
Fabricante de medicamentos: Takeda Pharmaceuticals America, Inc. (Updated: 2025-01-06)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
FEIBA® (complejo coagulante anti-inhibidor)
polvo liofilizado para solución, para administración intravenosa
Aprobación inicial en EE. UU.: 1986
ADVERTENCIA: EVENTOS EMBÓLICOS Y TROMBÓTICOS
Consulte la información completa de prescripción para obtener la advertencia completa en recuadro.
- Se han notificado eventos tromboembólicos durante la vigilancia postcomercialización, particularmente después de la administración de dosis altas o en pacientes con factores de riesgo trombótico.
- Controle a los pacientes que reciben FEIBA para detectar signos y síntomas de eventos tromboembólicos (5.1, 6.2).
INDICACIONES Y USO
FEIBA es un complejo coagulante anti-inhibidor indicado para su uso en pacientes con hemofilia A y B con inhibidores para:
- Control y prevención de episodios hemorrágicos.
- Manejo perioperatorio.
- Profilaxis de rutina para prevenir o reducir la frecuencia de episodios hemorrágicos.
FEIBA no está indicado para el tratamiento de episodios hemorrágicos resultantes de deficiencias del factor de coagulación en ausencia de inhibidores del factor VIII o del factor IX. (1)
POSOLOGÍA Y ADMINISTRACIÓN
Para uso intravenoso después de la reconstitución únicamente (2)
- Cada vial de FEIBA contiene la cantidad indicada de actividad de derivación del inhibidor del factor VIII en unidades. (2.1)
| Tipo de hemorragia | Dosis (unidad/kg) | Frecuencia/Duración |
|---|---|---|
| Control y prevención de hemorragias | 50 – 100 | Determinado por el tipo de episodio hemorrágico |
| Manejo perioperatorio | 50 – 100 | Determinado por el tipo de intervención quirúrgica |
| Profilaxis de rutina | 85 | Día por medio |
La velocidad máxima de inyección o infusión no debe superar las 10 unidades por kg de peso corporal por minuto. (2.3)
PRESENTACIONES Y CONCENTRACIONES
FEIBA está disponible como un polvo liofilizado en viales de dosis única que contienen nominalmente 500, 1000 o 2500 unidades por vial. (3)
CONTRAINDICACIONES
ADVERTENCIAS Y PRECAUCIONES
- FEIBA puede causar eventos tromboembólicos después de dosis superiores a 200 unidades por kg por día y en pacientes con factores de riesgo trombótico. Controle a los pacientes que reciben FEIBA para detectar signos y síntomas de eventos tromboembólicos. (5.1)
- Pueden producirse anafilaxia y reacciones de hipersensibilidad graves. Si aparecen síntomas, interrumpa el tratamiento con FEIBA y administre el tratamiento adecuado. (5.2)
- FEIBA está hecho de plasma humano y puede contener agentes infecciosos, p. ej., virus, la enfermedad de Creutzfeldt-Jakob variante (vCJD) y, teóricamente, el agente de la enfermedad de Creutzfeldt-Jacob (CJD). (5.3)
REACCIONES ADVERSAS
- Las reacciones adversas más comunes observadas en >5% de los sujetos fueron anemia, diarrea, hemartrosis, anticuerpo de superficie de la hepatitis B positivo, náuseas y vómitos. (6.1)
- Las reacciones adversas graves a los medicamentos son la hipersensibilidad y los eventos tromboembólicos, incluido el accidente cerebrovascular, la embolia pulmonar y la trombosis venosa profunda. (5.1, 5.2, 6)
Para notificar SOSPECHAS DE REACCIONES ADVERSAS, comuníquese con Takeda Pharmaceuticals U.S.A., Inc. al 1-877-TAKEDA-7 (1-877-825-3327) o con la FDA al 1-800-FDA-1088 o www.fda.gov/medwatch.
INTERACCIONES MEDICAMENTOSAS
Considere la posibilidad de eventos trombóticos cuando se utilizan antifibrinolíticos sistémicos como el ácido tranexámico y el ácido aminocaproico. No se recomienda el uso de antifibrinolíticos dentro de aproximadamente 6 a 12 horas después de la administración de FEIBA. (7)
Consulte el punto 17 para obtener INFORMACIÓN PARA EL PACIENTE.
Revisado: 12/2024
Tabla de contenido
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDO*
ADVERTENCIA: EVENTOS EMBÓLICOS Y TROMBÓTICOS
1 INDICACIONES Y USO
2 POSOLOGÍA Y ADMINISTRACIÓN
2.1 Dosis
2.2 Preparación y Reconstitución
2.3 Administración
3 FORMAS Y CONCENTRACIONES FARMACÉUTICAS
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Eventos Embólicos y Trombóticos
5.2 Reacciones de Hipersensibilidad
5.3 Transmisión de Agentes Infecciosos
5.4 Presencia de Isoaglutininas e Interferencia con las Pruebas de Laboratorio
6 REACCIONES ADVERSAS
6.1 Experiencia en Ensayos Clínicos
6.2 Experiencia Postcomercialización
7 INTERACCIONES MEDICAMENTOSAS
7.1 Medicamentos Concomitantes
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.4 Uso Pediátrico
8.5 Uso Geriátrico
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de Acción
12.2 Farmacodinamia
12.3 Farmacocinética
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
14 ESTUDIOS CLÍNICOS
15 REFERENCIAS
16 PRESENTACIÓN/ALMACENAMIENTO Y MANEJO
17 INFORMACIÓN PARA EL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información completa de prescripción no se enumeran.
ADVERTENCIA EN EL RECUADRO
ADVERTENCIA: EVENTOS EMBÓLICOS Y TROMBÓTICOS
- Se han notificado eventos tromboembólicos durante la vigilancia postcomercialización tras la infusión de FEIBA, particularmente después de la administración de dosis altas o en pacientes con factores de riesgo trombótico.
- Controle a los pacientes que reciben FEIBA para detectar signos y síntomas de eventos tromboembólicos.
1 INDICACIONES Y USO
FEIBA es un complejo coagulante anti-inhibidor indicado para su uso en pacientes con hemofilia A y B con inhibidores para:
- Control y prevención de episodios hemorrágicos
- Manejo perioperatorio
- Profilaxis rutinaria para prevenir o reducir la frecuencia de episodios hemorrágicos.
FEIBA no está indicado para el tratamiento de episodios hemorrágicos resultantes de deficiencias de factores de coagulación en ausencia de inhibidores del factor VIII de la coagulación o del factor IX de la coagulación.
2 DOSIS Y ADMINISTRACIÓN
Para uso intravenoso solo después de la reconstitución.
2.1 Dosis
La Tabla 1 proporciona una guía para la dosificación de FEIBA.
| Dose (unit/kg) | Frecuencia de Dosis (horas) | Duración del Tratamiento | |
|---|---|---|---|
| Control y Prevención de Hemorragias | |||
| Hemorragia Articular | 50 – 100 | 12 | Hasta que el dolor y las discapacidades agudas mejoren. |
| Hemorragia de la Membrana Mucosa | 50 – 100 | 6 | Al menos 1 día o hasta que la hemorragia se resuelva. |
| Hemorragia de Tejidos Blandos (ej., hemorragia retroperitoneal) |
100 | 12 | Hasta la resolución del sangrado. |
| Otra Hemorragia Grave (ej., hemorragias del SNC) |
100 | 6 – 12 | Hasta la resolución del sangrado. |
| Manejo Perioperatorio | |||
| Preoperatorio | 50 – 100 | Dosis única | Inmediatamente antes de la cirugía. |
| Postoperatorio | 50 – 100 | 6 – 12 | Hasta que se logre la resolución del sangrado y la cicatrización. |
| Profilaxis de Rutina | |||
| 85 | Cada otro día | ||
- La dosis y la duración del tratamiento dependen de la ubicación y la extensión del sangrado, y del estado clínico del paciente. Es necesario un control cuidadoso de la terapia de reemplazo en casos de cirugía mayor o episodios de sangrado que pongan en peligro la vida.
- Cada vial de FEIBA contiene la cantidad etiquetada de actividad de bypass del inhibidor del factor VIII en unidades.
- Base la dosis y la frecuencia de FEIBA en la respuesta clínica individual. La respuesta clínica al tratamiento con FEIBA puede variar según el paciente y puede no correlacionarse con el título de inhibidor del paciente.
- Registre el nombre del paciente y el número de lote del producto para mantener un vínculo entre el paciente y el lote del producto.
- No exceda una dosis única de 100 unidades por kg de peso corporal o una dosis diaria de 200 unidades por kg de peso corporal [see Warnings and Precautions (5.1)].
2.2 Preparación y Reconstitución
- Utilice una técnica aséptica durante todo el proceso de reconstitución.
- Si el paciente usa más de un vial por inyección, reconstituya cada vial de acuerdo con las siguientes instrucciones.
- Deje que los viales de FEIBA y agua estéril para inyección (diluyente) alcancen la temperatura ambiente, si están refrigerados.
- Retire las tapas de plástico de los viales de concentrado y diluyente.
- Limpie los tapones de ambos viales con un hisopo con alcohol estéril y déjelos secar antes de usarlos.
- Abra el paquete del dispositivo BAXJECT II Hi-Flow retirando la tapa por completo sin tocar el interior (Fig. A). No retire el dispositivo del paquete. No toque la punta transparente.
- Coloque el vial de diluyente sobre una superficie plana y sólida. Dé la vuelta al paquete e inserte la punta de plástico transparente a través del tapón del diluyente presionando hacia abajo (Fig. B).
- Sujete el paquete del dispositivo BAXJECT II Hi-Flow por los bordes y retire el paquete del dispositivo (Fig. C). No retire la tapa protectora azul del dispositivo BAXJECT II Hi-Flow. No toque la punta morada.
- Dé la vuelta al sistema, de modo que el vial de diluyente quede arriba. Inserte rápidamente la punta morada del dispositivo BAXJECT II Hi-Flow completamente en el vial de FEIBA. El vacío atraerá el diluyente hacia el vial de FEIBA (Fig. D). La conexión de los dos viales debe hacerse rápidamente para cerrar la vía de fluido abierta creada por la primera inserción de la punta en el vial de diluyente.
- Gire suavemente (no agite) el vial hasta que FEIBA se disuelva por completo. Asegúrese de que FEIBA se haya disuelto por completo; de lo contrario, el material activo no pasará a través del filtro del dispositivo. La solución reconstituida debe inspeccionarse visualmente para detectar partículas antes de la administración. La solución debe desecharse si no es transparente o está descolorida.
- Después de que se complete la reconstitución de FEIBA, la inyección o infusión debe comenzar inmediatamente y debe completarse dentro de las tres horas siguientes a la reconstitución. No refrigerar después de la reconstitución. Deseche la porción no utilizada.
Figura A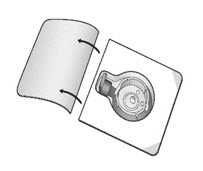 |
Figura B |
Figura C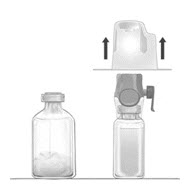 |
Figura D |
2.3 Administración
Solo para inyección intravenosa o infusión intravenosa después de la reconstitución.
- Inspeccione visualmente la solución reconstituida de FEIBA para detectar partículas y decoloración antes de la administración. La apariencia de la solución debe ser incolora a ligeramente amarillenta. No lo use si se observan partículas o decoloración.
- Enjuague las vías de acceso venoso con solución salina isotónica antes y después de la infusión de FEIBA. No administrar en el mismo tubo o contenedor con otros medicamentos.
- Use jeringas Luer lock de plástico porque las proteínas como FEIBA tienden a adherirse a la superficie de las jeringas de vidrio.
- Retire la tapa protectora azul del dispositivo BAXJECT II Hi-Flow. Conecte firmemente la jeringa al dispositivo BAXJECT II Hi-Flow (NO ASPIRE AIRE EN LA JERINGA) girando la jeringa en el sentido de las agujas del reloj hasta la posición de parada. Se recomienda encarecidamente el uso de una jeringa Luer lock para garantizar una conexión firme entre la jeringa y el dispositivo BAXJECT II Hi-Flow (Fig. E).
- Invierta el sistema para que el producto FEIBA disuelto quede arriba. Aspire el producto disuelto con cuidado en la jeringa tirando del émbolo lentamente para evitar la formación de espuma (Fig. F).
- Asegúrese de que se mantenga la conexión firme entre el dispositivo BAXJECT II Hi-Flow y la jeringa.
- Desconecte la jeringa.
- Coloque una aguja adecuada e inyecte o infunda por vía intravenosa a una velocidad que no exceda las 10 unidades por kg de peso corporal por minuto. Se puede usar una bomba de jeringa para controlar la velocidad de administración. Para un paciente con un peso corporal de 75 kg, esto corresponde a una velocidad de infusión de hasta 15 ml por minuto.
Figura E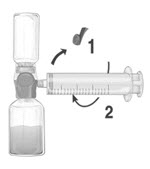 |
Figura F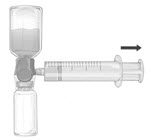 |
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
FEIBA está disponible como un polvo liofilizado en viales de vidrio de dosis única que contienen nominalmente 500, 1000 ó 2500 unidades por vial.
4 CONTRAINDICACIONES
- Reacciones anafilácticas conocidas o reacciones de hipersensibilidad graves a FEIBA o a cualquiera de sus componentes, incluyendo factores del sistema generador de quinina.
- Coagulación intravascular diseminada (CID).
- Trombosis o embolia aguda (incluyendo infarto de miocardio).
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Eventos Embólicos y Trombóticos
Pueden producirse eventos tromboembólicos (incluida la trombosis venosa, la embolia pulmonar, el infarto de miocardio y el accidente cerebrovascular) con FEIBA, particularmente después de la administración de dosis altas (superiores a 200 unidades por kg por día) y/o en pacientes con factores de riesgo trombótico [véase Reacciones adversas (6)].
Los pacientes con CID, enfermedad aterosclerótica avanzada, lesión por aplastamiento, septicemia o tratamiento concomitante con factor VIIa recombinante tienen un mayor riesgo de desarrollar eventos trombóticos debido al factor tisular circulante o a una coagulopatía predisponente. El beneficio potencial del tratamiento con FEIBA debe sopesarse frente al riesgo potencial de estos eventos tromboembólicos.
Controle a los pacientes que reciben más de 100 unidades por kg de peso corporal de FEIBA para detectar el desarrollo de CID, isquemia coronaria aguda y signos y síntomas de otros eventos tromboembólicos. Si se producen signos o síntomas clínicos, como dolor o presión en el pecho, dificultad para respirar, alteración de la conciencia, la visión o el habla, hinchazón y/o dolor en las extremidades o el abdomen, interrumpa la infusión e inicie las medidas diagnósticas y terapéuticas adecuadas.
No se ha establecido la seguridad y eficacia de FEIBA para el sangrado irruptivo en pacientes que reciben emicizumab. Se informaron casos de microangiopatía trombótica (MAT) en un ensayo clínico donde los sujetos recibieron FEIBA como parte de un régimen de tratamiento para el sangrado irruptivo después del tratamiento con emicizumab.6 Considere los beneficios y los riesgos con FEIBA si se considera necesario para los pacientes que reciben profilaxis con emicizumab. Si se requiere tratamiento con FEIBA para pacientes que reciben emicizumab, el médico tratante de hemofilia debe controlar estrechamente los signos y síntomas de MAT. En los estudios clínicos de FEIBA no se ha informado microangiopatía trombótica (MAT).
5.2 Reacciones de Hipersensibilidad
Pueden producirse reacciones de hipersensibilidad y alérgicas, incluidas reacciones anafilactoides graves, después de la infusión de FEIBA. Los síntomas incluyen urticaria, angioedema, manifestaciones gastrointestinales, broncoespasmo e hipotensión. Estas reacciones pueden ser graves y sistémicas (p. ej., anafilaxia con urticaria y angioedema, broncoespasmo y shock circulatorio). También se han notificado otras reacciones a la infusión, como escalofríos, pirexia e hipertensión. Si se producen signos y síntomas de reacciones alérgicas graves, interrumpa inmediatamente la administración de FEIBA y proporcione atención de apoyo adecuada.
5.3 Transmisión de Agentes Infecciosos
Debido a que FEIBA se elabora a partir de plasma humano, puede conllevar un riesgo de transmisión de agentes infecciosos, p. ej., virus y el agente de la enfermedad de Creutzfeldt-Jakob variante (vCJD), y teóricamente, el agente de la enfermedad de Creutzfeldt-Jakob (CJD). El riesgo se ha minimizado mediante la selección de donantes de plasma para la exposición previa a ciertos virus, mediante la prueba de la presencia de ciertas infecciones víricas actuales y mediante la inactivación y eliminación de ciertos virus durante el proceso de fabricación [véase Descripción (11)]. A pesar de estas medidas, el producto aún puede transmitir potencialmente agentes patógenos humanos. También existe la posibilidad de que aún puedan estar presentes agentes infecciosos desconocidos.
Todas las infecciones que un médico considere que posiblemente hayan sido transmitidas por este producto deben ser notificadas por el médico u otros proveedores de atención médica a Takeda Pharmaceuticals U.S.A., Inc. al 1-877-TAKEDA-7 (1-877-825-3327) (en los EE. UU.) y/o a FDA MedWatch (1-800-FDA-1088 o www.fda.gov/medwatch).
5.4 Presencia de Isohemaglutininas e Interferencia con las Pruebas de Laboratorio
FEIBA contiene isohemaglutininas del grupo sanguíneo (anti-A y anti-B). La transmisión pasiva de anticuerpos a antígenos eritrocíticos, p. ej., A, B, D, puede interferir con algunas pruebas serológicas para anticuerpos de eritrocitos, como la prueba de antiglobulina (prueba de Coombs).
6 REACCIONES ADVERSAS
Las reacciones adversas más frecuentes observadas en >5% de los sujetos en el ensayo de profilaxis fueron anemia, diarrea, hemartrosis, anticuerpo de superficie del virus de la hepatitis B positivo, náuseas y vómitos.
Las reacciones adversas graves observadas con FEIBA son reacciones de hipersensibilidad y eventos tromboembólicos, incluyendo accidente cerebrovascular, embolia pulmonar y trombosis venosa profunda.
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se realizan en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no se pueden comparar directamente con las tasas en los ensayos clínicos de otro fármaco y pueden no reflejar las tasas observadas en la práctica clínica.
La evaluación de seguridad de FEIBA se basa en la revisión de los datos de dos ensayos clínicos prospectivos en los que se utilizó FEIBA para el tratamiento de episodios hemorrágicos agudos y un ensayo prospectivo que comparó el uso de FEIBA como profilaxis versus tratamiento a demanda.
Las reacciones adversas notificadas en dos ensayos clínicos prospectivos en los que se utilizó FEIBA para el tratamiento de episodios hemorrágicos agudos fueron escalofríos, dolor torácico, molestias torácicas, mareos, disgeusia, disnea, hipoestesia, aumento del título de inhibidores (respuesta anamnésica), náuseas, pirexia y somnolencia. Específicamente, el primer ensayo fue un ensayo multicéntrico, aleatorizado, doble ciego en 15 sujetos con hemofilia A con inhibidores de los factores VIII. El segundo ensayo fue un estudio multicéntrico de FEIBA realizado en 44 sujetos con hemofilia A con inhibidores, 3 sujetos con hemofilia B con inhibidores y 2 sujetos con inhibidor adquirido del factor VIII. De las 489 infusiones utilizadas para tratar hemorragias agudas durante el segundo ensayo, 18 (3,7%) causaron reacciones transitorias menores de escalofríos, fiebre, náuseas, mareos y disgeusia. De 49 sujetos, 10 (20%) tuvieron un aumento en sus títulos de inhibidores después del tratamiento con FEIBA. Cinco de estos sujetos (50%) tuvieron aumentos de diez veces o más, y 3 (30%) de estos sujetos recibieron concentrados de factor VIII o IX dentro de las 2 semanas previas al tratamiento con FEIBA. Estos aumentos anamnésicos no se asociaron con una disminución de la eficacia de FEIBA.
La Tabla 2 enumera las reacciones adversas en >5% de los sujetos notificadas en el ensayo prospectivo aleatorizado de profilaxis que comparó la profilaxis con FEIBA con el tratamiento a demanda en 36 sujetos con hemofilia A y B con inhibidores de los factores VIII o IX3. La población del ensayo incluyó 33 (92%) sujetos con hemofilia A y 3 (8,3%) sujetos con hemofilia B. Cuatro (11%) sujetos tenían entre 7 y menos de 12 años de edad, 5 (14%) tenían entre 12 y menos de 16 años de edad, y 27 (75%) tenían 16 años de edad o más. Un total de 29 (80,6%) sujetos eran caucásicos, 3 (8,3%) asiáticos, 2 (5,6%) negros/afroamericanos y 2 (5,6%) otros. Los sujetos recibieron un total de 4513 infusiones (3131 para profilaxis y 1382 a demanda).
| Clasificación de órganos por sistemas MedDRA | Reacción adversa | Número de RA | Número de sujetos | Porcentaje de sujetos (N=36) |
|---|---|---|---|---|
| Trastornos sanguíneos y del sistema linfático | Anemia | 2 | 2 | 5.6 |
| Trastornos gastrointestinales | Diarrea | 2 | 2 | 5.6 |
| Náuseas | 2 | 2 | 5.6 | |
| Vómitos | 2 | 2 | 5.6 | |
| Exploraciones complementarias | Anticuerpo de superficie del virus de la hepatitis B positivo | 4 | 4 | 11.1 |
| Trastornos del sistema musculoesquelético y del tejido conjuntivo | Hemartrosis | 5 | 3 | 8.3 |
La Tabla 3 lista las reacciones adversas y los eventos adversos relacionados con la infusión notificados en >5% de los sujetos en cualquiera de los tres grupos (50% de reducción de volumen, aumento de la velocidad a 4 U/kg/min; 50% de reducción de volumen, aumento de la velocidad a 10 U/kg/min; 50% de reducción de volumen general) del ensayo cruzado prospectivo aleatorizado que evalúa la tolerabilidad y la seguridad de la infusión de volumen reducido de FEIBA a la velocidad de infusión estándar de 2 U/kg/min y a velocidades aumentadas de 4 y 10 U/kg/min en un total de 33 sujetos tratados con hemofilia A congénita con inhibidores.
| Clasificación de órganos del sistema MedDRA | Reacción adversa | 50% de reducción de volumen; aumento de la velocidad a 4 U/kg/min (N=30) n (%); m |
50% de reducción de volumen; aumento de la velocidad a 10 U/kg/min (N=28) n (%); m |
General 50% de reducción de volumen* (N=30) n (%); m |
|
|---|---|---|---|---|---|
| n=Número de sujetos, m=número de eventos | |||||
|
|||||
| Reacciones adversas (RAs) | Trastornos del sistema nervioso | Cefalea | 0 | 0 | 2 (6.7%); 2 |
| EAs relacionados con la infusión | Trastornos del sistema nervioso | Cefalea | 0 | 0 | 2 (6.7%); 2 |
6.2 Experiencia postcomercialización
Las siguientes reacciones adversas se han identificado durante el uso posterior a la aprobación de FEIBA. Debido a que la notificación postcomercialización de reacciones adversas es voluntaria y proviene de una población de tamaño incierto, no siempre es posible estimar de forma fiable la frecuencia de estas reacciones o establecer una relación causal con la exposición al producto.
Trastornos de la sangre y del sistema linfático: coagulación intravascular diseminada
Trastornos cardíacos: taquicardia, rubor
Trastornos respiratorios, torácicos y mediastínicos: broncoespasmo, sibilancias
Trastornos gastrointestinales: malestar abdominal
Trastornos de la piel y del tejido subcutáneo: prurito
Trastornos generales y alteraciones en el lugar de administración: malestar general, sensación de calor, dolor en el lugar de inyección
Pruebas complementarias: aumento del dímero D de fibrina
7 INTERACCIONES MEDICAMENTOSAS
7.1 Medications concomitantes
Considere la posibilidad de eventos trombóticos cuando se utilizan antifibrinolíticos sistémicos como el ácido tranexámico y el ácido aminocaproico durante el tratamiento con FEIBA. No se han realizado estudios adecuados y bien controlados del uso combinado o secuencial de FEIBA y antifibrinolíticos de factor VIIa recombinante, o emicizumab. No se recomienda el uso de antifibrinolíticos dentro de aproximadamente 6 a 12 horas después de la administración de FEIBA.
La experiencia clínica de un ensayo clínico con emicizumab sugiere que puede existir una posible interacción medicamentosa con emicizumab cuando se utilizó FEIBA como parte de un régimen de tratamiento para hemorragias irruptivas6 [ver Advertencias y precauciones (5.1)].
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de Riesgo
No hay datos sobre el uso de FEIBA en mujeres embarazadas que informen sobre un riesgo asociado al fármaco. No hay estudios adecuados y bien controlados en mujeres embarazadas. No se han realizado estudios de reproducción animal con FEIBA. Tampoco se sabe si FEIBA puede causar daño fetal cuando se administra a una mujer embarazada o puede afectar la capacidad reproductiva.
En la población general de EE. UU., el riesgo de fondo estimado de defectos congénitos importantes y aborto espontáneo en embarazos clínicamente reconocidos es del 2-4 % y del 15-20 %, respectivamente.
No hay información disponible sobre el efecto de FEIBA en el trabajo de parto y el parto.
8.2 Lactancia
Resumen de Riesgo
No hay información sobre la presencia de FEIBA en la leche materna, el efecto en el niño amamantado o los efectos sobre la producción de leche.
Los beneficios para el desarrollo y la salud de la lactancia materna deben considerarse junto con la necesidad clínica de la madre de FEIBA y cualquier posible efecto adverso en el niño amamantado por FEIBA o por la afección subyacente.
8.4 Uso Pediátrico
La seguridad y eficacia de FEIBA se han evaluado en nueve sujetos pediátricos tratados en el ensayo de profilaxis de rutina, incluidos 4 sujetos de ≥7 a <12 años de edad y 5 sujetos de ≥12 a <16 años de edad. La dosificación para todos los sujetos pediátricos se basó en el peso corporal. Se administraron un total de 576 infusiones para el tratamiento de 223 episodios de hemorragia (504 infusiones para episodios de hemorragia articular, 72 infusiones para episodios de hemorragia muscular y de tejidos blandos). En 223 (100%) de los episodios, se logró la hemostasia con una o más infusiones. La eficacia hemostática se calificó como excelente o buena en la mayoría (96,9%) de los episodios de hemorragia en ambos regímenes a las 24 horas posteriores a la infusión. La tasa de episodios de hemorragia anualizada mediana (ABR) para niños de ≥7 a <12 años de edad fue de 7,7 hemorragias por paciente por año, en comparación con 39 para los sujetos tratados con terapia a demanda [ver Estudios Clínicos (14)].
La seguridad y eficacia de FEIBA no se han evaluado en recién nacidos.
8.5 Uso en Geriatría
La seguridad y eficacia de FEIBA no se han evaluado en sujetos de ≥65 años de edad.
11 DESCRIPCIÓN
FEIBA (Complejo Coagulante Anti-Inhibidor) es una fracción de plasma humano estéril liofilizada con actividad de bypass del inhibidor del factor VIII que se reconstituye para administración intravenosa. La actividad de bypass del inhibidor del factor VIII se expresa en unidades arbitrarias. Una unidad de actividad se define como la cantidad de FEIBA que acorta el aPTT del plasma de referencia del inhibidor del factor VIII de alto título al 50% del valor en blanco.
FEIBA contiene principalmente factores II, IX y X no activados y principalmente factor VII activado. Contiene unidades aproximadamente iguales de actividad de bypass del inhibidor del factor VIII y factores del complejo de protrombina. Además, la preparación contiene de 1 a 6 unidades de antígeno coagulante del factor VIII (FVIII C:Ag) por mL. El producto contiene trazas de factores del sistema generador de quinina. No contiene heparina. FEIBA reconstituido contiene 4 mg de citrato trisódico y 8 mg de cloruro de sodio por mL.
FEIBA se fabrica a partir de grandes grupos de plasma humano. La detección de agentes potencialmente infecciosos comienza con el proceso de selección de donantes y continúa durante la recolección y preparación del plasma. Cada donación de plasma individual utilizada en la fabricación de FEIBA se recolecta en establecimientos de sangre aprobados por la FDA y se analiza mediante pruebas serológicas autorizadas por la FDA para el antígeno de superficie de la hepatitis B (HBsAg) y para anticuerpos contra el virus de la inmunodeficiencia humana (VIH-1/VIH-2) y el virus de la hepatitis C (VHC). Se analizan mini-grupos del plasma y se obtienen resultados negativos para la presencia de VIH-1 y VHC mediante pruebas de ácido nucleico (NAT) autorizadas por la FDA.
Para reducir el riesgo de transmisión viral, el proceso de fabricación de FEIBA incluye dos pasos dedicados e independientes de eliminación/inactivación de virus, a saber, nanofiltración de 35 nm y un proceso de tratamiento con vapor de calor. Además, la adsorción de DEAE-Sephadex contribuye al perfil de seguridad viral de FEIBA.
In vitro se han utilizado estudios de aumento para validar la capacidad del proceso de fabricación para eliminar e inactivar virus. La Tabla 4 resume los resultados de los estudios de eliminación viral para FEIBA.
| Virus Type | Enveloped RNA | Enveloped DNA | Non-enveloped RNA | Non-enveloped DNA | |||
|---|---|---|---|---|---|---|---|
| Virus Family | Retroviridae | Flaviviridae | Herpesviridae | Picornaviridae | Parvoviridae | ||
|
|||||||
| Virus* | HIV-1 | BVDV | WNV | PRV | HAV | B19V† | MMV |
| DEAE Sephadex Adsorption | 3.2 | 1.8 | ND | 2.5 | 1.5 | 1.7 | 1.2 |
| 35 nm Nanofiltration | >5.3 | 2.1 | 4.7 | >5.7 | 2.6 | 0.2‡ | 1.0 |
| Vapor-Heat Treatment | >5.9 | >5.6 | >8.1 | >6.7 | >5.2 | 3.5 | 0.9‡ |
| Overall virus reduction factor (log10) | >14.4 | >9.5 | >12.8 | >14.9 | >9.3 | 5.2 | 2.2 |
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
El mecanismo de acción de FEIBA todavía es objeto de debate científico. FEIBA contiene múltiples componentes, principalmente los factores II, IX y X no activados y principalmente el factor VII activado. Estos factores pueden interactuar con los factores de coagulación plasmática y las plaquetas para aumentar la generación de trombina deteriorada de pacientes hemofílicos con inhibidores, lo que lleva a la hemostasia.
12.2 Farmacodinamia
La evaluación de laboratorio de la coagulación no se correlaciona necesariamente con la eficacia hemostática de FEIBA ni la predice. La actividad de derivación del inhibidor del factor VIII de FEIBA se ha demostrado in vitro así como in vivo. FEIBA puede acortar el tiempo de tromboplastina parcial activada (aPTT) y aumentar la generación de trombina en plasma que contiene inhibidor del factor VIII.4,5
12.3 Farmacocinética
FEIBA se compone de diferentes factores de coagulación, con diferentes vidas medias para los componentes individuales. Las propiedades farmacocinéticas de FEIBA no se han estudiado formalmente en humanos.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
No se han realizado estudios a largo plazo en animales para evaluar el potencial carcinogénico de FEIBA ni estudios para determinar la genotoxicidad o el efecto de FEIBA sobre la fertilidad. Se completó una evaluación del potencial carcinogénico de FEIBA para demostrar un riesgo carcinogénico mínimo derivado del uso del producto.
14 ESTUDIOS CLÍNICOS
Control y Prevención de Episodios Hemorrágicos
La eficacia de FEIBA en el tratamiento de episodios hemorrágicos ha sido demostrada por dos ensayos clínicos prospectivos.1,2
El primer ensayo fue un ensayo multicéntrico, aleatorizado, doble ciego que comparó el efecto de FEIBA y un concentrado de complejo de protrombina no activado en 15 sujetos con hemofilia A e inhibidores del factor VIII. Los criterios de inclusión fueron antecedentes de inhibidores de alto título, estado de alta respuesta, más de 1 episodio hemorrágico por mes en el año anterior y ausencia de signos de insuficiencia hepática. Se trataron un total de 150 episodios hemorrágicos, incluidos 117 articulares, 20 musculoesqueléticos y 4 mucocutáneos. Se utilizó uniformemente una dosis única de 88 unidades por kg de peso corporal para los tratamientos con FEIBA. Se permitió un segundo tratamiento para hemorragias musculares después de 12 horas y 6 horas después de hemorragias mucocutáneas, si fuera necesario.
Se pidió a los sujetos e investigadores que calificaran la eficacia hemostática en función de una escala de eficaz, parcialmente eficaz, no eficaz o no seguro. Los criterios para la evaluación de la eficacia fueron la gravedad del dolor, la mejoría subjetiva, la circunferencia del músculo o la articulación, la restricción de la movilidad articular, el cese de la hemorragia abierta, el inicio de una hemorragia recurrente y la cantidad y la naturaleza de los analgésicos. FEIBA fue eficaz en el 41% y parcialmente eficaz en el 25% de los episodios (es decir, una eficacia combinada del 66%), mientras que el concentrado de complejo de protrombina se calificó como eficaz en el 25% y parcialmente eficaz en el 21% de los episodios (es decir, una eficacia combinada del 46%).
El segundo ensayo con FEIBA fue un ensayo multicéntrico, aleatorizado y prospectivo. Este ensayo se realizó en 44 sujetos con hemofilia A e inhibidores, 3 sujetos con hemofilia B e inhibidores y 2 sujetos con inhibidor adquirido del factor VIII. Se diseñó para evaluar la eficacia de FEIBA en el tratamiento de episodios hemorrágicos articulares, de membranas mucosas, musculocutáneos y de emergencia, como hemorragias del sistema nervioso central y hemorragias quirúrgicas. Los criterios de inclusión utilizados fueron edad >4 años, antecedentes de título de inhibidor ≥4 Unidades de Bethesda (UB) y sin enfermedad hepática crónica. Se excluyeron los sujetos que tenían antecedentes de eventos tromboembólicos o reacciones alérgicas a FEIBA.
Se inscribieron cuarenta y nueve (49) sujetos con títulos de inhibidores superiores a 5 UB de nueve centros de hemofilia cooperantes. Los sujetos fueron tratados con 50 unidades por kg de peso corporal, repetidas a intervalos de 12 horas (intervalos de 6 horas en hemorragias de membrana mucosa), si fuera necesario. Se administraron un total de 489 infusiones para el tratamiento de 165 episodios hemorrágicos (102 articulares, 33 musculares y de tejidos blandos, 20 de membrana mucosa y 10 de emergencia, incluidos 3 hemorragias del sistema nervioso central y 4 procedimientos quirúrgicos). La hemorragia se controló en 153 episodios (93%). En 130 (78%) de los episodios, se logró la hemostasia con una o más infusiones en 36 horas. De estos, el 36% se controló con una infusión en 12 horas. Un 14% adicional de los episodios respondió después de más de 36 horas.
Profilaxis Rutinaria
En un ensayo clínico multicéntrico, abierto, prospectivo y aleatorizado que comparó a sujetos que recibieron FEIBA para profilaxis con sujetos que recibieron FEIBA para tratamiento a demanda, se analizaron 36 sujetos con hemofilia A y B e inhibidores del factor VIII o IX en el análisis por intención de tratar. La población del estudio incluyó 29 (80,6%) caucásicos, 3 (8,3%) asiáticos, 2 (5,6%) negros/afroamericanos y 2 (5,6%) otros. Los criterios de inclusión fueron sujetos con antecedentes de inhibidores de alto título o de bajo título refractarios al aumento de la dosis de factor VIII o IX, rango de edad entre 4 y 65 años, y sujetos que recibían agentes de derivación con ≥12 hemorragias en los 12 meses anteriores a la inclusión en el ensayo. Se excluyeron los sujetos con antecedentes de eventos tromboembólicos, enfermedad hepática sintomática o un recuento plaquetario <100.000 por mL, y aquellos que recibían inducción de tolerancia inmunitaria o profilaxis rutinaria.
Los sujetos fueron aleatorizados para recibir 12 meses de tratamiento profiláctico o a demanda con FEIBA. Diecisiete sujetos aleatorizados al brazo de profilaxis recibieron 85 unidades por kg de FEIBA cada dos días. Diecinueve sujetos aleatorizados al brazo a demanda recibieron FEIBA para el tratamiento de episodios hemorrágicos agudos según la dosis y el régimen de dosificación recomendados. Las articulaciones diana se definieron como ≥4 episodios hemorrágicos en 6 meses. En este ensayo, los tobillos, las rodillas, los codos y las caderas fueron las localizaciones de las articulaciones diana. Las articulaciones diana preexistentes no se consideraron como nuevas articulaciones diana.
La eficacia hemostática para el tratamiento de hemorragias agudas se evaluó a las 6 y 24 horas según una escala de cuatro puntos preespecificada de excelente, buena, regular o ninguna. Una evaluación de “ninguna” se consideró un fracaso del tratamiento. Los criterios para la evaluación de la eficacia fueron el alivio del dolor, el cese de la hemorragia y el número de infusiones necesarias para tratar una hemorragia.
Se notificaron un total de 825 episodios hemorrágicos, incluidos 196 que ocurrieron durante la profilaxis y 629 que ocurrieron durante la terapia a demanda. La mayoría (78%) de los 794 episodios hemorrágicos que se calificaron en cuanto a eficacia se trataron con 1 o 2 infusiones. La eficacia hemostática se calificó como excelente o buena para el 74% de los episodios hemorrágicos calificados a las 6 horas posteriores a la infusión y para el 87% de los episodios hemorrágicos a las 24 horas posteriores a la infusión. Un total de 19 (2,4%) hemorragias se calificaron como “ninguna” a las 6 horas posteriores a la infusión; 1 hemorragia (0,1%) se calificó como “ninguna” a las 24 horas.
La eficacia hemostática para la profilaxis rutinaria se evaluó en comparación con los sujetos que recibieron terapia a demanda.
La mediana general de la tasa anual de hemorragias (TAH) para el brazo a demanda fue de 28,7 en comparación con 7,9 para el brazo de profilaxis, lo que representa una reducción del 72% en la mediana de la TAH con profilaxis. Cuando se analizó por sitio (por ejemplo, articular, no articular) y causa de la hemorragia (por ejemplo, espontánea, traumática), el tratamiento profiláctico con FEIBA dio como resultado una reducción de más del 50% en la TAH. Hubo menos sujetos en el brazo de profilaxis que desarrollaron nuevas articulaciones diana (7 nuevas articulaciones diana en 5 sujetos tratados con profilaxis en comparación con 23 nuevas articulaciones diana en 11 sujetos en el brazo a demanda). Se desarrollaron articulaciones diana en dos sujetos en el brazo a demanda y tres en el brazo de profilaxis que no tenían articulaciones diana notificadas al inicio del ensayo. Un total de 3 de 17 (18%) sujetos no tuvieron episodios hemorrágicos con profilaxis. En el brazo a demanda, todos los sujetos experimentaron un episodio hemorrágico.
La TAH por categoría de edad entre los regímenes a demanda y de profilaxis se proporciona en la Tabla 5. Un sujeto adolescente con profilaxis tuvo una mayor tasa de hemorragia, posiblemente debido al aumento de la actividad física después de la inclusión en el estudio.
| Categoría de edad | Según necesidad: Número de sujetos |
Según necesidad: Mediana de ABR |
Profilaxis: Número de sujetos |
Profilaxis: Mediana de ABR |
|---|---|---|---|---|
| Niños (≥7 a <12 años) |
2 | 39.3 | 2 | 7.7 |
| Adolescentes (≥12 a <16 años) |
2 | 30.9 | 3 | 27.5 |
| Adultos (≥16 años) |
15 | 23.9 | 12 | 6.9 |
15 REFERENCIAS
- Sjamsoedin LJ, Heijnen L, Mauser-Bunschoten EP, van Geijlswijk JL, van Houwelingen H, van Asten P, Sixma JJ. El efecto del Concentrado de Complejo de Protombina Activada (FEIBA) en el sangrado articular y muscular en pacientes con Hemofilia A y anticuerpos contra el Factor VIII. N Engl J Med. 1981;305(13): 717-721.
- Hilgartner MW, Knatterud GL. El uso del producto de Actividad de Derivación del Inhibidor del Factor Ocho (FEIBA IMMUNO) para el tratamiento de episodios hemorrágicos en hemofílicos con inhibidores. Blood. 1983;61(1): 36-40.
- Antunes SV, Tangada S, Stasyshyn O, Mamonov V, Phillips J, Guzman-Becerra N, Grigorian A, Ewenstein B, Wong WY. Comparación aleatorizada de regímenes profilácticos y a demanda con FEIBA NF en el tratamiento de la hemofilia A y B con inhibidores. Haemophilia. 2013; DOI 10.1111/hae.12246.
- Turecek PL, Varadi K, Gritsch H, Auer W, Picler L, Eder G, Schwarz HP. Factor Xa y protrombina: mecanismo de acción de FEIBA. Vox Sanguinis 1999;77 Suppl 1:72-79.
- Turecek PL, Varadi K, Gritch H, Schwarz HP. FEIBA: Modo de acción Haemophilia 2004;10: Suppl. 2:3-9
- Oldenburg et al. Profilaxis con Emicizumab en la Hemofilia A con Inhibidores. N Engl J Med 2017:377:809-818.
16 SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
Cómo se Suministra/Presentación
FEIBA está disponible en viales de dosis única en las siguientes concentraciones de dosis nominales:
| Concentración Nominal | Código de Color | Rango de Potencia del Factor VIII | Kit NDC | Volumen de Agua Estéril |
|---|---|---|---|---|
| 500 units | Naranja | 350-650 units per vial | 64193-426-02 | 10 mL |
| 1000 units | Verde | 700-1300 units per vial | 64193-424-02 | 20 mL |
| 2500 units | Morado | 1750-3250 units per vial | 64193-425-02 | 50 mL |
El número de unidades de actividad de bypass del inhibidor del factor VIII se indica en la etiqueta de cada vial.
FEIBA se envasa con un volumen adecuado (10 mL, 20 mL o 50 mL) de agua estéril para inyección, U.S.P., un dispositivo de transferencia sin aguja BAXJECT II Hi-Flow y un prospecto.
No fabricado con látex de caucho natural.
Almacenamiento y Manipulación
- Almacenar a una temperatura de 2°C a 25°C (36°F a 77°F)
- Almacenar en el envase original para protegerlo de la luz.
- No congelar.
- Antes de la preparación y reconstitución, deje que los viales de FEIBA y agua estéril para inyección (diluyente) alcancen la temperatura ambiente, si están refrigerados.
17 INFORMACIÓN DE ASESORAMIENTO AL PACIENTE
Informar a los pacientes:
- sobre los signos y síntomas de trombosis, como dolor o presión en el pecho, dificultad para respirar, alteración de la conciencia, la visión o el habla, hinchazón y/o dolor en las extremidades o el abdomen. Aconsejar a los pacientes que busquen atención médica inmediata si se produce alguno de estos síntomas.
- sobre los signos y síntomas de reacciones de hipersensibilidad, como urticaria, angioedema, manifestaciones gastrointestinales, broncoespasmo e hipotensión. Aconsejar a los pacientes que interrumpan el uso del producto si se producen estos síntomas y que busquen tratamiento de emergencia inmediato.
- que debido a que FEIBA se fabrica a partir de sangre humana, puede conllevar un riesgo de transmisión de agentes infecciosos, por ejemplo, virus, el agente de la enfermedad de Creutzfeldt-Jakob variante (vCJD) y, teóricamente, el agente de la enfermedad de Creutzfeldt-Jakob (CJD).
- si están en terapia profiláctica con emicizumab y necesitan FEIBA para tratar un episodio de hemorragia irruptiva, entonces deben ser controlados por su médico tratante de hemofilia, preferiblemente en el centro de tratamiento de hemofilia (HTC).
- que informen de cualquier reacción adversa o problema después de la administración de FEIBA a su médico tratante de hemofilia.
Para inscribirse en el Sistema de Notificación al Paciente confidencial de toda la industria, llame al 1-888-873-2838.
SECCIÓN NO CLASIFICADA DE SPL
Takeda Pharmaceuticals U.S.A., Inc.
Cambridge, MA 02142
Licencia de EE. UU. N.º 1898
FEIBA es una marca registrada de Baxalta Incorporated.
Takeda y ![]() son marcas registradas de Takeda Pharmaceutical Company Limited.
son marcas registradas de Takeda Pharmaceutical Company Limited.
©2024 Takeda Pharmaceutical Company Limited. Todos los derechos reservados.
PANEL PRINCIPAL DE VISUALIZACIÓN – Caja del Kit
20 mL liofilizado
NDC 64193-424-02
Complejo Coagulante Anti-Inhibidor
FEIBA
Nanofiltrado y Calentado por Vapor
Para uso intravenoso después de la reconstitución
No contiene conservantes
Dosis y administración: Ver el prospecto adjunto.
Almacenamiento: 2°C a 25°C. Almacenar en el envase original para protegerlo de la luz.
Reconstitución: Usar 20 mL de Agua estéril para inyección (API), USP; No refrigerar después de la reconstitución; Usar dentro de las 3 horas posteriores a la
reconstitución.
No congelar
Rx solamente
Takeda
Incluye
Dispositivo de transferencia sin aguja BAXJECT II Hi-Flow

PRINCIPAL PANEL DE PRESENTACIÓN – Etiqueta de vial de 20 mL
20 mL tamaño, liofilizado
NDC 64193-324-01
Complejo Coagulante Anti-Inhibidor
FEIBA
Nanofiltrado & Calentado por Vapor
Para uso intravenoso después de la reconstitución
No contiene conservantes
Dosis y Administración: Ver el prospecto adjunto.
Almacenamiento: 36°F a 77°F (2°C a 25°C).
Reconstitución: Usar 20 mL de Agua estéril para inyección (API), USP; No refrigerar
después de la reconstitución; Usar dentro de las 3 horas posteriores a la reconstitución.
Rx solamente
0754401

PANEL PRINCIPAL DE VISUALIZACIÓN – Caja del Kit
Frasco de dosis única
NDC 64193-425-02
Anti-Inhibitor Coagulant Complex
FEIBA
Nanofiltrado y tratado con vapor
Para uso intravenoso después de la reconstitución
No contiene conservantes
Dosis y administración: Consulte el prospecto adjunto.
Almacenamiento: 36°F a 77°F (2°C a 25°C).
Almacenar en el envase original para protegerlo de la luz.
Reconstitución: Use 50 mL sWFI, U.S.P.; No refrigerar después de la reconstitución.; Usar dentro de las 3 horas posteriores a la
reconstitución.
No congelar
Rx Only
Takeda
Incluye
BAXJECT II Hi-Flow
Dispositivo de transferencia sin aguja

ETIQUETA PRINCIPAL DEL PANEL – Vial de 50 mL
Presentación de 50 mL, desecado
NDC 64193-325-01
Anti-Inhibitor Coagulant Complex
FEIBA
Nanofiltrado y Tratado con Vapor
Para uso intravenoso después de la reconstitución
No contiene conservantes
Dosis y Administración: Consultar el prospecto adjunto.
Almacenamiento: 36°F a 77°F (2°C a 25°C).
Reconstitución: Usar 50 mL de sWFI, U.S.P.; No refrigerar
después de la reconstitución.; Usar dentro de las 3 horas posteriores a la reconstitución.
Rx only
0754404

PANEL PRINCIPAL DE VISUALIZACIÓN – Caja del Kit
Vial de dosis única
NDC 64193-426-02
Complejo Coagulante Anti-Inhibidor
FEIBA
Nanofiltrado y Calentado con Vapor
Para uso intravenoso después de la reconstitución
No contiene conservantes
Dosis y administración: Ver el prospecto adjunto.
Almacenamiento: 2°C a 25°C (36°F a 77°F). Almacenar en el envase original para proteger
de la luz.
Reconstitución: Usar 10 mL de Agua estéril para inyección (API), USP; No refrigerar después de la reconstitución; Usar dentro de
3 horas de la reconstitución.
No congelar
Rx solamente
Takeda
Incluye
Dispositivo de transferencia sin aguja BAXJECT II Hi-Flow

Panel de visualización principal – Etiqueta de frasco de 10 mL
Vial de dosis única
NDC 64193-326-01
Complejo Coagulante Anti-Inhibidor
FEIBA
Nanofiltrado y Calentado con Vapor
Para uso intravenoso después de la reconstitución
Sin conservante
Dosis y administración: Ver el prospecto adjunto.
Almacenamiento: 36°F a 77°F (2°C a 25°C).
Reconstitución: Usar 10 mL de Agua estéril para inyección (API), USP; No refrigerar después de la
reconstitución; Usar dentro de las 3 horas posteriores a la reconstitución.
Rx solamente
Takeda
0754406
