
Fabricante de medicamentos: Cordavis Limited (Updated: 2023-11-16)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
HUMIRA® (adalimumab) inyección, para uso subcutáneo
Aprobación inicial en EE. UU.: 2002
ADVERTENCIA: INFECCIONES GRAVES Y TUMORES MALIGNOS
Consulte la información de prescripción completa para ver la advertencia completa en recuadro.
INFECCIONES GRAVES (5.1, 6.1):
-
Mayor riesgo de infecciones graves que provocan hospitalización o muerte, como tuberculosis (TB), sepsis bacteriana, infecciones fúngicas invasivas (como histoplasmosis) e infecciones por otros patógenos oportunistas.
-
Suspenda HUMIRA si un paciente desarrolla una infección grave o sepsis durante el tratamiento.
-
Realice una prueba de TB latente; si es positiva, comience el tratamiento para la TB antes de comenzar con HUMIRA.
- Controle a todos los pacientes para detectar la TB activa durante el tratamiento, incluso si la prueba inicial de TB latente es negativa.
TUMORES MALIGNOS (5.2):
-
Se han notificado casos de linfoma y otros tumores malignos, algunos mortales, en niños y adolescentes tratados con bloqueadores del TNF, incluido HUMIRA.
- Se han producido casos poscomercialización de linfoma hepatoesplénico de células T (HSTCL), un tipo raro de linfoma de células T, en adolescentes y adultos jóvenes con enfermedad inflamatoria intestinal tratados con bloqueadores del TNF, incluido HUMIRA.
INDICACIONES Y USO
HUMIRA es un bloqueador del factor de necrosis tumoral (TNF) indicado para:
- Artritis reumatoide (AR) (1.1): reducir los signos y síntomas, inducir una respuesta clínica importante, inhibir la progresión del daño estructural y mejorar la función física en pacientes adultos con AR activa de moderada a grave.
- Artritis idiopática juvenil (AIJ) (1.2): reducir los signos y síntomas de la AIJ poliarticular activa de moderada a grave en pacientes de 2 años de edad o mayores.
- Artritis psoriásica (APs) (1.3): reducir los signos y síntomas, inhibir la progresión del daño estructural y mejorar la función física en pacientes adultos con APs activa.
- Espondilitis anquilosante (EA) (1.4): reducir los signos y síntomas en pacientes adultos con EA activa.
- Enfermedad de Crohn (EC) (1.5): tratamiento de la enfermedad de Crohn activa de moderada a grave en adultos y pacientes pediátricos de 6 años de edad o mayores.
-
Colitis ulcerosa (CU) (1.6): tratamiento de la colitis ulcerosa activa de moderada a grave en adultos y pacientes pediátricos de 5 años de edad o mayores.
Limitaciones de uso: No se ha establecido la eficacia en pacientes que han perdido la respuesta o que no toleraron los bloqueadores del TNF. - Psoriasis en placas (Ps) (1.7): tratamiento de pacientes adultos con psoriasis en placas crónica de moderada a grave que son candidatos a terapia sistémica o fototerapia, y cuando otras terapias sistémicas son médicamente menos apropiadas.
- Hidradenitis supurativa (HS) (1.8): tratamiento de la hidradenitis supurativa de moderada a grave en pacientes de 12 años de edad o mayores.
- Uveítis (UV) (1.9): tratamiento de la uveítis no infecciosa intermedia, posterior y panuveítis en adultos y pacientes pediátricos de 2 años de edad o mayores.
DOSIFICACIÓN Y ADMINISTRACIÓN
- Administrar mediante inyección subcutánea (2)
Artritis reumatoide, artritis psoriásica, espondilitis anquilosante (2.1):
-
Adultos: 40 mg cada dos semanas.
- Algunos pacientes con AR que no reciben metotrexato pueden beneficiarse de un aumento de la dosis a 40 mg cada semana u 80 mg cada dos semanas.
Artritis idiopática juvenil o uveítis pediátrica (2.2):
| Peso pediátrico 2 años de edad o mayores |
Dosis recomendada |
| 10 kg (22 lb) a menos de 15 kg (33 lb) | 10 mg cada dos semanas |
| 15 kg (33 lb) a menos de 30 kg (66 lb) | 20 mg cada dos semanas |
| 30 kg (66 lb) y mayores | 40 mg cada dos semanas |
Enfermedad de Crohn (2.3):
- Adultos: 160 mg el día 1 (administrados en un día o divididos en dos días consecutivos); 80 mg el día 15; y 40 mg en días alternos a partir del día 29.
- Pacientes pediátricos de 6 años de edad y mayores:
| Peso pediátrico |
Dosis recomendada | |
| Días 1 y 15 | A partir del día 29 | |
| 17 kg (37 lb) a menos de 40 kg (88 lb) |
Día 1: 80 mg Día 15: 40 mg |
20 mg en días alternos |
| 40 kg (88 lb) o más |
Día 1: 160 mg (dosis única o dividida en dos días consecutivos) Día 15: 80 mg |
40 mg en días alternos |
Colitis ulcerosa (2.4):
- Adultos: 160 mg el día 1 (administrados en un día o divididos en dos días consecutivos), 80 mg el día 15 y 40 mg en días alternos a partir del día 29. Suspenda la administración en pacientes sin evidencia de remisión clínica a las ocho semanas (día 57).
- Pacientes pediátricos de 5 años de edad y mayores:
| Peso pediátrico |
Dosis recomendada | |
| Días 1 a 15 | A partir del día 29* | |
| 20 kg (44 lb) a menos de 40 kg (88 lb) |
Día 1: 80 mg Día 8: 40 mg Día 15: 40 mg |
40 mg en días alternos o 20 mg cada semana |
| 40 kg (88 lb) o más | Día 1: 160 mg (dosis única o dividida en dos días consecutivos) Día 8: 80 mg Día 15: 80 mg |
80 mg en días alternos o 40 mg cada semana |
| * Continúe con la dosis pediátrica recomendada en pacientes que cumplan 18 años de edad y que estén bien controlados con su régimen de HUMIRA. | ||
Psoriasis en placas o uveítis en adultos (2.5):
- Adultos: 80 mg de dosis inicial, seguidos de 40 mg en días alternos a partir de una semana después de la dosis inicial.
Hidradenitis supurativa (2.6):
-
Adultos:
◦ Día 1: 160 mg (administrados en un día o divididos en dos días consecutivos)
◦ Día 15: 80 mg
◦ Día 29 y dosis posteriores: 40 mg cada semana u 80 mg en días alternos - Adolescentes de 12 años de edad y mayores:
| Peso del adolescente |
Dosis recomendada |
| 30 kg (66 lb) a menos de 60 kg (132 lb) |
Día 1: 80 mg Día 8 y dosis posteriores: 40 mg en días alternos |
| 60 kg (132 lb) o más |
Día 1: 160 mg (administrados en un día o divididos en dos días consecutivos) Día 15: 80 mg Día 29 y dosis posteriores: 40 mg cada semana u 80 mg en días alternos |
INFORMACIÓN PRESCRIPTIVA DESTACADA
Inyección:
- Pluma precargada de dosis única (HUMIRA Pen): 80 mg/0.8 mL, 40 mg/0.8 mL y 40 mg/0.4 mL (3)
- Jeringa de vidrio precargada de dosis única: 80 mg/0.8 mL, 40 mg/0.8 mL, 40 mg/0.4 mL, 20 mg/0.4 mL, 20 mg/0.2 mL, 10 mg/0.2 mL, 10 mg/0.1 mL (3)
- Vial de vidrio de dosis única para uso institucional únicamente: 40 mg/0.8 mL (3)
CONTRAINDICACIONES
Ninguna (4)
ADVERTENCIAS Y PRECAUCIONES
- Infecciones graves: No comience a tomar HUMIRA durante una infección activa. Si se desarrolla una infección, controle cuidadosamente y suspenda HUMIRA si la infección se agrava. (5.1)
- Infecciones fúngicas invasivas: En el caso de pacientes que desarrollen una enfermedad sistémica con HUMIRA, considere la terapia antimicótica empírica para aquellos que residen o viajan a regiones donde las micosis son endémicas. (5.1)
- Tumores malignos: La incidencia de tumores malignos fue mayor en pacientes tratados con HUMIRA que en los controles (5.2)
- Anafilaxia o reacciones de hipersensibilidad graves pueden ocurrir (5.3)
- Reactivación del virus de la hepatitis B: Controle a los portadores del VHB durante el tratamiento y varios meses después. Si se produce la reactivación, suspenda HUMIRA y comience el tratamiento antiviral. (5.4)
- Enfermedad desmielinizante: Puede producirse una exacerbación o una nueva aparición. (5.5)
- Citopenias, pancitopenia: Aconseje a los pacientes que busquen atención médica inmediata si desarrollan síntomas y considere suspender HUMIRA. (5.6)
- Insuficiencia cardíaca: Puede producirse un empeoramiento o una nueva aparición. (5.8)
- Síndrome similar al lupus: Suspenda HUMIRA si se desarrolla el síndrome. (5.9)
REACCIONES ADVERSAS
Las reacciones adversas más comunes (>10%) son: infecciones (p. ej., de las vías respiratorias superiores, sinusitis), reacciones en el lugar de la inyección, dolor de cabeza y erupción cutánea. (6.1)
Para informar de SOSPECHAS DE REACCIONES ADVERSAS, póngase en contacto con AbbVie Inc. en el 1-800-633-9110 o con la FDA en el 1-800-FDA-1088 o en www.fda.gov/medwatch
INTERACCIONES FARMACOLÓGICAS
Consulte 17 para obtener INFORMACIÓN DE ASESORAMIENTO AL PACIENTE y la Guía del Medicamento.
Revisado: 11/2023
Tabla de contenido
FULL PRESCRIBING INFORMATION: CONTENTS*
ADVERTENCIA: INFECCIONES GRAVES Y TUMORES MALIGNOS
1 INDICACIONES Y USO
1.1 Artritis Reumatoide
1.2 Artritis Idiopática Juvenil
1.3 Artritis Psoriásica
1.4 Espondilitis Anquilosante
1.5 Enfermedad de Crohn
1.6 Colitis Ulcerosa
1.7 Psoriasis en Placas
1.8 Hidradenitis Supurativa
1.9 Uveítis
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Artritis Reumatoide, Artritis Psoriásica y Espondilitis Anquilosante
2.2 Artritis Idiopática Juvenil o Uveítis Pediátrica
2.3 Enfermedad de Crohn
2.4 Colitis Ulcerosa
2.5 Psoriasis en Placas o Uveítis en Adultos
2.6 Hidradenitis Supurativa
2.7 Monitoreo para Evaluar la Seguridad
2.8 Consideraciones Generales para la Administración
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Infecciones Graves
5.2 Tumores Malignos
5.3 Reacciones de Hipersensibilidad
5.4 Reactivación del Virus de la Hepatitis B
5.5 Reacciones Neurológicas
5.6 Reacciones Hematológicas
5.7 Mayor Riesgo de Infección Cuando se Usa con Anakinra
5.8 Insuficiencia Cardíaca
5.9 Autoinmunidad
5.10 Vacunas
5.11 Mayor Riesgo de Infección Cuando se Usa con Abatacept
6 REACCIONES ADVERSAS
6.1 Experiencia en Ensayos Clínicos
6.2 Inmunogenicidad
6.3 Experiencia Posterior a la Comercialización
7 INTERACCIONES CON MEDICAMENTOS
7.1 Metotrexato
7.2 Productos Biológicos
7.3 Vacunas Vivas
7.4 Sustratos del Citocromo P450
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.4 Uso Pediátrico
8.5 Uso Geriátrico
10 SOBREDOSIS
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de Acción
12.2 Farmacodinamia
12.3 Farmacocinética
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
14 ESTUDIOS CLÍNICOS
14.1 Artritis Reumatoide
14.2 Artritis Idiopática Juvenil
14.3 Artritis Psoriásica
14.4 Espondilitis Anquilosante
14.5 Enfermedad de Crohn en Adultos
14.6 Enfermedad de Crohn Pediátrica
14.7 Colitis Ulcerosa en Adultos
14.8 Colitis Ulcerosa Pediátrica
14.9 Psoriasis en Placas
14.10 Hidradenitis Supurativa
14.11 Uveítis en Adultos
14.12 Uveítis Pediátrica
15 REFERENCIAS
16 CÓMO SE SUMINISTRA/ALMACENAMIENTO Y MANIPULACIÓN
17 INFORMACIÓN DE ASESORAMIENTO AL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información de prescripción completa no se enumeran.
ADVERTENCIA EN CAJA
ADVERTENCIA: INFECCIONES GRAVES Y MALIGNIDAD
INFECCIONES GRAVES
Los pacientes tratados con HUMIRA tienen un mayor riesgo de desarrollar infecciones graves que pueden llevar a la hospitalización o la muerte [ver Advertencias y precauciones (5.1)]. La mayoría de los pacientes que desarrollaron estas infecciones estaban tomando inmunosupresores concomitantes como metotrexato o corticosteroides.
Suspenda HUMIRA si un paciente desarrolla una infección grave o sepsis.
Las infecciones reportadas incluyen:
-
Tuberculosis activa (TB), incluida la reactivación de la TB latente. Los pacientes con TB con frecuencia se han presentado con enfermedad diseminada o extrapulmonar. Examine a los pacientes para detectar TB latente antes de usar HUMIRA y durante la terapia. Inicie el tratamiento para la TB latente antes de usar HUMIRA.
-
Infecciones fúngicas invasivas, incluidas histoplasmosis, coccidioidomicosis, candidiasis, aspergilosis, blastomicosis y neumocistosis. Los pacientes con histoplasmosis u otras infecciones fúngicas invasivas pueden presentarse con enfermedad diseminada, en lugar de localizada. Las pruebas de antígeno y anticuerpos para histoplasmosis pueden ser negativas en algunos pacientes con infección activa. Considere la terapia antifúngica empírica en pacientes con riesgo de infecciones fúngicas invasivas que desarrollan enfermedad sistémica grave.
- Infecciones bacterianas, virales y otras debido a patógenos oportunistas, incluidas Legionella y Listeria.
Considere cuidadosamente los riesgos y beneficios del tratamiento con HUMIRA antes de iniciar la terapia en pacientes con infección crónica o recurrente.
Monitoree a los pacientes de cerca para detectar el desarrollo de signos y síntomas de infección durante y después del tratamiento con HUMIRA, incluido el posible desarrollo de TB en pacientes que dieron negativo para la infección latente por TB antes de iniciar la terapia [ver Advertencias y precauciones (5.1) y Reacciones adversas (6.1)].
MALIGNIDAD
Se han reportado linfoma y otras malignidades, algunas fatales, en niños y adolescentes tratados con bloqueadores del TNF, incluido HUMIRA [ver Advertencias y precauciones (5.2)]. Los casos posteriores a la comercialización de linfoma de células T hepatosplénico (HSTCL), un tipo raro de linfoma de células T, se han reportado en pacientes tratados con bloqueadores del TNF, incluido HUMIRA. Estos casos han tenido un curso de enfermedad muy agresivo y han sido fatales. La mayoría de los casos reportados de bloqueadores del TNF han ocurrido en pacientes con enfermedad de Crohn o colitis ulcerosa y la mayoría fueron en adolescentes y hombres jóvenes adultos. Casi todos estos pacientes habían recibido tratamiento con azatioprina o 6-mercaptopurina (6-MP) concomitantemente con un bloqueador del TNF en o antes del diagnóstico. No está claro si la aparición de HSTCL está relacionada con el uso de un bloqueador del TNF o un bloqueador del TNF en combinación con estos otros inmunosupresores [ver Advertencias y precauciones (5.2)].
1 INDICACIONES Y USO
1.1 Artritis Reumatoide
HUMIRA está indicado para reducir los signos y síntomas, inducir una respuesta clínica mayor, inhibir la progresión del daño estructural y mejorar la función física en pacientes adultos con artritis reumatoide activa de moderada a grave. HUMIRA se puede usar solo o en combinación con metotrexato u otros fármacos antirreumáticos modificadores de la enfermedad (FARME) no biológicos.
1.2 Artritis Idiopática Juvenil
HUMIRA está indicado para reducir los signos y síntomas de la artritis idiopática juvenil poliarticular activa de moderada a grave en pacientes de 2 años de edad o mayores. HUMIRA se puede usar solo o en combinación con metotrexato.
1.3 Artritis Psoriásica
HUMIRA está indicado para reducir los signos y síntomas, inhibir la progresión del daño estructural y mejorar la función física en pacientes adultos con artritis psoriásica activa. HUMIRA se puede usar solo o en combinación con FARME no biológicos.
1.4 Espondilitis Anquilosante
HUMIRA está indicado para reducir los signos y síntomas en pacientes adultos con espondilitis anquilosante activa.
1.5 Enfermedad de Crohn
HUMIRA está indicado para el tratamiento de la enfermedad de Crohn activa de moderada a grave en adultos y pacientes pediátricos de 6 años de edad o mayores.
1.6 Colitis Ulcerosa
HUMIRA está indicado para el tratamiento de la colitis ulcerosa activa de moderada a grave en adultos y pacientes pediátricos de 5 años de edad o mayores.
Limitaciones de Uso
No se ha establecido la eficacia de HUMIRA en pacientes que han perdido la respuesta o que fueron intolerantes a los bloqueadores del TNF [ver Estudios Clínicos (14.7, 14.8)].
1.7 Psoriasis en Placas
HUMIRA está indicado para el tratamiento de pacientes adultos con psoriasis en placas crónica de moderada a grave que son candidatos a terapia sistémica o fototerapia, y cuando otras terapias sistémicas son médicamente menos apropiadas. HUMIRA solo debe administrarse a pacientes que serán monitoreados de cerca y que tendrán visitas de seguimiento regulares con un médico [ver Advertencias y Precauciones (5)].
1.8 Hidradenitis Supurativa
HUMIRA está indicado para el tratamiento de la hidradenitis supurativa de moderada a grave en pacientes de 12 años de edad o mayores.
1.9 Uveítis
HUMIRA está indicado para el tratamiento de la uveítis intermedia, posterior y panuveítis no infecciosa en adultos y pacientes pediátricos de 2 años de edad o mayores.
2 DOSIS Y ADMINISTRACIÓN
2.1 Artritis Reumatoide, Artritis Psoriásica y Espondilitis Anquilosante
La dosis subcutánea recomendada de HUMIRA para pacientes adultos con artritis reumatoide (AR), artritis psoriásica (AP) o espondilitis anquilosante (EA) es de 40 mg administrados cada dos semanas. El metotrexato (MTX), otros fármacos antirreumáticos modificadores de la enfermedad (FARME) no biológicos, los glucocorticoides, los fármacos antiinflamatorios no esteroideos (AINE) y/o los analgésicos pueden continuarse durante el tratamiento con HUMIRA. En el tratamiento de la AR, algunos pacientes que no toman MTX concomitante pueden obtener un beneficio adicional al aumentar la dosis de HUMIRA a 40 mg cada semana o 80 mg cada dos semanas.
2.2 Artritis Idiopática Juvenil o Uveítis Pediátrica
La dosis subcutánea recomendada de HUMIRA para pacientes de 2 años de edad o mayores con artritis idiopática juvenil poliarticular (AIJ) o uveítis pediátrica se basa en el peso, como se muestra a continuación. El MTX, los glucocorticoides, los AINE y/o los analgésicos pueden continuarse durante el tratamiento con HUMIRA.
| Peso Pediátrico (2 años de edad o mayores) |
Dosis Recomendada |
| 10 kg (22 lbs) a menos de 15 kg (33 lbs) | 10 mg cada dos semanas |
| 15 kg (33 lbs) a menos de 30 kg (66 lbs) | 20 mg cada dos semanas |
| 30 kg (66 lbs) y mayor | 40 mg cada dos semanas |
HUMIRA no se ha estudiado en pacientes con AIJ poliarticular o uveítis pediátrica menores de 2 años de edad o en pacientes con un peso inferior a 10 kg.
2.3 Enfermedad de Crohn
Adultos
La dosis subcutánea recomendada de HUMIRA para pacientes adultos con enfermedad de Crohn (EC) es de 160 mg inicialmente en el Día 1 (administrados en un día o divididos en dos días consecutivos), seguido de 80 mg dos semanas después (Día 15). Dos semanas después (Día 29) comience una dosis de 40 mg cada dos semanas. Los aminosalicilatos y/o los corticosteroides pueden continuarse durante el tratamiento con HUMIRA. La azatioprina, la 6-mercaptopurina (6-MP) [ver Advertencias y precauciones (5.2)] o el MTX pueden continuarse durante el tratamiento con HUMIRA si es necesario.
Pediatría
La dosis subcutánea recomendada de HUMIRA para pacientes pediátricos de 6 años de edad o mayores con enfermedad de Crohn (EC) se basa en el peso corporal, como se muestra a continuación:
| Peso Pediátrico |
Dosis Recomendada | |
| Días 1 a 15 | A partir del Día 29 | |
| 17 kg (37 lbs) a menos de 40 kg (88 lbs) |
Día 1: 80 mg Día 15: 40 mg |
20 mg cada dos semanas |
| 40 kg (88 lbs) y mayor | Día 1: 160 mg (dosis única o dividida en dos días consecutivos) Día 15: 80 mg |
40 mg cada dos semanas |
2.4 Colitis ulcerosa
Adultos
La dosis subcutánea recomendada de HUMIRA para pacientes adultos con colitis ulcerosa es de 160 mg inicialmente en el Día 1 (administrada en un día o dividida en dos días consecutivos), seguida de 80 mg dos semanas después (Día 15). Dos semanas después (Día 29) continúe con una dosis de 40 mg cada dos semanas.
Suspenda HUMIRA en pacientes adultos sin evidencia de remisión clínica a las ocho semanas (Día 57) de tratamiento. Los aminosalicilatos y/o corticosteroides pueden continuarse durante el tratamiento con HUMIRA. La azatioprina y la 6-mercaptopurina (6-MP) [ver Advertencias y precauciones (5.2)] pueden continuarse durante el tratamiento con HUMIRA si es necesario.
Pediatría
La dosis subcutánea recomendada de HUMIRA para pacientes pediátricos de 5 años de edad o mayores con colitis ulcerosa se basa en el peso corporal como se muestra a continuación:
| Peso pediátrico | Dosis recomendada | |
| Días 1 a 15 | A partir del Día 29* | |
| 20 kg (44 lbs) a menos de 40 kg (88 lbs) |
Día 1: 80 mg Día 8: 40 mg Día 15: 40 mg |
40 mg cada dos semanas o 20 mg cada semana |
| 40 kg (88 lbs) y mayor | Día 1: 160 mg (dosis única o dividida en dos días consecutivos) Día 8: 80 mg Día 15: 80 mg |
80 mg cada dos semanas o 40 mg cada semana |
| * Continúe la dosis pediátrica recomendada en pacientes que cumplen 18 años de edad y que están bien controlados con su régimen de HUMIRA. | ||
2.5 Psoriasis en placas o uveítis en adultos
La dosis subcutánea recomendada de HUMIRA para pacientes adultos con psoriasis en placas (Ps) o uveítis (UV) es una dosis inicial de 80 mg, seguida de 40 mg administrada cada dos semanas a partir de una semana después de la dosis inicial. El uso de HUMIRA en Ps crónica moderada a grave más allá de un año no se ha evaluado en estudios clínicos controlados.
2.6 Hidradenitis supurativa
Adultos
La dosis subcutánea recomendada de HUMIRA para pacientes adultos con hidradenitis supurativa (HS) es una dosis inicial de 160 mg (administrada en un día o dividida en dos días consecutivos), seguida de 80 mg dos semanas después (Día 15). Comience la dosificación semanal de 40 mg u 80 mg cada dos semanas dos semanas después (Día 29).
Adolescentes
La dosis subcutánea recomendada de HUMIRA para pacientes adolescentes de 12 años de edad o mayores que pesan al menos 30 kg con hidradenitis supurativa (HS) se basa en el peso corporal como se muestra a continuación [ver Uso en poblaciones específicas (8.4) y Farmacología clínica (12.3)]:
| Peso corporal del adolescente Pacientes (12 años de edad o mayores) |
Dosis recomendada |
| 30 kg (66 lbs) a menos de 60 kg (132 lbs) |
|
| 60 kg (132 lbs) y mayor |
|
2.7 Monitorización para evaluar la seguridad
Antes de iniciar HUMIRA y periódicamente durante el tratamiento, evalúe a los pacientes para detectar tuberculosis activa y realice pruebas para detectar infección latente [ver Advertencias y precauciones (5.1)].
2.8 Consideraciones generales para la administración
HUMIRA está destinado a ser utilizado bajo la guía y supervisión de un médico. Un paciente puede autoinyectarse HUMIRA o un cuidador puede inyectar HUMIRA utilizando el bolígrafo HUMIRA o la jeringa precargada si un médico determina que es apropiado, y con seguimiento médico, según sea necesario, después de una capacitación adecuada en la técnica de inyección subcutánea.
HUMIRA se puede sacar del refrigerador durante 15 a 30 minutos antes de inyectarlo para permitir que el líquido alcance la temperatura ambiente. No retire la tapa o la cubierta mientras se permite que alcance la temperatura ambiente. Inspeccione cuidadosamente la solución en el bolígrafo HUMIRA, la jeringa precargada o el vial de uso institucional de dosis única en busca de partículas y decoloración antes de la administración subcutánea. Si se observan partículas y decoloraciones, no utilice el producto. HUMIRA no contiene conservantes; por lo tanto, deseche las porciones no utilizadas del medicamento que queden en la jeringa. NOTA: Indique a los pacientes sensibles al látex que no manipulen la cubierta de la aguja del bolígrafo HUMIRA de 40 mg/0.8 mL y la jeringa precargada de 40 mg/0.8 mL, 20 mg/0.4 mL y 10 mg/0.2 mL porque puede contener látex de caucho natural [ver Cómo se suministra/Almacenamiento y manipulación (16)].
Indique a los pacientes que utilizan el bolígrafo HUMIRA o la jeringa precargada que inyecten la cantidad total en la jeringa, de acuerdo con las instrucciones proporcionadas en las Instrucciones de uso [ver Instrucciones de uso].
Las inyecciones deben realizarse en sitios separados en el muslo o el abdomen. Rote los sitios de inyección y no administre inyecciones en áreas donde la piel esté sensible, magullada, roja o dura.
Si se olvida una dosis, administre la dosis lo antes posible. Después de eso, reanude la dosificación a la hora programada habitual.
El vial de uso institucional de dosis única HUMIRA es solo para administración en un entorno institucional, como un hospital, consultorio médico o clínica. Retire la dosis utilizando una aguja y jeringa estériles y administre de inmediato por un profesional sanitario en un entorno institucional. Administre solo una dosis por vial. El vial no contiene conservantes; por lo tanto, deseche las porciones no utilizadas.
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
HUMIRA es una solución transparente e incolora disponible como:
-
Pluma (HUMIRA Pen)
Inyección: 80 mg/0.8 mL en una pluma de dosis única.
Inyección: 40 mg/0.8 mL en una pluma de dosis única.
Inyección: 40 mg/0.4 mL en una pluma de dosis única. -
Jeringa precargada
Inyección: 80 mg/0.8 mL en una jeringa de vidrio precargada de dosis única.
Inyección: 40 mg/0.8 mL en una jeringa de vidrio precargada de dosis única.
Inyección: 40 mg/0.4 mL en una jeringa de vidrio precargada de dosis única.
Inyección: 20 mg/0.4 mL en una jeringa de vidrio precargada de dosis única.
Inyección: 20 mg/0.2 mL en una jeringa de vidrio precargada de dosis única.
Inyección: 10 mg/0.2 mL en una jeringa de vidrio precargada de dosis única.
Inyección: 10 mg/0.1 mL en una jeringa de vidrio precargada de dosis única. -
Vial de uso institucional de dosis única
Inyección: 40 mg/0.8 mL en un vial de vidrio de dosis única, para uso institucional únicamente.
4 CONTRAINDICACIONES
Ninguna.
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Infecciones graves
Los pacientes tratados con HUMIRA tienen un mayor riesgo de desarrollar infecciones graves que afectan a varios sistemas y sitios de órganos que pueden provocar hospitalización o muerte. Se han notificado infecciones oportunistas debidas a bacterias, micobacterias, hongos invasivos, virus, parásitos u otros patógenos oportunistas, incluida la aspergilosis, blastomicosis, candidiasis, coccidioidomicosis, histoplasmosis, legionelosis, listeriosis, neumocistosis y tuberculosis con los bloqueadores del TNF. Los pacientes a menudo han presentado enfermedad diseminada en lugar de localizada.
El uso concomitante de un bloqueador del TNF y abatacept o anakinra se asoció con un mayor riesgo de infecciones graves en pacientes con artritis reumatoide (AR); por lo tanto, no se recomienda el uso concomitante de HUMIRA y estos productos biológicos en el tratamiento de pacientes con AR [ver Advertencias y precauciones (5.7, 5.11) e Interacciones medicamentosas (7.2)].
No se debe iniciar el tratamiento con HUMIRA en pacientes con una infección activa, incluidas las infecciones localizadas. Los pacientes de 65 años o más, los pacientes con afecciones concomitantes y/o los pacientes que toman inmunosupresores concomitantes (como corticosteroides o metotrexato) pueden tener un mayor riesgo de infección. Considere los riesgos y beneficios del tratamiento antes de iniciar la terapia en pacientes:
- con infección crónica o recurrente;
- que han estado expuestos a la tuberculosis;
- con antecedentes de una infección oportunista;
- que han residido o viajado a áreas de tuberculosis endémica o micosis endémicas, como histoplasmosis, coccidioidomicosis o blastomicosis; o
- con afecciones subyacentes que pueden predisponerlos a la infección.
Tuberculosis
Se han notificado casos de reactivación de la tuberculosis e infecciones nuevas por tuberculosis en pacientes que reciben HUMIRA, incluidos pacientes que previamente recibieron tratamiento para la tuberculosis latente o activa. Los informes incluyeron casos de tuberculosis pulmonar y extrapulmonar (es decir, diseminada). Evalúe a los pacientes para detectar factores de riesgo de tuberculosis y realice pruebas de infección latente antes de iniciar HUMIRA y periódicamente durante la terapia.
Se ha demostrado que el tratamiento de la infección latente por tuberculosis antes de la terapia con bloqueadores del TNF reduce el riesgo de reactivación de la tuberculosis durante la terapia. Antes de iniciar HUMIRA, evalúe si es necesario el tratamiento para la tuberculosis latente; y considere una induración de ≥ 5 mm un resultado positivo de la prueba cutánea de tuberculina, incluso para pacientes previamente vacunados con Bacilo de Calmette-Guérin (BCG).
Considere la terapia antituberculosa antes de iniciar HUMIRA en pacientes con antecedentes de tuberculosis latente o activa en quienes no se puede confirmar un curso de tratamiento adecuado, y para pacientes con una prueba negativa para tuberculosis latente pero que tienen factores de riesgo de infección por tuberculosis. A pesar del tratamiento profiláctico para la tuberculosis, se han producido casos de tuberculosis reactivada en pacientes tratados con HUMIRA. Se recomienda la consulta con un médico con experiencia en el tratamiento de la tuberculosis para ayudar en la decisión de si es apropiado iniciar la terapia antituberculosa para un paciente individual.
Considere seriamente la tuberculosis en el diagnóstico diferencial en pacientes que desarrollan una nueva infección durante el tratamiento con HUMIRA, especialmente en pacientes que previamente o recientemente han viajado a países con una alta prevalencia de tuberculosis, o que han tenido contacto cercano con una persona con tuberculosis activa.
Monitoreo
Monitoree de cerca a los pacientes para detectar el desarrollo de signos y síntomas de infección durante y después del tratamiento con HUMIRA, incluido el desarrollo de tuberculosis en pacientes que dieron negativo en la prueba de infección latente por tuberculosis antes de iniciar la terapia. Las pruebas de infección latente por tuberculosis también pueden ser falsamente negativas mientras se está en terapia con HUMIRA.
Suspenda HUMIRA si un paciente desarrolla una infección grave o sepsis. Para un paciente que desarrolla una nueva infección durante el tratamiento con HUMIRA, monitórelo de cerca, realice una evaluación diagnóstica rápida y completa apropiada para un paciente inmunocomprometido e inicie la terapia antimicrobiana apropiada.
Infecciones fúngicas invasivas
Si los pacientes desarrollan una enfermedad sistémica grave y residen o viajan a regiones donde las micosis son endémicas, considere la infección fúngica invasiva en el diagnóstico diferencial. Las pruebas de antígeno y anticuerpos para la histoplasmosis pueden ser negativas en algunos pacientes con infección activa. Considere la terapia antifúngica empírica apropiada, teniendo en cuenta tanto el riesgo de infección fúngica grave como los riesgos de la terapia antifúngica, mientras se realiza una evaluación diagnóstica. Para ayudar en el manejo de estos pacientes, considere la consulta con un médico con experiencia en el diagnóstico y tratamiento de infecciones fúngicas invasivas.
5.2 Neoplasias malignas
Considere los riesgos y beneficios del tratamiento con bloqueadores del TNF, incluido HUMIRA, antes de iniciar la terapia en pacientes con una neoplasia maligna conocida que no sea un cáncer de piel no melanoma (NMSC) tratado con éxito o cuando se considere continuar con un bloqueador del TNF en pacientes que desarrollan una neoplasia maligna.
Neoplasias malignas en adultos
En las porciones controladas de los ensayos clínicos de algunos bloqueadores del TNF, incluido HUMIRA, se han observado más casos de malignidades entre los pacientes adultos tratados con bloqueadores del TNF en comparación con los pacientes adultos tratados con control. Durante las porciones controladas de 39 ensayos clínicos globales de HUMIRA en pacientes adultos con artritis reumatoide (AR), artritis psoriásica (APs), espondilitis anquilosante (EA), enfermedad de Crohn (EC), colitis ulcerosa (CU), psoriasis en placas (Ps), hidradenitis supurativa (HS) y uveítis (UV), se observaron malignidades, distintas del cáncer de piel no melanoma (basal y de células escamosas), a una tasa (intervalo de confianza del 95%) de 0,7 (0,48, 1,03) por 100 años-paciente entre 7973 pacientes tratados con HUMIRA frente a una tasa de 0,7 (0,41, 1,17) por 100 años-paciente entre 4848 pacientes tratados con control (duración media del tratamiento de 4 meses para los pacientes tratados con HUMIRA y 4 meses para los pacientes tratados con control). En 52 ensayos clínicos globales controlados y no controlados de HUMIRA en pacientes adultos con AR, APs, EA, EC, CU, Ps, HS y UV, las malignidades más frecuentemente observadas, distintas del linfoma y el NMSC, fueron de mama, colon, próstata, pulmón y melanoma. Las malignidades en los pacientes tratados con HUMIRA en las porciones controladas y no controladas de los estudios fueron similares en tipo y número a lo que se esperaría en la población general de EE. UU. según la base de datos SEER (ajustada por edad, sexo y raza).1
En ensayos controlados de otros bloqueadores del TNF en pacientes adultos con mayor riesgo de malignidades (es decir, pacientes con EPOC con antecedentes significativos de tabaquismo y pacientes tratados con ciclofosfamida con granulomatosis de Wegener), una mayor proporción de malignidades se produjo en el grupo de bloqueadores del TNF en comparación con el grupo de control.
Cáncer de piel no melanoma
Durante las porciones controladas de 39 ensayos clínicos globales de HUMIRA en pacientes adultos con AR, APs, EA, EC, CU, Ps, HS y UV, la tasa (intervalo de confianza del 95%) de NMSC fue de 0,8 (0,52, 1,09) por 100 años-paciente entre los pacientes tratados con HUMIRA y 0,2 (0,10, 0,59) por 100 años-paciente entre los pacientes tratados con control. Examine a todos los pacientes, y en particular a los pacientes con antecedentes médicos de terapia inmunosupresora prolongada previa o pacientes con psoriasis con antecedentes de tratamiento con PUVA para la presencia de NMSC antes y durante el tratamiento con HUMIRA.
Linfoma y leucemia
En las porciones controladas de los ensayos clínicos de todos los bloqueadores del TNF en adultos, se han observado más casos de linfoma entre los pacientes tratados con bloqueadores del TNF en comparación con los pacientes tratados con control. En las porciones controladas de 39 ensayos clínicos globales de HUMIRA en pacientes adultos con AR, APs, EA, EC, CU, Ps, HS y UV, se produjeron 2 linfomas entre 7973 pacientes tratados con HUMIRA frente a 1 entre 4848 pacientes tratados con control. En 52 ensayos clínicos globales controlados y no controlados de HUMIRA en pacientes adultos con AR, APs, EA, EC, CU, Ps, HS y UV con una duración media de aproximadamente 0,7 años, incluidos 24 605 pacientes y más de 40 215 años-paciente de HUMIRA, la tasa observada de linfomas fue de aproximadamente 0,11 por 100 años-paciente. Esto es aproximadamente 3 veces más alto de lo esperado en la población general de EE. UU. según la base de datos SEER (ajustada por edad, sexo y raza).1 Las tasas de linfoma en los ensayos clínicos de HUMIRA no se pueden comparar con las tasas de linfoma en los ensayos clínicos de otros bloqueadores del TNF y pueden no predecir las tasas observadas en una población de pacientes más amplia. Los pacientes con AR y otras enfermedades inflamatorias crónicas, particularmente aquellos con enfermedad altamente activa y/o exposición crónica a terapias inmunosupresoras, pueden tener un riesgo mayor (hasta varias veces) que la población general de desarrollar linfoma, incluso en ausencia de bloqueadores del TNF. Se han notificado casos postcomercialización de leucemia aguda y crónica en asociación con el uso de bloqueadores del TNF en AR y otras indicaciones. Incluso en ausencia de terapia con bloqueadores del TNF, los pacientes con AR pueden tener un riesgo mayor (aproximadamente 2 veces) que la población general de desarrollar leucemia.
Malignidades en pacientes pediátricos y adultos jóvenes
Se han notificado malignidades, algunas fatales, entre niños, adolescentes y adultos jóvenes que recibieron tratamiento con bloqueadores del TNF (inicio de la terapia ≤ 18 años de edad), de los cuales HUMIRA es un miembro. Aproximadamente la mitad de los casos fueron linfomas, incluidos el linfoma de Hodgkin y el linfoma no Hodgkin. Los demás casos representaron una variedad de diferentes malignidades e incluyeron malignidades raras generalmente asociadas con la inmunosupresión y malignidades que no se observan generalmente en niños y adolescentes. Las malignidades ocurrieron después de una mediana de 30 meses de terapia (rango de 1 a 84 meses). La mayoría de los pacientes estaban recibiendo inmunosupresores concomitantes. Estos casos se notificaron después de la comercialización y se derivan de una variedad de fuentes, incluidos los registros y los informes espontáneos posteriores a la comercialización.
Se han notificado casos postcomercialización de linfoma de células T hepatosplénico (HSTCL), un tipo raro de linfoma de células T, en pacientes tratados con bloqueadores del TNF, incluido HUMIRA. Estos casos han tenido un curso de enfermedad muy agresivo y han sido fatales. La mayoría de los casos notificados de bloqueadores del TNF se han producido en pacientes con enfermedad de Crohn o colitis ulcerosa y la mayoría fueron en adolescentes y adultos jóvenes. Casi todos estos pacientes habían recibido tratamiento con los inmunosupresores azatioprina o 6-mercaptopurina (6-MP) de forma concomitante con un bloqueador del TNF en o antes del diagnóstico. No está claro si la aparición de HSTCL está relacionada con el uso de un bloqueador del TNF o un bloqueador del TNF en combinación con estos otros inmunosupresores. El riesgo potencial con la combinación de azatioprina o 6-mercaptopurina y HUMIRA debe considerarse cuidadosamente.
5.3 Reacciones de hipersensibilidad
Se han notificado anafilaxia y edema angioneurótico tras la administración de HUMIRA. Si se produce una reacción anafiláctica u otra reacción alérgica grave, interrumpa inmediatamente la administración de HUMIRA e instituya la terapia adecuada. En los ensayos clínicos de HUMIRA, se han observado reacciones de hipersensibilidad (p. ej., erupción cutánea, reacción anafilactoide, reacción medicamentosa fija, reacción medicamentosa no especificada, urticaria).
5.4 Reactivación del virus de la hepatitis B
El uso de bloqueadores del TNF, incluido HUMIRA, puede aumentar el riesgo de reactivación del virus de la hepatitis B (VHB) en pacientes que son portadores crónicos de este virus. En algunos casos, la reactivación del VHB que ocurre en conjunto con la terapia con bloqueadores del TNF ha sido fatal. La mayoría de estos informes han ocurrido en pacientes que reciben concomitantemente otros medicamentos que suprimen el sistema inmunitario, lo que también puede contribuir a la reactivación del VHB. Evalúe a los pacientes en riesgo de infección por VHB para obtener evidencia previa de infección por VHB antes de iniciar la terapia con bloqueadores del TNF. Tenga precaución al prescribir bloqueadores del TNF para pacientes identificados como portadores de VHB. No hay datos suficientes disponibles sobre la seguridad o eficacia del tratamiento de pacientes que son portadores de VHB con terapia antiviral en combinación con terapia con bloqueadores del TNF para prevenir la reactivación del VHB. Para los pacientes que son portadores de VHB y requieren tratamiento con bloqueadores del TNF, controle de cerca a estos pacientes para detectar signos clínicos y de laboratorio de infección activa por VHB durante toda la terapia y durante varios meses después de la finalización de la terapia. En pacientes que desarrollan reactivación del VHB, suspenda HUMIRA e inicie una terapia antiviral eficaz con tratamiento de apoyo adecuado. No se conoce la seguridad de reanudar la terapia con bloqueadores del TNF después de que la reactivación del VHB esté controlada. Por lo tanto, tenga precaución al considerar la reanudación de la terapia con HUMIRA en esta situación y controle a los pacientes de cerca.
5.5 Reacciones neurológicas
El uso de agentes bloqueadores del TNF, incluido HUMIRA, se ha asociado con casos raros de aparición de nuevos síntomas clínicos o exacerbación de los mismos y/o evidencia radiográfica de enfermedad desmielinizante del sistema nervioso central, incluida la esclerosis múltiple (EM) y la neuritis óptica, y enfermedad desmielinizante periférica, incluido el síndrome de Guillain-Barré. Tenga precaución al considerar el uso de HUMIRA en pacientes con trastornos desmielinizantes preexistentes o de reciente aparición del sistema nervioso central o periférico; se debe considerar la interrupción de HUMIRA si se desarrolla alguno de estos trastornos. Existe una asociación conocida entre la uveítis intermedia y los trastornos desmielinizantes centrales.
5.6 Reacciones hematológicas
Se han notificado informes raros de pancitopenia, incluida la anemia aplásica, con agentes bloqueadores del TNF. Las reacciones adversas del sistema hematológico, incluida la citopenia médicamente significativa (por ejemplo, trombocitopenia, leucopenia) se han notificado con poca frecuencia con HUMIRA. La relación causal de estos informes con HUMIRA sigue sin estar clara. Avise a todos los pacientes que busquen atención médica inmediata si desarrollan signos y síntomas sugestivos de discrasias sanguíneas o infección (por ejemplo, fiebre persistente, hematomas, sangrado, palidez) mientras están tomando HUMIRA. Considere la interrupción de la terapia con HUMIRA en pacientes con anomalías hematológicas significativas confirmadas.
5.7 Mayor riesgo de infección cuando se usa con anakinra
El uso concomitante de anakinra (un antagonista de la interleucina-1) y otro bloqueador del TNF se asoció con una mayor proporción de infecciones graves y neutropenia y ningún beneficio adicional en comparación con el bloqueador del TNF solo en pacientes con AR. Por lo tanto, no se recomienda la combinación de HUMIRA y anakinra [ver Interacciones medicamentosas (7.2)].
5.8 Insuficiencia cardíaca
Se han notificado casos de empeoramiento de la insuficiencia cardíaca congestiva (ICC) y aparición de nueva ICC con bloqueadores del TNF. También se han observado casos de empeoramiento de la ICC con HUMIRA. HUMIRA no se ha estudiado formalmente en pacientes con ICC; sin embargo, en ensayos clínicos de otro bloqueador del TNF, se observó una tasa más alta de reacciones adversas graves relacionadas con la ICC. Tenga precaución al usar HUMIRA en pacientes que tienen insuficiencia cardíaca y mónitorelos cuidadosamente.
5.9 Autoinmunidad
El tratamiento con HUMIRA puede provocar la formación de autoanticuerpos y, en raras ocasiones, el desarrollo de un síndrome similar al lupus. Si un paciente desarrolla síntomas sugestivos de un síndrome similar al lupus después del tratamiento con HUMIRA, suspenda el tratamiento [ver Reacciones adversas (6.1)].
5.10 Inmunizaciones
En un ensayo clínico controlado con placebo en pacientes con AR, no se detectó ninguna diferencia en la respuesta de anticuerpos anti-neumococo entre los grupos de tratamiento con HUMIRA y placebo cuando la vacuna neumocócica polisacárida y la vacuna contra la influenza se administraron de forma concurrente con HUMIRA. Proporciones similares de pacientes desarrollaron niveles protectores de anticuerpos anti-influenza entre los grupos de tratamiento con HUMIRA y placebo; sin embargo, los títulos en conjunto a los antígenos de la influenza fueron moderadamente más bajos en los pacientes que recibieron HUMIRA. Se desconoce la importancia clínica de esto. Los pacientes que toman HUMIRA pueden recibir vacunas concurrentes, excepto las vacunas vivas. No hay datos disponibles sobre la transmisión secundaria de la infección por vacunas vivas en pacientes que reciben HUMIRA.
Se recomienda que los pacientes pediátricos, si es posible, se pongan al día con todas las inmunizaciones de acuerdo con las pautas de inmunización actuales antes de iniciar la terapia con HUMIRA. Los pacientes que toman HUMIRA pueden recibir vacunas concurrentes, excepto las vacunas vivas.
La seguridad de administrar vacunas vivas o atenuadas en bebés expuestos a HUMIRA in útero es desconocida. Se deben considerar los riesgos y beneficios antes de vacunar (vivas o atenuadas) a los bebés expuestos [ver Uso en poblaciones específicas (8.1, 8.4)].
5.11 Riesgo aumentado de infección cuando se usa con abatacept
En ensayos controlados, la administración concomitante de bloqueadores del TNF y abatacept se asoció con una mayor proporción de infecciones graves que el uso de un bloqueador del TNF solo; la terapia combinada, en comparación con el uso de un bloqueador del TNF solo, no ha demostrado una mejora del beneficio clínico en el tratamiento de la AR. Por lo tanto, no se recomienda la combinación de abatacept con bloqueadores del TNF, incluido HUMIRA [ver Interacciones medicamentosas (7.2)].
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas clínicamente significativas se describen en otros lugares de la etiqueta:
- Infecciones graves [ver Advertencias y precauciones (5.1)]
- Neoplasias malignas [ver Advertencias y precauciones (5.2)]
- Reacciones de hipersensibilidad [ver Advertencias y precauciones (5.3)]
- Reactivación del virus de la hepatitis B [ver Advertencias y precauciones (5.4)]
- Reacciones neurológicas [ver Advertencias y precauciones (5.5)]
- Reacciones hematológicas [ver Advertencias y precauciones (5.6)]
- Insuficiencia cardíaca [ver Advertencias y precauciones (5.8)]
- Autoinmunidad [ver Advertencias y precauciones (5.9)]
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se llevan a cabo en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un medicamento no se pueden comparar directamente con las tasas en los ensayos clínicos de otro medicamento y es posible que no reflejen las tasas observadas en la práctica.
La reacción adversa más común con HUMIRA fueron las reacciones en el lugar de la inyección. En los ensayos controlados con placebo, el 20% de los pacientes tratados con HUMIRA desarrollaron reacciones en el lugar de la inyección (eritema y/o picazón, hemorragia, dolor o hinchazón), en comparación con el 14% de los pacientes que recibieron placebo. La mayoría de las reacciones en el lugar de la inyección se describieron como leves y generalmente no requirieron la interrupción del medicamento.
La proporción de pacientes que interrumpieron el tratamiento debido a reacciones adversas durante la parte doble ciego y controlada con placebo de los estudios en pacientes con AR (es decir, estudios RA-I, RA-II, RA-III y RA-IV) fue del 7% para los pacientes que recibieron HUMIRA y del 4% para los pacientes tratados con placebo. Las reacciones adversas más comunes que llevaron a la interrupción de HUMIRA en estos estudios de AR fueron reacción de brote clínico (0.7%), erupción cutánea (0.3%) y neumonía (0.3%).
Infecciones
En las partes controladas de los 39 ensayos clínicos globales de HUMIRA en pacientes adultos con AR, PsA, AS, CD, CU, Ps, HS y UV, la tasa de infecciones graves fue de 4.3 por 100 pacientes-años en 7973 pacientes tratados con HUMIRA en comparación con una tasa de 2.9 por 100 pacientes-años en 4848 pacientes tratados con control. Las infecciones graves observadas incluyeron neumonía, artritis séptica, infecciones protésicas y postquirúrgicas, erisipela, celulitis, diverticulitis y pielonefritis [ver Advertencias y precauciones (5.1)].
Tuberculosis e infecciones oportunistas
En 52 ensayos clínicos controlados y no controlados en AR, PsA, AS, CD, CU, Ps, HS y UV que incluyeron a 24,605 pacientes tratados con HUMIRA, la tasa de tuberculosis activa informada fue de 0.20 por 100 pacientes-años y la tasa de conversión positiva de PPD fue de 0.09 por 100 pacientes-años. En un subgrupo de 10,113 pacientes tratados con HUMIRA en Estados Unidos y Canadá, la tasa de tuberculosis activa informada fue de 0.05 por 100 pacientes-años y la tasa de conversión positiva de PPD fue de 0.07 por 100 pacientes-años. Estos ensayos incluyeron informes de tuberculosis miliar, linfática, peritoneal y pulmonar. La mayoría de los casos de tuberculosis ocurrieron dentro de los primeros ocho meses después del inicio de la terapia y pueden reflejar la recrudescencia de la enfermedad latente. En estos ensayos clínicos globales, se han informado casos de infecciones oportunistas graves a una tasa general de 0.05 por 100 pacientes-años. Algunos casos de infecciones oportunistas graves y tuberculosis han sido fatales [ver Advertencias y precauciones (5.1)].
Autoanticuerpos
En los ensayos controlados de artritis reumatoide, el 12% de los pacientes tratados con HUMIRA y el 7% de los pacientes tratados con placebo que tenían títulos de ANA negativos al inicio desarrollaron títulos positivos en la semana 24. Dos pacientes de 3046 tratados con HUMIRA desarrollaron signos clínicos sugestivos de síndrome similar al lupus de inicio nuevo. Los pacientes mejoraron después de la interrupción de la terapia. Ningún paciente desarrolló nefritis lúpica o síntomas del sistema nervioso central. Se desconoce el impacto del tratamiento a largo plazo con HUMIRA en el desarrollo de enfermedades autoinmunes.
Elevaciones de las enzimas hepáticas
Se han informado casos de reacciones hepáticas graves, incluyendo insuficiencia hepática aguda, en pacientes que reciben bloqueadores del TNF. En ensayos controlados de fase 3 de HUMIRA (40 mg SC cada dos semanas) en pacientes con AR, PsA y AS con una duración del período de control que varía de 4 a 104 semanas, las elevaciones de ALT ≥ 3 x LSN ocurrieron en el 3.5% de los pacientes tratados con HUMIRA y el 1.5% de los pacientes tratados con control. Dado que muchos de estos pacientes en estos ensayos también estaban tomando medicamentos que causan elevaciones de las enzimas hepáticas (por ejemplo, AINE, MTX), no está claro cuál es la relación entre HUMIRA y las elevaciones de las enzimas hepáticas. En un ensayo controlado de fase 3 de HUMIRA en pacientes con AIJ poliarticular de 4 a 17 años, las elevaciones de ALT ≥ 3 x LSN ocurrieron en el 4.4% de los pacientes tratados con HUMIRA y el 1.5% de los pacientes tratados con control (las elevaciones de ALT fueron más comunes que las de AST); las elevaciones de las pruebas de las enzimas hepáticas fueron más frecuentes entre aquellos tratados con la combinación de HUMIRA y MTX que entre aquellos tratados solo con HUMIRA. En general, estas elevaciones no llevaron a la interrupción del tratamiento con HUMIRA. No se produjeron elevaciones de ALT ≥ 3 x LSN en el estudio abierto de HUMIRA en pacientes con AIJ poliarticular de 2 a <4 años.
En ensayos controlados de Fase 3 de HUMIRA (dosis iniciales de 160 mg y 80 mg, u 80 mg y 40 mg en los Días 1 y 15, respectivamente, seguidas de 40 mg cada dos semanas) en pacientes adultos con enfermedad de Crohn con una duración del período de control que oscila entre 4 y 52 semanas, las elevaciones de ALT ≥ 3 x ULN ocurrieron en el 0,9% de los pacientes tratados con HUMIRA y el 0,9% de los pacientes tratados con control. En el ensayo de Fase 3 de HUMIRA en pacientes pediátricos con enfermedad de Crohn que evaluó la eficacia y seguridad de dos regímenes de dosis de mantenimiento basados en el peso corporal después de la terapia de inducción basada en el peso corporal hasta 52 semanas de tratamiento, las elevaciones de ALT ≥ 3 x ULN ocurrieron en el 2,6% (5/192) de los pacientes, de los cuales 4 estaban recibiendo inmunosupresores concomitantes en el momento de la línea de base; ninguno de estos pacientes interrumpió el tratamiento debido a anomalías en las pruebas de ALT. En ensayos controlados de Fase 3 de HUMIRA (dosis iniciales de 160 mg y 80 mg en los Días 1 y 15, respectivamente, seguidas de 40 mg cada dos semanas) en pacientes adultos con UC con una duración del período de control que oscila entre 1 y 52 semanas, las elevaciones de ALT ≥ 3 x ULN ocurrieron en el 1,5% de los pacientes tratados con HUMIRA y el 1,0% de los pacientes tratados con control. En el ensayo controlado de Fase 3 de HUMIRA en pacientes con colitis ulcerosa pediátrica (N=93), que evaluó la eficacia y seguridad de una dosis de mantenimiento de 0,6 mg/kg (máximo de 40 mg) cada dos semanas (N=31) y una dosis de mantenimiento de 0,6 mg/kg (máximo de 40 mg) cada semana (N=32), después de dosis de inducción basadas en el peso corporal de 2,4 mg/kg (máximo de 160 mg) en la Semana 0 y la Semana 1, y 1,2 mg/kg (máximo de 80 mg) en la Semana 2 (N=63), o una dosis de inducción de 2,4 mg/kg (máximo de 160 mg) en la Semana 0, placebo en la Semana 1, y 1,2 mg/kg (máximo de 80 mg) en la Semana 2 (N=30), las elevaciones de ALT ≥ 3 X ULN ocurrieron en el 1,1% (1/93) de los pacientes. En ensayos controlados de Fase 3 de HUMIRA (dosis inicial de 80 mg y luego 40 mg cada dos semanas) en pacientes con Ps con una duración del período de control que oscila entre 12 y 24 semanas, las elevaciones de ALT ≥ 3 x ULN ocurrieron en el 1,8% de los pacientes tratados con HUMIRA y el 1,8% de los pacientes tratados con control. En ensayos controlados de HUMIRA (dosis iniciales de 160 mg en la Semana 0 y 80 mg en la Semana 2, seguidas de 40 mg cada semana a partir de la Semana 4), en sujetos con HS con una duración del período de control que oscila entre 12 y 16 semanas, las elevaciones de ALT ≥ 3 x ULN ocurrieron en el 0,3% de los sujetos tratados con HUMIRA y el 0,6% de los sujetos tratados con control. En ensayos controlados de HUMIRA (dosis iniciales de 80 mg en la Semana 0 seguidas de 40 mg cada dos semanas a partir de la Semana 1) en pacientes adultos con uveítis con una exposición de 165,4 PYs y 119,8 PYs en pacientes tratados con HUMIRA y tratados con control, respectivamente, las elevaciones de ALT ≥ 3 x ULN ocurrieron en el 2,4% de los pacientes tratados con HUMIRA y el 2,4% de los pacientes tratados con control.
Otras reacciones adversas
Estudios clínicos de artritis reumatoide
Los datos descritos a continuación reflejan la exposición a HUMIRA en 2468 pacientes, incluidos 2073 expuestos durante 6 meses, 1497 expuestos durante más de un año y 1380 en estudios adecuados y bien controlados (Estudios RA-I, RA-II, RA-III y RA-IV). HUMIRA se estudió principalmente en ensayos controlados con placebo y en estudios de seguimiento a largo plazo con una duración de hasta 36 meses. La población tenía una edad media de 54 años, el 77% eran mujeres, el 91% eran caucásicas y tenían artritis reumatoide activa de moderada a grave. La mayoría de los pacientes recibieron 40 mg de HUMIRA cada dos semanas [ver Estudios clínicos (14.1)].
La Tabla 1 resume las reacciones notificadas con una tasa de al menos el 5% en los pacientes tratados con HUMIRA 40 mg cada dos semanas en comparación con el placebo y con una incidencia superior al placebo. En el Estudio RA-III, los tipos y frecuencias de reacciones adversas en la extensión de etiqueta abierta del segundo año fueron similares a las observadas en la parte de doble ciego de un año.
| HUMIRA 40 mg subcutáneo Cada dos semanas |
Placebo | |
| (N=705) | (N=690) | |
| Reacción adversa (Término preferido) | ||
| Respiratorio | ||
| Infección de las vías respiratorias superiores | 17% | 13% |
| Sinusitis | 11% | 9% |
| Síndrome gripal | 7% | 6% |
| Gastrointestinal | ||
| Náuseas | 9% | 8% |
| Dolor abdominal | 7% | 4% |
| Pruebas de laboratorio* | ||
| Prueba de laboratorio anormal | 8% | 7% |
| Hipercolesterolemia | 6% | 4% |
| Hiperlipidemia | 7% | 5% |
| Hematuria | 5% | 4% |
| Fosfatasa alcalina aumentada | 5% | 3% |
| Otros | ||
| Dolor de cabeza | 12% | 8% |
| Erupción cutánea | 12% | 6% |
| Lesión accidental | 10% | 8% |
| Reacción en el sitio de inyección ** | 8% | 1% |
| Dolor de espalda | 6% | 4% |
| Infección del tracto urinario | 8% | 5% |
| Hipertensión | 5% | 3% |
| * Las anormalidades en las pruebas de laboratorio se reportaron como reacciones adversas en los ensayos europeos ** No incluye eritema, picazón, hemorragia, dolor o hinchazón en el sitio de inyección |
||
Reacciones adversas menos frecuentes en estudios clínicos de artritis reumatoide
Otras reacciones adversas graves infrecuentes que no aparecen en las secciones de Advertencias y precauciones o Reacciones adversas que ocurrieron con una incidencia inferior al 5% en pacientes tratados con HUMIRA en estudios de AR fueron:
Cuerpo como un todo: Dolor en la extremidad, dolor pélvico, cirugía, dolor en el tórax
Sistema cardiovascular: Arritmia, fibrilación auricular, dolor en el pecho, trastorno de la arteria coronaria, paro cardíaco, encefalopatía hipertensiva, infarto de miocardio, palpitaciones, derrame pericárdico, pericarditis, síncope, taquicardia
Sistema digestivo: Colecistitis, colelitiasis, esofagitis, gastroenteritis, hemorragia gastrointestinal, necrosis hepática, vómitos
Sistema endocrino: Trastorno de la paratiroides
Sistema hemopoyético y linfático: Agranulocitosis, policitemia
Trastornos metabólicos y nutricionales: Deshidratación, curación anormal, cetosis, paraproteinemia, edema periférico
Sistema musculoesquelético: Artritis, trastorno óseo, fractura ósea (no espontánea), necrosis ósea, trastorno articular, calambres musculares, miastenia, artritis piógena, sinovitis, trastorno tendinoso
Neoplasia: Adenoma
Sistema nervioso: Confusión, parestesia, hematoma subdural, temblor
Sistema respiratorio: Asma, broncoespasmo, disnea, disminución de la función pulmonar, derrame pleural
Sentidos especiales: Catarata
Trombosis: Trombosis de la pierna
Sistema urogenital: Cistitis, cálculo renal, trastorno menstrual
Estudios clínicos de artritis idiopática juvenil
En general, las reacciones adversas en los pacientes tratados con HUMIRA en los ensayos de artritis idiopática juvenil poliarticular (AIAJ) (Estudios JIA-I y JIA-II) [ver Estudios clínicos (14.2)] fueron similares en frecuencia y tipo a las observadas en pacientes adultos [ver Advertencias y precauciones (5), Reacciones adversas (6)]. Los hallazgos importantes y las diferencias con los adultos se analizan en los siguientes párrafos.
En el Estudio JIA-I, HUMIRA se estudió en 171 pacientes de 4 a 17 años de edad, con AIAJ poliarticular. Las reacciones adversas graves notificadas en el estudio incluyeron neutropenia, faringitis estreptocócica, aumento de las aminotransferasas, herpes zóster, miositis, metrorragia y apendicitis. Se observaron infecciones graves en el 4% de los pacientes en aproximadamente 2 años desde el inicio del tratamiento con HUMIRA e incluyeron casos de herpes simple, neumonía, infección del tracto urinario, faringitis y herpes zóster.
En el Estudio JIA-I, el 45% de los pacientes experimentaron una infección mientras recibían HUMIRA con o sin MTX concomitante en las primeras 16 semanas de tratamiento. Los tipos de infecciones notificadas en pacientes tratados con HUMIRA fueron generalmente similares a las que se observan comúnmente en pacientes con AIAJ poliarticular que no reciben tratamiento con bloqueadores del TNF. Tras el inicio del tratamiento, las reacciones adversas más frecuentes que se produjeron en esta población de pacientes tratados con HUMIRA fueron el dolor en el lugar de la inyección y la reacción en el lugar de la inyección (19% y 16%, respectivamente). Un evento adverso menos comúnmente notificado en pacientes que recibieron HUMIRA fue el granuloma anular, que no condujo a la interrupción del tratamiento con HUMIRA.
En las primeras 48 semanas de tratamiento en el Estudio JIA-I, se observaron reacciones de hipersensibilidad no graves en aproximadamente el 6% de los pacientes e incluyeron principalmente reacciones de hipersensibilidad alérgica localizada y erupción alérgica.
En el Estudio JIA-I, el 10% de los pacientes tratados con HUMIRA que tenían anticuerpos anti-ADNdc negativos en la línea de base desarrollaron títulos positivos después de 48 semanas de tratamiento. Ningún paciente desarrolló signos clínicos de autoinmunidad durante el ensayo clínico.
Aproximadamente el 15% de los pacientes tratados con HUMIRA desarrollaron elevaciones leves a moderadas de la creatincinasa (CPK) en el Estudio JIA-I. Se observaron elevaciones que superaban 5 veces el límite superior de la normalidad en varios pacientes. Las concentraciones de CPK disminuyeron o volvieron a la normalidad en todos los pacientes. La mayoría de los pacientes pudieron continuar con HUMIRA sin interrupción.
En el Estudio JIA-II, HUMIRA se estudió en 32 pacientes de 2 a <4 años de edad o de 4 años de edad o más que pesaban <15 kg con AIAJ poliarticular. El perfil de seguridad para esta población de pacientes fue similar al perfil de seguridad observado en pacientes de 4 a 17 años de edad con AIAJ poliarticular.
En el Estudio JIA-II, el 78% de los pacientes experimentaron una infección mientras recibían HUMIRA. Estas incluyeron nasofaringitis, bronquitis, infección del tracto respiratorio superior, otitis media, y en su mayoría fueron de gravedad leve a moderada. Se observaron infecciones graves en el 9% de los pacientes que recibieron HUMIRA en el estudio e incluyeron caries dentales, gastroenteritis por rotavirus y varicela.
En el Estudio JIA-II, se observaron reacciones alérgicas no graves en el 6% de los pacientes e incluyeron urticaria intermitente y erupción cutánea, todas de gravedad leve.
Estudios clínicos de artritis psoriásica y espondilitis anquilosante
HUMIRA se ha estudiado en 395 pacientes con artritis psoriásica (APs) en dos ensayos controlados con placebo y en un estudio de etiqueta abierta y en 393 pacientes con espondilitis anquilosante (EA) en dos estudios controlados con placebo [ver Estudios clínicos (14.3, 14.4)]. El perfil de seguridad para los pacientes con APs y EA tratados con HUMIRA 40 mg cada dos semanas fue similar al perfil de seguridad observado en pacientes con AR, Estudios HUMIRA RA-I a IV.
Estudios clínicos de enfermedad de Crohn
Adultos: El perfil de seguridad de HUMIRA en 1478 pacientes adultos con enfermedad de Crohn de cuatro estudios controlados con placebo y dos estudios de extensión de etiqueta abierta [ver Estudios clínicos (14.5)] fue similar al perfil de seguridad observado en pacientes con AR.
Pacientes pediátricos de 6 a 17 años: El perfil de seguridad de HUMIRA en 192 pacientes pediátricos de un estudio doble ciego (Estudio PCD-I) y un estudio de extensión de etiqueta abierta [ver Estudios clínicos (14.6)] fue similar al perfil de seguridad observado en pacientes adultos con enfermedad de Crohn.
Durante la fase de inducción de etiqueta abierta de 4 semanas del Estudio PCD-I, las reacciones adversas más comunes que ocurrieron en la población pediátrica tratada con HUMIRA fueron dolor en el sitio de inyección y reacción en el sitio de inyección (6% y 5%, respectivamente).
Un total del 67% de los niños experimentaron una infección mientras recibían HUMIRA en el Estudio PCD-I. Estos incluyeron infección del tracto respiratorio superior y nasofaringitis.
Un total del 5% de los niños experimentaron una infección grave mientras recibían HUMIRA en el Estudio PCD-I. Estos incluyeron infección viral, sepsis relacionada con el dispositivo (catéter), gastroenteritis, influenza H1N1 e histoplasmosis diseminada.
En el Estudio PCD-I, se observaron reacciones alérgicas en el 5% de los niños, todas no graves y principalmente reacciones localizadas.
Estudios clínicos de colitis ulcerosa
Adultos: El perfil de seguridad de HUMIRA en 1010 pacientes adultos con colitis ulcerosa (CU) de dos estudios controlados con placebo y un estudio de extensión de etiqueta abierta [ver Estudios clínicos (14.7)] fue similar al perfil de seguridad observado en pacientes con AR.
Pacientes pediátricos de 5 a 17 años: El perfil de seguridad de HUMIRA en 93 pacientes pediátricos con colitis ulcerosa de un estudio doble ciego y un estudio de extensión de etiqueta abierta [ver Estudios clínicos (14.8)] fue similar al perfil de seguridad observado en pacientes adultos con colitis ulcerosa.
Estudios clínicos de psoriasis en placas
HUMIRA se ha estudiado en 1696 sujetos con psoriasis en placas (Ps) en estudios controlados con placebo y de extensión de etiqueta abierta [ver Estudios clínicos (14.9)]. El perfil de seguridad para los sujetos con Ps tratados con HUMIRA fue similar al perfil de seguridad observado en los sujetos con AR con las siguientes excepciones. En las partes controladas con placebo de los ensayos clínicos en sujetos con Ps, los sujetos tratados con HUMIRA tuvieron una mayor incidencia de artralgia en comparación con los controles (3% vs. 1%).
Estudios clínicos de hidradenitis supurativa
HUMIRA se ha estudiado en 727 sujetos con hidradenitis supurativa (HS) en tres estudios controlados con placebo y un estudio de extensión de etiqueta abierta [ver Estudios clínicos (14.10)]. El perfil de seguridad para los sujetos con HS tratados con HUMIRA semanalmente fue consistente con el perfil de seguridad conocido de HUMIRA.
El brote de HS, definido como ≥25% de aumento desde el inicio en el recuento de abscesos y nódulos inflamatorios y con un mínimo de 2 lesiones adicionales, se documentó en 22 (22%) de los 100 sujetos que fueron retirados del tratamiento con HUMIRA después del punto de tiempo de eficacia primaria en dos estudios.
Estudios clínicos de uveítis
HUMIRA se ha estudiado en 464 pacientes adultos con uveítis (UV) en estudios controlados con placebo y de extensión de etiqueta abierta y en 90 pacientes pediátricos con uveítis (Estudio PUV-I) [ver Estudios clínicos (14.11, 14.12)] . El perfil de seguridad para los pacientes con UV tratados con HUMIRA fue similar al perfil de seguridad observado en los pacientes con AR.
6.2 Inmunogenicidad
Al igual que con todas las proteínas terapéuticas, existe la posibilidad de inmunogenicidad. La detección de la formación de anticuerpos depende en gran medida de la sensibilidad y especificidad del ensayo. Además, la incidencia observada de positividad de anticuerpos (incluidos los anticuerpos neutralizantes) en un ensayo puede verse influenciada por varios factores, incluida la metodología del ensayo, el manejo de las muestras, el momento de la recolección de las muestras, los medicamentos concomitantes y la enfermedad subyacente. Por estas razones, la comparación de la incidencia de anticuerpos en los estudios descritos a continuación con la incidencia de anticuerpos en otros estudios o con otros productos de adalimumab puede ser engañosa.
Hay dos ensayos que se han utilizado para medir los anticuerpos anti-adalimumab. Con el ELISA, los anticuerpos contra adalimumab solo se pudieron detectar cuando las concentraciones séricas de adalimumab fueron < 2 mcg/mL. El ensayo ECL puede detectar títulos de anticuerpos anti-adalimumab independientemente de las concentraciones de adalimumab en las muestras de suero. La incidencia del desarrollo de anticuerpos anti-adalimumab (AAA) en pacientes tratados con HUMIRA se presenta en la Tabla 2.
| Indicaciones | Duración del estudio | Incidencia de anticuerpos anti-adalimumab por ELISA (n/N) | Incidencia de anticuerpos anti-adalimumab por ensayo ECL (n/N) | ||
| En todos los pacientes que recibieron adalimumab | En pacientes con concentraciones séricas de adalimumab < 2 mcg/mL | ||||
| Artritis reumatoidea |
6 a 12 meses | 5% (58/1062) | NR | NA | |
| Artritis Idiopática Juvenil (AIJ) | De 4 a 17 años de edadb | 48 semanas | 16% (27/171) | NR | NA |
| De 2 a 4 años de edad o ≥ 4 años de edad y con un peso < 15 kg | 24 semanas | 7% (1/15)c | NR | NA | |
| Artritis Psoriásicad | 48 semanase | 13% (24/178) | NR | NA | |
| Espondilitis Anquilosante | 24 semanas | 9% (16/185) | NR | NA | |
| Enfermedad de Crohn en Adultos | 56 semanas | 3% (7/269) | 8% (7/86) | NA | |
| Enfermedad de Crohn en Pediatría | 52 semanas | 3% (6/182) | 10% (6/58) | NA | |
| Colitis Ulcerosa en Adultos | 52 semanas | 5% (19/360) | 21% (19/92) | NA | |
| Colitis Ulcerosa en Pediatría | 52 semanas | 3% (3/100) | 13% (3/23) | 33% (33/100)i | |
| Psoriasis en Placasf | Hasta 52 semanasg | 8% (77/920) | 21% (77/372) | NA | |
| Hidradenitis Supurativa | 36 semanas | 7% (30/461) | 28% (58/207)h | 61% (272/445)j | |
| Uveítis No Infecciosa | 52 semanas | 5% (12/249) | 21% (12/57) | 40% (99/249)k | |
n: número de pacientes con anticuerpos anti-adalimumab; NR: no reportado; NA: No aplicable (no realizado)
a En pacientes que reciben metotrexato (MTX) concomitante, la incidencia de anticuerpos anti-adalimumab fue del 1% en comparación con el 12% con HUMIRA en monoterapia
b En pacientes que reciben MTX concomitante, la incidencia de anticuerpos anti-adalimumab fue del 6% en comparación con el 26% con HUMIRA en monoterapia
c Este paciente recibió MTX concomitante
d En pacientes que reciben MTX concomitante, la incidencia de desarrollo de anticuerpos fue del 7% en comparación con el 1% en AR
e Sujetos inscritos después de completar 2 estudios previos de 24 semanas o 12 semanas de tratamientos.
f En pacientes con psoriasis en placas que estaban en monoterapia con HUMIRA y posteriormente se retiraron del tratamiento, la tasa de anticuerpos contra adalimumab después del retratamiento fue similar a la tasa observada antes del retiro
g Un estudio de Fase 2 de 12 semanas y un estudio de Fase 3 de 52 semanas
h Entre los sujetos en los 2 estudios de Fase 3 que interrumpieron el tratamiento con HUMIRA por hasta 24 semanas y en quienes los niveles séricos de adalimumab posteriormente disminuyeron a <2 mcg/mL (aproximadamente el 22% del total de sujetos estudiados)
i No se observó ninguna asociación aparente entre el desarrollo de anticuerpos y la seguridad. La asociación del desarrollo de anticuerpos y el resultado de la eficacia no se evaluó debido al número limitado de sujetos en cada grupo de tratamiento estratificado por el título de anticuerpos anti-adalimumab.
j No se observó ninguna asociación aparente entre el desarrollo de anticuerpos y la seguridad
k No se observó ninguna correlación del desarrollo de anticuerpos con la seguridad o la eficacia de los resultados
Artritis reumatoide y artritis psoriásica: Los pacientes en los estudios RA-I, RA-II y RA-III se analizaron en múltiples puntos de tiempo para detectar anticuerpos contra adalimumab utilizando el ELISA durante el período de 6 a 12 meses. No se observó ninguna correlación aparente del desarrollo de anticuerpos con las reacciones adversas. Con la monoterapia, los pacientes que reciben dosis cada dos semanas pueden desarrollar anticuerpos con más frecuencia que aquellos que reciben dosis semanales. En pacientes que reciben la dosis recomendada de 40 mg cada dos semanas como monoterapia, la respuesta ACR 20 fue menor entre los pacientes con anticuerpos positivos que entre los pacientes con anticuerpos negativos. Se desconoce la inmunogenicidad a largo plazo de HUMIRA.
6.3 Experiencia postcomercialización
Las siguientes reacciones adversas se han identificado durante el uso posterior a la aprobación de HUMIRA. Debido a que estas reacciones se informan voluntariamente de una población de tamaño incierto, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal con la exposición a HUMIRA.
Trastornos gastrointestinales: Diverticulitis, perforaciones del intestino grueso, incluidas perforaciones asociadas con diverticulitis y perforaciones apendiculares asociadas con apendicitis, pancreatitis
Trastornos generales y condiciones del sitio de administración: Pirexia
Trastornos hepatobiliares: Insuficiencia hepática, hepatitis
Trastornos del sistema inmunitario: Sarcoidosis
Neoplasias benignas, malignas e indeterminadas (incluidos quistes y pólipos): Carcinoma de células de Merkel (carcinoma neuroendocrino de la piel)
Trastornos del sistema nervioso: Trastornos desmielinizantes (por ejemplo, neuritis óptica, síndrome de Guillain-Barré), accidente cerebrovascular
Trastornos respiratorios: Enfermedad pulmonar intersticial, incluida la fibrosis pulmonar, embolia pulmonar
Reacciones cutáneas: Síndrome de Stevens-Johnson, vasculitis cutánea, eritema multiforme, psoriasis nueva o que empeora (todos los subtipos, incluida la pustulosa y la palmoplantar), alopecia, reacción cutánea liquenoide
Trastornos vasculares: Vasculitis sistémica, trombosis venosa profunda
7 INTERACCIONES MEDICAMENTOSAS
7.1 Metotrexato
HUMIRA se ha estudiado en pacientes con artritis reumatoide (AR) que toman metotrexato (MTX) concomitante. Aunque el MTX redujo la depuración aparente de adalimumab, los datos no sugieren la necesidad de ajustar la dosis de HUMIRA o MTX [ver Farmacología clínica (12.3)].
7.2 Productos biológicos
En estudios clínicos en pacientes con AR, se ha observado un mayor riesgo de infecciones graves con la combinación de bloqueadores del TNF con anakinra o abatacept, sin beneficio adicional; por lo tanto, no se recomienda el uso de HUMIRA con abatacept o anakinra en pacientes con AR [ver Advertencias y precauciones (5.7, 5.11)]. También se ha observado una mayor tasa de infecciones graves en pacientes con AR tratados con rituximab que recibieron tratamiento posterior con un bloqueador del TNF. No hay información suficiente sobre el uso concomitante de HUMIRA y otros productos biológicos para el tratamiento de AR, PsA, AS, CD, UC, Ps, HS y UV. No se recomienda la administración concomitante de HUMIRA con otros DMARD biológicos (por ejemplo, anakinra y abatacept) u otros bloqueadores del TNF en base al posible aumento del riesgo de infecciones y otras posibles interacciones farmacológicas.
7.3 Vacunas vivas
Evite el uso de vacunas vivas con HUMIRA [ver Advertencias y precauciones (5.10)].
7.4 Sustratos del citocromo P450
La formación de enzimas CYP450 puede verse suprimida por el aumento de las concentraciones de citocinas (por ejemplo, TNFα, IL-6) durante la inflamación crónica. Es posible que una molécula que antagoniza la actividad de las citocinas, como el adalimumab, influya en la formación de enzimas CYP450. Al iniciar o suspender HUMIRA en pacientes que están siendo tratados con sustratos del CYP450 con un índice terapéutico estrecho, se recomienda controlar el efecto (por ejemplo, warfarina) o la concentración del fármaco (por ejemplo, ciclosporina o teofilina) y la dosis individual del producto farmacéutico puede ajustarse según sea necesario.
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de riesgos
Los estudios disponibles sobre el uso de adalimumab durante el embarazo no establecen de forma fiable una asociación entre adalimumab y los defectos congénitos importantes. Se dispone de datos clínicos del Registro de Embarazos de HUMIRA de la Organización de Especialistas en Información Teratológica (OTIS)/MotherToBaby en mujeres embarazadas con artritis reumatoide (AR) o enfermedad de Crohn (EC). Los resultados del registro mostraron una tasa del 10% de defectos congénitos importantes con el uso de adalimumab en el primer trimestre del embarazo en mujeres con AR o EC y una tasa del 7,5% de defectos congénitos importantes en la cohorte de comparación con la misma enfermedad. La falta de un patrón de defectos congénitos importantes es tranquilizadora y las diferencias entre los grupos de exposición pueden haber afectado a la aparición de defectos congénitos (véanse Datos).
El adalimumab se transfiere activamente a través de la placenta durante el tercer trimestre del embarazo y puede afectar a la respuesta inmunitaria en el lactante expuesto en el útero (véanse Consideraciones clínicas). En un estudio de desarrollo embrio-fetal perinatal realizado en monos cynomolgus, no se observaron daños fetales ni malformaciones con la administración intravenosa de adalimumab durante la organogénesis y posteriormente en la gestación, a dosis que produjeron exposiciones hasta aproximadamente 373 veces la dosis máxima recomendada en humanos (DMRH) de 40 mg subcutáneos sin metotrexato (véanse Datos).
Se desconoce el riesgo de fondo estimado de defectos congénitos importantes y abortos espontáneos para las poblaciones indicadas. Todos los embarazos tienen un riesgo de fondo de defectos congénitos, pérdida u otros resultados adversos. En la población general de EE. UU., el riesgo de fondo estimado de defectos congénitos importantes y abortos espontáneos en embarazos clínicamente reconocidos es del 2-4% y del 15-20%, respectivamente.
Consideraciones clínicas
Riesgo materno y embrionario/fetal asociado a la enfermedad
Los datos publicados sugieren que el riesgo de resultados adversos en el embarazo en mujeres con AR o enfermedad inflamatoria intestinal (EII) está asociado con una mayor actividad de la enfermedad. Los resultados adversos del embarazo incluyen el parto prematuro (antes de las 37 semanas de gestación), los lactantes con bajo peso al nacer (menos de 2500 g) y los pequeños para la edad gestacional al nacer.
Reacciones adversas fetales/neonatales
Los anticuerpos monoclonales se transportan cada vez más a través de la placenta a medida que avanza el embarazo, y la mayor cantidad se transfiere durante el tercer trimestre (véanse Datos). Deben considerarse los riesgos y beneficios antes de administrar vacunas vivas o vivas atenuadas a los lactantes expuestos a HUMIRA en el útero [véase Uso en poblaciones específicas (8.4)].
Datos
Datos humanos
Un registro prospectivo de cohorte de exposición al embarazo realizado por OTIS/MotherToBaby en EE. UU. y Canadá entre 2004 y 2016 comparó el riesgo de defectos congénitos importantes en recién nacidos vivos de 221 mujeres (69 con AR, 152 con EC) tratadas con adalimumab durante el primer trimestre y 106 mujeres (74 con AR, 32 con EC) no tratadas con adalimumab.
La proporción de defectos congénitos importantes entre los recién nacidos vivos en las cohortes tratadas y no tratadas con adalimumab fue del 10% (8,7% AR, 10,5% EC) y del 7,5% (6,8% AR, 9,4% EC), respectivamente. La falta de un patrón de defectos congénitos importantes es tranquilizadora y las diferencias entre los grupos de exposición pueden haber afectado a la aparición de defectos congénitos. Este estudio no puede establecer de forma fiable si existe una asociación entre adalimumab y los defectos congénitos importantes debido a las limitaciones metodológicas del registro, como el pequeño tamaño de la muestra, la naturaleza voluntaria del estudio y el diseño no aleatorizado.
En un estudio clínico independiente realizado en diez mujeres embarazadas con EII tratadas con HUMIRA, se midieron las concentraciones de adalimumab en el suero materno, así como en la sangre del cordón umbilical (n=10) y en el suero del lactante (n=8) el día del parto. La última dosis de HUMIRA se administró entre 1 y 56 días antes del parto. Las concentraciones de adalimumab fueron de 0,16-19,7 µg/mL en la sangre del cordón umbilical, 4,28-17,7 µg/mL en el suero del lactante y 0-16,1 µg/mL en el suero materno. En todos los casos menos uno, la concentración de adalimumab en la sangre del cordón umbilical fue mayor que la concentración en el suero materno, lo que sugiere que adalimumab atraviesa activamente la placenta. Además, un lactante presentó concentraciones séricas en cada uno de los siguientes momentos: 6 semanas (1,94 µg/mL), 7 semanas (1,31 µg/mL), 8 semanas (0,93 µg/mL) y 11 semanas (0,53 µg/mL), lo que sugiere que adalimumab puede detectarse en el suero de los lactantes expuestos en el útero durante al menos 3 meses desde el nacimiento.
Datos en animales
En un estudio de desarrollo embrio-fetal perinatal, se administró adalimumab a monas cynomolgus embarazadas desde los días 20 a 97 de gestación a dosis que produjeron exposiciones hasta 373 veces superiores a las alcanzadas con la DMRH sin metotrexato (sobre la base del AUC con dosis IV maternas de hasta 100 mg/kg/semana). El adalimumab no provocó daños en los fetos ni malformaciones.
8.2 Lactancia
Resumen de riesgos
Datos limitados de casos clínicos en la literatura publicada describen la presencia de adalimumab en la leche materna a dosis infantiles del 0,1% al 1% de la concentración sérica materna. Los datos publicados sugieren que se espera que la exposición sistémica de un lactante amamantado sea baja porque el adalimumab es una molécula grande y se degrada en el tracto gastrointestinal. Sin embargo, se desconocen los efectos de la exposición local en el tracto gastrointestinal. No hay informes de efectos adversos del adalimumab en el lactante amamantado ni efectos sobre la producción de leche. Deben considerarse los beneficios del desarrollo y la salud de la lactancia materna junto con la necesidad clínica de HUMIRA de la madre y cualquier posible efecto adverso en el niño amamantado por HUMIRA o por la condición materna subyacente.
8.4 Uso pediátrico
Se ha establecido la seguridad y eficacia de HUMIRA para:
- reducir los signos y síntomas de la AIJ poliarticular activa de moderada a grave en pacientes pediátricos de 2 años de edad y mayores.
- el tratamiento de la enfermedad de Crohn activa de moderada a grave en pacientes pediátricos de 6 años de edad y mayores.
- el tratamiento de la colitis ulcerosa activa de moderada a grave en pacientes pediátricos de 5 años de edad y mayores.
- el tratamiento de la hidradenitis supurativa de moderada a grave en pacientes de 12 años de edad y mayores.
- el tratamiento de la uveítis no infecciosa intermedia, posterior y panuveítis en pacientes pediátricos de 2 años de edad y mayores.
Debido a su inhibición del TNFα, HUMIRA administrado durante el embarazo podría afectar la respuesta inmune en el recién nacido y el lactante in utero. Los datos de ocho lactantes expuestos a HUMIRA in utero sugieren que el adalimumab atraviesa la placenta [ver Uso en poblaciones específicas (8.1)]. Se desconoce la importancia clínica de las concentraciones elevadas de adalimumab en lactantes. Se desconoce la seguridad de la administración de vacunas vivas o vivas atenuadas en lactantes expuestos. Los riesgos y beneficios deben considerarse antes de vacunar (vacunas vivas o vivas atenuadas) a los lactantes expuestos.
Se han notificado casos poscomercialización de linfoma, incluido el linfoma hepatoesplénico de células T y otras neoplasias malignas, algunas mortales, en niños, adolescentes y adultos jóvenes que recibieron tratamiento con bloqueadores del TNF, incluido HUMIRA [ver Advertencias y precauciones (5.2)].
Artritis idiopática juvenil
En el estudio JIA-I, se demostró que HUMIRA reduce los signos y síntomas de la AIJ poliarticular activa en pacientes de 4 a 17 años de edad [ver Estudios clínicos (14.2)]. En el estudio JIA-II, el perfil de seguridad para pacientes de 2 a <4 años de edad fue similar al perfil de seguridad para pacientes de 4 a 17 años de edad con AIJ poliarticular [ver Reacciones adversas (6.1)]. HUMIRA no se ha estudiado en pacientes con AIJ poliarticular menores de 2 años de edad ni en pacientes con un peso inferior a 10 kg.
La seguridad de HUMIRA en pacientes en los ensayos de AIJ poliarticular fue generalmente similar a la observada en adultos con ciertas excepciones [ver Reacciones adversas (6.1)].
No se ha establecido la seguridad y eficacia de HUMIRA en pacientes pediátricos con AIJ menores de 2 años de edad.
Enfermedad de Crohn pediátrica
Se ha establecido la seguridad y eficacia de HUMIRA para el tratamiento de la enfermedad de Crohn activa de moderada a grave en pacientes pediátricos de 6 años de edad y mayores. El uso de HUMIRA para esta indicación está respaldado por evidencia de estudios adecuados y bien controlados en adultos con datos adicionales de un estudio clínico aleatorizado, doble ciego, de 52 semanas de duración de dos concentraciones de dosis de HUMIRA en 192 pacientes pediátricos (de 6 a 17 años de edad) [ver Reacciones adversas (6.1), Farmacología clínica (12.2, 12.3), Estudios clínicos (14.6)]. El perfil de reacciones adversas en pacientes de 6 a 17 años de edad fue similar al de los adultos.
No se ha establecido la seguridad y eficacia de HUMIRA en pacientes pediátricos con enfermedad de Crohn menores de 6 años de edad.
Colitis ulcerosa pediátrica
Se ha establecido la seguridad y eficacia de HUMIRA para el tratamiento de la colitis ulcerosa activa de moderada a grave en pacientes pediátricos de 5 años de edad y mayores. El uso de HUMIRA para esta indicación está respaldado por evidencia de estudios adecuados y bien controlados en adultos con datos adicionales de un estudio clínico aleatorizado, doble ciego, de 52 semanas de duración de dos concentraciones de dosis de HUMIRA en 93 pacientes pediátricos (de 5 a 17 años de edad) [ver Reacciones adversas (6.1), Farmacología clínica (12.3), Estudios clínicos (14.8)]. El perfil de reacciones adversas en pacientes de 5 a 17 años de edad fue similar al de los adultos.
No se ha establecido la eficacia de HUMIRA en pacientes que han perdido la respuesta o que no toleraron los bloqueadores del TNF.
No se ha establecido la seguridad y eficacia de HUMIRA en pacientes pediátricos con colitis ulcerosa menores de 5 años de edad.
Uveítis pediátrica
Se ha establecido la seguridad y eficacia de HUMIRA para el tratamiento de la uveítis no infecciosa en pacientes pediátricos de 2 años de edad y mayores. El uso de HUMIRA está respaldado por evidencia de estudios adecuados y bien controlados de HUMIRA en adultos y un estudio clínico controlado, aleatorizado, 2:1 en 90 pacientes pediátricos [ver Estudios clínicos (14.12)]. No se ha establecido la seguridad y eficacia de HUMIRA en pacientes pediátricos con uveítis menores de 2 años de edad.
Hidradenitis supurativa
El uso de HUMIRA en pacientes pediátricos de 12 años de edad y mayores para la HS está respaldado por evidencia de estudios adecuados y bien controlados de HUMIRA en pacientes adultos con HS. El modelado y la simulación farmacocinéticos poblacionales adicionales predijeron que la dosificación de HUMIRA basada en el peso en pacientes pediátricos de 12 años de edad y mayores puede proporcionar una exposición generalmente similar a la de los pacientes adultos con HS. El curso de la HS es lo suficientemente similar en pacientes adultos y adolescentes como para permitir la extrapolación de datos de pacientes adultos a pacientes adolescentes. La dosis recomendada en pacientes pediátricos de 12 años de edad o mayores se basa en el peso corporal [ver Dosis y administración (2.6), Farmacología clínica (12.3) y Estudios clínicos (14.10)].
No se ha establecido la seguridad y eficacia de HUMIRA en pacientes menores de 12 años con HS.
8.5 Uso geriátrico
Un total de 519 pacientes con AR de 65 años de edad y mayores, incluidos 107 pacientes de 75 años de edad y mayores, recibieron HUMIRA en los estudios clínicos de AR-I a IV. No se observaron diferencias generales en la eficacia entre estos pacientes y los pacientes más jóvenes. La frecuencia de infecciones graves y neoplasias malignas entre los pacientes tratados con HUMIRA de 65 años de edad y mayores fue mayor que para los menores de 65 años. Considere los beneficios y riesgos de HUMIRA en pacientes de 65 años de edad y mayores. En pacientes tratados con HUMIRA, controle de cerca el desarrollo de infección o neoplasia maligna [ver Advertencias y precauciones (5.1, 5.2)].
10 SOBREDOSIS
Se han administrado dosis de hasta 10 mg/kg a pacientes en ensayos clínicos sin evidencia de toxicidades limitantes de la dosis. En caso de sobredosis, se recomienda monitorizar al paciente para detectar cualquier signo o síntoma de reacciones o efectos adversos e instaurar inmediatamente el tratamiento sintomático adecuado.
11 DESCRIPCIÓN
Adalimumab es un bloqueador del factor de necrosis tumoral. Adalimumab es un anticuerpo monoclonal recombinante humano IgG1 creado utilizando tecnología de visualización de fagos, lo que da como resultado un anticuerpo con regiones variables de cadena pesada y ligera derivadas de humanos y regiones constantes de IgG1:k humanas. Adalimumab se produce mediante tecnología de ADN recombinante en un sistema de expresión de células de mamíferos (ovario de hámster chino (CHO)) y se purifica mediante un proceso que incluye pasos específicos de inactivación y eliminación viral. Consta de 1330 aminoácidos y tiene un peso molecular de aproximadamente 148 kilodaltons.
HUMIRA (adalimumab) inyección se suministra como una solución estéril, libre de conservantes para administración subcutánea. El producto farmacéutico se suministra como un bolígrafo prellenado de dosis única (HUMIRA Pen), como una jeringa de vidrio prellenada de 1 mL de dosis única o como un vial de dosis única para uso institucional. Dentro del bolígrafo se encuentra una jeringa de vidrio prellenada de 1 mL de dosis única. La solución de HUMIRA es transparente e incolora, con un pH de aproximadamente 5.2.
Cada jeringa prellenada o bolígrafo prellenado de 80 mg/0.8 mL administra 0.8 mL (80 mg) de producto farmacéutico. Cada 0.8 mL de HUMIRA contiene adalimumab (80 mg), manitol (33.6 mg), polisorbato 80 (0.8 mg) y agua para inyección, USP.
Cada jeringa prellenada o bolígrafo prellenado de 40 mg/0.4 mL administra 0.4 mL (40 mg) de producto farmacéutico. Cada 0.4 mL de HUMIRA contiene adalimumab (40 mg), manitol (16.8 mg), polisorbato 80 (0.4 mg) y agua para inyección, USP.
Cada jeringa prellenada, bolígrafo prellenado o vial de dosis única para uso institucional de 40 mg/0.8 mL administra 0.8 mL (40 mg) de producto farmacéutico. Cada 0.8 mL de HUMIRA contiene adalimumab (40 mg), ácido cítrico monohidratado (1.04 mg), fosfato disódico dihidratado (1.22 mg), manitol (9.6 mg), fosfato monosódico dihidratado (0.69 mg), polisorbato 80 (0.8 mg), cloruro de sodio (4.93 mg), citrato de sodio (0.24 mg) y agua para inyección, USP. Se agrega hidróxido de sodio según sea necesario para ajustar el pH.
Cada jeringa prellenada de 20 mg/0.2 mL administra 0.2 mL (20 mg) de producto farmacéutico. Cada 0.2 mL de HUMIRA contiene adalimumab (20 mg), manitol (8.4 mg), polisorbato 80 (0.2 mg) y agua para inyección, USP.
Cada jeringa prellenada de 20 mg/0.4 mL administra 0.4 mL (20 mg) de producto farmacéutico. Cada 0.4 mL de HUMIRA contiene adalimumab (20 mg), ácido cítrico monohidratado (0.52 mg), fosfato disódico dihidratado (0.61 mg), manitol (4.8 mg), fosfato monosódico dihidratado (0.34 mg), polisorbato 80 (0.4 mg), cloruro de sodio (2.47 mg), citrato de sodio (0.12 mg) y agua para inyección, USP. Se agrega hidróxido de sodio según sea necesario para ajustar el pH.
Cada jeringa prellenada de 10 mg/0.1 mL administra 0.1 mL (10 mg) de producto farmacéutico. Cada 0.1 mL de HUMIRA contiene adalimumab (10 mg), manitol (4.2 mg), polisorbato 80 (0.1 mg) y agua para inyección, USP.
Cada jeringa prellenada de 10 mg/0.2 mL administra 0.2 mL (10 mg) de producto farmacéutico. Cada 0.2 mL de HUMIRA contiene adalimumab (10 mg), ácido cítrico monohidratado (0.26 mg), fosfato disódico dihidratado (0.31 mg), manitol (2.4 mg), fosfato monosódico dihidratado (0.17 mg), polisorbato 80 (0.2 mg), cloruro de sodio (1.23 mg), citrato de sodio (0.06 mg) y agua para inyección, USP. Se agrega hidróxido de sodio según sea necesario para ajustar el pH.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
Adalimumab se une específicamente al TNF-alfa y bloquea su interacción con los receptores de TNF de superficie celular p55 y p75. Adalimumab también lisa las células que expresan TNF de superficie in vitro en presencia de complemento. Adalimumab no se une ni inactiva la linfotoxina (TNF-beta). El TNF es una citocina natural que participa en las respuestas inflamatorias e inmunitarias normales. Se encuentran concentraciones elevadas de TNF en el líquido sinovial de pacientes con AR, AAI, APs y AE, y desempeñan un papel importante tanto en la inflamación patológica como en la destrucción articular que son características de estas enfermedades. También se encuentran concentraciones aumentadas de TNF en las placas de psoriasis. En Ps, el tratamiento con HUMIRA puede reducir el grosor epidérmico y la infiltración de células inflamatorias. Se desconoce la relación entre estas actividades farmacodinámicas y el mecanismo o los mecanismos por los que HUMIRA ejerce sus efectos clínicos.
Adalimumab también modula las respuestas biológicas que son inducidas o reguladas por el TNF, incluidos los cambios en las concentraciones de moléculas de adhesión responsables de la migración de leucocitos (ELAM-1, VCAM-1 e ICAM-1 con una IC50 de 1-2 X 10-10M).
12.2 Farmacodinamia
Después del tratamiento con HUMIRA, se observó una disminución en las concentraciones de reactantes de fase aguda de la inflamación (proteína C reactiva [PCR] y velocidad de sedimentación globular [VSG]) y citocinas séricas (IL-6) en comparación con el valor inicial en pacientes con artritis reumatoide. También se observó una disminución en las concentraciones de PCR en pacientes con enfermedad de Crohn, colitis ulcerosa e hidradenitis supurativa. Las concentraciones séricas de metaloproteinasas de matriz (MMP-1 y MMP-3) que producen la remodelación tisular responsable de la destrucción del cartílago también disminuyeron después de la administración de HUMIRA.
Para pacientes pediátricos de 5 a 17 años con colitis ulcerosa, la dosis recomendada de HUMIRA se basa en las relaciones dosis/exposición-eficacia modeladas y los datos farmacocinéticos. No se prevén diferencias clínicamente relevantes en la eficacia entre la dosis más alta estudiada administrada en el ensayo clínico (semanas 0 a 52 en el estudio PUC-I) [ver Estudios clínicos (14.8)] y la dosis recomendada [ver Dosificación y administración (2.4)].
12.3 Farmacocinética
La farmacocinética de adalimumab fue lineal en el rango de dosis de 0,5 a 10 mg/kg después de la administración de una dosis intravenosa única (HUMIRA no está aprobado para uso intravenoso). Después de la administración subcutánea cada dos semanas y cada semana de 20, 40 y 80 mg, las concentraciones séricas mínimas medias de adalimumab en estado estacionario aumentaron aproximadamente proporcionalmente a la dosis en pacientes con AR. La semivida terminal media fue de aproximadamente 2 semanas, con un rango de 10 a 20 días en los diferentes estudios. Los sujetos sanos y los pacientes con AR mostraron una farmacocinética similar de adalimumab.
Se estima que la exposición a adalimumab en pacientes tratados con 80 mg cada dos semanas es comparable a la de los pacientes tratados con 40 mg cada semana.
Absorción
La biodisponibilidad absoluta media de adalimumab después de una dosis subcutánea única de 40 mg fue del 64%. El tiempo medio para alcanzar la concentración máxima fue de 5,5 días (131 ± 56 horas) y la concentración sérica máxima fue de 4,7 ± 1,6 mcg/mL en sujetos sanos después de una administración subcutánea única de 40 mg de HUMIRA.
Distribución
El volumen de distribución (Vss) osciló entre 4,7 y 6,0 L después de la administración intravenosa de dosis que oscilaron entre 0,25 y 10 mg/kg en pacientes con AR.
Eliminación
La farmacocinética de dosis única de adalimumab en pacientes con AR se determinó en varios estudios con dosis intravenosas que oscilaron entre 0,25 y 10 mg/kg. El aclaramiento sistémico de adalimumab es de aproximadamente 12 mL/hora. En estudios a largo plazo con dosificación de más de dos años, no hubo evidencia de cambios en el aclaramiento con el tiempo en pacientes con AR.
Población de pacientes
Artritis reumatoide y espondilitis anquilosante: En pacientes que recibieron 40 mg de HUMIRA cada dos semanas, las concentraciones mínimas medias de adalimumab en estado estacionario fueron de aproximadamente 5 mcg/mL y de 8 a 9 mcg/mL, sin y con tratamiento concomitante con MTX, respectivamente. Las concentraciones de adalimumab en el líquido sinovial de cinco pacientes con artritis reumatoide oscilaron entre el 31 y el 96% de las del suero. La farmacocinética de adalimumab en pacientes con AE fue similar a la de los pacientes con AR.
Artritis psoriásica: En pacientes que recibieron 40 mg cada dos semanas, las concentraciones mínimas medias de adalimumab en estado estacionario fueron de 6 a 10 mcg/mL y de 8,5 a 12 mcg/mL, sin y con tratamiento concomitante con MTX, respectivamente.
Psoriasis en placas: La concentración mínima media de adalimumab en estado estacionario fue de aproximadamente 5 a 6 mcg/mL durante el tratamiento con HUMIRA de 40 mg cada dos semanas.
Uveítis en adultos: La concentración media de adalimumab en estado estacionario fue de aproximadamente 8 a 10 mcg/mL durante el tratamiento con HUMIRA de 40 mg cada dos semanas.
Hidradenitis supurativa en adultos: Las concentraciones mínimas de adalimumab fueron de aproximadamente 7 a 8 mcg/mL en la semana 2 y la semana 4, respectivamente, después de recibir 160 mg en la semana 0 seguidos de 80 mg en la semana 2. Las concentraciones mínimas medias en estado estacionario en la semana 12 hasta la semana 36 fueron de aproximadamente 7 a 11 mcg/mL durante el tratamiento con HUMIRA de 40 mg cada semana.
Adultos con enfermedad de Crohn: Las concentraciones mínimas medias de adalimumab fueron de aproximadamente 12 mcg/mL en la semana 2 y la semana 4 después de recibir 160 mg en la semana 0 seguidos de 80 mg en la semana 2. Las concentraciones mínimas medias en estado estacionario fueron de 7 mcg/mL en la semana 24 y la semana 56 durante el tratamiento con HUMIRA de 40 mg cada dos semanas.
Colitis ulcerosa en adultos: Las concentraciones mínimas medias de adalimumab fueron de aproximadamente 12 mcg/mL en la semana 2 y la semana 4 después de recibir 160 mg en la semana 0 seguidos de 80 mg en la semana 2. Las concentraciones mínimas medias en estado estacionario fueron de aproximadamente 8 mcg/mL y 15 mcg/mL en la semana 52 después de recibir una dosis de HUMIRA de 40 mg cada dos semanas y 40 mg cada semana, respectivamente.
Efectos de los anticuerpos contra el fármaco en la farmacocinética
Artritis Reumatoide: Se identificó una tendencia hacia un mayor aclaramiento aparente de adalimumab en presencia de anticuerpos anti-adalimumab.
Colitis Ulcerosa Pediátrica: Los anticuerpos contra adalimumab mediante ensayo ECL se asociaron con concentraciones reducidas de adalimumab en suero en pacientes pediátricos con colitis ulcerosa de moderada a grave actividad.
Hidradenitis Supurativa: En sujetos con HS de moderada a grave, los anticuerpos contra adalimumab se asociaron con concentraciones reducidas de adalimumab en suero. En general, la magnitud de la reducción en las concentraciones de adalimumab en suero es mayor con títulos crecientes de anticuerpos contra adalimumab.
Poblaciones Específicas
Pacientes de edad avanzada: Se observó un aclaramiento más bajo con el aumento de la edad en pacientes con AR de 40 a >75 años.
Pacientes pediátricos:
Artritis Idiopática Juvenil:
- 4 años a 17 años de edad: Las concentraciones mínimas en estado estacionario de adalimumab fueron de 6,8 mcg/mL y 10,9 mcg/mL en pacientes con un peso <30 kg que recibieron 20 mg de HUMIRA por vía subcutánea cada dos semanas como monoterapia o con MTX concomitante, respectivamente. Las concentraciones mínimas en estado estacionario de adalimumab fueron de 6,6 mcg/mL y 8,1 mcg/mL en pacientes con un peso ≥30 kg que recibieron 40 mg de HUMIRA por vía subcutánea cada dos semanas como monoterapia o con tratamiento concomitante con MTX, respectivamente.
- 2 años a <4 años de edad o 4 años de edad y mayores con un peso <15 kg: Las concentraciones mínimas en estado estacionario de adalimumab fueron de 6,0 mcg/mL y 7,9 mcg/mL en pacientes que recibieron HUMIRA por vía subcutánea cada dos semanas como monoterapia o con tratamiento concomitante con MTX, respectivamente.
Hidradenitis Supurativa Pediátrica: Se predice que las concentraciones de adalimumab en pacientes adolescentes con HS que reciben los regímenes de dosificación recomendados serán similares a las observadas en sujetos adultos con HS según el modelado farmacocinético poblacional y la simulación.
Enfermedad de Crohn Pediátrica: Las concentraciones medias ± DE de adalimumab fueron de 15,7±6,5 mcg/mL en la semana 4 después de 160 mg en la semana 0 y 80 mg en la semana 2, y de 10,5±6,0 mcg/mL en la semana 52 después de la dosificación de 40 mg cada dos semanas en pacientes con un peso ≥ 40 kg. Las concentraciones medias ± DE de adalimumab fueron de 10,6±6,1 mcg/mL en la semana 4 después de la dosificación de 80 mg en la semana 0 y 40 mg en la semana 2, y de 6,9±3,6 mcg/mL en la semana 52 después de la dosificación de 20 mg cada dos semanas en pacientes con un peso < 40 kg.
Colitis Ulcerosa Pediátrica: La concentración mínima en estado estacionario de adalimumab fue de 5,0±3,3 mcg/mL en la semana 52 después de la administración subcutánea de 0,6 mg/kg (máximo de 40 mg) cada dos semanas en pacientes pediátricos con UC de 5 a 17 años de edad. En los pacientes que recibieron 0,6 mg/kg (máximo de 40 mg) cada semana, la concentración mínima en estado estacionario media fue de 15,7±5,6 mcg/mL en la semana 52 en pacientes pediátricos con UC de 5 a 17 años de edad.
Pacientes masculinos y femeninos: No se observaron diferencias farmacocinéticas relacionadas con el sexo después de la corrección por el peso corporal del paciente. Los sujetos sanos y los pacientes con artritis reumatoide mostraron farmacocinéticas similares de adalimumab.
Pacientes con insuficiencia renal o hepática: No hay datos farmacocinéticos disponibles en pacientes con insuficiencia hepática o renal.
Concentraciones de factor reumatoide o PCR: Se predijeron aumentos menores en el aclaramiento aparente en pacientes con AR que recibieron dosis inferiores a la dosis recomendada y en pacientes con AR con concentraciones altas de factor reumatoide o PCR. Es probable que estos aumentos no sean clínicamente importantes.
Estudios de interacción medicamentosa:
Metotrexato: El MTX redujo el aclaramiento aparente de adalimumab después de la administración única y múltiple en un 29% y un 44%, respectivamente, en pacientes con AR [ver Interacciones medicamentosas (7.1)].
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
No se han realizado estudios a largo plazo en animales con HUMIRA para evaluar el potencial carcinogénico o su efecto sobre la fertilidad.
14 ESTUDIOS CLÍNICOS
14.1 Artritis Reumatoide
La eficacia y seguridad de HUMIRA se evaluaron en cinco estudios aleatorizados, doble ciego en pacientes ≥18 años de edad con artritis reumatoide (AR) activa diagnosticada según los criterios del Colegio Estadounidense de Reumatología (ACR). Los pacientes tenían al menos 6 articulaciones inflamadas y 9 articulaciones dolorosas. HUMIRA se administró por vía subcutánea en combinación con metotrexato (MTX) (12,5 a 25 mg, estudios RA-I, RA-III y RA-V) o como monoterapia (estudios RA-II y RA-V) o con otros fármacos antirreumáticos modificadores de la enfermedad (DMARD) (estudio RA-IV).
El estudio RA-I evaluó a 271 pacientes que habían fracasado en la terapia con al menos un DMARD pero no más de cuatro y que no habían respondido adecuadamente al MTX. Se administraron dosis de 20, 40 u 80 mg de HUMIRA o placebo cada dos semanas durante 24 semanas.
El estudio RA-II evaluó a 544 pacientes que habían fracasado en la terapia con al menos un DMARD. Se administraron dosis de placebo, 20 o 40 mg de HUMIRA como monoterapia cada dos semanas o semanalmente durante 26 semanas.
El estudio RA-III evaluó a 619 pacientes que no habían respondido adecuadamente al MTX. Los pacientes recibieron placebo, 40 mg de HUMIRA cada dos semanas con inyecciones de placebo en semanas alternas, o 20 mg de HUMIRA semanalmente durante un máximo de 52 semanas. El estudio RA-III tuvo un punto final primario adicional a las 52 semanas de inhibición de la progresión de la enfermedad (detectada por los resultados de rayos X). Tras la finalización de las primeras 52 semanas, 457 pacientes se inscribieron en una fase de extensión de etiqueta abierta en la que se administraron 40 mg de HUMIRA cada dos semanas durante un máximo de 5 años.
El estudio RA-IV evaluó la seguridad en 636 pacientes que eran DMARD-naive o que se les permitió permanecer en su terapia reumatológica preexistente siempre que la terapia fuera estable durante un mínimo de 28 días. Los pacientes fueron aleatorizados a 40 mg de HUMIRA o placebo cada dos semanas durante 24 semanas.
El estudio RA-V evaluó a 799 pacientes con AR de moderada a gravemente activa de menos de 3 años de duración que tenían ≥18 años de edad y eran naive a MTX. Los pacientes fueron aleatorizados para recibir MTX (optimizado a 20 mg/semana para la semana 8), HUMIRA 40 mg cada dos semanas o terapia combinada HUMIRA/MTX durante 104 semanas. Los pacientes fueron evaluados para detectar signos y síntomas, y para la progresión radiográfica del daño articular. La duración media de la enfermedad entre los pacientes inscritos en el estudio fue de 5 meses. La dosis media de MTX alcanzada fue de 20 mg.
Respuesta clínica
El porcentaje de pacientes tratados con HUMIRA que lograron respuestas ACR 20, 50 y 70 en los estudios RA-II y III se muestra en la Tabla 3.
| Estudio RA-II Monoterapia (26 semanas) |
Estudio RA-III Combinación de metotrexato (24 y 52 semanas) |
||||
| Respuesta | Placebo | HUMIRA | HUMIRA | Placebo/MTX | HUMIRA/MTX |
| 40 mg cada | 40 mg semanalmente | 40 mg cada | |||
| dos semanas | dos semanas | ||||
| N=110 | N=113 | N=103 | N=200 | N=207 | |
| ACR20 | |||||
| Mes 6 | 19% | 46%* | 53%* | 30% | 63%* |
| Mes 12 | NA | NA | NA | 24% | 59%* |
| ACR50 | |||||
| Mes 6 | 8% | 22%* | 35%* | 10% | 39%* |
| Mes 12 | NA | NA | NA | 10% | 42%* |
| ACR70 | |||||
| Mes 6 | 2% | 12%* | 18%* | 3% | 21%* |
| Mes 12 | NA | NA | NA | 5% | 23%* |
| * p<0.01, HUMIRA vs. placebo | |||||
Los resultados del Estudio RA-I fueron similares al Estudio RA-III; los pacientes que recibieron HUMIRA 40 mg cada dos semanas en el Estudio RA-I también lograron tasas de respuesta ACR 20, 50 y 70 del 65%, 52% y 24%, respectivamente, en comparación con las respuestas placebo del 13%, 7% y 3%, respectivamente, a los 6 meses (p<0,01).
Los resultados de los componentes de los criterios de respuesta ACR para los Estudios RA-II y RA-III se muestran en la Tabla 4. Las tasas de respuesta ACR y la mejora en todos los componentes de la respuesta ACR se mantuvieron hasta la semana 104. Durante los 2 años del Estudio RA-III, el 20% de los pacientes con HUMIRA que recibieron 40 mg cada dos semanas lograron una respuesta clínica importante, definida como el mantenimiento de una respuesta ACR 70 durante un período de 6 meses. Las respuestas ACR se mantuvieron en proporciones similares de pacientes durante hasta 5 años con tratamiento continuo con HUMIRA en la parte de etiqueta abierta del Estudio RA-III.
| Estudio RA-II | Estudio RA-III | |||||||
| Parámetro (mediana) | Placebo N=110 |
HUMIRAa N=113 |
Placebo/MTX N=200 |
HUMIRAa/MTX N=207 |
||||
| Línea de base | Sem 26 | Línea de base | Sem 26 | Línea de base | Sem 24 | Línea de base | Sem 24 | |
| Número de articulaciones sensibles (0-68) | 35 | 26 | 31 | 16* | 26 | 15 | 24 | 8* |
| Número de articulaciones inflamadas (0-66) | 19 | 16 | 18 | 10* | 17 | 11 | 18 | 5* |
| Evaluación global del médicob | 7.0 | 6.1 | 6.6 | 3.7* | 6.3 | 3.5 | 6.5 | 2.0* |
| Evaluación global del pacienteb | 7.5 | 6.3 | 7.5 | 4.5* | 5.4 | 3.9 | 5.2 | 2.0* |
| Dolorb | 7.3 | 6.1 | 7.3 | 4.1* | 6.0 | 3.8 | 5.8 | 2.1* |
| Índice de discapacidad (HAQ)c | 2.0 | 1.9 | 1.9 | 1.5* | 1.5 | 1.3 | 1.5 | 0.8* |
| PCR (mg/dL) | 3.9 | 4.3 | 4.6 | 1.8* | 1.0 | 0.9 | 1.0 | 0.4* |
| a 40 mg HUMIRA administrados cada dos semanas b Escala analógica visual; 0 = mejor, 10 = peor c Índice de discapacidad del Cuestionario de Evaluación de la Salud; 0 = mejor, 3 = peor, mide la capacidad del paciente para realizar lo siguiente: vestirse/arreglarse, levantarse, comer, caminar, alcanzar, agarrar, mantener la higiene y mantener la actividad diaria * p<0,001, HUMIRA vs. placebo, basado en el cambio medio desde la línea de base |
||||||||
La evolución temporal de la respuesta ACR 20 para el Estudio RA-III se muestra en la Figura 1.
En el Estudio RA-III, el 85% de los pacientes con respuestas ACR 20 en la semana 24 mantuvieron la respuesta en la semana 52. La evolución temporal de la respuesta ACR 20 para el Estudio RA-I y el Estudio RA-II fue similar.
Figura 1. Respuestas ACR 20 del Estudio RA-III durante 52 semanas

En el Estudio RA-IV, el 53% de los pacientes tratados con HUMIRA 40 mg cada dos semanas más el tratamiento estándar tuvieron una respuesta ACR 20 en la semana 24 en comparación con el 35% en el grupo placebo más el tratamiento estándar (p<0,001). No se observaron reacciones adversas únicas relacionadas con la combinación de HUMIRA (adalimumab) y otros DMARD.
En el Estudio RA-V con pacientes con AR de inicio reciente y sin tratamiento previo con MTX, el tratamiento combinado con HUMIRA más MTX condujo a un mayor porcentaje de pacientes que alcanzaron respuestas ACR que la monoterapia con MTX o la monoterapia con HUMIRA en la semana 52, y las respuestas se mantuvieron en la semana 104 (ver Tabla 5).
| Respuesta | MTXb N=257 |
HUMIRAc N=274 |
HUMIRA/MTX N=268 |
| ACR20 Semana 52 Semana 104 |
63% 56% |
54% 49% |
73% 69% |
| ACR50 Semana 52 Semana 104 |
46% 43% |
41% 37% |
62% 59% |
| ACR70 Semana 52 Semana 104 |
27% 28% |
26% 28% |
46% 47% |
| Respuesta clínica mayor a | 28% | 25% | 49% |
| a La respuesta clínica mayor se define como alcanzar una respuesta ACR70 durante un período continuo de seis meses b p<0,05, HUMIRA/MTX vs. MTX para ACR 20 p<0,001, HUMIRA/MTX vs. MTX para ACR 50 y 70, y respuesta clínica mayor c p<0,001, HUMIRA/MTX vs. HUMIRA |
|||
En la semana 52, todos los componentes individuales de los criterios de respuesta ACR para el Estudio RA-V mejoraron en el grupo HUMIRA/MTX y las mejoras se mantuvieron hasta la semana 104.
Respuesta radiográfica
En el Estudio RA-III, el daño articular estructural se evaluó radiográficamente y se expresó como cambio en la puntuación total de Sharp (TSS) y sus componentes, la puntuación de erosión y la puntuación de estrechamiento del espacio articular (JSN), en el mes 12 en comparación con el inicio. En el inicio, la TSS mediana fue de aproximadamente 55 en los grupos placebo y 40 mg cada dos semanas. Los resultados se muestran en la Tabla 6. Los pacientes tratados con HUMIRA/MTX demostraron una menor progresión radiográfica que los pacientes que recibieron solo MTX en la semana 52.
| Placebo/MTX | HUMIRA/MTX 40 mg cada dos semanas |
Placebo/MTX- HUMIRA/MTX (95% Intervalo de confianza *) |
Valor p** | |
| Puntuación total de Sharp | 2,7 | 0,1 | 2,6 (1,4, 3,8) | <0,001 |
| Puntuación de erosión | 1,6 | 0,0 | 1,6 (0,9, 2,2) | <0,001 |
| Puntuación de JSN | 1,0 | 0,1 | 0,9 (0,3, 1,4) | 0,002 |
| *Intervalos de confianza del 95% para las diferencias en las puntuaciones de cambio entre MTX y HUMIRA. **Basado en el análisis de rango |
||||
En la extensión de etiqueta abierta del Estudio RA-III, el 77% de los pacientes originales tratados con cualquier dosis de HUMIRA fueron evaluados radiográficamente a los 2 años. Los pacientes mantuvieron la inhibición del daño estructural, según lo medido por el TSS. El cincuenta y cuatro por ciento no tuvo progresión del daño estructural, según se define por un cambio en el TSS de cero o menos. El cincuenta y cinco por ciento (55%) de los pacientes tratados originalmente con 40 mg de HUMIRA cada dos semanas han sido evaluados radiográficamente a los 5 años. Los pacientes continuaron con la inhibición del daño estructural, con un 50% que no mostró progresión del daño estructural, definido por un cambio en el TSS de cero o menos.
En el Estudio RA-V, el daño articular estructural se evaluó como en el Estudio RA-III. Se observó una mayor inhibición de la progresión radiográfica, según lo evaluado por los cambios en el TSS, la puntuación de erosión y el JSN, en el grupo de combinación HUMIRA/MTX en comparación con el grupo de monoterapia con MTX o HUMIRA en la semana 52, así como en la semana 104 (ver Tabla 7).
| MTXa N=257 |
HUMIRAa,b N=274 |
HUMIRA/MTX N=268 |
||
| 52 Semanas | Puntuación total de Sharp | 5.7 (4.2, 7.3) | 3.0 (1.7, 4.3) | 1.3 (0.5, 2.1) |
| Puntuación de erosión | 3.7 (2.7, 4.8) | 1.7 (1.0, 2.4) | 0.8 (0.4, 1.2) | |
| Puntuación de JSN | 2.0 (1.2, 2.8) | 1.3 (0.5, 2.1) | 0.5 (0.0, 1.0) | |
| 104 Semanas | Puntuación total de Sharp | 10.4 (7.7, 13.2) | 5.5 (3.6, 7.4) | 1.9 (0.9, 2.9) |
| Puntuación de erosión | 6.4 (4.6, 8.2) | 3.0 (2.0, 4.0) | 1.0 (0.4, 1.6) | |
| Puntuación de JSN | 4.1 (2.7, 5.4) | 2.6 (1.5, 3.7) | 0.9 (0.3, 1.5) | |
| * media (intervalo de confianza del 95%) a p<0.001, HUMIRA/MTX vs. MTX en las semanas 52 y 104 y para HUMIRA/MTX vs. HUMIRA en la semana 104 b p<0.01, para HUMIRA/MTX vs. HUMIRA en la semana 52 |
||||
Respuesta de la función física
En los estudios RA-I a IV, HUMIRA mostró una mejora significativamente mayor que el placebo en el índice de discapacidad del Cuestionario de Evaluación de la Salud (HAQ-DI) desde el inicio hasta el final del estudio, y una mejora significativamente mayor que el placebo en los resultados de salud evaluados por la Encuesta de Salud de Forma Corta (SF 36). Se observó una mejora tanto en el Resumen del Componente Físico (PCS) como en el Resumen del Componente Mental (MCS).
En el estudio RA-III, la mejora media (IC del 95%) en el HAQ-DI desde el inicio en la semana 52 fue de 0,60 (0,55, 0,65) para los pacientes con HUMIRA y de 0,25 (0,17, 0,33) para los pacientes con placebo/MTX (p<0,001). El sesenta y tres por ciento de los pacientes tratados con HUMIRA lograron una mejora de 0,5 o mayor en el HAQ-DI en la semana 52 en la parte de doble ciego del estudio. El ochenta y dos por ciento de estos pacientes mantuvieron esa mejora hasta la semana 104 y una proporción similar de pacientes mantuvo esta respuesta hasta la semana 260 (5 años) de tratamiento abierto. La mejora media en el SF-36 se mantuvo hasta el final de la medición en la semana 156 (3 años).
En el estudio RA-V, el HAQ-DI y el componente físico del SF-36 mostraron una mayor mejora (p<0,001) para el grupo de terapia combinada HUMIRA/MTX en comparación con el grupo de monoterapia con MTX o el grupo de monoterapia con HUMIRA en la semana 52, que se mantuvo hasta la semana 104.
14.2 Artritis Idiopática Juvenil
La seguridad y eficacia de HUMIRA se evaluaron en dos estudios (Estudios JIA-I y JIA-II) en pacientes con artritis idiopática juvenil (AIJ) poliarticular activa.
Estudio JIA-I
La seguridad y eficacia de HUMIRA se evaluaron en un estudio multicéntrico, aleatorizado, de retirada, doble ciego, de grupos paralelos en 171 pacientes de 4 a 17 años de edad con AIJ poliarticular. En el estudio, los pacientes se estratificaron en dos grupos: tratados con MTX o no tratados con MTX. Todos los pacientes debían mostrar signos de enfermedad activa moderada o grave a pesar del tratamiento previo con AINE, analgésicos, corticosteroides o DMARD. Los pacientes que habían recibido tratamiento previo con cualquier DMARD biológico fueron excluidos del estudio.
El estudio incluyó cuatro fases: una fase de introducción abierta (OL-LI; 16 semanas), una fase de retirada aleatorizada doble ciego (DB; 32 semanas), una fase de extensión abierta (OLE-BSA; hasta 136 semanas) y una fase de dosis fija abierta (OLE-FD; 16 semanas). En las tres primeras fases del estudio, HUMIRA se administró en función de la superficie corporal a una dosis de 24 mg/m2 hasta una dosis corporal total máxima de 40 mg por vía subcutánea (SC) cada dos semanas. En la fase OLE-FD, los pacientes fueron tratados con 20 mg de HUMIRA SC cada dos semanas si su peso era inferior a 30 kg y con 40 mg de HUMIRA SC cada dos semanas si su peso era de 30 kg o superior. Los pacientes permanecieron con dosis estables de AINE o prednisona (≤0,2 mg/kg/día o 10 mg/día como máximo).
Los pacientes que demostraron una respuesta ACR Pediátrica 30 al final de la fase OL-LI fueron aleatorizados a la fase doble ciego (DB) del estudio y recibieron HUMIRA o placebo cada dos semanas durante 32 semanas o hasta que se produjo un brote de la enfermedad. El brote de la enfermedad se definió como un empeoramiento ≥30% desde el inicio en ≥3 de los 6 criterios principales del ACR Pediátrico, ≥2 articulaciones activas y una mejora de >30% en no más de 1 de los 6 criterios. Después de 32 semanas o en el momento del brote de la enfermedad durante la fase DB, los pacientes fueron tratados en la fase de extensión abierta en función del régimen de BSA (OLE-BSA), antes de pasar a un régimen de dosis fija en función del peso corporal (fase OLE-FD).
Respuesta clínica del estudio JIA-I
Al final de la fase OL-LI de 16 semanas, el 94% de los pacientes del estrato de MTX y el 74% de los pacientes del estrato sin MTX fueron respondedores al ACR Pediátrico 30. En la fase DB, significativamente menos pacientes que recibieron HUMIRA experimentaron un brote de la enfermedad en comparación con el placebo, tanto sin MTX (43% vs. 71%) como con MTX (37% vs. 65%). Más pacientes tratados con HUMIRA continuaron mostrando respuestas ACR Pediátrica 30/50/70 en la semana 48 en comparación con los pacientes tratados con placebo. Las respuestas del ACR Pediátrico se mantuvieron hasta dos años en la fase OLE en los pacientes que recibieron HUMIRA durante todo el estudio.
Estudio JIA-II
HUMIRA se evaluó en un estudio abierto, multicéntrico, en 32 pacientes de 2 a <4 años de edad o de 4 años de edad o más que pesaban <15 kg con AIJ poliarticular de moderada a gravemente activa. La mayoría de los pacientes (97%) recibieron al menos 24 semanas de tratamiento con HUMIRA a una dosis de 24 mg/m2 hasta un máximo de 20 mg cada dos semanas como una única inyección SC hasta una duración máxima de 120 semanas. Durante el estudio, la mayoría de los pacientes utilizaron MTX concomitante, con menos informes de uso de corticosteroides o AINE. El objetivo principal del estudio fue la evaluación de la seguridad [véase Reacciones adversas (6.1)].
14.3 Artritis Psoriásica
La seguridad y eficacia de HUMIRA se evaluaron en dos estudios aleatorizados, doble ciego, controlados con placebo en 413 pacientes con artritis psoriásica (APs). Tras la finalización de ambos estudios, 383 pacientes se inscribieron en un estudio de extensión abierto, en el que se administró 40 mg de HUMIRA cada dos semanas.
El estudio PsA-I incluyó a 313 pacientes adultos con APs de moderada a gravemente activa (>3 articulaciones inflamadas y >3 articulaciones dolorosas) que habían tenido una respuesta inadecuada a la terapia con AINE en una de las siguientes formas: (1) afectación interfalángica distal (DIP) (N=23); (2) artritis poliarticular (ausencia de nódulos reumatoides y presencia de psoriasis en placas) (N=210); (3) artritis mutilans (N=1); (4) APs asimétrica (N=77); o (5) similar a la AS (N=2). Los pacientes que recibían terapia con MTX (158 de 313 pacientes) en el momento de la inscripción (dosis estable de ≤30 mg/semana durante >1 mes) podían continuar con el MTX a la misma dosis. Se administraron dosis de HUMIRA 40 mg o placebo cada dos semanas durante el período de doble ciego de 24 semanas del estudio.
En comparación con el placebo, el tratamiento con HUMIRA dio lugar a mejoras en las medidas de la actividad de la enfermedad (véanse las tablas 8 y 9). Entre los pacientes con APs que recibieron HUMIRA, las respuestas clínicas fueron evidentes en algunos pacientes en el momento de la primera visita (dos semanas) y se mantuvieron hasta las 88 semanas en el estudio abierto en curso. Se observaron respuestas similares en pacientes con cada uno de los subtipos de artritis psoriásica, aunque se inscribieron pocos pacientes con los subtipos de artritis mutilans y similares a la espondilitis anquilosante. Las respuestas fueron similares en los pacientes que sí o no recibían terapia concomitante con MTX en el momento de la inclusión.
Los pacientes con afectación psoriásica de al menos el tres por ciento de la superficie corporal (BSA) fueron evaluados para las respuestas del Índice de Área y Severidad Psoriásica (PASI). A las 24 semanas, las proporciones de pacientes que lograron una mejora del 75% o 90% en el PASI fueron del 59% y el 42%, respectivamente, en el grupo HUMIRA (N=69), en comparación con el 1% y el 0%, respectivamente, en el grupo placebo (N=69) (p<0.001). Las respuestas PASI fueron evidentes en algunos pacientes en el momento de la primera visita (dos semanas). Las respuestas fueron similares en pacientes que sí o no estaban recibiendo terapia concomitante con MTX al inicio del estudio.
| Placebo N=162 |
HUMIRA* N=151 |
|
| ACR20 Semana 12 Semana 24 |
14% 15% |
58% 57% |
| ACR50 Semana 12 Semana 24 |
4% 6% |
36% 39% |
| ACR70 Semana 12 Semana 24 |
1% 1% |
20% 23% |
| * p<0.001 para todas las comparaciones entre HUMIRA y placebo | ||
| Placebo N=162 |
HUMIRA* N=151 |
|||
| Parámetro: mediana | Línea de base | 24 semanas | Línea de base | 24 semanas |
| Número de articulaciones sensiblesa | 23.0 | 17.0 | 20.0 | 5.0 |
| Número de articulaciones inflamadasb | 11.0 | 9.0 | 11.0 | 3.0 |
| Evaluación global del médicoc | 53.0 | 49.0 | 55.0 | 16.0 |
| Evaluación global del pacientec | 49.5 | 49.0 | 48.0 | 20.0 |
| Dolorc | 49.0 | 49.0 | 54.0 | 20.0 |
| Índice de discapacidad (HAQ) d | 1.0 | 0.9 | 1.0 | 0.4 |
| PCR (mg/dL)e | 0.8 | 0.7 | 0.8 | 0.2 |
| * p<0.001 para las comparaciones de HUMIRA vs. placebo basadas en cambios medianos a Escala 0-78 b Escala 0-76 c Escala analógica visual; 0=mejor, 100=peor d Índice de Discapacidad del Cuestionario de Evaluación de la Salud; 0=mejor, 3=peor; mide la capacidad del paciente para realizar lo siguiente: vestirse/arreglarse, levantarse, comer, caminar, alcanzar, agarrar, mantener la higiene y mantener la actividad diaria. e Rango normal: 0-0.287 mg/dL |
||||
Se observaron resultados similares en un estudio adicional de 12 semanas en 100 pacientes con artritis psoriásica moderada a grave que habían tenido una respuesta subóptima a la terapia con DMARD, como se manifestó por ≥3 articulaciones sensibles y ≥3 articulaciones inflamadas al momento de la inscripción.
Respuesta radiográfica
Se evaluaron los cambios radiográficos en los estudios de PsA. Se obtuvieron radiografías de manos, muñecas y pies al inicio y a la semana 24 durante el período de doble ciego cuando los pacientes estaban en HUMIRA o placebo y a la semana 48 cuando todos los pacientes estaban en HUMIRA de etiqueta abierta. Los lectores cegados al grupo de tratamiento utilizaron una puntuación total modificada de Sharp (mTSS), que incluía las articulaciones interfalángicas distales (es decir, no idéntica a la TSS utilizada para la artritis reumatoide), para evaluar las radiografías.
Los pacientes tratados con HUMIRA demostraron una mayor inhibición de la progresión radiográfica en comparación con los pacientes tratados con placebo y este efecto se mantuvo a las 48 semanas (ver Tabla 10).
| Placebo N=141 |
HUMIRA N=133 |
||
| Semana 24 | Semana 24 | Semana 48 | |
| Media inicial | 22.1 | 23.4 | 23.4 |
| Cambio medio ± DE | 0.9 ± 3.1 | -0.1 ± 1.7 | -0.2 ± 4.9* |
| * <0.001 para la diferencia entre HUMIRA, Semana 48 y Placebo, Semana 24 (análisis primario) | |||
14.4 Espondilitis Anquilosante
La seguridad y eficacia de HUMIRA 40 mg cada dos semanas se evaluó en 315 pacientes adultos en un estudio aleatorizado, doble ciego, controlado con placebo de 24 semanas en pacientes con espondilitis anquilosante (EA) activa que habían tenido una respuesta inadecuada a los glucocorticoides, AINE, analgésicos, metotrexato o sulfasalazina. La EA activa se definió como pacientes que cumplieron al menos dos de los siguientes tres criterios: (1) una puntuación del índice de actividad de la enfermedad de Bath AS (BASDAI) ≥4 cm, (2) una puntuación de la escala analógica visual (VAS) para el dolor de espalda total ≥ 40 mm, y (3) rigidez matutina ≥ 1 hora. El período ciego fue seguido por un período de etiqueta abierta durante el cual los pacientes recibieron HUMIRA 40 mg cada dos semanas por vía subcutánea durante un máximo de 28 semanas adicionales.
La mejora en las medidas de la actividad de la enfermedad se observó por primera vez en la semana 2 y se mantuvo hasta las 24 semanas, como se muestra en la Figura 2 y la Tabla 11.
Las respuestas de los pacientes con anquilosis espinal total (n = 11) fueron similares a las de los que no tenían anquilosis total.
Figura 2. Respuesta ASAS 20 por visita, estudio AS-I

A las 12 semanas, las respuestas ASAS 20/50/70 se lograron en el 58%, 38% y 23%, respectivamente, de los pacientes que recibieron HUMIRA, en comparación con el 21%, 10% y 5%, respectivamente, de los pacientes que recibieron placebo (p <0,001). Se observaron respuestas similares en la semana 24 y se mantuvieron en los pacientes que recibieron HUMIRA de etiqueta abierta hasta las 52 semanas.
Una mayor proporción de pacientes tratados con HUMIRA (22%) logró un bajo nivel de actividad de la enfermedad a las 24 semanas (definido como un valor <20 [en una escala de 0 a 100 mm] en cada uno de los cuatro parámetros de respuesta ASAS) en comparación con los pacientes tratados con placebo (6%).
| Placebo N=107 |
HUMIRA N=208 |
|||
| Línea de base media |
Semana 24 media |
Línea de base media |
Semana 24 media |
|
| Criterios de respuesta ASAS 20* | ||||
| Evaluación global del paciente de la actividad de la enfermedada* | 65 | 60 | 63 | 38 |
| Dolor de espalda total* | 67 | 58 | 65 | 37 |
| Inflamaciónb* | 6.7 | 5.6 | 6.7 | 3.6 |
| BASFIc* | 56 | 51 | 52 | 34 |
| Puntuación BASDAId* | 6.3 | 5.5 | 6.3 | 3.7 |
| Puntuación BASMIe* | 4.2 | 4.1 | 3.8 | 3.3 |
| Tragus a la pared (cm) | 15.9 | 15.8 | 15.8 | 15.4 |
| Flexión lumbar (cm) | 4.1 | 4.0 | 4.2 | 4.4 |
| Rotación cervical (grados) | 42.2 | 42.1 | 48.4 | 51.6 |
| Flexión lateral lumbar (cm) | 8.9 | 9.0 | 9.7 | 11.7 |
| Distancia intermaleolar (cm) | 92.9 | 94.0 | 93.5 | 100.8 |
| CRPf* | 2.2 | 2.0 | 1.8 | 0.6 |
| a Porcentaje de sujetos con al menos un 20% y una mejora de 10 unidades medida en una Escala Visual Analógica (EVA) con 0 = “ninguna” y 100 = “grave” b media de las preguntas 5 y 6 de BASDAI (definido en ‘d’) c Índice Funcional de Espondilitis Anquilosante de Bath d Índice de Actividad de la Enfermedad de Espondilitis Anquilosante de Bath e Índice de Metrología de Espondilitis Anquilosante de Bath f Proteína C Reactiva (mg/dL) * estadísticamente significativo para las comparaciones entre HUMIRA y placebo en la semana 24 |
||||
Un segundo estudio aleatorizado, multicéntrico, doble ciego, controlado con placebo de 82 pacientes con espondilitis anquilosante mostró resultados similares.
Los pacientes tratados con HUMIRA lograron una mejora desde el inicio en la puntuación del Cuestionario de Calidad de Vida de la Espondilitis Anquilosante (ASQoL) (-3.6 vs. -1.1) y en la puntuación del Resumen del Componente Físico (PCS) de la Encuesta de Salud de Forma Corta (SF-36) (7.4 vs. 1.9) en comparación con los pacientes tratados con placebo en la semana 24.
14.5 Enfermedad de Crohn en Adultos
La seguridad y eficacia de múltiples dosis de HUMIRA se evaluaron en pacientes adultos con enfermedad de Crohn de moderada a gravemente activa, CD, (Índice de Actividad de la Enfermedad de Crohn (CDAI) ≥ 220 y ≤ 450) en estudios aleatorizados, doble ciego, controlados con placebo. Se permitieron dosis estables concomitantes de aminosalicilatos, corticosteroides y/o agentes inmunomoduladores, y el 79% de los pacientes continuaron recibiendo al menos uno de estos medicamentos.
La inducción de la remisión clínica (definida como CDAI < 150) se evaluó en dos estudios. En el Estudio CD-I, 299 pacientes naive a bloqueadores del TNF se aleatorizaron a uno de cuatro grupos de tratamiento: el grupo placebo recibió placebo en las semanas 0 y 2, el grupo 160/80 recibió 160 mg de HUMIRA en la semana 0 y 80 mg en la semana 2, el grupo 80/40 recibió 80 mg en la semana 0 y 40 mg en la semana 2, y el grupo 40/20 recibió 40 mg en la semana 0 y 20 mg en la semana 2. Los resultados clínicos se evaluaron en la semana 4.
En el segundo estudio de inducción, Estudio CD-II, 325 pacientes que habían perdido la respuesta a, o eran intolerantes a, la terapia previa con infliximab se aleatorizaron para recibir 160 mg de HUMIRA en la semana 0 y 80 mg en la semana 2, o placebo en las semanas 0 y 2. Los resultados clínicos se evaluaron en la semana 4.
El mantenimiento de la remisión clínica se evaluó en el Estudio CD-III. En este estudio, 854 pacientes con enfermedad activa recibieron HUMIRA de forma abierta, 80 mg en la semana 0 y 40 mg en la semana 2. Los pacientes fueron luego aleatorizados en la semana 4 a 40 mg de HUMIRA cada dos semanas, 40 mg de HUMIRA cada semana o placebo. La duración total del estudio fue de 56 semanas. Los pacientes en respuesta clínica (disminución en CDAI ≥70) en la semana 4 fueron estratificados y analizados por separado de aquellos que no estaban en respuesta clínica en la semana 4.
Inducción de la Remisión Clínica
Un mayor porcentaje de los pacientes tratados con 160/80 mg de HUMIRA logró la inducción de la remisión clínica en comparación con el placebo en la semana 4, independientemente de si los pacientes eran naive a bloqueadores del TNF (CD-I), o habían perdido la respuesta o eran intolerantes al infliximab (CD-II) (ver Tabla 12).
| CD-I | CD-II | |||
| Placebo N=74 |
HUMIRA 160/80 mg N=76 |
Placebo N=166 |
HUMIRA 160/80 mg N=159 |
|
| Semana 4 | ||||
| Remisión clínica | 12% | 36%* | 7% | 21%* |
| Respuesta clínica | 34% | 58%** | 34% | 52%** |
| La remisión clínica es la puntuación de CDAI < 150; la respuesta clínica es una disminución en CDAI de al menos 70 puntos. * p<0.001 para HUMIRA vs. comparación por pares de proporciones de placebo ** p<0.01 para HUMIRA vs. comparación por pares de proporciones de placebo |
||||
Mantenimiento de la remisión clínica
En el Estudio CD-III en la Semana 4, el 58% (499/854) de los pacientes tuvieron una respuesta clínica y fueron evaluados en el análisis primario. En las Semanas 26 y 56, mayores proporciones de pacientes que tuvieron una respuesta clínica en la Semana 4 lograron la remisión clínica en el grupo de mantenimiento de HUMIRA 40 mg cada dos semanas en comparación con los pacientes en el grupo de mantenimiento con placebo (ver Tabla 13). El grupo que recibió terapia con HUMIRA cada semana no demostró tasas de remisión significativamente más altas en comparación con el grupo que recibió HUMIRA cada dos semanas.
| Placebo | 40 mg HUMIRA cada dos semanas |
|
| N=170 | N=172 | |
| Semana 26 | ||
| Remisión clínica | 17% | 40%* |
| Respuesta clínica | 28% | 54%* |
| Semana 56 | ||
| Remisión clínica | 12% | 36%* |
| Respuesta clínica | 18% | 43%* |
| La remisión clínica es la puntuación de CDAI < 150; la respuesta clínica es una disminución de la puntuación de CDAI de al menos 70 puntos. *p<0.001 para las comparaciones por pares de proporciones de HUMIRA vs. placebo |
||
14.6 Enfermedad de Crohn pediátrica
Se llevó a cabo un estudio clínico aleatorizado, doble ciego, de 52 semanas de 2 concentraciones de dosis de HUMIRA (Estudio PCD-I) en 192 pacientes pediátricos (de 6 a 17 años de edad) con enfermedad de Crohn de moderada a gravemente activa (definida como puntuación del Índice de Actividad de la Enfermedad de Crohn Pediátrica (PCDAI) > 30). Los pacientes inscritos habían tenido durante el período de dos años anterior una respuesta inadecuada a los corticosteroides o un inmunomodulador (es decir, azatioprina, 6-mercaptopurina o metotrexato). Los pacientes que habían recibido previamente un bloqueador del TNF pudieron inscribirse si habían perdido previamente la respuesta o tenían intolerancia a ese bloqueador del TNF.
Los pacientes recibieron terapia de inducción de etiqueta abierta a una dosis basada en su peso corporal (≥40 kg y <40 kg). Los pacientes que pesaban ≥40 kg recibieron 160 mg (en la semana 0) y 80 mg (en la semana 2). Los pacientes que pesaban <40 kg recibieron 80 mg (en la semana 0) y 40 mg (en la semana 2). En la semana 4, los pacientes dentro de cada categoría de peso corporal (≥40 kg y <40 kg) se asignaron aleatoriamente 1:1 a uno de dos regímenes de dosis de mantenimiento (dosis alta y dosis baja). La dosis alta fue de 40 mg cada dos semanas para pacientes que pesaban ≥40 kg y 20 mg cada dos semanas para pacientes que pesaban <40 kg. La dosis baja fue de 20 mg cada dos semanas para pacientes que pesaban ≥40 kg y 10 mg cada dos semanas para pacientes que pesaban <40 kg.
Se permitieron dosis estables concomitantes de corticosteroides (dosis de prednisona ≤40 mg/día o equivalente) e inmunomoduladores (azatioprina, 6-mercaptopurina o metotrexato) durante todo el estudio.
En la semana 12, los pacientes que experimentaron un brote de la enfermedad (aumento en el PCDAI de ≥ 15 desde la semana 4 y PCDAI absoluto > 30) o que no respondieron (no lograron una disminución en el PCDAI de ≥ 15 desde el inicio durante 2 visitas consecutivas al menos 2 semanas de diferencia) se les permitió aumentar la dosis (es decir, cambiar de la dosificación ciega cada dos semanas a la dosificación ciega cada semana); los pacientes que aumentaron la dosis se consideraron fracasos del tratamiento.
En el inicio, el 38% de los pacientes recibían corticosteroides y el 62% de los pacientes recibían un inmunomodulador. El cuarenta y cuatro por ciento (44%) de los pacientes habían perdido previamente la respuesta o eran intolerantes a un bloqueador del TNF. La puntuación media del PCDAI al inicio fue de 40.
De los 192 pacientes en total, 188 pacientes completaron el período de inducción de 4 semanas, 152 pacientes completaron 26 semanas de tratamiento y 124 pacientes completaron 52 semanas de tratamiento. El cincuenta y un por ciento (51%) (48/95) de los pacientes en el grupo de dosis de mantenimiento baja aumentaron la dosis y el 38% (35/93) de los pacientes en el grupo de dosis de mantenimiento alta aumentaron la dosis.
En la semana 4, el 28% (52/188) de los pacientes estaban en remisión clínica (definida como PCDAI ≤ 10).
Las proporciones de pacientes en remisión clínica (definida como PCDAI ≤ 10) y respuesta clínica (definida como reducción en el PCDAI de al menos 15 puntos desde el inicio) se evaluaron en las semanas 26 y 52.
Tanto en la semana 26 como en la 52, la proporción de pacientes en remisión clínica y respuesta clínica fue numéricamente mayor en el grupo de dosis alta en comparación con el grupo de dosis baja (Tabla 14). El régimen de mantenimiento recomendado es de 20 mg cada dos semanas para pacientes que pesan < 40 kg y 40 mg cada dos semanas para pacientes que pesan ≥ 40 kg. La dosificación semanal no es el régimen de dosificación de mantenimiento recomendado [ver Dosificación y administración (2.3)].
| Dosis de mantenimiento baja† (20 o 10 mg cada dos semanas) N = 95 |
Dosis de mantenimiento alta# (40 o 20 mg cada dos semanas) N = 93 |
|
| Semana 26 | ||
| Remisión clínica‡ | 28% | 39% |
| Respuesta clínica§ | 48% | 59% |
| Semana 52 | ||
| Remisión clínica‡ | 23% | 33% |
| Respuesta clínica§ | 28% | 42% |
| †La dosis de mantenimiento baja fue de 20 mg cada dos semanas para pacientes que pesaban ≥ 40 kg y 10 mg cada dos semanas para pacientes que pesaban < 40 kg. #La dosis de mantenimiento alta fue de 40 mg cada dos semanas para pacientes que pesaban ≥ 40 kg y 20 mg cada dos semanas para pacientes que pesaban < 40 kg. ‡Remisión clínica definida como PCDAI ≤ 10. §Respuesta clínica definida como reducción en el PCDAI de al menos 15 puntos desde el inicio. |
||
14.7 Colitis ulcerosa en adultos
La seguridad y eficacia de HUMIRA se evaluaron en pacientes adultos con colitis ulcerosa de moderada a gravemente activa (puntuación de Mayo de 6 a 12 en una escala de 12 puntos, con una subpuntuación de endoscopia de 2 a 3 en una escala de 0 a 3) a pesar del tratamiento concomitante o previo con inmunosupresores como corticosteroides, azatioprina o 6-MP en dos estudios clínicos aleatorizados, doble ciego, controlados con placebo (Estudios UC-I y UC-II). Ambos estudios reclutaron pacientes naive a bloqueadores del TNF, pero el Estudio UC-II también permitió la entrada de pacientes que perdieron la respuesta o fueron intolerantes a los bloqueadores del TNF. El cuarenta por ciento (40%) de los pacientes inscritos en el Estudio UC-II había usado previamente otro bloqueador del TNF.
Se permitieron dosis estables concomitantes de aminosalicilatos e inmunosupresores. En los Estudios UC-I y II, los pacientes estaban recibiendo aminosalicilatos (69%), corticosteroides (59%) y/o azatioprina o 6-MP (37%) al inicio del estudio. En ambos estudios, el 92% de los pacientes recibieron al menos uno de estos medicamentos.
La inducción de la remisión clínica (definida como una puntuación de Mayo ≤ 2 sin subpuntuaciones individuales > 1) en la semana 8 se evaluó en ambos estudios. La remisión clínica en la semana 52 y la remisión clínica sostenida (definida como remisión clínica en las semanas 8 y 52) se evaluaron en el Estudio UC-II.
En el Estudio UC-I, 390 pacientes naive a bloqueadores del TNF se asignaron aleatoriamente a uno de tres grupos de tratamiento para el análisis de eficacia primaria. El grupo placebo recibió placebo en las semanas 0, 2, 4 y 6. El grupo 160/80 recibió 160 mg de HUMIRA en la semana 0 y 80 mg en la semana 2, y el grupo 80/40 recibió 80 mg de HUMIRA en la semana 0 y 40 mg en la semana 2. Después de la semana 2, los pacientes en ambos grupos de tratamiento con HUMIRA recibieron 40 mg cada dos semanas.
En el Estudio UC-II, 518 pacientes se asignaron aleatoriamente para recibir HUMIRA 160 mg en la semana 0, 80 mg en la semana 2 y 40 mg cada dos semanas a partir de la semana 4 hasta la semana 50, o placebo a partir de la semana 0 y cada dos semanas hasta la semana 50. Se permitió la reducción gradual de los corticosteroides a partir de la semana 8.
En ambos Estudios UC-I y UC-II, un mayor porcentaje de los pacientes tratados con 160/80 mg de HUMIRA en comparación con los pacientes tratados con placebo lograron la inducción de la remisión clínica. En el Estudio UC-II, un mayor porcentaje de los pacientes tratados con 160/80 mg de HUMIRA en comparación con los pacientes tratados con placebo lograron la remisión clínica sostenida (remisión clínica en las semanas 8 y 52) (Tabla 15).
| Estudio UC-I | Estudio UC-II | |||||
| Placebo N=130 |
HUMIRA 160/80 mg N=130 |
Diferencia de tratamiento (IC del 95%) |
Placebo N=246 |
HUMIRA 160/80 mg N=248 |
Diferencia de tratamiento (IC del 95%) |
|
| Inducción de la remisión clínica (Remisión clínica en la semana 8) | 9.2% | 18.5% | 9.3%* (0.9%, 17.6%) |
9.3% | 16.5% | 7.2%* (1.2%, 12.9%) |
| Remisión clínica sostenida (Remisión clínica en las semanas 8 y 52) | N/A | N/A | N/A | 4.1% | 8.5% | 4.4%* (0.1%, 8.6%) |
| La remisión clínica se define como una puntuación de Mayo ≤ 2 sin subpuntuaciones individuales > 1. IC=Intervalo de confianza * p<0.05 para HUMIRA vs. comparación por pares de proporciones con placebo |
||||||
En el estudio UC-I, no se observó diferencia estadísticamente significativa en la remisión clínica entre el grupo de HUMIRA 80/40 mg y el grupo de placebo en la Semana 8.
En el estudio UC-II, el 17.3% (43/248) del grupo de HUMIRA estaba en remisión clínica en la Semana 52 en comparación con el 8.5% (21/246) del grupo de placebo (diferencia de tratamiento: 8.8%; intervalo de confianza del 95% (IC): [2.8%, 14.5%]; p<0.05).
En el subgrupo de pacientes en el estudio UC-II con uso previo de bloqueadores del TNF, la diferencia de tratamiento para la inducción de remisión clínica pareció ser menor que la observada en toda la población del estudio, y las diferencias de tratamiento para la remisión clínica sostenida y la remisión clínica en la Semana 52 parecieron ser similares a las observadas en toda la población del estudio. El subgrupo de pacientes con uso previo de bloqueadores del TNF logró la inducción de remisión clínica en un 9% (9/98) en el grupo de HUMIRA versus un 7% (7/101) en el grupo de placebo, y la remisión clínica sostenida en un 5% (5/98) en el grupo de HUMIRA versus un 1% (1/101) en el grupo de placebo. En el subgrupo de pacientes con uso previo de bloqueadores del TNF, el 10% (10/98) estaba en remisión clínica en la Semana 52 en el grupo de HUMIRA versus el 3% (3/101) en el grupo de placebo.
14.8 Colitis Ulcerosa Pediátrica
La seguridad y eficacia de HUMIRA se evaluaron en un ensayo multicéntrico, aleatorizado y doble ciego (Estudio PUC-I, NCT02065557) en 93 pacientes pediátricos de 5 a 17 años de edad con colitis ulcerosa moderada a grave (puntuación de Mayo de 6 a 12 con puntuación de endoscopia de 2 a 3 puntos, confirmada por endoscopia leída centralmente) que tuvieron una respuesta inadecuada o intolerancia al tratamiento con corticosteroides y/o un inmunomodulador (es decir, azatioprina, 6-mercaptopurina o metotrexato). Quince de los 93 pacientes (16%) en el estudio tenían experiencia previa con un bloqueador del TNF. A los pacientes que recibieron corticosteroides al inicio se les permitió reducir su terapia con corticosteroides después de la Semana 4.
Setenta y siete pacientes fueron inicialmente asignados al azar en una proporción de 3:2 para recibir tratamiento a ciegas con una de dos dosis de HUMIRA. Los pacientes en ambos grupos de dosis recibieron 2.4 mg/kg (máximo de 160 mg) en la Semana 0, 1.2 mg/kg (máximo de 80 mg) en la Semana 2 y 0.6 mg/kg (máximo de 40 mg) en las Semanas 4 y 6. El grupo de dosis más alta también recibió una dosis adicional de 2.4 mg/kg (máximo de 160 mg) en la Semana 1. Después de una modificación en el diseño del estudio, se inscribieron 16 pacientes adicionales que recibieron tratamiento a ciegas con HUMIRA en la dosis más alta.
En la Semana 8, 62 pacientes que demostraron respuesta clínica según la Puntuación Mayo Parcial (PMS; un subconjunto de la puntuación Mayo sin componente endoscópico y definido como una disminución en la PMS ≥ 2 puntos y ≥ 30% desde el inicio) fueron asignados al azar en partes iguales para recibir tratamiento a ciegas con HUMIRA 0.6 mg/kg (máximo de 40 mg) cada dos semanas (grupo de dosis más baja) o 0.6 mg/kg (máximo de 40 mg) cada semana (grupo de dosis más alta). Antes de una modificación en el diseño del estudio, se asignaron al azar 12 pacientes adicionales que demostraron respuesta clínica según la PMS para recibir placebo.
No se esperan diferencias clínicamente relevantes en la eficacia entre la dosis más alta estudiada durante el ensayo PUC-I de 52 semanas y la dosis recomendada de HUMIRA [consulte la Dosificación y Administración (2.4), Farmacología Clínica (12.2)].
Los pacientes que cumplieron los criterios de recaída de la enfermedad en o después de la Semana 12 fueron asignados al azar para recibir una dosis de reinducción de 2.4 mg/kg (máximo de 160 mg) o una dosis de 0.6 mg/kg (máximo de 40 mg) y luego continuaron con la dosis a la que fueron asignados en la Semana 8.
Los criterios de valoración co-primarios del estudio fueron la remisión clínica según la PMS (definida como PMS ≤ 2 y ninguna subpuntuación individual > 1) en la Semana 8, y la remisión clínica según la Puntuación Mayo (definida como Puntuación Mayo ≤ 2 y ninguna subpuntuación individual > 1) en la Semana 52 en pacientes que lograron respuesta clínica según la PMS en la Semana 8. Los criterios de valoración secundarios incluyeron la respuesta según la Puntuación Mayo (definida como una disminución en la Puntuación Mayo de ≥ 3 puntos y ≥ 30% desde el inicio) en los respondedores según la PMS en la Semana 8, la mejora endoscópica (definida como una subpuntuación de endoscopia Mayo ≤ 1) en la Semana 52 en los respondedores según la PMS en la Semana 8, y la remisión según la Puntuación Mayo en la Semana 52 en los remitentes según la PMS en la Semana 8.
Resultados en la Semana 8
En la Semana 8, la remisión según la PMS se logró en el 60% [28/47; intervalo de confianza del 95% (IC): (44%, 74%)] de los pacientes en el grupo de dosis más alta (sin incluir a los 16 pacientes que recibieron dosis más alta a ciegas) y en el 43% [13/30; IC del 95%: (25%, 63%)] de los pacientes en el grupo de dosis más baja. Los resultados del grupo de dosis más alta son representativos de los resultados esperados con la dosis recomendada [consulte la Dosificación y Administración (2.4), Farmacología Clínica (12.2)].
Resultados en la Semana 52
En la Semana 52, se evaluaron los criterios de valoración en la población de pacientes que recibieron placebo a ciegas, HUMIRA 0.6 mg/kg (máximo de 40 mg) cada dos semanas, o HUMIRA 0.6 mg/kg (máximo de 40 mg) cada semana entre la Semana 8 y la Semana 52 (Tabla 16).
|
|
Placeboa
n/N (%), IC del 95% |
HUMIRA Máximo de 40 mg (0.6 mg/kg) cada dos semanasb n/N (%), IC del 95% |
HUMIRA Máximo de 40 mg (0.6 mg/kg) cada semanac n/N (%), IC del 95% |
| Remisión clínica en la semana 8 de los respondedores a PMS | 4/12 (33%) (10%, 65%) |
9/31 (29%) (14%, 48%) |
14/31 (45%) (27%, 64%) |
| Respuesta clínica en la semana 8 de los respondedores a PMS | 4/12 (33%) (10%, 65%) |
19/31 (61%) (42%, 78%) |
21/31 (68%) (49%, 83%) |
| Mejora endoscópica en la semana 8 de los respondedores a PMS | 4/12 (33%) (10%, 65%) |
12/31 (39%) (22%, 58%) |
16/31 (52%) (33%, 70%) |
| Remisión clínica en la semana 8 de los remisores a PMS | 3/8 (38%) (9%, 76%) |
9/21 (43%) (22%, 66%) |
10/22 (45%) (24%, 68%) |
| IC=Intervalo de confianza a Doce pacientes que demostraron respuesta clínica según PMS en la semana 8 fueron aleatorizados para recibir placebo. Hay limitaciones para la interpretabilidad de los datos del placebo debido al pequeño tamaño de la muestra. b La dosificación cada dos semanas estudiada durante el ensayo PUC-I de 52 semanas es una dosis más baja que la dosis recomendada de HUMIRA [ver Dosificación y administración (2.4)]. c No se anticipan diferencias clínicamente relevantes en la eficacia entre la dosis más alta estudiada administrada durante el ensayo PUC-I de 52 semanas y la dosis recomendada de HUMIRA. Nota: Los pacientes con valores faltantes en la semana 52 o que fueron aleatorizados para recibir tratamiento de reinducción o mantenimiento debido a un brote de la enfermedad se consideraron no respondedores para los puntos finales de la semana 52. |
|||
14.9 Psoriasis en placas
La seguridad y eficacia de HUMIRA se evaluaron en estudios aleatorizados, doble ciego, controlados con placebo en 1696 sujetos adultos con psoriasis en placas crónica de moderada a grave (Ps) que eran candidatos a terapia sistémica o fototerapia.
El estudio Ps-I evaluó a 1212 sujetos con Ps crónica con ≥10% de superficie corporal (BSA) involucrada, Evaluación Global del Médico (PGA) de al menos gravedad moderada de la enfermedad e Índice de Área y Severidad de la Psoriasis (PASI) ≥12 dentro de tres períodos de tratamiento. En el período A, los sujetos recibieron placebo o HUMIRA a una dosis inicial de 80 mg en la semana 0 seguida de una dosis de 40 mg cada dos semanas a partir de la semana 1. Después de 16 semanas de terapia, los sujetos que lograron al menos una respuesta PASI 75 en la semana 16, definida como una mejora en la puntuación PASI de al menos 75% en relación con la línea de base, ingresaron al período B y recibieron 40 mg de HUMIRA de etiqueta abierta cada dos semanas. Después de 17 semanas de terapia de etiqueta abierta, los sujetos que mantuvieron al menos una respuesta PASI 75 en la semana 33 y fueron originalmente aleatorizados para recibir terapia activa en el período A fueron re-aleatorizados en el período C para recibir 40 mg de HUMIRA cada dos semanas o placebo durante 19 semanas adicionales. En todos los grupos de tratamiento, la puntuación PASI media de referencia fue de 19 y la puntuación de Evaluación Global del Médico de referencia osciló entre “moderada” (53%) a “grave” (41%) a “muy grave” (6%).
El estudio Ps-II evaluó a 99 sujetos aleatorizados a HUMIRA y 48 sujetos aleatorizados a placebo con psoriasis en placas crónica con ≥10% de BSA involucrada y PASI ≥12. Los sujetos recibieron placebo o una dosis inicial de 80 mg de HUMIRA en la semana 0 seguida de 40 mg cada dos semanas a partir de la semana 1 durante 16 semanas. En todos los grupos de tratamiento, la puntuación PASI media de referencia fue de 21 y la puntuación PGA de referencia osciló entre “moderada” (41%) a “grave” (51%) a “muy grave” (8%).
Los estudios Ps-I y II evaluaron la proporción de sujetos que lograron una enfermedad “clara” o “mínima” en la escala PGA de 6 puntos y la proporción de sujetos que lograron una reducción en la puntuación PASI de al menos 75% (PASI 75) desde la línea de base en la semana 16 (ver Tabla 17 y 18).
Además, el estudio Ps-I evaluó la proporción de sujetos que mantuvieron una PGA de enfermedad “clara” o “mínima” o una respuesta PASI 75 después de la semana 33 y en o antes de la semana 52.
| HUMIRA 40 mg cada dos semanas | Placebo | |
| N = 814 | N = 398 | |
| PGA: Claro o mínimo* | 506 (62%) | 17 (4%) |
| PASI 75 | 578 (71%) | 26 (7%) |
| * Claro = sin elevación de la placa, sin escamas, más o menos hiperpigmentación o coloración rosa o roja difusa Mínimo = posible pero difícil de determinar si hay una ligera elevación de la placa por encima de la piel normal, más o menos sequedad superficial con algo de coloración blanca, más o menos hasta la coloración roja |
||
| HUMIRA 40 mg cada dos semanas | Placebo | |
| N = 99 | N = 48 | |
| PGA: Claro o mínimo* | 70 (71%) | 5 (10%) |
| PASI 75 | 77 (78%) | 9 (19%) |
| * Claro = sin elevación de la placa, sin escamas, más o menos hiperpigmentación o coloración rosada o roja difusa Mínimo = posible pero difícil de determinar si hay una ligera elevación de la placa por encima de la piel normal, más o menos sequedad superficial con algo de coloración blanca, más o menos hasta la coloración roja |
||
Además, en el Estudio Ps-I, los sujetos que recibieron HUMIRA y mantuvieron un PASI 75 fueron re-aleatorizados a HUMIRA (N = 250) o placebo (N = 240) en la semana 33. Después de 52 semanas de tratamiento con HUMIRA, más sujetos que recibieron HUMIRA mantuvieron la eficacia en comparación con los sujetos que fueron re-aleatorizados a placebo en función del mantenimiento de un PGA de “claro” o “mínimo” de la enfermedad (68% vs. 28%) o un PASI 75 (79% vs. 43%).
Un total de 347 respondedores estables participaron en una evaluación de retirada y re-tratamiento en un estudio de extensión de etiqueta abierta. El tiempo medio hasta la recaída (descenso a PGA “moderado” o peor) fue de aproximadamente 5 meses. Durante el período de retirada, ningún sujeto experimentó una transformación a psoriasis pustulosa o eritrodérmica. Un total de 178 sujetos que recayeron reiniciaron el tratamiento con 80 mg de HUMIRA, luego 40 mg cada dos semanas a partir de la semana 1. En la semana 16, el 69% (123/178) de los sujetos tuvieron una respuesta de PGA “claro” o “mínimo”.
Un estudio aleatorizado, doble ciego (Estudio Ps-III) comparó la eficacia y seguridad de HUMIRA frente a placebo en 217 sujetos adultos. Los sujetos del estudio tenían que tener psoriasis en placas crónica de al menos gravedad moderada en la escala PGA, afectación de las uñas de al menos gravedad moderada en una escala de 5 puntos de Evaluación Global del Médico de la Psoriasis de las Uñas (PGA-F), una puntuación del Índice de Gravedad de la Psoriasis de las Uñas Modificado (mNAPSI) para la uña diana de ≥ 8, y o bien una afectación del BSA de al menos el 10% o una afectación del BSA de al menos el 5% con una puntuación mNAPSI total para todas las uñas de ≥ 20. Los sujetos recibieron una dosis inicial de 80 mg de HUMIRA seguida de 40 mg cada dos semanas (comenzando una semana después de la dosis inicial) o placebo durante 26 semanas, seguido de un tratamiento con HUMIRA de etiqueta abierta durante 26 semanas adicionales. Este estudio evaluó la proporción de sujetos que lograron una evaluación “clara” o “mínima” con al menos una mejora de 2 grados en la escala PGA-F y la proporción de sujetos que lograron al menos una mejora del 75% desde el inicio en la puntuación mNAPSI (mNAPSI 75) en la semana 26.
En la semana 26, una mayor proporción de sujetos del grupo HUMIRA que del grupo placebo logró el punto final PGA-F. Además, una mayor proporción de sujetos del grupo HUMIRA que del grupo placebo logró mNAPSI 75 en la semana 26 (ver Tabla 19).
| Punto final | HUMIRA 40 mg cada dos semanas* N=109 |
Placebo N=108 |
| PGA-F: ≥2-grado de mejora y claro o mínimo | 49% | 7% |
| mNAPSI 75 | 47% | 3% |
| *Los sujetos recibieron 80 mg de HUMIRA en la semana 0, seguidos de 40 mg cada dos semanas a partir de la semana 1. | ||
14.10 Hidradenitis Suppurativa
Dos estudios aleatorizados, doble ciego, controlados con placebo (Estudios HS-I y II) evaluaron la seguridad y eficacia de HUMIRA en un total de 633 sujetos adultos con hidradenitis supurativa (HS) de moderada a grave con enfermedad en estadio II o III de Hurley y con al menos 3 abscesos o nódulos inflamatorios. En ambos estudios, los sujetos recibieron placebo o HUMIRA a una dosis inicial de 160 mg en la semana 0, 80 mg en la semana 2 y 40 mg cada semana a partir de la semana 4 y continuaron hasta la semana 11. Los sujetos utilizaron lavado antiséptico tópico diariamente. Se permitió el uso concomitante de antibióticos orales en el Estudio HS-II.
Ambos estudios evaluaron la respuesta clínica a la hidradenitis supurativa (HiSCR) en la semana 12. HiSCR se definió como al menos una reducción del 50% en el recuento total de abscesos y nódulos inflamatorios sin aumento en el recuento de abscesos y sin aumento en el recuento de fístulas drenantes en relación con el valor inicial (ver Tabla 18). La reducción del dolor en la piel relacionado con HS se evaluó utilizando una escala de calificación numérica en pacientes que ingresaron al estudio con una puntuación inicial de referencia de 3 o más en una escala de 11 puntos.
En ambos estudios, una mayor proporción de sujetos tratados con HUMIRA que con placebo lograron HiSCR (ver Tabla 20).
| Estudio HS I | Estudio HS II* | |||
| Placebo | Humira 40 mg semanal | Placebo | Humira 40 mg semanal | |
| Respuesta clínica a la hidradenitis supurativa (HiSCR) | N = 154 40 (26%) |
N = 153 64 (42%) |
N=163 45 (28%) |
N=163 96 (59%) |
| *19.3% de los sujetos en el Estudio HS-II continuaron la terapia antibiótica oral de referencia durante el estudio. | ||||
En ambos estudios, desde la semana 12 hasta la semana 35 (período B), los sujetos que habían recibido HUMIRA fueron re-aleatorizados a 1 de 3 grupos de tratamiento (HUMIRA 40 mg cada semana, HUMIRA 40 mg cada dos semanas o placebo). Los sujetos que habían sido aleatorizados a placebo fueron asignados a recibir HUMIRA 40 mg cada semana (Estudio HS-I) o placebo (Estudio HS-II).
Durante el período B, se documentó un brote de HS, definido como un aumento ≥25% desde el inicio en el recuento de abscesos y nódulos inflamatorios y con un mínimo de 2 lesiones adicionales, en 22 (22%) de los 100 sujetos que fueron retirados del tratamiento con HUMIRA después del punto de tiempo de eficacia primaria en dos estudios.
14.11 Uveítis en Adultos
La seguridad y eficacia de HUMIRA se evaluaron en pacientes adultos con uveítis intermedia, posterior y panuveítis no infecciosa, excluyendo pacientes con uveítis anterior aislada, en dos estudios aleatorizados, doble ciego, controlados con placebo (UV I y II). Los pacientes recibieron placebo o HUMIRA a una dosis inicial de 80 mg seguida de 40 mg cada dos semanas a partir de una semana después de la dosis inicial. El criterio de valoración principal de eficacia en ambos estudios fue el ´tiempo hasta el fracaso del tratamiento´.
El fracaso del tratamiento fue un resultado multicomponente definido como el desarrollo de nuevas lesiones coriorretinianas inflamatorias y/o vasculares retinianas inflamatorias, un aumento en el grado de células de la cámara anterior (AC) o el grado de neblina vítrea (VH) o una disminución en la mejor agudeza visual corregida (BCVA).
El estudio UV I evaluó a 217 pacientes con uveítis activa mientras estaban siendo tratados con corticosteroides (prednisona oral a una dosis de 10 a 60 mg/día). Todos los pacientes recibieron una dosis estandarizada de prednisona 60 mg/día al inicio del estudio seguida de un programa de reducción obligatorio, con la interrupción completa de los corticosteroides para la semana 15.
El estudio UV II evaluó a 226 pacientes con uveítis inactiva mientras estaban siendo tratados con corticosteroides (prednisona oral de 10 a 35 mg/día) al inicio para controlar su enfermedad. Los pacientes posteriormente se sometieron a un programa de reducción obligatorio, con la interrupción completa de los corticosteroides para la semana 19.
Respuesta Clínica
Los resultados de ambos estudios demostraron una reducción estadísticamente significativa del riesgo de fracaso del tratamiento en pacientes tratados con HUMIRA en comparación con los pacientes que recibieron placebo. En ambos estudios, todos los componentes del criterio de valoración principal contribuyeron de forma acumulativa a la diferencia general entre los grupos de HUMIRA y placebo (Tabla 21).
| UV I | UV II | |||||
| Placebo (N = 107) |
HUMIRA (N = 110) |
HR [95% CI]a |
Placebo (N = 111) |
HUMIRA (N = 115) |
HR [95% CI]a |
|
| Fracasob n (%) | 84 (78.5) | 60 (54.5) | 0.50 [0.36, 0.70] |
61 (55.0) | 45 (39.1) | 0.57 [0.39, 0.84] |
| Tiempo Mediano hasta el Fracaso (Meses) [95% CI] |
3.0 [2.7, 3.7] |
5.6 [3.9, 9.2] |
N/A | 8.3 [4.8, 12.0] |
NEc | N/A |
| ª HR de HUMIRA versus placebo de la regresión de riesgos proporcionales con el tratamiento como factor. b El fracaso del tratamiento en o después de la semana 6 en el estudio UV I, o en o después de la semana 2 en el estudio UV II, se contó como evento. Los sujetos que abandonaron el estudio fueron censurados en el momento de la deserción. c NE = no estimable. Menos de la mitad de los sujetos en riesgo tuvieron un evento. |
||||||
14.12 Uveítis Pediátrica
La seguridad y eficacia de HUMIRA se evaluaron en un estudio aleatorizado, doble ciego, controlado con placebo de 90 pacientes pediátricos de 2 a < 18 años de edad con uveítis no infecciosa activa asociada a JIA (PUV-I). Los pacientes recibieron placebo o 20 mg de adalimumab (si < 30 kg) o 40 mg de adalimumab (si ≥ 30 kg) cada dos semanas en combinación con una dosis de metotrexato. Se permitieron dosis concomitantes de corticosteroides al inicio del estudio, seguidas de una reducción obligatoria de los corticosteroides tópicos dentro de los 3 meses.
El criterio de valoración principal fue el “tiempo hasta el fracaso del tratamiento”. Los criterios que determinaron el fracaso del tratamiento fueron el empeoramiento o la falta de mejora sostenida de la inflamación ocular, o el empeoramiento de las comorbilidades oculares.
Respuesta Clínica
HUMIRA redujo significativamente el riesgo de fracaso del tratamiento en un 75% en relación con el placebo (HR = 0,25 [IC del 95%: 0,12, 0,49]) (Tabla 22).
| Placebo (N=30) |
HUMIRA (N=60) |
HR (IC del 95%)ª | |
| Fracaso (n[%]) | 18 (60%) | 16 (26,7%) | 0,25 (0,12, 0,49) |
| Tiempo mediano hasta el Fracaso (Semanas) (IC del 95%)ᵇ |
24,1 (12,4, 81,0) |
NEᶜ | |
| a HR de adalimumab frente a placebo de la regresión de riesgos proporcionales con el tratamiento como factor. b Estimado en base a la curva de Kaplan-Meier. c NE = no estimable. Menos de la mitad de los sujetos en riesgo tuvieron un evento. |
|||
Figura 4: Curvas de Kaplan-Meier que resumen el tiempo hasta el fracaso del tratamiento (Estudio PUV-I)

Estudio PUV-I
Nota: P = Placebo (Número en riesgo); H = HUMIRA (Número en riesgo).
15 REFERENCIAS
- National Cancer Institute. Surveillance, Epidemiology, and End Results Database (SEER) Program. SEER Incidence Crude Rates, 17 Registries, 2000-2007.
16 CÓMO SE SUMINISTRA/ALMACENAMIENTO Y MANEJO
HUMIRA® (adalimumab) se suministra como una solución estéril, transparente e incolora sin conservantes para administración subcutánea. Las siguientes configuraciones de empaque están disponibles.
-
HUMIRA Pen Carton – 40 mg/0.4 mL
HUMIRA se suministra en una caja que contiene dos preparaciones de alcohol y dos bandejas de dosis. Cada bandeja de dosis consta de un bolígrafo de dosis única, que contiene una jeringa de vidrio precargada de 1 mL con una pared delgada fija, aguja de ½ pulgada, que proporciona 40 mg/0.4 mL de HUMIRA. La cubierta de la aguja negra no está hecha con látex de caucho natural. El número NDC es 83457-554-02 -
HUMIRA Pen Carton – 80 mg/0.8 mL
HUMIRA se suministra en una caja que contiene dos preparaciones de alcohol y dos bandejas de dosis. Cada bandeja de dosis consta de un bolígrafo de dosis única, que contiene una jeringa de vidrio precargada de 1 mL con una pared delgada fija, aguja de ½ pulgada, que proporciona 80 mg/0.8 mL de HUMIRA. La cubierta de la aguja negra no está hecha con látex de caucho natural. El número NDC es 83457-124-02. -
Prefilled Syringe Carton – 40 mg/0.4 mL
HUMIRA se suministra en una caja que contiene dos preparaciones de alcohol y dos bandejas de dosis. Cada bandeja de dosis consta de una jeringa de vidrio precargada de 1 mL de dosis única con una pared delgada fija, aguja de ½ pulgada, que proporciona 40 mg/0.4 mL de HUMIRA. La cubierta de la aguja negra no está hecha con látex de caucho natural. El número NDC es 83457-243-02. -
Prefilled Syringe Carton – 20 mg/0.2 mL
HUMIRA se suministra en una caja que contiene dos preparaciones de alcohol y dos bandejas de dosis. Cada bandeja de dosis consta de una jeringa de vidrio precargada de 1 mL de dosis única con una pared delgada fija, aguja de ½ pulgada, que proporciona 20 mg/0.2 mL de HUMIRA. La cubierta de la aguja negra no está hecha con látex de caucho natural. El número NDC es 83457-616-02. -
Prefilled Syringe Carton – 10 mg/0.1 mL
HUMIRA se suministra en una caja que contiene dos preparaciones de alcohol y dos bandejas de dosis. Cada bandeja de dosis consta de una jeringa de vidrio precargada de 1 mL de dosis única con una pared delgada fija, aguja de ½ pulgada, que proporciona 10 mg/0.1 mL de HUMIRA. La cubierta de la aguja negra no está hecha con látex de caucho natural. El número NDC es 83457-817-02.
Almacenamiento y estabilidad
No use después de la fecha de vencimiento en el contenedor. HUMIRA debe refrigerarse a 36°F a 46°F (2°C a 8°C). NO CONGELAR. No use si se congela, incluso si se ha descongelado.
Guarde en la caja original hasta el momento de la administración para proteger de la luz.
Si es necesario, por ejemplo, cuando viaja, HUMIRA se puede almacenar a temperatura ambiente hasta un máximo de 77°F (25°C) durante un período de hasta 14 días, con protección de la luz. HUMIRA debe desecharse si no se usa dentro del período de 14 días. Registre la fecha en que HUMIRA se retira por primera vez del refrigerador en los espacios proporcionados en la caja y la bandeja de dosis.
No almacene HUMIRA en calor o frío extremos.
17 INFORMACIÓN PARA EL PACIENTE
Avise al paciente o cuidador que lea la etiqueta del paciente aprobada por la FDA (Guía de medicamentos y Instrucciones de uso).
Infecciones
Informe a los pacientes que HUMIRA puede disminuir la capacidad de su sistema inmunitario para combatir infecciones. Indique a los pacientes la importancia de comunicarse con su médico si desarrollan algún síntoma de infección, incluida la tuberculosis, las infecciones fúngicas invasivas y la reactivación de las infecciones por el virus de la hepatitis B [ver Advertencias y precauciones (5.1, 5.2, 5.4)].
Neoplasias
Oriente a los pacientes sobre el riesgo de neoplasias mientras reciben HUMIRA [ver Advertencias y precauciones (5.2)]
Reacciones de hipersensibilidad
Avise a los pacientes que busquen atención médica inmediata si experimentan algún síntoma de reacciones de hipersensibilidad graves. Avise a los pacientes sensibles al látex que la tapa de la aguja del bolígrafo HUMIRA de 40 mg/0,8 mL y la jeringa precargada de 40 mg/0,8 mL, 20 mg/0,4 mL y 10 mg/0,2 mL puede contener látex de caucho natural [ver Advertencias y precauciones (5.3), Cómo se suministra/Almacenamiento y manipulación (16)].
Otras condiciones médicas
Avise a los pacientes que informen cualquier signo de condiciones médicas nuevas o que empeoren, como insuficiencia cardíaca congestiva, enfermedad neurológica, trastornos autoinmunitarios o citopenias. Avise a los pacientes que informen cualquier síntoma sugestivo de una citopenia, como moretones, sangrado o fiebre persistente [ver Advertencias y precauciones (5.5, 5.6, 5.8, 5.9)].
Instrucciones sobre la técnica de inyección
Informe a los pacientes que la primera inyección debe realizarse bajo la supervisión de un profesional sanitario cualificado. Si un paciente o cuidador va a administrar HUMIRA, instrúyalo en las técnicas de inyección y evalúe su capacidad para inyectar por vía subcutánea para garantizar la administración adecuada de HUMIRA [ver Instrucciones de uso].
Para los pacientes que usarán el bolígrafo HUMIRA, dígales que:
- Escucharán un ‘clic’ fuerte cuando se presione el botón activador de color ciruela. El clic fuerte significa el inicio de la inyección.
- Deben mantener el bolígrafo HUMIRA presionado contra la piel estirada y apretada hasta que se inyecte todo el medicamento. Esto puede tardar hasta 15 segundos.
- Sabrán que la inyección ha terminado cuando el marcador amarillo aparezca completamente en la ventana de visualización y deje de moverse.
Indique a los pacientes que deseche las agujas y jeringas usadas o el bolígrafo usado en un contenedor de eliminación de objetos punzantes aprobado por la FDA inmediatamente después de su uso. Indique a los pacientes que no deseche las agujas y jeringas sueltas o el bolígrafo en la basura de su hogar. Indique a los pacientes que si no tienen un contenedor de eliminación de objetos punzantes aprobado por la FDA, pueden usar un contenedor doméstico que esté hecho de plástico resistente, que se pueda cerrar con una tapa hermética y resistente a las perforaciones sin que los objetos punzantes puedan salir, que sea vertical y estable durante el uso, que sea resistente a las fugas y que esté correctamente etiquetado para advertir sobre los residuos peligrosos dentro del contenedor.
Indique a los pacientes que cuando su contenedor de eliminación de objetos punzantes esté casi lleno, deberán seguir las pautas de su comunidad para la forma correcta de desechar su contenedor de eliminación de objetos punzantes. Indique a los pacientes que puede haber leyes estatales o locales con respecto a la eliminación de agujas y jeringas usadas. Refiera a los pacientes al sitio web de la FDA en http://www.fda.gov/safesharpsdisposal para obtener más información sobre la eliminación segura de objetos punzantes y para obtener información específica sobre la eliminación de objetos punzantes en el estado en el que viven.
Indique a los pacientes que no deseche su contenedor de eliminación de objetos punzantes usado en la basura de su hogar a menos que las pautas de su comunidad lo permitan. Indique a los pacientes que no reciclen su contenedor de eliminación de objetos punzantes usado.
AbbVie Inc.
North Chicago, IL 60064, EE. UU.
Número de licencia de EE. UU. 1889
20082819 11/2023
Fabricado para:
Cordavis Trading Limited
Dublín, Irlanda
Guía de medicación
| GUÍA DEL MEDICAMENTO HUMIRA® (Hu-MARE-ah) (adalimumab) inyección, para uso subcutáneo |
||
| Lea la Guía del Medicamento que viene con HUMIRA antes de empezar a usarlo y cada vez que renueve su receta. Es posible que haya nueva información. Esta Guía del Medicamento no sustituye las conversaciones con su médico sobre su enfermedad o tratamiento. | ||
| ¿Cuál es la información más importante que debo saber sobre HUMIRA? HUMIRA es un medicamento que afecta su sistema inmunitario. HUMIRA puede disminuir la capacidad de su sistema inmunitario para combatir infecciones. Se han producido infecciones graves en personas que toman HUMIRA. Estas infecciones graves incluyen tuberculosis (TB) e infecciones causadas por virus, hongos o bacterias que se han propagado por todo el cuerpo. Algunas personas han muerto a causa de estas infecciones.
No debe empezar a tomar HUMIRA si tiene algún tipo de infección a menos que su médico le diga que puede hacerlo.
|
||
|
|
|
Después de empezar a tomar HUMIRA, llame a su médico inmediatamente si tiene una infección o cualquier signo de infección. |
||
Cáncer
|
||
| ¿Qué es HUMIRA? HUMIRA es un medicamento llamado bloqueador del factor de necrosis tumoral (TNF). HUMIRA se utiliza:
|
||
| ¿Qué debo informarle a mi médico antes de tomar HUMIRA? Es posible que HUMIRA no sea adecuado para usted. Antes de comenzar a tomar HUMIRA, informe a su médico sobre todas sus afecciones médicas, incluso si:
Informe a su médico sobre todos los medicamentos que toma, incluidos los medicamentos recetados y de venta libre, las vitaminas y los suplementos a base de hierbas.
Lleve consigo una lista de sus medicamentos para mostrársela a su médico y farmacéutico cada vez que reciba un nuevo medicamento. |
||
¿Cómo debo tomar HUMIRA?
|
||
| ¿Cuáles son los posibles efectos secundarios de HUMIRA? HUMIRA puede causar efectos secundarios graves, que incluyen: Consulte “¿Cuál es la información más importante que debo saber sobre HUMIRA?” |
||
|
||
|
|
|
|
||
|
|
|
|
||
|
|
|
|
||
|
||
|
|
|
|
||
|
||
|
|
|
|
|
|
|
||
| Llame a su médico u obtenga atención médica de inmediato si presenta alguno de los síntomas mencionados anteriormente. Es posible que se suspenda su tratamiento con HUMIRA.
Los efectos secundarios más comunes de HUMIRA incluyen:
|
||
| Estos no son todos los posibles efectos secundarios de HUMIRA. Informe a su médico si tiene algún efecto secundario que le moleste o que no desaparezca. Pídale a su médico o farmacéutico más información. Llame a su médico para que le aconseje acerca de los efectos secundarios. Puede informar los efectos secundarios a la FDA al 1-800-FDA-1088. |
||
¿Cómo debo almacenar HUMIRA?
Mantenga HUMIRA, los suministros de inyección y todos los demás medicamentos fuera del alcance de los niños. |
||
| Información general sobre el uso seguro y eficaz de HUMIRA. En ocasiones, los medicamentos se recetan para fines distintos de los que se enumeran en una Guía del Medicamento. No utilice HUMIRA para una afección para la que no se le recetó. No le dé HUMIRA a otras personas, incluso si tienen la misma afección. Podría perjudicarles. Esta Guía del Medicamento resume la información más importante sobre HUMIRA. Si desea más información, hable con su médico. Puede pedirle a su farmacéutico o médico información sobre HUMIRA que esté escrita para profesionales de la salud. |
||
| ¿Cuáles son los ingredientes de HUMIRA? Ingrediente activo: adalimumab Pluma de HUMIRA 40 mg/0.8 ml, jeringa precargada de HUMIRA 40 mg/0.8 ml, jeringa precargada de HUMIRA 20 mg/0.4 ml, jeringa precargada de HUMIRA 10 mg/0.2 ml y vial de HUMIRA 40 mg/0.8 ml para uso institucional: Ingredientes inactivos: ácido cítrico monohidratado, fosfato disódico dihidratado, manitol, fosfato monosódico dihidratado, polisorbato 80, cloruro de sodio, citrato de sodio y agua para inyección. Se añade hidróxido de sodio según sea necesario para ajustar el pH. Pluma de HUMIRA 80 mg/0.8 ml, jeringa precargada de HUMIRA 80 mg/0.8 ml, pluma de HUMIRA 40 mg/0.4 ml, jeringa precargada de HUMIRA 40 mg/0.4 ml, jeringa precargada de HUMIRA 20 mg/0.2 ml y jeringa precargada de HUMIRA 10 mg/0.1 ml: Ingredientes inactivos: manitol, polisorbato 80 y agua para inyección. Fabricado por: AbbVie Inc., North Chicago, IL 60064, EE. UU. Fabricado para: Cordavis Trading Limited, Dublín, Irlanda |
||
| Esta Guía del Medicamento ha sido aprobada por la Administración de Alimentos y Medicamentos de los Estados Unidos. 20082821 |
Revisado: 11/2023 | |
INSTRUCCIONES DE USO
| INSTRUCCIONES DE USO HUMIRA® (Hu-MARE-ah) (adalimumab) 40 mg/0.4 mL Pluma de dosis única |
|
Antes de inyectar: Su proveedor de atención médica debe mostrarle cómo usar HUMIRA antes de que lo use por primera vez. Llame a su proveedor de atención médica o al 1-800-4HUMIRA (1-800-448-6472) si necesita ayuda. |
|
| Información importante que necesita saber antes de inyectar HUMIRA | |
| No use la pluma y llame a su proveedor de atención médica o farmacéutico si: | |
|
|
| Mantenga las tapas puestas hasta justo antes de su inyección. ¿Cómo debo almacenar HUMIRA? |
|
|
|
|
Mantenga HUMIRA, los suministros de inyección y todos los demás medicamentos fuera del alcance de los niños. Lea las instrucciones en todas las páginas antes de usar la pluma de HUMIRA |
|
 |
Saque HUMIRA del refrigerador. Deje HUMIRA a temperatura ambiente durante 15 a 30 minutos antes de inyectar.
|
 |
Verifique la fecha de vencimiento en la etiqueta de la pluma. No use la pluma si la fecha de vencimiento ha pasado. Coloque lo siguiente en una superficie limpia y plana:
Lave y seque sus manos. |
 |
Elija un sitio de inyección:
Limpie el sitio de inyección en movimientos circulares con la toallita con alcohol.
|
 |
Sostenga la pluma con la Tapa Gris n.º 1 hacia arriba. Verifique la ventana.
|
 |
Retire la Tapa Gris n.º 1 hacia arriba. Tire la tapa.
Retire la Tapa de color ciruela n.º 2 hacia arriba. |
 |
Apriete la piel en el lugar de la inyección para formar un área elevada y manténgala firmemente hasta que se complete la inyección. Apunte la flecha blanca hacia el lugar de la inyección. Coloque la funda blanca de la aguja en línea recta (ángulo de 90°) contra el lugar de la inyección. Sostenga la pluma para que pueda ver la ventana de inspección. No presione el botón activador color ciruela hasta que esté listo para inyectar. |
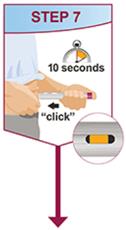 |
Es importante que empuje firmemente la pluma hacia abajo hasta que toque el lugar de la inyección antes de comenzar la inyección. Siga presionando hacia abajo para evitar que la pluma se mueva de la piel durante la inyección. Presione el botón activador color ciruela y cuente lentamente hasta 10 segundos.
|
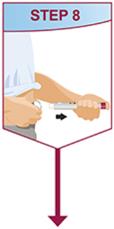 |
Cuando se complete la inyección, retire lentamente la pluma de la piel. La funda blanca de la aguja cubrirá la punta de la aguja.
Si hay más de unas pocas gotas de líquido en el lugar de la inyección, llame al 1-800-4HUMIRA (1-800-448-6472) para obtener ayuda.
|
 |
¿Cómo debo desechar la pluma de HUMIRA usada?
|
| Las tapas de la pluma, la toallita con alcohol, la bola de algodón o la gasa, la bandeja de dosis y el envase pueden tirarse a la basura doméstica. | |
| Preguntas sobre el uso de la pluma de HUMIRA
¿Qué ocurre si no he recibido formación presencial de un profesional sanitario?
¿Cómo sé cuándo se ha completado la inyección?
¿Qué debo hacer si hay más de unas pocas gotas de líquido en el lugar de la inyección?
¿Qué ocurre si no tengo un contenedor de eliminación de objetos punzocortantes aprobado por la FDA o un contenedor doméstico adecuado?
|
|
| Mantenga siempre la pluma y el contenedor de objetos punzocortantes fuera del alcance de los niños. | |
 |
Lleve un registro de las fechas y los lugares de las inyecciones. Para recordar cuándo debe tomar HUMIRA, marque su calendario con antelación. |
 |
|
| Este Instructions for Use ha sido aprobado por la U.S. Food and Drug Administration. | Revised 11/2023 |
| Manufactured by AbbVie Inc. North Chicago, IL 60064 U.S.A. US License Number 1889 20082820 Manufactured for: Cordavis Trading Limited, Dublin, Ireland |
|
INSTRUCCIONES DE USO
| INSTRUCCIONES DE USO | |
| HUMIRA® (Hu-MARE-ah) (adalimumab) Paquetes que contienen 80 mg/0.8 mL Pluma de dosis única |
|
| Antes de la inyección: Su proveedor de atención médica debe mostrarle cómo usar HUMIRA antes de que lo use por primera vez. Llame a su proveedor de atención médica o al 1-800-4HUMIRA (1-800-448-6472) si necesita ayuda. |
|
| Información importante que necesita saber antes de inyectar HUMIRA | |
| No use la pluma y llame a su proveedor de atención médica o farmacéutico si: | |
|
|
| Mantenga las tapas puestas hasta justo antes de su inyección.
¿Cómo debo almacenar HUMIRA? |
|
|
|
|
Mantenga HUMIRA, los suministros de inyección y todos los demás medicamentos fuera del alcance de los niños. Lea las instrucciones en todas las páginas antes de usar la pluma de HUMIRA |
|
 |
Saque HUMIRA del refrigerador. Deje HUMIRA a temperatura ambiente durante 15 a 30 minutos antes de inyectar.
|
|
Verifique la fecha de vencimiento en la etiqueta de la pluma. No use la pluma si la fecha de vencimiento ha pasado. Coloque lo siguiente en una superficie limpia y plana:
Lave y seque sus manos. |
|
Elija un sitio de inyección:
Limpie el sitio de inyección en movimientos circulares con la toallita con alcohol.
|
 |
Sostenga la pluma con la Tapa Gris n.º 1 hacia arriba. Verifique la ventana.
|
|
Retire la Tapa Gris n.º 1 hacia arriba. Tire la tapa.
Retire la Tapa de color ciruela n.º 2 hacia arriba. |
 |
Apriete la piel en el lugar de la inyección para formar un área levantada y sujétela firmemente hasta que se complete la inyección. Apunte la flecha blanca hacia el lugar de la inyección. Coloque la funda blanca de la aguja en línea recta (ángulo de 90°) contra el lugar de la inyección. Sostenga la pluma de modo que pueda ver la ventana de inspección. No presione el botón activador color ciruela hasta que esté listo para inyectar. |
 |
Es importante que empuje la pluma firmemente hacia abajo hasta que toque el lugar de la inyección antes de comenzar la inyección. Presione el botón activador color ciruela y cuente lentamente hasta 15 segundos.
|
|
Cuando se complete la inyección, retire lentamente la pluma de la piel. La funda blanca de la aguja cubrirá la punta de la aguja.
Si hay más de unas pocas gotas de líquido en el lugar de la inyección, llame al 1-800-4HUMIRA (1-800-448-6472) para obtener ayuda.
|
|
¿Cómo debo desechar la pluma de HUMIRA usada?
|
| Las tapas de la pluma, la toallita con alcohol, la bolita de algodón o la gasa, la bandeja de dosis y el embalaje pueden tirarse a la basura doméstica. | |
| Preguntas sobre el uso de la pluma de HUMIRA
¿Qué sucede si no he recibido formación presencial de un profesional sanitario?
¿Cómo sé cuándo se ha completado la inyección?
¿Qué debo hacer si hay más de unas pocas gotas de líquido en el lugar de la inyección?
¿Qué sucede si no tengo un contenedor de desechos punzocortantes aprobado por la FDA ni un contenedor doméstico adecuado?
|
|
| Mantenga siempre la pluma y el contenedor de desechos punzocortantes fuera del alcance de los niños. | |
|
Lleve un registro de las fechas y los lugares de las inyecciones. Para recordar cuándo debe tomar HUMIRA, marque su calendario con antelación. |
 |
|
| Estas instrucciones de uso han sido aprobadas por la Administración de Alimentos y Medicamentos de los Estados Unidos. | Revisado en 11/2023 |
| Fabricado por AbbVie Inc. North Chicago, IL 60064 U.S.A. Número de licencia de EE. UU. 1889 20081409 Fabricado para: Cordavis Trading Limited, Dublín, Irlanda |
|
INSTRUCCIONES DE USO
| INSTRUCCIONES DE USO | |
|
HUMIRA® (Hu-MARE-ah) (adalimumab) 80 mg/0.8 mL, 40 mg/0.4 mL, 20 mg/0.2 mL y 10 mg/0.1 mL Jeringa precargada de dosis única |
|
Antes de la inyección: Su proveedor de atención médica debe mostrarle cómo usar HUMIRA antes de que lo use por primera vez. Llame a su proveedor de atención médica o al 1-800-4HUMIRA (1-800-448-6472) si necesita ayuda. |
|
| Información importante que debe saber antes de la inyección de HUMIRA | |
| No use la jeringa precargada y llame a su proveedor de atención médica o farmacéutico si: | |
|
|
| Mantenga la cubierta de la aguja puesta hasta justo antes de su inyección. ¿Cómo debo almacenar HUMIRA? |
|
|
|
|
Mantenga HUMIRA, los suministros de inyección y todos los demás medicamentos fuera del alcance de los niños. Lea las instrucciones en todas las páginas antes de usar la jeringa precargada de dosis única de HUMIRA |
|
|
Saque HUMIRA del refrigerador. Deje HUMIRA a temperatura ambiente durante 15 a 30 minutos antes de la inyección.
|
 |
Verifique la fecha de vencimiento en la etiqueta de la jeringa precargada. No use la jeringa precargada si la fecha de vencimiento ha pasado. Coloque lo siguiente en una superficie limpia y plana:
Lave y seque sus manos. |
 |
Elija un sitio de inyección:
Limpie el sitio de inyección en movimientos circulares con la toallita con alcohol.
|
 |
Sostenga la jeringa precargada con una mano. Retire suavemente la cubierta de la aguja en línea recta con la otra mano.
|
 |
Sostenga la jeringa precargada con la aguja hacia arriba.
|
 |
Sostenga el cuerpo de la jeringa precargada con una mano entre el pulgar y los dedos índice. Sostenga la jeringa precargada en su mano como un lápiz. No tire hacia atrás del émbolo en ningún momento. |
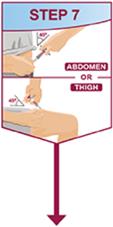 |
Apriete suavemente el área de la piel limpia en el lugar de la inyección con la otra mano. Mantenga la piel firme. Inserte la aguja en la piel en un ángulo de aproximadamente 45 grados con un movimiento rápido y similar a una flecha.
Empuje lentamente el émbolo hasta el final hasta que se inyecte todo el líquido y la jeringa precargada esté vacía. |
 |
Cuando se complete la inyección, retire lentamente la aguja de la piel manteniendo la jeringa precargada en el mismo ángulo. Después de completar la inyección, coloque una bola de algodón o una gasa en la piel del lugar de la inyección.
|
 |
¿Cómo debo desechar la jeringa precargada HUMIRA usada?
|
| La cubierta de la aguja, la toallita con alcohol, la bola de algodón o la gasa, la bandeja de dosis y el embalaje se pueden colocar en la basura de su hogar. | |
|
Preguntas sobre el uso de la jeringa precargada de dosis única HUMIRA ¿Qué pasa si no he recibido capacitación en persona de un profesional de la salud?
¿Qué pasa si no tengo un contenedor de eliminación de objetos punzantes aprobado por la FDA o un contenedor doméstico adecuado?
|
|
| Siempre mantenga la jeringa precargada y el contenedor de eliminación de objetos punzantes fuera del alcance de los niños. | |
|
Lleve un registro de las fechas y ubicaciones de sus inyecciones. Para ayudarlo a recordar cuándo tomar HUMIRA, marque su calendario con anticipación. |
 |
|
| Estas Instrucciones de uso han sido aprobadas por la Administración de Alimentos y Medicamentos de los Estados Unidos. | Revisado 11/2023 |
| Fabricado por AbbVie Inc. North Chicago, IL 60064 EE. UU. Número de licencia de EE. UU. 1889 20082850 Fabricado para: Cordavis Trading Limited, Dublín, Irlanda |
|
PANEL DE VISUALIZACIÓN PRINCIPAL
Panel de visualización principal
NDC: 83457-554-02
2 PLUMAS PRELLENAS DE DOSIS ÚNICA
HUMIRA® PEN
adalimumab
40 mg/0.4 mL
SOLO PARA USO SUBCUTÁNEO
AGUJA DE 29 GAUGE
ATENCIÓN FARMACÉUTICO: Cada paciente debe recibir la Guía de medicamentos adjunta.
La caja completa debe dispensarse como una unidad.
Devolver a la farmacia si el sello de la bandeja de dosis está roto o falta.
ESTA CAJA CONTIENE:
- 2 bandejas de dosis (cada una contiene 1 pluma prellenada de dosis única con aguja fija de ½ pulgada de calibre 29)
- 2 preparaciones de alcohol • 1 Guía de medicamentos • 1 folleto informativo • 1 Instrucciones de uso
Fabricado para:
cordavis™
HUMIRA.COM
Solo con receta médica
abbvie

PANEL PRINCIPAL DE EXHIBICIÓN
NDC: 83457-124-02
2 PLUMAS PRELLENADAS DE DOSIS ÚNICA
HUMIRA® PEN
adalimumab
80 mg/0.8 mL
PARA USO SUBCUTÁNEO ÚNICAMENTE
AGUJA DE CALIBRE 29
ATENCIÓN FARMACÉUTICO: Cada paciente debe recibir la Guía del Medicamento adjunta.
La cubierta de la aguja para la jeringa no está hecha con látex de caucho natural.
Toda la caja debe dispensarse como una unidad.
Devuelva a la farmacia si el sello de la bandeja de la dosis está roto o falta.
ESTA CAJA CONTIENE:
- 2 bandejas de dosis (cada una con 1 pluma precargada de dosis única con aguja fija de calibre 29 y ½ pulgada de longitud)
- 2 preparaciones de alcohol • 1 Guía del Medicamento • 1 prospecto • 1 Instrucciones de Uso
Fabricado para:
cordavis™
HUMIRA.COM
Rx only
abbvie

PANEL DE VISUALIZACIÓN PRINCIPAL
NDC: 83457-817-02
2 JERINGAS PRELLENAS DE DOSIS ÚNICA
HUMIRA®
adalimumab
10 mg/0.1 mL
SOLO PARA USO SUBCUTÁNEO
AGUJA DE 29 GAUGE
ATENCIÓN FARMACÉUTICO: Cada paciente debe recibir la Guía del Medicamento adjunta.
La cubierta de la aguja para la jeringa no está hecha con látex de caucho natural.
Toda la caja debe dispensarse como una unidad.
Devolver a la farmacia si el sello de la bandeja de dosis está roto o falta.
ESTA CAJA CONTIENE:
- 2 bandejas de dosis (cada una contiene 1 pluma prellenada de dosis única con aguja fija de 29 gauge ½ pulgada de longitud)
- 2 preparaciones de alcohol • 1 Guía del Medicamento • 1 folleto informativo
- 1 Instrucciones de uso
Fabricado para:
cordavis™
HUMIRA.COM
Solo con receta médica
abbvie

PANEL DE VISUALIZACIÓN PRINCIPAL
NDC: 83457-616-02
2 JERINGAS PRELLENAS DE DOSIS ÚNICA
HUMIRA®
adalimumab
20 mg/0.2 mL
SOLO PARA USO SUBCUTÁNEO
AGUJA DE 29 GAUGE
ATENCIÓN FARMACÉUTICO: Cada paciente debe recibir la Guía del Medicamento adjunta.
La cubierta de la aguja para la jeringa no está hecha con látex de caucho natural.
Todo el cartón debe dispensarse como una unidad.
Devuelva a la farmacia si el sello de la bandeja de dosis está roto o falta.
ESTE CARTÓN CONTIENE:
- 2 bandejas de dosis (cada una contiene 1 pluma prellenada de dosis única con aguja fija de 29 gauge ½ pulgada de longitud)
- 2 preparaciones de alcohol • 1 Guía del Medicamento • 1 folleto de información
- 1 Instrucciones de uso
Fabricado para:
cordavis™
HUMIRA.COM
Solo con receta médica
abbvie

PANEL DE VISUALIZACIÓN PRINCIPAL
NDC: 83457-243-02
2 JERINGAS PRELLENAS DE DOSIS ÚNICA
HUMIRA®
adalimumab
40 mg/0.4 mL
SOLO PARA USO SUBCUTÁNEO
AGUJAS DE 29 GAUGE
ATENCIÓN FARMACÉUTICO: Cada paciente debe recibir la Guía de Medicamentos adjunta.
La cubierta de la aguja para la jeringa no está hecha con látex de caucho natural
ESTA CAJA CONTIENE:
- 2 bandejas de dosis (cada una contiene 1 pluma prellenada de dosis única con aguja fija de 29 gauge ½ pulgada de longitud)
- 2 preparaciones de alcohol • 1 Guía de medicamentos • 1 folleto informativo
- 1 Instrucciones de uso
Fabricado para:
cordavis™
HUMIRA.COM
Solo con receta médica
abbvie
