Fabricante de medicamentos: Janssen Biotech, Inc. (Updated: 2025-01-28)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
DARZALEX
®(daratumumab) inyección, para uso intravenoso
Aprobación inicial en EE. UU.: 2015
INDICACIONES Y USO
DARZALEX es un anticuerpo citolítico dirigido a CD38 indicado para el tratamiento de pacientes adultos con mieloma múltiple:
- en combinación con lenalidomida y dexametasona en pacientes de nuevo diagnóstico que no son candidatos a trasplante autólogo de células madre y en pacientes con mieloma múltiple recidivante o refractario que han recibido al menos un tratamiento previo
- en combinación con bortezomib, melfalán y prednisona en pacientes de nuevo diagnóstico que no son candidatos a trasplante autólogo de células madre
- en combinación con bortezomib, talidomida y dexametasona en pacientes de nuevo diagnóstico que son candidatos a trasplante autólogo de células madre
- en combinación con bortezomib y dexametasona en pacientes que han recibido al menos un tratamiento previo
- en combinación con carfilzomib y dexametasona en pacientes con mieloma múltiple recidivante o refractario que han recibido de una a tres líneas de tratamiento previas
- en combinación con pomalidomida y dexametasona en pacientes que han recibido al menos dos tratamientos previos, incluyendo lenalidomida y un inhibidor del proteasoma
- como monoterapia, en pacientes que han recibido al menos tres líneas de tratamiento previas incluyendo un inhibidor del proteasoma (IP) y un agente inmunomodulador o que son doblemente refractarios a un IP y un agente inmunomodulador. (
1)
POSOLOGÍA Y ADMINISTRACIÓN
- Premedicar con corticosteroides, antipiréticos y antihistamínicos. (
2.3) - Diluir y administrar como infusión intravenosa. (
2.5) - La dosis recomendada es de 16 mg/kg de peso corporal real. Consulte la información completa de prescripción para los medicamentos utilizados en combinación y el programa. (
2.2) - Administrar medicamentos posinfusión. (
2.3)
PRESENTACIONES Y CONCENTRACIONES
CONTRAINDICACIONES
Pacientes con antecedentes de hipersensibilidad grave a daratumumab o a cualquiera de los componentes de la formulación. (
4)
ADVERTENCIAS Y PRECAUCIONES
-
Reacciones relacionadas con la infusión: Interrumpir la infusión de DARZALEX para las reacciones relacionadas con la infusión de cualquier gravedad. Suspender permanentemente la infusión en caso de reacciones anafilácticas o reacciones relacionadas con la infusión que pongan en peligro la vida e instaurar atención de emergencia adecuada. (
2.4,
5.1)
-
Interferencia con la prueba cruzada y la detección de anticuerpos de eritrocitos: Tipificar y cribar a los pacientes antes de comenzar el tratamiento. Informar a los bancos de sangre que un paciente ha recibido DARZALEX. (
5.2,
7.1)
-
Neutropenia: Controlar periódicamente los recuentos sanguíneos completos durante el tratamiento. Controlar a los pacientes con neutropenia para detectar signos de infección. Puede ser necesario retrasar la dosis para permitir la recuperación de los neutrófilos. (
5.3)
-
Trombocitopenia: Controlar periódicamente los recuentos sanguíneos completos durante el tratamiento. Puede ser necesario retrasar la dosis para permitir la recuperación de las plaquetas. (
5.4)
-
Toxicidad embriofetal: Puede causar daño fetal. Advertir a las mujeres embarazadas sobre el riesgo potencial para el feto y aconsejar a las mujeres en edad fértil que utilicen un método anticonceptivo eficaz. (
5.6,
8.1,
8.3)
REACCIONES ADVERSAS
Las reacciones adversas más frecuentemente notificadas (incidencia ≥20%) son: infección de las vías respiratorias superiores, neutropenia, reacciones relacionadas con la infusión, trombocitopenia, diarrea, estreñimiento, anemia, neuropatía sensitiva periférica, fatiga, edema periférico, náuseas, tos, pirexia, disnea y astenia. (
6.1)
Para notificar SOSPECHAS DE REACCIONES ADVERSAS, póngase en contacto con Janssen Biotech, Inc. al 1-800-526-7736 (1-800-JANSSEN) o con la FDA al 1-800-FDA-1088 o
www.fda.gov/medwatch.
Consulte la sección 17 para obtener información sobre el asesoramiento al paciente y el etiquetado del paciente aprobado por la FDA.
Revisado: 1/2025
Tabla de contenido
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDO*
1 INDICACIONES Y USO
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Información Importante sobre la Dosificación
2.2 Dosis Recomendada
2.3 Medicamentos Concomitantes Recomendados
2.4 Modificaciones de la Dosis para las Reacciones Adversas
2.5 Preparación y Administración
3 FORMAS Y CONCENTRACIONES FARMACÉUTICAS
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Reacciones Relacionadas con la Infusión
5.2 Interferencia con las Pruebas Serológicas
5.3 Neutropenia
5.4 Trombocitopenia
5.5 Interferencia con la Determinación de la Respuesta Completa
5.6 Toxicidad Embrio-Fetal
6 REACCIONES ADVERSAS
6.1 Experiencia en Ensayos Clínicos
6.2 Experiencia Postcomercialización
7 INTERACCIONES MEDICAMENTOSAS
7.1 Efectos de Daratumumab en las Pruebas de Laboratorio
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.3 Mujeres y Hombres en Edad Reproductiva
8.4 Uso Pediátrico
8.5 Uso Geriátrico
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de Acción
12.2 Farmacodinamia
12.3 Farmacocinética
12.6 Inmunogenicidad
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
14 ESTUDIOS CLÍNICOS
14.1 Mieloma Múltiple de Nuevo Diagnóstico
14.2 Mieloma Múltiple Recidivante/Refractario
15 REFERENCIAS
16 PRESENTACIÓN/ALMACENAMIENTO Y MANEJO
17 INFORMACIÓN PARA EL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información completa de prescripción no se enumeran.
1 INDICACIONES Y USO
DARZALEX está indicado para el tratamiento de pacientes adultos con mieloma múltiple:
- en combinación con lenalidomida y dexametasona en pacientes de nuevo diagnóstico que no son candidatos a trasplante autólogo de células madre y en pacientes con mieloma múltiple recidivante o refractario que han recibido al menos un tratamiento previo.
- en combinación con bortezomib, melfalán y prednisona en pacientes de nuevo diagnóstico que no son candidatos a trasplante autólogo de células madre.
- en combinación con bortezomib, talidomida y dexametasona en pacientes de nuevo diagnóstico que son candidatos a trasplante autólogo de células madre.
- en combinación con bortezomib y dexametasona en pacientes que han recibido al menos un tratamiento previo.
- en combinación con carfilzomib y dexametasona en pacientes con mieloma múltiple recidivante o refractario que han recibido de una a tres líneas de tratamiento previas.
- en combinación con pomalidomida y dexametasona en pacientes que han recibido al menos dos tratamientos previos incluyendo lenalidomida y un inhibidor del proteasoma.
- como monoterapia, en pacientes que han recibido al menos tres líneas de tratamiento previas incluyendo un inhibidor del proteasoma (PI) y un agente inmunomodulador o que son doblemente refractarios a un PI y un agente inmunomodulador.
2 DOSIS Y ADMINISTRACIÓN
2.1 Información Importante sobre la Dosificación
- Administrar medicamentos preinfusión y postinfusión
[ver
Dosificación y Administración (2.3)]
.
- Administrar únicamente como infusión intravenosa después de la dilución en inyección de Cloruro de Sodio al 0,9%
[ver
Dosificación y Administración (2.5)].
- DARZALEX debe ser administrado por un profesional sanitario, con acceso inmediato a equipo de emergencia y apoyo médico adecuado para controlar las reacciones relacionadas con la infusión si se producen
[ver
Advertencias y Precauciones (5.1)].
- Tipificar y cribar a los pacientes antes de comenzar el tratamiento con DARZALEX
[ver
Advertencias y Precauciones (5.2)]
.
2.2 Dosis Recomendada
Monoterapia y en combinación con Lenalidomida (D-Rd) o Pomalidomida (D-Pd) y Dexametasona
El régimen de dosificación de DARZALEX en la Tabla 1 es para terapia combinada (regímenes de ciclo de 4 semanas) y monoterapia de la siguiente manera:
– terapia combinada con lenalidomida y dexametasona en dosis bajas para pacientes de nuevo diagnóstico no elegibles para trasplante autólogo de células madre (ASCT) y en pacientes con mieloma múltiple recidivante/refractario
– terapia combinada con pomalidomida y dexametasona en dosis bajas para pacientes con mieloma múltiple recidivante/refractario
– monoterapia para pacientes con mieloma múltiple recidivante/refractario.
La dosis recomendada de DARZALEX es de 16 mg/kg de peso corporal real administrada como infusión intravenosa según el siguiente régimen de dosificación:
| Semanas | Régimen |
|---|---|
| Semanas 1 a 8 | semanal (total de 8 dosis) |
| Semanas 9 a 24 * |
cada dos semanas (total de 8 dosis) |
| Semana 25 en adelante hasta la progresión de la enfermedad † |
cada cuatro semanas |
Para obtener instrucciones sobre la dosificación de los agentes combinados administrados con DARZALEX,
ver
Estudios Clínicos (14) e información de prescripción del fabricante.
En combinación con Bortezomib, Melfalán y Prednisona (D-VMP)
El régimen de dosificación de DARZALEX en la Tabla 2 es para terapia combinada con bortezomib, melfalán y prednisona (régimen de ciclo de 6 semanas) para pacientes con mieloma múltiple de nuevo diagnóstico no elegibles para ASCT.
La dosis recomendada de DARZALEX es de 16 mg/kg de peso corporal real administrada como infusión intravenosa según el siguiente régimen de dosificación:
| Semanas | Régimen |
|---|---|
| Semanas 1 a 6 | semanal (total de 6 dosis) |
| Semanas 7 a 54 * |
cada tres semanas (total de 16 dosis) |
| Semana 55 en adelante hasta la progresión de la enfermedad † |
cada cuatro semanas |
Para obtener instrucciones sobre la dosificación de los agentes combinados administrados con DARZALEX, consulte
Estudios Clínicos (14.1).
En combinación con bortezomib, talidomida y dexametasona (D-VTd)
El régimen de dosificación de DARZALEX en la Tabla 3 es para terapia combinada con bortezomib, talidomida y dexametasona (régimen de ciclo de 4 semanas) para pacientes con mieloma múltiple de nuevo diagnóstico elegibles para ASCT.
La dosis recomendada de DARZALEX es de 16 mg/kg de peso corporal real administrada como infusión intravenosa de acuerdo con el siguiente régimen de dosificación:
| Fase del tratamiento | Semanas | Programa |
|---|---|---|
| Inducción | Semanas 1 a 8 | semanal (total de 8 dosis) |
| Semanas 9 a 16 * |
cada dos semanas (total de 4 dosis) | |
| Interrumpir para quimioterapia de alta dosis y ASCT | ||
| Consolidación | Semanas 1 a 8 † |
cada dos semanas (total de 4 dosis) |
Para obtener instrucciones sobre la dosificación de los agentes combinados administrados con DARZALEX,
consulte
Estudios Clínicos (14.1) y la información de prescripción del fabricante.
En combinación con bortezomib y dexametasona (D-Vd)
El régimen de dosificación de DARZALEX en la Tabla 4 es para terapia combinada con bortezomib y dexametasona (ciclo de 3 semanas) para pacientes con mieloma múltiple recidivante/refractario.
La dosis recomendada de DARZALEX es de 16 mg/kg de peso corporal real administrada como infusión intravenosa de acuerdo con el siguiente régimen de dosificación:
| Semanas | Programa |
|---|---|
| Semanas 1 a 9 | semanal (total de 9 dosis) |
| Semanas 10 a 24 * |
cada tres semanas (total de 5 dosis) |
| Semana 25 en adelante hasta la progresión de la enfermedad † |
cada cuatro semanas |
Para obtener instrucciones sobre la dosificación de los agentes combinados administrados con DARZALEX, consulte
Estudios Clínicos (14.2) y la información de prescripción del fabricante
.
En combinación con Carfilzomib y Dexametasona (DKd)
La dosis recomendada de DARZALEX cuando se administra en combinación con carfilzomib y dexametasona (ciclo de 4 semanas) para pacientes con mieloma múltiple recidivante/refractario se proporciona en la Tabla 5.
| Semanas | Dosis de DARZALEX * |
Programa |
|---|---|---|
| Semana 1 | 8 mg/kg | días 1 y 2 (total de 2 dosis) |
| Semanas 2 a 8 | 16 mg/kg | semanal (total de 7 dosis) |
| Semanas 9 a 24 † |
16 mg/kg | cada dos semanas (total de 8 dosis) |
| Semana 25 en adelante hasta la progresión de la enfermedad ‡ |
16 mg/kg | cada cuatro semanas |
Para obtener instrucciones sobre la dosificación de los agentes combinados administrados con DARZALEX, consulte
Estudios Clínicos (14.1) y la información de prescripción del fabricante
.
Velocidades de Infusión
Administre DARZALEX por vía intravenosa a la velocidad de infusión que se describe a continuación en la Tabla 6. Considere la escalada incremental de la velocidad de infusión solo en ausencia de reacciones relacionadas con la infusión.
La dosis recomendada de 16 mg/kg que se administrará el Día 1 cuando DARZALEX se administra como monoterapia o en combinación se puede dividir en dos días consecutivos, de modo que se administre una dosis de 8 mg/kg el Día 1 y el Día 2, respectivamente.
| Volumen de dilución | Velocidad inicial (primera hora) | Incremento de velocidad | Velocidad máxima | |
|---|---|---|---|---|
|
||||
| Infusión de la semana 1 | ||||
| Opción 1 (infusión de dosis única) | ||||
| Día 1 de la semana 1 (16 mg/kg) | 1000 mL | 50 mL/hora | 50 mL/hora cada hora | 200 mL/hora |
| Opción 2 (infusión de dosis dividida) | ||||
| Día 1 de la semana 1 (8 mg/kg) | 500 mL | 50 mL/hora | 50 mL/hora cada hora | 200 mL/hora |
| Día 2 de la semana 1 (8 mg/kg) | 500 mL | 50 mL/hora | 50 mL/hora cada hora | 200 mL/hora |
| Semana 2 (16 mg/kg)† | 500 mL | 50 mL/hora | 50 mL/hora cada hora | 200 mL/hora |
| Semana 3 en adelante (16 mg/kg)‡ | 500 mL | 100 mL/hora | 50 mL/hora cada hora | 200 mL/hora |
2.3 Medicamentos concomitantes recomendados
Medicación preinfusión
Administre los siguientes medicamentos preinfusión de 1 a 3 horas antes de cada infusión de DARZALEX:
- Corticosteroide (de acción prolongada o intermedia)
Monoterapia:
Administre metilprednisolona 100 mg (o equivalente) por vía intravenosa. Después de la segunda infusión, considere reducir la dosis a 60 mg (o equivalente) administrada por vía oral o intravenosa.
En combinación:
Administre dexametasona 20 mg (o equivalente) por vía oral o intravenosa.
Cuando la dexametasona es el corticosteroide específico del régimen de fondo, la dosis de dexametasona que forma parte del régimen de fondo servirá como premedicación en los días de infusión de DARZALEX
[véase
Estudios clínicos (14)].
No administre corticosteroides específicos del régimen de fondo (p. ej., prednisona) en los días de infusión de DARZALEX cuando los pacientes hayan recibido dexametasona (o equivalente) como premedicación.
- Acetaminofeno 650 mg a 1000 mg por vía oral
- Difenhidramina 25 mg a 50 mg (o equivalente) por vía oral o intravenosa.
Medicación posinfusión
Administre los siguientes medicamentos posinfusión:
-
Monoterapia:
Administre metilprednisolona 20 mg (o una dosis equivalente de un corticosteroide de acción intermedia o prolongada) por vía oral durante 2 días a partir del día siguiente a la administración de DARZALEX.
En combinación:
Considere la administración de metilprednisolona oral a una dosis inferior o igual a 20 mg (o una dosis equivalente de un corticosteroide de acción intermedia o prolongada) a partir del día siguiente a la administración de una infusión de DARZALEX.
Si se administra un corticosteroide específico del régimen de fondo (p. ej., dexametasona, prednisona) el día después de la infusión de DARZALEX, es posible que no se necesiten corticosteroides adicionales[véase
Para pacientes con antecedentes de enfermedad pulmonar obstructiva crónica, considere recetar broncodilatadores de acción corta y larga y corticosteroides inhalados. Después de las cuatro primeras infusiones de DARZALEX, considere suspender estos medicamentos posinfusión adicionales si el paciente no experimenta una reacción importante relacionada con la infusión.
2.4 Modificaciones de la dosis para las reacciones adversas
No se recomiendan reducciones de la dosis de DARZALEX. Considere la suspensión de DARZALEX para permitir la recuperación del recuento de células sanguíneas en caso de mielosupresión
[véase
Advertencias y precauciones (5.3,
5.4)]
.
Para obtener información sobre los medicamentos administrados en combinación con DARZALEX, consulte la información de prescripción del fabricante.
Reacciones relacionadas con la infusión
Para las reacciones relacionadas con la infusión de cualquier grado/gravedad, interrumpa inmediatamente la infusión de DARZALEX y maneje los síntomas. El manejo de las reacciones relacionadas con la infusión puede requerir además una reducción en la velocidad de infusión o la interrupción del tratamiento con DARZALEX como se describe a continuación
[véase
Advertencias y precauciones (5.1)]
.
- Grado 1–2 (leve a moderado): Una vez que los síntomas de la reacción desaparezcan, reanude la infusión a no más de la mitad de la velocidad a la que se produjo la reacción. Si el paciente no experimenta más síntomas de reacción, la escalada de la velocidad de infusión puede reanudarse en incrementos e intervalos según sea clínicamente apropiado hasta la velocidad máxima de 200 mL/hora (Tabla 6).
- Grado 3 (grave): Una vez que los síntomas de la reacción desaparezcan, considere reiniciar la infusión a no más de la mitad de la velocidad a la que se produjo la reacción. Si el paciente no experimenta síntomas adicionales, reanude la escalada de la velocidad de infusión en incrementos e intervalos como se describe en la Tabla 6. Repita el procedimiento anterior en caso de recurrencia de síntomas de Grado 3. Suspenda permanentemente DARZALEX tras la tercera aparición de una reacción relacionada con la infusión de Grado 3 o superior.
- Grado 4 (potencialmente mortal): Suspenda permanentemente DARZALEX.
2.5 Preparación y administración
Preparación
DARZALEX es solo para una sola dosis.
Prepare la solución para infusión utilizando una técnica aséptica de la siguiente manera:
- Calcule la dosis (mg), el volumen total (mL) de la solución de DARZALEX necesaria y el número de viales de DARZALEX necesarios en función del peso corporal real del paciente.
- Hay disponibles viales de DARZALEX de la misma concentración con diferentes NDCs que se pueden mezclar en la misma bolsa de infusión
[véase
Presentación/Almacenamiento y manipulación (16)]
.
- Compruebe que la solución de DARZALEX sea incolora o de color amarillo pálido. No la utilice si presenta partículas opacas, decoloración u otras partículas extrañas.
- Extraiga un volumen de inyección de cloruro de sodio al 0,9 % de la bolsa/contenedor de infusión que sea igual al volumen requerido de la solución de DARZALEX.
- Extraiga la cantidad necesaria de solución de DARZALEX y diluya hasta el volumen adecuado añadiéndola a la bolsa/contenedor de infusión que contiene inyección de cloruro de sodio al 0,9 % como se especifica en la Tabla 6
[véase
Posología y administración (2.2)]
. Las bolsas/contenedores de infusión deben estar hechos de cloruro de polivinilo (PVC), polipropileno (PP), polietileno (PE) o mezcla de poliolefina (PP+PE). Diluya en condiciones asépticas adecuadas. Deseche cualquier porción no utilizada que quede en el vial.
- Invierta suavemente la bolsa/contenedor para mezclar la solución. No agite.
- Los productos farmacéuticos parenterales deben inspeccionarse visualmente para detectar la presencia de partículas y decoloración antes de su administración, siempre que la solución y el contenedor lo permitan. La solución diluida puede desarrollar partículas proteínicas muy pequeñas, traslúcidas o blancas, ya que el daratumumab es una proteína. No la utilice si observa partículas visiblemente opacas, decoloración o partículas extrañas.
- Si no se utiliza inmediatamente, almacene la solución diluida refrigerada hasta 24 horas a 2 °C a 8 °C (36 °F a 46 °F) y/o a temperatura ambiente hasta 15 horas a 15 °C a 25 °C (59 °F a 77 °F). El almacenamiento a temperatura ambiente incluye el tiempo de infusión. Proteger de la luz durante el almacenamiento. No congelar.
Administración
- Si se almacena en el refrigerador, deje que la solución alcance la temperatura ambiente. Administre la solución diluida mediante infusión intravenosa utilizando un equipo de infusión equipado con un regulador de flujo y con un filtro de polietersulfona (PES) estéril, no pirogénico, de baja unión a proteínas en línea (tamaño de poro de 0,22 micrómetros o 0,2 micrómetros). Los equipos de administración deben estar hechos de poliuretano (PU), polibutadieno (PBD), PVC, PP o PE.
- No almacene ninguna porción no utilizada de la solución de infusión para su reutilización. Cualquier producto o material de desecho no utilizado debe eliminarse de acuerdo con los requisitos locales.
- No infunda DARZALEX de forma concomitante en la misma vía intravenosa con otros agentes.
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
DARZALEX es una solución incolora a amarillo pálido, sin conservantes, disponible como:
Inyección:
- 100 mg/5 mL (20 mg/mL) en un vial de dosis única.
- 400 mg/20 mL (20 mg/mL) en un vial de dosis única.
4 CONTRAINDICACIONES
DARZALEX está contraindicado en pacientes con antecedentes de hipersensibilidad grave (p. ej., reacciones anafilácticas) a daratumumab o a cualquiera de los componentes de la formulación
[ver
Advertencias y precauciones (5.1)]
.
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Reacciones relacionadas con la infusión
DARZALEX puede causar reacciones graves y/o importantes relacionadas con la infusión, incluidas reacciones anafilácticas. Estas reacciones pueden ser potencialmente mortales y se han notificado desenlaces mortales
[ver
.
En los ensayos clínicos (monoterapia y combinación: N=2066), se produjeron reacciones relacionadas con la infusión en el 37 % de los pacientes con la infusión de la semana 1 (16 mg/kg), el 2 % con la infusión de la semana 2 y, de forma acumulativa, el 6 % con las infusiones posteriores. Menos del 1 % de los pacientes presentaron una reacción relacionada con la infusión de grado 3/4 en la semana 2 o en infusiones posteriores. El tiempo medio hasta el inicio fue de 1,5 horas (intervalo: 0 a 73 horas). La incidencia de modificación de la infusión debido a reacciones fue del 36 %. Las duraciones medias de las infusiones de 16 mg/kg para la semana 1, la semana 2 y las infusiones posteriores fueron de aproximadamente 7, 4 y 3 horas, respectivamente. Casi todas las reacciones se produjeron durante la infusión o en las 4 horas siguientes a la finalización de DARZALEX. Antes de la introducción de la medicación posterior a la infusión en los ensayos clínicos, las reacciones relacionadas con la infusión se produjeron hasta 48 horas después de la infusión.
Se han producido reacciones graves, como broncospasmo, hipoxia, disnea, hipertensión, taquicardia, cefalea, edema laríngeo, edema pulmonar y reacciones adversas oculares, como derrame coroideo, miopía aguda y glaucoma de ángulo estrecho agudo. Los signos y síntomas pueden incluir síntomas respiratorios, como congestión nasal, tos, irritación de garganta, así como escalofríos, vómitos y náuseas. Los signos y síntomas menos frecuentes fueron sibilancias, rinitis alérgica, pirexia, malestar torácico, prurito, hipotensión y visión borrosa
[ver
.
Cuando la dosificación de DARZALEX se interrumpió en el contexto de ASCT (CASSIOPEIA) durante una mediana de 3,75 meses (intervalo: 2,4 a 6,9 meses), tras la reiniciación de DARZALEX, la incidencia de reacciones relacionadas con la infusión fue del 11 % para la primera infusión después de ASCT. La velocidad de infusión/volumen de dilución utilizado tras la reiniciación fue el utilizado para la última infusión de DARZALEX antes de la interrupción para ASCT. Las reacciones relacionadas con la infusión que se produjeron al reiniciar DARZALEX después de ASCT fueron consistentes en términos de síntomas y gravedad (grado 3 o 4: <1 %) con las notificadas en estudios anteriores en la semana 2 o en infusiones posteriores.
En EQUULEUS, a los pacientes que recibieron tratamiento combinado (n=97) se les administró la primera dosis de 16 mg/kg en la semana 1 dividida en dos días, es decir, 8 mg/kg el día 1 y el día 2, respectivamente. La incidencia de reacciones relacionadas con la infusión de cualquier grado fue del 42 %, con un 36 % de los pacientes que experimentaron reacciones relacionadas con la infusión el día 1 de la semana 1, un 4 % el día 2 de la semana 1 y un 8 % con infusiones posteriores. El tiempo medio hasta el inicio de una reacción fue de 1,8 horas (intervalo: 0,1 a 5,4 horas). La incidencia de interrupciones de la infusión debido a reacciones fue del 30 %. Las duraciones medias de las infusiones fueron de 4,2 horas para el día 1 de la semana 1, 4,2 horas para el día 2 de la semana 1 y 3,4 horas para las infusiones posteriores.
Premedique a los pacientes con antihistamínicos, antipiréticos y corticosteroides. Vigile a los pacientes con frecuencia durante toda la infusión
[ver
Posología y administración (2.3)]
. Interrumpa la infusión de DARZALEX para reacciones de cualquier gravedad e instaurar un tratamiento médico según sea necesario. Suspenda permanentemente el tratamiento con DARZALEX si se produce una reacción anafiláctica o una reacción potencialmente mortal (grado 4) e instaurar los cuidados de urgencia adecuados. Para los pacientes con reacciones de grado 1, 2 o 3, reduzca la velocidad de infusión al reiniciar la infusión
[ver
Posología y administración (2.4)]
.
Para reducir el riesgo de reacciones retardadas relacionadas con la infusión, administre corticosteroides orales a todos los pacientes después de las infusiones de DARZALEX
[ver
Posología y administración (2.3)]
. Los pacientes con antecedentes de enfermedad pulmonar obstructiva crónica pueden necesitar medicamentos adicionales después de la infusión para controlar las complicaciones respiratorias. Considere la posibilidad de prescribir broncodilatadores de acción corta y larga duración y corticosteroides inhalados para pacientes con enfermedad pulmonar obstructiva crónica
[ver
Posología y administración (2.3)]
.
Se han producido reacciones adversas oculares, como miopía aguda y estrechamiento del ángulo de la cámara anterior debido a derrames ciliocoroideos con potencial de aumento de la presión intraocular o glaucoma, con la infusión de DARZALEX. Si se producen síntomas oculares, interrumpa la infusión de DARZALEX y busque una evaluación oftalmológica inmediata antes de reiniciar DARZALEX.
5.2 Interferencia con las pruebas serológicas
El daratumumab se une a CD38 en los glóbulos rojos (GR) y produce una prueba de antiglobulina indirecta (prueba de Coombs indirecta) positiva. La prueba de antiglobulina indirecta positiva mediada por daratumumab puede persistir hasta 6 meses después de la última infusión de daratumumab. El daratumumab unido a los GR enmascara la detección de anticuerpos contra antígenos menores en el suero del paciente
[ver
. La determinación del grupo sanguíneo ABO y Rh del paciente no se ve afectada
[ver
Interacciones medicamentosas (7.1)]
.
Notifique a los centros de transfusión sanguínea sobre esta interferencia con las pruebas serológicas e informe a los bancos de sangre de que un paciente ha recibido DARZALEX. Tipifique y cribe a los pacientes antes de comenzar el tratamiento con DARZALEX
[ver
Posología y administración (2.1)]
.
5.3 Neutropenia
DARZALEX puede aumentar la neutropenia inducida por la terapia de fondo
[ver
Controle los recuentos sanguíneos completos periódicamente durante el tratamiento de acuerdo con la información de prescripción del fabricante para las terapias de fondo. Vigile a los pacientes con neutropenia para detectar signos de infección. Considere la posibilidad de suspender DARZALEX hasta que se recuperen los neutrófilos.
5.4 Trombocitopenia
DARZALEX puede aumentar la trombocitopenia inducida por la terapia de fondo
[ver
Controle periódicamente los recuentos sanguíneos completos durante el tratamiento de acuerdo con la información de prescripción del fabricante para las terapias de fondo. Considere la posibilidad de suspender DARZALEX hasta la recuperación de las plaquetas.
5.5 Interferencia con la Determinación de la Respuesta Completa
Daratumumab es un anticuerpo monoclonal kappa IgG humano que puede detectarse tanto en la electroforesis de proteínas séricas (SPE) como en los ensayos de inmunofijación (IFE) utilizados para el control clínico de la M-proteína endógena
[ver
Interacciones Medicamentosas (7.1)]
. Esta interferencia puede afectar la determinación de la respuesta completa y de la progresión de la enfermedad en algunos pacientes con proteína de mieloma kappa IgG.
5.6 Toxicidad Embrio-Fetal
Basándose en el mecanismo de acción, DARZALEX puede causar daño fetal cuando se administra a una mujer embarazada. DARZALEX puede causar depleción de las células inmunitarias fetales y disminución de la densidad ósea. Avise a las mujeres embarazadas del riesgo potencial para el feto. Aconseje a las mujeres con potencial reproductivo que utilicen un método anticonceptivo eficaz durante el tratamiento con DARZALEX y durante 3 meses después de la última dosis
[ver
Uso en Poblaciones Específicas (8.1,
8.3)]
.
La combinación de DARZALEX con lenalidomida, pomalidomida o talidomida está contraindicada en mujeres embarazadas, ya que la lenalidomida, la pomalidomida y la talidomida pueden causar defectos de nacimiento y la muerte del feto. Consulte la información de prescripción de lenalidomida, pomalidomida o talidomida sobre el uso durante el embarazo.
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas clínicamente significativas se describen en otras partes del etiquetado:
- Reacciones relacionadas con la infusión
[ver
Advertencias y precauciones (5.1)]
. - Neutropenia
[ver
Advertencias y precauciones (5.3)]
. - Trombocitopenia
[ver
Advertencias y precauciones (5.4)]
.
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se realizan en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no se pueden comparar directamente con las tasas en los ensayos clínicos de otro fármaco y pueden no reflejar las tasas observadas en la práctica.
Los datos de seguridad que se describen a continuación reflejan la exposición a DARZALEX (16 mg/kg) en 2459 pacientes con mieloma múltiple, incluidos 2303 pacientes que recibieron DARZALEX en combinación con regímenes de fondo y 156 pacientes que recibieron DARZALEX como monoterapia. En esta población de seguridad agrupada, las reacciones adversas más comunes (≥20%) fueron infección de las vías respiratorias superiores, neutropenia, reacciones relacionadas con la infusión, trombocitopenia, diarrea, estreñimiento, anemia, neuropatía sensitiva periférica, fatiga, edema periférico, náuseas, tos, pirexia, disnea y astenia.
Mieloma múltiple de nuevo diagnóstico no elegible para trasplante autólogo de células madre
Tratamiento combinado con lenalidomida y dexametasona (DRd)
La seguridad de DARZALEX en combinación con lenalidomida y dexametasona se evaluó en MAIA
[ver
Las reacciones adversas descritas en la Tabla 7 reflejan la exposición a DARZALEX durante una duración media del tratamiento de 25,3 meses (rango: 0,1 a 40,44 meses) para daratumumab-lenalidomida-dexametasona (DRd) y de 21,3 meses (rango: 0,03 a 40,64 meses) para lenalidomida-dexametasona (Rd).
Las reacciones adversas graves con una incidencia 2% mayor en el brazo DRd en comparación con el brazo Rd fueron neumonía (DRd 15% vs Rd 8%), bronquitis (DRd 4% vs Rd 2%) y deshidratación (DRd 2% vs Rd <1%).
| Sistema orgánico Reacción adversa |
DRd (N=364) | Rd (N=365) | ||||
|---|---|---|---|---|---|---|
| Todos los grados (%) | Grado 3 (%) | Grado 4 (%) | Todos los grados (%) | Grado 3 (%) | Grado 4 (%) | |
| Clave: D=daratumumab, Rd=lenalidomida-dexametasona. | ||||||
|
||||||
| Trastornos gastrointestinales | ||||||
| Diarrea | 57 | 7 | 0 | 46 | 4 | 0 |
| Estreñimiento | 41 | 1 | <1 | 36 | <1 | 0 |
| Náuseas | 32 | 1 | 0 | 23 | 1 | 0 |
| Vómitos | 17 | 1 | 0 | 12 | <1 | 0 |
| Infecciones | ||||||
| Infección del tracto respiratorio superior | 52 | 2 | <1 | 36 | 2 | <1 |
| Bronquitis | 29 | 3 | 0 | 21 | 1 | 0 |
| Neumonía | 26 | 14 | 1 | 14 | 7 | 1 |
| Infección del tracto urinario | 18 | 2 | 0 | 10 | 2 | 0 |
| Trastornos generales y condiciones en el sitio de administración | ||||||
| Reacciones relacionadas con la infusión | 41 | 2 | <1 | 0 | 0 | 0 |
| Edema periférico | 41 | 2 | 0 | 33 | 1 | 0 |
| Fatiga | 40 | 8 | 0 | 28 | 4 | 0 |
| Astenia | 32 | 4 | 0 | 25 | 3 | <1 |
| Pirexia | 23 | 2 | 0 | 18 | 2 | 0 |
| Escalofríos | 13 | 0 | 0 | 2 | 0 | 0 |
| Trastornos musculoesqueléticos y del tejido conjuntivo | ||||||
| Dolor de espalda | 34 | 3 | <1 | 26 | 3 | <1 |
| Espasmos musculares | 29 | 1 | 0 | 22 | 1 | 0 |
| Trastornos respiratorios, torácicos y mediastínicos | ||||||
| Disnea | 32 | 3 | <1 | 20 | 1 | 0 |
| Tos | 30 | <1 | 0 | 18 | 0 | 0 |
| Trastornos del sistema nervioso | ||||||
| Neuropatía sensitiva periférica | 24 | 1 | 0 | 15 | 0 | 0 |
| Cefalea | 19 | 1 | 0 | 11 | 0 | 0 |
| Parestesia | 16 | 0 | 0 | 8 | 0 | 0 |
| Trastornos del metabolismo y la nutrición | ||||||
| Disminución del apetito | 22 | 1 | 0 | 15 | <1 | <1 |
| Hiperglucemia | 14 | 6 | 1 | 8 | 3 | 1 |
| Hipocalcemia | 14 | 1 | <1 | 9 | 1 | 1 |
| Trastornos vasculares | ||||||
| Hipertensión | 13 | 6 | <1 | 7 | 4 | 0 |
Anormalidades de laboratorio que empeoran durante el tratamiento desde el valor basal que se indican en la Tabla 8.
| DRd (N=364) | Rd (N=365) | |||||
|---|---|---|---|---|---|---|
| Todos los grados (%) |
Grado 3 (%) |
Grado 4 (%) |
Todos los grados (%) |
Grado 3 (%) |
Grado 4 (%) |
|
| Clave: D=daratumumab, Rd=lenalidomida-dexametasona. | ||||||
| Leucopenia | 90 | 30 | 5 | 82 | 20 | 4 |
| Neutropenia | 91 | 39 | 17 | 77 | 28 | 11 |
| Linfopenia | 84 | 41 | 11 | 75 | 36 | 6 |
| Trombocitopenia | 67 | 6 | 3 | 58 | 7 | 4 |
| Anemia | 47 | 13 | 0 | 57 | 24 | 0 |
Tratamiento combinado con bortezomib, melfalán y prednisona
La seguridad de DARZALEX en combinación con bortezomib, melfalán y prednisona se evaluó en ALCYONE
[véase
Las reacciones adversas descritas en la Tabla 9 reflejan la exposición a DARZALEX durante una duración media del tratamiento de 14,7 meses (rango: 0 a 25,8 meses) para daratumumab, bortezomib, melfalán y prednisona (D-VMP) y de 12 meses (rango: 0,1 a 14,9 meses) para VMP.
Las reacciones adversas graves con una incidencia al menos un 2% mayor en el brazo D-VMP en comparación con el brazo VMP fueron neumonía (D-VMP 11% vs VMP 4%), infección del tracto respiratorio superior (D-VMP 5% vs VMP 1%) y edema pulmonar (D-VMP 2% vs VMP 0%).
| Sistema orgánico Reacción adversa |
D-VMP (N=346) | VMP (N=354) | ||||
|---|---|---|---|---|---|---|
| Todos los grados (%) | Grado 3 (%) | Grado 4 (%) | Todos los grados (%) | Grado 3 (%) | Grado 4 (%) | |
| Clave: D=daratumumab, VMP=bortezomib-melfalán-prednisona. | ||||||
|
||||||
| Infecciones | ||||||
| Infección del tracto respiratorio superior | 48 | 5 | 0 | 28 | 3 | 0 |
| Neumonía | 16 | 12 | < 1 | 6 | 5 | < 1 |
| Trastornos generales y alteraciones en el lugar de administración | ||||||
| Reacciones relacionadas con la infusión | 28 | 4 | 1 | 0 | 0 | 0 |
| Edema periférico | 21 | 1 | < 1 | 14 | 1 | 0 |
| Trastornos respiratorios, torácicos y mediastínicos | ||||||
| Tos | 16 | < 1 | 0 | 8 | < 1 | 0 |
| Disnea | 13 | 2 | 1 | 5 | 1 | 0 |
| Trastornos vasculares | ||||||
| Hipertensión | 10 | 4 | < 1 | 3 | 2 | 0 |
Anormalidades de laboratorio que empeoran durante el tratamiento desde el valor basal que se indican en la Tabla 10.
| D-VMP (N=346) | VMP (N=354) | |||||
|---|---|---|---|---|---|---|
| Todos los grados (%) |
Grado 3 (%) |
Grado 4 (%) |
Todos los grados (%) |
Grado 3 (%) |
Grado 4 (%) |
|
| Clave: D=daratumumab, VMP=bortezomib-melfalán-prednisona. | ||||||
| Trombocitopenia | 88 | 27 | 11 | 88 | 26 | 16 |
| Neutropenia | 86 | 34 | 10 | 87 | 32 | 11 |
| Linfopenia | 85 | 46 | 12 | 83 | 44 | 9 |
| Anemia | 47 | 18 | 0 | 50 | 21 | 0 |
Mieloma Múltiple de Nuevo Diagnóstico Elegible para Trasplante Autólogo de Células Madre
Tratamiento Combinado con Bortezomib, Talidomida y Dexametasona (DVTd)
La seguridad de DARZALEX en combinación con bortezomib, talidomida y dexametasona se evaluó en CASSIOPEIA
[véase
Las reacciones adversas descritas en la Tabla 11 reflejan la exposición a DARZALEX hasta el día 100 después del trasplante. La duración media del tratamiento de inducción/TACA/consolidación fue de 8,9 meses (rango: 7,0 a 12,0 meses) para DVTd y 8,7 meses (rango: 6,4 a 11,5 meses) para VTd.
Las reacciones adversas graves con una incidencia 2% mayor en el brazo DVTd en comparación con el brazo VTd fueron bronquitis (DVTd 2% vs VTd <1%) y neumonía (DVTd 6% vs VTd 4%).
| Sistema orgánico Reacción adversa |
DVTd (N=536) | VTd (N=538) | ||||
|---|---|---|---|---|---|---|
| Todos los grados (%) | Grado 3 (%) | Grado 4 (%) | Todos los grados (%) | Grado 3 (%) | Grado 4 (%) | |
| Clave: D=daratumumab, VTd=bortezomib-talidomida-dexametasona. Nota: Las toxicidades relacionadas con el laboratorio de hematología se excluyeron y se informaron por separado en la tabla siguiente. |
||||||
|
||||||
| Trastornos generales y alteraciones en el lugar de administración | ||||||
| Reacciones relacionadas con la infusión | 35 | 3 | <1 | 0 | 0 | 0 |
| Pirexia | 26 | 2 | <1 | 21 | 2 | 0 |
| Trastornos gastrointestinales | ||||||
| Náuseas | 30 | 4 | 0 | 24 | 2 | <1 |
| Vómitos | 16 | 2 | 0 | 10 | 2 | 0 |
| Infecciones | ||||||
| Infección del tracto respiratorio superior | 27 | 1 | 0 | 17 | 1 | 0 |
| Bronquitis | 20 | 1 | 0 | 13 | 1 | 0 |
| Trastornos respiratorios, torácicos y mediastínicos | ||||||
| Tos | 17 | 0 | 0 | 9 | 0 | 0 |
| Trastornos vasculares | ||||||
| Hipertensión | 10 | 4 | 0 | 5 | 2 | 0 |
| DVTd (N=536) | VTd (N=538) | |||||
|---|---|---|---|---|---|---|
| Todos los grados (%) |
Grado 3 (%) |
Grado 4 (%) |
Todos los grados (%) |
Grado 3 (%) |
Grado 4 (%) |
|
| Clave: D=daratumumab, VTd=bortezomib-talidomida-dexametasona. | ||||||
| Linfopenia | 95 | 44 | 15 | 91 | 37 | 10 |
| Leucopenia | 82 | 14 | 10 | 57 | 6 | 9 |
| Trombocitopenia | 81 | 9 | 5 | 58 | 8 | 3 |
| Neutropenia | 63 | 19 | 14 | 41 | 10 | 9 |
| Anemia | 36 | 4 | 0 | 35 | 5 | 0 |
Mieloma Múltiple en recaída/refractario
Tratamiento combinado con lenalidomida y dexametasona
La seguridad de DARZALEX en combinación con lenalidomida y dexametasona se evaluó en POLLUX
[véase
Las reacciones adversas descritas en la Tabla 13 reflejan la exposición a DARZALEX durante una duración media del tratamiento de 13,1 meses (rango: 0 a 20,7 meses) para daratumumab-lenalidomida-dexametasona (DRd) y de 12,3 meses (rango: 0,2 a 20,1 meses) para lenalidomida-dexametasona (Rd).
Se produjeron reacciones adversas graves en el 49% de los pacientes del brazo DRd en comparación con el 42% en el brazo Rd. Las reacciones adversas graves con una incidencia al menos un 2% mayor en el brazo DRd en comparación con el brazo Rd fueron neumonía (DRd 12% vs Rd 10%), infección del tracto respiratorio superior (DRd 7% vs Rd 4%), gripe y pirexia (DRd 3% vs Rd 1% para cada una).
Las reacciones adversas provocaron la interrupción del tratamiento en el 7% (n=19) de los pacientes del brazo DRd frente al 8% (n=22) en el brazo Rd.
| Reacción adversa | DRd (N=283) | Rd (N=281) | ||||
|---|---|---|---|---|---|---|
| Todos los grados (%) |
Grado 3 (%) |
Grado 4 (%) |
Todos los grados (%) |
Grado 3 (%) |
Grado 4 (%) |
|
| Clave: D=daratumumab, Rd=lenalidomida-dexametasona. | ||||||
|
||||||
| Infecciones | ||||||
| Infección del tracto respiratorio superior | 65 | 6 | < 1 | 51 | 4 | 0 |
| Trastornos generales y alteraciones en el lugar de administración | ||||||
| Reacciones relacionadas con la infusión | 48 | 5 | 0 | 0 | 0 | 0 |
| Fatiga | 35 | 6 | < 1 | 28 | 2 | 0 |
| Pirexia | 20 | 2 | 0 | 11 | 1 | 0 |
| Trastornos gastrointestinales | ||||||
| Diarrea | 43 | 5 | 0 | 25 | 3 | 0 |
| Náuseas | 24 | 1 | 0 | 14 | 0 | 0 |
| Vómitos | 17 | 1 | 0 | 5 | 1 | 0 |
| Trastornos respiratorios, torácicos y mediastínicos | ||||||
| Tos | 30 | 0 | 0 | 15 | 0 | 0 |
| Disnea | 21 | 3 | < 1 | 12 | 1 | 0 |
| Trastornos musculoesqueléticos y del tejido conjuntivo | ||||||
| Espasmos musculares | 26 | 1 | 0 | 19 | 2 | 0 |
| Trastornos del sistema nervioso | ||||||
| Cefalea | 13 | 0 | 0 | 7 | 0 | 0 |
Anormalidades de laboratorio que empeoran durante el tratamiento desde el valor basal que se indican en la Tabla 14.
| DRd (N=283) | Rd (N=281) | |||||
|---|---|---|---|---|---|---|
| Todos los grados (%) |
Grado 3 (%) |
Grado 4 (%) |
Todos los grados (%) |
Grado 3 (%) |
Grado 4 (%) |
|
| Clave: D=daratumumab, Rd=lenalidomida-dexametasona. | ||||||
| Linfopenia | 95 | 42 | 10 | 87 | 32 | 6 |
| Neutropenia | 92 | 36 | 17 | 87 | 32 | 8 |
| Trombocitopenia | 73 | 7 | 6 | 67 | 10 | 5 |
| Anemia | 52 | 13 | 0 | 57 | 19 | 0 |
Tratamiento combinado con bortezomib y dexametasona
La seguridad de DARZALEX en combinación con bortezomib y dexametasona se evaluó en CASTOR
[véase
Las reacciones adversas descritas en la Tabla 15 reflejan la exposición a DARZALEX durante una duración media del tratamiento de 6,5 meses (rango: 0 a 14,8 meses) para daratumumab-bortezomib-dexametasona (DVd) y de 5,2 meses (rango: 0,2 a 8,0 meses) para el brazo de bortezomib-dexametasona (Vd).
Se produjeron reacciones adversas graves en el 42 % de los pacientes del brazo DVd en comparación con el 34 % en el brazo Vd. Las reacciones adversas graves con una incidencia al menos un 2 % mayor en el brazo DVd en comparación con el brazo Vd fueron infección del tracto respiratorio superior (DVd 5 % frente a Vd 2 %), diarrea y fibrilación auricular (DVd 2 % frente a Vd 0 % para cada una).
Las reacciones adversas provocaron la interrupción del tratamiento en el 7 % (n=18) de los pacientes del brazo DVd frente al 9 % (n=22) en el brazo Vd.
| Reacción adversa | DVd (N=243) | Vd (N=237) | ||||
|---|---|---|---|---|---|---|
| Todos los grados (%) |
Grado 3 (%) |
Grado 4 (%) |
Todos los grados (%) |
Grado 3 (%) |
Grado 4 (%) |
|
| Clave: D=daratumumab, Vd=bortezomib-dexametasona. | ||||||
|
||||||
| Trastornos del sistema nervioso | ||||||
| Neuropatía sensitiva periférica | 47 | 5 | 0 | 38 | 6 | < 1 |
| Trastornos generales y alteraciones en el lugar de administración | ||||||
| Reacciones relacionadas con la infusión | 45 | 9 | 0 | 0 | 0 | 0 |
| Edema periférico | 22 | 1 | 0 | 13 | 0 | 0 |
| Pirexia | 16 | 1 | 0 | 11 | 1 | 0 |
| Infecciones | ||||||
| Infección del tracto respiratorio superior | 44 | 6 | 0 | 30 | 3 | < 1 |
| Trastornos gastrointestinales | ||||||
| Diarrea | 32 | 3 | < 1 | 22 | 1 | 0 |
| Vómitos | 11 | 0 | 0 | 4 | 0 | 0 |
| Trastornos respiratorios, torácicos y mediastínicos | ||||||
| Tos | 27 | 0 | 0 | 14 | 0 | 0 |
| Disnea | 21 | 4 | 0 | 11 | 1 | 0 |
Las anormalidades de laboratorio que empeoran durante el tratamiento se enumeran en la Tabla 16.
| DVd (N=243) | Vd (N=237) | |||||
|---|---|---|---|---|---|---|
| Todos los grados (%) |
Grado 3 (%) |
Grado 4 (%) |
Todos los grados (%) |
Grado 3 (%) |
Grado 4 (%) |
|
| Clave: D=daratumumab, Vd=bortezomib-dexametasona. | ||||||
| Trombocitopenia | 90 | 28 | 19 | 85 | 22 | 13 |
| Linfopenia | 89 | 41 | 7 | 81 | 24 | 3 |
| Neutropenia | 58 | 12 | 3 | 40 | 5 | < 1 |
| Anemia | 48 | 13 | 0 | 56 | 14 | 0 |
Tratamiento combinado con Carfilzomib y Dexametasona dos veces por semana (20/56 mg/m
2)
La seguridad de DARZALEX en combinación con carfilzomib y dexametasona dos veces por semana se evaluó en CANDOR
[véase
Estudios Clínicos (14.2)]
. Las reacciones adversas descritas en la Tabla 17 reflejan la exposición a DARZALEX durante una duración media del tratamiento de 16,1 meses (rango: 0,1 a 23,7 meses) para el grupo daratumumab-carfilzomib-dexametasona (DKd) y una duración media del tratamiento de 9,3 meses (rango: 0,1 a 22,4 meses) para el grupo carfilzomib-dexametasona (Kd).
Se produjeron reacciones adversas graves en el 56 % de los pacientes que recibieron DARZALEX en combinación con Kd y en el 46 % de los pacientes que recibieron Kd. Las reacciones adversas graves más frecuentes notificadas en el brazo DKd en comparación con el brazo Kd fueron neumonía (DKd 14 % vs Kd 9 %), pirexia (DKd 4,2 % vs Kd 2,0 %), gripe (DKd 3,9 % vs Kd 1,3 %), sepsis (DKd 3,9 % vs Kd 1,3 %), anemia (DKd 2,3 % vs Kd 0,7 %), bronquitis (DKd 1,9 % vs Kd 0 %) y diarrea (DKd 1,6 % vs Kd 0 %). Se produjeron reacciones adversas mortales en los 30 días posteriores a la última dosis de cualquier tratamiento del estudio en el 10 % de los 308 pacientes que recibieron DARZALEX en combinación con Kd frente al 5 % de los 153 pacientes que recibieron Kd. La reacción adversa mortal más frecuente fue la infección (4,5 % vs 2,6 %).
La interrupción permanente de DARZALEX debido a una reacción adversa se produjo en el 9 % de los pacientes. Las reacciones adversas (>1 %) que provocaron la interrupción permanente de DARZALEX incluyeron neumonía.
Las reacciones relacionadas con la infusión que se produjeron el día de administración de cualquier dosis de DARZALEX o al día siguiente se produjeron en el 18 % de los pacientes y las que se produjeron el día de administración de la primera dosis de DARZALEX o al día siguiente se produjeron en el 12 %.
| Reacción adversa | DKd (N=308) | Kd (N=153) | ||
|---|---|---|---|---|
| Todos los grados | Grados 3 o 4 | Todos los grados | Grados 3 o 4 | |
| (%) | (%) | (%) | (%) | |
| Clave: D=daratumumab; Kd=carfilzomib-dexametasona. | ||||
|
||||
| Trastornos generales y alteraciones en el lugar de administración | ||||
| Reacciones relacionadas con la infusión * |
41 | 12 | 28 | 5 |
| Fatiga † |
32 | 11 | 28 | 8 |
| Pirexia | 20 | 1.9 | 15 | 0.7 |
| Infecciones | ||||
| Infección del tracto respiratorio ‡ |
40 § |
7 | 29 | 3.3 |
| Neumonía | 18 § |
13 | 12 | 9 |
| Bronquitis | 17 | 2.6 | 12 | 1.3 |
| Trastornos de la sangre y del sistema linfático | ||||
| Trombocitopenia ¶ |
37 | 25 | 30 | 16 |
| Anemia # |
33 | 17 | 31 | 14 |
| Trastornos gastrointestinales | ||||
| Diarrea | 32 | 3.9 | 14 | 0.7 |
| Náuseas | 18 | 0 | 13 | 0.7 |
| Trastornos vasculares | ||||
| Hipertensión | 31 | 18 | 28 | 13 |
| Trastornos respiratorios, torácicos y mediastínicos | ||||
| Tos Þ |
21 | 0 | 21 | 0 |
| Disnea | 20 | 3.9 | 22 | 2.6 |
| Trastornos psiquiátricos | ||||
| Insomnio | 18 | 3.9 | 11 | 2 |
| Trastornos musculoesqueléticos y del tejido conjuntivo | ||||
| Dolor de espalda | 16 | 1.9 | 10 | 1.3 |
Reacciones adversas que ocurren con una frecuencia de < 15%
- Trastornos de la sangre y del sistema linfático: neutropenia, linfopenia, leucopenia, neutropenia febril
- Trastornos cardíacos: fibrilación auricular
- Trastornos gastrointestinales: vómitos, estreñimiento
- Trastornos generales y alteraciones en el lugar de administración: edema periférico, astenia, escalofríos
- Infecciones: influenza, infección del tracto urinario, sepsis, shock séptico
- Trastornos del metabolismo y de la nutrición: disminución del apetito, hiperglucemia, hipocalcemia, deshidratación
- Trastornos musculoesqueléticos y del tejido conjuntivo: espasmos musculares, artralgia, dolor torácico musculoesquelético
- Trastornos del sistema nervioso: dolor de cabeza, mareos, neuropatía sensitiva periférica, parestesia, síndrome de encefalopatía posterior reversible
- Trastornos respiratorios, torácicos y mediastínicos: edema pulmonar
- Trastornos de la piel y del tejido subcutáneo: erupción, prurito
Tratamiento combinado con carfilzomib y dexametasona una vez por semana (20/70 mg/m
2)
La seguridad de DARZALEX en combinación con carfilzomib y dexametasona una vez por semana se evaluó en EQUULEUS
[véase
Estudios clínicos (14.2)]
. Las reacciones adversas descritas en la Tabla 18 reflejan la exposición a DARZALEX durante una duración media del tratamiento de 19,8 meses (rango: 0,3 a 34,5 meses).
Se notificaron reacciones adversas graves en el 48% de los pacientes. Las reacciones adversas graves más frecuentes fueron neumonía (4,7%), infección de las vías respiratorias superiores (4,7%), carcinoma de células basales (4,7%), influenza (3,5%), deterioro general del estado de salud físico (3,5%) e hipercalcemia (3,5%). Las reacciones adversas mortales en los 30 días posteriores a la última dosis de cualquier tratamiento del estudio se produjeron en el 3,5% de los pacientes que murieron por deterioro general del estado de salud físico, insuficiencia multiorgánica secundaria a aspergilosis pulmonar y progresión de la enfermedad.
La interrupción permanente de DARZALEX debido a una reacción adversa se produjo en el 8% de los pacientes. Ninguna reacción adversa que provocó la interrupción permanente de DARZALEX se produjo en más de un paciente.
Las reacciones relacionadas con la infusión que ocurrieron el día de la administración de cualquier dosis de DARZALEX o al día siguiente ocurrieron en el 44% de los pacientes. Para los pacientes que recibieron la dosis inicial dividida de DARZALEX, las reacciones relacionadas con la infusión que ocurrieron en el 36% y el 4% el primer y segundo día de administración de DARZALEX, respectivamente.
| Reacción adversa | DKd (N=85) | ||
|---|---|---|---|
| Todos los grados (%) | Grados 3 o 4 (%) | ||
| Clave: D=daratumumab; Kd=carfilzomib-dexametasona. | |||
|
|||
| Trastornos de la sangre y del sistema linfático | |||
| Trombocitopenia * |
68 | 32 | |
| Anemia † |
52 | 21 | |
| Neutropenia ‡ |
31 | 21 | |
| Linfopenia § |
29 | 25 | |
| Trastornos generales y condiciones en el sitio de administración | ||
| Fatiga ¶ |
54 | 18 |
| Reacciones relacionadas con la infusión # |
53 | 12 |
| Pirexia | 37 | 1.2 |
| Infecciones | ||
| Infección del tracto respiratorio Þ |
53 | 3.5 |
| Bronquitis | 19 | 0 |
| Nasofaringitis | 18 | 0 |
| Influenza | 17 | 3.5 |
| Trastornos gastrointestinales | ||
| Náuseas | 42 | 1.2 |
| Vómitos | 40 | 1.2 |
| Diarrea | 38 | 2.4 |
| Estreñimiento | 17 | 0 |
| Trastornos respiratorios, torácicos y mediastínicos | ||
| Disnea | 35 | 3.5 |
| Tos ß |
33 | 0 |
| Trastornos vasculares | ||
| Hipertensión | 33 | 20 |
| Trastornos psiquiátricos | ||
| Insomnio | 33 | 4.7 |
| Trastornos del sistema nervioso | ||
| Cefalea | 27 | 1.2 |
| Trastornos musculoesqueléticos y del tejido conjuntivo | ||
| Dolor de espalda | 25 | 0 |
| Dolor en extremidades | 15 | 0 |
Reacciones adversas que ocurren con una frecuencia de < 15%
- Trastornos de la sangre y del sistema linfático: leucopenia, neutropenia febril
- Trastornos cardíacos: fibrilación auricular
- Trastornos gastrointestinales: pancreatitis
- Trastornos generales y alteraciones en el lugar de administración: edema periférico, escalofríos
- Infecciones: neumonía, infección del tracto urinario, sepsis, shock séptico
- Trastornos del metabolismo y de la nutrición: disminución del apetito, hiperglucemia, deshidratación, hipocalcemia
- Trastornos musculoesqueléticos y del tejido conjuntivo: espasmos musculares, dolor torácico musculoesquelético, artralgia
- Trastornos del sistema nervioso: mareos, parestesia, neuropatía sensitiva periférica
- Trastornos de la piel y del tejido subcutáneo: prurito, erupción
Tratamiento combinado con pomalidomida y dexametasona
La seguridad de DARZALEX en combinación con pomalidomida y dexametasona se evaluó en EQUULEUS
[ver
. Las reacciones adversas descritas en la Tabla 19 reflejan la exposición a DARZALEX, pomalidomida y dexametasona (DPd) durante una duración media del tratamiento de 6 meses (rango: 0,03 a 16,9 meses).
La incidencia general de reacciones adversas graves fue del 49 %. Las reacciones adversas graves notificadas en ≥5 % de los pacientes incluyeron neumonía (7 %). Las reacciones adversas provocaron la interrupción del tratamiento en el 13 % de los pacientes.
| Reacción adversa | DPd (N=103) | ||
|---|---|---|---|
| Todos los grados (%) |
Grado 3 (%) |
Grado 4 (%) |
|
| Clave: D=daratumumab, Pd=pomalidomida-dexametasona. | |||
|
|||
| Trastornos generales y alteraciones en el lugar de administración | |||
| Fatiga | 50 | 10 | 0 |
| Reacciones relacionadas con la infusión | 50 | 4 | 0 |
| Pirexia | 25 | 1 | 0 |
| Escalofríos | 20 | 0 | 0 |
| Edema periférico | 17 | 4 | 0 |
| Astenia | 15 | 0 | 0 |
| Dolor torácico no cardíaco | 15 | 0 | 0 |
| Dolor | 11 | 0 | 0 |
| Infecciones | |||
| Infección de las vías respiratorias superiores | 50 | 4 | 1 |
| Neumonía | 15 | 8 | 2 |
| Trastornos respiratorios, torácicos y mediastínicos | |||
| Tos | 43 | 1 | 0 |
| Disnea | 33 | 6 | 1 |
| Congestión nasal | 16 | 0 | 0 |
| Trastornos gastrointestinales | |||
| Diarrea | 38 | 3 | 0 |
| Estreñimiento | 33 | 0 | 0 |
| Náuseas | 30 | 0 | 0 |
| Vómitos | 21 | 2 | 0 |
| Trastornos musculoesqueléticos y del tejido conjuntivo | |||
| Espasmos musculares | 26 | 1 | 0 |
| Dolor de espalda | 25 | 6 | 0 |
| Artralgia | 22 | 2 | 0 |
| Dolor en extremidades | 15 | 0 | 0 |
| Dolor óseo | 13 | 4 | 0 |
| Dolor torácico musculoesquelético | 13 | 2 | 0 |
| Trastornos psiquiátricos | |||
| Insomnio | 23 | 2 | 0 |
| Ansiedad | 13 | 0 | 0 |
| Trastornos del sistema nervioso | |||
| Mareos | 21 | 2 | 0 |
| Temblor | 19 | 3 | 0 |
| Cefalea | 17 | 0 | 0 |
| Trastornos del metabolismo y la nutrición | |||
| Hipokalemia | 16 | 3 | 0 |
| Hiperglucemia | 13 | 5 | 1 |
| Disminución del apetito | 11 | 0 | 0 |
Las anormalidades de laboratorio que empeoran durante el tratamiento se enumeran en la Tabla 20.
| DPd (N=103) |
|||
|---|---|---|---|
| Todos los grados (%) |
Grado 3 (%) |
Grado 4 (%) |
|
| Clave: D=daratumumab, Pd=pomalidomida-dexametasona. | |||
| Neutropenia | 95 | 36 | 46 |
| Linfopenia | 94 | 45 | 26 |
| Trombocitopenia | 75 | 10 | 10 |
| Anemia | 57 | 30 | 0 |
Monoterapia
La seguridad de DARZALEX se evaluó en 156 pacientes adultos con mieloma múltiple recidivante y refractario en tres ensayos clínicos abiertos. Los pacientes recibieron DARZALEX 16 mg/kg. La duración media de la exposición fue de 3,3 meses (rango: 0,03 a 20,04 meses).
Se notificaron reacciones adversas graves en 51 (33%) pacientes. Las reacciones adversas graves más frecuentes fueron neumonía (6%), deterioro general del estado de salud físico (3%) y pirexia (3%).
Las reacciones adversas provocaron un retraso en el tratamiento en 24 (15%) pacientes, con mayor frecuencia por infecciones. Las reacciones adversas provocaron la interrupción del tratamiento en 6 (4%) pacientes.
Las reacciones adversas que se produjeron en al menos el 10% de los pacientes se presentan en la Tabla 21. La Tabla 22 describe las anormalidades de laboratorio de grado 3-4 notificadas a una tasa de ≥10%.
| Reacción adversa | DARZALEX (N=156) |
||
|---|---|---|---|
| Todos los grados (%) |
Grado 3 (%) |
Grado 4 (%) |
|
| Trastornos generales y condiciones en el sitio de administración | |||
| Reacción relacionada con la infusión | 48 | 3 | 0 |
| Fatiga | 39 | 2 | 0 |
| Pirexia | 21 | 1 | 0 |
| Escalofríos | 10 | 0 | 0 |
| Trastornos gastrointestinales | |||
| Náuseas | 27 | 0 | 0 |
| Diarrea | 16 | 1 | 0 |
| Estreñimiento | 15 | 0 | 0 |
| Vómitos | 14 | 0 | 0 |
| Trastornos musculoesqueléticos y del tejido conjuntivo | |||
| Dolor de espalda | 23 | 2 | 0 |
| Artralgia | 17 | 0 | 0 |
| Dolor en extremidades | 15 | 1 | 0 |
| Dolor torácico musculoesquelético | 12 | 1 | 0 |
| Trastornos respiratorios, torácicos y mediastínicos | |||
| Tos | 21 | 0 | 0 |
| Congestión nasal | 17 | 0 | 0 |
| Disnea | 15 | 1 | 0 |
| Infecciones | |||
| Infección de las vías respiratorias superiores | 20 | 1 | 0 |
| Nasofaringitis | 15 | 0 | 0 |
| Neumonía | 11 | 6 | 0 |
| Trastornos del metabolismo y de la nutrición | |||
| Disminución del apetito | 15 | 1 | 0 |
| Trastornos del sistema nervioso | |||
| Cefalea | 12 | 1 | 0 |
| Trastornos vasculares | |||
| Hipertensión | 10 | 5 | 0 |
| Daratumumab 16 mg/kg (N=156) | |||
|---|---|---|---|
| Todos los grados (%) |
Grado 3 (%) |
Grado 4 (%) |
|
| Linfopenia | 72 | 30 | 10 |
| Neutropenia | 60 | 17 | 3 |
| Trombocitopenia | 48 | 10 | 8 |
| Anemia | 45 | 19 | 0 |
Reactivación del virus del herpes zóster
Se recomendó profilaxis para la reactivación del virus del herpes zóster para pacientes en algunos ensayos clínicos de DARZALEX. En estudios de monoterapia, se notificó herpes zóster en el 3% de los pacientes. En los estudios de terapia combinada, se notificó herpes zóster en el 2-5% de los pacientes que recibieron DARZALEX.
Infecciones
Las infecciones de grado 3 o 4 se notificaron de la siguiente manera:
- Estudios en pacientes con enfermedad recidivante/refractaria: DVd: 21% vs. Vd: 19%; DRd: 28% vs. Rd: 23%; DPd: 28%; DKd
1: 37%, Kd
1: 29%; DKd
2: 21% - Estudios en pacientes de nuevo diagnóstico: D-VMP: 23%, VMP: 15%; DRd: 32%, Rd: 23%; DVTd: 22%; VTd: 20%.
La neumonía fue la infección grave (grado 3 o 4) más frecuentemente notificada en todos los estudios. En los estudios controlados activos, las interrupciones del tratamiento debido a infecciones se produjeron en el 1-4% de los pacientes.
Las infecciones mortales (grado 5) se notificaron de la siguiente manera:
- Estudios en pacientes con enfermedad recidivante/refractaria: DVd: 1%, Vd: 2%; DRd: 2%, Rd: 1%; DPd: 2%; DKd
1: 5%, Kd
1: 3%; DKd
2: 0% - Estudios en pacientes de nuevo diagnóstico: D-VMP: 1%, VMP: 1%; DRd: 2%, Rd: 2%; DVTd: 0%, VTd: 0%.
Las infecciones mortales fueron generalmente poco frecuentes y equilibradas entre los regímenes que contenían DARZALEX y los brazos de control activo. Las infecciones mortales se debieron principalmente a neumonía y sepsis.
Reactivación del virus de la hepatitis B (VHB)
Se ha notificado reactivación del virus de la hepatitis B en menos del 1% de los pacientes (incluidos casos mortales) tratados con DARZALEX en ensayos clínicos.
Otra experiencia en ensayos clínicos
Se han notificado las siguientes reacciones adversas tras la administración de daratumumab e hialuronidasa para inyección subcutánea:
Trastornos del sistema nervioso: Síncope
6.2 Experiencia postcomercialización
Las siguientes reacciones adversas se han identificado durante el uso posterior a la aprobación de daratumumab. Debido a que estas reacciones se notifican voluntariamente a partir de una población de tamaño incierto, no siempre es posible estimar de forma fiable su frecuencia o establecer una relación causal con la exposición al fármaco.
Trastornos del sistema inmunitario: Reacción anafiláctica, IRR (incluidas muertes)
Trastornos gastrointestinales: Pancreatitis
Infecciones: Citomegalovirus, Listeriosis
7 INTERACCIONES MEDICAMENTOSAS
7.1 Efectos de Daratumumab en las Pruebas de Laboratorio
Interferencia con las pruebas de antiglobulina indirecta (prueba de Coombs indirecta)
Daratumumab se une a CD38 en los glóbulos rojos y interfiere con las pruebas de compatibilidad, incluyendo la detección de anticuerpos y la prueba cruzada. Los métodos de mitigación de la interferencia de Daratumumab incluyen el tratamiento de los glóbulos rojos reactivos con ditiotreitol (DTT) para interrumpir la unión de daratumumab
[ver
o genotipado. Dado que el sistema del grupo sanguíneo Kell también es sensible al tratamiento con DTT, suministre unidades K-negativas después de descartar o identificar aloanticuerpos utilizando glóbulos rojos tratados con DTT.
Si se requiere una transfusión de emergencia, administre glóbulos rojos compatibles con ABO/RhD no cruzados según las prácticas del banco de sangre local.
Interferencia con la electroforesis de proteínas séricas y las pruebas de inmunofijación
Daratumumab puede detectarse en la electroforesis de proteínas séricas (SPE) y los ensayos de inmunofijación (IFE) utilizados para el control de las inmunoglobulinas monoclonales de la enfermedad (proteína M). Pueden producirse resultados falsos positivos en los ensayos SPE e IFE para pacientes con proteína de mieloma IgG kappa, lo que afecta a la evaluación inicial de las respuestas completas según los criterios del Grupo de Trabajo Internacional del Mieloma (IMWG). En pacientes con respuesta parcial muy buena persistente, donde se sospecha interferencia de daratumumab, considere el uso de un ensayo IFE específico de daratumumab aprobado por la FDA para distinguir daratumumab de cualquier proteína M endógena restante en el suero del paciente, para facilitar la determinación de una respuesta completa.
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de riesgos
DARZALEX puede causar daño fetal cuando se administra a una mujer embarazada. La evaluación de los riesgos asociados con los productos de daratumumab se basa en el mecanismo de acción y los datos de modelos animales con eliminación del antígeno diana CD38
(ver
. No hay datos disponibles sobre el uso de DARZALEX en mujeres embarazadas para evaluar el riesgo asociado al fármaco de defectos congénitos importantes, aborto espontáneo o resultados adversos maternos o fetales. No se han realizado estudios de reproducción animal.
Se desconoce el riesgo de fondo estimado de defectos congénitos importantes y aborto espontáneo para la población indicada. Todos los embarazos tienen un riesgo de fondo de defectos congénitos, pérdida u otros resultados adversos. En la población general de EE. UU., el riesgo de fondo estimado de defectos congénitos importantes y aborto espontáneo en embarazos clínicamente reconocidos es del 2% al 4% y del 15% al 20%, respectivamente.
La combinación de DARZALEX y lenalidomida, pomalidomida o talidomida está contraindicada en mujeres embarazadas, porque la lenalidomida, la pomalidomida y la talidomida pueden causar defectos congénitos y la muerte del feto. La lenalidomida, la pomalidomida y la talidomida solo están disponibles a través de un programa REMS. Consulte la información de prescripción de lenalidomida, pomalidomida o talidomida sobre el uso durante el embarazo.
Consideraciones clínicas
Reacciones adversas fetales/neonatales
Los anticuerpos monoclonales de inmunoglobulina G1 (IgG1) se transfieren a través de la placenta. En función de su mecanismo de acción, DARZALEX puede causar la depleción de células inmunitarias fetales CD38 positivas y la disminución de la densidad ósea. Aplazar la administración de vacunas vivas a recién nacidos y lactantes expuestos a DARZALEX
in utero hasta que se complete una evaluación hematológica.
Datos
Datos en animales
Los ratones que fueron modificados genéticamente para eliminar toda la expresión de CD38 (ratones con eliminación de CD38) tuvieron una densidad ósea reducida al nacer que se recuperó a los 5 meses de edad. Los datos de estudios que utilizan modelos animales con eliminación de CD38 también sugieren la participación de CD38 en la regulación de las respuestas inmunitarias humorales (ratones), la tolerancia inmunitaria fetomaterna (ratones) y el desarrollo embrionario temprano (ranas).
8.2 Lactancia
Resumen de riesgos
No hay datos sobre la presencia de daratumumab en la leche humana, los efectos en el lactante o los efectos sobre la producción de leche. Se sabe que la inmunoglobulina G materna está presente en la leche humana. Los datos publicados sugieren que los anticuerpos en la leche materna no entran en la circulación neonatal y infantil en cantidades sustanciales. Debido al potencial de reacciones adversas graves en el lactante cuando se administra DARZALEX con lenalidomida, pomalidomida o talidomida, se aconseja a las mujeres que no amamanten durante el tratamiento con DARZALEX. Consulte la información de prescripción de lenalidomida, pomalidomida o talidomida para obtener información adicional.
8.3 Mujeres y hombres con potencial reproductivo
DARZALEX puede causar daño fetal cuando se administra a una mujer embarazada
[ver
Uso en poblaciones específicas (8.1)]
.
Prueba de embarazo
Con la combinación de DARZALEX con lenalidomida, pomalidomida o talidomida, consulte el etiquetado de lenalidomida, pomalidomida o talidomida para conocer los requisitos de las pruebas de embarazo antes de iniciar el tratamiento en mujeres con potencial reproductivo.
Anticoncepción
Aconseje a las mujeres con potencial reproductivo que utilicen métodos anticonceptivos eficaces durante el tratamiento con DARZALEX y durante 3 meses después de la última dosis. Además, consulte el etiquetado de lenalidomida, pomalidomida o talidomida para obtener recomendaciones adicionales sobre anticoncepción.
8.4 Uso pediátrico
No se ha establecido la seguridad y eficacia de DARZALEX en pacientes pediátricos.
La seguridad y la eficacia de DARZALEX en combinación con quimioterapia se evaluaron, pero no se establecieron, en un único ensayo abierto (DELPHINUS; NCT03384654) en 34 pacientes pediátricos (de 2 a <17 años de edad) con leucemia linfoblástica aguda o linfoma linfoblástico recidivantes/refractarios. No se observaron nuevas señales de seguridad en estos pacientes pediátricos. Los parámetros farmacocinéticos en estos pacientes pediátricos estuvieron dentro del rango de valores observados previamente en adultos con mieloma múltiple que recibieron la misma dosis según el peso corporal.
8.5 Uso geriátrico
De los 2459 pacientes que recibieron DARZALEX a la dosis recomendada, el 38% tenían entre 65 y 74 años, y el 15% tenían 75 años o más. No se observaron diferencias generales en la eficacia entre estos pacientes y los pacientes más jóvenes. La incidencia de reacciones adversas graves fue mayor en los pacientes mayores que en los más jóvenes
[ver
. Entre los pacientes con mieloma múltiple recidivante y refractario (n=1213), las reacciones adversas graves que ocurrieron con mayor frecuencia en pacientes de 65 años o mayores fueron neumonía y sepsis. Dentro del grupo DKd en CANDOR, las reacciones adversas mortales ocurrieron en el 14% de los pacientes de 65 años o mayores en comparación con el 6% de los pacientes menores de 65 años. Entre los pacientes con mieloma múltiple recién diagnosticado que no son elegibles para el trasplante autólogo de células madre (n=710), la reacción adversa grave que ocurrió con mayor frecuencia en pacientes de 75 años o mayores fue la neumonía.
11 DESCRIPCIÓN
Daratumumab es un anticuerpo monoclonal humano IgG1κ que se une al antígeno CD38. Se produce en células de ovario de hámster chino (CHO) utilizando tecnología de ADN recombinante. El peso molecular de daratumumab es aproximadamente 148 kDa.
DARZALEX
®(daratumumab) inyección se suministra como una solución incolora a amarillo pálido sin conservantes para uso intravenoso en un vial de dosis única. El pH es 5.5.
Cada vial de dosis única de 20 mL de DARZALEX contiene (NDC 57894-502-20) 400 mg de daratumumab, ácido acético glacial (3.7 mg), manitol (510 mg), polisorbato 20 (8 mg), trihidrato de acetato de sodio (59.3 mg), cloruro de sodio (70.1 mg) y Agua para inyección, USP.
Cada vial de dosis única de 5 mL de DARZALEX contiene (NDC 57894-502-05) 100 mg de daratumumab, ácido acético glacial (0.9 mg), manitol (127.5 mg), polisorbato 20 (2 mg), trihidrato de acetato de sodio (14.8 mg), cloruro de sodio (17.5 mg) y Agua para inyección, USP.
Cada vial de dosis única de 20 mL de DARZALEX contiene (NDC 57894-505-20) 400 mg de daratumumab, L-histidina (7 mg), monohidrato de clorhidrato de L-histidina (32.6 mg), L-metionina (20 mg), polisorbato 20 (8 mg), sorbitol (1093 mg) y Agua para inyección, USP.
Cada vial de dosis única de 5 mL de DARZALEX contiene (NDC 57894-505-05) 100 mg de daratumumab, L-histidina (1.8 mg), monohidrato de clorhidrato de L-histidina (8.2 mg), L-metionina (5 mg), polisorbato 20 (2 mg), sorbitol (273.3 mg) y Agua para inyección, USP.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
CD38 es una glicoproteína transmembrana (48 kDa) expresada en la superficie de células hematopoyéticas, incluyendo el mieloma múltiple y otros tipos de células y tejidos, y tiene múltiples funciones, tales como adhesión mediada por receptores, señalización y modulación de la actividad de ciclasa e hidrolasa. Daratumumab es un anticuerpo monoclonal humano IgG1κ (mAb) que se une a CD38 e inhibe el crecimiento de células tumorales que expresan CD38 induciendo apoptosis directamente a través de la reticulación mediada por Fc, así como por lisis de células tumorales mediada por el sistema inmunitario a través de citotoxicidad celular dependiente del complemento (CDC), citotoxicidad celular dependiente de anticuerpos (ADCC) y fagocitosis celular dependiente de anticuerpos (ADCP). Un subconjunto de células supresoras derivadas de mieloides (CD38+MDSCs), células T reguladoras (CD38+T
regs) y células B (CD38+B
regs) se reducen con daratumumab.
12.2 Farmacodinamia
Las células NK expresan CD38 y son susceptibles a la lisis celular mediada por daratumumab. Se observaron disminuciones en los recuentos absolutos y los porcentajes de células NK totales (CD16+CD56+) y activadas (CD16+CD56
dim) NK en sangre periférica completa y médula ósea con el tratamiento con DARZALEX.
12.3 Farmacocinética
El área bajo la curva concentración-tiempo (AUC) de daratumumab aumenta más que proporcionalmente en un rango de dosis de 1 a 24 mg/kg (0,06 a 1,5 veces la dosis recomendada aprobada) como monoterapia o de 1 a 16 mg/kg (0,06 a 1 vez la dosis recomendada aprobada) como terapia combinada.
Después de la administración de la dosis recomendada aprobada de DARZALEX como monoterapia o en terapia combinada, la concentración máxima sérica media (C
max) fue aproximadamente 2,7 a 3 veces mayor al final de la administración semanal en comparación con la primera dosis. La concentración sérica media ± desviación estándar (DE) en el valle (C
min) al final de la administración semanal fue de 573 ± 332 µg/mL cuando DARZALEX se administró como monoterapia y de 502 ± 196 a 607 ± 231 µg/mL cuando DARZALEX se administró como terapia combinada. La administración fraccionada de la primera dosis dio como resultado un perfil farmacocinético diferente en el primer día en comparación con la administración única; sin embargo, se predijeron y observaron concentraciones similares de C
max y C
min después de la administración de la segunda dosis fraccionada en el Día 2 de la Semana 1.
Cuando DARZALEX se administró como monoterapia, se alcanzó el estado estacionario de daratumumab aproximadamente a los 5 meses del período de dosificación cada 4 semanas (en la infusión número 21
st). En estado estacionario, la relación de acumulación media ± DE de daratumumab para C
max fue de 1,6 ± 0,5.
Distribución
El volumen de distribución de daratumumab fue de 4,7 ± 1,3 L como monoterapia y de 4,4 ± 1,5 L como terapia combinada después de la administración de la dosis aprobada.
Eliminación
El aclaramiento de daratumumab disminuyó con el aumento de la dosis y con la administración múltiple. Se estimó que el aclaramiento lineal medio ± DE fue de 171,4 ± 95,3 mL/día y la semivida terminal media ± DE estimada asociada con el aclaramiento lineal fue de 18 ± 9 días después de la administración de la dosis recomendada aprobada de DARZALEX como monoterapia. La semivida terminal fue similar cuando DARZALEX se administró como terapia combinada.
Poblaciones específicas
No se observaron diferencias clínicamente significativas en la farmacocinética de daratumumab como monoterapia o como terapia combinada en función del sexo, la edad (31 a 93 años), la insuficiencia hepática leve [bilirrubina total de 1 a 1,5 veces el límite superior de la normalidad (LSN) o aminotransferasa aspártica (AST) >LSN] y moderada (bilirrubina total de 1,5 a 3 veces el LSN y cualquier AST), o insuficiencia renal [aclaramiento de creatinina (CLcr) de 15 a 89 mL/min]. Se desconoce el efecto de la insuficiencia hepática grave (bilirrubina total >3 veces el LSN y cualquier AST) en la farmacocinética de daratumumab.
12.6 Inmunogenicidad
La incidencia observada de anticuerpos anti-fármaco (ADA, incluyendo anticuerpos neutralizantes) depende en gran medida de la sensibilidad y especificidad del ensayo. Las diferencias en los métodos de ensayo impiden comparaciones significativas de la incidencia de ADA en los estudios descritos a continuación con la incidencia de ADA en otros estudios, incluidos los de daratumumab u otros productos de daratumumab.
Con un tratamiento medio con DARZALEX que osciló entre 3,3 y 48 meses en 10 ensayos clínicos de pacientes con mieloma múltiple tratados con DARZALEX como monoterapia o como terapias combinadas, la incidencia del desarrollo de anticuerpos anti-daratumumab fue del 0,6% (14/2179) y 12 pacientes dieron positivo para anticuerpos neutralizantes. Debido a la baja incidencia de anticuerpos anti-fármaco, se desconoce el efecto de estos anticuerpos en la farmacocinética, farmacodinamia, seguridad o eficacia de los productos de daratumumab.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogenicidad, Mutagénesis, Deterioro de la Fertilidad
No se han realizado estudios de carcinogenicidad o genotoxicidad con daratumumab. No se han realizado estudios en animales para evaluar los posibles efectos de daratumumab en la reproducción o el desarrollo, o para determinar los posibles efectos sobre la fertilidad en hombres o mujeres.
14 ESTUDIOS CLÍNICOS
14.1 Mieloma Múltiple Recién Diagnosticado
Tratamiento Combinado con Lenalidomida y Dexametasona en Pacientes No Elegibles para Trasplante Autólogo de Células Madre
MAIA (NCT02252172), un ensayo abierto, aleatorizado y con control activo, comparó el tratamiento con DARZALEX 16 mg/kg en combinación con lenalidomida y dosis bajas de dexametasona (DRd) con el tratamiento con lenalidomida y dosis bajas de dexametasona (Rd) en pacientes con mieloma múltiple recién diagnosticado no elegibles para trasplante autólogo de células madre. La lenalidomida (25 mg una vez al día por vía oral en los días 1 a 21 de ciclos repetidos de 28 días [4 semanas]) se administró con dosis bajas de dexametasona oral o intravenosa de 40 mg/semana (o una dosis reducida de 20 mg/semana para pacientes >75 años o con un índice de masa corporal [IMC] <18,5). En los días de infusión de DARZALEX, la dosis de dexametasona se administró como medicación previa a la infusión. El tratamiento se continuó en ambos brazos hasta la progresión de la enfermedad o toxicidad inaceptable.
Un total de 737 pacientes fueron aleatorizados: 368 al brazo DRd y 369 al brazo Rd. Las características demográficas y de la enfermedad basales fueron similares entre los dos grupos de tratamiento. La mediana de edad fue de 73 (rango: 45 a 90) años, con un 44% de los pacientes ≥75 años de edad. El cincuenta y dos por ciento (52%) de los pacientes eran hombres, el 92% blancos, el 4% negros o afroamericanos y el 1% asiáticos. El tres por ciento (3%) de los pacientes reportaron una etnia hispana o latina. Treinta y cuatro (34%) tenían una puntuación de rendimiento del Eastern Cooperative Oncology Group (ECOG) de 0, el 50% tenía una puntuación ECOG de 1 y el 17% tenía una puntuación ECOG de ≥2. El veintisiete por ciento tenía enfermedad en estadio I del Sistema Internacional de Estadificación (ISS), el 43% en estadio II del ISS y el 29% en estadio III del ISS. La eficacia se evaluó mediante la supervivencia libre de progresión (SLP) según los criterios del International Myeloma Working Group (IMWG).
MAIA demostró una mejora en la Supervivencia Libre de Progresión (SLP) en el brazo DRd en comparación con el brazo Rd; la mediana de la SLP no se había alcanzado en el brazo DRd y fue de 31,9 meses en el brazo Rd (hazard ratio [HR]=0,56; IC del 95%: 0,43, 0,73; p<0,0001), lo que representa una reducción del 44% en el riesgo de progresión de la enfermedad o muerte en pacientes tratados con DRd. Después de una mediana de seguimiento de 64 meses, la mediana de la SLP fue de 61,9 meses (IC del 95%: 54,8, NE) en el brazo DRd y de 34,4 meses (IC del 95%: 29,6, 39,2) en el brazo Rd.
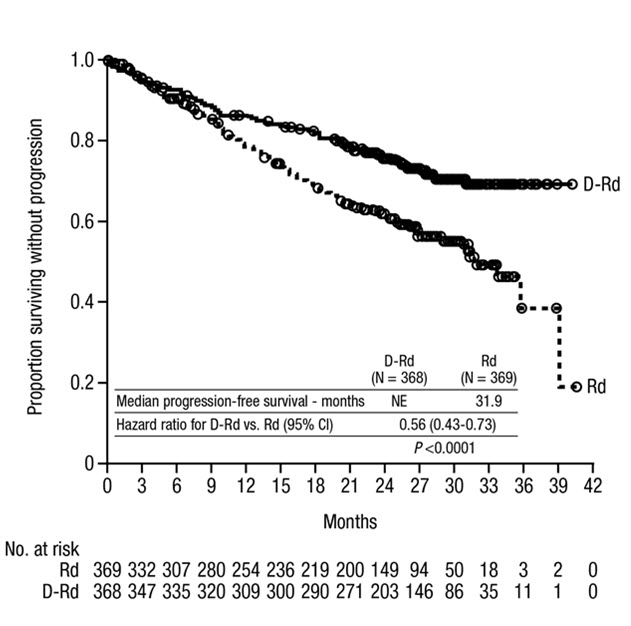
Después de una mediana de seguimiento de 56 meses, MAIA demostró una mejora en la supervivencia general (OS) en el brazo DRd en comparación con el brazo Rd (HR=0.68; IC del 95%: 0.53, 0.86; p=0.0013), lo que representa una reducción del 32% en el riesgo de muerte en pacientes tratados en el brazo DRd. La mediana de OS no se alcanzó para ninguno de los brazos.
Figura 2: Curva de Kaplan-Meier de OS en MAIA
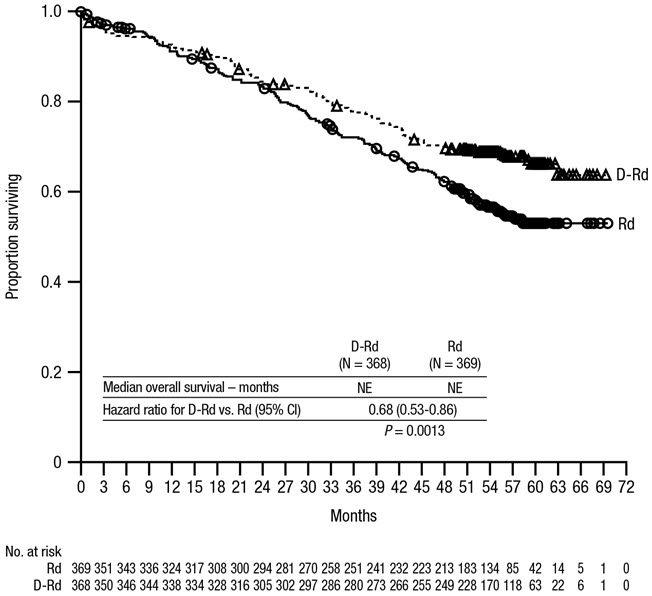
Los resultados adicionales de eficacia de MAIA se presentan en la Tabla 23.
| DRd (N=368) | Rd (N=369) | |
|---|---|---|
| DRd=daratumumab-lenalidomide-dexamethasone; Rd=lenalidomide-dexamethasone; MRD=minimal residual disease; CI=confidence interval | ||
| Respuesta general (sCR+CR+VGPR+PR) n(%) | 342 (92.9%) | 300 (81.3%) |
| p-value | <0.0001 | |
| Stringent complete response (sCR) | 112 (30.4%) | 46 (12.5%) |
| Complete response (CR) | 63 (17.1%) | 46 (12.5%) |
| Very good partial response (VGPR) | 117 (31.8%) | 104 (28.2%) |
| Partial response (PR) | 50 (13.6%) | 104 (28.2%) |
| CR or better (sCR + CR) | 175 (47.6%) | 92 (24.9%) |
| p-value | <0.0001 | |
| VGPR or better (sCR + CR + VGPR) | 292 (79.3%) | 196 (53.1%) |
| p-value | <0.0001 | |
| MRD negativity rate | 89 (24.2%) | 27 (7.3%) |
| 95% CI (%) | (19.9%, 28.9%) | (4.9%, 10.5%) |
| p-value | <0.0001 | |
| MRD negativity rate in patients with CR or better | ||
| Number of patients with CR or better | N=175 | N=92 |
| MRD negativity rate n(%) | 89 (50.9%) | 27 (29.3%) |
| 95% CI (%) | (43.2%, 58.5%) | (20.3%, 39.8%) |
En los pacientes que respondieron al tratamiento, la mediana del tiempo hasta la respuesta fue de 1.05 meses (rango: 0.2 a 12.1 meses) en el grupo DRd y de 1.05 meses (rango: 0.3 a 15.3 meses) en el grupo Rd. La mediana de la duración de la respuesta no se había alcanzado en el grupo DRd y fue de 34.7 meses (IC del 95%: 30.8, no estimable) en el grupo Rd.
Tratamiento de combinación con bortezomib, melfalán y prednisona (VMP) en pacientes no elegibles para trasplante autólogo de células madre
ALCYONE (NCT02195479), un ensayo abierto, aleatorizado y con control activo, comparó el tratamiento con DARZALEX 16 mg/kg en combinación con bortezomib, melfalán y prednisona (D-VMP) con el tratamiento con VMP en pacientes con mieloma múltiple recién diagnosticado que no eran elegibles para un trasplante autólogo de células madre. El bortezomib se administró mediante inyección subcutánea (SC) a una dosis de 1.3 mg/m
2 de superficie corporal dos veces por semana en las Semanas 1, 2, 4 y 5 durante el primer ciclo de 6 semanas (Ciclo 1; 8 dosis), seguido de administraciones una vez por semana en las Semanas 1, 2, 4 y 5 durante ocho ciclos más de 6 semanas (Ciclos 2 a 9; 4 dosis por ciclo). El melfalán a 9 mg/m
2 y la prednisona a 60 mg/m
2 se administraron por vía oral en los Días 1 a 4 de los nueve ciclos de 6 semanas (Ciclos 1 a 9). DARZALEX se continuó hasta la progresión de la enfermedad o una toxicidad inaceptable.
Se aleatorizó a un total de 706 pacientes: 350 al grupo D-VMP y 356 al grupo VMP. Las características demográficas y de la enfermedad iniciales fueron similares entre los dos grupos de tratamiento. La mediana de edad fue de 71 años (rango: 40 a 93), con un 30% de los pacientes ≥75 años de edad. La mayoría eran blancos (85%), mujeres (54%), el 25% tenía una puntuación de rendimiento ECOG de 0, el 50% tenía una puntuación de rendimiento ECOG de 1 y el 25% tenía una puntuación de rendimiento ECOG de 2. El diecinueve por ciento de los pacientes tenía enfermedad en Estadio I del ISS, el 42% en Estadio II del ISS y el 38% en Estadio III del ISS. La eficacia se evaluó mediante la PFS según los criterios del IMWG y la supervivencia global (OS).
ALCYONE demostró una mejora en la PFS en el grupo D-VMP en comparación con el grupo VMP (HR=0.50; IC del 95%: 0.38, 0.65; p<0.0001), lo que representa una reducción del 50% en el riesgo de progresión de la enfermedad o muerte en pacientes tratados con D-VMP. Después de una mediana de seguimiento de 40 meses, la mediana de la PFS fue de 36.4 meses (IC del 95%: 32.1, 45.9) en el grupo D-VMP y de 19.3 meses (IC del 95%: 18.0, 20.4) en el grupo VMP.
|
| Figura 3: Curva de Kaplan-Meier de la PFS en ALCYONE* |
|
|
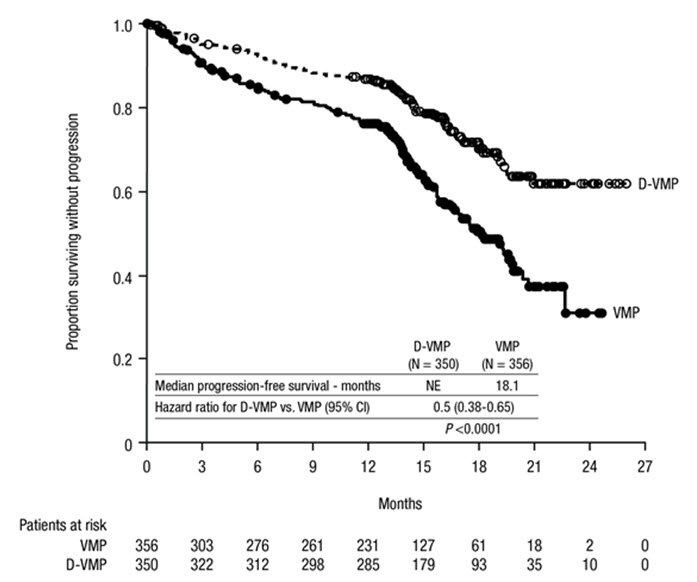
Después de una mediana de seguimiento de 40 meses, ALCYONE demostró una mejora en la supervivencia global (OS) en el grupo D-VMP en comparación con el grupo VMP (HR=0.60; IC del 95%: 0.46, 0.80; p=0.0003), lo que representa una reducción del 40% en el riesgo de muerte en los pacientes tratados en el grupo D-VMP. No se alcanzó la mediana de la OS para ninguno de los grupos.
Después de una mediana de seguimiento de 87 meses, la mediana de la OS fue de 83 meses (IC del 95%: 72.5, NE) en el grupo D-VMP y de 53.6 meses (IC del 95%: 46.3, 60.9) en el grupo VMP.
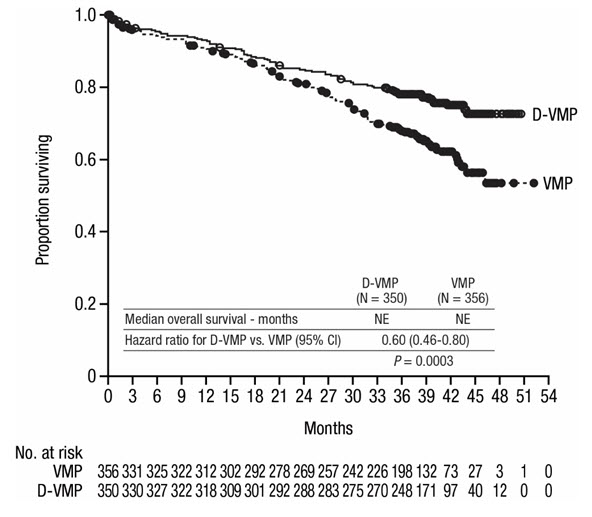
Resultados adicionales de eficacia de ALCYONE se presentan en la Tabla 24.
| D-VMP (N=350) | VMP (N=356) | |
|---|---|---|
| D-VMP = daratumumab-bortezomib-melphalan-prednisone; VMP = bortezomib-melphalan-prednisone; MRD = minimal residual disease; CI = confidence interval | ||
| Respuesta global (sCR+CR+VGPR+PR) n(%) * |
318 (90.9%) | 263 (73.9%) |
| Valor p † |
<0.0001 | |
| Respuesta completa estricta (sCR) | 63 (18.0%) | 25 (7.0%) |
| Respuesta completa (CR) | 86 (24.6%) | 62 (17.4%) |
| Respuesta parcial muy buena (VGPR) | 100 (28.6%) | 90 (25.3%) |
| Respuesta parcial (PR) | 69 (19.7%) | 86 (24.2%) |
| Tasa de negatividad de MRD *,‡n(%) |
78 (22.3%) | 22 (6.2%) |
| 95% CI (%) | (18.0, 27.0) | (3.9, 9.2) |
| Valor p § |
<0.0001 | |
| Tasa de negatividad de MRD en pacientes con CR o mejor ‡ |
||
| Número de pacientes con CR o mejor | N=149 | N=87 |
| Tasa de negatividad de MRD n(%) | 74 (49.7%) | 22 (25.3%) |
| 95% CI (%) | (41.4, 58.0) | (16.6, 35.7) |
En los pacientes respondedores, el tiempo mediano de respuesta fue de 0,79 meses (rango: 0,4 a 15,5 meses) en el grupo D-VMP y de 0,82 meses (rango: 0,7 a 12,6 meses) en el grupo VMP. La duración mediana de la respuesta no se había alcanzado en el grupo D-VMP y fue de 21,3 meses (rango: 0,5+, 23,7+) en el grupo VMP.
Tratamiento combinado con Bortezomib, Talidomida y Dexametasona en pacientes elegibles para trasplante autólogo de células madre (ASCT)
CASSIOPEIA (NCT02541383), un ensayo aleatorio, abierto, controlado activamente comparó el tratamiento de inducción y consolidación con DARZALEX 16 mg/kg en combinación con bortezomib, talidomida y dexametasona (DVTd) con el tratamiento con bortezomib, talidomida y dexametasona (VTd) en pacientes con mieloma múltiple de novo elegibles para ASCT. La fase de consolidación del tratamiento comenzó un mínimo de 30 días después del ASCT, cuando el paciente se había recuperado suficientemente y el injerto se había completado. El ensayo se limitó a pacientes de 65 años o menos. El bortezomib se administró por inyección subcutánea (SC) o intravenosa (IV) a una dosis de 1,3 mg/m
2de área de superficie corporal dos veces por semana durante dos semanas (Días 1, 4, 8 y 11) de ciclos de tratamiento de inducción de 28 días (4 semanas) repetidos (Ciclos 1–4) y dos ciclos de consolidación (Ciclos 5 y 6) después del ASCT después del Ciclo 4. La talidomida se administró por vía oral a 100 mg diarios durante los seis ciclos de bortezomib. La dexametasona (oral o intravenosa) se administró a 40 mg en los Días 1, 2, 8, 9, 15, 16, 22 y 23 de los Ciclos 1 y 2, y a 40 mg en los Días 1–2 y 20 mg en los días de dosificación posteriores (Días 8, 9, 15, 16) de los Ciclos 3–4. La dexametasona 20 mg se administró en los Días 1, 2, 8, 9, 15, 16 en los Ciclos 5 y 6. En los días de la infusión de DARZALEX, la dosis de dexametasona se administró por vía intravenosa como medicamento preinfusión.
Se aleatorizaron un total de 1.085 pacientes: 543 al brazo DVTd y 542 al brazo VTd. Las características demográficas y de la enfermedad en la línea de base fueron similares entre los dos grupos de tratamiento. La edad mediana fue de 58 años (rango: 22 a 65 años). La mayoría eran hombres (59%), el 48% tenía un puntaje de rendimiento ECOG de 0, el 42% tenía un puntaje de rendimiento ECOG de 1 y el 10% tenía un puntaje de rendimiento ECOG de 2. El 40% tenía la Etapa ISS I, el 45% tenía la Etapa ISS II y el 15% tenía la Etapa ISS III de la enfermedad.
La eficacia se evaluó mediante la tasa de respuesta completa estricta (sCR) en el día 100 postrasplante, la tasa de respuesta completa (CR) en el día 100 postrasplante y la supervivencia libre de progresión (PFS).
| DVTd (N = 543) | VTd (N = 542) | |
|---|---|---|
| D-VTd = daratumumab-bortezomib-talidomida-dexametasona; VTd = bortezomib-talidomida-dexametasona | ||
| Respuesta global (sCR + CR + VGPR + PR) n (%) | 503 (92,6%) | 487 (89,9%) |
| Respuesta completa estricta (sCR) | 157 (28,9%) | 110 (20,3%) |
| Valor p | 0,0010 | |
| Respuesta completa (CR) | 54 (9,9%) | 31 (5,7%) |
| Respuesta parcial muy buena (VGPR) | 242 (44,6%) | 282 (52,0%) |
| Respuesta parcial (PR) | 50 (9,2%) | 64 (11,8%) |
CASSIOPEIA demostró una mejora en la PFS en el brazo DVTd en comparación con el brazo VTd; con un seguimiento mediano de 18,8 meses, la PFS mediana no se había alcanzado en ninguno de los brazos. El tratamiento con DVTd resultó en una reducción del riesgo de progresión o muerte en un 53% en comparación con el VTd solo (HR = 0,47; IC 95%: 0,33, 0,67; p < 0,0001).
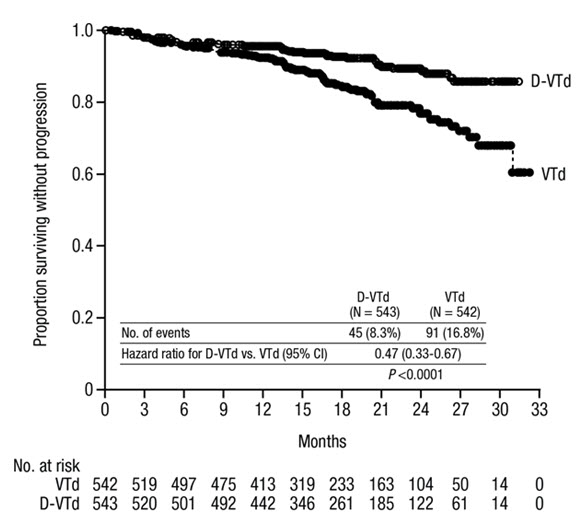
14.2 Mieloma Múltiple Recidivante/Resistente al Tratamiento
Tratamiento Combinado con Lenalidomida y Dexametasona
POLLUX (NCT02076009), un ensayo abierto, aleatorizado y con control activo, comparó el tratamiento con DARZALEX 16 mg/kg en combinación con lenalidomida y dosis bajas de dexametasona (DRd) con el tratamiento con lenalidomida y dosis bajas de dexametasona (Rd) en pacientes con mieloma múltiple que habían recibido al menos un tratamiento previo. La lenalidomida (25 mg una vez al día por vía oral en los días 1 a 21 de ciclos repetidos de 28 días [4 semanas]) se administró con dosis bajas de dexametasona oral o intravenosa de 40 mg/semana (o una dosis reducida de 20 mg/semana para pacientes >75 años o IMC <18.5). En los días de infusión de DARZALEX, se administraron 20 mg de la dosis de dexametasona como medicación previa a la infusión y el resto se administró el día después de la infusión. Para los pacientes con una dosis reducida de dexametasona, la dosis completa de 20 mg se administró como medicación previa a la infusión de DARZALEX. Los ajustes de dosis para lenalidomida y dexametasona se aplicaron de acuerdo con la información de prescripción del fabricante. El tratamiento se continuó en ambos brazos hasta la progresión de la enfermedad o toxicidad inaceptable.
Un total de 569 pacientes fueron aleatorizados; 286 al brazo DRd y 283 al brazo Rd. Las características demográficas y de la enfermedad basales fueron similares entre el brazo de DARZALEX y el brazo de control. La mediana de edad de los pacientes fue de 65 años (rango de 34 a 89 años), el 11% tenía ≥75 años, el 59% eran hombres; 69% blancos, 18% asiáticos y 3% afroamericanos. Los pacientes habían recibido una mediana de 1 línea de tratamiento previa. El sesenta y tres por ciento (63%) de los pacientes habían recibido un trasplante autólogo previo de células madre (ASCT). La mayoría de los pacientes (86%) recibieron un PI previo, el 55% de los pacientes habían recibido un agente inmunomodulador previo, incluido el 18% de los pacientes que habían recibido lenalidomida previamente; y el 44% de los pacientes habían recibido tanto un PI como un agente inmunomodulador previos. Al inicio del estudio, el 27% de los pacientes eran resistentes a la última línea de tratamiento. El dieciocho por ciento (18%) de los pacientes eran resistentes solo a un PI y el 21% eran resistentes a bortezomib. La eficacia se evaluó mediante la PFS según los criterios del IMWG.
POLLUX demostró una mejora en la PFS en el brazo DRd en comparación con el brazo Rd (HR=0,37; IC del 95%: 0,27, 0,52; p<0,0001), lo que representa una reducción del 63% en el riesgo de progresión de la enfermedad o muerte en pacientes tratados con DRd. Después de una mediana de seguimiento de 55 meses, la mediana de la PFS fue de 45,0 meses (IC del 95%: 34,1, 53,9) en el brazo DRd y fue de 17,5 meses (IC del 95%: 13,9, 20,8) en el brazo Rd.
|
| Figura 6: Curva de Kaplan-Meier de la PFS en POLLUX* |
|
|
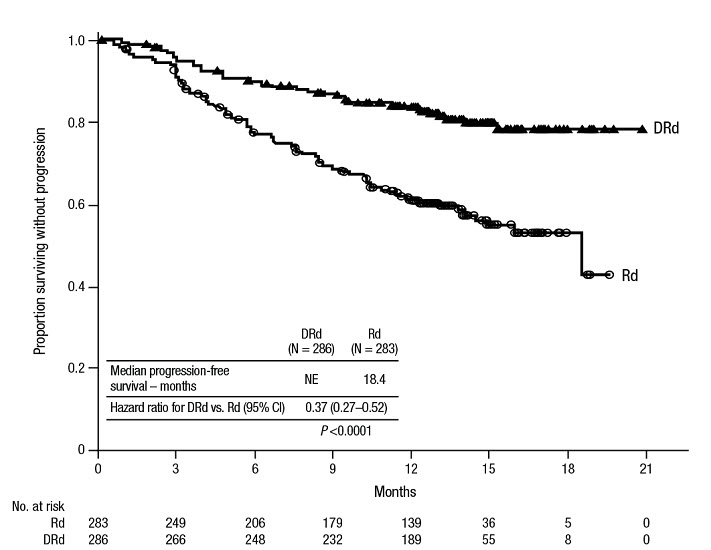
Después de una mediana de seguimiento de 80 meses, POLLUX demostró una mejora en la supervivencia general (SG) en el brazo DRd en comparación con el brazo Rd (HR=0,73; IC del 95%: 0,58, 0,91; p=0,0044), lo que representa una reducción del 27% en el riesgo de muerte en pacientes tratados en el brazo DRd. La mediana de la SG fue de 67,6 meses en el brazo DRd y de 51,8 meses en el brazo Rd.
Figura 7: Curva de Kaplan-Meier de la SG en POLLUX
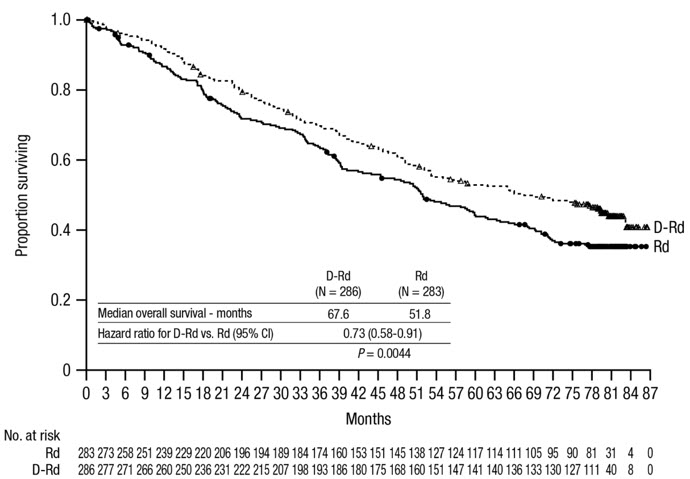
Los resultados de eficacia adicionales de POLLUX se presentan en la Tabla 26.
| DRd (N=286) | Rd (N=283) | |
|---|---|---|
| DRd = daratumumab- lenalidomida-dexametasona; Rd = lenalidomida-dexametasona | ||
| Respuesta general (sCR+CR+VGPR+PR) | 261 (91.3%) | 211 (74.6%) |
| Valor p | <0.0001 | |
| Respuesta completa estricta (sCR) | 51 (17.8%) | 20 (7.1%) |
| Respuesta completa (CR) | 70 (24.5%) | 33 (11.7%) |
| Respuesta parcial muy buena (VGPR) | 92 (32.2%) | 69 (24.4%) |
| Respuesta parcial (PR) | 48 (16.8%) | 89 (31.4%) |
En los respondedores, el tiempo mediano de respuesta fue de 1 mes (rango: 0,9 a 13 meses) en el grupo DRd y de 1,1 meses (rango: 0,9 a 10 meses) en el grupo Rd. La duración mediana de la respuesta no se había alcanzado en el grupo DRd (rango: 1+ a 19,8+ meses) y fue de 17,4 meses (rango: 1,4 a 18,5+ meses) en el grupo Rd.
Tratamiento de combinación con Bortezomib y Dexametasona
CASTOR (NCT02136134), un ensayo clínico de fase 3 abierto, aleatorizado y controlado con un tratamiento activo, comparó el tratamiento con DARZALEX 16 mg/kg en combinación con bortezomib y dexametasona (DVd), con el tratamiento con bortezomib y dexametasona (Vd) en pacientes con mieloma múltiple que habían recibido al menos un tratamiento previo. El bortezomib se administró por inyección SC o IV a una dosis de 1,3 mg/m
2 de superficie corporal dos veces por semana durante dos semanas (Días 1, 4, 8 y 11) de ciclos de tratamiento repetidos de 21 días (3 semanas), para un total de 8 ciclos. La dexametasona se administró por vía oral a una dosis de 20 mg los Días 1, 2, 4, 5, 8, 9, 11 y 12 de cada uno de los 8 ciclos de bortezomib (80 mg/semana durante dos de cada tres semanas del ciclo de bortezomib) o una dosis reducida de 20 mg/semana para pacientes > 75 años, IMC < 18,5, diabetes mellitus mal controlada o intolerancia previa a la terapia esteroidea. En los días de la infusión de DARZALEX, se administró una dosis de 20 mg de dexametasona como medicación previa a la infusión. Para pacientes con una dosis reducida de dexametasona, se administró la dosis completa de 20 mg como medicación previa a la infusión de DARZALEX. El bortezomib y la dexametasona se administraron durante 8 ciclos de tres semanas en ambos brazos de tratamiento; mientras que DARZALEX se administró hasta la progresión de la enfermedad. Sin embargo, la dexametasona 20 mg se continuó como medicación previa a la infusión de DARZALEX en el brazo DVd. Los ajustes de dosis de bortezomib y dexametasona se aplicaron de acuerdo con la información de prescripción del fabricante.
Se aleatorizaron un total de 498 pacientes; 251 al brazo DVd y 247 al brazo Vd. Las características demográficas y de la enfermedad en la línea de base fueron similares entre el brazo de DARZALEX y el brazo de control. La edad media de los pacientes fue de 64 años (rango 30 a 88 años); el 12% eran ≥ 75 años, el 57% eran hombres; el 87% eran blancos, el 5% asiáticos y el 4% afroamericanos. Los pacientes habían recibido una mediana de 2 líneas de tratamiento previo y el 61% de los pacientes habían recibido un trasplante autólogo de células madre (ASCT) previo. El sesenta y nueve por ciento (69%) de los pacientes habían recibido un PI previo (el 66% recibió bortezomib) y el 76% de los pacientes recibieron un agente inmunomodulador (el 42% recibió lenalidomida). En la línea de base, el 32% de los pacientes eran refractarios a la última línea de tratamiento y las proporciones de pacientes refractarios a cualquier tratamiento previo específico estaban en general bien equilibradas entre los grupos de tratamiento. El treinta y tres por ciento (33%) de los pacientes eran refractarios solo a un agente inmunomodulador, con el 24% de los pacientes en el brazo DVd y el 33% de los pacientes en el brazo Vd refractarios a lenalidomida, respectivamente. La eficacia se evaluó por PFS basada en los criterios IMWG.
CASTOR demostró una mejora en PFS en el brazo DVd en comparación con el brazo Vd (HR = 0,39; IC del 95%: 0,28, 0,53; p < 0,0001), lo que representa una reducción del 61% en el riesgo de progresión de la enfermedad o muerte para los pacientes tratados con DVd en comparación con Vd. Después de un seguimiento mediano de 50 meses, la PFS mediana fue de 16,7 meses (IC del 95%: 13,1, 19,4) en el brazo DVd y de 7,1 meses (IC del 95%: 6,2, 7,7) en el brazo Vd.
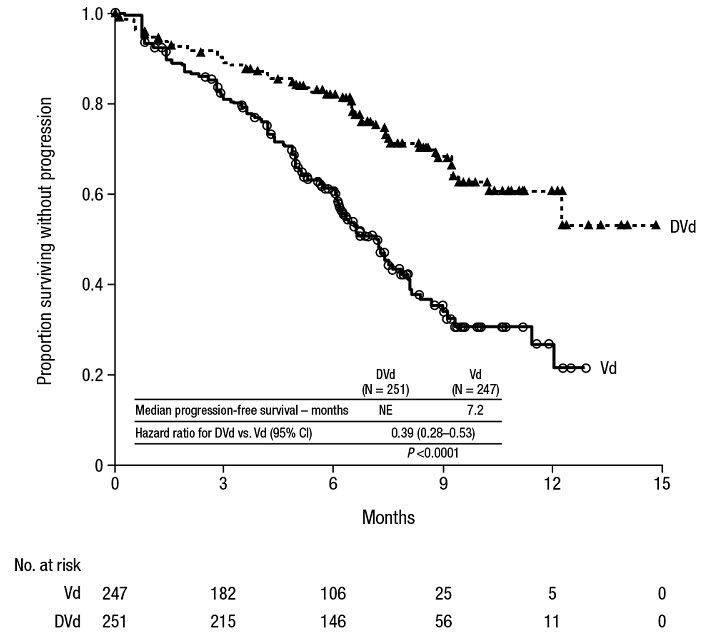
Después de una mediana de seguimiento de 73 meses, CASTOR demostró una mejora en la supervivencia general (SG) en el brazo DVd en comparación con el brazo Vd (HR=0,74; IC del 95%: 0,59, 0,92; p=0,0075), lo que representa una reducción del 26% en el riesgo de muerte en pacientes tratados en el brazo DVd. La mediana de la SG fue de 49,6 meses en el brazo DVd y de 38,5 meses en el brazo Vd.
Figura 9: Curva de Kaplan-Meier de la SG en CASTOR
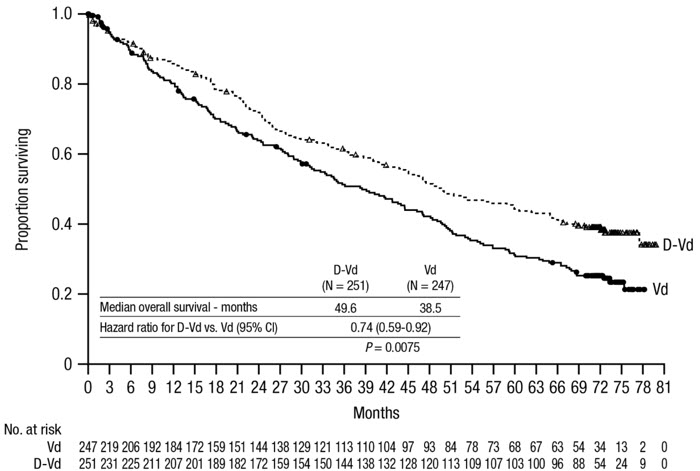
Los resultados adicionales de eficacia de CASTOR se presentan en la Tabla 27.
| DVd (N=251) | Vd (N=247) | |
|---|---|---|
| DVd = daratumumab-bortezomib-dexametasona; Vd = bortezomib-dexametasona | ||
| Respuesta general (sCR+CR+VGPR+PR) | 199 (79.3%) | 148 (59.9%) |
| Valor p † |
<0.0001 | |
| Respuesta completa estricta (sCR) | 11 (4.4%) | 5 (2.0%) |
| Respuesta completa (CR) | 35 (13.9%) | 16 (6.5%) |
| Respuesta parcial muy buena (VGPR) | 96 (38.2%) | 47 (19.0%) |
| Respuesta parcial (PR) | 57 (22.7%) | 80 (32.4%) |
En los pacientes que respondieron al tratamiento, la mediana del tiempo hasta la respuesta fue de 0.8 meses (rango: 0.7 a 4 meses) en el grupo DVd y de 1.5 meses (rango: 0.7 a 5 meses) en el grupo Vd. La mediana de la duración de la respuesta no se había alcanzado en el grupo DVd (rango: 1.4+ a 14.1+ meses) y fue de 7.9 meses (1.4+ a 12+ meses) en el grupo Vd.
Tratamiento combinado con carfilzomib dos veces por semana (20/56 mg/m
2) y dexametasona
CANDOR (NCT03158688) fue un ensayo aleatorizado, abierto, multicéntrico que evaluó la combinación de DARZALEX con carfilzomib dos veces por semana y dexametasona (DKd) versus carfilzomib dos veces por semana y dexametasona (Kd) en pacientes con mieloma múltiple recidivante o refractario que habían recibido al menos 1 a 3 líneas de tratamiento previas. Los pacientes que presentaban las siguientes condiciones fueron excluidos del ensayo: asma persistente moderada o grave conocida en los últimos 2 años, enfermedad pulmonar obstructiva crónica (EPOC) conocida con un FEV1 <50% del valor normal previsto e insuficiencia cardíaca congestiva activa. La aleatorización se estratificó según el ISS (estadio 1 o 2 vs estadio 3) en la selección, la exposición previa a inhibidores del proteasoma (sí vs no), el número de líneas de tratamiento previas (1 vs ≥2) o el tratamiento previo con anticuerpos contra el antígeno de diferenciación de cúmulos 38 (CD38) (sí vs no).
DARZALEX se administró por vía intravenosa a una dosis de 8 mg/kg en el Ciclo 1 los Días 1 y 2. Posteriormente, DARZALEX se administró por vía intravenosa a una dosis de 16 mg/kg los Días 8, 15 y 22 del Ciclo 1; los Días 1, 8, 15 y 22 del Ciclo 2; los Días 1 y 15 de los Ciclos 3 a 6; y el Día 1 de cada ciclo de 28 días hasta la progresión de la enfermedad. Carfilzomib se administró por vía intravenosa a una dosis de 20 mg/m
2en el Ciclo 1 los Días 1 y 2; a una dosis de 56 mg/m
2 en el Ciclo 1 los Días 8, 9, 15 y 16; y a una dosis de 56 mg/m
2 los Días 1, 2, 8, 9, 15 y 16 de cada ciclo de 28 días a partir de entonces. Se administraron 20 mg de dexametasona por vía oral o intravenosa los Días 1, 2, 8, 9, 15 y 16 y luego 40 mg por vía oral o intravenosa el Día 22 de cada ciclo de 28 días. Para los pacientes >75 años con una dosis reducida de dexametasona de 20 mg, la dosis completa de 20 mg se administró como medicación previa a la infusión de DARZALEX los días en que se administró DARZALEX. La dosificación de dexametasona se dividió en los días en que se administró carfilzomib en ambos brazos del estudio. El tratamiento se continuó en ambos brazos hasta la progresión de la enfermedad o una toxicidad inaceptable.
Se aleatorizó a un total de 466 pacientes; 312 al brazo DKd y 154 al brazo Kd. Las características demográficas y de la enfermedad basales fueron similares entre los brazos. La mediana de edad fue de 64 años (rango de 29 a 84 años), el 9% tenía ≥75 años, el 58% eran hombres; el 79% blancos, el 14% asiáticos y el 2% negros. Los pacientes habían recibido una mediana de 2 líneas de tratamiento previas y el 58% de los pacientes habían recibido un trasplante autólogo previo de células madre (ASCT). La mayoría de los pacientes (92%) recibieron un PI previo y, de ellos, el 34% eran refractarios al PI, incluido el régimen. El cuarenta y dos por ciento (42%) de los pacientes habían recibido lenalidomida previamente y, de ellos, el 33% eran refractarios a un régimen que contenía lenalidomida.
La eficacia se evaluó mediante la evaluación del IRC de la PFS según los criterios de respuesta del IMWG. Los resultados de eficacia se presentan en la Figura 10. CANDOR demostró una mejora en la PFS en el brazo DKd en comparación con el brazo Kd; la mediana de la PFS no se había alcanzado en el brazo DKd y fue de 15.8 meses en el brazo Kd (hazard ratio [HR]=0.63; IC del 95%: 0.46, 0.85; p=0.0014), lo que representa una reducción del 37% en el riesgo de progresión de la enfermedad o muerte para los pacientes tratados con DKd versus Kd.
Figura 10: Curva de Kaplan-Meier de la PFS en CANDOR
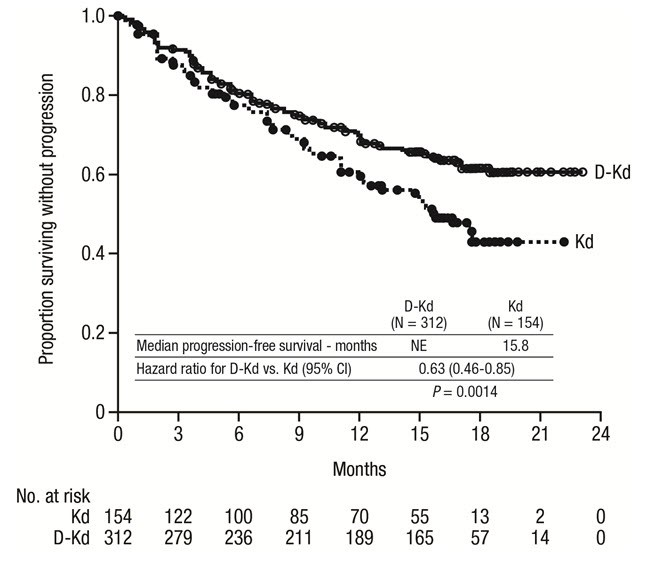
Los resultados adicionales de eficacia de CANDOR se presentan en la Tabla 28.
| DKd (N=312) | Kd (N=154) | |
|---|---|---|
| DKd = daratumumab-carfilzomib-dexametasona; Kd = carfilzomib-dexametasona; MRD [-] CR=enfermedad residual mínima; IC=intervalo de confianza | ||
| Respuesta general (sCR+CR+VGPR+PR) n(%) | 263 (84%) | 115 (75%) |
| IC del 95% (%) | (80, 88) | (67, 81) |
| valor p *(unilateral) |
0.0040 | |
| Respuesta completa (CR) | 89 (28%) | 16 (10%) |
| Respuesta parcial muy buena (VGPR) | 127 (41%) | 59 (38%) |
| Respuesta parcial (PR) | 47 (15%) | 40 (26%) |
| Tasa de MRD [-] CR a los 12 meses n(%) † |
39 (12%) | 2 (1.3%) |
| IC del 95% (%) | (9, 17) | (0.2, 4.6) |
| p-value *(1-sided) |
<0.0001 | |
| MRD [-] CR † |
43 (14%) | 5 (3.2%) |
La mediana del tiempo hasta la respuesta fue de 1 mes (rango: 1 a 14 meses) en el grupo DKd y de 1 mes (rango: 1 a 10 meses) en el grupo Kd. La mediana de la duración de la respuesta no se había alcanzado en el grupo DKd y fue de 16.6 meses (IC del 95%: 13.9, no estimable) en el grupo Kd.
Tratamiento combinado con carfilzomib una vez por semana (20/70 mg/m
2) y dexametasona
EQUULEUS (NCT01998971) fue un ensayo abierto, de múltiples cohortes, que evaluó la combinación de DARZALEX con carfilzomib una vez por semana y dexametasona en pacientes con mieloma múltiple recidivante o refractario que habían recibido al menos 1 a 3 líneas de tratamiento previas. Los pacientes que presentaban las siguientes características fueron excluidos del ensayo: asma persistente moderada o grave conocida en los últimos 2 años, enfermedad pulmonar obstructiva crónica (EPOC) conocida con un FEV1 <50% del valor normal previsto o insuficiencia cardíaca congestiva activa (definida como Clase III-IV de la New York Heart Association).
A diez pacientes se les administró DARZALEX a una dosis de 16 mg/kg por vía intravenosa en el Ciclo 1, Día 1 y a los pacientes restantes se les administró DARZALEX a una dosis de 8 mg/kg por vía intravenosa en el Ciclo 1, Días 1 y 2. Posteriormente, se administró DARZALEX por vía intravenosa a una dosis de 16 mg/kg en los Días 8, 15 y 22 del Ciclo 1; Días 1, 8, 15 y 22 del Ciclo 2; Días 1 y 15 de los Ciclos 3 a 6; y luego el Día 1 para los ciclos restantes de cada ciclo de 28 días. Carfilzomib se administró por vía intravenosa una vez por semana a una dosis de 20 mg/m
2 en el Día 1 del Ciclo 1 y se aumentó a una dosis de 70 mg/m
2 en los Días 8 y 15 del Ciclo 1, y los Días 1, 8 y 15 de cada ciclo posterior de 28 días. En los Ciclos 1 y 2, se administraron 20 mg de dexametasona por vía oral o intravenosa en los Días 1, 2, 8, 9, 15, 16, 22 y 23; en los ciclos 3 a 6, se administraron 20 mg de dexametasona por vía oral o intravenosa en los Días 1, 2, 15 y 16 y a una dosis de 40 mg en los Días 8 y 22; y en los ciclos 7 y posteriores, se administraron 20 mg de dexametasona por vía oral o intravenosa en los Días 1 y 2 y a una dosis de 40 mg en los Días 8, 15 y 22. Para los pacientes >75 años de edad, se administraron 20 mg de dexametasona por vía oral o intravenosa semanalmente después de la primera semana. El tratamiento continuó hasta la progresión de la enfermedad o una toxicidad inaceptable.
En el ensayo EQUULEUS se inscribieron 85 pacientes. La mediana de edad de los pacientes fue de 66 años (rango: 38 a 85 años), con un 9% de pacientes ≥75 años de edad; el 54% eran hombres; el 80% eran blancos, el 3.5% eran negros y el 3.5% eran asiáticos. Los pacientes del estudio habían recibido una mediana de 2 líneas de tratamiento previas. El setenta y tres por ciento (73%) de los pacientes habían recibido ASCT previo. Todos los pacientes recibieron bortezomib previamente y el 95% de los pacientes recibieron lenalidomida previamente. El cincuenta y nueve por ciento (59%) de los pacientes eran refractarios a lenalidomida y el 29% de los pacientes eran refractarios tanto a un PI como a un IMiD.
Los resultados de eficacia se basaron en la tasa de respuesta global utilizando los criterios del IMWG. Los resultados de eficacia se presentan en la Tabla 29. La mediana del tiempo hasta la respuesta fue de 0.95 meses (rango: 0.9, 14.3). La mediana de la duración de la respuesta fue de 28 meses (IC del 95%: 20.5, no estimable).
| N=85 | |
|---|---|
| ORR = sCR+CR+VGPR+PR IC = intervalo de confianza |
|
| Tasa de respuesta global (ORR) | 69 (81%) |
| 95% IC (%) | (71, 89) |
| Respuesta completa estricta (sCR) | 18 (21%) |
| Respuesta completa (CR) | 12 (14%) |
| Respuesta parcial muy buena (VGPR) | 28 (33%) |
| Respuesta parcial (PR) | 11 (13%) |
Tratamiento combinado con pomalidomida y dexametasona
EQUULEUS (NCT01998971) fue un ensayo abierto en el que 103 pacientes con mieloma múltiple que habían recibido previamente un IP y un agente inmunomodulador, recibieron 16 mg/kg de DARZALEX en combinación con pomalidomida y dexametasona a dosis bajas hasta la progresión de la enfermedad. La pomalidomida (4 mg una vez al día por vía oral en los días 1 a 21 de ciclos repetidos de 28 días [4 semanas]) se administró con una dosis baja de dexametasona oral o intravenosa de 40 mg/semana (dosis reducida de 20 mg/semana para pacientes >75 años o IMC <18,5). En los días de infusión de DARZALEX, se administraron 20 mg de la dosis de dexametasona como medicación previa a la infusión y el resto se administró el día después de la infusión. Para los pacientes con una dosis reducida de dexametasona, la dosis completa de 20 mg se administró como medicación previa a la infusión de DARZALEX.
La mediana de edad de los pacientes fue de 64 años (rango: 35 a 86 años), con un 8% de pacientes ≥75 años de edad. Los pacientes del estudio habían recibido una mediana de 4 líneas de tratamiento previas. El setenta y cuatro por ciento (74%) de los pacientes habían recibido previamente ASCT. El noventa y ocho por ciento (98%) de los pacientes recibieron tratamiento previo con bortezomib y el 33% de los pacientes recibieron carfilzomib previamente. Todos los pacientes recibieron tratamiento previo con lenalidomida, y el 98% de los pacientes fueron tratados previamente con la combinación de bortezomib y lenalidomida. El ochenta y nueve por ciento (89%) de los pacientes eran refractarios a la lenalidomida y el 71% refractarios al bortezomib; el 64% de los pacientes eran refractarios al bortezomib y la lenalidomida.
Los resultados de eficacia se basaron en la tasa de respuesta global determinada por el Comité de Revisión Independiente utilizando los criterios del IMWG (véase la
Tabla 30).
| N=103 | |
|---|---|
| ORR = sCR+CR+VGPR+PR IC = Intervalo de Confianza |
|
| Tasa de respuesta global (ORR) | 61 (59.2%) |
| 95% IC (%) | (49.1, 68.8) |
| Respuesta completa estricta (sCR) | 8 (7.8%) |
| Respuesta completa (CR) | 6 (5.8%) |
| Respuesta parcial muy buena (VGPR) | 29 (28.2%) |
| Respuesta parcial (PR) | 18 (17.5%) |
La mediana del tiempo hasta la respuesta fue de 1 mes (rango: 0,9 a 2,8 meses). La mediana de la duración de la respuesta fue de 13,6 meses (rango: 0,9+ a 14,6+ meses).
Monoterapia
SIRIUS (NCT01985126) fue un ensayo abierto que evaluó la monoterapia con DARZALEX en pacientes con mieloma múltiple recidivante o refractario que habían recibido al menos 3 líneas de tratamiento previas, incluyendo un inhibidor del proteasoma y un agente inmunomodulador, o que eran doblemente refractarios a un inhibidor del proteasoma y un agente inmunomodulador. En 106 pacientes, se administró DARZALEX 16 mg/kg con medicación previa y posterior a la infusión. El tratamiento continuó hasta que se produjo una toxicidad inaceptable o la progresión de la enfermedad.
La mediana de edad de los pacientes fue de 63,5 años (rango: 31 a 84 años), el 49% eran hombres y el 79% eran blancos. Los pacientes habían recibido una mediana de 5 líneas de tratamiento previas. El ochenta por ciento de los pacientes habían recibido previamente un trasplante autólogo de células madre (ASCT). Los tratamientos previos incluyeron bortezomib (99%), lenalidomida (99%), pomalidomida (63%) y carfilzomib (50%). Al inicio del estudio, el 97% de los pacientes eran refractarios a la última línea de tratamiento, el 95% eran refractarios tanto a un inhibidor del proteasoma (IP) como a un agente inmunomodulador, y el 77% eran refractarios a los agentes alquilantes.
Los resultados de eficacia se basaron en la tasa de respuesta global determinada por la evaluación del Comité de Revisión Independiente utilizando los criterios del IMWG (véase la
Tabla 31).
| N=106 | |
|---|---|
| ORR = sCR+CR+VGPR+PR IC = Intervalo de confianza |
|
| Tasa de respuesta global (ORR) | 31 (29.2%) |
| 95% IC (%) | (20.8, 38.9) |
| Respuesta completa estricta (sCR) | 3 (2.8%) |
| Respuesta completa (CR) | 0 |
| Respuesta parcial muy buena (VGPR) | 10 (9.4%) |
| Respuesta parcial (PR) | 18 (17.0%) |
La mediana del tiempo hasta la respuesta fue de 1 mes (rango: 0.9 a 5.6 meses). La mediana de la duración de la respuesta fue de 7.4 meses (rango: 1.2 a 13.1+ meses).
El estudio GEN501 (NCT00574288) fue un ensayo abierto de escalada de dosis que evaluó la monoterapia con DARZALEX en pacientes con mieloma múltiple recidivante o refractario que habían recibido al menos 2 terapias citorreductoras diferentes. En 42 pacientes, se administró DARZALEX 16 mg/kg con medicación previa y posterior a la infusión. El tratamiento continuó hasta que se produjo una toxicidad inaceptable o una progresión de la enfermedad.
La mediana de edad de los pacientes fue de 64 años (rango: 44 a 76 años), el 64% eran hombres y el 76% eran blancos. Los pacientes del estudio habían recibido una mediana de 4 líneas de tratamiento previas. El setenta y cuatro por ciento de los pacientes habían recibido ASCT previamente. Las terapias previas incluyeron bortezomib (100%), lenalidomida (95%), pomalidomida (36%) y carfilzomib (19%). Al inicio del estudio, el 76% de los pacientes eran refractarios a la última línea de tratamiento, el 64% de los pacientes eran refractarios tanto a un PI como a un agente inmunomodulador, y el 60% de los pacientes eran refractarios a los agentes alquilantes.
La tasa de respuesta global fue del 36% (IC del 95%: 21.6, 52.0%) con 1 RC y 3 RMVG. La mediana del tiempo hasta la respuesta fue de 1 mes (rango: 0.5 a 3.2 meses). La mediana de la duración de la respuesta no fue estimable (rango: 2.2 a 13.1+ meses).
15 REFERENCIAS
- Chapuy, CI, RT Nicholson, MD Aguad, et al., 2015, Resolving the daratumumab interference with blood compatibility testing, Transfusion, 55:1545–1554 (accessible at http://onlinelibrary.wiley.com/doi/10.1111/trf.13069/epdf).
16 SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
Cómo se Suministra
DARZALEX
®(daratumumab) inyección es una solución incolora a amarillo pálido, sin conservantes para infusión intravenosa.
NDC 57894-502-05 y NDC 57894-505-05 cada uno contiene un vial de dosis única de 100 mg/5 mL (20 mg/mL)
NDC 57894-502-20 y NDC 57894-505-20 cada uno contiene un vial de dosis única de 400 mg/20 mL (20 mg/mL)
Almacenamiento y Estabilidad
Conservar en nevera entre 2°C y 8°C (36°F y 46°F).
No congelar ni agitar. Proteger de la luz. Este producto no contiene conservantes.
17 INFORMACIÓN DE ASESORAMIENTO AL PACIENTE
Aconseje al paciente que lea el etiquetado del paciente aprobado por la FDA (Información para el paciente).
Reacciones relacionadas con la infusión
Aconseje a los pacientes que busquen atención médica inmediata ante cualquiera de los siguientes signos y síntomas de reacciones relacionadas con la infusión: picazón, goteo o obstrucción nasal; fiebre, escalofríos, náuseas, vómitos, irritación de garganta, tos, dolor de cabeza, mareos o aturdimiento, taquicardia, malestar torácico, sibilancias, dificultad para respirar o falta de aliento, picazón y visión borrosa
[ver
Advertencias y precauciones (5.1)]
.
Neutropenia
Aconseje a los pacientes que se pongan en contacto con su proveedor de atención médica si tienen fiebre
[ver
Advertencias y precauciones (5.3)]
.
Trombocitopenia
Aconseje a los pacientes que se pongan en contacto con su proveedor de atención médica si notan signos de hematomas o sangrado
[ver
Advertencias y precauciones (5.4)]
.
Interferencia con las pruebas de laboratorio
Aconseje a los pacientes que informen a sus proveedores de atención médica, incluido el personal de los centros de transfusión de sangre, que están tomando DARZALEX, en caso de una transfusión planificada
[ver
Advertencias y precauciones (5.2)]
.
Aconseje a los pacientes que DARZALEX puede afectar los resultados de algunas pruebas utilizadas para determinar la respuesta completa en algunos pacientes y que pueden ser necesarias pruebas adicionales para evaluar la respuesta
[ver
Advertencias y precauciones (5.5)]
.
Reactivación del virus de la hepatitis B (VHB)
Aconseje a los pacientes que informen a los proveedores de atención médica si alguna vez han tenido o podrían tener una infección por hepatitis B y que DARZALEX podría causar que el virus de la hepatitis B se vuelva activo nuevamente
[ver
.
Toxicidad embriofetal
Avise a las mujeres embarazadas del posible peligro para el feto. Aconseje a las mujeres en edad fértil que informen a su proveedor de atención médica sobre un embarazo conocido o sospechoso
[ver
Advertencias y precauciones (5.6),
Uso en poblaciones específicas (8.1,
8.3)]
.
Aconseje a las mujeres en edad fértil que eviten quedar embarazadas durante el tratamiento con DARZALEX y durante 3 meses después de la última dosis
[ver
Uso en poblaciones específicas (8.1,
8.3)]
.
Aconseje a los pacientes que la lenalidomida, la pomalidomida o la talidomida tienen el potencial de causar daño fetal y tienen requisitos específicos con respecto a la anticoncepción, las pruebas de embarazo, la donación de sangre y esperma, y la transmisión en el esperma. La lenalidomida, la pomalidomida y la talidomida solo están disponibles a través de un programa REMS
[ver
Uso en poblaciones específicas (8.1,
8.3)]
.
Intolerancia hereditaria a la fructosa (IHF)
DARZALEX contiene sorbitol. Avise a los pacientes con IHF de los riesgos relacionados con el sorbitol
[ver
.
SECCIÓN NO CLASIFICADA DE SPL
Fabricado por:
Janssen Biotech, Inc.
Horsham, PA 19044, EE. UU.
Número de licencia de EE. UU. 1864
Para información de patentes: www.janssenpatents.com
© 2015–2021 Janssen Pharmaceutical Companies
PROSPECTO PARA EL PACIENTE
| Esta información para el paciente ha sido aprobada por la Administración de Alimentos y Medicamentos de los EE. UU. | Revisado: 11/2022 | ||
| INFORMACIÓN PARA EL PACIENTE DARZALEX ®(Dar’-zah-lex) (daratumumab) inyección, para uso intravenoso |
|||
| DARZALEX se puede usar con otros medicamentos llamados lenalidomida, talidomida o pomalidomida. También debe leer la Guía del medicamento que viene con lenalidomida, talidomida o pomalidomida si usa DARZALEX con estos medicamentos. |
|||
| ¿Qué es DARZALEX? DARZALEX es un medicamento recetado que se usa para tratar a adultos con mieloma múltiple:
No se sabe si DARZALEX es seguro y eficaz en niños. |
|||
No reciba DARZALEX:
|
|||
Antes de recibir DARZALEX, informe a su proveedor de atención médica sobre todas sus afecciones médicas, incluso si:
Informe a su proveedor de atención médica sobre todos los medicamentos que toma, incluidos los medicamentos recetados y de venta libre, las vitaminas y los suplementos herbales. |
|||
¿Cómo recibiré DARZALEX?
|
|||
| ¿Cuáles son los posibles efectos secundarios de DARZALEX? DARZALEX puede causar reacciones graves, incluyendo:
|
|||
|
|
|
|
Los efectos secundarios más comunes de DARZALEX incluyen: |
|||
|
|
|
|
| Estos no son todos los posibles efectos secundarios de DARZALEX. Llame a su médico para obtener asesoramiento médico sobre los efectos secundarios. Puede informar los efectos secundarios a la FDA al 1-800-FDA-1088. |
|||
| Información general sobre el uso seguro y eficaz de DARZALEX. Los medicamentos a veces se recetan para fines distintos de los que se enumeran en un prospecto de información para el paciente. Puede pedirle a su farmacéutico o proveedor de atención médica información sobre DARZALEX que esté escrita para profesionales de la salud. |
|||
| ¿Cuáles son los ingredientes de DARZALEX? Ingrediente activo: daratumumab Ingredientes inactivos: pueden incluir ácido acético glacial, L-histidina, monohidrato de clorhidrato de L-histidina, L-metionina, manitol, polisorbato 20, trihidrato de acetato de sodio, cloruro de sodio, sorbitol y agua para inyección. Fabricado por: Janssen Biotech, Inc., Horsham, PA 19044, EE. UU.; Número de licencia de EE. UU. 1864 Para obtener información sobre patentes: www.janssenpatents.com © 2015–2021 Janssen Pharmaceutical Companies Para obtener más información, llame al 1-800-526-7736 o visite www.DARZALEX.com. |
|||
PRINCIPAL PANEL DE PRESENTACIÓN – Vial de 100 mg/5 mL en caja
NDC 57894-502-05
DARZALEX
®
(daratumumab)
Inyección
100 mg/5 mL
(20 mg/mL)
Sólo para infusión intravenosa
Diluir antes de usar
Rx only
Vial de dosis única.
Deseche la porción no utilizada
janssen

PANEL PRINCIPAL DE EXHIBICIÓN – Cartón de vial de 100 mg/5 mL – 57894-505
NDC 57894-505-05
DARZALEX
®
(daratumumab)
Injection
100 mg/5 mL
(20 mg/mL)
Para Infusión Intravenosa Solamente
Diluir Antes de Usar
Rx solamente
Un vial de 5 mL
Vial de dosis única.
Deseche la porción no utilizada.
janssen