
Fabricante de medicamentos: sanofi-aventis U.S. LLC (Updated: 2024-09-27)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
DUPIXENT® (dupilumab) inyección, para uso subcutáneo
Aprobación inicial en EE. UU.: 2017
CAMBIOS MAYORES RECIENTES
| Indicaciones y uso, Sinusitis crónica con pólipos nasales (1.3) | 09/2024 |
| Indicaciones y uso, Esofagitis eosinofílica (1.4) | 01/2024 |
| Indicaciones y uso, Enfermedad pulmonar obstructiva crónica (1.6) | 09/2024 |
| Dosis y administración, 100 mg Q2W en pacientes pediátricos de 6 a 11 años de edad con asma (2.4) | Eliminado 04/2024 |
| Dosis y administración, Sinusitis crónica con pólipos nasales (2.5) | 09/2024 |
| Dosis y administración, Esofagitis eosinofílica (2.6) | 01/2024 |
| Dosis y administración, Enfermedad pulmonar obstructiva crónica (2.8) | 09/2024 |
| Advertencias y precauciones (5.2, 5.4) | 09/2024 |
INDICACIONES Y USO
DUPIXENT es un antagonista del receptor alfa de la interleucina-4 indicado:
Dermatitis atópica
para el tratamiento de pacientes adultos y pediátricos de 6 meses de edad o mayores con DA moderada a grave cuya enfermedad no está adecuadamente controlada con terapias tópicas de prescripción o cuando esas terapias no son aconsejables. DUPIXENT se puede usar con o sin corticosteroides tópicos. (1.1)
Asma
como tratamiento de mantenimiento adicional de pacientes adultos y pediátricos de 6 años de edad o mayores con asma moderada a grave caracterizada por un fenotipo eosinofílico o con asma dependiente de corticosteroides orales. (1.2)
Limitaciones de uso: No para el alivio del broncoespasmo agudo o el estado asmático. (1.2)
Sinusitis crónica con pólipos nasales
como tratamiento de mantenimiento adicional en pacientes adultos y pediátricos de 12 años de edad o mayores con sinusitis crónica con pólipos nasales (CRSwNP) inadecuadamente controlada. (1.3)
Esofagitis eosinofílica
para el tratamiento de pacientes adultos y pediátricos de 1 año de edad o mayores, con un peso de al menos 15 kg, con esofagitis eosinofílica (EoE). (1.4)
Prúrigo nodular
para el tratamiento de pacientes adultos con prúrigo nodular (PN). (1.5)
Enfermedad pulmonar obstructiva crónica
como tratamiento de mantenimiento adicional de pacientes adultos con enfermedad pulmonar obstructiva crónica (EPOC) inadecuadamente controlada y un fenotipo eosinofílico. (1.6)
Limitaciones de uso: No para el alivio del broncoespasmo agudo. (1.6)
DOSIS Y ADMINISTRACIÓN
Dermatitis atópica
Dosis en adultos (2.3):
- La dosis recomendada es una dosis inicial de 600 mg (dos inyecciones de 300 mg), seguida de 300 mg cada dos semanas (Q2W).
Dosis en pacientes pediátricos de 6 meses a 5 años de edad (2.3):
| Peso corporal | Dosis inicial y subsiguiente |
|---|---|
| 5 a menos de 15 kg | 200 mg (una inyección de 200 mg) cada 4 semanas (Q4W) |
| 15 a menos de 30 kg | 300 mg (una inyección de 300 mg) cada 4 semanas (Q4W) |
Dosis en pacientes pediátricos de 6 a 17 años de edad (2.3):
| Peso corporal | Dosis de carga inicial | Dosis subsiguiente* |
|---|---|---|
|
||
| 15 a menos de 30 kg | 600 mg (dos inyecciones de 300 mg) | 300 mg Q4W |
| 30 a menos de 60 kg | 400 mg (dos inyecciones de 200 mg) | 200 mg Q2W |
| 60 kg o más | 600 mg (dos inyecciones de 300 mg) | 300 mg Q2W |
Asma
Dosificación en pacientes adultos y pediátricos de 12 años o más (2.4):
| Dosis de carga inicial | Dosificación posterior |
|---|---|
| 400 mg (dos inyecciones de 200 mg) | 200 mg cada 2 semanas (Q2W) |
| O | |
| 600 mg (dos inyecciones de 300 mg) | 300 mg cada 2 semanas (Q2W) |
| Dosificación para pacientes con asma dependiente de corticosteroides orales o con dermatitis atópica moderada a grave como comorbilidad o adultos con sinusitis crónica con pólipos nasales como comorbilidad | |
| 600 mg (dos inyecciones de 300 mg) | 300 mg cada 2 semanas (Q2W) |
Dosificación en pacientes pediátricos de 6 a 11 años de edad (2.4):
| Peso corporal | Dosis inicial y dosificación posterior |
|---|---|
| 15 a menos de 30 kg | 300 mg cada cuatro semanas (Q4W) |
| ≥30 kg | 200 mg cada dos semanas (Q2W) |
Para pacientes pediátricos de 6 a 11 años de edad con asma y dermatitis atópica moderada a grave como comorbilidad, siga la dosificación recomendada según la Tabla 2, que incluye una dosis de carga inicial. (2.3)
Sinusitis crónica con pólipos nasales (2.5):
- La dosificación recomendada para pacientes adultos y pediátricos de 12 años de edad o más es de 300 mg administrados cada dos semanas (Q2W).
Esofagitis eosinofílica (2.6):
| Peso corporal | Dosificación recomendada en pacientes adultos y pediátricos de 1 año o más, que pesen al menos 15 kg |
|---|---|
| 15 a menos de 30 kg | 200 mg cada dos semanas (Q2W) |
| 30 a menos de 40 kg | 300 mg cada dos semanas (Q2W) |
| 40 kg o más | 300 mg cada semana (QW) |
Prurigo Nodularis (2.7):
- La dosis recomendada para pacientes adultos es una dosis inicial de 600 mg (dos inyecciones de 300 mg), seguida de 300 mg administrados cada dos semanas (Q2W).
Enfermedad Pulmonar Obstructiva Crónica (2.8):
- La dosis recomendada para pacientes adultos es de 300 mg administrados cada dos semanas (Q2W).
FORMAS Y FUERZAS DE DOSIFICACIÓN
CONTRAINDICACIONES
Hipersensibilidad conocida a dupilumab o a cualquier excipiente de DUPIXENT. (4)
ADVERTENCIAS Y PRECAUCIONES
- Hipersensibilidad: Se han producido reacciones de hipersensibilidad, incluida la anafilaxia, el suero, el angioedema, la urticaria, la erupción cutánea, el eritema nodoso y el eritema multiforme. Suspenda DUPIXENT en caso de reacción de hipersensibilidad. (5.1)
- Conjuntivitis y queratitis: Avise a los pacientes que informen a su proveedor de atención médica sobre la aparición o el empeoramiento de los síntomas oculares. Considere la posibilidad de realizar un examen oftalmológico, según corresponda. (5.2)
- Condiciones eosinofílicas: Esté atento a la erupción vasculítica, el empeoramiento de los síntomas pulmonares y/o la neuropatía, especialmente al reducir los corticosteroides orales. (5.3)
- Reducción de la dosis de corticosteroides: No suspenda abruptamente los corticosteroides sistémicos, tópicos o inhalados al iniciar DUPIXENT. Disminuya los esteroides gradualmente, si corresponde. (5.5)
- Artralgia: Avise a los pacientes que informen a su proveedor de atención médica sobre la aparición o el empeoramiento de los síntomas articulares. Si los síntomas persisten o empeoran, considere la posibilidad de una evaluación reumatológica y/o la suspensión de DUPIXENT. (5.7)
- Infecciones parasitarias (helmintos): Trate las infecciones por helmintos preexistentes antes de iniciar DUPIXENT. Si los pacientes se infectan mientras reciben DUPIXENT y no responden al tratamiento antihelmíntico, suspenda DUPIXENT hasta que la infección se resuelva. (5.8)
- Vacunaciones: Evite el uso de vacunas vivas. (5.9)
REACCIONES ADVERSAS
Las reacciones adversas más comunes son:
- Dermatitis atópica (incidencia ≥1%): reacciones en el sitio de inyección, conjuntivitis, blefaritis, herpes oral, queratitis, prurito ocular, otra infección por virus del herpes simple, ojo seco y eosinofilia. (6.1)
- Asma (incidencia ≥1%): reacciones en el sitio de inyección, dolor orofaríngeo y eosinofilia. (6.1)
- Rinosinusitis crónica con pólipos nasales (incidencia ≥1%): reacciones en el sitio de inyección, eosinofilia, insomnio, dolor de muelas, gastritis, artralgia y conjuntivitis. (6.1)
- Esofagitis eosinofílica (incidencia ≥2%): reacciones en el sitio de inyección, infecciones del tracto respiratorio superior, artralgia e infecciones virales por herpes. (6.1)
- Prurigo nodularis (incidencia ≥2%): nasofaringitis, conjuntivitis, infección por herpes, mareos, mialgia y diarrea. (6.1)
- Enfermedad pulmonar obstructiva crónica (incidencia ≥2%): infección viral, dolor de cabeza, nasofaringitis, dolor de espalda, diarrea, artralgia, infección del tracto urinario, reacciones de administración local, rinitis, eosinofilia, dolor de muelas y gastritis. (6.1)
Para informar sobre REACCIONES ADVERSAS SOSPECHOSAS, comuníquese con Regeneron al 1-844-387-4936 o con la FDA al 1-800-FDA-1088 o www.fda.gov/medwatch.
Ver 17 para INFORMACIÓN PARA EL PACIENTE y el etiquetado del paciente aprobado por la FDA.
Revisado: 9/2024
Tabla de Contenido
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICACIONES Y USO
1.1 Dermatitis Atópica
1.2 Asma
1.3 Rinosinusitis Crónica con Pólipos Nasales
1.4 Esofagitis Eosinofílica
1.5 Prurigo Nodular
1.6 Enfermedad Pulmonar Obstructiva Crónica
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Instrucciones Importantes de Administración
2.2 Vacunación Antes del Tratamiento
2.3 Dosis Recomendada para la Dermatitis Atópica
2.4 Dosis Recomendada para el Asma
2.5 Dosis Recomendada para la Rinosinusitis Crónica con Pólipos Nasales
2.6 Dosis Recomendada para la Esofagitis Eosinofílica
2.7 Dosis Recomendada para el Prurigo Nodular
2.8 Dosis Recomendada para la Enfermedad Pulmonar Obstructiva Crónica
2.9 Dosis Olvidadas
2.10 Preparación para el Uso
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Hipersensibilidad
5.2 Conjuntivitis y Queratitis
5.3 Afecciones Eosinofílicas
5.4 Síntomas Agudos de Asma o Enfermedad Pulmonar Obstructiva Crónica o Enfermedad Aguda en Deterioro
5.5 Riesgo Asociado con la Reducción Brusca de la Dosis de Corticosteroides
5.6 Pacientes con Asma Comórbida
5.7 Artralgia
5.8 Infecciones Parasitarias (Helmintos)
5.9 Vacunas
6 REACCIONES ADVERSAS
6.1 Experiencia en Ensayos Clínicos
6.2 Experiencia Posterior a la Comercialización
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.4 Uso Pediátrico
8.5 Uso Geriátrico
10 SOBREDOSIS
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de Acción
12.2 Farmacodinamia
12.3 Farmacocinética
12.6 Inmunogenicidad
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
14 ESTUDIOS CLÍNICOS
14.1 Dermatitis Atópica
14.2 Asma
14.3 Rinosinusitis Crónica con Pólipos Nasales
14.4 Esofagitis Eosinofílica
14.5 Prurigo Nodular
14.6 Enfermedad Pulmonar Obstructiva Crónica
16 CÓMO SE SUMINISTRA/ALMACENAMIENTO Y MANIPULACIÓN
17 INFORMACIÓN DE ASESORAMIENTO AL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información de prescripción completa no se enumeran.
1 INDICACIONES Y USO
1.1 Dermatitis atópica
DUPIXENT está indicado para el tratamiento de pacientes adultos y pediátricos de 6 meses de edad y mayores con dermatitis atópica (DA) de moderada a grave cuya enfermedad no se controla adecuadamente con terapias tópicas de venta con receta o cuando dichas terapias no son recomendables. DUPIXENT puede usarse con o sin corticosteroides tópicos.
1.2 Asma
DUPIXENT está indicado como tratamiento de mantenimiento complementario en pacientes adultos y pediátricos de 6 años de edad y mayores con asma de moderada a grave caracterizada por un fenotipo eosinofílico o con asma dependiente de corticosteroides orales [consulte Estudios clínicos (14)].
1.3 Rinosinusitis crónica con pólipos nasales
DUPIXENT está indicado como tratamiento de mantenimiento complementario en pacientes adultos y pediátricos de 12 años de edad y mayores con rinosinusitis crónica con pólipos nasales (RSCcPN) inadecuadamente controlada.
1.4 Esofagitis eosinofílica
DUPIXENT está indicado para el tratamiento de pacientes adultos y pediátricos de 1 año de edad y mayores, con un peso de al menos 15 kg, con esofagitis eosinofílica (EoE).
1.5 Prurigo nodular
DUPIXENT está indicado para el tratamiento de pacientes adultos con prurigo nodular (PN).
1.6 Enfermedad pulmonar obstructiva crónica
DUPIXENT está indicado como tratamiento de mantenimiento complementario en pacientes adultos con enfermedad pulmonar obstructiva crónica (EPOC) inadecuadamente controlada y un fenotipo eosinofílico.
2 DOSIS Y ADMINISTRACIÓN
2.1 Instrucciones importantes de administración
DUPIXENT se administra mediante inyección subcutánea.
DUPIXENT está destinado a ser utilizado bajo la supervisión de un profesional sanitario. Proporcione una formación adecuada a los pacientes y/o cuidadores sobre la preparación y administración de DUPIXENT antes de su uso, de acuerdo con las “Instrucciones de uso”.
Uso de la pluma precargada o la jeringa precargada
La pluma precargada de DUPIXENT está destinada a su uso en pacientes adultos y pediátricos de 2 años de edad o mayores.
La jeringa precargada de DUPIXENT está destinada a su uso en pacientes adultos y pediátricos de 6 meses de edad o mayores.
Un cuidador o un paciente de 12 años de edad o mayor puede inyectar DUPIXENT utilizando la jeringa precargada o la pluma precargada. En pacientes pediátricos de 12 a 17 años de edad, administre DUPIXENT bajo la supervisión de un adulto. En pacientes pediátricos de 6 meses a menos de 12 años de edad, administre DUPIXENT por un cuidador.
Instrucciones de administración
Para los pacientes con AD, asma y PN que toman una dosis inicial de 600 mg, administre cada una de las dos inyecciones de DUPIXENT de 300 mg en diferentes lugares de inyección.
Para los pacientes con AD y asma que toman una dosis inicial de 400 mg, administre cada una de las dos inyecciones de DUPIXENT de 200 mg en diferentes lugares de inyección.
Administre la inyección subcutánea en el muslo o el abdomen, excepto en las 2 pulgadas (5 cm) alrededor del ombligo. También se puede utilizar la parte superior del brazo si un cuidador administra la inyección.
Rote el lugar de inyección con cada inyección. NO inyecte DUPIXENT en la piel que esté sensible, dañada, magullada o con cicatrices.
Las “Instrucciones de uso” de DUPIXENT contienen instrucciones más detalladas sobre la preparación y administración de DUPIXENT [ver Instrucciones de uso].
2.2 Vacunación antes del tratamiento
Considere la posibilidad de completar todas las vacunas apropiadas para la edad, según las recomendaciones actuales de las directrices de inmunización, antes de iniciar el tratamiento con DUPIXENT [ver Advertencias y precauciones (5.9)].
2.3 Dosis recomendada para la dermatitis atópica
Dosis en adultos
La dosis recomendada de DUPIXENT para pacientes adultos es una dosis inicial de 600 mg (dos inyecciones de 300 mg), seguida de 300 mg cada dos semanas (Q2W).
Dosis en pacientes pediátricos de 6 meses a 5 años de edad
La dosis recomendada de DUPIXENT para pacientes pediátricos de 6 meses a 5 años de edad se especifica en la Tabla 1.
| Peso corporal | Dosis inicial* y posterior |
|---|---|
|
|
| 5 a menos de 15 kg | 200 mg (una inyección de 200 mg) cada 4 semanas (Q4W) |
| 15 a menos de 30 kg | 300 mg (una inyección de 300 mg) cada 4 semanas (Q4W) |
Dosis en pacientes pediátricos de 6 a 17 años de edad
La dosis recomendada de DUPIXENT para pacientes pediátricos de 6 a 17 años de edad se especifica en la Tabla 2.
| Peso corporal | Dosis de carga inicial | Dosis posterior |
|---|---|---|
| 15 a menos de 30 kg | 600 mg (dos inyecciones de 300 mg) | 300 mg cada 4 semanas (Q4W) |
| 30 a menos de 60 kg | 400 mg (dos inyecciones de 200 mg) | 200 mg cada dos semanas (Q2W) |
| 60 kg o más | 600 mg (dos inyecciones de 300 mg) | 300 mg cada dos semanas (Q2W) |
2.4 Dosificación Recomendada para el Asma
Dosificación en pacientes adultos y pediátricos de 12 años o más
La dosis recomendada de DUPIXENT para pacientes adultos y pediátricos de 12 años de edad o más se especifica en la Tabla 3.
| Dosis de Carga Inicial | Dosificación Subsiguiente |
|---|---|
| 400 mg (dos inyecciones de 200 mg) | 200 mg cada 2 semanas (Q2W) |
| O | |
| 600 mg (dos inyecciones de 300 mg) | 300 mg cada 2 semanas (Q2W) |
| Dosificación para pacientes con asma dependiente de corticosteroides orales o con dermatitis atópica moderada a grave concomitante o adultos con sinusitis crónica concomitante con pólipos nasales | |
| 600 mg (dos inyecciones de 300 mg) | 300 mg cada 2 semanas (Q2W) |
Dosificación en pacientes pediátricos de 6 a 11 años de edad
La dosis recomendada de DUPIXENT para pacientes pediátricos de 6 a 11 años de edad se especifica en la Tabla 4.
| Peso Corporal | Dosificación Inicial* y Subsiguiente |
|---|---|
|
|
| 15 a menos de 30 kg | 300 mg cada cuatro semanas (Q4W) |
| ≥30 kg | 200 mg cada dos semanas (Q2W) |
Para pacientes pediátricos de 6 a 11 años de edad con asma y AD moderada a grave concomitante, siga la dosis recomendada según la Tabla 2, que incluye una dosis de carga inicial [ver Dosificación y Administración (2.3)].
2.5 Dosificación Recomendada para la Sinusitis Crónica con Pólipos Nasales
La dosis recomendada de DUPIXENT para pacientes adultos y pediátricos de 12 años de edad o más es de 300 mg administrados cada dos semanas (Q2W).
2.6 Dosificación Recomendada para la Esofagitis Eosinofílica
La dosis recomendada de DUPIXENT para pacientes adultos y pediátricos de 1 año de edad o más, que pesen al menos 15 kg, se especifica en la Tabla 5.
| Peso Corporal | Dosificación Recomendada |
|---|---|
| 15 a menos de 30 kg | 200 mg cada dos semanas (Q2W) |
| 30 a menos de 40 kg | 300 mg cada dos semanas (Q2W) |
| 40 kg o más | 300 mg cada semana (QW) |
2.7 Dosis recomendada para prurigo nodular
La dosis recomendada de DUPIXENT para pacientes adultos es una dosis inicial de 600 mg (dos inyecciones de 300 mg) seguida de 300 mg administrados cada dos semanas (Q2W).
2.8 Dosis recomendada para la enfermedad pulmonar obstructiva crónica
La dosis recomendada de DUPIXENT para pacientes adultos es de 300 mg administrados cada dos semanas (Q2W).
2.9 Dosis olvidadas
Si se olvida una dosis semanal, administre la dosis lo antes posible y comience un nuevo programa semanal a partir de la fecha de la última dosis administrada.
Si se olvida una dosis cada dos semanas, administre la inyección dentro de los 7 días siguientes a la dosis olvidada y luego reanude el programa original del paciente. Si la dosis olvidada no se administra dentro de los 7 días, espere hasta la siguiente dosis del programa original.
Si se olvida una dosis cada 4 semanas, administre la inyección dentro de los 7 días siguientes a la dosis olvidada y luego reanude el programa original del paciente. Si la dosis olvidada no se administra dentro de los 7 días, administre la dosis, comenzando un nuevo programa basado en esta fecha.
2.10 Preparación para el uso
Antes de la inyección, retire DUPIXENT del refrigerador y deje que DUPIXENT alcance la temperatura ambiente (45 minutos para la jeringa precargada de 300 mg/2 mL o el bolígrafo precargado, y 30 minutos para la jeringa precargada de 200 mg/1.14 mL o el bolígrafo precargado) sin quitar la tapa de la aguja. Después de retirarlo del refrigerador, DUPIXENT debe usarse dentro de los 14 días o desecharse.
Inspeccione DUPIXENT visualmente en busca de partículas y decoloración antes de la administración. DUPIXENT es una solución transparente a ligeramente opalescente, incolora a amarillo pálido. No lo use si el líquido contiene partículas visibles, está descolorido o turbio (que no sea transparente a ligeramente opalescente, incoloro a amarillo pálido). DUPIXENT no contiene conservantes; por lo tanto, deseche cualquier producto no utilizado que quede en la jeringa precargada o el bolígrafo precargado.
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
DUPIXENT es una solución transparente a ligeramente opalescente, incolora a amarillo pálido en una:
Jeringa precargada de dosis única con protector de aguja como:
- Inyección: 300 mg/2 mL
- Inyección: 200 mg/1.14 mL
Pluma precargada de dosis única como:
- Inyección: 300 mg/2 mL
- Inyección: 200 mg/1.14 mL
4 CONTRAINDICACIONES
DUPIXENT está contraindicado en pacientes con hipersensibilidad conocida a dupilumab o a cualquier excipiente de DUPIXENT [ver Advertencias y precauciones (5.1)].
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Hipersensibilidad
Se han notificado reacciones de hipersensibilidad, incluida la anafilaxia, el suero o reacciones similares al suero, el angioedema, la urticaria generalizada, la erupción cutánea, el eritema nodoso y el eritema multiforme. Si se produce una reacción de hipersensibilidad clínicamente significativa, instituya la terapia adecuada y suspenda DUPIXENT [ver Reacciones adversas (6.1, 6.2) y Farmacología clínica (12.6)].
5.2 Conjuntivitis y queratitis
Se han notificado reacciones adversas de conjuntivitis y queratitis en los ensayos clínicos.
La conjuntivitis y la queratitis ocurrieron con más frecuencia en sujetos con AD que recibieron DUPIXENT en comparación con aquellos que recibieron placebo. La conjuntivitis fue el trastorno ocular más frecuentemente notificado. La mayoría de los sujetos con conjuntivitis o queratitis se recuperaron o se estaban recuperando durante el período de tratamiento [ver Reacciones adversas (6.1)].
Entre los sujetos con asma, las frecuencias de conjuntivitis y queratitis fueron similares entre DUPIXENT y placebo [ver Reacciones adversas (6.1)].
En sujetos adultos con CRSwNP, la frecuencia de conjuntivitis fue del 2% en el grupo DUPIXENT en comparación con el 1% en el grupo placebo en el grupo de seguridad de 24 semanas; estos sujetos se recuperaron. No se notificaron casos de queratitis en el programa de desarrollo de CRSwNP [ver Reacciones adversas (6.1)].
Entre los sujetos con EoE, no hubo informes de conjuntivitis y queratitis en el grupo DUPIXENT en los ensayos controlados con placebo [ver Reacciones adversas (6.1)].
En sujetos con PN, la frecuencia de conjuntivitis fue del 4% en el grupo DUPIXENT en comparación con el 1% en el grupo placebo; estos sujetos se recuperaron o se estaban recuperando durante el período de tratamiento. No se notificaron casos de queratitis en el programa de desarrollo de PN [ver Reacciones adversas (6.1)].
Entre los sujetos con EPOC, la frecuencia de conjuntivitis y queratitis fue del 1,4% y el 0,1% en el grupo DUPIXENT y del 1% y el 0% en el grupo placebo, respectivamente [ver Reacciones adversas (6.1)].
También se han notificado eventos adversos de conjuntivitis y queratitis con DUPIXENT en entornos de poscomercialización, predominantemente en pacientes con AD. Algunos pacientes informaron alteraciones visuales (por ejemplo, visión borrosa) asociadas con conjuntivitis o queratitis.
Avise a los pacientes o sus cuidadores que informen a su proveedor de atención médica sobre la aparición o el empeoramiento de los síntomas oculares. Considere la posibilidad de realizar un examen oftalmológico a los pacientes que desarrollen conjuntivitis que no se resuelva después del tratamiento estándar o signos y síntomas sugestivos de queratitis, según corresponda [ver Reacciones adversas (6.1)].
5.3 Condiciones eosinofílicas
Los pacientes que están siendo tratados por asma pueden presentar eosinofilia sistémica grave, a veces presentando características clínicas de neumonía eosinofílica o vasculitis consistente con granulomatosis eosinofílica con poliangiitis, afecciones que a menudo se tratan con terapia corticosteroide sistémica. Estos eventos pueden estar asociados con la reducción de la terapia con corticosteroides orales. Los profesionales de la salud deben estar atentos a la erupción vasculítica, el empeoramiento de los síntomas pulmonares, las complicaciones cardíacas y/o la neuropatía que se presentan en sus pacientes con eosinofilia. Se notificaron casos de neumonía eosinofílica en sujetos adultos que participaron en el programa de desarrollo del asma y se han notificado casos de vasculitis consistente con granulomatosis eosinofílica con poliangiitis con DUPIXENT en sujetos adultos que participaron en el programa de desarrollo del asma, así como en sujetos adultos con asma comórbida en el programa de desarrollo de CRSwNP. No se ha establecido una asociación causal entre DUPIXENT y estas afecciones.
5.4 Síntomas agudos de asma o enfermedad pulmonar obstructiva crónica o enfermedad aguda en deterioro
DUPIXENT no debe utilizarse para tratar los síntomas agudos o las exacerbaciones agudas del asma o la EPOC. No use DUPIXENT para tratar el broncoespasmo agudo o el estado asmático. Los pacientes deben buscar atención médica si su asma o EPOC permanece sin control o empeora después del inicio del tratamiento con DUPIXENT.
5.5 Riesgo asociado con la reducción abrupta de la dosis de corticosteroides
No suspenda abruptamente los corticosteroides sistémicos, tópicos o inhalados al iniciar la terapia con DUPIXENT. Las reducciones en la dosis de corticosteroides, si son apropiadas, deben ser graduales y realizarse bajo la supervisión directa de un profesional de la salud. La reducción de la dosis de corticosteroides puede estar asociada con síntomas de abstinencia sistémica y/o desenmascarar afecciones previamente suprimidas por la terapia con corticosteroides sistémicos.
5.6 Pacientes con asma comórbida
Avise a los pacientes con asma comórbida que no ajusten ni detengan sus tratamientos para el asma sin consultar con sus médicos.
5.7 Artralgia
Se ha notificado artralgia con el uso de DUPIXENT, y algunos pacientes informan alteraciones de la marcha o disminución de la movilidad asociada con síntomas articulares; algunos casos dieron lugar a hospitalización [ver Reacciones adversas (6.1)]. En los informes de poscomercialización, la aparición de artralgia fue variable, desde días hasta meses después de la primera dosis de DUPIXENT. Los síntomas de algunos pacientes se resolvieron mientras continuaban el tratamiento con DUPIXENT y otros pacientes se recuperaron o se estaban recuperando después de la suspensión de DUPIXENT.
Aconseje a los pacientes que informen a su médico sobre la aparición o el empeoramiento de los síntomas articulares. Si los síntomas persisten o empeoran, considere una evaluación reumatológica y/o la interrupción de DUPIXENT.
5.8 Infecciones parasitarias (Helmintos)
Los pacientes con infecciones por helmintos conocidas fueron excluidos de la participación en los estudios clínicos. Se desconoce si DUPIXENT influirá en la respuesta inmunitaria contra las infecciones por helmintos.
Trate a los pacientes con infecciones por helmintos preexistentes antes de iniciar el tratamiento con DUPIXENT. Si los pacientes se infectan mientras reciben tratamiento con DUPIXENT y no responden al tratamiento antihelmíntico, suspenda el tratamiento con DUPIXENT hasta que la infección se resuelva. Se notificaron reacciones adversas de infecciones por helmintos (5 casos de enterobiasis y 1 caso de ascariasis) en sujetos pediátricos de 6 a 11 años que participaron en el programa de desarrollo del asma pediátrico [ver Reacciones adversas (6.1)].
5.9 Vacunaciones
Considere completar todas las vacunas apropiadas para la edad, según lo recomendado por las pautas de inmunización actuales, antes de iniciar el tratamiento con DUPIXENT. Evite el uso de vacunas vivas durante el tratamiento con DUPIXENT. Se desconoce si la administración de vacunas vivas durante el tratamiento con DUPIXENT afectará la seguridad o la eficacia de estas vacunas. Hay datos limitados disponibles sobre la coadministración de DUPIXENT con vacunas no vivas [ver Farmacología clínica (12.2)].
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas clínicamente significativas se describen en otras secciones del prospecto:
- Hipersensibilidad [ver Advertencias y precauciones (5.1)]
- Conjuntivitis y queratitis [ver Advertencias y precauciones (5.2)]
- Artralgia [ver Advertencias y precauciones (5.7)]
- Infecciones parasitarias (por helmintos) [ver Advertencias y precauciones (5.8)]
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se llevan a cabo en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un medicamento no pueden compararse directamente con las tasas en los ensayos clínicos de otro medicamento y pueden no reflejar las tasas observadas en la práctica.
Adultos con dermatitis atópica
Tres ensayos aleatorizados, doble ciego, controlados con placebo, multicéntricos (SOLO 1, SOLO 2 y CHRONOS) y un ensayo de determinación de dosis (AD-1021) evaluaron la seguridad de DUPIXENT en sujetos con DA de moderada a grave. La población de seguridad tenía una edad media de 38 años; el 41 % de los sujetos eran mujeres, el 67 % eran de raza blanca, el 24 % eran asiáticos y el 6 % eran de raza negra; en cuanto a las enfermedades concomitantes, el 48 % de los sujetos tenían asma, el 49 % tenían rinitis alérgica, el 37 % tenían alergia alimentaria y el 27 % tenían conjuntivitis alérgica. En estos 4 ensayos, 1472 sujetos fueron tratados con inyecciones subcutáneas de DUPIXENT, con o sin corticosteroides tópicos (CET) concomitantes.
Un total de 739 sujetos fueron tratados con DUPIXENT durante al menos 1 año en el programa de desarrollo para la DA de moderada a grave.
SOLO 1, SOLO 2 y AD-1021 compararon la seguridad de la monoterapia con DUPIXENT con placebo hasta la semana 16. CHRONOS comparó la seguridad de DUPIXENT + CET con placebo + CET hasta la semana 52.
AD-1225 es un ensayo de extensión abierto (OLE) multicéntrico que evaluó la seguridad a largo plazo de dosis repetidas de DUPIXENT durante 260 semanas de tratamiento en adultos con DA de moderada a grave que habían participado previamente en ensayos controlados de DUPIXENT o habían sido seleccionados para SOLO 1 o SOLO 2. Los datos de seguridad en AD-1225 reflejan la exposición a DUPIXENT 200 mg SC, 300 mg SC y 300 mg cada dos semanas en 2677 sujetos, incluyendo 2254 expuestos durante al menos 52 semanas, 1224 expuestos durante al menos 100 semanas, 561 expuestos durante al menos 148 semanas y 179 expuestos durante al menos 260 semanas.
Semanas 0 a 16 (SOLO 1, SOLO 2, CHRONOS y AD-1021)
En los ensayos de monoterapia con DUPIXENT (SOLO 1, SOLO 2 y AD-1021) hasta la semana 16, la proporción de sujetos que interrumpieron el tratamiento debido a eventos adversos fue del 1,9 % tanto en el grupo de DUPIXENT 300 mg cada dos semanas como en el grupo de placebo. La Tabla 6 resume las reacciones adversas que ocurrieron con una tasa de al menos el 1 % en los grupos de monoterapia con DUPIXENT 300 mg cada dos semanas, y en el grupo de DUPIXENT + CET, todas con una tasa más alta que en sus respectivos grupos de comparación durante las primeras 16 semanas de tratamiento.
| Reacción adversa | Monoterapia con DUPIXENT* | DUPIXENT + CET† | ||
|---|---|---|---|---|
| DUPIXENT 300 mg cada dos semanas‡ |
Placebo | DUPIXENT 300 mg cada dos semanas‡ + CET |
Placebo + CET | |
| N=529 n (%) |
N=517 n (%) |
N=110 n (%) |
N=315 n (%) |
|
|
||||
| Reacción en el lugar de la inyección | 51 (10) | 28 (5) | 11 (10) | 18 (6) |
| Conjuntivitis§ | 51 (10) | 12 (2) | 10 (9) | 15 (5) |
| Blepharitis | 2 (<1) | 1 (<1) | 5 (5) | 2 (1) |
| Herpes oral | 20 (4) | 8 (2) | 3 (3) | 5 (2) |
| Queratitis¶ | 1 (<1) | 0 | 4 (4) | 0 |
| Prurito ocular | 3 (1) | 1 (<1) | 2 (2) | 2 (1) |
| Otra infección por el virus del herpes simple# | 10 (2) | 6 (1) | 1 (1) | 1 (<1) |
| Ojo seco | 1 (<1) | 0 | 2 (2) | 1 (<1) |
Seguridad hasta la semana 52 (CHRONOS)
En el ensayo de DUPIXENT con TCS concomitante (CHRONOS) hasta la semana 52, la proporción de sujetos que interrumpieron el tratamiento debido a eventos adversos fue del 1.8 % en el grupo de DUPIXENT 300 mg cada 2 semanas + TCS y del 7.6 % en el grupo de placebo + TCS. Dos sujetos interrumpieron DUPIXENT debido a reacciones adversas: dermatitis atópica (1 sujeto) y dermatitis exfoliativa (1 sujeto).
El perfil de seguridad de DUPIXENT + TCS hasta la semana 52 fue generalmente consistente con el perfil de seguridad observado en la semana 16.
Seguridad hasta las 260 semanas (AD-1225)
El perfil de seguridad a largo plazo observado en este ensayo hasta las 260 semanas fue generalmente consistente con el perfil de seguridad de DUPIXENT observado en estudios controlados.
Sujetos pediátricos de 12 a 17 años de edad con dermatitis atópica
Se evaluó la seguridad de DUPIXENT en un ensayo de 250 sujetos pediátricos de 12 a 17 años de edad con DA de moderada a grave (AD-1526). El perfil de seguridad de DUPIXENT en estos sujetos hasta la semana 16 fue similar al perfil de seguridad observado en adultos con DA.
Se evaluó la seguridad a largo plazo de DUPIXENT en un estudio de extensión abierto en sujetos pediátricos de 12 a 17 años de edad con DA de moderada a grave (AD-1434). El perfil de seguridad de DUPIXENT en sujetos seguidos hasta la semana 52 fue similar al perfil de seguridad observado en la semana 16 en AD-1526. El perfil de seguridad a largo plazo de DUPIXENT observado en sujetos pediátricos de 12 a 17 años de edad fue consistente con el observado en adultos con DA.
Sujetos pediátricos de 6 a 11 años de edad con dermatitis atópica
Se evaluó la seguridad de DUPIXENT con TCS concomitante en un ensayo de 367 sujetos pediátricos de 6 a 11 años de edad con DA grave (AD-1652). El perfil de seguridad de DUPIXENT + TCS en estos sujetos hasta la semana 16 fue similar al perfil de seguridad de los ensayos en sujetos adultos y pediátricos de 12 a 17 años de edad con DA.
Se evaluó la seguridad a largo plazo de DUPIXENT ± TCS en un estudio de extensión abierto de 368 sujetos pediátricos de 6 a 11 años de edad con DA (AD-1434). Entre los sujetos que ingresaron a este estudio, 110 (30 %) tenían DA moderada y 72 (20 %) tenían DA grave al momento de la inscripción en AD-1434. El perfil de seguridad de DUPIXENT ± TCS en sujetos seguidos hasta la semana 52 fue similar al perfil de seguridad observado hasta la semana 16 en AD-1652. El perfil de seguridad a largo plazo de DUPIXENT ± TCS observado en sujetos pediátricos de 6 a 11 años de edad fue consistente con el observado en sujetos adultos y pediátricos de 12 a 17 años de edad con DA [ver Uso en poblaciones específicas (8.4)].
Sujetos pediátricos de 6 meses a 5 años de edad con dermatitis atópica
Se evaluó la seguridad de DUPIXENT con TCS concomitante en un ensayo de 161 sujetos pediátricos de 6 meses a 5 años de edad con DA de moderada a grave (AD-1539). El perfil de seguridad de DUPIXENT + TCS en estos sujetos hasta la semana 16 fue similar al perfil de seguridad de los ensayos en adultos y sujetos pediátricos de 6 a 17 años de edad con DA.
Se evaluó la seguridad a largo plazo de DUPIXENT ± TCS en un estudio de extensión abierto de 180 sujetos pediátricos de 6 meses a 5 años de edad con DA (AD-1434). La mayoría de los sujetos fueron tratados con DUPIXENT 300 mg cada 4 semanas. El perfil de seguridad de DUPIXENT ± TCS en sujetos seguidos hasta la semana 52 fue similar al perfil de seguridad observado hasta la semana 16 en AD-1539. El perfil de seguridad a largo plazo de DUPIXENT ± TCS observado en sujetos pediátricos de 6 meses a 5 años de edad fue consistente con el observado en adultos y sujetos pediátricos de 6 a 17 años de edad con DA. Además, se reportó enfermedad de manos, pies y boca en 9 (5 %) sujetos pediátricos y papiloma cutáneo en 4 (2 %) sujetos pediátricos tratados con DUPIXENT ± TCS. Estos casos no condujeron a la interrupción del fármaco del estudio [ver Uso en poblaciones específicas (8.4)].
Dermatitis atópica con afectación de manos o pies
Se evaluó la seguridad de DUPIXENT en un ensayo de 16 semanas, multicéntrico, aleatorizado, doble ciego, de grupos paralelos, controlado con placebo (Liberty-AD-HAFT) en 133 sujetos adultos y pediátricos de 12 a 17 años de edad con dermatitis atópica con afectación de manos o pies de moderada a grave [ver Estudios clínicos (14)]. En este ensayo, 67 sujetos recibieron DUPIXENT y 66 sujetos recibieron placebo. Los sujetos tratados con DUPIXENT recibieron la dosis recomendada en función de su edad y peso corporal [ver Dosificación y administración (2.3)]. El perfil de seguridad de DUPIXENT en estos sujetos hasta la semana 16 fue consistente con el perfil de seguridad de los estudios en sujetos adultos y pediátricos de 6 meses de edad y mayores con DA de moderada a grave.
Asma
Adultos y sujetos pediátricos de 12 años de edad y mayores con asma
Un total de 2888 adultos y sujetos pediátricos de 12 a 17 años de edad con asma (AS) de moderada a grave fueron evaluados en 3 ensayos aleatorizados, controlados con placebo, multicéntricos, de 24 a 52 semanas de duración (DRI12544, QUEST y VENTURE). De estos, 2678 tenían antecedentes de 1 o más exacerbaciones graves en el año anterior a la inscripción a pesar del uso regular de dosis medias a altas de corticosteroides inhalados más un(os) controlador(es) adicional(es) (DRI12544 y QUEST). Se inscribió a un total de 210 sujetos con asma dependiente de corticosteroides orales que recibían altas dosis de corticosteroides inhalados más hasta dos controladores adicionales (VENTURE). La población de seguridad (DRI12544 y QUEST) tenía entre 12 y 87 años de edad, de los cuales el 63 % eran mujeres y el 82 % eran blancos. Se administró DUPIXENT 200 mg o 300 mg por vía subcutánea cada 2 semanas, después de una dosis inicial de 400 mg o 600 mg, respectivamente.
En DRI12544 y QUEST, la proporción de sujetos que interrumpieron el tratamiento debido a eventos adversos fue del 4% del grupo placebo, 3% del grupo DUPIXENT 200 mg Q2W y 6% del grupo DUPIXENT 300 mg Q2W.
La Tabla 7 resume las reacciones adversas que ocurrieron a una tasa de al menos 1% en sujetos tratados con DUPIXENT y a una tasa mayor que en sus respectivos grupos de comparación en DRI12544 y QUEST.
| Reacción adversa | DRI12544 y QUEST | ||
|---|---|---|---|
| DUPIXENT 200 mg Q2W |
DUPIXENT 300 mg Q2W |
Placebo | |
| N=779 n (%) |
N=788 n (%) |
N=792 n (%) |
|
|
|||
| Reacciones en el sitio de inyección* | 111 (14%) | 144 (18%) | 50 (6%) |
| Dolor orofaríngeo | 13 (2%) | 19 (2%) | 7 (1%) |
| Eosinophilia† | 17 (2%) | 16 (2%) | 2 (<1%) |
Las reacciones en el lugar de inyección fueron más comunes con la dosis de carga (inicial).
El perfil de seguridad de DUPIXENT hasta la semana 52 fue generalmente consistente con el perfil de seguridad observado en la semana 24.
Pacientes pediátricos de 6 a 11 años de edad con asma
Se evaluó la seguridad de DUPIXENT en 405 pacientes pediátricos de 6 a 11 años de edad con asma de moderada a grave (VOYAGE). El perfil de seguridad de DUPIXENT en estos pacientes hasta la semana 52 fue similar al perfil de seguridad de los estudios en pacientes adultos y pediátricos de 12 años de edad y mayores con asma de moderada a grave con la adición de infecciones por helmintos. Se notificaron infecciones por helmintos en el 2,2 % (6 pacientes) del grupo de DUPIXENT y en el 0,7 % (1 paciente) del grupo de placebo. La mayoría de los casos fueron enterobiasis, notificadas en el 1,8 % (5 pacientes) del grupo de DUPIXENT y en ninguno del grupo de placebo. Hubo un caso de ascariasis en el grupo de DUPIXENT. Todos los casos de infección por helmintos fueron de leves a moderados y los pacientes se recuperaron con tratamiento antihelmíntico sin suspender el tratamiento con DUPIXENT.
Rinosinusitis crónica con pólipos nasales
Se evaluó a un total de 722 pacientes adultos con rinosinusitis crónica con pólipos nasales (RSCcPN) en 2 ensayos multicéntricos, aleatorizados y controlados con placebo, de 24 a 52 semanas de duración (SINUS-24 y SINUS-52). El conjunto de seguridad consistió en datos de las primeras 24 semanas de tratamiento de ambos estudios.
En el conjunto de seguridad, la proporción de pacientes adultos que suspendieron el tratamiento debido a eventos adversos fue del 5 % en el grupo de placebo y del 2 % en el grupo de DUPIXENT 300 mg cada 2 semanas.
La Tabla 8 resume las reacciones adversas que ocurrieron a una tasa de al menos el 1 % en pacientes adultos tratados con DUPIXENT y a una tasa mayor que en su respectivo grupo de comparación en SINUS-24 y SINUS-52.
| Reacción adversa | SINUS-24 y SINUS-52 | |
|---|---|---|
| DUPIXENT 300 mg cada 2 semanas |
Placebo | |
| N=440 n (%) |
N=282 n (%) |
|
|
||
| Reacciones en el lugar de inyección* | 28 (6%) | 12 (4%) |
| Conjuntivitis† | 7 (2%) | 2 (1%) |
| Artralgia | 14 (3%) | 5 (2%) |
| Gastritis | 7 (2%) | 2 (1%) |
| Insomnio | 6 (1%) | 0 (<1%) |
| Eosinofilia | 5 (1%) | 1 (<1%) |
| Dolor de muelas | 5 (1%) | 1 (<1%) |
El perfil de seguridad de DUPIXENT hasta la semana 52 fue generalmente consistente con el perfil de seguridad observado en la semana 24.
Esofagitis Eosinofílica
Adultos y Sujetos Pediátricos de 12 Años de Edad y Mayores con EeE
Un total de 239 sujetos adultos y pediátricos de 12 años de edad y mayores, con un peso de al menos 40 kg, con EeE fueron evaluados en un ensayo aleatorizado, doble ciego, de grupos paralelos, multicéntrico, controlado con placebo, que incluyó dos períodos de tratamiento de 24 semanas (Estudio EoE-1 Partes A y B) y recibieron DUPIXENT 300 mg SC cada dos semanas o placebo [ver Estudios Clínicos (14.4)].
La proporción de sujetos que interrumpieron el tratamiento debido a eventos adversos fue del 2% en el grupo placebo y del 2% en el grupo DUPIXENT 300 mg SC cada dos semanas.
La Tabla 9 resume las reacciones adversas que ocurrieron a una tasa de al menos el 2% en los sujetos tratados con DUPIXENT y a una tasa mayor que en su respectivo grupo de comparación en las Partes A y B.
| Estudio EoE-1 Partes A y B | ||
|---|---|---|
| Reacción Adversa | DUPIXENT 300 mg SC cada dos semanas N=122 n (%) |
Placebo N=117 n (%) |
|
||
| Reacciones en el sitio de inyección* | 46 (38%) | 39 (33%) |
| Infecciones de las vías respiratorias superiores† | 22 (18%) | 12 (10%) |
| Artralgia | 3 (2%) | 1 (1%) |
| Infecciones por el virus del herpes‡ | 3 (2%) | 1 (1%) |
El perfil de seguridad de DUPIXENT en 72 sujetos pediátricos de 12 a 17 años de edad, con un peso de al menos 40 kg, y adultos en las Partes A y B fue similar.
Sujetos pediátricos de 1 a 11 años de edad, con un peso de al menos 15 kg, con EoE
Un total de 61 sujetos pediátricos de 1 a 11 años de edad, con un peso de al menos 15 kg, con EoE fueron evaluados en un ensayo aleatorizado, ciego, en grupo paralelo, multicéntrico, que incluyó un período inicial de tratamiento controlado con placebo de 16 semanas (Estudio EoE-2 Parte A) y un período de tratamiento activo prolongado de 36 semanas (Estudio EoE-2 Parte B). Los sujetos de la Parte A recibieron un régimen de dosificación basado en el peso de DUPIXENT o placebo [véase Estudios clínicos (14.4)]. Todos los sujetos de la Parte B completaron la Parte A y recibieron tratamiento activo con regímenes de dosificación basados en el peso de DUPIXENT en la Parte B (N = 47).
El perfil de seguridad de DUPIXENT hasta la Semana 16 del Estudio EoE-2 Parte A fue generalmente similar al perfil de seguridad en adultos y sujetos pediátricos de 12 años de edad y mayores con EoE. En la Parte B, se informó una infección por helmintos en un sujeto tratado con DUPIXENT.
Prurigo Nodularis
Un total de 309 sujetos adultos con prurigo nodularis (PN) fueron evaluados en dos ensayos aleatorizados, doble ciego, controlados con placebo, multicéntricos de 24 semanas (PRIME y PRIME2). El grupo de seguridad incluyó datos del período de tratamiento de 24 semanas y del período de seguimiento de 12 semanas de ambos ensayos.
La proporción de sujetos que suspendieron el tratamiento debido a eventos adversos fue del 3% en el grupo de placebo y del 0% en el grupo de DUPIXENT 300 mg Q2W.
La población de seguridad tenía una edad media de 49 años; el 65% de los sujetos eran mujeres, el 56% eran blancos, el 34% eran asiáticos y el 6% eran negros o afroamericanos. Los sujetos con condiciones comórbidas incluyeron el 43% de los sujetos con antecedentes de atopia (definida como tener un historial médico de DA, rinitis alérgica / rinoconjuntivitis alérgica, asma o alergia alimentaria), el 8% de los sujetos con antecedentes de hipotiroidismo y el 9% de los sujetos con antecedentes de diabetes mellitus tipo 2.
La Tabla 10 resume las reacciones adversas que ocurrieron en una tasa de al menos el 2% en los sujetos tratados con DUPIXENT y a una tasa más alta que en su grupo comparador respectivo en PRIME y PRIME2.
| Reacción adversa | PRIME y PRIME2 | |
|---|---|---|
| DUPIXENT 300 mg Q2W |
Placebo | |
| N = 152 n (%) |
N = 157 n (%) |
|
|
||
| Nasofaringitis* | 8 (5%) | 3 (2%) |
| Conjuntivitis† | 6 (4%) | 2 (1%) |
| Infección por herpes‡ | 5 (3%) | 0% |
| Mareo§ | 5 (3%) | 2 (1%) |
| Mialgia¶ | 5 (3%) | 2 (1%) |
| Diarrea | 4 (3%) | 1 (1%) |
Enfermedad Pulmonar Obstructiva Crónica
Un total de 1874 sujetos adultos con enfermedad pulmonar obstructiva crónica (EPOC) inadecuadamente controlada y un fenotipo eosinofílico fueron evaluados en dos ensayos aleatorizados, doble ciego, multicéntricos, de grupos paralelos, controlados con placebo con un período de tratamiento de 52 semanas (BOREAS y NOTUS) [ver Estudios Clínicos (14.6)]. De los sujetos aleatorizados, 1872 recibieron al menos una dosis de DUPIXENT 300 mg o placebo por vía subcutánea cada 2 semanas (Q2W). La seguridad de DUPIXENT se evaluó en la población de seguridad agrupada de BOREAS y NOTUS, que consistió en 938 sujetos adultos tratados con DUPIXENT. De los sujetos tratados con DUPIXENT, el 98% utilizó la terapia triple inhalada al inicio del estudio (que comprende un corticosteroide inhalado, un beta-agonista de acción prolongada y un antagonista muscarínico de acción prolongada), y el 97% tenía bronquitis crónica.
La Tabla 11 resume las reacciones adversas que ocurrieron en al menos el 2% de los sujetos tratados con DUPIXENT y a una tasa mayor que el placebo en los ensayos BOREAS y NOTUS.
| Adverse Reaction | BOREAS and NOTUS | |
|---|---|---|
| DUPIXENT 300 mg Q2W |
Placebo | |
| N=938 n (%) |
N=934 n (%) |
|
| Viral Infection* | 133 (14.2) | 115 (12.3) |
| Headache | 73 (7.8) | 62 (6.6) |
| Nasopharyngitis | 73 (7.8) | 69 (7.4) |
| Back Pain | 42 (4.5) | 29 (3.1) |
| Diarrhea* | 35 (3.7) | 30 (3.2) |
| Arthralgia | 29 (3.1) | 25 (2.7) |
| Urinary Tract Infection | 28 (3.0) | 18 (1.9) |
| Local Administration Reaction* | 26 (2.8) | 6 (0.6) |
| Injection Site Reaction | 11 (1.2) | 2 (0.2) |
| Rhinitis | 24 (2.6) | 17 (1.8) |
| Eosinophilia† | 22 (2.3) | 7 (0.7) |
| Toothache | 20 (2.1) | 11 (1.2) |
| Gastritis | 19 (2) | 7 (0.7) |
Reacción adversa menos frecuente en sujetos con EPOC: colecistitis
En sujetos adultos con EPOC, se notificó colecistitis en 6 sujetos (0,6 %) en el grupo de DUPIXENT en comparación con 1 sujeto (0,1 %) en el grupo de placebo. Entre estos sujetos, se notificó colecistitis grave en 4 (0,4 %) del grupo de DUPIXENT en comparación con el 0 % del grupo de placebo.
Reacciones adversas específicas para DA, asma, CRSwNP, EoE, NP y EPOC
Conjuntivitis y queratitis
En sujetos adultos con DA, se notificó conjuntivitis en el 10 % (34 por 100 sujetos-año) en el grupo de dosis de 300 mg cada 2 semanas y en el 2 % del grupo de placebo (8 por 100 sujetos-año) durante el período de tratamiento de 16 semanas de los ensayos de monoterapia (SOLO 1, SOLO 2 y AD-1021). Durante el período de tratamiento de 52 semanas del ensayo de DA con tratamiento concomitante (CHRONOS), se notificó conjuntivitis en el 16 % del grupo de DUPIXENT 300 mg cada 2 semanas + corticosteroides tópicos (CST) (20 por 100 sujetos-año) y en el 9 % del grupo de placebo + CST (10 por 100 sujetos-año). Durante el ensayo OLE a largo plazo con datos de hasta 260 semanas (AD-1225), se notificó conjuntivitis en el 21 % del grupo de DUPIXENT (12 por 100 sujetos-año).
En los ensayos de monoterapia con DUPIXENT para la DA (SOLO 1, SOLO 2 y AD-1021) hasta la semana 16, se notificó queratitis en <1 % del grupo de DUPIXENT (1 por 100 sujetos-año) y en el 0 % del grupo de placebo (0 por 100 sujetos-año). En el ensayo de DA de 52 semanas con DUPIXENT + corticosteroides tópicos (CST) (CHRONOS), se notificó queratitis en el 4 % del grupo de DUPIXENT + CST (4 por 100 sujetos-año) y en el 2 % del grupo de placebo + CST (2 por 100 sujetos-año). La conjuntivitis y la queratitis se produjeron con mayor frecuencia en sujetos con DA que recibieron DUPIXENT. La conjuntivitis fue el trastorno ocular notificado con mayor frecuencia. Durante el ensayo OLE a largo plazo con datos de hasta 260 semanas (AD-1225), se notificó queratitis en el 3 % del grupo de DUPIXENT (1 por 100 sujetos-año). La mayoría de los sujetos con conjuntivitis o queratitis se recuperaron o se estaban recuperando durante el período de tratamiento.
Entre los sujetos con asma, la frecuencia de conjuntivitis y queratitis fue similar entre DUPIXENT y placebo.
En sujetos adultos con CRSwNP, la frecuencia de conjuntivitis fue del 2 % en el grupo de DUPIXENT en comparación con el 1 % en el grupo de placebo en el grupo de seguridad de 24 semanas; estos sujetos se recuperaron.
En el estudio de CRSwNP de 52 semanas (SINUS-52), la frecuencia de conjuntivitis fue del 3 % en los sujetos adultos con DUPIXENT y del 1 % en los sujetos con placebo; todos estos sujetos se recuperaron. No hubo casos de queratitis notificados en el programa de desarrollo de CRSwNP [ver Advertencias y precauciones (5.2)].
Entre los sujetos con EoE, no hubo notificaciones de conjuntivitis ni queratitis en el grupo de DUPIXENT en los ensayos controlados con placebo [ver Advertencias y precauciones (5.2)]. En el período de extensión de tratamiento activo de 36 semanas del estudio EoE-2, parte B, se notificó conjuntivitis en el 4 % de los sujetos pediátricos tratados con DUPIXENT con EoE.
Entre los sujetos con NP, la frecuencia de conjuntivitis fue del 4 % en el grupo de DUPIXENT en comparación con el 1 % en el grupo de placebo; todos estos sujetos se recuperaron o se estaban recuperando durante el período de tratamiento. No hubo casos de queratitis notificados en el programa de desarrollo de NP [ver Advertencias y precauciones (5.2)].
Entre los sujetos con EPOC, la frecuencia de conjuntivitis y queratitis fue del 1,4 % y el 0,1 % en el grupo de DUPIXENT y del 1 % y el 0 % en el grupo de placebo, respectivamente [ver Advertencias y precauciones (5.2)].
Eczema herpético y herpes zóster
La tasa de eczema herpético fue similar en los grupos de placebo y DUPIXENT en los ensayos de DA. Las tasas se mantuvieron estables hasta las 260 semanas en el ensayo OLE a largo plazo (AD-1225).
Se notificó herpes zóster en <1 % de los grupos de DUPIXENT (1 por 100 sujetos-año) y en <1 % del grupo de placebo (1 por 100 sujetos-año) en los ensayos de monoterapia de DA de 16 semanas. En el ensayo de DA de 52 semanas con DUPIXENT + CST, se notificó herpes zóster en el 1 % del grupo de DUPIXENT + CST (1 por 100 sujetos-año) y en el 2 % del grupo de placebo + CST (2 por 100 sujetos-año). Durante el ensayo OLE a largo plazo con datos de hasta 260 semanas (AD-1225), el 2,0 % de los sujetos tratados con DUPIXENT notificaron herpes zóster (0,94 por 100 sujetos-año de seguimiento). Entre los sujetos con asma, la frecuencia de herpes zóster fue similar entre DUPIXENT y placebo. Entre los sujetos con CRSwNP o EoE no se notificaron casos de herpes zóster ni eczema herpético.
Entre los sujetos con NP, se notificó herpes zóster y herpes zóster oftálmico en <1 % del grupo de DUPIXENT (1 por 100 sujetos-año) y en el 0 % del grupo de placebo.
Entre los sujetos con EPOC, se notificó herpes zóster en el 0,9 % del grupo de DUPIXENT y en el 0,2 % del grupo de placebo. Se notificó herpes zóster oftálmico en el 0,1 % del grupo de DUPIXENT y en el 0,2 % del grupo de placebo.
Reacciones de hipersensibilidad
Se notificaron reacciones de hipersensibilidad en <1 % de los sujetos tratados con DUPIXENT. Estas incluyeron anafilaxia, enfermedad del suero o reacciones similares a la enfermedad del suero, urticaria generalizada, erupción cutánea, eritema nudoso y eritema multiforme [ver Contraindicaciones (4), Advertencias y precauciones (5.1) y Farmacología clínica (12.6)].
Eosinófilos
Los sujetos tratados con DUPIXENT con DA, asma, CRSwNP y EPOC tuvieron un mayor aumento inicial del recuento de eosinófilos en sangre con respecto al valor inicial en comparación con los sujetos tratados con placebo. En sujetos adultos con DA (SOLO 1, SOLO 2 y AD-1021), los aumentos medios y medianos de los eosinófilos en sangre desde el valor inicial hasta la semana 4 fueron de 100 y 0 células/mcl, respectivamente. En sujetos pediátricos <6 años con DA, los aumentos medios y medianos desde el valor inicial hasta la semana 4 fueron de 478 y 90 células/mcl, respectivamente.
En sujetos adultos y pediátricos de 12 años de edad y mayores con asma (DRI12544 y QUEST), los aumentos medios y medianos en los eosinófilos en sangre desde el inicio hasta la semana 4 fueron de 130 y 10 células/mcL, respectivamente. En sujetos de 6 a 11 años de edad con asma (VOYAGE), los aumentos medios y medianos en los eosinófilos en sangre desde el inicio hasta la semana 12 fueron de 124 y 0 células/mcL, respectivamente.
En sujetos adultos con CRSwNP (SINUS-24 y SINUS-52), los aumentos medios y medianos en los eosinófilos en sangre desde el inicio hasta la semana 16 fueron de 150 y 50 células/mcL, respectivamente.
En sujetos con EPOC (BOREAS y NOTUS), los aumentos medios y medianos en los eosinófilos en sangre desde el inicio hasta la semana 8 fueron de 60 y 0 células/mcL, respectivamente.
No se observó un aumento desde el inicio en el recuento de eosinófilos en sangre en sujetos adultos y pediátricos de 12 años de edad y mayores con EoE tratados con DUPIXENT en comparación con placebo (Estudio EoE-1). En sujetos pediátricos de 1 a 11 años de edad con EoE (Estudio EoE-2 Parte A), los recuentos de eosinófilos en sangre fueron generalmente consistentes con los observados en el Estudio EoE-1.
En sujetos con PN (PRIME y PRIME2), la disminución media y mediana en los eosinófilos en sangre desde el inicio hasta la semana 4 fueron de 9 y 10 células/mcL, respectivamente.
En los ensayos para la indicación de EPOC, la eosinofilia emergente del tratamiento (≥500 células/mcL) fue mayor en DUPIXENT (41,7%) que en el grupo placebo (39,4%); ninguno de los casos se asoció con síntomas clínicos, y la eosinofilia emergente del tratamiento (≥1000 células/mcL) fue mayor en DUPIXENT (13,6%) que en el grupo placebo (8,1%).
En los ensayos para las indicaciones de DA, asma y CRSwNP, la incidencia de eosinofilia emergente del tratamiento (≥500 células/mcL) fue similar en los grupos de DUPIXENT y placebo.
En los ensayos para la indicación de PN, la incidencia de eosinofilia emergente del tratamiento (≥500 células/mcL) fue menor en DUPIXENT que en el grupo placebo.
La eosinofilia emergente del tratamiento (≥5000 células/mcL) se notificó en <3% de los sujetos tratados con DUPIXENT y en <0,5% de los sujetos tratados con placebo (SOLO 1, SOLO 2 y AD-1021; DRI12544, QUEST y VOYAGE; SINUS-24 y SINUS-52; PRIME y PRIME2; BOREAS y NOTUS). Los recuentos de eosinófilos en sangre disminuyeron hasta casi el valor inicial o se mantuvieron por debajo de los niveles iniciales (PRIME y PRIME2; BOREAS y NOTUS) durante el tratamiento del estudio. En el estudio AD-1539, se notificó eosinofilia emergente del tratamiento (≥5000 células/mcL) en el 8% de los sujetos tratados con DUPIXENT y en el 0% de los sujetos tratados con placebo [ver Advertencias y precauciones (5.3)].
Eventos tromboembólicos cardiovasculares
En el ensayo controlado con placebo de 1 año de duración en sujetos adultos y pediátricos de 12 años de edad y mayores con asma (QUEST), se notificaron eventos tromboembólicos cardiovasculares (muertes cardiovasculares, infartos de miocardio no mortales y accidentes cerebrovasculares no mortales) en 1 (0,2%) del grupo de DUPIXENT 200 mg cada 2 semanas, 4 (0,6%) del grupo de DUPIXENT 300 mg cada 2 semanas y 2 (0,3%) del grupo placebo.
En el ensayo controlado con placebo de 1 año de duración en sujetos con DA (CHRONOS), se notificaron eventos tromboembólicos cardiovasculares (muertes cardiovasculares, infartos de miocardio no mortales y accidentes cerebrovasculares no mortales) en 1 (0,9%) del grupo de DUPIXENT + TCS 300 mg cada 2 semanas, 0 (0,0%) del grupo de DUPIXENT + TCS 300 mg cada semana y 1 (0,3%) del grupo placebo + TCS.
En el ensayo controlado con placebo de 24 semanas de duración en sujetos adultos con CRSwNP (SINUS-24), se notificaron eventos tromboembólicos cardiovasculares (muertes cardiovasculares, infartos de miocardio no mortales y accidentes cerebrovasculares no mortales) en 1 (0,7%) del grupo de DUPIXENT y 0 (0,0%) del grupo placebo.
En el ensayo controlado con placebo de 1 año de duración en sujetos adultos con CRSwNP (SINUS-52), no se notificaron casos de eventos tromboembólicos cardiovasculares (muertes cardiovasculares, infartos de miocardio no mortales y accidentes cerebrovasculares no mortales) en ningún brazo de tratamiento.
En el ensayo controlado con placebo de 24 semanas de duración en sujetos con EoE (Estudio EoE-1 Partes A y B), no se notificaron casos de eventos tromboembólicos cardiovasculares (muertes cardiovasculares, infartos de miocardio no mortales y accidentes cerebrovasculares no mortales) en ningún brazo de tratamiento.
6.2 Experiencia poscomercialización
Las siguientes reacciones adversas se han identificado durante el uso poscomercialización de DUPIXENT. Debido a que estas reacciones se notifican de forma voluntaria a partir de una población de tamaño incierto, no siempre es posible estimar de forma fiable su frecuencia o establecer una relación causal con la exposición al fármaco.
Trastornos del sistema inmunitario: angioedema [ver Advertencias y precauciones (5.1)]
Trastornos de la piel y del tejido subcutáneo: Reacciones cutáneas faciales, como eritema, erupción cutánea, descamación, edema, pápulas, prurito, ardor y dolor
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Registro de Exposición al Embarazo
Existe un registro de exposición al embarazo que monitorea los resultados del embarazo en mujeres expuestas a DUPIXENT durante el embarazo.
Los profesionales de la salud y los pacientes pueden llamar al 1-877-311-8972 o visitar https://mothertobaby.org/ongoing-study/dupixent/ para inscribirse o para obtener información sobre el registro.
Resumen de Riesgos
Los datos disponibles de informes de casos y series de casos con el uso de DUPIXENT en mujeres embarazadas no han identificado un riesgo asociado al fármaco de defectos de nacimiento mayores, aborto espontáneo o resultados maternos o fetales adversos. Se sabe que los anticuerpos IgG humanos cruzan la barrera placentaria; por lo tanto, DUPIXENT puede transmitirse de la madre al feto en desarrollo. Existen efectos adversos en los resultados maternos y fetales asociados con el asma en el embarazo (ver Consideraciones Clínicas). En un estudio de desarrollo prenatal y postnatal mejorado, no se observaron efectos adversos en el desarrollo en la descendencia nacida de monos preñados después de la administración subcutánea de un anticuerpo homólogo contra el receptor alfa de la interleucina-4 (IL-4Rα) durante la organogénesis hasta el parto a dosis de hasta 10 veces la dosis humana máxima recomendada (MRHD) (ver Datos).
El riesgo de fondo de defectos de nacimiento mayores y aborto espontáneo para las poblaciones indicadas es desconocido. Todos los embarazos tienen un riesgo de fondo de defectos de nacimiento, pérdida u otros resultados adversos. En la población general de EE. UU., el riesgo de fondo estimado de defectos de nacimiento mayores y aborto espontáneo en embarazos clínicamente reconocidos es del 2% al 4% y del 15% al 20%, respectivamente.
Consideraciones Clínicas
Riesgo Materno y/o Embriofetal Asociado a la Enfermedad
En mujeres con asma mal controlada o moderadamente controlada, la evidencia demuestra que existe un mayor riesgo de preeclampsia en la madre y prematuridad, bajo peso al nacer y pequeño para la edad gestacional en el neonato. El nivel de control del asma debe controlarse estrechamente en las mujeres embarazadas y el tratamiento debe ajustarse según sea necesario para mantener un control óptimo.
Datos
Datos de Animales
En un estudio de toxicidad de desarrollo prenatal y postnatal mejorado, monos cynomolgus preñados recibieron dosis subcutáneas semanales de anticuerpo homólogo contra IL-4Rα hasta 10 veces la MRHD (en una base de mg/kg de 100 mg/kg/semana) desde el comienzo de la organogénesis hasta el parto. No se observaron efectos adversos relacionados con el tratamiento sobre la toxicidad embrio-fetal o las malformaciones, o sobre el desarrollo morfológico, funcional o inmunológico en los bebés desde el nacimiento hasta los 6 meses de edad.
8.2 Lactancia
Resumen de Riesgos
No hay datos sobre la presencia de dupilumab en la leche materna, los efectos en el lactante amamantado o los efectos sobre la producción de leche. Se sabe que la IgG materna está presente en la leche materna. Se desconocen los efectos de la exposición gastrointestinal local y la exposición sistémica limitada a dupilumab en el lactante amamantado. Los beneficios para el desarrollo y la salud de la lactancia materna deben considerarse junto con la necesidad clínica de la madre de DUPIXENT y cualquier posible efecto adverso en el niño amamantado por DUPIXENT o por la condición materna subyacente.
8.4 Uso Pediátrico
Dermatitis Atópica
La seguridad y eficacia de DUPIXENT se han establecido en pacientes pediátricos de 6 meses de edad o mayores con AD moderada a grave, cuya enfermedad no está adecuadamente controlada con terapias tópicas de prescripción o cuando esas terapias no son aconsejables [ver Estudios Clínicos (14.1)].
El uso de DUPIXENT en este grupo de edad está respaldado por datos de los siguientes ensayos clínicos:
- AD-1526 que incluyó 251 sujetos pediátricos de 12 a 17 años de edad con AD moderada a grave. De los 251 sujetos, 82 fueron tratados con DUPIXENT 200 mg Q2W (<60 kg) o 300 mg Q2W (≥60 kg) y 85 fueron tratados con placebo coincidente
- AD-1652 que incluyó 367 sujetos pediátricos de 6 a 11 años de edad con AD grave. De los 367 sujetos, 120 fueron tratados con DUPIXENT 300 mg Q4W + TCS (15 a <30 kg) o 200 mg Q2W + TCS (≥30 kg) y 123 fueron tratados con placebo coincidente + TCS
- AD-1539 que incluyó 162 sujetos pediátricos de 6 meses a 5 años de edad con AD moderada a grave. De los 162 sujetos, 83 fueron tratados con DUPIXENT 200 mg Q4W + TCS (5 a <15 kg) o 300 mg Q4W + TCS (15 a <30 kg) y 79 sujetos fueron asignados para ser tratados con placebo coincidente + TCS
- AD-1434, un estudio de extensión de etiqueta abierta que reclutó a 275 sujetos pediátricos de 12 a 17 años de edad tratados con DUPIXENT ± TCS, 368 sujetos pediátricos de 6 a 11 años de edad tratados con DUPIXENT ± TCS y 180 sujetos pediátricos de 6 meses a 5 años de edad tratados con DUPIXENT ± TCS
- Liberty-AD-HAFT que incluyó 27 sujetos pediátricos de 12 a 17 años de edad con dermatitis atópica con afectación moderada a grave de manos y/o pies tratados con DUPIXENT (N=14) o placebo coincidente (N=13)
La seguridad y la eficacia fueron generalmente consistentes entre los pacientes pediátricos y adultos [ver Reacciones adversas (6.1) y Estudios clínicos (14.1)]. Además, se informó enfermedad mano-pie-boca en 9 (5%) sujetos pediátricos y papiloma cutáneo se informó en 4 (2%) sujetos pediátricos de 6 meses a 5 años de edad tratados con DUPIXENT ± TCS en AD-1434. Estos casos no llevaron a la interrupción del fármaco del estudio [ver Reacciones adversas (6.1)].
No se ha establecido la seguridad y la eficacia en pacientes pediátricos menores de 6 meses de edad con AD.
Asma
La seguridad y la eficacia de DUPIXENT para un tratamiento de mantenimiento adicional en pacientes con asma moderada a grave caracterizada por un fenotipo eosinofílico o con asma dependiente de corticosteroides orales se han establecido en pacientes pediátricos de 6 años de edad y mayores. El uso de DUPIXENT para esta indicación está respaldado por evidencia de estudios adecuados y bien controlados en pacientes adultos y pediátricos de 6 años y mayores [ver Estudios clínicos (14.2)].
Sujetos pediátricos de 12 a 17 años de edad:
Un total de 107 sujetos pediátricos de 12 a 17 años de edad con asma moderada a grave fueron incluidos en QUEST y recibieron 200 mg (N=21) o 300 mg (N=18) DUPIXENT (o placebo coincidente 200 mg [N=34] o 300 mg [N=34]) Q2W. Las exacerbaciones del asma y la función pulmonar se evaluaron tanto en sujetos pediátricos de 12 a 17 años de edad como en adultos. Para ambas dosis de 200 mg y 300 mg Q2W, se observaron mejoras en FEV1 (cambio medio de LS desde el inicio en la semana 12) (0,36 L y 0,27 L, respectivamente). Para la dosis de 200 mg Q2W, los sujetos tuvieron una reducción en la tasa de exacerbaciones graves que fue consistente con los adultos. La exposición a dupilumab fue mayor en sujetos pediátricos de 12 a 17 años de edad que en adultos en el nivel de dosis respectivo, lo que se debió principalmente a la diferencia en el peso corporal [ver Farmacología clínica (12.3)].
El perfil de eventos adversos en sujetos pediátricos de 12 a 17 años de edad fue generalmente similar al de los adultos [ver Reacciones adversas (6.1)].
Sujetos pediátricos de 6 a 11 años de edad:
Un total de 408 sujetos pediátricos de 6 a 11 años de edad con asma moderada a grave fueron incluidos en VOYAGE, que evaluó dosis de 100 mg Q2W o 200 mg Q2W. Se demostró una mejora en las exacerbaciones del asma y la función pulmonar [ver Estudios clínicos (14.2)]. La eficacia de DUPIXENT 300 mg Q4W en sujetos de 6 a 11 años de edad con peso corporal de 15 a <30 kg se extrapoló de la eficacia de 100 mg Q2W en VOYAGE con el apoyo de análisis farmacocinéticos poblacionales que muestran niveles de exposición a fármacos más altos con 300 mg Q4W [ver Farmacología clínica (12.3)]. Los sujetos que completaron el período de tratamiento del estudio VOYAGE pudieron participar en el estudio de extensión de etiqueta abierta (LTS14424). Dieciocho sujetos (≥15 a <30 kg) de 365 sujetos fueron expuestos a 300 mg Q4W en este estudio, y el perfil de seguridad en estos dieciocho sujetos fue consistente con el observado en VOYAGE. La seguridad adicional para DUPIXENT 300 mg Q4W se basa en la información de seguridad disponible de la indicación pediátrica de AD [ver Reacciones adversas (6.1) y Farmacología clínica (12.3)].
No se ha establecido la seguridad y la eficacia en pacientes pediátricos menores de 6 años de edad con asma.
CRSwNP
La seguridad y la eficacia de DUPIXENT para el tratamiento de mantenimiento adicional en pacientes con sinusitis crónica con pólipos nasales (CRSwNP) inadecuadamente controlada se han establecido en pacientes pediátricos de 12 años de edad y mayores. El uso de DUPIXENT para esta indicación está respaldado por evidencia de estudios adecuados y bien controlados de DUPIXENT como tratamiento de mantenimiento adicional en adultos con CRSwNP inadecuadamente controlada (SINUS-24 y SINUS-52) con los siguientes datos adicionales:
- Datos farmacocinéticos (PK) de pacientes adultos y pediátricos de 12 años de edad y mayores con asma moderada a grave y pacientes adultos con CRSwNP inadecuadamente controlada
- Datos de seguridad en pacientes pediátricos de 12 años de edad y mayores con asma moderada a grave [ver Reacciones adversas (6.1), Farmacología clínica (12.3) y Estudios clínicos (14.3)]
No se ha establecido la seguridad y la eficacia en pacientes pediátricos menores de 12 años de edad con CRSwNP.
EoE
La seguridad y eficacia de DUPIXENT para el tratamiento de la EoE se han establecido en sujetos pediátricos de 1 año de edad o mayores, con un peso de al menos 15 kg. El uso de DUPIXENT en esta población está respaldado por un estudio adecuado y bien controlado en adultos y 72 sujetos pediátricos de 12 a 17 años de edad (Estudio EoE-1), un estudio clínico en 61 sujetos pediátricos de 1 a 11 años de edad (Estudio EoE-2), y datos farmacocinéticos en sujetos adultos y pediátricos de 1 a 17 años de edad. La seguridad de DUPIXENT en sujetos pediátricos de 1 a 17 años de edad fue similar a la de los adultos [ver Reacciones adversas (6.1), Farmacología clínica (12.3), y Estudios clínicos (14.4)].
No se ha establecido la seguridad y eficacia en pacientes pediátricos menores de 1 año de edad, o con un peso inferior a 15 kg, con EoE.
8.5 Uso en Geriatría
De los 1539 sujetos con AD expuestos a DUPIXENT en un estudio de rango de dosis y ensayos controlados con placebo, 70 sujetos tenían 65 años o más. Los ensayos clínicos de DUPIXENT en AD no incluyeron un número suficiente de sujetos de 65 años o más para determinar si responden de manera diferente a los sujetos más jóvenes [ver Farmacología clínica (12.3)].
De los 1977 sujetos con asma expuestos a DUPIXENT, un total de 240 sujetos tenían 65 años o más. La eficacia y seguridad en este grupo de edad fue similar a la de la población general del estudio.
De los 440 sujetos con CRSwNP expuestos a DUPIXENT, un total de 79 sujetos tenían 65 años o más. La eficacia y seguridad en este grupo de edad fueron similares a la de la población general del estudio.
Los estudios clínicos de DUPIXENT en EoE no incluyeron un número suficiente de sujetos de 65 años o más para determinar si responden de manera diferente a los sujetos adultos más jóvenes.
De los 152 sujetos con PN expuestos a DUPIXENT, un total de 37 tenían 65 años o más, incluyendo 8 sujetos de 75 años o más. Los ensayos clínicos no incluyeron un número suficiente de sujetos de 65 años de edad o más para determinar si responden de manera diferente a los sujetos adultos más jóvenes.
De los 1874 sujetos con EPOC aleatorizados en ensayos clínicos de DUPIXENT, un total de 1072 tenían 65 años o más, mientras que 244 sujetos tenían 75 años o más. No se han observado diferencias generales en la seguridad o eficacia de DUPIXENT entre los sujetos de 65 años de edad o más y los sujetos adultos más jóvenes.
10 SOBREDOSIS
No existe un tratamiento específico para la sobredosis de DUPIXENT. En caso de sobredosis, comuníquese con el Centro de Control de Envenenamiento (1-800-222-1222) para obtener las recomendaciones más recientes y controle al paciente para detectar cualquier signo o síntoma de reacciones adversas e instituya inmediatamente el tratamiento sintomático apropiado.
11 DESCRIPCIÓN
Dupilumab, un antagonista del receptor alfa de la interleucina-4, es un anticuerpo monoclonal humano de la subclase IgG4 que se une a la subunidad IL-4Rα e inhibe la señalización de IL-4 e IL-13. Dupilumab tiene un peso molecular aproximado de 147 kDa.
Dupilumab se produce mediante tecnología de ADN recombinante en cultivo de suspensión de células ováricas de hámster chino.
DUPIXENT (dupilumab) Injection se suministra como una solución estéril, sin conservantes, transparente a ligeramente opalescente, incolora a amarillo pálido para inyección subcutánea. DUPIXENT se proporciona como una jeringa precargada de dosis única con protector de aguja o una pluma precargada de dosis única en una jeringa de vidrio transparente de tipo 1 siliconizada. La tapa de la aguja no está hecha con látex de caucho natural.
Cada jeringa precargada de 300 mg o pluma precargada administra 300 mg de dupilumab en 2 mL que también contiene clorhidrato de L-arginina (10,5 mg), L-histidina (6,2 mg), polisorbato 80 (4 mg), acetato de sodio (2 mg), sacarosa (100 mg) y agua para inyección, pH 5,9.
Cada jeringa precargada de 200 mg o pluma precargada administra 200 mg de dupilumab en 1,14 mL que también contiene clorhidrato de L-arginina (12 mg), L-histidina (3,5 mg), polisorbato 80 (2,3 mg), acetato de sodio (1,2 mg), sacarosa (57 mg) y agua para inyección, pH 5,9.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
Dupilumab es un anticuerpo monoclonal humano IgG4 que inhibe la señalización de la interleucina-4 (IL-4) y la interleucina-13 (IL-13) al unirse específicamente a la subunidad IL-4Rα compartida por los complejos receptores IL-4 e IL-13. Dupilumab inhibe la señalización de IL-4 a través del receptor tipo I y la señalización de IL-4 e IL-13 a través del receptor tipo II.
La inflamación impulsada por IL-4 e IL-13 es un componente importante en la patogénesis del asma, la AD, la CRSwNP, la EoE, la PN y la EPOC. Múltiples tipos de células que expresan IL-4Rα (por ejemplo, mastocitos, eosinófilos, macrófagos, linfocitos, células epiteliales, células caliciformes) y mediadores inflamatorios (por ejemplo, histamina, eicosanoides, leucotrienos, citoquinas, quimioquinas) están involucrados en la inflamación. El bloqueo de IL-4Rα con dupilumab inhibe las respuestas inflamatorias inducidas por citoquinas IL-4 e IL-13, incluida la liberación de citoquinas proinflamatorias, quimioquinas, óxido nítrico e IgE. El mecanismo de acción de dupilumab no se ha establecido definitivamente.
12.2 Farmacodinamia
De acuerdo con la inhibición de la señalización de IL-4 e IL-13, el tratamiento con dupilumab disminuyó ciertos biomarcadores de inflamación. En sujetos con asma, el óxido nítrico exhalado fraccional (FeNO) y las concentraciones circulantes de eotaxina-3, IgE total, IgE específica de alérgenos, TARC y periostina disminuyeron en relación con el placebo. Las reducciones en estos biomarcadores fueron comparables para los regímenes de 300 mg Q2W y 200 mg Q2W. Estos marcadores estuvieron cerca de la supresión máxima después de 2 semanas de tratamiento, excepto para la IgE, que disminuyó más lentamente. Estos efectos se mantuvieron durante todo el tratamiento. La reducción porcentual mediana desde el inicio en las concentraciones de IgE total con tratamientos con dupilumab fue del 52% en la semana 24 (DRI12544) y del 70% en la semana 52 (QUEST). Para FeNO, la reducción porcentual media desde el inicio en la semana 2 fue del 35% y del 24% en DRI12544 y QUEST, respectivamente, y en la población general de seguridad, el nivel medio de FeNO disminuyó a 20 ppb.
Respuesta de anticuerpos a vacunas no vivas durante el tratamiento con DUPIXENT
En un estudio clínico, los sujetos adultos con AD fueron tratados una vez por semana durante 16 semanas con 300 mg de DUPIXENT (el doble de la frecuencia de dosificación recomendada). Después de 12 semanas de administración, los sujetos recibieron una vacuna Tdap y una vacuna de polisacárido meningocócica. Las respuestas de anticuerpos al toxoide tetánico y al polisacárido meningocócico del serogrupo C se evaluaron 4 semanas después. Las respuestas de anticuerpos tanto al toxoide tetánico como al polisacárido meningocócico del serogrupo C fueron similares en los sujetos tratados con DUPIXENT y los tratados con placebo. No se evaluaron las respuestas de anticuerpos a los otros componentes activos de ambas vacunas. Tampoco se evaluaron las respuestas de anticuerpos a otras vacunas no vivas.
12.3 Farmacocinética
La farmacocinética de dupilumab es similar en sujetos con AD, asma, CRSwNP, EoE, PN y EPOC.
Absorción
Después de una dosis subcutánea (SC) inicial de 600 mg, 400 mg o 300 mg, dupilumab alcanzó concentraciones máximas medias ± DE (Cmax) de 70.1±24.1 mcg/mL, 41.8±12.4 mcg/mL o 30.5±9.39 mcg/mL, respectivamente, aproximadamente 1 semana después de la dosis. Las concentraciones en estado estacionario se lograron en la semana 16 después de la administración de una dosis inicial de 600 mg y una dosis de 300 mg semanal o Q2W, o una dosis inicial de 400 mg y una dosis de 200 mg Q2W, o 300 mg Q2W sin una dosis de carga. En los ensayos clínicos, las concentraciones mínimas medias ± DE en estado estacionario oscilaron entre 55.3±34.3 mcg/mL y 80.2±35.3 mcg/mL para 300 mg administrados Q2W, entre 173±75.9 mcg/mL y 195±71.7 mcg/mL para 300 mg administrados semanalmente, y entre 29.2±18.7 y 36.5±22.2 mcg/mL para 200 mg administrados Q2W.
La biodisponibilidad de dupilumab después de una dosis SC es similar entre los sujetos con AD, asma, CRSwNP, EoE, PN y EPOC, y oscila entre el 61% y el 64%.
Eliminación
La vía metabólica de dupilumab no se ha caracterizado. Como un anticuerpo monoclonal humano IgG4, se espera que dupilumab se degrade en pequeños péptidos y aminoácidos a través de vías catabólicas de la misma manera que la IgG endógena. Después de la última dosis en estado estacionario de 300 mg QW, 300 mg Q2W, 200 mg Q2W, 300 mg Q4W o 200 mg Q4W dupilumab, los tiempos medianos hasta la concentración no detectable (<78 ng/mL) oscilaron entre 9 y 13 semanas en adultos y sujetos pediátricos de 12 a 17 años de edad. Los análisis farmacocinéticos poblacionales indican que los tiempos medianos hasta la concentración no detectable son aproximadamente 1.5 veces (hasta 19 semanas) y 2.5 veces (hasta 32 semanas) más largos en sujetos pediátricos de 6 a 11 años de edad y sujetos pediátricos de 6 meses a 5 años de edad, respectivamente.
Linealidad de la dosis
Dupilumab exhibió farmacocinética no lineal mediada por el objetivo con exposiciones que aumentaron de manera mayor que proporcional a la dosis. La exposición sistémica aumentó 30 veces cuando la dosis aumentó 8 veces después de una sola dosis de dupilumab de 75 mg a 600 mg (es decir, 0.25 veces a 2 veces la dosis recomendada).
Peso
Las concentraciones mínimas de dupilumab fueron más bajas en los sujetos con mayor peso corporal.
Inmunogenicidad
El desarrollo de anticuerpos contra dupilumab se asoció con concentraciones séricas más bajas de dupilumab. Algunos sujetos que tenían títulos altos de anticuerpos tampoco tenían concentraciones séricas detectables de dupilumab.
Poblaciones específicas
Edad
Basado en el análisis farmacocinético poblacional, la edad no afectó la depuración de dupilumab en adultos y en sujetos pediátricos de 6 a 17 años de edad. En sujetos pediátricos de 6 meses a 5 años de edad, la depuración aumentó con la edad.
Pacientes geriátricos
No se observaron diferencias generales en la farmacocinética de dupilumab entre los sujetos adultos mayores y los más jóvenes.
Pacientes pediátricos
Dermatitis atópica
Para los sujetos pediátricos de 12 a 17 años de edad con AD que recibieron dosis cada dos semanas (Q2W) con 200 mg (<60 kg) o 300 mg (≥60 kg), la concentración media ± DE en estado estacionario de dupilumab fue de 54,5±27,0 mcg/mL.
Para los sujetos pediátricos de 6 a 11 años de edad con AD que recibieron dosis cada dos semanas (Q2W) con 200 mg (≥30 kg) o dosis cada cuatro semanas (Q4W) con 300 mg (<30 kg), la concentración media ± DE en estado estacionario fue de 86,0±34,6 mcg/mL y 98,7±33,2 mcg/mL, respectivamente.
Para los sujetos pediátricos de 6 meses a 5 años de edad con AD que recibieron dosis cada cuatro semanas (Q4W) con 300 mg (≥15 a <30 kg) o 200 mg (≥5 a <15 kg), la concentración media ± DE en estado estacionario fue de 110±42,8 mcg/mL y 109±50,8 mcg/mL, respectivamente.
Asma
Un total de 107 sujetos pediátricos de 12 a 17 años de edad con asma fueron inscritos en QUEST. Las concentraciones medias ± DE en estado estacionario de dupilumab fueron de 107±51,6 mcg/mL y 46,7±26,9 mcg/mL, respectivamente, para 300 mg o 200 mg administrados Q2W.
En VOYAGE, la farmacocinética de dupilumab se investigó en 270 sujetos con asma de moderada a grave después de la administración subcutánea de 100 mg Q2W (para 91 sujetos pediátricos con un peso <30 kg) o 200 mg Q2W (para 179 sujetos pediátricos con un peso ≥30 kg). La concentración media ± DE en estado estacionario fue de 58,4±28,0 mcg/mL y 85,1±44,9 mcg/mL, respectivamente. La simulación de una dosis subcutánea de 300 mg Q4W en sujetos pediátricos de 6 a 11 años de edad con un peso corporal de ≥15 a <30 kg dio como resultado concentraciones en estado estacionario predichas (98,7±41,0 mcg/mL) y concentraciones promedio más altas que las concentraciones en estado estacionario observadas y las concentraciones promedio de 100 mg Q2W (<30 kg).
CRSwNP
No se han realizado estudios clínicos en pacientes pediátricos de 12 años de edad o mayores con CRSwNP. Se espera que las exposiciones a dupilumab sean comparables entre adultos y pacientes pediátricos de 12 años de edad o mayores a la dosis recomendada para CRSwNP (300 mg cada 2 semanas).
Esofagitis eosinofílica
En el Estudio EoE-1, la farmacocinética de dupilumab se investigó en 35 sujetos pediátricos de 12 a 17 años de edad, con un peso de al menos 40 kg, con EoE, que recibieron 300 mg QW. La concentración media ± DE en estado estacionario de dupilumab fue de 227±95,3 mcg/mL.
En el Estudio EoE-2 Parte A, la farmacocinética de dupilumab se investigó en 20 sujetos pediátricos de 1 a 11 años de edad con EoE que recibieron los siguientes regímenes de dosificación basados en el peso: ≥15 a <30 kg (200 mg Q2W) y ≥30 a <40 kg (300 mg Q2W). En la semana 16, la concentración media ± DE en estado estacionario de dupilumab fue de 174±66,2 mcg/mL.
Se espera que la exposición sistémica en sujetos pediátricos de 1 a 11 años de edad con un peso corporal ≥40 kg que reciben 300 mg QW sea comparable a la de los sujetos adultos y pediátricos de 12 años de edad o mayores con un peso corporal ≥40 kg. Se espera que la exposición sistémica en sujetos pediátricos de 12 a 17 años de edad con un peso corporal <40 kg que reciben 300 mg Q2W sea comparable a la de los sujetos pediátricos de 6 a 11 años de edad.
Estudios de interacción medicamentosa
No se espera un efecto de dupilumab en la PK de los medicamentos administrados conjuntamente. Basado en el análisis poblacional, los medicamentos comúnmente administrados conjuntamente no tuvieron ningún efecto en la farmacocinética de DUPIXENT en sujetos con asma de moderada a grave.
Sustratos del citocromo P450
Los efectos de dupilumab en la farmacocinética de midazolam (metabolizado por CYP3A4), warfarina (metabolizado por CYP2C9), omeprazol (metabolizado por CYP2C19), metoprolol (metabolizado por CYP2D6) y cafeína (metabolizado por CYP1A2) se evaluaron en un estudio con 12-13 sujetos evaluables con AD (una dosis de carga SC de 600 mg seguida de 300 mg SC semanalmente durante seis semanas). No se observaron cambios clínicamente significativos en el AUC. El efecto más grande se observó para metoprolol (CYP2D6) con un aumento en el AUC del 29%.
12.6 Inmunogenicidad
La incidencia observada de anticuerpos contra el fármaco depende en gran medida de la sensibilidad y la especificidad del ensayo. Las diferencias en los métodos de ensayo excluyen comparaciones significativas de la incidencia de anticuerpos contra el fármaco en los estudios descritos a continuación con la incidencia de anticuerpos contra el fármaco en otros estudios, incluidos los de DUPIXENT o de otros productos de dupilumab.
Dermatitis atópica
Aproximadamente el 6% de los sujetos con AD que recibieron DUPIXENT 300 mg Q2W durante 52 semanas desarrollaron anticuerpos contra DUPIXENT; aproximadamente el 2% exhibió respuestas persistentes de ADA, y aproximadamente el 1% tuvo anticuerpos neutralizantes. Se observaron resultados similares en sujetos pediátricos de 6 meses a 11 años de edad con AD que recibieron DUPIXENT 200 mg Q2W, 200 mg Q4W o 300 mg Q4W.
Aproximadamente el 16% de los sujetos pediátricos de 12 a 17 años de edad con AD que recibieron DUPIXENT 300 mg o 200 mg Q2W durante 16 semanas desarrollaron anticuerpos contra DUPIXENT; aproximadamente el 3% exhibió respuestas persistentes de ADA, y aproximadamente el 5% tuvo anticuerpos neutralizantes.
Asma
Aproximadamente el 5% de los sujetos con asma que recibieron DUPIXENT 300 mg Q2W durante 52 semanas desarrollaron anticuerpos contra DUPIXENT; aproximadamente el 2% exhibió respuestas ADA persistentes y aproximadamente el 2% tuvo anticuerpos neutralizantes. Se observaron resultados similares en sujetos pediátricos de 6 a 11 años de edad con asma que recibieron DUPIXENT 100 mg Q2W o 200 mg Q2W hasta por 52 semanas.
Aproximadamente el 9% de los sujetos con asma que recibieron DUPIXENT 200 mg Q2W durante 52 semanas desarrollaron anticuerpos contra DUPIXENT; aproximadamente el 4% exhibió respuestas ADA persistentes y aproximadamente el 4% tuvo anticuerpos neutralizantes.
Rinosinusitis crónica con pólipos nasales
Aproximadamente el 5% de los sujetos adultos con CRSwNP que recibieron DUPIXENT 300 mg Q2W durante 52 semanas desarrollaron anticuerpos contra DUPIXENT; aproximadamente el 2% exhibió respuestas ADA persistentes y aproximadamente el 3% tuvo anticuerpos neutralizantes.
Esofagitis eosinofílica
Aproximadamente el 1% de los sujetos con EoE que recibieron DUPIXENT 300 mg QW durante 52 semanas desarrollaron anticuerpos contra DUPIXENT; ningún sujeto exhibió respuestas ADA persistentes o tuvo anticuerpos neutralizantes. Se observaron resultados similares en sujetos pediátricos de 1 a 11 años de edad con EoE que recibieron DUPIXENT 200 mg Q2W o 300 mg Q2W durante 52 semanas.
Prurigo nodular
Aproximadamente el 8% de los sujetos con PN que recibieron DUPIXENT 300 mg Q2W durante 24 semanas desarrollaron anticuerpos contra DUPIXENT; aproximadamente el 1% exhibió respuestas ADA persistentes y aproximadamente el 3% tuvo anticuerpos neutralizantes.
Enfermedad pulmonar obstructiva crónica
Aproximadamente el 8% de los sujetos con EPOC que recibieron DUPIXENT 300 mg Q2W durante 52 semanas desarrollaron anticuerpos contra DUPIXENT; aproximadamente el 3% exhibió respuestas ADA persistentes y aproximadamente el 3% tuvo anticuerpos neutralizantes.
Los títulos de anticuerpos detectados en los sujetos que recibieron DUPIXENT fueron en su mayoría bajos. En los sujetos que recibieron DUPIXENT, el desarrollo de anticuerpos de alto título contra DUPIXENT se asoció con concentraciones séricas más bajas de dupilumab [ver Farmacología clínica (12.3)].
Dos sujetos adultos con AD que experimentaron respuestas de anticuerpos de alto título desarrollaron suero o reacciones similares al suero durante la terapia con DUPIXENT [ver Advertencias y precauciones (5.1)].
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No se han realizado estudios en animales para evaluar el potencial carcinogénico o mutagénico de dupilumab.
No se observaron efectos sobre los parámetros de fertilidad, como los órganos reproductivos, la duración del ciclo menstrual o el análisis de esperma, en ratones sexualmente maduros a los que se administró por vía subcutánea un anticuerpo homólogo contra IL-4Rα en dosis de hasta 200 mg/kg/semana.
14 ESTUDIOS CLÍNICOS
14.1 Dermatitis atópica
Adultos con dermatitis atópica
Tres ensayos aleatorizados, doble ciego, controlados con placebo (SOLO 1 (NCT02277743), SOLO 2 (NCT02277769) y CHRONOS (NCT02260986)) reclutaron un total de 2119 sujetos adultos de 18 años de edad o mayores con DA moderada a grave no controlada adecuadamente con medicamentos tópicos. La gravedad de la enfermedad se definió mediante una puntuación de Evaluación Global del Investigador (IGA) ≥3 en la evaluación general de las lesiones de DA en una escala de gravedad de 0 a 4, una puntuación del Índice de Área y Severidad del Eczema (EASI) ≥16 en una escala de 0 a 72 y una superficie corporal mínima involucrada ≥10%. Al inicio del estudio, el 59% de los sujetos eran hombres, el 67% eran blancos, el 52% de los sujetos tenían una puntuación IGA inicial de 3 (DA moderada) y el 48% de los sujetos tenían una puntuación IGA inicial de 4 (DA grave). La puntuación EASI media inicial fue de 33 y la puntuación media semanal de la Escala de Calificación Numérica de Picor Máximo (NRS) fue de 7 en una escala de 0 a 10.
En los tres ensayos, los sujetos del grupo DUPIXENT recibieron inyecciones subcutáneas de DUPIXENT 600 mg en la semana 0, seguidas de 300 mg cada dos semanas (Q2W). En los ensayos de monoterapia (SOLO 1 y SOLO 2), los sujetos recibieron DUPIXENT o placebo durante 16 semanas.
En el ensayo de terapia concomitante (CHRONOS), los sujetos recibieron DUPIXENT o placebo con corticosteroides tópicos (TCS) concomitantes y, según fuera necesario, inhibidores tópicos de la calcineurina solo para áreas problemáticas, como la cara, el cuello, las áreas intertriginosas y genitales durante 52 semanas.
Los tres ensayos evaluaron el criterio de valoración principal, el cambio desde el inicio del estudio hasta la semana 16 en la proporción de sujetos con una IGA 0 (clara) o 1 (casi clara) y al menos una mejora de 2 puntos. Otros criterios de valoración incluyeron la proporción de sujetos con EASI-75 (mejora de al menos el 75% en la puntuación EASI desde el inicio del estudio) y la reducción del picor, definida como al menos una mejora de 4 puntos en la NRS de picor máximo desde el inicio del estudio hasta la semana 16.
Respuesta clínica en la semana 16 (SOLO 1, SOLO 2 y CHRONOS)
Los resultados de los ensayos de monoterapia con DUPIXENT (SOLO 1 y SOLO 2) y el ensayo de DUPIXENT con TCS concomitantes (CHRONOS) se presentan en la Tabla 12.
| SOLO 1 | SOLO 2 | CHRONOS | ||||
|---|---|---|---|---|---|---|
| DUPIXENT 300 mg Q2W |
Placebo | DUPIXENT 300 mg Q2W |
Placebo | DUPIXENT 300 mg Q2W + TCS |
Placebo + TCS | |
|
||||||
| Número de sujetos aleatorizados (FAS)* | 224 | 224 | 233 | 236 | 106 | 315 |
| IGA 0 o 1†,‡ | 38% | 10% | 36% | 9% | 39% | 12% |
| EASI-75‡ | 51% | 15% | 44% | 12% | 69% | 23% |
| EASI-90‡ | 36% | 8% | 30% | 7% | 40% | 11% |
| Número de sujetos con puntuación NRS de picor máximo inicial ≥4 | 213 | 212 | 225 | 221 | 102 | 299 |
| NRS de picor máximo (≥4 puntos de mejora)‡ | 41% | 12% | 36% | 10% | 59% | 20% |
| Figura 1: Proporción de sujetos adultos de 18 años de edad o mayores con AD moderada a grave con ≥4 puntos de mejora en la NRS de pico de prurito en los estudios SOLO 1* y SOLO 2* (FAS)† | |
|---|---|
| SOLO 1 | SOLO 2 |
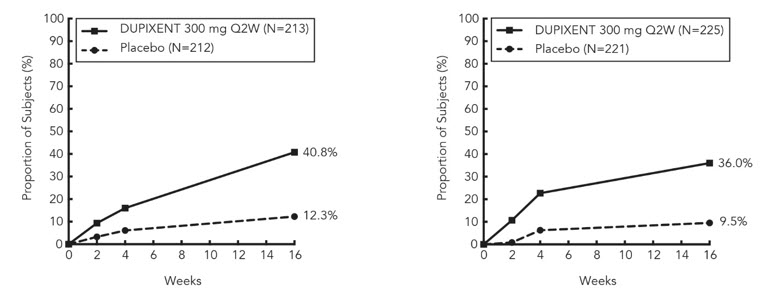 |
|
En CHRONOS, de los 421 sujetos, 353 habían estado en el estudio durante 52 semanas al momento del análisis de datos. De estos 353 sujetos, los respondedores en la semana 52 representan una mezcla de sujetos que mantuvieron su eficacia desde la semana 16 (por ejemplo, el 53% de los respondedores de DUPIXENT IGA 0 o 1 en la semana 16 siguieron siendo respondedores en la semana 52) y sujetos que no fueron respondedores en la semana 16 que luego respondieron al tratamiento (por ejemplo, el 24% de los no respondedores de DUPIXENT IGA 0 o 1 en la semana 16 se convirtieron en respondedores en la semana 52). Los resultados de los análisis de apoyo de los 353 sujetos en el ensayo de DUPIXENT con TCS concomitante (CHRONOS) se presentan en la Tabla 13.
| DUPIXENT 300 mg Q2W + TCS |
Placebo + TCS | |
|---|---|---|
|
||
| Número de sujetos* | 89 | 264 |
| Respondedor†,‡ en la semana 16 y 52 | 22% | 7% |
| Respondedor en la semana 16 pero no respondedor en la semana 52 | 20% | 7% |
| No respondedor en la semana 16 y respondedor en la semana 52 | 13% | 6% |
| No respondedor en la semana 16 y 52 | 44% | 80% |
| Tasa general de respondedores†,‡ en la semana 52 | 36% | 13% |
Los efectos del tratamiento en subgrupos (peso, edad, género, raza y tratamiento previo, incluidos los inmunosupresores) en SOLO 1, SOLO 2 y CHRONOS fueron generalmente consistentes con los resultados en la población de estudio general.
En SOLO 1, SOLO 2 y CHRONOS, un tercer brazo de tratamiento aleatorizado de DUPIXENT 300 mg QW no demostró un beneficio adicional del tratamiento en comparación con DUPIXENT 300 mg Q2W.
Los sujetos en SOLO 1 y SOLO 2 que tuvieron un IGA 0 o 1 con una reducción de ≥ 2 puntos fueron re-aleatorizados en SOLO CONTINUE (NCT02395133). SOLO CONTINUE evaluó múltiples regímenes de dosis de monoterapia de DUPIXENT para mantener la respuesta al tratamiento. El estudio incluyó sujetos aleatorizados para continuar con DUPIXENT 300 mg Q2W (62 sujetos) o cambiar a placebo (31 sujetos) durante 36 semanas. Las respuestas de IGA 0 o 1 en la semana 36 fueron las siguientes: 33 (53%) en el grupo Q2W y 3 (10%) en el grupo placebo.
Sujetos pediátricos de 12 a 17 años con dermatitis atópica
La eficacia de la monoterapia de DUPIXENT en sujetos pediátricos de 12 a 17 años se evaluó en un ensayo multicéntrico, aleatorizado, doble ciego y controlado con placebo (AD-1526; NCT03054428) en 251 sujetos pediátricos de 12 a 17 años, con DA moderada a grave definida por un puntaje IGA ≥ 3 (escala de 0 a 4), un puntaje EASI ≥ 16 (escala de 0 a 72) y una participación mínima de BSA de ≥ 10%. Los sujetos elegibles inscritos en este ensayo tuvieron una respuesta insuficiente previa a la medicación tópica.
Los sujetos en el grupo DUPIXENT con un peso inicial de < 60 kg recibieron una dosis inicial de 400 mg en la semana 0, seguida de 200 mg Q2W durante 16 semanas. Los sujetos con un peso inicial de ≥ 60 kg recibieron una dosis inicial de 600 mg en la semana 0, seguida de 300 mg Q2W durante 16 semanas. Los sujetos podían recibir tratamiento de rescate a discreción del investigador. Los sujetos que recibieron tratamiento de rescate se consideraron no respondedores.
En AD-1526, la edad media fue de 14.5 años, el peso mediano fue de 59.4 kg, el 41% de los sujetos eran mujeres, el 63% eran blancos, el 15% eran asiáticos y el 12% eran negros. Al inicio, el 46% de los sujetos tenían un puntaje IGA de 3 (DA moderada), el 54% tenían un puntaje IGA de 4 (DA grave), la participación media de BSA era del 57% y el 42% habían recibido inmunosupresores sistémicos previos. Además, al inicio, el puntaje medio EASI era de 36 y el promedio semanal de Peak Pruritus NRS era 8 en una escala de 0-10. En general, el 92% de los sujetos tenían al menos una condición alérgica comórbida; el 66% tenían rinitis alérgica, el 54% tenían asma y el 61% tenían alergias alimentarias.
El punto final primario fue la proporción de sujetos con un IGA 0 (claro) o 1 (casi claro) y al menos una mejora de 2 puntos desde el inicio hasta la semana 16. Otros resultados evaluados incluyeron la proporción de sujetos con EASI-75 o EASI-90 (mejora de al menos 75% o 90% en EASI desde el inicio, respectivamente), y la reducción del picor medida por Peak Pruritus NRS (mejora de ≥ 4 puntos).
Los resultados de eficacia en la semana 16 para AD-1526 se presentan en la Tabla 14.
| DUPIXENT† 200 mg (<60 kg) o 300 mg (≥60 kg) Q2W N=82* |
Placebo N=85* |
|
|---|---|---|
|
||
| IGA 0 o 1‡,§ | 24% | 2% |
| EASI-75§ | 42% | 8% |
| EASI-90§ | 23% | 2% |
| Peak Pruritus NRS (mejora de ≥ 4 puntos)§ | 37% | 5% |
Una mayor proporción de sujetos aleatorizados a DUPIXENT logró una mejora en el NRS de pico de prurito en comparación con placebo (definido como ≥4 puntos de mejora en la semana 4). Ver Figura 2.
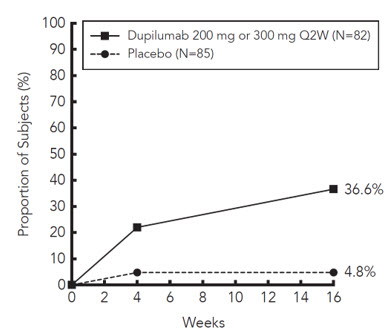
Sujetos pediátricos de 6 a 11 años con dermatitis atópica
La eficacia y seguridad del uso de DUPIXENT concomitantemente con TCS en sujetos pediátricos se evaluó en un ensayo multicéntrico, aleatorizado, doble ciego y controlado con placebo (AD-1652; NCT03345914) en 367 sujetos de 6 a 11 años de edad, con AD definida por una puntuación IGA de 4 (escala de 0 a 4), una puntuación EASI ≥ 21 (escala de 0 a 72) y una participación mínima de BSA de ≥ 15%. Los sujetos elegibles inscritos en este ensayo tuvieron una respuesta insuficiente previa al tratamiento tópico. La inscripción se estratificó según el peso basal (< 30 kg; ≥ 30 kg).
Los sujetos en el grupo DUPIXENT Q4W + TCS recibieron una dosis inicial de 600 mg el día 1, seguida de 300 mg Q4W desde la semana 4 hasta la semana 12, independientemente del peso. Los sujetos en el grupo DUPIXENT Q2W + TCS con un peso basal de < 30 kg recibieron una dosis inicial de 200 mg el día 1, seguida de 100 mg Q2W desde la semana 2 hasta la semana 14, y los sujetos con un peso basal de ≥ 30 kg recibieron una dosis inicial de 400 mg el día 1, seguida de 200 mg Q2W desde la semana 2 hasta la semana 14. A los sujetos se les permitió recibir tratamiento de rescate a discreción del investigador. Los sujetos que recibieron tratamiento de rescate se consideraron no respondedores.
En AD-1652, la edad media era de 8,5 años, el peso mediano era de 29,8 kg, el 50% de los sujetos eran mujeres, el 69% eran blancos, el 17% eran negros y el 8% eran asiáticos. Al inicio, la participación media de BSA era del 58% y el 17% habían recibido previamente inmunosupresores sistémicos no esteroideos. Además, al inicio la puntuación media EASI era de 37,9 y la puntuación promedio semanal del peor prurito diario era de 7,8 en una escala de 0-10. En general, el 92% de los sujetos tenían al menos una condición alérgica coexistente; el 64% tenía alergias alimentarias, el 63% tenía otras alergias, el 60% tenía rinitis alérgica y el 47% tenía asma.
El punto final primario era la proporción de sujetos con una IGA 0 (clara) o 1 (casi clara) en la semana 16. Otros resultados evaluados incluyeron la proporción de sujetos con EASI-75 o EASI-90 (mejora de al menos 75% o 90% en EASI desde el inicio, respectivamente) y la reducción del prurito medida por la puntuación NRS de prurito pico (mejora de ≥ 4 puntos).
La Tabla 15 presenta los resultados por estratos de peso basal para los regímenes de dosis aprobados.
| DUPIXENT 300 mg Q4W† + TCS |
Placebo + TCS | DUPIXENT 200 mg Q2W‡ + TCS |
Placebo + TCS | |
|---|---|---|---|---|
| (N = 61) | (N = 61) | (N = 59) | (N = 62) | |
| < 30 kg | < 30 kg | ≥ 30 kg | ≥ 30 kg | |
|
||||
| IGA 0 o 1§,¶ | 30% | 13% | 39% | 10% |
| EASI-75¶ | 75% | 28% | 75% | 26% |
| EASI-90¶ | 46% | 7% | 36% | 8% |
| Puntuación NRS de prurito pico (mejora de ≥ 4 puntos)¶ | 54% | 12% | 61% | 13% |
Una mayor proporción de sujetos aleatorizados a DUPIXENT + TCS logró una mejora en el NRS de Picor Máximo en comparación con placebo + TCS (definido como una mejora de ≥ 4 puntos en la Semana 16). Vea Figura 3.
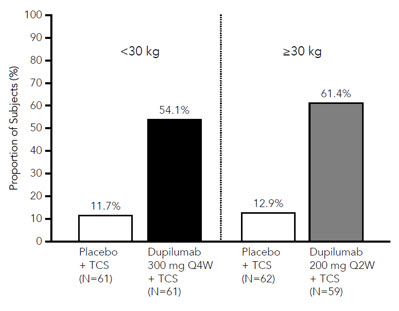
Sujetos Pediátricos de 6 Meses a 5 Años con Dermatitis Atópica
La eficacia y seguridad del uso de DUPIXENT concomitantemente con TCS en sujetos pediátricos se evaluó en un ensayo multicéntrico, aleatorizado, doble ciego, controlado con placebo (AD-1539; NCT03346434) en 162 sujetos de 6 meses a 5 años de edad, con DA moderada a grave definida por una puntuación IGA ≥ 3 (escala de 0 a 4), una puntuación EASI ≥ 16 (escala de 0 a 72) y una participación mínima de BSA de ≥ 10%. Los sujetos elegibles inscritos en este ensayo tuvieron una respuesta inadecuada previa al tratamiento tópico. La inscripción se estratificó por el peso basal (≥ 5 a < 15 kg y ≥ 15 a < 30 kg).
Los sujetos en el grupo DUPIXENT Q4W + TCS con un peso basal de ≥ 5 a < 15 kg recibieron una dosis inicial de 200 mg en el Día 1, seguida de 200 mg Q4W desde la Semana 4 hasta la Semana 12, y los sujetos con un peso basal de ≥ 15 a < 30 kg recibieron una dosis inicial de 300 mg en el Día 1, seguida de 300 mg Q4W desde la Semana 4 hasta la Semana 12. Los sujetos podían recibir tratamiento de rescate a discreción del investigador. Los sujetos que recibieron tratamiento de rescate se consideraron no respondedores.
En AD-1539, la edad media era de 3,8 años, el peso mediano era de 16,5 kg, el 39% de los sujetos eran mujeres, el 69% eran blancos, el 19% eran negros y el 6% eran asiáticos. Al inicio, la participación media de BSA era del 58%, y el 29% de los sujetos habían recibido inmunosupresores sistémicos previos. Además, al inicio, la puntuación media EASI era de 34,1 y la puntuación media semanal de la peor picazón / comezón diaria era de 7,6 en una escala de 0-10. En general, el 81,4% de los sujetos tenían al menos una condición alérgica coexistente; el 68,3% tenían alergias alimentarias, el 52,8% tenían otras alergias, el 44,1% tenían rinitis alérgica y el 25,5% tenían asma.
El punto final primario era la proporción de sujetos con una IGA 0 (clara) o 1 (casi clara) en la Semana 16. Otros resultados evaluados incluyeron la proporción de sujetos con EASI-75 o EASI-90 (mejora de al menos 75% o 90% en EASI desde el inicio, respectivamente), y la reducción de la picazón medida por el NRS de la Peor Picazón / Comezón (mejora de ≥ 4 puntos).
Los resultados de eficacia en la Semana 16 para AD-1539 se presentan en la Tabla 16.
| DUPIXENT + TCS 200 mg (5 a < 15 kg) o 300 mg (15 a < 30 kg) Q4W† |
Placebo + TCS | Diferencia vs. Placebo (95 % IC) | |
|---|---|---|---|
| (N = 83)* | (N = 79)* | ||
| IC = intervalo de confianza | |||
|
|||
| IGA 0 o 1‡,§ | 28% | 4% | 24% (13%, 34%) |
| EASI-75§ | 53% | 11% | 42% (29%, 55%) |
| EASI-90§ | 25% | 3% | 23% (12%, 33%) |
| NRS de la Peor Picazón / Comezón (mejora de ≥ 4 puntos)§ | 48% | 9% | 39% (26%, 52%) |
Dermatitis atópica con afectación de manos y/o pies
La eficacia y seguridad de DUPIXENT se evaluaron en un ensayo clínico aleatorizado, doble ciego, paralelo, controlado con placebo de 16 semanas, multicéntrico (Liberty-AD-HAFT; NCT04417894) en 133 sujetos adultos y pediátricos de 12 a 17 años de edad con dermatitis atópica con afectación moderada a grave de manos y/o pies, definida por un diagnóstico establecido de dermatitis atópica y un cribado para descartar la dermatitis de contacto irritante y alérgica mediante la historia y las pruebas de parche adecuadas, y por un puntaje IGA (manos y pies) ≥3 (escala de 0 a 4) y un puntaje de la Escala Numérica de Calificación de Picazón Máxima (NRS) de manos y pies para la intensidad máxima de la picazón ≥4 (escala de 0 a 10). Cincuenta y tres (53) por ciento (N = 70/133) de los sujetos también tenían dermatitis atópica moderada a grave fuera de las manos o los pies (IGA global ≥3). Los sujetos elegibles tuvieron una respuesta insuficiente previa o intolerancia al tratamiento de la dermatitis de manos y/o pies con medicamentos tópicos para AD. En este ensayo, 67 sujetos recibieron DUPIXENT y 66 sujetos recibieron placebo. Los sujetos tratados con DUPIXENT recibieron la dosis recomendada en función de su edad y peso corporal [ver Dosificación y administración (2.3) ] . A los sujetos no se les permitió el uso concomitante de tratamientos tópicos para AD en las manos y los pies durante el ensayo, pero se les permitió el uso de tratamientos tópicos para AD en otras partes del cuerpo con ciertas restricciones.
En Liberty-AD-HAFT, el 38% de los sujetos eran hombres, el 80% eran blancos, el 13% eran asiáticos y el 5% eran negros o afroamericanos. En cuanto a la etnia, el 4% se identificó como hispano o latino y el 96% se identificó como no hispano o latino. Setenta y dos (72) por ciento (N = 96/133) de los sujetos tenían un puntaje IGA (manos y pies) basal de 3 (dermatitis atópica con afectación moderada de manos y/o pies) y el 28% (N = 37/133) de los sujetos tenían un puntaje IGA (manos y pies) basal de 4 (dermatitis atópica con afectación grave de manos y/o pies). El puntaje promedio semanal basal de la Escala NRS de Picazón Máxima de manos y pies era 7.1.
El punto final primario fue la proporción de sujetos con un puntaje IGA de manos y pies de 0 (claro) o 1 (casi claro) en la semana 16. El punto final secundario clave fue la reducción de la picazón medida por la Escala NRS de Picazón Máxima de manos y pies (mejora ≥4 puntos).
Los resultados de eficacia en la semana 16 para Liberty-AD-HAFT se presentan en la Tabla 17.
| DUPIXENT 200/300 mg Q2W† |
Placebo | Diferencia vs. Placebo (95 % IC) | |
|---|---|---|---|
| (N = 67)* | (N = 66)* | ||
| IC = intervalo de confianza | |||
|
|||
| IGA (manos y pies) 0 o 1‡,§ | 40% | 17% | 24% (9%, 38%) |
| Mejora (reducción) de la Escala NRS de Picazón Máxima promedio semanal de manos y pies ≥4§ | 52% | 14% | 39% (24%, 53%) |
14.2 Asma
El programa de desarrollo del asma para pacientes de 12 años o más incluyó tres ensayos aleatorizados, doble ciego, controlados con placebo, de grupos paralelos y multicéntricos (DRI12544 (NCT01854047), QUEST (NCT02414854) y VENTURE (NCT02528214)) de 24 a 52 semanas de duración del tratamiento que reclutaron un total de 2888 sujetos. Los sujetos inscritos en DRI12544 y QUEST debían tener antecedentes de 1 o más exacerbaciones de asma que requirieron tratamiento con corticosteroides sistémicos o visita al departamento de emergencias u hospitalización para el tratamiento del asma en el año anterior a la entrada al ensayo. Los sujetos inscritos en VENTURE requirieron dependencia de corticosteroides orales diarios además del uso regular de corticosteroides inhalados de alta dosis más un controlador adicional. En los 3 ensayos, los sujetos se inscribieron sin requerir un recuento mínimo de eosinófilos en sangre basal. En QUEST y VENTURE, se excluyeron los sujetos con un nivel de eosinófilos en sangre de cribado de >1500 células/mcL (<1.3%). DUPIXENT se administró como complemento al tratamiento de fondo del asma. Los sujetos continuaron con la terapia de fondo para el asma durante toda la duración de los estudios, excepto en VENTURE, en el que la dosis de OCS se redujo gradualmente como se describe a continuación.
DRI12544
DRI12544 fue un estudio de rango de dosis de 24 semanas que incluyó 776 sujetos adultos (de 18 años de edad o más). DUPIXENT en comparación con placebo se evaluó en sujetos adultos con asma de moderada a grave con un corticosteroide inhalado de dosis media o alta y un agonista beta de acción prolongada. Los sujetos se asignaron aleatoriamente para recibir 200 mg (N=150) o 300 mg (N=157) de DUPIXENT cada dos semanas (Q2W) o 200 mg (N=154) o 300 mg (N=157) de DUPIXENT cada 4 semanas después de una dosis inicial de 400 mg, 600 mg o placebo (N=158), respectivamente. El criterio de valoración principal fue el cambio medio desde el inicio hasta la semana 12 en FEV1 (L) en sujetos con eosinófilos en sangre basal ≥300 células/mcL. Otros criterios de valoración incluyeron el porcentaje de cambio desde el inicio en FEV1 y la tasa anualizada de eventos de exacerbación grave del asma durante el período de tratamiento controlado con placebo de 24 semanas. Los resultados se evaluaron en la población general y en subgrupos basados en el recuento de eosinófilos en sangre basal (≥300 células/mcL y <300 células/mcL). Otros criterios de valoración secundarios adicionales incluyeron las tasas de respuesta en el Cuestionario de control del asma informado por el paciente (ACQ-5) y las puntuaciones del Cuestionario de calidad de vida del asma, versión estandarizada (AQLQ(S)).
QUEST
QUEST fue un estudio de 52 semanas que incluyó 1902 sujetos adultos y pediátricos (de 12 años de edad o más). DUPIXENT en comparación con placebo se evaluó en 107 sujetos pediátricos de 12 a 17 años de edad y 1795 sujetos adultos con asma de moderada a grave con un corticosteroide inhalado (ICS) de dosis media o alta y un mínimo de uno y hasta dos medicamentos controladores adicionales. Los sujetos se asignaron aleatoriamente para recibir 200 mg (N=631) o 300 mg (N=633) de DUPIXENT Q2W (o placebo coincidente para 200 mg [N=317] o 300 mg [N=321] Q2W) después de una dosis inicial de 400 mg, 600 mg o placebo, respectivamente. Los criterios de valoración principales fueron la tasa anualizada de eventos de exacerbación grave durante el período controlado con placebo de 52 semanas y el cambio desde el inicio en FEV1 pre-broncodilatador en la semana 12 en la población general (sin restricciones por el recuento mínimo de eosinófilos en sangre basal). Otros criterios de valoración secundarios adicionales incluyeron las tasas de exacerbación grave anualizadas y FEV1 en sujetos con diferentes niveles basales de eosinófilos en sangre, así como las tasas de respuesta en las puntuaciones ACQ-5 y AQLQ(S).
VENTURE
VENTURE fue un estudio de reducción de corticosteroides orales de 24 semanas en 210 sujetos adultos y pediátricos de 15 años de edad o más con asma que requirieron corticosteroides orales diarios además del uso regular de corticosteroides inhalados de alta dosis más un controlador adicional. Después de optimizar la dosis de OCS durante el período de selección, los sujetos recibieron 300 mg de DUPIXENT (N=103) o placebo (N=107) una vez Q2W durante 24 semanas después de una dosis inicial de 600 mg o placebo. Los sujetos continuaron recibiendo su medicamento actual para el asma durante el estudio; sin embargo, su dosis de OCS se redujo cada 4 semanas durante la fase de reducción de OCS (semana 4-20), siempre que se mantuviera el control del asma. El criterio de valoración principal fue la reducción porcentual de la dosis de corticosteroides orales en las semanas 20 a 24 en comparación con la dosis basal, mientras se mantenía el control del asma en la población general (sin restricciones por el recuento mínimo de eosinófilos en sangre basal). Otros criterios de valoración secundarios adicionales incluyeron la tasa anualizada de eventos de exacerbación grave durante el período de tratamiento y la tasa de respuesta en las puntuaciones ACQ-5 y AQLQ(S).
Las características demográficas y basales de estos 3 ensayos se proporcionan en la Tabla 18 a continuación.
| Parámetro | DRI12544 (N=776) |
QUEST (N=1902) |
VENTURE (N=210) |
|---|---|---|---|
| ICS = corticosteroide inhalado; FEV1 = Volumen espiratorio forzado en 1 segundo; AD = dermatitis atópica; NP = pólipos nasales; AR = rinitis alérgica; FeNO = fracción de óxido nítrico exhalado | |||
| Edad media (años) (DE) | 49 (13) | 48 (15) | 51 (13) |
| % Femenino | 63 | 63 | 61 |
| % Blanco | 78 | 83 | 94 |
| Duración del asma (años), media (± DE) | 22 (15) | 21 (15) | 20 (14) |
| Nunca fumó (%) | 77 | 81 | 81 |
| Exacerbaciones medias en el año anterior (± DE) | 2.2 (2.1) | 2.1 (2.2) | 2.1 (2.2) |
| Uso de ICS de alta dosis (%) | 50 | 52 | 89 |
| FEV1 pre-dosis (L) en la línea de base (± DE) | 1.84 (0.54) | 1.78 (0.60) | 1.58 (0.57) |
| Porcentaje medio predicho de FEV1 en la línea de base (%) (± DE) | 61 (11) | 58 (14) | 52 (15) |
| % Reversibilidad (± DE) | 27 (15) | 26 (22) | 19 (23) |
| Historia médica atópica % General (AD %, NP %, AR %) |
73 (8, 11, 62) |
78 (10, 13, 69) |
72 (8, 21, 56) |
| FeNO medio ppb (± DE) | 39 (35) | 35 (33) | 38 (31) |
| IgE total medio UI/mL (± DE) | 435 (754) | 432 (747) | 431 (776) |
| Recuento medio de eosinófilos en sangre en la línea de base (± DE) células/mcL | 350 (430) | 360 (370) | 350 (310) |
Exacerbaciones en sujetos con asma
DRI12544 y QUEST evaluaron la frecuencia de exacerbaciones graves del asma definidas como deterioro del asma que requirió el uso de corticosteroides sistémicos durante al menos 3 días u hospitalización o visita a la sala de emergencias debido a asma que requirió corticosteroides sistémicos. En la población de análisis primario (sujetos con recuento de eosinófilos en sangre basal de ≥300 células/mcL en DRI12544 y la población general en QUEST), los sujetos que recibieron DUPIXENT 200 mg o 300 mg Q2W tuvieron reducciones significativas en la tasa de exacerbaciones del asma en comparación con el placebo. En la población general en QUEST, la tasa de exacerbaciones graves fue de 0,46 y 0,52 para DUPIXENT 200 mg Q2W y 300 mg Q2W, respectivamente, en comparación con las tasas de placebo coincidentes de 0,87 y 0,97. La razón de tasas de exacerbaciones graves en comparación con el placebo fue de 0,52 (IC del 95%: 0,41, 0,66) y 0,54 (IC del 95%: 0,43, 0,68) para DUPIXENT 200 mg Q2W y 300 mg Q2W, respectivamente. Los resultados en sujetos con recuentos de eosinófilos en sangre basal ≥300 células/mcL en DRI12544 y QUEST se muestran en la Tabla 19.
Las tasas de respuesta por eosinófilos en sangre basal y FeNO basal para QUEST se muestran para la población general en la Figura 4 y la Figura 5, respectivamente. La elevación de FeNO puede ser un marcador del fenotipo de asma eosinofílico cuando está respaldado por datos clínicos. Los análisis de subgrupos preespecificados de DRI12544 y QUEST demostraron que hubo mayores reducciones en las exacerbaciones graves en sujetos con niveles más altos de eosinófilos en sangre basal (≥150 células/mcL) o FeNO (≥25 ppb). En QUEST, las reducciones en las exacerbaciones fueron significativas en el subgrupo de sujetos con eosinófilos en sangre basal ≥150 células/mcL. En sujetos con recuento de eosinófilos en sangre basal <150 células/mcL y FeNO <25 ppb, se observaron tasas de exacerbación grave similares entre DUPIXENT y placebo.
En QUEST, la razón de tasas estimada de exacerbaciones que llevaron a hospitalizaciones y/o visitas a la sala de emergencias versus placebo fue de 0,53 (IC del 95%: 0,28, 1,03) y 0,74 (IC del 95%: 0,32, 1,70) con DUPIXENT 200 mg o 300 mg Q2W, respectivamente.
| Ensayo | Tratamiento | EOS en sangre basal ≥300 células/mcL (población de análisis primario, DRI12544) |
||
|---|---|---|---|---|
| N | Tasa (IC del 95%) |
Razón de tasas (IC del 95%) |
||
| DRI12544 | DUPIXENT 200 mg Q2W |
65 | 0,30 (0,13, 0,68) |
0,29 (0,11, 0,76) |
| DUPIXENT 300 mg Q2W |
64 | 0,20 (0,08, 0,52) |
0,19 (0,07, 0,56) |
|
| Placebo | 68 | 1,04 (0,57, 1,90) |
||
| QUEST | DUPIXENT 200 mg Q2W |
264 | 0,37 (0,29, 0,48) |
0,34 (0,24, 0,48) |
| Placebo | 148 | 1,08 (0,85, 1,38) |
||
| DUPIXENT 300 mg Q2W |
277 | 0,40 (0,32, 0,51) |
0,33 (0,23, 0,45) |
|
| Placebo | 142 | 1,24 (0,97, 1,57) |
||
Figura 4: Riesgo relativo en la tasa de eventos anualizada de exacerbaciones graves en todo el recuento de eosinófilos en sangre basal (células/mcL) en sujetos con asma de moderada a grave (QUEST)
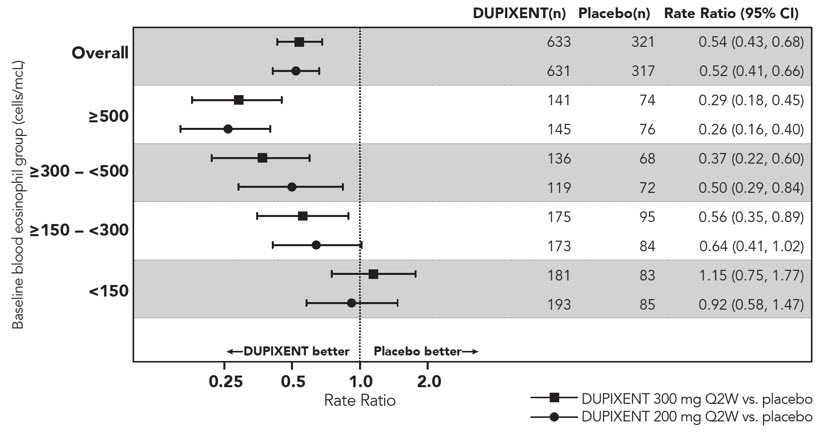
Figura 5: Riesgo relativo en la tasa de eventos anualizada de exacerbaciones graves en todo el grupo de FeNO basal (ppb) en sujetos con asma de moderada a grave (QUEST)
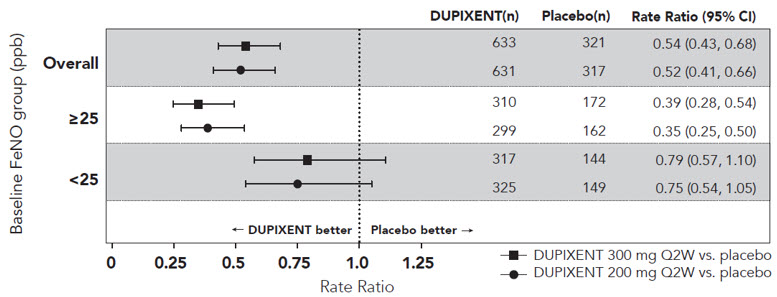
El tiempo hasta la primera exacerbación fue más largo para los sujetos que recibieron DUPIXENT en comparación con el placebo en QUEST (Figura 6).
Función pulmonar en sujetos con asma
Se observaron aumentos significativos en el FEV1 pre-broncodilatador en la semana 12 para DRI12544 y QUEST en las poblaciones de análisis primarias (sujetos con recuento de eosinófilos en sangre basal de ≥300 células/mcL en DRI12544 y la población general en QUEST). En la población general en QUEST, el cambio medio de LS del FEV1 desde el inicio fue de 0,32 L (21%) y 0,34 L (23%) para DUPIXENT 200 mg Q2W y 300 mg Q2W, respectivamente, en comparación con las medias de placebo coincidentes de 0,18 L (12%) y 0,21 L (14%). La diferencia media del tratamiento frente a placebo fue de 0,14 L (IC del 95%: 0,08, 0,19) y 0,13 L (IC del 95%: 0,08, 0,18) para DUPIXENT 200 mg Q2W y 300 mg Q2W, respectivamente. Los resultados en sujetos con recuentos de eosinófilos en sangre basal ≥300 células/mcL en DRI12544 y QUEST se muestran en la Tabla 20.
Las mejoras en el FEV1 por eosinófilos en sangre basal y FeNO basal para QUEST se muestran en las Figuras 7 y 8, respectivamente. El análisis de subgrupos de DRI12544 y QUEST demostró una mayor mejora en los sujetos con eosinófilos en sangre basal más altos (≥150 células/mcL) o FeNO (≥25 ppb). En los sujetos con recuento de eosinófilos en sangre basal <150 células/mcL y FeNO <25 ppb, se observaron diferencias similares en el FEV1 entre DUPIXENT y placebo.
Los cambios medios en el FEV1 a lo largo del tiempo en QUEST se muestran en la Figura 9.
| Ensayo | Tratamiento | EOS en sangre basal ≥300 células/mcL (población de análisis primaria, DRI12544) |
||
|---|---|---|---|---|
| N | Cambio medio de LS desde el inicio L (%) |
Diferencia media de LS frente a placebo (IC del 95%) |
||
| DRI12544 | DUPIXENT 200 mg Q2W |
65 | 0,43 (25,9) | 0,26 (0,11, 0,40) |
| DUPIXENT 300 mg Q2W |
64 | 0,39 (25,8) | 0,21 (0,06, 0,36) |
|
| Placebo | 68 | 0,18 (10,2) | ||
| QUEST | DUPIXENT 200 mg Q2W |
264 | 0,43 (29,0) | 0,21 (0,13, 0,29) |
| Placebo | 148 | 0,21 (15,6) | ||
| DUPIXENT 300 mg Q2W |
277 | 0,47 (32,5) | 0,24 (0,16, 0,32) |
|
| Placebo | 142 | 0,22 (14,4) | ||
Figura 7: Diferencia de media LS en el cambio desde el inicio hasta la semana 12 en comparación con el placebo en FEV1 pre-broncodilatador en función de los recuentos de eosinófilos en sangre al inicio (células/mcL) en sujetos con asma de moderada a grave (QUEST)
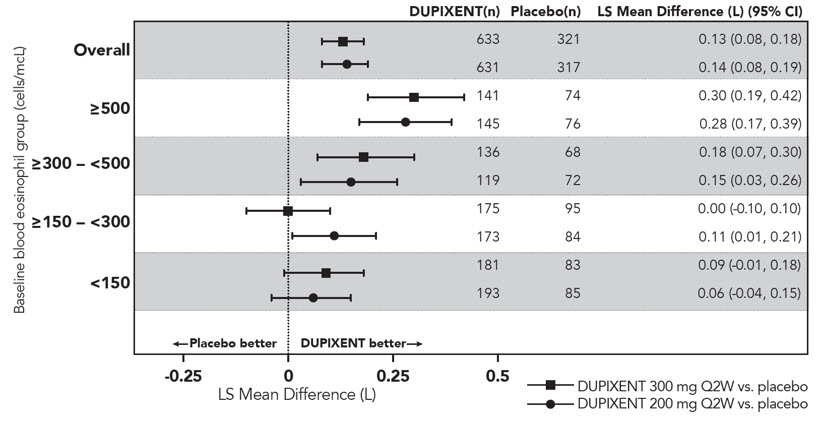
Figura 8: Diferencia de media LS en el cambio desde el inicio hasta la semana 12 en comparación con el placebo en FEV1 pre-broncodilatador en función de FeNO al inicio (ppb) en sujetos con asma de moderada a grave (QUEST)
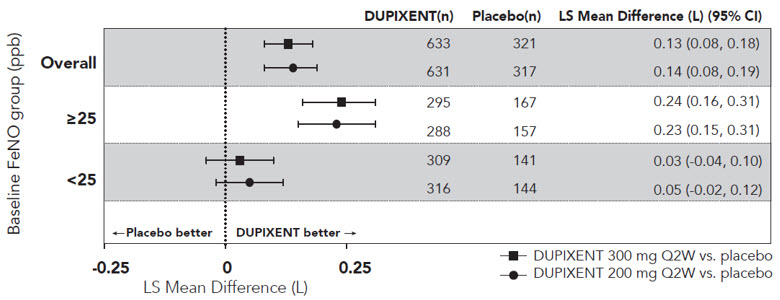
Figura 9: Cambio medio desde el inicio en FEV1 pre-broncodilatador (L) a lo largo del tiempo en sujetos con asma de moderada a grave con eosinófilos en sangre al inicio ≥300 células/mcL (QUEST)
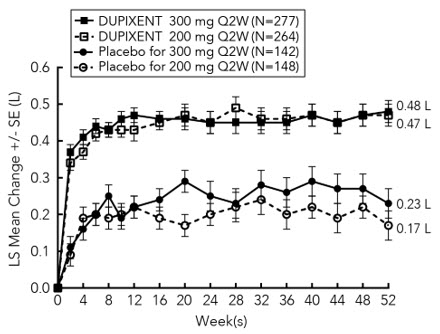
Puntos finales secundarios adicionales en el ensayo de asma (QUEST)
ACQ-5 y AQLQ(S) se evaluaron en QUEST a las 52 semanas. La tasa de respuesta se definió como una mejora en la puntuación de 0,5 o más (rango de escala 0-6 para ACQ-5 y 1-7 para AQLQ(S)).
- La tasa de respuesta de ACQ-5 para DUPIXENT 200 mg y 300 mg Q2W en la población general fue del 69% frente al 62% del placebo (odds ratio 1,37; IC del 95%: 1,01, 1,86) y del 69% frente al 63% del placebo (odds ratio 1,28; IC del 95%: 0,94, 1,73), respectivamente; y las tasas de respuesta de AQLQ(S) fueron del 62% frente al 54% del placebo (odds ratio 1,61; IC del 95%: 1,17, 2,21) y del 62% frente al 57% del placebo (odds ratio 1,33; IC del 95%: 0,98, 1,81), respectivamente.
- La tasa de respuesta de ACQ-5 para DUPIXENT 200 mg y 300 mg Q2W en sujetos con eosinófilos en sangre al inicio ≥300 células/mcL fue del 75% frente al 67% del placebo (odds ratio: 1,46; IC del 95%: 0,90, 2,35) y del 71% frente al 64% del placebo (odds ratio: 1,39; IC del 95%: 0,88, 2,19), respectivamente; y las tasas de respuesta de AQLQ(S) fueron del 71% frente al 55% del placebo (odds ratio: 2,02; IC del 95%: 1,24, 3,32) y del 65% frente al 55% del placebo (odds ratio: 1,79; IC del 95%: 1,13, 2,85), respectivamente.
Reducción de corticosteroides orales en el ensayo de asma (VENTURE)
VENTURE evaluó el efecto de DUPIXENT en la reducción del uso de corticosteroides orales de mantenimiento. La dosis media de corticosteroides orales al inicio fue de 12 mg en el grupo placebo y de 11 mg en el grupo que recibió DUPIXENT. El punto final primario fue la reducción porcentual desde el inicio de la dosis final de corticosteroides orales en la semana 24, manteniendo el control del asma.
En comparación con el placebo, los sujetos que recibieron DUPIXENT lograron mayores reducciones en la dosis diaria de mantenimiento de corticosteroides orales, manteniendo el control del asma. La reducción porcentual media de la dosis diaria de OCS desde el inicio fue del 70% (mediana del 100%) en los sujetos que recibieron DUPIXENT (IC del 95%: 60%, 80%) en comparación con el 42% (mediana del 50%) en los sujetos que recibieron placebo (IC del 95%: 33%, 51%). Se observaron reducciones del 50% o más en la dosis de OCS en 82 (80%) sujetos que recibieron DUPIXENT en comparación con 57 (53%) en los que recibieron placebo. La proporción de sujetos con una dosis final media inferior a 5 mg en la semana 24 fue del 72% para DUPIXENT y del 37% para el placebo (odds ratio 4,48 IC del 95%: 2,39, 8,39). Un total de 54 (52%) sujetos que recibieron DUPIXENT frente a 31 (29%) sujetos en el grupo placebo tuvieron una reducción del 100% en su dosis de OCS.
En este ensayo de 24 semanas, las exacerbaciones del asma (definidas como un aumento temporal de la dosis de corticosteroides orales durante al menos 3 días) fueron menores en los sujetos que recibieron DUPIXENT en comparación con los que recibieron placebo (tasa anualizada 0,65 y 1,60 para el grupo DUPIXENT y placebo, respectivamente; tasa ratio 0,41 [IC del 95% 0,26, 0,63]) y la mejora en FEV1 pre-broncodilatador desde el inicio hasta la semana 24 fue mayor en los sujetos que recibieron DUPIXENT en comparación con los que recibieron placebo (diferencia de media LS para DUPIXENT frente a placebo de 0,22 L [IC del 95%: 0,09 a 0,34 L]). Los efectos sobre la función pulmonar y sobre la reducción de los esteroides orales y las exacerbaciones fueron similares independientemente de los niveles de eosinófilos en sangre al inicio. El ACQ-5 y el AQLQ(S) también se evaluaron en VENTURE y mostraron mejoras similares a las de QUEST.
Sujetos pediátricos de 6 a 11 años de edad con asma
La eficacia y seguridad de DUPIXENT en sujetos pediátricos se evaluó en un estudio multicéntrico, aleatorizado, doble ciego, controlado con placebo de 52 semanas (VOYAGE; NCT02948959) en 408 sujetos de 6 a 11 años de edad, con asma de moderada a grave con un ICS de dosis media o alta y un segundo medicamento controlador o un ICS de dosis alta solo. Los sujetos debían tener antecedentes de 1 o más exacerbaciones del asma que requirieron tratamiento con corticosteroides sistémicos o visita al servicio de urgencias u hospitalización para el tratamiento del asma en el año anterior a la entrada en el ensayo. Los sujetos se aleatorizaron a DUPIXENT (N=273) o placebo coincidente (N=135) cada dos semanas en función del peso corporal <30 kg (100 mg Q2W) o ≥30 kg (200 mg Q2W). La eficacia de DUPIXENT 300 mg Q4W se extrapoló de la eficacia de 100 mg Q2W en VOYAGE con el apoyo de análisis farmacocinéticos poblacionales que mostraron niveles de exposición al fármaco más altos con 300 mg Q4W [ver Uso pediátrico (8.4) y Farmacocinética (12.3)].
El punto final primario fue la tasa anualizada de eventos de exacerbación grave del asma durante el período de 52 semanas controlado con placebo. Las exacerbaciones graves del asma se definieron como un deterioro del asma que requirió el uso de corticosteroides sistémicos durante al menos 3 días o hospitalización o visita a urgencias debido al asma que requirió corticosteroides sistémicos. El punto final secundario clave fue el cambio desde el inicio en el porcentaje de FEV1 pre-broncodilatador predicho en la semana 12. Los puntos finales secundarios adicionales incluyeron el cambio medio desde el inicio y las tasas de respuesta en las puntuaciones de ACQ-7-IA (Cuestionario de control del asma-7-administrado por el entrevistador) y PAQLQ(S)-IA (Cuestionario de calidad de vida del asma pediátrico con entrevista estandarizada de actividades administrada por el entrevistador).
La demografía y las características al inicio de VOYAGE se proporcionan en la Tabla 21 a continuación.
| Parámetro | VOYAGE (N=408) |
|---|---|
| ICS = corticosteroide inhalado; FEV1 = volumen espiratorio forzado en 1 segundo; AD = dermatitis atópica; AR = rinitis alérgica; FeNO = fracción de óxido nítrico exhalado | |
| Edad media (años) (DE) | 9 (2) |
| % Femenino | 36 |
| % Blanco | 88 |
| Peso corporal medio (kg) | 36 |
| Exacerbaciones medias en el año anterior (± DE) | 2.4 (2.2) |
| Uso de ICS de alta dosis (%) | 44 |
| FEV1 (L) previo a la dosis al inicio (± DE) | 1.48 (0.41) |
| Porcentaje medio predicho de FEV1 (%) (±DE) | 78 (15) |
| Porcentaje medio de reversibilidad (± DE) | 20 (21) |
| Historia médica atópica % General (AD %, AR %) |
92 (36, 82) |
| FeNO medio ppb (± DE) | 28 (24) |
| % de sujetos con FeNO ppb ≥20 | 50 |
| Mediana total de IgE UI/mL (±DE) | 792 (1093) |
| Recuento medio de eosinófilos al inicio (± DE) células/mcL | 502 (395) |
DUPIXENT redujo significativamente la tasa anualizada de eventos de exacerbación grave del asma durante el período de tratamiento de 52 semanas en comparación con el placebo en poblaciones con un fenotipo eosinofílico, como lo indica el aumento de eosinófilos en sangre y/o la población con FeNO elevado. Los análisis de subgrupos para los resultados del tratamiento con DUPIXENT basados en el nivel de eosinófilos basal o el nivel de FeNO basal fueron similares a los ensayos pediátricos (de 12 a 17 años de edad) y para adultos y se describen para la población de asma adulta y pediátrica (de 12 a 17 años de edad) anterior. En sujetos con recuento de eosinófilos en sangre basal <150 células/mcL y FeNO <20 ppb, se observaron tasas de exacerbación grave del asma similares entre DUPIXENT y el placebo.
Se observaron mejoras significativas en el porcentaje predicho de FEV1 pre-broncodilatador en la semana 12. Se observaron mejoras significativas en el porcentaje predicho de FEV1 tan pronto como en la semana 2 y se mantuvieron hasta la semana 52 en VOYAGE (Figura 10).
Los resultados de eficacia para VOYAGE se presentan en la Tabla 22.
| Tratamiento | EOS ≥300 células/mcL* | ||
|---|---|---|---|
| Tasa anualizada de exacerbaciones graves durante 52 semanas | |||
| N | Tasa (IC del 95%) |
Razón de tasas (IC del 95%) |
|
| DUPIXENT 100 mg Q2W† (<30 kg)/ 200 mg Q2W (≥30 kg) |
175 | 0.24 (0.16, 0.35) |
0.35 (0.22, 0.56) |
| Placebo | 84 | 0.67 (0.47, 0.95) |
|
| Cambio medio desde el inicio en el porcentaje predicho de FEV1 en la semana 12 | |||
| N | LS media Δ desde el inicio | Diferencia de LS media vs. Placebo (IC del 95%) | |
| DUPIXENT 100 mg Q2W† (<30 kg)/ 200 mg Q2W (≥30 kg) |
168 | 10.15 | 5.32 (1.76, 8.88) |
| Placebo | 80 | 4.83 | |
Figura 10: Cambio medio desde el basal en el porcentaje previsto de FEV1 previo al broncodilatador (L) a lo largo del tiempo en sujetos pediátricos de 6 a 11 años de edad en VOYAGE (Eosinófilos en sangre basal ≥300 células/mcL)
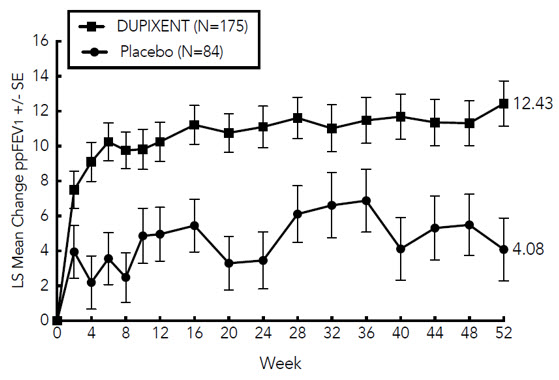
También se observaron mejoras en ACQ-7-IA y PAQLQ(S)-IA en la semana 24 y se mantuvieron en la semana 52. Se observaron tasas de respuesta más altas para ACQ-7-IA y PAQLQ(S)-IA en comparación con el placebo en la semana 24. La tasa de respuesta se definió como una mejora en la puntuación de 0.5 o más (rango de escala 0-6 para ACQ-7-IA y 1-7 para PAQLQ(S)-IA). En el subgrupo de sujetos con un recuento de eosinófilos en sangre basal ≥300 células/mcL, DUPIXENT condujo a una mayor proporción de sujetos con una respuesta en ACQ-7-IA (80.6% frente al 64.3% del placebo) con un OR de 2.79 (95% CI: 1.43, 5.44), y en PAQLQ(S)-IA (72.8% frente al 63.0% del placebo) con un OR de 1.84 (95% CI: 0.92, 3.65) en la semana 24.
14.3 Rinossinusite crónica com pólipos nasais
A eficácia do DUPIXENT como tratamento de manutenção adicional em adultos com rinossinusite crónica com pólipos nasais (CRSwNP) não controlada adequadamente foi avaliada em dois estudos randomizados, duplo-cegos, de grupo paralelo, multicêntricos, controlados por placebo (SINUS-24 (NCT02912468) e SINUS-52 (NCT02898454)) em 724 adultos com 18 anos de idade ou mais, com corticosteroides intranasais de fundo (INCS). Estes estudos incluíram sujetos com CRSwNP apesar de cirurgia prévia naso-sinusal ou tratamento com, ou que eram inelegíveis para receber ou intolerantes a, corticosteroides sistémicos nos últimos 2 anos. Sujetos com rinossinusite crónica sem pólipos nasais não foram incluídos nestes ensaios. O resgate com corticosteroides sistémicos ou cirurgia foi permitido durante os estudos a critério do investigador. Em SINUS-24, um total de 276 sujetos foram randomizados para receber 300 mg de DUPIXENT (N=143) ou placebo (N=133) a cada duas semanas durante 24 semanas. Em SINUS-52, 448 sujetos foram randomizados para receber 300 mg de DUPIXENT (N=150) a cada duas semanas durante 52 semanas, 300 mg de DUPIXENT (N=145) a cada duas semanas até a semana 24 seguido de 300 mg de DUPIXENT a cada 4 semanas até a semana 52, ou placebo (N=153). Todos os sujetos apresentaram evidência de opacificação sinusal na tomografia computadorizada dos seios da face Lund Mackay (LMK) e 73% a 90% dos sujetos tinham opacificação de todos os seios. Os sujetos foram estratificados com base nas suas histórias de cirurgia prévia e co-morbidade de asma/doença respiratória exacerbada por antiinflamatórios não esteroides (NSAID-ERD). Um total de 63% dos sujetos relatou cirurgia sinusal anterior, com um número médio de 2,0 cirurgias anteriores, 74% usou corticosteroides sistémicos nos últimos 2 anos com um número médio de 1,6 cursos de corticosteroides sistémicos nos últimos 2 anos, 59% tinha co-morbidade de asma e 28% tinha NSAID-ERD.
Os pontos finais de eficácia co-primários foram a mudança desde o basal até a semana 24 na pontuação bilateral endoscópica de pólipos nasais (NPS; escala 0-8) classificada por leitores cegos centrais, e a mudança desde o basal até a semana 24 na pontuação de congestão/obstrução nasal média ao longo de 28 dias (NC; escala 0-3), conforme determinada pelos sujetos usando um diário diário. Para NPS, os pólipos de cada lado do nariz foram classificados numa escala categórica (0 = sem pólipos; 1 = pólipos pequenos no meato médio que não atingem abaixo da borda inferior do turbinal médio; 2 = pólipos que atingem abaixo da borda inferior do turbinal médio; 3 = pólipos grandes que atingem a borda inferior do turbinal inferior ou pólipos mediais ao turbinal médio; 4 = pólipos grandes que causam obstrução completa da cavidade nasal inferior). A pontuação total era a soma das pontuações da direita e da esquerda. A congestão nasal foi classificada diariamente pelos sujetos numa escala categórica de 0 a 3 (0 = sem sintomas; 1 = sintomas leves; 2 = sintomas moderados; 3 = sintomas graves).
Em ambos os estudos, os pontos finais secundários chave na semana 24 incluíram a mudança desde o basal em: pontuação da tomografia computadorizada dos seios da face LMK, perda diária do olfato e teste de resultado sino-nasal de 22 itens (SNOT-22). A pontuação da tomografia computadorizada dos seios da face LMK avaliou a opacificação de cada seio utilizando uma escala de 0 a 2 (0 = normal; 1 = opacificação parcial; 2 = opacificação total), obtendo uma pontuação máxima de 12 por lado e uma pontuação máxima total de 24 (pontuações mais altas indicam mais opacificação). A perda do olfato foi classificada reflexivamente pelo sujeito todas as manhãs numa escala de 0 a 3 (0 = sem sintomas, 1 = sintomas leves, 2 = sintomas moderados, 3 = sintomas graves). SNOT-22 inclui 22 itens que avaliam os sintomas e o impacto dos sintomas associados ao CRSwNP, com cada item pontuado de 0 (sem problema) a 5 (problema tão ruim quanto pode ser) com uma pontuação global que varia de 0 a 110. SNOT-22 teve um período de recordação de 2 semanas. Nos resultados de eficácia agregados, foi avaliada a redução na proporção de sujetos resgatados com corticosteroides sistémicos e/ou cirurgia naso-sinusal (até a semana 52).
A demografia e as características basais destes 2 ensaios são fornecidas na Tabela 23 abaixo.
| Parâmetro | SINUS-24 (N=276) |
SINUS-52 (N=448) |
|---|---|---|
| SD = desvio padrão; AM = manhã; NPS = pontuação de pólipos nasais; SNOT-22 = teste de resultado sino-nasal de 22 itens; NSAID-ERD = asma/doença respiratória exacerbada por antiinflamatórios não esteroides | ||
|
||
| Média de idade (anos) (SD) | 50 (13) | 52 (12) |
| % Homens | 57 | 62 |
| Duração média de CRSwNP (anos) (SD) | 11 (9) | 11 (10) |
| Sujetos com ≥1 cirurgia anterior (%) | 72 | 58 |
| Sujetos con uso de corticosteroides sistémicos en los 2 años anteriores (%) | 65 | 80 |
| Media de NPS endoscópica bilateral* (DE), rango 0-8 | 5.8 (1.3) | 6.1 (1.2) |
| Media de la puntuación de congestión nasal (NC)* (DE), rango 0-3 | 2.4 (0.6) | 2.4 (0.6) |
| Media de la puntuación total de la TC de los senos LMK* (DE), rango 0-24 | 19 (4.4) | 18 (3.8) |
| Media de la puntuación de pérdida del olfato *(AM), (DE) rango 0-3 | 2.7 (0.5) | 2.8 (0.5) |
| Media de la puntuación total de SNOT-22* (DE), rango 0-110 | 49.4 (20.2) | 51.9 (20.9) |
| Media de eosinófilos en sangre (células/mcL) (DE) | 440 (330) | 430 (350) |
| Media de IgE total IU/mL (DE) | 212 (276) | 240 (342) |
| Historia médica atópica % General |
75 | 82 |
| Asma (%) | 58 | 60 |
| NSAID-ERD (%) | 30 | 27 |
Respuesta clínica (SINUS-24 y SINUS-52)
Los resultados de los criterios de valoración primarios en los estudios de CRSwNP se presentan en la Tabla 24.
| SINUS-24 | SINUS-52 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Placebo (n=133) |
DUPIXENT 300 mg Q2W (n=143) |
Diferencia de media LS frente a placebo (IC del 95%) |
Placebo (n=153) |
DUPIXENT 300 mg Q2W (n=295) |
Diferencia de media LS frente a placebo (IC del 95%) |
|||||
| Criterios de valoración primarios en la semana 24 | ||||||||||
| Puntuaciones | Media basal | Cambio de media LS | Media basal | Cambio de media LS | Media basal | Cambio de media LS | Media basal | Cambio de media LS | ||
| Una reducción en la puntuación indica mejoría. NPS = puntuación de pólipos nasales; NC = congestión/obstrucción nasal |
||||||||||
| NPS | 5.86 | 0.17 | 5.64 | -1.89 | -2.06 (-2.43, -1.69) |
5.96 | 0.10 | 6.18 | -1.71 | -1.80 (-2.10, -1.51) |
| NC | 2.45 | -0.45 | 2.26 | -1.34 | -0.89 (-1.07, -0.71) |
2.38 | -0.38 | 2.46 | -1.25 | -0.87 (-1.03, -0.71) |
Se observó una eficacia estadísticamente significativa en SINUS-52 con respecto a la mejora en la puntuación endoscópica bilateral de NPS en la semana 24 y la semana 52 (ver Figura 11).
Figura 11: Cambio medio de LS desde el inicio en la puntuación de pólipos nasales bilaterales (NPS) hasta la semana 52 en sujetos de 18 años de edad o mayores con CRSwNP (SINUS-52 – Población ITT)
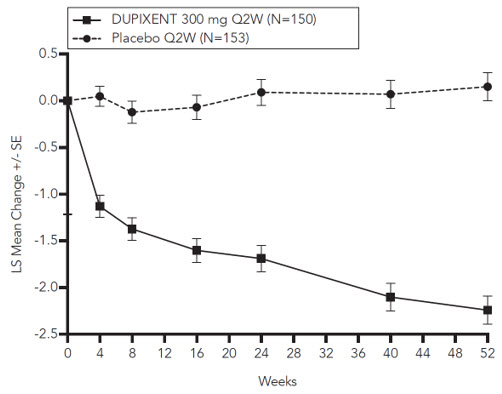
Se observaron resultados similares en SINUS-24 en la semana 24. En el período posterior al tratamiento, cuando los sujetos estaban fuera de DUPIXENT, el efecto del tratamiento disminuyó con el tiempo (ver Figura 12).
Figura 12: Cambio medio de LS desde el inicio en la puntuación de pólipos nasales bilaterales (NPS) hasta la semana 48 en sujetos de 18 años de edad o mayores con CRSwNP (SINUS-24 – Población ITT)
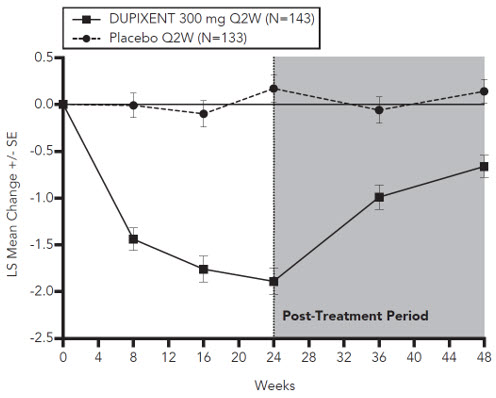
En la semana 52, la diferencia media de LS para la congestión nasal en el grupo DUPIXENT versus placebo fue -0,98 (IC del 95% -1,17, -0,79). En ambos estudios, se observaron mejoras significativas en la congestión nasal ya en la primera evaluación en la semana 4. La diferencia media de LS para la congestión nasal en la semana 4 en el grupo DUPIXENT versus placebo fue -0,41 (IC del 95%: -0,52, -0,30) en SINUS-24 y -0,37 (IC del 95%: -0,46, -0,27) en SINUS-52.
Se observó una disminución significativa en la puntuación de la tomografía computarizada de los senos LMK. La diferencia media de LS para la puntuación de la tomografía computarizada de los senos LMK en la semana 24 en el grupo DUPIXENT versus placebo fue -7,44 (IC del 95%: -8,35, -6,53) en SINUS-24 y -5,13 (IC del 95%: -5,80, -4,46) en SINUS-52. En la semana 52, en SINUS-52, la diferencia media de LS para la puntuación de la tomografía computarizada de los senos LMK en el grupo DUPIXENT versus placebo fue -6,94 (IC del 95%: -7,87, -6,01).
Dupilumab mejoró significativamente la pérdida del olfato en comparación con el placebo. La diferencia media de LS para la pérdida del olfato en la semana 24 en el grupo DUPIXENT versus placebo fue -1,12 (IC del 95%: -1,31, -0,93) en SINUS-24 y -0,98 (IC del 95%: -1,15, -0,81) en SINUS-52. En la semana 52, la diferencia media de LS para la pérdida del olfato en el grupo DUPIXENT versus placebo fue -1,10 (IC del 95% -1,31, -0,89). En ambos estudios, se observaron mejoras significativas en la gravedad diaria de la pérdida del olfato ya en la primera evaluación en la semana 4.
Dupilumab disminuyó significativamente los síntomas sinonasales medidos por SNOT-22 en comparación con el placebo. La diferencia media de LS para SNOT-22 en la semana 24 en el grupo DUPIXENT versus placebo fue -21,12 (IC del 95%: -25,17, -17,06) en SINUS-24 y -17,36 (IC del 95%: -20,87, -13,85) en SINUS-52. En la semana 52, la diferencia media de LS en el grupo DUPIXENT versus placebo fue -20,96 (IC del 95% -25,03, -16,89).
En el análisis agrupado preespecificado ajustado por multiplicidad de dos estudios, el tratamiento con DUPIXENT dio como resultado una reducción significativa del uso de corticosteroides sistémicos y la necesidad de cirugía sinonasal en comparación con el placebo (HR de 0,24; IC del 95%: 0,17, 0,35) (ver Figura 13). La proporción de sujetos que requirieron corticosteroides sistémicos se redujo en un 74% (HR de 0,26; IC del 95%: 0,18, 0,38). El número total de cursos de corticosteroides sistémicos por año se redujo en un 75% (RR de 0,25; IC del 95%: 0,17, 0,37). La proporción de sujetos que requirieron cirugía se redujo en un 83% (HR de 0,17; IC del 95%: 0,07, 0,46).
Figura 13: Curva de Kaplan Meier para el tiempo hasta el primer uso de corticosteroides sistémicos y/o cirugía sinonasal durante el período de tratamiento en sujetos de 18 años de edad o mayores con CRSwNP (SINUS-24 y SINUS-52 agrupados – Población ITT)
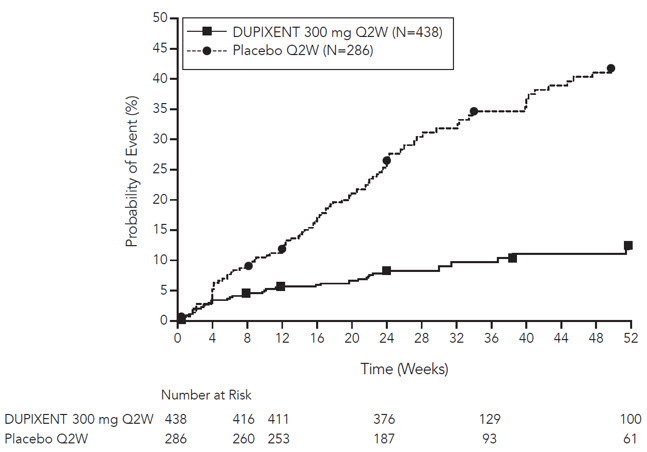
Los efectos de DUPIXENT en los criterios de valoración primarios de NPS y congestión nasal y el criterio de valoración secundario clave de la puntuación de la tomografía computarizada de los senos LMK fueron consistentes en sujetos con cirugía previa y sin cirugía previa.
En sujetos con asma comórbida, las mejoras en el FEV1 prebroncodilatador fueron similares a las de los sujetos en el programa de asma.
14.4 Esofagitis eosinofílica
Sujetos adultos y pediátricos de 12 años de edad o mayores con EoE
Se llevó a cabo un solo ensayo aleatorizado, doble ciego, de grupos paralelos, multicéntrico, controlado con placebo, que incluyó dos períodos de tratamiento de 24 semanas (Estudio EoE-1 Partes A y B) en sujetos adultos y pediátricos de 12 años de edad o mayores, con un peso de al menos 40 kg, con EoE (NCT03633617). En ambas partes, los sujetos fueron aleatorizados para recibir 300 mg de DUPIXENT cada semana o placebo. Los sujetos elegibles tenían ≥15 eosinófilos intraepiteliales por campo de alto poder (eos/hpf) después de un curso de tratamiento con un inhibidor de la bomba de protones (IBP) ya sea antes o durante el período de selección y síntomas de disfagia medidos por el Cuestionario de síntomas de disfagia (DSQ). En el inicio, el 43% de los sujetos en la Parte A y el 37% de los sujetos en la Parte B tenían antecedentes de dilataciones esofágicas previas.
Las características demográficas y de línea de base fueron similares en las Partes A y B. Se inscribieron un total de 81 sujetos (61 adultos y 20 sujetos pediátricos) en la Parte A y 159 sujetos (107 adultos y 52 sujetos pediátricos) en la Parte B. La edad media en años fue de 32 años (rango de 13 a 62 años) en la Parte A y de 28 años (rango de 12 a 66 años) en la Parte B. La mayoría de los sujetos fueron hombres (60% en la Parte A y 68% en la Parte B) y blancos (96% en la Parte A y 90% en la Parte B). La puntuación media de DSQ de línea de base (DE) fue de 33,6 (12,4) en la Parte A y de 37,2 (10,7) en la Parte B.
Los criterios de valoración de eficacia coprimarios en las Partes A y B fueron la (1) proporción de sujetos que lograron la remisión histológica definida como un recuento máximo de eosinófilos intraepiteliales esofágicos de ≤6 eos/hpf en la semana 24; y (2) el cambio absoluto en la puntuación de DSQ autoinformada desde el inicio hasta la semana 24.
Los resultados de eficacia para las Partes A y B se presentan en la Tabla 25.
| Estudio EoE-1 Parte A | Estudio EoE-1 Parte B | |||||
|---|---|---|---|---|---|---|
| DUPIXENT 300 mg QW* | Placebo* | Diferencia vs. Placebo (IC del 95%)* |
DUPIXENT 300 mg QW* | Placebo* | Diferencia vs. Placebo (IC del 95%)* |
|
| N = 42 | N = 39 | N = 80 | N = 79 | |||
|
||||||
| Variables de resultado primarias | ||||||
| Proporción de sujetos que alcanzan la remisión histológica (conteo máximo de eosinófilos intraepiteliales esofágicos ≤6 eos/hpf), n (%) | 25 (59.5) |
2 (5.1) |
57.0 (40.9, 73.1) |
47 (58.8) |
5 (6.3) |
53.5 (41.2, 65.8) |
| Cambio absoluto desde el inicio en la puntuación DSQ (0-84†), media LS (EE) | -21.9 (2.5) |
-9.6 (2.8) |
-12.3 (-19.1, -5.5) |
-23.8 (1.9) |
-13.9 (1.9) |
-9.9 (-14.8, -5.0) |
En las partes A y B, una mayor proporción de sujetos aleatorizados a DUPIXENT alcanzó la remisión histológica (recuento máximo de eosinófilos intraepiteliales esofágicos ≤ 6 eos/hpf) en comparación con el placebo. El tratamiento con DUPIXENT también resultó en una mejora significativa en el cambio medio LS en la puntuación DSQ en comparación con el placebo en la semana 24. Los resultados de los análisis basados en anclas que incorporaron las perspectivas de los sujetos indicaron que la mejora observada en la disfagia de las partes A y B representa una mejora clínicamente significativa dentro del sujeto.
Sujetos pediátricos de 1 a 11 años de edad, con un peso de al menos 15 kg, con EoE
La eficacia y seguridad de DUPIXENT se evaluó en sujetos pediátricos de 1 a 11 años de edad, con un peso de al menos 15 kg, con EoE en un ensayo clínico aleatorizado, ciego, paralelo, multicéntrico (Estudio EoE-2 Partes A y B; NCT04394351). Los sujetos elegibles tenían ≥ 15 eosinófilos intraepiteliales por campo de alta potencia (eos/hpf) a pesar de un tratamiento con un inhibidor de la bomba de protones (IPP) ya sea antes o durante el período de selección y un historial de signos y síntomas de EoE. La Parte A evaluó regímenes de dosificación basados en el peso de DUPIXENT, 200 mg cada 2 semanas (≥ 15 a < 30 kg) y 300 mg cada 2 semanas (≥ 30 a < 60 kg), o placebo en 61 sujetos durante el período de tratamiento de 16 semanas.
La dosis recomendada de 300 mg semanales para sujetos pediátricos de 1 a 11 años de edad con un peso de ≥ 40 kg se basa en datos farmacocinéticos modelados para proporcionar exposiciones comparables a la dosis de 300 mg semanales en adultos y sujetos pediátricos de 12 años de edad y mayores con un peso de ≥ 40 kg con EoE [véase Dosificación y administración (2.6) y Farmacocinética (12.3) ].
Cuarenta y siete sujetos que completaron la Parte A se evaluaron en el período de tratamiento activo extendido de 36 semanas (Estudio EoE-2 Parte B). Todos los sujetos en la Parte B fueron tratados con los regímenes de dosificación basados en el peso de DUPIXENT descritos para la Parte A.
De los sujetos totales evaluados en la Parte A, la edad media era de 8 años, el peso mediano era de 28 kg y el 75% eran varones. El 7% se identificó como hispano o latino; el 85% se identificó como blanco, el 12% como negro, el 2% como asiático y el 2% se identificó como otro subgrupo racial.
El punto final de eficacia primario en la Parte A fue la proporción de sujetos que alcanzó la remisión histológica definida como un recuento máximo de eosinófilos intraepiteliales esofágicos de ≤ 6 eos/hpf en la semana 16.
Los resultados de eficacia de la Parte A se presentan en la Tabla 26.
| DUPIXENT* N = 32 |
Placebo N = 29 |
Diferencia vs Placebo (95% CI) |
|
|---|---|---|---|
|
|||
| Proporción de sujetos que alcanzaron la remisión histológica (recuento máximo de eosinófilos intraepiteliales esofágicos ≤ 6 eos/hpf), n (%)† | 21 (65.6) |
1 (3.4) |
62.0 (44.00, 79.95) |
En la Parte B, la remisión histológica se alcanzó en la semana 52 en 17/32 sujetos tratados con DUPIXENT en las Partes A y B y en 8/15 sujetos tratados con placebo en la Parte A y DUPIXENT en la Parte B.
En el Estudio EoE-2 Parte A, un resultado reportado por el observador, el Cuestionario de Signos/Síntomas de EoE Pediátrico para Cuidadores (PESQ-C), se utilizó para medir los signos de EoE. Se observó una mayor disminución en la proporción de días con 1 o más signos de EoE (basado en el PESQ-C) en los sujetos tratados con DUPIXENT en comparación con el placebo después de 16 semanas de tratamiento.
14.5 Prurigo nodularis
El programa de desarrollo del prurigo nodularis (PN) incluyó dos ensayos aleatorizados, doble ciego, controlados con placebo, multicéntricos, de grupos paralelos de 24 semanas (PRIME (NCT04183335) y PRIME 2 (NCT04202679)) en 311 sujetos adultos de 18 años de edad o más con prurito (WI-NRS ≥ 7 en una escala de 0 a 10) y más de o igual a 20 lesiones nodulares. PRIME y PRIME 2 evaluaron el efecto de DUPIXENT en la mejora del prurito así como su efecto en las lesiones de PN.
En estos dos ensayos, los sujetos recibieron ya sea DUPIXENT subcutáneo 600 mg (dos inyecciones de 300 mg) en el día 1, seguido de 300 mg una vez cada dos semanas (Q2W) durante 24 semanas, o placebo equivalente.
En estos ensayos, la edad media fue de 49.5 años, el peso mediano fue de 71 kg, el 65% de los sujetos eran mujeres, el 57% eran blancos, el 6% eran negros y el 34% eran asiáticos. En el punto de partida, la escala media Worst Itch-Numeric Rating Scale (WI-NRS) fue de 8.5, el 66% tenía de 20 a 100 nódulos (moderado) y el 34% tenía más de 100 nódulos (grave). El once por ciento (11%) de los sujetos estaba tomando dosis estables de antidepresivos en el punto de partida y se les instruyó a continuar tomando estos medicamentos durante el ensayo. El cuarenta y tres por ciento (43%) tenía un historial de atopia (definido como tener un historial médico de AD, rinitis alérgica/rinoconjuntivitis, asma o alergia alimentaria).
La WI-NRS consta de un solo ítem, calificado en una escala de 0 (“sin prurito”) a 10 (“prurito imaginable más grave”). Los sujetos fueron solicitados a calificar la intensidad de su prurito más grave (picazón) en las últimas 24 horas utilizando esta escala. La Evaluación Global del Investigador para Prurigo Nodularis-Etapa (IGA PN-S) es una escala que mide el número aproximado de nódulos utilizando una escala de 5 puntos de 0 (claro) a 4 (grave).
La eficacia se evaluó con la proporción de sujetos con mejora (reducción) en la WI-NRS en ≥4 puntos, la proporción de sujetos con IGA PN-S 0 o 1 (equivalente a 0-5 nódulos), y la proporción de sujetos que lograron una respuesta tanto en la WI-NRS como en la IGA PN-S según los criterios descritos anteriormente.
Los resultados de eficacia de PRIME y PRIME2 se presentan en la Tabla 27 y en las Figuras 14, 15 y 16.
| PRIME | PRIME2 | |||||
|---|---|---|---|---|---|---|
| Placebo (N=76) |
DUPIXENT 300 mg Q2W (N=75) |
Diferencia (95% CI) para DUPIXENT vs. Placebo | Placebo (N=82) |
DUPIXENT 300 mg Q2W (N=78) |
Diferencia (95% CI) para DUPIXENT vs. Placebo | |
| Proporción de sujetos con tanto una mejora (reducción) en la WI-NRS en ≥4 puntos desde el punto de partida hasta la semana 24 y un IGA PN-S 0 o 1 en la semana 24* | 9.2% | 38.7% | 29.6% (16.4, 42.8) |
8.5% | 32.1% | 25.5% (13.1, 37.9) |
| Proporción de sujetos con mejora (reducción) en la WI-NRS en ≥4 puntos desde el punto de partida en la semana 24* | 18.4% | 60.0% | 42.7% (27.8, 57.7) |
19.5% | 57.7% | 42.6% (29.1, 56.1) |
| Proporción de sujetos con IGA PN-S 0 o 1 en la semana 24* | 18.4% | 48.0% | 28.3% (13.4, 43.2) |
15.9% | 44.9% | 30.8% (16.4, 45.2) |
| Proporción de sujetos con mejora (reducción) en la WI-NRS en ≥4 puntos desde el punto de partida en la semana 12* | 15.8%† | 44.0%† | 29.2% (14.5, 43.8)† |
22.0% | 37.2% | 16.8% (2.3, 31.2) |
Figura 14: Proporción de sujetos adultos con PN con mejora de ≥ 4 puntos en el WI-NRS y IGA PN-S 0 o 1 a lo largo del tiempo en PRIME y PRIME2
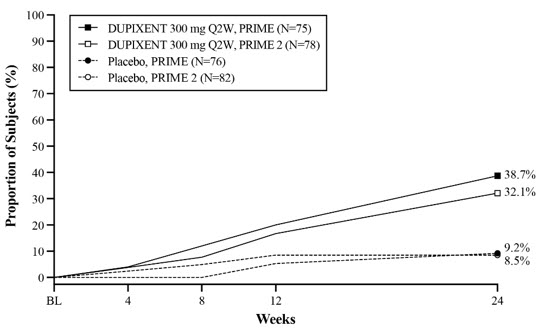
Figura 15: Proporción de sujetos adultos con PN con mejora de ≥ 4 puntos en el WI-NRS a lo largo del tiempo en PRIME y PRIME2
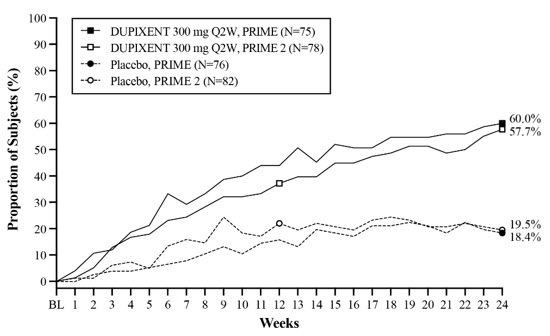
Figura 16: Proporción de sujetos adultos con IGA PN-S 0 o 1 a lo largo del tiempo en PRIME y PRIME 2
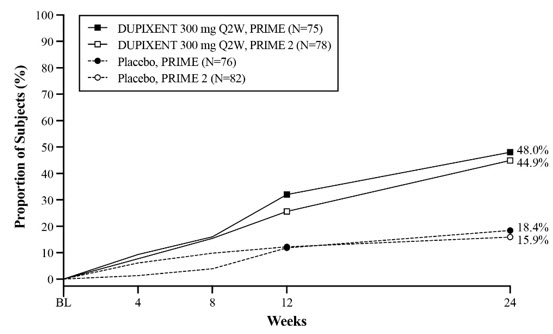
Los datos de eficacia no mostraron efecto diferencial del tratamiento en los subgrupos demográficos.
14.6 Enfermedad pulmonar obstructiva crónica
La eficacia de DUPIXENT como tratamiento de mantenimiento adicional en pacientes adultos con EPOC no controlada adecuadamente y fenotipo eosinofílico se evaluó en dos ensayos aleatorizados, doble ciego, multicéntricos, en grupos paralelos, controlados con placebo (BOREAS [NCT03930732] y NOTUS [NCT04456673]) de 52 semanas de duración. Los dos ensayos incluyeron un total de 1874 sujetos adultos con EPOC.
Ambos ensayos incluyeron sujetos con un diagnóstico de EPOC con limitación del flujo aéreo de moderada a grave (relación FEV1/FVC post-broncodilatador <0,7 y FEV1 post-broncodilatador del 30% al 70% previsto) y un recuento mínimo de eosinófilos en sangre de 300 células/mcL en la selección. La inclusión en el ensayo requirió un historial de exacerbaciones de al menos 2 moderadas o 1 grave en el año anterior a pesar de recibir el tratamiento de mantenimiento triple consistente en un antagonista muscarínico de acción prolongada (LAMA), un agonista beta de acción prolongada (LABA) y un corticoesteroide inhalado (ICS), y síntomas de tos productiva crónica durante al menos 3 meses en el año pasado. Más del 95% de los sujetos en cada ensayo tenían bronquitis crónica. Los sujetos también tuvieron un puntaje de disnea del Consejo Médico de Investigación (MRC) ≥2 (rango 0-4). Las exacerbaciones de la EPOC se definieron como un empeoramiento clínicamente significativo de los síntomas de la EPOC, incluidos aumentos en la disnea, sibilancias, tos, volumen de esputo y/o aumento en la purulencia del esputo. La gravedad de la exacerbación se definió como moderada si se requería tratamiento con corticoesteroides sistémicos y/o antibióticos, o grave si resultaron en hospitalización o observación durante más de 24 horas en un departamento de emergencias o un centro de atención urgente.
En ambos ensayos, los sujetos se aleatorizaron para recibir DUPIXENT 300 mg por vía subcutánea cada dos semanas (Q2W) o placebo además de su tratamiento de mantenimiento de fondo durante 52 semanas.
La demografía y las características basales de las poblaciones de ensayo BOREAS y NOTUS se proporcionan en la Tabla 28 a continuación.
| Parámetro | BOREAS (N = 939) |
NOTUS (N = 935) |
|---|---|---|
| ICS = corticoesteroide inhalado; LAMA = antagonista muscarínico de acción prolongada; LABA = agonista beta de acción prolongada; FEV1 = volumen espiratorio forzado en 1 segundo; FVC = capacidad vital forzada |
||
|
||
| Edad media (años) (± DE) | 65,1 (8,1) | 65,0 (8,3) |
| Varones (%) | 66,0 | 67,6 |
| Blancos, N (%) | 790 (84,1) | 838 (89,6) |
| Asiáticos, N (%) | 134 (14,3) | 10 (1,1) |
| Negros, N (%) | 5 (0,5) | 12 (1,3) |
| Indios americanos o nativos de Alaska, N (%) | 7 (0,7) | 48 (5,1) |
| Otros/múltiples, N (%) | 3 (0,3) | 27 (2,9) |
| Etnicidad hispana/latina, N (%) | 261 (27,8) | 300 (32,1) |
| Historial medio de tabaquismo (años-paquete) (± DE) | 40,5 (23,4) | 40,3 (27,2) |
| Fumadores actuales (%) | 30,0 | 29,5 |
| Bronquitis crónica (%) | 95,0 | 99,9 |
| Emfisema (%) | 32,6 | 30,4 |
ICS/LAMA/LABA (%)
Exacerbaciones en sujetos adultos con EPOC
El criterio de valoración principal para los ensayos BOREAS y NOTUS fue la tasa anualizada de exacerbaciones moderadas o graves de la EPOC durante el período de tratamiento de 52 semanas. En ambos ensayos, DUPIXENT demostró una reducción significativa en la tasa anualizada de exacerbaciones moderadas o graves de la EPOC en comparación con el placebo cuando se añadió a la terapia de mantenimiento de fondo (véase Tabla 29).
| Ensayo | Tratamiento (N) |
Tasa (exacerbaciones/año) | Razón de tasas frente a placebo (IC del 95%) |
|---|---|---|---|
| BOREAS | DUPIXENT 300 mg Q2W (N=468) |
0.78 | 0.71 (0.58, 0.86) |
| Placebo (N=471) |
1.10 | ||
| NOTUS | DUPIXENT 300 mg Q2W (N=470) |
0.86 | 0.66 (0.54, 0.82) |
| Placebo (N=465) |
1.30 | ||
Treatment with DUPIXENT decreased the risk of a moderate to severe COPD exacerbation as measured by time to first exacerbation when compared with placebo in BOREAS (HR: 0.80; 95% CI: 0.66, 0.98) and NOTUS (HR: 0.71; 95% CI: 0.57, 0.89).
Lung Function in Adult Subjects with COPD
In both trials (BOREAS and NOTUS), DUPIXENT demonstrated numerical improvement in post-bronchodilator FEV1 at Weeks 12 and 52 compared to placebo when added to background maintenance therapy (see Table 30 and Figure 17). Significant improvements of similar magnitude were observed in change from baseline in pre-bronchodilator FEV1 at Weeks 12 and 52 in subjects treated with DUPIXENT compared to placebo across both trials.
| Trial | Treatment (N) |
LS Mean Change from baseline mL | LS Mean Difference vs. placebo mL (95% CI) |
|---|---|---|---|
|
|||
| Post-bronchodilator FEV1 at Week 12 | |||
| BOREAS | DUPIXENT 300 mg Q2W (N=468) |
158 | 74 (31, 117) |
| Placebo (N=471) |
84 | ||
| NOTUS | DUPIXENT 300 mg Q2W (N=470) |
134 | 68 (26, 110) |
| Placebo (N=465) |
67 | ||
| Post-bronchodilator FEV1 at Week 52 | |||
| BOREAS | DUPIXENT 300 mg Q2W (N=468) |
138 | 79 (34, 124) |
| Placebo (N=471) |
58 | ||
| NOTUS* | DUPIXENT 300 mg Q2W (N=362) |
127 | 67 (16, 119) |
| Placebo (N=359) |
59 | ||
| Figure 17: LS Mean Change from Baseline in Post-Bronchodilator FEV1 (mL) Over Time in BOREAS and NOTUS* Trials for COPD | |
|---|---|
| BOREAS | NOTUS |
|
|
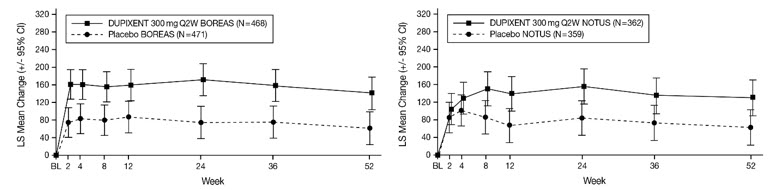 |
|
Health-Related Quality of Life
In both trials (BOREAS and NOTUS), the St. George’s Respiratory Questionnaire (SGRQ) total score responder rate (defined as the proportion of subjects with SGRQ improvement from baseline of at least 4 points) at Week 52 was evaluated. In BOREAS, the responder rate was 51% for subjects treated with DUPIXENT versus 43% for placebo (N=939, odds ratio: 1.44; 95% CI: 1.10, 1.89). In NOTUS, the responder rate was 51% for subjects treated with DUPIXENT versus 47% for placebo (N=721, odds ratio: 1.16; 95% CI: 0.86, 1.58).
16 FORMA DE SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
Presentación
DUPIXENT (dupilumab) Inyección es una solución transparente a ligeramente opalescente, incolora a amarillo pálido, que se suministra en jeringas precargadas de dosis única con protector de aguja o bolígrafos precargados.
La jeringa precargada con protector de aguja está diseñada para administrar:
- 300 mg de DUPIXENT en 2 mL de solución (NDC 0024-5914-00)
- 200 mg de DUPIXENT en 1.14 mL de solución (NDC 0024-5918-00)
El bolígrafo precargado está diseñado para administrar:
- 300 mg de DUPIXENT en 2 mL de solución (NDC 0024-5915-00)
- 200 mg de DUPIXENT en 1.14 mL de solución (NDC 0024-5919-00)
DUPIXENT está disponible en cajas que contienen 2 jeringas precargadas con protector de aguja o 2 bolígrafos precargados.
| Tamaño del envase | Jeringa precargada de 300 mg/2 mL con protector de aguja | Jeringa precargada de 200 mg/1.14 mL con protector de aguja |
|---|---|---|
| Envase de 2 jeringas | NDC 0024-5914-01 | NDC 0024-5918-01 |
| Tamaño del envase | Bolígrafo precargado de 300 mg/2 mL | Bolígrafo precargado de 200 mg/1.14 mL |
|---|---|---|
| Envase de 2 bolígrafos | NDC 0024-5915-02 | NDC 0024-5919-02 |
Almacenamiento y manipulación
DUPIXENT es estéril y sin conservantes. Deseche cualquier porción no utilizada.
Almacenar refrigerado a 2°C a 8°C (36°F a 46°F) en la caja original para protegerlo de la luz.
Si es necesario, DUPIXENT puede mantenerse a temperatura ambiente hasta 25°C (77°F) durante un máximo de 14 días. No almacenar por encima de 25°C (77°F). Después de retirarlo del refrigerador, DUPIXENT debe utilizarse en un plazo de 14 días o desecharse.
No exponer DUPIXENT al calor o la luz solar directa.
NO congelar. NO agitar.
17 INFORMACIÓN PARA EL PACIENTE
Aconseje al paciente que lea la información para el paciente aprobada por la FDA (Información para el paciente e Instrucciones de uso).
Registro de embarazos
Existe un registro de exposición durante el embarazo que realiza un seguimiento de los resultados del embarazo en mujeres expuestas a DUPIXENT durante el embarazo. Anime a las pacientes a participar y aconséjelas sobre cómo pueden inscribirse en el registro [ver Uso en poblaciones específicas (8.1)].
Instrucciones de administración
Capacite adecuadamente a los pacientes y/o cuidadores sobre la técnica de inyección subcutánea adecuada, incluida la técnica aséptica, y la preparación y administración de DUPIXENT antes de su uso. Aconseje a los pacientes que sigan las recomendaciones de eliminación de objetos punzocortantes [ver Posología y administración (2.1) e Instrucciones de uso].
Hipersensibilidad
Aconseje a los pacientes que interrumpan el tratamiento con DUPIXENT y que busquen atención médica inmediata si experimentan cualquier síntoma de reacciones de hipersensibilidad sistémica [ver Advertencias y precauciones (5.1)].
Conjuntivitis y queratitis
Aconseje a los pacientes que consulten a su médico si desarrollan síntomas oculares nuevos o si estos empeoran [ver Advertencias y precauciones (5.2)].
Enfermedades eosinofílicas
Aconseje a los pacientes que informen a su médico si presentan características clínicas de neumonía eosinofílica o vasculitis compatibles con granulomatosis eosinofílica con poliangitis [ver Advertencias y precauciones (5.3)].
No indicado para los síntomas agudos del asma o la enfermedad pulmonar obstructiva crónica ni para el deterioro agudo de la enfermedad
Informe a los pacientes que DUPIXENT no trata los síntomas agudos ni las exacerbaciones agudas del asma o la EPOC. Informe a los pacientes que deben buscar atención médica si su asma o EPOC no se controla o empeora después del inicio del tratamiento con DUPIXENT [ver Advertencias y precauciones (5.4)].
Reducción de la dosis de corticosteroides
Informe a los pacientes que no deben interrumpir el tratamiento con corticosteroides sistémicos o inhalados, excepto bajo la supervisión directa de un médico. Informe a los pacientes que la reducción de la dosis de corticosteroides puede estar asociada a síntomas de abstinencia sistémica y/o desenmascarar enfermedades previamente suprimidas por el tratamiento con corticosteroides sistémicos [ver Advertencias y precauciones (5.5)].
Pacientes con asma comórbida
Aconseje a los pacientes con asma comórbida que no ajusten ni interrumpan su tratamiento para el asma sin consultar a sus médicos [ver Advertencias y precauciones (5.6)].
Artralgia
Aconseje a los pacientes que informen a su médico de la aparición o el empeoramiento de los síntomas articulares [ver Advertencias y precauciones (5.7)].
Infecciones parasitarias (por helmintos)
Aconseje a los pacientes que informen a su médico si presentan características clínicas compatibles con una infección por helmintos [ver Advertencias y precauciones (5.8)].
Vacunaciones
Aconseje a los pacientes que no se recomienda la vacunación con vacunas vivas inmediatamente antes ni durante el tratamiento con DUPIXENT. Indique a los pacientes que informen a su médico de que están tomando DUPIXENT antes de una posible vacunación [ver Advertencias y precauciones (5.9)].
SECCIÓN NO CLASIFICADA DE SPL
Fabricado por:
Regeneron Pharmaceuticals, Inc.
Tarrytown, NY 10591
Licencia de EE. UU. No. 1760
Comercializado por:
sanofi-aventis U.S. LLC (Bridgewater, NJ 08807) y
Regeneron Pharmaceuticals, Inc. (Tarrytown, NY 10591)
Para información de patentes: www.dupixent-patents.com
DUPIXENT® es una marca registrada de Sanofi Biotechnology
© 2024 Regeneron Pharmaceuticals, Inc. / sanofi-aventis U.S. LLC. Todos los derechos reservados.
INSERTO PARA EL PACIENTE
| Esta información para el paciente ha sido aprobada por la Administración de Alimentos y Medicamentos de los Estados Unidos. | Revisado: Septiembre de 2024 | |||
| Información para el paciente DUPIXENT® (DU-pix-ent) (dupilumab) inyección, para uso subcutáneo |
||||
| ¿Qué es DUPIXENT? DUPIXENT es un medicamento recetado que se usa: |
||||
|
||||
|
||||
| No use DUPIXENT si es alérgico a dupilumab o a cualquiera de los ingredientes de DUPIXENT. Consulte el final de este folleto de información para el paciente para obtener una lista completa de los ingredientes de DUPIXENT. | ||||
Antes de usar DUPIXENT, informe a su proveedor de atención médica sobre todas sus afecciones médicas, incluso si:
Informe a su proveedor de atención médica sobre todos los medicamentos que toma, incluidos los medicamentos recetados y de venta libre, las vitaminas y los suplementos herbales.
No cambie ni deje de tomar sus otros medicamentos, incluidos los medicamentos con corticosteroides u otros medicamentos para el asma, sin hablar con su proveedor de atención médica. Esto puede hacer que otros síntomas que estaban controlados por esos medicamentos vuelvan a aparecer. |
||||
¿Cómo debo usar DUPIXENT?
|
||||
| ¿Cuáles son los posibles efectos secundarios de DUPIXENT? DUPIXENT puede causar efectos secundarios graves, que incluyen:
|
||||
|
|
|
||
|
||||
|
|
|||
Los efectos secundarios más comunes de DUPIXENT incluyen: |
||||
|
|
|||
Los siguientes efectos secundarios adicionales se han reportado con DUPIXENT:
Informe a su proveedor de atención médica si tiene algún efecto secundario que le moleste o que no desaparezca. |
||||
¿Cómo debo guardar DUPIXENT?
Mantenga DUPIXENT y todos los medicamentos fuera del alcance de los niños. |
||||
| Información general sobre el uso seguro y eficaz de DUPIXENT. Los medicamentos a veces se recetan para fines distintos de los que se enumeran en un folleto de información para el paciente. No use DUPIXENT para una afección para la que no fue recetado. No le dé DUPIXENT a otras personas, incluso si tienen los mismos síntomas que usted. Puede hacerles daño. Puede pedirle a su farmacéutico o proveedor de atención médica información sobre DUPIXENT que esté escrita para profesionales de la salud. |
||||
| ¿Cuáles son los ingredientes de DUPIXENT? Ingrediente activo: dupilumab Ingredientes inactivos: clorhidrato de L-arginina, L-histidina, polisorbato 80, acetato de sodio, sacarosa y agua para inyección. Fabricado por: Regeneron Pharmaceuticals, Inc., Tarrytown, NY 10591 U.S. License No. 1760 Comercializado por: sanofi-aventis U.S. LLC (Bridgewater, NJ 08807) y Regeneron Pharmaceuticals, Inc. (Tarrytown, NY 10591) DUPIXENT® es una marca registrada de Sanofi Biotechnology / © 2024 Regeneron Pharmaceuticals, Inc. / sanofi-aventis U.S. LLC. Todos los derechos reservados. Para obtener más información sobre DUPIXENT, visite www.DUPIXENT.com o llame al 1-844-DUPIXENT (1-844-387-4936). |
||||
INSTRUCCIONES DE USO DE DUPIXENT® (DU-pix-ent) (dupilumab) inyección, para uso subcutáneo Jeringa precargada de dosis única con protector de aguja
Lea estas Instrucciones de Uso antes de usar la jeringa precargada de DUPIXENT. No se inyecte usted mismo ni a otra persona hasta que le hayan mostrado cómo inyectar DUPIXENT. Su proveedor de atención médica puede mostrarle a usted o a su cuidador cómo preparar e inyectar una dosis de DUPIXENT antes de que intente hacerlo usted mismo por primera vez. Guarde estas instrucciones para futuras consultas. Llame a su proveedor de atención médica si tiene alguna pregunta.
Este dispositivo es una jeringa precargada de dosis única (denominada “jeringa DUPIXENT” en estas instrucciones). Contiene 300 mg de DUPIXENT para inyección debajo de la piel (inyección subcutánea).
Las partes de la jeringa DUPIXENT se muestran a continuación:
| Información importante | |
|---|---|
|
|
¿Cómo debo almacenar DUPIXENT?
|
|
Paso 1: Retire
Retire la jeringa DUPIXENT de la caja sujetando la parte central del cuerpo de la jeringa.
 No retire la tapa de la aguja hasta que esté listo para inyectar.
No retire la tapa de la aguja hasta que esté listo para inyectar.
No utilice la jeringa DUPIXENT si se ha caído sobre una superficie dura o está dañada.
Paso 2: Prepare
Asegúrese de tener lo siguiente:
- la jeringa precargada DUPIXENT
- 1 toallita con alcohol*
- 1 bola de algodón o gasa*
- un contenedor para objetos punzantes* (Ver Paso 13)
*Artículos no incluidos en la caja

Paso 3: Compruebe
Cuando reciba sus jeringas DUPIXENT, compruebe siempre que:
- tiene el medicamento y la dosis correctos.
- la fecha de caducidad de la jeringa precargada de dosis única no ha pasado.
No utilice la jeringa DUPIXENT si la fecha de caducidad ha pasado.
Paso 4: Inspeccione
Observe el medicamento a través de la ventana de visualización de la jeringa DUPIXENT:
Compruebe si el líquido es transparente e incoloro a amarillo pálido.
Nota: Es posible que vea una burbuja de aire, esto es normal.
No utilice la jeringa DUPIXENT si el líquido está descolorido o turbio, o si contiene copos o partículas visibles.
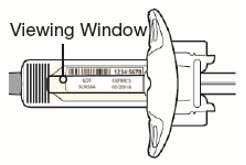
Paso 5: Espere 45 minutos
Coloque la jeringa DUPIXENT sobre una superficie plana y deje que se caliente naturalmente a temperatura ambiente durante al menos 45 minutos.
No caliente la jeringa DUPIXENT.
No exponga la jeringa DUPIXENT a la luz solar directa.
No guarde las jeringas DUPIXENT a temperatura ambiente durante más de 14 días. Deseche (elimine) cualquier jeringa DUPIXENT que haya estado a temperatura ambiente durante más de 14 días.
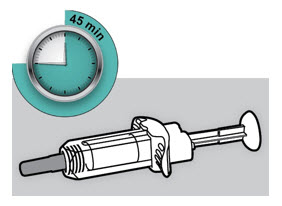
Paso 6: Elija su lugar de inyección
- Puede inyectarse en el muslo o el estómago, excepto en las 2 pulgadas (5 cm) alrededor del ombligo.
- Si un cuidador le inyecta la dosis, también puede utilizar la zona externa de la parte superior del brazo.
- Elija un lugar diferente cada vez que se inyecte DUPIXENT.
No inyecte en la piel que esté sensible, dañada, magullada o con cicatrices.
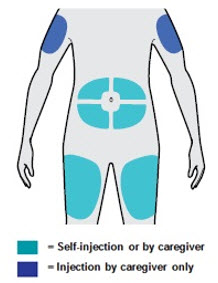
Paso 7: Limpie
Lávese las manos.
Limpie el lugar de inyección con una toallita con alcohol.
Deje que la piel se seque antes de inyectar.
No vuelva a tocar el lugar de inyección ni sople sobre él antes de la inyección.
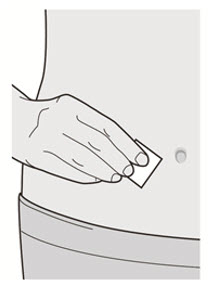
Paso 8: Retire la tapa de la aguja
Sujete la jeringa DUPIXENT en la parte central del cuerpo de la jeringa con la aguja apuntando hacia afuera y retire la tapa de la aguja.
No vuelva a colocar la tapa de la aguja.
No toque la aguja.
Inyecte su medicamento inmediatamente después de retirar la tapa de la aguja.
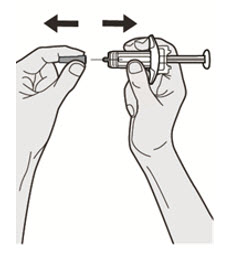
Paso 9: Pellizque
Pellizque un pliegue de piel en el lugar de inyección (muslo o estómago, excepto las 2 pulgadas alrededor del ombligo, o la zona externa de la parte superior del brazo si se lo inyecta un cuidador). La figura siguiente muestra un ejemplo de cómo pellizcar un pliegue de piel en el estómago.
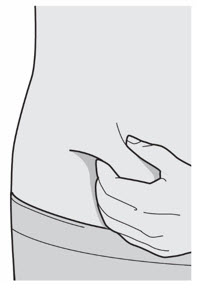
Paso 10: Inserte
Inserte la aguja completamente en el pliegue de la piel con un ángulo de aproximadamente 45°.
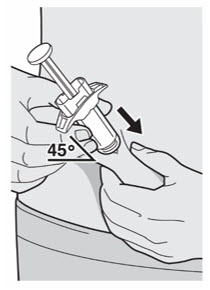
Paso 11: Empuje
Relaje el pellizco.
Empuje la varilla del émbolo hacia abajo lenta y constantemente hasta el final, hasta que la jeringa DUPIXENT esté vacía.
Nota: Sentirá cierta resistencia. Esto es normal.
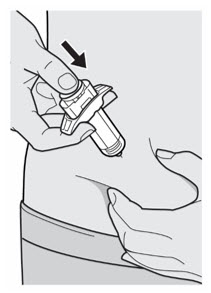
Paso 12: Suelte y retire
Levante el pulgar para soltar la varilla del émbolo hasta que la aguja quede cubierta por el protector de la aguja y luego retire la jeringa del lugar de inyección.
Presione ligeramente una bola de algodón o gasa sobre el lugar de inyección si ve sangre.
No vuelva a colocar la tapa de la aguja.
No se frote la piel después de la inyección.
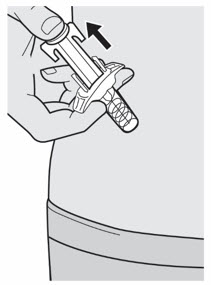
Deposite las agujas, las jeringas DUPIXENT y las tapas de las agujas usadas en un contenedor de eliminación de objetos punzantes aprobado por la FDA inmediatamente después de su uso.
No deseche (tire) las agujas, las jeringas DUPIXENT y las tapas de las agujas en la basura de su hogar.
Si no tiene un contenedor de eliminación de objetos punzantes aprobado por la FDA, puede usar un contenedor doméstico que sea:
- hecho de plástico resistente,
- se puede cerrar con una tapa hermética y resistente a las perforaciones, sin que los objetos punzantes puedan salir,
- vertical y estable durante el uso,
- a prueba de fugas, y
- adecuadamente etiquetado para advertir sobre los residuos peligrosos dentro del contenedor
Cuando su contenedor de eliminación de objetos punzantes esté casi lleno, deberá seguir las pautas de su comunidad para la forma correcta de desechar su contenedor de eliminación de objetos punzantes. Puede haber leyes estatales o locales sobre cómo debe desechar las agujas y jeringas usadas.
Para obtener más información sobre la eliminación segura de objetos punzantes y para obtener información específica sobre la eliminación de objetos punzantes en el estado en el que vive, visite el sitio web de la FDA en: http://www.fda.gov/safesharpsdisposal
No deseche su contenedor de eliminación de objetos punzantes usado en la basura de su hogar a menos que las pautas de su comunidad lo permitan. No recicle su contenedor de eliminación de objetos punzantes usado.
No vuelva a colocar la tapa de la aguja.
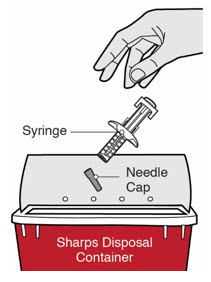

Fabricado por: Regeneron Pharmaceuticals, Inc. Tarrytown, NY 10591
Licencia de EE. UU. No. 1760
Comercializado por: sanofi-aventis U.S. LLC (Bridgewater, NJ 08807) y Regeneron Pharmaceuticals, Inc. (Tarrytown, NY 10591)
DUPIXENT® es una marca registrada de Sanofi Biotechnology
© 2023 Regeneron Pharmaceuticals, Inc. / sanofi-aventis U.S. LLC. Todos los derechos reservados.
Estas Instrucciones de uso han sido aprobadas por la Administración de Alimentos y Medicamentos de los Estados Unidos.
Revisado: Julio de 2023
INSTRUCCIONES DE USO DE DUPIXENT® (DU-pix-ent) (dupilumab) inyección, para uso subcutáneo Jeringa precargada de dosis única con protector de aguja
Lea estas Instrucciones de Uso antes de usar la jeringa precargada de DUPIXENT. No se inyecte usted mismo ni a otra persona hasta que le hayan mostrado cómo inyectar DUPIXENT. Su profesional sanitario puede mostrarle a usted o a su cuidador cómo preparar e inyectar una dosis de DUPIXENT antes de que intente hacerlo usted mismo por primera vez. Guarde estas instrucciones para futuras consultas. Llame a su profesional sanitario si tiene alguna pregunta.
Este dispositivo es una jeringa precargada de dosis única (denominada “jeringa DUPIXENT” en estas instrucciones). Contiene 200 mg de DUPIXENT para inyección bajo la piel (inyección subcutánea).
Las partes de la jeringa DUPIXENT se muestran a continuación:
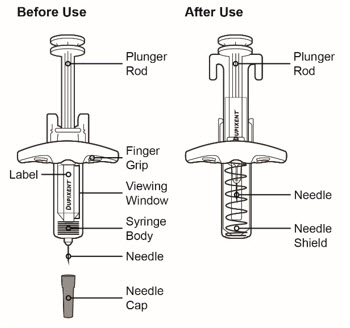
| Información importante | |
|---|---|
|
|
¿Cómo debo guardar DUPIXENT?
|
|
Paso 1: Retire
Retire la jeringa DUPIXENT de la caja sujetando la parte central del cuerpo de la jeringa.
 No retire la tapa de la aguja hasta que esté listo para inyectarse.
No retire la tapa de la aguja hasta que esté listo para inyectarse.
No utilice la jeringa DUPIXENT si se ha caído sobre una superficie dura o está dañada.
Paso 2: Prepare
Asegúrese de tener lo siguiente:
- la jeringa precargada DUPIXENT
- 1 toallita con alcohol*
- 1 bola de algodón o gasa*
- un contenedor para objetos punzantes* (Consulte el Paso 13)
*Artículos no incluidos en la caja

Paso 3: Compruebe
Cuando reciba sus jeringas DUPIXENT, compruebe siempre que:
- tiene el medicamento y la dosis correctos.
- la fecha de caducidad de la jeringa precargada de dosis única no ha pasado.
No utilice la jeringa DUPIXENT si la fecha de caducidad ha pasado.
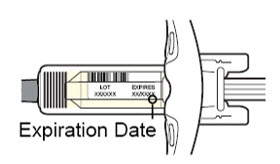
Paso 4: Inspeccione
Observe el medicamento a través de la ventana de visualización de la jeringa DUPIXENT:
Compruebe si el líquido es transparente e incoloro a amarillo pálido.
Nota: Es posible que vea una burbuja de aire, esto es normal.
No utilice la jeringa DUPIXENT si el líquido está descolorido o turbio, o si contiene copos o partículas visibles.
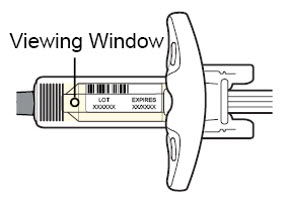
Paso 5: Espere 30 minutos
Coloque la jeringa DUPIXENT sobre una superficie plana y deje que se caliente a temperatura ambiente de forma natural durante al menos 30 minutos.
No caliente la jeringa DUPIXENT.
No exponga la jeringa DUPIXENT a la luz solar directa.
No guarde las jeringas DUPIXENT a temperatura ambiente durante más de 14 días. Deseche (elimine) cualquier jeringa DUPIXENT que haya estado a temperatura ambiente durante más de 14 días.
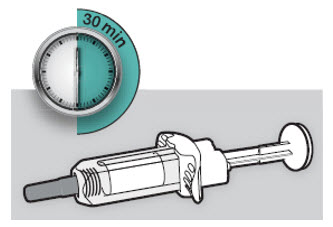
Paso 6: Elija el lugar de la inyección
- Puede inyectarse en el muslo o el estómago, excepto en las 2 pulgadas (5 cm) alrededor del ombligo.
- Si un cuidador le inyecta la dosis, también puede utilizar la zona externa de la parte superior del brazo.
- Elija un lugar diferente cada vez que se inyecte DUPIXENT.
No se inyecte en la piel que esté sensible, dañada, magullada o con cicatrices.
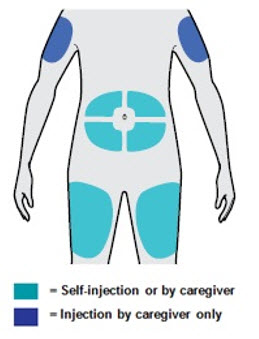
Paso 7: Limpie
Lávese las manos.
Limpie el lugar de la inyección con una toallita con alcohol.
Deje que la piel se seque antes de inyectarse.
No vuelva a tocar el lugar de la inyección ni sople sobre él antes de la inyección.
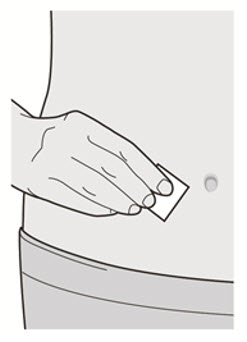
Paso 8: Retire la tapa de la aguja
Sujete la jeringa DUPIXENT en la parte central del cuerpo de la jeringa con la aguja apuntando hacia afuera y retire la tapa de la aguja.
No vuelva a colocar la tapa de la aguja.
No toque la aguja.
Inyéctese el medicamento inmediatamente después de retirar la tapa de la aguja.
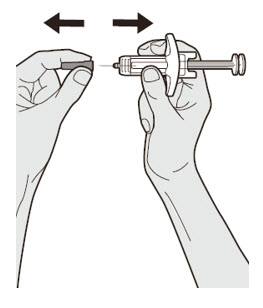
Paso 9: Pellizque
Pellizque un pliegue de piel en el lugar de la inyección (muslo o estómago, excepto 2 pulgadas alrededor del ombligo, o zona externa de la parte superior del brazo si se lo inyecta un cuidador). La figura siguiente muestra un ejemplo de cómo pellizcar un pliegue de piel en el estómago.
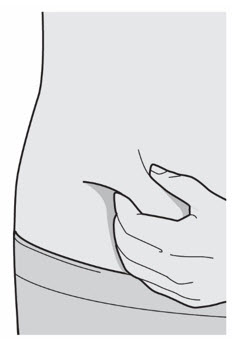
Paso 10: Inserte
Inserte la aguja completamente en el pliegue de la piel con un ángulo de aproximadamente 45°.
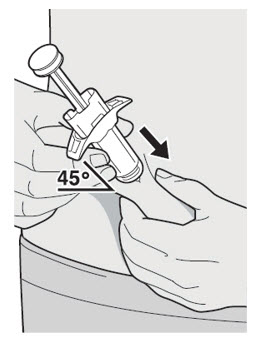
Paso 11: Empuje
Relaje el pellizco.
Empuje el émbolo hacia abajo lenta y constantemente hasta que llegue al final, hasta que la jeringa DUPIXENT esté vacía.
Nota: Sentirá cierta resistencia. Esto es normal.
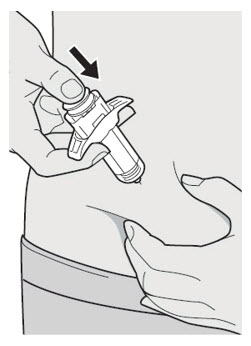
Paso 12: Suelte y retire
Levante el pulgar para soltar el émbolo hasta que la aguja quede cubierta por la protección de la aguja y luego retire la jeringa del lugar de la inyección.
Presione ligeramente una bola de algodón o gasa sobre el lugar de la inyección si ve sangre.
No vuelva a colocar la tapa de la aguja.
No se frote la piel después de la inyección.
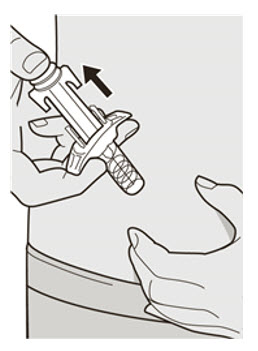
Deposite las agujas usadas, las jeringas DUPIXENT y las tapas de las agujas en un contenedor de eliminación de objetos punzantes aprobado por la FDA inmediatamente después de su uso.
No deseche (tire) las agujas, las jeringas DUPIXENT y las tapas de las agujas en la basura de su hogar.
Si no tiene un contenedor de eliminación de objetos punzantes aprobado por la FDA, puede usar un contenedor doméstico que sea:
- hecho de plástico resistente,
- se puede cerrar con una tapa hermética y resistente a las perforaciones, sin que los objetos punzantes puedan salir,
- vertical y estable durante el uso,
- a prueba de fugas, y
- adecuadamente etiquetado para advertir sobre los residuos peligrosos dentro del contenedor
Cuando su contenedor de eliminación de objetos punzantes esté casi lleno, deberá seguir las pautas de su comunidad para la forma correcta de desechar su contenedor de eliminación de objetos punzantes. Puede haber leyes estatales o locales sobre cómo debe desechar las agujas y jeringas usadas.
Para obtener más información sobre la eliminación segura de objetos punzantes y para obtener información específica sobre la eliminación de objetos punzantes en el estado en el que vive, visite el sitio web de la FDA en: http://www.fda.gov/safesharpsdisposal
No deseche su contenedor de eliminación de objetos punzantes usado en la basura de su hogar a menos que las pautas de su comunidad lo permitan. No recicle su contenedor de eliminación de objetos punzantes usado.
No vuelva a colocar la tapa de la aguja.
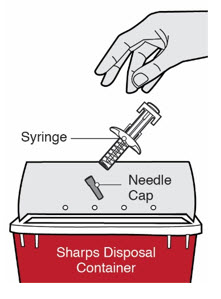

Fabricado por: Regeneron Pharmaceuticals, Inc. Tarrytown, NY 10591
Licencia de EE. UU. No. 1760
Comercializado por: sanofi-aventis U.S. LLC (Bridgewater, NJ 08807) y Regeneron Pharmaceuticals, Inc. (Tarrytown, NY 10591)
DUPIXENT® es una marca registrada de Sanofi Biotechnology
© 2023 Regeneron Pharmaceuticals, Inc. / sanofi-aventis U.S. LLC. Todos los derechos reservados.
Estas Instrucciones de uso han sido aprobadas por la Administración de Alimentos y Medicamentos de los Estados Unidos.
Revisado: Julio de 2023
INSTRUCCIONES DE USO
Instrucciones de uso
DUPIXENT® (DU-pix-ent)
(dupilumab)
inyección, para uso subcutáneo
Pluma precargada de dosis única (300 mg/2 mL)
Estas instrucciones de uso contienen información sobre cómo inyectar DUPIXENT.
Lea estas instrucciones de uso antes de usar la pluma precargada de DUPIXENT. No se inyecte usted mismo ni a otra persona hasta que le hayan mostrado cómo inyectar DUPIXENT. Su profesional sanitario puede mostrarle a usted o a su cuidador cómo preparar e inyectar una dosis de DUPIXENT antes de que intente hacerlo usted mismo por primera vez. Guarde estas instrucciones de uso. Llame a su profesional sanitario si tiene alguna pregunta.
Esta pluma precargada de DUPIXENT solo está indicada para su uso en adultos y niños de 2 años de edad o mayores.
Esta pluma precargada de DUPIXENT es un dispositivo de dosis única. Contiene 300 mg de DUPIXENT para inyección bajo la piel (inyección subcutánea).
Las partes de la pluma precargada de DUPIXENT se muestran a continuación:
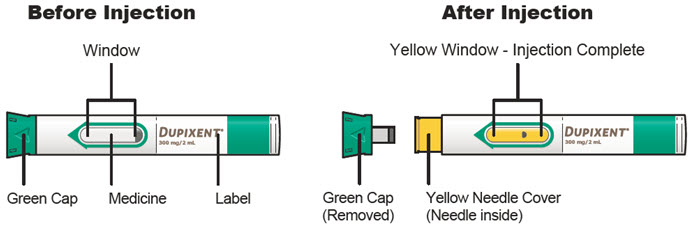
Información importante
|
Almacenamiento de DUPIXENT
|
| A. Prepárese para inyectar | |
|---|---|
| A1. Reúna los materiales | |
| Busque una superficie de trabajo limpia y plana. Asegúrese de tener los siguientes materiales: |

|
| A2. Compruebe la pluma | |
|
 |
| A3. Mire la etiqueta | |
|
 |
|
 |

| A4. Compruebe el medicamento | |
|---|---|
|
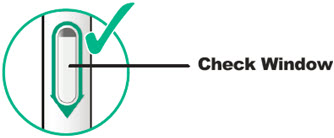 |

| A5. Espere 45 minutos | |
|---|---|
|
 |

| B. Elija y prepare su sitio de inyección | |
|---|---|
| B1. Lávese bien las manos con agua y jabón | |
 |
|
| B2. Elija un sitio de inyección | |
|
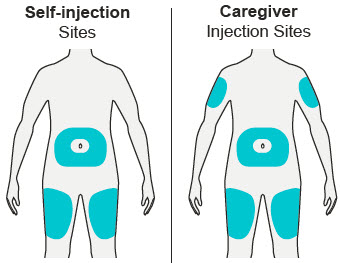
|


| B3. Prepare el sitio de inyección | |
|---|---|
|
 |
| C. Administre la inyección | |
| C1. Retire la tapa verde | |
|
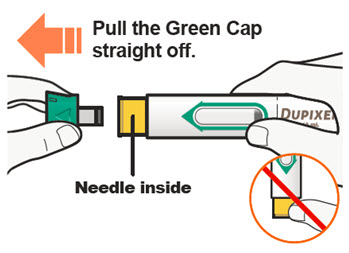 |


| C2. Pellizque la piel y coloque | ||
|---|---|---|
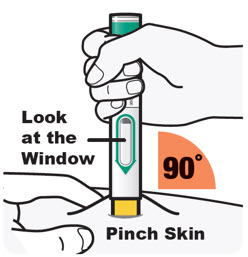 |
||
Pellizque la piel antes y durante la inyección.
|
||
| C3. Presione hacia abajo ➜ Observe que la ventana se vuelve completamente amarilla ➜ Luego cuente hasta 5 | ||
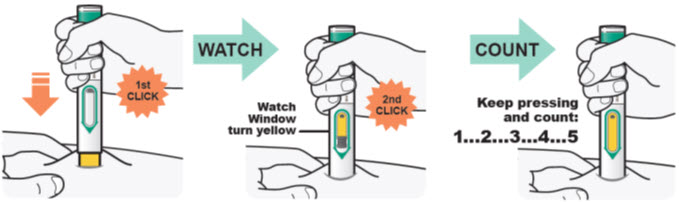 |
||
Presione y mantenga el bolígrafo precargado DUPIXENT firmemente contra la piel hasta que no pueda ver la cubierta amarilla de la aguja.
|
Siga presionando el bolígrafo precargado DUPIXENT contra la piel y observe la ventana:
|
Siga presionando el bolígrafo precargado DUPIXENT contra la piel y cuente hasta 5 para asegurarse de recibir la dosis completa. |
| No es necesario pellizcar la piel para adultos y niños de 12 años o más. | |
|---|---|
| C4. Retire | |
|
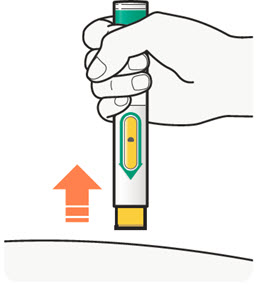 |


| D. Deseche el bolígrafo precargado DUPIXENT usado | |
|---|---|
| Estas Instrucciones de uso han sido aprobadas por la Administración de Alimentos y Medicamentos de los Estados Unidos. | Revisado: Julio de 2023 |
| Cómo desechar (tirar) el bolígrafo precargado DUPIXENT:
Coloque los bolígrafos precargados DUPIXENT usados y las tapas verdes en un contenedor de eliminación de objetos punzantes aprobado por la FDA inmediatamente después de su uso.
Si no tiene un contenedor de eliminación de objetos punzantes aprobado por la FDA, puede usar un contenedor doméstico que sea:
Cuando su contenedor de eliminación de objetos punzantes esté casi lleno, deberá seguir las pautas de su comunidad para la forma correcta de desechar su contenedor de eliminación de objetos punzantes. Puede haber leyes estatales o locales sobre cómo debe desechar los bolígrafos precargados DUPIXENT usados.
|
|
|
|
|
|
Fabricado por: Regeneron Pharmaceuticals, Inc. Tarrytown, NY 10591 Licencia de EE. UU. No. 1760 Comercializado por: sanofi-aventis U.S. LLC (Bridgewater, NJ 08807) y Regeneron Pharmaceuticals, Inc. (Tarrytown, NY 10591) DUPIXENT® es una marca registrada de Sanofi Biotechnology © 2023 Regeneron Pharmaceuticals, Inc. / sanofi-aventis U.S. LLC. Todos los derechos reservados. |
|



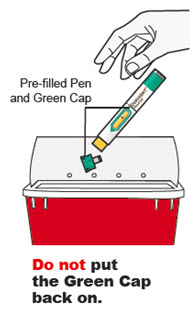

INSTRUCCIONES DE USO
DUPIXENT® (DU-pix-ent)
(dupilumab)
inyección, para uso subcutáneo
Pluma precargada de dosis única (200 mg/1.14 mL)
Estas Instrucciones de uso contienen información sobre cómo inyectar DUPIXENT.
Lea estas Instrucciones de uso antes de usar la pluma precargada de DUPIXENT. No se inyecte a sí mismo ni a otra persona hasta que le hayan mostrado cómo inyectar DUPIXENT. Su proveedor de atención médica puede mostrarle a usted o a su cuidador cómo preparar e inyectar una dosis de DUPIXENT antes de que intente hacerlo usted mismo por primera vez. Conserve estas Instrucciones de uso. Llame a su proveedor de atención médica si tiene alguna pregunta.
Esta pluma precargada de DUPIXENT solo debe usarse en adultos y niños mayores de 2 años.
Esta pluma precargada de DUPIXENT es un dispositivo de dosis única. Contiene 200 mg de DUPIXENT para inyección debajo de la piel (inyección subcutánea).
Las partes de la pluma precargada de DUPIXENT se muestran a continuación:
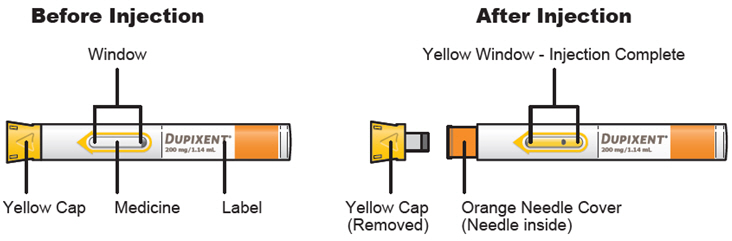
Información importante
|
Almacenamiento de DUPIXENT
|
| A. Prepárese para la inyección | |
|---|---|
| A1. Reúna los suministros | |
| Busque una superficie de trabajo limpia y plana. Asegúrese de tener los siguientes suministros: |
|
| A2. Revise la pluma | |
|
  |
| A3. Mire la etiqueta | |
|
 |
|
 |

| A4. Revise el medicamento | |
|---|---|
|
 |

| A5. Espere 30 minutos | |
|---|---|
|
 |

| B. Elija y prepare el lugar de inyección | |
|---|---|
| B1. Lávese bien las manos con agua y jabón | |
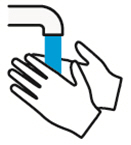 |
|
| B2. Elija un lugar de inyección | |
|
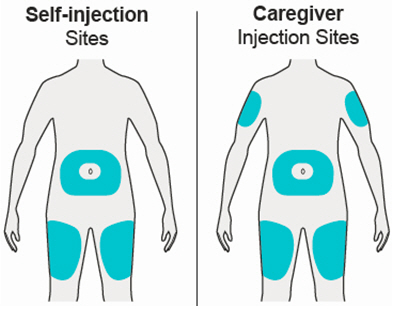
|


| B3. Prepare el lugar de inyección | |
|---|---|
|
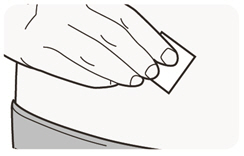 |
| C. Aplique la inyección | |
| C1. Retire la tapa amarilla | |
|
 |


| C2. Pellizque la piel y coloque | ||
|---|---|---|
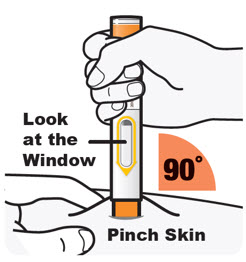 |
||
Pellizque la piel antes y durante la inyección.
|
||
| C3. Presione hacia abajo ➜ Observe cómo la ventana se vuelve completamente amarilla ➜ Luego cuente hasta 5 | ||
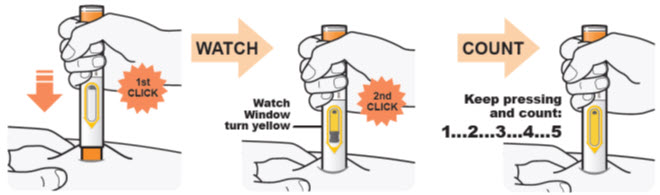 |
||
Presione y sostenga la pluma precargada DUPIXENT firmemente contra la piel hasta que no pueda ver el protector de aguja naranja.
|
Siga presionando la pluma precargada DUPIXENT contra la piel y observe la ventana:
|
Siga presionando la pluma precargada DUPIXENT contra la piel y cuente hasta 5 para asegurarse de recibir la dosis completa. |
| No es necesario pellizcar la piel en el caso de adultos y niños mayores de 12 años. | |
|---|---|
| C4. Retirar | |
|
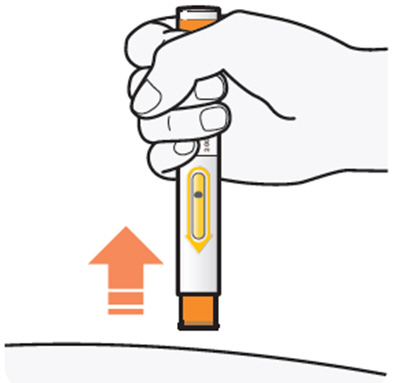 |


| D. Deseche la pluma precargada DUPIXENT usada | |
|---|---|
| Estas Instrucciones de uso han sido aprobadas por la Administración de Alimentos y Medicamentos de los Estados Unidos. | Revisión: julio de 2023 |
| Cómo desechar (tirar) la pluma precargada DUPIXENT:
Coloque las plumas precargadas DUPIXENT y las tapas amarillas usadas en un contenedor de desechos de objetos punzocortantes aprobado por la FDA inmediatamente después de su uso.
Si no tiene un contenedor de desechos de objetos punzocortantes aprobado por la FDA, puede utilizar un contenedor doméstico que sea:
Cuando su contenedor de desechos de objetos punzocortantes esté casi lleno, deberá seguir las pautas de su comunidad para la eliminación correcta del mismo. Puede haber leyes estatales o locales sobre cómo desechar las plumas precargadas DUPIXENT usadas. Para obtener más información sobre la eliminación segura de objetos punzocortantes e información específica sobre la eliminación de objetos punzocortantes en el estado en el que vive, visite el sitio web de la FDA en: http://www.fda.gov/safesharpsdisposal
|
|
|
|
|
|
Fabricado por: Regeneron Pharmaceuticals, Inc. Tarrytown, NY 10591 U.S. License No. 1760 Comercializado por: sanofi-aventis U.S. LLC (Bridgewater, NJ 08807) y Regeneron Pharmaceuticals, Inc. (Tarrytown, NY 10591) DUPIXENT® es una marca registrada de Sanofi Biotechnology © 2023 Regeneron Pharmaceuticals, Inc. / sanofi-aventis U.S. LLC. Todos los derechos reservados. |
|



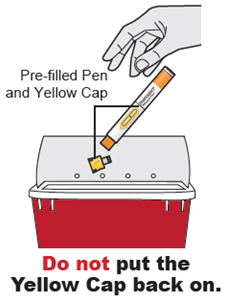

INSTRUCCIONES DE USO DUPIXENT® (DU-pix-ent) (dupilumab) inyección, para uso subcutáneo Jeringa precargada de dosis única con protector de aguja
Lea estas Instrucciones de uso antes de usar la jeringa precargada de DUPIXENT. No inyecte al niño hasta que le hayan mostrado cómo inyectar DUPIXENT. Su proveedor de atención médica puede mostrarle cómo preparar e inyectar una dosis de DUPIXENT antes de que intente hacerlo usted mismo por primera vez. Conserve estas instrucciones para usarlas en el futuro. Llame a su proveedor de atención médica si tiene alguna pregunta.
Este dispositivo es una jeringa precargada de dosis única (denominada “jeringa de DUPIXENT” en estas instrucciones). Contiene 100 mg de DUPIXENT para inyección debajo de la piel (inyección subcutánea).
Las partes de la jeringa de DUPIXENT se muestran a continuación:
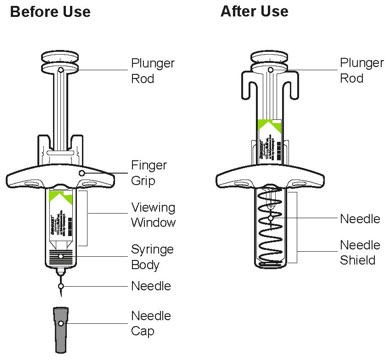
| Información importante | |
|
|
¿Cómo debo almacenar DUPIXENT?
|
|
Paso 1: Retirar
Retire la jeringa DUPIXENT del envase sujetando la parte central del cuerpo de la jeringa.
 No retire la tapa de la aguja hasta que esté listo para inyectar.
No retire la tapa de la aguja hasta que esté listo para inyectar.
 No use la jeringa DUPIXENT si se le ha caído sobre una superficie dura o si está dañada.
No use la jeringa DUPIXENT si se le ha caído sobre una superficie dura o si está dañada.
Paso 2: Preparar
Asegúrese de tener lo siguiente:
- la jeringa precargada DUPIXENT
- 1 toallita con alcohol*
- 1 bola de algodón o gasa*
- un contenedor para desechar objetos punzocortantes* (consulte el Paso 13)
*Artículos no incluidos en el envase

Paso 3: Revisar
Cuando reciba las jeringas DUPIXENT, compruebe siempre que:
- tenga el medicamento y la dosis correctos.
- la fecha de caducidad de la jeringa precargada de dosis única no haya pasado.
 No use la jeringa DUPIXENT si ha pasado la fecha de caducidad.
No use la jeringa DUPIXENT si ha pasado la fecha de caducidad.
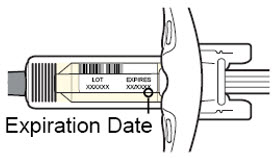
Paso 4: Inspeccionar
Observe el medicamento a través de la ventana de visualización de la jeringa DUPIXENT:
Compruebe si el líquido es transparente e incoloro o amarillo pálido.
Nota: Es posible que vea una burbuja de aire, esto es normal.
 No use la jeringa DUPIXENT si el líquido está descolorido o turbio, o si contiene escamas o partículas visibles.
No use la jeringa DUPIXENT si el líquido está descolorido o turbio, o si contiene escamas o partículas visibles.
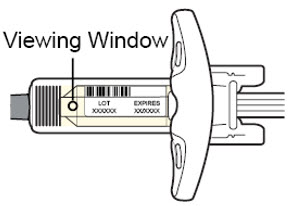
Paso 5: Espere 30 minutos
Coloque la jeringa DUPIXENT sobre una superficie plana y deje que se caliente a temperatura ambiente durante al menos 30 minutos.
 No caliente la jeringa DUPIXENT.
No caliente la jeringa DUPIXENT.
 No exponga la jeringa DUPIXENT a la luz solar directa.
No exponga la jeringa DUPIXENT a la luz solar directa.
 No mantenga las jeringas DUPIXENT a temperatura ambiente durante más de 14 días. Deseche cualquier jeringa DUPIXENT que haya estado a temperatura ambiente durante más de 14 días.
No mantenga las jeringas DUPIXENT a temperatura ambiente durante más de 14 días. Deseche cualquier jeringa DUPIXENT que haya estado a temperatura ambiente durante más de 14 días.
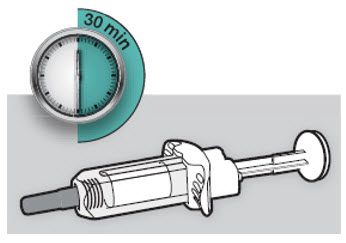
Paso 6: Elegir el lugar de inyección
- Puede inyectarse en el muslo, la zona externa de la parte superior del brazo o el estómago, excepto en los 5 cm (2 pulgadas) alrededor del ombligo.
- Elija un lugar diferente cada vez que se inyecte DUPIXENT.
 No se inyecte en la piel que esté sensible, dañada, amoratada o con cicatrices.
No se inyecte en la piel que esté sensible, dañada, amoratada o con cicatrices.
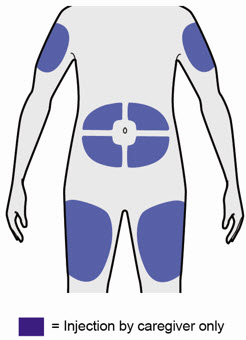
Paso 7: Limpiar
Lávese las manos.
Limpie el lugar de la inyección con una toallita con alcohol.
Deje que la piel se seque antes de inyectar.
 No vuelva a tocar el lugar de la inyección ni sople sobre él antes de la inyección.
No vuelva a tocar el lugar de la inyección ni sople sobre él antes de la inyección.
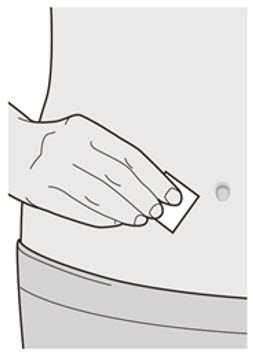
Paso 8: Retirar la tapa de la aguja
Sujete la jeringa DUPIXENT por la parte central del cuerpo de la jeringa con la aguja apuntando en dirección contraria a usted y retire la tapa de la aguja.
 No vuelva a colocar la tapa de la aguja.
No vuelva a colocar la tapa de la aguja.
 No toque la aguja.
No toque la aguja.
Inyecte el medicamento inmediatamente después de retirar la tapa de la aguja.
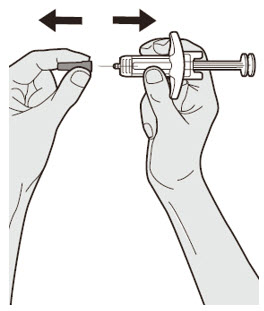
Paso 9: Pellizcar
Pellizque un pliegue de piel en el lugar de la inyección (muslo o estómago, excepto a 5 cm del ombligo, o la zona externa de la parte superior del brazo). La siguiente figura muestra un ejemplo de cómo pellizcar un pliegue de piel en el estómago.
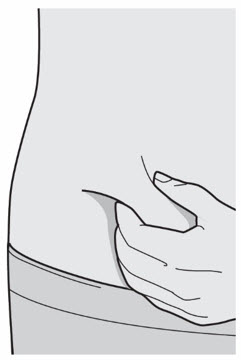
Paso 10: Insertar
Inserte la aguja completamente en el pliegue de la piel en un ángulo de aproximadamente 45°.
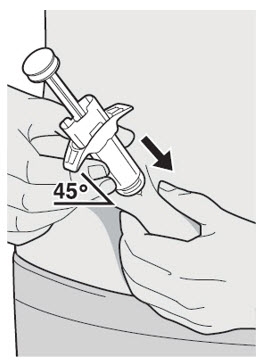
Paso 11: Empujar
Deje de pellizcar.
Empuje el émbolo hacia abajo de forma lenta y constante hasta que la jeringa DUPIXENT esté vacía.
Nota: Sentirá algo de resistencia. Esto es normal.
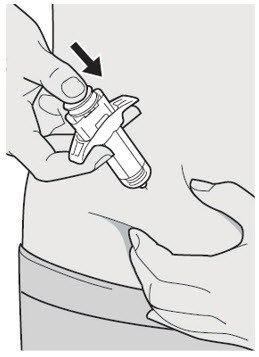
Paso 12: Soltar y retirar
Levante el pulgar para soltar el émbolo hasta que la aguja quede cubierta por el protector de la aguja y, a continuación, retire la jeringa del lugar de la inyección.
Presione ligeramente una bola de algodón o una gasa sobre el lugar de la inyección si ve sangre.
 No vuelva a colocar la tapa de la aguja.
No vuelva a colocar la tapa de la aguja.
 No frote la piel después de la inyección.
No frote la piel después de la inyección.
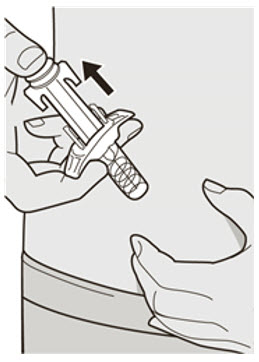
Coloque las agujas, jeringas DUPIXENT y tapas de agujas usadas en un contenedor de eliminación de objetos punzocortantes aprobado por la FDA inmediatamente después de su uso.
 No deseche (tire) las agujas, jeringas DUPIXENT y tapas de agujas en la basura doméstica.
No deseche (tire) las agujas, jeringas DUPIXENT y tapas de agujas en la basura doméstica.
Si no tiene un contenedor de eliminación de objetos punzocortantes aprobado por la FDA, puede usar un contenedor doméstico que sea:
- hecho de plástico resistente,
- se puede cerrar con una tapa ajustada y resistente a perforaciones, sin que puedan salir objetos punzocortantes,
- vertical y estable durante su uso,
- a prueba de fugas, y
- debidamente etiquetado para advertir sobre residuos peligrosos dentro del contenedor
Cuando su contenedor de eliminación de objetos punzocortantes esté casi lleno, deberá seguir las pautas de su comunidad para la forma correcta de desecharlo. Puede haber leyes estatales o locales sobre cómo debe desechar las agujas y jeringas usadas.
Para obtener más información sobre la eliminación segura de objetos punzocortantes y para obtener información específica sobre la eliminación de objetos punzocortantes en el estado en el que vive, visite el sitio web de la FDA en: http://www.fda.gov/safesharpsdisposal
No deseche el contenedor de objetos punzocortantes usados en la basura doméstica a menos que las pautas de su comunidad lo permitan. No recicle su contenedor de objetos punzocortantes usados.
 No vuelva a colocar la tapa de la aguja.
No vuelva a colocar la tapa de la aguja.
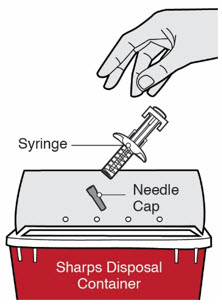

Fabricado por: Regeneron Pharmaceuticals, Inc. Tarrytown, NY 10591
Licencia de EE. UU. n.° 1760
Comercializado por: sanofi-aventis U.S. LLC (Bridgewater, NJ 08807) y Regeneron Pharmaceuticals,
Inc. (Tarrytown, NY 10591)
DUPIXENT® es una marca registrada de Sanofi Biotechnology
© 2023 Regeneron Pharmaceuticals, Inc. / sanofi-aventis U.S. LLC. Todos los derechos reservados.
Estas instrucciones de uso han sido aprobadas por la Administración de Alimentos y Medicamentos de los Estados Unidos.
Revisado: julio de 2023
PANEL PRINCIPAL DE PRESENTACIÓN – Jeringa de 300 mg/2 mL Caja – 5914
NDC 0024-5914-01
Rx Only
DUPIXENT®
(dupilumab)
Injection
300 mg/2 mL (150 mg/mL)
Para uso subcutáneo únicamente.
300 mg/2 mL
2
Dosis única
Jeringas precargadas
con protector de aguja
Mantener fuera del alcance de los niños. No usar después de la
fecha de caducidad. No usar si el sello está roto o dañado.
Almacenar refrigerado a 36° a 46°F (2° a 8°C) en el envase original.
Dosificación y administración: Consulte el prospecto para obtener información sobre la dosificación y las instrucciones de uso.
sanofi | REGENERON

PANEL PRINCIPAL DE VISUALIZACIÓN – Cartón de la jeringa de 200 mg/1,14 ml
NDC 0024-5918-01
Rx Only
DUPIXENT®
(dupilumab)
Injection
200 mg/1.14 mL (175 mg/mL)
Para uso subcutáneo únicamente.
200 mg/1.14 mL
2
Jeringas precargadas de dosis única
con protector de aguja
Mantener fuera del alcance de los niños. No usar después de la
fecha de caducidad. No usar si el sello está roto o dañado.
Almacenar refrigerado a 36° a 46°F (2° a 8°C) en el envase original.
Dosificación y administración: Consulte el prospecto para obtener información sobre la dosificación y las instrucciones de uso.
sanofi | REGENERON

PANEL PRINCIPAL DE PRESENTACIÓN – Cartón de pluma precargada de 300 mg/2 mL
NDC 0024-5915-02
Rx Only
DUPIXENT®
(dupilumab)
Inyección
Pluma precargada de dosis única
300 mg/2 mL (150 mg/mL)
Para uso subcutáneo únicamente.
2
Plumas precargadas
de dosis única
Mantener fuera del alcance de los niños. No usar
después de la fecha de caducidad. No usar si el sello está roto
o dañado.
Conservar en nevera entre 36° y 46°F (2° y 8°C) en el envase original.
NO congelar. NO calentar.
Dosificación y administración: Consultar el prospecto para obtener información sobre la dosificación
y las instrucciones de uso.
300 mg/2 mL
sanofi | REGENERON

PANEL PRINCIPAL DE EXHIBICIÓN – Cartón de pluma precargada de 200 mg/1,14 ml
NDC 0024-5919-02
Rx Only
DUPIXENT®
(dupilumab)
Inyección
Pluma precargada de dosis única
200 mg/1.14 mL (175 mg/mL)
Para uso subcutáneo únicamente.
2
Plumas precargadas
de dosis única
Mantener fuera del alcance de los niños. No usar después de la fecha de caducidad. No usar si el sello está roto o dañado.
Conservar en nevera entre 36° y 46°F (2° y 8°C) en
el envase original. No congelar. No calentar.
Dosificación y administración: Consulte el prospecto
para obtener información sobre la dosificación e instrucciones de uso.
200 mg/1.14 mL
sanofi | REGENERON

PANEL PRINCIPAL DE EXHIBICIÓN – Caja con jeringa de 100 mg/0.67 mL
NDC 0024-5911-02
Rx Only
DUPIXENT®
(dupilumab)
Inyección
100 mg/0.67 mL (150 mg/mL)
Sólo para uso subcutáneo.
2
Jeringas precargadas de
dosis única
con protector de aguja
Mantener fuera del alcance de los niños. No utilizar después de la fecha de caducidad. No utilizar si el precinto está roto o dañado.
Conservar en nevera entre 36° y 46°F (2° y 8°C) en el envase original.
Dosificación y administración: Consultar el prospecto para obtener información sobre la dosificación y las instrucciones de uso.
100 mg/0.67 mL
sanofi | REGENERON
