Fabricante de medicamentos: Takeda Pharmaceuticals America, Inc. (Updated: 2024-06-06)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
ENTYVIO (vedolizumab) para inyección, para uso intravenoso
ENTYVIO (vedolizumab) inyección, para uso subcutáneo
ENTYVIO PEN (vedolizumab) inyección, para uso subcutáneo
Aprobación inicial en EE. UU.: 2014
CAMBIOS RECIENTES IMPORTANTES
INDICACIONES Y USO
DOSIFICACIÓN Y ADMINISTRACIÓN
Información importante sobre la administración
- Antes de iniciar ENTYVIO, actualice las inmunizaciones de acuerdo con las pautas de inmunización actuales. (2.1, 5.5)
- Administración intravenosa: ENTYVIO debe administrarse por vía intravenosa por un profesional sanitario. (2.1)
- Inyección subcutánea: La jeringa precargada de ENTYVIO y ENTYVIO PEN están diseñadas para uso subcutáneo. Un paciente puede autoinyectarse o un cuidador puede inyectar después de recibir una capacitación adecuada sobre la técnica correcta de inyección subcutánea. (2.1)
Dosificación recomendada (2.2)
- Semana 0: 300 mg infundidos por vía intravenosa durante aproximadamente 30 minutos.
- Semana 2: 300 mg infundidos por vía intravenosa durante aproximadamente 30 minutos.
-
Semana 6: Los pacientes pueden permanecer en terapia intravenosa con ENTYVIO o cambiar a inyección subcutánea después de recibir dos dosis intravenosas de ENTYVIO administradas en la semana 0 y la semana 2.
- Infusion intravenosa: 300 mg infundidos durante aproximadamente 30 minutos y luego cada ocho semanas a partir de entonces.
- Inyección subcutánea: 108 mg por vía subcutánea una vez cada dos semanas.
- Suspenda ENTYVIO en pacientes que no muestren evidencia de beneficio terapéutico para la semana 14.
- Los pacientes que actualmente reciben y responden a la terapia intravenosa con ENTYVIO después de la semana 6 también pueden cambiar a la inyección subcutánea. Administre la primera dosis subcutánea en lugar de la próxima infusión intravenosa programada y cada dos semanas a partir de entonces.
Instrucciones de preparación y administración:
FORMAS DE DOSIFICACIÓN Y FUERZAS
Infusion intravenosa
- Para inyección: 300 mg de vedolizumab en un vial de dosis única. (3)
Inyección subcutánea
CONTRAINDICACIONES
Pacientes que han tenido una reacción de hipersensibilidad grave o severa conocida a ENTYVIO o a cualquiera de sus excipientes. (4)
ADVERTENCIAS Y PRECAUCIONES
- Reacciones relacionadas con la infusión y reacciones de hipersensibilidad: Suspenda ENTYVIO e inicie el tratamiento apropiado si se producen reacciones graves. (5.1)
- Infecciones: No se recomienda el tratamiento con ENTYVIO en pacientes con infecciones activas y graves hasta que las infecciones estén controladas. Considere la posibilidad de suspender ENTYVIO en pacientes que desarrollen una infección grave mientras están en tratamiento con ENTYVIO. (5.2)
- Leucoencefalopatía multifocal progresiva (LMP): Aunque es poco probable, no se puede descartar el riesgo de LMP. Controle a los pacientes en busca de cualquier signo o síntoma neurológico nuevo o que empeore. (5.3)
REACCIONES ADVERSAS
- Las reacciones adversas más comunes (incidencia ≥3% y ≥1% mayor que el placebo) son: nasofaringitis, dolor de cabeza, artralgia, náuseas, pirexia, infección del tracto respiratorio superior, fatiga, tos, bronquitis, influenza, dolor de espalda, erupción cutánea, prurito, sinusitis, dolor orofaríngeo y dolor en las extremidades. (6.1)
- Las reacciones adversas con ENTYVIO subcutáneo son similares a las informadas con ENTYVIO intravenoso, con la excepción de las reacciones en el sitio de inyección informadas con ENTYVIO subcutáneo. (6.1)
Para informar sobre REACCIONES ADVERSAS SOSPECHOSAS, comuníquese con Takeda Pharmaceuticals U.S.A., Inc. al 1-877-TAKEDA-7 (1-877-825-3327) o con la FDA al 1-800-FDA-1088 o www.fda.gov/medwatch.
Consulte 17 para obtener INFORMACIÓN PARA EL PACIENTE y la Guía de medicamentos.
Revisado: 5/2024
Tabla de Contenido
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDO*
1 INDICACIONES Y USO
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Información Importante de Administración
2.2 Dosis Recomendada en Adultos con Colitis Ulcerosa y Enfermedad de Crohn
2.3 Instrucciones de Preparación y Administración para Infusión Intravenosa
2.4 Instrucciones de Preparación y Administración para Inyección Subcutánea
3 FORMAS Y FUERZAS DE DOSIFICACIÓN
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Reacciones Relacionadas con la Infusión y Reacciones de Hipersensibilidad
5.2 Infecciones
5.3 Leucoencefalopatía Multifocal Progresiva
5.4 Lesión Hepática
5.5 Vacunas Vivas y Orales
6 REACCIONES ADVERSAS
6.1 Experiencia en Ensayos Clínicos
6.2 Experiencia Postcomercialización
7 INTERACCIONES MEDICAMENTOSAS
7.1 Productos de Natalizumab
7.2 Bloqueadores del TNF
7.3 Sustratos del CYP450
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.4 Uso Pediátrico
8.5 Uso Geriátrico
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de Acción
12.2 Farmacodinamia
12.3 Farmacocinética
12.6 Inmunogenicidad
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
14 ESTUDIOS CLÍNICOS
14.1 Estudios Clínicos en Colitis Ulcerosa
14.2 Estudios Clínicos en Enfermedad de Crohn
16 CÓMO SE SUMINISTRA/ALMACENAMIENTO Y MANEJO
17 INFORMACIÓN PARA EL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información completa de prescripción no están listadas.
1 INDICACIONES Y USO
ENTYVIO está indicado en adultos para el tratamiento de:
- colitis ulcerosa (CU) activa de moderada a grave.
- enfermedad de Crohn (EC) activa de moderada a grave.
2 DOSIS Y ADMINISTRACIÓN
2.1 Información Importante sobre la Administración
Antes de iniciar el tratamiento con ENTYVIO, actualice las inmunizaciones de acuerdo con las pautas de inmunización actuales [ver Advertencias y precauciones (5.5)].
Administración intravenosa
- ENTYVIO debe ser administrado por un profesional sanitario preparado para manejar reacciones de hipersensibilidad, incluida la anafilaxia, si ocurren [ver Advertencias y precauciones (5.1)]. Deben estar disponibles medidas de monitorización y apoyo médico apropiadas para su uso inmediato. Observe a los pacientes durante la infusión y hasta que la infusión esté completa.
- Reconstituya y diluya el polvo liofilizado de ENTYVIO antes de la administración como una infusión intravenosa de 30 minutos [ver Dosificación y administración (2.3)].
Inyección subcutánea
- La jeringa precargada de ENTYVIO y el ENTYVIO PEN están destinados para uso subcutáneo bajo la guía y supervisión de un profesional sanitario.
- Los pacientes pueden autoinyectarse o los cuidadores pueden inyectar ENTYVIO por vía subcutánea utilizando la jeringa precargada de ENTYVIO o el ENTYVIO PEN después de recibir capacitación en la técnica de inyección subcutánea. Proporcione capacitación adecuada a los pacientes y/o cuidadores sobre la técnica de inyección subcutánea de ENTYVIO.
2.2 Dosificación recomendada en adultos con colitis ulcerosa y enfermedad de Crohn
- Semana 0: Administrar ENTYVIO 300 mg por infusión intravenosa durante aproximadamente 30 minutos [ver Dosificación y administración (2.3)].
- Semana 2: Administrar ENTYVIO 300 mg por infusión intravenosa durante aproximadamente 30 minutos.
-
Semana 6: Los pacientes pueden permanecer en terapia intravenosa con ENTYVIO o cambiar a inyección subcutánea después de recibir dos dosis intravenosas de ENTYVIO administradas en la semana 0 y la semana 2.
- Infusión intravenosa: Administrar ENTYVIO 300 mg por infusión intravenosa durante aproximadamente 30 minutos y luego cada ocho semanas a partir de entonces.
- Inyección subcutánea: Administrar ENTYVIO 108 mg por vía subcutánea una vez cada 2 semanas.
- Suspenda el tratamiento en los pacientes que no muestren evidencia de beneficio terapéutico para la semana 14.
2.3 Instrucciones de preparación y administración para la infusión intravenosa
Instrucciones de reconstitución
- Retire la tapa abatible del vial de dosis única y limpie con una torunda de alcohol. Reconstituya el vial de ENTYVIO que contiene el polvo liofilizado con 4,8 mL de Agua estéril para inyección, Solución inyectable de cloruro de sodio al 0,9% o Solución inyectable de Ringer lactato, a temperatura ambiente (20 °C a 25 °C [68 °F a 77 °F]), utilizando una jeringa con una aguja de calibre 21 a 25.
- Inserte la aguja de la jeringa en el vial a través del centro del tapón y dirija el flujo de Agua estéril para inyección, Solución inyectable de cloruro de sodio al 0,9% o Solución inyectable de Ringer lactato, hacia la pared de vidrio del vial para evitar una espuma excesiva.
- Gire suavemente el vial durante al menos 15 segundos para disolver el polvo liofilizado. No agite ni invierta vigorosamente.
- Deje reposar la solución hasta por 20 minutos a temperatura ambiente para permitir la reconstitución y que se asiente cualquier espuma; el vial se puede girar e inspeccionar para verificar la disolución durante este tiempo. Si no se disuelve completamente después de 20 minutos, permita otros 10 minutos para la disolución. No utilice el vial si el producto farmacéutico no se disuelve en 30 minutos.
- Inspeccione visualmente la solución reconstituida de ENTYVIO para detectar partículas y decoloración antes de la dilución. La solución debe ser clara u opalescente, incolora a amarillo pardusco claro y libre de partículas visibles. No administre la solución reconstituida que muestre un color no característico o que contenga partículas.
- Una vez disuelto, invierta suavemente el vial tres veces.
- Inmediatamente, retire 5 mL (300 mg) de la solución reconstituida de ENTYVIO utilizando una jeringa con una aguja de calibre 21 a 25. Deseche cualquier porción restante de la solución reconstituida en el vial.
Instrucciones de dilución
Agregue los 5 mL (300 mg) de la solución reconstituida de ENTYVIO a 250 mL de Solución inyectable de cloruro de sodio al 0,9% o Solución inyectable de Ringer lactato, y mezcle suavemente la bolsa de infusión. No agregue otros productos medicinales a la solución de infusión preparada o al equipo de infusión intravenosa. Una vez reconstituida y diluida, utilice la solución de infusión lo antes posible.
Deseche cualquier porción no utilizada de la solución de infusión.
Instrucciones de administración
Después de que la infusión esté completa, enjuague con 30 mL de Solución inyectable de cloruro de sodio al 0,9% o Solución inyectable de Ringer lactato.
Almacenamiento y estabilidad
Las condiciones de almacenamiento específicas y la duración de la solución reconstituida en el vial y la solución diluida en la bolsa de infusión se detallan en Tabla 1.
No congele la solución reconstituida en el vial o la solución diluida en la bolsa de infusión.
| Solución | Condiciones de almacenamiento | |
|---|---|---|
| Refrigeración (2°C a 8°C [36°F a 46°F]) |
Temperatura ambiente (20°C a 25°C [68°F a 77°F]) |
|
|
||
| Solución reconstituida (en Agua Estéril para Inyección, Inyección de Cloruro de Sodio al 0.9%, o Inyección de Ringer Lactato, dentro del vial) |
8 horas | Usar inmediatamente después de la reconstitución |
| Solución diluida (en Inyección de Cloruro de Sodio al 0.9%) |
24 horas*,† | 12 horas* |
| Solución diluida (en Inyección de Ringer Lactato) |
6 horas* | Usar inmediatamente después de la dilución |
El tiempo de almacenamiento combinado de la solución reconstituida de ENTYVIO en el vial y la solución diluida en la bolsa de infusión con Inyección de Cloruro de Sodio al 0.9%, es un total de 12 horas a temperatura ambiente (20°C a 25°C [68°F a 77°F]) o 24 horas refrigerado (2°C a 8°C [36°F a 46°F]). Este tiempo de almacenamiento combinado puede incluir hasta ocho horas de la solución reconstituida en el vial a 2°C a 8°C.
El tiempo de almacenamiento combinado de la solución reconstituida de ENTYVIO en el vial y la solución diluida en la bolsa de infusión con Inyección de Ringer Lactato, es un total de seis horas refrigerado (2°C a 8°C [36°F a 46°F]).
2.4 Instrucciones de preparación y administración para la inyección subcutánea
- Inspeccione la solución visualmente para detectar partículas y decoloración antes de la administración. ENTYVIO en jeringa precargada o ENTYVIO PEN debe ser una solución clara a moderadamente opalescente, incolora a ligeramente amarilla. No use jeringas precargadas de ENTYVIO o ENTYVIO PEN con partículas visibles o decoloración.
- Administre cada inyección subcutánea en una ubicación anatómica diferente (como los muslos, cualquier cuadrante del abdomen o la parte superior de los brazos) que la inyección anterior. La administración de ENTYVIO en la parte posterior de la parte superior del brazo solo puede ser realizada por un profesional de la salud o un cuidador. No inyecte en lunares, cicatrices, moretones o áreas donde la piel esté sensible, eritematosa o indurada.
Dosis subcutánea omitida
Si el tratamiento con ENTYVIO subcutáneo se interrumpe o si se omite una dosis programada(s) de ENTYVIO subcutáneo, inyecte la siguiente dosis subcutánea lo antes posible y luego cada 2 semanas a partir de entonces.
En caso de administración incompleta de la dosis (es decir, el paciente intenta administrar la dosis con ENTYVIO PEN, sin embargo, no está seguro si se administró una dosis completa), indique al paciente que llame a su farmacia o proveedor de atención médica.
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
Infusion intravenosa
- Para inyección: 300 mg de vedolizumab como un pastel liofilizado blanco a blanquecino en un vial de dosis única para reconstitución.
Inyección subcutánea
- Inyección: 108 mg/0.68 mL de vedolizumab como una solución transparente a moderadamente opalescente, incolora a ligeramente amarilla en una jeringa precargada de dosis única con dispositivo de seguridad para la aguja.
- Inyección: 108 mg/0.68 mL de vedolizumab como una solución transparente a moderadamente opalescente, incolora a ligeramente amarilla en un bolígrafo precargado de dosis única (ENTYVIO PEN).
4 CONTRAINDICACIONES
ENTYVIO está contraindicado en pacientes que han tenido una reacción de hipersensibilidad grave o severa conocida a ENTYVIO o a cualquiera de sus excipientes (como disnea, broncoespasmo, urticaria, rubefacción, erupción cutánea y aumento de la frecuencia cardíaca) [consulte Advertencias y precauciones (5.1)].
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Reacciones relacionadas con la infusión y reacciones de hipersensibilidad
Se han notificado reacciones relacionadas con la infusión y reacciones de hipersensibilidad, incluida la anafilaxia, disnea, broncoespasmo, urticaria, rubor, erupción cutánea y aumento de la presión arterial y la frecuencia cardíaca [ver Reacciones adversas (6.1, 6.2)]. Estas reacciones pueden ocurrir con la primera o las siguientes infusiones de ENTYVIO y pueden variar en su tiempo de inicio desde durante la infusión o hasta varias horas después de la infusión.
Si se produce anafilaxia u otras reacciones graves relacionadas con la infusión o de hipersensibilidad, suspenda la administración de ENTYVIO inmediatamente e inicie el tratamiento adecuado.
5.2 Infecciones
Los pacientes tratados con ENTYVIO tienen un mayor riesgo de desarrollar infecciones [ver Reacciones adversas (6.1)]. Las infecciones más comúnmente notificadas en los ensayos clínicos que ocurrieron a una tasa mayor con ENTYVIO que con placebo involucraron la mucosa respiratoria superior y nasal (por ejemplo, nasofaringitis, infección del tracto respiratorio superior). También se han notificado infecciones graves en pacientes tratados con ENTYVIO, incluido el absceso anal, la sepsis (algunas fatales), la tuberculosis, la sepsis por salmonella, la meningitis por Listeria, la giardiasis y la colitis por citomegalovirus.
ENTYVIO no se recomienda en pacientes con infecciones activas graves hasta que las infecciones estén controladas. Considere suspender el tratamiento en pacientes que desarrollen una infección grave mientras están en tratamiento con ENTYVIO. Tenga precaución al considerar el uso de ENTYVIO en pacientes con antecedentes de infecciones graves recurrentes. Considere la posibilidad de realizar una prueba de detección de tuberculosis (TB) de acuerdo con la práctica local. Para la leucoencefalopatía multifocal progresiva (PML), [ver Advertencias y precauciones (5.3)].
5.3 Leucoencefalopatía multifocal progresiva
La PML, una infección oportunista rara y a menudo fatal del sistema nervioso central (SNC), se ha notificado con inmunosupresores sistémicos, incluido otro antagonista del receptor de integrina. La PML es causada por el virus de John Cunningham (JC) y generalmente solo ocurre en pacientes que están inmunocomprometidos. Se ha notificado un caso de PML en un paciente tratado con ENTYVIO con múltiples factores contribuyentes en el entorno de postcomercialización (por ejemplo, infección por el virus de la inmunodeficiencia humana [VIH] con un recuento de CD4 de 300 células/mm3 y inmunosupresión previa y concomitante). Aunque es poco probable, no se puede descartar un riesgo de PML.
Controle a los pacientes que reciben ENTYVIO para detectar cualquier aparición nueva o empeoramiento de los signos y síntomas neurológicos. Los signos y síntomas típicos asociados con la PML son diversos, progresan durante días o semanas e incluyen debilidad progresiva en un lado del cuerpo o torpeza de las extremidades, alteración de la visión y cambios en el pensamiento, la memoria y la orientación que conducen a confusión y cambios de personalidad. La progresión de los déficits generalmente conduce a la muerte o discapacidad grave durante semanas o meses. Si se sospecha PML, suspenda la dosificación con ENTYVIO y consulte a un neurólogo; si se confirma, suspenda la dosificación de forma permanente.
5.4 Lesión hepática
Ha habido informes de elevaciones de transaminasas y/o bilirrubina en pacientes que reciben ENTYVIO. En general, la combinación de elevaciones de transaminasas y bilirrubina elevada sin evidencia de obstrucción generalmente se reconoce como un predictor importante de lesión hepática grave que puede conducir a la muerte o la necesidad de un trasplante de hígado en algunos pacientes. ENTYVIO debe suspenderse en pacientes con ictericia u otra evidencia de lesión hepática significativa [ver Reacciones adversas (6.1)].
5.5 Vacunas vivas y orales
Antes de iniciar el tratamiento con ENTYVIO, todos los pacientes deben ponerse al día con todas las inmunizaciones de acuerdo con las pautas de inmunización actuales [ver Dosificación y administración (2.1)]. Los pacientes que reciben ENTYVIO pueden recibir vacunas no vivas (por ejemplo, inyección de vacuna contra la influenza) y pueden recibir vacunas vivas si los beneficios superan los riesgos. No hay datos sobre la transmisión secundaria de la infección por vacunas vivas en pacientes que reciben ENTYVIO [ver Reacciones adversas (6.1)].
6 REACCIONES ADVERSAS
Los siguientes temas también se discuten en detalle en la sección de Advertencias y precauciones:
- Reacciones relacionadas con la infusión y reacciones de hipersensibilidad [ver Advertencias y precauciones (5.1)]
- Infecciones [ver Advertencias y precauciones (5.2)]
- Leucoencefalopatía multifocal progresiva [ver Advertencias y precauciones (5.3)]
- Lesión hepática [ver Advertencias y precauciones (5.4)]
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se llevan a cabo en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no se pueden comparar directamente con las tasas en los ensayos clínicos de otro fármaco y pueden no reflejar las tasas observadas en la práctica.
Los datos descritos a continuación reflejan la exposición a ENTYVIO intravenoso en 3.326 pacientes y voluntarios sanos en ensayos clínicos, incluidos 1.396 expuestos durante más de un año y 835 expuestos durante más de dos años.
Infusión intravenosa
Los datos de seguridad descritos en Tabla 2 se derivan de cuatro ensayos controlados de fase 3 (Ensayos UC I y II y Ensayos CD I y III); se incluyen datos de pacientes adultos que recibieron tratamiento con ENTYVIO intravenoso de etiqueta abierta en las semanas 0 y 2 (antes de ingresar al Ensayo UC II y al Ensayo CD III) y de las semanas 6 a 52 (no respondedores en la semana 6 del Ensayo UC I y el Ensayo CD I) [ver Estudios clínicos (14.1, 14.2)].
En estos ensayos, 1.434 pacientes recibieron ENTYVIO 300 mg por vía intravenosa durante un máximo de 52 semanas y 297 pacientes recibieron placebo durante un máximo de 52 semanas. De estos, 769 pacientes tenían colitis ulcerosa y 962 pacientes tenían enfermedad de Crohn. Los pacientes estuvieron expuestos durante una duración media de 259 días (Ensayos UC I y II) y 247 días (Ensayos CD I y III).
Se informaron reacciones adversas en el 52% de los pacientes tratados con ENTYVIO intravenoso y en el 45% de los pacientes tratados con placebo (Ensayos UC I y II: 49% con ENTYVIO y 37% con placebo; Ensayos CD I y III: 55% con ENTYVIO y 47% con placebo). Se informaron reacciones adversas graves en el 7% de los pacientes tratados con ENTYVIO intravenoso en comparación con el 4% de los pacientes tratados con placebo (Ensayos UC I y II: 8% con ENTYVIO y 7% con placebo; Ensayos CD I y III: 12% con ENTYVIO y 9% con placebo).
Las reacciones adversas más comunes (informadas por ≥3% de los pacientes tratados con ENTYVIO intravenoso en el grupo combinado de Ensayos UC I y II y Ensayos CD I y III y ≥1% más altas que en el grupo combinado de placebo) fueron nasofaringitis, dolor de cabeza, artralgia, náuseas, pirexia, infección del tracto respiratorio superior, fatiga, tos, bronquitis, influenza, dolor de espalda, erupción cutánea, prurito, sinusitis, dolor orofaríngeo y dolor en las extremidades (Tabla 2).
| Reacción adversa | ENTYVIO IV† (N=1434) |
Placebo‡ (N=297) |
|---|---|---|
|
||
| Nasofaringitis | 13% | 7% |
| Dolor de cabeza | 12% | 11% |
| Artralgia | 12% | 10% |
| Náuseas | 9% | 8% |
| Pirexia | 9% | 7% |
| Infección del tracto respiratorio superior | 7% | 6% |
| Fatiga | 6% | 3% |
| Tos | 5% | 3% |
| Bronquitis | 4% | 3% |
| Influenza | 4% | 2% |
| Dolor de espalda | 4% | 3% |
| Erupción cutánea | 3% | 2% |
| Prurito | 3% | 1% |
| Sinusitis | 3% | 1% |
| Dolor orofaríngeo | 3% | 1% |
| Dolor en las extremidades | 3% | 1% |
Los datos de seguridad para los pacientes (n=279) en los ensayos I y II de UC y los ensayos I y III de CD que recibieron ENTYVIO intravenoso en las semanas 0 y 2 y luego fueron asignados aleatoriamente a placebo en la semana 6 durante un máximo de 52 semanas, y para los pacientes (n=416) en el ensayo II de CD, un ensayo de 10 semanas de enfermedad de Crohn, son similares a los que se enumeran en Tabla 2.
Reacciones relacionadas con la infusión y reacciones de hipersensibilidad
Se han notificado reacciones graves relacionadas con la infusión y reacciones de hipersensibilidad, incluida la anafilaxia, después de la administración intravenosa de ENTYVIO en ensayos clínicos [ver Advertencias y precauciones (5.1)]. En los ensayos I y II de UC y los ensayos I y III de CD, se notificó un caso de anafilaxia [uno de 1.434 pacientes tratados con ENTYVIO intravenoso (0,07 %)] por un paciente con enfermedad de Crohn durante la segunda infusión (los síntomas notificados fueron disnea, broncoespasmo, urticaria, rubor, erupción cutánea y aumento de la presión arterial y la frecuencia cardíaca) y se controló con la interrupción de la infusión y el tratamiento con antihistamínicos e hidrocortisona intravenosa.
En los ensayos I y II de UC y los ensayos I y III de CD, el 4 % de los pacientes tratados con ENTYVIO intravenoso y el 3 % de los pacientes tratados con placebo experimentaron una reacción relacionada con la infusión (IRR). Las IRR más frecuentes observadas en los pacientes tratados con ENTYVIO intravenoso (notificadas más de dos veces) fueron náuseas, dolor de cabeza, prurito, mareos, fatiga, reacción relacionada con la infusión, pirexia, urticaria y vómitos (cada una de estas reacciones adversas ocurrió en <1 % en todos los pacientes tratados con ENTYVIO intravenoso) y ninguna reacción adversa individual notificada ocurrió a una tasa superior al 1 %. Estas reacciones generalmente ocurrieron dentro de las primeras dos horas después de la infusión y se resolvieron sin tratamiento o después del tratamiento con antihistamínicos y/o hidrocortisona IV. Menos del 1 % de los pacientes tratados con ENTYVIO intravenoso tuvieron IRR evaluadas por el investigador como graves, y las IRR que requirieron la interrupción del tratamiento del estudio ocurrieron en <1 %.
En los ensayos clínicos, para los pacientes con IRR leves o reacciones de hipersensibilidad, se les permitió a los médicos pretratar con tratamiento médico estándar (p. ej., antihistamínicos, hidrocortisona y/o acetaminofén) antes de la próxima infusión.
Infecciones
En los ensayos I y II de UC y los ensayos I y III de CD, la tasa de infecciones fue de 0,85 por paciente-año en los pacientes tratados con ENTYVIO intravenoso y de 0,7 por paciente-año en los pacientes tratados con placebo [ver Advertencias y precauciones (5.2)]. Las infecciones consistieron principalmente en nasofaringitis, infección del tracto respiratorio superior, sinusitis e infección del tracto urinario. El dos por ciento de los pacientes interrumpieron ENTYVIO intravenoso debido a infecciones.
En los ensayos I y II de UC y los ensayos I y III de CD, la tasa de infecciones graves fue de 0,07 por paciente-año en los pacientes tratados con ENTYVIO intravenoso y de 0,06 por paciente-año en los pacientes tratados con placebo. Las infecciones graves fueron más comunes en pacientes con enfermedad de Crohn que en pacientes con colitis ulcerosa, y los abscesos anales fueron la reacción adversa grave más frecuentemente notificada en pacientes con enfermedad de Crohn. Durante más de 48 meses, no hubo un aumento en la tasa de infecciones graves.
En ensayos de extensión a largo plazo controlados y de etiqueta abierta en adultos tratados con ENTYVIO intravenoso, se han notificado infecciones graves, incluido el absceso anal, la sepsis (algunas fatales), la tuberculosis, la sepsis por salmonella, la meningitis por Listeria, la giardiasis y la colitis por citomegalovirus.
En los ensayos I y II de UC y los ensayos I y III de CD, se notificó sepsis, incluida la sepsis bacteriana y el shock séptico, en cuatro de 1.434 (0,3 %) pacientes tratados con ENTYVIO intravenoso y en dos de 297 pacientes tratados con placebo (0,7 %). Durante estos ensayos, dos pacientes con enfermedad de Crohn tratados con ENTYVIO intravenoso murieron debido a sepsis o shock séptico notificados; ambos pacientes tenían comorbilidades significativas y un curso hospitalario complicado que contribuyó a las muertes. En un ensayo de extensión a largo plazo de etiqueta abierta, se notificaron casos adicionales de sepsis (algunas fatales), incluida la sepsis bacteriana y el shock séptico. La tasa de sepsis en pacientes con colitis ulcerosa o enfermedad de Crohn que recibieron ENTYVIO intravenoso fue de dos por 1.000 paciente-años.
En los ensayos clínicos, todos los pacientes fueron examinados para detectar tuberculosis. Se diagnosticó un caso de tuberculosis pulmonar latente durante los ensayos controlados con ENTYVIO intravenoso. Se diagnosticaron casos adicionales de tuberculosis pulmonar durante el ensayo de etiqueta abierta. Todos estos casos observados ocurrieron fuera de los Estados Unidos (EE. UU.) y ninguno de los pacientes tuvo manifestaciones extrapulmonares.
Lesión hepática
Ha habido informes de elevaciones de transaminasas y/o bilirrubina en pacientes que reciben ENTYVIO intravenoso [ver Advertencias y precauciones (5.4)]. En los ensayos I y II de UC y los ensayos I y III de CD, tres pacientes notificaron reacciones adversas graves de hepatitis, que se manifestaron como transaminasas elevadas con o sin bilirrubina elevada y síntomas compatibles con hepatitis (p. ej., malestar general, náuseas, vómitos, dolor abdominal, anorexia). Estas reacciones adversas ocurrieron después de dos a cinco dosis intravenosas de ENTYVIO; sin embargo, según la información del informe de casos, no está claro si las reacciones indicaron una etiología inducida por fármacos o autoinmune. Todos los pacientes se recuperaron después de la interrupción de la terapia, algunos requirieron tratamiento con corticosteroides. En los ensayos controlados, la incidencia de elevaciones de ALT y AST ≥3× ULN fue <2 % en los pacientes tratados con ENTYVIO intravenoso y en los pacientes tratados con placebo. En el ensayo de etiqueta abierta, se observó un caso adicional de hepatitis grave.
Neoplasias
En los ensayos I y II de UC y los ensayos I y III de CD, se notificaron neoplasias (excluyendo displasia y carcinoma de células basales) en seis de 1.434 (0,4 %) pacientes tratados con ENTYVIO intravenoso, incluido el cáncer de colon (n=2), el carcinoma de células transicionales (n=1), el cáncer de mama (n=1), el tumor carcinoide del apéndice (n=1) y el carcinoma de células escamosas (n=1). Se notificó una neoplasia en uno de 297 (0,3 %) pacientes tratados con placebo (carcinoma de células escamosas).
Las neoplasias (excluyendo displasia y carcinoma de células basales) observadas durante el ensayo de extensión a largo plazo de etiqueta abierta en curso incluyeron linfoma de células B, cáncer de mama, cáncer de colon, neoplasia hepática maligna, neoplasia pulmonar maligna, melanoma maligno, cáncer de pulmón de carcinoma neuroendocrino primario, cáncer renal y carcinoma de células escamosas. En general, el número de neoplasias en los ensayos clínicos fue pequeño; sin embargo, la exposición a largo plazo fue limitada.
Inyección subcutánea después de dos dosis intravenosas de ENTYVIO
ENTYVIO se administró como una inyección subcutánea en pacientes adultos con colitis ulcerosa y enfermedad de Crohn en ensayos clínicos doble ciego controlados con placebo (Ensayo SC UC y Ensayo SC CD, respectivamente). Los pacientes que lograron una respuesta clínica después de dos dosis de ENTYVIO administradas como una infusión intravenosa en la semana 0 y la semana 2 fueron asignados aleatoriamente 2:1 en la semana 6 a ENTYVIO como una inyección subcutánea (N=106) o placebo (N=56) (Ensayo SC UC) y como una inyección subcutánea (N=275) o placebo (N=134) (Ensayo SC CD) [ver Estudios clínicos (14.1, 14.2)].
El perfil de seguridad durante un máximo de 52 semanas de tratamiento total fue similar entre los pacientes que cambiaron a ENTYVIO como una inyección subcutánea en los ensayos clínicos SC UC y SC CD y los pacientes en los ensayos clínicos UC y CD que recibieron ENTYVIO como una infusión intravenosa (Tabla 2) excepto por las reacciones en el sitio de inyección, que se informaron con ENTYVIO subcutáneo. Las reacciones en el sitio de inyección con ENTYVIO subcutáneo se informaron en el 10% (11/106) de los pacientes en el Ensayo SC UC, incluyendo eritema en el sitio de inyección, erupción cutánea, prurito, hinchazón, hematomas y hematoma. Las reacciones en el sitio de inyección con ENTYVIO subcutáneo se informaron en el 3% (8/275) de los pacientes en el Ensayo SC CD, incluyendo eritema en el sitio de inyección, prurito, urticaria, dolor, erupción cutánea y edema.
Vacunas vivas y orales
No hay datos sobre la transmisión secundaria de la infección por vacunas vivas en pacientes que reciben ENTYVIO.
En un estudio controlado con placebo de voluntarios sanos, 61 sujetos recibieron una sola dosis intravenosa de ENTYVIO 750 mg (2,5 veces la dosis recomendada), y 62 sujetos recibieron placebo seguido de vacunación intramuscular con antígeno de superficie de la hepatitis B y vacuna oral contra el cólera. Después de la vacunación intramuscular con tres dosis de antígeno de superficie de la hepatitis B recombinante, aquellos tratados con ENTYVIO intravenoso no tuvieron tasas más bajas de inmunidad protectora contra el virus de la hepatitis B. Sin embargo, aquellos expuestos a ENTYVIO intravenoso tuvieron tasas de seroconversión y títulos de anticuerpos contra el cólera más bajos en relación con el placebo después de recibir las dos dosis de una vacuna oral contra el cólera inactivada. El impacto en otras vacunas orales y en las vacunas nasales en pacientes es desconocido.
6.2 Experiencia postcomercialización
Las siguientes reacciones adversas se han identificado durante el uso posterior a la aprobación de ENTYVIO. Debido a que estas reacciones se informan voluntariamente de una población de tamaño incierto, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal con la exposición al fármaco.
Trastornos del sistema inmunitario: Anafilaxia [ver Advertencias y precauciones (5.1)].
Trastornos del sistema gastrointestinal: Pancreatitis aguda.
Trastornos respiratorios, torácicos y mediastínicos: Enfermedad pulmonar intersticial, neumonitis.
7 INTERACCIONES MEDICAMENTOSAS
7.1 Productos de Natalizumab
Debido al potencial de riesgo aumentado de PML y otras infecciones, evite el uso concomitante de ENTYVIO con productos de natalizumab.
7.2 Bloqueadores de TNF
Debido al potencial de riesgo aumentado de infecciones, evite el uso concomitante de ENTYVIO con bloqueadores de TNF.
7.3 Sustratos de CYP450
La formación de enzimas CYP450 puede ser suprimida por niveles aumentados de ciertas citoquinas (por ejemplo, IL-6, IL-10, TNFα, IFN) durante la inflamación crónica. Por lo tanto, el uso de ENTYVIO puede normalizar la formación de enzimas CYP450 al modular la enfermedad subyacente. Al iniciar o suspender ENTYVIO en pacientes tratados con sustratos de CYP450, controle las concentraciones del fármaco u otros parámetros terapéuticos, y ajuste la dosis del sustrato de CYP según sea necesario. Consulte la información de prescripción de sustratos de CYP específicos.
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de Riesgos
Los datos disponibles del Registro de Embarazo de ENTYVIO de la Organización de Especialistas en Información de Teratología (OTIS)/MotherToBaby, la literatura publicada y la farmacovigilancia en mujeres embarazadas no han identificado de manera confiable un riesgo asociado a ENTYVIO de defectos de nacimiento mayores, aborto espontáneo o resultados adversos maternos o fetales (ver Datos). Existen riesgos para la madre y el feto asociados con la enfermedad inflamatoria intestinal durante el embarazo (ver Consideraciones Clínicas).
No se observó daño fetal en estudios de reproducción animal con administración intravenosa de vedolizumab a conejos y monos a niveles de dosis 20 veces la dosis humana recomendada (ver Datos).
El riesgo de fondo de defectos de nacimiento mayores y aborto espontáneo para las poblaciones indicadas es desconocido. Todos los embarazos tienen un riesgo de fondo de defectos de nacimiento, pérdida u otros resultados adversos. En la población general de los EE. UU., el riesgo de fondo estimado de defectos de nacimiento mayores y aborto espontáneo en embarazos clínicamente reconocidos es del 2 al 4% y el aborto espontáneo es del 15 al 20%, respectivamente.
Consideraciones Clínicas
Riesgo Materno y Embrión/Fetal Asociado a la Enfermedad
Los datos publicados sugieren que el riesgo de resultados adversos del embarazo en mujeres con enfermedad inflamatoria intestinal (EII) está asociado con un aumento de la actividad de la enfermedad. Los resultados adversos del embarazo incluyen parto prematuro (antes de las 37 semanas de gestación), bebés con bajo peso al nacer (menos de 2.500 g) y pequeños para la edad gestacional al nacer.
Reacciones Adversas Fetales/Neonatales
ENTYVIO administrado durante el embarazo podría afectar las respuestas inmunitarias en el recién nacido y el lactante expuestos en el útero. La importancia clínica de los bajos niveles de ENTYVIO en los lactantes expuestos en el útero es desconocida. La seguridad de administrar vacunas vivas o atenuadas en lactantes expuestos es desconocida.
Datos
Datos Humanos
El registro de exposición al embarazo de vedolizumab realizado por el estudio OTIS/MotherToBaby en los Estados Unidos y Canadá recopiló datos observacionales prospectivos entre 2015 y 2022 para evaluar el riesgo de defectos de nacimiento mayores en bebés nacidos vivos de mujeres con colitis ulcerosa (CU) o enfermedad de Crohn (EC) tratadas con vedolizumab durante el embarazo. El estudio comparó pacientes embarazadas con CU o EC expuestas a vedolizumab con pacientes embarazadas con CU o EC tratadas con otros productos biológicos. El registro incluyó 99 mujeres (58 con CU, 41 con EC) tratadas con vedolizumab durante el embarazo y 76 mujeres (27 con CU, 49 con EC) expuestas a otros productos biológicos durante el embarazo.
La proporción de defectos de nacimiento mayores entre los bebés nacidos vivos en pacientes con CU o EC tratadas con vedolizumab y pacientes con CU o EC tratadas con otros productos biológicos fue del 7,4% (7/94) y del 5,6% (4/71), respectivamente. En general, no hubo evidencia de un mayor riesgo de defectos de nacimiento estructurales mayores (RR ajustado 1,07, IC del 95%: 0,33, 3,52).
Las limitaciones metodológicas del registro, incluido el pequeño tamaño de la muestra y el diseño no aleatorizado, dieron como resultado una capacidad limitada para estimar el riesgo de defectos de nacimiento mayores y otros resultados maternos e infantiles. Las conclusiones del registro de embarazo fueron consistentes con la literatura publicada y la farmacovigilancia.
Datos Animales
Se ha realizado un estudio de reproducción en conejas embarazadas a dosis intravenosas únicas de hasta 100 mg/kg administradas el día 7 de gestación (aproximadamente 20 veces la dosis humana recomendada) y no ha revelado evidencia de deterioro de la fertilidad o daño al feto debido a vedolizumab. Un estudio de desarrollo pre y postnatal en monos no mostró evidencia de ningún efecto adverso en el desarrollo pre y postnatal a dosis intravenosas de hasta 100 mg/kg (aproximadamente 20 veces la dosis humana recomendada).
8.2 Lactancia
Resumen de Riesgos
Los datos de un estudio clínico de lactancia muestran la presencia de vedolizumab en la leche materna. La dosis diaria media calculada para el lactante fue de 0,02 mg/kg/día por vía oral (ver Datos). Se espera que la exposición sistémica en un lactante amamantado sea baja porque los anticuerpos monoclonales se degradan en gran medida en el tracto gastrointestinal. No hay datos sobre los efectos de vedolizumab en el lactante amamantado, o los efectos sobre la producción de leche. Los beneficios para el desarrollo y la salud de la lactancia materna deben considerarse junto con la necesidad clínica de la madre de ENTYVIO y cualquier posible efecto adverso en el lactante amamantado de ENTYVIO o de la condición materna subyacente.
Datos
Se realizó un estudio de lactancia con leche materna en 9 mujeres adultas lactantes que estaban siendo tratadas por colitis ulcerosa activa o enfermedad de Crohn con ENTYVIO intravenoso cada 8 semanas después de alcanzar el estado estable y completar la fase de inducción (administración de ENTYVIO a las 0, 2 y 6 semanas). Las concentraciones medias de ENTYVIO en la leche materna oscilaron entre 0,03 y 0,26 mcg/mL. La dosis diaria media calculada para el lactante por vía oral fue de 0,02 mg/kg/día calculada como producto de la concentración media durante el intervalo de dosificación de 8 semanas y el consumo estandarizado de leche de 150 mL/kg/día.
8.4 Uso Pediátrico
La seguridad y eficacia de ENTYVIO en pacientes pediátricos no se han establecido.
8.5 Uso Geriátrico
Los ensayos clínicos de ENTYVIO no incluyeron un número suficiente de pacientes de 65 años o más (72 pacientes con enfermedad de Crohn o colitis ulcerosa de 65 años o más fueron tratados con ENTYVIO durante los ensayos controlados de fase 3) para determinar si responden de manera diferente a los pacientes adultos más jóvenes. Sin embargo, no se observaron diferencias generales en la seguridad o eficacia entre estos pacientes y los pacientes adultos más jóvenes, y otras experiencias clínicas informadas no han identificado diferencias en las respuestas entre los pacientes ancianos y los pacientes más jóvenes.
11 DESCRIPCIÓN
Vedolizumab, un antagonista de los receptores de integrinas, es un anticuerpo monoclonal IgG1 humanizado producido en células ovarias de hámster chino que se une a la integrina humana α4β7. ENTYVIO tiene un peso molecular aproximado de 147 kilodaltons.
ENTYVIO intravenoso
ENTYVIO (vedolizumab) para inyección se suministra como un pastel liofilizado estéril, blanco a casi blanco, sin conservantes, para infusión intravenosa. Tras la reconstitución con 4,8 mL de Agua Estéril para Inyección, USP, Inyección de Cloruro de Sodio al 0,9%, USP, o Inyección de Ringer Lactato, USP, la concentración resultante es de 60 mg/mL con un volumen suministrable de 5 mL (300 mg) y el pH resultante es aproximadamente 6,3.
Cada vial de dosis única contiene 300 mg de vedolizumab, hidrocloruro de arginina (131,7 mg), histidina (23 mg), monohidrocloruro de histidina (21,4 mg), polisorbato 80 (3 mg) y sacarosa (500 mg).
ENTYVIO subcutáneo
ENTYVIO (vedolizumab) inyección se suministra como una solución estéril, clara a moderadamente opalescente, incolora a ligeramente amarilla, sin conservantes, para administración subcutánea.
Cada jeringa precargada de dosis única o pluma precargada de dosis única (ENTYVIO PEN) contiene 108 mg de vedolizumab, hidrocloruro de arginina (17,77 mg), ácido cítrico monohidratado (0,18 mg), histidina (3,86 mg), monohidrocloruro de histidina (1,86 mg), polisorbato 80 (1,35 mg), citrato sódico dihidrato (4,71 mg) y Agua Estéril para Inyección, USP, a un pH de 6,5.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
Vedolizumab es un anticuerpo monoclonal humanizado que se une específicamente a la integrina α4β7 y bloquea la interacción de la integrina α4β7 con la molécula de adhesión celular de dirección mucosa-1 (MAdCAM-1) e inhibe la migración de los linfocitos T de memoria a través del endotelio hacia el tejido parenquimatoso gastrointestinal inflamado. Vedolizumab no se une ni inhibe la función de las integrinas α4β1 y αEβ7 y no antagoniza la interacción de las integrinas α4 con la molécula de adhesión celular vascular-1 (VCAM-1).
La integrina α4β7 se expresa en la superficie de un subconjunto discreto de linfocitos T de memoria que migran preferentemente hacia el tracto gastrointestinal. MAdCAM-1 se expresa principalmente en las células endoteliales del intestino y juega un papel fundamental en la localización de los linfocitos T al tejido linfático del intestino. La interacción de la integrina α4β7 con MAdCAM-1 se ha implicado como un importante contribuyente a la inflamación crónica que es una característica de la colitis ulcerosa y la enfermedad de Crohn.
12.2 Farmacodinamia
En ensayos clínicos con ENTYVIO intravenoso en dosis que van de 0,2 a 10 mg/kg (que incluyen dosis fuera de la dosis recomendada), se observó la saturación de los receptores α4β7 en subconjuntos de linfocitos circulantes involucrados en la vigilancia inmunitaria del intestino.
En ensayos clínicos con ENTYVIO intravenoso en dosis que van de 0,2 a 10 mg/kg y de 180 a 750 mg (que incluyen dosis fuera de la dosis recomendada) en sujetos sanos y en pacientes con colitis ulcerosa o enfermedad de Crohn, vedolizumab no elevó los neutrófilos, basófilos, eosinófilos, linfocitos T auxiliares y citotóxicos, linfocitos T auxiliares de memoria totales, monocitos o células asesinas naturales.
Se observó una reducción de la inflamación gastrointestinal en las muestras de biopsia rectal de pacientes con colitis ulcerosa de fase 2 expuestos a ENTYVIO durante cuatro o seis semanas en comparación con el control con placebo, según la evaluación histopatológica.
En un estudio de 14 sujetos sanos, ENTYVIO no afectó los recuentos de células linfocíticas CD4+, los recuentos de células linfocíticas CD8+ o las razones CD4+:CD8+ en el LCR [ver Farmacología clínica (12.3)].
12.3 Farmacocinética
Se observaron farmacocinéticas similares en pacientes con colitis ulcerosa y enfermedad de Crohn que recibieron 300 mg de ENTYVIO como una infusión intravenosa de 30 minutos en las semanas 0, 2 y 6, y luego cada ocho semanas hasta la semana 52 (Tabla 3).
| Población de pacientes | Semanas 0, 2 y 6 ENTYVIO 300 mg intravenoso | Después de la semana 6 a la 52 ENTYVIO 300 mg intravenoso cada 8 semanas |
|---|---|---|
| Concentración sérica en valle en la semana 6 (mcg/mL) | Concentración sérica en valle en la semana 46† (mcg/mL) |
|
| Colitis ulcerosa | 26,3 ± 12,9 (N=210) |
11,2 ± 7,2 (N=77) |
| Enfermedad de Crohn | 27,4 ± 19,2 (N=198) |
13,0 ± 9,1 (N=72) |
En pacientes con colitis ulcerosa y enfermedad de Crohn, que recibieron 300 mg de ENTYVIO como infusión intravenosa de 30 minutos en las semanas 0 y 2, seguido de 108 mg de ENTYVIO como inyección subcutánea cada 2 semanas a partir de la semana 6, las concentraciones séricas mínimas promedio en estado estacionario fueron 35.8 mcg/mL (DE ± 15.2) y 31.4 mcg/mL (DE ± 14.7), respectivamente.
La biodisponibilidad de vedolizumab después de una inyección subcutánea de dosis única de 108 mg en relación con una infusión intravenosa de dosis única de 300 mg en sujetos sanos fue aproximadamente del 75%. Después de una inyección subcutánea de dosis única de 108 mg en sujetos sanos, la Tmax mediana fue de 7 días con un rango de 3 a 14 días y la Cmax media fue de 15.4 mcg/mL (DE ± 3.2).
El aclaramiento de vedolizumab depende de vías lineales y no lineales; el aclaramiento no lineal disminuye con el aumento de las concentraciones. Los análisis farmacocinéticos poblacionales indicaron que el aclaramiento lineal fue de aproximadamente 0.16 L/día, la vida media sérica fue de aproximadamente 26 días y el volumen de distribución fue de aproximadamente 5 L.
Vedolizumab no se detectó en el líquido cefalorraquídeo (LCR) de 14 sujetos sanos a las cinco semanas después de una administración intravenosa única de 450 mg de ENTYVIO (1.5 veces la dosis recomendada).
Poblaciones específicas
El análisis farmacocinético poblacional mostró que la gravedad del estado de la enfermedad, el peso corporal, el tratamiento previo con terapia con bloqueador del TNF, la edad (18 a 78 años), la albúmina sérica, los inmunomoduladores coadministrados (incluida la azatioprina, 6-mercaptopurina, metotrexato) y los aminosalicilatos coadministrados no tuvieron un efecto clínicamente significativo en la farmacocinética de ENTYVIO.
La farmacocinética de vedolizumab en pacientes con insuficiencia renal o hepática no se ha estudiado.
12.6 Inmunogenicidad
La incidencia observada de anticuerpos contra el fármaco depende en gran medida de la sensibilidad y especificidad del ensayo. Las diferencias en los métodos de ensayo excluyen comparaciones significativas de la incidencia de anticuerpos contra el fármaco en los estudios descritos a continuación con la incidencia de anticuerpos contra el fármaco en otros estudios, incluidos los de ENTYVIO o de otros productos de vedolizumab.
Adultos tratados con ENTYVIO intravenoso
La incidencia de anticuerpos contra el fármaco a ENTYVIO intravenoso utilizando un método de electroquimioluminiscencia (ECL) tolerante a los fármacos para pacientes en los ensayos I y II de UC y los ensayos I y III de CD que recibieron tratamiento continuo con ENTYVIO administrado como infusión intravenosa durante 52 semanas fue del 6% (86 de 1.427 pacientes tratados con ENTYVIO en total). De los 86 pacientes que dieron positivo para anticuerpos anti-vedolizumab, 20 pacientes fueron persistentemente positivos (en dos o más visitas de estudio consecutivas) y 56 desarrollaron anticuerpos neutralizantes contra vedolizumab.
Entre los pacientes tratados con ENTYVIO que desarrollaron anticuerpos anti-vedolizumab persistentes, 14/20 pacientes tuvieron concentraciones séricas mínimas de vedolizumab que se redujeron marcadamente o fueron indetectables y 15/20 pacientes no lograron la remisión clínica en la semana 52 en los ensayos I y II de UC y los ensayos I y III de CD. Debido a la baja ocurrencia de anticuerpos anti-vedolizumab persistentes (1%; 20/1.427), el efecto de estos anticuerpos en la seguridad y eficacia de ENTYVIO en estos estudios no se ha caracterizado completamente.
Adultos tratados con ENTYVIO subcutáneo
La incidencia de anticuerpos contra el fármaco a ENTYVIO utilizando un método de ECL tolerante a los fármacos para pacientes en el ensayo SC UC y el ensayo SC CD que recibieron tratamiento continuo durante 52 semanas fue del 3.4% (13 de 381 pacientes tratados con ENTYVIO subcutáneo en total). De los 13 pacientes que dieron positivo para anticuerpos anti-vedolizumab, 7 pacientes fueron persistentemente positivos (en dos o más visitas de estudio consecutivas) y 7 pacientes desarrollaron anticuerpos neutralizantes contra vedolizumab. Dos de los 7 pacientes con enfermedad de Crohn y ninguno de los 6 pacientes con colitis ulcerosa que tuvieron anticuerpos anti-vedolizumab positivos lograron la remisión clínica en la semana 52. No hay datos suficientes para evaluar el efecto de los anticuerpos contra el fármaco en la farmacocinética, la eficacia y la seguridad de ENTYVIO en los ensayos SC UC y SC CD.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
No se han realizado estudios a largo plazo en animales para evaluar el potencial carcinogénico de vedolizumab. No se han realizado estudios para evaluar la posible alteración de la fertilidad o el potencial mutagénico de vedolizumab.
14 ESTUDIOS CLÍNICOS
14.1 Estudios Clínicos en Colitis Ulcerosa
Administración Intravenosa
La seguridad y eficacia de ENTYVIO intravenoso se evaluaron en dos ensayos aleatorizados, doble ciego, controlados con placebo (Ensayos UC I y II) en pacientes adultos con colitis ulcerosa (UC) moderada a gravemente activa, definida como una puntuación de Mayo de 6 a 12 con una subpuntuación de endoscopia de dos o tres. La puntuación de Mayo varía de 0 a 12 y tiene cuatro subescalas que se puntúan de cero (normal) a tres (más grave): frecuencia de las heces, sangrado rectal, hallazgos en la endoscopia y evaluación global del médico. Una subpuntuación de endoscopia de dos se define por eritema marcado, falta de patrón vascular, friabilidad y erosiones; una subpuntuación de endoscopia de tres se define por sangrado espontáneo y ulceración.
Los pacientes inscritos en los EE. UU. habían tenido en el período de cinco años anterior una respuesta inadecuada o intolerancia a la terapia inmunomoduladora (es decir, azatioprina o 6-mercaptopurina) y/o una respuesta inadecuada, pérdida de respuesta o intolerancia a un bloqueador del TNF. Fuera de los EE. UU., el tratamiento previo con corticosteroides fue suficiente para la entrada si en el período de cinco años anterior los pacientes fueron dependientes de corticosteroides (es decir, incapaces de reducir con éxito los corticosteroides sin un regreso de los síntomas de la UC) o tuvieron una respuesta inadecuada o intolerancia a los corticosteroides.
Los pacientes que habían recibido natalizumab alguna vez en el pasado, y los pacientes que habían recibido un bloqueador del TNF en los 60 días anteriores fueron excluidos de la inscripción. No se permitió el uso concomitante de natalizumab o un bloqueador del TNF.
Ensayo UC I – Intravenoso
En el Ensayo UC I, 374 pacientes fueron aleatorizados de forma doble ciego (3:2) para recibir ENTYVIO 300 mg o placebo mediante infusión intravenosa en la semana 0 y la semana 2. Las evaluaciones de eficacia se realizaron en la semana 6. Se permitieron dosis estables concomitantes de aminosalicilatos, corticosteroides (dosis de prednisona ≤30 mg/día o equivalente) e inmunomoduladores (azatioprina o 6-mercaptopurina) hasta la semana 6.
En la línea de base, los pacientes recibieron corticosteroides (54%), inmunomoduladores (azatioprina o 6-mercaptopurina) (30%) y/o aminosalicilatos (74%). El 39% de los pacientes tuvieron una respuesta inadecuada, pérdida de respuesta o intolerancia a la terapia con bloqueador del TNF. El 18% de los pacientes tuvieron una respuesta inadecuada, incapacidad para reducir o intolerancia al tratamiento previo con corticosteroides solo (es decir, no habían recibido inmunomoduladores o bloqueadores del TNF previos). La puntuación de Mayo media en la línea de base fue de 9 en el grupo ENTYVIO y de 8 en el grupo placebo.
En el Ensayo UC I, un mayor porcentaje de pacientes tratados con ENTYVIO intravenoso en comparación con los pacientes tratados con placebo lograron una respuesta clínica en la semana 6 (definida en Tabla 4). Un mayor porcentaje de pacientes tratados con ENTYVIO intravenoso en comparación con los pacientes tratados con placebo también lograron una remisión clínica en la semana 6 (definida en Tabla 4). Además, un mayor porcentaje de pacientes tratados con ENTYVIO tuvo una mejora en la apariencia endoscópica de la mucosa en la semana 6 (definida en Tabla 4).
| Criterio de valoración | Placebo N=149 |
ENTYVIO IV N=225 |
p-valor | Diferencia de tratamiento y IC del 95% |
|---|---|---|---|---|
|
||||
| Respuesta clínica* en la semana 6 | 26% | 47% | <0.001 | 22% (12%, 32%) |
| Remisión clínica† en la semana 6 | 5% | 17% | 0.001 | 12% (5%, 18%) |
| Mejora en la apariencia endoscópica de la mucosa‡ en la semana 6 | 25% | 41% | 0.001 | 16% (6%, 26%) |
UC Trial II – Intravenoso
Para ser aleatorizados a un tratamiento en UC Trial II, los pacientes debían haber recibido ENTYVIO intravenoso y estar en respuesta clínica en la semana 6. Los pacientes podrían haber venido de UC Trial I o de un grupo que recibió ENTYVIO de forma abierta.
En UC Trial II, 373 pacientes fueron aleatorizados de forma doble ciego (1:1:1) a uno de los siguientes regímenes a partir de la semana 6: ENTYVIO intravenoso 300 mg cada ocho semanas, ENTYVIO intravenoso 300 mg cada cuatro semanas o placebo cada cuatro semanas. Las evaluaciones de eficacia se realizaron en la semana 52. Se permitieron los aminosalicilatos y los corticosteroides concomitantes hasta la semana 52. Los inmunomoduladores concomitantes (azatioprina o 6-mercaptopurina) se permitieron fuera de los EE. UU., pero no se permitieron más allá de la semana 6 en los EE. UU.
En la semana 6, los pacientes estaban recibiendo corticosteroides (61%), inmunomoduladores (azatioprina o 6-mercaptopurina) (32%) y aminosalicilatos (75%). El 32% de los pacientes tuvieron una respuesta inadecuada, pérdida de respuesta o intolerancia a una terapia con bloqueador del TNF. En la semana 6, la puntuación mediana de Mayo fue de 8 en el grupo de ENTYVIO cada ocho semanas, el grupo de ENTYVIO cada cuatro semanas y el grupo placebo. Los pacientes que habían logrado una respuesta clínica en la semana 6 y estaban recibiendo corticosteroides debían comenzar un régimen de reducción de corticosteroides en la semana 6.
En UC Trial II, un porcentaje mayor de pacientes en los grupos tratados con ENTYVIO intravenoso en comparación con placebo lograron la remisión clínica en la semana 52 y mantuvieron la respuesta clínica (respuesta clínica en las semanas 6 y 52) (Tabla 5). Además, un porcentaje mayor de pacientes en los grupos tratados con ENTYVIO intravenoso en comparación con placebo estuvieron en remisión clínica en las semanas 6 y 52, y tuvieron una mejora del aspecto endoscópico de la mucosa en la semana 52 (Tabla 5). En el subgrupo de pacientes que lograron una respuesta clínica en la semana 6 y estaban recibiendo medicación con corticosteroides en la línea de base, una mayor proporción de pacientes en los grupos tratados con ENTYVIO intravenoso en comparación con placebo suspendieron los corticosteroides y estuvieron en remisión clínica en la semana 52 (Tabla 5).
El régimen de dosificación de ENTYVIO cada cuatro semanas no demostró un beneficio clínico adicional sobre el régimen de dosificación cada ocho semanas. El régimen de dosificación cada cuatro semanas no es el régimen de dosificación recomendado [ver Dosificación y administración (2.2)].
| Criterio de valoración | Placebo† N=126 |
ENTYVIO IV Cada 8 Semanas N=122 |
p-valor | Diferencia de tratamiento y IC del 95% |
|---|---|---|---|---|
|
||||
| Remisión clínica en la semana 52 | 16% | 42% | <0.001 | 26% (15%, 37%) |
| Respuesta clínica en las semanas 6 y 52 | 24% | 57% | <0.001 | 33% (21%, 45%) |
| Mejora del aspecto endoscópico de la mucosa‡ en la semana 52 | 20% | 52% | <0.001 | 32% (20%, 44%) |
| Remisión clínica en las semanas 6 y 52 | 9% | 21% | 0.008 | 12% (3%, 21%) |
| Remisión clínica sin corticosteroides§ | 14%§ | 31%§ | 0.012 | 18% (4%, 31%) |
Administración Subcutánea
Ensayo SC UC – Subcutáneo
La seguridad y eficacia de ENTYVIO subcutáneo se evaluó en un ensayo aleatorizado, doble ciego, controlado con placebo (Ensayo SC UC; NCT02611830) en pacientes adultos con colitis ulcerosa de moderada a gravemente activa, definida como una puntuación de Mayo de 6 a 12 con una subpuntuación de endoscopia de dos o tres. La puntuación de Mayo basal fue de 9 a 12 en aproximadamente el 62% y de 6 a 8 en aproximadamente el 38% de la población total del ensayo.
El ensayo incluyó pacientes que habían experimentado una respuesta inadecuada, pérdida de respuesta o intolerancia a al menos uno de los siguientes: al menos un régimen de 12 semanas de azatioprina o 6-mercaptopurina, inducción con un bloqueador del TNF o corticosteroides. Se permitió a los pacientes usar dosis estables concomitantes de aminosalicilatos orales, corticosteroides orales (prednisona ≤30 mg/día o budesonida ≤9 mg/día), azatioprina o 6-mercaptopurina, probióticos y/o antidiarreicos. Las terapias biológicas concomitantes, el tratamiento rectal con ácido 5-aminosalicílico o enemas/supositorios de corticosteroides estaban prohibidos.
Todos los pacientes recibieron ENTYVIO intravenoso 300 mg en forma abierta en la semana 0 y la semana 2. Para poder ser aleatorizados al tratamiento en el Ensayo SC UC, los pacientes debían tener una respuesta clínica en la semana 6. Un total de 162 pacientes fueron aleatorizados en la semana 6 de forma doble ciego (2:1) a ENTYVIO 108 mg administrado por inyección subcutánea o placebo cada 2 semanas. Las evaluaciones de eficacia se realizaron en la semana 52.
A partir de la semana 6, los pacientes que estaban recibiendo corticosteroides debían comenzar un régimen de reducción de corticosteroides.
En el momento de la aleatorización a la fase doble ciego (semana 6), los pacientes estaban recibiendo corticosteroides (51%), inmunomoduladores (azatioprina o 6-mercaptopurina) (33%) y aminosalicilatos (80%). El 37% de los pacientes habían tenido una respuesta inadecuada, pérdida de respuesta o intolerancia a una terapia con bloqueador del TNF antes de la inscripción.
Los pacientes en la fase doble ciego tenían una edad media de 39 años (rango de 18 a 69 años); el 61% eran hombres; el 83% se identificó como blanco, el 17% como asiático y <1% se identificó como otro grupo racial.
El criterio de valoración principal fue la proporción de pacientes en remisión clínica, definida como una puntuación de Mayo ≤2 puntos y ninguna subpuntuación individual >1 punto en la semana 52. Los criterios de valoración secundarios incluyeron la proporción de pacientes con mejoría del aspecto endoscópico de la mucosa en la semana 52 y la respuesta clínica tanto en la semana 6 como en la semana 52 (ver Tabla 6).
| Criterio de Valoración | Placebo† | ENTYVIO 108 mg SC Cada 2 Semanas‡ | Estimación§ de la Diferencia de Tratamiento vs. Placebo (IC del 95%) |
|---|---|---|---|
|
|||
| Remisión Clínica¶ en la Semana 52 | |||
| Población Total | N=56 14% |
N=106 46% |
32 (20, 45)# |
| Fallo previo del bloqueador del TNF | N=20 10% |
N=40 35% |
|
| Sin fallo previo del bloqueador del TNF | N=36 17% |
N=66 53% |
|
| Mejora del Aspecto Endoscópico de la Mucosa en la Semana 52Þ | |||
| Población Total | N=56 21% |
N=106 57% |
36 (22, 49)# |
| Fallo previo del bloqueador del TNF | N=20 10% |
N=40 48% |
|
| Sin fallo previo del bloqueador del TNF | N=36 28% |
N=66 62% |
|
| Respuesta clínica en las semanas 6 y 52ß | |||
| Población total | N=56 29% |
N=106 64% |
36 (21, 51)# |
| Fallo previo del bloqueador de TNF | N=20 20% |
N=40 68% |
|
| Sin fallo previo del bloqueador de TNF | N=36 33% |
N=66 62% |
|
El mantenimiento de la remisión en la semana 52 en el subgrupo de pacientes que estaban en remisión en la semana 6, fue del 64% (16/25) en el grupo tratado con ENTYVIO en comparación con el 20% (3/15) en el grupo placebo. La diferencia en el tratamiento fue del 44% (IC del 95%: 9%, 69%).
14.2 Estudios clínicos en la enfermedad de Crohn
Administración intravenosa
La seguridad y eficacia de ENTYVIO intravenoso se evaluaron en tres ensayos clínicos aleatorizados, doble ciego, controlados con placebo (Ensayos CD I, II y III) en pacientes adultos con enfermedad de Crohn (CD) moderada a gravemente activa (puntuación del Índice de Actividad de la Enfermedad de Crohn [CDAI] de 220 a 450).
Los pacientes inscritos en los EE. UU. habían tenido en el período de cinco años anterior una respuesta inadecuada o intolerancia a la terapia inmunomoduladora (es decir, azatioprina, 6-mercaptopurina o metotrexato) y/o una respuesta inadecuada, pérdida de respuesta o intolerancia a uno o más bloqueadores del TNF. Fuera de los EE. UU., el tratamiento previo con corticosteroides fue suficiente para la entrada si en el período de cinco años anterior los pacientes fueron dependientes de corticosteroides (es decir, incapaces de reducir con éxito los corticosteroides sin un regreso de los síntomas de la CD) o tuvieron una respuesta inadecuada o intolerancia a los corticosteroides.
Los pacientes que habían recibido natalizumab alguna vez en el pasado y los pacientes que habían recibido un bloqueador del TNF en los últimos 30 a 60 días fueron excluidos de la inscripción. No se permitió el uso concomitante de natalizumab o un bloqueador del TNF.
Ensayo CD I – Intravenoso
En el Ensayo CD I, 368 pacientes fueron aleatorizados de forma doble ciego (3:2) para recibir ENTYVIO 300 mg o placebo mediante infusión intravenosa en la semana 0 y la semana 2. Las evaluaciones de eficacia se realizaron en la semana 6. Se permitieron dosis estables concomitantes de aminosalicilatos, corticosteroides (dosis de prednisona ≤30 mg/día o equivalente) e inmunomoduladores (azatioprina, 6-mercaptopurina o metotrexato) hasta la semana 6.
En la línea de base, los pacientes estaban recibiendo corticosteroides (49%), inmunomoduladores (azatioprina, 6-mercaptopurina o metotrexato) (35%) y/o aminosalicilatos (46%). El cuarenta y ocho por ciento de los pacientes tuvieron una respuesta inadecuada, pérdida de respuesta o intolerancia a la terapia con bloqueador del TNF. El diecisiete por ciento de los pacientes tuvieron una respuesta inadecuada, incapacidad para reducir o intolerancia al tratamiento previo con corticosteroides solo (es decir, no habían recibido inmunomoduladores o bloqueadores del TNF previos). La puntuación media de CDAI en la línea de base fue de 324 en el grupo de ENTYVIO intravenoso y 319 en el grupo placebo.
En el Ensayo CD I, un porcentaje significativamente mayor de pacientes tratados con ENTYVIO intravenoso logró la remisión clínica (definida como CDAI ≤150) en comparación con el placebo en la semana 6 (Tabla 7). Sin embargo, la diferencia en el porcentaje de pacientes que demostraron respuesta clínica (definida como una disminución de ≥100 puntos en la puntuación de CDAI desde la línea de base) no fue estadísticamente significativa en la semana 6.
Ensayo CD II – Intravenoso
En comparación con el Ensayo CD I, el Ensayo CD II inscribió un mayor número de pacientes que en el período de cinco años anterior habían tenido una respuesta inadecuada, pérdida de respuesta o intolerancia a uno o más bloqueadores del TNF (76%); esta fue la población de análisis principal. En el Ensayo CD II, 416 pacientes fueron aleatorizados de forma doble ciego (1:1) para recibir ENTYVIO intravenoso 300 mg o placebo en las semanas 0, 2 y 6. Las evaluaciones de eficacia se realizaron en las semanas 6 y 10. Se permitieron aminosalicilatos, corticosteroides e inmunomoduladores concomitantes (azatioprina, 6-mercaptopurina o metotrexato) hasta la semana 10.
En la línea de base, los pacientes estaban recibiendo corticosteroides (54%), inmunomoduladores (azatioprina, 6-mercaptopurina o metotrexato) (34%) y aminosalicilatos (31%). La puntuación media de CDAI en la línea de base fue de 317 en el grupo ENTYVIO y 301 en el grupo placebo.
Para el criterio de valoración principal (remisión clínica en la semana 6), el tratamiento con ENTYVIO intravenoso no produjo una mejora estadísticamente significativa con respecto al placebo (Tabla 7). Los criterios de valoración secundarios, incluidas las evaluaciones en la semana 10, no se probaron porque el criterio de valoración principal no fue estadísticamente significativo.
| Criterio de valoración | Placebo | ENTYVIO IV | p-valor | Diferencia en el tratamiento y IC del 95% |
|---|---|---|---|---|
|
||||
| Ensayo CD I: Remisión clínica* en la semana 6 |
7% (10/148) |
15% (32/220) |
0.041† | 8% (1%, 14%) |
| Ensayo CD II‡: Remisión clínica* en la semana 6 |
12% (19/157) |
15% (24/158) |
NS§ | 3% (-5%, 11%) |
CD Trial III – Intravenoso
Para ser aleatorizado a tratamiento en CD Trial III, los pacientes debían haber recibido ENTYVIO intravenoso y estar en respuesta clínica (definida como una disminución ≥70 puntos en la puntuación CDAI desde el inicio) en la semana 6. Los pacientes podrían haber venido de CD Trial I o de un grupo que recibió ENTYVIO intravenoso de forma abierta.
En CD Trial III, 461 pacientes fueron aleatorizados de forma doble ciego (1:1:1) a uno de los siguientes regímenes a partir de la semana 6: ENTYVIO intravenoso 300 mg cada ocho semanas, ENTYVIO intravenoso 300 mg cada cuatro semanas o placebo cada cuatro semanas. Las evaluaciones de eficacia se realizaron en la semana 52. Se permitieron aminosalicilatos y corticosteroides concomitantes hasta la semana 52. Se permitieron inmunomoduladores concomitantes (azatioprina, 6-mercaptopurina o metotrexato) fuera de los EE. UU., pero no se permitieron más allá de la semana 6 en los EE. UU.
En la semana 6, los pacientes estaban recibiendo corticosteroides (59%), inmunomoduladores (azatioprina, 6-mercaptopurina o metotrexato) (31%) y aminosalicilatos (41%). El cincuenta y uno por ciento de los pacientes tuvieron una respuesta inadecuada, pérdida de respuesta o intolerancia a una terapia con bloqueador del TNF. En la semana 6, la puntuación CDAI mediana fue de 322 en el grupo de ENTYVIO intravenoso cada ocho semanas, 316 en el grupo de ENTYVIO intravenoso cada cuatro semanas y 315 en el grupo placebo. Los pacientes que habían logrado una respuesta clínica (≥70 de disminución en la puntuación CDAI desde el inicio) en la semana 6 y estaban recibiendo corticosteroides debían comenzar un régimen de reducción de corticosteroides en la semana 6.
En CD Trial III, un porcentaje mayor de pacientes en los grupos tratados con ENTYVIO intravenoso en comparación con placebo estuvieron en remisión clínica (definida como una puntuación CDAI ≤150) en la semana 52. Un porcentaje mayor de pacientes en los grupos tratados con ENTYVIO intravenoso en comparación con placebo tuvieron una respuesta clínica (definida como una disminución ≥100 en la puntuación CDAI desde el inicio) en la semana 52 (Tabla 8). En el subgrupo de pacientes que estaban recibiendo corticosteroides al inicio y que estaban en respuesta clínica en la semana 6 (definida como una disminución ≥70 en la puntuación CDAI desde el inicio), una mayor proporción de pacientes en los grupos tratados con ENTYVIO intravenoso en comparación con placebo suspendieron los corticosteroides en la semana 52 y estuvieron en remisión clínica en la semana 52 (Tabla 8).
El régimen de dosificación de ENTYVIO cada cuatro semanas no demostró un beneficio clínico adicional sobre el régimen de dosificación cada ocho semanas. El régimen de dosificación cada cuatro semanas no es el régimen de dosificación recomendado [ver Dosificación y administración (2.2)].
| Criterio de valoración | Placebo† N=153 |
ENTYVIO IV Cada 8 Semanas N=154 |
Valor p | Diferencia de tratamiento y IC del 95% |
|---|---|---|---|---|
|
||||
| Remisión clínica‡ en la semana 52 | 22% | 39% | 0.001 | 17% (7%, 28%) |
| Respuesta clínica§ en la semana 52 | 30% | 44% | 0.013 | 13% (3%, 24%) |
| Remisión clínica sin corticosteroides¶ | 16%¶ | 32%¶ | 0.015 | 16% (3%, 29%) |
Administración Subcutánea
Ensayo SC CD – Subcutáneo
La seguridad y eficacia de ENTYVIO subcutáneo se evaluó en un ensayo aleatorizado, doble ciego, controlado con placebo (Ensayo SC CD; NCT02611817) en pacientes adultos con enfermedad de Crohn de moderada a gravemente activa, definida como una puntuación CDAI de 220 a 450. En la línea de base, la puntuación CDAI mediana fue de 316 (rango: 198 a 559).
El ensayo incluyó pacientes que habían experimentado una respuesta inadecuada, pérdida de respuesta o intolerancia a al menos uno de los siguientes: corticosteroides, inmunomoduladores (azatioprina, 6-mercaptopurina o metotrexato) o bloqueadores del TNF (incluidos los no respondedores primarios). Se permitió a los pacientes usar dosis estables concomitantes de aminosalicilatos orales, corticosteroides orales (prednisona ≤30 mg/día, budesonida ≤9 mg/día o esteroide equivalente), inmunomoduladores, probióticos, antidiarreicos y/o antibióticos. Las terapias biológicas concomitantes, el tratamiento rectal con ácido 5-aminosalicílico o enemas/supositorios de corticosteroides estaban prohibidos.
Todos los pacientes recibieron ENTYVIO intravenoso de 300 mg en etiqueta abierta en la semana 0 y la semana 2. Para poder ser aleatorizados al tratamiento en el Ensayo SC CD, los pacientes debían tener una respuesta clínica (definida como una disminución ≥70 puntos en la puntuación CDAI desde la línea de base) en la semana 6. Un total de 409 pacientes fueron aleatorizados en la semana 6 de forma doble ciego (2:1) a ENTYVIO 108 mg administrado por inyección subcutánea o placebo cada 2 semanas. Las evaluaciones de eficacia se realizaron en la semana 52.
A partir de la semana 6, los pacientes que estaban recibiendo corticosteroides debían comenzar un régimen de reducción de corticosteroides.
En el momento de la aleatorización a la fase doble ciego (semana 6), los pacientes estaban recibiendo corticosteroides (45%), inmunomoduladores (32%) y aminosalicilatos (45%). El cincuenta y uno por ciento de los pacientes habían tenido una respuesta inadecuada, pérdida de respuesta o intolerancia a una terapia con bloqueador del TNF antes de la inscripción.
Los pacientes en la fase doble ciego tenían una edad media de 38 años (rango de 18 a 76 años); el 55% eran hombres; el 91% se identificó como blanco, el 6% como asiático y el 3% se identificó como otro grupo racial.
El criterio de valoración principal fue la proporción de pacientes con remisión clínica (puntuación CDAI ≤150) en la semana 52 (ver Tabla 9).
| Criterio de valoración | Placebo† | ENTYVIO SC 108 mg Cada 2 semanas |
Estimación‡ de la diferencia de tratamiento (IC del 95%) Vedolizumab SC vs. Placebo |
|---|---|---|---|
| Remisión clínica§ en la semana 52 | |||
|
|||
| Población total | N=134 34% |
N=275 48% |
14 (4, 24)¶ |
| Fallo/exposición previa al bloqueador del TNF | N=71 27% |
N=168 48% |
|
| Sin fallo/exposición previa al bloqueador del TNF | N=63 43% |
N=107 49% |
|
Entre los pacientes que usaban corticosteroides orales en la línea de base (semana 0) y lograron una respuesta clínica en la semana 6, el 45% (43/95) tratados con ENTYVIO subcutáneo en comparación con el 18% (8/44) tratados con placebo suspendieron los corticosteroides y estaban en remisión clínica en la semana 52. Este resultado no fue estadísticamente significativo bajo el procedimiento de prueba múltiple preespecificado.
16 FORMA DE SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
Presentación
Infusion intravenosa
ENTYVIO® (vedolizumab) para inyección para infusión intravenosa se suministra en viales de vidrio estériles de dosis única, que contienen 300 mg de vedolizumab como un pastel liofilizado de blanco a blanquecino.
- ENTYVIO: vial de dosis única de 300 mg en caja individual: NDC 64764-300-20
Inyección subcutánea
ENTYVIO (vedolizumab) inyección para uso subcutáneo está disponible en una jeringa precargada o un bolígrafo precargado como una solución transparente a moderadamente opalescente e incolora a ligeramente amarilla.
La jeringa precargada ENTYVIO desechable de dosis única y el bolígrafo precargado ENTYVIO desechable de dosis única (ENTYVIO PEN) están compuestos por una jeringa de vidrio larga de 1 mL con una aguja fija de calibre 27 de pared delgada, ½ pulgada. La jeringa tiene una cubierta de aguja de goma encapsulada en una carcasa de plástico y un tapón de goma. No está hecho con látex de caucho natural.
- ENTYVIO: jeringa precargada de dosis única de 108 mg/0,68 en una caja individual: NDC 64764-107-11
- ENTYVIO PEN: bolígrafo precargado de dosis única de 108 mg/0,68 en una caja individual: NDC 64764-108-21
Almacenamiento y manipulación
- Refrigere los viales, jeringas precargadas y bolígrafos precargados ENTYVIO sin abrir a 2 °C a 8 °C (36 °F a 46 °F).
- Si es necesario, la jeringa precargada ENTYVIO o el bolígrafo precargado ENTYVIO se pueden dejar fuera del refrigerador en el paquete original a temperatura ambiente hasta 25 °C (77 °F) durante un máximo de 7 días (por ejemplo, cuando viaja). No use la jeringa precargada ENTYVIO o el bolígrafo precargado ENTYVIO si se deja fuera del refrigerador por más de 7 días.
- No congele el vial, la jeringa precargada o el bolígrafo precargado ENTYVIO. No use el vial, la jeringa precargada o el bolígrafo precargado ENTYVIO si se ha congelado.
- No agite la jeringa precargada ENTYVIO o el bolígrafo precargado ENTYVIO.
- Conserve en el paquete original para protegerlo de la luz hasta el momento de su uso.
17 INFORMACIÓN PARA EL PACIENTE
Aconseje al paciente que lea el etiquetado del paciente aprobado por la FDA (Guía de medicamentos e Instrucciones de uso).
Reacciones relacionadas con la infusión e hipersensibilidad
Indique a los pacientes que informen inmediatamente si experimentan síntomas compatibles con una reacción de hipersensibilidad durante o después de una infusión de ENTYVIO [ver Advertencias y precauciones (5.1)].
Infecciones
Informe a los pacientes que pueden tener más probabilidades de desarrollar infecciones cuando toman ENTYVIO. Indique a los pacientes que le digan a su proveedor de atención médica si desarrollan algún signo o síntoma de infección [ver Advertencias y precauciones (5.2)].
Leucoencefalopatía multifocal progresiva
Informe a los pacientes que la leucoencefalopatía multifocal progresiva (PML) ha ocurrido en pacientes que recibieron algunos productos antagonistas del receptor de integrina e inmunosupresores sistémicos. Indique a los pacientes que informen inmediatamente si experimentan cualquier aparición nueva o empeoramiento de los signos y síntomas neurológicos, ya que estos podrían ser indicativos de PML [ver Advertencias y precauciones (5.3)].
Lesión hepática
Informe a los pacientes que se han producido niveles elevados de transaminasas con o sin bilirrubina elevada en pacientes que recibieron ENTYVIO. Indique a los pacientes que informen rápidamente cualquier síntoma que pueda indicar lesión hepática, incluida la fatiga, la anorexia, el dolor en el cuadrante superior derecho del abdomen, la orina oscura o la ictericia [ver Advertencias y precauciones (5.4)].
Técnica de dosificación subcutánea
Proporcione orientación a los pacientes y cuidadores sobre la técnica de administración subcutánea adecuada y cómo usar correctamente la jeringa precargada de dosis única ENTYVIO o el bolígrafo precargado de dosis única ENTYVIO [ver Instrucciones de uso].
SECCIÓN NO CLASIFICADA SPL
Fabricado por:
Takeda Pharmaceuticals U.S.A., Inc.
Cambridge, MA 02142
Licencia de EE. UU. No. 1898
ENTYVIO es una marca registrada de Millennium Pharmaceuticals Inc. y se utiliza bajo licencia de Takeda Pharmaceuticals U.S.A., Inc.
Todos los demás nombres de marcas comerciales son propiedad de sus respectivos dueños.
©2024 Takeda Pharmaceuticals U.S.A., Inc.
VMB245 R14
Guía de medicación
| GUÍA DE MEDICAMENTO | ||
|---|---|---|
| ENTYVIO® (en ti’ vee oh) (vedolizumab) para inyección, para uso intravenoso |
ENTYVIO® (en ti’ vee oh) (vedolizumab) inyección, para uso subcutáneo |
ENTYVIO® (en ti’ vee oh) PEN (vedolizumab) inyección, para uso subcutáneo |
| Esta Guía de Medicamento ha sido aprobada por la Administración de Alimentos y Medicamentos de los Estados Unidos | VMB245 R11 Revisado: 5/2024 | |
| ¿Cuál es la información más importante que debo conocer sobre ENTYVIO? ENTYVIO puede causar efectos secundarios graves, que incluyen:
Consulte “¿Cuáles son los posibles efectos secundarios de ENTYVIO?“ para obtener más información sobre los efectos secundarios. |
||
| ¿Qué es ENTYVIO? ENTYVIO es un medicamento recetado que se usa en adultos para el tratamiento de:
No se sabe si ENTYVIO es seguro y eficaz en niños menores de 18 años. |
||
| ¿Quién no debe recibir ENTYVIO? No reciba ENTYVIO si usted ha tenido una reacción alérgica a ENTYVIO o a alguno de los ingredientes de ENTYVIO. Vea el final de esta Guía de Medicamento para obtener una lista completa de los ingredientes de ENTYVIO. |
||
Antes de recibir ENTYVIO, informe a su proveedor de atención médica sobre todas sus condiciones médicas, incluso si usted:
Informe a su proveedor de atención médica sobre todos los medicamentos que toma, incluidos medicamentos con receta y sin receta, vitaminas y suplementos herbales. |
||
| ¿Cómo debo usar ENTYVIO? Cuando se administra en una vena (por vía intravenosa):
Cuando se administra debajo de la piel (por vía subcutánea):
|
||
| ¿Cuáles son los posibles efectos secundarios de ENTYVIO? ENTYVIO puede causar efectos secundarios graves, consulte “¿Cuál es la información más importante que debo saber sobre ENTYVIO?“ Los efectos secundarios más comunes de ENTYVIO incluyen: resfriado común, dolor de cabeza, dolor en las articulaciones, náuseas, fiebre, infecciones de la nariz y la garganta, cansancio, tos, bronquitis, gripe, dolor de espalda, sarpullido, picazón, sinusitis, dolor de garganta, dolor en las extremidades y con inyecciones debajo de la piel: dolor, hinchazón, picazón, urticaria, hematomas, sarpullido o enrojecimiento en el lugar de la inyección. Estos no son todos los posibles efectos secundarios de ENTYVIO. Llame a su médico para obtener asesoramiento médico sobre los efectos secundarios. Puede informar los efectos secundarios a la FDA al 1-800-FDA-1088. |
||
¿Cómo debo almacenar ENTYVIO?
Mantenga ENTYVIO y todos los medicamentos fuera del alcance de los niños. |
||
| Información general sobre el uso seguro y eficaz de ENTYVIO. Los medicamentos a veces se recetan para fines distintos de los que se enumeran en una Guía del medicamento. No use ENTYVIO para una afección para la que no fue recetado. No le dé ENTYVIO a otras personas, incluso si tienen los mismos síntomas que usted. Puede perjudicarlos. Puede preguntarle a su farmacéutico o proveedor de atención médica información sobre ENTYVIO que esté escrita para profesionales de la salud. |
||
| ¿Cuáles son los ingredientes de ENTYVIO? Ingrediente activo: vedolizumab Ingredientes inactivos en el vial para infusión intravenosa: hidrocloruro de arginina, histidina, monoclorhidrato de histidina, polisorbato 80 y sacarosa Ingredientes inactivos en la jeringa precargada o la pluma precargada para inyección subcutánea: hidrocloruro de arginina, ácido cítrico monohidratado, histidina, monoclorhidrato de histidina, polisorbato 80, citrato de sodio dihidratado y agua estéril para inyección Fabricado por: Takeda Pharmaceuticals U.S.A., Inc. Cambridge, MA 02142 Licencia estadounidense n.° 1898 ENTYVIO es una marca comercial registrada de Millennium Pharmaceuticals Inc. y Takeda Pharmaceuticals U.S.A., Inc. la utiliza bajo licencia. Todos los demás nombres comerciales son propiedad de sus respectivos dueños. ©2024 Takeda Pharmaceuticals U.S.A., Inc. Para obtener más información, visite www.ENTYVIO.com o llame al 1-877-TAKEDA7 (1-877-825-3327). |
||
INSTRUCCIONES DE USO
ENTYVIO® (en ti’ vee oh) PEN
(vedolizumab)
inyección, para uso subcutáneo
Pluma precargada de dosis única
Estas instrucciones de uso contienen información sobre cómo inyectarse ENTYVIO.
Su pluma precargada de dosis única de ENTYVIO
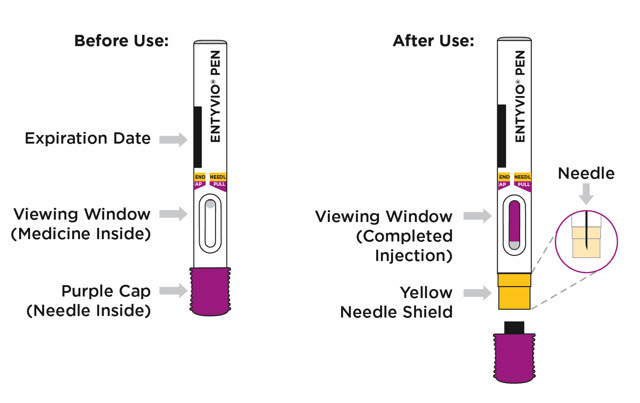
Información importante que debe saber antes de inyectarse ENTYVIO:
|
Almacenamiento de ENTYVIO
- Guarde su pluma precargada en el refrigerador entre 36 °F y 46 °F (2 °C y 8 °C).
- Su pluma precargada se puede dejar en su caja a temperatura ambiente hasta 77 °F (25 °C) durante un máximo de 7 días (por ejemplo, cuando viaja). No use la pluma precargada si se deja fuera del refrigerador por más de 7 días.
- No congele la pluma precargada.
- No deje la pluma precargada a la luz solar directa.
- Deseche la pluma precargada en un contenedor de eliminación de objetos punzantes aprobado por la FDA si se ha dejado fuera del refrigerador por más de 7 días, se ha congelado o se ha dejado a la luz solar directa. Consulte Paso 14 para obtener instrucciones sobre cómo desechar la pluma precargada.
- Siempre mantenga las plumas ENTYVIO PEN, el contenedor de eliminación de objetos punzantes y todos los medicamentos fuera del alcance de los niños.
| U.S. License No. 1898 | ||
| Preparación de sus suministros | ||
| Paso 1. Retire la caja de ENTYVIO PEN del refrigerador | ||
Tome 1 caja de pluma precargada del refrigerador y verifique la fecha de vencimiento en la caja (consulte Figura A).
|
 Figura A Figura A |
|
| Paso 2. Espere 30 minutos | ||
Espere 30 minutos y deje que la pluma precargada alcance la temperatura ambiente (consulte Figura B).
|
 Figura B Figura B |
|
| Paso 3. Reúna los suministros | ||
Busque una superficie limpia y plana, como una mesa. Reúna los suministros que no están en la caja de la pluma precargada (consulte Figura C).
|
 Figura C Figura C |
|
| Preparación para inyectar ENTYVIO | ||
| Paso 4. Lávese las manos | ||
| Lávese las manos con jabón y agua limpia (consulte Figura D). |  Figura D Figura D |
|
| Paso 5. Retire la pluma precargada de la bandeja | ||
Despegue el papel de la bandeja y levante la pluma precargada directamente (consulte Figura E).
|
 Figura E Figura E |
|
| Paso 6. Inspeccione la pluma precargada | ||
Verifique la fecha de vencimiento (EXP) impresa en la pluma precargada y el medicamento en la ventana de visualización de la pluma precargada (consulte Figura F). El medicamento debe ser incoloro a amarillo pálido. Es normal ver burbujas de aire. Inspeccione la pluma precargada para detectar cualquier daño.
|
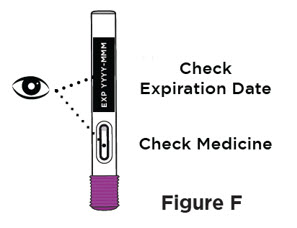 |
|
| Paso 7. Elija el sitio de inyección | ||
Elija un sitio de inyección en su piel desnuda de uno de los siguientes (consulte Figura G):
|
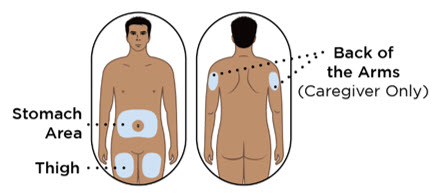 Figura G Figura G |
|
| Paso 8. Limpie el sitio de inyección | ||
Limpie el sitio de inyección con una almohadilla con alcohol (vea Figura H). Deje que su piel se seque.
|
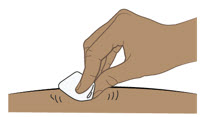 Figure H Figure H |
|
| Continúe con Paso 9 ➔ | ||
| Inyectando ENTYVIO | ||
| Paso 9. Retire la tapa morada y deséchela | ||
Cuando esté listo para inyectar, tire de la tapa morada hacia arriba y deséchela inmediatamente en el contenedor de eliminación de objetos punzantes (vea Figura I).
|
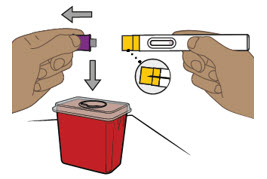 Figure I Figure I |
|
| Paso 10. Coloque la pluma precargada en el sitio de inyección | ||
|
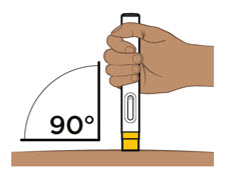 Figure J Figure J |
|
| Paso 11. Comience a inyectar ENTYVIO | Paso 12. Complete la inyección de ENTYVIO | |
Presione la pluma precargada hacia abajo y manténgala presionada durante al menos 10 segundos (vea Figura K).
|
Continúe sosteniendo la pluma precargada con presión constante hasta que la ventana de visualización se haya llenado de morado para asegurarse de que ha recibido su dosis completa (vea Figura L).
|
|
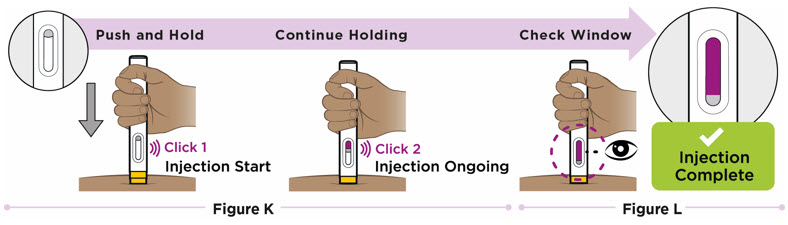 |
||
| Paso 13. Levante la pluma precargada de la piel | ||
Levante la pluma precargada del sitio de inyección (vea Figura M). El protector de aguja amarillo se bajará y se bloqueará sobre la aguja.
|
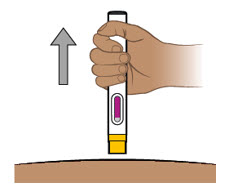 Figure M Figure M |
|
| Paso 14. Deseche la pluma precargada | ||
Deseche la pluma precargada usada en un contenedor de eliminación de objetos punzantes aprobado por la FDA inmediatamente después de su uso (vea Figura N). No recicle ni deseche la pluma precargada en la basura de su hogar.
Si tiene alguna pregunta o inquietud sobre su ENTYVIO PEN, llame a su proveedor de atención médica. También puede llamar al 877-825-3327 o visitar www.ENTYVIO.com para obtener más información. |
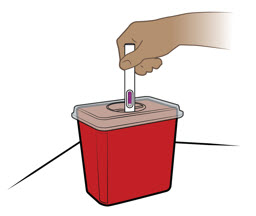 Figure N Figure N |
|
Estas Instrucciones de uso han sido aprobadas por la Administración de Alimentos y Medicamentos de los Estados Unidos.
Revisado: 5/2024
INSTRUCCIONES DE USO
ENTYVIO® (en ti’ vee oh)
(vedolizumab)
Inyección, para uso subcutáneo
Jeringa precargada de dosis única
Estas Instrucciones de uso contienen información sobre cómo inyectar ENTYVIO.
Su jeringa precargada de dosis única de ENTYVIO
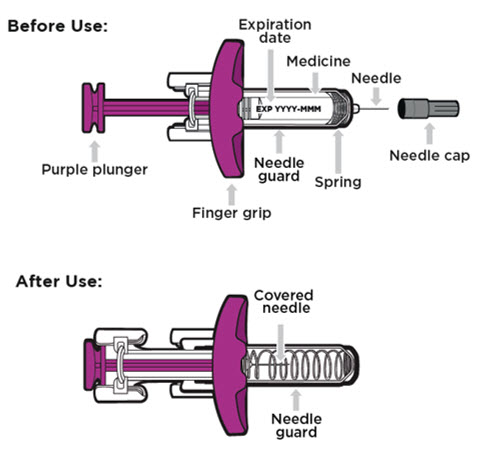
Información importante que necesita saber antes de inyectar ENTYVIO:
|
Almacenamiento de ENTYVIO
- Guarde su jeringa precargada en el refrigerador entre 36 °F y 46 °F (2 °C y 8 °C).
- Su jeringa precargada se puede dejar en su caja a temperatura ambiente hasta 77 °F (25 °C) durante un máximo de 7 días (por ejemplo, cuando viaja). No use la jeringa precargada si se deja fuera del refrigerador por más de 7 días.
- No congele la jeringa precargada.
- No deje la jeringa precargada a la luz solar directa.
- Deseche la jeringa precargada en un contenedor de eliminación de objetos punzantes aprobado por la FDA si se ha dejado fuera del refrigerador por más de 7 días, se ha congelado o se ha dejado a la luz solar directa. Consulte Paso 14 para obtener instrucciones sobre cómo desechar la jeringa precargada.
- Siempre mantenga las jeringas precargadas de ENTYVIO, el contenedor de eliminación de objetos punzantes y todos los medicamentos fuera del alcance de los niños.
| U.S. License No. 1898 | ||
| Preparación de sus suministros | ||
| Paso 1. Retire la caja de la jeringa precargada de ENTYVIO del refrigerador | ||
Tome 1 caja de jeringa precargada del refrigerador y verifique la fecha de vencimiento en la caja (consulte Figura A).
|
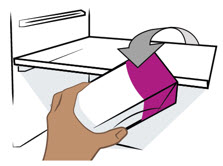 Figura A Figura A |
|
| Paso 2. Espere 30 minutos | ||
Espere 30 minutos y deje que la jeringa precargada alcance la temperatura ambiente (consulte Figura B).
|
 Figura B Figura B |
|
| Paso 3. Reúna los suministros | ||
Busque una superficie limpia y plana, como una mesa. Reúna los suministros que no están en la caja de la jeringa precargada (consulte Figura C).
|
 Figura C Figura C |
|
| Preparación para inyectar ENTYVIO | ||
| Paso 4. Lávese las manos | ||
| Lávese las manos con agua y jabón (consulte Figura D). |  Figura D Figura D |
|
| Paso 5. Retire la jeringa precargada de la bandeja | ||
Despegue el papel de la bandeja y levante la jeringa precargada hacia arriba (consulte Figura E).
|
 Figura E Figura E |
|
| Paso 6. Inspeccione la jeringa precargada | ||
Verifique la fecha de vencimiento (EXP) impresa en la jeringa precargada y el medicamento en la jeringa precargada (consulte Figura F). El medicamento debe ser incoloro a amarillo pálido. Es normal ver burbujas de aire. Inspeccione la jeringa precargada para detectar cualquier daño.
|
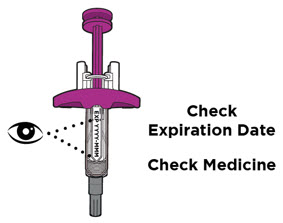 Figura F Figura F |
|
| Paso 7. Elija el sitio de inyección | ||
Seleccione un lugar de inyección en su piel descubierta de una de las siguientes (ver Figura G):
|
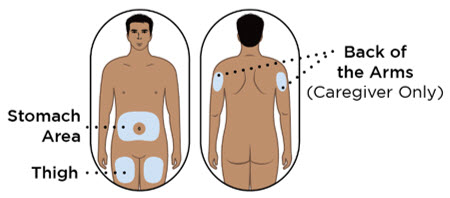 Figura G Figura G |
|
| Paso 8. Limpie el lugar de la inyección | ||
Limpie el lugar de la inyección con una almohadilla de alcohol (ver Figura H). Deje que su piel se seque.
|
 Figura H Figura H |
|
| Continúe a Paso 9 ➔ | ||
| Inyectando ENTYVIO | ||
| Paso 9. Retire el capuchón de la aguja y deséchelo | ||
Cuando esté listo para inyectar, retire el capuchón de la aguja hacia fuera y deséchelo inmediatamente en el recipiente para desechos de punciones (ver Figura I). Puede ver una gota de líquido en el extremo de la aguja. Esto es normal.
|
 Figura I Figura I |
|
| Paso 10. Apriete la piel | ||
Con el capuchón de la aguja quitado, sostenga la jeringa precargada con una mano y apriete la piel alrededor del lugar de la inyección con la otra mano (ver Figura J).
|
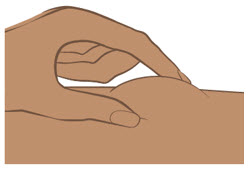 Figura J Figura J |
|
| Paso 11. Inserte la jeringa precargada en un ángulo de 45 grados | ||
Inserte la aguja a aproximadamente un ángulo de 45 grados hasta que esté totalmente dentro de la piel pellizcada (ver Figura K).
|
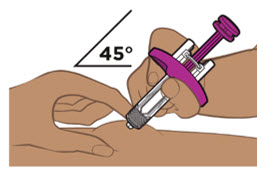 Figura K Figura K |
|
| Paso 12. Empuje el émbolo hacia abajo | ||
Empuje el émbolo completamente hacia abajo hasta que se inyecte todo el medicamento (ver Figura L).
|
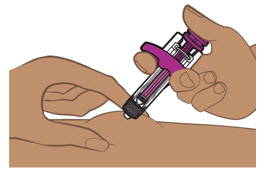 Figura L Figura L |
|
| Paso 13.quite el pulgar del émbolo | ||
Quitar el pulgar del émbolo para permitir que el protector de la aguja cubra la aguja (ver Figura M).
|
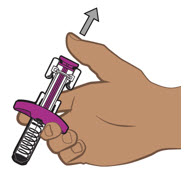 Figura M Figura M |
|
| Paso 14. Deseche (elimine) la jeringa precargada | ||
Deseche (elimine) la jeringa precargada usada en un contenedor de eliminación de objetos punzantes aprobado por la FDA inmediatamente después de su uso (vea Figura N). No recicle ni deseche la jeringa precargada en la basura de su hogar.
Si tiene alguna pregunta o inquietud sobre su jeringa precargada ENTYVIO, llame a su proveedor de atención médica. También puede llamar al 877-825-3327 o visitar www.ENTYVIO.com para obtener más información. |
 Figure N Figure N |
|
Estas Instrucciones de uso han sido aprobadas por la Administración de Alimentos y Medicamentos de los Estados Unidos.
Revisado: 5/2024
Panel de visualización principal – Etiqueta de frasco de 300 mg
NDC 64764-300-20 Sólo con receta
Entyvio
vedolizumab
para inyección
300 mg por vial*
Para uso intravenoso solamente
Debe reconstitutirse
y diluirse antes de usarse
Vial de dosis única – Desechar la porción no utilizada

PANEL DE EXHIBICIÓN PRINCIPAL – Cartón de vial de 300 mg
NDC 64764-300-20 Rx only
Entyvio
vedolizumab
para inyección
300 mg por vial*
ATENCIÓN: Cada paciente debe
recibir la Guía del Medicamento adjunta.
Para uso intravenoso únicamente
Debe reconstituirse y diluirse antes
de su uso
Vial de dosis única – Deseche la porción no utilizada
Takeda

PANEL PRINCIPAL DE PRESENTACIÓN – Bandeja para bolígrafos de 108 mg/0,68 mL Caja
NDC 64764-108-21 Rx only
ENTYVIO® PEN
(vedolizumab)
inyección
108 mg/0.68 mL
Para Uso Subcutáneo Solamente
1 Pluma precargada de dosis única
ATENCIÓN FARMACÉUTICO: Cada
paciente debe recibir la
Guía de Medicamentos adjunta.
PANEL DE EXHIBICIÓN PRINCIPAL – Bandeja de jeringa de 108 mg/0,68 mL de cartón
NDC 64764-107-11 Rx only
ENTYVIO®
(vedolizumab)
injection
108 mg/0.68 mL
Para uso subcutáneo únicamente
1 Jeringa precargada de dosis única
ATENCIÓN FARMACÉUTICO: Cada paciente
debe recibir la Guía del medicamento adjunta.