Fabricante de medicamentos: Regeneron Pharmaceuticals, Inc. (Updated: 2024-10-10)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
EYLEA® (aflibercept) inyección, para uso intravítreo
Aprobación inicial en EE. UU.: 2011
CAMBIOS RECIENTES IMPORTANTES
| Advertencias y precauciones (5.1) | 12/2023 |
INDICACIONES Y USO
EYLEA es un inhibidor del factor de crecimiento endotelial vascular (VEGF) indicado para el tratamiento de pacientes con:
DOSIFICACIÓN Y ADMINISTRACIÓN
-
Degeneración macular neovascular (húmeda) relacionada con la edad (DMAE)
- La dosis recomendada para EYLEA es de 2 mg (0,05 mL de solución de 40 mg/mL) administrada por inyección intravítrea cada 4 semanas (aproximadamente cada 28 días, mensualmente) durante los primeros 3 meses, seguida de 2 mg (0,05 mL de solución de 40 mg/mL) por inyección intravítrea una vez cada 8 semanas (2 meses). (2.5)
- Aunque EYLEA puede dosificarse con la misma frecuencia que 2 mg cada 4 semanas (aproximadamente cada 25 días, mensualmente), no se demostró una eficacia adicional en la mayoría de los pacientes cuando EYLEA se dosificó cada 4 semanas en comparación con cada 8 semanas. Algunos pacientes pueden necesitar una dosificación cada 4 semanas (mensualmente) después de las primeras 12 semanas (3 meses). (2.5)
- Aunque no es tan eficaz como el régimen de dosificación recomendado cada 8 semanas, los pacientes también pueden tratarse con una dosis cada 12 semanas después de un año de terapia eficaz. Los pacientes deben ser evaluados regularmente. (2.5)
-
Edema macular después de la oclusión de la vena retiniana (OVR)
- La dosis recomendada para EYLEA es de 2 mg (0,05 mL de solución de 40 mg/mL) administrada por inyección intravítrea una vez cada 4 semanas (aproximadamente cada 25 días, mensualmente). (2.6)
-
Edema macular diabético (EMD) y retinopatía diabética (RD)
- La dosis recomendada para EYLEA es de 2 mg (0,05 mL de solución de 40 mg/mL) administrada por inyección intravítrea cada 4 semanas (aproximadamente cada 28 días, mensualmente) durante las primeras 5 inyecciones, seguida de 2 mg (0,05 mL de solución de 40 mg/mL) por inyección intravítrea una vez cada 8 semanas (2 meses). (2.7, 2.8)
- Aunque EYLEA puede dosificarse con la misma frecuencia que 2 mg cada 4 semanas (aproximadamente cada 25 días, mensualmente), no se demostró una eficacia adicional en la mayoría de los pacientes cuando EYLEA se dosificó cada 4 semanas en comparación con cada 8 semanas. Algunos pacientes pueden necesitar una dosificación cada 4 semanas (mensualmente) después de las primeras 20 semanas (5 meses). (2.7, 2.8)
-
Retinopatía del prematuro (ROP)
- La dosis recomendada para EYLEA es de 0,4 mg (0,01 mL o 10 microlitros de solución de 40 mg/mL) administrada por inyección intravítrea. El tratamiento puede administrarse bilateralmente el mismo día. Las inyecciones pueden repetirse en cada ojo. El intervalo de tratamiento entre las dosis inyectadas en el mismo ojo debe ser de al menos 10 días. (2.9)
FORMAS DE DOSIFICACIÓN Y FUERZAS
CONTRAINDICACIONES
ADVERTENCIAS Y PRECAUCIONES
- La endoftalmitis, los desprendimientos de retina y la vasculitis retiniana con o sin oclusión pueden ocurrir después de las inyecciones intravítreas. Los pacientes y/o cuidadores deben recibir instrucciones para informar cualquier signo y/o síntoma sugestivo de endoftalmitis, desprendimiento de retina o vasculitis retiniana sin demora y deben ser manejados adecuadamente. (5.1)
- Se han observado aumentos en la presión intraocular dentro de los 60 minutos posteriores a una inyección intravítrea. (5.2)
- En los bebés con ROP, el tratamiento con EYLEA requerirá períodos prolongados de monitoreo de ROP (5.3)
- Existe un riesgo potencial de eventos tromboembólicos arteriales después del uso intravítreo de inhibidores del VEGF. (5.4)
REACCIONES ADVERSAS
Las reacciones adversas más comunes (≥5%) informadas en pacientes que recibieron EYLEA fueron hemorragia conjuntival, dolor ocular, catarata, desprendimiento vítreo, flotadores vítreos y aumento de la presión intraocular. (6.1)
Para informar las REACCIONES ADVERSAS SOSPECHOSAS, comuníquese con Regeneron al 1-855-395-3248 o con la FDA al 1-800-FDA-1088 o www.fda.gov/medwatch.
Ver 17 para INFORMACIÓN PARA EL PACIENTE.
Revisado: 10/2024
Tabla de Contenido
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICACIONES Y USO
1.1 Neovascular (Wet) Age-Related Macular Degeneration (AMD)
1.2 Macular Edema Following Retinal Vein Occlusion (RVO)
1.3 Diabetic Macular Edema (DME)
1.4 Diabetic Retinopathy (DR)
1.5 Retinopathy of Prematurity (ROP)
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Important Injection Instructions
2.2 Preparación para la administración – Jeringa precargada
2.3 Preparación para la administración – Vial
2.4 Injection Procedure for Adults
2.5 Neovascular (Wet) Age-Related Macular Degeneration (AMD)
2.6 Macular Edema Following Retinal Vein Occlusion (RVO)
2.7 Diabetic Macular Edema (DME)
2.8 Diabetic Retinopathy (DR)
2.9 Retinopathy of Prematurity (ROP)
3 FORMAS FARMACÉUTICAS Y CONCENTRACIONES
4 CONTRAINDICACIONES
4.1 Ocular or Periocular Infections
4.2 Active Intraocular Inflammation
4.3 Hypersensitivity
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Endophthalmitis, Retinal Detachments, and Retinal Vasculitis with or without Occlusion
5.2 Increase in Intraocular Pressure
5.3 Extended Monitoring and Additional Treatment in ROP
5.4 Thromboembolic Events
6 REACCIONES ADVERSAS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.3 Mujeres y hombres en edad fértil
8.4 Uso pediátrico
8.5 Uso geriátrico
10 SOBREDOSIS
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.6 Immunogenicity
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 ESTUDIOS CLÍNICOS
14.1 Neovascular (Wet) Age-Related Macular Degeneration (AMD)
14.2 Macular Edema Following Central Retinal Vein Occlusion (CRVO)
14.3 Macular Edema Following Branch Retinal Vein Occlusion (BRVO)
14.4 Diabetic Macular Edema (DME)
14.5 Diabetic Retinopathy (DR)
14.6 Retinopathy of Prematurity (ROP)
16 CÓMO SUMINISTRARLO/ALMACENAMIENTO Y MANIPULACIÓN
16.1 Cómo suministrarlo
16.2 Almacenamiento y manipulación
17 INFORMACIÓN DE ASESORAMIENTO AL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información de prescripción completa no se enumeran.
1 INDICACIONES Y USO
EYLEA está indicado para el tratamiento de:
2 DOSIS Y ADMINISTRACIÓN
2.1 Instrucciones Importantes de Inyección
Para inyección intravítrea oftálmica. EYLEA solo debe ser administrado por un médico cualificado.
Jeringa precargada: Se necesita una aguja de inyección estéril de 30-gauge × ½-pulgada, pero no se proporciona.
Vial: Se necesita una aguja de filtro estéril de 5 micrones (18-gauge × 1½-pulgada), una jeringa Luer lock de 1 mL y una aguja de inyección estéril de 30-gauge × ½-pulgada.
EYLEA está disponible envasado de la siguiente manera:
- Jeringa precargada
- Kit de vial con componentes de inyección (aguja de filtro, jeringa, aguja de inyección)
2.2 Preparación para la Administración – Jeringa precargada
La jeringa de vidrio precargada de EYLEA es estéril y para un solo uso en un solo ojo. No use la jeringa precargada de EYLEA para el tratamiento de la ROP.
La jeringa precargada debe inspeccionarse visualmente antes de la administración. No la use si se observan partículas, turbidez o decoloración, o si el envase está abierto o dañado. La apariencia de la tapa de la jeringa en la jeringa precargada puede variar (por ejemplo, color y diseño). No la use si alguna parte de la jeringa precargada está dañada o si la tapa de la jeringa está separada de la conexión Luer.
La inyección intravítrea debe realizarse con una aguja de inyección de 30-gauge × ½-pulgada (no proporcionada).
La jeringa precargada contiene más de la dosis recomendada de 2 mg de aflibercept (equivalente a 50 microlitros). El exceso de volumen debe desecharse antes de la administración.
DESCRIPCIÓN DE LA JERINGA PRECARGADA – Figura 1:
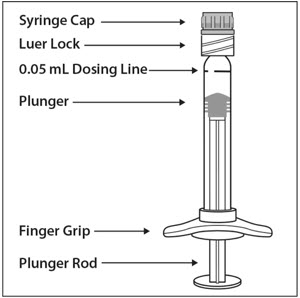
Utilice una técnica aséptica para llevar a cabo los siguientes pasos:
1. PREPARAR
Cuando esté listo para administrar EYLEA, abra la caja y retire el blíster esterilizado. Abra cuidadosamente el blíster esterilizado asegurándose de la esterilidad de su contenido. Mantenga la jeringa en la bandeja estéril hasta que esté listo para el montaje.
2. RETIRAR LA JERINGA
Utilizando una técnica aséptica, retire la jeringa del blíster esterilizado.
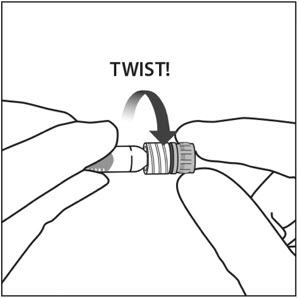
3. GIRAR LA TAPA DE LA JERINGA
Gire (no rompa) la tapa de la jeringa sujetando la jeringa con una mano y la tapa de la jeringa con el pulgar y el índice de la otra mano (ver Figura 2).
Nota: Para evitar comprometer la esterilidad del producto, no tire del émbolo.
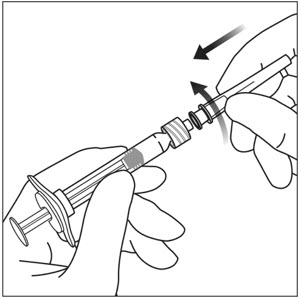
4. FIJAR LA AGUJA
Utilizando una técnica aséptica, gire firmemente una aguja de inyección de 30-gauge × ½-pulgada en la punta de la jeringa Luer lock (ver Figura 3).
Nota: Cuando esté listo para administrar EYLEA, retire el protector de plástico de la aguja.
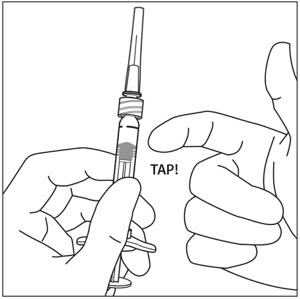
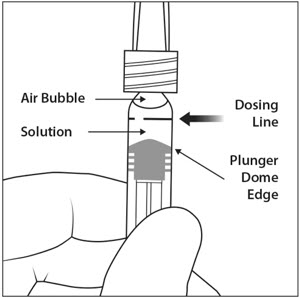
5. DESALOJAR LAS BURBUJAS DE AIRE
Sosteniendo la jeringa con la aguja apuntando hacia arriba, compruebe la jeringa para ver si hay burbujas. Si hay burbujas, golpee suavemente la jeringa con el dedo hasta que las burbujas suban a la superficie (ver Figura 4).
6. EXPULSAR EL AIRE Y AJUSTAR LA DOSIS
Para eliminar todas las burbujas y expulsar el exceso de fármaco, presione lentamente el émbolo hasta alinear el borde del domo del émbolo (ver Figura 5a) con la línea de dosificación negra de la jeringa (equivalente a 50 microlitros) (ver Figura 5b).
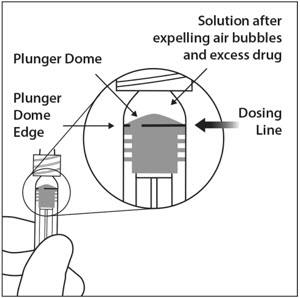
7. La jeringa precargada es para un solo uso en un solo ojo. Después de la inyección, cualquier producto no utilizado debe desecharse.
2.3 Preparación para la Administración – Vial
EYLEA debe inspeccionarse visualmente antes de la administración. Si se observan partículas, turbidez o decoloración, el vial no debe utilizarse.
El vial de vidrio es para un solo uso en un solo ojo.
Utilice una técnica aséptica para llevar a cabo los siguientes pasos de preparación:
Prepárese para la inyección intravítrea con los siguientes dispositivos médicos de un solo uso:
- una aguja de filtro estéril de 5 micras (18-gauge × 1½-pulgada)
- una jeringa estéril Luer lock de 1 mL con marcas para medir 0,05 mL para adultos o 0,01 mL para bebés prematuros con ROP
- una aguja de inyección estéril (30-gauge × ½-pulgada)
- Retire la tapa de plástico protectora del vial (ver Figura 6).
Figura 6: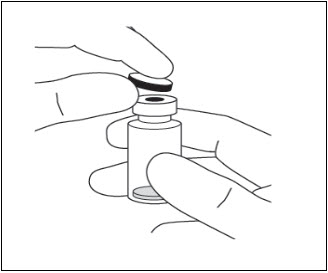
- Limpie la parte superior del vial con una toallita con alcohol (ver Figura 7).
Figura 7: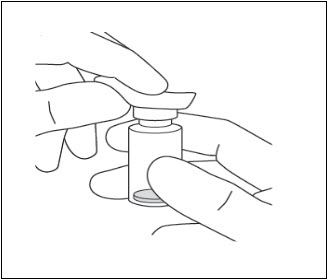
- Retire la aguja de filtro de 18-gauge × 1½-pulgada, 5 micras y la jeringa de 1 mL de su embalaje. Conecte la aguja de filtro a la jeringa girándola en la punta de la jeringa Luer lock (ver Figura 8).
- Introduzca la aguja de filtro en el centro del tapón del vial hasta que la aguja esté completamente insertada en el vial y la punta toque el fondo o el borde inferior del vial.
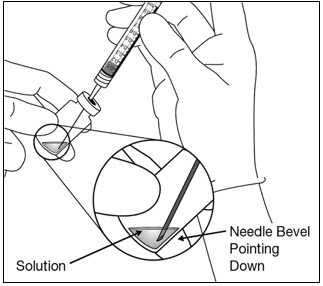
- Utilizando una técnica aséptica, retire todo el contenido del vial de EYLEA en la jeringa, manteniendo el vial en posición vertical, ligeramente inclinado para facilitar la extracción completa. Para evitar la introducción de aire, asegúrese de que el bisel de la aguja de filtro esté sumergido en el líquido. Continúe inclinando el vial durante la extracción manteniendo el bisel de la aguja de filtro sumergido en el líquido (ver Figura 9a y Figura 9b).
Figura 9a: Figura 9b: 
2.2 Preparación para la Administración
- Retire la tapa del vial de EYLEA (ver Figura 8).
- Retire la tapa de la jeringa precargada de EYLEA (ver Figura 9).
- Conecte la aguja de filtro a la jeringa precargada (ver Figura 9).
- Inserte la aguja de filtro en el vial de EYLEA. Asegúrese de que la aguja de filtro esté completamente insertada en el vial.
- Tire lentamente del émbolo de la jeringa hasta que el vial esté completamente vacío. Asegúrese de que el émbolo se tire lo suficiente hacia atrás al vaciar el vial para vaciar completamente la aguja de filtro.
- Retire la aguja de filtro de la jeringa y deseche la aguja de filtro de forma adecuada. Nota: La aguja de filtro no debe utilizarse para la inyección intravítrea.
- Retire la aguja de inyección de 30-gauge × ½-pulgada de su embalaje y conecte la aguja de inyección a la jeringa girando firmemente la aguja de inyección en la punta de la jeringa Luer lock (ver Figura 10).
- Cuando esté listo para administrar EYLEA, retire el protector de plástico de la aguja.
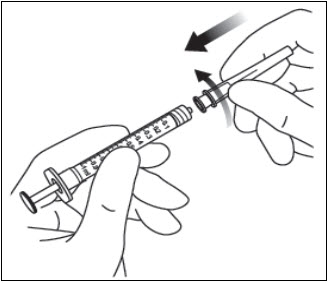
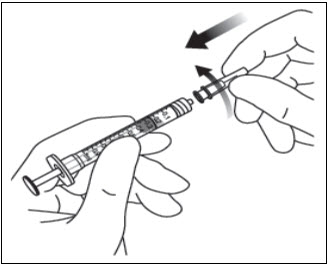
- Sosteniendo la jeringa con la aguja apuntando hacia arriba, revise la jeringa para ver si hay burbujas. Si hay burbujas, golpee suavemente la jeringa con el dedo hasta que las burbujas suban a la superficie (ver Figura 11).
Figura 11: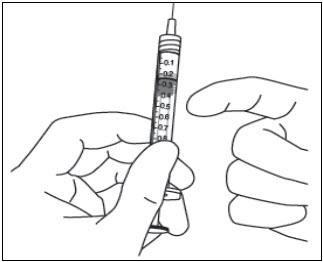
Administración en Adultos:
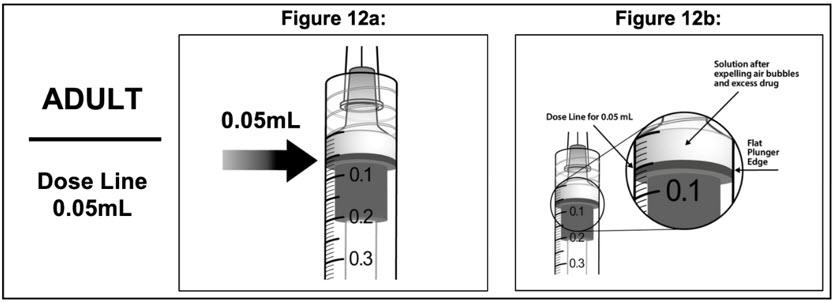
- Para eliminar todas las burbujas y expulsar el exceso de medicamento, presione LENTAMENTE el émbolo de la jeringa hasta que el borde del émbolo se alinee con la línea que marca 0.05 mL en la jeringa (ver Figura 12a y Figura 12b).
2.4 Procedimiento de Inyección para Adultos
El procedimiento de inyección intravítrea debe llevarse a cabo en condiciones asépticas controladas, que incluyen la desinfección quirúrgica de las manos y el uso de guantes estériles, una sábana estéril y un espéculo de párpados estéril (o equivalente). Se debe administrar anestesia adecuada y un microbicida de amplio espectro tópico antes de la inyección.
Jeringa precargada: Inyecte presionando el émbolo con cuidado y con presión constante. No aplique presión adicional una vez que el émbolo haya llegado al fondo de la jeringa. Puede quedar un pequeño volumen residual en la jeringa después de que se haya inyectado una dosis completa. Esto es normal. No administre ninguna solución residual que se observe en la jeringa.
Inmediatamente después de la inyección intravítrea, los pacientes deben ser monitoreados para detectar una elevación de la presión intraocular. El monitoreo adecuado puede consistir en una verificación de la perfusión de la cabeza del nervio óptico o tonometría. Si es necesario, debe estar disponible una aguja de paracentesis estéril.
Después de la inyección intravítrea, se debe instruir a los pacientes y/o cuidadores para que informen cualquier signo y/o síntoma sugestivo de endoftalmitis o desprendimiento de retina (por ejemplo, dolor ocular, enrojecimiento del ojo, fotofobia, visión borrosa) sin demora [ver Información para el Paciente (17)].
Cada jeringa precargada o vial estéril solo debe utilizarse para el tratamiento de un solo ojo. Si el ojo contralateral requiere tratamiento, se debe utilizar una nueva jeringa precargada o vial estéril y el campo estéril, la jeringa, los guantes, las sábanas, el espéculo de párpados, el filtro y las agujas de inyección deben cambiarse antes de administrar EYLEA al otro ojo.
Después de la inyección, cualquier producto no utilizado debe desecharse.
2.5 Degeneración Macular Asociada a la Edad (DMAE) Neovascular (Húmeda)
La dosis recomendada para EYLEA es de 2 mg (0.05 mL de solución de 40 mg/mL) administrada mediante inyección intravítrea cada 4 semanas (aproximadamente cada 28 días, mensualmente) durante las primeras 12 semanas (3 meses), seguida de 2 mg (0.05 mL de solución de 40 mg/mL) mediante inyección intravítrea una vez cada 8 semanas (2 meses). Aunque EYLEA puede dosificarse con la misma frecuencia que 2 mg cada 4 semanas (aproximadamente cada 25 días, mensualmente), no se demostró una eficacia adicional en la mayoría de los pacientes cuando EYLEA se dosificó cada 4 semanas en comparación con cada 8 semanas [ver Estudios Clínicos (14.1)]. Algunos pacientes pueden necesitar una dosificación cada 4 semanas (mensualmente) después de las primeras 12 semanas (3 meses). Aunque no es tan eficaz como el régimen de dosificación recomendado cada 8 semanas, los pacientes también pueden tratarse con una dosis cada 12 semanas después de un año de terapia eficaz. Los pacientes deben ser evaluados regularmente.
2.6 Edema Macular Posterior a la Oclusión de la Vena Retiniana (OVR)
La dosis recomendada para EYLEA es de 2 mg (0.05 mL de solución de 40 mg/mL) administrada mediante inyección intravítrea una vez cada 4 semanas (aproximadamente cada 25 días, mensualmente) [ver Estudios Clínicos (14.2), (14.3)].
2.7 Edema Macular Diabético (EMD)
La dosis recomendada para EYLEA es de 2 mg (0.05 mL de solución de 40 mg/mL) administrada mediante inyección intravítrea cada 4 semanas (aproximadamente cada 28 días, mensualmente) durante las primeras 5 inyecciones, seguida de 2 mg (0.05 mL de solución de 40 mg/mL) mediante inyección intravítrea una vez cada 8 semanas (2 meses). Aunque EYLEA puede dosificarse con la misma frecuencia que 2 mg cada 4 semanas (aproximadamente cada 25 días, mensualmente), no se demostró una eficacia adicional en la mayoría de los pacientes cuando EYLEA se dosificó cada 4 semanas en comparación con cada 8 semanas [ver Estudios Clínicos (14.4)]. Algunos pacientes pueden necesitar una dosificación cada 4 semanas (mensualmente) después de las primeras 20 semanas (5 meses).
2.8 Retinopatía Diabética (RD)
La dosis recomendada para EYLEA es de 2 mg (0,05 mL de solución de 40 mg/mL) administrada por inyección intravítrea cada 4 semanas (aproximadamente cada 28 días, mensualmente) durante las primeras 5 inyecciones, seguida de 2 mg (0,05 mL de solución de 40 mg/mL) por inyección intravítrea una vez cada 8 semanas (2 meses). Aunque EYLEA puede administrarse con una frecuencia de 2 mg cada 4 semanas (aproximadamente cada 25 días, mensualmente), no se demostró una eficacia adicional en la mayoría de los pacientes cuando EYLEA se administró cada 4 semanas en comparación con cada 8 semanas [ver Estudios Clínicos (14.5)]. Algunos pacientes pueden necesitar una dosificación cada 4 semanas (mensualmente) después de las primeras 20 semanas (5 meses).
2.9 Retinopatía del Prematuro (ROP)
Dosificación Recomendada
La dosis recomendada para EYLEA es de 0,4 mg (0,01 mL o 10 microlitros de solución de 40 mg/mL) administrada por inyección intravítrea. El tratamiento se inicia con una sola inyección por ojo elegible y puede administrarse bilateralmente el mismo día. Las inyecciones pueden repetirse en cada ojo. El intervalo de tratamiento entre las dosis inyectadas en el mismo ojo debe ser de al menos 10 días [ver Farmacología Clínica (12.3) y Estudios Clínicos (14.6)].
Instrucciones de Administración en Bebés Prematuros con ROP
El procedimiento de inyección intravítrea debe llevarse a cabo en condiciones asépticas controladas, que incluyen la desinfección quirúrgica de las manos y el uso de guantes estériles, una sábana estéril y un espéculo de párpados estéril (o equivalente). Se debe administrar anestesia adecuada y un microbicida de amplio espectro tópico antes de la inyección.
Inmediatamente después de la inyección intravítrea, los pacientes deben ser monitoreados para detectar una elevación de la presión intraocular. El monitoreo adecuado puede consistir en una verificación de la perfusión de la cabeza del nervio óptico o la tonometría. Si es necesario, debe estar disponible una aguja de paracentesis estéril.
Después de la inyección intravítrea, los pacientes y/o cuidadores deben recibir instrucciones para informar cualquier signo y/o síntoma sugestivo de endoftalmitis o desprendimiento de retina (por ejemplo, dolor ocular, enrojecimiento del ojo, fotofobia, visión borrosa) sin demora [ver Información para el Paciente (17)].
Cada vial estéril solo debe usarse para el tratamiento de un solo ojo. No use la jeringa precargada de EYLEA para el tratamiento de ROP. Si el ojo contralateral requiere tratamiento, se debe usar un nuevo vial estéril y el campo estéril, la jeringa, los guantes, las sábanas, el espéculo de párpados, el filtro y las agujas de inyección deben cambiarse antes de administrar EYLEA al otro ojo.
Siga los pasos 1-10 enumerados en 2.3 Preparación para la Administración – Vial.
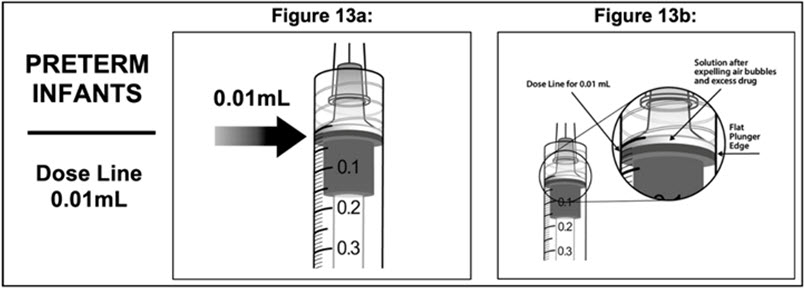
11 . Para eliminar todas las burbujas y expulsar el exceso de medicamento, deprima LENTAMENTE la varilla del émbolo de modo que el borde del émbolo se alinee con la línea que marca 0,01 mL en la jeringa (ver Figura 13a y Figura 13b).
Para el tratamiento de ROP, la aguja de inyección debe insertarse en el ojo a 1 mm del limbo con la aguja en ángulo para evitar el lente y evitar la retina.
Después de la inyección, cualquier producto no utilizado debe desecharse.
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
EYLEA es una solución transparente, incolora a amarillo pálido disponible como:
- Inyección: 2 mg (0.05 mL de una solución de 40 mg/mL) en una jeringa de vidrio precargada de dosis única
- Inyección: 2 mg (0.05 mL de una solución de 40 mg/mL) en un vial de vidrio de dosis única
4 CONTRAINDICACIONES
4.1 Infecciones oculares o perioculares
EYLEA está contraindicado en pacientes con infecciones oculares o perioculares.
4.2 Inflamación intraocular activa
EYLEA está contraindicado en pacientes con inflamación intraocular activa.
4.3 Hipersensibilidad
EYLEA está contraindicado en pacientes con hipersensibilidad conocida a aflibercept o a alguno de los excipientes de EYLEA. Las reacciones de hipersensibilidad pueden manifestarse como erupción cutánea, prurito, urticaria, reacciones anafilácticas/anafilactoides graves o inflamación intraocular grave.
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Endophthalmitis, Retinal Detachments, and Retinal Vasculitis with or without Occlusion
Las inyecciones intravítreas, incluidas las de EYLEA, se han asociado con endophthalmitis y desprendimientos de retina [ver Reacciones adversas (6.1)] y, con menos frecuencia, vasculitis retiniana con o sin oclusión [ver Reacciones adversas (6.2)]. Siempre se debe utilizar una técnica de inyección aséptica adecuada al administrar EYLEA. Se debe indicar a los pacientes y/o cuidadores que informen de inmediato cualquier signo o síntoma que sugiera endophthalmitis, desprendimiento de retina o vasculitis retiniana, y que se les debe tratar de forma adecuada [ver Dosificación y administración (2.4) e Información de orientación al paciente (17)].
5.2 Aumento de la presión intraocular
Se han observado aumentos agudos de la presión intraocular en los 60 minutos siguientes a la inyección intravítrea, incluso con EYLEA [ver Reacciones adversas (6.1)]. También se han notificado aumentos sostenidos de la presión intraocular después de la administración repetida de dosis intravítreas de inhibidores del factor de crecimiento endotelial vascular (VEGF). La presión intraocular y la perfusión de la cabeza del nervio óptico deben controlarse y manejarse adecuadamente [ver Dosificación y administración (2.4)].
5.3 Control prolongado y tratamiento adicional en la ROP
La reactivación de la angiogénesis anormal y la tortuosidad pueden ocurrir después del tratamiento con EYLEA. Los bebés deben ser monitoreados de cerca después de la inyección con EYLEA hasta que la vascularización de la retina se haya completado o hasta que el examinador esté seguro de que la reactivación de la ROP no ocurrirá. En los bebés con ROP, el tratamiento con EYLEA requerirá períodos prolongados de monitoreo de la ROP y pueden ser necesarias inyecciones adicionales de EYLEA y/o tratamientos con láser.
5.4 Eventos tromboembólicos
Existe un riesgo potencial de eventos tromboembólicos arteriales (ETA) después del uso intravítreo de inhibidores del VEGF, incluido EYLEA. Los ETA se definen como accidente cerebrovascular no mortal, infarto de miocardio no mortal o muerte vascular (incluidas las muertes por causa desconocida). La incidencia de eventos tromboembólicos notificados en los estudios de DMAE húmeda durante el primer año fue del 1,8 % (32 de 1824) en el grupo combinado de pacientes tratados con EYLEA en comparación con el 1,5 % (9 de 595) en los pacientes tratados con ranibizumab; hasta la semana 96, la incidencia fue del 3,3 % (60 de 1824) en el grupo de EYLEA en comparación con el 3,2 % (19 de 595) en el grupo de ranibizumab. La incidencia en los estudios de EMD desde el inicio hasta la semana 52 fue del 3,3 % (19 de 578) en el grupo combinado de pacientes tratados con EYLEA en comparación con el 2,8 % (8 de 287) en el grupo de control; desde el inicio hasta la semana 100, la incidencia fue del 6,4 % (37 de 578) en el grupo combinado de pacientes tratados con EYLEA en comparación con el 4,2 % (12 de 287) en el grupo de control. No se notificaron eventos tromboembólicos en los pacientes tratados con EYLEA en los primeros seis meses de los estudios de OVR.
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas potencialmente graves se describen en otras partes de la etiqueta:
- Hipersensibilidad [ver Contraindicaciones (4.3)]
- Endoftalmitis, desprendimientos de retina y vasculitis retiniana con o sin oclusión [ver Advertencias y precauciones (5.1)]
- Aumento de la presión intraocular [ver Advertencias y precauciones (5.2)]
- Eventos tromboembólicos [ver Advertencias y precauciones (5.4)]
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se llevan a cabo en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no se pueden comparar directamente con las tasas en otros ensayos clínicos del mismo o de otro fármaco y pueden no reflejar las tasas observadas en la práctica.
Un total de 2980 pacientes adultos tratados con EYLEA constituyeron la población de seguridad en ocho estudios de fase 3. Entre ellos, 2379 pacientes fueron tratados con la dosis recomendada de 2 mg. Las reacciones adversas graves relacionadas con el procedimiento de inyección se han producido en <0,1% de las inyecciones intravítreas con EYLEA, incluida la endoftalmitis y el desprendimiento de retina. Las reacciones adversas más comunes (≥5%) notificadas en pacientes que recibieron EYLEA fueron hemorragia conjuntival, dolor ocular, catarata, desprendimiento vítreo, cuerpos flotantes vítreos y aumento de la presión intraocular.
Degeneración macular asociada a la edad (DMAE) neovascular (húmeda)
Los datos descritos a continuación reflejan la exposición a EYLEA en 1824 pacientes con DMAE húmeda, incluidos 1223 pacientes tratados con la dosis de 2 mg, en 2 estudios clínicos controlados doble ciego (VIEW1 y VIEW2) durante 24 meses (con control activo en el año 1) [ver Estudios clínicos (14.1)].
Los datos de seguridad observados en el grupo EYLEA en un estudio de fase 2 doble ciego de 52 semanas fueron consistentes con estos resultados.
Tabla 1: Reacciones adversas más comunes (≥1%) en estudios de DMAE húmeda
Reacciones adversas Línea de base a la semana 52 Línea de base a la semana 96 EYLEA
(N=1824)Control activo (ranibizumab)
(N=595)EYLEA
(N=1824)Control (ranibizumab)
(N=595)Hemorragia conjuntival 25% 28% 27% 30% Dolor ocular 9% 9% 10% 10% Catarata 7% 7% 13% 10% Desprendimiento vítreo 6% 6% 8% 8% Cuerpos flotantes vítreos 6% 7% 8% 10% Aumento de la presión intraocular 5% 7% 7% 11% Hiperemia ocular 4% 8% 5% 10% Defecto del epitelio corneal 4% 5% 5% 6% Desprendimiento del epitelio pigmentario de la retina 3% 3% 5% 5% Dolor en el sitio de inyección 3% 3% 3% 4% Sensación de cuerpo extraño en los ojos 3% 4% 4% 4% Aumento de la lagrimación 3% 1% 4% 2% Visión borrosa 2% 2% 4% 3% Inflamación intraocular 2% 3% 3% 4% Desgarro del epitelio pigmentario de la retina 2% 1% 2% 2% Hemorragia en el sitio de inyección 1% 2% 2% 2% Edema de párpados 1% 2% 2% 3% Edema corneal 1% 1% 1% 1% Desprendimiento de retina <1% <1% 1% 1% Las reacciones adversas graves menos frecuentes notificadas en <1% de los pacientes tratados con EYLEA fueron hipersensibilidad, desgarro de la retina y endoftalmitis.
Edema macular después de la oclusión de la vena retiniana (OVR)
Los datos descritos a continuación reflejan una exposición de 6 meses a EYLEA con una dosis mensual de 2 mg en 218 pacientes después de la oclusión de la vena central de la retina (OVCR) en 2 estudios clínicos (COPERNICUS y GALILEO) y 91 pacientes después de la oclusión de la vena retiniana de rama (OVRB) en un estudio clínico (VIBRANT) [ver Estudios clínicos (14.2), (14.3)].
Tabla 2: Reacciones adversas más frecuentes (≥1%) en los estudios de OVR
Reacciones adversas OVCR OVRB EYLEA
(N=218)Control
(N=142)EYLEA
(N=91)Control
(N=92)Dolor ocular 13% 5% 4% 5% Hemorragia conjuntival 12% 11% 20% 4% Presión intraocular aumentada 8% 6% 2% 0% Defecto del epitelio corneal 5% 4% 2% 0% Cuerpos flotantes vítreos 5% 1% 1% 0% Hiperemia ocular 5% 3% 2% 2% Sensación de cuerpo extraño en los ojos 3% 5% 3% 0% Desprendimiento vítreo 3% 4% 2% 0% Lagrimeo aumentado 3% 4% 3% 0% Dolor en el sitio de inyección 3% 1% 1% 0% Visión borrosa 1% <1% 1% 1% Inflamación intraocular 1% 1% 0% 0% Catarata <1% 1% 5% 0% Edema de párpados <1% 1% 1% 0% Las reacciones adversas menos frecuentes notificadas en <1% de los pacientes tratados con EYLEA en los estudios de CRVO fueron edema corneal, desgarro de la retina, hipersensibilidad y endoftalmitis.
Edema macular diabético (DME) y retinopatía diabética (DR)
Los datos descritos a continuación reflejan la exposición a EYLEA en 578 pacientes con DME tratados con la dosis de 2 mg en 2 estudios clínicos controlados doble ciego (VIVID y VISTA) desde el inicio hasta la semana 52 y desde el inicio hasta la semana 100 [ver Estudios clínicos (14.4)].
Tabla 3: Reacciones adversas más frecuentes (≥1%) en los estudios de DME
Reacciones adversas Inicio hasta la semana 52 Inicio hasta la semana 100 EYLEA
(N=578)Control
(N=287)EYLEA
(N=578)Control
(N=287)Hemorragia conjuntival 28% 17% 31% 21% Dolor ocular 9% 6% 11% 9% Catarata 8% 9% 19% 17% Moscas volantes 6% 3% 8% 6% Defecto del epitelio corneal 5% 3% 7% 5% Presión intraocular aumentada 5% 3% 9% 5% Hiperemia ocular 5% 6% 5% 6% Desprendimiento vítreo 3% 3% 8% 6% Sensación de cuerpo extraño en los ojos 3% 3% 3% 3% Lagrimeo aumentado 3% 2% 4% 2% Visión borrosa 2% 2% 3% 4% Inflamación intraocular 2% <1% 3% 1% Dolor en el sitio de inyección 2% <1% 2% <1% Edema de párpados <1% 1% 2% 1% Retinopatía del prematuro (ROP)
Los datos descritos a continuación reflejan la exposición a EYLEA en 168 bebés prematuros con ROP asignados aleatoriamente a EYLEA y tratados con la dosis de 0,4 mg en 2 estudios clínicos (BUTTERFLEYE y FIREFLEYE/FIREFLEYE NEXT) desde el momento de la primera administración hasta las 52 semanas de edad cronológica [ver Estudios clínicos (14.6)]. Las reacciones adversas establecidas para las indicaciones en adultos se consideran aplicables a los bebés prematuros con ROP, aunque no todas se observaron en los estudios clínicos.
Tabla 4: Reacciones adversas en estudios de ROP *
Reacciones adversas Desde el inicio hasta las 52 semanas de edad cronológica Desde el inicio hasta las 52 semanas de edad cronológica BUTTERFLEYE FIREFLEYE/FIREFLEYE NEXT EYLEA
(N=93)Láser
(N=27)EYLEA
(N=75)Láser
(N=38)Desprendimiento de retina 6% 7% 5% 5% Hemorragia conjuntival† 5% 0% 9% 0% Presión intraocular aumentada 0% 0% 4% 0% Defecto del epitelio corneal 1% 0% 0% 0% Edema de párpados 0% 4% 3% 8% Edema corneal 0% 0% 1% 3% Opacidades lenticulares 0% 0% 1% 0% 6.2 Experiencia postcomercialización
Las siguientes reacciones adversas se han identificado durante el uso posterior a la aprobación de aflibercept. Debido a que estas reacciones se informan voluntariamente de una población de tamaño incierto, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal con la exposición al fármaco.
Trastornos oculares:
- Vasculitis retiniana y vasculitis retiniana oclusiva relacionada con la inyección intravítrea con aflibercept (informada a una tasa de 0,6 y 0,2 por millón de inyecciones, respectivamente, según la experiencia postcomercialización desde noviembre de 2011 hasta noviembre de 2023).
- Escleritis.
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de Riesgos
No se han realizado estudios adecuados y bien controlados con EYLEA en mujeres embarazadas. Aflibercept produjo efectos adversos embrio-fetales en conejos, incluyendo malformaciones externas, viscerales y esqueléticas. No se identificó un nivel de efecto adverso fetal no observado (NOAEL). En la dosis más baja que se demostró que produce efectos adversos embrio-fetales, las exposiciones sistémicas (basadas en el AUC para aflibercept libre) fueron aproximadamente 6 veces más altas que los valores de AUC observados en humanos después de un solo tratamiento intravítreo a la dosis clínica recomendada [ver Datos de animales].
Los estudios de reproducción en animales no siempre son predictivos de la respuesta humana, y no se sabe si EYLEA puede causar daño fetal cuando se administra a una mujer embarazada. Con base en el mecanismo de acción anti-VEGF para aflibercept [ver Farmacología clínica (12.1)], el tratamiento con EYLEA puede representar un riesgo para el desarrollo embrio-fetal humano. EYLEA debe usarse durante el embarazo solo si el beneficio potencial justifica el riesgo potencial para el feto.
Todos los embarazos tienen un riesgo de fondo de defectos de nacimiento, pérdida u otros resultados adversos. El riesgo de fondo de defectos de nacimiento mayores y aborto espontáneo para la población indicada es desconocido. En la población general de los EE. UU., el riesgo de fondo estimado de defectos de nacimiento mayores y aborto espontáneo en embarazos clínicamente reconocidos es del 2-4% y del 15-20%, respectivamente.
Datos
En dos estudios de desarrollo embrio-fetal, aflibercept produjo efectos adversos embrio-fetales cuando se administró cada tres días durante la organogénesis a conejas embarazadas a dosis intravenosas ≥3 mg por kg, o cada seis días durante la organogénesis a dosis subcutáneas ≥0,1 mg por kg.
Los efectos adversos embrio-fetales incluyeron un aumento de la incidencia de pérdida postimplantación y malformaciones fetales, incluyendo anasarca, hernia umbilical, hernia diafragmática, gastrosquisis, paladar hendido, ectrodactilia, atresia intestinal, espina bífida, encefalomeningocele, defectos cardíacos y de vasos sanguíneos mayores, y malformaciones esqueléticas (vértebras fusionadas, esternón y costillas; arcos y costillas vertebrales supernumerarios; y osificación incompleta). El nivel de efecto adverso materno no observado (NOAEL) en estos estudios fue de 3 mg por kg. Aflibercept produjo malformaciones fetales en todas las dosis evaluadas en conejos y no se identificó el NOAEL fetal. En la dosis más baja que se demostró que produce efectos adversos embrio-fetales en conejos (0,1 mg por kg), la exposición sistémica (AUC) de aflibercept libre fue aproximadamente 6 veces más alta que la exposición sistémica (AUC) observada en pacientes adultos después de una sola dosis intravítrea de 2 mg.
8.2 Lactancia
Resumen de Riesgos
No hay información sobre la presencia de aflibercept en la leche materna, los efectos del fármaco en el lactante amamantado o los efectos del fármaco en la producción/excreción de leche. Debido a que muchos fármacos se excretan en la leche materna, y debido a que existe la posibilidad de absorción y daño al crecimiento y desarrollo del lactante, no se recomienda EYLEA durante la lactancia.
Los beneficios para el desarrollo y la salud de la lactancia materna deben considerarse junto con la necesidad clínica de la madre de EYLEA y cualquier posible efecto adverso en el niño amamantado por EYLEA.
8.3 Mujeres y Hombres en Edad Reproductiva
Anticoncepción
Se recomienda a las mujeres en edad reproductiva que utilicen métodos anticonceptivos eficaces antes de la dosis inicial, durante el tratamiento y durante al menos 3 meses después de la última inyección intravítrea de EYLEA.
Infertilidad
No hay datos sobre los efectos de EYLEA en la fertilidad humana. Aflibercept afectó adversamente los sistemas reproductivos femenino y masculino en monos cynomolgus cuando se administró por inyección intravenosa a una dosis aproximadamente 1500 veces más alta que el nivel sistémico observado en pacientes adultos con una dosis intravítrea de 2 mg. No se identificó un nivel de efecto adverso no observado (NOAEL). Estos hallazgos fueron reversibles dentro de las 20 semanas posteriores a la interrupción del tratamiento [ver Toxicología no clínica (13.1)].
8.4 Uso Pediátrico
La seguridad y eficacia de EYLEA se han demostrado en dos estudios clínicos de bebés prematuros con ROP.
Estos dos estudios aleatorizaron a bebés prematuros entre el tratamiento inicial con EYLEA o láser. La eficacia de cada tratamiento está respaldada por la demostración de un curso clínico que fue mejor de lo que se hubiera esperado sin tratamiento [ver Dosificación y administración (2.9), Reacciones adversas (6.1), Farmacología clínica (12.3) y Estudios clínicos (14.6)].
8.5 Uso Geriátrico
En los estudios clínicos, aproximadamente el 76% (2049/2701) de los pacientes aleatorizados al tratamiento con EYLEA tenían ≥65 años de edad y aproximadamente el 46% (1250/2701) tenían ≥75 años de edad. No se observaron diferencias significativas en la eficacia o la seguridad con el aumento de la edad en estos estudios.
10 SOBREDOSIS
La sobredosis con un volumen de inyección aumentado puede aumentar la presión intraocular. Por lo tanto, en caso de sobredosis, se debe controlar la presión intraocular y, si el médico tratante lo considera necesario, se debe iniciar el tratamiento adecuado.
11 DESCRIPCIÓN
Aflibercept es una proteína de fusión recombinante que consiste en porciones de los dominios extracelulares de los receptores humanos del VEGF 1 y 2 fusionados a la porción Fc de la IgG1 humana formulada como una solución isoosmótica para administración intravítrea. Aflibercept es una glicoproteína dimérica con un peso molecular de proteína de 97 kilodaltons (kDa) y contiene glicosilación, que constituye un 15% adicional de la masa molecular total, lo que da como resultado un peso molecular total de 115 kDa. Aflibercept se produce en células recombinantes de ovario de hámster chino (CHO).
EYLEA (aflibercept) Injection es una solución estéril, clara e incolora a amarillo pálido. EYLEA no contiene conservante antimicrobiano y se suministra como una solución acuosa estéril para inyección intravítrea en una jeringa de vidrio precargada de dosis única o un vial de vidrio de dosis única diseñado para administrar 0.05 mL (50 microlitros) de solución que contiene 2 mg de aflibercept en polisorbato 20 (0.015 mg), cloruro de sodio (0.117 mg), fosfato monosódico monohidratado (0.055 mg), fosfato disódico heptahidratado (0.027 mg), sacarosa (2.5 mg) y agua para inyección con un pH de 6.2.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
El factor de crecimiento endotelial vascular A (VEGF-A) y el factor de crecimiento placentario (PlGF) son miembros de la familia de factores angiogénicos VEGF que pueden actuar como factores mitogénicos, quimiotácticos y de permeabilidad vascular para las células endoteliales. El VEGF actúa a través de dos tirosina quinasa receptores, VEGFR-1 y VEGFR-2, presentes en la superficie de las células endoteliales. El PlGF se une solo al VEGFR-1, que también está presente en la superficie de los leucocitos. La activación de estos receptores por el VEGF-A puede resultar en neovascularización y permeabilidad vascular.
Aflibercept actúa como un receptor señuelo soluble que se une al VEGF-A y al PlGF, y por lo tanto puede inhibir la unión y activación de estos receptores VEGF cognados.
12.2 Farmacodinámica
Degeneración macular asociada a la edad neovascular (húmeda) (DMAE)
En los estudios clínicos, las medidas anatómicas de la actividad de la enfermedad mejoraron de manera similar en todos los grupos de tratamiento desde el inicio hasta la semana 52. Los datos anatómicos no se utilizaron para influir en las decisiones de tratamiento durante el primer año.
Edema macular tras oclusión de la vena retiniana (RVO)
Se observaron reducciones en el grosor medio de la retina en COPERNICUS, GALILEO y VIBRANT en la semana 24 en comparación con el inicio. Los datos anatómicos no se utilizaron para influir en las decisiones de tratamiento [ver Estudios clínicos (14.2), (14.3)].
Edema macular diabético (EMD)
Se observaron reducciones en el grosor medio de la retina en VIVID y VISTA en las semanas 52 y 100 en comparación con el inicio. Los datos anatómicos no se utilizaron para influir en las decisiones de tratamiento con EYLEA [ver Estudios clínicos (14.4)].
12.3 Farmacocinética
EYLEA se administra intravítreamente para ejercer efectos locales en el ojo. En pacientes con DMAE húmeda, RVO o EMD, después de la administración intravítrea de EYLEA, se espera que una fracción de la dosis administrada se una al VEGF endógeno en el ojo para formar un complejo aflibercept:VEGF inactivo. Una vez absorbido en la circulación sistémica, el aflibercept se presenta en el plasma como aflibercept libre (no unido al VEGF) y una forma más predominante e inactiva estable con el VEGF endógeno circulante (es decir, el complejo aflibercept:VEGF).
Absorción/Distribución
Después de la administración intravítrea de 2 mg por ojo de EYLEA a pacientes con DMAE húmeda, RVO y EMD, la Cmax media de aflibercept libre en el plasma fue de 0,02 mcg/mL (rango: 0 a 0,054 mcg/mL), 0,05 mcg/mL (rango: 0 a 0,081 mcg/mL) y 0,03 mcg/mL (rango: 0 a 0,076 mcg/mL), respectivamente, y se alcanzó en 1 a 3 días. Las concentraciones de aflibercept libre en el plasma fueron indetectables dos semanas después de la dosificación en todos los pacientes. El aflibercept no se acumuló en el plasma cuando se administró en dosis repetidas intravítreamente cada 4 semanas. Se estima que después de la administración intravítrea de 2 mg a los pacientes, la concentración plasmática máxima media de aflibercept libre es más de 100 veces menor que la concentración de aflibercept requerida para unir al VEGF sistémico en un 50%.
El volumen de distribución de aflibercept libre después de la administración intravenosa (I.V.) de aflibercept se ha determinado que es aproximadamente 6 L.
Metabolismo/Eliminación
Aflibercept es una proteína terapéutica y no se han realizado estudios de metabolismo de fármacos. Se espera que el aflibercept se elimine tanto a través de la disposición mediada por el objetivo a través de la unión al VEGF endógeno libre como a través del metabolismo por proteólisis. La vida media de eliminación terminal (t1/2) de aflibercept libre en el plasma fue de aproximadamente 5 a 6 días después de la administración I.V. de dosis de 2 a 4 mg/kg de aflibercept.
Poblaciones específicas
Pacientes pediátricos
Se evaluó la farmacocinética de aflibercept en recién nacidos prematuros con ROP a una dosis de 0,4 mg de aflibercept (por ojo) administrado de forma unilateral o bilateral. En el estudio BUTTERFLEYE, las concentraciones medias de aflibercept libre en el plasma disminuyeron de un máximo de 0,583 mcg/mL en el día 1 a 0,0406 mcg/mL en el día 28 en pacientes tratados bilateralmente.
En el estudio FIREFLEYE, las concentraciones medias de aflibercept libre en el plasma de todos los pacientes (combinación de administración bilateral y unilateral) disminuyeron de un máximo de 0,481 mcg/mL en el día 1 a 0,13 mcg/mL en el día 28. Las concentraciones de aflibercept libre en el plasma disminuyeron posteriormente a valores inferiores o cercanos al límite inferior de cuantificación en aproximadamente 8 semanas.
Después de una dosis IVT de aflibercept 0,4 mg por ojo administrada bilateralmente a intervalos de 10 o 14 días, la estimación farmacocinética poblacional de la razón de acumulación máxima media de aflibercept libre en el plasma fue de aproximadamente 2,0 y 1,4. No se espera acumulación de aflibercept libre en el plasma para dosis IVT de 0,4 mg por ojo administradas bilateralmente a intervalos de dosificación de 21 días o más.
Insuficiencia renal
El análisis farmacocinético de un subgrupo de pacientes (n = 492) en un estudio de DMAE húmeda, de los cuales el 43% tenía insuficiencia renal (leve n = 120, moderada n = 74 y grave n = 16), no reveló diferencias con respecto a las concentraciones plasmáticas de aflibercept libre después de la administración intravítrea cada 4 u 8 semanas. Se observaron resultados similares en pacientes en un estudio de RVO y en pacientes en un estudio de EMD. No se necesita un ajuste de la dosis basado en el estado de insuficiencia renal para pacientes con DMAE húmeda, RVO o EMD.
12.6 Inmunogenicidad
Al igual que con todas las proteínas terapéuticas, existe la posibilidad de una respuesta inmunitaria en los pacientes tratados con EYLEA. La inmunogenicidad de EYLEA se evaluó en muestras de suero. Los datos de inmunogenicidad reflejan el porcentaje de pacientes cuyos resultados de las pruebas se consideraron positivos para anticuerpos contra EYLEA en inmunoensayos. La detección de una respuesta inmunitaria depende en gran medida de la sensibilidad y la especificidad de los ensayos utilizados, el manejo de las muestras, el momento de la recolección de las muestras, los medicamentos concomitantes y la enfermedad subyacente. Por estas razones, la comparación de la incidencia de anticuerpos contra EYLEA con la incidencia de anticuerpos contra otros productos puede ser engañosa.
En los estudios de AMD húmeda, RVO y DME, la incidencia previa al tratamiento de inmunorreactividad a EYLEA fue de aproximadamente 1% a 3% en todos los grupos de tratamiento. Después de la dosificación con EYLEA durante 24-100 semanas, se detectaron anticuerpos contra EYLEA en un rango de porcentaje similar de pacientes. De manera similar, después de la dosificación unilateral o bilateral en estudios pediátricos de ROP, se detectaron anticuerpos contra EYLEA en menos del 1% de los pacientes.
No hubo diferencias en la eficacia o la seguridad entre los pacientes con o sin inmunorreactividad.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
No se han realizado estudios sobre el potencial mutagénico o carcinogénico de aflibercept. Los efectos sobre la fertilidad masculina y femenina se evaluaron como parte de un estudio de 6 meses en monos con administración intravenosa de aflibercept en dosis semanales que van de 3 a 30 mg por kg. Se observaron menstruaciones ausentes o irregulares asociadas con alteraciones en los niveles hormonales reproductivos femeninos y cambios en la morfología y motilidad de los espermatozoides en todos los niveles de dosis. Además, las hembras mostraron una disminución del peso ovárico y uterino acompañado de un desarrollo lúteo comprometido y una reducción de los folículos en maduración. Estos cambios se correlacionaron con la atrofia uterina y vaginal. No se identificó un Nivel de Efecto Adverso No Observado (NOAEL). La administración intravenosa de la dosis más baja de aflibercept evaluada en monos (3 mg por kg) resultó en una exposición sistémica (AUC) para aflibercept libre que fue aproximadamente 1500 veces mayor que la exposición sistémica observada en pacientes adultos después de una dosis intravítrea de 2 mg. Todos los cambios fueron reversibles dentro de las 20 semanas posteriores a la suspensión del tratamiento.
13.2 Toxicología y/o Farmacología Animal
Se observaron erosiones y ulceraciones del epitelio respiratorio en los cornetes nasales en monos tratados con aflibercept intravítreamente a dosis intravítreas de 2 o 4 mg por ojo. En el NOAEL de 0,5 mg por ojo en monos, la exposición sistémica (AUC) fue 56 veces mayor que la exposición observada en pacientes adultos después de una dosis intravítrea de 2 mg y 2 veces mayor en función de Cmax en comparación con los valores correspondientes observados en bebés prematuros de FIREFLEYE. No se observaron efectos similares en otros estudios clínicos [ver Estudios Clínicos (14)].
14 ESTUDIOS CLÍNICOS
14.1 Degeneración macular asociada a la edad (DMAE) neovascular (húmeda)
La seguridad y eficacia de EYLEA se evaluaron en dos estudios aleatorizados, multicéntricos, doble ciego y controlados con activo en pacientes con DMAE húmeda. Un total de 2412 pacientes fueron tratados y evaluables para la eficacia (1817 con EYLEA) en los dos estudios (VIEW1 y VIEW2). En cada estudio, hasta la semana 52, los pacientes fueron asignados aleatoriamente en una proporción de 1:1:1:1 a 1 de 4 regímenes de dosificación: 1) EYLEA administrado 2 mg cada 8 semanas después de 3 dosis mensuales iniciales (EYLEA 2Q8); 2) EYLEA administrado 2 mg cada 4 semanas (EYLEA 2Q4); 3) EYLEA 0.5 mg administrado cada 4 semanas (EYLEA 0.5Q4); y 4) ranibizumab administrado 0.5 mg cada 4 semanas (ranibizumab 0.5 mg Q4). Las visitas especificadas en el protocolo se realizaron cada 28±3 días. Las edades de los pacientes oscilaron entre 49 y 99 años, con una media de 76 años.
En ambos estudios, el criterio de valoración principal de eficacia fue la proporción de pacientes que mantuvieron la visión, definida como la pérdida de menos de 15 letras de agudeza visual en la semana 52 en comparación con la línea de base. Tanto los grupos EYLEA 2Q8 como EYLEA 2Q4 mostraron tener una eficacia clínicamente equivalente al grupo ranibizumab 0.5 mg Q4 en el año 1.
Los resultados detallados del análisis de los estudios VIEW1 y VIEW2 se muestran en la Tabla 5 y la Figura 14 a continuación.
Tabla 5: Resultados de eficacia en la semana 52 (Conjunto de análisis completo con LOCF) en los estudios VIEW1 y VIEW2
VIEW1 VIEW2 EYLEA
2 mg Q8 semanas *EYLEA
2 mg Q4 semanasranibizu-mab
0.5 mg Q4 semanasEYLEA
2 mg Q8 semanas *EYLEA
2 mg Q4 semanasranibizu-mab
0.5 mg Q4 semanasConjunto de análisis completo N=301 N=304 N=304 N=306 N=309 N=291 BCVA = Mejor agudeza visual corregida; IC = Intervalo de confianza; ETDRS = Estudio de retinopatía diabética de tratamiento temprano; LOCF = Última observación llevada hacia adelante (los valores de línea de base no se llevan hacia adelante); los intervalos de confianza del 95.1% se presentaron para ajustar la evaluación de seguridad realizada durante el estudio Resultados de eficacia Proporción de pacientes que mantuvieron la agudeza visual (%)
(<15 letras de pérdida de BCVA)94% 95% 94% 95% 95% 95% Diferencia† (%)
(95.1% IC)0.6
(-3.2, 4.4)1.3
(-2.4, 5.0)0.6
(-2.9, 4.0)-0.3
(-4.0, 3.3)Cambio medio en BCVA medido por la puntuación de letras ETDRS desde la línea de base 7.9 10.9 8.1 8.9 7.6 9.4 Diferencia† en la media de LS
(95.1% IC)0.3
(-2.0, 2.5)3.2
(0.9, 5.4)-0.9
(-3.1, 1.3)-2.0
(-4.1, 0.2)Número de pacientes que ganaron al menos 15 letras de visión desde la línea de base (%) 92
(31%)114
(38%)94
(31%)96
(31%)91
(29%)99
(34%)Diferencia† (%)
(95.1% IC)-0.4
(-7.7, 7.0)6.6
(-1.0, 14.1)-2.6
(-10.2, 4.9)-4.6
(-12.1, 2.9)Los efectos del tratamiento en subgrupos evaluables (por ejemplo, edad, sexo, raza, agudeza visual basal) en cada estudio fueron en general consistentes con los resultados en las poblaciones generales.
Figura 14: Cambio medio en la agudeza visual desde el inicio hasta la semana 96* en los estudios VIEW1 y VIEW2
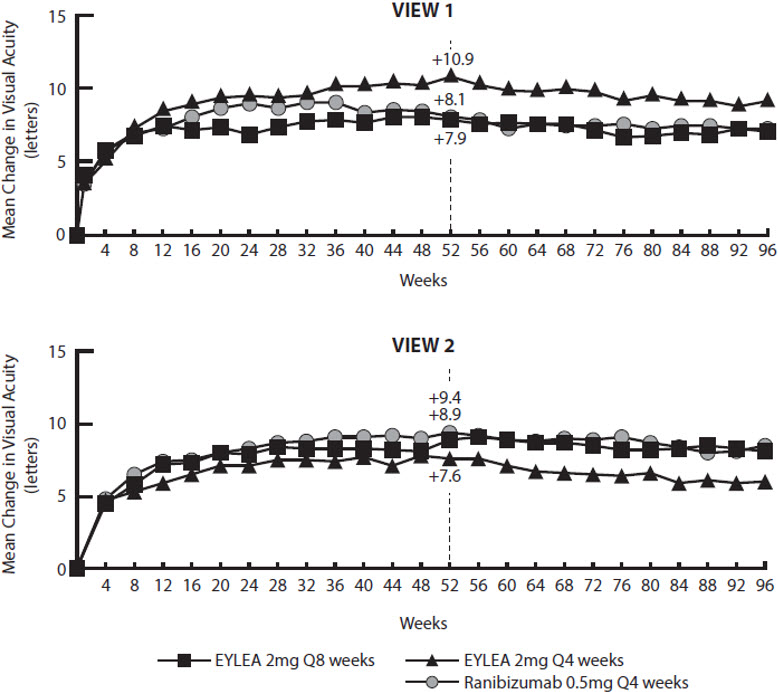
*Los programas de dosificación de los pacientes se individualizaron de las semanas 52 a 96 utilizando un régimen de dosificación modificado de 12 semanas.
Los estudios VIEW1 y VIEW2 tuvieron una duración de 96 semanas. Sin embargo, después de 52 semanas, los pacientes ya no siguieron un programa de dosificación fijo. Entre la semana 52 y la semana 96, los pacientes continuaron recibiendo el fármaco y la concentración de dosis a la que fueron asignados inicialmente en un programa de dosificación modificado de 12 semanas (dosis al menos cada 12 semanas y dosis adicionales según sea necesario). Por lo tanto, durante el segundo año de estos estudios no hubo un brazo de comparación de control activo.
14.2 Edema macular después de la oclusión venosa central de la retina (OVCR)
La seguridad y eficacia de EYLEA se evaluaron en dos estudios aleatorizados, multicéntricos, doble ciego y controlados con placebo en pacientes con edema macular después de la OVCR. Un total de 358 pacientes fueron tratados y evaluables para la eficacia (217 con EYLEA) en los dos estudios (COPERNICUS y GALILEO). En ambos estudios, los pacientes fueron asignados aleatoriamente en una proporción de 3:2 a 2 mg de EYLEA administrados cada 4 semanas (2Q4), o inyecciones simuladas (grupo control) administradas cada 4 semanas durante un total de 6 inyecciones. Las visitas especificadas en el protocolo se realizaron cada 28±7 días. Las edades de los pacientes oscilaron entre los 22 y los 89 años, con una media de 64 años.
En ambos estudios, el criterio de valoración principal de eficacia fue la proporción de pacientes que ganaron al menos 15 letras en la AVBC en comparación con el inicio. En la semana 24, el grupo de EYLEA 2 mg Q4 fue superior al grupo control para el criterio de valoración principal.
Los resultados del análisis de los estudios COPERNICUS y GALILEO se muestran en la Tabla 6 y la Figura 15 a continuación.
Tabla 6: Resultados de eficacia en la semana 24 (Conjunto de análisis completo con LOCF) en los estudios COPERNICUS y GALILEO
COPERNICUS GALILEO Control EYLEA
2 mg Q4 semanasControl EYLEA
2 mg Q4 semanasN=73 N=114 N=68 N=103 - *
- La diferencia es EYLEA 2 mg Q4 semanas menos Control
- †
- La diferencia y el IC se calculan utilizando la prueba de Cochran-Mantel-Haenszel (CMH) ajustada por factores basales; se presentaron intervalos de confianza del 95,1% para ajustar las múltiples evaluaciones realizadas durante el estudio
- ‡
- p<0,01 en comparación con Control
- §
- Media LS e IC basados en un modelo ANCOVA
Resultados de eficacia Proporción de pacientes que ganaron al menos 15 letras en la AVBC desde el inicio (%) 12% 56% 22% 60% Diferencia ponderada *, † (%)
(95,1% IC)44,8%‡
(32,9, 56,6)38,3%‡
(24,4, 52,1)Cambio medio en la AVBC medido por la puntuación de letras ETDRS desde el inicio (DE) -4,0
(18,0)17,3
(12,8)3,3
(14,1)18,0
(12,2)Diferencia en la media LS *, §
(95,1% IC)21,7‡
(17,3, 26,1)14,7‡
(10,7, 18,7)Figura 15: Cambio medio en la AVBC medida por la puntuación de letras ETDRS desde el inicio hasta la semana 24 en los estudios COPERNICUS y GALILEO
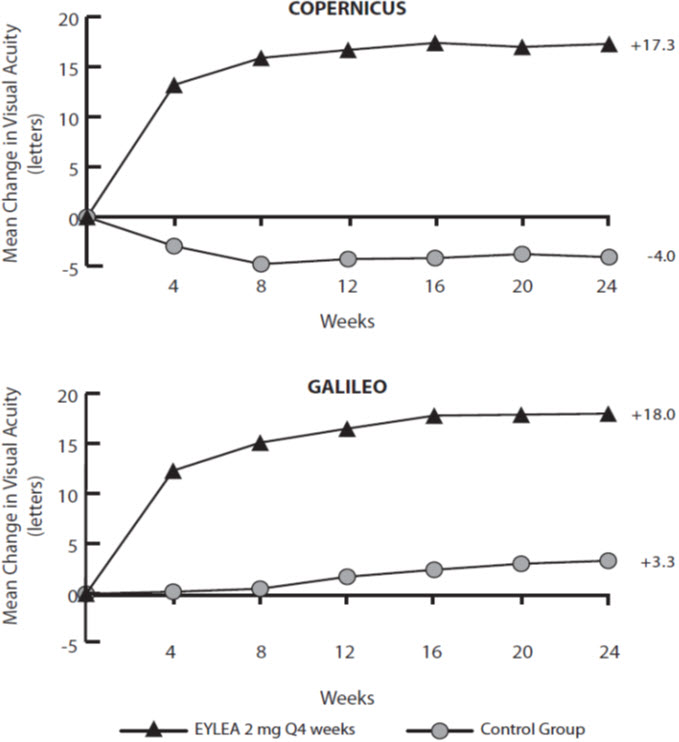
Los efectos del tratamiento en subgrupos evaluables (por ejemplo, edad, sexo, raza, agudeza visual basal, estado de perfusión retiniana y duración de la CRVO) en cada estudio y en el análisis combinado fueron en general consistentes con los resultados en las poblaciones generales.
14.3 Edema macular tras la oclusión de la vena retiniana rama (OVRR)
La seguridad y eficacia de EYLEA se evaluaron en un estudio controlado, aleatorizado, multicéntrico, doble ciego de 24 semanas en pacientes con edema macular tras OVRR. Un total de 181 pacientes fueron tratados y evaluables para la eficacia (91 con EYLEA) en el estudio VIBRANT. En el estudio, los pacientes fueron asignados aleatoriamente en una proporción de 1:1 a 2 mg de EYLEA administrados cada 4 semanas (2Q4) o fotocoagulación láser administrada en el inicio y posteriormente según fuera necesario (grupo control). Las visitas especificadas en el protocolo se realizaron cada 28±7 días. Las edades de los pacientes oscilaron entre 42 y 94 años, con una media de 65 años.
En el estudio VIBRANT, el criterio de valoración principal de eficacia fue la proporción de pacientes que ganaron al menos 15 letras en la AVBC en la semana 24 en comparación con el inicio. En la semana 24, el grupo EYLEA 2 mg Q4 fue superior al grupo control para el criterio de valoración principal.
Los resultados detallados del análisis del estudio VIBRANT se muestran en la Tabla 7 y la Figura 16 a continuación.
Tabla 7: Resultados de eficacia en la semana 24 (Conjunto de análisis completo con LOCF) en el estudio VIBRANT
VIBRANT Control EYLEA
2 mg Q4 semanasN=90 N=91 - *
- La diferencia es EYLEA 2 mg Q4 semanas menos Control
- †
- La diferencia y el IC se calculan utilizando el esquema de ponderación de Mantel-Haenszel ajustado por región (América del Norte frente a Japón) y categoría de AVBC basal (> 20/200 y ≤ 20/200)
- ‡
- p<0,01 en comparación con Control
- §
- Media LS y IC basados en un modelo ANCOVA
Resultados de eficacia Proporción de pacientes que ganaron al menos 15 letras en la AVBC desde el inicio (%) 26,7% 52,7% Diferencia ponderada *, † (%)
(IC del 95%)26,6%‡
(13,0, 40,1)Cambio medio en la AVBC medida por la puntuación de letras ETDRS desde el inicio (DE) 6,9
(12,9)17,0
(11,9)Diferencia en la media LS *, §
(IC del 95%)10,5‡
(7,1, 14,0)Figura 16: Cambio medio en la AVBC medida por la puntuación de letras ETDRS desde el inicio hasta la semana 24 en el estudio VIBRANT
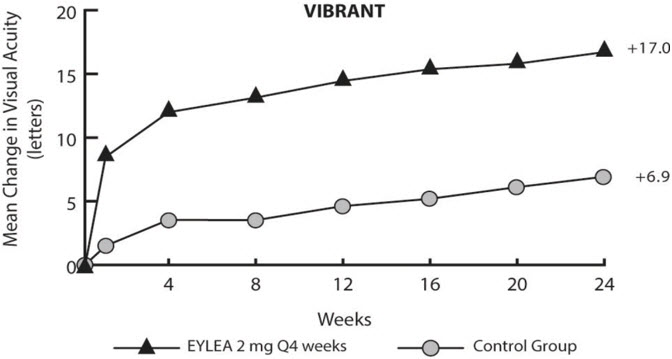
Los efectos del tratamiento en subgrupos evaluables (por ejemplo, edad, sexo y estado de perfusión retiniana basal) en el estudio fueron en general consistentes con los resultados en las poblaciones generales.
14.4 Edema macular diabético (EMD)
La seguridad y eficacia de EYLEA se evaluaron en dos estudios aleatorizados, multicéntricos, doble ciego y controlados en pacientes con EMD. Un total de 862 pacientes aleatorizados y tratados fueron evaluables para la eficacia. Las visitas especificadas en el protocolo se realizaron cada 28±7 días. Las edades de los pacientes oscilaron entre los 23 y los 87 años, con una media de 63 años.
De ellos, 576 fueron aleatorizados a grupos de EYLEA en los dos estudios (VIVID y VISTA). En cada estudio, los pacientes fueron asignados aleatoriamente en una proporción de 1:1:1 a 1 de 3 regímenes de dosificación: 1) EYLEA administrado 2 mg cada 8 semanas después de 5 inyecciones mensuales iniciales (EYLEA 2Q8); 2) EYLEA administrado 2 mg cada 4 semanas (EYLEA 2Q4); y 3) fotocoagulación láser macular (en el inicio y luego según sea necesario). A partir de la semana 24, los pacientes que cumplían un umbral preestablecido de pérdida de visión eran elegibles para recibir tratamiento adicional: los pacientes de los grupos de EYLEA podían recibir láser y los pacientes del grupo de láser podían recibir EYLEA.
En ambos estudios, el criterio de valoración principal de eficacia fue el cambio medio desde el inicio en la AVBC en la semana 52 medido por la puntuación de letras ETDRS. La eficacia de ambos grupos de EYLEA 2Q8 y EYLEA 2Q4 fue estadísticamente superior al grupo control. Esta mejora estadísticamente superior en la AVBC se mantuvo en la semana 100 en ambos estudios.
Los resultados del análisis de los estudios VIVID y VISTA se muestran en la Tabla 8 y la Figura 17 a continuación.
Tabla 8: Resultados de eficacia en las semanas 52 y 100 (Conjunto de análisis completo con LOCF) en los estudios VIVID y VISTA
VIVID VISTA EYLEA
2 mg Q8 semanas *EYLEA
2 mg Q4 semanasControl EYLEA
2 mg Q8 semanas *EYLEA
2 mg Q4 semanasControl Conjunto de análisis completo N=135 N=136 N=132 N=151 N=154 N=154 - *
- Después del inicio del tratamiento con 5 inyecciones mensuales
- †
- Media LS e IC basados en un modelo ANCOVA con la medida de AVBC basal como covariable y un factor para el grupo de tratamiento. Además, los factores de estratificación especificados en el protocolo se incluyeron en el modelo
- ‡
- La diferencia es el grupo EYLEA menos el grupo control
- §
- p<0.01 en comparación con el control
- ¶
- La diferencia con el intervalo de confianza (IC) y la prueba estadística se calcula utilizando el esquema de ponderación de Mantel-Haenszel ajustado por los factores de estratificación especificados en el protocolo
Resultados de eficacia en la semana 52 Cambio medio en la AVBC medida por la puntuación de letras ETDRS desde el inicio (DE) 10.7
(9.3)10.5
(9.6)1.2
(10.6)10.7
(8.2)12.5
(9.5)0.2
(12.5)Diferencia†, ‡ en la media LS
(97.5% IC)9.1§
(6.3, 11.8)9.3§
(6.5, 12.0)10.5§
(7.7, 13.2)12.2§
(9.4, 15.0)Proporción de pacientes que ganaron al menos 15 letras en la AVBC desde el inicio (%) 33.3% 32.4% 9.1% 31.1% 41.6% 7.8% Diferencia Ajustada‡, ¶ (%)
(IC del 97.5%)24.2%§
(13.5, 34.9)23.3%§
(12.6, 33.9)23.3%§
(13.5, 33.1)34.2%§
(24.1, 44.4)Resultados de Eficacia en la Semana 100 Cambio medio en la AVBC medida por la puntuación de letras ETDRS desde el inicio (DE) 9.4
(10.5)11.4
(11.2)0.7
(11.8)11.1
(10.7)11.5
(13.8)0.9
(13.9)Diferencia†, ‡ en la media de LS
(IC del 97.5%)8.2§
(5.2, 11.3)10.7§
(7.6, 13.8)10.1§
(7.0, 13.3)10.6§
(7.1, 14.2)Proporción de pacientes que ganaron al menos 15 letras en la AVBC desde el inicio (%) 31.1% 38.2% 12.1% 33.1% 38.3% 13.0% Diferencia Ajustada‡, ¶ (%)
(IC del 97.5%)19.0%§
(8.0, 29.9)26.1%§
(14.8, 37.5)20.1%§
(9.6, 30.6)25.8%§
(15.1, 36.6)Figura 17: Cambio medio en la AVBC medida por la puntuación de letras ETDRS desde el inicio hasta la semana 100 en los estudios VIVID y VISTA
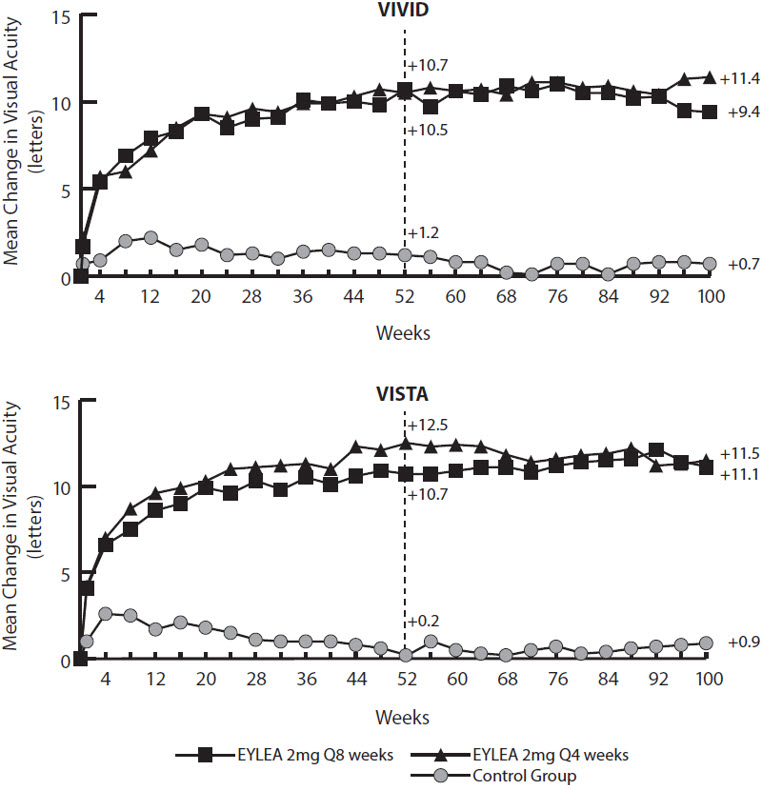
Los efectos del tratamiento en el subgrupo de pacientes que habían sido tratados previamente con un inhibidor del VEGF antes de participar en el estudio fueron similares a los observados en los pacientes que no habían recibido tratamiento previo con un inhibidor del VEGF.
Los efectos del tratamiento en los subgrupos evaluables (por ejemplo, edad, sexo, raza, HbA1c basal, agudeza visual basal, terapia previa con anti-VEGF) en cada estudio fueron en general consistentes con los resultados en las poblaciones generales.
14.5 Retinopatía diabética (RD)
Los datos de eficacia y seguridad de EYLEA en la retinopatía diabética (RD) se derivan de los estudios VIVID, VISTA y PANORAMA.
VIVID Y VISTA
En los estudios VIVID y VISTA, un resultado de eficacia fue el cambio en la escala de gravedad de la retinopatía diabética del Estudio de Tratamiento Temprano de la Retinopatía Diabética (ETDRS-DRSS). La puntuación ETDRS-DRSS se evaluó al inicio y aproximadamente cada 6 meses a partir de entonces durante la duración de los estudios [ver Estudios clínicos (14.4)].
Todos los pacientes inscritos tenían RD y DME al inicio. La mayoría de los pacientes inscritos en estos estudios (77%) tenían retinopatía diabética no proliferativa (NPDR) moderada a grave según el ETDRS-DRSS. En la semana 100, la proporción de pacientes que mejoraron al menos 2 pasos en el ETDRS-DRSS fue significativamente mayor en ambos grupos de tratamiento con EYLEA (2Q4 y 2Q8) en comparación con el grupo control.
Los resultados del análisis del ETDRS-DRSS en la semana 100 en los estudios VIVID y VISTA se muestran en la Tabla 9 a continuación.
Tabla 9: Proporción de pacientes que lograron una mejora ≥2 pasos desde el inicio en la puntuación ETDRS-DRSS en la semana 100 en los estudios VIVID y VISTA
VIVID VISTA EYLEA
2 mg Q8 semanas *EYLEA
2 mg Q4 semanasControl EYLEA
2 mg Q8 semanas *EYLEA
2 mg Q4 semanasControl Pacientes evaluables† N=101 N=97 N=99 N=148 N=153 N=150 Los valores ETDRS-DRSS no graduables posteriores al inicio se trataron como faltantes y se imputaron utilizando los últimos valores ETDRS-DRSS graduables (incluidos los valores iniciales si todos los valores posteriores al inicio fueron faltantes o no graduables) - *
- Después del inicio del tratamiento con 5 inyecciones mensuales
- †
- El número de pacientes evaluables incluyó a todos los pacientes que tenían datos ETDRS-DRSS válidos al inicio
- ‡
- La diferencia con el intervalo de confianza (IC) se calculó utilizando el esquema de ponderación de Mantel-Haenszel ajustado por los factores de estratificación especificados en el protocolo
- §
- La diferencia es EYLEA menos el grupo control
- ¶
- p<0.01 en comparación con el control
Número de pacientes con una mejora ≥2 pasos en ETDRS-DRSS desde 32 27 7 56 58 24 Inicio (%) (32%) (28%) (7%) (38%) (38%) (16%) Diferencia‡, § (%) 24%¶ 21%¶ 22%¶ 22%¶ (97.5% IC) (12, 36) (9, 33) (11, 33) (11, 33) Los resultados de los subgrupos evaluables (por ejemplo, edad, sexo, raza, HbA1c basal, agudeza visual basal) sobre la proporción de pacientes que lograron una mejora de ≥2 pasos en la ETDRS-DRSS desde el inicio hasta la semana 100 fueron, en general, consistentes con los de la población general.
PANORAMA
El estudio PANORAMA evaluó la seguridad y eficacia de EYLEA en un estudio aleatorizado, multicéntrico, doble ciego y controlado en pacientes con retinopatía diabética no proliferativa de moderada a severa (NPDR) (ETDRS-DRSS de 47 o 53), sin DME central (CI-DME). Un total de 402 pacientes aleatorizados fueron evaluables para la eficacia. Las visitas especificadas en el protocolo se realizaron cada 28±7 días durante las primeras 5 visitas, luego cada 8 semanas (56±7 días). Las edades de los pacientes oscilaron entre 25 y 85 años, con una media de 55,7 años.
Los pacientes fueron asignados aleatoriamente en una proporción de 1:1:1 a 1 de 3 regímenes de dosificación: 1) 3 inyecciones iniciales mensuales de EYLEA 2 mg seguidas de una inyección después de 8 semanas y luego una inyección cada 16 semanas (EYLEA 2Q16); 2) 5 inyecciones mensuales de EYLEA 2 mg seguidas de una inyección cada 8 semanas (EYLEA 2Q8); y 3) tratamiento simulado.
El criterio de valoración principal de eficacia fue la proporción de pacientes que mejoraron en ≥2 pasos en la DRSS desde el inicio hasta la semana 24 en los grupos combinados de EYLEA y en la semana 52 en los grupos 2Q16 y 2Q8 individualmente frente al simulado. Un criterio de valoración secundario clave fue la proporción de pacientes que desarrollaron el criterio de valoración compuesto de retinopatía diabética proliferativa o neovascularización del segmento anterior hasta la semana 52.
En la semana 52, la eficacia en los grupos 2Q16 y 2Q8 fue superior al grupo simulado (véase Tabla 10 y Tabla 11). La proporción de pacientes con una mejora de ≥2 pasos a lo largo del tiempo se muestra en la Figura 18.
Tabla 10: Proporción de pacientes que lograron una mejora de ≥2 pasos desde el inicio en la puntuación ETDRS-DRSS en las semanas 24 y 52 en PANORAMA
PANORAMA Semana 24 Semana 52 EYLEA
CombinadoControl
(simulado)EYLEA
2Q16EYLEA
2Q8Control
(simulado)Conjunto de análisis completo N=269 N=133 N=135 N=134 N=133 Los valores ETDRS-DRSS no graduables después del inicio se trataron como perdidos y se imputaron utilizando los últimos valores ETDRS-DRSS graduables (incluidos los valores iniciales si todos los valores posteriores al inicio estaban perdidos o no eran graduables) - *
- La diferencia es el grupo EYLEA menos el simulado
- †
- La diferencia con CI se calculó utilizando el esquema de ponderación de Mantel-Haenszel ajustado por la variable de estratificación DRSS basal
- ‡
- p<0,01 en comparación con Control. El valor p se calculó utilizando una prueba de Cochran-Mantel-Haenszel de 2 colas ajustada por la variable de estratificación DRSS basal.
Proporción de pacientes con una mejora de ≥2 pasos en ETDRS-DRSS desde el inicio (%) 58% 6% 65% 80% 15% Diferencia ajustada* (%)
(95% CI)†52%‡
(45, 60)50%‡
(40, 60)65%‡
(56, 74)Figura 18: Proporción de pacientes que lograron una mejora ≥2 pasos desde el inicio en la puntuación ETDRS-DRSS hasta la semana 52 en PANORAMA
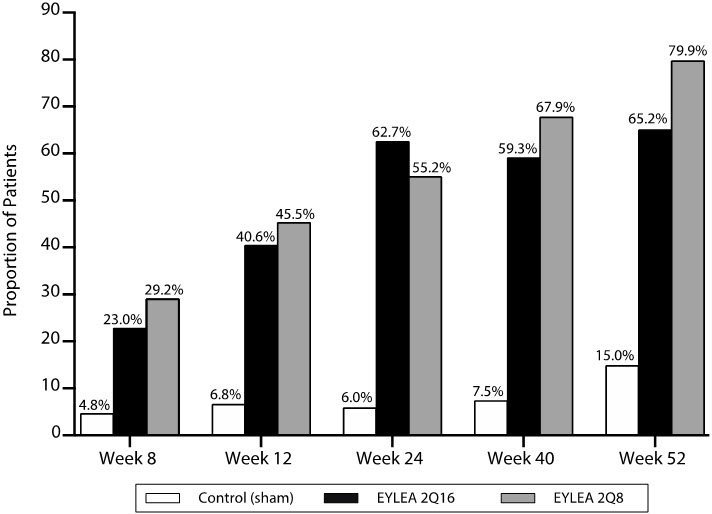
Tabla 11: Efecto de EYLEA en el empeoramiento de la retinopatía diabética en PANORAMA hasta la semana 52
EYLEA
2Q16EYLEA
2Q8Control
(Sham)Conjunto de análisis completo N=135 N=134 N=133 PDR = Retinopatía diabética proliferativa; ASNV = Neovascularización del segmento anterior Punto final compuesto de desarrollar PDR o ASNV* Tasa de eventos† 4.0%‡ 2.4%‡ 20.1% Razón de riesgo 0.15 0.12 Desarrollo de retinopatía diabética proliferativa§ Tasa de eventos† 1.6%‡ 0.0%‡ 11.9% Razón de riesgo 0.11 0.00 14.6 Retinopatía del prematuro (ROP)
Los datos de eficacia y seguridad de EYLEA en ROP se derivan de dos estudios (BUTTERFLEYE y FIREFLEYE/FIREFLEYE NEXT). BUTTERFLEYE fue un estudio de 52 semanas. FIREFLEYE incluyó 24 semanas de tratamiento y seguimiento. FIREFLEYE NEXT fue un seguimiento observacional de FIREFLEYE hasta la semana 52.
Tanto los estudios BUTTERFLEYE como FIREFLEYE evaluaron la eficacia, seguridad y tolerabilidad de EYLEA en estudios aleatorizados, de 2 brazos, abiertos, de grupos paralelos. Los estudios se realizaron en bebés prematuros con ROP, proporcionando una comparación entre el tratamiento con EYLEA y la terapia de fotocoagulación con láser (láser). Cada ojo elegible recibió el tratamiento de estudio asignado en la línea de base. El re-tratamiento y/o el tratamiento de rescate se administraron si fuera necesario según criterios preestablecidos. El tratamiento de rescate podría incluir potencialmente el tratamiento alternativo (EYLEA o láser). El re-tratamiento con aflibercept, si fuera necesario, se administró hasta 2 veces en un ojo en particular, con al menos 28 días entre inyecciones consecutivas.
Los pacientes elegibles tenían una edad gestacional máxima al nacer de 32 semanas o un peso al nacer máximo de 1500 g, tenían que pesar > 800 g el día del tratamiento y tenían ROP sin tratamiento clasificado de acuerdo con la Clasificación Internacional para la Retinopatía del Prematuro (IC-ROP 2005) en al menos un ojo con uno de los siguientes hallazgos retinianos:
- ROP Zona 1 Estadio 1+, 2+, 3 o 3+, o
- ROP Zona II Estadio 2+ o 3+, o
- AP-ROP (retinopatía posterior agresiva)
El criterio de valoración principal de eficacia de cada estudio fue la proporción de pacientes con ausencia de ROP activa y resultados estructurales desfavorables (desprendimiento de retina, arrastre macular, pliegue macular, opacidad retrolental) en la semana 52 de edad cronológica.
En BUTTERFLEYE, los pacientes se asignaron aleatoriamente en una proporción de 3:1 para recibir 1 de 2 regímenes de tratamiento: 1) EYLEA 0.4 mg en la línea de base y, si fuera necesario, hasta 2 inyecciones adicionales y 2) fotocoagulación con láser en cada ojo en la línea de base y, si fuera necesario, re-tratamiento. En FIREFLEYE, los pacientes se asignaron aleatoriamente a los mismos dos tratamientos, pero en una proporción de 2:1. El tratamiento de rescate se administró si fuera necesario, según criterios preestablecidos. En ambos estudios, más del 92% de todos los pacientes tratados en el grupo de aflibercept recibieron inyecciones bilaterales durante el estudio.
Los resultados de la semana 52 de edad cronológica en los estudios BUTTERFLEYE y FIREFLEYE/FIREFLEYE NEXT se muestran en la Tabla 12 a continuación.
La proporción de pacientes sin reactivaciones clínicamente significativas de ROP que tampoco desarrollaron resultados estructurales desfavorables fue mayor en cada brazo de cada estudio de lo que se habría esperado en los bebés que no habían recibido tratamiento. Ningún ensayo demostró superioridad de un brazo en comparación con el otro brazo. Ningún ensayo demostró inferioridad de un brazo en comparación con el otro brazo.
Tabla 12: Resultados de eficacia en la semana 52 de edad cronológica en los estudios BUTTERFLEYE y FIREFLEYE/FIREFLEYE NEXT
BUTTERFLEYE* FIREFLEYE/FIREFLEYE NEXT* EYLEA 0.4 mg Láser EYLEA 0.4 mg Láser Conjunto de análisis completo† N=93 N=27 N=75 N=38 Resultados de eficacia - *
- En caso de tratamiento bilateral, el éxito se logró solo si ambos ojos cumplieron con el criterio de valoración principal. El intervalo de tratamiento entre 2 dosis inyectadas en el mismo ojo tuvo que ser de al menos 28 días.
- †
- Incluyó pacientes que fueron tanto aleatorizados como tratados de los estudios BUTTERFLEYE y FIREFLEYE/FIREFLEYE NEXT. Esta fue la población de análisis principal como se definió en los Planes de Análisis Estadístico.
- ‡
- La diferencia con el intervalo de confianza (IC) se calculó utilizando el esquema de ponderación de Mantel-Haenszel ajustado por el estado de ROP de referencia. Criterio de éxito: Límite inferior del 95.1% de IC por encima del -5%.
Proporción de pacientes con ausencia de ROP activa y resultados estructurales desfavorables (%) 79.6% 77.8% 78.7% 81.6% Diferencia ajustada‡ (%)
(95.1% IC)1.81% (-15.7, 19.3) -1.88% (-17.0, 13.2) 16 FORMA DE SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
16.1 Cómo se suministra
Cada jeringa precargada o vial es una solución transparente, incolora a amarillo pálido y es solo para uso en un solo ojo. EYLEA se suministra en las siguientes presentaciones [ver Dosificación y administración (2.1), (2.2), (2.3), (2.4) y (2.9)]. Deseche la porción no utilizada.
NÚMERO NDC TIPO DE CAJA CONTENIDO DE LA CAJA 61755-005-01 Jeringa precargada un blíster que contiene una EYLEA
2 mg (0,05 mL de una solución de 40 mg/mL) jeringa de vidrio estéril, de dosis única, precargada
una Información de prescripción61755-005-02 Kit de vial con componentes de inyección un vial de EYLEA de 2 mg (0,05 mL de una solución de 40 mg/mL) de dosis única
una aguja de filtro de 18-gauge × 1½-pulgadas, 5-micras, para extraer el contenido del vial
una aguja de inyección de 30-gauge × ½-pulgada para inyección intravítrea
una jeringa de 1-mL para administración
una Información de prescripción16.2 Almacenamiento y manipulación
Refrigere EYLEA a 2°C a 8°C (36°F a 46°F). No congelar. No usar después de la fecha estampada en la caja y la etiqueta del contenedor. Almacenar en la caja original hasta el momento de su uso para protegerlo de la luz. No abra la bandeja blíster sellada hasta el momento de su uso.
17 INFORMACIÓN PARA EL PACIENTE
En los días posteriores a la administración de EYLEA, los pacientes corren el riesgo de desarrollar endoftalmitis, desprendimiento de retina o vasculitis retiniana con o sin oclusión. Si el ojo se enrojece, se vuelve sensible a la luz, duele o desarrolla un cambio en la visión, aconseje a los pacientes y/o cuidadores que busquen atención médica inmediata de un oftalmólogo [ver Advertencias y precauciones (5.1)].
Los pacientes pueden experimentar alteraciones visuales temporales después de una inyección intravítrea de EYLEA y de los exámenes oculares asociados [ver Reacciones adversas (6)]. Aconseje a los pacientes que no conduzcan ni utilicen maquinaria hasta que la función visual se haya recuperado suficientemente. En los bebés con retinopatía del prematuro, el tratamiento con EYLEA requerirá períodos prolongados de control de la retinopatía del prematuro.
SECCIÓN NO CLASIFICADA DE SPL
Fabricado por:
Regeneron Pharmaceuticals, Inc.
777 Old Saw Mill River Road
Tarrytown, NY 10591-6707
Número de Licencia de EE. UU. 1760
Para información de patentes: https://www.regeneron.com/downloads/us-patent-products.pdf
EYLEA es una marca registrada de Regeneron Pharmaceuticals, Inc.
© 2024, Regeneron Pharmaceuticals, Inc.
Todos los derechos reservados.
Fecha de revisión: Octubre de 2024
PANEL PRINCIPAL DE VISUALIZACIÓN – Vial Cartón de 2 mg/0.05 mL
NDC 61755-005-02
EYLEA®
(aflibercept) Inyección
Para inyección intravítrea2 mg (0.05 mL de una solución de 40 mg/mL)
Vial de dosis únicaContenido del cartón: Cada cartón de EYLEA contiene
- un vial de vidrio de 3 mL de dosis única de EYLEA
- una aguja de filtro de 18-gauge x 1½-pulgada, 5-micrón
para la extracción del contenido del vial (la aguja de filtro no
se debe utilizar para la inyección intravítrea) - una aguja de 30-gauge x ½-pulgada para inyección intravítrea
- una jeringa de plástico de 1 mL para la administración
- una Información de Prescripción
Rx ONLY

PANEL PRINCIPAL DE VISUALIZACIÓN – Jeringa Cartón 2 mg/0.05 mL
NDC 61755-005-01
Rx ONLYEYLEA®
(aflibercept) Injection
Para inyección intravítrea2 mg (0.05 mL de una solución de 40 mg/mL)
Jeringa precargada de dosis única
Contenido del envase:
- un blíster que contiene
una jeringa de vidrio precargada estéril, de dosis única - una Información de prescripción
REGENERON
 Tags: PD-1
Tags: PD-1