
Fabricante de medicamentos: Amneal Pharmaceuticals LLC (Updated: 2023-11-25)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
Initial U.S. Approval: 2001
INDICACIONES Y USO
Las tabletas de mesilato de imatinib son un inhibidor de la cinasa indicado para el tratamiento de:
- Pacientes adultos y pediátricos recién diagnosticados con leucemia mieloide crónica con cromosoma Filadelfia positivo (LMC Ph+) en fase crónica. (1.1)
- Pacientes con leucemia mieloide crónica con cromosoma Filadelfia positivo (LMC Ph+) en crisis blástica (CB), fase acelerada (FA) o en fase crónica (FC) después del fracaso del tratamiento con interferón alfa. (1.2)
- Pacientes adultos con leucemia linfoblástica aguda con cromosoma Filadelfia positivo (LLA Ph+) recidivante o refractaria. (1.3)
- Pacientes pediátricos con leucemia linfoblástica aguda con cromosoma Filadelfia positivo (LLA Ph+) recién diagnosticada en combinación con quimioterapia. (1.4)
- Pacientes adultos con síndromes mielodisplásicos/mieloproliferativos (SMD/SMP) asociados con reordenamientos del gen del receptor del factor de crecimiento derivado de plaquetas (PDGFR). (1.5)
- Pacientes adultos con mastocitosis sistémica agresiva (MSA) sin la mutación D816V c-Kit o con estado mutacional c-Kit desconocido. (1.6)
- Pacientes adultos con síndrome hipereosinofílico (SHE) y/o leucemia eosinofílica crónica (LEC) que tienen la cinasa de fusión FIP1L1-PDGFRα (análisis mutacional o demostración de deleción del alelo CHIC2 por hibridación in situ fluorescente [FISH]) y para pacientes con SHE y/o LEC que son negativos o desconocidos para la cinasa de fusión FIP1L1-PDGFRα. (1.7)
- Pacientes adultos con dermatofibrosarcoma protuberans (DFSP) irresecable, recurrente y/o metastásico. (1.8)
- Pacientes con tumores del estroma gastrointestinal (GIST) malignos irresecables y/o metastásicos positivos para Kit (CD117). (1.9)
- Tratamiento adyuvante de pacientes adultos después de la resección de GIST positivo para Kit (CD117). (1.10)
DOSIFICACIÓN Y ADMINISTRACIÓN
- Adultos con LMC Ph+ FC (2.2): 400 mg/día
- Adultos con LMC Ph+ FA o CB (2.2): 600 mg/día
- Pediatría con LMC Ph+ FC (2.3): 340 mg/m2/día
- Adultos con LLA Ph+ (2.4): 600 mg/día
- Pediatría con LLA Ph+ (2.5): 340 mg/m2/día
- Adultos con SMD/SMP (2.6): 400 mg/día
- Adultos con MSA (2.7): 100 mg/día o 400 mg/día
- Adultos con SHE/LEC (2.8): 100 mg/día o 400 mg/día
- Adultos con DFSP (2.9): 800 mg/día
- Adultos con GIST metastásico y/o irresecable (2.10): 400 mg/día
- Tratamiento adyuvante de adultos con GIST (2.11): 400 mg/día
- Pacientes con insuficiencia hepática leve a moderada (2.12): 400 mg/día
- Pacientes con insuficiencia hepática grave (2.12): 300 mg/día
Todas las dosis de las tabletas de mesilato de imatinib deben tomarse con una comida y un vaso grande de agua. Las dosis de 400 mg o 600 mg deben administrarse una vez al día, mientras que una dosis de 800 mg debe administrarse como 400 mg dos veces al día. Las tabletas de mesilato de imatinib pueden disolverse en agua o zumo de manzana para los pacientes que tengan dificultad para tragar. La dosificación diaria de 800 mg o más debe lograrse utilizando la tableta de 400 mg para reducir la exposición al hierro.
FORMAS FARMACÉUTICAS Y CONCENTRACIONES
Tabletas (ranuradas): 100 mg y 400 mg (3)
CONTRAINDICACIONES
Ninguna. (4)
ADVERTENCIAS Y PRECAUCIONES
- Se ha producido edema y retención grave de líquidos. Pese a los pacientes con regularidad y controle el aumento de peso rápido e inesperado interrumpiendo el medicamento y administrando diuréticos. (5.1, 6.1)
- Se han producido citopenias, en particular anemia, neutropenia y trombocitopenia. Controlar con reducción de la dosis, interrupción de la dosis o suspensión del tratamiento. Realice recuentos sanguíneos completos semanalmente durante el primer mes, dos veces por semana durante el segundo mes y periódicamente a partir de entonces. (5.2)
- Se han notificado casos de insuficiencia cardíaca congestiva grave y disfunción ventricular izquierda, especialmente en pacientes con comorbilidades y factores de riesgo. Controle y trate a los pacientes con enfermedad cardíaca o factores de riesgo de insuficiencia cardíaca. (5.3)
- Puede producirse hepatotoxicidad grave, incluso mortal. Evalúe la función hepática antes de iniciar el tratamiento y mensualmente a partir de entonces o según esté clínicamente indicado. Controle la función hepática cuando se combine con quimioterapia que se sabe que está asociada con la disfunción hepática. (5.4)
- Se han notificado hemorragias de grado 3/4 en estudios clínicos en pacientes con LMC recién diagnosticada y con GIST. Las localizaciones tumorales gastrointestinales pueden ser la fuente de hemorragias gastrointestinales en el GIST. (5.5)
- Se han notificado perforaciones gastrointestinales (GI), algunas mortales. (5.6)
- El shock cardiogénico/disfunción ventricular izquierda se ha asociado con el inicio del mesilato de imatinib en pacientes con enfermedades asociadas a niveles elevados de eosinófilos (p. ej., HES, SMD/SPM y ASM). (5.7)
- Se han notificado reacciones dermatológicas ampollosas (p. ej., eritema multiforme y síndrome de Stevens-Johnson) con el uso de mesilato de imatinib. (5.8)
- Se ha notificado hipotiroidismo en pacientes tiroidectomizados en tratamiento de sustitución con levotiroxina. Controle estrechamente los niveles de TSH en estos pacientes. (5.9)
- Se pueden producir daños fetales cuando se administra a una mujer embarazada. Informe a las mujeres sobre los posibles daños para el feto y sobre la necesidad de utilizar métodos anticonceptivos eficaces. (5.10, 8.1)
- Se ha notificado retraso del crecimiento en niños y preadolescentes que reciben mesilato de imatinib. Se recomienda un estrecho seguimiento del crecimiento en niños en tratamiento con mesilato de imatinib. (5.11, 6.2)
- Síndrome de lisis tumoral. Se recomienda una estrecha vigilancia. (5.12)
- Se han recibido informes de accidentes de tráfico en pacientes que recibían mesilato de imatinib. Advierta a los pacientes que tengan cuidado al conducir un coche o manejar maquinaria. (5.13)
- Toxicidad renal. Puede producirse una disminución de la función renal en pacientes que reciben mesilato de imatinib. Evalúe la función renal al inicio y durante el tratamiento, prestando atención a los factores de riesgo de disfunción renal. (5.14)
REACCIONES ADVERSAS
Las reacciones adversas notificadas con mayor frecuencia (superiores o iguales al 30%) son edema, náuseas, vómitos, calambres musculares, dolor musculoesquelético, diarrea, erupción cutánea, fatiga y dolor abdominal. (6.1)
Para notificar REACCIONES ADVERSAS SOSPECHOSAS, póngase en contacto con Amneal Pharmaceuticals en el teléfono 1-877-835-5472 o con la FDA en el teléfono 1-800-FDA-1088 o en www.fda.gov/medwatch.
INTERACCIONES FARMACOLÓGICAS
- Los inductores del CYP3A4 pueden disminuir la Cmáx. y el área bajo la curva (AUC) del mesilato de imatinib. (2.12, 7.1, 12.3)
- Los inhibidores del CYP3A4 pueden aumentar la Cmáx. y el AUC del mesilato de imatinib. (7.2, 12.3)
- El mesilato de imatinib es un inhibidor del CYP3A4 y el CYP2D6 que puede aumentar la Cmáx. y el AUC de otros fármacos. (7.3, 7.4, 12.3)
- Los pacientes que requieran anticoagulación deben recibir heparina de bajo peso molecular o estándar y no warfarina. (7.3)
Consulte la sección 17 para obtener INFORMACIÓN DE ASESORAMIENTO AL PACIENTE.
Revisado: 11/2023
Tabla de Contenido
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDO*
1 INDICACIONES Y USO
1.1 Leucemia mieloide crónica de novo positiva para Filadelfia (Ph+ LMC)
1.2 Ph+ LMC en crisis blástica (CB), fase acelerada (FA) o fase crónica (FC) después de la terapia con interferón-alfa (IFN)
1.3 Pacientes adultos con leucemia linfoblástica aguda positiva para Filadelfia (Ph+ LLA)
1.4 Pacientes pediátricos con
leucemia linfoblástica aguda positiva para Filadelfia (Ph+ LLA)
1.5 Enfermedades mielodisplásicas/mieloproliferativas (EMD/EMP)
1.6 Mastocitosis sistémica agresiva (MSA)
1.7 Síndrome hipereosinofílico (SHE) y/o leucemia eosinofílica crónica (LEC)
1.8 Dermatofibrosarcoma protuberans (DFSP)
1.9 Tumores estromales gastrointestinales (TSG) Kit+
1.10 Tratamiento adyuvante
de TSG
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Administración del medicamento
2.2 Pacientes adultos con Ph+ LMC FC, FA, o CB
2.3 Pacientes pediátricos
con Ph+ LMC FC
2.4 Pacientes adultos con Ph+ LLA
2.5 Pacientes pediátricos con
Ph+ LLA
2.6 Pacientes adultos con EMD/EMP
2.7 Pacientes adultos con MSA
2.8 Pacientes adultos con SHE/LEC
2.9 Pacientes adultos con DFSP
2.10 Pacientes adultos con TSG metastásico y/o irresecable
2.11 Pacientes adultos con TSG adyuvante
2.12 Pautas de modificación de la dosis
2.13 Ajuste de la dosis para hepatotoxicidad y reacciones adversas no hematológicas
2.14 Ajuste de la dosis para reacciones adversas hematológicas
3 FORMAS FARMACÉUTICAS Y FUERZAS
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Retención de líquidos y edema
5.2 Toxicidad hematológica
5.3 Insuficiencia cardíaca congestiva y disfunción ventricular izquierda
5.4 Hepatotoxicidad
5.5 Hemorragia
5.6 Trastornos gastrointestinales
5.7 Toxicidad cardíaca hipereosinofílica
5.8 Toxicidades dermatológicas
5.9 Hipotiroidismo
5.10 Toxicidad embrio-fetal
5.11 Retraso del crecimiento en niños y adolescentes
5.12 Síndrome de lisis tumoral
5.13 Deterioros relacionados con la conducción y el uso de maquinaria
5.14 Toxicidad renal
6 REACCIONES ADVERSAS
6.1 Experiencia en ensayos clínicos
6.2 Experiencia postcomercialización
7 INTERACCIONES MEDICAMENTOSAS
7.1 Agentes que inducen el metabolismo del CYP3A
7.2 Agentes que inhiben el metabolismo del CYP3A
7.3 Interacciones con medicamentos metabolizados por CYP3A4
7.4 Interacciones con medicamentos metabolizados por CYP2D6
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.3 Mujeres y hombres en edad fértil
8.4 Uso pediátrico
8.5 Uso geriátrico
8.6 Insuficiencia hepática
8.7 Insuficiencia renal
10 SOBREDOSIS
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
12.3 Farmacocinética
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, mutagénesis, deterioro de la fertilidad
13.2 Toxicología y/o farmacología animal
14 ESTUDIOS CLÍNICOS
14.1 Leucemia mieloide crónica
14.2 LMC pediátrica
14.3 Leucemia linfoblástica aguda
14.4 LLA pediátrica
14.5 Enfermedades mielodisplásicas/mieloproliferativas
14.6 Mastocitosis sistémica agresiva
14.7 Síndrome hipereosinofílico/leucemia eosinofílica crónica
14.8 Dermatofibrosarcoma protuberans
14.9 Tumores estromales gastrointestinales
15 REFERENCIAS
16 FORMA DE SUMINISTRO/ALMACENAMIENTO Y MANEJO
17 INFORMACIÓN PARA EL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información completa de prescripción no están enumeradas.
1 INDICACIONES Y USO
1.1 Leucemia mieloide crónica de novo positiva para el cromosoma Filadelfia (Ph+ LMC)
Pacientes adultos y pediátricos de nuevo diagnóstico con leucemia mieloide crónica positiva para el cromosoma Filadelfia (Ph+ LMC) en fase crónica.
1.2 Ph+ LMC en crisis blástica (CB), fase acelerada (FA) o fase crónica (FC) después de la terapia con interferón-alfa (IFN)
Pacientes con leucemia mieloide crónica positiva para el cromosoma Filadelfia en crisis blástica, fase acelerada o en fase crónica después del fracaso de la terapia con interferón-alfa.
1.3 Pacientes adultos con Ph+ leucemia linfoblástica aguda (LLA)
Pacientes adultos con leucemia linfoblástica aguda positiva para el cromosoma Filadelfia (Ph+ LLA) recidivante o refractaria.
1.4 Pacientes pediátricos con
Ph+ leucemia linfoblástica aguda (LLA)
Pacientes pediátricos con leucemia linfoblástica aguda positiva para el cromosoma Filadelfia (Ph+ LLA) de nuevo diagnóstico en combinación con quimioterapia.
1.5 Enfermedades mielodisplásicas/mieloproliferativas (EMD/EMP)
Pacientes adultos con enfermedades mielodisplásicas/mieloproliferativas asociadas con reordenamientos del gen del receptor del factor de crecimiento derivado de plaquetas (PDGFR).
1.6 Mastocitosis sistémica agresiva (MSA)
Pacientes adultos con mastocitosis sistémica agresiva sin la mutación D816V c-Kit o con estado mutacional c-Kit desconocido.
1.7 Síndrome hipereosinofílico (SHE) y/o leucemia eosinofílica crónica (LEC)
Pacientes adultos con síndrome hipereosinofílico y/o leucemia eosinofílica crónica que tienen la quinasa de fusión FIP1L1-PDGFRα (análisis mutacional o demostración de la deleción del alelo CHIC2 mediante hibridación in situ fluorescente [FISH]) y para pacientes con SHE y/o LEC que son negativos o desconocidos para la quinasa de fusión FIP1L1-PDGFRα.
1.8 Dermatofibrosarcoma protuberans (DFSP)
Pacientes adultos con dermatofibrosarcoma protuberans irresecable, recurrente y/o metastásico.
1.9 Tumores estromales gastrointestinales (TEG) Kit+
Pacientes con tumores estromales gastrointestinales malignos irresecables y/o metastásicos positivos para Kit (CD117).
1.10 Tratamiento adyuvante
de TEG
Tratamiento adyuvante de pacientes adultos después de la resección completa macroscópica de TEG positivos para Kit (CD117).
2 DOSIS Y ADMINISTRACIÓN
2.1 Administración del fármaco
La dosis prescrita debe administrarse por vía oral, con una comida y un vaso grande de agua. Las dosis de 400 mg o 600 mg deben administrarse una vez al día, mientras que una dosis de 800 mg debe administrarse como 400 mg dos veces al día.
Para pacientes que no puedan tragar las tabletas recubiertas con película, las tabletas pueden dispersarse en un vaso de agua o jugo de manzana. El número requerido de tabletas debe colocarse en el volumen apropiado de bebida (aproximadamente 50 mL para una tableta de 100 mg y 200 mL para una tableta de 400 mg) y mezclarse con una cuchara. La suspensión debe administrarse inmediatamente después de la desintegración completa de la(s) tableta(s).
Para la dosificación diaria de 800 mg y más, la dosificación debe realizarse utilizando la tableta de 400 mg para reducir la exposición al hierro.
El tratamiento puede continuar siempre que no haya evidencia de enfermedad progresiva o toxicidad inaceptable.
2.2 Pacientes adultos con LMC CP, AP o BC Ph +
La dosis recomendada de tabletas de imatinib mesilato es de 400 mg/día para pacientes adultos en fase crónica de LMC y de 600 mg/día para pacientes adultos en fase acelerada o crisis blástica.
En LMC, un aumento de la dosis de 400 mg a 600 mg en pacientes adultos con enfermedad en fase crónica, o de 600 mg a 800 mg (dado como 400 mg dos veces al día) en pacientes adultos en fase acelerada o crisis blástica puede considerarse en ausencia de reacción adversa grave al fármaco y neutropenia o trombocitopenia grave no relacionada con la leucemia en las siguientes circunstancias: progresión de la enfermedad (en cualquier momento), falta de lograr una respuesta hematológica satisfactoria después de al menos 3 meses de tratamiento, falta de lograr una respuesta citogenética después de 6 a 12 meses de tratamiento, o pérdida de una respuesta hematológica o citogenética previamente lograda.
2.3 Pacientes pediátricos con LMC CP Ph +
La dosis recomendada de tabletas de imatinib mesilato para niños con LMC Ph + recién diagnosticado es de 340 mg/m2/día (no debe exceder 600 mg). El tratamiento con tabletas de imatinib mesilato puede administrarse como una dosis diaria o la dosis diaria puede dividirse en dos – una porción dosificada por la mañana y una porción por la noche. No hay experiencia con el tratamiento con tabletas de imatinib mesilato en niños menores de 1 año de edad.
2.4 Pacientes adultos con ALL Ph +
La dosis recomendada de tabletas de imatinib mesilato es de 600 mg/día para pacientes adultos con ALL Ph + recidivante/refractario.
2.5 Pacientes pediátricos con ALL Ph +
La dosis recomendada de tabletas de imatinib mesilato que se debe administrar en combinación con quimioterapia a niños con ALL Ph + recién diagnosticado es de 340 mg/m2/día (no debe exceder 600 mg). El tratamiento con tabletas de imatinib mesilato puede administrarse como una dosis diaria.
2.6 Pacientes adultos con MDS/MPD
Determinar el estado de las reordenaciones del gen PDGFRb antes de iniciar el tratamiento.
La dosis recomendada de tabletas de imatinib mesilato es de 400 mg/día para pacientes adultos con MDS/MPD.
2.7 Pacientes adultos con ASM
Determinar el estado de la mutación D816V c-Kit antes de iniciar el tratamiento.
La dosis recomendada de tabletas de imatinib mesilato es de 400 mg/día para pacientes adultos con ASM sin la mutación D816V c-Kit. Si el estado mutacional de c-Kit no se conoce o no está disponible, el tratamiento con tabletas de imatinib mesilato 400 mg/día puede considerarse para pacientes con ASM que no responden satisfactoriamente a otras terapias. Para pacientes con ASM asociada con eosinofilia, una enfermedad hematológica clonal relacionada con la fusión quinasa FIP1L1-PDGFRα, se recomienda una dosis inicial de 100 mg/día. Un aumento de la dosis de 100 mg a 400 mg para estos pacientes puede considerarse en ausencia de reacciones adversas al fármaco si las evaluaciones demuestran una respuesta insuficiente al tratamiento.
2.8 Pacientes adultos con HES/CEL
La dosis recomendada de tabletas de imatinib mesilato es de 400 mg/día para pacientes adultos con HES/CEL. Para pacientes con HES/CEL que demuestren la fusión quinasa FIP1L1-PDGFRα, se recomienda una dosis inicial de 100 mg/día. Un aumento de la dosis de 100 mg a 400 mg para estos pacientes puede considerarse en ausencia de reacciones adversas al fármaco si las evaluaciones demuestran una respuesta insuficiente al tratamiento.
2.9 Pacientes adultos con DFSP
La dosis recomendada de tabletas de imatinib mesilato es de 800 mg/día para pacientes adultos con DFSP.
2.10 Pacientes adultos con GIST metastásico y/o irresecable
La dosis recomendada de tabletas de imatinib mesilato es de 400 mg/día para pacientes adultos con GIST maligno irresecable y/o metastásico. Un aumento de la dosis hasta 800 mg diarios (dado como 400 mg dos veces al día) puede considerarse, según lo indique clínicamente, en pacientes que muestren signos o síntomas claros de progresión de la enfermedad a una dosis más baja y en ausencia de reacciones adversas graves al fármaco.
2.11 Pacientes adultos con GIST adyuvante
La dosis recomendada de imatinib mesylate tablets es de 400 mg/día para el tratamiento adyuvante de pacientes adultos después de la resección macroscópica completa de GIST. En ensayos clínicos, se estudió un año de imatinib mesylate tablets y tres años de imatinib mesylate tablets. En la población de pacientes definida en el Estudio 2, se recomiendan tres años de imatinib mesylate tablets [ver Estudios clínicos (14.8)]. Se desconoce la duración óptima del tratamiento con imatinib mesylate tablets.
2.12 Pautas de modificación de la dosis
Inductores potentes del CYP3A4 concomitantes: Debe evitarse el uso concomitante de inductores potentes del CYP3A4 (p. ej., dexametasona, fenitoína, carbamazepina, rifampicina, rifabutina, rifampicina, fenobarbital). Si los pacientes deben recibir un inductor potente del CYP3A4 de forma conjunta, según los estudios farmacocinéticos, la dosis de imatinib mesylate tablets debe aumentarse al menos un 50 % y la respuesta clínica debe controlarse cuidadosamente [ver Interacciones farmacológicas (7.1)].
Insuficiencia hepática: Los pacientes con insuficiencia hepática leve y moderada no requieren un ajuste de la dosis y deben ser tratados con la dosis recomendada. Se debe utilizar una reducción del 25 % en la dosis recomendada para pacientes con insuficiencia hepática grave [ver Uso en poblaciones específicas (8.6)].
Insuficiencia renal: Los pacientes con insuficiencia renal moderada (aclaramiento de creatinina [CrCl] = 20 a 39 ml/min) deben recibir una reducción del 50 % en la dosis inicial recomendada y las dosis futuras pueden aumentarse según se tolere. No se recomiendan dosis superiores a 600 mg en pacientes con insuficiencia renal leve (CrCl = 40 a 59 ml/min). Para los pacientes con insuficiencia renal moderada, no se recomiendan dosis superiores a 400 mg.
Imatinib debe utilizarse con precaución en pacientes con insuficiencia renal grave. Una dosis de 100 mg/día fue tolerada en dos pacientes con insuficiencia renal grave [ver Advertencias y precauciones (5.3), Uso en poblaciones específicas (8.7)].
2.13 Ajuste de la dosis por hepatotoxicidad y reacciones adversas no hematológicas
Si se producen elevaciones de la bilirrubina superiores a 3 veces el límite superior normal institucional (LSNI) o de las transaminasas hepáticas superiores a 5 veces el LSNI, se debe suspender la administración de imatinib mesylate tablets hasta que los niveles de bilirrubina hayan vuelto a ser inferiores a 1,5 veces el LSNI y los niveles de transaminasas a menos de 2,5 veces el LSNI. En adultos, el tratamiento con imatinib mesylate tablets puede continuarse a una dosis diaria reducida (es decir, de 400 mg a 300 mg, de 600 mg a 400 mg o de 800 mg a 600 mg). En niños, las dosis diarias pueden reducirse en las mismas circunstancias de 340 mg/m2/día a 260 mg/m2/día.
Si se desarrolla una reacción adversa no hematológica grave (como hepatotoxicidad grave o retención de líquidos grave), se debe suspender la administración de imatinib mesylate tablets hasta que el acontecimiento se haya resuelto. Posteriormente, el tratamiento puede reanudarse según corresponda en función de la gravedad inicial del acontecimiento.
2.14 Ajuste de la dosis por reacciones adversas hematológicas
Se recomienda la reducción de la dosis o las interrupciones del tratamiento para la neutropenia grave y la trombocitopenia, como se indica en la Tabla 1.
Tabla 1: Ajustes de la dosis para la neutropenia y la trombocitopenia
|
ASM asociada a eosinofilia |
RAN inferior a 1,0 x 109/l |
1. Suspender imatinib mesylate tablets hasta que el RAN sea superior o igual a 1,5 x 109/l y las plaquetas superiores o iguales a 75 x 109/l 2. Reanudar el tratamiento con imatinib mesylate tablets a la dosis anterior (es decir, la dosis antes de la reacción adversa grave) |
|
SHE/CEL con quinasa de fusión FIP1L1-PDGFRα |
RAN inferior a 1,0 x 109/l |
1. Suspender imatinib mesylate tablets hasta que el RAN sea superior o igual a 1,5 x 109/l y las plaquetas superiores o iguales a 75 x 109/l 2. Reanudar el tratamiento con imatinib mesylate tablets a la dosis anterior (es decir, la dosis antes de la reacción adversa grave) |
|
Chronic Phase CML (starting dose MDS/MPD, ASM and HES/CEL GIST (starting dose 400 mg)
|
ANC less than 1.0 x 109/L |
1. Suspenda las tabletas de mesilato de imatinib hasta que el ANC sea mayor o igual a 1.5 x 109/L y las plaquetas sean mayores o iguales a 75 x 109/L 2. Reanude el tratamiento con tabletas de mesilato de imatinib a la dosis inicial original de 400 mg 3. Si se repite un ANC inferior a 1.0 x 109/L y/o plaquetas inferiores a 50 x 109/L, repita el paso 1 y reanude las tabletas de mesilato de imatinib a una dosis reducida de 300 mg |
|
Ph+ CML : Accelerated Phase and (starting dose 600 mg) |
ANC less than 0.5 x 109/L |
1. Compruebe si la citopenia está relacionada con la leucemia (aspirado de médula ósea o biopsia) 2. Si la citopenia no está relacionada con la leucemia, reduzca la dosis de tabletas de mesilato de imatinib a 400 mg 3. Si la citopenia persiste durante 2 semanas, reduzca aún más a 300 mg 4. Si la citopenia persiste durante 4 semanas y sigue sin estar relacionada con la leucemia, suspenda las tabletas de mesilato de imatinib hasta que el ANC sea superior o igual a 1 x 109/L y las plaquetas sean superiores o iguales a 20 x 109/L y luego reanude el tratamiento con 300 mg |
|
DFSP |
ANC less than 1.0 x 109/L |
1. Suspenda las tabletas de mesilato de imatinib hasta que el ANC sea mayor o igual a 1.5 x 109/L y las plaquetas sean mayores o iguales a 75 x 109/L 2. Reanude el tratamiento con tabletas de mesilato de imatinib a 600 mg 3. En caso de que se repita un ANC inferior a 1.0 x 109/L y/o plaquetas inferiores a 50 x 109/L, repita el paso 1 y reanude las tabletas de mesilato de imatinib a una dosis reducida de 400 mg |
|
Pediatric newly diagnosed chronic |
ANC less than 1.0 x 109/L |
1. Suspenda las tabletas de mesilato de imatinib hasta que el ANC sea mayor o igual a 1.5 x 109/L y las plaquetas sean mayores o iguales a 75 x 109/L 2. Reanude el tratamiento con tabletas de mesilato de imatinib a la dosis anterior (es decir, la dosis antes de la reacción adversa grave) 3. En caso de que se repita un ANC inferior a 1.0 x 109/L y/o plaquetas inferiores a 50 x 109/L, repita el paso 1 y reanude las tabletas de mesilato de imatinib a una dosis reducida de 260 mg/m2 |
|
Abbreviations: ANC, absolute neutrophil count; ASM, aggressive systemic mastocytosis; CEL, chronic eosinophilic leukemia; CML, chronic myeloid leukemia; DFSP, dermatofibrosarcoma protuberans; HES, hypereosinophilic syndrome; MDS/MPD, myelodysplastic/myeloproliferative diseases; PDGFR, platelet-derived growth factor receptor; Ph+ CML, Philadelphia chromosome positive chronic myeloid leukemia; Ph+ ALL, Philadelphia chromosome positive acute lymphoblastic leukemia. |
||
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
Comprimidos recubiertos con película de 100 mg:
Comprimidos de color marrón, redondos, ranurados, recubiertos con película, con borde biselado, marcados con “AN” en el lado ranurado y “794” en el otro lado.
Comprimidos recubiertos con película de 400 mg:
Comprimidos de color marrón, ovalados, ranurados, recubiertos con película, con borde biselado, marcados con “AN” en el lado ranurado y “795” en el otro lado.
4 CONTRAINDICACIONES
Ninguna.
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Retención de líquidos y edema
El mesilato de imatinib a menudo se asocia con edema y, en ocasiones, con retención grave de líquidos [ver Reacciones adversas (6.1)]. Pese y controle a los pacientes regularmente para detectar signos y síntomas de retención de líquidos. Investigue cuidadosamente el aumento de peso rápido inesperado y proporcione el tratamiento adecuado. La probabilidad de edema aumentó con dosis más altas de mesilato de imatinib y una edad superior a 65 años en los estudios de LMC. Se informó edema superficial grave en el 1,5% de los pacientes con LMC recién diagnosticados que tomaron mesilato de imatinib y en el 2% al 6% de otros pacientes adultos con LMC que tomaron mesilato de imatinib. Además, se informaron otras reacciones graves de retención de líquidos (por ejemplo, derrame pleural, derrame pericárdico, edema pulmonar y ascitis) en el 1,3% de los pacientes con LMC recién diagnosticados que tomaron mesilato de imatinib y en el 2% al 6% de otros pacientes adultos con LMC que tomaron mesilato de imatinib. Se informó retención grave de líquidos en el 9% al 13,1% de los pacientes que tomaron mesilato de imatinib para GIST [ver Reacciones adversas (6.1)]. En un ensayo aleatorizado en pacientes con LMC Ph+ recién diagnosticada en fase crónica que comparó mesilato de imatinib y nilotinib, se produjo retención grave de líquidos (grado 3 o 4) en el 2,5% de los pacientes que recibieron mesilato de imatinib y en el 3,9% de los pacientes que recibieron nilotinib 300 mg dos veces al día. Se observaron derrames (incluido derrame pleural, derrame pericárdico, ascitis) o edema pulmonar en el 2,1% (ninguno fue de grado 3 o 4) de los pacientes en el brazo de mesilato de imatinib y en el 2,2% (0,7% de grado 3 o 4) de los pacientes en el brazo de nilotinib 300 mg dos veces al día.
5.2 Toxicidad hematológica
El tratamiento con mesilato de imatinib se asocia con anemia, neutropenia y trombocitopenia. Realice hemogramas completos semanalmente durante el primer mes, quincenalmente durante el segundo mes y periódicamente a partir de entonces según esté clínicamente indicado (por ejemplo, cada 2 o 3 meses). En la LMC, la aparición de estas citopenias depende de la etapa de la enfermedad y es más frecuente en pacientes con LMC en fase acelerada o crisis blástica que en pacientes con LMC en fase crónica. En pacientes pediátricos con LMC, las toxicidades más frecuentes observadas fueron citopenias de grado 3 o 4, incluida la neutropenia, la trombocitopenia y la anemia. Estas generalmente ocurren dentro de los primeros meses de terapia [ver Dosificación y administración (2.14)].
5.3 Insuficiencia cardíaca congestiva y disfunción ventricular izquierda
Se ha informado insuficiencia cardíaca congestiva y disfunción ventricular izquierda en pacientes que toman mesilato de imatinib. Las reacciones adversas cardíacas fueron más frecuentes en pacientes de edad avanzada o con comorbilidades, incluida la historia médica previa de enfermedad cardíaca. En un estudio internacional aleatorizado de fase 3 en 1106 pacientes con LMC Ph+ recién diagnosticada en fase crónica, se observó insuficiencia cardíaca grave y disfunción ventricular izquierda en el 0,7% de los pacientes que tomaron mesilato de imatinib en comparación con el 0,9% de los pacientes que tomaron IFN + Ara-C. En otro ensayo aleatorizado con pacientes con LMC Ph+ recién diagnosticada en fase crónica que comparó mesilato de imatinib y nilotinib, se observó insuficiencia cardíaca en el 1,1% de los pacientes en el brazo de mesilato de imatinib y en el 2,2% de los pacientes en el brazo de nilotinib 300 mg dos veces al día y la insuficiencia cardíaca grave (grado 3 o 4) ocurrió en el 0,7% de los pacientes en cada grupo. Controle cuidadosamente a los pacientes con enfermedad cardíaca o factores de riesgo para enfermedad cardíaca o antecedentes de insuficiencia renal. Evalúe y trate a cualquier paciente con signos o síntomas compatibles con insuficiencia cardíaca o renal.
5.4 Hepatotoxicidad
La hepatotoxicidad, en ocasiones grave, puede ocurrir con el mesilato de imatinib [ver Reacciones adversas (6.1)]. Se han notificado casos de insuficiencia hepática mortal y lesión hepática grave que requieren trasplante de hígado con el uso a corto y largo plazo de mesilato de imatinib. Controle la función hepática (transaminasas, bilirrubina y fosfatasa alcalina) antes de iniciar el tratamiento y mensualmente, o según esté clínicamente indicado. Maneje las anormalidades de laboratorio con la interrupción del mesilato de imatinib y/o la reducción de la dosis [ver Dosificación y administración (2.13)]. Cuando el mesilato de imatinib se combina con quimioterapia, se ha observado toxicidad hepática en forma de elevación de las transaminasas e hiperbilirrubinemia. Además, ha habido informes de insuficiencia hepática aguda. Se recomienda el control de la función hepática.
5.5 Hemorragia
En un ensayo de mesilato de imatinib versus IFN+Ara-C en pacientes con LMC recién diagnosticada, el 1,8% de los pacientes tuvo hemorragia de grado 3/4. En los estudios de GIST irresecable o metastásico de fase 3, 211 pacientes (12,9%) informaron hemorragia de grado 3/4 en cualquier sitio. En el estudio de GIST irresecable o metastásico de fase 2, 7 pacientes (5%) tuvieron un total de 8 hemorragias de grado 3/4 de CTC; gastrointestinal (GI) (3 pacientes), intratumoral (3 pacientes) o ambas (1 paciente). Los sitios tumorales gastrointestinales pueden haber sido la fuente de las hemorragias GI. En un ensayo aleatorizado en pacientes con LMC Ph+ recién diagnosticada en fase crónica que comparó mesilato de imatinib y nilotinib, se produjo hemorragia GI en el 1,4% de los pacientes en el brazo de mesilato de imatinib y en el 2,9% de los pacientes en el brazo de nilotinib 300 mg dos veces al día. Ninguno de estos eventos fue de grado 3 o 4 en el brazo de mesilato de imatinib; el 0,7% fue de grado 3 o 4 en el brazo de nilotinib 300 mg dos veces al día. Además, se ha informado ectasia vascular antral gástrica en la experiencia postcomercialización.
5.6 Trastornos Gastrointestinales
El mesilato de imatinib a veces se asocia con irritación gastrointestinal. El mesilato de imatinib debe tomarse con alimentos y un vaso grande de agua para minimizar este problema. Se han reportado casos raros, incluidas muertes, de perforación gastrointestinal.
5.7 Toxicidad Cardiaca Hipereosinofílica
En pacientes con síndrome hipereosinofílico con infiltración oculta de células HES dentro del miocardio, se han asociado casos de shock cardiogénico/disfunción ventricular izquierda con degranulación de células HES tras el inicio del tratamiento con mesilato de imatinib. Se informó que la condición era reversible con la administración de esteroides sistémicos, medidas de soporte circulatorio y la suspensión temporal del mesilato de imatinib.
La enfermedad mielodisplásica/mieloproliferativa y la mastocitosis sistémica pueden estar asociadas con niveles altos de eosinófilos. Considere realizar un ecocardiograma y determinar la troponina sérica en pacientes con HES/CEL, y en pacientes con MDS/MPD o ASM asociado con niveles altos de eosinófilos. Si alguno de los dos es anormal, considere el uso profiláctico de esteroides sistémicos (1 a 2 mg/kg) durante una a dos semanas de forma concomitante con mesilato de imatinib al inicio del tratamiento.
5.8 Toxicidades Dermatológicas
Se han reportado reacciones dermatológicas bulbosas, incluyendo eritema multiforme y síndrome de Stevens-Johnson, con el uso de mesilato de imatinib. En algunos casos de reacciones dermatológicas bulbosas, incluyendo eritema multiforme y síndrome de Stevens-Johnson reportados durante la vigilancia postcomercialización, se observó una reacción dermatológica recurrente tras la reexposición. Varios informes postcomercialización extranjeros han descrito casos en los que los pacientes toleraron la reintroducción del tratamiento con mesilato de imatinib después de la resolución o mejora de la reacción bulbular. En estos casos, el mesilato de imatinib se reanudó a una dosis más baja que la que provocó la reacción y algunos pacientes también recibieron tratamiento concomitante con corticosteroides o antihistamínicos.
5.9 Hipotiroidismo
Se han reportado casos clínicos de hipotiroidismo en pacientes tiroidectomizados que reciben reemplazo de levotiroxina durante el tratamiento con mesilato de imatinib. Monitorear los niveles de TSH en estos pacientes.
5.10 Toxicidad Embriofetal
El mesilato de imatinib puede causar daño fetal cuando se administra a una mujer embarazada. El mesilato de imatinib fue teratógeno en ratas cuando se administró durante la organogénesis a dosis aproximadamente iguales a la dosis máxima humana de 800 mg/día basada en el área de superficie corporal (BSA). Se observó una pérdida significativa postimplantación en ratas hembras a las que se administró mesilato de imatinib a dosis aproximadamente la mitad de la dosis máxima humana de 800 mg/día basada en BSA. Avise a las pacientes sexualmente activas con potencial reproductivo que usen métodos anticonceptivos efectivos (métodos que resulten en tasas de embarazo inferiores al 1%) cuando usen mesilato de imatinib y durante 14 días después de dejar de usar mesilato de imatinib. Si este medicamento se usa durante el embarazo o si la paciente queda embarazada mientras toma este medicamento, informe a la paciente sobre el posible peligro para el feto [ver Uso en poblaciones específicas (8.1)].
5.11 Retraso del Crecimiento en Niños y Adolescentes
Se ha reportado retraso del crecimiento en niños y preadolescentes que reciben mesilato de imatinib. Se desconocen los efectos a largo plazo del tratamiento prolongado con mesilato de imatinib sobre el crecimiento en niños. Por lo tanto, monitorear el crecimiento en niños bajo tratamiento con mesilato de imatinib [ver Reacciones adversas (6.1)].
5.12 Síndrome de Lisis Tumoral
Se han reportado casos de Síndrome de Lisis Tumoral (TLS), incluidos casos fatales, en pacientes con CML, GIST, ALL y leucemia eosinofílica que reciben mesilato de imatinib. Los pacientes en riesgo de TLS son aquellos con tumores que tienen una alta tasa de proliferación o una alta carga tumoral antes del tratamiento. Monitorear estos pacientes de cerca y tomar las precauciones apropiadas. Debido a la posible aparición de TLS, corregir la deshidratación clínicamente significativa y tratar los niveles altos de ácido úrico antes de iniciar el mesilato de imatinib.
5.13 Deterioros Relacionados con la Conducción y el Uso de Maquinaria
Se han reportado accidentes de vehículos motorizados en pacientes que reciben mesilato de imatinib. Avise a los pacientes que pueden experimentar efectos secundarios, como mareos, visión borrosa o somnolencia durante el tratamiento con mesilato de imatinib. Recomiende precaución al conducir un automóvil o operar maquinaria.
5.14 Toxicidad Renal
Puede ocurrir una disminución de la función renal en pacientes que reciben mesilato de imatinib. Los valores medianos estimados de la tasa de filtración glomerular (eGFR) en pacientes con mesilato de imatinib 400 mg diarios para CML de nuevo diagnóstico (cuatro ensayos aleatorizados) y GIST maligno (un ensayo de un solo brazo) disminuyeron de un valor basal de 85 mL/min/1,73 m2 (N=1,190) a 75 mL/min/1,73 m2 a los 12 meses (N=1,082) y 69 mL/min/1,73 m2 a los 60 meses (N=549). Evaluar la función renal antes de iniciar el mesilato de imatinib y monitorear durante el tratamiento, prestando atención a los factores de riesgo de disfunción renal, como la insuficiencia renal preexistente, la diabetes mellitus, la hipertensión y la insuficiencia cardíaca congestiva.
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas graves se describen en otras partes del etiquetado:
- Retención de líquidos y edema [ver Advertencias y precauciones (5.1)]
- Toxicidad hematológica [ver Advertencias y precauciones (5.2)]
- Insuficiencia cardíaca congestiva y disfunción ventricular izquierda [ver Advertencias y precauciones (5.3)]
- Hepatotoxicidad [ver Advertencias y precauciones (5.4)]
- Hemorragia [ver Advertencias y precauciones (5.5)]
- Trastornos gastrointestinales [ver Advertencias y precauciones (5.6)]
- Toxicidad cardíaca hipereosinofílica [ver Advertencias y precauciones (5.7)]
- Toxicidades dermatológicas [ver Advertencias y precauciones (5.8)]
- Hipotiroidismo [ver Advertencias y precauciones (5.9)]
- Retraso del crecimiento en niños y adolescentes [ver Advertencias y precauciones (5.11)]
- Síndrome de lisis tumoral [ver Advertencias y precauciones (5.12)]
- Deterioros relacionados con la conducción y el uso de maquinaria [ver Advertencias y precauciones (5.13)]
- Toxicidad renal [ver Advertencias y precauciones (5.14)]
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se llevan a cabo en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no se pueden comparar directamente con las tasas en los ensayos clínicos de otro fármaco y pueden no reflejar las tasas observadas en la práctica.
Leucemia mieloide crónica
La mayoría de los pacientes tratados con mesilato de imatinib experimentaron reacciones adversas en algún momento. El mesilato de imatinib se suspendió debido a reacciones adversas relacionadas con el fármaco en el 2,4% de los pacientes que recibieron mesilato de imatinib en el ensayo aleatorizado de pacientes recién diagnosticados con LMC Ph+ en fase crónica que comparaba mesilato de imatinib versus IFN+Ara-C, y en el 12,5% de los pacientes que recibieron mesilato de imatinib en el ensayo aleatorizado de pacientes recién diagnosticados con LMC Ph+ en fase crónica que comparaba mesilato de imatinib y nilotinib. El mesilato de imatinib se suspendió debido a reacciones adversas relacionadas con el fármaco en el 4% de los pacientes en fase crónica después del fracaso de la terapia con interferón alfa, en el 4% de los pacientes en fase acelerada y en el 5% de los pacientes en crisis blástica.
Las reacciones adversas relacionadas con el fármaco más frecuentemente reportadas fueron edema, náuseas y vómitos, calambres musculares, dolor musculoesquelético, diarrea y erupción cutánea (Tabla 2 y Tabla 3 para LMC recién diagnosticada, Tabla 4 para otros pacientes con LMC). El edema fue más frecuente en la región periorbitaria o en las extremidades inferiores y se controló con diuréticos, otras medidas de apoyo o reduciendo la dosis de mesilato de imatinib [ver Dosificación y administración (2.13)]. La frecuencia de edema superficial grave fue del 1,5% al 6%.
Una variedad de reacciones adversas representan retención de líquidos local o general, incluyendo derrame pleural, ascitis, edema pulmonar y aumento rápido de peso con o sin edema superficial. Estas reacciones parecen estar relacionadas con la dosis, fueron más comunes en los estudios de fase acelerada y crisis blástica (donde la dosis fue de 600 mg/día), y son más comunes en los ancianos. Estas reacciones generalmente se controlaron interrumpiendo el tratamiento con mesilato de imatinib y utilizando diuréticos u otras medidas de apoyo apropiadas. Estas reacciones pueden ser graves o potencialmente mortales.
Las reacciones adversas, independientemente de la relación con el fármaco del estudio, que se reportaron en al menos el 10% de los pacientes tratados con mesilato de imatinib se muestran en las Tablas 2, 3 y 4.
Tabla 2: Reacciones adversas independientemente de la relación con el fármaco del estudio reportadas en el ensayo clínico de LMC recién diagnosticada en el estudio de mesilato de imatinib versus IFN+Ara-C (mayor o igual al 10% de pacientes tratados con mesilato de imatinib)(1)
|
Todos los grados |
Grados CTC* 3/4 |
|||
|
Término preferido |
Mesilato de imatinib |
IFN+Ara−C |
Mesilato de imatinib |
IFN+Ara−C |
|
Retención de líquidos |
61.7 |
11.1 |
2.5 |
0.9 |
|
− Edema superficial |
59.9 |
9.6 |
1.5 |
0.4 |
|
− Otras reacciones de retención de líquidos2 |
6.9 |
1.9 |
1.3 |
0.6 |
|
Náuseas |
49.5 |
61.5 |
1.3 |
5.1 |
|
Calambres musculares |
49.2 |
11.8 |
2.2 |
0.2 |
|
Dolor musculoesquelético |
47.0 |
44.8 |
5.4 |
8.6 |
|
Diarrea |
45.4 |
43.3 |
3.3 |
3.2 |
|
Erupción cutánea y términos relacionados |
40.1 |
26.1 |
2.9 |
2.4 |
|
Fatiga |
38.8 |
67.0 |
1.8 |
25.1 |
|
Dolor de cabeza |
37.0 |
43.3 |
0.5 |
3.8 |
|
Dolor en las articulaciones |
31.4 |
38.1 |
2.5 |
7.7 |
|
Dolor abdominal |
36.5 |
25.9 |
4.2 |
3.9 |
|
Nasofaringitis |
30.5 |
8.8 |
0 |
0.4 |
|
Hemorragia |
28.9 |
21.2 |
1.8 |
1.7 |
|
– Hemorragia gastrointestinal |
1.6 |
1.1 |
0.5 |
0.2 |
|
– Hemorragia del SNC |
0.2 |
0.4 |
0 |
0.4 |
|
Mialgia |
24.1 |
38.8 |
1.5 |
8.3 |
|
Vómitos |
22.5 |
27.8 |
2.0 |
3.4 |
|
Dispepsia |
18.9 |
8.3 |
0 |
0.8 |
|
Tos |
20.0 |
23.1 |
0.2 |
0.6 |
|
Dolor faringolaríngeo |
18.1 |
11.4 |
0.2 |
0 |
|
Infección de las vías respiratorias superiores |
21.2 |
8.4 |
0.2 |
0.4 |
|
Mareo |
19.4 |
24.4 |
0.9 |
3.8 |
|
Fiebre |
17.8 |
42.6 |
0.9 |
3.0 |
|
Aumento de peso |
15.6 |
2.6 |
2.0 |
0.4 |
|
Insomnio |
14.7 |
18.6 |
0 |
2.3 |
|
Depresión |
14.9 |
35.8 |
0.5 |
13.1 |
|
Influenza |
13.8 |
6.2 |
0.2 |
0.2 |
|
Dolor óseo |
11.3 |
15.6 |
1.6 |
3.4 |
|
Estreñimiento |
11.4 |
14.4 |
0.7 |
0.2 |
|
Sinusitis |
11.4 |
6.0 |
0.2 |
0.2 |
|
Abreviaturas: CML, leucemia mieloide crónica; SNC, sistema nervioso central; CTC, criterios de terminología común; GI, gastrointestinal; IFN, interferón alfa. *Criterios de terminología común del NCI para eventos adversos, versión 3.0. (1)Se enumeran todas las reacciones adversas que ocurren en más del 10% de los pacientes tratados con mesilato de imatinib, independientemente de la relación sospechosa con el tratamiento. (2)Otras reacciones de retención de líquidos incluyen derrame pleural, ascitis, edema pulmonar, derrame pericárdico, anasarca, edema agravado y retención de líquidos no especificada de otra manera. |
||||
Tabla 3: Reacciones adversas no hematológicas más frecuentemente notificadas (independientemente de la relación con el fármaco del estudio) en pacientes con CML-CP Ph+ de nueva aparición en el estudio de Imatinib Mesilato versus nilotinib (mayor o igual al 10% en los grupos de Imatinib Mesilato 400 mg una vez al día o nilotinib 300 mg dos veces al día) Análisis de 60 mesesa
|
Pacientes con CML-CP Ph+ de nueva aparición |
|||||
|
Imatinib Mesilato 400 mg una vez al día N=280 |
nilotinib 300 mg dos veces al día N=279 |
Imatinib Mesilato 400 mg una vez al día N=280 |
nilotinib 300 mg dos veces al día N=279 |
||
|
Sistema corporal y término preferido |
Todos los grados (%) |
Grados CTCb 3/4 (%) |
|||
|
Trastornos de la piel y del tejido subcutáneo |
Erupción cutánea |
19 |
38 |
2 |
<1 |
|
Prurito |
7 |
21 |
0 |
<1 |
|
|
Alopecia |
7 |
13 |
0 |
0 |
|
|
Piel seca |
6 |
12 |
0 |
0 |
|
|
Trastornos gastrointestinales |
Náuseas |
41 |
22 |
2 |
2 |
|
Estreñimiento |
8 |
20 |
0 |
<1 |
|
|
Diarrea |
46 |
19 |
4 |
1 |
|
|
Vómitos |
27 |
15 |
<1 |
<1 |
|
|
Dolor abdominal superior |
14 |
18 |
<1 |
1 |
|
|
Dolor abdominal |
12 |
15 |
0 |
2 |
|
|
Dispepsia |
12 |
10 |
0 |
0 |
|
|
Trastornos del sistema nervioso |
Dolor de cabeza |
23 |
32 |
<1 |
3 |
|
Mareos |
11 |
12 |
<1 |
<1 |
|
|
Trastornos generales y condiciones del lugar de administración |
Fatiga |
20 |
23 |
1 |
1 |
|
Fiebre |
13 |
14 |
0 |
<1 |
|
|
Astenia |
12 |
14 |
0 |
<1 |
|
|
Edema periférico |
20 |
9 |
0 |
<1 |
|
|
Edema facial |
14 |
<1 |
<1 |
0 |
|
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Mialgia |
19 |
19 |
<1 |
<1 |
|
Artralgia |
17 |
22 |
<1 |
<1 |
|
|
Espasmos musculares |
34 |
12 |
1 |
0 |
|
|
Dolor en la extremidad |
16 |
15 |
<1 |
<1 |
|
|
Dolor de espalda |
17 |
19 |
1 |
1 |
|
|
Trastornos respiratorios, torácicos y mediastínicos |
Tos |
13 |
17 |
0 |
0 |
|
Dolor orofaríngeo |
6 |
12 |
0 |
0 |
|
|
Disnea |
6 |
11 |
<1 |
2 |
|
|
Infecciones e infestaciones |
Nasofaringitis |
21 |
27 |
0 |
0 |
|
Infección de las vías respiratorias altas |
14 |
17 |
0 |
<1 |
|
|
Influenza |
9 |
13 |
0 |
0 |
|
|
Gastroenteritis |
10 |
7 |
<1 |
0 |
|
|
Trastornos oculares |
Edema de los párpados |
19 |
1 |
<1 |
0 |
|
Edema periorbitario |
15 |
<1 |
0 |
0 |
|
|
Trastornos psiquiátricos |
Insomnio |
9 |
11 |
0 |
0 |
|
Trastorno vascular |
Hipertensión |
4 |
10 |
<1 |
1 |
|
Abreviatura: Ph+ CML-CP, leucemia mieloide crónica positiva para el cromosoma Filadelfia-fase crónica. aExcluyendo anomalías de laboratorio. bCriterios de terminología común del NCI para eventos adversos, versión 3.0. |
|||||
Tabla 4: Reacciones adversas independientemente de la relación con el fármaco del estudio reportadas en otros ensayos clínicos de CML (mayores o iguales al 10% de todos los pacientes en cualquier ensayo)(1)
|
Crisis blástica mieloide |
Fase acelerada |
Fase crónica, fracaso de IFN |
||||
|
Término preferido |
Todos los grados |
Grado 3/4 |
Todos los grados |
Grado 3/4 |
Todos los grados |
Grado 3/4 |
|
Retención de líquidos |
72 |
11 |
76 |
6 |
69 |
4 |
|
– Edema superficial |
66 |
6 |
74 |
3 |
67 |
2 |
|
– Otra retención de líquidos |
||||||
|
Reacciones (2) |
22 |
6 |
15 |
4 |
7 |
2 |
|
Náuseas |
71 |
5 |
73 |
5 |
63 |
3 |
|
Calambres musculares |
28 |
1 |
47 |
0.4 |
62 |
2 |
|
Vómitos |
54 |
4 |
58 |
3 |
36 |
2 |
|
Diarrea |
43 |
4 |
57 |
5 |
48 |
3 |
|
Hemorragia |
53 |
19 |
49 |
11 |
30 |
2 |
|
– Hemorragia del SNC |
9 |
7 |
3 |
3 |
2 |
1 |
|
– Hemorragia GI |
8 |
4 |
6 |
5 |
2 |
0.4 |
|
Dolor Musculoesquelético |
42 |
9 |
49 |
9 |
38 |
2 |
|
Fatiga |
30 |
4 |
46 |
4 |
48 |
1 |
|
Erupción Cutánea |
36 |
5 |
47 |
5 |
47 |
3 |
|
Pirexia |
41 |
7 |
41 |
8 |
21 |
2 |
|
Artralgia |
25 |
5 |
34 |
6 |
40 |
1 |
|
Cefalea |
27 |
5 |
32 |
2 |
36 |
0.6 |
|
Dolor Abdominal |
30 |
6 |
33 |
4 |
32 |
1 |
|
Aumento de Peso |
5 |
1 |
17 |
5 |
32 |
7 |
|
Tos |
14 |
0.8 |
27 |
0.9 |
20 |
0 |
|
Dispepsia |
12 |
0 |
22 |
0 |
27 |
0 |
|
Mialgia |
9 |
0 |
24 |
2 |
27 |
0.2 |
|
Nasofaringitis |
10 |
0 |
17 |
0 |
22 |
0.2 |
|
Astenia |
18 |
5 |
21 |
5 |
15 |
0.2 |
|
Disnea |
15 |
4 |
21 |
7 |
12 |
0.9 |
|
Upper Respiratory Tract |
||||||
|
Infection |
3 |
0 |
12 |
0.4 |
19 |
0 |
|
Anorexia |
14 |
2 |
17 |
2 |
7 |
0 |
|
Sudoración nocturna |
13 |
0.8 |
17 |
1 |
14 |
0.2 |
|
Estreñimiento |
16 |
2 |
16 |
0.9 |
9 |
0.4 |
|
Mareo |
12 |
0.4 |
13 |
0 |
16 |
0.2 |
|
Faringitis |
10 |
0 |
12 |
0 |
15 |
0 |
|
Insomnio |
10 |
0 |
14 |
0 |
14 |
0.2 |
|
Prurito |
8 |
1 |
14 |
0.9 |
14 |
0.8 |
|
Hipokalemia |
13 |
4 |
9 |
2 |
6 |
0.8 |
|
Neumonía |
13 |
7 |
10 |
7 |
4 |
1 |
|
Ansiedad |
8 |
0.8 |
12 |
0 |
8 |
0.4 |
|
Toxicidad hepática |
10 |
5 |
12 |
6 |
6 |
3 |
|
Rigidez |
10 |
0 |
12 |
0.4 |
10 |
0 |
|
Dolor de pecho |
7 |
2 |
10 |
0.4 |
11 |
0.8 |
|
Influenza |
0.8 |
0.4 |
6 |
0 |
11 |
0.2 |
|
Sinusitis |
4 |
0.4 |
11 |
0.4 |
9 |
0.4 |
|
Abbreviations: CML, chronic myeloid leukemia; IFN, Interferon-alpha. (1) Todas las reacciones adversas que ocurren en más del 10% de los pacientes se enumeran independientemente de la relación sospechosa con el tratamiento. (2) Otras reacciones de retención de líquidos incluyen derrame pleural, ascitis, edema pulmonar, derrame pericárdico, anasarca, edema agravado y retención de líquidos no especificada de otra manera. |
||||||
Anormalidades hematológicas y bioquímicas de laboratorio
Las citopenias, y en particular la neutropenia y la trombocitopenia, fueron un hallazgo constante en todos los estudios, con una frecuencia mayor a dosis iguales o superiores a 750 mg (estudio de fase 1). La aparición de citopenias en pacientes con LMC también dependía de la etapa de la enfermedad.
En pacientes con LMC recién diagnosticada, las citopenias fueron menos frecuentes que en los demás pacientes con LMC (ver Tablas 5, 6 y 7). La frecuencia de neutropenia y trombocitopenia de grado 3 o 4 fue entre 2 y 3 veces mayor en la crisis blástica y la fase acelerada en comparación con la fase crónica (ver Tablas 4 y 5). La duración media de los episodios de neutropenia y trombocitopenia varió de 2 a 3 semanas, y de 2 a 4 semanas, respectivamente.
Estas reacciones generalmente se pueden controlar con una reducción de la dosis o una interrupción del tratamiento con imatinib mesilato, pero pueden requerir la interrupción permanente del tratamiento.
Tabla 5: Anormalidades de laboratorio en el ensayo clínico de LMC recién diagnosticada (Imatinib mesilato versus IFN+Ara-C)
|
Imatinib mesilato |
IFN+Ara−C |
|||
|
Grados CTC |
Grado 3 |
Grado 4 |
Grado 3 |
Grado 4 |
|
Parámetros hematológicos* |
||||
|
− Neutropenia* |
13.1 |
3.6 |
20.8 |
4.5 |
|
− Trombocitopenia* |
8.5 |
0.4 |
15.9 |
0.6 |
|
− Anemia |
3.3 |
1.1 |
4.1 |
0.2 |
|
Parámetros bioquímicos |
||||
|
− Creatinina elevada |
0 |
0 |
0.4 |
0 |
|
− Bilirrubina elevada |
0.9 |
0.2 |
0.2 |
0 |
|
− Fosfatasa alcalina elevada |
0.2 |
0 |
0.8 |
0 |
|
− SGOT (AST)/SGPT (ALT) elevada |
4.7 |
0.5 |
7.1 |
0.4 |
|
Abreviaturas: LMC, leucemia mieloide crónica; IFN, interferón-alfa; SGOT, transaminasa glutámico-oxalacética sérica ahora se conoce como aspartato aminotransferasa (AST); SGPT, transaminasa glutámico-pirúvica sérica ahora se conoce como alanina aminotransferasa (ALT). *p menor que 0.001 (diferencia en las anormalidades de grado 3 más 4 entre los dos grupos de tratamiento). |
||||
Tabla 6: Porcentaje de incidencia de anormalidades de laboratorio clínicamente relevantes de Grado 3/4* en el ensayo clínico de CML de nuevo diagnóstico (Imatinib Mesylate versus nilotinib)
|
Imatinib Mesylate 400 mg una vez al día N=280 (%) |
nilotinib 300 mg dos veces al día N=279 (%) |
|
|
Parámetros hematológicos |
||
|
Trombocitopenia |
9 |
10 |
|
Neutropenia |
22 |
12 |
|
Anemia |
6 |
4 |
|
Parámetros bioquímicos |
||
|
Liasa elevada |
4 |
9 |
|
Hiperglucemia |
<1 |
7 |
|
Hipofosfatemia |
10 |
8 |
|
Bilirrubina (total) elevada |
<1 |
4 |
|
SGPT (ALT) elevada |
3 |
4 |
|
Hiperkalemia |
1 |
2 |
|
Hiponatremia |
<1 |
1 |
|
Hipokalemia |
2 |
<1 |
|
SGOT (AST) elevada |
1 |
1 |
|
Albúmina disminuida |
<1 |
0 |
|
Hipocalcemia |
<1 |
<1 |
|
Fosfatasa alcalina elevada |
<1 |
0 |
|
Creatinina elevada |
<1 |
0 |
|
Abreviaturas: CML, leucemia mieloide crónica; SGOT, transaminasa glutámico-oxalacética sérica ahora se conoce como aspartato aminotransferasa (AST); SGPT, transaminasa glutámico-pirúvica sérica ahora se conoce como alanina aminotransferasa (ALT). *Criterios comunes de terminología del NCI para eventos adversos, versión 3.0. |
||
Tabla 7: Anormalidades de laboratorio en otros ensayos clínicos de LMC
|
Crisis blástica mieloide |
Fase acelerada |
Fase crónica, fracaso de IFN 400 mg |
||||
|
Grados de CTC(1) |
Grado 3 |
Grado 4 |
Grado 3 |
Grado 4 |
Grado 3 |
Grado 4 |
|
Parámetros hematológicos |
||||||
|
− Neutropenia |
16 |
48 |
23 |
36 |
27 |
9 |
|
− Trombocitopenia |
30 |
33 |
31 |
13 |
21 |
<1 |
|
− Anemia |
42 |
11 |
34 |
7 |
6 |
1 |
|
Parámetros bioquímicos |
||||||
|
− Creatinina elevada |
1.5 |
0 |
1.3 |
0 |
0.2 |
0 |
|
− Bilirrubina elevada |
3.8 |
0 |
2.1 |
0 |
0.6 |
0 |
|
− Fosfatasa alcalina elevada |
4.6 |
0 |
5.5 |
0.4 |
0.2 |
0 |
|
− SGOT (AST) elevada |
1.9 |
0 |
3.0 |
0 |
2.3 |
0 |
|
− SGPT (ALT) elevada |
2.3 |
0.4 |
4.3 |
0 |
2.1 |
0 |
|
Abreviaturas: CML, leucemia mieloide crónica; CTC, criterios de terminología común; IFN, interferón-alfa; SGOT, transaminasa glutámico-oxalacética sérica ahora se conoce como aspartato aminotransferasa (AST); SGPT, transaminasa glutámico-pirúvica sérica ahora se conoce como alanina aminotransferasa (ALT). (1)Grados CTC: neutropenia (Grado 3 mayor o igual a 0,5 a 1,0 x 109/L, Grado 4 menor que 0,5 x 109/L), trombocitopenia (Grado 3 mayor o igual a 10 a 50 x 109/L, Grado 4 menor que 10 x 109/L), anemia (hemoglobina mayor o igual a 65 a 80 g/L, Grado 4 menor que 65 g/L), creatinina elevada (Grado 3 mayor que 3 a 6 x límite superior del rango normal [ULN], Grado 4 mayor que 6 x ULN), bilirrubina elevada (Grado 3 mayor que 3 a 10 x ULN, Grado 4 mayor que 10 x ULN), fosfatasa alcalina elevada (Grado 3 mayor que 5 a 20 x ULN, Grado 4 mayor que 20 x ULN), SGOT o SGPT elevados (Grado 3 mayor que 5 a 20 x ULN, Grado 4 mayor que 20 x ULN). |
||||||
Hepatotoxicidad
Un aumento severo de las transaminasas o la bilirrubina ocurrió en aproximadamente el 5% de los pacientes con LMC (ver Tablas 6 y 7) y generalmente se manejó con reducción o interrupción de la dosis (la duración mediana de estos episodios fue de aproximadamente 1 semana). El tratamiento se suspendió permanentemente debido a anormalidades en los laboratorios hepáticos en menos del 1.0% de los pacientes con LMC. Un paciente, que tomaba acetaminofén regularmente por fiebre, murió de insuficiencia hepática aguda. En el ensayo de fase 2 de GIST, se observaron elevaciones de grado 3 o 4 de SGPT (ALT) en el 6.8% de los pacientes y elevaciones de grado 3 o 4 de SGOT (AST) en el 4.8% de los pacientes. Se observó un aumento de la bilirrubina en el 2.7% de los pacientes.
Reacciones adversas en la población pediátrica
Terapia de un solo agente
El perfil de seguridad general de los pacientes pediátricos tratados con imatinib mesilato en 93 niños estudiados fue similar al encontrado en estudios con pacientes adultos, excepto que el dolor musculoesquelético fue menos frecuente (20.5%) y no se reportó edema periférico. Las náuseas y los vómitos fueron las reacciones adversas individuales más comúnmente reportadas con una incidencia similar a la observada en pacientes adultos. La mayoría de los pacientes experimentaron reacciones adversas en algún momento durante el estudio. La incidencia de eventos de Grado 3/4 en todos los tipos de reacciones adversas fue del 75%; los eventos con la mayor incidencia de Grado 3/4 en pacientes pediátricos con LMC estaban principalmente relacionados con la mielosupresión.
En combinación con quimioterapia de múltiples agentes
Se inscribieron pacientes pediátricos y jóvenes adultos con ALL de muy alto riesgo, definidos como aquellos con una supervivencia libre de eventos esperada a 5 años de menos del 45%, después de la terapia de inducción en un protocolo piloto de grupo cooperativo multicéntrico no aleatorizado. La población de estudio incluyó pacientes con una mediana de edad de 10 años (1 a 21 años), el 61% de los cuales eran varones, el 75% eran blancos, el 7% eran negros y el 6% eran asiáticos / isleños del Pacífico. Los pacientes con ALL Ph + (n = 92) fueron asignados a recibir imatinib mesilato y se trataron en 5 cohortes sucesivas. La exposición a imatinib mesilato se incrementó sistemáticamente en cohortes sucesivas mediante una introducción más temprana y una duración más prolongada.
La seguridad de imatinib mesilato administrado en combinación con quimioterapia intensiva se evaluó comparando la incidencia de eventos adversos de grado 3 y 4, neutropenia (menos de 750 / mcL) y trombocitopenia (menos de 75,000 / mcL) en los 92 pacientes con ALL Ph + en comparación con 65 pacientes con ALL Ph – inscritos en el ensayo que no recibieron imatinib mesilato. También se evaluó la seguridad comparando la incidencia de eventos adversos en ciclos de terapia administrados con o sin imatinib mesilato. El protocolo incluyó hasta 18 ciclos de terapia. Los pacientes estuvieron expuestos a un total acumulado de 1,425 ciclos de terapia, 778 con imatinib mesilato y 647 sin imatinib mesilato. Las reacciones adversas que se reportaron con una incidencia de 5% o más en pacientes con ALL Ph + en comparación con ALL Ph – o con una incidencia de 1% o más en ciclos de terapia que incluyeron imatinib mesilato se presentan en la Tabla 8.
|
Tabla 8: Reacciones adversas reportadas con mayor frecuencia en pacientes tratados con el fármaco en estudio (mayor que 5%) o en ciclos con el fármaco en estudio (mayor que 1%) |
||||
|
Evento adverso Eventos adversos de Grado 3 y 4 |
Incidencia por paciente ALL Ph + con Imatinib Mesilato |
Incidencia por paciente ALL Ph – Sin Imatinib Mesilato |
Por paciente Incidencia por ciclo Con Imatinib Mesilato * |
Por paciente Incidencia por ciclo Sin Imatinib Mesilato ** |
|
N = 92 n (%) |
N = 65 n (%) |
N = 778 n (%) |
N = 647 n (%) |
|
|
Náuseas y/o vómitos |
15 (16) |
6 (9) |
28 (4) |
8 (1) |
|
Hipopotasemia |
31 (34) |
16 (25) |
72 (9) |
32 (5) |
|
Neumonitis |
7 (8) |
1 (1) |
7 (1) |
1 (< 1) |
|
Derrame pleural |
6 (7) |
0 |
6 (1) |
0 |
Dolor abdominal
8 (9)
2 (3)
9 (1)
3 (< 1)
Anorexia
10 (11)
3 (5)
19 (2)
4 (1)
Hemorragia
11 (12)
4 (6)
17 (2)
8 (1)
Hipoxia
8 (9)
2 (3)
12 (2)
2 (< 1)
Mialgia
5 (5)
0
4 (1)
1 (< 1)
Estomatitis
15 (16)
8 (12)
22 (3)
14 (2)
Diarrea
8 (9)
3 (5)
12 (2)
3 (< 1)
Erupción/Trastorno de la piel
4 (4)
0
5 (1)
0
Infección
49 (53)
32 (49)
131 (17)
92 (14)
Hepática (transaminasas y/o bilirrubina)
52 (57)
38 (58)
172 (22)
113 (17)
Hipotensión
10 (11)
5 (8)
16 (2)
6 (1)
Mielosupresión
Neutropenia (< 750/mcL)
92 (100)
63 (97)
556 (71)
218 (34)
Trombocitopenia (< 75,000/mcL)
90 (92)
63 (97)
431 (55)
329 (51)
Abbreviations: Ph+ ALL, Philadelphia chromosome positive acute lymphoblastic leukemia; Ph- ALL, Philadelphia chromosome negative acute lymphoblastic leukemia.
*Defined as the frequency of adverse events (AEs) per patient per treatment cycles that included imatinib mesylate (includes patients with Ph+ ALL that received cycles with imatinib mesylate).
**Defined as the frequency of AEs per patient per treatment cycles that did not include imatinib mesylate (includes patients with Ph+ ALL that received cycles without imatinib mesylate as well as all patients with Ph- ALL who did not receive imatinib mesylate in any treatment cycle).
Reacciones adversas en otras subpoblaciones
En pacientes de edad avanzada (mayores de 65 años), con la excepción del edema, donde fue más frecuente, no hubo evidencia de un aumento en la incidencia o gravedad de las reacciones adversas. En las mujeres hubo un aumento en la frecuencia de neutropenia, así como edema superficial de Grado 1/2, dolor de cabeza, náuseas, escalofríos, vómitos, erupción cutánea y fatiga. No se observaron diferencias relacionadas con la raza, pero los subgrupos eran demasiado pequeños para una evaluación adecuada.
Leucemia linfoblástica aguda
Las reacciones adversas fueron similares para Ph+ ALL que para Ph+ CML. Las reacciones adversas relacionadas con el fármaco más frecuentemente reportadas en los estudios de Ph+ ALL fueron náuseas y vómitos leves, diarrea, mialgia, calambres musculares y erupción cutánea. El edema superficial fue un hallazgo común en todos los estudios y se describió principalmente como edemas periorbitarios o de las extremidades inferiores. Estos edemas se reportaron como eventos de Grado 3/4 en el 6.3% de los pacientes y pueden manejarse con diuréticos, otras medidas de apoyo o, en algunos pacientes, reduciendo la dosis de imatinib mesilato.
Enfermedades mielodisplásicas/mieloproliferativas
Las reacciones adversas, independientemente de la relación con el fármaco del estudio, que se reportaron en al menos el 10% de los pacientes tratados con imatinib mesilato para MDS/MPD en el estudio de Fase 2, se muestran en la Tabla 9.
Tabla 9: Reacciones adversas independientemente de la relación con el fármaco del estudio reportadas (más de un paciente) en pacientes con MPD en el estudio de Fase 2 (mayor o igual al 10% de todos los pacientes) Todos los grados
|
Término preferido |
N=7 |
|
Náuseas |
4 (57.1) |
|
Diarrea |
3 (42.9) |
|
Anemia |
2 (28.6) |
|
Fatiga |
2 (28.6) |
|
Calambres musculares |
3 (42.9) |
|
Artralgia |
2 (28.6) |
|
Edema periorbitario |
2 (28.6) |
|
Abreviatura: MPD, Enfermedad mieloproliferativa. |
|
Mastocitosis Sistémica Agresiva
Todos los pacientes con mastocitosis sistémica agresiva (ASM) experimentaron al menos una reacción adversa en algún momento. Las reacciones adversas más frecuentes fueron diarrea, náuseas, ascitis, calambres musculares, disnea, fatiga, edema periférico, anemia, prurito, erupción cutánea e infección del tracto respiratorio inferior. Ninguno de los 5 pacientes del estudio de fase 2 con ASM suspendió el mesilato de imatinib debido a reacciones adversas relacionadas con el fármaco o valores de laboratorio anormales.
Síndrome Hipereosinofílico y Leucemia Eosinofílica Crónica
El perfil de seguridad en la población de pacientes con HES/CEL no parece ser diferente del perfil de seguridad del mesilato de imatinib observado en otras poblaciones con malignidad hematológica, como la LMC Ph+. Todos los pacientes experimentaron al menos una reacción adversa, siendo las más comunes los trastornos gastrointestinales, cutáneos y musculoesqueléticos. Las anomalías hematológicas también fueron frecuentes, con casos de leucopenia, neutropenia, linfopenia y anemia de grado 3 de la CTC.
Dermatofibrosarcoma Protuberans
Las reacciones adversas, independientemente de la relación con el fármaco del estudio, que se notificaron en al menos el 10% de los 12 pacientes tratados con mesilato de imatinib para DFSP en el estudio de fase 2 se muestran en la Tabla 10.
Tabla 10: Reacciones adversas independientemente de la relación con el fármaco del estudio notificadas en pacientes con DFSP en el estudio de fase 2 (mayor o igual al 10% de todos los pacientes) Todos los grados
|
Término preferido |
N=12 |
|
Náuseas |
5 (41.7) |
|
Diarrea |
3 (25.0) |
|
Vómitos |
3 (25.0) |
|
Edema periorbitario |
4 (33.3) |
|
Edema facial |
2 (16.7) |
|
Erupción cutánea |
3 (25.0) |
|
Fatiga |
5 (41.7) |
|
Edema periférico |
4 (33.3) |
|
Pirexia |
2 (16.7) |
|
Edema ocular |
4 (33.3) |
|
Lagrimeo aumentado |
3 (25.0) |
|
Disnea de esfuerzo |
2 (16.7) |
|
Anemia |
3 (25.0) |
|
Rinitis |
2 (16.7) |
|
Anorexia |
2 (16.7) |
| Abreviatura: DFSP, dermatofibrosarcoma protuberans. | |
Las anormalidades de laboratorio clínicamente relevantes o graves en los 12 pacientes tratados con imatinib mesilato para DFSP en el estudio de fase 2 se presentan en la Tabla 11.
Tabla 11: Anormalidades de laboratorio reportadas en pacientes con DFSP en el estudio de fase 2
|
N=12 |
||
|
Grados CTC(1) |
Grado 3 % |
Grado 4 % |
|
Parámetros hematológicos |
||
|
– Anemia |
17 |
0 |
|
– Trombocitopenia |
17 |
0 |
|
– Neutropenia |
0 |
8 |
|
Parámetros bioquímicos |
||
|
– Creatinina elevada |
0 |
8 |
|
Abreviatura: CTC, criterios de terminología común. (1)Grados CTC: neutropenia (Grado 3 mayor o igual a 0,5 a 1,0 x 109/L, Grado 4 menor que 0,5 x 109/L), trombocitopenia (Grado 3 mayor o igual a 10 a 50 x 109/L, Grado 4 menor que 10 x 109/L), anemia (Grado 3 mayor o igual a 65 a 80 g/L, Grado 4 menor que 65 g/L), creatinina elevada (Grado 3 mayor que 3 a 6 x límite superior del rango normal [ULN], Grado 4 mayor que 6 x ULN). |
||
Tumores Estromales Gastrointestinales
GIST metastásico maligno y/o irresecable
En los ensayos de Fase 3, la mayoría de los pacientes tratados con imatinib mesilato experimentaron reacciones adversas en algún momento. Las reacciones adversas más frecuentes fueron edema, fatiga, náuseas, dolor abdominal, diarrea, erupción cutánea, vómitos, mialgia, anemia y anorexia. El fármaco se suspendió por reacciones adversas en un total de 89 pacientes (5,4%). El edema superficial, con mayor frecuencia periorbital o edema de las extremidades inferiores, se controló con diuréticos, otras medidas de apoyo o reduciendo la dosis de imatinib mesilato [ver Dosificación y administración (2.13)]. Se observó edema grave (CTC Grado 3/4) en 182 pacientes (11,1%).
Las reacciones adversas, independientemente de la relación con el fármaco del estudio, que se informaron en al menos el 10% de los pacientes tratados con imatinib mesilato se muestran en la Tabla 12.
En general, la incidencia de todas las reacciones adversas y la incidencia de reacciones adversas graves (CTC Grado 3 y superior) fueron similares entre los dos brazos de tratamiento, excepto por el edema, que se informó con mayor frecuencia en el grupo de 800 mg.
Tabla 12: Número (%) de pacientes con reacciones adversas independientemente de la relación con el fármaco del estudio donde la frecuencia es mayor o igual al 10% en cualquier grupo (conjunto de análisis completo) en los ensayos clínicos de Fase 3 de GIST metastásico maligno y/o irresecable
|
Imatinib 400 mg N = 818 |
Imatinib 800 mg N = 822 |
|||
|
Término informado o especificado |
Todos los grados % |
Grados 3/4/5 % |
Todos los grados % |
Grados 3/4/5 % |
|
Edema |
76.7 |
9.0 |
86.1 |
13.1 |
|
Fatiga/letargo, malestar, astenia |
69.3 |
11.7 |
74.9 |
12.2 |
|
Náuseas |
58.1 |
9.0 |
64.5 |
7.8 |
|
Dolor abdominal/calambres |
57.2 |
13.8 |
55.2 |
11.8 |
|
Diarrea |
56.2 |
8.1 |
58.2 |
8.6 |
|
Erupción cutánea/descamación |
38.1 |
7.6 |
49.8 |
8.9 |
|
Vómitos |
37.4 |
9.2 |
40.6 |
7.5 |
|
Mialgia |
32.2 |
5.6 |
30.2 |
3.8 |
|
Anemia |
32.0 |
4.9 |
34.8 |
6.4 |
|
Anorexia |
31.1 |
6.6 |
35.8 |
4.7 |
|
Other GI toxicity |
25.2 |
8.1 |
28.1 |
6.6 |
|
Headache |
22.0 |
5.7 |
19.7 |
3.6 |
|
Other pain (excluding tumor related pain) |
20.4 |
5.9 |
20.8 |
5.0 |
|
Other dermatology/skin toxicity |
17.6 |
5.9 |
20.1 |
5.7 |
|
Leukopenia |
17.0 |
0.7 |
19.6 |
1.6 |
|
Other constitutional symptoms |
16.7 |
6.4 |
15.2 |
4.4 |
|
Cough |
16.1 |
4.5 |
14.5 |
3.2 |
|
Infection (without neutropenia) |
15.5 |
6.6 |
16.5 |
5.6 |
|
Pruritus |
15.4 |
5.4 |
18.9 |
4.3 |
|
Other neurological toxicity |
15.0 |
6.4 |
15.2 |
4.9 |
|
Cough |
16.1 |
4.5 |
14.5 |
3.2 |
|
Infection (without neutropenia) |
15.5 |
6.6 |
16.5 |
5.6 |
|
Pruritus |
15.4 |
5.4 |
18.9 |
4.3 |
|
Other neurological toxicity |
15.0 |
6.4 |
15.2 |
4.9 |
|
Constipation |
14.8 |
5.1 |
14.4 |
4.1 |
|
Other renal/genitourinary toxicity |
14.2 |
6.5 |
13.6 |
5.2 |
|
Arthralgia (joint pain) |
13.6 |
4.8 |
12.3 |
3.0 |
|
Dyspnea (shortness of breath) |
13.6 |
6.8 |
14.2 |
5.6 |
|
Fiebre en ausencia de neutropenia (ANC < 1.0 x 109/L) |
13.2 |
4.9 |
12.9 |
3.4 |
|
Sudoración |
12.7 |
4.6 |
8.5 |
2.8 |
|
Otra hemorragia |
12.3 |
6.7 |
13.3 |
6.1 |
|
Aumento de peso |
12.0 |
1.0 |
10.6 |
0.6 |
|
Alopecia |
11.9 |
4.3 |
14.8 |
3.2 |
|
Dispepsia/acidez estomacal |
11.5 |
0.6 |
10.9 |
0.5 |
|
Neutropenia/granulocitopenia |
11.5 |
3.1 |
16.1 |
4.1 |
|
Escalofríos/temblores |
11.0 |
4.6 |
10.2 |
3.0 |
|
Mareos/vértigo |
11.0 |
4.8 |
10.0 |
2.8 |
|
Aumento de la creatinina |
10.8 |
0.4 |
10.1 |
0.6 |
|
Flatulencia |
10.0 |
0.2 |
10.1 |
0.1 |
|
Estomatitis/faringitis (mucositis oral/faríngea) |
9.2 |
5.4 |
10.0 |
4.3 |
|
Linfopenia |
6.0 |
0.7 |
10.1 |
1.9 |
|
Abreviaturas: ANC, recuento absoluto de neutrófilos; GI, gastrointestinal; GIST, tumores estromales gastrointestinales. |
||||
No se informaron ni evaluaron anormalidades clínicamente relevantes o graves de los valores de laboratorio hematológicos o bioquímicos de rutina en los ensayos de fase 3 de GIST. Las anormalidades graves de los valores de laboratorio informadas en el ensayo de fase 2 de GIST se presentan en la Tabla 13.
Tabla 13: Anormalidades de laboratorio en el ensayo de fase 2 de GIST metastásico y/o irresecable
|
400 mg (n = 73) % |
600 mg (n = 74) % |
|||
|
Grados CTC1 |
Grado 3 |
Grado 4 |
Grado 3 |
Grado 4 |
|
Parámetros hematológicos |
||||
|
− Anemia |
3 |
0 |
8 |
1 |
|
− Trombocitopenia |
0 |
0 |
1 |
0 |
|
− Neutropenia |
7 |
3 |
8 |
3 |
|
Parámetros bioquímicos |
||||
|
− Creatinina elevada |
0 |
0 |
3 |
0 |
|
− Albúmina reducida |
3 |
0 |
4 |
0 |
|
− Bilirrubina elevada |
1 |
0 |
1 |
3 |
|
− Fosfatasa alcalina elevada |
0 |
0 |
3 |
0 |
|
− SGOT (AST) elevado |
4 |
0 |
3 |
3 |
|
− SGPT (ALT) elevado |
6 |
0 |
7 |
1 |
|
Abreviaturas: CTC, criterios de terminología común; GIST, tumores estromales gastrointestinales; SGOT, transaminasa glutámica oxalacética sérica ahora se conoce como aspartato aminotransferasa (AST); SGPT, transaminasa glutámica pirúvica sérica ahora se conoce como alanina aminotransferasa (ALT). 1Grados CTC: neutropenia (Grado 3 mayor o igual a 0,5 a 1,0 x 109/L, Grado 4 menor que 0,5 x 109/L), trombocitopenia (Grado 3 mayor o igual a 10 a 50 x 109/L, Grado 4 menor que 10 x 109/L), anemia (Grado 3 mayor o igual a 65 a 80 g/L, Grado 4 menor que 65 g/L), creatinina elevada (Grado 3 mayor que 3 a 6 x límite superior del rango normal [ULN], Grado 4 mayor que 6 x ULN), bilirrubina elevada (Grado 3 mayor que 3 a 10 x ULN, Grado 4 mayor que 10 x ULN), fosfatasa alcalina, SGOT o SGPT elevados (Grado 3 mayor que 5 a 20 x ULN, Grado 4 mayor que 20 x ULN), albúmina (Grado 3 menor que 20 g/L). |
||||
Tratamiento Adyuvante de GIST
En el Estudio 1, la mayoría de los pacientes tratados con imatinib mesilato y placebo experimentaron al menos una reacción adversa en algún momento. Las reacciones adversas más frecuentes fueron similares a las reportadas en otros estudios clínicos en otras poblaciones de pacientes e incluyen diarrea, fatiga, náuseas, edema, disminución de la hemoglobina, erupción cutánea, vómitos y dolor abdominal. No se reportaron nuevas reacciones adversas en el contexto del tratamiento adyuvante de GIST que no se hubieran reportado previamente en otras poblaciones de pacientes, incluidos pacientes con GIST metastásico irresecable y/o maligno. El fármaco se suspendió por reacciones adversas en 57 pacientes (17%) y 11 pacientes (3%) de los pacientes tratados con imatinib mesilato y placebo, respectivamente. El edema, las alteraciones gastrointestinales (náuseas, vómitos, distensión abdominal y diarrea), la fatiga, la hemoglobina baja y la erupción cutánea fueron las reacciones adversas más frecuentes en el momento de la suspensión.
En el Estudio 2, la suspensión del tratamiento debido a reacciones adversas se produjo en 15 pacientes (8%) y 27 pacientes (14%) de los brazos de tratamiento de 12 meses y 36 meses con imatinib mesilato, respectivamente. Como en ensayos anteriores, las reacciones adversas más comunes fueron diarrea, fatiga, náuseas, edema, disminución de la hemoglobina, erupción cutánea, vómitos y dolor abdominal.
Las reacciones adversas, independientemente de la relación con el fármaco del estudio, que se reportaron en al menos el 5% de los pacientes tratados con imatinib mesilato se muestran en la Tabla 14 (Estudio 1) y la Tabla 15 (Estudio 2). No hubo muertes atribuibles al tratamiento con imatinib mesilato en ninguno de los ensayos.
Tabla 14: Reacciones adversas independientemente de la relación con el fármaco del estudio reportadas en el Estudio 1 (mayor o igual al 5% de los pacientes tratados con imatinib mesilato)(1)
|
Término Preferido |
Todos los Grados CTC |
Grado CTC 3* y Superior |
||
|
Imatinib mesilato (n = 337) |
Placebo (n = 345) |
Imatinib mesilato (n = 337) |
Placebo (n = 345) |
|
|
% |
% |
% |
% |
|
|
Diarrea |
59.3 |
29.3 |
3.0 |
1.4 |
|
Fatiga |
57.0 |
40.9 |
2.1 |
1.2 |
|
Náuseas |
53.1 |
27.8 |
2.4 |
1.2 |
|
Edema periorbital |
47.2 |
14.5 |
1.2 |
0 |
|
Hemoglobina Disminuida |
46.9 |
27.0 |
0.6 |
0 |
|
Edema periférico |
26.7 |
14.8 |
0.3 |
0 |
|
Erupción cutánea (Exfoliativa) |
26.1 |
12.8 |
2.7 |
0 |
|
Vómitos |
25.5 |
13.9 |
2.4 |
0.6 |
|
Dolor abdominal |
21.1 |
22.3 |
3.0 |
1.4 |
|
Dolor de cabeza |
19.3 |
20.3 |
0.6 |
0 |
|
Dispepsia |
17.2 |
13.0 |
0.9 |
0 |
|
Anorexia |
16.9 |
8.7 |
0.3 |
0 |
|
Aumento de peso |
16.9 |
11.6 |
0.3 |
0 |
|
Enzimas hepáticas (ALT) aumentadas |
16.6 |
13.0 |
2.7 |
0 |
|
Espasmos musculares |
16.3 |
3.3 |
0 |
0 |
|
Conteo de neutrófilos disminuido |
16.0 |
6.1 |
3.3 |
0.9 |
|
Artralgia |
15.1 |
14.5 |
0 |
0.3 |
|
Conteo de glóbulos blancos disminuido |
14.5 |
4.3 |
0.6 |
0.3 |
|
Estreñimiento |
12.8 |
17.7 |
0 |
0.3 |
|
Mareo |
12.5 |
10.7 |
0 |
0.3 |
|
Enzimas hepáticas (AST) aumentadas |
12.2 |
7.5 |
2.1 |
0 |
|
Mialgia |
12.2 |
11.6 |
0 |
0.3 |
|
Creatinina en sangre aumentada |
11.6 |
5.8 |
0 |
0.3 |
|
Tos |
11.0 |
11.3 |
0 |
0 |
|
Prurito |
11.0 |
7.8 |
0.9 |
0 |
|
Pérdida de peso |
10.1 |
5.2 |
0 |
0 |
|
Hiperglucemia |
9.8 |
11.3 |
0.6 |
1.7 |
|
Insomnia |
9.8 |
7.2 |
0.9 |
0 |
|
Lacrimation increased |
9.8 |
3.8 |
0 |
0 |
|
Alopecia |
9.5 |
6.7 |
0 |
0 |
|
Flatulence |
8.9 |
9.6 |
0 |
0 |
|
Rash |
8.9 |
5.2 |
0.9 |
0 |
|
Abdominal distension |
7.4 |
6.4 |
0.3 |
0.3 |
|
Back pain |
7.4 |
8.1 |
0.6 |
0 |
|
Pain in extremity |
7.4 |
7.2 |
0.3 |
0 |
|
Hypokalemia |
7.1 |
2.0 |
0.9 |
0.6 |
|
Depression |
6.8 |
6.4 |
0.9 |
0.6 |
|
Facial edema |
6.8 |
1.2 |
0.3 |
0 |
|
Blood alkaline phosphatase increased |
6.5 |
7.5 |
0 |
0 |
|
Dry skin |
6.5 |
5.2 |
0 |
0 |
|
Dysgeusia |
6.5 |
2.9 |
0 |
0 |
|
Abdominal pain upper |
6.2 |
6.4 |
0.3 |
0 |
|
Neuropathy peripheral |
5.9 |
6.4 |
0 |
0 |
|
Hypocalcemia |
5.6 |
1.7 |
0.3 |
0 |
|
Leukopenia |
5.0 |
2.6 |
0.3 |
0 |
|
Recuento plaquetario disminuido |
5.0 |
3.5 |
0 |
0 |
|
Estomatitis |
5.0 |
1.7 |
0.6 |
0 |
|
Infección del tracto respiratorio superior |
5.0 |
3.5 |
0 |
0 |
|
Visión borrosa |
5.0 |
2.3 |
0 |
0 |
|
Abreviaturas: CTC, criterios de terminología común; GIST, tumores estromales gastrointestinales; SGOT, transaminasa glutámica oxaloacética sérica ahora se conoce como aspartato aminotransferasa (AST); SGPT, transaminasa glutámica pirúvica sérica ahora se conoce como alanina aminotransferasa (ALT). *NCI Common Terminology Criteria for Adverse Events, versión 3.0. (1)Se enumeran todas las reacciones adversas que ocurren en más del 5% de los pacientes, independientemente de la relación sospechosa con el tratamiento. Un paciente con múltiples ocurrencias de una reacción adversa se cuenta solo una vez en la categoría de reacción adversa. |
||||
Tabla 15: Reacciones adversas independientemente de la relación con el fármaco del estudio por término preferido Todos los grados y grados 3/4 (mayores o iguales al 5% de los pacientes tratados con imatinib mesilato) Estudio 2(1)
|
Término preferido |
Todos los grados CTC |
Grados CTC 3 y superiores |
||
|
Imatinib mesilato 12 meses (N = 194) |
Imatinib mesilato 36 meses (N = 198) |
Imatinib mesilato 12 meses (N = 194) |
Imatinib mesilato 36 meses (N = 198) |
|
|
% |
% |
% |
% |
|
|
Pacientes con al menos un AE |
99.0 |
100.0 |
20.1 |
32.8 |
|
Hemoglobina disminuida |
72.2 |
80.3 |
0.5 |
0.5 |
|
Edema periorbitario |
59.3 |
74.2 |
0.5 |
1.0 |
|
Lactato deshidrogenasa sanguínea aumentada |
43.3 |
60.1 |
0 |
0 |
|
Diarrea |
43.8 |
54.0 |
0.5 |
2.0 |
|
Náuseas |
44.8 |
51.0 |
1.5 |
0.5 |
|
Espasmos musculares |
30.9 |
49.0 |
0.5 |
1.0 |
|
Fatiga |
48.5 |
48.5 |
1.0 |
0.5 |
|
Recuento de glóbulos blancos disminuido |
34.5 |
47.0 |
2.1 |
3.0 |
|
Dolor |
25.8 |
45.5 |
1.0 |
3.0 |
|
Creatinina sanguínea aumentada |
30.4 |
44.4 |
0 |
0 |
|
Edema periférico |
33.0 |
40.9 |
0.5 |
1.0 |
|
Dermatitis |
29.4 |
38.9 |
2.1 |
1.5 |
|
Aspartate aminotransferase increased |
30.9 |
37.9 |
1.5 |
3.0 |
|
Alanine aminotransferase increased |
28.9 |
34.3 |
2.1 |
3.0 |
|
Neutrophil count decreased |
24.2 |
33.3 |
4.6 |
5.1 |
|
Hypoproteinemia |
23.7 |
31.8 |
0 |
0 |
|
Infection |
13.9 |
27.8 |
1.5 |
2.5 |
|
Weight increased |
13.4 |
26.8 |
0 |
0.5 |
|
Pruritus |
12.9 |
25.8 |
0 |
0 |
|
Flatulence |
19.1 |
24.7 |
1.0 |
0.5 |
|
Vomiting |
10.8 |
22.2 |
0.5 |
1.0 |
|
Dyspepsia |
17.5 |
21.7 |
0.5 |
1.0 |
|
Hypoalbuminemia |
11.9 |
21.2 |
0 |
0 |
|
Edema |
10.8 |
19.7 |
0 |
0.5 |
|
Abdominal distension |
11.9 |
19.2 |
0.5 |
0 |
|
Headache |
8.2 |
18.2 |
0 |
0 |
|
Lacrimation increased |
18.0 |
17.7 |
0 |
0 |
|
Arthralgia |
8.8 |
17.2 |
0 |
1.0 |
|
Blood alkaline phosphatase increased |
10.8 |
16.7 |
0 |
0.5 |
|
Disnea |
6.2 |
16.2 |
0.5 |
1.5 |
|
Mialgia |
9.3 |
15.2 |
0 |
1.0 |
|
Recuento plaquetario disminuido |
11.3 |
14.1 |
0 |
0 |
|
Bilirrubina sanguínea aumentada |
11.3 |
13.1 |
0 |
0 |
|
Disgeusia |
9.3 |
12.6 |
0 |
0 |
|
Parestesia |
5.2 |
12.1 |
0 |
0.5 |
|
Visión borrosa |
10.8 |
11.1 |
1.0 |
0.5 |
|
Alopecia |
11.3 |
10.6 |
0 |
0 |
|
Disminución del apetito |
9.8 |
10.1 |
0 |
0 |
|
Estreñimiento |
8.8 |
9.6 |
0 |
0 |
|
Pirexia |
6.2 |
9.6 |
0 |
0 |
|
Depresión |
3.1 |
8.1 |
0 |
0 |
|
Dolor abdominal |
2.6 |
7.6 |
0 |
0 |
|
Conjuntivitis |
5.2 |
7.6 |
0 |
0 |
|
Reacción de fotosensibilidad |
3.6 |
7.1 |
0 |
0 |
|
Mareo |
4.6 |
6.6 |
0.5 |
0 |
|
Hemorragia |
3.1 |
6.6 |
0 |
0 |
|
Piel seca |
6.7 |
6.1 |
0.5 |
0 |
|
Nasofaringitis |
1.0 |
6.1 |
0 |
0.5 |
|
Palpitaciones |
5.2 |
5.1 |
0 |
0 |
|
Abreviaturas: AE, evento adverso; CTC, criterios de terminología común. Un paciente con múltiples ocurrencias de una reacción adversa se cuenta solo una vez en la categoría de reacción adversa. |
||||
Reacciones adversas de múltiples ensayos clínicos
Trastornos cardíacos:
Estimado del 1% al 10%: palpitaciones, derrame pericárdico
Estimado del 0,1% al 1%: insuficiencia cardíaca congestiva, taquicardia, edema pulmonar
Estimado del 0,01% al 0,1%: arritmia, fibrilación auricular, paro cardíaco, infarto de miocardio, angina de pecho
Trastornos vasculares:
Estimado del 1% al 10%: rubor, hemorragia
Estimado del 0,1% al 1%: hipertensión, hipotensión, frialdad periférica, fenómeno de Raynaud, hematoma, hematoma subdural
Investigaciones:
Estimado del 1% al 10%: aumento de la creatina fosfoquinasa (CPK) en sangre, aumento de la amilasa en sangre
Estimado del 0,1% al 1%: aumento de la lactato deshidrogenasa (LDH) en sangre
Trastornos de la piel y del tejido subcutáneo:
Estimado del 1% al 10%: piel seca, alopecia, edema facial, eritema, reacción de fotosensibilidad, trastorno de las uñas, púrpura
Estimado del 0,1% al 1%: dermatitis exfoliativa, erupción bullosa, psoriasis, erupción pustular, contusión, aumento de la sudoración, urticaria, equimosis, aumento de la tendencia a los hematomas, hipotricosis, hipopigmentación de la piel, hiperpigmentación de la piel, onicoclasis, foliculitis, petequias, eritema multiforme, paniculitis (incluida la eritema nodoso)
Estimado del 0,01% al 0,1%: erupción vesicular, síndrome de Stevens-Johnson, pustulosis exantemática aguda generalizada, dermatosis neutrofílica febril aguda (síndrome de Sweet), decoloración de las uñas, angioedema, vasculitis leucocitoclástica
Trastornos gastrointestinales:
Estimado del 1% al 10%: distensión abdominal, reflujo gastroesofágico, boca seca, gastritis
Estimado del 0,1% al 1%: úlcera gástrica, estomatitis, ulceración bucal, eructos, melena, esofagitis, ascitis, hematemesis, queilitis, disfagia, pancreatitis
Estimado del 0,01% al 0,1%: colitis, íleo, enfermedad inflamatoria intestinal
Trastornos generales y condiciones en el lugar de administración:
Estimado del 1% al 10%: debilidad, anasarca, escalofríos
Estimado del 0,1% al 1%: malestar
Trastornos de la sangre y del sistema linfático:
Estimado del 1% al 10%: pancitopenia, neutropenia febril, linfopenia, eosinofilia
Estimado del 0,1% al 1%: trombocitemia, depresión de la médula ósea, linfadenopatía
Estimado del 0,01% al 0,1%: anemia hemolítica, anemia aplásica
Trastornos hepatobiliares:
Estimado del 0,1% al 1%: hepatitis, ictericia
Estimado del 0,01% al 0,1%: insuficiencia hepática y necrosis hepática1
Trastornos del sistema inmunitario:
Estimado del 0,01% al 0,1%: angioedema
Infecciones e infestaciones:
Estimado del 0,1% al 1%: sepsis, herpes simple, herpes zóster, celulitis, infección del tracto urinario, gastroenteritis
Estimado del 0,01% al 0,1%: infección micótica
Trastornos del metabolismo y la nutrición:
Estimado del 1% al 10%: disminución del peso, disminución del apetito
Estimado del 0,1% al 1%: deshidratación, gota, aumento del apetito, hiperuricemia, hipercalcemia, hiperglucemia, hiponatremia, hiperpotasemia, hipomagnesemia
Trastornos musculoesqueléticos y del tejido conjuntivo:
Estimado del 1% al 10%: hinchazón de las articulaciones
Estimado del 0,1% al 1%: rigidez articular y muscular, debilidad muscular, artritis
Trastornos del sistema nervioso/psiquiátricos:
Estimado del 1% al 10%: parestesia, hipestesia
Estimado del 0,1% al 1%: síncope, neuropatía periférica, somnolencia, migraña, deterioro de la memoria, disminución de la libido, ciática, síndrome de piernas inquietas, temblor
Estimado del 0,01% al 0,1%: aumento de la presión intracraneal1, estado confusional, convulsiones, neuritis óptica
Trastornos renales y urinarios:
Estimado del 0,1% al 1%: insuficiencia renal aguda, aumento de la frecuencia urinaria, hematuria, dolor renal
Trastornos del aparato reproductor y de las mamas:
Estimado del 0,1% al 1%: agrandamiento de las mamas, menorragia, disfunción sexual, ginecomastia, disfunción eréctil, menstruación irregular, dolor en el pezón, edema escrotal
Trastornos respiratorios, torácicos y mediastínicos:
Estimado del 1% al 10%: epistaxis
Estimado del 0,1% al 1%: derrame pleural
Estimado del 0,01% al 0,1%: neumonitis intersticial, fibrosis pulmonar, dolor pleurítico, hipertensión pulmonar, hemorragia pulmonar
Trastornos endocrinos:
Estimado del 0,1%–1%: hipotiroidismo, hipertiroidismo
Trastornos del ojo, el oído y el laberinto:
Estimado del 1% al 10%: conjuntivitis, visión borrosa, edema orbital, hemorragia conjuntival, ojo seco
Estimado del 0,1% al 1%: vértigo, tinnitus, irritación ocular, dolor ocular, hemorragia escleral, hemorragia retiniana, blefaritis, edema macular, pérdida de audición, catarata
Estimado del 0,01% al 0,1%: papiledema1, glaucoma
1Incluyendo algunas muertes.
6.2 Experiencia postcomercialización
Las siguientes reacciones adversas adicionales se han identificado durante el uso posterior a la aprobación de imatinib mesilato. Debido a que estas reacciones se notifican voluntariamente de una población de tamaño incierto, no siempre es posible estimar de forma fiable su frecuencia o establecer una relación causal con la exposición al fármaco.
Trastornos de la sangre y del sistema linfático: microangiopatía trombótica
Trastornos cardíacos: pericarditis, taponamiento cardíaco1
Trastornos oculares: hemorragia vítrea
Trastornos gastrointestinales: íleo/obstrucción intestinal, hemorragia tumoral/necrosis tumoral, perforación GI1 [ver Advertencias y precauciones (5.6)], diverticulitis, ectasia vascular antral gástrica
Infecciones: reactivación del virus de la hepatitis B1
Trastornos musculoesqueléticos y del tejido conjuntivo: osteonecrosis, rabdomiólisis/miopatía, retraso del crecimiento en niños, dolor musculoesquelético al suspender el tratamiento (incluyendo mialgia, dolor en las extremidades, artralgia, dolor óseo)
Trastornos del sistema nervioso: edema cerebral1
Trastornos de la reproducción: cuerpo lúteo hemorrágico/quiste ovárico hemorrágico
Trastornos respiratorios, torácicos y mediastínicos: insuficiencia respiratoria aguda1, enfermedad pulmonar intersticial
Trastornos de la piel y del tejido subcutáneo: queratosis liquenoide, liquen plano, necrólisis epidérmica tóxica, síndrome de eritrodisestesia palmar-plantar, erupción medicamentosa con eosinofilia y síntomas sistémicos (DRESS), pseudoporfiria, pénfigo
Trastornos vasculares: trombosis/embolia, shock anafiláctico
1Incluyendo algunas muertes.
7 INTERACCIONES MEDICAMENTOSAS
7.1 Agentes que inducen el metabolismo de CYP3A
La administración concomitante de mesilato de imatinib e inductores fuertes de CYP3A4 puede reducir la exposición total a imatinib; considere agentes alternativos [ver Farmacología clínica (12.3)].
7.2 Agentes que inhiben el metabolismo de CYP3A
La administración concomitante de mesilato de imatinib e inhibidores fuertes de CYP3A4 puede resultar en un aumento significativo de la exposición a imatinib. El jugo de toronja también puede aumentar las concentraciones plasmáticas de imatinib; evite el jugo de toronja [ver Farmacología clínica (12.3)].
7.3 Interacciones con medicamentos metabolizados por CYP3A4
El mesilato de imatinib aumentará la concentración plasmática de medicamentos metabolizados por CYP3A4 (por ejemplo, triazolo-benzodiazepinas, bloqueadores de los canales de calcio de dihidropiridina, ciertos inhibidores de la HMG-CoA reductasa, etc.). Tenga precaución al administrar mesilato de imatinib con sustratos de CYP3A4 que tienen un estrecho margen terapéutico.
Debido a que la warfarina se metaboliza por CYP2C9 y CYP3A4, use heparina de bajo peso molecular o estándar en lugar de warfarina en pacientes que requieren anticoagulación [ver Farmacología clínica (12.3)].
7.4 Interacciones con medicamentos metabolizados por CYP2D6
Tenga precaución al administrar mesilato de imatinib con sustratos de CYP2D6 que tienen un estrecho margen terapéutico.
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de riesgos
El mesilato de imatinib puede causar daño fetal cuando se administra a una mujer embarazada, según datos humanos y animales. No hay estudios clínicos sobre el uso de mesilato de imatinib en mujeres embarazadas. Se han recibido informes posteriores a la comercialización de abortos espontáneos y anomalías congénitas en mujeres que han estado expuestas al mesilato de imatinib durante el embarazo. Los estudios de reproducción en ratas han demostrado que el mesilato de imatinib indujo teratogenicidad y un aumento en la incidencia de anomalías congénitas después de la exposición prenatal al mesilato de imatinib en dosis iguales a la dosis humana máxima recomendada de 800 mg / día basada en el área de superficie corporal. Aconsejar a las mujeres que eviten el embarazo cuando toman mesilato de imatinib. Si este fármaco se usa durante el embarazo o si la paciente se queda embarazada mientras toma este fármaco, informar a la paciente del peligro potencial para el feto.
El riesgo de fondo de defectos de nacimiento mayores y abortos espontáneos en la población indicada no se conoce; sin embargo, en la población general de los EE. UU., el riesgo de fondo estimado de defectos de nacimiento mayores en embarazos clínicamente reconocidos es del 2% al 4% y el de abortos espontáneos es del 15% al 20%.
Datos
Datos animales
En estudios de desarrollo embriofetal en ratas y conejos, los animales embarazados recibieron dosis orales de mesilato de imatinib de hasta 100 mg / kg / día y 60 mg / kg / día, respectivamente, durante el período de organogénesis.
En ratas, el mesilato de imatinib fue teratogénico a 100 mg / kg / día (aproximadamente igual a la dosis humana máxima de 800 mg / día basada en el área de superficie corporal), el número de fetos con encefalocele y exencefalia fue mayor que los valores históricos de control y estos hallazgos se asociaron con huesos craneales faltantes o subdesarrollados. Se asociaron pesos corporales fetales medios más bajos con una osificación esquelética retardada.
En conejos, a dosis 1,5 veces mayores que la dosis humana máxima de 800 mg / día basada en el área de superficie corporal, no se observaron efectos en los parámetros reproductivos con respecto a los sitios de implantación, el número de fetos vivos, la proporción de sexos o el peso fetal. Los exámenes de los fetos no revelaron ningún cambio morfológico relacionado con el fármaco.
En un estudio de desarrollo prenatal y postnatal en ratas, las ratas embarazadas recibieron dosis orales de mesilato de imatinib durante la gestación (organogénesis) y la lactancia de hasta 45 mg / kg / día. Cinco animales desarrollaron un flujo vaginal rojo en el grupo de 45 mg / kg / día en los días 14 o 15 de la gestación, cuya importancia es desconocida ya que todas las hembras produjeron camadas viables y ninguna tuvo un aumento de la pérdida post-implantación. Otros efectos maternos observados solo en la dosis de 45 mg / kg / día (aproximadamente la mitad de la dosis humana máxima de 800 mg / día basada en el área de superficie corporal) incluyeron un número mayor de crías nacidas muertas y crías que murieron entre los días 0 y 4 después del parto. En la descendencia F1 en este mismo nivel de dosis, los pesos corporales medios se redujeron desde el nacimiento hasta el sacrificio final y el número de camadas que alcanzaron el criterio de separación del prepucio disminuyó ligeramente. No hubo otros efectos significativos en los parámetros de desarrollo o en las pruebas de comportamiento. La fertilidad F1 no se vio afectada, pero se observaron efectos reproductivos a 45 mg / kg / día, incluyendo un número mayor de reabsorciones y un número menor de fetos viables. El nivel sin efecto observable (NOEL) tanto para los animales maternos como para la generación F1 fue de 15 mg / kg / día.
8.2 Lactancia
Resumen de riesgos
La imatinib y su metabolito activo se excretan en la leche humana. Debido al potencial de reacciones adversas graves en los lactantes amamantados por el mesilato de imatinib, aconsejar a una mujer lactante que no amamante durante el tratamiento y durante 1 mes después de la última dosis.
Datos humanos
Según datos de 3 mujeres que amamantan y toman mesilato de imatinib, la relación leche:plasma es de aproximadamente 0,5 para la imatinib y de aproximadamente 0,9 para el metabolito activo. Considerando la concentración combinada de imatinib y metabolito activo, un lactante amamantado podría recibir hasta un 10% de la dosis terapéutica materna basada en el peso corporal.
8.3 Mujeres y hombres con potencial reproductivo
Los informes humanos posteriores a la comercialización y los estudios en animales han demostrado que el mesilato de imatinib es perjudicial para el feto en desarrollo[ver Uso en poblaciones específicas (8.1)].
Pruebas de embarazo
Comprobar el estado de embarazo en mujeres con potencial reproductivo antes de iniciar el tratamiento con mesilato de imatinib.
Anticoncepción
Mujeres
Aconsejar a las pacientes mujeres con potencial reproductivo que utilicen anticoncepción eficaz (métodos que resultan en tasas de embarazo inferiores al 1%) cuando usen mesilato de imatinib durante el tratamiento y durante catorce días después de interrumpir el tratamiento con mesilato de imatinib [ver Uso en poblaciones específicas (8.1)].
Infertilidad
No se ha estudiado el riesgo de infertilidad en mujeres o hombres con potencial reproductivo en humanos. En un estudio en ratas, la fertilidad en machos y hembras no se vio afectada [ver Toxicología no clínica (13)].
8.4 Uso pediátrico
La seguridad y eficacia del mesilato de imatinib se han demostrado en pacientes pediátricos con LMC Ph + en fase crónica recién diagnosticada y ALL Ph + [ver Estudios clínicos (14.2, 14.4)]. No hay datos en niños menores de 1 año de edad.
8.5 Uso geriátrico
En los estudios clínicos de LMC, aproximadamente el 20 % de los pacientes eran mayores de 65 años. En el estudio de pacientes con LMC recién diagnosticada, el 6 % de los pacientes eran mayores de 65 años. La frecuencia de edema fue mayor en pacientes mayores de 65 años en comparación con pacientes más jóvenes; no se observaron otras diferencias en el perfil de seguridad [ver Advertencias y precauciones (5.1)]. La eficacia de imatinib mesilato fue similar en pacientes mayores y jóvenes.
En el estudio de GIST irresecable o metastásico, el 16 % de los pacientes eran mayores de 65 años. No se observaron diferencias evidentes en el perfil de seguridad o eficacia en pacientes mayores de 65 años en comparación con pacientes más jóvenes, pero el pequeño número de pacientes no permite un análisis formal.
En el estudio adyuvante de GIST, 221 pacientes (31 %) eran mayores de 65 años. No se observaron diferencias en el perfil de seguridad en pacientes mayores de 65 años en comparación con pacientes más jóvenes, con la excepción de una mayor frecuencia de edema. La eficacia de imatinib mesilato fue similar en pacientes mayores de 65 años y pacientes más jóvenes.
8.6 Insuficiencia hepática
El efecto de la insuficiencia hepática en la farmacocinética tanto de imatinib como de su principal metabolito, CGP74588, se evaluó en 84 pacientes con cáncer con diversos grados de insuficiencia hepática a dosis de imatinib que oscilaban entre 100 mg y 800 mg.
La insuficiencia hepática leve y moderada no influye en la exposición a imatinib y CGP74588. En pacientes con insuficiencia hepática grave, la Cmáx. y el área bajo la curva (AUC) de imatinib aumentaron en un 63 % y un 45 % y la Cmáx. y el AUC de CGP74588 aumentaron en un 56 % y un 55 %, en relación con los pacientes con función hepática normal [ver Farmacología clínica (12.3)]. Reduzca la dosis en un 25 % para pacientes con insuficiencia hepática grave [ver Dosificación y administración (2.12)].
Tabla 16: Clasificación de la función hepática
|
Prueba de función hepática |
Normal (n=14) |
Leve (n=30) |
Moderada (n=20) |
Grave (n=20) |
|
Bilirrubina total |
menor o igual al LSN |
mayor de 1.0 a 1.5 veces el LSN |
mayor de 1.5 a 3 veces el LSN |
mayor de 3 a 10 veces el LSN |
|
SGOT |
menor o igual al LSN |
mayor que el LSN (puede ser normal si la bilirrubina total es mayor que el LSN) |
Cualquiera |
Cualquiera |
|
Abreviatura: SGOT, transaminasa glutámico-oxalacética sérica ahora se conoce como aspartato aminotransferasa (AST); LSN, límite superior normal para la institución. |
||||
8.7 Insuficiencia renal
El efecto de la insuficiencia renal en la farmacocinética de imatinib se evaluó en 59 pacientes con cáncer y diversos grados de insuficiencia renal a dosis únicas y en estado estacionario de imatinib que oscilaron entre 100 y 800 mg/día. La exposición media a imatinib (AUC normalizada por dosis) en pacientes con insuficiencia renal leve y moderada aumentó de 1.5 a 2 veces en comparación con los pacientes con función renal normal. No hay datos suficientes en pacientes con insuficiencia renal grave [ver Farmacología clínica (12.3)]. Son necesarias reducciones de dosis para pacientes con insuficiencia renal moderada y grave [ver Dosificación y administración (2.12)].
Tabla 17: Clasificación de la función renal
|
Disfunción renal |
Pruebas de función renal |
|
Leve |
CrCL = 40 a 59 mL/min |
|
Moderada |
CrCL = 20 a 39 mL/min |
|
Grave |
CrCL = menos de 20 mL/min |
|
Abreviatura: CrCL, aclaramiento de creatinina. |
|
10 SOBREDOSIS
La experiencia con dosis superiores a 800 mg es limitada. Se han notificado casos aislados de sobredosis de imatinib mesilato. En caso de sobredosis, observe al paciente y administre el tratamiento de apoyo adecuado.
Sobredosis en adultos
1.200 a 1.600 mg (duración variable entre 1 y 10 días): Náuseas, vómitos, diarrea, erupción eritematosa, edema, hinchazón, fatiga, espasmos musculares, trombocitopenia, pancitopenia, dolor abdominal, dolor de cabeza, disminución del apetito.
1.800 a 3.200 mg (hasta 3.200 mg diarios durante 6 días): Debilidad, mialgia, aumento de la CPK, aumento de la bilirrubina, dolor gastrointestinal.
6.400 mg (dosis única): Un caso en la literatura informó de un paciente que experimentó náuseas, vómitos, dolor abdominal, pirexia, hinchazón facial, disminución del recuento de neutrófilos, aumento de las transaminasas.
8 a 10 g (dosis única): Se han notificado vómitos y dolor gastrointestinal.
Un paciente con crisis blástica mieloide experimentó elevaciones de grado 1 de la creatinina sérica, ascitis de grado 2 y niveles elevados de transaminasas hepáticas, y elevaciones de grado 3 de la bilirrubina después de tomar inadvertidamente 1.200 mg de imatinib mesilato al día durante 6 días. La terapia se interrumpió temporalmente y se produjo una reversión completa de todas las anomalías en el plazo de una semana. El tratamiento se reanudó a una dosis de 400 mg al día sin recurrencia de reacciones adversas. Otro paciente desarrolló calambres musculares graves después de tomar 1.600 mg de imatinib mesilato al día durante 6 días. La resolución completa de los calambres musculares se produjo tras la interrupción de la terapia y el tratamiento se reanudó posteriormente. Otro paciente al que se le recetaron 400 mg al día, tomó 800 mg de imatinib mesilato el día 1 y 1.200 mg el día 2. La terapia se interrumpió, no se produjeron reacciones adversas y el paciente reanudó la terapia.
Sobredosis pediátrica
Un niño de 3 años expuesto a una dosis única de 400 mg experimentó vómitos, diarrea y anorexia; y otro niño de 3 años expuesto a una dosis única de 980 mg experimentó disminución del recuento de glóbulos blancos (WBC) y diarrea.
11 DESCRIPCIÓN
Imatinib es un inhibidor de la quinasa de molécula pequeña. Las tabletas de imatinib mesilato recubiertas con película se suministran como tabletas de 100 mg y 400 mg para administración oral. Cada tableta de 100 mg contiene 119,5 mg de imatinib mesilato, equivalente a 100 mg de imatinib base libre. Cada tableta de 400 mg contiene 478 mg de imatinib mesilato, equivalente a 400 mg de imatinib base libre. El imatinib mesilato se designa químicamente como 4-[(4-metil-1-piperazinil)metil]-N-[4-metil-3-[[4-(3-piridinil)-2-pirimidinil]amino]-fenil]benzamida metanosulfonato y su fórmula estructural es:
El imatinib mesilato es un polvo cristalino de blanco a blanquecino a marrón o amarillento. Su fórmula molecular es C29H31N7O • CH4SO3 y su peso molecular es 589,7 g/mol. El imatinib mesilato es soluble en tampones acuosos menores o iguales a pH 5,5, pero es muy poco soluble a insoluble en tampones acuosos neutros/alcalinos. En solventes no acuosos, la sustancia farmacológica es libremente soluble a muy poco soluble en dimetilsulfóxido, metanol y etanol, pero es insoluble en n-octanol, acetona y acetonitrilo.
Ingredientes inactivos: dióxido de silicio coloidal, crospovidona, estearato de magnesio, celulosa microcristalina, povidona. Recubrimiento de la tableta: óxido de hierro rojo, óxido de hierro amarillo, hipromelosa, polietilenglicol, talco y dióxido de titanio.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
El mesilato de imatinib es un inhibidor de la proteína tirosina quinasa que inhibe la tirosina quinasa BCR-ABL, la tirosina quinasa anormal constitutiva creada por la anomalía del cromosoma Filadelfia en la LMC. El imatinib inhibe la proliferación e induce la apoptosis en líneas celulares positivas a BCR-ABL, así como en células leucémicas frescas de pacientes con leucemia mieloide crónica positiva al cromosoma Filadelfia. El imatinib inhibe la formación de colonias en ensayos que utilizan muestras de sangre periférica y médula ósea ex vivo de pacientes con LMC.
In vivo, el imatinib inhibe el crecimiento tumoral de células mieloides murinas transfectadas con BCR-ABL, así como de líneas de leucemia positivas a BCR-ABL derivadas de pacientes con LMC en crisis blástica.
El imatinib también es un inhibidor de las tirosina quinasas receptoras para el factor de crecimiento derivado de plaquetas (PDGF) y el factor de células madre (SCF), c-Kit, e inhibe los eventos celulares mediados por PDGF y SCF. In vitro, el imatinib inhibe la proliferación e induce la apoptosis en células GIST, que expresan una mutación activadora de c-Kit.
12.3 Farmacocinética
La farmacocinética del mesilato de imatinib se ha evaluado en estudios en sujetos sanos y en estudios farmacocinéticos poblacionales en más de 900 pacientes. La farmacocinética del mesilato de imatinib es similar en pacientes con LMC y GIST.
Absorción y distribución
El imatinib se absorbe bien después de la administración oral, alcanzándose la Cmax entre 2 y 4 horas después de la dosis. La biodisponibilidad absoluta media es del 98%. El AUC media del imatinib aumenta proporcionalmente con el aumento de las dosis, que van de 25 mg a 1.000 mg. No hay cambios significativos en la farmacocinética del imatinib con la administración repetida, y la acumulación es de 1,5 a 2,5 veces en estado estacionario cuando el mesilato de imatinib se administra una vez al día. A concentraciones clínicamente relevantes de imatinib, la unión a las proteínas plasmáticas en experimentos in vitro es de aproximadamente el 95%, principalmente a la albúmina y a la α1-glucoproteína ácida.
Eliminación
Metabolismo
El CYP3A4 es la principal enzima responsable del metabolismo del imatinib. Otras enzimas del citocromo P450, como CYP1A2, CYP2D6, CYP2C9 y CYP2C19, desempeñan un papel menor en su metabolismo. El principal metabolito activo circulante en humanos es el derivado de piperazina N-desmetilado, formado predominantemente por CYP3A4. Muestra una potencia in vitro similar a la del imatinib original. El AUC plasmático de este metabolito es de aproximadamente el 15% del AUC del imatinib. La unión a proteínas plasmáticas del metabolito N-desmetilado CGP74588 es similar a la del compuesto original. Los estudios con microsomas hepáticos humanos demostraron que el mesilato de imatinib es un potente inhibidor competitivo de CYP2C9, CYP2D6 y CYP3A4/5 con valores de Ki de 27, 7,5 y 8 μM, respectivamente.
Excreción
La eliminación del imatinib se produce principalmente en las heces, principalmente en forma de metabolitos. Basándose en la recuperación del compuesto(s) después de una dosis oral de imatinib marcado con 14C, aproximadamente el 81% de la dosis se eliminó en 7 días, en las heces (68% de la dosis) y en la orina (13% de la dosis). El imatinib sin cambios representó el 25% de la dosis (5% en orina, 20% en heces), el resto fueron metabolitos.
Tras la administración oral en voluntarios sanos, las semividas de eliminación del imatinib y su principal metabolito activo, el derivado N-desmetilado (CGP74588), son de aproximadamente 18 y 40 horas, respectivamente.
Típicamente, se espera que el aclaramiento del imatinib en un paciente de 50 años que pesa 50 kg sea de 8 L/h, mientras que para un paciente de 50 años que pesa 100 kg el aclaramiento aumentará a 14 L/h. La variabilidad interindividual del 40% en el aclaramiento no justifica el ajuste inicial de la dosis en función del peso corporal y/o la edad, pero indica la necesidad de un seguimiento estrecho de la toxicidad relacionada con el tratamiento.
Poblaciones específicas
Insuficiencia hepática
El efecto de la insuficiencia hepática en la farmacocinética tanto del imatinib como de su principal metabolito, CGP74588, se evaluó en 84 pacientes con cáncer y diferentes grados de insuficiencia hepática [ver Uso en poblaciones específicas (8.6)] a dosis de imatinib que iban de 100 mg a 800 mg. La exposición tanto al imatinib como al CGP74588 fue comparable entre cada uno de los grupos con insuficiencia hepática leve y moderada y el grupo normal. Los pacientes con insuficiencia hepática grave tienden a tener una mayor exposición tanto al imatinib como a su metabolito que los pacientes con función hepática normal. En estado estacionario, la Cmax/dosis media y el AUC/dosis media del imatinib aumentaron en aproximadamente un 63% y un 45%, respectivamente, en pacientes con insuficiencia hepática grave en comparación con los pacientes con función hepática normal. La Cmax/dosis media y el AUC/dosis media del CGP74588 aumentaron en aproximadamente un 56% y un 55%, respectivamente, en pacientes con insuficiencia hepática grave en comparación con los pacientes con función hepática normal. Es necesario reducir la dosis en pacientes con insuficiencia hepática grave [ver Dosificación y administración (2.12)].
Insuficiencia renal
Se evaluó el efecto del deterioro renal en la farmacocinética de imatinib en 59 pacientes con cáncer con diversos grados de deterioro renal [ver Uso en poblaciones específicas (8.7)] a dosis únicas y en estado estacionario de imatinib que oscilan entre 100 y 800 mg/día. La exposición media a imatinib (AUC normalizada por la dosis) en pacientes con deterioro renal leve y moderado aumentó de 1,5 a 2 veces en comparación con los pacientes con función renal normal. Las AUC no aumentaron para dosis superiores a 600 mg en pacientes con deterioro renal leve. Las AUC no aumentaron para dosis superiores a 400 mg en pacientes con deterioro renal moderado. Dos pacientes con deterioro renal grave recibieron una dosis de 100 mg/día y sus exposiciones fueron similares a las observadas en pacientes con función renal normal que recibieron 400 mg/día. Son necesarias reducciones de la dosis para pacientes con deterioro renal moderado y grave [ver Dosificación y administración (2.12)].
Uso pediátrico
Al igual que en los pacientes adultos, imatinib se absorbió rápidamente después de la administración oral en pacientes pediátricos, con una Cmax de 2 a 4 horas. El aclaramiento oral aparente fue similar a los valores de los adultos (11,0 L/h/m2 en niños frente a 10,0 L/h/m2 en adultos), al igual que la semivida (14,8 horas en niños frente a 17,1 horas en adultos). La dosificación en niños a 260 mg/m2 y 340 mg/m2 logró un AUC similar a la dosis de 400 mg en adultos. La comparación de AUC en el día 8 frente al día 1 a niveles de dosis de 260 mg/m2 y 340 mg/m2 reveló una acumulación de fármacos de 1,5 y 2,2 veces, respectivamente, después de la dosificación repetida una vez al día. La AUC media de imatinib no aumentó proporcionalmente con el aumento de la dosis.
Basándose en el análisis farmacocinético poblacional agrupado en pacientes pediátricos con trastornos hematológicos (LMC, ALL Ph+ u otros trastornos hematológicos tratados con imatinib), el aclaramiento de imatinib aumenta con el aumento de la BSA. Después de corregir el efecto de la BSA, otros datos demográficos, como la edad, el peso corporal y el índice de masa corporal, no tuvieron efectos clínicamente significativos en la exposición a imatinib. El análisis confirmó que la exposición a imatinib en pacientes pediátricos que recibieron 260 mg/m2 una vez al día (sin exceder los 400 mg una vez al día) o 340 mg/m2 una vez al día (sin exceder los 600 mg una vez al día) fueron similares a las de los pacientes adultos que recibieron imatinib 400 mg o 600 mg una vez al día.
Interacciones medicamentosas
Agentes que inducen el metabolismo del CYP3A
El pretratamiento de voluntarios sanos con dosis múltiples de rifampicina seguido de una dosis única de mesilato de imatinib, aumentó el aclaramiento oral de la dosis de mesilato de imatinib en 3,8 veces, lo que disminuyó significativamente (p menor que 0,05) la Cmax y el AUC medias.
Se observaron hallazgos similares en pacientes que recibieron de 400 a 1.200 mg/día de mesilato de imatinib de forma concomitante con fármacos antiepilépticos inductores de enzimas (EIAED) (por ejemplo, carbamazepina, oxcarbamazepina, fenitoína, fosfenitoína, fenobarbital y primidona). La AUC media normalizada por la dosis para imatinib en los pacientes que recibieron EIAED disminuyó un 73% en comparación con los pacientes que no recibieron EIAED.
La administración concomitante de mesilato de imatinib y hierba de San Juan condujo a una reducción del 30% en el AUC de imatinib.
Considere agentes terapéuticos alternativos con un menor potencial de inducción enzimática en pacientes cuando esté indicada la rifampicina u otros inductores del CYP3A4. Se han administrado dosis de mesilato de imatinib de hasta 1.200 mg/día (600 mg dos veces al día) a pacientes que reciben inductores fuertes del CYP3A4 de forma concomitante [ver Dosificación y administración (2.12)].
Agentes que inhiben el metabolismo del CYP3A
Hubo un aumento significativo en la exposición a imatinib (la Cmax y el AUC medias aumentaron un 26% y un 40%, respectivamente) en sujetos sanos cuando el mesilato de imatinib se administró de forma conjunta con una dosis única de ketoconazol (un inhibidor del CYP3A4). Se recomienda precaución al administrar mesilato de imatinib con inhibidores fuertes del CYP3A4 (por ejemplo, ketoconazol, itraconazol, claritromicina, atazanavir, indinavir, nefazodona, nelfinavir, ritonavir, saquinavir, telithromicina y voriconazol). El zumo de pomelo también puede aumentar las concentraciones plasmáticas de imatinib y debe evitarse.
Interacciones con fármacos metabolizados por el CYP3A4
El mesilato de imatinib aumenta la Cmax y el AUC medias de simvastatina (sustrato del CYP3A4) 2 y 3,5 veces, respectivamente, lo que sugiere una inhibición del CYP3A4 por el mesilato de imatinib. Se recomienda especial precaución al administrar mesilato de imatinib con sustratos del CYP3A4 que tienen un estrecho margen terapéutico (por ejemplo, alfentanilo, ciclosporina, diergotamina, ergotamina, fentanilo, pimozida, quinidina, sirolimus o tacrolimus).
El mesilato de imatinib aumentará la concentración plasmática de otros fármacos metabolizados por el CYP3A4 (por ejemplo, triazolo-benzodiazepinas, bloqueadores de los canales de calcio de dihidropiridina, ciertos inhibidores de la HMG-CoA reductasa, etc.).
Debido a que la warfarina se metaboliza por el CYP2C9 y el CYP3A4, los pacientes que requieren anticoagulación deben recibir heparina de bajo peso molecular o estándar en lugar de warfarina.
Interacciones con fármacos metabolizados por el CYP2D6
El mesilato de imatinib aumentó la Cmax y el AUC medias de metoprolol en aproximadamente un 23%, lo que sugiere que el mesilato de imatinib tiene un efecto inhibidor débil sobre el metabolismo mediado por el CYP2D6. Sin embargo, no es necesario ajustar la dosis, pero se recomienda precaución al administrar mesilato de imatinib con sustratos del CYP2D6 que tienen un estrecho margen terapéutico.
Interacciones con el paracetamol
In vitro, el mesilato de imatinib inhibe la vía del O-glucurónido del paracetamol (Ki 58,5 μM). La administración conjunta de mesilato de imatinib (400 mg/día durante 8 días) con paracetamol (1.000 mg en dosis única en el día 8) en pacientes con LMC no produjo ningún cambio en la farmacocinética del paracetamol. La farmacocinética del mesilato de imatinib no se alteró en presencia de una dosis única de paracetamol. No existen datos farmacocinéticos ni de seguridad sobre el uso concomitante de mesilato de imatinib a dosis superiores a 400 mg/día o el uso crónico concomitante de paracetamol y mesilato de imatinib.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
En el estudio de carcinogenicidad en ratas de 2 años, la administración de imatinib a 15, 30 y 60 mg/kg/día resultó en una reducción estadísticamente significativa en la longevidad de los machos a 60 mg/kg/día y las hembras a mayor o igual a 30 mg/kg/día. Los órganos diana para los cambios neoplásicos fueron los riñones (túbulos renales y pelvis renal), la vejiga urinaria, la uretra, la glándula prepucial y clitoriana, el intestino delgado, las glándulas paratiroides, las glándulas suprarrenales y el estómago no glandular. No se observaron lesiones neoplásicas a: 30 mg/kg/día para los riñones, la vejiga urinaria, la uretra, el intestino delgado, las glándulas paratiroides, las glándulas suprarrenales y el estómago no glandular, y 15 mg/kg/día para la glándula prepucial y clitoriana. El papiloma/carcinoma de la glándula prepucial/clitoriana se observó a 30 y 60 mg/kg/día, lo que representa aproximadamente 0,5 a 4 o 0,3 a 2,4 veces la exposición diaria humana (basada en AUC) a 400 mg/día u 800 mg/día, respectivamente, y 0,4 a 3,0 veces la exposición diaria en niños (basada en AUC) a 340 mg/m2. El adenoma/carcinoma del túbulo renal, los neoplasmas de células transicionales de la pelvis renal, los papilomas de células transicionales de la vejiga urinaria y la uretra, los adenocarcinomas del intestino delgado, los adenomas de las glándulas paratiroides, los tumores medulares benignos y malignos de las glándulas suprarrenales y los papilomas/carcinomas del estómago no glandular se observaron a 60 mg/kg/día. No se conoce la relevancia de estos hallazgos en el estudio de carcinogenicidad en ratas para los humanos. Se obtuvieron efectos genotóxicos positivos para imatinib en un ensayo de células de mamíferos in vitro (ovario de hámster chino) para clastogenicidad (aberraciones cromosómicas) en presencia de activación metabólica. Dos intermediarios del proceso de fabricación, que también están presentes en el producto final, son positivos para la mutagénesis en el ensayo de Ames. Uno de estos intermediarios también fue positivo en el ensayo de linfoma de ratón. Imatinib no fue genotóxico cuando se probó en un ensayo de células bacterianas in vitro (prueba de Ames), un ensayo de células de mamíferos in vitro (linfoma de ratón) y un ensayo de micronúcleos de rata in vivo.
En un estudio de fertilidad, las ratas macho fueron dosificadas durante 70 días antes del apareamiento y las ratas hembra fueron dosificadas 14 días antes del apareamiento y hasta el día 6 de gestación. El peso testicular y epididimario y el porcentaje de espermatozoides móviles disminuyeron a 60 mg/kg, aproximadamente tres cuartas partes de la dosis clínica máxima de 800 mg/día basada en BSA. Esto no se observó a dosis menores o iguales a 20 mg/kg (una cuarta parte de la dosis humana máxima de 800 mg). La fertilidad de las ratas macho y hembra no se vio afectada.
La fertilidad no se vio afectada en el estudio preclínico de fertilidad y desarrollo embrionario temprano, aunque se observaron pesos testiculares y epididimarios más bajos, así como una reducción en el número de espermatozoides móviles en las ratas macho de dosis alta. En el estudio preclínico pre y postnatal en ratas, la fertilidad en la descendencia de primera generación tampoco se vio afectada por el mesilato de imatinib.
13.2 Toxicología y/o Farmacología Animal
Toxicidades por uso a largo plazo
Es importante considerar las posibles toxicidades sugeridas por los estudios en animales, específicamente, la toxicidad hepática, renal y cardíaca y la inmunosupresión. Se observó toxicidad hepática grave en perros tratados durante 2 semanas, con enzimas hepáticas elevadas, necrosis hepatocelular, necrosis de los conductos biliares e hiperplasia de los conductos biliares. Se observó toxicidad renal en monos tratados durante 2 semanas, con mineralización focal y dilatación de los túbulos renales y nefrosis tubular. Se observó un aumento del nitrógeno ureico en sangre (BUN) y la creatinina en varios de estos animales. Se observó un aumento de la tasa de infecciones oportunistas con el tratamiento crónico con imatinib en estudios con animales de laboratorio. En un estudio de 39 semanas con monos, el tratamiento con imatinib resultó en un empeoramiento de las infecciones por malaria normalmente suprimidas en estos animales. Se observó linfopenia en animales (como en humanos). Se identificaron toxicidades adicionales a largo plazo en un estudio de 2 años con ratas. El examen histopatológico de las ratas tratadas que murieron en el estudio reveló miocardiopatía (ambos sexos), nefropatía progresiva crónica (hembras) y papiloma de la glándula prepucial como las principales causas de muerte o razones para el sacrificio. Las lesiones no neoplásicas observadas en este estudio de 2 años que no se identificaron en estudios preclínicos anteriores fueron el sistema cardiovascular, el páncreas, los órganos endocrinos y los dientes. Los cambios más importantes incluyeron hipertrofia y dilatación cardíaca, lo que condujo a signos de insuficiencia cardíaca en algunos animales.
14 ESTUDIOS CLÍNICOS
14.1 Leucemia mieloide crónica
Fase crónica, recién diagnosticada:
Se ha llevado a cabo un estudio aleatorio de fase 3, abierto, multicéntrico e internacional (imatimib mesilato frente a IFN + Ara-C) en pacientes con leucemia mieloide crónica (LMC) en fase crónica recién diagnosticada con cromosoma Filadelfia positivo (Ph +). Este estudio comparó el tratamiento con imatimib mesilato de un solo agente o una combinación de interferón alfa (IFN) más citarabina (Ara-C). Los pacientes podían cambiar al brazo de tratamiento alternativo si no mostraban una respuesta hematológica completa (RHC) a los 6 meses, una respuesta citogenética mayor (RCM) a los 12 meses o si perdían una RHC o RCM. Los pacientes con un aumento de glóbulos blancos o una intolerancia grave al tratamiento también podían cambiar al brazo de tratamiento alternativo con el permiso del comité de monitorización del estudio (SMC). En el brazo de imatimib mesilato, los pacientes fueron tratados inicialmente con 400 mg diarios. Se permitieron aumentos de dosis de 400 mg diarios a 600 mg diarios y luego de 600 mg diarios a 800 mg diarios. En el brazo de IFN, los pacientes fueron tratados con una dosis objetivo de IFN de 5 MIU / m2 / día por vía subcutánea en combinación con Ara-C 20 mg / m2 / día por vía subcutánea durante 10 días / mes.
Un total de 1.106 pacientes fueron aleatorizados en 177 centros de 16 países, 553 en cada brazo. Las características basales estaban bien equilibradas entre los dos brazos. La edad mediana era de 51 años (rango, 18 a 70 años), con un 21,9% de pacientes de 60 años o más. Había un 59% de hombres y un 41% de mujeres; un 89,9% de pacientes caucásicos y un 4,7% de pacientes negros. En el punto de corte para este análisis (7 años después de que se hubiera reclutado al último paciente), la duración mediana del tratamiento de primera línea fue de 82 y 8 meses en el brazo de imatimib mesilato y el brazo de IFN, respectivamente. La duración mediana del tratamiento de segunda línea con imatimib mesilato fue de 64 meses. El sesenta por ciento de los pacientes aleatorizados a imatimib mesilato todavía están recibiendo tratamiento de primera línea. En estos pacientes, la dosis promedio de imatimib mesilato fue de 403 mg ± 57 mg. En general, en los pacientes que reciben imatimib mesilato de primera línea, la dosis diaria promedio administrada fue de 406 mg ± 76 mg. Debido a las interrupciones y los cambios de tratamiento, solo el 2% de los pacientes aleatorizados a IFN todavía estaban en tratamiento de primera línea. En el brazo de IFN, la retirada del consentimiento (14%) fue la razón más frecuente para la interrupción del tratamiento de primera línea, y la razón más frecuente para cambiar al brazo de imatimib mesilato fue la intolerancia grave al tratamiento (26%) y la progresión (14%).
El punto final de eficacia primario del estudio fue la supervivencia libre de progresión (SLP). La progresión se definió como cualquiera de los siguientes eventos: progresión a fase acelerada o crisis blástica (FA / CB), muerte, pérdida de RHC o RCM, o en pacientes que no alcanzaron una RHC un aumento de glóbulos blancos a pesar de un manejo terapéutico adecuado. El protocolo especificaba que el análisis de la progresión compararía la población con intención de tratar (ITT): los pacientes aleatorizados para recibir imatimib mesilato se compararon con los pacientes aleatorizados para recibir IFN. Los pacientes que cambiaron de tratamiento antes de la progresión no se censuraron en el momento del cambio, y los eventos que ocurrieron en estos pacientes después del cambio se atribuyeron al tratamiento aleatorizado original. La tasa estimada de supervivencia libre de progresión a 84 meses en la población ITT fue del 81,2% [95% CI: 78, 85] en el brazo de imatimib mesilato y del 60,6% [56, 65] en el brazo de IFN (p menor que 0,0001, prueba de log-rank), (Figura 1). Con 7 años de seguimiento, hubo 93 (16,8%) eventos de progresión en el brazo de imatimib mesilato: 37 (6,7%) progresión a FA / CB, 31 (5,6%) pérdida de RCM, 15 (2,7%) pérdida de RHC o aumento de glóbulos blancos y 10 (1,8%) muertes no relacionadas con LMC. En contraste, hubo 165 (29,8%) eventos en el brazo de IFN + Ara-C, de los cuales 130 ocurrieron durante el tratamiento de primera línea con IFN – Ara – C. La tasa estimada de pacientes libres de progresión a fase acelerada (FA) o crisis blástica (CB) a 84 meses fue del 92,5% [90, 95] en el brazo de imatimib mesilato en comparación con el 85,1%, [82, 89] (p menor o igual a 0,001) en el brazo de IFN, (Figura 2). Las tasas anuales de cualquier evento de progresión han disminuido con el tiempo en el tratamiento. La probabilidad de permanecer libre de progresión a 60 meses fue del 95% para los pacientes que estaban en respuesta citogenética completa (RCC) con respuesta molecular (reducción mayor o igual a 3 log en los transcritos BCR – ABL medidos por reacción en cadena de la polimerasa de transcriptasa inversa cuantitativa) a los 12 meses, en comparación con el 89% para los pacientes en RCC pero sin una respuesta molecular mayor y el 70% en los pacientes que no estaban en RCC en este punto de tiempo (p menor que 0,001).
Figura 1: Supervivencia Libre de Progresión (Principio ITT)
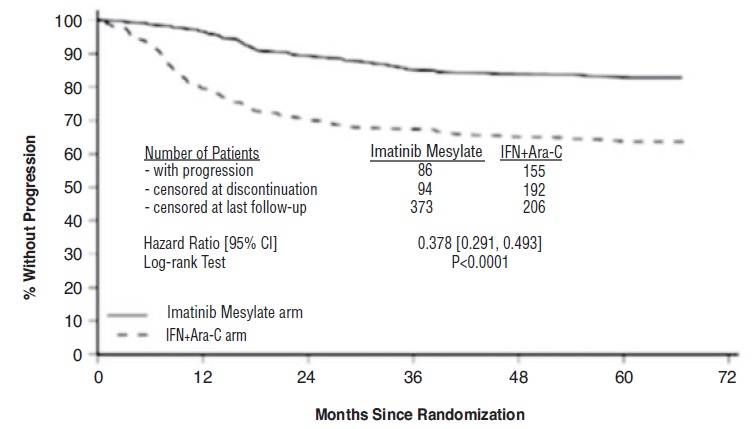
Figura 2: Tiempo hasta la Progresión a FA o CB (Principio ITT)

Un total de 71 (12,8%) y 85 (15,4%) pacientes murieron en el grupo de imatimib mesilato y el grupo de IFN + Ara – C, respectivamente. A los 84 meses, la supervivencia global estimada es del 86,4% (83, 90) frente al 83,3% (80, 87) en el grupo aleatorizado de imatimib mesilato y el grupo de IFN + Ara – C, respectivamente (p = 0,073 prueba de log – rank). La razón de riesgo es de 0,750 con un IC 95% de 0,547 a 1,028. Este punto final de tiempo hasta el evento puede verse afectado por la alta tasa de cambio de tratamiento de IFN + Ara – C a imatimib mesilato. La respuesta citogenética mayor, la respuesta hematológica, la evaluación de la enfermedad residual mínima (respuesta molecular), el tiempo hasta la fase acelerada o la crisis blástica y la supervivencia fueron los puntos finales secundarios principales. Los datos de respuesta se muestran en la Tabla 18. La respuesta hematológica completa, la respuesta citogenética mayor y la RCC también fueron estadísticamente significativamente más altas en el brazo de imatimib mesilato en comparación con el brazo de IFN + Ara – C (no se consideraron datos de cambio de tratamiento para la evaluación de las respuestas). El tiempo mediano hasta la RCC en los 454 respondedores fue de 6 meses (rango, 2 a 64 meses, percentiles 25th a 75th = 3 a 11 meses), con un 10% de las respuestas observadas solo después de 22 meses de tratamiento.
Tabla 18: Respuesta en el Estudio de LMC Recién Diagnosticada (Datos de 84 Meses)
|
Mejor Tasa de Respuesta |
Mesilato de Imatinib |
IFN+Ara−C |
|
n = 553 |
n = 553 |
|
|
Respuesta Hematológica1 |
||
|
CHR Rate n (%) |
||
|
534 (96.6%)* |
313 (56.6%)* |
|
|
[95% CI] |
||
|
[94.7%, 97.9%] |
[52.4%, 60.8%] |
|
|
Respuesta Citogenética2 |
||
|
Respuesta Citogenética Mayor n (%) |
||
|
472 (85.4%)* |
93 (16.8%)* |
|
|
[95% CI] |
||
|
[82.1%, 88.2%] |
[13.8%, 20.2%] |
|
|
No Confirmada3 |
||
|
88.6%* |
23.3%* |
|
|
Respuesta Citogenética Completa n (%) |
||
|
413 (74.7%)* |
36 (6.5%)* |
|
|
[95% CI] |
||
|
[70.8, 78.3] |
[4.6, 8.9] |
|
|
No Confirmada3 |
||
|
82.5%* |
11.6%* |
|
|
*p menor que 0.001, prueba exacta de Fischer. 1Criterios de respuesta hematológica (todas las respuestas deben confirmarse después de mayor o igual a 4 semanas): WBC menor que 10 x 109/L, plaquetas menor que 450 x 109/L, mielocito + metamielocito menor que 5% en sangre, sin blastos ni promielocitos en sangre, sin afectación extramedular. 2Criterios de respuesta citogenética (confirmados después de mayor o igual a 4 semanas): completa (0% de metafases Ph+) o parcial (1% a 35%). Una respuesta mayor (0% a 35%) combina respuestas completas y parciales. 3Respuesta citogenética no confirmada se basa en una sola evaluación citogenética de médula ósea, por lo tanto, las respuestas citogenéticas completas o parciales no confirmadas pueden haber tenido una respuesta citogenética menor en una evaluación posterior de médula ósea. |
||
La respuesta molecular se definió de la siguiente manera: en la sangre periférica, después de 12 meses de terapia, reducción de mayor o igual a 3 logaritmos en la cantidad de transcripciones BCR-ABL (medidas mediante la prueba de PCR cuantitativa en tiempo real con transcriptasa inversa) sobre una línea de base estandarizada. La respuesta molecular solo se evaluó en un subconjunto de pacientes que tuvieron una CCyR a los 12 meses o más tarde (N = 333). La tasa de respuesta molecular en pacientes que tuvieron una CCyR en el brazo de imatinib mesilato fue del 59% a los 12 meses y del 72% a los 24 meses.
Se utilizaron escalas de modificadores de respuesta biológica específicas del tratamiento, funcionales y físicas del instrumento FACT-BRM (Evaluación Funcional del Cáncer – Modificador de Respuesta Biológica) para evaluar los efectos generales informados por los pacientes de la toxicidad del interferón en 1.067 pacientes con LMC en fase crónica. Después de un mes de terapia hasta 6 meses de terapia, hubo una disminución del 13% al 21% en el índice mediano desde la línea de base en pacientes tratados con IFN, consistente con un aumento de los síntomas de toxicidad por IFN. No hubo cambios aparentes desde la línea de base en el índice mediano para los pacientes tratados con imatinib mesilato.
Se llevó a cabo un ensayo aleatorizado, multicéntrico, de etiqueta abierta (imatinib mesilato versus nilotinib) para determinar la eficacia del imatinib mesilato versus nilotinib en pacientes adultos con LMC-CP Ph+ confirmada citogenéticamente. Los pacientes estaban dentro de los 6 meses del diagnóstico y no habían sido tratados previamente para LMC-CP, excepto por hidroxiurea y/o anagrelida.
La eficacia se basó en un total de 846 pacientes: 283 pacientes en el grupo de imatinib mesilato 400 mg una vez al día, 282 pacientes en el grupo de nilotinib 300 mg dos veces al día, 281 pacientes en el grupo de nilotinib 400 mg dos veces al día.
La edad media fue de 46 años en el grupo de imatinib mesilato y de 47 años en ambos grupos de nilotinib, con un 12%, 13% y 10% de pacientes mayores o iguales a 65 años en los grupos de tratamiento de imatinib mesilato 400 mg una vez al día, nilotinib 300 mg dos veces al día y nilotinib 400 mg dos veces al día, respectivamente. Hubo ligeramente más pacientes masculinos que femeninos en todos los grupos (56%, 56% y 62% en los grupos de tratamiento de imatinib mesilato 400 mg una vez al día, nilotinib 300 mg dos veces al día y nilotinib 400 mg dos veces al día, respectivamente). Más del 60% de todos los pacientes eran caucásicos y el 25% eran asiáticos.
El análisis de datos primario se realizó cuando los 846 pacientes completaron 12 meses de tratamiento o lo interrumpieron antes. Los análisis posteriores se realizaron cuando los pacientes completaron 24, 36, 48 y 60 meses de tratamiento o lo interrumpieron antes. El tiempo medio de tratamiento fue de aproximadamente 61 meses en los tres grupos de tratamiento.
El criterio de valoración principal de eficacia fue la respuesta molecular mayor (MMR) a los 12 meses después del inicio de la medicación del estudio. La MMR se definió como menos o igual al 0,1% de BCR-ABL/ABL % según la escala internacional medida por RQ-PCR, lo que corresponde a una reducción mayor o igual a 3 log de la transcripción de BCR-ABL desde la línea de base estandarizada. Los criterios de valoración de eficacia se resumen en la Tabla 19.
Doce pacientes en el brazo de imatinib mesilato progresaron a fase acelerada o crisis blásticas (7 pacientes dentro de los primeros 6 meses, 2 pacientes dentro de los 6 a 12 meses, 2 pacientes dentro de los 12 a 18 meses y 1 paciente dentro de los 18 a 24 meses), mientras que dos pacientes en el brazo de nilotinib progresaron a fase acelerada o crisis blásticas (ambos dentro de los primeros 6 meses de tratamiento).
Tabla 19: Eficacia (MMR y CCyR) de Imatinib Mesilato Comparado con Nilotinib en LMC-CP Ph+ de Nuevo Diagnóstico
|
Imatinib Mesilato 400 mg una vez al día |
nilotinib 300 mg dos veces al día |
||
|
N = 283 |
N = 282 |
||
|
MMR a los 12 meses (IC del 95%) |
22% (17.6, 27.6) |
44% (38.4, 50.3) |
|
|
Valor Pa |
<0.0001 |
||
|
CCyRb a los 12 meses (IC del 95%) |
65% (59.2, 70.6) |
80% (75.0, 84.6) |
|
|
MMR a los 24 meses (IC del 95%) |
38% (31.8, 43.4) |
62% (55.8, 67.4) |
|
|
CCyRb a los 24 meses (IC del 95%) |
77% (71.7, 81.8) |
87% (82.4, 90.6) |
|
|
Abreviaturas: CCyR, respuesta citogenética completa; MMR, respuesta molecular mayor; LMC-CP Ph+, leucemia mieloide crónica positiva para el cromosoma Filadelfia – fase crónica. a Prueba CMH estratificada por grupo de riesgo de Sokal. b CCyR: 0% de metafases Ph+. Las respuestas citogenéticas se basaron en el porcentaje de metafases Ph-positivas entre mayor o igual a 20 células en metafase en cada muestra de médula ósea. |
|||
Al cabo de 60 meses, el MMR fue alcanzado por el 60% de los pacientes con imatinib mesilato y el 77% de los pacientes con nilotinib.
No se alcanzó la mediana de supervivencia global en ninguno de los grupos. En el momento del análisis final de 60 meses, la tasa de supervivencia estimada fue del 91,7% para los pacientes con imatinib mesilato y del 93,7% para los pacientes con nilotinib.
Fase crónica tardía de LMC y LMC en estadio avanzado: Se llevaron a cabo tres estudios internacionales, de etiqueta abierta, de un solo brazo, de fase 2 para determinar la seguridad y eficacia del imatinib mesilato en pacientes con LMC Ph+: 1) en la fase crónica después del fracaso de la terapia con IFN, 2) en la fase acelerada de la enfermedad o 3) en la crisis blástica mieloide. Aproximadamente el 45% de los pacientes eran mujeres y el 6% eran negros. En los estudios clínicos, el 38% al 40% de los pacientes tenían 60 años o más y el 10% al 12% de los pacientes tenían 70 años o más.
Fase crónica, tratamiento previo con interferón-alfa: 532 pacientes fueron tratados con una dosis inicial de 400 mg; se permitió la escalada de dosis a 600 mg. Los pacientes se distribuyeron en tres categorías principales según su respuesta al interferón previo: fracaso en lograr (dentro de los 6 meses) o pérdida de una respuesta hematológica completa (29%), fracaso en lograr (dentro de un año) o pérdida de una respuesta citogenética mayor (35%), o intolerancia al interferón (36%). Los pacientes habían recibido una mediana de 14 meses de tratamiento previo con IFN en dosis de 25 x 106 unidades/semana o más y estaban todos en la fase crónica tardía, con un tiempo mediano desde el diagnóstico de 32 meses. La eficacia se evaluó en base a la tasa de respuesta hematológica y mediante exámenes de médula ósea para evaluar la tasa de respuesta citogenética mayor (hasta el 35% de metafases Ph+) o CCyR (0% de metafases Ph+). La duración mediana del tratamiento fue de 29 meses, con el 81% de los pacientes tratados durante 24 meses o más (máximo = 31,5 meses). Los resultados de eficacia se presentan en la Tabla 20. Las tasas de respuesta citogenética mayor confirmada fueron más altas en pacientes con intolerancia al IFN (66%) y fracaso citogenético (64%), que en pacientes con fracaso hematológico (47%). Se logró una respuesta hematológica en el 98% de los pacientes con fracaso citogenético, en el 94% de los pacientes con fracaso hematológico y en el 92% de los pacientes intolerantes al IFN.
Fase acelerada: Se inscribieron 235 pacientes con enfermedad en fase acelerada. Estos pacientes cumplieron con uno o más de los siguientes criterios: 15% o más – menos del 30% de blastos en PB o BM; 30% o más de blastos + promielocitos en PB o BM; 20% o más de basófilos en PB; y menos de 100 x 109/L de plaquetas. Los primeros 77 pacientes se iniciaron con 400 mg, y los 158 pacientes restantes se iniciaron con 600 mg.
La eficacia se evaluó principalmente en base a la tasa de respuesta hematológica, reportada como respuesta hematológica completa, no evidencia de leucemia (es decir, la limpieza de blastos de la médula y la sangre, pero sin una recuperación completa de la sangre periférica como en las respuestas completas) o el retorno a la fase crónica de LMC. También se evaluaron las respuestas citogenéticas. La duración mediana del tratamiento fue de 18 meses, con el 45% de los pacientes tratados durante 24 meses o más (máximo = 35 meses). Los resultados de eficacia se presentan en la Tabla 20. Las tasas de respuesta en la LMC en fase acelerada fueron más altas en el grupo de dosis de 600 mg que en el grupo de 400 mg: respuesta hematológica (75% frente a 64%), respuesta citogenética mayor confirmada y no confirmada (31% frente a 19%).
Crisis blástica mieloide: Se inscribieron 260 pacientes con crisis blástica mieloide. Estos pacientes tenían 30% o más de blastos en PB o BM y/o afectación extramedular que no fuera el bazo o el hígado; 95 (37%) habían recibido quimioterapia previa para el tratamiento de la fase acelerada o la crisis blástica (“pacientes pretratados”), mientras que 165 (63%) no lo habían recibido (“pacientes no tratados”). Los primeros 37 pacientes se iniciaron con 400 mg; los 223 pacientes restantes se iniciaron con 600 mg.
La eficacia se evaluó principalmente en base a la tasa de respuesta hematológica, reportada como respuesta hematológica completa, no evidencia de leucemia o el retorno a la fase crónica de LMC utilizando los mismos criterios que para el estudio en la fase acelerada. También se evaluaron las respuestas citogenéticas. La duración mediana del tratamiento fue de 4 meses, con el 21% de los pacientes tratados durante 12 meses o más y el 10% durante 24 meses o más (máximo = 35 meses). Los resultados de eficacia se presentan en la Tabla 20. La tasa de respuesta hematológica fue más alta en pacientes no tratados que en pacientes tratados (36% frente a 22%, respectivamente) y en el grupo que recibió una dosis inicial de 600 mg en lugar de 400 mg (33% frente a 16%). La tasa de respuesta citogenética mayor confirmada y no confirmada también fue más alta en el grupo de dosis de 600 mg que en el grupo de 400 mg (17% frente a 8%).
Tabla 20: Respuesta en estudios de leucemia mieloide crónica
|
Fase crónica con fracaso de IFN (n = 532) 400 mg |
Fase acelerada (n = 235) 600 mg n = 158 400 mg n = 77 % de pacientes [IC 95%] |
Crisis blástica mieloide (n = 260) 600 mg n = 223 400 mg n = 37 |
|
|
Respuesta hematológica1 |
95% [92,3 a 96,3] |
71% [64,8 a 76,8] |
31% [25,2 a 36,8] |
|
Respuesta hematológica completa (RHC) |
95% |
38% |
7% |
|
No Evidence of Leukemia (NEL) |
No aplicable |
13% |
5% |
|
Return to Chronic Phase (RTC) |
No aplicable |
20% |
18% |
|
Major Cytogenetic Response2 |
60% [55.3 to 63.8] |
21% [16.2 to 27.1] |
7% [4.5 to 11.2] |
|
(Unconfirmed3) |
(65%) |
(27%) |
(15%) |
|
Complete4 (Unconfirmed3) |
39% (47%) |
16% (20%) |
2% (7%) |
|
Abbreviations: BM=bone marrow, PB=peripheral blood 1Hematologic response criteria (all responses to be confirmed after greater than or equal to 4 weeks): CHR:Chronic phase study [WBC less than 10 x 109/L, platelet less than 450 x 109/L, myelocytes + metamyelocytes less than 5% in blood, no blasts and promyelocytes in blood, basophils less than 20%, no extramedullary involvement] and in the accelerated and blast crisis studies [absolute neutrophil count (ANC) greater than or equal to 1.5 x 109/L, platelets greater than or equal to 100 x 109/L, no blood blasts, BM blasts less than 5% and no extramedullary disease]. NEL: Same criteria as for CHR but ANC greater than or equal to 1 x 109/L and platelets greater than or equal to 20 x 109/L (accelerated and blast crisis studies). RTC: less than 15% blasts BM and PB, less than 30% blasts + promyelocytes in BM and PB, less than 20% basophils in PB, no extramedullary disease other than spleen and liver (accelerated and blast crisis studies). 2Cytogenetic response criteria (confirmed after greater than or equal to 4 weeks): complete (0% Ph+ metaphases) or partial (1% to 35%). A major response (0% to 35%) combines both complete and partial responses. 3Unconfirmed cytogenetic response is based on a single bone marrow cytogenetic evaluation, therefore unconfirmed complete or partial cytogenetic responses might have had a lesser cytogenetic response on a subsequent bone marrow evaluation. 4Complete cytogenetic response confirmed by a second bone marrow cytogenetic evaluation performed at least 1 month after the initial bone marrow study. |
|||
El tiempo medio de respuesta hematológica fue de 1 mes. En la fase crónica tardía de la LMC, con un tiempo medio desde el diagnóstico de 32 meses, se estimó que el 87,8% de los pacientes que lograron una respuesta citogenética mayor (MCyR) mantuvieron su respuesta 2 años después de lograr su respuesta inicial. Después de 2 años de tratamiento, se estimó que el 85,4% de los pacientes estaban libres de progresión a la fase acelerada (AP) o la fase blástica (BC), y la supervivencia global estimada fue del 90,8% [88,3, 93,2]. En la fase acelerada, la duración mediana de la respuesta hematológica fue de 28,8 meses para los pacientes con una dosis inicial de 600 mg (16,5 meses para 400 mg). Se estimó que el 63,8% de los pacientes que lograron una MCyR todavía estaban en respuesta 2 años después de lograr la respuesta inicial. La supervivencia mediana fue de 20,9 [13,1, 34,4] meses para el grupo de 400 mg y no se alcanzó todavía para el grupo de 600 mg (p = 0,0097). Se estimó que el 46,2% [34,7, 57,7] frente al 65,8% [58,4, 73,3] de los pacientes seguían vivos después de 2 años de tratamiento en los grupos de dosis de 400 mg frente a 600 mg, respectivamente. En la crisis blástica, la duración mediana estimada de la respuesta hematológica es de 10 meses. Se estimó que el 27,2% [16,8, 37,7] de los respondientes hematológicos mantuvieron su respuesta 2 años después de lograr su respuesta inicial. La supervivencia mediana fue de 6,9 [5,8, 8,6] meses, y se estimó que el 18,3% [13,4, 23,3] de todos los pacientes con crisis blástica seguían vivos 2 años después del inicio del estudio.
Los resultados de eficacia fueron similares en hombres y mujeres y en pacientes menores y mayores de 65 años. Se observaron respuestas en pacientes negros, pero el número de pacientes negros era demasiado pequeño para permitir una comparación cuantitativa.
14.2 Leucemia mieloide crónica pediátrica
Un total de 51 pacientes pediátricos con LMC en fase crónica recién diagnosticada y sin tratamiento previo se inscribieron en un ensayo clínico de fase 2, abierto, multicéntrico y de un solo brazo. Los pacientes fueron tratados con imatinib mesilato 340 mg/m2/día, sin interrupciones en ausencia de toxicidad limitante de la dosis. Se observó una respuesta hematológica completa (CHR) en el 78% de los pacientes después de 8 semanas de tratamiento. La tasa de respuesta citogenética completa (CCyR) fue del 65%, comparable a los resultados observados en adultos. Además, se observó una respuesta citogenética parcial (PCyR) en el 16%. La mayoría de los pacientes que lograron una CCyR desarrollaron la CCyR entre los meses 3 y 10, con un tiempo medio de respuesta basado en la estimación de Kaplan-Meier de 6,74 meses. Se permitió que los pacientes fueran retirados del tratamiento del protocolo para someterse a un tratamiento alternativo, incluyendo el trasplante de células madre hematopoyéticas. Treinta y un niños recibieron trasplante de células madre. De los 31 niños, 5 fueron trasplantados después de la progresión de la enfermedad en el estudio y 1 se retiró del estudio durante la primera semana de tratamiento y recibió el trasplante aproximadamente 4 meses después de la retirada. Veinticinco niños se retiraron del tratamiento del protocolo para someterse a un trasplante de células madre después de recibir una mediana de 9 cursos de veintiocho días (rango, 4 a 24). De los 25 pacientes, 13 (52%) tenían CCyR y 5 (20%) tenían PCyR al final del tratamiento del protocolo.
Un estudio abierto, de un solo brazo, inscribió a 14 pacientes pediátricos con LMC en fase crónica Ph + recurrente después del trasplante de células madre o resistente a la terapia con interferón alfa. Estos pacientes no habían recibido previamente imatinib mesilato y tenían edades que oscilaban entre los 3 y los 20 años; 3 tenían entre 3 y 11 años, 9 tenían entre 12 y 18 años y 2 tenían más de 18 años. Los pacientes fueron tratados con dosis de 260 mg/m2/día (n = 3), 340 mg/m2/día (n = 4), 440 mg/m2/día (n = 5) y 570 mg/m2/día (n = 2). En los 13 pacientes para los que se dispone de datos citogenéticos, 4 lograron una respuesta citogenética mayor, 7 lograron una CCyR y 2 tuvieron una respuesta citogenética mínima.
En un segundo estudio, 2 de 3 pacientes con LMC en fase crónica Ph + resistente a la terapia con interferón alfa lograron una CCyR con dosis de 242 y 257 mg/m2/día.
14.3 Leucemia linfoblástica aguda
Se estudiaron un total de 48 pacientes con leucemia linfoblástica aguda positiva para el cromosoma Filadelfia (Ph + ALL) con enfermedad recidivante/refractaria, de los cuales 43 recibieron la dosis recomendada de imatinib mesilato de 600 mg/día. Además, 2 pacientes con Ph + ALL recidivante/refractaria recibieron imatinib mesilato 600 mg/día en un estudio de fase 1.
Las tasas de respuesta hematológica y citogenética confirmadas y no confirmadas para los 43 pacientes del estudio de fase 2 de Ph + ALL recidivante/refractaria y para los 2 pacientes del estudio de fase 1 se muestran en la Tabla 21. La duración mediana de la respuesta hematológica fue de 3,4 meses y la duración mediana de la MCyR fue de 2,3 meses.
Tabla 21: Efecto de Imatinib Mesilato en Ph + ALL Recidivante/Refractaria
|
Estudio de Fase 2 (N = 43) n(%) |
Estudio de Fase 1 (N = 2) n(%) |
|
|
CHR |
8 (19) |
2 (100) |
|
NEL |
5 (12) |
|
|
RTC/PHR |
11 (26) |
|
|
MCyR |
15 (35) |
|
|
CCyR |
9 (21) |
PCyR
6 (14)
14.4 Leucemia linfoblástica aguda pediátrica
Pacientes pediátricos y jóvenes adultos con ALL de muy alto riesgo, definidos como aquellos con una supervivencia libre de eventos (EFS) esperada a 5 años inferior al 45%, fueron inscritos después de la terapia de inducción en un protocolo piloto de grupo cooperativo multicéntrico, no aleatorizado.
Se evaluó la seguridad y eficacia del mesilato de imatinib (340 mg/m2/día) en combinación con quimioterapia intensiva en un subgrupo de pacientes con ALL Ph+. El protocolo incluyó quimioterapia intensiva y trasplante de células madre hematopoyéticas después de 2 cursos de quimioterapia para pacientes con un donante familiar HLA compatible apropiado. Hubo 92 pacientes elegibles con ALL Ph+ inscritos. La mediana de edad fue de 9,5 años (1 a 21 años: 2,2% entre 1 y menos de 2 años, 56,5% entre 2 y menos de 12 años, 34,8% entre 12 y menos de 18 años y 6,5% entre 18 y 21 años). El 64% eran varones, el 75% eran blancos, el 9% eran asiáticos / isleños del Pacífico y el 5% eran negros. En 5 cohortes sucesivas de pacientes, la exposición al mesilato de imatinib se aumentó sistemáticamente mediante una introducción más temprana y una duración prolongada. La cohorte 1 recibió la menor intensidad y la cohorte 5 recibió la mayor intensidad de exposición al mesilato de imatinib.
Hubo 50 pacientes con ALL Ph+ asignados a la cohorte 5, todos los cuales recibieron mesilato de imatinib más quimioterapia; 30 fueron tratados exclusivamente con quimioterapia y mesilato de imatinib y 20 recibieron quimioterapia más mesilato de imatinib y luego se sometieron a trasplante de células madre hematopoyéticas, seguido de un tratamiento posterior con mesilato de imatinib. Los pacientes de la cohorte 5 tratados con quimioterapia recibieron exposición continua diaria al mesilato de imatinib a partir del primer curso de quimioterapia posterior a la inducción y continuando durante los ciclos de mantenimiento 1 a 4 de quimioterapia. Durante los ciclos de mantenimiento 5 a 12, el mesilato de imatinib se administró durante 28 días de cada ciclo de 56 días. Los pacientes que se sometieron a trasplante de células madre hematopoyéticas recibieron 42 días de mesilato de imatinib antes del HSCT y 28 semanas (196 días) de mesilato de imatinib después del período inmediatamente posterior al trasplante. La EFS estimada a 4 años de los pacientes de la cohorte 5 fue del 70% (95% CI: 54, 81). El tiempo de seguimiento mediano para la EFS en el corte de datos de la cohorte 5 fue de 40,5 meses.
14.5 Enfermedades mielodisplásicas / mieloproliferativas
Se realizó un ensayo clínico de fase 2, abierto, multicéntrico, para probar el mesilato de imatinib en diversas poblaciones de pacientes que sufren de enfermedades potencialmente mortales asociadas con las proteínas tirosina cinasas Abl, Kit o PDGFR. Este estudio incluyó a 7 pacientes con MDS/MPD. Estos pacientes fueron tratados con mesilato de imatinib 400 mg diarios. Las edades de los pacientes inscritos oscilaron entre 20 y 86 años. Además, se informó de 24 pacientes más con MDS/MPD de 2 a 79 años en 12 informes de casos publicados y un estudio clínico. Estos pacientes también recibieron mesilato de imatinib a una dosis de 400 mg diarios, con la excepción de tres pacientes que recibieron dosis más bajas. De la población total de 31 pacientes tratados para MDS/MPD, 14 (45%) lograron una respuesta hematológica completa y 12 (39%) una respuesta citogenética mayor (incluyendo 10 con una CCyR). Dieciséis pacientes tuvieron una translocación, que involucra el cromosoma 5q33 o 4q12, resultando en un reordenamiento del gen PDGFR. Todos estos pacientes respondieron hematológicamente (13 completamente). La respuesta citogenética se evaluó en 12 de los 14 pacientes, todos los cuales respondieron (10 pacientes completamente). Solo 1 (7%) de los 14 pacientes sin una translocación asociada con el reordenamiento del gen PDGFR logró una respuesta hematológica completa y ninguno logró una respuesta citogenética mayor. Un paciente más con un reordenamiento del gen PDGFR en recaída molecular después del trasplante de médula ósea respondió molecularmente. La duración mediana del tratamiento fue de 12,9 meses (0,8 a 26,7) en los 7 pacientes tratados dentro del estudio de fase 2 y osciló entre 1 semana y más de 18 meses en los pacientes que respondieron en la literatura publicada. Los resultados se presentan en la Tabla 22. Las duraciones de respuesta de los pacientes del estudio de fase 2 oscilaron de 141+ días a 457+ días.
Tabla 22: Respuesta en MDS/MPD
|
Número de pacientes N |
Respuesta Hematológica Completa N (%) |
Respuesta Citogenética Mayor N (%) |
|
|
Población general |
31 |
14 (45) |
12 (39) |
|
Translocación del cromosoma 5 |
14 |
11 (79) |
11 (79) |
|
Translocación del cromosoma 4 |
2 |
2 (100) |
1 (50) |
|
Otros / sin translocación |
14 |
1 (7) |
0 |
Molecular Relapse
1
NE
NE
Abbreviations: NE, not evaluable; MDS/MPD, myelodysplastic/myeloproliferative disease.
14.6 Mastocitosis Sistémica Agresiva
Se llevó a cabo un estudio de fase 2, multicéntrico, abierto, que evaluó el mesilato de imatinib en diversas poblaciones de pacientes con enfermedades potencialmente mortales asociadas con las tirosina quinasas proteicas Abl, Kit o PDGFR. Este estudio incluyó a 5 pacientes con ASM tratados con 100 mg a 400 mg de mesilato de imatinib al día. Estos 5 pacientes tenían entre 49 y 74 años de edad. Además de estos 5 pacientes, 10 informes de casos publicados y series de casos describen el uso de mesilato de imatinib en 23 pacientes adicionales con ASM de 26 a 85 años que también recibieron de 100 mg a 400 mg de mesilato de imatinib al día.
Se evaluaron las anormalidades citogenéticas en 20 de los 28 pacientes con ASM tratados con mesilato de imatinib de los informes publicados y en el estudio de fase 2. Siete de estos 20 pacientes tenían la quinasa de fusión FIP1L1-PDGFRα (o deleción CHIC2). Los pacientes con esta anormalidad citogenética eran predominantemente hombres y tenían eosinofilia asociada con su enfermedad de mastocitos sistémica. Dos pacientes tenían una mutación Kit en la región yuxtamembranosa (una Phe522Cys y una K509I) y cuatro pacientes tenían una mutación D816V c-Kit (no considerada sensible al mesilato de imatinib), uno con CML concomitante.
De los 28 pacientes tratados por ASM, 8 (29%) lograron una respuesta hematológica completa y 9 (32%) una respuesta hematológica parcial (PHR) (61% de tasa de respuesta general). La duración media del tratamiento con mesilato de imatinib para los 5 pacientes con ASM en el estudio de fase 2 fue de 13 meses (rango, 1,4 a 22,3 meses) y entre 1 mes y más de 30 meses en los pacientes que respondieron descritos en la literatura médica publicada. En la Tabla 23 se proporciona un resumen de las tasas de respuesta al mesilato de imatinib en ASM. Las duraciones de respuesta de los pacientes de la literatura oscilaron entre 1+ y 30+ meses.
Tabla 23: Respuesta en ASM
|
Anormalidad Citogenética |
Número de Pacientes N |
Respuesta Hematológica Completa N (%) |
Respuesta Hematológica Parcial N (%) |
|
Quinasa de Fusión FIP1L1-PDGFRα (o Deleción CHIC2) |
7 |
7 (100) |
0 |
|
Mutación Yuxtamembranosa |
2 |
0 |
2 (100) |
|
Anormalidad Citogenética Desconocida o No Detectada |
15 |
0 |
7 (44) |
|
Mutación D816V |
4 |
1* (25) |
0 |
|
Total |
28 |
8 (29) |
9 (32) |
|
Abbreviations: ASM, mastocitosis sistémica agresiva; PDGFR, receptor del factor de crecimiento derivado de plaquetas. * El paciente tenía leucemia mieloide crónica (CML) y (ASM) concomitantes. |
|||
No se ha demostrado que el mesilato de imatinib sea eficaz en pacientes con formas menos agresivas de mastocitosis sistémica (SM). Por lo tanto, no se recomienda el uso del mesilato de imatinib en pacientes con mastocitosis cutánea, mastocitosis sistémica indolente (SM latente o mastocitosis medular ósea aislada), SM con una enfermedad hematológica clonal no mastocitaria asociada, leucemia de mastocitos, sarcoma de mastocitos o mastocitoma extracutáneo. Los pacientes que albergan la mutación D816V de c-Kit no son sensibles al mesilato de imatinib y no deben recibir mesilato de imatinib.
14.7 Síndrome hipereosinofílico/Leucemia eosinofílica crónica
Se realizó un estudio de fase 2, multicéntrico, abierto, que evaluó el mesilato de imatinib en diversas poblaciones de pacientes con enfermedades potencialmente mortales asociadas a las tirosina quinasas proteicas Abl, Kit o PDGFR. Este estudio incluyó a 14 pacientes con síndrome hipereosinofílico/leucemia eosinofílica crónica (HES/CEL). Los pacientes con HES fueron tratados con 100 mg a 1.000 mg de mesilato de imatinib al día. Las edades de estos pacientes oscilaron entre los 16 y los 64 años. Se informó de otros 162 pacientes con HES/CEL de 11 a 78 años en 35 informes de casos y series de casos publicados. Estos pacientes recibieron mesilato de imatinib a dosis de 75 mg a 800 mg al día. Las tasas de respuesta hematológica se resumen en la Tabla 24. Las duraciones de la respuesta para los pacientes de la literatura oscilaron entre 6+ semanas y 44 meses.
Tabla 24: Respuesta en HES/CEL
|
Anomalía citogenética |
Número de pacientes |
Respuesta hematológica completa Respuesta N (%) |
Respuesta hematológica parcial Respuesta N (%) |
|
Quinasa de fusión FIP1L1-PDGFRα positiva |
61 |
61 (100) |
0 |
|
Quinasa de fusión FIP1L1-PDGFRα negativa |
56 |
12 (21) |
9 (16) |
|
Anomalía citogenética desconocida |
59 |
34 (58) |
7 (12) |
|
Total |
176 |
107 (61) |
23 (13) |
| Abreviaturas: CEL, leucemia eosinofílica crónica; HES, síndrome hipereosinofílico; PDGFR, receptor del factor de crecimiento derivado de plaquetas. | |||
14.8 Dermatofibrosarcoma Protuberans
El dermatofibrosarcoma protuberans (DFSP) es un sarcoma de tejidos blandos cutáneo. Se caracteriza por una translocación de los cromosomas 17 y 22 que da como resultado la fusión del gen alfa 1 del colágeno tipo 1 y el gen PDGF B.
Se llevó a cabo un estudio de fase 2, multicéntrico, abierto, que probó el mesilato de imatinib en una población diversa de pacientes con enfermedades potencialmente mortales asociadas con las tirosina quinasas de proteínas Abl, Kit o PDGFR. Este estudio incluyó a 12 pacientes con DFSP que fueron tratados con mesilato de imatinib 800 mg diarios (rango de edad, de 23 a 75 años). El DFSP era metastásico, localmente recurrente después de la resección quirúrgica inicial y no se consideró susceptible de cirugía adicional en el momento de la inclusión en el estudio. Se informan otros 6 pacientes con DFSP tratados con mesilato de imatinib en 5 informes de casos publicados, con edades comprendidas entre los 18 meses y los 49 años. Por lo tanto, la población total tratada para DFSP comprende 18 pacientes, 8 de ellos con enfermedad metastásica. Los pacientes adultos informados en la literatura publicada fueron tratados con 400 mg (4 casos) u 800 mg (1 caso) de mesilato de imatinib diarios. Un solo paciente pediátrico recibió 400 mg/m2/diario, posteriormente aumentado a 520 mg/m2/diario. Diez pacientes tenían la reordenación del gen PDGF B, 5 no tenían citogenética disponible y 3 tenían anomalías citogenéticas complejas. Las respuestas al tratamiento se describen en la Tabla 25.
Tabla 25: Respuesta en DFSP
|
Número de pacientes (n = 18) |
% |
|
|
Respuesta completa |
7 |
39 |
|
Respuesta parcial * |
8 |
44 |
|
Total de respondedores |
15 |
83 |
|
* 5 pacientes quedaron libres de enfermedad por cirugía. |
||
Doce de estos 18 pacientes lograron una respuesta completa (7 pacientes) o quedaron libres de enfermedad mediante cirugía después de una respuesta parcial (5 pacientes, incluido un niño) para una tasa de respuesta completa total del 67%. Otros 3 pacientes lograron una respuesta parcial, para una tasa de respuesta general del 83%. De los 8 pacientes con enfermedad metastásica, cinco respondieron (62%), tres de ellos completamente (37%). Para los 10 pacientes del estudio con la reordenación del gen PDGF B, hubo 4 respuestas completas y 6 respuestas parciales. La duración media de la respuesta en el estudio de fase 2 fue de 6,2 meses, con una duración máxima de 24,3 meses, mientras que en la literatura publicada osciló entre 4 semanas y más de 20 meses.
14.9 Tumores estromales gastrointestinales
GIST metastásico maligno e irresecable y/o maligno
Se realizaron dos estudios de fase 3 multinacionales, aleatorizados y de etiqueta abierta en pacientes con GIST maligno irresecable o metastásico. Los dos diseños de estudio fueron similares, lo que permitió un análisis combinado predefinido de seguridad y eficacia. Se inscribieron un total de 1.640 pacientes en los dos estudios y se aleatorizaron 1:1 para recibir 400 mg u 800 mg por vía oral diariamente de forma continua hasta la progresión de la enfermedad o la toxicidad inaceptable. Los pacientes del grupo de tratamiento de 400 mg diarios que experimentaron progresión de la enfermedad pudieron cambiar para recibir tratamiento con 800 mg diarios. Los estudios fueron diseñados para comparar las tasas de respuesta, la supervivencia libre de progresión y la supervivencia general entre los grupos de dosis. La edad media al ingreso del paciente fue de 60 años. Los hombres representaron el 58% de los pacientes inscritos. Todos los pacientes tenían un diagnóstico patológico de GIST maligno irresecable y/o metastásico positivo para CD117.
El objetivo principal de los dos estudios fue evaluar la supervivencia libre de progresión (SLP) con un objetivo secundario de supervivencia general (SG) en un estudio o supervivencia general con un objetivo secundario de SLP en el otro estudio. Se realizó un análisis planificado de la SG y la SLP de los conjuntos de datos combinados de estos dos estudios. Los resultados de este análisis combinado se muestran en la Tabla 26.
|
Tabla 26: Supervivencia general, supervivencia libre de progresión y tasas de respuesta tumoral en los ensayos de fase 3 de GIST |
||
|
Mesilato de imatinib 400 mg N = 818 |
Mesilato de imatinib 800 mg N = 822 |
|
|
Supervivencia libre de progresión (meses) |
||
|
Mediana |
18.9 |
23.2 |
|
IC del 95% |
17.4 a 21.2 |
20.8 a 24.9 |
|
Supervivencia general (meses) |
49.0 |
48.7 |
|
IC del 95% |
45.3 a 60.0 |
45.3 a 51.6 |
|
Mejor respuesta tumoral general |
||
|
Respuesta completa |
43 (5.3%) |
41 (5.0%) |
|
Respuesta parcial |
377 (46.1%) |
402 (48.9%) |
|
Abreviatura: GIST, tumores estromales gastrointestinales. |
||
El seguimiento mediano para los estudios combinados fue de 37.5 meses. No se observaron diferencias en la supervivencia general entre los grupos de tratamiento (p = 0.98). Los pacientes que cruzaron después de la progresión de la enfermedad del grupo de tratamiento de 400 mg/día al grupo de tratamiento de 800 mg/día (n = 347) tuvieron una mediana de 3.4 meses y una exposición media de 7.7 meses al mesilato de imatinib después del cruce.
Se realizó un estudio de fase 2 abierto, multinacional en pacientes con GIST maligno metastásico o irresecable positivo para Kit (CD117). En este estudio, se inscribieron 147 pacientes y se asignaron aleatoriamente para recibir 400 mg o 600 mg por vía oral todos los días durante un máximo de 36 meses. El resultado principal del estudio fue la tasa de respuesta objetiva. Se requirió que los tumores fueran medibles al ingreso en al menos un sitio de la enfermedad, y la caracterización de la respuesta se basó en los criterios del Southwestern Oncology Group (SWOG). No hubo diferencias en las tasas de respuesta entre los 2 grupos de dosis. La tasa de respuesta fue del 68.5% para el grupo de 400 mg y del 67.6% para el grupo de 600 mg. El tiempo mediano hasta la respuesta fue de 12 semanas (el rango fue de 3 a 98 semanas) y la duración mediana estimada de la respuesta es de 118 semanas (IC del 95%: 86, no alcanzado).
Tratamiento adyuvante del GIST
En el contexto adyuvante, se investigó el mesilato de imatinib en un ensayo aleatorizado, doble ciego, controlado con placebo, multicéntrico en el que participaron 713 pacientes (Estudio 1). Los pacientes se asignaron aleatoriamente uno a uno al mesilato de imatinib a 400 mg/día o placebo coincidente durante 12 meses. Las edades de estos pacientes oscilaron entre los 18 y los 91 años. Se incluyeron pacientes que tenían un diagnóstico histológico de GIST primario, que expresaban proteína KIT mediante inmunoquímica y un tamaño tumoral mayor o igual a 3 cm en la dimensión máxima con resección macroscópica completa del GIST primario entre 14 y 70 días antes del registro.
La supervivencia libre de recurrencia (SLR) se definió como el tiempo transcurrido desde la fecha de aleatorización hasta la fecha de recurrencia o muerte por cualquier causa. En un análisis intermedio planificado, el seguimiento mediano fue de 15 meses en pacientes sin un evento de SLR; hubo 30 eventos de SLR en el brazo de mesilato de imatinib de 12 meses en comparación con 70 eventos de SLR en el brazo de placebo con una razón de riesgo de 0.398 (IC del 95%: 0.259, 0.610), p menor que 0.0001. Después del análisis intermedio de SLR, 79 de los 354 pacientes inicialmente asignados aleatoriamente al brazo de placebo fueron elegibles para cruzar al brazo de mesilato de imatinib de 12 meses. Setenta y dos de estos 79 pacientes posteriormente cruzaron al tratamiento con mesilato de imatinib. En un análisis actualizado, el seguimiento mediano para los pacientes sin un evento de SLR fue de 50 meses. Hubo 74 (21%) eventos de SLR en el brazo de mesilato de imatinib de 12 meses en comparación con 98 (28%) eventos en el brazo de placebo con una razón de riesgo de 0.718 (IC del 95%: 0.531 a 0.971) (Figura 3). El seguimiento mediano para la SO en pacientes que aún viven fue de 61 meses. Hubo 26 (7%) y 33 (9%) muertes en los brazos de mesilato de imatinib de 12 meses y placebo, respectivamente, con una razón de riesgo de 0.816 (IC del 95%: 0.488 a 1.365).
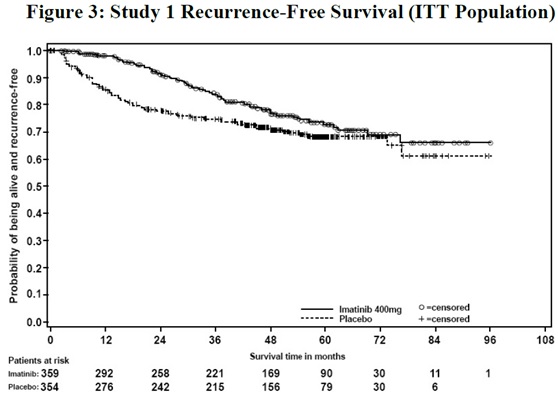
Un segundo ensayo aleatorizado, multicéntrico, abierto, de fase 3 en el contexto adyuvante (Estudio 2) comparó 12 meses de tratamiento con mesilato de imatinib con 36 meses de tratamiento con mesilato de imatinib a 400 mg/día en pacientes adultos con GIST positivo para KIT (CD117) después de la resección quirúrgica con uno de los siguientes: diámetro tumoral mayor de 5 cm y recuento mitótico mayor de 5/50 campos de gran aumento (HPF), o diámetro tumoral mayor de 10 cm y cualquier recuento mitótico, o tumor de cualquier tamaño con recuento mitótico mayor de 10/50 HPF, o tumores que se rompieron en la cavidad peritoneal. Hubo un total de 397 pacientes aleatorizados en el ensayo con 199 pacientes en el brazo de tratamiento de 12 meses y 198 pacientes en el brazo de tratamiento de 36 meses. La edad media fue de 61 años (rango, 22 a 84 años).
La SLR se definió como el tiempo transcurrido desde la fecha de aleatorización hasta la fecha de recurrencia o muerte por cualquier causa. El seguimiento mediano para los pacientes sin un evento de SLR fue de 42 meses. Hubo 84 (42%) eventos de SLR en el brazo de tratamiento de 12 meses y 50 (25%) eventos de SLR en el brazo de tratamiento de 36 meses. Treinta y seis meses de tratamiento con mesilato de imatinib prolongaron significativamente la SLR en comparación con 12 meses de tratamiento con mesilato de imatinib con una razón de riesgo de 0.46 (IC del 95%: 0.32, 0.65), p menor que 0.0001 (Figura 4).
El seguimiento mediano para la supervivencia general (SO) en pacientes que aún viven fue de 48 meses. Hubo 25 (13%) muertes en el brazo de tratamiento de 12 meses y 12 (6%) muertes en el brazo de tratamiento de 36 meses. Treinta y seis meses de tratamiento con mesilato de imatinib prolongaron significativamente la SO en comparación con 12 meses de tratamiento con mesilato de imatinib con una razón de riesgo de 0.45 (IC del 95%: 0.22, 0.89), p = 0.0187 (Figura 5).
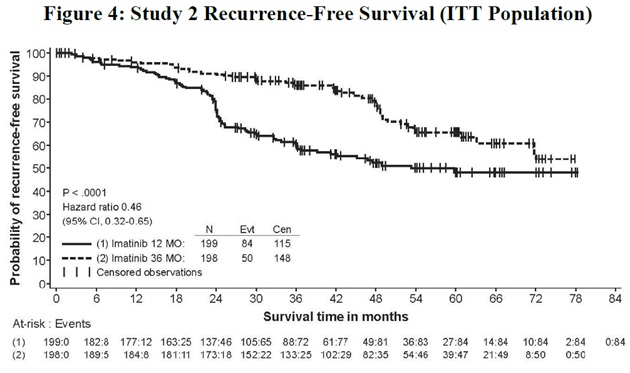
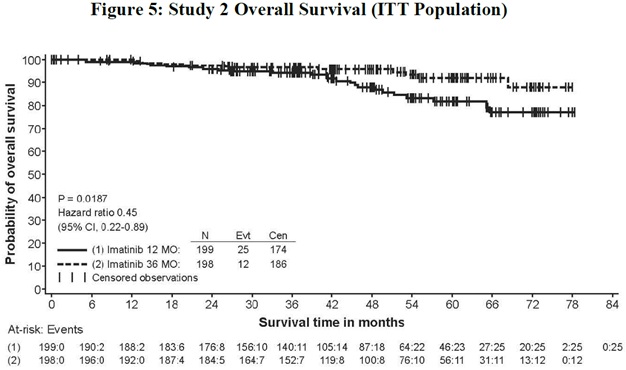
15 REFERENCIAS
Drogas Peligrosas de OSHA. OSHA. [Accedido el 20 de septiembre de 2013, desde http://www.osha.gov/SLTC/hazardousdrugs/index.html]
16 FORMA DE SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
Cada tableta recubierta con película contiene mesilato de imatinib equivalente a 100 mg o 400 mg de imatinib base libre.
Las tabletas de mesilato de imatinib, 100 mg, se suministran como tabletas marrones, redondas, ranuradas, recubiertas con película y con borde biselado, grabadas con “AN” en el lado ranurado y “794” en el otro lado.
Frascos de 30 NDC 65162-794-03
Frascos de 90 NDC 65162-794-09
Las tabletas de mesilato de imatinib, 400 mg, se suministran como tabletas marrones, ovaladas, ranuradas, recubiertas con película y con borde biselado, grabadas con “AN” en el lado ranurado y “795” en el otro lado.
Frascos de 30 NDC 65162-795-03
Frascos de 90 NDC 65162-795-09
Almacenamiento y manipulación
Conservar a una temperatura de 20° a 25°C (68° a 77°F); se permiten excursiones de temperatura entre 15° y 30°C (59° y 86°F) [ver USP Temperatura ambiente controlada]. Proteger de la humedad.
Dispensar en un recipiente hermético según se define en la USP.
No triture las tabletas de mesilato de imatinib. Evite el contacto directo de las tabletas trituradas con la piel o las membranas mucosas. Si se produce dicho contacto, lave bien la zona afectada como se indica en las referencias. Evite la exposición a las tabletas trituradas.
17 INFORMACIÓN PARA EL PACIENTE
Dosificación y Administración
Aconseje a los pacientes que tomen imatinib mesilato exactamente como se lo recetaron, que no cambien su dosis ni que dejen de tomar imatinib mesilato a menos que su médico se lo indique. Si el paciente omitió una dosis de imatinib mesilato, debe tomar la siguiente dosis programada a su hora habitual. El paciente no debe tomar dos dosis al mismo tiempo. Aconseje a los pacientes que tomen imatinib mesilato con una comida y un vaso grande de agua [ver Dosificación y Administración (2.1)].
Retención de líquidos y edema
Informe a los pacientes de la posibilidad de desarrollar edema y retención de líquidos. Aconseje a los pacientes que se pongan en contacto con su proveedor de atención médica si se produce un aumento de peso rápido inesperado [ver Advertencias y precauciones (5.1)].
Hepatotoxicidad
Informe a los pacientes de la posibilidad de desarrollar anomalías en la función hepática y toxicidad hepática grave. Aconseje a los pacientes que se pongan en contacto con su proveedor de atención médica de inmediato si se presentan signos de insuficiencia hepática, como ictericia, anorexia, sangrado o hematomas [ver Advertencias y precauciones (5.4)].
Embarazo y lactancia
Aconseje a los pacientes que informen a su médico si están embarazadas o creen que pueden estarlo. Aconseje a las mujeres en edad fértil que eviten quedar embarazadas mientras toman imatinib mesilato. Las pacientes en edad fértil que toman imatinib mesilato deben utilizar métodos anticonceptivos altamente efectivos durante el tratamiento y durante catorce días después de suspender el tratamiento con imatinib mesilato [ver Uso en poblaciones específicas (8.3)]. Evite la lactancia durante el tratamiento y durante 1 mes después de la última dosis [ver Uso en poblaciones específicas (8.2)].
Interacciones medicamentosas
Imatinib mesilato y ciertos otros medicamentos, como warfarina, eritromicina y fenitoína, incluidos los medicamentos de venta libre, como los productos herbales, pueden interactuar entre sí. Aconseje a los pacientes que le digan a su médico si están tomando o planean tomar suplementos de hierro. Evite el jugo de toronja y otros alimentos que se sabe que inhiben el CYP3A4 mientras toma imatinib mesilato [ver Interacciones medicamentosas (7)].
Pediátrico
Aconseje a los pacientes que se ha informado de retraso en el crecimiento en niños y preadolescentes que reciben imatinib mesilato. Se desconocen los efectos a largo plazo del tratamiento prolongado con imatinib mesilato sobre el crecimiento en niños. Por lo tanto, controle de cerca el crecimiento de los niños que reciben tratamiento con imatinib mesilato [ver Advertencias y precauciones (5.11)].
Conducir y usar máquinas
Aconseje a los pacientes que pueden experimentar efectos secundarios, como mareos, visión borrosa o somnolencia durante el tratamiento con imatinib mesilato. Por lo tanto, advierta a los pacientes sobre conducir un automóvil o operar maquinaria [ver Advertencias y precauciones (5.13)].
Distribuido por:
Amneal Pharmaceuticals LLC
Bridgewater, NJ 08807
Rev. 11-2023-05
PANEL DE VISUALIZACIÓN PRINCIPAL
NDC 65162-794-03
Imatinib Mesylate Tablets, 100 mg
Rx only
30 Tablets
Amneal Pharmaceuticals LLC

NDC 65162-795-03
Imatinib Mesylate Tablets, 400 mg
Rx only
30 Tablets
Amneal Pharmaceuticals LLC
