Fabricante de medicamentos: Alembic Pharmaceuticals Inc. (Updated: 2023-02-17)
DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
Tabletas de ERLOTINIB, para uso oral
Aprobación inicial de los EE. UU.: 2004
INDICACIONES Y USO
Las tabletas de erlotinib son un inhibidor de la kinasa indicado para: (1)
- El tratamiento de pacientes con cáncer de pulmón de células no pequeñas (NSCLC) metastásico cuyos tumores tienen deleciones del exón 19 del receptor del factor de crecimiento epidérmico (EGFR) o mutaciones de sustitución del exón 21 (L858R) detectadas por una prueba aprobada por la FDA que reciben tratamiento de primera línea, mantenimiento o segunda línea o superior después de la progresión después de al menos un régimen de quimioterapia anterior. (1.1)
- Tratamiento de primera línea de pacientes con cáncer de páncreas localmente avanzado, irresecable o metastásico, en combinación con gemcitabina. (1.2)
Limitaciones de uso: (1)
- No se ha establecido la seguridad y eficacia de las tabletas de erlotinib en pacientes con NSCLC cuyos tumores tienen otras mutaciones de EGFR. (1.1)
- No se recomiendan las tabletas de erlotinib para uso en combinación con quimioterapia a base de platino. (1.1)
DOSIS Y ADMINISTRACIÓN
- NSCLC: 150 mg por vía oral, con el estómago vacío, una vez al día. (2.2)
- Cáncer de páncreas: 100 mg por vía oral, con el estómago vacío, una vez al día. (2.3)
FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
Tabletas: 25 mg, 100 mg y 150 mg (3) (3)
CONTRAINDICACIONES
Ninguna. (4) (4)
ADVERTENCIAS Y PRECAUCIONES
- Enfermedad pulmonar intersticial (ILD): Ocurre en el 1,1% de los pacientes. Suspenda erlotinib por el inicio agudo de síntomas pulmonares inexplicables nuevos o progresivos, como disnea, tos y fiebre. Suspenda erlotinib si se diagnostica ILD. (5.1)
- Insuficiencia renal: Controle la función renal y los electrolitos, particularmente en pacientes con riesgo de deshidratación. Suspenda erlotinib por toxicidad renal severa. (5.2)
- Hepatotoxicidad: Ocurre con o sin insuficiencia hepática, incluida la insuficiencia hepática y el síndrome hepatorrenal: Controle las pruebas hepáticas periódicas. Suspenda o interrumpa erlotinib por pruebas hepáticas graves o que empeoran. (5.3)
- Perforaciones gastrointestinales: Suspenda erlotinib. (5.4)
- Trastornos ampollosos y exfoliativos de la piel: Suspenda erlotinib. (5.5)
- Accidente cerebrovascular (ACV): El riesgo de ACV aumenta en pacientes con cáncer de páncreas. (5.6)
- Anemia hemolítica microangiopática (MAHA): El riesgo de MAHA aumenta en pacientes con cáncer de páncreas. (5.7)
- Trastornos oculares: Suspenda erlotinib por perforación corneal, ulceración o queratitis grave persistente. (5.8)
- Hemorragia en pacientes que toman warfarina: Controle regularmente el INR en pacientes que toman warfarina u otros anticoagulantes derivados de la cumarina. (5.9)
- Toxicidad embriofetal: Puede causar daño fetal. Informe a las mujeres en edad reproductiva del riesgo potencial para el feto y que deben usar un método anticonceptivo eficaz. (5.10, 8.1, 8.3)
REACCIONES ADVERSAS
Las reacciones adversas más comunes (≥ 20%) con erlotinib de un análisis agrupado en pacientes con NSCLC en todas las líneas de terapia aprobadas, con y sin mutaciones de EGFR, y en pacientes con cáncer de páncreas fueron erupción cutánea, diarrea, anorexia, fatiga, disnea, tos, náuseas y vómitos. (6.1) (6)
Para informar REACCIONES ADVERSAS SOSPECHADAS, comuníquese con Alembic Pharmaceuticals Limited al 1-866-210-9797 o con la FDA al 1-800-FDA-1088 o http://www.fda.gov/medwatch (6)
INTERACCIONES FARMACOLÓGICAS
- Los inhibidores de CYP3A4 o un inhibidor combinado de CYP3A4 y CYP1A2 aumentan las concentraciones plasmáticas de erlotinib. Evite el uso concomitante. Si no es posible, reduzca la dosis de erlotinib. (2.4, 7)
- Los inductores de CYP3A4 disminuyen las concentraciones plasmáticas de erlotinib. Evite el uso concomitante. Si no es posible, aumente la dosis de erlotinib. (2.4, 7)
- El tabaquismo y los inductores de CYP1A2 disminuyen las concentraciones plasmáticas de erlotinib. Evite el uso concomitante. Si no es posible, aumente la dosis de erlotinib. (2.4, 7)
- Los medicamentos que aumentan el pH gástrico disminuyen las concentraciones plasmáticas de erlotinib. Para los inhibidores de la bomba de protones, evite el uso concomitante si es posible. Para los antagonistas de los receptores H-2, tome erlotinib 10 horas después de la dosis del antagonista de los receptores H-2. Para el uso con antiácidos, separe la dosis por varias horas. (2.4, 7)
USO EN POBLACIONES ESPECÍFICAS
Lactancia: No amamantar (8.2) (8)
Consulte la sección 17 para obtener INFORMACIÓN DE ORIENTACIÓN AL PACIENTE.
Revisado: 2/2023
Tabla de Contenido
INFORMACIÓN DE PRESCRIPCIÓN COMPLETA: CONTENIDO*
1 INDICACIONES Y USO
1.1 Cáncer de Pulmón de Células No Pequeñas (NSCLC)
1.2 Cáncer de Páncreas
2 DOSIS Y ADMINISTRACIÓN
2.1 Selección de Pacientes con NSCLC Metastásico
2.2 Dosis Recomendada – NSCLC
2.3 Dosis Recomendada – Cáncer de Páncreas
2.4 Modificaciones de Dosis
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Enfermedad Pulmonar Intersticial (ILD)
5.2 Insuficiencia Renal
5.3 Hepatotoxicidad con o sin Insuficiencia Hepática
5.4 Perforación Gastrointestinal
5.5 Trastornos Cutáneos Ampollosos y Exfoliativos
5.6 Accidente Cerebrovascular
5.7 Anemia Hemolítica Microangiopática con Trombocitopenia
5.8 Trastornos Oculares
5.9 Hemorragia en Pacientes Tomando Warfarina
5.10 Toxicidad Embrio-fetal
6 REACCIONES ADVERSAS
6.1 Experiencia en Ensayos Clínicos
6.2 Experiencia Post-Comercialización
7 INTERACCIONES MEDICAMENTOSAS
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.3 Mujeres y Hombres con Potencial Reproductivo
8.4 Uso Pediátrico
8.5 Uso Geriátrico
8.6 Insuficiencia Hepática
10 SOBREDOSIS
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de Acción
12.3 Farmacocinética
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
14 ESTUDIOS CLÍNICOS
14.1 Cáncer de Pulmón de Células No Pequeñas (NSCLC) – Tratamiento de Primera Línea de Pacientes con Mutaciones EGFR
14.2 NSCLC – Falta de Eficacia de Erlotinib en el Tratamiento de Mantenimiento de Pacientes sin Mutaciones EGFR
14.3 NSCLC – Tratamiento de Mantenimiento o Tratamiento de Segunda/Tercera Línea
14.4 NSCLC – Falta de Eficacia de Erlotinib Administrado Simultáneamente con Quimioterapia
14.5 Cáncer de Páncreas – Erlotinib Administrado Simultáneamente con Gemcitabina
16 PRESENTACIÓN/ALMACENAMIENTO Y MANIPULACIÓN
17 INFORMACIÓN DE ASESORAMIENTO PARA EL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información de prescripción completa no están enumeradas.
1 INDICACIONES Y USO
1.1 Cáncer de Pulmón de Células No Pequeñas (NSCLC)
Los comprimidos de erlotinib están indicados para:
- El tratamiento de pacientes con cáncer de pulmón de células no pequeñas (NSCLC) metastásico cuyos tumores tienen deleciones del exón 19 del receptor del factor de crecimiento epidérmico (EGFR) o mutaciones de sustitución del exón 21 (L858R) detectadas por una prueba aprobada por la FDA que reciben tratamiento de primera línea, mantenimiento o segunda línea o superior después de la progresión después de al menos un régimen de quimioterapia previo [ver Estudios Clínicos (14.1, 14.3)].
Limitaciones de uso:
- No se ha establecido la seguridad y eficacia de los comprimidos de erlotinib en pacientes con NSCLC cuyos tumores tienen otras mutaciones de EGFR [ver Estudios Clínicos (14.1, 14.2)].
- No se recomienda el uso de los comprimidos de erlotinib en combinación con quimioterapia a base de platino [ver Estudios Clínicos (14.4)].
1.2 Cáncer de Páncreas
El comprimido de erlotinib en combinación con gemcitabina está indicado para el tratamiento de primera línea de pacientes con cáncer de páncreas localmente avanzado, irresecable o metastásico [ver Estudios Clínicos (14.5)].
2 POSOLOGÍA Y ADMINISTRACIÓN
2.1 Selección de pacientes con NSCLC metastásico
Seleccione a los pacientes para el tratamiento del NSCLC metastásico con comprimidos de erlotinib en función de la presencia de deleciones del exón 19 del EGFR o mutaciones de sustitución del exón 21 (L858R) en muestras de tumor o plasma [Ver Estudios Clínicos (14.1, 14.2)]. Si estas mutaciones no se detectan en una muestra de plasma, analice el tejido tumoral si está disponible. La información sobre las pruebas aprobadas por la FDA para la detección de mutaciones de EGFR en NSCLC está disponible en: http://www.fda.gov/CompanionDiagnostics.
2.2 Dosis recomendada – NSCLC
La dosis diaria recomendada de comprimidos de erlotinib para NSCLC es de 150 mg tomados con el estómago vacío, es decir, al menos una hora antes o dos horas después de la ingestión de alimentos. El tratamiento debe continuar hasta que se produzca progresión de la enfermedad o toxicidad inaceptable.
2.3 Dosis recomendada – Cáncer de páncreas
La dosis diaria recomendada de comprimidos de erlotinib para el cáncer de páncreas es de 100 mg tomados una vez al día en combinación con gemcitabina. Tome los comprimidos de erlotinib con el estómago vacío, es decir, al menos una hora antes o dos horas después de la ingestión de alimentos. El tratamiento debe continuar hasta que se produzca progresión de la enfermedad o toxicidad inaceptable [ver Estudios Clínicos (14.5)].
2.4 Modificaciones de dosis
| Reacciones adversas |
||
| Pulmonar† |
Enfermedad pulmonar intersticial (ILD) | Descontinuar las tabletas de erlotinib |
| Durante la evaluación diagnóstica de posible ILD | Suspender las tabletas de erlotinib* |
|
| Hepático† |
Toxicidad hepática grave que no mejora significativamente o se resuelve en tres semanas | Descontinuar las tabletas de erlotinib |
| En pacientes con insuficiencia hepática preexistente u obstrucción biliar por duplicación de la bilirrubina o triplicación de los valores de transaminasas sobre el valor inicial | Suspender las tabletas de erlotinib* y considerar la discontinuación | |
| En pacientes sin insuficiencia hepática preexistente por niveles de bilirrubina total mayores a 3 veces el límite superior normal o transaminasas mayores a 5 veces el límite superior normal | Suspender las tabletas de erlotinib* y considerar la discontinuación | |
| Renal† |
Para toxicidad renal grave (grado 3 a 4 de CTCAE) | Suspender las tabletas de erlotinib* y considerar la discontinuación |
| Gastrointestinal† |
Perforación gastrointestinal | Descontinuar las tabletas de erlotinib |
| Para diarrea grave persistente que no responde al tratamiento médico (p. ej., loperamida) | Suspender las tabletas de erlotinib* |
|
| Cutánea† |
Condiciones cutáneas ampollosas, vesiculosas o exfoliativas graves | Descontinuar las tabletas de erlotinib |
| Para erupciones cutáneas graves que no responden al tratamiento médico | Suspender las tabletas de erlotinib* |
|
| Ocular† |
Perforación corneal o ulceración grave | Descontinuar las tabletas de erlotinib |
| Para queratitis de grado 3 a 4 (versión 4.0 de NCI-CTC) o de grado 2 que dura más de 2 semanas | Suspender las tabletas de erlotinib* |
|
| Para trastornos oculares agudos/que empeoran, como dolor ocular | Suspender las tabletas de erlotinib* y considerar la discontinuación | |
| Interacciones con otros medicamentos |
||
| Inhibidores de CYP3A4‡ |
Si ocurren reacciones graves con el uso concomitante de inhibidores potentes de CYP3A4 [como atazanavir, claritromicina, indinavir, itraconazol, ketoconazol, nefazodona, nelfinavir, ritonavir, saquinavir, telitromicina, troleandomicina (TAO), voriconazol, o jugo de toronja o pomelo] o cuando se usa concomitantemente con un inhibidor tanto de CYP3A4 como de CYP1A2 (p. ej., ciprofloxacina) | Reducir las tabletas de erlotinib en disminuciones de 50 mg; evitar el uso concomitante si es posible |
| Inductores de CYP3A4‡ |
Uso concomitante con inductores de CYP3A4, como rifampicina, rifabutina, rifapentina, fenitoína, carbamazepina, fenobarbital o hierba de San Juan | Aumentar las tabletas de erlotinib en incrementos de 50 mg a intervalos de 2 semanas hasta un máximo de 450 mg según se tolere. Evitar el uso concomitante si es posible |
| Fumar cigarrillos concurrentemente ठ|
Fumar cigarrillos concurrentemente | Aumentar las tabletas de erlotinib en incrementos de 50 mg a intervalos de 2 semanas hasta un máximo de 300 mg. Reducir inmediatamente la dosis de tabletas de erlotinib a la dosis recomendada (150 mg o 100 mg diarios) al cesar de fumar |
| Inhibidores de la bomba de protones | La separación de las dosis puede no eliminar la interacción ya que los inhibidores de la bomba de protones afectan el pH del tracto gastrointestinal superior por un período prolongado | Evitar el uso concomitante si es posible |
| Antagonistas del receptor H2 | Si se requiere tratamiento con un antagonista del receptor H2 como la ranitidina, separar las dosis. | Las tabletas de erlotinib se deben tomar 10 horas después de la dosis del antagonista del receptor H2 y por lo menos 2 horas antes de la siguiente dosis del antagonista del receptor H2 |
| Antiácidos | No se ha evaluado el efecto de los antiácidos sobre la farmacocinética de erlotinib. | La dosis de antiácido y la dosis de tabletas de erlotinib deben estar separadas por varias horas, si es necesario un antiácido |
† Para información adicional, ver Advertencias y precauciones (5).
* Reduzca las tabletas de erlotinib en decrementos de 50 mg al reiniciar el tratamiento después de retener el tratamiento por una toxicidad limitante de la dosis que se haya resuelto a la línea de base o grado ≤ 1.
‡ Para información adicional, ver Interacciones medicamentosas (7).
§ Para información adicional, ver Farmacología Clínica (12.3).
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
Las tabletas de erlotinib están disponibles en las siguientes concentraciones:
Las tabletas de erlotinib de 25 mg son tabletas redondas, biconvexas, recubiertas con una película blanca y grabadas con “L55” en un lado y lisas en el otro lado.
Las tabletas de erlotinib de 100 mg son tabletas redondas, biconvexas, recubiertas con una película blanca y grabadas con “L630” en un lado y lisas en el otro lado.
Las tabletas de erlotinib de 150 mg son tabletas redondas, biconvexas, recubiertas con una película blanca y grabadas con “L631” en un lado y lisas en el otro lado.
4 CONTRAINDICACIONES
Ninguna.
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Enfermedad pulmonar intersticial (EPI)
Pueden ocurrir casos graves de EPI, incluyendo casos fatales, con el tratamiento con erlotinib. La incidencia general de EPI en aproximadamente 32,000 pacientes tratados con erlotinib en estudios no controlados y estudios con quimioterapia concurrente fue de aproximadamente 1.1%. En pacientes con EPI, el inicio de los síntomas ocurrió entre 5 días a más de 9 meses (mediana 39 días) después de iniciar la terapia con erlotinib.
Suspenda erlotinib en caso de aparición aguda de síntomas pulmonares nuevos o progresivos inexplicables como disnea, tos y fiebre, en espera de una evaluación diagnóstica. Si se confirma EPI, suspenda permanentemente erlotinib [ver Dosificación y Administración (2.4)].
5.2 Insuficiencia renal
Pueden ocurrir síndrome hepatorrenal, insuficiencia renal aguda grave, incluyendo casos fatales, e insuficiencia renal con el tratamiento con erlotinib. La insuficiencia renal puede surgir por exacerbación de deterioro hepático basal subyacente o deshidratación severa. La incidencia agrupada de insuficiencia renal grave en los 3 estudios de monoterapia de cáncer de pulmón fue de 0.5% en los brazos de erlotinib y 0.8% en los brazos de control. La incidencia de insuficiencia renal en el estudio de cáncer de páncreas fue de 1.4% en el brazo de erlotinib más gemcitabina y 0.4% en el brazo de control. Suspenda erlotinib en pacientes que desarrollen insuficiencia renal grave hasta que se resuelva la toxicidad renal. Realice monitoreo periódico de la función renal y electrolitos séricos durante el tratamiento con erlotinib [ver Reacciones Adversas (6.1) y Dosificación y Administración (2.4)].
5.3 Hepatotoxicidad con o sin insuficiencia hepática
Pueden ocurrir insuficiencia hepática y síndrome hepatorrenal, incluyendo casos fatales, con el tratamiento con erlotinib en pacientes con función hepática normal; el riesgo de toxicidad hepática se incrementa en pacientes con deterioro hepático basal. En estudios clínicos en los que se excluyeron pacientes con deterioro hepático moderado a severo, la incidencia agrupada de insuficiencia hepática en los 3 estudios de monoterapia de cáncer de pulmón fue del 0.4% en los brazos de erlotinib y 0% en los brazos de control. La incidencia de insuficiencia hepática en el estudio de cáncer de páncreas fue de 0.4% en el brazo de erlotinib más gemcitabina y 0.4% en el brazo de control. En un estudio farmacocinético en 15 pacientes con deterioro hepático moderado (Child-Pugh B) asociado con una carga tumoral hepática significativa, 10 de estos 15 pacientes murieron dentro de los 30 días posteriores a la última dosis de erlotinib. Un paciente murió de síndrome hepatorrenal, 1 paciente murió de insuficiencia hepática rápidamente progresiva y los 8 pacientes restantes murieron de enfermedad progresiva. Seis de los 10 pacientes que murieron tenían bilirrubina total basal > 3 x LSN.
Realice pruebas hepáticas periódicas (transaminasas, bilirrubina y fosfatasa alcalina) durante el tratamiento con erlotinib. Se requiere una mayor frecuencia de monitoreo de la función hepática en pacientes con deterioro hepático preexistente u obstrucción biliar. Suspenda erlotinib en pacientes sin deterioro hepático preexistente con niveles de bilirrubina total superiores a 3 veces el límite superior normal o transaminasas superiores a 5 veces el límite superior normal. Suspenda erlotinib en pacientes con deterioro hepático preexistente u obstrucción biliar con duplicación de los valores de bilirrubina o triplicación de los valores de transaminasas con respecto al inicio. Suspenda erlotinib en pacientes cuyos resultados anormales de las pruebas hepáticas que cumplan con los criterios anteriores no mejoren significativamente o se resuelvan dentro de las tres semanas [ver Dosificación y Administración (2.4) y Farmacología Clínica (12.3)].
5.4 Perforación gastrointestinal
Puede ocurrir perforación gastrointestinal, incluidos casos fatales, con el tratamiento con erlotinib. Los pacientes que reciben agentes antiangiogénicos, corticosteroides, AINE o quimioterapia basada en taxanos concomitantes, o que tienen antecedentes de ulceración péptica o enfermedad diverticular, pueden tener un mayor riesgo de perforación [ver Reacciones Adversas (6.1, 6.2)]. La incidencia agrupada de perforación gastrointestinal en los 3 estudios de monoterapia de cáncer de pulmón fue del 0.2% en los brazos de erlotinib y 0.1% en los brazos de control. La incidencia de perforación gastrointestinal en el estudio de cáncer de páncreas fue de 0.4% en el brazo de erlotinib más gemcitabina y 0% en el brazo de control. Suspenda permanentemente erlotinib en pacientes que desarrollen perforación gastrointestinal [ver Dosificación y Administración (2.4)].
5.5 Trastornos cutáneos ampollosos y exfoliativos
Pueden ocurrir afecciones cutáneas ampollosas, con ampollas y exfoliativas, incluidos casos sugestivos de síndrome de Stevens-Johnson/necrólisis epidérmica tóxica, que en algunos casos fueron fatales, con el tratamiento con erlotinib [ver Reacciones Adversas (6.1, 6.2)]. La incidencia agrupada de trastornos cutáneos ampollosos y exfoliativos en los 3 estudios de monoterapia de cáncer de pulmón fue del 1.2% en los brazos de erlotinib y 0% en los brazos de control. La incidencia de trastornos cutáneos ampollosos y exfoliativos en el estudio de cáncer de páncreas fue de 0.4% en el brazo de erlotinib más gemcitabina y 0% en el brazo de control. Suspenda el tratamiento con erlotinib si el paciente desarrolla condiciones graves ampollosas, con ampollas o exfoliativas [ver Dosificación y Administración (2.4)].
5.6 Accidente cerebrovascular
En el ensayo de carcinoma de páncreas, siete pacientes en el grupo de erlotinib/gemcitabina desarrollaron accidentes cerebrovasculares (incidencia: 2.5%). Uno de estos fue hemorrágico y fue el único evento fatal. En comparación, en el grupo de placebo/gemcitabina no hubo accidentes cerebrovasculares. La incidencia agrupada de accidente cerebrovascular en los 3 estudios de monoterapia de cáncer de pulmón fue del 0.6% en los brazos de erlotinib y no fue mayor que la observada en los brazos de control.
5.7 Anemia hemolítica microangiopática con trombocitopenia
La incidencia agrupada de anemia hemolítica microangiopática con trombocitopenia en los 3 estudios de monoterapia de cáncer de pulmón fue del 0% en los brazos de erlotinib y 0.1% en los brazos de control. La incidencia de anemia hemolítica microangiopática con trombocitopenia en el estudio de cáncer de páncreas fue del 1.4% en el brazo de erlotinib más gemcitabina y 0% en el brazo de control.
5.8 Trastornos oculares
Pueden ocurrir disminución de la producción de lágrimas, crecimiento anormal de las pestañas, queratoconjuntivitis seca o queratitis con el tratamiento con erlotinib y pueden conducir a perforación o ulceración corneal [ver Reacciones Adversas (6.1) y (6.2)]. La incidencia agrupada de trastornos oculares en los 3 estudios de monoterapia de cáncer de pulmón fue del 17.8% en los brazos de erlotinib y 4% en los brazos de control. La incidencia de trastornos oculares en el estudio de cáncer de páncreas fue del 12.8% en el brazo de erlotinib más gemcitabina y 11.4% en el brazo de control. Interrumpa o suspenda la terapia con erlotinib si los pacientes presentan trastornos oculares agudos o que empeoran, como dolor ocular [ver Dosificación y Administración (2.4)].
5.9 Hemorragia en pacientes que toman warfarina
Pueden ocurrir hemorragias graves y fatales asociadas con elevaciones del índice internacional normalizado (INR) cuando se administran simultáneamente erlotinib y warfarina. Monitoree regularmente el tiempo de protrombina y el INR durante el tratamiento con erlotinib en pacientes que toman warfarina u otros anticoagulantes derivados de la cumarina [ver Reacciones Adversas (6.1) e Interacciones con otros medicamentos (7)].
5.10 Toxicidad embriofetal
Con base en datos en animales y su mecanismo de acción, erlotinib puede causar daño fetal cuando se administra a una mujer embarazada. Cuando se administró durante la organogénesis, la administración de erlotinib resultó en letalidad embriofetal y aborto en conejos a exposiciones aproximadamente 3 veces la exposición a la dosis diaria humana recomendada de 150 mg. Informe a las mujeres embarazadas sobre el riesgo potencial para el feto.
Aconseje a las mujeres en edad fértil que utilicen un método anticonceptivo eficaz durante la terapia y durante un mes después de la última dosis de erlotinib [ver Uso en Poblaciones Específicas (8.1) y (8.3), Farmacología Clínica (12.1)].
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas graves, que pueden incluir muertes, se discuten con mayor detalle en otras secciones del etiquetado:
- Enfermedad pulmonar intersticial (EPI) [ver Advertencias y precauciones (5.1)]
- Insuficiencia renal [ver Advertencias y precauciones (5.2)]
- Hepatotoxicidad con o sin insuficiencia hepática [ver Advertencias y precauciones (5.3)]
- Perforación gastrointestinal [ver Advertencias y precauciones (5.4)]
- Trastornos ampollosos y exfoliativos de la piel [ver Advertencias y precauciones (5.5)]
- Accidente cerebrovascular [ver Advertencias y precauciones (5.6)]
- Anemia hemolítica microangiopática con trombocitopenia [ver Advertencias y precauciones (5.7)]
- Trastornos oculares [ver Advertencias y precauciones (5.8)]
- Hemorragia en pacientes que toman warfarina [ver Advertencias y precauciones (5.9)]
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se realizan en condiciones ampliamente variadas, las tasas de reacciones adversas observadas en los ensayos clínicos de un medicamento no se pueden comparar directamente con las tasas en los ensayos clínicos de otro medicamento y pueden no reflejar las tasas observadas en la práctica.
La evaluación de seguridad de erlotinib se basa en más de 1200 pacientes con cáncer que recibieron erlotinib como monoterapia, más de 300 pacientes que recibieron erlotinib 100 o 150 mg más gemcitabina, y 1228 pacientes que recibieron erlotinib de forma concurrente con otras quimioterapias. Las reacciones adversas más comunes con erlotinib son erupción cutánea y diarrea, generalmente con inicio durante el primer mes de tratamiento. La incidencia de erupción cutánea y diarrea de los estudios clínicos de erlotinib para el tratamiento de NSCLC y cáncer de páncreas fue del 70% para erupción cutánea y 42% para diarrea.
Cáncer de pulmón de células no pequeñas
Tratamiento de primera línea de pacientes con mutaciones de EGFR
Las reacciones adversas más frecuentes (≥ 30%) en pacientes tratados con erlotinib fueron diarrea, astenia, erupción cutánea, tos, disnea y disminución del apetito. En pacientes tratados con erlotinib, la mediana de tiempo hasta el inicio de la erupción cutánea fue de 15 días y la mediana de tiempo hasta el inicio de la diarrea fue de 32 días.
Las reacciones adversas de Grado 3 a 4 más frecuentes en pacientes tratados con erlotinib fueron erupción cutánea y diarrea.
En el 37% de los pacientes tratados con erlotinib se produjeron interrupciones o reducciones de dosis debido a reacciones adversas, y el 14,3% de los pacientes tratados con erlotinib suspendieron el tratamiento debido a reacciones adversas. En pacientes tratados con erlotinib, las reacciones adversas notificadas con más frecuencia que llevaron a la modificación de la dosis fueron erupción cutánea (13%), diarrea (10%) y astenia (3,6%).
Las reacciones adversas comunes en el Estudio 1, que ocurrieron en al menos el 10% de los pacientes que recibieron erlotinib o quimioterapia y un aumento de ≥ 5% en el grupo tratado con erlotinib, se clasifican según los Criterios comunes de toxicidad del Instituto Nacional del Cáncer para eventos adversos versión 3.0 (NCI-CTCAE v3.0) Grado en la Tabla 1. La mediana de duración del tratamiento con erlotinib fue de 9,6 meses en el Estudio 1.
Tabla 1: Reacciones adversas con una tasa de incidencia ≥ 10% y un aumento de ≥ 5% en el grupo tratado con erlotinib (Estudio 1)
| Reacción adversa |
Erlotinib N = 84 |
Quimioterapia† N = 83 |
||
| Todos los grados % |
Grados 3 a 4 % |
Todos los grados % |
Grados 3 a 4 % |
|
| Erupción cutánea‡ |
85 | 14 | 5 | 0 |
| Diarrea | 62 | 5 | 21 | 1 |
| Tos | 48 | 1 | 40 | 0 |
| Disnea | 45 | 8 | 30 | 4 |
| Piel seca | 21 | 1 | 2 | 0 |
| Dolor de espalda | 19 | 2 | 5 | 0 |
| Dolor de pecho | 18 | 1 | 12 | 0 |
| Conjuntivitis | 18 | 0 | 0 | 0 |
| Inflamación mucosa | 18 | 1 | 6 | 0 |
| Prurito | 16 | 0 | 1 | 0 |
| Paroniquia | 14 | 0 | 0 | 0 |
| Artralgia | 13 | 1 | 6 | 1 |
| Dolor musculoesquelético | 11 | 1 | 1 | 0 |
† Quimioterapia basada en platino (cisplatino o carboplatino con gemcitabina o docetaxel).
‡ Erupción cutánea como un término compuesto incluye erupción cutánea, acné, foliculitis, eritema, dermatitis acneiforme, dermatitis, síndrome de eritrodisestesia palmar-plantar, erupción exfoliativa, erupción eritematosa, erupción prurítica, toxicidad cutánea, eczema, erupción folicular, úlcera cutánea.
Toxicidad hepática: Un paciente tratado con erlotinib experimentó insuficiencia hepática fatal y otros cuatro pacientes experimentaron anomalías en las pruebas hepáticas de grado 3 a 4 en el Estudio 1 [ver Advertencias y precauciones (5.3)].
Tratamiento de mantenimiento
Las reacciones adversas, independientemente de la causalidad, que ocurrieron en al menos el 3% de los pacientes tratados con erlotinib en monoterapia a 150 mg y al menos un 3% más a menudo que en el grupo de placebo en el ensayo de mantenimiento aleatorizado (Estudio 3) se resumen por Grado NCI-CTCAE v3.0 en la Tabla 2.
Las reacciones adversas más comunes en pacientes que recibieron erlotinib en monoterapia a 150 mg fueron erupción cutánea y diarrea. Se produjo erupción cutánea y diarrea de grado 3 a 4 en el 9% y el 2%, respectivamente, en pacientes tratados con erlotinib. La erupción cutánea y la diarrea resultaron en la interrupción del estudio en el 1% y el 0,5% de los pacientes tratados con erlotinib, respectivamente. Se necesitó reducción de dosis o interrupción por erupción cutánea y diarrea en el 5% y el 3% de los pacientes, respectivamente. En pacientes tratados con erlotinib, la mediana de tiempo hasta la aparición de la erupción cutánea fue de 10 días y la mediana de tiempo hasta la aparición de la diarrea fue de 15 días.
Tabla 2: Estudio de mantenimiento de NSCLC: Reacciones adversas que ocurren con una tasa de incidencia ≥ 10% y un aumento de ≥ 5% en el grupo de erlotinib en monoterapia en comparación con el grupo de placebo (Estudio 3)
| Reacción adversa |
Erlotinib N = 433 |
PLACEBO N = 445 |
||||
| Cualquier grado |
Grado 3 |
Grado 4 |
Cualquier grado |
Grado 3 |
Grado 4 |
|
| % |
% |
% |
% |
% |
% |
|
| Erupción† |
60 | 9 | 0 | 9 | 0 | 0 |
| Diarrea | 20 | 2 | 0 | 4 | 0 | 0 |
†Erupción como término compuesto incluye: erupción, acné, dermatitis acneiforme, fisuras de la piel, eritema, erupción papular, erupción generalizada, erupción pruriginosa, exfoliación de la piel, urticaria, dermatitis, eccema, erupción exfoliativa, dermatitis exfoliativa, furúnculo, erupción macular, erupción pustular, hiperpigmentación de la piel, reacción cutánea, úlcera cutánea.
Se observaron anomalías en las pruebas hepáticas, incluidas elevaciones de ALT, de grado 2 o mayor en el 3% de los pacientes tratados con erlotinib y en el 1% de los pacientes tratados con placebo. Se observaron elevaciones de bilirrubina de grado 2 y superiores en el 5% de los pacientes tratados con erlotinib y en <1% en el grupo placebo [ver Dosificación y administración (2.4) y Advertencias y precauciones (5.3)].
Tratamiento de segunda/tercera línea
Las reacciones adversas, independientemente de la causalidad, que ocurrieron en al menos el 10% de los pacientes tratados con erlotinib en monoterapia a 150 mg y al menos un 5% más a menudo que en el grupo placebo en el ensayo aleatorizado de pacientes con NSCLC se resumen por grado NCI-CTC v2.0 en la Tabla 3.
Las reacciones adversas más comunes en esta población de pacientes fueron erupción cutánea y diarrea. La erupción y la diarrea de grado 3 a 4 ocurrieron en el 9% y el 6%, respectivamente, en los pacientes tratados con erlotinib. La erupción cutánea y la diarrea provocaron la interrupción del estudio en el 1% de los pacientes tratados con erlotinib. El seis por ciento y el uno por ciento de los pacientes necesitaron reducción de la dosis por erupción y diarrea, respectivamente. La mediana de tiempo hasta la aparición de la erupción fue de 8 días y la mediana de tiempo hasta la aparición de la diarrea fue de 12 días.
Tabla 3: Estudio de NSCLC de 2ª/3ª línea: Reacciones adversas que ocurren con una tasa de incidencia ≥ 10% y un aumento de ≥ 5% en el grupo de erlotinib en monoterapia en comparación con el grupo placebo (Estudio 4)
| Reacción Adversa |
Erlotinib 150 mg N=485 |
Placebo N=242 |
||||
| Cualquier Grado |
Grado 3 |
Grado 4 |
Cualquier Grado |
Grado 3 |
Grado 4 |
|
| % |
% |
% |
% |
% |
% |
|
| Rash† |
75 | 8 | <1 | 17 | 0 | 0 |
| Diarrea | 54 | 6 | <1 | 18 | <1 | 0 |
| Anorexia | 52 | 8 | 1 | 38 | 5 | <1 |
| Fatiga | 52 | 14 | 4 | 45 | 16 | 4 |
| Disnea | 41 | 17 | 11 | 35 | 15 | 11 |
| Náuseas | 33 | 3 | 0 | 24 | 2 | 0 |
| Infección | 24 | 4 | 0 | 15 | 2 | 0 |
| Estomatitis | 17 | <1 | 0 | 3 | 0 | 0 |
| Prurito | 13 | <1 | 0 | 5 | 0 | 0 |
| Sequedad de la piel | 12 | 0 | 0 | 4 | 0 | 0 |
| Conjuntivitis | 12 | <1 | 0 | 2 | <1 | 0 |
| Queratoconjuntivitis seca | 12 | 0 | 0 | 3 | 0 | 0 |
†Rash como término compuesto incluye: rash, síndrome de eritrodisestesia palmar-plantar, acné, trastorno de la piel, trastorno de la pigmentación, eritema, úlcera cutánea, dermatitis exfoliativa, rash papular, descamación de la piel.
Se observaron anomalías en las pruebas de función hepática [incluido el aumento de alanina aminotransferasa (ALT), aspartato aminotransferasa (AST) y bilirrubina] en pacientes que recibieron erlotinib en monoterapia de 150 mg. Estas elevaciones fueron principalmente transitorias o asociadas con metástasis hepáticas. Las elevaciones de ALT de Grado 2 [> 2,5 a 5 veces el límite superior de lo normal (LSN)] ocurrieron en el 4% y < 1% de los pacientes tratados con erlotinib y placebo, respectivamente. No se observaron elevaciones de Grado 3 (> 5 a 20 x LSN) en pacientes tratados con erlotinib. La dosis de erlotinib debe interrumpirse o discontinuarse si los cambios en la función hepática son graves [ver Dosificación y Administración (2.4)].
Cáncer de páncreas-Erlotinib administrado simultáneamente con gemcitabina
Este fue un estudio aleatorizado, doble ciego, controlado con placebo de erlotinib (150 mg o 100 mg diarios) o placebo más gemcitabina (1000 mg/m2 por infusión intravenosa) en pacientes con cáncer de páncreas localmente avanzado, irresecable o metastásico (Estudio 5). La población de seguridad comprendió 282 pacientes en el grupo de erlotinib (259 en la cohorte de 100 mg y 23 en la cohorte de 150 mg) y 280 pacientes en el grupo de placebo (256 en la cohorte de 100 mg y 24 en la cohorte de 150 mg).
Las reacciones adversas que ocurrieron en al menos el 10% de los pacientes tratados con erlotinib 100 mg más gemcitabina en el ensayo aleatorizado de pacientes con cáncer de páncreas (Estudio 5) se clasificaron según NCI-CTC v2.0 en la Tabla 4.
Las reacciones adversas más comunes en pacientes con cáncer de páncreas que recibieron erlotinib 100 mg más gemcitabina fueron fatiga, erupción cutánea, náuseas, anorexia y diarrea. En el brazo de erlotinib más gemcitabina, se informó erupción cutánea y diarrea de Grado 3 a 4 en el 5% de los pacientes, respectivamente. La mediana de tiempo hasta la aparición de la erupción y la diarrea fue de 10 días y 15 días, respectivamente. La erupción y la diarrea resultaron en reducciones de dosis en el 2% de los pacientes y resultaron en la interrupción del estudio en hasta el 1% de los pacientes que recibieron erlotinib más gemcitabina. Las reacciones adversas graves (≥ Grado 3 NCI-CTC) en el grupo de erlotinib más gemcitabina con incidencias <5% incluyeron síncope, arritmias, íleo, pancreatitis, anemia hemolítica, incluida la anemia hemolítica microangiopática con trombocitopenia, infarto de miocardio/isquemia, accidentes cerebrovasculares, incluida hemorragia cerebral, e insuficiencia renal [ver Advertencias y Precauciones (5)].
La cohorte de 150 mg se asoció con una tasa más alta de ciertas reacciones adversas específicas de clase, incluida la erupción, y requirió una reducción o interrupción de la dosis más frecuente.
Tabla 4: Reacciones adversas que ocurren con una tasa de incidencia ≥ 10% y un aumento de ≥ 5% en pacientes con cáncer de páncreas tratados con erlotinib: cohorte de 100 mg (Estudio 5)
| Reacción Adversa |
Erlotinib + Gemcitabina 1000 mg/m2 IV N=259 |
Placebo + Gemcitabina 1000 mg/m2 IV N=256 |
||||
|---|---|---|---|---|---|---|
| Cualquier Grado |
Grado 3 |
Grado 4 |
Cualquier Grado |
Grado 3 |
Grado 4 |
|
| % |
% |
% |
% |
% |
% |
|
| Rash† |
70 | 5 | 0 | 30 | 1 | 0 |
| Diarrea | 48 | 5 | <1 | 36 | 2 | 0 |
| Disminución del peso | 39 | 2 | 0 | 29 | <1 | 0 |
| Infección* |
39 | 13 | 3 | 30 | 9 | 2 |
| Pirexia | 36 | 3 | 0 | 30 | 4 | 0 |
| Estomatitis | 22 | <1 | 0 | 12 | 0 | 0 |
| Depresión | 19 | 2 | 0 | 14 | <1 | 0 |
| Tos | 16 | 0 | 0 | 11 | 0 | 0 |
| Dolor de cabeza | 15 | <1 | 0 | 10 | 0 | 0 |
* Las infecciones como término compuesto incluyen infecciones con patógenos no especificados, así como trastornos infecciosos bacterianos (incluyendo clamidiales, rickettsiales, micobacterianos y micoplásmicos), parasitarios (incluyendo helmínticos, ectoparasitarios y protozoarios), virales y fúngicos.
† Rash como término compuesto incluye: rash, síndrome de eritrodisestesia palmo-plantar, trastorno de la pigmentación, dermatitis acneiforme, foliculitis, reacción de fotosensibilidad, síndrome de Stevens-Johnson, urticaria, rash eritematoso, trastorno de la piel, úlcera de la piel.
Diez pacientes (4%) en el grupo de erlotinib/gemcitabina y tres pacientes (1%) en el grupo de placebo/gemcitabina desarrollaron trombosis venosa profunda. La incidencia general de eventos trombóticos de grado 3 o 4, incluida la trombosis venosa profunda, fue del 11% para erlotinib más gemcitabina y del 9% para placebo más gemcitabina.
Las incidencias de anormalidades en las pruebas hepáticas (≥ Grado 2) en el Estudio 5 se proporcionan en la Tabla 5 [ver Dosificación y Administración (2.4) y Advertencias y Precauciones (5.3)].
Tabla 5: Anormalidades en las Pruebas Hepáticas en Pacientes con Cáncer de Páncreas: Cohorte de 100 mg (Estudio 5)
| Erlotinib + Gemcitabine 1000 mg/m2 IV N=259 |
Placebo + Gemcitabine 1000 mg/m2 IV N=256 |
|||||
| Grado 2 |
Grado 3 |
Grado 4 |
Grado 2 |
Grado 3 |
Grado 4 |
|
| Bilirrubina | 17% | 10% | <1% | 11% | 10% | 3% |
| ALT | 31% | 13% | <1% | 22% | 9% | 0% |
| AST | 24% | 10% | <1% | 19% | 9% | 0% |
Indicaciones para NSCLC y páncreas: Reacciones adversas seleccionadas de baja frecuencia
Trastornos gastrointestinales
Se han reportado casos de hemorragia gastrointestinal (incluidas muertes), algunos asociados con la administración concomitante de warfarina o AINE [ver Advertencias y precauciones (5.9) e Interacciones farmacológicas (7)]. Estas reacciones adversas se reportaron como hemorragia por úlcera péptica (gastritis, úlceras gastroduodenales), hematemesis, hematoquecia, melena y hemorragia por posible colitis.
6.2 Experiencia posterior a la comercialización
Las siguientes reacciones adversas se han identificado durante el uso posterior a la aprobación de erlotinib. Debido a que estas reacciones se informan voluntariamente de una población de tamaño incierto, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal con la exposición al fármaco.
Trastornos musculoesqueléticos y del tejido conectivo: miopatía, incluyendo rabdomiólisis, en combinación con terapia con estatinas
Trastornos oculares: inflamación ocular incluyendo uveítis
7 INTERACCIONES MEDICAMENTOSAS
Inhibidores de CYP3A4
La administración conjunta de erlotinib con un inhibidor potente de CYP3A4 o un inhibidor combinado de CYP3A4 y CYP1A2 aumentó la exposición a erlotinib. Erlotinib se metaboliza principalmente por CYP3A4 y en menor medida por CYP1A2. Una mayor exposición a erlotinib puede aumentar el riesgo de toxicidad relacionada con la exposición [ver Farmacología Clínica (12.3)].
Evite administrar conjuntamente erlotinib con inhibidores potentes de CYP3A4 (por ejemplo, boceprevir, claritromicina, conivaptan, indinavir, itraconazol, ketoconazol, lopinavir/ritonavir, nefazodona, nelfinavir, posaconazol, ritonavir, saquinavir, telitromicina, voriconazol, pomelo o jugo de pomelo) o un inhibidor combinado de CYP3A4 y CYP1A2 (por ejemplo, ciprofloxacino). Reduzca la dosis de erlotinib cuando se administre conjuntamente con un inhibidor potente de CYP3A4 o un inhibidor combinado de CYP3A4 y CYP1A2 si la administración conjunta es inevitable [ver Dosis y Administración (2.4)].
Inductores de CYP3A4
El tratamiento previo con un inductor de CYP3A4 antes de erlotinib disminuyó la exposición a erlotinib [ver Farmacología Clínica (12.3)]. Aumente la dosis de erlotinib si la administración conjunta con inductores de CYP3A4 (por ejemplo, carbamazepina, fenitoína, rifampicina, rifabutina, rifapentina, fenobarbital y hierba de San Juan) es inevitable [ver Dosis y Administración (2.4)].
Inductores de CYP1A2 y Tabaquismo
Fumar cigarrillos disminuyó la exposición a erlotinib. Evite fumar tabaco (inductor de CYP1A2) y evite el uso concomitante de erlotinib con inductores moderados de CYP1A2 (por ejemplo, teriflunomida, rifampicina o fenitoína). Aumente la dosis de erlotinib en pacientes que fuman tabaco o cuando la administración conjunta con inductores moderados de CYP1A2 es inevitable [ver Dosis y Administración (2.4) y Farmacología Clínica (12.3)].
Medicamentos que Aumentan el pH Gástrico
La administración conjunta de erlotinib con inhibidores de la bomba de protones (por ejemplo, omeprazol) y antagonistas de los receptores H-2 (por ejemplo, ranitidina) disminuyó la exposición a erlotinib [ver Farmacología Clínica (12.3)]. Para los inhibidores de la bomba de protones, evite el uso concomitante si es posible. Para los antagonistas de los receptores H-2 y antiácidos, modifique el esquema de dosificación [ver Dosis y Administración (2.4)]. Aumentar la dosis de erlotinib cuando se administra conjuntamente con agentes que elevan el pH gástrico no es probable que compense la pérdida de exposición.
Anticoagulantes
Se han reportado interacciones con anticoagulantes derivados de la cumarina, incluida la warfarina, que conducen a un aumento del Índice Internacional Normalizado (INR) y reacciones adversas hemorrágicas, que en algunos casos fueron fatales, en pacientes que recibieron erlotinib. Monitoree regularmente el tiempo de protrombina o el INR en pacientes que toman anticoagulantes derivados de la cumarina. No se recomienda la modificación de la dosis de erlotinib [ver Advertencias y Precauciones (5.9) y Reacciones Adversas (6.1)].
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de riesgos
Según datos en animales y su mecanismo de acción, erlotinib puede causar daño fetal cuando se administra a una mujer embarazada. Los datos limitados disponibles sobre el uso de erlotinib en mujeres embarazadas no son suficientes para informar un riesgo de defectos congénitos importantes o aborto espontáneo. Cuando se administró durante la organogénesis, la administración de erlotinib resultó en letalidad embriofetal y aborto en conejos a exposiciones aproximadamente 3 veces la exposición a la dosis diaria humana recomendada de 150 mg. Informe a las mujeres embarazadas del riesgo potencial para el feto.
En la población general de EE. UU., el riesgo estimado de defectos congénitos importantes y aborto espontáneo en embarazos clínicamente reconocidos es del 2 al 4 % y del 15 al 20 %, respectivamente.
Datos
Datos en animales
Se ha demostrado que erlotinib causa toxicidad materna que resulta en letalidad embriofetal y aborto en conejos cuando se administra durante el período de organogénesis en dosis que resultan en concentraciones plasmáticas de fármaco aproximadamente 3 veces las alcanzadas con la dosis recomendada en humanos (AUC a dosis diaria de 150 mg). Durante el mismo período, no hubo aumento en la incidencia de letalidad embriofetal o aborto en conejos o ratas a dosis que resultan en exposiciones aproximadamente iguales a las de los humanos a la dosis diaria recomendada. En un estudio independiente de fertilidad, las ratas hembra tratadas con 30 mg/m2/día o 60 mg/m2/día (0.3 o 0.7 veces la dosis diaria recomendada, sobre una base de mg/m2) de erlotinib tuvieron un aumento en las reabsorciones tempranas que resultó en una disminución en el número de fetos vivos.
No se observaron efectos teratogénicos en conejos o ratas dosificados con erlotinib durante la organogénesis en dosis de hasta 600 mg/m2/día en el conejo (3 veces la concentración plasmática del fármaco observada en humanos a 150 mg/día) y hasta 60 mg/m2/día en la rata (0.7 veces la dosis recomendada de 150 mg/día sobre una base de mg/m2).
8.2 Lactancia
Resumen de riesgos
No hay datos sobre la presencia de erlotinib en la leche humana, o los efectos de erlotinib en el lactante o en la producción de leche. Debido al potencial de reacciones adversas graves en lactantes por erlotinib, que incluyen enfermedad pulmonar intersticial, hepatotoxicidad, trastornos cutáneos ampollosos y exfoliativos, anemia hemolítica microangiopática con trombocitopenia, trastornos oculares y diarrea. Aconseje a las mujeres en período de lactancia que no amamanten durante el tratamiento con erlotinib y durante 2 semanas después de la dosis final.
8.3 Mujeres y hombres con capacidad reproductiva
Anticoncepción
Mujeres
Erlotinib puede causar daño fetal cuando se administra a una mujer embarazada [ver Uso en poblaciones específicas (8.1)]. Aconseje a las mujeres con potencial reproductivo que utilicen anticoncepción efectiva durante el tratamiento con erlotinib y durante un mes después de la última dosis de erlotinib.
8.4 Uso pediátrico
No se ha establecido la seguridad y eficacia de erlotinib en pacientes pediátricos.
En un ensayo abierto, multicéntrico, 25 pacientes pediátricos (mediana de edad 14 años, rango de 3 a 20 años) con ependimoma recurrente o refractario fueron aleatorizados (1:1) a erlotinib o etopósido. Trece pacientes recibieron erlotinib a una dosis de 85 mg/m2/día por vía oral hasta la progresión de la enfermedad, muerte, solicitud del paciente, decisión del investigador de suspender el fármaco del estudio o toxicidad intolerable. Cuatro pacientes aleatorizados a etopósido también recibieron erlotinib después de la progresión de la enfermedad. El ensayo se terminó prematuramente por falta de eficacia; no se observaron respuestas objetivas en estos 17 pacientes tratados con erlotinib.
No se identificaron nuevos eventos adversos en la población pediátrica.
Según el análisis de farmacocinética poblacional realizado en 105 pacientes pediátricos (de 2 a 21 años) con cáncer, las estimaciones de la media geométrica de CL/F/BSA (clearance aparente normalizado al área de superficie corporal) fueron comparables entre los tres grupos de edad: 2 a 6 años (n = 29), 7 a 16 años (n = 59) y 17 a 21 años (n = 17).
8.5 Uso geriátrico
De los 1297 sujetos en estudios clínicos de erlotinib para el tratamiento de NSCLC y cáncer de páncreas, el 40 % tenían 65 años o más, mientras que el 10 % tenían 75 años o más. No se observaron diferencias generales en seguridad o eficacia entre los sujetos de 65 años o más y los menores de 65 años.
8.6 Insuficiencia hepática
La insuficiencia hepática y el síndrome hepatorrenal, incluidos los casos mortales, pueden ocurrir con el tratamiento con erlotinib en pacientes con función hepática normal; el riesgo de toxicidad hepática aumenta en pacientes con deterioro hepático basal [ver Advertencias y precauciones (5.3), Reacciones adversas (6.1, 6.2) y Dosificación y administración]. Monitoree a los pacientes con insuficiencia hepática (bilirrubina total mayor que el límite superior normal (ULN) o Child-Pugh A, B y C) durante la terapia con erlotinib. El tratamiento con erlotinib debe usarse con mayor monitoreo en pacientes con bilirrubina total mayor que 3 x ULN [ver Advertencias y precauciones (5.3), Reacciones adversas (6.1, 6.2) y Dosificación y administración (2.4)].
10 SOBREDOSIS
Suspender erlotinib en pacientes con una sobredosis o sospecha de sobredosis e instituir tratamiento sintomático.
11 DESCRIPCIÓN
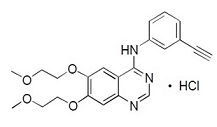
Erlotinib, un inhibidor de kinasa, es una quinazolinamina con el nombre químico N-(3-etinilfenil)-6,7-bis(2-metoxietoxi)-4-quinazolinamina. La tableta de erlotinib contiene erlotinib como la sal de hidrocloruro que tiene la siguiente fórmula estructural:
El hidrocloruro de erlotinib tiene la fórmula molecular C22H23N3O4.HCl y un peso molecular de 429.9. La molécula tiene un pKa de 5.42 a 25°C. Es libremente soluble en ácido fórmico, muy ligeramente soluble en N,N-Dimetilformamida y prácticamente insoluble en agua.
Las tabletas de erlotinib para administración oral están disponibles en tres concentraciones de dosis que contienen hidrocloruro de erlotinib (27.3 mg, 109.3 mg y 163.9 mg) equivalentes a 25 mg, 100 mg y 150 mg de erlotinib y los siguientes ingredientes inactivos: lactosa monohidrato, celulosa microcristalina, glicolato sódico de almidón, estearato de magnesio y lauril sulfato de sodio. Los ingredientes inactivos del recubrimiento pelicular son hipromelosa, hidroxipropilcelulosa, dióxido de titanio y polietilenglicol 400.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
El receptor del factor de crecimiento epidérmico (EGFR) se expresa en la superficie celular tanto de células normales como cancerosas. En algunas células tumorales, la señalización a través de este receptor juega un papel en la supervivencia y proliferación de las células tumorales, independientemente del estado mutacional de EGFR. Erlotinib inhibe de forma reversible la actividad quinasa de EGFR, evitando la autofosforilación de los residuos de tirosina asociados con el receptor y, por lo tanto, inhibiendo la señalización posterior. La afinidad de unión de erlotinib por las mutaciones de EGFR de deleción del exón 19 o del exón 21 (L858R) es mayor que su afinidad por el receptor de tipo salvaje. La inhibición de otros receptores de tirosina quinasa por erlotinib no se ha caracterizado completamente.
12.3 Farmacocinética
Absorción
Erlotinib se absorbe aproximadamente un 60% después de la administración oral. Los niveles plasmáticos máximos ocurren 4 horas después de la dosificación.
Efecto de los alimentos
Los alimentos aumentaron la biodisponibilidad de erlotinib a aproximadamente el 100%.
Distribución
Erlotinib se une en un 93% a la albúmina plasmática y a la glicoproteína ácida alfa-1 (AAG).
Erlotinib tiene un volumen aparente de distribución de 232 litros.
Eliminación
Erlotinib se elimina con una vida media de 36,2 horas en pacientes que reciben el régimen de erlotinib en monoterapia de 2a/3a línea. Por lo tanto, el tiempo para alcanzar la concentración plasmática en estado estacionario sería de 7 a 8 días.
Metabolismo
Erlotinib se metaboliza principalmente por CYP3A4 y en menor medida por CYP1A2, y por la isoforma extrahepática CYP1A1, in vitro.
Excreción
Después de una dosis oral de 100 mg, se recuperó el 91% de la dosis: 83% en heces (1% de la dosis como compuesto original) y 8% en orina (0,3% de la dosis como compuesto original).
Poblaciones específicas
Ni la edad, el peso corporal ni el sexo tuvieron un efecto clínicamente significativo sobre la exposición sistémica a erlotinib en pacientes con NSCLC que recibieron erlotinib en monoterapia para el tratamiento de 2a/3a línea o para el tratamiento de mantenimiento, y en pacientes con cáncer de páncreas que recibieron erlotinib más gemcitabina. Se desconoce la farmacocinética de erlotinib en pacientes con función renal comprometida.
Pacientes con insuficiencia hepática
La evidencia in vitro e in vivo sugiere que erlotinib se elimina principalmente por el hígado. Sin embargo, la exposición a erlotinib fue similar en pacientes con insuficiencia hepática moderada (Child-Pugh B) en comparación con pacientes con función hepática adecuada, incluidos pacientes con cáncer de hígado primario o metástasis hepáticas.
Pacientes fumadores de cigarrillos
En un ensayo de farmacocinética de dosis única en voluntarios sanos, fumar cigarrillos (inductor moderado de CYP1A2) aumentó el aclaramiento de erlotinib y disminuyó el AUC0-inf de erlotinib en un 64% (IC del 95%: 46 a 76%) en fumadores actuales en comparación con ex fumadores o personas que nunca habían fumado. En un ensayo de NSCLC, los fumadores actuales alcanzaron concentraciones plasmáticas mínimas de erlotinib en estado estacionario que fueron aproximadamente 2 veces menores que las de los ex fumadores o los pacientes que nunca habían fumado. Este efecto se acompañó de un aumento del 24% en el aclaramiento plasmático aparente de erlotinib. En otro estudio realizado en pacientes con NSCLC que eran fumadores actuales, los análisis farmacocinéticos en estado estacionario indicaron un aumento proporcional a la dosis en la exposición a erlotinib cuando la dosis de erlotinib se aumentó de 150 mg a 300 mg. [ver Dosificación y administración (2.4), Interacciones farmacológicas (7) e Información de asesoramiento al paciente (17)].
Estudios de interacción farmacológica
La coadministración de gemcitabina no tuvo efecto sobre el aclaramiento plasmático de erlotinib.
Inhibidores de CYP3A4
La coadministración con un potente inhibidor de CYP3A4, ketoconazol, aumentó el AUC de erlotinib en un 67%. La coadministración con un inhibidor combinado de CYP3A4 y CYP1A2, ciprofloxacino, aumentó la exposición a erlotinib [AUC] en un 39% y aumentó la concentración máxima de erlotinib [Cmax] en un 17%. [ver Modificaciones de la dosis (2.4), Interacciones farmacológicas (7)].
Inductores de CYP3A4
El pretratamiento con el inductor de CYP3A4 rifampicina, durante 7 a 11 días antes de erlotinib, disminuyó el AUC de erlotinib en un 58% a 80% [ver Modificaciones de la dosis (2.4), Interacciones farmacológicas (7)].
Inductores de CYP1A2 o tabaquismo
Ver la sección Poblaciones específicas [ver Modificaciones de la dosis (2.4), Interacciones farmacológicas (7)].
Medicamentos que aumentan el pH gástrico
La solubilidad de erlotinib depende del pH y disminuye a medida que aumenta el pH. Cuando se coadministró un inhibidor de la bomba de protones (omeprazol) con erlotinib, la exposición a erlotinib [AUC] disminuyó en un 46% y la concentración máxima de erlotinib [Cmax] disminuyó en un 61%. Cuando se administró erlotinib 2 horas después de una dosis de 300 mg de un antagonista del receptor H-2 (ranitidina), el AUC de erlotinib se redujo en un 33% y la Cmax de erlotinib se redujo en un 54%. Cuando se administró erlotinib con ranitidina 150 mg dos veces al día (al menos 10 horas después de la dosis anterior de ranitidina por la noche y 2 horas antes de la dosis matutina de ranitidina), el AUC de erlotinib disminuyó en un 15% y la Cmax de erlotinib disminuyó en un 17% [ver Modificaciones de la dosis (2.4), Interacciones farmacológicas (7)].
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
Se realizaron estudios de carcinogenicidad de dos años en ratones y ratas con erlotinib a dosis orales de hasta 60 mg/kg/día en ratones, 5 mg/kg/día en ratas hembra y 10 mg/kg/día en ratas macho. Los estudios fueron negativos para hallazgos carcinogénicos. La exposición en ratones a la dosis más alta probada fue aproximadamente 10 veces la exposición en humanos a la dosis de erlotinib de 150 mg/día. La dosis más alta evaluada en ratas macho resultó en exposiciones que fueron el doble de las de los humanos y las exposiciones a la dosis más alta probada en ratas hembra fueron ligeramente más bajas que las de los humanos.
Erlotinib no causó daño genético en una serie de ensayos in vitro (mutación bacteriana, aberración cromosómica de linfocitos humanos y mutación de células de mamíferos) y en la prueba de micronúcleos de médula ósea de ratón in vivo.
Erlotinib no perjudicó la fertilidad en ratas macho o hembra.
14 ESTUDIOS CLÍNICOS
14.1 Cáncer de pulmón de células no pequeñas (NSCLC) – Tratamiento de primera línea de pacientes con mutaciones EGFR
Estudio 1
La seguridad y eficacia de erlotinib como monoterapia para el tratamiento de primera línea de pacientes con NSCLC metastásico que contienen deleciones del exón 19 de EGFR o mutaciones de sustitución del exón 21 (L858R) se demostró en el Estudio 1, un ensayo clínico aleatorizado, abierto, realizado en Europa. Ciento setenta y cuatro (174) pacientes blancos fueron aleatorizados 1:1 para recibir 150 mg de erlotinib una vez al día hasta la progresión de la enfermedad (n = 86) o cuatro ciclos de quimioterapia estándar basada en doblete de platino (n = 88); los regímenes de quimioterapia estándar fueron cisplatino más gemcitabina, cisplatino más docetaxel, carboplatino más gemcitabina y carboplatino más docetaxel. La principal medida de resultado de eficacia fue la supervivencia libre de progresión (PFS) según lo evaluado por el investigador. La aleatorización se estratificó por mutación EGFR (deleción del exón 19 o sustitución del exón 21 (L858R)) y el estado funcional del Eastern Cooperative Oncology Group (ECOG PS) (0 frente a 1 frente a 2). El estado de la mutación EGFR para la selección e inscripción de pacientes se determinó mediante un ensayo de ensayos clínicos (CTA). Las muestras tumorales de 134 pacientes (69 pacientes del brazo de erlotinib y 65 pacientes del brazo de quimioterapia) se analizaron retrospectivamente mediante el diagnóstico complementario aprobado por la FDA, cobas® EGFR Mutation Test.
Los datos demográficos de referencia de la población general del estudio fueron: mujeres (72 %), blancos (99 %), edad ≥65 años (51 %), ECOG PS 1 (53 %), con ECOG PS 0 (33 %) y ECOG PS 2 (14 %), fumadores actuales (11 %), exfumadores (20 %) y nunca fumadores (69 %). Las características de la enfermedad fueron 93% en estadio IV y 7% en estadio IIIb con derrame pleural según la clasificación de la American Joint Commission on Cancer (AJCC, 6a edición), 93% de adenocarcinoma, 66% de deleciones de mutación del exón 19 y 34% de mutación puntual del exón 21 (L858R) por CTA.
Se demostró una mejora estadísticamente significativa en la PFS determinada por el investigador (basada en RECIST 1 o progresión clínica) para los pacientes aleatorizados a erlotinib en comparación con los aleatorizados a quimioterapia (ver Tabla 6 y Figura 1). Se observaron resultados similares para PFS (basados en RECIST 1) para el subgrupo evaluado por un comité de revisión independiente (aproximadamente el 75% de los pacientes evaluados en el Estudio 1) y en el subgrupo de 134 pacientes (77% de la población del Estudio 1) con mutaciones EGFR confirmadas por cobas® EGFR Mutation Test.
Un análisis de la supervivencia global (OS) especificado en el protocolo realizado en el momento del análisis final de PFS no mostró diferencias estadísticamente significativas entre los brazos de erlotinib y quimioterapia. En el momento del corte de datos, el 84% de los pacientes en el brazo de quimioterapia habían recibido al menos un tratamiento posterior, de los cuales el 97% recibió un inhibidor de la tirosina quinasa EGFR. En el brazo de erlotinib, el 66% de los pacientes había recibido al menos un tratamiento posterior.
Tabla 6: Resultados de eficacia (Estudio 1)
| Parámetro de eficacia |
Erlotinib (N = 86) |
Quimioterapia (N = 88) |
|---|---|---|
| Supervivencia libre de progresión |
||
| Número de progresiones o muertes | 71 (83%) | 63 (72%) |
| PFS mediana en meses (IC del 95%) | 10.4 (8.7, 12.9) | 5.2 (4.6, 6) |
| Razón de riesgo (HR) (IC del 95%)(1) |
0.34 (0.23, 0.49) | |
| Valor p (prueba de log-rank no estratificada) | < 0.001 | |
| Supervivencia global |
||
| Número de muertes (%) | 55 (64%) | 54 (61%) |
| OS mediana en meses (IC del 95%) | 22.9 (17, 26.8) | 19.5 (17.3, 28.4) |
| Razón de riesgo (HR) (IC del 95%)1 |
0.93 (0.64, 1.35) | |
| Respuesta objetiva |
||
| Tasa de respuesta objetiva (IC del 95%) | 65% (54.1%, 75.1%) | 16% (9%, 25.3%) |
(1) Modelo de regresión de Cox no estratificado.
Figura 1: Curvas de Kaplan-Meier de PFS evaluada por el investigador en el Estudio 1
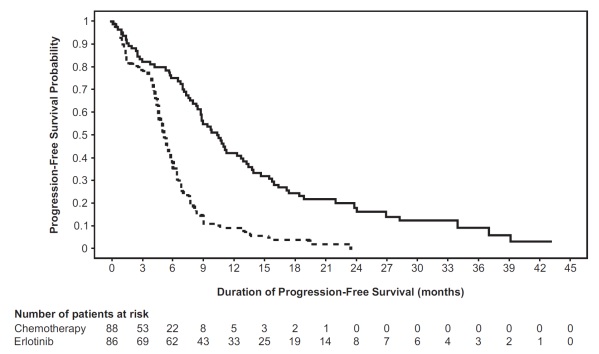
En análisis exploratorios de subgrupos basados en el subtipo de mutación EGFR, el HR para PFS fue 0.27 (IC del 95%: 0.17 a 0.43) en pacientes con deleciones del exón 19 y 0.52 (IC del 95%: 0.29 a 0.95) en pacientes con sustitución del exón 21 (L858R). El HR para OS fue 0.94 (IC del 95%: 0.57 a 1.54) en el subgrupo de deleción del exón 19 y 0.99 (IC del 95%: 0.56 a 1.76) en el subgrupo de sustitución del exón 21 (L858R).
14.2 CPCNP – Falta de eficacia de erlotinib en el tratamiento de mantenimiento de pacientes sin mutaciones de EGFR
La falta de eficacia de erlotinib para el tratamiento de mantenimiento de pacientes con CPCNP sin mutaciones activadoras de EGFR se demostró en el Estudio 2. El Estudio 2 fue un ensayo aleatorizado, multicéntrico, controlado con placebo de 643 pacientes con CPCNP avanzado sin una deleción del exón 19 de EGFR o una mutación L858R del exón 21 que no habían experimentado progresión de la enfermedad después de cuatro ciclos de quimioterapia basada en platino. Los pacientes fueron aleatorizados 1:1 para recibir 150 mg de erlotinib o placebo por vía oral una vez al día (322 erlotinib, 321 placebo) hasta la progresión de la enfermedad o toxicidad inaceptable. Después de la progresión con la terapia inicial, los pacientes eran elegibles para ingresar a una fase abierta. Las características basales fueron las siguientes: mediana de edad de 61 años (35% ≥ 65 años), 75% hombres, 77% blancos, 21% asiáticos, 28% ECOG PS 0, 72% ECOG PS 1, 16% nunca fumadores, 58% fumadores actuales, 57% adenocarcinoma, 35% carcinoma de células escamosas, 22% enfermedad en estadio IIIB no susceptible de tratamiento de modalidad combinada y 78% enfermedad en estadio IV. El cincuenta por ciento de los pacientes aleatorizados a erlotinib ingresaron a la fase abierta y recibieron quimioterapia, mientras que el 77% de los pacientes aleatorizados a placebo ingresaron a la fase abierta y recibieron erlotinib.
El principal resultado de eficacia fue la supervivencia general (SG). La mediana de SG fue de 9,7 meses en el brazo de erlotinib y de 9,5 meses en el brazo de placebo; el cociente de riesgo para la SG fue de 1,02 (IC del 95%: 0,85; 1,22). La mediana de SLP fue de 3 meses en el brazo de erlotinib y de 2,8 meses en el brazo de placebo; el cociente de riesgo para la SLP fue de 0,94 (IC del 95%: 0,8; 1,11).
14.3 CPCNP – Tratamiento de mantenimiento o tratamiento de segunda/tercera línea
Dos ensayos aleatorizados, doble ciego, controlados con placebo, los Estudios 3 y 4, examinaron la eficacia y seguridad de erlotinib administrado a pacientes con CPCNP metastásico como terapia de mantenimiento después del tratamiento inicial con quimioterapia (Estudio 3) o con progresión de la enfermedad después del tratamiento inicial con quimioterapia (Estudio 4). No se requirió la determinación del estado de mutación de EGFR para la inscripción.
Estudio 3
La eficacia y seguridad de erlotinib como tratamiento de mantenimiento del CPCNP se demostraron en el Estudio 3, un ensayo aleatorizado, doble ciego, controlado con placebo realizado en 26 países, en 889 pacientes con CPCNP metastásico cuya enfermedad no progresó durante la quimioterapia de primera línea basada en platino. Los pacientes fueron aleatorizados 1:1 para recibir 150 mg de erlotinib o placebo por vía oral una vez al día (438 erlotinib, 451 placebo) hasta la progresión de la enfermedad o toxicidad inaceptable. El objetivo principal del estudio fue determinar si la administración de erlotinib después de la quimioterapia estándar basada en platino en el tratamiento del CPCNP resultó en una supervivencia libre de progresión (SLP) mejorada en comparación con el placebo, en todos los pacientes o en pacientes con tumores positivos para inmunohistoquímica (IHC) de EGFR.
Los datos demográficos basales de la población general del estudio fueron los siguientes: hombres (74%), edad < 65 años (66%), ECOG PS 1 (69%), ECOG PS 0 (31%), blancos (84%), asiáticos (15%), fumadores actuales (55%), ex fumadores (27%) y nunca fumadores (17%). Las características de la enfermedad fueron las siguientes: Estadio IV (75%), Estadio IIIb con derrame (25%) según la clasificación de AJCC (6a edición) con subtipos histológicos de adenocarcinoma, incluido bronquioloalveolar (45%), escamoso (40%) y de células grandes (5%); y EGFR IHC positivo (70%), negativo (14%), indeterminado (4%) y faltante (12%).
Tabla 7: Resultados de eficacia (Estudio 3): (Población ITT) 1
| Parámetro de eficacia |
Erlotinib (N = 438) |
Placebo (N = 451) |
| Supervivencia libre de progresión (SLP) según la evaluación del investigador |
||
| Número de progresiones o muertes (%) | 349 (80%) | 400 (89%) |
| SLP mediana en meses (IC del 95%) | 2,8 (2,8; 3,1) | 2,6 (1,9; 2,7) |
| Hazard ratio (IC del 95%)(2) |
0,71 (0,62; 0,82) | |
| Valor de p (prueba de log-rank estratificada) (2,3) |
p < 0,0001 | |
| Supervivencia general (SG) |
||
| Número de muertes | 298 (68%) | 350 (78%) |
| SG mediana en meses (IC del 95%) | 12 (10,6; 13,9) | 11 (9,9; 12,1) |
| Hazard ratio (IC del 95%) (2) |
0,81 (0,7; 0,95) | |
| Valor de p (prueba de log-rank estratificada) (3) |
0,0088 | |
(1) Los pacientes con EP antes de la aleatorización fueron excluidos del análisis de SLP y TPP.
(2) Modelo de regresión de Cox univariado.
(3) Prueba de log-rank no estratificada.
La Figura 2 muestra las curvas de Kaplan-Meier para la supervivencia general (población ITT).
Figura 2: Curvas de Kaplan-Meier para la supervivencia general de los pacientes por grupo de tratamiento en el Estudio 3
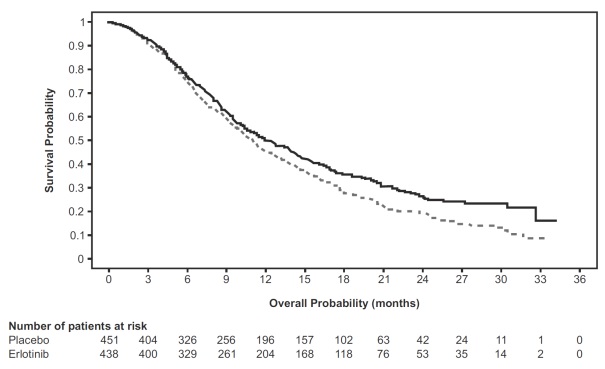
Nota: HR es de un modelo de regresión de Cox univariado.
Estudio 4
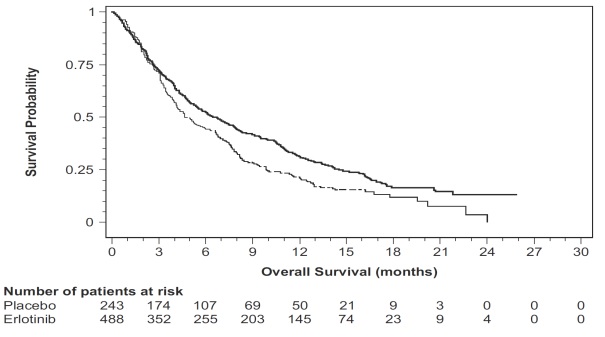
La eficacia y seguridad del erlotinib como agente único se evaluó en el Estudio 4, un ensayo aleatorizado, doble ciego y controlado con placebo en 731 pacientes con NSCLC localmente avanzado o metastásico después del fracaso de al menos un régimen de quimioterapia. Los pacientes fueron aleatorizados 2:1 para recibir erlotinib 150 mg o placebo (488 erlotinib, 243 placebo) por vía oral una vez al día hasta la progresión de la enfermedad o toxicidad inaceptable. Las medidas de eficacia incluyeron supervivencia global, tasa de respuesta y supervivencia libre de progresión (PFS). También se examinó la duración de la respuesta. El objetivo primario fue la supervivencia. El estudio se realizó en 17 países.
Los datos demográficos basales de la población general del estudio fueron los siguientes: hombres (65%), blancos (78%), asiáticos (12%), negros (4%), edad < 65 años (62%), ECOG PS 1 (53%), ECOG PS 0 (13%), ECOG PS 2 (25%), ECOG PS 3 (9%), fumadores actuales o exfumadores (75%), nunca fumadores (20%) y exposición a terapia previa con platino (93%). Las características del tumor fueron las siguientes: adenocarcinoma (50%), escamoso (30%), célula grande indiferenciada (9%) y mixto de células no pequeñas (2%).
Los resultados del estudio se muestran en la Tabla 8.
Tabla 8: Resultados de eficacia (Estudio 4)
| Parámetro de eficacia | Erlotinib (N = 488) |
Placebo (N = 243) |
| Supervivencia global (OS) | ||
| Número de muertes | 378 (77%) | 209 (86%) |
| Mediana de OS en meses (IC del 95%) | 6.7 (5.5, 7.8) | 4.7 (4.1, 6.3) |
| Hazard Ratio (IC del 95%) (1) | 0.73 (0.61, 0.86) | |
| valor p (prueba log-rank estratificada) (2) | p < 0.001 | |
| Supervivencia libre de progresión (PFS) | ||
| Número de progresiones o muertes (%) | 402 (82%) | 211 (87%) |
| Mediana de PFS en meses (IC del 95%) | 2.3 (1.9, 3.3) | 1.8 (1.8, 1.9) |
| Hazard Ratio (IC del 95%) 1 | 0.59 (0.5, 0.7) | |
| Respuesta objetiva | ||
| Tasa de respuesta objetiva (IC del 95%) | 8.9% (6.4, 12) | 0.9% (0.1, 3.4) |
(1) Modelo de regresión de Cox con las siguientes covariables: estado de rendimiento ECOG, número de regímenes previos, platino previo, mejor respuesta a quimioterapia previa.
(2) Prueba de log-rank de dos lados estratificada por estado de rendimiento ECOG, número de regímenes previos, platino previo, mejor respuesta a quimioterapia previa.
La Figura 3 muestra las curvas de Kaplan-Meier para la supervivencia global.
Figura 3: Curvas de Kaplan-Meier para la supervivencia global de los pacientes por grupo de tratamiento en el Estudio 4
14.4 NSCLC – Falta de eficacia del erlotinib administrado simultáneamente con quimioterapia
Los resultados de dos ensayos multicéntricos, controlados con placebo y aleatorizados en más de 1000 pacientes realizados en pacientes de primera línea con NSCLC localmente avanzado o metastásico no mostraron beneficio clínico con la administración concurrente de erlotinib con quimioterapia basada en platino [carboplatino y paclitaxel (erlotinib, N = 526) o gemcitabina y cisplatino (erlotinib, N = 580)].
14.5 Cáncer de páncreas – Erlotinib administrado simultáneamente con gemcitabina
La eficacia y seguridad del erlotinib en combinación con gemcitabina como tratamiento de primera línea se evaluó en el Estudio 5, un ensayo aleatorizado, doble ciego y controlado con placebo en 569 pacientes con cáncer de páncreas localmente avanzado, irresecable o metastásico. Los pacientes fueron aleatorizados 1:1 para recibir erlotinib (100 mg o 150 mg) o placebo una vez al día en un horario continuo más gemcitabina por infusión intravenosa (1000 mg/m2, Ciclo 1 – Días 1, 8, 15, 22, 29, 36 y 43 de un ciclo de 8 semanas; Ciclo 2 y ciclos subsiguientes – Días 1, 8 y 15 de un ciclo de 4 semanas [la dosis y el cronograma aprobados para el cáncer de páncreas, ver el prospecto de gemcitabina]). El erlotinib o el placebo se tomaron por vía oral una vez al día hasta la progresión de la enfermedad o una toxicidad inaceptable. El objetivo primario fue la supervivencia. Los objetivos secundarios incluyeron la tasa de respuesta y la supervivencia libre de progresión (PFS). También se examinó la duración de la respuesta. El estudio se realizó en 18 países. Un total de 285 pacientes fueron aleatorizados para recibir gemcitabina más erlotinib (261 pacientes en la cohorte de 100 mg y 24 pacientes en la cohorte de 150 mg) y 284 pacientes fueron aleatorizados para recibir gemcitabina más placebo (260 pacientes en la cohorte de 100 mg y 24 pacientes en la cohorte de 150 mg). Demasiado pocos pacientes fueron tratados en la cohorte de 150 mg para sacar conclusiones.
En la cohorte de 100 mg, los datos demográficos basales de la población general del estudio fueron los siguientes: hombres (52%), blancos (88%), asiáticos (7%), negros (2%), edad < 65 años (53%), ECOG PS 1 (51%), ECOG PS 0 (32%) y ECOG PS 2 (17%). Había una proporción ligeramente mayor de mujeres en el brazo de erlotinib (51%) en comparación con el brazo de placebo (44%). La mediana de tiempo desde el diagnóstico inicial hasta la aleatorización fue aproximadamente de 1 mes. La mayoría de los pacientes (76%) tenían metástasis a distancia al inicio del estudio y el 24% tenía enfermedad localmente avanzada.
Los resultados del estudio se muestran en la Tabla 9.
Tabla 9: Resultados de eficacia: Cohorte de erlotinib 100 mg (Estudio 5)
| Parámetro de eficacia |
Erlotinib + Gemcitabina (N = 261) |
Placebo + Gemcitabina (N = 260) |
|---|---|---|
| Supervivencia general (SG) |
||
| Número de muertes | 250 | 254 |
| Mediana de SG en meses (IC del 95%) | 6.5 (6, 7.4) | 6 (5.1, 6.7) |
| Hazard ratio (IC del 95%)(1) |
0.81 (0.68, 0.97) | |
| Valor p (prueba de log-rank estratificada) (2) |
0.028 | |
| Supervivencia libre de progresión (SLP) |
||
| Número de progresiones o muertes (%) | 225 | 232 |
| Mediana de SLP en meses (IC del 95%) | 3.8 (3.6, 4.9) | 3.6 (3.3, 3.8) |
| Hazard ratio (IC del 95%) (1) |
0.76 (0.64, 0.92) | |
| Respuesta objetiva |
||
| Tasa de respuesta objetiva (IC del 95%) | 8.6% (5.4, 12.9) | 7.9% (4.8, 12) |
(1) Modelo de regresión de Cox con las siguientes covariables: estado funcional ECOG y extensión de la enfermedad.
(2) Prueba de log-rank de dos colas estratificada por estado funcional ECOG y extensión de la enfermedad.
La supervivencia se evaluó en la población por intención de tratar. La Figura 4 representa las curvas de Kaplan-Meier para la supervivencia general en la cohorte de 100 mg. Los análisis primarios de supervivencia y SLP fueron pruebas de log-rank de dos colas estratificadas por estado funcional ECOG y extensión de la enfermedad.
Figura 4: Curvas de Kaplan-Meier para la supervivencia general: cohorte de 100 mg en el Estudio 5
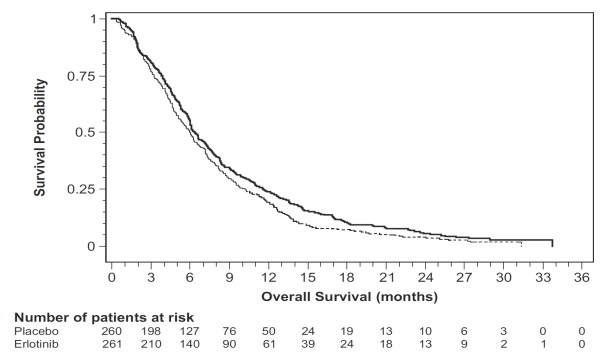
Nota: El HR es del modelo de regresión de Cox con las siguientes covariables: estado funcional ECOG y extensión de la enfermedad. El valor p es de la prueba de log-rank de dos colas estratificada por estado funcional ECOG y extensión de la enfermedad.
16 FORMA DE SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
Los comprimidos de erlotinib de 25 mg son redondos, biconvexos, comprimidos recubiertos con película blanca grabados con “L55” en un lado y lisos en el otro lado.
Frasco de 30 comprimidos con cierre resistente a niños, NDC 62332-565-30
Frasco de 90 comprimidos con cierre resistente a niños, NDC 62332-565-90
Los comprimidos de erlotinib de 100 mg son redondos, biconvexos, comprimidos recubiertos con película blanca grabados con “L630” en un lado y lisos en el otro lado.
Frasco de 30 comprimidos con cierre resistente a niños, NDC 62332-566-30
Frasco de 90 comprimidos con cierre resistente a niños, NDC 62332-566-90
Los comprimidos de erlotinib de 150 mg son redondos, biconvexos, comprimidos recubiertos con película blanca grabados con “L631” en un lado y lisos en el otro lado.
Frasco de 30 comprimidos con cierre resistente a niños, NDC 62332-567-30
Frasco de 90 comprimidos con cierre resistente a niños, NDC 62332-567-90
Almacenar a 25°C (77°F); se permiten excursiones de 15° a 30°C (59° a 86°F). Ver USP Temperatura Ambiente Controlada.
17 INFORMACIÓN DE ASESORAMIENTO AL PACIENTE
Sarpullido, trastornos cutáneos ampollosos y exfoliativos
- Informe a los pacientes que pueden ocurrir o empeorar reacciones cutáneas en áreas expuestas al sol mientras toman comprimidos de erlotinib, y la intervención proactiva puede incluir una crema emoliente sin alcohol y el uso de protector solar o evitar la exposición al sol. Informe a los pacientes que se ha reportado hiperpigmentación o piel seca, con o sin fisuras digitales en la piel, y en la mayoría de los casos se asociaron con sarpullido [ver Reacciones Adversas (6.1)].
- Informe a los pacientes que los comprimidos de erlotinib pueden aumentar el riesgo de trastornos cutáneos ampollosos y exfoliativos y que deben buscar atención médica inmediata para reacciones cutáneas graves [ver Advertencias y Precauciones (5.5)].
Diarrea
Informe a los pacientes que la diarrea generalmente se puede manejar con loperamida y que deben comunicarse con su proveedor de atención médica en caso de diarrea grave o persistente [ver Reacciones Adversas (6.1)].
Enfermedad pulmonar intersticial
Informe a los pacientes sobre el riesgo de ILD grave o mortal, incluida la neumonitis. Aconseje a los pacientes que se comuniquen con su proveedor de atención médica de inmediato para informar sobre la aparición o el empeoramiento de la falta de aire o tos inexplicables [ver Dosificación y Administración (2.4) y Advertencias y Precauciones (5.1)].
Insuficiencia renal
Informe a los pacientes sobre el riesgo de desarrollar insuficiencia renal. Informe a los pacientes sobre la necesidad de que el proveedor de atención médica controle la función renal y los electrolitos [ver Advertencias y Precauciones (5.2)].
Hepatotoxicidad
Aconseje a los pacientes que informen inmediatamente signos o síntomas de hepatotoxicidad [ver Advertencias y Precauciones (5.3)].
Perforaciones gastrointestinales
Informe a los pacientes que los comprimidos de erlotinib pueden aumentar el riesgo de perforación gastrointestinal o fístula y que busquen atención médica inmediata para dolor abdominal intenso [ver Dosificación y Administración (2.4) y Advertencias y Precauciones (5.4)].
Accidente cerebrovascular
Informe a los pacientes sobre el riesgo de accidente cerebrovascular y que busquen atención médica inmediata [ver Dosificación y Administración (2.4) y Advertencias y Precauciones (5.6)].
Trastornos oculares
Aconseje a los pacientes que se comuniquen de inmediato con su proveedor de atención médica si desarrollan signos o síntomas oculares, lagrimeo, sensibilidad a la luz, visión borrosa, dolor ocular, ojo rojo o cambios en la visión [ver Dosificación y Administración (2.4) y Advertencias y Precauciones (5.8)].
Hemorragia en pacientes que toman warfarina
Informe a los pacientes que reciben warfarina sobre la necesidad de controlar el INR u otros anticoagulantes derivados de la cumarina [ver Advertencias y Precauciones (5.9) e Interacciones Medicamentosas (7)].
Trastornos del cabello y las uñas
Informe a los pacientes que se han reportado trastornos del cabello y las uñas, incluido hirsutismo y uñas quebradizas y sueltas [ver Reacciones Adversas (6.1)].
Toxicidad embriofetal
- Informe a las mujeres embarazadas y a las mujeres en edad fértil sobre el riesgo potencial para el feto. Aconseje a las mujeres en edad fértil que informen a su proveedor de atención médica sobre un embarazo conocido o sospechado [ver Advertencias y Precauciones (5.10), Uso en Poblaciones Específicas (8.1)].
- Aconseje a las mujeres en edad fértil que utilicen un método anticonceptivo eficaz durante el tratamiento con comprimidos de erlotinib y durante 1 mes después de la última dosis [ver Uso en Poblaciones Específicas (8.3)].
Lactancia
- Aconseje a las mujeres que no amamanten durante el tratamiento con comprimidos de erlotinib y durante 2 semanas después de la dosis final [ver Uso en Poblaciones Específicas (8.2)].
Tabaquismo
- Aconseje a los pacientes que se comuniquen con su proveedor de atención médica para cualquier cambio en el estado de tabaquismo y que es posible que sea necesario ajustar la dosis de los comprimidos de erlotinib si fuman [ver Interacciones Medicamentosas (7) y Farmacología Clínica (12.3)]
- Aconseje a los pacientes que dejen de fumar [ver Farmacología Clínica (12.3)].
Llame a su médico para obtener asesoramiento médico sobre los efectos secundarios. Puede informar los efectos secundarios a la FDA al 1-800-FDA-1088.
Para obtener más información, llame al 1-866-210-9797.
Las marcas enumeradas son marcas comerciales de sus respectivos propietarios.
Fabricado por:
Alembic Pharmaceuticals Limited
Formulation Division II, Survey No. 84,
87 And 88, Panelav, Taluka Halol,
Panchmahal, Gujarat 389350, India
Fabricado para:
Alembic Pharmaceuticals, Inc.
Bedminster, NJ 07921, USA
Revisado: 02/2023
ETIQUETA DEL ENVASE. PANEL DE VISUALIZACIÓN PRINCIPAL 25 mg
NDC 62332-565-30
Tabletas de Erlotinib
25 mg
Rx solamente
30 Tabletas
Alembic

ETIQUETA DEL ENVASE. PANEL DE VISUALIZACIÓN PRINCIPAL 100 mg
NDC 62332-566-30
Tabletas de Erlotinib
100 mg
Rx only
30 Tabletas
Alembic

ETIQUETA DEL ENVASE. PANEL DE VISUALIZACIÓN PRINCIPAL 150 mg
NDC 62332-567-30
Tabletas de Erlotinib
150 mg
Solo con receta
30 Tabletas
Alembic