. En pacientes con CCR avanzada (n=20), a la dosis de 5 mg dos veces al día en estado alimentado, la media geométrica (CV %) de Cmax y AUC0–24 fueron 27,8 (79 %) ng/mL y 265 (77 %) ng·h/mL, respectivamente. La media geométrica (CV %) del aclaramiento y el volumen de distribución aparente fueron 38 (80 %) L/h y 160 (105 %) L, respectivamente.
Metabolismo y eliminación
La semivida plasmática de INLYTA oscila entre 2,5 y 6,1 horas. El axitinib se metaboliza principalmente en el hígado por CYP3A4/5 y en menor medida por CYP1A2, CYP2C19 y UGT1A1. Después de la administración oral de una dosis radiactiva de 5 mg de axitinib, aproximadamente el 41 % de la radiactividad se recuperó en las heces y aproximadamente el 23 % se recuperó en la orina. El axitinib sin cambios, que representa el 12 % de la dosis, fue el componente principal identificado en las heces. No se detectó axitinib sin cambios en la orina; los metabolitos del ácido carboxílico y el sulfóxido representaron la mayor parte de la radiactividad en la orina. En plasma, el metabolito N-glucurónido representó el componente radiactivo predominante (50 % de la radiactividad circulante) y el axitinib sin cambios y el metabolito sulfóxido representaron aproximadamente el 20 % de la radiactividad circulante.
Los metabolitos del sulfóxido y el N-glucurónido muestran una potencia in vitro aproximadamente ≥400 veces menor contra VEGFR-2 en comparación con el axitinib.
Interacciones medicamentosas
Efectos de otros medicamentos sobre INLYTA
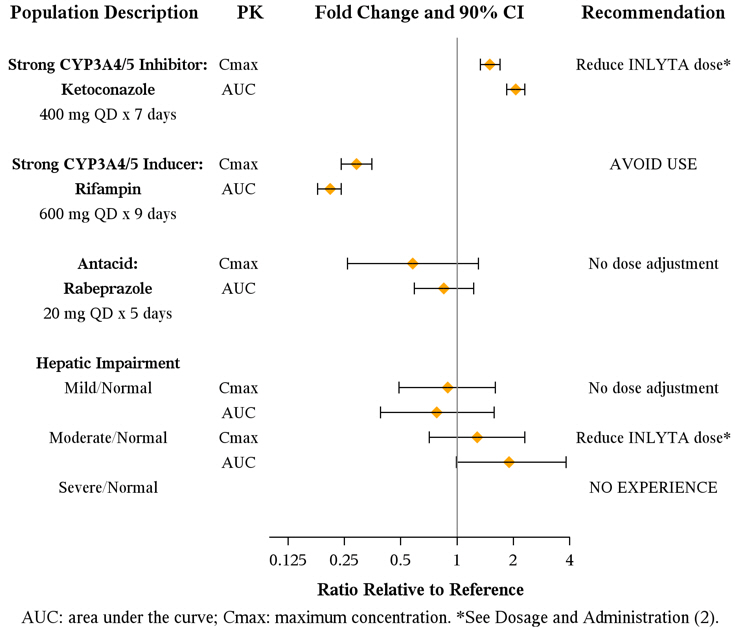
El axitinib se metaboliza principalmente en el hígado por CYP3A4/5. Además, la solubilidad acuosa del axitinib depende del pH, y un pH más alto produce una menor solubilidad. Los efectos de un inhibidor potente de CYP3A4/5, un inductor potente de CYP3A4/5 y un antiácido sobre la farmacocinética del axitinib se presentan en la Figura 1 [ver Posología y administración (2.2) y Interacciones medicamentosas (7.1, 7.2)].
Figura 1. Impacto de los medicamentos coadministrados y la insuficiencia hepática en la farmacocinética del axitinib
Efectos de INLYTA sobre otros medicamentos
Los estudios in vitro demostraron que el axitinib tiene el potencial de inhibir CYP1A2 y CYP2C8. Sin embargo, la coadministración de axitinib con paclitaxel, un sustrato de CYP2C8, no aumentó las concentraciones plasmáticas de paclitaxel en los pacientes.
Los estudios in vitro indicaron que el axitinib no inhibe CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5 o UGT1A1 a concentraciones plasmáticas terapéuticas. Los estudios in vitro en hepatocitos humanos indicaron que el axitinib no induce CYP1A1, CYP1A2 o CYP3A4/5.
El axitinib es un inhibidor del transportador de eflujo P-glucoproteína (P-gp) in vitro. Sin embargo, no se espera que INLYTA inhiba P-gp a concentraciones plasmáticas terapéuticas.
Poblaciones específicas
Pacientes con insuficiencia hepática
Los efectos de la insuficiencia hepática en la farmacocinética de axitinib se presentan en la Figura 1 [ver Posología y administración (2.2), Advertencias y precauciones (5.12), Uso en poblaciones específicas (8.6)].
Pacientes con insuficiencia renal
Se realizó un análisis farmacocinético poblacional (basado en la función renal preexistente) en 590 voluntarios sanos y pacientes, incluidos cinco con insuficiencia renal grave (15 mL/min ≤CLcr <29 mL/min), 64 con insuficiencia renal moderada (30 mL/min ≤CLcr <59 mL/min) y 139 con insuficiencia renal leve (60 mL/min ≤CLcr <89 mL/min). La insuficiencia renal leve a grave no tuvo efectos significativos en la farmacocinética de axitinib. Solo se dispone de datos de un paciente con enfermedad renal en etapa terminal [ver Uso en poblaciones específicas (8.7)].
Otros factores intrínsecos
Los análisis farmacocinéticos poblacionales indican que no hay efectos clínicamente relevantes de la edad, el sexo, la raza, el peso corporal, el área de superficie corporal, el genotipo UGT1A1 o el genotipo CYP2C19 en el aclaramiento de axitinib.
INLYTA en combinación con avelumab
Cuando se administraron 5 mg de INLYTA en combinación con 10 mg/kg de avelumab, las exposiciones respectivas de INLYTA y avelumab fueron comparables a las de los agentes únicos. No hubo evidencia que sugiriera un cambio clínicamente relevante del aclaramiento de avelumab con el tiempo en pacientes con CCR avanzada.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogenicidad, Mutagénesis, Deterioro de la Fertilidad
No se han realizado estudios de carcinogenicidad con axitinib.
Axitinib no fue mutagénico en una prueba in vitro de reversión de mutación bacteriana (Ames) y no fue clastogénico en la prueba in vitro de aberración cromosómica en linfocitos humanos. Axitinib fue genotóxico en la prueba in vivo de micronúcleos en médula ósea de ratón.
INLYTA tiene el potencial de afectar la función reproductiva y la fertilidad en humanos. En estudios de toxicología de dosis repetidas, se observaron hallazgos en el tracto reproductor masculino en los testículos/epidídimo (disminución del peso del órgano, atrofia o degeneración, disminución del número de células germinales, hipospermia o formas anormales de espermatozoides, reducción de la densidad y el recuento de espermatozoides) a ≥15 mg/kg/dosis administrados por vía oral dos veces al día en ratones (aproximadamente 7 veces la exposición sistémica (AUC) en pacientes a la dosis inicial recomendada) y ≥1,5 mg/kg/dosis administrados por vía oral dos veces al día en perros (aproximadamente 0,1 veces el AUC en pacientes a la dosis inicial recomendada). Los hallazgos en el tracto reproductor femenino en ratones y perros incluyeron signos de madurez sexual retrasada, reducción o ausencia de cuerpos lúteos, disminución del peso uterino y atrofia uterina a ≥5 mg/kg/dosis (aproximadamente 1,5 o 0,3 veces el AUC en pacientes a la dosis inicial recomendada en comparación con ratones y perros, respectivamente).
En un estudio de fertilidad en ratones, axitinib no afectó el apareamiento ni la tasa de fertilidad cuando se administró por vía oral dos veces al día a los machos a ninguna dosis probada hasta 50 mg/kg/dosis después de al menos 70 días de administración (aproximadamente 57 veces el AUC en pacientes a la dosis inicial recomendada). En ratones hembra, se observó una reducción de la fertilidad y la viabilidad embrionaria en todas las dosis probadas (≥15 mg/kg/dosis administradas por vía oral dos veces al día) después de al menos 15 días de tratamiento con axitinib (aproximadamente 10 veces el AUC en pacientes a la dosis inicial recomendada).
14 ESTUDIOS CLÍNICOS
14.1 RCC avanzado de primera línea
INLYTA en combinación con Avelumab
La eficacia y seguridad de INLYTA en combinación con avelumab se demostró en el ensayo JAVELIN Renal 101 (NCT02684006), un estudio aleatorizado, multicéntrico, abierto, de INLYTA en combinación con avelumab en 886 pacientes con RCC avanzado no tratado, independientemente de la expresión de PD-L1 tumoral [población por intención de tratar (ITT)]. Se excluyeron los pacientes con enfermedades o afecciones autoinmunitarias que requirieran inmunosupresión sistémica.
La aleatorización se estratificó según el estado de rendimiento del Eastern Cooperative Oncology Group (ECOG) (0 vs. 1) y la región (Estados Unidos vs. Canadá/Europa occidental vs. el resto del mundo). Los pacientes fueron aleatorizados (1:1) a uno de los siguientes brazos de tratamiento:
- •
- INLYTA 5 mg dos veces al día por vía oral se administró en combinación con avelumab 10 mg/kg en infusión intravenosa cada 2 semanas (N=442). Los pacientes que toleraron INLYTA 5 mg dos veces al día sin eventos adversos relacionados con INLYTA de grado 2 o superior durante 2 semanas consecutivas pudieron aumentar a 7 mg y posteriormente a 10 mg dos veces al día. INLYTA podría interrumpirse o reducirse a 3 mg dos veces al día y posteriormente a 2 mg dos veces al día para controlar la toxicidad.
- •
- Sunitinib 50 mg una vez al día por vía oral durante 4 semanas seguidas de 2 semanas de descanso (N=444) hasta la progresión radiográfica o clínica o toxicidad inaceptable.
El tratamiento con INLYTA y avelumab continuó hasta la progresión de la enfermedad definida por RECIST v1.1 mediante la evaluación de Revisión Central Independiente Ciega (BICR) o toxicidad inaceptable. La administración de INLYTA y avelumab se permitió más allá de la progresión de la enfermedad definida por RECIST si el paciente era clínicamente estable y se consideraba que estaba obteniendo un beneficio clínico por parte del investigador. La evaluación del estado tumoral se realizó al inicio, después de la aleatorización a las 6 semanas, luego cada 6 semanas hasta los 18 meses después de la aleatorización, y cada 12 semanas hasta la progresión de la enfermedad confirmada documentada por BICR.
Las características basales fueron una mediana de edad de 61 años (rango: 27 a 88), el 38% de los pacientes tenían 65 años o más, el 75% eran hombres, el 75% eran blancos y el estado de rendimiento ECOG fue 0 (63%) o 1 (37%), respectivamente. La distribución de los pacientes por grupos de riesgo de la base de datos internacional de carcinoma de células renales metastásicas (IMDC) fue del 21% favorable, 62% intermedio y 16% pobre.
Las principales medidas de resultado de eficacia fueron la supervivencia libre de progresión (SLP), evaluada por un BICR utilizando RECIST v1.1 y la supervivencia general (SG) en pacientes con tumores PD-L1 positivos utilizando un ensayo de ensayo clínico (nivel de expresión de PD-L1 ≥1%). Dado que la SLP fue estadísticamente significativa en pacientes con tumores PD-L1 positivos [HR 0,61 (IC del 95%: 0,48, 0,79)], se probó luego en la población ITT y también se demostró una mejora estadísticamente significativa en la SLP en la población ITT.
Con una mediana de seguimiento de supervivencia general de 19 meses, los datos de supervivencia general fueron inmaduros con un 27% de muertes en la población ITT.
Los resultados de eficacia se presentan en la Tabla 10 y la Figura 2.
| Variables de eficacia (basadas en la evaluación BICR) | INLYTA más avelumab (N=422) |
Sunitinib (N=444) |
|---|---|---|
| BICR: Revisión Central Independiente Ciega; IC: Intervalo de confianza; NE: No estimable. | ||
|
||
|
Supervivencia libre de progresión (SLP) |
||
|
Eventos (%) |
180 (41) |
216 (49) |
|
Mediana en meses (IC del 95%) |
13.8 (11.1, NE) |
8.4 (6.9, 11.1) |
|
Razón de riesgo (IC del 95%) |
0.69 (0.56, 0.84) |
|
|
Valor p bilateral* |
0.0002 |
|
|
Tasa de respuesta objetiva confirmada (TRO) |
||
|
Tasa de respuesta objetiva n (%) |
227 (51.4) |
114 (25.7) |
|
(IC del 95%) |
(46.6, 56.1) |
(21.7, 30.0) |
|
Respuesta completa (RC) n (%) |
15 (3.4) |
8 (1.8) |
|
Respuesta parcial (RP) n (%) |
212 (48) |
106 (24) |
Figura 2. Estimaciones de K-M para la SLP según la evaluación del BICR – ITT
INLYTA en combinación con Pembrolizumab
La eficacia de INLYTA en combinación con pembrolizumab se investigó en KEYNOTE-426 (NCT02853331), un ensayo aleatorizado, multicéntrico y abierto realizado en 861 pacientes que no habían recibido terapia sistémica para RCC avanzado. Los pacientes fueron reclutados independientemente del estado de expresión tumoral de PD-L1. Los pacientes con enfermedad autoinmune activa que requirieran inmunosupresión sistémica en los últimos 2 años fueron inelegibles. La aleatorización se estratificó por las categorías de riesgo del Consorcio Internacional de la Base de Datos de RCC metastásico (IMDC) (favorable versus intermedio versus pobre) y la región geográfica (América del Norte versus Europa Occidental versus “Resto del mundo”).
Los pacientes fueron aleatorizados (1:1) a uno de los siguientes brazos de tratamiento:
- •
- INLYTA 5 mg por vía oral, dos veces al día en combinación con pembrolizumab 200 mg por vía intravenosa cada 3 semanas hasta 24 meses. Los pacientes que toleraron INLYTA 5 mg dos veces al día durante 2 ciclos consecutivos (6 semanas) pudieron aumentar a 7 mg y posteriormente a 10 mg dos veces al día. INLYTA podría interrumpirse o reducirse a 3 mg dos veces al día y posteriormente a 2 mg dos veces al día para controlar la toxicidad.
- •
- Sunitinib 50 mg por vía oral, una vez al día durante 4 semanas y luego sin tratamiento durante 2 semanas.
El tratamiento con INLYTA y pembrolizumab continuó hasta la progresión de la enfermedad definida por RECIST v1.1 o una toxicidad inaceptable. La administración de INLYTA y pembrolizumab se permitió más allá de la progresión de la enfermedad definida por RECIST si el paciente era clínicamente estable y se consideraba que estaba obteniendo un beneficio clínico por parte del investigador. La evaluación del estado tumoral se realizó al inicio, después de la aleatorización en la semana 12, luego cada 6 semanas hasta la semana 54, y luego cada 12 semanas a partir de entonces.
Las características de la población del estudio fueron: edad media de 62 años (rango: 26 a 90); 38% de edad 65 o mayor; 73% hombres; 79% blancos y 16% asiáticos; 20% y 80% de los pacientes tenían una KPS basal de 70 a 80 y 90 a 100, respectivamente; y la distribución de los pacientes por categorías de riesgo IMDC fue del 31% favorable, 56% intermedio y 13% pobre.
Las principales medidas de resultado de eficacia fueron la SO y la SLP según la evaluación del BICR de acuerdo con RECIST v1.1, modificada para seguir un máximo de 10 lesiones diana y un máximo de 5 lesiones diana por órgano. Las medidas de resultado de eficacia adicionales incluyeron la TEO, según la evaluación del BICR. Se demostró una mejora estadísticamente significativa en la SO en el primer análisis interino preespecificado en pacientes aleatorizados a INLYTA en combinación con pembrolizumab en comparación con sunitinib. El ensayo también demostró mejoras estadísticamente significativas en la SLP y la TEO.
Se realizó un análisis actualizado de la SO cuando se observaron 418 muertes según el número previsto de muertes para el análisis final preespecificado. La Tabla 11 y la Figura 3 resumen los resultados de eficacia de KEYNOTE-426.
| Variable principal | INLYTA y Pembrolizumab N=432 |
Sunitinib N=429 |
||
|---|---|---|---|---|
| IC: intervalo de confianza; NR: no alcanzado; TEO: tasa de respuesta objetiva; SO: supervivencia general; SLP: supervivencia libre de progresión. | ||||
|
||||
|
SO |
||||
|
Número de pacientes con evento (%) |
59 (14%) |
97 (23%) |
||
|
Mediana en meses (IC del 95%) |
NR (NR, NR) |
NR (NR, NR) |
||
|
Razón de riesgo* (IC del 95%) |
0,53 (0,38, 0,74) |
|||
|
Valor p † |
<0,0001 ‡ |
|||
|
Tasa de SO a 12 meses |
90% (86, 92) |
78% (74, 82) |
||
|
Supervivencia global actualizada |
||
|
Número de pacientes con evento (%) |
193 (45%) |
225 (52%) |
|
Mediana en meses (IC del 95%) |
45.7 (43.6, NR) |
40.1 (34.3, 44.2) |
|
Hazard ratio* (IC del 95%) |
0.73 (0.60, 0.88) |
|
|
SPT |
||
|
Número de pacientes con evento (%) |
183 (42%) |
213 (50%) |
|
Mediana en meses (IC del 95%) |
15.1 (12.6, 17.7) |
11.0 (8.7, 12.5) |
|
Hazard ratio* (IC del 95%) |
0.69 (0.56, 0.84) |
|
|
p-Value † |
0.0001§ |
|
|
TGR |
||
|
Tasa de respuesta global confirmada (IC del 95%) |
59% (54, 64) |
36% (31, 40) |
|
Tasa de respuesta completa |
6% |
2% |
|
Tasa de respuesta parcial |
53% |
34% |
|
p-Value¶ |
<0.0001 |
|
Figura 3. Curva de Kaplan-Meier para la supervivencia general en KEYNOTE-426
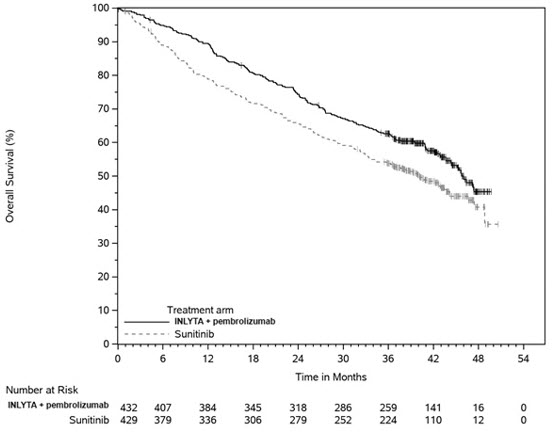
En un análisis exploratorio, el análisis actualizado de la SG en pacientes con riesgo favorable, intermedio, intermedio/pobre y pobre del IMDC demostró una HR de 1,17 (IC del 95%: 0,76, 1,80), 0,67 (IC del 95%: 0,52, 0,86), 0,64 (IC del 95%: 0,52, 0,80) y 0,51 (IC del 95%: 0,32, 0,81), respectivamente.
14.2 RCC avanzado de segunda línea
La seguridad y la eficacia de INLYTA se evaluaron en un estudio multicéntrico de fase 3, aleatorizado y abierto. Los pacientes (N=723) con RCC avanzado cuya enfermedad había progresado durante o después del tratamiento con 1 terapia sistémica previa, incluidos regímenes que contenían sunitinib, bevacizumab, temsirolimus o citocinas, se asignaron aleatoriamente (1:1) para recibir INLYTA (N=361) o sorafenib (N=362). La supervivencia libre de progresión (SLP) se evaluó mediante un comité de revisión central independiente y ciego. Otros criterios de valoración incluyeron la tasa de respuesta objetiva (TRO) y la supervivencia general (SG).
De los pacientes inscritos en este estudio, 389 pacientes (54%) habían recibido 1 terapia previa basada en sunitinib, 251 pacientes (35%) habían recibido 1 terapia previa basada en citocinas (interleucina-2 o interferón-alfa), 59 pacientes (8%) habían recibido 1 terapia previa basada en bevacizumab y 24 pacientes (3%) habían recibido 1 terapia previa basada en temsirolimus. Las características demográficas y de la enfermedad iniciales fueron similares entre los grupos de INLYTA y sorafenib con respecto a la edad (mediana de 61 años), el sexo (72% hombres), la raza (75% blancos, 21% asiáticos), el estado de rendimiento del Eastern Cooperative Oncology Group (ECOG) (55% 0, 45% 1) y la histología (99% de células claras).
Hubo una ventaja estadísticamente significativa para INLYTA sobre sorafenib para el criterio de valoración de SLP (véanse la Tabla 12 y la Figura 4). No hubo diferencias estadísticamente significativas entre los brazos en la SG.
| Criterio de valoración/Población del estudio | INLYTA | Sorafenib | HR (IC del 95%) | Valor p |
|---|---|---|---|---|
| IC: Intervalo de confianza; HR: Razón de riesgos (INLYTA/sorafenib); ITT: Intención de tratar; TRO: Tasa de respuesta objetiva; NS: No significativo; SG: Supervivencia general; SLP: Supervivencia libre de progresión | ||||
|
||||
|
ITT general |
N= 361 |
N = 362 |
||
|
6,7 (6,3, 8,6) |
4,7 (4,6, 5,6) |
0,67 (0,54, 0,81) |
<0,0001‡ |
|
|
SG mediana en meses |
20,1 (16,7, 23,4) |
19,2 (17,5, 22,3) |
0,97 (0,80, 1,17) |
NS |
|
TRO % (IC del 95%) |
19,4 (15,4, 23,9) |
9,4 (6,6, 12,9) |
2,06§ (1,41, 3,00) |
–¶ |
|
SLP por tratamiento previo |
||||
|
Subgrupo refractario a sunitinib |
N=194 |
N=195 |
||
|
Mediana, meses (IC del 95%) |
4.8 (4.5, 6.4) |
3.4 (2.8, 4.7) |
0.74 (0.57, 0.96) |
–¶ |
|
Subgrupo refractario a citocinas |
N=126 |
N=125 |
||
|
Mediana, meses (IC del 95%) |
12.1 (10.1, 13.9) |
6.5 (6.3, 8.3) |
0.46 (0.32, 0.68) |
–¶ |
Figura 4. Curva de Kaplan-Meier para la supervivencia libre de progresión según la evaluación independiente (población por intención de tratar)
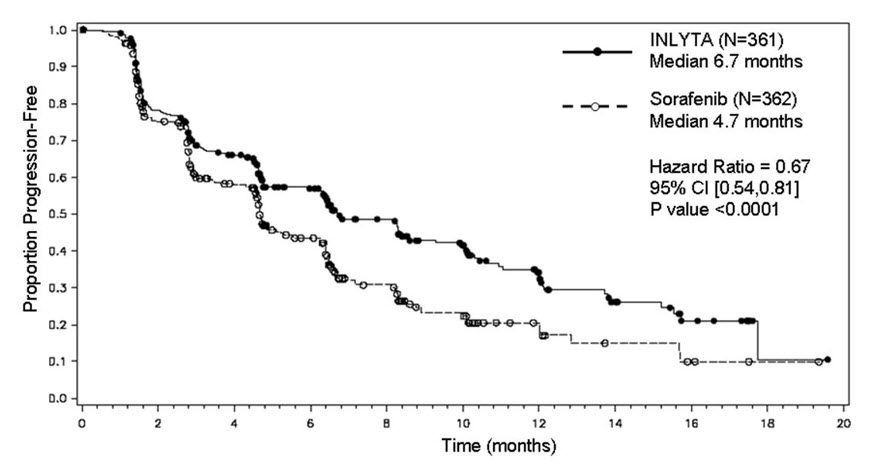
16 SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
Las tabletas de INLYTA se suministran de la siguiente manera:
- •
- Las tabletas de 1 mg son tabletas ovaladas recubiertas con película roja, con la inscripción “Pfizer” en una cara y “1 XNB” en la otra; disponibles en frascos de 180: NDC 0069-0145-01.
- •
- Las tabletas de 5 mg son tabletas triangulares recubiertas con película roja, con la inscripción “Pfizer” en una cara y “5 XNB” en la otra; disponibles en frascos de 60: NDC 0069-0151-11.
- •
- Almacenar a 20°C a 25°C (68°F a 77°F); se permiten excursiones de 15°C a 30°C (59°F a 86°F) [ver USP Temperatura Ambiente Controlada].
17 INFORMACIÓN DE ASESORAMIENTO AL PACIENTE
Aconseje al paciente que lea el etiquetado del paciente aprobado por la FDA (Información para el paciente).
Hipertensión
Avise a los pacientes que puede desarrollarse hipertensión durante el tratamiento con INLYTA y que la presión arterial debe controlarse regularmente durante el tratamiento [ver Advertencias y precauciones (5.1)].
Eventos tromboembólicos arteriales/venosos
Avise a los pacientes que se han observado eventos tromboembólicos arteriales y venosos durante el tratamiento con INLYTA y que informen a su médico si experimentan síntomas que sugieran eventos tromboembólicos [ver Advertencias y precauciones (5.2, 5.3)].
Hemorragia
Avise a los pacientes que INLYTA puede aumentar el riesgo de sangrado y que informen a su médico de inmediato sobre cualquier episodio de sangrado [ver Advertencias y precauciones (5.4)].
Insuficiencia cardíaca
Avise a los pacientes que puede desarrollarse insuficiencia cardíaca durante el tratamiento con INLYTA y que los signos o síntomas de insuficiencia cardíaca deben controlarse regularmente durante el tratamiento [ver Advertencias y precauciones (5.5)].
Trastornos gastrointestinales
Avise a los pacientes que pueden desarrollarse trastornos gastrointestinales como diarrea, náuseas, vómitos y estreñimiento durante el tratamiento con INLYTA y que busquen atención médica inmediata si experimentan dolor abdominal persistente o intenso, ya que se han notificado casos de perforación y fístula gastrointestinal en pacientes que toman INLYTA [ver Advertencias y precauciones (5.6) y Reacciones adversas (6.1)].
Función tiroidea anormal
Avise a los pacientes que puede desarrollarse una función tiroidea anormal durante el tratamiento con INLYTA y que informen a su médico si se presentan síntomas de función tiroidea anormal [ver Advertencias y precauciones (5.7)].
Cicatrización deficiente de heridas
Avise a los pacientes que INLYTA puede afectar la cicatrización de heridas. Avise a los pacientes que informen a su proveedor de atención médica sobre cualquier procedimiento quirúrgico planificado [ver Advertencias y precauciones (5.8)].
Síndrome de leucoencefalopatía posterior reversible
Avise a los pacientes que informen a su médico si tienen un empeoramiento de la función neurológica compatible con RPLS (dolor de cabeza, convulsiones, letargo, confusión, ceguera y otros trastornos visuales y neurológicos) [ver Advertencias y precauciones (5.9)].
Hepatotoxicidad
Informe a los pacientes sobre los signos y síntomas de hepatotoxicidad. Avise a los pacientes que se comuniquen con su proveedor de atención médica de inmediato si presentan signos o síntomas de hepatotoxicidad [ver Advertencias y precauciones (5.11)].
Eventos cardiovasculares adversos importantes
Avise a los pacientes que reciben INLYTA en combinación con avelumab que se comuniquen con su proveedor de atención médica de inmediato si presentan signos o síntomas de eventos cardiovasculares, que incluyen, entre otros, malestar torácico nuevo o que empeora, disnea o edema periférico [ver Advertencias y precauciones (5.13)].
Toxicidad embriofetal
Avise a las mujeres que informen a su proveedor de atención médica si están embarazadas o quedan embarazadas. Informe a las pacientes mujeres sobre el riesgo para el feto y la posible pérdida del embarazo [ver Uso en poblaciones específicas (8.1)].
Avise a las mujeres en edad fértil que usen métodos anticonceptivos eficaces durante el tratamiento con INLYTA y durante 1 semana después de la última dosis.
Avise a los pacientes varones con parejas femeninas en edad fértil que usen métodos anticonceptivos eficaces durante el tratamiento y durante 1 semana después de la última dosis [ver Advertencias y precauciones (5.14) y Uso en poblaciones específicas (8.3)].
Cuando INLYTA se usa en combinación con avelumab o pembrolizumab, consulte la información completa de prescripción de avelumab o pembrolizumab para obtener información sobre el embarazo y la anticoncepción.
Lactancia
Avise a los pacientes que no deben amamantar mientras toman INLYTA y durante 2 semanas después de recibir la última dosis [ver Uso en poblaciones específicas (8.2)].
Cuando INLYTA se usa en combinación con avelumab o pembrolizumab, consulte la información completa de prescripción de avelumab o pembrolizumab para obtener información sobre la lactancia.
Infertilidad
Avise a los hombres y mujeres en edad fértil que INLYTA puede afectar la fertilidad [ver Uso en poblaciones específicas (8.3)].
Medicamentos concomitantes
Avise a los pacientes que informen a su médico sobre todos los medicamentos concomitantes, vitaminas o suplementos dietéticos y herbales.
SECCIÓN NO CLASIFICADA SPL
El etiquetado de este producto puede haberse actualizado. Para obtener la información de prescripción más reciente, visite www.pfizer.com.

LAB-0561-8.0
PROSPECTO PARA EL PACIENTE
|
INFORMACIÓN PARA EL PACIENTE |
|||
|
Información importante: Si su proveedor de atención médica le receta INLYTA para que lo tome con avelumab o pembrolizumab, lea también la Guía de medicamentos para avelumab o pembrolizumab. |
|||
|
¿Qué es INLYTA?
No se sabe si INLYTA es seguro y eficaz en niños. |
|||
|
Antes de tomar INLYTA, informe a su proveedor de atención médica sobre todas sus afecciones médicas, incluso si:
Para las mujeres, informe a su proveedor de atención médica si:
Para los hombres con parejas femeninas que pueden quedar embarazadas:
Informe a su proveedor de atención médica sobre todos los medicamentos que toma, incluidos los medicamentos recetados y de venta libre, las vitaminas y los suplementos herbales. INLYTA y ciertos otros medicamentos pueden afectarse mutuamente causando efectos secundarios graves. |
|||
|
¿Cómo debo tomar INLYTA?
|
|||
|
¿Qué debo evitar mientras tomo INLYTA?
|
|||
|
¿Cuáles son los posibles efectos secundarios de INLYTA?
|
|||
|
|
||
|
|||
- o
- hemorragia inesperada o hemorragia que dura mucho tiempo, como:
- •
- sangrado inusual de las encías
- •
- sangrado menstrual o vaginal más abundante de lo normal
- •
- sangrado intenso o que no puede controlar
- •
- orina rosada o marrón
- •
- heces rojas o negras (parecidas al alquitrán)
- •
- moretones que aparecen sin causa conocida o que aumentan de tamaño
- •
- tos con sangre o coágulos de sangre
- •
- vómitos con sangre o vómitos con aspecto de “posos de café”
- o
- dolor, hinchazón o dolor en las articulaciones inesperados
- o
- dolores de cabeza, mareos o debilidad
- •
- Insuficiencia cardíaca. Su proveedor de atención médica debe revisarlo para detectar signos o síntomas de insuficiencia cardíaca regularmente durante el tratamiento con INLYTA. La insuficiencia cardíaca puede ser grave y, a veces, puede provocar la muerte. Informe a su proveedor de atención médica si presenta alguno de los siguientes síntomas durante su tratamiento con INLYTA:
- o
- cansancio
- o
- hinchazón del área del estómago (abdomen), piernas o tobillos
- o
- falta de aliento
- o
- venas del cuello prominentes
- •
-
Rotura en el estómago o la pared intestinal (perforación). Una rotura en el estómago o la pared intestinal puede ser grave y, a veces, puede provocar la muerte. Busque ayuda médica de inmediato si presenta los siguientes síntomas:
- o
- dolor intenso en el área del estómago (abdominal) o dolor en el área del estómago que no desaparece
- o
- vómitos con sangre
- o
- heces rojas o negras
- •
- Problemas de la glándula tiroides. Su proveedor de atención médica debe realizar análisis de sangre para controlar la función de su glándula tiroides antes y durante su tratamiento con INLYTA. Informe a su proveedor de atención médica si presenta alguno de los siguientes síntomas durante su tratamiento con INLYTA:
- o
- cansancio que empeora o que no desaparece
- o
- sensación de calor o frío
- o
- su voz se profundiza
- o
- aumento o pérdida de peso
- o
- pérdida de cabello
- o
- calambres y dolores musculares
- •
-
Riesgo de problemas de cicatrización de heridas. Las heridas pueden no cicatrizar correctamente durante el tratamiento con INLYTA. Informe a su proveedor de atención médica si planea someterse a alguna cirugía antes de comenzar o durante el tratamiento con INLYTA.
- o
- Debe dejar de tomar INLYTA al menos 2 días antes de la cirugía programada.
- o
- Su proveedor de atención médica debe indicarle cuándo puede volver a tomar INLYTA después de la cirugía.
- •
- Síndrome de leucoencefalopatía posterior reversible (RPLS). Puede producirse una afección llamada síndrome de leucoencefalopatía posterior reversible (RPLS) durante el tratamiento con INLYTA. Llame a su proveedor de atención médica de inmediato si presenta:
- o
- dolor de cabeza
- o
- convulsiones
- o
- debilidad
- o
- confusión
- o
- presión arterial alta
- o
- ceguera o cambio en la visión
- o
- problemas para pensar
- •
- Proteína en la orina. Su proveedor de atención médica debe controlar la proteína en su orina antes y durante su tratamiento con INLYTA. Si desarrolla proteína en la orina, su proveedor de atención médica puede disminuir su dosis de INLYTA o suspender su tratamiento.
- •
-
Problemas hepáticos. Su proveedor de atención médica le realizará análisis de sangre antes y durante su tratamiento con INLYTA. Su proveedor de atención médica puede retrasar o suspender su tratamiento con INLYTA si desarrolla problemas hepáticos graves.
Informe a su proveedor de atención médica de inmediato si presenta alguno de los siguientes síntomas:
- o
- coloración amarillenta de la piel o el blanco de los ojos
- o
- náuseas o vómitos intensos
- o
- dolor en el lado derecho del área del estómago (abdomen)
- o
- orina oscura (color té)
- o
- sangrado o formación de moretones con más facilidad de lo normal
- •
- Problemas cardíacos. Cuando se usa INLYTA con el medicamento avelumab, pueden producirse problemas cardíacos graves que pueden provocar la muerte. Su proveedor de atención médica le controlará los problemas cardíacos durante su tratamiento con INLYTA. Informe a su proveedor de atención médica de inmediato u obtenga ayuda médica si presenta alguno de los siguientes síntomas:
- o
- hinchazón del área del estómago, piernas, manos, pies o tobillos
- o
- falta de aliento
- o
- náuseas o vómitos
- o
- dolor o molestia torácica nueva o que empeora, incluido dolor o presión
- o
- aumento de peso
- o
- dolor o molestia en los brazos, la espalda, el cuello o la mandíbula
- o
- sudor frío repentino
- o
- sensación de mareo o aturdimiento
Los efectos secundarios más comunes de INLYTA con avelumab incluyen:
|
|
||
|
Los efectos secundarios más comunes de INLYTA con pembrolizumab incluyen: |
|||
|
|
||
|
Los efectos secundarios más comunes de INLYTA cuando se usa solo incluyen: |
|||
|
|
||
|
INLYTA puede causar problemas de fertilidad en hombres y mujeres, lo que puede afectar su capacidad para tener un hijo. Hable con su proveedor de atención médica si esto le preocupa. |
|||
|
¿Cómo debo guardar INLYTA? Guarde INLYTA a temperatura ambiente entre 68°F y 77°F (20°C y 25°C). Mantenga INLYTA y todos los medicamentos fuera del alcance de los niños. |
|||
|
Información general sobre el uso seguro y eficaz de INLYTA. |
|||
|
¿Cuáles son los ingredientes de INLYTA? |
|||
 |
LAB-0439-8.0 |
||
|
Para obtener más información, visite www.inlyta.com o llame al 877-0744-5675 |
|||
Esta información para el paciente ha sido aprobada por la Administración de Alimentos y Medicamentos de los EE. UU. Revisado: 07/2024
PANEL DE VISUALIZACIÓN PRINCIPAL – Etiqueta de la botella de tabletas de 1 mg
Pfizer
NDC 0069-0145-01
Inlyta®
(axitinib) tablets
1 mg
180 Tablets
Rx only

PANEL PRINCIPAL DE EXHIBICIÓN – Etiqueta de la botella de tabletas de 5 mg
Pfizer
NDC 0069-0151-11
Inlyta®
(axitinib) tabletas
5 mg
60 Tabletas
Rx only