
Fabricante de medicamentos: Novartis Pharmaceuticals Corporation (Updated: 2024-07-22)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
Consulte la información de prescripción completa de GILENYA.
GILENYA (fingolimod) cápsulas, para administración oral
Aprobación inicial en EE. UU.: 2010
CAMBIOS IMPORTANTES RECIENTES
INDICACIONES Y USO
GILENYA es un modulador del receptor de esfingosina 1-fosfato indicado para el tratamiento de las formas recurrentes de esclerosis múltiple (EM), que incluyen el síndrome clínicamente aislado, la enfermedad recurrente-remitente y la enfermedad secundaria progresiva activa, en pacientes de 10 años de edad y mayores. (1)
DOSIFICACIÓN Y ADMINISTRACIÓN
- Se requieren evaluaciones antes de iniciar GILENYA. (2.1)
- Dosis recomendada para adultos y pacientes pediátricos (de 10 años de edad y mayores) que pesan más de 40 kg: 0.5 mg por vía oral una vez al día, con o sin alimentos. (2.2, 2.3)
- Dosis recomendada para pacientes pediátricos (de 10 años de edad y mayores) que pesan 40 kg o menos: 0.25 mg por vía oral una vez al día, con o sin alimentos. (2.2, 2.3)
- Monitoreo de la primera dosis (incluido el reinicio después de una interrupción superior a 14 días y aumentos de dosis):
- Observar a todos los pacientes para detectar bradicardia durante al menos 6 horas; controlar el pulso y la presión arterial cada hora. Se requieren electrocardiogramas (ECG) antes de la administración y al final del período de observación. (2.4)
- Monitorear hasta la resolución si la frecuencia cardíaca es < 45 latidos por minuto (lpm) en adultos, < 55 lpm en pacientes de 12 años de edad y mayores, o < 60 lpm en pacientes pediátricos de 10 a menos de 12 años de edad, bloqueo auriculoventricular (AV), o si la frecuencia cardíaca más baja después de la dosis es al final del período de observación. (2.4)
- Monitorear la bradicardia sintomática con ECG hasta que se resuelva. Continuar durante la noche si se requiere intervención; repetir el monitoreo de la primera dosis para la segunda dosis. (2.4)
- Observar a los pacientes durante la noche si tienen un mayor riesgo de bradicardia sintomática, bloqueo cardíaco, intervalo QTc prolongado o si toman medicamentos con riesgo conocido de torsades de pointes. (2.4, 7.1)
CONTRAINDICACIONES
- Infarto de miocardio reciente, angina inestable, accidente cerebrovascular, ataque isquémico transitorio (AIT), insuficiencia cardíaca descompensada con hospitalización o insuficiencia cardíaca de clase III/IV. (4)
- Antecedentes de bloqueo AV de segundo grado Mobitz tipo II o de tercer grado o síndrome del seno enfermo, a menos que el paciente tenga un marcapasos. (4)
- Intervalo QTc basal ≥ 500 mseg. (4)
- Arritmias cardíacas que requieren tratamiento antiarrítmico con medicamentos antiarrítmicos de clase Ia o clase III. (4)
- Hipersensibilidad a fingolimod o a sus excipientes. (4)
ADVERTENCIAS Y PRECAUCIONES
- Infecciones: GILENYA puede aumentar el riesgo. Obtenga un hemograma completo (CBC) antes de iniciar el tratamiento. Monitoree la infección durante el tratamiento y durante 2 meses después de la interrupción. No comience en pacientes con infecciones activas. (5.2)
- Leucoencefalopatía multifocal progresiva (LMP): Suspenda GILENYA ante el primer signo o síntoma que sugiera LMP. (5.3)
- Edema macular: Aumenta el riesgo de edema macular. Obtenga una evaluación inicial del fondo de ojo, incluida la mácula, cerca del inicio del tratamiento con GILENYA. Realice una evaluación del fondo de ojo, incluida la mácula, de 3 a 4 meses después del inicio del tratamiento, periódicamente mientras esté en tratamiento y en cualquier momento que haya un cambio en la visión. Considere suspender GILENYA si se desarrolla edema macular. La diabetes mellitus y la uveítis aumentan el riesgo. (5.4)
- Daño hepático: Obtenga los resultados de las enzimas hepáticas antes del inicio y periódicamente durante el tratamiento. Monitoree de cerca a los pacientes con insuficiencia hepática grave. Suspenda si hay evidencia de daño hepático sin otra causa. (5.5, 8.6, 12.3)
- Síndrome de encefalopatía posterior reversible (SEPR): Si se sospecha, suspenda GILENYA. (5.6)
- Efectos respiratorios: Evaluar cuando esté clínicamente indicado. (5.7)
- Riesgo fetal: Puede causar daño fetal. Advierta a las mujeres en edad fértil sobre el riesgo potencial para el feto y que usen un método anticonceptivo eficaz durante el tratamiento y durante 2 meses después de suspender GILENYA. (5.8, 8.1, 8.3)
- Aumento grave de la discapacidad después de suspender GILENYA: Monitoree el desarrollo de un aumento grave de la discapacidad después de la interrupción y comience el tratamiento adecuado según sea necesario. (5.9)
- EM tumefactiva: Considérelo cuando ocurra una recaída grave de la EM durante el tratamiento o después de la interrupción. Obtenga imágenes y comience el tratamiento según sea necesario. (5.10)
- Aumento de la presión arterial (PA): Monitoree la PA durante el tratamiento. (5.11)
- Tumores malignos: Se recomienda un examen de la piel antes o poco después del inicio del tratamiento y periódicamente a partir de entonces. Las lesiones cutáneas sospechosas deben ser evaluadas. (5.12)
REACCIONES ADVERSAS
Reacciones adversas más comunes (incidencia ≥ 10 % y mayor que placebo): dolor de cabeza, elevación de las transaminasas hepáticas, diarrea, tos, influenza, sinusitis, dolor de espalda, dolor abdominal y dolor en las extremidades. (6.1)
Para informar de SOSPECHAS DE REACCIONES ADVERSAS, póngase en contacto con Novartis Pharmaceuticals Corporation al 1-888-669-6682 o con la FDA al 1-800-FDA-1088 o www.fda.gov/medwatch.
INTERACCIONES MEDICAMENTOSAS
Consulte la sección 17 para obtener INFORMACIÓN DE ASESORAMIENTO AL PACIENTE y la Guía del Medicamento.
Revisado: 6/2024
Tabla de Contenido
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDO*
1 INDICACIONES Y USO
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Evaluación previa al inicio de GILENYA
2.2 Instrucciones importantes de administración
2.3 Dosis recomendada
2.4 Monitoreo de la primera dosis
2.5 Monitoreo después de la reiniciación de la terapia tras la interrupción
3 FORMAS Y FUERZAS DE DOSIFICACIÓN
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Bradiarritmia y bloqueos auriculoventriculares
5.2 Infecciones
5.3 Leucoencefalopatía multifocal progresiva
5.4 Edema macular
5.5 Lesión hepática
5.6 Síndrome de encefalopatía posterior reversible
5.7 Efectos respiratorios
5.8 Riesgo fetal
5.9 Aumento severo de la discapacidad después de suspender GILENYA
5.10 Esclerosis múltiple tumefactiva
5.11 Aumento de la presión arterial
5.12 Neoplasias
5.13 Efectos del sistema inmunitario tras la interrupción de GILENYA
5.14 Reacciones de hipersensibilidad
6 REACCIONES ADVERSAS
6.1 Experiencia en ensayos clínicos
6.2 Experiencia postcomercialización
7 INTERACCIONES MEDICAMENTOSAS
7.1 Fármacos que prolongan el QT
7.2 Ketoconazol
7.3 Vacunas
7.4 Terapias antineoplásicas, inmunosupresoras o inmunomoduladoras
7.5 Fármacos que disminuyen la frecuencia cardíaca o la conducción auriculoventricular (por ejemplo, betabloqueantes o diltiazem)
7.6 Interacción con pruebas de laboratorio
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.3 Mujeres y hombres en edad fértil
8.4 Uso pediátrico
8.5 Uso geriátrico
8.6 Insuficiencia hepática
8.7 Insuficiencia renal
10 SOBREDOSIS
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
12.2 Farmacodinamia
12.3 Farmacocinética
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, mutagénesis, deterioro de la fertilidad
13.2 Toxicología y/o farmacología animal
14 ESTUDIOS CLÍNICOS
14.1 Adultos
14.2 Pacientes pediátricos (de 10 a menos de 18 años de edad)
16 FORMA DE SUMINISTRO/ALMACENAMIENTO Y MANEJO
16.1 Forma de suministro
16.2 Almacenamiento y manejo
17 INFORMACIÓN PARA EL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información completa de prescripción no están enumeradas.
1 INDICACIONES Y USO
GILENYA está indicado para el tratamiento de las formas recurrentes de esclerosis múltiple (EM), que incluyen el síndrome clínicamente aislado, la enfermedad remitente-recurrente y la enfermedad progresiva secundaria activa, en pacientes de 10 años de edad o mayores.
2 DOSIS Y ADMINISTRACIÓN
2.1 Evaluación previa al inicio de GILENYA
Evaluación cardíaca
Obtenga una evaluación cardíaca en pacientes con ciertas condiciones preexistentes [ver Advertencias y precauciones (5.1)].
Antes de comenzar el tratamiento, determine si los pacientes están tomando medicamentos que podrían disminuir la frecuencia cardíaca o la conducción auriculoventricular (AV) [ver Dosis y administración (2.4), Interacciones medicamentosas (7.5)].
Recuento sanguíneo completo (CBC)
Revise los resultados de un CBC reciente [ver Advertencias y precauciones (5.2), Interacciones medicamentosas (7.6)].
Niveles de transaminasas séricas (ALT y AST) y bilirrubina total
Antes de comenzar el tratamiento con GILENYA (es decir, dentro de los 6 meses), obtenga los niveles de transaminasas séricas [alanina aminotransferasa (ALT) y aspartato aminotransferasa (AST)] y bilirrubina total [ver Advertencias y precauciones (5.5)].
Evaluación oftalmológica
Obtenga una evaluación basal del fondo de ojo, incluida la mácula, cerca del inicio del tratamiento con GILENYA [ver Advertencias y precauciones (5.4)].
Examen de la piel
Obtenga un examen de la piel basal antes o poco después del inicio de GILENYA. Si se observa una lesión cutánea sospechosa, debe evaluarse de inmediato [ver Advertencias y precauciones (5.12)].
Medicamentos previos
Si los pacientes están tomando terapias antineoplásicas, inmunosupresoras o inmunomoduladoras, o si hay antecedentes de uso previo de estos medicamentos, considere posibles efectos inmunosupresores aditivos no deseados antes de iniciar el tratamiento con GILENYA [ver Advertencias y precauciones (5.2), Interacciones medicamentosas (7.4)].
Vacunaciones
Analice a los pacientes para detectar anticuerpos contra el virus de la varicela zóster (VZV) antes de iniciar GILENYA; se recomienda la vacunación contra el VZV en pacientes seronegativos antes de comenzar el tratamiento con GILENYA [ver Advertencias y precauciones (5.2)]. Se recomienda que los pacientes pediátricos, si es posible, completen todas las inmunizaciones de acuerdo con las pautas de inmunización actuales antes de iniciar la terapia con GILENYA.
2.2 Instrucciones importantes de administración
Los pacientes que inician GILENYA y aquellos que reinician el tratamiento después de la interrupción durante más de 14 días requieren monitoreo de la primera dosis. Este monitoreo también se recomienda cuando se aumenta la dosis en pacientes pediátricos [ver Dosis y administración (2.4, 2.5)].
GILENYA se puede tomar con o sin alimentos.
2.3 Dosis recomendada
En adultos y pacientes pediátricos de 10 años de edad o más que pesan más de 40 kg, la dosis recomendada de GILENYA es de 0.5 mg por vía oral una vez al día.
En pacientes pediátricos de 10 años de edad o más que pesan menos de o igual a 40 kg, la dosis recomendada de GILENYA es de 0.25 mg por vía oral una vez al día.
Las dosis de fingolimod superiores a 0.5 mg se asocian con una mayor incidencia de reacciones adversas sin beneficio adicional.
2.4 Monitoreo de la primera dosis
El inicio del tratamiento con GILENYA da como resultado una disminución de la frecuencia cardíaca, para la cual se recomienda el monitoreo [ver Advertencias y precauciones (5.1), Farmacología clínica (12.2)]. Antes de la dosificación y al final del período de observación, obtenga un electrocardiograma (ECG) en todos los pacientes.
Monitoreo de las primeras 6 horas
Administre la primera dosis de GILENYA en un entorno en el que estén disponibles los recursos para manejar adecuadamente la bradicardia sintomática. Monitoree a todos los pacientes durante 6 horas después de la primera dosis para detectar signos y síntomas de bradicardia con medición horaria del pulso y la presión arterial.
Monitoreo adicional después del monitoreo de 6 horas
Continúe monitoreando hasta que la anormalidad se resuelva si alguna de las siguientes está presente (incluso en ausencia de síntomas) después de 6 horas:
- la frecuencia cardíaca 6 horas después de la dosis es inferior a 45 latidos por minuto (lpm) en adultos, inferior a 55 lpm en pacientes pediátricos de 12 años de edad o más, o inferior a 60 lpm en pacientes pediátricos de 10 u 11 años de edad;
- la frecuencia cardíaca 6 horas después de la dosis está en el valor más bajo después de la dosis, lo que sugiere que el efecto farmacodinámico máximo en el corazón puede no haberse producido;
- el ECG 6 horas después de la dosis muestra un nuevo bloqueo auriculoventricular (AV) de segundo grado o superior.
Si se produce bradicardia sintomática después de la dosis, inicie la administración adecuada, comience el monitoreo continuo del ECG y continúe monitoreando hasta que los síntomas se hayan resuelto si no se requiere tratamiento farmacológico. Si se requiere tratamiento farmacológico, continúe monitoreando durante la noche y repita el monitoreo de 6 horas después de la segunda dosis.
Monitoreo nocturno
Se debe instituir el monitoreo continuo del ECG durante la noche en una instalación médica:
- en pacientes que requieren intervención farmacológica para la bradicardia sintomática. En estos pacientes, la estrategia de monitoreo de la primera dosis debe repetirse después de la segunda dosis de GILENYA;
- en pacientes con algunas condiciones cardíacas y cerebrovasculares preexistentes [ver Advertencias y precauciones (5.1)];
- en pacientes con un intervalo QTc prolongado antes de la dosificación o durante la observación de 6 horas, o con riesgo adicional de prolongación del QT, o en terapia concomitante con medicamentos que prolongan el QT con un riesgo conocido de torsades de pointes [ver Advertencias y precauciones (5.1), Interacciones medicamentosas (7.1)];
- en pacientes que reciben terapia concomitante con medicamentos que disminuyen la frecuencia cardíaca o la conducción AV [ver Interacciones medicamentosas (7.5)].
2.5 Monitoreo después de la reiniciación de la terapia tras la interrupción
Al reiniciar GILENYA después de la interrupción durante más de 14 días después del primer mes de tratamiento, realice el monitoreo de la primera dosis, porque los efectos sobre la frecuencia cardíaca y la conducción AV pueden reaparecer al reintroducir el tratamiento con GILENYA [ver Dosis y administración (2.4)]. Las mismas precauciones (monitoreo de la primera dosis) que para la dosificación inicial son aplicables. Dentro de las primeras 2 semanas de tratamiento, se recomiendan los procedimientos de la primera dosis después de una interrupción de 1 día o más; durante las semanas 3 y 4 de tratamiento, se recomiendan los procedimientos de la primera dosis después de una interrupción del tratamiento de más de 7 días.
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
GILENYA está disponible como:
- Cápsulas duras de 0.25 mg con cuerpo y tapa opacos color marfil, con la impresión radial negra “FTY 0.25 mg” en la tapa y una banda radial negra en el cuerpo de la cápsula.
- Cápsulas duras de 0.5 mg con cuerpo opaco blanco y tapa amarillo brillante con la impresión “FTY 0.5 mg” en la tapa y 2 bandas radiales impresas en el cuerpo de la cápsula con tinta amarilla.
4 CONTRAINDICACIONES
GILENYA está contraindicado en pacientes que han:
- en los últimos 6 meses experimentado infarto de miocardio, angina inestable, accidente cerebrovascular, ataque isquémico transitorio (AIT), insuficiencia cardíaca descompensada que requiera hospitalización o insuficiencia cardíaca de clase III/IV
- antecedentes o presencia de bloqueo AV de segundo grado tipo Mobitz II o de tercer grado o síndrome de seno enfermo, a menos que el paciente tenga un marcapasos funcional [ver Advertencias y precauciones (5.1)]
- un intervalo QTc basal ≥ 500 mseg
- arritmias cardíacas que requieren tratamiento antiarrítmico con fármacos antiarrítmicos de clase Ia o clase III
- tenido una reacción de hipersensibilidad a fingolimod o a cualquiera de los excipientes de GILENYA. Las reacciones observadas incluyen erupción cutánea, urticaria y angioedema al inicio del tratamiento [ver Advertencias y precauciones (5.14)].
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Bradicardia y Bloqueos Auriculoventriculares
Debido al riesgo de bradicardia y bloqueos AV, los pacientes deben ser monitoreados durante el inicio del tratamiento con GILENYA [ver Dosis y Administración (2.4)].
Reducción de la Frecuencia Cardíaca
Después de la primera dosis de GILENYA, la disminución de la frecuencia cardíaca comienza dentro de una hora. En el Día 1, la disminución máxima de la frecuencia cardíaca generalmente ocurre dentro de las 6 horas y se recupera, aunque no a los niveles basales, de 8 a 10 horas después de la dosis. Debido a la variación diurna fisiológica, hay un segundo período de disminución de la frecuencia cardíaca dentro de las 24 horas posteriores a la primera dosis. En algunos pacientes, la disminución de la frecuencia cardíaca durante el segundo período es más pronunciada que la disminución observada en las primeras 6 horas. Las frecuencias cardíacas por debajo de 40 lpm en adultos y por debajo de 50 lpm en pacientes pediátricos ocurrieron raramente. En ensayos clínicos controlados en pacientes adultos, se informaron reacciones adversas de bradicardia sintomática después de la primera dosis en el 0.6% de los pacientes que recibieron GILENYA 0.5 mg y en el 0.1% de los pacientes que recibieron placebo. Los pacientes que experimentaron bradicardia generalmente fueron asintomáticos, pero algunos pacientes experimentaron hipotensión, mareos, fatiga, palpitaciones y/o dolor en el pecho que generalmente se resolvieron dentro de las primeras 24 horas de tratamiento.
Los pacientes con algunas afecciones preexistentes (por ejemplo, enfermedad cardíaca isquémica, antecedentes de infarto de miocardio, insuficiencia cardíaca congestiva, antecedentes de paro cardíaco, enfermedad cerebrovascular, hipertensión no controlada, antecedentes de bradicardia sintomática, antecedentes de síncope recurrente, apnea del sueño grave no tratada, bloqueo AV, bloqueo sinoauricular) pueden tolerar mal la bradicardia inducida por GILENYA o experimentar trastornos del ritmo graves después de la primera dosis de GILENYA. Antes del tratamiento con GILENYA, estos pacientes deben tener una evaluación cardíaca realizada por un médico capacitado adecuadamente para realizar dicha evaluación, y si se tratan con GILENYA, deben ser monitoreados durante la noche con ECG continuo en un centro médico después de la primera dosis.
Dado que el inicio del tratamiento con GILENYA, los resultados muestran una disminución de la frecuencia cardíaca y pueden prolongar el intervalo QT, los pacientes con un intervalo QTc prolongado (> 450 mseg adultos y hombres pediátricos, > 470 mseg mujeres adultas o > 460 mseg mujeres pediátricas) antes de la dosificación o durante la observación de 6 horas, o con riesgo adicional de prolongación del QT (por ejemplo, hipokalemia, hipomagnesemia, síndrome de QT largo congénito) o en terapia concomitante con medicamentos que prolongan el QT con un riesgo conocido de torsades de pointes (por ejemplo, citalopram, clorpromazina, haloperidol, metadona, eritromicina) deben ser monitoreados durante la noche con ECG continuo en un centro médico.
Después de la segunda dosis, puede ocurrir una mayor disminución de la frecuencia cardíaca en comparación con la frecuencia cardíaca antes de la segunda dosis, pero este cambio es de menor magnitud que el observado después de la primera dosis. Con la dosificación continua, la frecuencia cardíaca vuelve a la línea de base dentro de 1 mes de tratamiento crónico. Los datos clínicos indican que los efectos de GILENYA sobre la frecuencia cardíaca son máximos después de la primera dosis, aunque los efectos más leves sobre la frecuencia cardíaca pueden persistir durante, en promedio, de 2 a 4 semanas después del inicio de la terapia, momento en el que la frecuencia cardíaca generalmente vuelve a la línea de base. Los médicos deben continuar estando atentos a los informes de los pacientes sobre los síntomas cardíacos.
Bloqueos Auriculoventriculares
El inicio del tratamiento con GILENYA ha provocado retrasos transitorios en la conducción AV. En ensayos clínicos controlados en pacientes adultos, el bloqueo AV de primer grado después de la primera dosis ocurrió en el 4.7% de los pacientes que recibieron GILENYA y en el 1.6% de los pacientes que recibieron placebo. En un estudio de 697 pacientes con datos disponibles de monitoreo Holter de 24 horas después de su primera dosis (N = 351 que recibieron GILENYA y N = 346 que recibieron placebo), los bloqueos AV de segundo grado (Tipos I de Mobitz [Wenckebach] o bloqueos AV 2:1) ocurrieron en el 4% (N = 14) de los pacientes que recibieron GILENYA y el 2% (N = 7) de los pacientes que recibieron placebo. De los 14 pacientes que recibieron GILENYA, 7 pacientes tuvieron bloqueo AV 2:1 (5 pacientes dentro de las primeras 6 horas después de la dosis y 2 pacientes después de 6 horas después de la dosis). Todos los bloqueos AV de segundo grado en placebo fueron del Tipo I de Mobitz y ocurrieron después de las primeras 12 horas después de la dosis. Las anomalías de la conducción fueron generalmente transitorias y asintomáticas, y se resolvieron dentro de las primeras 24 horas de tratamiento, pero ocasionalmente requirieron tratamiento con atropina o isoproterenol.
Experiencia Postcomercialización
En el entorno postcomercialización, se ha observado bloqueo AV de tercer grado y bloqueo AV con escape de unión durante el período de observación de 6 horas de la primera dosis con GILENYA. Se han producido eventos aislados de inicio tardío, incluida la asistolia transitoria y la muerte inexplicable, dentro de las 24 horas posteriores a la primera dosis. Estos eventos se vieron afectados por medicamentos concomitantes y/o enfermedades preexistentes, y la relación con GILENYA es incierta. También se informaron casos de síncope después de la primera dosis de GILENYA.
5.2 Infecciones
Riesgo de Infecciones
GILENYA causa una reducción dependiente de la dosis en el recuento de linfocitos periféricos al 20%–30% de los valores basales debido a la secuestración reversible de los linfocitos en los tejidos linfoides. Por lo tanto, GILENYA puede aumentar el riesgo de infecciones, algunas de naturaleza grave [ver Farmacología Clínica (12.2)]. Se han producido infecciones potencialmente mortales y fatales en asociación con GILENYA.
Antes de iniciar el tratamiento con GILENYA, debe estar disponible un CBC reciente (es decir, dentro de los 6 meses o después de la interrupción de la terapia previa). Considere suspender el tratamiento con GILENYA si un paciente desarrolla una infección grave y vuelva a evaluar los beneficios y los riesgos antes de reiniciar la terapia. Debido a que la eliminación de fingolimod después de la interrupción puede tardar hasta 2 meses, continúe monitoreando las infecciones durante todo este período. Indique a los pacientes que reciben GILENYA que informen los síntomas de infecciones a un médico. Los pacientes con infecciones agudas o crónicas activas no deben comenzar el tratamiento hasta que la(s) infección(es) se resuelva.
En los ensayos controlados con placebo de EM en pacientes adultos, la tasa general de infecciones (72%) con GILENYA fue similar al placebo. Sin embargo, la bronquitis, el herpes zóster, la influenza, la sinusitis y la neumonía fueron más comunes en los pacientes tratados con GILENYA. Las infecciones graves ocurrieron a una tasa del 2.3% en el grupo GILENYA versus el 1.6% en el grupo placebo.
En el entorno postcomercialización, se han notificado infecciones graves con patógenos oportunistas, incluidos virus (por ejemplo, virus de John Cunningham [JCV], virus del herpes simple 1 y 2, virus varicela zóster), hongos (por ejemplo, criptococos) y bacterias (por ejemplo, micobacterias atípicas) con GILENYA. Los pacientes con síntomas y signos compatibles con cualquiera de estas infecciones deben someterse a una evaluación diagnóstica inmediata y al tratamiento adecuado.
Infecciones por virus del herpes
En ensayos controlados con placebo en pacientes adultos, la tasa de infecciones herpéticas fue del 9% en pacientes que recibieron GILENYA 0.5 mg y del 7% en placebo.
Dos pacientes murieron por infecciones herpéticas durante los ensayos controlados. Una muerte se debió a herpes zóster primario diseminado y la otra a encefalitis por herpes simple. En ambos casos, los pacientes estaban tomando una dosis de 1.25 mg de fingolimod (superior a la dosis recomendada de 0.5 mg) y habían recibido terapia con corticosteroides de alta dosis para tratar posibles recaídas de EM.
Se han producido eventos graves y potencialmente mortales de varicela zóster diseminada e infecciones por herpes simple, incluidos casos de encefalitis e insuficiencia multiorgánica, con GILENYA en el entorno postcomercialización. Incluya las infecciones herpéticas diseminadas en el diagnóstico diferencial de los pacientes que reciben GILENYA y presentan una recaída de EM atípica o insuficiencia multiorgánica.
Se han notificado casos de sarcoma de Kaposi en el entorno postcomercialización. El sarcoma de Kaposi es un trastorno angioproliferativo que se asocia con la infección por el virus del herpes humano 8 (HHV-8). Los pacientes con síntomas o signos compatibles con el sarcoma de Kaposi deben ser derivados para una evaluación y manejo diagnóstico inmediato.
Infecciones criptocócicas
Se han notificado infecciones criptocócicas, incluidos casos de meningitis criptocócica fatal e infecciones criptocócicas diseminadas, con GILENYA en el entorno postcomercialización. Las infecciones criptocócicas generalmente han ocurrido después de aproximadamente 2 años de tratamiento con GILENYA, pero pueden ocurrir antes. Se desconoce la relación entre el riesgo de infección criptocócica y la duración del tratamiento. Los pacientes con síntomas y signos compatibles con una infección criptocócica deben someterse a una evaluación diagnóstica inmediata y al tratamiento.
Tratamiento previo y concomitante con terapias antineoplásicas, inmunosupresoras o inmunomoduladoras
En estudios clínicos, los pacientes que recibieron GILENYA no recibieron tratamiento concomitante con terapias antineoplásicas, inmunosupresoras no corticosteroides o inmunomoduladoras utilizadas para el tratamiento de la EM. Se esperaría que el uso concomitante de GILENYA con cualquiera de estas terapias, y también con corticosteroides, aumentara el riesgo de inmunosupresión [ver Interacciones medicamentosas (7.4)].
Al cambiar a GILENYA de medicamentos inmunomoduladores o inmunosupresores, considere la duración de sus efectos y su modo de acción para evitar efectos inmunosupresores aditivos no deseados.
Pruebas de anticuerpos contra el virus varicela zóster/vacunación
Los pacientes sin un historial confirmado por un profesional de la salud de varicela o sin documentación de un ciclo completo de vacunación contra el VZV deben someterse a pruebas de anticuerpos contra el VZV antes de iniciar GILENYA. Se recomienda la vacunación contra el VZV de los pacientes con anticuerpos negativos antes de comenzar el tratamiento con GILENYA, después de lo cual se debe posponer el inicio del tratamiento con GILENYA durante 1 mes para permitir que se produzca el efecto completo de la vacunación [ver Interacciones medicamentosas (7.3), Uso en poblaciones específicas (8.4)].
Infección por el virus del papiloma humano
Se han notificado infecciones por el virus del papiloma humano (VPH), incluidos papilomas, displasia, verrugas y cáncer relacionado con el VPH, en pacientes tratados con GILENYA en el entorno postcomercialización. Se debe considerar la vacunación contra el VPH antes de iniciar el tratamiento con GILENYA, teniendo en cuenta las recomendaciones de vacunación. Se recomienda la detección del cáncer, incluida la prueba de Papanicolaou (Pap), según el estándar de atención para los pacientes que utilizan una terapia inmunosupresora.
5.3 Leucoencefalopatía multifocal progresiva
Se han producido casos de leucoencefalopatía multifocal progresiva (LMP) en pacientes con EM que recibieron GILENYA en el entorno postcomercialización. La LMP es una infección viral oportunista del cerebro causada por el virus JC (JCV) que normalmente solo ocurre en pacientes inmunodeprimidos y que generalmente conduce a la muerte o discapacidad grave. La LMP se ha producido en pacientes que no habían sido tratados previamente con natalizumab, que tiene una asociación conocida con la LMP, no estaban tomando ningún otro medicamento inmunosupresor o inmunomodulador de forma concomitante y no tenían ninguna condición médica sistémica en curso que resultara en una función del sistema inmunitario comprometida. Una duración del tratamiento más larga aumenta el riesgo de LMP en pacientes tratados con GILENYA; la mayoría de los casos se han producido en pacientes tratados con GILENYA durante al menos 18 meses.
Al primer signo o síntoma sugestivo de LMP, suspenda GILENYA y realice una evaluación diagnóstica adecuada. Los síntomas típicos asociados con la LMP son diversos, progresan de días a semanas e incluyen debilidad progresiva en un lado del cuerpo o torpeza de las extremidades, alteración de la visión y cambios en el pensamiento, la memoria y la orientación que conducen a confusión y cambios de personalidad.
Las imágenes de resonancia magnética (IRM) pueden ser evidentes antes de los signos o síntomas clínicos. Se han notificado casos de LMP, diagnosticados en función de los hallazgos de IRM y la detección de ADN del JCV en el líquido cefalorraquídeo en ausencia de signos o síntomas clínicos específicos de la LMP, en pacientes tratados con medicamentos para la EM asociados con la LMP, incluido GILENYA. Muchos de estos pacientes posteriormente se volvieron sintomáticos con LMP. Por lo tanto, la monitorización con IRM para detectar signos que puedan ser compatibles con la LMP puede ser útil, y cualquier hallazgo sospechoso debe conducir a una investigación adicional para permitir un diagnóstico temprano de la LMP, si está presente. Se ha notificado una menor mortalidad y morbilidad relacionadas con la LMP después de la interrupción de otro medicamento para la EM asociado con la LMP en pacientes con LMP que inicialmente eran asintomáticos en comparación con los pacientes con LMP que tenían signos y síntomas clínicos característicos en el momento del diagnóstico. Se desconoce si estas diferencias se deben a la detección temprana y la interrupción del tratamiento de la EM o a las diferencias en la enfermedad en estos pacientes.
Si se confirma la PML, se debe interrumpir el tratamiento con GILENYA.
Se ha notificado el síndrome inflamatorio de reconstitución inmune (IRIS) en pacientes tratados con moduladores del receptor S1P, incluido GILENYA, que desarrollaron PML y posteriormente interrumpieron el tratamiento. El IRIS se presenta como un deterioro clínico en el estado del paciente que puede ser rápido, puede provocar complicaciones neurológicas graves o la muerte, y a menudo se asocia con cambios característicos en la resonancia magnética. El tiempo de aparición del IRIS en pacientes con PML fue generalmente dentro de unos pocos meses después de la interrupción del modulador del receptor S1P. Se debe realizar un seguimiento para detectar el desarrollo de IRIS y el tratamiento adecuado de la inflamación asociada.
5.4 Edema macular
Los moduladores del receptor S1P, incluido GILENYA, se han asociado con un mayor riesgo de edema macular. Obtenga una evaluación inicial del fondo de ojo, incluida la mácula, cerca del inicio del tratamiento con GILENYA. Realice un examen del fondo de ojo, incluida la mácula, de 3 a 4 meses después de comenzar el tratamiento, periódicamente mientras esté en terapia, y en cualquier momento en que haya un cambio en la visión.
Se produjo un aumento dependiente de la dosis en el riesgo de edema macular en el programa de desarrollo clínico de GILENYA.
En estudios de 2 años, doble ciego, controlados con placebo en pacientes adultos con esclerosis múltiple, el edema macular con o sin síntomas visuales ocurrió en el 1,5% de los pacientes (11/799) tratados con fingolimod 1,25 mg, el 0,5% de los pacientes (4/783) tratados con GILENYA 0,5 mg y el 0,4% de los pacientes (3/773) tratados con placebo. El edema macular ocurrió predominantemente durante los primeros 3 a 4 meses de terapia. Estos ensayos clínicos excluyeron a pacientes con diabetes mellitus, un factor de riesgo conocido para el edema macular (ver más abajo Edema macular en pacientes con antecedentes de uveítis o diabetes mellitus). Los síntomas del edema macular incluyeron visión borrosa y disminución de la agudeza visual. El examen oftalmológico de rutina detectó edema macular en algunos pacientes sin síntomas visuales. El edema macular generalmente se resolvió parcial o completamente con o sin tratamiento después de la interrupción del medicamento. Algunos pacientes tuvieron una pérdida residual de la agudeza visual incluso después de la resolución del edema macular. El edema macular también se ha notificado en pacientes que toman GILENYA en el entorno poscomercialización, generalmente dentro de los primeros 6 meses de tratamiento.
No se ha evaluado la continuación de GILENYA en pacientes que desarrollan edema macular. El edema macular durante un período prolongado (es decir, 6 meses) puede provocar una pérdida visual permanente. Considere interrumpir GILENYA si se desarrolla edema macular; esta decisión debe incluir una evaluación de los beneficios y riesgos potenciales para el paciente individual. No se ha evaluado el riesgo de recurrencia después de la reexposición.
Edema macular en pacientes con antecedentes de uveítis o diabetes mellitus
Los pacientes con antecedentes de uveítis y los pacientes con diabetes mellitus tienen un mayor riesgo de edema macular durante la terapia con GILENYA. En la experiencia combinada de ensayos clínicos en pacientes adultos con todas las dosis de fingolimod, la tasa de edema macular fue mayor en pacientes con EM con antecedentes de uveítis en comparación con aquellos sin antecedentes de uveítis (aproximadamente 20% versus 0,6%, respectivamente). GILENYA no se ha probado en pacientes con EM con diabetes mellitus.
5.5 Lesión hepática
Se ha producido una lesión hepática clínicamente significativa en pacientes tratados con GILENYA en el entorno poscomercialización. Los signos de lesión hepática, que incluyen enzimas hepáticas séricas marcadamente elevadas y bilirrubina total elevada, se han producido tan pronto como diez días después de la primera dosis y también se han notificado después del uso prolongado. Se han notificado casos de insuficiencia hepática aguda que requieren trasplante de hígado.
En ensayos clínicos controlados con placebo de 2 años en pacientes adultos, la elevación de las enzimas hepáticas (ALT, AST y GGT) a 3 veces el límite superior de lo normal (ULN) o más ocurrió en el 14% de los pacientes tratados con GILENYA 0,5 mg y el 3% de los pacientes con placebo. Las elevaciones 5 veces el ULN o más ocurrieron en el 4,5% de los pacientes con GILENYA y el 1% de los pacientes con placebo. La mayoría de las elevaciones ocurrieron dentro de los 6 a 9 meses. En los ensayos clínicos, GILENYA se suspendió si la elevación superó las 5 veces el ULN. Los niveles de transaminasas séricas volvieron a la normalidad dentro de aproximadamente 2 meses después de la interrupción de GILENYA. La recurrencia de las elevaciones de las transaminasas hepáticas ocurrió con la reexposición en algunos pacientes.
Antes de comenzar el tratamiento con GILENYA (dentro de los 6 meses), obtenga los niveles de transaminasas séricas (ALT y AST) y bilirrubina total. Obtenga los niveles de transaminasas y bilirrubina total periódicamente hasta dos meses después de la interrupción de GILENYA.
Los pacientes deben ser monitoreados para detectar signos y síntomas de cualquier lesión hepática. Mida los niveles de transaminasas hepáticas y bilirrubina de inmediato en pacientes que reporten síntomas que puedan indicar lesión hepática, incluida fatiga nueva o que empeora, anorexia, dolor en el cuadrante superior derecho del abdomen, orina oscura o ictericia. En este contexto clínico, si se encuentra que el paciente tiene una alanina aminotransferasa (ALT) mayor que tres veces el rango de referencia con bilirrubina total sérica mayor que dos veces el rango de referencia, el tratamiento con GILENYA debe interrumpirse. El tratamiento no debe reanudarse si no se puede establecer una etiología alternativa plausible para los signos y síntomas, porque estos pacientes tienen riesgo de lesión hepática grave inducida por medicamentos.
Debido a que la exposición a GILENYA se duplica en pacientes con insuficiencia hepática grave, estos pacientes deben ser monitoreados de cerca, ya que el riesgo de reacciones adversas es mayor [ver Uso en poblaciones específicas (8.6), Farmacología clínica (12.3)].
5.6 Síndrome de encefalopatía posterior reversible
Se han notificado casos raros de síndrome de encefalopatía posterior reversible (PRES) en pacientes adultos que reciben GILENYA. Los síntomas notificados incluyeron inicio repentino de cefalea intensa, alteración del estado mental, trastornos visuales y convulsiones. Los síntomas de PRES suelen ser reversibles, pero pueden evolucionar a un accidente cerebrovascular isquémico o hemorragia cerebral. El retraso en el diagnóstico y el tratamiento puede provocar secuelas neurológicas permanentes. Si se sospecha PRES, se debe interrumpir el tratamiento con GILENYA.
5.7 Efectos respiratorios
Se observaron reducciones dependientes de la dosis en el volumen espiratorio forzado en 1 segundo (FEV1) y la capacidad de difusión pulmonar para el monóxido de carbono (DLCO) en pacientes tratados con GILENYA tan pronto como 1 mes después del inicio del tratamiento. En ensayos controlados con placebo de 2 años en pacientes adultos, la reducción desde el inicio en el porcentaje de valores predichos para FEV1 en el momento de la última evaluación con el fármaco fue del 2,8% para GILENYA 0,5 mg y del 1,0% para placebo. Para DLCO, la reducción desde el inicio en el porcentaje de valores predichos en el momento de la última evaluación con el fármaco fue del 3,3% para GILENYA 0,5 mg y del 0,5% para placebo. Los cambios en FEV1 parecen ser reversibles después de la interrupción del tratamiento. No hay información suficiente para determinar la reversibilidad de la disminución de DLCO después de la interrupción del fármaco. En ensayos controlados con placebo de EM en pacientes adultos, se notificó disnea en el 9% de los pacientes que recibieron GILENYA 0,5 mg y en el 7% de los pacientes que recibieron placebo. Varios pacientes interrumpieron el tratamiento con GILENYA debido a disnea inexplicable durante los estudios de extensión (no controlados). GILENYA no se ha probado en pacientes con EM con función respiratoria comprometida.
Se debe realizar una evaluación espirométrica de la función respiratoria y una evaluación de DLCO durante el tratamiento con GILENYA si está clínicamente indicado.
5.8 Riesgo fetal
Según los hallazgos de estudios en animales, GILENYA puede causar daño fetal cuando se administra a una mujer embarazada. En estudios de reproducción en animales realizados en ratas y conejos, se observó toxicidad para el desarrollo con la administración de fingolimod a dosis inferiores a la dosis humana recomendada. Avise a las mujeres embarazadas y a las mujeres en edad fértil del posible riesgo para el feto. Debido a que se necesitan aproximadamente 2 meses para eliminar GILENYA del cuerpo, avise a las mujeres en edad fértil que utilicen métodos anticonceptivos eficaces para evitar el embarazo durante y durante los 2 meses posteriores a la interrupción del tratamiento con GILENYA [ver Uso en poblaciones específicas (8.1, 8.3)].
5.9 Aumento grave de la discapacidad después de interrumpir GILENYA
Se ha notificado un aumento grave de la discapacidad acompañado de múltiples lesiones nuevas en la RMN después de la interrupción de GILENYA en la fase de postcomercialización. Los pacientes en la mayoría de estos casos notificados no volvieron al estado funcional que tenían antes de interrumpir GILENYA. El aumento de la discapacidad generalmente ocurrió dentro de las 12 semanas posteriores a la interrupción de GILENYA, pero se notificó hasta 24 semanas después de la interrupción de GILENYA.
Controle a los pacientes para detectar el desarrollo de un aumento grave de la discapacidad después de la interrupción de GILENYA e inicie el tratamiento adecuado según sea necesario.
Después de interrumpir GILENYA en el contexto de la PML, controle el desarrollo del síndrome inflamatorio de reconstitución inmunitaria (PML-IRIS) [ver Advertencias y precauciones (5.3)].
5.10 Esclerosis múltiple tumefactiva
Se han observado recaídas de EM con lesiones desmielinizantes tumefactivas en las imágenes durante el tratamiento con GILENYA y después de la interrupción de GILENYA en la fase de postcomercialización. La mayoría de los casos notificados de EM tumefactiva en pacientes que reciben GILENYA se han producido dentro de los primeros 9 meses después del inicio de GILENYA, pero la EM tumefactiva puede ocurrir en cualquier momento durante el tratamiento. También se han notificado casos de EM tumefactiva dentro de los primeros 4 meses después de la interrupción de GILENYA. Se debe considerar la EM tumefactiva cuando se produce una recaída grave de EM durante el tratamiento con GILENYA, especialmente durante el inicio, o después de la interrupción de GILENYA, lo que lleva a una evaluación por imágenes y al inicio del tratamiento adecuado.
5.11 Aumento de la presión arterial
En ensayos clínicos controlados de EM en adultos, los pacientes tratados con GILENYA 0,5 mg tuvieron un aumento promedio sobre placebo de aproximadamente 3 mmHg en la presión sistólica y aproximadamente 2 mmHg en la presión diastólica, detectado por primera vez después de aproximadamente 1 mes del inicio del tratamiento y que persistió con el tratamiento continuo. La hipertensión se notificó como una reacción adversa en el 8% de los pacientes que recibieron GILENYA 0,5 mg y en el 4% de los pacientes que recibieron placebo. Se debe controlar la presión arterial (PA) durante el tratamiento con GILENYA.
5.12 Neoplasias
Neoplasias cutáneas
El riesgo de neoplasias cutáneas (incluido el carcinoma basocelular (CBC), el carcinoma de células escamosas (CCE) y el melanoma) está aumentado en los pacientes tratados con moduladores del receptor S1P. El uso de GILENYA se ha asociado con un mayor riesgo de CBC y melanoma.
En ensayos controlados con placebo de dos años en pacientes adultos, la incidencia de CBC fue del 2% en los pacientes que recibieron GILENYA 0,5 mg y del 1% en los pacientes que recibieron placebo [ver Reacciones adversas (6.1)]. Se han notificado melanoma, carcinoma basocelular, carcinoma de células escamosas, sarcoma de Kaposi [ver Advertencias y precauciones (5.2)] y carcinoma de células de Merkel con GILENYA en la fase de postcomercialización.
Se recomienda realizar exámenes de la piel antes o poco después del inicio del tratamiento y periódicamente a partir de entonces para todos los pacientes, particularmente aquellos con factores de riesgo para el cáncer de piel. Se aconseja a los proveedores y pacientes que monitoreen las lesiones cutáneas sospechosas. Si se observa una lesión cutánea sospechosa, debe evaluarse de inmediato. Como es habitual para los pacientes con mayor riesgo de cáncer de piel, la exposición a la luz solar y la luz ultravioleta debe limitarse usando ropa protectora y usando un protector solar con un alto factor de protección. No se recomienda la fototerapia concomitante con radiación UV-B o fotoquimioterapia PUVA en pacientes que toman GILENYA.
Linfoma
Se han presentado casos de linfoma, incluidos los tipos de células T y células B y el linfoma del SNC, en pacientes que reciben GILENYA. La tasa de notificación de linfoma no Hodgkin con GILENYA es mayor que la esperada en la población general ajustada por edad, sexo y región. También se ha notificado linfoma de células T cutáneo (incluido el micosis fungoide) con GILENYA en la poscomercialización.
5.13 Efectos del sistema inmunitario después de la interrupción de GILENYA
Fingolimod permanece en la sangre y tiene efectos farmacodinámicos, incluida la disminución del recuento de linfocitos, hasta 2 meses después de la última dosis de GILENYA. El recuento de linfocitos generalmente vuelve al rango normal dentro de 1 a 2 meses de suspender la terapia [ver Farmacología clínica (12.2)]. Debido a los efectos farmacodinámicos continuos de fingolimod, el inicio de otros medicamentos durante este período justifica las mismas consideraciones necesarias para la administración concomitante (por ejemplo, riesgo de efectos inmunosupresores aditivos) [ver Interacciones medicamentosas (7.4)].
5.14 Reacciones de hipersensibilidad
Se han notificado reacciones de hipersensibilidad, incluida la erupción cutánea, la urticaria y el angioedema, con GILENYA en la poscomercialización. GILENYA está contraindicado en pacientes con antecedentes de hipersensibilidad a fingolimod o a cualquiera de sus excipientes [ver Contraindicaciones (4)].
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas graves se describen en otra parte del etiquetado:
- Bradiarritmia y bloqueos auriculoventriculares [see Warnings and Precautions (5.1)]
- Infecciones [see Warnings and Precautions (5.2)]
- Leucoencefalopatía multifocal progresiva [see Warnings and Precautions (5.3)]
- Edema macular [see Warnings and Precautions (5.4)]
- Daño hepático [see Warnings and Precautions (5.5)]
- Síndrome de encefalopatía posterior reversible [see Warnings and Precautions (5.6)]
- Efectos respiratorios [see Warnings and Precautions (5.7)]
- Riesgo fetal [see Warnings and Precautions (5.8)]
- Aumento grave de la discapacidad después de suspender GILENYA [see Warnings and Precautions (5.9)]
- Esclerosis múltiple tumefactiva [see Warnings and Precautions (5.10)]
- Presión arterial elevada [see Warnings and Precautions (5.11)]
- Tumores malignos [see Warnings and Precautions (5.12)]
- Efectos sobre el sistema inmunitario después de la suspensión de GILENYA [see Warnings and Precautions (5.13)]
- Reacciones de hipersensibilidad [see Warnings and Precautions (5.14)]
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se realizan bajo condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un medicamento no se pueden comparar directamente con las tasas en los ensayos clínicos de otro medicamento y pueden no reflejar las tasas observadas en la práctica.
Adultos
En ensayos clínicos (Estudios 1, 2 y 3), un total de 1212 pacientes con formas recurrentes de esclerosis múltiple recibieron GILENYA 0.5 mg. Esto incluyó a 783 pacientes que recibieron GILENYA 0.5 mg en los ensayos controlados con placebo de 2 años de duración (Estudios 1 y 3) y 429 pacientes que recibieron GILENYA 0.5 mg en el ensayo controlado con activo de 1 año de duración (Estudio 2). La exposición general en los ensayos controlados fue equivalente a 1716 personas-año. Aproximadamente 1000 pacientes recibieron al menos 2 años de tratamiento con GILENYA 0.5 mg. En todos los estudios clínicos, incluidos los estudios de extensión no controlados, la exposición a GILENYA 0.5 mg fue de aproximadamente 4119 personas-año.
En los ensayos controlados con placebo, las reacciones adversas más frecuentes (incidencia ≥ 10 % y mayor que con placebo) para GILENYA 0.5 mg fueron dolor de cabeza, elevación de las transaminasas hepáticas, diarrea, tos, influenza, sinusitis, dolor de espalda, dolor abdominal y dolor en las extremidades. Los eventos adversos que llevaron a la suspensión del tratamiento y que ocurrieron en más del 1 % de los pacientes que tomaban GILENYA 0.5 mg fueron elevaciones de las transaminasas séricas (4.7 % en comparación con el 1 % con placebo) y carcinoma basocelular (1 % en comparación con el 0.5 % con placebo).
La Tabla 1 enumera las reacciones adversas en estudios clínicos en adultos que ocurrieron en ≥ 1 % de los pacientes tratados con GILENYA y con una tasa ≥ 1 % mayor que con placebo.
| Abreviaturas: ALT, alanina transaminasa; AST, aspartato transferasa; GGT, gamma-glutamil transferasa. | ||
| Reacciones adversas a medicamentos | GILENYA 0.5 mg N = 783 % |
Placebo N = 773 % |
| Infecciones | ||
|
Influenza |
11 | 8 |
|
Sinusitis |
11 | 8 |
|
Bronchitis |
8 | 5 |
|
Herpes zoster |
2 | 1 |
|
Tinea versicolor |
2 | < 1 |
| Trastornos cardíacos | ||
|
Bradicardia |
3 | 1 |
| Trastornos del sistema nervioso | ||
|
Dolor de cabeza |
25 | 24 |
|
Migraña |
6 | 4 |
| Trastornos gastrointestinales | ||
|
Náuseas |
13 | 12 |
|
Diarrea |
13 | 10 |
|
Dolor abdominal |
11 | 10 |
| Trastornos generales y afecciones en el sitio de administración | ||
|
Asthenia |
2 | 1 |
| Trastornos musculoesqueléticos y del tejido conjuntivo | ||
|
Dolor de espalda |
10 | 9 |
|
Dolor en las extremidades |
10 | 7 |
| Trastornos de la piel y del tejido subcutáneo | ||
|
Alopecia |
3 | 2 |
|
Actinic keratosis |
2 | 1 |
| Investigaciones | ||
|
Liver transaminase elevations (ALT/GGT/AST) |
15 | 4 |
|
Blood triglycerides increased |
3 | 1 |
| Trastornos respiratorios, torácicos y mediastínicos | ||
|
Cough |
12 | 11 |
|
Dyspnea |
9 | 7 |
| Trastornos oculares | ||
|
Vision blurred |
4 | 2 |
| Trastornos vasculares | ||
|
Hypertension |
8 | 4 |
| Trastornos de la sangre y del sistema linfático | ||
|
Lymphopenia |
7 | < 1 |
|
Leukopenia |
2 | < 1 |
| Neoplasias benignas, malignas y no especificadas (incluidos quistes y pólipos) |
||
|
Skin papilloma |
3 | 2 |
|
Basal cell carcinoma |
2 | 1 |
También se notificaron reacciones adversas de convulsiones, mareos, neumonía, eccema y prurito en los Estudios 1 y 3, pero no cumplieron los criterios de tasa de notificación para su inclusión en la Tabla 1 (la diferencia fue inferior al 1%).
Las reacciones adversas con GILENYA 0.5 mg en el Estudio 2, el estudio de 1 año de duración con control activo (frente a interferón beta-1a), fueron generalmente similares a las de los Estudios 1 y 3.
Eventos vasculares
Se notificaron eventos vasculares, incluidos accidentes cerebrovasculares isquémicos y hemorrágicos, y enfermedad oclusiva arterial periférica en ensayos clínicos previos a la comercialización en pacientes que recibieron dosis de GILENYA (1.25 mg a 5 mg) superiores a las recomendadas para su uso en la EM. Se han notificado eventos similares con GILENYA en el entorno poscomercialización, aunque no se ha establecido una relación causal.
Convulsiones
Se han notificado casos de convulsiones, incluido el estado epiléptico, con el uso de GILENYA en ensayos clínicos y en el entorno poscomercialización en adultos [ver Reacciones adversas (6.2)]. En los ensayos clínicos en adultos, la tasa de convulsiones fue del 0.9% en los pacientes tratados con GILENYA y del 0.3% en los pacientes tratados con placebo. Se desconoce si estos eventos estuvieron relacionados con los efectos de la esclerosis múltiple por sí sola, con GILENYA o con una combinación de ambos.
Pacientes pediátricos de 10 años de edad o mayores
En el ensayo pediátrico controlado (Estudio 4), el perfil de seguridad en pacientes pediátricos que recibieron GILENYA 0.25 mg o 0.5 mg al día fue similar al observado en pacientes adultos.
En el estudio pediátrico, se notificaron casos de convulsiones en el 5.6% de los pacientes tratados con GILENYA y en el 0.9% de los pacientes tratados con interferón beta-1a [ver Uso en poblaciones específicas (8.4)].
6.2 Experiencia poscomercialización
Las siguientes reacciones adversas se han identificado durante el uso poscomercialización de GILENYA. Debido a que estas reacciones se notifican voluntariamente a partir de una población de tamaño incierto, no siempre es posible estimar de forma fiable su frecuencia o establecer una relación causal con la exposición al fármaco.
Trastornos de la sangre y del sistema linfático: Anemia hemolítica y trombocitopenia
Trastornos hepatobiliares: Lesión hepática [ver Advertencias y precauciones (5.5)]
Infecciones: Infecciones, incluidas las infecciones criptocócicas [ver Advertencias y precauciones (5.2)], infección por el virus del papiloma humano (VPH), incluidos papilomas, displasia, verrugas y cáncer relacionado con el VPH [ver Advertencias y precauciones (5.2)], leucoencefalopatía multifocal progresiva [ver Advertencias y precauciones (5.3)]
Trastornos musculoesqueléticos y del tejido conjuntivo: Artralgia, mialgia
Trastornos del sistema nervioso: Síndrome de encefalopatía posterior reversible [ver Advertencias y precauciones (5.6)], convulsiones, incluido el estado epiléptico [ver Reacciones adversas (6.1)]
Neoplasias benignas, malignas y no especificadas (incluidos quistes y pólipos): melanoma, carcinoma de células de Merkel, linfoma cutáneo de células T (incluida la micosis fungoide), sarcoma de Kaposi, carcinoma de células escamosas [ver Advertencias y precauciones (5.12)]
Trastornos de la piel y del tejido subcutáneo: Hipersensibilidad [ver Advertencias y precauciones (5.14)]
7 INTERACCIONES MEDICAMENTOSAS
7.1 Fármacos que Prolongan el QT
GILENYA no se ha estudiado en pacientes tratados con fármacos que prolongan el intervalo QT. Los fármacos que prolongan el intervalo QT se han asociado con casos de torsades de pointes en pacientes con bradicardia. Dado que el inicio del tratamiento con GILENYA produce una disminución de la frecuencia cardíaca y puede prolongar el intervalo QT, los pacientes que toman fármacos que prolongan el QT con un riesgo conocido de torsades de pointes (por ejemplo, citalopram, clorpromazina, haloperidol, metadona, eritromicina) deben ser monitorizados durante la noche con ECG continuo en un centro médico [ver Posología y administración (2.4), Advertencias y precauciones (5.1)].
7.2 Ketoconazol
Los niveles sanguíneos de fingolimod y fingolimod-fosfato aumentan 1,7 veces cuando se utilizan de forma concomitante con ketoconazol. Los pacientes que utilizan GILENYA y ketoconazol sistémico de forma concomitante deben ser monitorizados estrechamente, ya que el riesgo de reacciones adversas es mayor.
7.3 Vacunas
GILENYA reduce la respuesta inmunitaria a la vacunación. La vacunación puede ser menos eficaz durante y hasta 2 meses después de la interrupción del tratamiento con GILENYA [ver Farmacología clínica (12.2)]. Evite el uso de vacunas atenuadas vivas durante y durante 2 meses después del tratamiento con GILENYA debido al riesgo de infección. Se recomienda que los pacientes pediátricos, si es posible, se pongan al día con todas las inmunizaciones de acuerdo con las directrices actuales de inmunización antes de iniciar el tratamiento con GILENYA.
7.4 Terapias Antineoplásicas, Inmunosupresoras o Inmunomoduladoras
Se espera que las terapias antineoplásicas, inmunomoduladoras o inmunosupresoras, (incluidos los corticosteroides) aumenten el riesgo de inmunosupresión, y debe considerarse el riesgo de efectos aditivos del sistema inmunitario si estas terapias se administran de forma concomitante con GILENYA. Al cambiar de fármacos con efectos inmunitarios prolongados, como natalizumab, teriflunomida o mitoxantrona, debe considerarse la duración y el modo de acción de estos fármacos para evitar efectos inmunosupresores aditivos no deseados al iniciar GILENYA [ver Advertencias y precauciones (5.2)].
7.5 Fármacos que Ralentizan la Frecuencia Cardíaca o la Conducción Auriculoventricular (por ejemplo, betabloqueantes o diltiazem)
La experiencia con GILENYA en pacientes que reciben terapia concomitante con fármacos que ralentizan la frecuencia cardíaca o la conducción AV (por ejemplo, betabloqueantes, digoxina o bloqueadores de los canales de calcio que ralentizan la frecuencia cardíaca, como diltiazem o verapamilo) es limitada. Dado que el inicio del tratamiento con GILENYA puede producir una disminución adicional de la frecuencia cardíaca, el uso concomitante de estos fármacos durante el inicio de GILENYA puede asociarse con bradicardia grave o bloqueo cardíaco. Solicite consejo al médico que le receta estos fármacos sobre la posibilidad de cambiar a fármacos que no ralentizan la frecuencia cardíaca o la conducción auriculoventricular antes de iniciar GILENYA. Los pacientes que no puedan cambiar deben tener una monitorización continua del ECG durante la noche después de la primera dosis [ver Posología y administración (2.4), Advertencias y precauciones (5.1)].
7.6 Interacción con las Pruebas de Laboratorio
Debido a que GILENYA reduce el recuento de linfocitos sanguíneos mediante la redistribución en los órganos linfoides secundarios, el recuento de linfocitos sanguíneos periféricos no puede utilizarse para evaluar el estado del subconjunto de linfocitos de un paciente tratado con GILENYA. Debe estar disponible un CBC reciente antes de iniciar el tratamiento con GILENYA.
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de Riesgos
Basándose en los hallazgos de estudios en animales, GILENYA puede causar daño fetal cuando se administra a una mujer embarazada. Los datos de los informes prospectivos al Registro de Embarazo de GILENYA (GPR) actualmente no son suficientes para permitir una evaluación adecuada del riesgo asociado al fármaco para defectos de nacimiento y aborto espontáneo en humanos.
En estudios orales realizados en ratas y conejos, fingolimod demostró toxicidad para el desarrollo, incluyendo un aumento en las malformaciones (ratas) y embrioletalidad, cuando se administró a animales preñados. En ratas, la dosis máxima sin efecto fue menor que la dosis humana recomendada de 0.5 mg/día en base a la superficie corporal (mg/m2). Las malformaciones viscerales fetales más comunes en ratas fueron el tronco arterial persistente y el defecto septal ventricular. Se sabe que el receptor afectado por fingolimod (receptor de esfingosina 1-fosfato) está involucrado en la formación vascular durante la embriogénesis (ver Datos). Advierta a las mujeres embarazadas sobre el riesgo potencial para un feto.
En la población general de los EE. UU., el riesgo de fondo estimado de defectos de nacimiento mayores y aborto espontáneo en embarazos clínicamente reconocidos es del 2% al 4% y del 15% al 20%, respectivamente. Se desconoce el riesgo de fondo de defectos de nacimiento mayores y aborto espontáneo para la población indicada.
Consideraciones Clínicas
En las mujeres que planean quedar embarazadas, GILENYA debe suspenderse 2 meses antes de la concepción planificada.
La posibilidad de un aumento severo en la discapacidad debe considerarse en las mujeres que interrumpen o están considerando interrumpir GILENYA debido al embarazo o al embarazo planificado. En muchos de los casos en los que se informó un aumento en la discapacidad después de suspender GILENYA, las pacientes habían suspendido GILENYA debido al embarazo o al embarazo planificado [ver Advertencias y Precauciones (5.9)].
Datos
Datos de Animales
Cuando fingolimod se administró por vía oral a ratas preñadas durante el período de organogénesis (0, 0.03, 0.1 y 0.3 mg/kg/día o 0, 1, 3 y 10 mg/kg/día), se observaron incidencias aumentadas de malformaciones fetales y muertes embrio fetales en todas las dosis excepto en la dosis más baja probada (0.03 mg/kg/día), que es menor que la dosis humana recomendada (RHD) en base a mg/m2. La administración oral a conejas preñadas durante la organogénesis (0, 0.5, 1.5 y 5 mg/kg/día) resultó en incidencias aumentadas de mortalidad embrio fetal y retraso del crecimiento fetal en las dosis medias y altas. La dosis sin efecto para estos efectos en conejos (0.5 mg/kg/día) es aproximadamente 20 veces la RHD en base a mg/m2.
Cuando fingolimod se administró por vía oral a ratas hembras durante el embarazo y la lactancia (0, 0.05, 0.15 y 0.5 mg/kg/día), la supervivencia de las crías disminuyó en todas las dosis y se observó un déficit neuroconductual (aprendizaje) en la descendencia en la dosis alta. La dosis de bajo efecto de 0.05 mg/kg/día es similar a la RHD en base a mg/m2.
8.2 Lactancia
Resumen de Riesgos
No hay datos sobre la presencia de fingolimod en la leche materna, los efectos en el lactante amamantado o los efectos del fármaco en la producción de leche. Fingolimod se excreta en la leche de ratas tratadas. Cuando un fármaco está presente en la leche animal, es probable que el fármaco esté presente en la leche humana. Los beneficios para el desarrollo y la salud de la lactancia materna deben considerarse junto con la necesidad clínica de la madre de GILENYA y cualquier efecto adverso potencial en el lactante amamantado de GILENYA o de la condición materna subyacente.
8.3 Mujeres y Hombres con Potencial Reproductivo
Prueba de Embarazo
El estado de embarazo de las mujeres con potencial reproductivo debe verificarse antes de comenzar el tratamiento con GILENYA [ver Uso en Poblaciones Específicas (8.1)].
Anticoncepción
Antes de iniciar el tratamiento con GILENYA, las mujeres con potencial reproductivo deben ser asesoradas sobre el potencial de un riesgo grave para el feto y la necesidad de una anticoncepción eficaz durante el tratamiento con GILENYA [ver Advertencias y Precauciones (5.8) y Uso en Poblaciones Específicas (8.1)]. Dado que se necesitan aproximadamente 2 meses para eliminar el compuesto del cuerpo después de suspender el tratamiento, el riesgo potencial para el feto puede persistir y las mujeres deben usar una anticoncepción eficaz durante este período [ver Advertencias y Precauciones (5.8, 5.13)].
8.4 Uso Pediátrico
La seguridad y eficacia de GILENYA para el tratamiento de las formas recurrentes de esclerosis múltiple en pacientes pediátricos de 10 a menos de 18 años de edad se establecieron en un estudio clínico aleatorizado, doble ciego en 215 pacientes (GILENYA n = 107; interferón (IFN) beta-1a intramuscular n = 108) [ver Estudios Clínicos (14.2)].
En el estudio pediátrico controlado, el perfil de seguridad en pacientes pediátricos (de 10 a menos de 18 años de edad) que recibieron GILENYA 0.25 mg o 0.5 mg diarios fue similar al observado en pacientes adultos. En el estudio pediátrico, se informaron casos de convulsiones en el 5.6% de los pacientes tratados con GILENYA y el 0.9% de los pacientes tratados con interferón beta-1a.
Se recomienda que los pacientes pediátricos, si es posible, completen todas las inmunizaciones de acuerdo con las pautas de inmunización actuales antes de iniciar la terapia con GILENYA.
No se ha establecido la seguridad y eficacia de GILENYA en pacientes pediátricos menores de 10 años.
Datos de Toxicidad Animal Juvenil
En un estudio en el que fingolimod (0.3, 1.5 o 7.5 mg/kg/día) se administró por vía oral a ratas jóvenes desde el destete hasta la madurez sexual, se observaron cambios en la densidad mineral ósea y deterioro neuroconductual persistente (inicio de sobresalto auditivo alterado) en todas las dosis. Se observó una maduración sexual tardía en las hembras en la dosis más alta probada y en los machos en todas las dosis. Los cambios óseos observados en ratas juveniles tratadas con fingolimod son consistentes con un papel informado de S1P en la regulación de la homeostasis mineral ósea.
Cuando fingolimod (0,5 o 5 mg/kg/día) se administró por vía oral a ratas desde el período neonatal hasta la madurez sexual, se observó una marcada disminución en la respuesta de anticuerpos dependiente de células T en ambas dosis. Este efecto no se había recuperado completamente a las 6 a 8 semanas después del final del tratamiento.
En general, no se identificó una dosis sin efecto para los efectos adversos en el desarrollo en animales jóvenes.
8.5 Uso en Geriatría
Los estudios clínicos de EM de GILENYA no incluyeron un número suficiente de pacientes de 65 años o más para determinar si responden de manera diferente a los pacientes más jóvenes. GILENYA debe usarse con precaución en pacientes de 65 años o más, reflejando la mayor frecuencia de disminución de la función hepática o renal, y de enfermedades concomitantes u otra terapia farmacológica.
8.6 Insuficiencia hepática
Debido a que la exposición a fingolimod, pero no a fingolimod-fosfato, se duplica en pacientes con insuficiencia hepática grave, los pacientes con insuficiencia hepática grave deben ser monitoreados de cerca, ya que el riesgo de reacciones adversas puede ser mayor [ver Advertencias y precauciones (5.5), Farmacología clínica (12.3)].
No es necesario ajustar la dosis en pacientes con insuficiencia hepática leve o moderada.
8.7 Insuficiencia renal
El nivel sanguíneo de algunos metabolitos de GILENYA aumenta (hasta 13 veces) en pacientes con insuficiencia renal grave [ver Farmacología clínica (12.3)]. La toxicidad de estos metabolitos no se ha explorado completamente. El nivel sanguíneo de estos metabolitos no se ha evaluado en pacientes con insuficiencia renal leve o moderada.
10 SOBREDOSIS
GILENYA puede inducir bradicardia y bloqueos de la conducción AV (incluido el bloqueo AV completo). La disminución de la frecuencia cardíaca generalmente comienza dentro de 1 hora de la primera dosis y es máxima dentro de las 6 horas en la mayoría de los pacientes [ver Advertencias y precauciones (5.1)]. En caso de sobredosis de GILENYA, observe a los pacientes durante la noche con monitoreo continuo del ECG en un centro médico y obtenga mediciones regulares de la presión arterial [ver Dosis y administración (2.4)].
Ni la diálisis ni el intercambio de plasma eliminan fingolimod del cuerpo.
11 DESCRIPCIÓN
Fingolimod es un modulador del receptor de esfingosina 1-fosfato.
Químicamente, fingolimod es clorhidrato de 2-amino-2-[2-(4-octilfenil)etil]propan-1,3-diol. Su estructura se muestra a continuación:
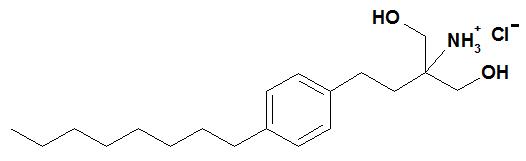
El clorhidrato de fingolimod es un polvo blanco a blanco prácticamente que es libremente soluble en agua y alcohol y soluble en propilenglicol. Tiene un peso molecular de 343,93 g/mol.
GILENYA se proporciona como cápsulas de gelatina dura de 0,25 mg y 0,5 mg para uso oral.
Cada cápsula de 0,25 mg contiene 0,28 mg de clorhidrato de fingolimod, equivalente a 0,25 mg de fingolimod.
Cada cápsula de 0,5 mg contiene 0,56 mg de clorhidrato de fingolimod, equivalente a 0,5 mg de fingolimod.
Cada cápsula de GILENYA de 0,25 mg contiene los siguientes ingredientes inactivos: gelatina, hidroxipropilbetadex, hidroxipropilcelulosa, estearato de magnesio, manitol, dióxido de titanio y óxido de hierro amarillo.
Cada cápsula de GILENYA de 0,5 mg contiene los siguientes ingredientes inactivos: gelatina, estearato de magnesio, manitol, dióxido de titanio y óxido de hierro amarillo.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
Fingolimod se metaboliza por la esfingosina quinasa al metabolito activo, fingolimod-fosfato. Fingolimod-fosfato es un modulador del receptor de esfingosina 1-fosfato, y se une con alta afinidad a los receptores de esfingosina 1-fosfato 1, 3, 4 y 5. Fingolimod-fosfato bloquea la capacidad de los linfocitos para salir de los ganglios linfáticos, reduciendo el número de linfocitos en sangre periférica. Se desconoce el mecanismo por el cual fingolimod ejerce efectos terapéuticos en la esclerosis múltiple, pero puede implicar la reducción de la migración de linfocitos al sistema nervioso central.
12.2 Farmacodinamia
Frecuencia cardíaca y ritmo
Fingolimod causa una reducción transitoria de la frecuencia cardíaca y la conducción AV al inicio del tratamiento [ver Advertencias y precauciones (5.1)].
La frecuencia cardíaca aumenta progresivamente después del primer día, volviendo a los valores basales dentro de 1 mes del inicio del tratamiento crónico.
Las respuestas autonómicas del corazón, incluida la variación diurna de la frecuencia cardíaca y la respuesta al ejercicio, no se ven afectadas por el tratamiento con fingolimod.
El tratamiento con fingolimod no está asociado con una disminución del gasto cardíaco.
Potencial para prolongar el intervalo QT
En un estudio exhaustivo del intervalo QT de dosis de 1,25 o 2,5 mg de fingolimod en estado estacionario, cuando aún estaba presente un efecto cronotrópico negativo de fingolimod, el tratamiento con fingolimod provocó una prolongación del QTc, con el límite superior del intervalo de confianza del 90% (IC) de 14,0 mseg. No hay una señal consistente de aumento de la incidencia de valores atípicos de QTc, ya sea absoluta o cambio desde el valor basal, asociado con el tratamiento con fingolimod. En los estudios de EM, no hubo una prolongación clínicamente relevante del intervalo QT, pero los pacientes con riesgo de prolongación del QT no se incluyeron en los estudios clínicos.
Sistema inmunitario
Efectos sobre el número de células inmunitarias en la sangre
En un estudio en el que 12 sujetos adultos recibieron GILENYA 0,5 mg al día, el recuento de linfocitos disminuyó a aproximadamente el 60% del valor basal dentro de las 4 a 6 horas posteriores a la primera dosis. Con la dosificación diaria continua, el recuento de linfocitos continuó disminuyendo durante un período de 2 semanas, alcanzando un recuento mínimo de aproximadamente 500 células/mcL o aproximadamente el 30% del valor basal. En un estudio controlado con placebo en 1272 pacientes con EM (de los cuales 425 recibieron fingolimod 0,5 mg al día y 418 recibieron placebo), el 18% (N = 78) de los pacientes que recibieron fingolimod 0,5 mg alcanzaron un mínimo de < 200 células/mcL en al menos 1 ocasión. Ningún paciente que recibió placebo alcanzó un mínimo de < 200 células/mcL. Los recuentos bajos de linfocitos se mantienen con la dosificación diaria crónica de GILENYA 0,5 mg al día.
La dosificación crónica de fingolimod conduce a una disminución leve en el recuento de neutrófilos a aproximadamente el 80% del valor basal. Los monocitos no se ven afectados por fingolimod.
Los aumentos en el recuento de linfocitos periféricos son evidentes dentro de los días de suspender el tratamiento con fingolimod y, por lo general, se alcanzan recuentos normales dentro de 1 a 2 meses.
Efecto sobre la respuesta de anticuerpos
GILENYA reduce la respuesta inmunitaria a la vacunación, como se evaluó en 2 estudios.
En el primer estudio, la inmunogenicidad de la inmunización con hemocianina de lapa clave (KLH) y la vacuna neumocócica polisacárida (PPV-23) se evaluó mediante títulos de IgM e IgG en un estudio en estado estacionario, aleatorizado y controlado con placebo en voluntarios adultos sanos. En comparación con el placebo, los títulos de IgM específicos del antígeno disminuyeron en un 91% y un 25% en respuesta a KLH y PPV-23, respectivamente, en sujetos que recibieron GILENYA 0,5 mg. De manera similar, los títulos de IgG disminuyeron en un 45% y un 50%, en respuesta a KLH y PPV-23, respectivamente, en sujetos que recibieron GILENYA 0,5 mg al día en comparación con el placebo. La tasa de respuesta para GILENYA 0,5 mg medida por el número de sujetos con un aumento > 4 veces en KLH IgG fue comparable al placebo y un 25% más baja para PPV-23 IgG, mientras que el número de sujetos con un aumento > 4 veces en KLH y PPV-23 IgM fue 75% y 40% más bajo, respectivamente, en comparación con el placebo. La capacidad de generar una reacción de hipersensibilidad retardada de tipo cutáneo a Candida y toxoides tetánicos disminuyó aproximadamente un 30% en sujetos que recibieron GILENYA 0,5 mg al día, en comparación con el placebo. Las respuestas inmunológicas se redujeron aún más con fingolimod 1,25 mg (una dosis superior a la recomendada en EM) [ver Advertencias y precauciones (5.2)].
En el segundo estudio, la inmunogenicidad de la vacuna contra la influenza estacional del hemisferio norte y el toxoide tetánico se evaluó en un estudio de 12 semanas en estado estacionario, aleatorizado y controlado con placebo de GILENYA 0,5 mg en pacientes adultos con esclerosis múltiple (n = 136). La tasa de respuesta 3 semanas después de la vacunación, definida como seroconversión o un aumento ≥ 4 veces en los anticuerpos dirigidos contra al menos 1 de las 3 cepas de influenza, fue del 54% para GILENYA 0,5 mg y del 85% en el grupo placebo. La tasa de respuesta 3 semanas después de la vacunación, definida como seroconversión o un aumento ≥ 4 veces en los anticuerpos dirigidos contra el toxoide tetánico fue del 40% para GILENYA 0,5 mg y del 61% en el grupo placebo.
Función pulmonar
Las dosis únicas de fingolimod ≥ 5 mg (10 veces la dosis recomendada) están asociadas con un aumento dependiente de la dosis en la resistencia de las vías respiratorias. En un estudio de 14 días de 0,5, 1,25 o 5 mg/día, fingolimod no se asoció con una oxigenación deteriorada o desaturación de oxígeno con el ejercicio o un aumento en la respuesta de las vías respiratorias a la metacolina. Los sujetos que recibieron tratamiento con fingolimod tuvieron una respuesta broncodilatadora normal a los beta-agonistas inhalados.
En un estudio controlado con placebo de 14 días en pacientes adultos con asma moderada, no se observó ningún efecto para GILENYA 0,5 mg (dosis recomendada en EM). Se observó una reducción del 10% en la FEV1 media a las 6 horas después de la dosificación en pacientes adultos que recibieron fingolimod 1,25 mg (una dosis superior a la recomendada para su uso en EM) en el día 10 del tratamiento. Fingolimod 1,25 mg se asoció con un aumento de 5 veces en el uso de beta-agonistas de acción corta de rescate.
12.3 Farmacocinética
Absorción
La Tmax de fingolimod es de 12 a 16 horas. La biodisponibilidad oral absoluta aparente es del 93%.
La ingesta de alimentos no altera la Cmax o (AUC) de fingolimod o fingolimod-fosfato. Por lo tanto, GILENYA se puede tomar sin tener en cuenta las comidas.
Tras la administración una vez al día, se alcanzan las concentraciones plasmáticas en estado estacionario en 1 a 2 meses y los niveles en estado estacionario son aproximadamente 10 veces mayores que con la dosis inicial.
Distribución
Fingolimod se distribuye en gran medida (86%) en los glóbulos rojos. Fingolimod-fosfato tiene una menor captación en las células sanguíneas de < 17%. Fingolimod y fingolimod-fosfato están unidos a proteínas en más del 99,7%. La unión de proteínas de fingolimod y fingolimod-fosfato no se altera por el deterioro renal o hepático.
Fingolimod se distribuye ampliamente en los tejidos corporales con un volumen de distribución de aproximadamente 1200 ± 260 L.
Metabolismo
La biotransformación de fingolimod en humanos se produce por 3 vías principales: por fosforilación estereoselectiva reversible al enantiómero farmacológicamente activo (S) de fingolimod-fosfato, por biotransformación oxidativa catalizada principalmente por el citocromo P450 4F2 (CYP4F2) y posiblemente otras isoenzimas CYP4F con posterior degradación similar a ácidos grasos a metabolitos inactivos, y por formación de análogos de ceramida farmacológicamente inactivos de fingolimod.
Inhibidores o inductores de CYP4F2 y posiblemente otras isoenzimas CYP4F pueden alterar la exposición de fingolimod o fingolimod-fosfato. Los estudios in vitro en hepatocitos indicaron que CYP3A4 puede contribuir al metabolismo de fingolimod en caso de inducción fuerte de CYP3A4.
Tras la administración oral única de [14C] fingolimod, los componentes relacionados con fingolimod más importantes en la sangre, según se juzga por su contribución al AUC hasta 816 horas después de la dosis de componentes total radiomarcados, son fingolimod en sí (23,3%), fingolimod-fosfato (10,3%) y metabolitos inactivos [metabolito de ácido carboxílico M3 (8,3%), metabolito de ceramida M29 (8,9%) y metabolito de ceramida M30 (7,3%)].
Eliminación
La depuración sanguínea de fingolimod es de 6,3 ± 2,3 L/h y la vida media terminal aparente promedio (t1/2) es de 6 a 9 días. Los niveles sanguíneos de fingolimod-fosfato disminuyen en paralelo con los de fingolimod en la fase terminal, dando vidas medias similares para ambos.
Después de la administración oral, aproximadamente el 81% de la dosis se excreta lentamente en la orina como metabolitos inactivos. Fingolimod y fingolimod-fosfato no se excretan intactos en la orina, pero son los componentes principales en las heces, con cantidades de cada uno que representan menos del 2,5% de la dosis.
Poblaciones específicas
Pacientes pediátricos
La concentración mediana de fingolimod-fosfato (fingolimod-P) en pacientes pediátricos con esclerosis múltiple de 10 a menos de 18 años fue de 1,10 ng/mL, en comparación con 1,35 ng/mL en pacientes adultos con esclerosis múltiple.
Pacientes geriátricos
El mecanismo de eliminación y los resultados de la farmacocinética poblacional sugieren que no sería necesario un ajuste de la dosis en pacientes ancianos. Sin embargo, la experiencia clínica en pacientes de más de 65 años es limitada.
Sexo
El sexo no tiene una influencia clínicamente significativa en la farmacocinética de fingolimod y fingolimod-fosfato.
Raza
No se puede evaluar adecuadamente el efecto de la raza en la farmacocinética de fingolimod y fingolimod-fosfato debido al bajo número de pacientes que se identificaron como negros o afroamericanos, asiáticos u otras razas en el programa clínico.
Deterioro renal
En pacientes adultos con deterioro renal grave, la Cmax y el AUC de fingolimod aumentan en un 32% y un 43%, respectivamente, y la Cmax y el AUC de fingolimod-fosfato aumentan en un 25% y un 14%, respectivamente, sin cambios en la vida media de eliminación aparente. Basado en estos hallazgos, la dosis de 0,5 mg de GILENYA es adecuada para uso en pacientes adultos con deterioro renal. Las dosis de 0,25 mg y 0,5 mg de GILENYA son adecuadas para uso en pacientes pediátricos con deterioro renal. La exposición sistémica de 2 metabolitos (M2 y M3) aumenta en 3 y 13 veces, respectivamente. La toxicidad de estos metabolitos no se ha caracterizado completamente.
No se ha realizado un estudio en pacientes con deterioro renal leve o moderado.
Deterioro hepático
En sujetos con deterioro hepático leve, moderado o grave (clase Child-Pugh A, B y C), no se observó cambio en la Cmax de fingolimod, pero el AUC0-∞ de fingolimod aumentó en un 12%, 44% y 103%, respectivamente. En pacientes con deterioro hepático grave (clase Child-Pugh C), la Cmax de fingolimod-fosfato disminuyó en un 22% y el AUC0-96 horas disminuyó en un 29%. La farmacocinética de fingolimod-fosfato no se evaluó en pacientes con deterioro hepático leve o moderado. La vida media de eliminación aparente de fingolimod no cambia en sujetos con deterioro hepático leve, pero se prolonga aproximadamente un 50% en pacientes con deterioro hepático moderado o grave.
Los pacientes con deterioro hepático grave (clase Child-Pugh C) deben ser monitoreados estrechamente, ya que el riesgo de reacciones adversas es mayor [ver Advertencias y Precauciones (5.5)].
No se necesita un ajuste de la dosis en pacientes con deterioro hepático leve o moderado (clase Child-Pugh A y B).
Interacciones medicamentosas
Ketoconazol
La administración conjunta de ketoconazol (un potente inhibidor de CYP3A y CYP4F) 200 mg dos veces al día en estado estacionario y una dosis única de fingolimod 5 mg condujo a un aumento del 70% en el AUC de fingolimod y fingolimod-fosfato. Los pacientes que usan GILENYA y ketoconazol sistémico concomitantemente deben ser monitoreados estrechamente, ya que el riesgo de reacciones adversas es mayor [ver Interacciones medicamentosas (7.2)].
Carbamazepina
La administración conjunta de carbamazepina (un potente inductor de enzimas CYP450) 600 mg dos veces al día en estado estacionario y una dosis única de fingolimod 2 mg disminuyó las concentraciones sanguíneas (AUC) de fingolimod y fingolimod-fosfato en aproximadamente un 40%. El impacto clínico de esta disminución es desconocido.
Otros potentes inductores de enzimas CYP450, como rifampicina, fenitoína, fenobarbital y hierba de San Juan, también pueden reducir el AUC de fingolimod y fingolimod-fosfato. El impacto clínico de esta disminución potencial es desconocido.
Potencial de fingolimod y fingolimod-fosfato para inhibir el metabolismo de medicamentos coadministrados
In vitro los estudios de inhibición utilizando microsomas hepáticos humanos agrupados y sustratos de sonda metabólica específicos demuestran que fingolimod tiene poca o ninguna capacidad para inhibir la actividad de las siguientes enzimas CYP: CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4 / 5 o CYP4A9 / 11 (solo fingolimod), y de manera similar, el fingolimod-fosfato tiene poca o ninguna capacidad para inhibir la actividad de CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 o CYP3A4 a concentraciones de hasta 3 órdenes de magnitud de concentraciones terapéuticas. Por lo tanto, es poco probable que fingolimod y fingolimod-fosfato reduzcan la eliminación de fármacos que se eliminan principalmente a través del metabolismo por las principales isoenzimas CYP descritas anteriormente.
Potencial de Fingolimod y Fingolimod-fosfato para inducir su propio metabolismo y / o el metabolismo de las comedicaciones
Se examinó fingolimod para su potencial para inducir CYP3A4, CYP1A2, CYP4F2 y MDR1 (P-glucoproteína) ARNm y CYP3A, CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19 y CYP4F2 actividad en hepatocitos humanos primarios. Fingolimod no indujo ARNm o actividad de las diferentes enzimas CYP y MDR1 con respecto al control del vehículo; por lo tanto, no se espera una inducción clínicamente relevante de las enzimas CYP o MDR1 probadas por fingolimod a concentraciones terapéuticas. También se examinó el fingolimod-fosfato para su potencial para inducir ARNm y / o actividad de CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP3A, CYP4F2, CYP4F3B y CYP4F12 humanos. No se espera que el fingolimod-fosfato tenga efectos de inducción clínicamente significativos en estas enzimas a dosis terapéuticas de fingolimod. In vitro los experimentos no proporcionaron una indicación de inducción de CYP por fingolimod-fosfato.
Transportadores
Basado en in vitro datos, no se espera que fingolimod, así como fingolimod-fosfato, inhiban la absorción de comedicaciones y / o biológicos transportados por los polipéptidos transportadores de aniones orgánicos OATP1B1, OATP1B3 o el polipéptido transportador de cotransporte de taurocolato de sodio (NTCP). De manera similar, no se espera que inhiban la eflujo de comedicaciones y / o biológicos transportados por la proteína de resistencia al cáncer de mama (BCRP), la bomba de exportación de sales biliares (BSEP), la proteína 2 asociada a la resistencia a múltiples fármacos (MRP2) o la P-glucoproteína (P-gp) a concentraciones terapéuticas.
Anticonceptivos orales
La coadministración de fingolimod 0,5 mg diarios con anticonceptivos orales (etinilestradiol y levonorgestrel) no provocó ningún cambio clínicamente significativo en la exposición a los anticonceptivos orales. La exposición a fingolimod y fingolimod-fosfato fue consistente con la de estudios previos. No se han realizado estudios de interacción con anticonceptivos orales que contengan otros progestágenos; sin embargo, no se espera un efecto de fingolimod en su exposición.
Ciclosporina
La farmacocinética de una dosis única de fingolimod no se alteró durante la coadministración con ciclosporina en estado estacionario, ni la farmacocinética en estado estacionario de la ciclosporina se alteró por fingolimod. Estos datos indican que GILENYA es poco probable que reduzca o aumente la eliminación de fármacos eliminados principalmente por CYP3A4. La inhibición potente de los transportadores MDR1 (P-gp), MRP2 y OATP-1B1 no influye en la disposición de fingolimod.
Isoproterenol, atropina, atenolol y diltiazem
La exposición a una dosis única de fingolimod y fingolimod-fosfato no se alteró por isoproterenol o atropina coadministrados. Del mismo modo, la farmacocinética de una dosis única de fingolimod y fingolimod-fosfato y la farmacocinética en estado estacionario de atenolol y diltiazem no se modificaron durante la coadministración de los 2 últimos fármacos individualmente con fingolimod.
Análisis de farmacocinética poblacional
Una evaluación de farmacocinética poblacional realizada en pacientes con EM no proporcionó evidencia de un efecto significativo de fluoxetina y paroxetina (inhibidores fuertes de CYP2D6) en las concentraciones predosis de fingolimod o fingolimod-fosfato. Además, las siguientes sustancias comúnmente coprescritas no tuvieron un efecto clínicamente relevante (< 20%) en las concentraciones predosis de fingolimod o fingolimod-fosfato: baclofeno, gabapentina, oxibutinina, amantadina, modafinilo, amitriptilina, pregabalina y corticosteroides.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
Se realizaron estudios de carcinogenicidad oral de fingolimod en ratones y ratas. En ratones, fingolimod se administró en dosis orales de 0, 0.025, 0.25 y 2.5 mg/kg/día durante un máximo de 2 años. La incidencia de linfoma maligno aumentó en machos y hembras a la dosis media y alta. La dosis más baja probada (0.025 mg/kg/día) es menor que la RHD de 0.5 mg/día en base al área de superficie corporal (mg/m2). En ratas, fingolimod se administró en dosis orales de 0, 0.05, 0.15, 0.5 y 2.5 mg/kg/día. No se observó un aumento en los tumores. La dosis más alta probada (2.5 mg/kg/día) es aproximadamente 50 veces la RHD en base a mg/m2.
Fingolimod fue negativo en una batería de ensayos in vitro (Ames, timidincinasa de linfoma de ratón, aberración cromosómica en células de mamíferos) e in vivo (micronúcleo en ratón y rata).
Cuando fingolimod se administró por vía oral (0, 1, 3 y 10 mg/kg/día) a ratas macho y hembra antes y durante el apareamiento, y continuando hasta el día 7 de gestación en las hembras, no se observó ningún efecto sobre la fertilidad hasta la dosis más alta probada (10 mg/kg), que es aproximadamente 200 veces la RHD en base a mg/m2.
13.2 Toxicología y/o Farmacología Animal
Se observó toxicidad pulmonar en 2 cepas diferentes de ratas y en perros y monos. Los hallazgos principales incluyeron un aumento en el peso del pulmón, asociado con hipertrofia del músculo liso, hiperdistensión de los alvéolos y/o aumento del colágeno. Se observó insuficiencia o falta de colapso pulmonar en la necropsia, generalmente correlacionada con cambios microscópicos, en todas las especies. En ratas y monos, se observó toxicidad pulmonar a todas las dosis orales probadas en estudios crónicos. Las dosis más bajas probadas en ratas (0.05 mg/kg/día en el estudio de carcinogenicidad de 2 años) y monos (0.5 mg/kg/día en el estudio de toxicidad de 39 semanas) son similares y aproximadamente 20 veces la RHD en base a mg/m2, respectivamente.
En el estudio oral de 52 semanas en monos, se observó dificultad respiratoria asociada con la administración de ketamina a dosis de 3 y 10 mg/kg/día; el animal más afectado se volvió hipóxico y requirió oxigenación. Como la ketamina no suele estar asociada con depresión respiratoria, este efecto se atribuyó a fingolimod. En un estudio posterior en ratas, se demostró que la ketamina potenciaba los efectos broncoconstrictores de fingolimod. Se desconoce la relevancia de estos hallazgos para los humanos.
14 ESTUDIOS CLÍNICOS
14.1 Adultos
La eficacia de GILENYA se demostró en 2 estudios que evaluaron dosis diarias de GILENYA 0,5 mg y 1,25 mg en pacientes con EM remitente-recurrente (EMRR). Ambos estudios incluyeron pacientes que habían experimentado al menos 2 recaídas clínicas durante los 2 años anteriores a la aleatorización o al menos 1 recaída clínica durante el año anterior a la aleatorización, y tenían una puntuación en la Escala Expandida del Estado de Discapacidad (EDSS) de 0 a 5,5. El estudio 1 fue un estudio aleatorizado, doble ciego, controlado con placebo de 2 años en pacientes con EMRR que no habían recibido ningún interferón-beta o acetato de glatiramer durante al menos los 3 meses anteriores y no habían recibido ningún natalizumab durante al menos los 6 meses anteriores. Las evaluaciones neurológicas se realizaron en la selección, cada 3 meses y en el momento de una recaída sospechosa. Las evaluaciones de IRM se realizaron en la selección, mes 6, mes 12 y mes 24. El criterio de valoración principal fue la tasa de recaídas anualizada.
La edad media fue de 37 años, la duración media de la enfermedad fue de 6,7 años y la puntuación media de EDSS en el inicio del estudio fue de 2,0. Los pacientes fueron aleatorizados para recibir GILENYA 0,5 mg (N = 425), 1,25 mg (N = 429) o placebo (N = 418) durante un máximo de 24 meses. El tiempo medio con el fármaco del estudio fue de 717 días con 0,5 mg, 715 días con 1,25 mg y 719 días con placebo.
La tasa de recaídas anualizada fue significativamente menor en los pacientes tratados con GILENYA que en los pacientes que recibieron placebo. El criterio de valoración secundario fue el tiempo hasta la progresión de la discapacidad confirmada a los 3 meses, medida por un aumento de al menos 1 punto desde el inicio del estudio en la EDSS (aumento de 0,5 puntos para los pacientes con una EDSS inicial de 5,5) sostenido durante 3 meses. El tiempo hasta el inicio de la progresión de la discapacidad confirmada a los 3 meses se retrasó significativamente con el tratamiento con GILENYA en comparación con el placebo. La dosis de 1,25 mg no produjo ningún beneficio adicional sobre la dosis de GILENYA 0,5 mg. Los resultados de este estudio se muestran en la Tabla 2 y la Figura 1.
| Abreviatura: IC, intervalo de confianza. Todos los análisis de los criterios de valoración clínicos fueron de intención de tratar. El análisis de IRM utilizó un conjunto de datos evaluable. ‡La razón de riesgos es una estimación del riesgo relativo de tener el evento de progresión de la discapacidad con GILENYA en comparación con el placebo. |
|||
| GILENYA 0,5 mg N = 425 |
Placebo N = 418 |
p-valor | |
| Criterios de valoración clínicos | |||
| Tasa de recaídas anualizada (criterio de valoración principal) | 0,18 | 0,40 | < 0,001 |
| Porcentaje de pacientes sin recaídas | 70% | 46% | < 0,001 |
| Razón de riesgos‡ de progresión de la discapacidad (IC del 95%) |
0,70 (0,52, 0,96) |
0,02 | |
| Criterio de valoración de IRM | |||
| Número medio (mediana) de lesiones nuevas o de nuevo aumento de tamaño T2 durante 24 meses | 2,5 (0) | 9,8 (5,0) | < 0,001 |
| Número medio (mediana) de lesiones T1 que realzan con Gd en el mes 24 | 0,2 (0) | 1,1 (0) | < 0,001 |
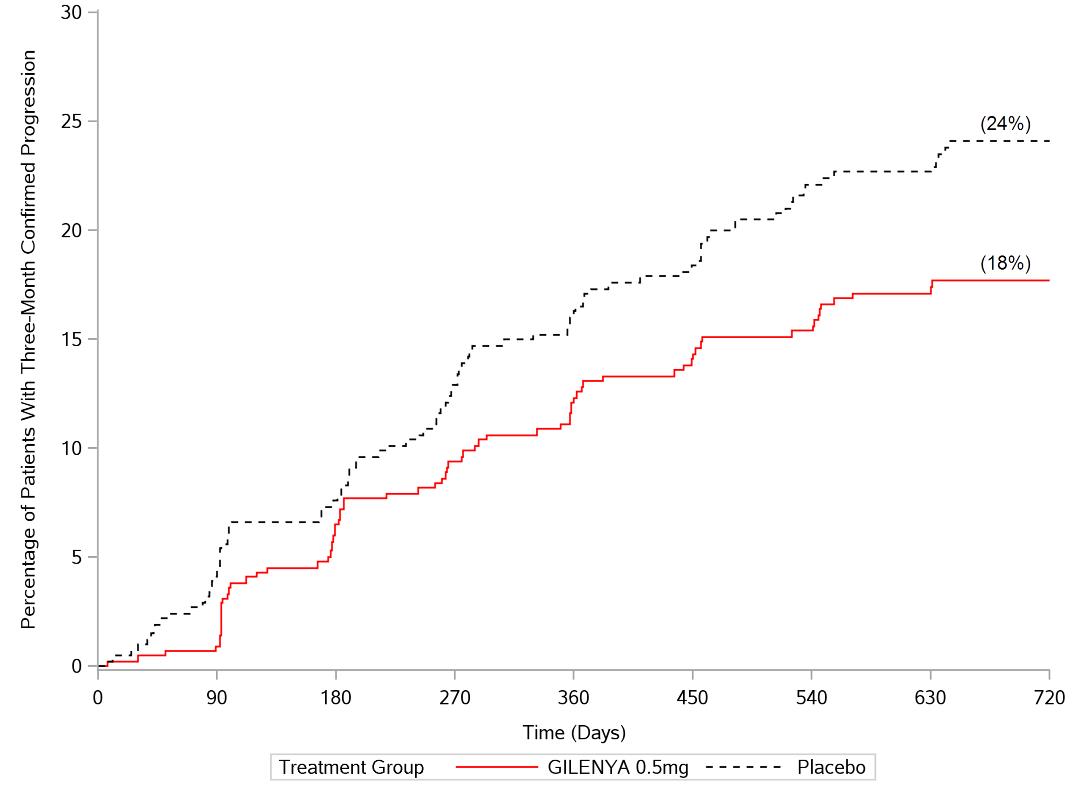
Figura 1: Tiempo hasta la progresión de la discapacidad confirmada a los 3 meses – Estudio 1 (población ITT)
El estudio 2 fue un estudio aleatorizado, doble ciego, doble ficticio, controlado con activo de 1 año en pacientes con RRMS que no habían recibido natalizumab en los 6 meses anteriores. Se permitió la terapia previa con interferón-beta o acetato de glatiramer hasta el momento de la aleatorización.
Se realizaron evaluaciones neurológicas en la selección, cada 3 meses y en el momento de las recaídas sospechosas. Se realizaron evaluaciones de resonancia magnética en la selección y en el mes 12. El criterio de valoración principal fue la tasa de recaídas anualizada.
La edad media fue de 36 años, la duración media de la enfermedad fue de 5,9 años y la puntuación media de EDSS en el inicio fue de 2,0. Los pacientes fueron aleatorizados para recibir GILENYA 0,5 mg (N = 431), 1,25 mg (N = 426) o interferón beta-1a, 30 mcg por vía intramuscular (IM) una vez por semana (N = 435) durante un máximo de 12 meses. El tiempo medio en el estudio con el fármaco fue de 365 días con GILENYA 0,5 mg, 354 días con 1,25 mg y 361 días con interferón beta-1a IM.
La tasa de recaídas anualizada fue significativamente menor en los pacientes tratados con GILENYA 0,5 mg que en los pacientes que recibieron interferón beta-1a IM. Los criterios de valoración secundarios clave fueron el número de lesiones T2 nuevas y de reciente aparición y el tiempo hasta el inicio de la progresión de la discapacidad confirmada a los 3 meses, medido por un aumento de al menos 1 punto desde el inicio en EDSS (aumento de 0,5 puntos para aquellos con EDSS inicial de 5,5) mantenido durante 3 meses. El número de lesiones T2 nuevas y de reciente aparición fue significativamente menor en los pacientes tratados con GILENYA que en los pacientes que recibieron interferón beta-1a IM. No hubo diferencias significativas en el tiempo hasta la progresión de la discapacidad confirmada a los 3 meses entre los pacientes tratados con GILENYA y los tratados con interferón beta-1a a los 12 meses. La dosis de 1,25 mg no produjo ningún beneficio adicional sobre la dosis de GILENYA 0,5 mg. Los resultados de este estudio se muestran en la Tabla 3.
| Abreviatura: IC, intervalo de confianza. Todos los análisis de los criterios de valoración clínicos fueron de intención de tratar. El análisis de resonancia magnética utilizó un conjunto de datos evaluable. ‡La razón de riesgos es una estimación del riesgo relativo de tener el evento de progresión de la discapacidad con GILENYA en comparación con el control. |
|||
| GILENYA 0,5 mg N = 429 |
Interferón beta-1a IM 30 mcg N = 431 |
p-valor | |
| Criterios de valoración clínicos | |||
| Tasa de recaídas anualizada (criterio de valoración principal) | 0,16 | 0,33 | < 0,001 |
| Porcentaje de pacientes sin recaída | 83% | 70% | < 0,001 |
| Razón de riesgos‡ de progresión de la discapacidad (IC del 95%) |
0,71 (0,42, 1,21) |
0,21 | |
| Criterio de valoración de resonancia magnética | |||
| Número medio (mediana) de lesiones T2 nuevas o de reciente aparición en 12 meses | 1,6 (0) | 2,6 (1,0) | 0,002 |
| Número medio (mediana) de lesiones T1 que realzan con Gd en el mes 12 | 0,2 (0) | 0,5 (0) | < 0,001 |
14.2 Pacientes pediátricos (de 10 a menos de 18 años de edad)
El estudio 4 (NCT 01892722) evaluó la eficacia de dosis orales diarias de GILENYA 0,25 mg o GILENYA 0,5 mg en pacientes pediátricos de 10 a menos de 18 años de edad con esclerosis múltiple remitente-recurrente. El estudio 4 fue un ensayo clínico aleatorizado, doble ciego, de 215 pacientes que comparó GILENYA con interferón beta-1a intramuscular. Se permitió la terapia previa con interferón-beta, fumarato de dimetilo o acetato de glatiramer hasta el momento de la aleatorización. El estudio incluyó pacientes que habían experimentado al menos 1 recaída clínica durante el año anterior o 2 recaídas durante los 2 años anteriores a la selección, o evidencia de 1 o más lesiones que mejoran con Gd en la resonancia magnética dentro de los 6 meses anteriores a la aleatorización, y tenían una puntuación EDSS de 0 a 5,5. Las evaluaciones neurológicas se programaron en la selección, cada 3 meses y en el momento de las recaídas sospechosas. Las evaluaciones de resonancia magnética se realizaron en la selección y cada 6 meses durante todo el estudio. El criterio de valoración principal fue la tasa de recaídas anualizada.
En la línea de base, la edad media fue de 16 años, la duración media de la enfermedad desde el primer síntoma fue de 1,5 años y la puntuación EDSS media fue de 1,5. Un paciente no recibió ningún fármaco del estudio y está excluido del análisis de eficacia. La duración media de la exposición al fármaco del estudio fue de 634 días en el grupo GILENYA (n = 107) y 547 días en el grupo interferón beta-1a (n = 107). En el grupo GILENYA, el 6,5% de los pacientes no completaron el estudio, en comparación con el 18,5% en el grupo interferón beta-1a.
El criterio de valoración principal, la tasa de recaídas anualizada (ARR), fue significativamente menor en los pacientes tratados con GILENYA (0,122) que en los pacientes que recibieron interferón beta-1a (0,675). La reducción relativa en ARR fue del 81,9%. La tasa anualizada del número de nuevas lesiones T2 o recién agrandadas hasta el mes 24 (criterio de valoración secundario clave) fue significativamente menor en los pacientes tratados con GILENYA, al igual que el número de lesiones T1 que mejoran con Gd por exploración hasta el mes 24.
La tabla 4 resume los resultados del estudio 4.
| Abreviaturas: IM, intramuscular; resonancia magnética, resonancia magnética; PO, por vía oral. Todos los análisis de los criterios de valoración clínicos se realizaron en el conjunto de análisis completo. Los análisis de resonancia magnética utilizaron el conjunto de datos evaluable. *Indica significancia estadística frente a interferón beta-1a IM a un nivel de 0,05 bilateral. |
||||
| GILENYA 0,25 mg o 0,5 mg PO N = 107 |
Interferón beta-1a 30 mcg IM N = 107 |
p-valor | Reducción relativa |
|
| Criterios de valoración clínicos | ||||
| Tasa de recaídas anualizada (criterio de valoración principal) | 0,122 | 0,675 | < 0,001* | 81,9% |
| Porcentaje de pacientes que permanecen libres de recaídas a los 24 meses | 86,0% | 45,8% | ||
| Criterios de valoración de resonancia magnética | ||||
| Tasa anualizada del número de nuevas lesiones T2 o recién agrandadas | 4,393 | 9,269 | < 0,001* | 52,6% |
| Número medio de lesiones T1 que mejoran con Gd por exploración hasta el mes 24 | 0,436 | 1,282 | < 0,001* | 66,0% |
16 FORMA DE SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
16.1 Cómo se suministra
Las cápsulas de GILENYA de 0,25 mg se suministran de la siguiente manera:
cápsulas duras de gelatina con cuerpo opaco de color marfil y tapa, con la impresión radial negra “FTY 0,25 mg” en la tapa y una banda radial negra en el cuerpo de la cápsula
Caja de 7 cápsulas que contiene 1 tarjeta blíster de 7 cápsulas por tarjeta blíster
NDC 0078-0965-89
Las cápsulas de GILENYA de 0,5 mg se suministran de la siguiente manera:
cápsulas duras de gelatina con cuerpo opaco blanco y tapa de color amarillo brillante con la impresión “FTY 0,5 mg” en la tapa y 2 bandas radiales impresas en el cuerpo de la cápsula con tinta amarilla.
Frasco de 30 cápsulas
NDC 0078-0607-15
16.2 Almacenamiento y manipulación
Las cápsulas de GILENYA deben almacenarse a 20ºC a 25ºC (68ºF a 77ºF); se permiten excursiones de 15ºC a 30ºC (59ºF a 86ºF). Proteger de la humedad.
17 INFORMACIÓN PARA EL PACIENTE
Aconseje al paciente que lea el etiquetado del paciente aprobado por la FDA (Guía de medicamentos).
Diga a los pacientes que no deben suspender GILENYA sin antes hablarlo con el médico que se lo recetó. Aconseje a los pacientes que se pongan en contacto con su médico si toman accidentalmente más GILENYA de lo prescrito.
Efectos cardíacos
Aconseje a los pacientes que la iniciación del tratamiento con GILENYA produce una disminución transitoria de la frecuencia cardíaca. Informe a los pacientes que deberán ser observados en el consultorio del médico u otra instalación durante al menos 6 horas después de la primera dosis, después de la reiniciación si el tratamiento se interrumpe o se suspende durante ciertos períodos, y después de que se aumente la dosis [ver Dosificación y administración (2.4), Advertencias y precauciones (5.1)].
Riesgo de infecciones
Informe a los pacientes que pueden tener un mayor riesgo de infecciones, algunas de las cuales podrían ser potencialmente mortales, cuando toman GILENYA, y que deben ponerse en contacto con su médico si desarrollan síntomas de infección. Aconseje a los pacientes que se debe evitar el uso de algunas vacunas durante el tratamiento con GILENYA y durante 2 meses después de la suspensión. Recomiende a los pacientes que retrasen el tratamiento con GILENYA hasta después de la vacunación contra el VZV si no han tenido varicela o una vacunación previa contra el VZV. Informe a los pacientes que el uso previo o concomitante de medicamentos que suprimen el sistema inmunitario puede aumentar el riesgo de infección [ver Advertencias y precauciones (5.2)].
Leucoencefalopatía multifocal progresiva
Informe a los pacientes que se han producido casos de leucoencefalopatía multifocal progresiva (LMP) en pacientes que recibieron GILENYA. Informe al paciente que la LMP se caracteriza por una progresión de déficits y generalmente conduce a la muerte o discapacidad grave en semanas o meses. Indique al paciente la importancia de ponerse en contacto con su médico si desarrolla algún síntoma sugestivo de LMP. Informe al paciente que los síntomas típicos asociados con la LMP son diversos, progresan durante días o semanas, e incluyen debilidad progresiva en un lado del cuerpo o torpeza de las extremidades, alteración de la visión y cambios en el pensamiento, la memoria y la orientación que conducen a confusión y cambios de personalidad [ver Advertencias y precauciones (5.3)].
Edema macular
Aconseje a los pacientes que GILENYA puede causar edema macular, y que deben realizarse un examen ocular cerca del inicio del tratamiento con GILENYA, que un profesional del cuidado de la visión les controle los ojos periódicamente mientras reciben terapia, y que se pongan en contacto con su proveedor de atención médica si experimentan algún cambio en su visión mientras toman GILENYA. Informe a los pacientes con diabetes mellitus o antecedentes de uveítis que su riesgo de edema macular es mayor [ver Advertencias y precauciones (5.4)].
Efectos hepáticos
Informe a los pacientes que GILENYA puede causar daño hepático. Aconseje a los pacientes que deben ponerse en contacto con su médico si tienen náuseas, vómitos, dolor abdominal, fatiga, anorexia o ictericia y/o orina oscura inexplicables [ver Advertencias y precauciones (5.5)].
Síndrome de encefalopatía posterior reversible
Aconseje a los pacientes que informen inmediatamente a su proveedor de atención médica de cualquier síntoma que implique la aparición repentina de dolor de cabeza intenso, alteración del estado mental, trastornos visuales o convulsiones. Informe a los pacientes que el tratamiento tardío podría conducir a secuelas neurológicas permanentes [ver Advertencias y precauciones (5.6)].
Efectos respiratorios
Aconseje a los pacientes que deben ponerse en contacto con su médico si experimentan disnea de nueva aparición o que empeora [ver Advertencias y precauciones (5.7)].
Riesgo fetal
- Aconseje a las mujeres embarazadas y a las mujeres en edad fértil sobre el riesgo potencial para un feto. Aconseje a las mujeres que informen a su proveedor de atención médica de un embarazo conocido o sospechoso [ver Advertencias y precauciones (5.8) y Uso en poblaciones específicas (8.1, 8.3)].
- Aconseje a las pacientes en edad fértil que utilicen métodos anticonceptivos eficaces durante el tratamiento con GILENYA y durante 2 meses después de la dosis final [ver Uso en poblaciones específicas (8.3)].
Aumento grave de la discapacidad después de suspender GILENYA
Informe a los pacientes que se ha notificado un aumento grave de la discapacidad después de la suspensión de GILENYA. Aconseje a los pacientes que se pongan en contacto con su médico si desarrollan un empeoramiento de los síntomas de la EM después de la suspensión de GILENYA [ver Advertencias y precauciones (5.9)].
Neoplasias malignas
Aconseje a los pacientes que el riesgo de carcinoma basocelular y melanoma aumenta con el uso de GILENYA y que se han notificado casos de carcinoma de células escamosas, carcinoma de células de Merkel y sarcoma de Kaposi. Aconseje a los pacientes que cualquier lesión cutánea sospechosa debe evaluarse con prontitud. Aconseje a los pacientes que limiten la exposición a la luz solar y a la luz ultravioleta usando ropa protectora y utilizando un protector solar con un alto factor de protección. Informe a los pacientes que también se ha producido linfoma en pacientes que reciben GILENYA [ver Advertencias y precauciones (5.12)].
Persistencia de los efectos de GILENYA después de la suspensión del medicamento
Aconseje a los pacientes que GILENYA permanece en la sangre y continúa teniendo efectos, incluido la disminución del recuento de linfocitos en sangre, hasta 2 meses después de la última dosis [ver Advertencias y precauciones (5.13)].
Reacciones de hipersensibilidad
Aconseje a los pacientes que GILENYA puede causar reacciones de hipersensibilidad, incluidas erupción cutánea, urticaria y angioedema. Aconseje a los pacientes que se pongan en contacto con su médico si tienen algún síntoma asociado con la hipersensibilidad [ver Advertencias y precauciones (5.14)].
Embarazo
Indique a los pacientes que si están embarazadas o planean quedar embarazadas mientras toman GILENYA deben informar a su médico [ver Uso en poblaciones específicas (8.1)].
GILENYA es una marca registrada de Novartis, AG.
Distribuido por:
Novartis Pharmaceuticals Corporation
East Hanover, New Jersey 07936
© Novartis
T2024-39
Guía de medicación
| This Medication Guide has been approved by the U.S. Food and Drug Administration. | Revised: June 2024 |
| GUÍA DEL MEDICAMENTO | |
| GILENYA® [je-LEN-yah] (fingolimod) cápsulas, para uso oral |
|
| Lea esta Guía del Medicamento antes de empezar a tomar GILENYA y cada vez que renueve su receta. Puede haber nueva información. Si usted es el padre de un niño que está siendo tratado con GILENYA, la siguiente información aplica a su hijo. Esta información no sustituye a las conversaciones con su médico sobre su enfermedad o su tratamiento. | |
| ¿Cuál es la información más importante que debo saber sobre GILENYA? | |
| GILENYA puede causar efectos secundarios graves, incluyendo: 1. Frecuencia cardíaca lenta (bradicardia o bradiarritmia) al empezar a tomar GILENYA. GILENYA puede hacer que su frecuencia cardíaca se ralentice, especialmente después de tomar la primera dosis. Le harán una prueba llamada electrocardiograma (ECG) para comprobar la actividad eléctrica de su corazón antes de que tome la primera dosis de GILENYA. Todos los adultos y niños estarán en observación por un profesional de la salud durante al menos 6 horas después de tomar su primera dosis de GILENYA. Los niños también deben estar en observación por un profesional de la salud durante al menos 6 horas después de tomar su primera dosis de 0.5 mg de GILENYA cuando cambien de la dosis de 0.25 mg. |
|
Su frecuencia cardíaca lenta suele volver a la normalidad en el plazo de 1 mes después de empezar a tomar GILENYA. Llame a su médico o acuda a la sala de emergencias del hospital más cercano de inmediato si tiene algún síntoma de frecuencia cardíaca lenta. Si olvida 1 o más dosis de GILENYA, es posible que deba estar en observación por un profesional de la salud cuando tome la siguiente dosis. Llame a su médico si olvida una dosis de GILENYA. Consulte “¿Cómo debo tomar GILENYA?”
|
|
| 2. Embarazo. Consulte a su médico antes de quedarse embarazada. Debe evitar quedarse embarazada mientras esté tomando GILENYA o en los dos meses siguientes a la interrupción del tratamiento debido al riesgo de que se produzcan daños en el bebé. | |
| 3. Infecciones. GILENYA puede aumentar el riesgo de sufrir infecciones graves que pueden poner en peligro la vida y causar la muerte. No debe recibir vacunas vivas durante el tratamiento con GILENYA ni durante los 2 meses siguientes a la interrupción del tratamiento. Consulte a su médico antes de vacunarse durante el tratamiento y durante los 2 meses siguientes al tratamiento con GILENYA. Si recibe una vacuna viva, puede contraer la infección que la vacuna pretendía prevenir. Es posible que las vacunas no funcionen tan bien cuando se administran durante el tratamiento con GILENYA.
Virus del papiloma humano (VPH). Debido al riesgo de infección por VPH, consulte a su médico para la realización de una citología vaginal de rutina. GILENYA reduce el número de glóbulos blancos (linfocitos) en la sangre. Esto suele volver a la normalidad en los 2 meses siguientes a la interrupción del tratamiento. Es posible que su médico le haga un análisis de sangre para comprobar sus glóbulos blancos antes de que empiece a tomar GILENYA. Llame a su médico de inmediato si tiene alguno de estos síntomas de infección durante el tratamiento con GILENYA y durante los 2 meses siguientes a su última dosis de GILENYA: |
|
|
|
| 4. Leucoencefalopatía multifocal progresiva (LMP). La LMP es una infección cerebral poco frecuente que suele provocar la muerte o una discapacidad grave. Si se produce la LMP, suele ocurrir en personas con sistemas inmunitarios debilitados, pero también se ha dado en personas que no los tienen debilitados. Los síntomas de la LMP empeoran con el paso de los días o de las semanas. Llame a su médico de inmediato si tiene algún síntoma nuevo o que empeore de LMP, que haya durado varios días, incluyendo: | |
|
|
| 5. Un problema con su visión llamado edema macular. El edema macular puede causar algunos de los mismos síntomas visuales que un ataque de esclerosis múltiple (EM) (neuritis óptica). Es posible que no note ningún síntoma con el edema macular. Si se produce edema macular, este suele comenzar en los primeros 3 a 4 meses después de empezar a tomar GILENYA, pero puede ocurrir en cualquier momento. Su médico debe examinarle la vista alrededor del momento en que empiece a tomar GILENYA, 3 a 4 meses después de empezar a tomar GILENYA, periódicamente mientras siga tomando GILENYA y cada vez que note cambios en la visión durante el tratamiento con GILENYA. El riesgo de sufrir edema macular es mayor si padece diabetes o ha tenido una inflamación del ojo llamada uveítis. Llame a su médico de inmediato si tiene alguno de los siguientes síntomas: |
|
|
|
| ¿Qué es GILENYA? GILENYA es un medicamento de venta con receta que se utiliza para tratar las formas recurrentes de esclerosis múltiple (EM), incluidos el síndrome clínicamente aislado, la enfermedad recurrente-remitente y la enfermedad secundaria progresiva activa, en adultos y niños de 10 años o mayores. Se desconoce si GILENYA es seguro y eficaz en niños menores de 10 años. |
|
| ¿Quién no debe tomar GILENYA? No tome GILENYA si usted: |
|
Consulte a su médico antes de tomar GILENYA si tiene alguna de estas afecciones, o si no sabe si tiene alguna de ellas. |
|
| ¿Qué debo decirle a mi médico antes de tomar GILENYA? Antes de tomar GILENYA, informe a su médico sobre todas sus afecciones médicas, incluso si tuvo o tiene ahora: |
|
|
|
| Informe a su médico de todos los medicamentos que toma o ha tomado recientemente, incluyendo los medicamentos con y sin receta, las vitaminas y los suplementos a base de hierbas. Especialmente, informe a su médico si toma medicamentos que afectan a su sistema inmunitario, incluidos los corticosteroides, o si los ha tomado en el pasado. Conozca los medicamentos que toma. Lleve consigo una lista de sus medicamentos para mostrársela a su médico y farmacéutico cuando reciba un nuevo medicamento. El uso de GILENYA junto con otros medicamentos puede afectar a ambos, provocando efectos secundarios graves. |
|
| ¿Cómo debo tomar GILENYA? | |
Consulte “¿Cuál es la información más importante que debo saber sobre GILENYA?” |
|
| ¿Cuáles son los posibles efectos secundarios de GILENYA? GILENYA puede causar efectos secundarios graves, que incluyen: |
|
|
|
| ◦ dolor de cabeza intenso repentino ◦ confusión repentina |
◦ pérdida repentina de la visión u otros cambios en la visión ◦ convulsiones |
|
|
| ◦ náuseas ◦ vómitos ◦ dolor de estómago ◦ cansancio |
◦ pérdida de apetito ◦ su piel o la parte blanca de sus ojos se vuelven amarillos ◦ orina oscura |
|
|
| Los efectos secundarios más comunes de GILENYA incluyen: | |
|
|
|
Dígale a su médico si tiene algún efecto secundario que le moleste o que no desaparezca. Estos no son todos los posibles efectos secundarios de GILENYA. Para obtener más información, consulte a su médico o farmacéutico. Llame a su médico para obtener asesoramiento médico sobre los efectos secundarios. Puede informar los efectos secundarios a la FDA al 1-800-FDA-1088. |
|
| ¿Cómo debo almacenar GILENYA? | |
|
|
| Información general sobre el uso seguro y eficaz de GILENYA. | |
| A veces, los medicamentos se recetan para fines distintos de los que se enumeran en una Guía del Medicamento. No use GILENYA para tratar una afección para la que no fue recetado. No le dé GILENYA a otras personas, incluso si tienen los mismos síntomas que usted. Podría perjudicarlos. Esta Guía del Medicamento resume la información más importante sobre GILENYA. Si desea obtener más información, hable con su médico. Puede pedirle a su médico o farmacéutico información sobre GILENYA que esté escrita para profesionales de la salud. | |
| ¿Cuáles son los ingredientes de GILENYA? | |
| Cápsulas de 0.25 mg Ingrediente activo: fingolimod Ingredientes inactivos: gelatina, hidroxipropilbetadex, hidroxipropilcelulosa, estearato de magnesio, manitol, dióxido de titanio, óxido de hierro amarillo. Cápsulas de 0.5 mg |
|
|
GILENYA es una marca registrada de Novartis, AG. Distribuido por: Novartis Pharmaceuticals Corporation, East Hanover, New Jersey 07936 |
|
|
© Novartis |
|
T2024-40
PANEL DE VISUALIZACIÓN PRINCIPAL
NDC 0078-0607-15
Rx only
GILENYA®
(fingolimod)
capsules
0.5 mg
Equivalent to 0.56 mg
fingolimod hydrochloride
30 Capsules
Dispense with enclosed Medication Guide.
NOVARTIS
PANEL DE VISUALIZACIÓN PRINCIPAL
NDC 0078-0965-89
GILENYA®
(fingolimod)
cápsulas
0.25 mg
Equivalente a 0.28 mg
clorhidrato de fingolimod
7 Cápsulas
Rx solamente
Este paquete incluye un suministro de 1 semana de cápsulas.
Dispense con la Guía de medicamentos adjunta.
NOVARTIS
