
Fabricante de medicamentos: Janssen Products, LP (Updated: 2024-08-09)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
ERLEADA
®(apalutamida) tabletas, para uso oral
Aprobación inicial en EE. UU. – 2018
CAMBIOS RECIENTES IMPORTANTES
| Dosificación y administración, Dosificación recomendada (
2.1) |
8/2024 |
| Dosificación y administración, Modificación de la dosis (
2.2) |
8/2024 |
| Dosificación y administración, Métodos alternativos de administración (
2.3) |
8/2024 |
| Advertencias y precauciones, Enfermedad pulmonar intersticial (ILD) (
5.6) |
8/2024 |
INDICACIONES Y USO
DOSIFICACIÓN Y ADMINISTRACIÓN
ERLEADA 240 mg administrado por vía oral una vez al día. Tragar las tabletas enteras. ERLEADA se puede tomar con o sin alimentos. (
2.1,
2.3)
Los pacientes también deben recibir un análogo de la hormona liberadora de gonadotropina (GnRH) de forma concurrente o deben haberse sometido a una orquiectomía bilateral. (
2.1)
CONTRAINDICACIONES
Ninguna. (
4)
ADVERTENCIAS Y PRECAUCIONES
- Se produjeron eventos cerebrovasculares y cardiovasculares isquémicos en pacientes que recibieron ERLEADA. Monitorear en busca de signos y síntomas de trastornos cerebrovasculares y enfermedad cardíaca isquémica. Optimizar el manejo de los factores de riesgo cardiovascular. (
5.1).
- Se produjeron fracturas en pacientes que recibieron ERLEADA. Evaluar a los pacientes para determinar el riesgo de fractura y tratar a los pacientes con agentes dirigidos a los huesos de acuerdo con las pautas establecidas. (
5.2)
- Se produjeron caídas en pacientes que recibieron ERLEADA con mayor incidencia en los ancianos. Evaluar a los pacientes para determinar el riesgo de caídas. (
5.3)
- Se produjeron convulsiones en el 0,4% de los pacientes que recibieron ERLEADA. Suspenda permanentemente ERLEADA en pacientes que desarrollen una convulsión durante el tratamiento. (
5.4)
- Reacciones adversas cutáneas graves (SCAR), incluido el síndrome de Stevens-Johnson/necrólisis epidérmica tóxica (SJS/TEN) y la reacción medicamentosa con eosinofilia y síntomas sistémicos (DRESS), se produjeron en pacientes tratados con ERLEADA. Interrompa ERLEADA si se desarrollan signos o síntomas de SCAR. Suspenda permanentemente si se confirman las SCAR. (
5.5)
- Enfermedad pulmonar intersticial (ILD)/neumonitis se produjo en pacientes tratados con ERLEADA. Suspenda ERLEADA en caso de sospecha de ILD/neumonitis. Suspenda permanentemente ERLEADA en pacientes con ILD/neumonitis grave o si no se identifican otras causas potenciales de ILD/neumonitis. (
2.2,
5.6)
- Toxicidad embrio-fetal: ERLEADA puede causar daño fetal. Aconseje a los hombres con parejas femeninas en edad fértil que utilicen métodos anticonceptivos eficaces. (
5.7,
8.1,
8.3)
REACCIONES ADVERSAS
Las reacciones adversas más comunes (≥10%) son fatiga, artralgia, erupción cutánea, disminución del apetito, caída, disminución del peso, hipertensión, sofocos, diarrea y fractura. (
6.1)
Para informar sobre REACCIONES ADVERSAS SOSPECHOSAS, comuníquese con Janssen Products, LP al 1-800-526-7736 (1-800-JANSSEN) o con la FDA al 1-800-FDA-1088 o
www.fda.gov/medwatch.
INTERACCIONES MEDICAMENTOSAS
El uso concomitante con medicamentos que son sustratos sensibles de CYP3A4, CYP2C19, CYP2C9, UGT, P-gp, BCRP u OATP1B1 puede provocar una pérdida de la actividad de estos medicamentos. (
7.2)
Ver 17 para INFORMACIÓN PARA EL PACIENTE y el etiquetado del paciente aprobado por la FDA.
Revisado: 8/2024
Tabla de Contenido
INFORMACIÓN COMPLETA PARA LA PRESCRIPCIÓN: CONTENIDO*
1 INDICACIONES Y USO
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Dosificación recomendada
2.2 Modificación de la dosis
2.3 Métodos alternativos de administración
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Eventos cardiovasculares isquémicos y cerebrovasculares
5.2 Fracturas
5.3 Caídas
5.4 Convulsiones
5.5 Reacciones adversas cutáneas graves
5.6 Enfermedad pulmonar intersticial (EPI)/Neumonitis
5.7 Toxicidad embriofetal
6 REACCIONES ADVERSAS
6.1 Experiencia de ensayos clínicos
6.2 Experiencia posterior a la comercialización
7 INTERACCIONES MEDICAMENTOSAS
7.1 Efecto de otros medicamentos sobre ERLEADA
7.2 Efecto de ERLEADA sobre otros medicamentos
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.3 Mujeres y hombres con potencial reproductivo
8.4 Uso pediátrico
8.5 Uso geriátrico
10 SOBREDOSIS
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
12.2 Farmacodinamia
12.3 Farmacocinética
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, mutagénesis, deterioro de la fertilidad
14 ESTUDIOS CLÍNICOS
16 CÓMO SE SUMINISTRA/ALMACENAMIENTO Y MANIPULACIÓN
17 INFORMACIÓN DE ASESORAMIENTO AL PACIENTE
- *
- Las secciones o subsecciones que se omiten de la información completa para la prescripción no se enumeran.
1 INDICACIONES Y USO
ERLEADA está indicado para el tratamiento de pacientes con
- Cáncer de próstata metastásico sensible a la castración (mCSPC)
- Cáncer de próstata no metastásico resistente a la castración (nmCRPC)
2 DOSIS Y ADMINISTRACIÓN
2.1 Dosis recomendada
La dosis recomendada de ERLEADA es de 240 mg administrados por vía oral una vez al día. Esto podría administrarse como un comprimido de 240 mg o cuatro comprimidos de 60 mg. Trague los comprimidos enteros. No triture ni parta los comprimidos. ERLEADA puede tomarse con o sin alimentos.
Los pacientes también deben recibir un análogo de la hormona liberadora de gonadotropina (GnRH) de forma concurrente o deben haberse sometido a una orquiectomía bilateral.
2.2 Modificación de la dosis
Si se producen reacciones adversas de Grado 3 o superior, u otras reacciones adversas intolerables, suspenda la administración de ERLEADA. Considere la posibilidad de interrumpir permanentemente el tratamiento con ERLEADA en caso de episodios cardiovasculares isquémicos y cerebrovasculares de Grado 3 o 4
[ver
Advertencias y precauciones (5.1)]
. Suspenda permanentemente el tratamiento con ERLEADA en caso de EPI/neumonitis grave o si no se identifican otras causas potenciales de EPI/neumonitis, o se confirman reacciones cutáneas graves (SCAR), o en caso de otras reacciones cutáneas de Grado 4
[ver
Advertencias y precauciones (5.5,
5.6)y
. Para otras reacciones adversas, cuando los síntomas mejoren a un grado inferior o igual a 1 o al grado original, reanude la administración de ERLEADA a la misma dosis o a una dosis reducida (180 mg o 120 mg), si está justificado.
2.3 Métodos alternativos de administración
Dispersar el (los) comprimido(s) en agua y administrar con zumo de naranja, puré de manzana o agua adicional
En el caso de pacientes que no puedan tragar los comprimidos enteros, la dosis recomendada de comprimidos de ERLEADA puede dispersarse en agua sin gas y luego administrarse con zumo de naranja, puré de manzana o agua adicional de la siguiente manera:
- Coloque la dosis completa prescrita de comprimidos de ERLEADA en una taza. No triture ni parta los comprimidos.
-
Para un comprimido de 240 mg: Añada unas 2 cucharaditas (10 ml) de agua sin gas para asegurarse de que el comprimido esté completamente sumergido en agua.
Para comprimidos de 60 mg (dosis prescrita de 240 mg, 180 mg o 120 mg): Añada unas 4 cucharaditas (20 ml) de agua sin gas para asegurarse de que los comprimidos estén completamente sumergidos en agua. - Espere 2 minutos hasta que el (los) comprimido(s) se haya(n) deshecho y extendido, luego remueva la mezcla.
- Añada 2 cucharadas (30 ml) de zumo de naranja, puré de manzana o agua adicional y remueva la mezcla.
- Trague la mezcla inmediatamente.
- Enjuague la taza con suficiente agua para asegurarse de que se toma toda la dosis y bébala inmediatamente.
No guarde ERLEADA mezclado con agua sin gas, zumo de naranja o puré de manzana para su uso posterior.
Administrar el (los) comprimido(s) a través de una sonda de alimentación
Los comprimidos de ERLEADA pueden administrarse a través de una sonda de alimentación de 8 French o superior de la siguiente manera:
-
Para un comprimido de 240 mg: Coloque el comprimido en el cilindro de la jeringa (utilice al menos una jeringa de 20 ml) y aspire 10 ml de agua sin gas en la jeringa.
Para comprimidos de 60 mg (dosis prescrita de 240 mg, 180 mg o 120 mg): Coloque la dosis completa prescrita de comprimidos de ERLEADA en el cilindro de la jeringa (utilice al menos una jeringa de 50 ml) y aspire 20 ml de agua sin gas en la jeringa. - Espere 10 minutos y luego agite enérgicamente para dispersar el contenido por completo.
- Administre inmediatamente a través de la sonda de alimentación.
- Vuelva a llenar la jeringa con agua sin gas y adminístrela. Repita la operación hasta que no queden residuos de comprimidos en la jeringa o en la sonda de alimentación.
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
Tabletas:
- 240 mg: gris azulado a gris, ovalada, recubierta con película y con “E240” grabado en un lado.
- 60 mg: ligeramente amarillenta a verde grisácea, oblonga, recubierta con película y con “AR 60” grabado en un lado.
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Eventos Cerebrovasculares e Isquémicos Cardiovasculares
Se produjeron eventos cerebrovasculares e isquémicos cardiovasculares, incluidos eventos que llevaron a la muerte, en pacientes que recibieron ERLEADA. Monitorear en busca de signos y síntomas de enfermedad cardíaca isquémica y trastornos cerebrovasculares. Optimizar el manejo de los factores de riesgo cardiovascular, como la hipertensión, la diabetes o la dislipidemia. Considere la posibilidad de suspender ERLEADA para eventos de Grado 3 y 4.
En un estudio aleatorizado (SPARTAN) de pacientes con nmCRPC, se produjeron eventos cardiovasculares isquémicos en el 3,7% de los pacientes tratados con ERLEADA y en el 2% de los pacientes tratados con placebo. En un estudio aleatorizado (TITAN) en pacientes con mCSPC, se produjeron eventos cardiovasculares isquémicos en el 4,4% de los pacientes tratados con ERLEADA y en el 1,5% de los pacientes tratados con placebo. En los estudios SPARTAN y TITAN, 4 pacientes (0,3%) tratados con ERLEADA y 2 pacientes (0,2%) tratados con placebo murieron por un evento cardiovascular isquémico.
En el estudio SPARTAN, los eventos cerebrovasculares ocurrieron en el 2,5% de los pacientes tratados con ERLEADA y en el 1% de los pacientes tratados con placebo
[ver
. En el estudio TITAN, los eventos cerebrovasculares ocurrieron en el 1,9% de los pacientes tratados con ERLEADA y en el 2,1% de los pacientes tratados con placebo. En los estudios SPARTAN y TITAN, 3 pacientes (0,2%) tratados con ERLEADA y 2 pacientes (0,2%) tratados con placebo murieron por un evento cerebrovascular.
Los pacientes con antecedentes de angina inestable, infarto de miocardio, insuficiencia cardíaca congestiva, accidente cerebrovascular o ataque isquémico transitorio dentro de los seis meses de la aleatorización fueron excluidos de los estudios SPARTAN y TITAN.
5.2 Fracturas
Se produjeron fracturas en pacientes que recibieron ERLEADA. Evaluar a los pacientes para determinar el riesgo de fractura. Monitorear y manejar a los pacientes en riesgo de fracturas de acuerdo con las pautas de tratamiento establecidas y considerar el uso de agentes dirigidos a los huesos.
En un estudio aleatorizado (SPARTAN) de pacientes con cáncer de próstata resistente a la castración no metastásico, se produjeron fracturas en el 12% de los pacientes tratados con ERLEADA y en el 7% de los pacientes tratados con placebo. Las fracturas de Grado 3-4 ocurrieron en el 2,7% de los pacientes tratados con ERLEADA y en el 0,8% de los pacientes tratados con placebo. El tiempo medio hasta el inicio de la fractura fue de 314 días (rango: 20 a 953 días) para los pacientes tratados con ERLEADA. La evaluación de densidad ósea de rutina y el tratamiento de la osteoporosis con agentes dirigidos a los huesos no se realizaron en el estudio SPARTAN.
En un estudio aleatorizado (TITAN) de pacientes con cáncer de próstata sensible a la castración metastásico, se produjeron fracturas en el 9% de los pacientes tratados con ERLEADA y en el 6% de los pacientes tratados con placebo. Las fracturas de Grado 3-4 fueron similares en ambos brazos en el 1,5%. El tiempo medio hasta el inicio de la fractura fue de 56 días (rango: 2 a 111 días) para los pacientes tratados con ERLEADA. La evaluación de densidad ósea de rutina y el tratamiento de la osteoporosis con agentes dirigidos a los huesos no se realizaron en el estudio TITAN.
5.3 Caídas
Se produjeron caídas en pacientes que recibieron ERLEADA con mayor frecuencia en los ancianos
[ver
Uso en poblaciones específicas (8.5)]
. Evaluar a los pacientes para determinar el riesgo de caídas.
En un estudio aleatorizado (SPARTAN), se produjeron caídas en el 16% de los pacientes tratados con ERLEADA en comparación con el 9% de los pacientes tratados con placebo. Las caídas no se asociaron con pérdida del conocimiento o convulsiones.
5.4 Convulsiones
Se produjeron convulsiones en pacientes que recibieron ERLEADA. Suspenda ERLEADA de forma permanente en pacientes que desarrollen una convulsión durante el tratamiento. Se desconoce si los medicamentos antiepilépticos evitarán las convulsiones con ERLEADA. Advierta a los pacientes sobre el riesgo de desarrollar una convulsión mientras reciben ERLEADA y de participar en cualquier actividad en la que la pérdida repentina del conocimiento pueda causarles daño a ellos mismos o a otros.
En dos estudios aleatorizados (SPARTAN y TITAN), cinco pacientes (0,4%) tratados con ERLEADA y un paciente tratado con placebo (0,1%) experimentaron una convulsión. La convulsión ocurrió de 159 a 650 días después del inicio de ERLEADA. Los pacientes con antecedentes de convulsiones, factores predisponentes para convulsiones o que recibían medicamentos conocidos por disminuir el umbral convulsivo o inducir convulsiones fueron excluidos. No hay experiencia clínica en la readministración de ERLEADA a pacientes que experimentaron una convulsión.
5.5 Reacciones adversas cutáneas graves
Se produjeron casos fatales y potencialmente mortales de reacciones adversas cutáneas graves (SCAR), incluido el síndrome de Stevens-Johnson/necrólisis epidérmica tóxica (SJS/TEN) y la reacción medicamentosa con eosinofilia y síntomas sistémicos (DRESS), en pacientes que recibieron ERLEADA
[ver
.
Monitorear a los pacientes para detectar el desarrollo de SCAR. Advierta a los pacientes sobre los signos y síntomas de SCAR (por ejemplo, un pródromo de fiebre, síntomas similares a la gripe, lesiones mucosas, erupción cutánea progresiva o linfadenopatía).
Si se sospecha una SCAR, interrumpa ERLEADA hasta que se haya determinado la etiología de la reacción. Se recomienda la consulta con un dermatólogo. Si se confirma una SCAR, o para otras reacciones cutáneas de grado 4, suspenda ERLEADA de forma permanente
[ver
Dosificación y administración (2.2)y
.
5.6 Enfermedad pulmonar intersticial (ILD)/Neumonitis
La enfermedad pulmonar intersticial (ILD) o neumonitis potencialmente mortal y mortal puede ocurrir en pacientes tratados con ERLEADA.
Se produjeron casos de ILD/neumonitis posteriores a la comercialización, incluidos casos mortales, en pacientes tratados con ERLEADA. En los ensayos clínicos (TITAN y SPARTAN, n=1327), el 0,8% de los pacientes tratados con ERLEADA experimentaron ILD/neumonitis, incluido el 0,2% que experimentó eventos de Grado 3
[ver
6.2)].
Controle a los pacientes para detectar síntomas nuevos o que empeoren que indiquen ILD/neumonitis (por ejemplo, disnea, tos, fiebre). Suspenda ERLEADA inmediatamente si se sospecha ILD/neumonitis.
Suspenda ERLEADA de forma permanente en pacientes con ILD/neumonitis grave o si no se identifican otras causas potenciales de ILD/neumonitis
[ver
Dosificación y administración (2.2)]
.
5.7 Toxicidad embrio-fetal
No se ha establecido la seguridad y eficacia de ERLEADA en mujeres. Con base en los hallazgos de animales y su mecanismo de acción, ERLEADA puede causar daño fetal y pérdida del embarazo cuando se administra a una mujer embarazada. En un estudio de reproducción animal, la administración oral de apalutamida a ratas embarazadas durante y después de la organogénesis provocó anomalías fetales y letalidad embrio-fetal a exposiciones maternas ≥ 2 veces la exposición clínica humana (AUC) a la dosis recomendada. Avise a los hombres con parejas femeninas en edad fértil que usen métodos anticonceptivos efectivos durante el tratamiento y durante 3 meses después de la última dosis de ERLEADA
[ver
Uso en poblaciones específicas (8.1,
8.3)y
.
6 REACCIONES ADVERSAS
Los siguientes se discuten con más detalle en otras secciones del etiquetado:
- Eventos cerebrovasculares e isquémicos cardiovasculares
[ver
Advertencias y precauciones (5.1)]
.
- Fracturas
[ver
Advertencias y precauciones (5.2)]
.
- Caídas
[ver
Advertencias y precauciones (5.3)]
.
- Convulsiones
[ver
Advertencias y precauciones (5.4)]
.
- Reacciones adversas cutáneas graves (SCAR)
[ver
Advertencias y precauciones (5.5)]
.
- Enfermedad pulmonar intersticial (ILD)
[ver
Advertencias y precauciones (5.6)]
.
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se llevan a cabo en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no se pueden comparar directamente con las tasas en los ensayos clínicos de otro fármaco y pueden no reflejar las tasas observadas en la práctica.
Las reacciones adversas más comunes (≥ 10%) que ocurrieron con más frecuencia en los pacientes tratados con ERLEADA (≥ 2% por encima del placebo) de los ensayos clínicos aleatorizados controlados con placebo (TITAN y SPARTAN) fueron fatiga, artralgia, erupción cutánea, disminución del apetito, caída, disminución del peso, hipertensión, sofocos, diarrea y fractura.
Cáncer de próstata metastásico sensible a la castración (mCSPC)
TITAN, un estudio clínico multicéntrico, aleatorizado (1:1), doble ciego, controlado con placebo, reclutó pacientes que tenían mCSPC. En este estudio, los pacientes recibieron ERLEADA a una dosis de 240 mg diarios o placebo. Todos los pacientes en el estudio TITAN recibieron un análogo de la hormona liberadora de gonadotropina (GnRH) concomitante o habían tenido orquiectomía bilateral previa. La duración media de la exposición fue de 20 meses (rango: 0 a 34 meses) en los pacientes que recibieron ERLEADA y 18 meses (rango: 0.1 a 34 meses) en los pacientes que recibieron placebo.
Diez pacientes (1.9%) que fueron tratados con ERLEADA murieron por reacciones adversas. Las razones de muerte fueron eventos cardiovasculares isquémicos (n=3), lesión renal aguda (n=2), paro cardiorrespiratorio (n=1), muerte súbita cardíaca (n=1), insuficiencia respiratoria (n=1), accidente cerebrovascular (n=1) y perforación de úlcera intestinal grande (n=1). ERLEADA se suspendió debido a reacciones adversas en el 8% de los pacientes, más comúnmente por erupción cutánea (2.3%). Las reacciones adversas que llevaron a la interrupción o reducción de la dosis de ERLEADA ocurrieron en el 23% de los pacientes; las más frecuentes (>1%) fueron erupción cutánea, fatiga e hipertensión. Las reacciones adversas graves ocurrieron en el 20% de los pacientes tratados con ERLEADA y el 20% en los pacientes que recibieron placebo.
La Tabla 1 muestra las reacciones adversas que ocurrieron en ≥10% en el brazo de ERLEADA en TITAN que ocurrieron con un aumento absoluto ≥2% en frecuencia en comparación con el placebo. La Tabla 2 muestra las anormalidades de laboratorio que ocurrieron en ≥15% de los pacientes, y con más frecuencia (>5%) en el brazo de ERLEADA en comparación con el placebo.
| ERLEADA N=524 |
Placebo N=527 |
|||
|---|---|---|---|---|
| Clase de sistema/órgano Reacción adversa |
Todos los grados % |
Grado 3–4 % |
Todos los grados % |
Grado 3–4 % |
|
||||
| Trastornos musculoesqueléticos y del tejido conjuntivo | ||||
| Artralgia | 17 | 0.4 | 15 | 0.9 |
| Trastornos de la piel y del tejido subcutáneo | ||||
| Erupción cutánea | 28 | 6 | 9 | 0.6 |
| Prurito | 11 | 0.2 | 4.6 | 0.2 |
| Trastornos vasculares | ||||
| Sofocos | 23 | 0 | 16 | 0 |
| Hipertensión | 18 | 8 | 16 | 9 |
Las reacciones adversas adicionales de interés que ocurrieron en menos del 10% de los pacientes tratados con ERLEADA incluyeron diarrea (9% versus 6% en placebo), espasmo muscular (3.1% versus 1.9% en placebo), disgeusia (3.2% versus 0.6% en placebo), hipotiroidismo (3.6% versus 0.6% en placebo) e ILD/neumonitis (1.1% versus 0.4% en placebo).
| ERLEADA N=524 |
Placebo N=527 |
|||
|---|---|---|---|---|
| Anormalidad de laboratorio | Todos los grados % |
Grado 3–4 % |
Todos los grados % |
Grado 3–4 % |
|
||||
| Hematología | ||||
| Glóbulos blancos disminuidos | 27 | 0.4 | 19 | 0.6 |
| Química | ||||
| Hipertrigliceridemia | 17 | 2.5 | 12 | 2.3 |
Cáncer de próstata resistente a la castración no metastásico (nmCRPC)
SPARTAN, un estudio clínico multicéntrico, aleatorizado (2:1), doble ciego, controlado con placebo, reclutó pacientes que tenían nmCRPC. En este estudio, los pacientes recibieron ERLEADA a una dosis de 240 mg diarios o un placebo. Todos los pacientes en el estudio SPARTAN recibieron un análogo de la hormona liberadora de gonadotropina (GnRH) concomitante o se sometieron a una orquiectomía bilateral. La duración media de la exposición fue de 33 meses (rango: 0,1 a 75 meses) en los pacientes que recibieron ERLEADA y de 11 meses (rango: 0,1 a 37 meses) en los pacientes que recibieron placebo.
Veinticuatro pacientes (3%) que fueron tratados con ERLEADA murieron por reacciones adversas. Las razones de muerte con ≥ 2 pacientes incluyeron infección (n=7), infarto de miocardio (n=3), evento cerebrovascular (n=2) y razón desconocida (n=3). ERLEADA se suspendió debido a reacciones adversas en el 11% de los pacientes, más comúnmente por erupción cutánea (3,2%). Las reacciones adversas que llevaron a la interrupción o reducción de la dosis de ERLEADA ocurrieron en el 33% de los pacientes; las más comunes (>1%) fueron erupción cutánea, diarrea, fatiga, náuseas, vómitos, hipertensión y hematuria. Las reacciones adversas graves ocurrieron en el 25% de los pacientes tratados con ERLEADA y en el 23% de los pacientes que recibieron placebo. Las reacciones adversas graves más frecuentes (>2%) fueron fractura (3,4%) en el brazo de ERLEADA y retención urinaria (3,8%) en el brazo de placebo.
La Tabla 3 muestra las reacciones adversas que ocurrieron en ≥10% en el brazo de ERLEADA en SPARTAN que ocurrieron con un aumento absoluto ≥2% en frecuencia en comparación con el placebo. La Tabla 4 muestra las anormalidades de laboratorio que ocurrieron en ≥15% de los pacientes, y con mayor frecuencia (>5%) en el brazo de ERLEADA en comparación con el placebo.
| ERLEADA N=803 |
Placebo N=398 |
|||
|---|---|---|---|---|
| Clase de sistema/órgano Reacción adversa |
Todos los grados % |
Grado 3–4 % |
Todos los grados % |
Grado 3–4 % |
|
||||
| Trastornos generales y condiciones del lugar de administración | ||||
| Fatiga | 39 | 1.4 | 28 | 0.3 |
| Trastornos musculoesqueléticos y del tejido conjuntivo | ||||
| Artralgia | 16 | 0 | 8 | 0 |
| Trastornos de la piel y del tejido subcutáneo | ||||
| Erupción cutánea | 25 | 5.2 | 6 | 0.3 |
| Trastornos del metabolismo y la nutrición | ||||
| Disminución del apetito | 12 | 0.1 | 9 | 0 |
| Edema periférico | 11 | 0 | 9 | 0 |
| Lesión, envenenamiento y complicaciones de procedimientos | ||||
| Caída | 16 | 1.7 | 9 | 0.8 |
| Fractura | 12 | 2.7 | 7 | 0.8 |
| Pruebas | ||||
| Pérdida de peso | 16 | 1.1 | 6 | 0.3 |
| Trastornos vasculares | ||||
| Hipertensión | 25 | 14 | 20 | 12 |
| Sofocación | 14 | 0 | 9 | 0 |
| Trastornos gastrointestinales | ||||
| Diarrea | 20 | 1.1 | 15 | 0.5 |
| Náuseas | 18 | 0 | 16 | 0 |
Las reacciones adversas adicionales clínicamente significativas que ocurrieron en menos del 10% de los pacientes tratados con ERLEADA incluyeron hipotiroidismo (8% versus 2% con placebo), prurito (6% versus 1.5% con placebo), insuficiencia cardíaca (2.2% versus 1% con placebo) e ILD/neumonitis (0.6% versus 0% con placebo).
| ERLEADA N=803 |
Placebo N=398 |
|||
|---|---|---|---|---|
| Anormalidad de laboratorio | Todos los grados % |
Grado 3–4 % |
Todos los grados % |
Grado 3–4 % |
|
||||
| Hematología | ||||
| Anemia | 70 | 0.4 | 64 | 0.5 |
| Leucopenia | 47 | 0.3 | 29 | 0 |
| Linfopenia | 41 | 1.8 | 21 | 1.6 |
| Química | ||||
| Hipercolesterolemia | 76 | 0.1 | 46 | 0 |
| Hiperglucemia | 70 | 2 | 59 | 1.0 |
| Hipertrigliceridemia | 67 | 1.6 | 49 | 0.8 |
| Hiperkalemia | 32 | 1.9 | 22 | 0.5 |
Erupción
En los datos combinados de dos estudios clínicos aleatorizados y controlados con placebo, SPARTAN y TITAN, la erupción asociada con ERLEADA se describió con mayor frecuencia como macular o maculopapular. Se informaron reacciones adversas de erupción en el 26% de los pacientes tratados con ERLEADA frente al 8% de los pacientes tratados con placebo. Se informaron erupciones de grado 3 (definidas como que cubren > 30% del área de superficie corporal [ASC]) con el tratamiento con ERLEADA (6%) frente a placebo (0,5%).
El inicio de la erupción ocurrió a una mediana de 83 días de tratamiento con ERLEADA. La erupción se resolvió en el 78% de los pacientes dentro de una mediana de 78 días desde el inicio de la erupción. La erupción se manejó comúnmente con antihistamínicos orales, corticosteroides tópicos y el 19% de los pacientes recibieron corticosteroides sistémicos. La reducción de la dosis o la interrupción de la dosis ocurrieron en el 14% y el 28% de los pacientes, respectivamente. De los pacientes que tuvieron interrupción de la dosis, el 59% experimentó recurrencia de la erupción al reintroducir ERLEADA.
Hipotiroidismo
En los datos combinados de dos estudios clínicos aleatorizados y controlados con placebo, SPARTAN y TITAN, se informó hipotiroidismo en el 8% de los pacientes tratados con ERLEADA y el 1,5% de los pacientes tratados con placebo según las evaluaciones de la hormona estimulante de la tiroides (TSH) cada 4 meses. El TSH elevado ocurrió en el 25% de los pacientes tratados con ERLEADA y el 7% de los pacientes tratados con placebo. La mediana de inicio fue en la primera evaluación programada. No hubo reacciones adversas de grado 3 o 4. Se inició la terapia de reemplazo tiroideo en el 4,9% de los pacientes tratados con ERLEADA. La terapia de reemplazo tiroideo, cuando esté clínicamente indicada, debe iniciarse o ajustarse la dosis
[ver
Interacciones medicamentosas (7.2)]
.
6.2 Experiencia post-comercialización
Las siguientes reacciones adversas adicionales se han identificado durante el uso posterior a la aprobación de ERLEADA. Debido a que estas reacciones se informan voluntariamente de una población de tamaño incierto, no siempre es posible estimar de manera confiable la frecuencia o establecer una relación causal con la exposición al fármaco.
Trastornos respiratorios, torácicos y mediastínicos:enfermedad pulmonar intersticial/neumonitis
[ver
Advertencias y precauciones (5.6)]
Trastornos de la piel y del tejido subcutáneo:síndrome de Stevens-Johnson/necrólisis epidérmica tóxica (SJS/TEN) y reacción medicamentosa con eosinofilia y síntomas sistémicos (DRESS).
7 INTERACCIONES MEDICAMENTOSAS
7.1 Efecto de Otros Medicamentos en ERLEADA
Inhibidores Fuertes de CYP2C8 o CYP3A4
Se predice que la coadministración de un inhibidor fuerte de CYP2C8 o CYP3A4 aumentará la exposición en estado estacionario de las fracciones activas (suma de apalutamida no unida más la N-desmetil-apalutamida no unida ajustada por potencia). No es necesario un ajuste inicial de la dosis, sin embargo, reduzca la dosis de ERLEADA en función de la tolerabilidad
[ver
Dosificación y Administración (2.2)]
. No se espera que los inhibidores leves o moderados de CYP2C8 o CYP3A4 afecten la exposición a apalutamida.
7.2 Efecto de ERLEADA en Otros Medicamentos
Sustratos de CYP3A4, CYP2C9, CYP2C19 y UGT
ERLEADA es un inductor fuerte de CYP3A4 y CYP2C19, y un inductor débil de CYP2C9 en humanos. El uso concomitante de ERLEADA con medicamentos que se metabolizan principalmente por CYP3A4, CYP2C19 o CYP2C9 puede resultar en una menor exposición a estos medicamentos. Se recomienda la sustitución de estos medicamentos cuando sea posible o evaluar la pérdida de actividad si se continúa con el medicamento. La administración concomitante de ERLEADA con medicamentos que son sustratos de la UDP-glucuronosil transferasa (UGT) puede resultar en una disminución de la exposición. Tenga precaución si los sustratos de UGT deben administrarse conjuntamente con ERLEADA y evalúe la pérdida de actividad
[ver
.
Sustratos de P-gp, BCRP u OATP1B1
Se demostró clínicamente que apalutamida es un inductor débil de la glicoproteína P (P-gp), la proteína de resistencia al cáncer de mama (BCRP) y el polipéptido transportador de aniones orgánicos 1B1 (OATP1B1). En estado estacionario, apalutamida redujo la exposición plasmática a fexofenadina (un sustrato de P-gp) y rosuvastatina (un sustrato de BCRP/OATP1B1). El uso concomitante de ERLEADA con medicamentos que son sustratos de P-gp, BCRP u OATP1B1 puede resultar en una menor exposición a estos medicamentos. Tenga precaución si los sustratos de P-gp, BCRP u OATP1B1 deben administrarse conjuntamente con ERLEADA y evalúe la pérdida de actividad si se continúa con el medicamento
[ver
.
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de riesgos
No se ha establecido la seguridad y eficacia de ERLEADA en mujeres. Según los hallazgos en animales y su mecanismo de acción, ERLEADA puede causar daño fetal y pérdida del embarazo cuando se administra a una mujer embarazada
[ver
. No hay datos disponibles sobre el uso de ERLEADA en mujeres embarazadas para informar sobre un riesgo asociado con el medicamento. En un estudio de reproducción animal, la administración oral de apalutamida a ratas embarazadas durante y después de la organogénesis provocó anomalías fetales y letalidad embriofetal en exposiciones maternas ≥ 2 veces la exposición clínica humana (AUC) a la dosis recomendada
(ver
.
Datos
Datos en animales
En un estudio piloto de toxicidad para el desarrollo embriofetal en ratas, la apalutamida causó toxicidad para el desarrollo cuando se administró en dosis orales de 25, 50 o 100 mg/kg/día durante y después del período de organogénesis (días gestacionales 6 a 20). Los hallazgos incluyeron letalidad embriofetal (resorciones) en dosis ≥50 mg/kg/día, disminución de la distancia anogenital fetal, glándula pituitaria deforme y variaciones esqueléticas (falanges no osificadas, costilla(s) toracolumbar(es) corta(s) supernumeraria(s) y hueso hioides pequeño, con osificación incompleta o deforme) a ≥25 mg/kg/día. Una dosis de 100 mg/kg/día causó toxicidad materna. Las dosis probadas en ratas dieron como resultado exposiciones sistémicas (AUC) de aproximadamente 2, 4 y 6 veces, respectivamente, el AUC en pacientes.
8.3 Mujeres y hombres con potencial reproductivo
Anticoncepción
Hombres
Según el mecanismo de acción y los hallazgos en un estudio de reproducción animal, se debe aconsejar a los pacientes varones con parejas femeninas con potencial reproductivo que utilicen un método anticonceptivo eficaz durante el tratamiento y durante los 3 meses posteriores a la última dosis de ERLEADA
[ver
Uso en poblaciones específicas (8.1)]
.
Infertilidad
Hombres
Según estudios en animales, ERLEADA puede afectar la fertilidad en hombres con potencial reproductivo
[ver
Toxicología no clínica (13.1)]
.
8.4 Uso pediátrico
No se ha establecido la seguridad y eficacia de ERLEADA en pacientes pediátricos.
8.5 Uso geriátrico
De los 1327 pacientes que recibieron ERLEADA en estudios clínicos, el 19 % de los pacientes eran menores de 65 años, el 41 % de los pacientes tenían entre 65 y 74 años y el 40 % tenían 75 años o más.
No se observaron diferencias generales en la eficacia entre pacientes mayores y jóvenes.
De los pacientes tratados con ERLEADA (n=1073), se produjeron reacciones adversas de grado 3 a 4 en el 39 % de los pacientes menores de 65 años, el 41 % de los pacientes de 65 a 74 años y el 49 % de los pacientes de 75 años o más. Las caídas en pacientes que recibieron ERLEADA con terapia de privación de andrógenos fueron elevadas en los ancianos, y ocurrieron en el 8 % de los pacientes menores de 65 años, el 10 % de los pacientes de 65 a 74 años y el 19 % de los pacientes de 75 años o más.
10 SOBREDOSIS
No se conoce un antídoto específico para la sobredosis de apalutamida. En caso de sobredosis, suspenda ERLEADA, tome medidas generales de apoyo hasta que la toxicidad clínica haya disminuido o se haya resuelto.
11 DESCRIPCIÓN
Apalutamida, el ingrediente activo de ERLEADA, es un inhibidor del receptor de andrógenos. Cada comprimido de ERLEADA contiene 60 mg o 240 mg de apalutamida. El nombre químico es (4 – [7 – (6 – Cian-5 – trifluorometilpiridin-3 – il) – 8 – oxo – 6 – tioxo – 5,7 – diazaspiro [3.4] oct – 5 – il] – 2 – fluoro – N – metilbenzamida). La apalutamida es un polvo blanco a ligeramente amarillo. La apalutamida es prácticamente insoluble en medios acuosos en un amplio rango de valores de pH.
El peso molecular es 477.44 y la fórmula molecular es C
21H
15F
4N
5O
2S. La fórmula estructural es:

ERLEADA
®(apalutamida) comprimidos están disponibles en comprimidos de 240 mg y comprimidos de 60 mg con los siguientes ingredientes inactivos:
- Comprimidos recubiertos de película de 240 mg: sílice anhidra coloidal, croscarmelosa sódica, hidroxipropil metilcelulosa – acetato succinato, celulosa microcristalina silificada y estearato de magnesio. El recubrimiento contiene monocaprilocaprato de glicerilo, óxido de hierro negro, alcohol polivinílico, talco, dióxido de titanio y copolímero injertado de alcohol vinílico.
- Comprimidos recubiertos de película de 60 mg: sílice anhidra coloidal, croscarmelosa sódica, hidroxipropil metilcelulosa – acetato succinato, estearato de magnesio, celulosa microcristalina y celulosa microcristalina silificada. El recubrimiento contiene óxido de hierro negro, óxido de hierro amarillo, polietilenglicol, alcohol polivinílico, talco y dióxido de titanio.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
Apalutamida es un inhibidor del receptor de andrógenos (AR) que se une directamente al dominio de unión al ligando del AR. Apalutamida inhibe la translocación nuclear del AR, inhibe la unión al ADN e impide la transcripción mediada por AR. Un metabolito principal, la N-desmetilapalutamida, es un inhibidor menos potente del AR y exhibió un tercio de la actividad de la apalutamida en un ensayo de reporter de transcripción
in vitro. La administración de apalutamida provocó una disminución de la proliferación de células tumorales y un aumento de la apoptosis, lo que condujo a una disminución del volumen tumoral en modelos de xenotrasplantes de ratón de cáncer de próstata.
12.2 Farmacodinamia
Apalutamida 240 mg diarios además de ADT en pacientes con mCSPC (TITAN) redujo el PSA a niveles indetectables (<0,2 ng/mL) en el 68% de los pacientes en comparación con el 32% de los pacientes que tomaron ADT solo.
Apalutamida 240 mg diarios además de ADT en pacientes con nmCRPC (SPARTAN) redujo el PSA a niveles indetectables (<0,2 ng/mL) en el 38% de los pacientes en comparación con ningún paciente (0%) que tomó ADT solo.
La relación exposición-respuesta y la evolución temporal de la respuesta farmacodinámica para la seguridad y la eficacia de la apalutamida no se han caracterizado completamente.
Electrofisiología cardíaca
El efecto de la apalutamida 240 mg una vez al día sobre el intervalo QTc se evaluó en un estudio QT dedicado de etiqueta abierta, no controlado, multicéntrico, de un solo brazo en 45 pacientes con CRPC. El cambio máximo medio del QTcF desde el inicio fue de 12,4 ms (IC superior del 90% de 2 caras: 16,0 ms). Un análisis de exposición-QT sugirió un aumento dependiente de la concentración en el QTcF para la apalutamida y su metabolito activo.
12.3 Farmacocinética
Los parámetros farmacocinéticos de la apalutamida se presentan como la media [desviación estándar (DE)] a menos que se especifique lo contrario. Apalutamida C
maxy el área bajo la curva de concentración (AUC) aumentaron proporcionalmente después de la administración repetida una vez al día de 30 a 480 mg (0,125 a 2 veces la dosis recomendada). Después de la administración de la dosis recomendada, la apalutamida alcanzó el estado estacionario después de 4 semanas y la relación de acumulación media fue de aproximadamente 5 veces. Apalutamida C
maxfue de 6,0 mcg/mL (1,7) y el AUC fue de 100 mcg∙h/mL (32) en estado estacionario. Las fluctuaciones diarias en las concentraciones plasmáticas de apalutamida fueron bajas, con una relación pico-valle media de 1,63. Se observó un aumento en la depuración aparente (CL/F) con la administración repetida, probablemente debido a la inducción del propio metabolismo de la apalutamida. El efecto de autoinducción probablemente alcanzó su máximo a la dosis recomendada porque la exposición a la apalutamida en el rango de dosis de 30 a 480 mg es proporcional a la dosis.
El metabolito activo principal N-desmetilapalutamida C
maxfue de 5,9 mcg/mL (1,0) y el AUC fue de 124 mcg∙h/mL (23) en estado estacionario después de la dosis recomendada. La N-desmetilapalutamida se caracterizó por un perfil de concentración-tiempo plano en estado estacionario con una relación pico-valle media de 1,27. La relación AUC metabolito/fármaco original media para la N-desmetilapalutamida después de la administración de dosis repetidas fue de 1,3. Con base en la exposición sistémica, la potencia relativa y las propiedades farmacocinéticas, la N-desmetilapalutamida probablemente contribuyó a la actividad clínica de la apalutamida.
Absorción
La biodisponibilidad oral absoluta media fue de aproximadamente el 100%. El tiempo medio para alcanzar la concentración plasmática máxima (t
max) fue de 2 horas (rango: 1 a 5 horas).
La administración oral de cuatro comprimidos de apalutamida de 60 mg dispersos en puré de manzana no produjo cambios clínicamente relevantes en C
maxy AUC en comparación con la administración de cuatro comprimidos intactos de 60 mg en ayunas.
Efecto de los alimentos
La administración de apalutamida a sujetos sanos en ayunas y con una comida rica en grasas (aproximadamente 500 a 600 calorías de grasa, 250 calorías de carbohidratos y 150 calorías de proteínas) no produjo cambios clínicamente relevantes en C
maxy AUC. El tiempo medio para alcanzar t
maxse retrasó aproximadamente 2 horas con los alimentos.
Distribución
El volumen de distribución aparente medio en estado estacionario de la apalutamida fue de aproximadamente 276 L.
La apalutamida fue del 96% y la N-desmetilapalutamida fue del 95% unida a las proteínas plasmáticas sin dependencia de la concentración.
Eliminación
El CL/F de la apalutamida fue de 1,3 L/h después de la administración de una sola dosis y aumentó a 2,0 L/h en estado estacionario después de la administración una vez al día, probablemente debido a la autoinducción del CYP3A4. La vida media efectiva media para la apalutamida en pacientes fue de aproximadamente 3 días en estado estacionario.
Metabolismo
El metabolismo es la principal vía de eliminación de la apalutamida. La apalutamida se metaboliza principalmente por CYP2C8 y CYP3A4 para formar el metabolito activo, N-desmetilapalutamida. La contribución de CYP2C8 y CYP3A4 en el metabolismo de la apalutamida se estima en un 58% y un 13% después de una dosis única, pero cambia a un 40% y un 37%, respectivamente, en estado estacionario.
La apalutamida representó el 45% y la N-desmetilapalutamida representó el 44% del AUC total después de una sola administración oral de apalutamida radiomarcada de 240 mg.
Excreción
Hasta 70 días después de una sola administración oral de apalutamida radiomarcada, se recuperó el 65% de la dosis en la orina (1,2% de la dosis como apalutamida sin cambios y 2,7% como N-desmetilapalutamida) y el 24% se recuperó en las heces (1,5% de la dosis como apalutamida sin cambios y 2% como N-desmetilapalutamida).
Poblaciones específicas
No se observaron diferencias clínicamente significativas en la farmacocinética de la apalutamida o la N-desmetil apalutamida en función de la edad (18-94 años), la raza (negro, asiático no japonés, japonés), el deterioro renal leve a moderado (eGFR 30-89 mL/min/1.73 m
2, estimado por la ecuación de modificación de la dieta en la enfermedad renal [MDRD]) o el deterioro hepático leve (Child-Pugh A) a moderado (Child-Pugh B).
Se desconoce el efecto del deterioro renal grave o la enfermedad renal en etapa terminal (eGFR ≤ 29 mL/min/1.73 m
2, MDRD) o el deterioro hepático grave (Child-Pugh C) en la farmacocinética de la apalutamida.
Interacciones farmacológicas
Efecto de otros fármacos en ERLEADA
Inhibidores potentes de CYP2C8
La C
maxde la apalutamida disminuyó un 21% mientras que el AUC aumentó un 68% tras la administración conjunta de ERLEADA en una dosis única de 240 mg con gemfibrozilo (un potente inhibidor de CYP2C8). Se prevé que el gemfibrozilo aumente la C
maxen estado estacionario de la apalutamida un 32% y el AUC un 44%. Para las fracciones activas (suma de la apalutamida no unida más la apalutamida N-desmetil no unida ajustada por potencia), se prevé que la C
maxen estado estacionario aumente un 19% y el AUC un 23%.
Inhibidores potentes de CYP3A4
La C
maxde la apalutamida disminuyó un 22% mientras que el AUC fue similar tras la administración conjunta de ERLEADA en una dosis única de 240 mg con itraconazol (un potente inhibidor de CYP3A4). Se prevé que el ketoconazol (un potente inhibidor de CYP3A4) aumente el AUC de la apalutamida en una dosis única un 24%, pero no tenga impacto en la C
max. Se prevé que el ketoconazol aumente la C
maxen estado estacionario de la apalutamida un 38% y el AUC un 51%. Para las fracciones activas, se prevé que la C
maxen estado estacionario aumente un 23% y el AUC un 28%.
Inductores de CYP3A4/CYP2C8
Se prevé que la rifampicina (un potente inductor de CYP3A4 y moderado de CYP2C8) disminuya la C
maxen estado estacionario de la apalutamida un 25% y el AUC un 34%. Para las fracciones activas, se prevé que la C
maxen estado estacionario disminuya un 15% y el AUC un 19%.
Agentes reductores de ácido
La apalutamida no es ionizable en las condiciones de pH fisiológico relevantes, por lo tanto, no se espera que los agentes reductores de ácido (por ejemplo, inhibidores de la bomba de protones, antagonistas de los receptores H
2, antiácidos) afecten la solubilidad y la biodisponibilidad de la apalutamida.
Fármacos que afectan a los transportadores
In vitro, la apalutamida y la N-desmetil apalutamida son sustratos de P-gp pero no de BCRP, OATP1B1 y OATP1B3. Debido a que la apalutamida se absorbe completamente después de la administración oral, P-gp no limita la absorción de la apalutamida y, por lo tanto, no se espera que la inhibición o inducción de P-gp afecte la biodisponibilidad de la apalutamida.
Efecto de ERLEADA en otros fármacos
Sustratos de CYP
In vitrolos estudios mostraron que la apalutamida y la N-desmetil apalutamida son inductores moderados a potentes de CYP3A4 y CYP2B6, son inhibidores moderados de CYP2B6 y CYP2C8, y son inhibidores débiles de CYP2C9, CYP2C19 y CYP3A4. La apalutamida y la N-desmetil apalutamida no afectan a CYP1A2 y CYP2D6 en concentraciones terapéuticamente relevantes.
La administración conjunta de ERLEADA con dosis orales únicas de sustratos sensibles de CYP resultó en una disminución del 92% en el AUC del midazolam (un sustrato de CYP3A4), una disminución del 85% en el AUC del omeprazol (un sustrato de CYP2C19) y una disminución del 46% en el AUC de la S-warfarina (un sustrato de CYP2C9). ERLEADA no causó cambios clínicamente significativos en la exposición a un sustrato de CYP2C8.
Sustratos de P-gp, BCRP y OATP1B1
La administración conjunta de ERLEADA con dosis orales únicas de sustratos de transportadores resultó en una disminución del 30% en el AUC de la fexofenadina (un sustrato de P-gp) y una disminución del 41% en el AUC de la rosuvastatina (un sustrato de BCRP/OATP1B1), pero no tuvo impacto en la C
max.
Sustratos de UGT
La apalutamida puede inducir UGT. La administración concomitante de ERLEADA con medicamentos que son sustratos de UGT puede resultar en una menor exposición a estos medicamentos.
Sustratos de OCT2, OAT1, OAT3 y MATEs
In vitro, la apalutamida y la N-desmetil apalutamida inhiben el transportador de cationes orgánicos 2 (OCT2), el transportador de aniones orgánicos 3 (OAT3) y las extrusiones de múltiples fármacos y toxinas (MATEs), y no inhiben el transportador de aniones orgánicos 1. No se prevé que la apalutamida cause cambios clínicamente significativos en la exposición a un sustrato de OAT3.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
En un estudio de carcinogenicidad de 2 años en ratas macho, se administró apalutamida por sonda gástrica a dosis de 5, 15 y 50 mg/kg/día. Apalutamida aumentó la incidencia de adenoma de células intersticiales de Leydig en los testículos a dosis ≥ 5 mg/kg/día (0,2 veces la exposición humana basada en AUC). Se considera que los hallazgos en los testículos están relacionados con la actividad farmacológica de apalutamida. Las ratas se consideran más sensibles que los humanos al desarrollo de tumores de células intersticiales en los testículos. La administración oral de apalutamida a ratones transgénicos rasH2 machos durante 6 meses no provocó un aumento de la incidencia de neoplasias a dosis de hasta 30 mg/kg/día.
Apalutamida no indujo mutaciones en la prueba de reversión de mutación bacteriana (Ames) y no fue genotóxico en ninguna de las pruebas de aberración cromosómica
in vitroo en la prueba de micronúcleos de médula ósea de rata
in vivoo en la prueba de cometa de rata
in vivo.
En estudios de toxicidad de dosis repetidas en ratas macho (hasta 26 semanas) y perros (hasta 39 semanas), se observó atrofia de la glándula prostática y las vesículas seminales, aspermia/hipospermia, degeneración tubular y/o hiperplasia o hipertrofia de las células intersticiales en el sistema reproductivo a ≥ 25 mg/kg/día en ratas (1,4 veces la exposición humana basada en AUC) y ≥ 2,5 mg/kg/día en perros (0,9 veces la exposición humana basada en AUC).
En un estudio de fertilidad en ratas macho, se observó una disminución en la concentración y motilidad de los espermatozoides, un aumento en la morfología anormal de los espermatozoides, tasas de copulación y fertilidad más bajas (al emparejarse con hembras no tratadas) junto con una reducción del peso de las glándulas sexuales secundarias y el epidídimo después de 4 semanas de dosificación a ≥ 25 mg/kg/día (0,8 veces la exposición humana basada en AUC). Se observó una reducción en el número de fetos vivos debido a un aumento de la pérdida pre y/o postimplantación después de 4 semanas de administración de 150 mg/kg/día (5,7 veces la exposición humana basada en AUC). Los efectos en las ratas macho fueron reversibles después de 8 semanas desde la última administración de apalutamida.
14 ESTUDIOS CLÍNICOS
La eficacia y seguridad de ERLEADA se establecieron en dos ensayos clínicos aleatorizados controlados con placebo.
TITAN (NCT02489318): Cáncer de próstata metastásico sensible a la castración (mCSPC)
TITAN fue un ensayo clínico aleatorizado, doble ciego, controlado con placebo y multinacional en el que 1052 pacientes con mCSPC fueron aleatorizados (1:1) para recibir ERLEADA por vía oral a una dosis de 240 mg una vez al día (N=525) o placebo una vez al día (N=527). Todos los pacientes en el ensayo TITAN recibieron un análogo de la GnRH concomitante o se habían sometido previamente a una orquiectomía bilateral. Los pacientes se estratificaron por puntuación de Gleason en el momento del diagnóstico, uso previo de docetaxel y región del mundo. Los pacientes con mCSPC de alto y bajo volumen fueron elegibles para el estudio. El alto volumen de la enfermedad se definió como metástasis que afectaban a las vísceras con 1 lesión ósea o la presencia de 4 o más lesiones óseas, al menos 1 de las cuales debía estar en una estructura ósea más allá de la columna vertebral y los huesos pélvicos.
Los siguientes datos demográficos de los pacientes y las características basales de la enfermedad estaban equilibrados entre los grupos de tratamiento. La mediana de edad fue de 68 años (rango 43–94) y el 23% de los pacientes tenían 75 años o más. La distribución racial fue 68% caucásica, 22% asiática y 2% negra. El sesenta y tres por ciento (63%) de los pacientes tenían enfermedad de alto volumen y el 37% tenían enfermedad de bajo volumen. El dieciséis por ciento (16%) de los pacientes se habían sometido previamente a cirugía, radioterapia de la próstata o ambas. La mayoría de los pacientes tenían una puntuación de Gleason de 8 o superior (67%). El sesenta y ocho por ciento (68%) de los pacientes recibieron tratamiento previo con un antiandrógeno (bicalutamida, flutamida o nilutamida). Todos los pacientes, excepto uno del grupo placebo, tenían una puntuación del estado funcional del Grupo Oncológico Cooperativo del Este (ECOG PS) de 0 o 1 al inicio del estudio.
Las principales medidas de resultado de eficacia del estudio fueron la supervivencia global (SG) y la supervivencia libre de progresión radiográfica (SLP). La supervivencia libre de progresión radiográfica se basó en la evaluación del investigador y se definió como el tiempo desde la aleatorización hasta la progresión radiográfica de la enfermedad o la muerte. La progresión radiográfica de la enfermedad se definió por la identificación de 2 o más nuevas lesiones óseas en una gammagrafía ósea con confirmación (criterios del Grupo de Trabajo 2 sobre Cáncer de Próstata) y/o progresión de la enfermedad en tejidos blandos.
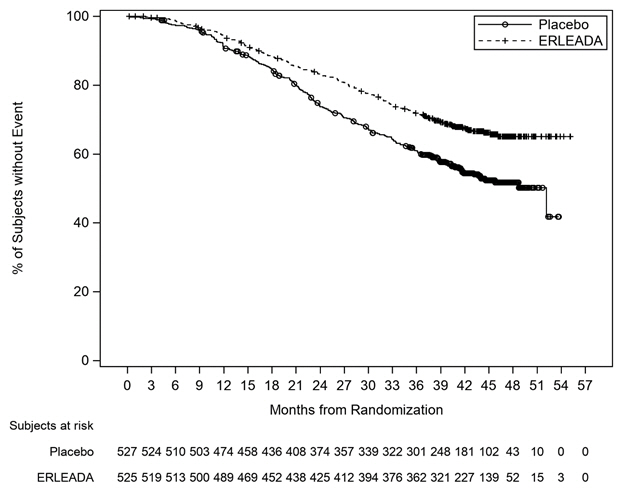
Se demostró una mejora estadísticamente significativa en la SG y la SLP en los pacientes aleatorizados para recibir ERLEADA en comparación con los pacientes aleatorizados para recibir placebo. Los resultados de la SG se basan en un análisis de eficacia intermedio preespecificado. Se realizó un análisis actualizado de la SG en el momento del análisis final del estudio, cuando se observaron 405 muertes. El tiempo medio de seguimiento fue de 44 meses. El 39% de los pacientes del grupo placebo pasaron a recibir ERLEADA. Los resultados de eficacia de TITAN se resumen en la Tabla 5 y las Figuras 1 y 2.
| Criterio de valoración | ERLEADA (N=525) |
Placebo (N=527) |
|---|---|---|
|
||
| Supervivencia global primaria | ||
| Muertes (%) | 83 (16%) | 117 (22%) |
| Mediana, meses (IC del 95%) | NE (NE, NE) | NE (NE, NE) |
| Hazard Ratio (IC del 95%) | 0,67 (0,51, 0,89) | |
| Valor p | 0,0053 | |
| Supervivencia global actualizada | ||
| Muertes (%) | 170 (32%) | 235 (45%) |
| Mediana, meses (IC del 95%) | NE (NE, NE) | 52 (42, NE) |
| Hazard Ratio (IC del 95%) | 0,65 (0,53, 0,79) | |
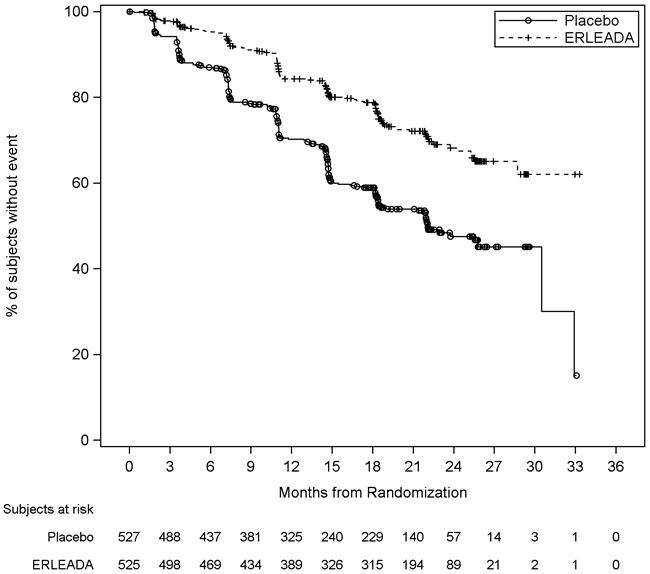
| Supervivencia libre de progresión radiográfica | ||
| Progresión de la enfermedad o muerte (%) | 134 (26%) | 231 (44%) |
| Mediana, meses (IC del 95%) | NE (NE, NE) | 22,1 (18, 33) |
| Hazard Ratio (IC del 95%) | 0,48 (0,39, 0,60) | |
| Valor p | <0,0001 | |
Se observó una mejora consistente en la rPFS en los siguientes subgrupos de pacientes: volumen de la enfermedad (alto vs bajo), uso previo de docetaxel (sí o no) y puntuación de Gleason en el momento del diagnóstico (≤7 vs. >7).
Se observó una mejora consistente en la OS en los siguientes subgrupos de pacientes: volumen de la enfermedad (alto vs bajo) y puntuación de Gleason en el momento del diagnóstico (≤7 vs. >7).
El tratamiento con ERLEADA dio lugar a un retraso estadísticamente significativo en el inicio de la quimioterapia citotóxica (HR = 0,39, IC del 95% = 0,27, 0,56; p < 0,0001).
| Figura 1: Gráfico de Kaplan-Meier de la supervivencia global (OS) actualizada; Población con mCSPC con intención de tratar (TITAN) |
 |
| Figura 2: Gráfico de Kaplan-Meier de la supervivencia libre de progresión radiográfica (rPFS); Población con mCSPC con intención de tratar (TITAN) |
 |
SPARTAN (NCT01946204): Cáncer de próstata no metastásico resistente a la castración (CnPRCm)
SPARTAN fue un ensayo clínico multicéntrico, doble ciego, aleatorizado (2:1), controlado con placebo en el que 1207 pacientes con CnPRCm fueron aleatorizados (2:1) para recibir ERLEADA por vía oral a una dosis de 240 mg una vez al día (N=806) o placebo una vez al día (N=401). Todos los pacientes del ensayo SPARTAN recibieron un análogo de la GnRH concomitante o se sometieron a una orquiectomía bilateral. Los pacientes se estratificaron según el tiempo de duplicación del antígeno prostático específico (TDA-PSA), el uso de agentes ahorradores de hueso y la enfermedad locorregional. Los pacientes debían tener un TDA-PSA ≤ 10 meses y confirmación de enfermedad no metastásica mediante revisión central independiente ciega (RCIC). Los resultados del PSA se cegaron y no se utilizaron para la interrupción del tratamiento. Los pacientes aleatorizados a cualquiera de los brazos interrumpieron el tratamiento debido a la progresión de la enfermedad radiográfica confirmada por RCIC, progresión solo locorregional, inicio de un nuevo tratamiento, toxicidad inaceptable o retirada.
Los siguientes datos demográficos de los pacientes y las características basales de la enfermedad estaban equilibrados entre los brazos de tratamiento. La mediana de edad fue de 74 años (rango 48–97) y el 26 % de los pacientes tenían 80 años o más. La distribución racial fue del 66 % de raza blanca, el 12 % de asiáticos y el 6 % de raza negra. El setenta y siete por ciento (77 %) de los pacientes de ambos brazos de tratamiento se habían sometido previamente a cirugía o radioterapia de próstata. La mayoría de los pacientes tenían una puntuación de Gleason de 7 o superior (78 %). El quince por ciento (15 %) de los pacientes presentaban ganglios linfáticos pélvicos de <2 cm al inicio del estudio. El setenta y tres por ciento (73 %) de los pacientes habían recibido tratamiento previo con un antiandrógeno; el 69 % de los pacientes habían recibido bicalutamida y el 10 % de los pacientes habían recibido flutamida. Todos los pacientes tenían una puntuación del estado funcional del Grupo Oncológico Cooperativo del Este (ECOG PS) de 0 o 1 al inicio del estudio.
La principal medida de resultado de eficacia del estudio fue la supervivencia libre de metástasis (SLM), definida como el tiempo desde la aleatorización hasta el momento de la primera evidencia de metástasis a distancia confirmada por RCIC, definida como nuevas lesiones óseas o de tejidos blandos o ganglios linfáticos agrandados por encima de la bifurcación ilíaca, o la muerte por cualquier causa, lo que ocurriera primero. Las variables principales de eficacia adicionales fueron el tiempo hasta la metástasis (TTM), la supervivencia libre de progresión (SLP), que también incluye la progresión locorregional, el tiempo hasta la progresión sintomática, la supervivencia global (SG) y el tiempo hasta el inicio de la quimioterapia citotóxica.
Se demostró una mejora estadísticamente significativa en la SLM y la SG en los pacientes aleatorizados para recibir ERLEADA en comparación con los pacientes aleatorizados para recibir placebo. La variable principal de eficacia (SLM) se vio respaldada por mejoras en el TTM y la SLP. El análisis final de la SG y el tiempo hasta el inicio de la quimioterapia citotóxica se realizó 32 meses después del análisis de la SLM, el TTM y la SLP. Los resultados de eficacia de SPARTAN se resumen en la Tabla 6 y las Figuras 3 y 4.
| Criterio de valoración | ERLEADA (N=806) |
Placebo (N=401) |
|---|---|---|
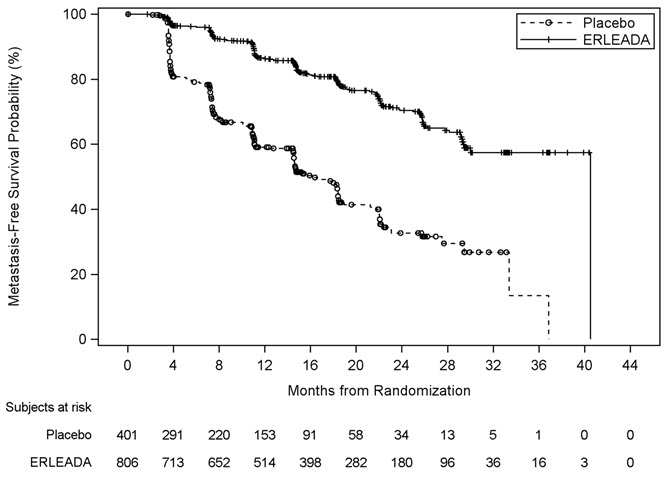
| Supervivencia libre de metástasis*,†,‡ | ||
| Número de eventos (%) | 184 (23%) | 194 (48%) |
| Mediana, meses (IC del 95%) | 40.5 (NE, NE) | 16.2 (15, 18) |
| Hazard Ratio (IC del 95%) | 0.28 (0.23, 0.35) | |
| Valor p | <0.0001 | |
| Tiempo hasta la metástasis*,† | ||
| Número de eventos (%) | 175 (22%) | 191 (48%) |
| Mediana, meses (IC del 95%) | 40.5 (NE, NE) | 16.6 (15, 18) |
| Hazard Ratio (IC del 95%) | 0.27 (0.22, 0.34) | |
| Valor p | <0.0001 | |
| Supervivencia libre de progresión*,† | ||
| Número de eventos (%) | 200 (25%) | 204 (51%) |
| Median, months (95% CI) | 40.5 (NE, NE) | 14.7 (14, 18) |
| Hazard Ratio (95% CI) | 0.29 (0.24, 0.36) | |
| p-value | <0.0001 | |
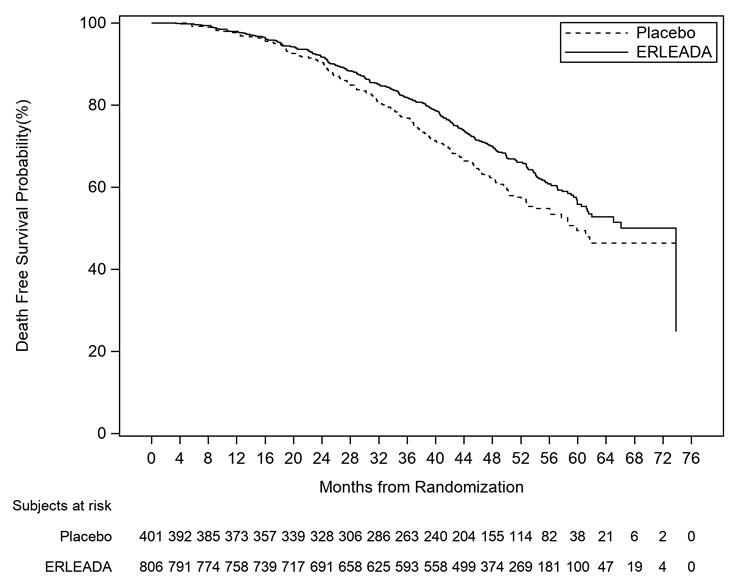
| Supervivencia global | ||
| Number of Events (%) | 274 (34%) | 154 (38%) |
| Median, months (95% CI) | 73.9 (61, NE) | 59.9 (53, NE) |
| Hazard Ratio (95% CI) | 0.78 (0.64, 0.96) | |
| p-value | 0.0161 | |
Se observaron resultados consistentes para la MFS en todos los subgrupos de pacientes, incluidos el PSADT (≤ 6 meses o > 6 meses), el uso de un agente ahorrador de hueso previo (sí o no) y la enfermedad locorregional (N0 o N1).
El tratamiento con ERLEADA dio lugar a un retraso estadísticamente significativo en el inicio de la quimioterapia citotóxica [HR = 0,63 (IC del 95%: 0,49, 0,81), p=0,0002].
| Figura 3: Curva de supervivencia libre de metástasis (MFS) de Kaplan-Meier en SPARTAN (nmCRPC) |
 |
| Figura 4: Curva de supervivencia global (SG) de Kaplan-Meier en SPARTAN (nmCRPC) |
 |
16 FORMA DE SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
ERLEADA
®(apalutamide) comprimidos están disponibles en las concentraciones y envases que se indican a continuación:
-
ERLEADA
®240 mg Comprimidos
Comprimidos recubiertos con película, de color gris azulado a gris, de forma ovalada, grabados con “E240” en una cara.
Número NDC 59676‐604‐30 – 30 comprimidos disponibles en frascos con un desecante de gel de sílice y con cierre de seguridad para niños.
-
ERLEADA
®60 mg Comprimidos
Comprimidos recubiertos con película, de color ligeramente amarillento a verde grisáceo, de forma oblonga, grabados con “AR 60” en una cara.
Número NDC 59676‐600‐12 – 120 comprimidos disponibles en frascos con un desecante de gel de sílice y con cierre de seguridad para niños.
Almacenamiento y manipulación
Conservar a una temperatura de 20 °C a 25 °C (68 °F a 77 °F); se permiten excursiones de temperatura de 15 °C a 30 °C (59 °F a 86 °F)
[ver USP Temperatura ambiente controlada].
Conservar en el envase original para protegerlo de la luz y la humedad. No deseche el desecante.
17 INFORMACIÓN PARA EL PACIENTE
Aconseje al paciente que lea la información para pacientes aprobada por la FDA (Información para pacientes e instrucciones de uso).
Eventos cerebrovasculares e isquémicos cardiovasculares
- Informe a los pacientes que ERLEADA se ha asociado con eventos cerebrovasculares e isquémicos cardiovasculares. Aconseje a los pacientes que busquen atención médica inmediata si se presenta algún síntoma que sugiera un evento cardiovascular o cerebrovascular
[consulte
Advertencias y precauciones (5.1)]
.
Caídas y fracturas
- Informe a los pacientes que ERLEADA se asocia con una mayor incidencia de caídas y fracturas
[consulte
Advertencias y precauciones (5.2,
5.3)]
.
Convulsiones
- Informe a los pacientes que ERLEADA se ha asociado con un mayor riesgo de convulsiones. Hable sobre las afecciones que pueden predisponer a las convulsiones y los medicamentos que pueden reducir el umbral convulsivo. Aconseje a los pacientes sobre el riesgo de realizar cualquier actividad en la que una pérdida repentina del conocimiento pueda causarles daños graves a ellos mismos o a otras personas. Informe a los pacientes que se comuniquen con su proveedor de atención médica de inmediato si experimentan una convulsión
[consulte
Advertencias y precauciones (5.4)]
.
Reacciones adversas cutáneas graves (SCAR)
- Informe a los pacientes que ERLEADA se ha asociado con SCAR (incluidos SJS/TEN y DRESS), que pueden ser mortales o potencialmente mortales. Aconseje a los pacientes que dejen de tomar ERLEADA y que se comuniquen con su proveedor de atención médica o que busquen atención médica de inmediato si experimentan signos o síntomas de SCAR
[consulte
Advertencias y precauciones (5.5)]
.
Enfermedad pulmonar intersticial (EPI)/neumonitis
- Informe a los pacientes sobre los riesgos de EPI/neumonitis mortales o potencialmente mortales. Aconseje a los pacientes que dejen de tomar ERLEADA y que se comuniquen con su proveedor de atención médica o que busquen atención médica de inmediato si presentan síntomas respiratorios nuevos o que empeoran
[consulte
Advertencias y precauciones (5.6)]
.
Erupción cutánea
- Informe a los pacientes que ERLEADA se asocia con erupciones cutáneas y que informen a su proveedor de atención médica si desarrollan una erupción cutánea
[consulte
6.2)]
.
Dosificación y administración
- Informe a los pacientes que reciben tratamiento concomitante con un análogo de la hormona liberadora de gonadotropina (GnRH) que deben mantener este tratamiento durante el curso del tratamiento con ERLEADA.
- Indique a los pacientes que tomen su dosis a la misma hora todos los días (una vez al día). ERLEADA puede tomarse con o sin alimentos. Cada comprimido debe tragarse entero. No triture ni parta los comprimidos
[consulte
Dosificación y administración (2.1)]
.
- Indique a los pacientes que no puedan tragar los comprimidos enteros que sigan las instrucciones para la concentración prescrita de los comprimidos de ERLEADA para conocer los métodos alternativos de administración, incluida la administración a través de una sonda de alimentación
[consulte
Dosificación y administración (2.3)]
.
- Informe a los pacientes que, en caso de que olviden una dosis diaria de ERLEADA, deben tomar su dosis normal tan pronto como sea posible el mismo día y volver a su horario normal al día siguiente. El paciente no debe tomar comprimidos adicionales para compensar la dosis olvidada
[consulte
Dosificación y administración (2.1)]
.
Toxicidad embriofetal
- Informe a los pacientes que ERLEADA puede ser perjudicial para el feto en desarrollo. Aconseje a los pacientes varones con parejas femeninas con potencial reproductivo que utilicen un método anticonceptivo eficaz durante el tratamiento y durante los 3 meses posteriores a la última dosis de ERLEADA. Aconseje a los pacientes varones que utilicen preservativo si mantienen relaciones sexuales con una mujer embarazada
[consulte
Advertencias y precauciones (5.7)]
.
Infertilidad
- Aconseje a los pacientes varones que ERLEADA puede afectar a la fertilidad y que no donen esperma durante el tratamiento ni durante los 3 meses posteriores a la última dosis de ERLEADA
[consulte
Uso en poblaciones específicas (8.3)]
.
SECCIÓN NO CLASIFICADA DE SPL
Fabricado para:
Janssen Products, LP
Horsham, PA 19044, USA
Para información de patentes: www.janssenpatents.com
© 2019, 2023 Janssen Pharmaceutical Companies
INSERTO PARA EL PACIENTE
| Esta información para el paciente ha sido aprobada por la Administración de Alimentos y Medicamentos de los Estados Unidos. | Revisado: 8/2024 | ||
| INFORMACIÓN PARA EL PACIENTE ERLEADA ® (er lee’dah) |
|||
| ¿Qué es ERLEADA? ERLEADA es un medicamento recetado que se usa para el tratamiento del cáncer de próstata:
No se sabe si ERLEADA es seguro y eficaz en mujeres. |
|||
Antes de tomar ERLEADA, informe a su proveedor de atención médica sobre todas sus afecciones médicas, incluso si:
Informe a su proveedor de atención médica sobre todos los medicamentos que toma, incluidos los medicamentos recetados y de venta libre, las vitaminas y los suplementos herbales. ERLEADA puede interactuar con muchos otros medicamentos. |
|||
¿Cómo debo tomar ERLEADA?
|
|||
| ¿Cuáles son los posibles efectos secundarios de ERLEADA? ERLEADA puede causar efectos secundarios graves, que incluyen:
|
|||
|
|
||
|
|||
|
|
||
|
|||
|
|
|
|
| Los efectos secundarios más comunes de ERLEADA incluyen: | |||
|
|
||
| Su proveedor de atención médica puede reducir su dosis, detener temporalmente o detener definitivamente el tratamiento con ERLEADA si tiene ciertos efectos secundarios. ERLEADA puede causar problemas de fertilidad en los hombres, lo que puede afectar la capacidad de engendrar hijos. Hable con su proveedor de atención médica si tiene preocupaciones sobre la fertilidad. Nodone esperma durante el tratamiento con ERLEADA y durante 3 meses después de la última dosis de ERLEADA. |
|||
¿Cómo debo almacenar ERLEADA?
Mantenga ERLEADA y todos los medicamentos fuera del alcance de los niños. |
|||
| Información general sobre el uso seguro y eficaz de ERLEADA. A veces, los medicamentos se recetan para fines distintos a los enumerados en un folleto de información para el paciente. No use ERLEADA para una afección para la que no fue prescrita. No le dé ERLEADA a otras personas, incluso si tienen los mismos síntomas que usted. Puede dañarlos. Puede pedir a su proveedor de atención médica o farmacéutico información sobre ERLEADA escrita para profesionales de la salud. |
|||
| ¿Cuáles son los ingredientes de ERLEADA? Ingrediente activo:apalutamida Ingredientes inactivos: 240 mg comprimidos recubiertos con película: sílice anhidra coloidal, croscarmelosa sódica, hidroxipropilmetilcelulosa-acetato succinato, celulosa microcristalina silificada y estearato de magnesio. El recubrimiento contiene monocaprilocaprato de glicerilo, óxido de hierro negro, alcohol polivinílico, talco, dióxido de titanio y copolímero injertado de alcohol vinílico. 60 mg comprimidos recubiertos con película: sílice anhidra coloidal, croscarmelosa sódica, hidroxipropilmetilcelulosa-acetato succinato, estearato de magnesio, celulosa microcristalina y celulosa microcristalina silificada. El recubrimiento contiene óxido de hierro negro, óxido de hierro amarillo, polietilenglicol, alcohol polivinílico, talco y dióxido de titanio. Fabricado para:Janssen Products, LP, Horsham, PA 19044, EE. UU. Para información de patentes: www.janssenpatents.com © 2019, 2023 Janssen Pharmaceutical Companies Para más información, llame a Janssen Products, LP al 1-800-526-7736 (1-800-JANSSEN) o vaya a www.erleada.com. |
|||
INSTRUCCIONES DE USO
| Estas Instrucciones de Uso han sido aprobadas por la Administración de Alimentos y Medicamentos de los Estados Unidos. | Revisado: 07/2024 | |
| INSTRUCCIONES DE USO ERLEADA ® ( er lee’dah) |
||
| Estas Instrucciones de Uso contienen información sobre cómo preparar y tomar o administrar una dosis de tabletas ERLEADA si no puede tragar las tabletas ERLEADA enteras o si tiene una sonda de alimentación. Lea estas Instrucciones de Uso antes de preparar y tomar o administrar la primera dosis de ERLEADA, y cada vez que reciba una nueva receta. Consulte con su médico o farmacéutico si tiene alguna pregunta.
Información importante que necesita saber antes de preparar una dosis de ERLEADA:
|
||
| Preparación y toma de ERLEADA si no puede tragar las tabletas enteras:
Preparación y toma de tabletas ERLEADA de 60 mg colocando las tabletas en agua no carbonatada y luego mezclándolas con jugo de naranja, puré de manzana o más agua no carbonatada: |
||
| Paso 1. | Coloque toda la dosis prescrita de tabletas de 60 mg en una taza.
No triture ni divida las tabletas. |
|
| Paso 2. | Agregue aproximadamente 4 cucharaditas (20 mL) de agua no carbonatada para asegurarse de que las tabletas estén completamente cubiertas de agua. | |
| Paso 3. | Espere 2 minutos hasta que las tabletas se desintegren y se dispersen, luego revuelva la mezcla. | |
| Paso 4. | Agregue 2 cucharadas (30 mL) de jugo de naranja, puré de manzana o agua no carbonatada a la taza y revuelva la mezcla. | |
| Paso 5. | Traga la mezcla inmediatamente. | |
| Paso 6. | Enjuague la taza con suficiente agua no carbonatada para asegurarse de que tome toda la dosis de ERLEADA y bébala inmediatamente. | |
| Noalmacene ERLEADA que esté mezclada con agua no carbonatada, jugo de naranja o puré de manzana para usarla más tarde. | ||
| Preparación y toma de la tableta ERLEADA de 240 mg colocando la tableta en agua no carbonatada y luego mezclándola con jugo de naranja, puré de manzana o más agua no carbonatada: | ||
| Paso 1. | Coloque la tableta entera de 240 mg en una taza.
No triture ni divida la tableta. |
|
| Paso 2. | Agregue aproximadamente 2 cucharaditas (10 mL) de agua no carbonatada para asegurarse de que la tableta esté completamente cubierta de agua. | |
| Paso 3. | Espere 2 minutos hasta que la tableta se desintegre y se disperse, luego revuelva la mezcla. | |
| Paso 4. | Agregue 2 cucharadas (30 mL) de jugo de naranja, puré de manzana o agua no carbonatada a la taza y revuelva la mezcla. | |
| Paso 5. | Traga la mezcla inmediatamente. | |
| Paso 6. | Enjuague la taza con suficiente agua no carbonatada para asegurarse de que tome toda la dosis de ERLEADA y bébala inmediatamente. | |
| Noalmacene ERLEADA que esté mezclada con agua no carbonatada, jugo de naranja o puré de manzana para usarla más tarde. | ||
| Preparación y administración de ERLEADA a través de una sonda de alimentación: Preparación y administración de tabletas ERLEADA de 60 mg a través de una sonda de alimentación de 8 French o más grande: |
||
| Paso 1. | Retire el émbolo de la jeringa (use una jeringa de al menos 50 mL). | |
| Paso 2. | Agregue toda la dosis prescrita de tabletas de 60 mg al cuerpo de la jeringa (barril) y coloque el émbolo de nuevo en la jeringa.
No triture ni divida las tabletas. |
|
| Paso 3. | Extraiga 20 mL de agua no carbonatada en la jeringa. | |
| Paso 4. | Espere 10 minutos y luego agite la jeringa muy bien (vigorosamente) para desintegrar las tabletas por completo. | |
| Paso 5. | Conecte la jeringa a la sonda de alimentación y administre la mezcla inmediatamente. | |
| Paso 6. | Aspire agua no carbonatada en la misma jeringa y enjuague a través del tubo de alimentación. Repita
Paso 6hasta que no queden trozos de tabletas en la jeringa o el tubo de alimentación. |
|
| Preparación y administración de la tableta de ERLEADA 240 mg a través de un tubo de alimentación de 8 French o más grande: | ||
| Paso 1. | Retire el émbolo de la jeringa (utilice al menos una jeringa de 20 mL). | |
| Paso 2. | Agregue una tableta de 240 mg al cuerpo de la jeringa (barril) y coloque el émbolo de nuevo en la jeringa.
No triture ni divida la tableta. |
|
| Paso 3. | Aspire 10 mL de agua no carbonatada en la jeringa. | |
| Paso 4. | Espere 10 minutos y luego agite la jeringa muy bien (vigorosamente) para romper la tableta por completo. | |
| Paso 5. | Conecte la jeringa al tubo de alimentación y administre la mezcla de inmediato. | |
| Paso 6. | Aspire agua no carbonatada en la misma jeringa y enjuague a través del tubo de alimentación. Repita
Paso 6hasta que no queden trozos de tabletas en la jeringa o el tubo de alimentación. |
|
¿Cómo debo almacenar ERLEADA?
Mantenga ERLEADA y todos los medicamentos fuera del alcance de los niños. Fabricado para:Janssen Products, LP, Horsham, PA 19044, EE. UU. |
||
ETIQUETA PRINCIPAL DEL ENVASE – Etiqueta del frasco de tabletas de 60 mg
NDC 59676-600-12
Erleada
®
(apalutamide) tablets
60 mg
Cada comprimido recubierto con película
contiene 60 mg de apalutamide.
Este envase es a prueba de niños.
Mantener fuera del alcance de los niños.
Rx solamente
120 comprimidos recubiertos con película
tablets
janssen

PANEL PRINCIPAL DE VISUALIZACIÓN – Etiqueta del frasco de tabletas de 240 mg
NDC 59676-604-30
Erleada
®
(apalutamide) tablets
240 mg
Cada comprimido recubierto con película contiene
240 mg de apalutamide.
Este envase es a prueba de niños.
Mantener fuera del alcance de los niños.
Rx solamente
30 comprimidos recubiertos con película
janssen
