Fabricante de medicamentos: Kyowa Kirin, Inc. (Updated: 2024-11-01)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
CRYSVITA® (burosumab-twza) inyección, para uso subcutáneo
Aprobación inicial en EE. UU.: 2018
INDICACIONES Y USO
CRYSVITA es un anticuerpo bloqueador del factor de crecimiento de fibroblastos 23 (FGF23) indicado para:
- El tratamiento de la hipofosfatemia ligada al cromosoma X (XLH) en pacientes adultos y pediátricos de 6 meses de edad o mayores. (1.1)
- El tratamiento de la hipofosfatemia relacionada con FGF23 en la osteomalacia inducida por tumores (TIO) asociada con tumores mesenquimales fosfatúricos que no se pueden resecar curativamente o localizar en pacientes adultos y pediátricos de 2 años de edad o mayores. (1.2)
DOSIFICACIÓN Y ADMINISTRACIÓN
Solo para uso subcutáneo (2)
-
XLH pediátrico (6 meses y mayores):
- Para pacientes que pesan menos de 10 kg, el régimen de dosis inicial es de 1 mg/kg de peso corporal redondeado al mg más cercano, administrado cada dos semanas (2.2)
- Para pacientes que pesan 10 kg o más, el régimen de dosis inicial es de 0,8 mg/kg de peso corporal redondeado al 10 mg más cercano, administrado cada dos semanas. La dosis inicial mínima es de 10 mg hasta una dosis máxima de 90 mg. (2.2)
La dosis se puede aumentar hasta aproximadamente 2 mg/kg (máximo 90 mg), administrada cada dos semanas para alcanzar un fósforo sérico normal. (2.2)
- XLH en adultos: El régimen de dosis es de 1 mg/kg de peso corporal redondeado al 10 mg más cercano hasta una dosis máxima de 90 mg administrada cada cuatro semanas. (2.3)
- TIO pediátrico (2 años y mayores): La dosis inicial es de 0,4 mg/kg de peso corporal redondeado al 10 mg más cercano cada 2 semanas. La dosis se puede aumentar hasta 2 mg/kg sin exceder los 180 mg, administrada cada dos semanas. (2.4)
- TIO en adultos: La dosis inicial es de 0,5 mg/kg cada cuatro semanas. La dosis se puede aumentar hasta 2 mg/kg sin exceder los 180 mg, administrada cada dos semanas. (2.5)
FORMAS Y FUERZAS DE DOSIFICACIÓN
Inyección: 10 mg/mL, 20 mg/mL o 30 mg/mL en un vial de dosis única (3)
CONTRAINDICACIONES
ADVERTENCIAS Y PRECAUCIONES
- Hipersensibilidad: Suspenda CRYSVITA si se producen reacciones de hipersensibilidad graves e inicie el tratamiento médico adecuado. (5.1)
- Hiperfosfatemia y riesgo de nefrocalcinosis: Para los pacientes que ya están tomando CRYSVITA, puede ser necesaria la interrupción de la dosis y/o la reducción de la dosis en función de los niveles de fósforo sérico del paciente. (5.2, 6.1)
- Reacciones en el sitio de inyección: La administración de CRYSVITA puede provocar reacciones locales en el sitio de inyección. Suspenda CRYSVITA si se producen reacciones graves en el sitio de inyección y administre el tratamiento médico adecuado. (5.3, 6.1)
REACCIONES ADVERSAS
- Las reacciones adversas más comunes (≥25% en el grupo CRYSVITA y > Control Activo) en pacientes pediátricos con XLH son: pirexia, reacción en el sitio de inyección, tos, vómitos, dolor en las extremidades, dolor de cabeza, absceso dental, caries dental. (6.1)
- Las reacciones adversas más comunes (>5% y en al menos 2 pacientes más que el placebo) en pacientes adultos con XLH son: dolor de espalda, dolor de cabeza, infección dental, síndrome de piernas inquietas, disminución de la vitamina D, mareos, estreñimiento, espasmos musculares, aumento del fósforo en sangre. (6.1)
- Las reacciones adversas más comunes (>10%) en pacientes con TIO son: absceso dental, espasmos musculares, mareos, estreñimiento, reacción en el sitio de inyección, erupción cutánea y dolor de cabeza. (6.1)
Para informar sobre REACCIONES ADVERSAS SOSPECHOSAS, comuníquese con Kyowa Kirin, Inc. al 1-844-768-3544 o con la FDA al 1-800-FDA-1088 o www.fda.gov/medwatch.
Ver 17 para INFORMACIÓN PARA EL PACIENTE.
Revisado: 3/2023
Tabla de Contenido
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDO*
1 INDICACIONES Y USO
1.1 Hipo fosfatemia ligada al cromosoma X
1.2 Osteomalacia inducida por tumor
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Información importante sobre la dosificación y la administración
2.2 Pacientes pediátricos con hipo fosfatemia ligada al cromosoma X (de 6 meses a menos de 18 años de edad)
2.3 Pacientes adultos con hipo fosfatemia ligada al cromosoma X (de 18 años de edad o mayores)
2.4 Pacientes pediátricos con osteomalacia inducida por tumor (de 2 años a menos de 18 años de edad)
2.5 Pacientes adultos con osteomalacia inducida por tumor (de 18 años de edad o mayores)
2.6 Dosis omitida
2.7 Suplementación con 25-hidroxivitamina D
2.8 Consideraciones generales para la administración subcutánea
3 FORMAS Y FUERZAS DE DOSIFICACIÓN
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Hipersensibilidad
5.2 Hiper fosfatemia y riesgo de nefrocalcinosis
5.3 Reacciones en el sitio de inyección
6 REACCIONES ADVERSAS
6.1 Experiencia en ensayos clínicos
6.2 Inmunogenicidad
6.3 Experiencia postcomercialización
7 INTERACCIONES MEDICAMENTOSAS
7.1 Fosfato oral y análogos de vitamina D activa
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.4 Uso pediátrico
8.5 Uso geriátrico
8.6 Insuficiencia renal
10 SOBREDOSIS
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
12.2 Farmacodinamia
12.3 Farmacocinética
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, mutagénesis, deterioro de la fertilidad
13.2 Toxicología y/o farmacología animal
14 ESTUDIOS CLÍNICOS
14.1 Hipo fosfatemia ligada al cromosoma X pediátrica
14.2 Hipo fosfatemia ligada al cromosoma X en adultos
14.3 Osteomalacia inducida por tumor
16 CÓMO SE SUMINISTRA/ALMACENAMIENTO Y MANEJO
17 INFORMACIÓN PARA EL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información completa de prescripción no están enumeradas.
1 INDICACIONES Y USO
1.1 Hipo fosfatemia ligada al cromosoma X
CRYSVITA está indicado para el tratamiento de la hipo fosfatemia ligada al cromosoma X (XLH) en pacientes adultos y pediátricos de 6 meses de edad o mayores.
1.2 Osteomalacia inducida por tumor
CRYSVITA está indicado para el tratamiento de la hipo fosfatemia relacionada con FGF23 en la osteomalacia inducida por tumor (TIO) asociada con tumores mesenquimales fosfatúricos que no pueden ser resecados curativamente o localizados en pacientes adultos y pediátricos de 2 años de edad o mayores.
2 DOSIS Y ADMINISTRACIÓN
2.1 Información Importante sobre la Dosis y la Administración
Suspenda el fosfato oral y/o los análogos de vitamina D activa (por ejemplo, calcitriol, paricalcitol, doxercalciferol, calcifediol) 1 semana antes de iniciar el tratamiento [ver Contraindicaciones (4)].
La concentración de fósforo sérico en ayunas debe estar por debajo del rango de referencia para la edad antes de iniciar el tratamiento [ver Contraindicaciones (4)].
CRYSVITA se administra mediante inyección subcutánea y debe ser administrado por un profesional sanitario.
El volumen máximo de CRYSVITA por inyección es de 1,5 mL. Si se requieren múltiples inyecciones, administre en diferentes sitios de inyección.
2.2 Pacientes pediátricos con hipofosfatemia ligada al cromosoma X (de 6 meses a menos de 18 años de edad)
Para los pacientes que pesan menos de 10 kg, la dosis inicial recomendada es de 1 mg/kg de peso corporal, redondeada al mg más cercano, administrada cada dos semanas.
Para los pacientes que pesan 10 kg o más, el régimen de dosis inicial recomendado es de 0,8 mg/kg de peso corporal, redondeado al 10 mg más cercano, administrado cada dos semanas. La dosis inicial mínima es de 10 mg hasta una dosis máxima de 90 mg.
Después de iniciar el tratamiento con CRYSVITA, mida el fósforo sérico en ayunas cada 4 semanas durante los primeros 3 meses de tratamiento y, a partir de entonces, según sea necesario. Si el fósforo sérico está por encima del límite inferior del rango de referencia para la edad y por debajo de 5 mg/dL, continúe el tratamiento con la misma dosis. Siga el programa de ajuste de dosis que se muestra a continuación para mantener el fósforo sérico dentro del rango de referencia para la edad.
Ajuste de la dosis
Vuelva a evaluar el nivel de fósforo sérico en ayunas 4 semanas después del ajuste de la dosis.
No ajuste CRYSVITA con más frecuencia que cada 4 semanas.
Aumento de la dosis:
Para los pacientes que pesan menos de 10 kg, si el fósforo sérico está por debajo del rango de referencia para la edad, la dosis puede aumentarse a 1,5 mg/kg, redondeada al mg más cercano, administrada cada dos semanas. Si se necesitan aumentos de dosis adicionales, la dosis puede aumentarse a la dosis máxima de 2 mg/kg, redondeada al mg más cercano, administrada cada dos semanas.
Para los pacientes que pesan 10 kg o más, si el fósforo sérico está por debajo del rango de referencia para la edad, la dosis puede aumentarse gradualmente hasta aproximadamente 2 mg/kg, administrada cada dos semanas (dosis máxima de 90 mg) de acuerdo con el programa de dosificación que se muestra en la Tabla 1.
| Peso corporal (kg) | Dosis inicial (mg) | Primer aumento de la dosis a (mg) | Segundo aumento de la dosis a (mg) |
|---|---|---|---|
| 10 – 14 | 10 | 15 | 20 |
| 15 – 18 | 10 | 20 | 30 |
| 19 – 31 | 20 | 30 | 40 |
| 32 – 43 | 30 | 40 | 60 |
| 44 – 56 | 40 | 60 | 80 |
| 57 – 68 | 50 | 70 | 90 |
| 69 – 80 | 60 | 90 | 90 |
| 81 – 93 | 70 | 90 | 90 |
| 94 – 105 | 80 | 90 | 90 |
| 106 y más | 90 | 90 | 90 |
Disminución de la dosis:
Si el fósforo sérico está por encima de 5 mg/dL, suspenda la siguiente dosis y vuelva a evaluar el nivel de fósforo sérico en 4 semanas. El paciente debe tener un fósforo sérico por debajo del rango de referencia para la edad para reiniciar CRYSVITA. Una vez que el fósforo sérico esté por debajo del rango de referencia para la edad, el tratamiento puede reiniciarse.
Para los pacientes que pesan menos de 10 kg, reinicie CRYSVITA a 0,5 mg/kg de peso corporal, redondeado al 1 mg más cercano, administrado cada dos semanas. Para los pacientes que pesan 10 kg o más, reinicie CRYSVITA de acuerdo con el programa de dosificación que se muestra en la Tabla 2.
| Dosis previa (mg) | Dosis de reiniciación (mg) |
|---|---|
| 10 | 5 |
| 15 | 10 |
| 20 | 10 |
| 30 | 10 |
| 40 | 20 |
| 50 | 20 |
| 60 | 30 |
| 70 | 30 |
| 80 | 40 |
| 90 | 40 |
Después de una disminución de la dosis, vuelva a evaluar el nivel de fósforo sérico 4 semanas después del ajuste de la dosis. Si el nivel permanece por debajo del rango de referencia para la edad después de la dosis de reiniciación, la dosis puede ajustarse como se describe en Aumento de la dosis.
2.3 Pacientes adultos con hipofosfatemia ligada al cromosoma X (18 años de edad o mayores)
El régimen de dosificación recomendado en adultos es de 1 mg/kg de peso corporal, redondeado al 10 mg más cercano hasta una dosis máxima de 90 mg, administrado cada cuatro semanas.
Después de iniciar el tratamiento con CRYSVITA, evalúe el fósforo sérico en ayunas mensualmente, medido 2 semanas después de la dosis, durante los primeros 3 meses de tratamiento, y después según sea apropiado. Si el fósforo sérico está dentro del rango normal, continúe con la misma dosis.
Disminución de la dosis
Vuelva a evaluar el nivel de fósforo sérico en ayunas 2 semanas después del ajuste de la dosis.
No ajuste CRYSVITA con más frecuencia que cada 4 semanas.
Si el fósforo sérico está por encima del rango normal, suspenda la siguiente dosis y vuelva a evaluar el nivel de fósforo sérico después de 4 semanas. El paciente debe tener un fósforo sérico por debajo del rango normal para poder reiniciar CRYSVITA. Una vez que el fósforo sérico esté por debajo del rango normal, el tratamiento puede reiniciarse a aproximadamente la mitad de la dosis inicial de inicio hasta una dosis máxima de 40 mg cada 4 semanas de acuerdo con el programa de dosificación que se muestra en la Tabla 3. Vuelva a evaluar el fósforo sérico 2 semanas después de cualquier cambio en la dosis.
| Dosis previa (mg) | Dosis de reiniciación (mg) |
|---|---|
| 40 | 20 |
| 50 | 20 |
| 60 | 30 |
| 70 | 30 |
| 80 y mayores | 40 |
2.4 Pacientes pediátricos con osteomalacia inducida por tumor (de 2 años a menos de 18 años de edad)
La dosis inicial recomendada para pediatría es de 0,4 mg/kg de peso corporal administrada cada 2 semanas, redondeada al 10 mg más cercano, hasta una dosis máxima de 2 mg/kg, sin exceder los 180 mg, administrada cada 2 semanas.
Después de iniciar el tratamiento con CRYSVITA, evalúe el fósforo sérico en ayunas mensualmente, medido 2 semanas después de la dosis, durante los primeros 3 meses de tratamiento, y posteriormente según sea apropiado. Si el fósforo sérico está dentro del rango de referencia para la edad, continúe con la misma dosis. Siga el programa de ajuste de dosis a continuación para mantener el fósforo sérico dentro del rango de referencia para la edad.
Ajuste de dosis
Reevalúe el nivel de fósforo sérico en ayunas 4 semanas después del ajuste de la dosis.
No ajuste CRYSVITA con más frecuencia que cada 4 semanas.
Aumento de dosis
Si el fósforo sérico está por debajo del rango de referencia para la edad, la dosis debe ajustarse de acuerdo con la Tabla 4 hasta la dosis máxima de 2 mg/kg cada 2 semanas. La dosis máxima no debe exceder los 180 mg.
| Peso corporal (kg) |
Dosis inicial (mg) |
Primer aumento de dosis a (mg) |
Segundo aumento de dosis a (mg) |
Tercer aumento de dosis* a (mg) |
|---|---|---|---|---|
|
||||
| 10 – 14 | 5 | 10 | 15 | 20 |
| 15 – 18 | 5 | 10 | 20 | 25 |
| 19 – 31 | 10 | 20 | 25 | 30 |
| 32 – 43 | 10 | 30 | 40 | 50 |
| 44 – 56 | 20 | 40 | 50 | 70 |
| 57 – 68 | 20 | 50 | 70 | 90 |
| 69 – 80 | 30 | 60 | 80 | 100 |
| 81 – 93 | 30 | 70 | 100 | 120 |
| 94 – 105 | 40 | 80 | 110 | 140 |
| 106 y más | 40 | 90 | 130 | 160 |
2.5 Pacientes adultos con osteomalacia inducida por tumor (18 años de edad o mayores)
La dosis inicial recomendada para adultos es de 0.5 mg/kg de peso corporal administrada cada 4 semanas, redondeada al 10 mg más cercano, hasta una dosis máxima de 2 mg/kg, sin exceder los 180 mg, administrada cada 2 semanas.
Después de iniciar el tratamiento con CRYSVITA, evalúe el fósforo sérico en ayunas mensualmente, medido 2 semanas después de la dosis, durante los primeros 3 meses de tratamiento, y después según sea apropiado. Si el fósforo sérico está dentro del rango normal, continúe con la misma dosis. Siga el programa de ajuste de dosis a continuación para mantener el fósforo sérico dentro del rango de referencia.
Ajuste de dosis
Reevalúe el nivel de fósforo sérico en ayunas 2 semanas después del ajuste de la dosis.
No ajuste CRYSVITA con más frecuencia que cada 4 semanas.
Aumento de dosis
Si el fósforo sérico está por debajo del rango normal, la dosis debe ajustarse de acuerdo con la Tabla 5 hasta la dosis máxima de 2 mg/kg, sin exceder los 180 mg, administrada cada 2 semanas. Para aquellos individuos que no alcanzan un fósforo sérico mayor que el límite inferior del rango normal, los médicos pueden considerar dividir la dosis total administrada cada 4 semanas y administrarla cada 2 semanas.
| Dosis inicial | Primer aumento de dosis‡ | Segundo aumento de dosis‡ | Tercer aumento de dosis‡ | Cuarto aumento de dosis | Quinto aumento de dosis (dosis máxima) |
|
|---|---|---|---|---|---|---|
|
||||||
| Si el fósforo sérico 2 semanas después del ajuste de la dosis está por debajo del límite inferior de lo normal | 0.5 mg/kg cada 4 semanas | Aumentar a: 1 mg/kg cada 4 semanas O 0.5 mg/kg cada 2 semanas |
Aumentar a: 1.5 mg/kg cada 4 semanas§ O 0.75 mg/kg cada 2 semanas |
Aumentar a: 2 mg/kg cada 4 semanas§ O 1 mg/kg cada 2 semanas |
Aumentar a: 1.5 mg/kg, sin exceder los 180 mg cada 2 semanas |
Aumentar a: 2 mg/kg, sin exceder los 180 mg cada 2 semanas |
2.5 Ajustes de Dosis
Reducción de la Dosis
Si el fósforo sérico está por encima del rango normal, suspenda la siguiente dosis y vuelva a evaluar el nivel de fósforo sérico en 4 semanas. El paciente debe tener el fósforo sérico por debajo del rango de referencia para reiniciar CRYSVITA. Una vez que el fósforo sérico esté por debajo del rango de referencia, el tratamiento puede reiniciarse a aproximadamente la mitad de la dosis inicial, hasta una dosis máxima de 180 mg administrada cada 2 semanas para adultos. Después de una reducción de la dosis, vuelva a evaluar el nivel de fósforo sérico 2 semanas después del ajuste de la dosis. Si el nivel permanece por debajo del rango de referencia después de la dosis de reinicio, la dosis puede ajustarse según lo descrito en la Tabla 5.
Interrupción de la Dosis
Si un paciente se somete a tratamiento del tumor subyacente (es decir, escisión quirúrgica o radioterapia), el tratamiento con CRYSVITA debe interrumpirse y el fósforo sérico debe volver a evaluarse después de que se haya completado el tratamiento. La dosis de CRYSVITA debe reiniciarse a la dosis inicial del paciente si el fósforo sérico permanece por debajo del límite inferior de lo normal. Siga el ajuste de la dosis según la Tabla 5 para mantener el fósforo sérico dentro del rango de referencia.
2.6 Dosis Olvidada
Si un paciente olvida una dosis, reanude CRYSVITA lo antes posible a la dosis prescrita. Para evitar dosis olvidadas, los tratamientos pueden administrarse 3 días antes o después de la fecha de tratamiento programada.
2.7 Suplementación con 25-Hidroxivitamina D
Controle los niveles de 25-hidroxivitamina D. Suplemente con colecalciferol o ergocalciferol para mantener los niveles de 25-hidroxivitamina D en el rango normal para la edad. No administre análogos activos de vitamina D durante el tratamiento con CRYSVITA [ver Contraindicaciones (4)].
2.8 Consideraciones Generales para la Administración Subcutánea
Los sitios de inyección deben rotarse con cada inyección administrada en una ubicación anatómica diferente (parte superior de los brazos, parte superior de los muslos, glúteos o cualquier cuadrante del abdomen) que la inyección anterior. No inyecte en lunares, cicatrices o áreas donde la piel esté sensible, magullada, roja, dura o no intacta. Si una dosis determinada en un día de dosificación requiere múltiples viales de CRYSVITA, el contenido de dos viales puede combinarse para la inyección. El volumen máximo de CRYSVITA por inyección es de 1,5 mL. Si se requieren múltiples inyecciones en un día de dosificación determinado, administre en diferentes sitios de inyección. Controle los signos de reacciones [ver Advertencias y Precauciones (5.3)].
Inspeccione visualmente CRYSVITA para detectar partículas y decoloración antes de la administración. CRYSVITA es una solución estéril, sin conservantes, transparente a ligeramente opalescente e incolora a amarillo marrón pálido para inyección subcutánea. No lo use si la solución está decolorada o turbia o si la solución contiene partículas o materia particulada extraña.
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
Inyección: 10 mg/mL, 20 mg/mL o 30 mg/mL solución transparente a ligeramente opalescente e incolora a amarillo-marrón pálido en un vial de dosis única.
4 CONTRAINDICACIONES
CRYSVITA está contraindicado:
- En el uso concomitante con fosfato oral y/o análogos de vitamina D activa (por ejemplo, calcitriol, paricalcitol, doxercalciferol, calcifediol) debido al riesgo de hiperfosfatemia [ver Advertencias y precauciones (5.2) y Interacciones medicamentosas (7.1)].
- Cuando el fósforo sérico está dentro o por encima del rango normal para la edad [ver Advertencias y precauciones (5.2)].
- En pacientes con insuficiencia renal grave o enfermedad renal en etapa terminal porque estas afecciones están asociadas con un metabolismo mineral anormal [ver Uso en población específica (8.6)].
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Hipersensibilidad
Se han notificado reacciones de hipersensibilidad (p. ej., erupción cutánea, urticaria) en pacientes con CRYSVITA. Suspenda CRYSVITA si se producen reacciones de hipersensibilidad graves e inicie el tratamiento médico adecuado [ver Reacciones adversas (6.1)].
5.2 Hiperfosfatemia y riesgo de nefrocalcinosis
Los aumentos en el fósforo sérico por encima del límite superior de lo normal pueden estar asociados con un mayor riesgo de nefrocalcinosis. Para los pacientes que ya están tomando CRYSVITA, puede ser necesaria la interrupción de la dosis y/o la reducción de la dosis en función de los niveles de fósforo sérico del paciente. Los pacientes con osteomalacia inducida por tumor que se someten a tratamiento del tumor subyacente deben interrumpir y ajustar la dosificación para prevenir la hiperfosfatemia [ver Dosificación y administración (2) y Reacciones adversas (6.1)].
5.3 Reacciones en el lugar de la inyección
La administración de CRYSVITA puede provocar reacciones locales en el lugar de la inyección. Suspenda CRYSVITA si se producen reacciones graves en el lugar de la inyección y administre el tratamiento médico adecuado [ver Reacciones adversas (6.1)].
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas se describen a continuación y en otras partes del etiquetado:
- Hipersensibilidad [ver Advertencias y precauciones (5.1)]
- Hiperfosfatemia y riesgo de nefrocalcinosis [ver Advertencias y precauciones (5.2)]
- Reacciones en el sitio de inyección [ver Advertencias y precauciones (5.3)]
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se llevan a cabo en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no se pueden comparar directamente con las tasas en los ensayos clínicos de otro fármaco y pueden no reflejar las tasas observadas en la práctica.
Reacciones adversas en pacientes pediátricos con XLH
CRYSVITA se estudió en tres estudios pediátricos de XLH. El estudio 1 es un estudio de fase 3 aleatorizado, abierto en pacientes con XLH de 1 a 12 años de edad, que fueron aleatorizados para recibir tratamiento con CRYSVITA o tratamiento con control activo de fosfato oral y vitamina D activa (CRYSVITA N = 29, Control activo N = 32). El estudio 2 es un estudio de fase 2 abierto en pacientes con XLH de 5 a 12 años de edad (N = 52). El estudio 3 es un estudio de fase 2 abierto en pacientes con XLH de 1 a menos de 5 años de edad (N = 13). En general, la población de pacientes fue de 1 a 12 años (edad media de 7,0 años), 49% hombres y 88% blancos.
En el estudio 1, los pacientes aleatorizados a CRYSVITA recibieron una dosis media de aproximadamente 0,90 mg/kg (rango 0,8-1,2 mg/kg) cada 2 semanas. Todos los pacientes de este grupo y el grupo de control activo completaron 64 semanas de tratamiento.
Las reacciones adversas que ocurrieron en ≥ 10% de los sujetos en el grupo de CRYSVITA, con mayor frecuencia que en los sujetos del grupo de control activo, durante el período de tratamiento de 64 semanas en el estudio 1 se muestran en la Tabla 6.
| Reacción adversa | CRYSVITA (N=29) n (%) |
Control activo (N=32) n (%) |
|---|---|---|
| n = número de pacientes con un evento; N = número total de pacientes que recibieron al menos una dosis de CRYSVITA o control activo | ||
|
||
| Fiebre | 16 (55) | 6 (19) |
| Reacción en el sitio de inyección* | 15 (52) | 0 (0) |
| Tos† | 15 (52) | 6 (19) |
| Vómitos | 12 (41) | 8 (25) |
| Dolor en la extremidad | 11 (38) | 10 (31) |
| Dolor de cabeza | 10 (34) | 6 (19) |
| Absceso dental‡ | 10 (34) | 4 (13) |
| Caries dental | 9 (31) | 2 (6) |
| Diarrea | 7 (24) | 2 (6) |
| Vitamina D disminuida§ | 7 (24) | 1 (3) |
| Estreñimiento | 5 (17) | 0 (0) |
| Erupción¶ | 4 (14) | 2 (6) |
| Náuseas | 3 (10) | 1 (3) |
En el Estudio 2, 26 de los pacientes recibieron CRYSVITA a una dosis media de 1,05 mg/kg (rango 0,4 – 2,0 mg/kg) cada 2 semanas en la Semana 64; los otros 26 pacientes recibieron CRYSVITA cada 4 semanas. La duración media de la exposición en el Estudio 2 fue de 124 semanas. En el Estudio 3, los pacientes recibieron CRYSVITA a una dosis media de 0,90 mg/kg (rango 0,8-1,2 mg/kg) cada 2 semanas en la Semana 40. La duración media de la exposición en el Estudio 3 fue de 45 semanas.
Las reacciones adversas que ocurrieron en más del 10% de los pacientes tratados con CRYSVITA de los Estudios 2 y 3 se muestran en la Tabla 7.
| Reacción adversa | Estudio 2 (N=52) n (%) |
Estudio 3 (N=13) n (%) |
Global (N=65) n (%) |
|---|---|---|---|
| n = número de pacientes con un evento; N = número total de pacientes que recibieron al menos una dosis de CRYSVITA | |||
|
|||
| Cefalea | 38 (73) | 1 (8) | 39 (60) |
| Reacción en el lugar de la inyección* | 35 (67) | 3 (23) | 38 (59) |
| Vómitos | 25 (48) | 6 (46) | 31 (48) |
| Fiebre | 23 (44) | 8 (62) | 31 (48) |
| Dolor en la extremidad | 24 (46) | 3 (23) | 27 (42) |
| Disminución de la vitamina D† | 19 (37) | 2 (15) | 21 (32) |
| Erupción‡ | 14 (27) | 1 (8) | 15 (23) |
| Dolor de muelas | 12 (23) | 2 (15) | 14 (22) |
| Mialgia | 9 (17) | 1 (8) | 10 (15) |
| Absceso dental | 8 (15) | 3 (23) | 11 (17) |
| Mareo§ | 8 (15) | 0 (0) | 8 (12) |
Reacciones de hipersensibilidad
En el Estudio 1 (N=29 para el brazo de CRYSVITA), las reacciones de hipersensibilidad más frecuentes fueron erupción cutánea (10%), erupción cutánea en el sitio de inyección (10%) y urticaria en el sitio de inyección (7%). En los Estudios 2 y 3 (N=65), las reacciones de hipersensibilidad más frecuentes fueron erupción cutánea (22%), erupción cutánea en el sitio de inyección (6%) y urticaria (5%).
Hiperfosfatemia
En los estudios pediátricos, no se informaron eventos de hiperfosfatemia.
Reacciones en el sitio de inyección (RSI)
En el Estudio 1 (N=29 para el brazo de CRYSVITA), el 52% de los pacientes tuvieron una reacción local en el sitio de inyección (por ejemplo, urticaria en el sitio de inyección, eritema, erupción cutánea, hinchazón, hematoma, dolor, prurito y hematoma) en el sitio de inyección de CRYSVITA. En los Estudios 2 y 3 (N=65), aproximadamente el 58% de los pacientes tuvieron una reacción local en el sitio de inyección en el sitio de inyección de CRYSVITA. Las reacciones en el sitio de inyección fueron generalmente leves en gravedad, ocurrieron dentro de 1 día de la inyección, duraron aproximadamente de 1 a 3 días, no requirieron tratamiento y se resolvieron en casi todos los casos.
Reacciones adversas en pacientes adultos con XLH
La seguridad de CRYSVITA en pacientes adultos con XLH se demostró en un estudio aleatorizado, doble ciego, controlado con placebo (Estudio 4) de 134 pacientes, de 20 a 63 años de edad (edad media de 41 años), de los cuales la mayoría eran blancos/caucásicos (81%) y mujeres (65%). Un total de 68 y 66 pacientes recibieron al menos una dosis de CRYSVITA o placebo, respectivamente. La dosis media de CRYSVITA fue de 0,95 mg/kg (rango de 0,3 a 1,2 mg/kg) por vía subcutánea cada 4 semanas. Las reacciones adversas notificadas en más del 5% de los pacientes tratados con CRYSVITA y en al menos 2 pacientes más que con placebo en la parte controlada con placebo de 24 semanas del Estudio 4 se muestran en la Tabla 8.
| Reacción adversa | CRYSVITA (N=68) n (%) |
Placebo (N=66) n (%) |
|---|---|---|
| n = número de pacientes con un evento; N = número total de pacientes que recibieron al menos una dosis de CRYSVITA o placebo | ||
|
||
| Dolor de espalda | 10 (15) | 6 (9) |
| Dolor de cabeza* | 9 (13) | 6 (9) |
| Infección dental† | 9 (13) | 6 (9) |
| Síndrome de piernas inquietas | 8 (12) | 5 (8) |
| Vitamina D disminuida‡ | 8 (12) | 3 (5) |
| Mareos | 7 (10) | 4 (6) |
| Espasmos musculares | 5 (7) | 2 (3) |
| Estreñimiento | 6 (9) | 0 (0) |
| Fósforo en sangre aumentado§ | 4 (6) | 0 (0) |
El estudio controlado con placebo de 24 semanas fue seguido por un período de tratamiento abierto de 24 semanas en el que todos los pacientes recibieron CRYSVITA por vía subcutánea cada 4 semanas. No se identificaron nuevas reacciones adversas en el período de extensión de etiqueta abierta.
Reacciones de hipersensibilidad
En el período de doble ciego del Estudio 4, aproximadamente el 6% de los pacientes en los grupos de tratamiento con CRYSVITA y placebo experimentaron un evento de hipersensibilidad. Los eventos fueron leves o moderados y no requirieron la interrupción del tratamiento.
Hiperfosfatemia
En el período de doble ciego del Estudio 4, el 7% de los pacientes en el grupo de tratamiento con CRYSVITA experimentaron hiperfosfatemia que cumplía con los criterios especificados en el protocolo para la reducción de la dosis (ya sea un solo fósforo sérico mayor que 5.0 mg/dL o fósforo sérico mayor que 4.5 mg/dL [el límite superior de lo normal] en dos ocasiones). La hiperfosfatemia se manejó con la reducción de la dosis. La dosis para todos los pacientes que cumplieron con los criterios especificados en el protocolo se redujo en un 50 por ciento. Un solo paciente requirió una segunda reducción de la dosis por hiperfosfatemia continua.
Reacciones en el sitio de inyección (ISR)
En el período de doble ciego del Estudio 4, aproximadamente el 12% de los pacientes en los grupos de tratamiento con CRYSVITA y placebo tuvieron una reacción local (por ejemplo, reacción en el sitio de inyección, eritema, erupción cutánea, hematomas, dolor, prurito y hematoma) en el sitio de la inyección. Las reacciones en el sitio de inyección generalmente fueron leves en gravedad, ocurrieron dentro de 1 día de la inyección, duraron aproximadamente de 1 a 3 días, no requirieron tratamiento y se resolvieron en casi todos los casos.
Síndrome de piernas inquietas (RLS)
En el período de doble ciego del Estudio 4, aproximadamente el 12% del grupo de tratamiento con CRYSVITA tuvo un empeoramiento del síndrome de piernas inquietas (RLS) de referencia o un nuevo inicio de RLS de gravedad leve a moderada; estos eventos no llevaron a la interrupción de la dosis. También se ha informado RLS no grave en otros estudios de XLH en adultos con dosis repetidas; en un caso, el empeoramiento del RLS de referencia llevó a la interrupción del medicamento y la posterior resolución del evento.
Estenosis espinal
La estenosis espinal es frecuente en adultos con XLH y se ha informado compresión de la médula espinal. En los estudios de fase 2 y fase 3 de CRYSVITA en adultos con XLH (total N=176), un total de 7 pacientes se sometieron a cirugía espinal. La mayoría de estos casos parecían involucrar la progresión de una estenosis espinal preexistente. Se desconoce si la terapia con CRYSVITA exacerba la estenosis espinal o la compresión de la médula espinal.
Reacciones adversas en pacientes con TIO
La seguridad de CRYSVITA en pacientes con TIO se demostró en dos estudios clínicos de un solo brazo (Estudio 6 y Estudio 7) que reclutaron un total de 27 pacientes. Catorce pacientes eran hombres, y los pacientes tenían entre 33 y 73 años de edad. La dosis media de CRYSVITA fue de 0.77 mg/kg cada 4 semanas y la duración media de la exposición fue de 121 semanas.
Las reacciones adversas informadas en pacientes adultos con TIO en los datos agrupados del Estudio 6 y el Estudio 7 se muestran en la Tabla 9.
| Reacción adversa | General (N=27) n (%) |
|---|---|
|
|
| Absceso dental* | 5 (19) |
| Espasmos musculares | 5 (19) |
| Mareos | 4 (15) |
| Estreñimiento | 4 (15) |
| Reacción en el sitio de inyección† | 4 (15) |
| Erupción‡ | 4 (15) |
| Dolor de cabeza | 3 (11) |
| Deficiencia de vitamina D | 2 (7) |
| Hiperfosfatemia | 2 (7) |
| Síndrome de piernas inquietas | 2 (7) |
6.1 Reacciones adversas
Reacciones de hipersensibilidad
En los datos agrupados de los estudios 6 y 7, el 22% de los pacientes experimentaron una reacción de hipersensibilidad. Las reacciones de hipersensibilidad más frecuentes fueron eczema (11%) y erupción cutánea (11%). Los eventos fueron de gravedad leve o moderada.
Hiperfosfatemia
En los datos agrupados de los estudios 6 y 7, 2 pacientes (7%) experimentaron hiperfosfatemia, que se controló con la reducción de la dosis.
Reacciones en el lugar de la inyección
La frecuencia de reacciones en el lugar de la inyección fue del 15% (reacción en el lugar de la inyección, dolor en el lugar de la inyección e hinchazón en el lugar de la inyección). Las reacciones en el lugar de la inyección fueron generalmente de gravedad leve, no requirieron tratamiento y se resolvieron en todos los casos.
Síndrome de piernas inquietas
En los datos agrupados de los estudios 6 y 7, 2 pacientes (7%) experimentaron síntomas de síndrome de piernas inquietas, que fueron leves y no requirieron interrupción del tratamiento.
6.2 Inmunogenicidad
Al igual que con todas las proteínas terapéuticas, existe la posibilidad de inmunogenicidad. La detección de la formación de anticuerpos depende en gran medida de la sensibilidad y la especificidad del ensayo. Además, la incidencia observada de positividad de anticuerpos (incluidos los anticuerpos neutralizantes) en un ensayo puede verse influenciada por varios factores, incluida la metodología del ensayo, el manejo de las muestras, el momento de la recolección de las muestras, los medicamentos concomitantes y la enfermedad subyacente. Por estas razones, la comparación de la incidencia de anticuerpos a burosumab-twza en los estudios descritos a continuación con la incidencia de anticuerpos en otros estudios o con otros productos puede ser engañosa.
En los estudios clínicos de XLH, ninguno (0/13) de los pacientes de 1 a 4 años, el 19% (10/52) de los pacientes de 5 a 12 años y el 15% (20/131) de los pacientes adultos dieron positivo para anticuerpos antidroga (ADA) después de recibir CRYSVITA. Entre estos, tres pacientes de 5 a 12 años dieron positivo para anticuerpos neutralizantes. La presencia de ADA no se asoció con cambios clínicamente relevantes en la farmacocinética, farmacodinamia, eficacia y seguridad de burosumab en pacientes con XLH.
En un estudio clínico de TIO, el 14% (2/14) de los pacientes adultos dieron positivo para ADA después de recibir CRYSVITA. Ninguno de los pacientes con ADA positivo dio positivo para anticuerpos neutralizantes. En otro estudio clínico de TIO, ninguno de los 13 pacientes adultos dio positivo para ADA después de recibir CRYSVITA.
6.3 Experiencia postcomercialización
Las siguientes reacciones adversas se han identificado durante el uso post-aprobación de CRYSVITA. Debido a que estas reacciones se reportan voluntariamente de una población de tamaño incierto, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal con la exposición al fármaco.
Investigaciones: Se ha reportado un aumento del fósforo en sangre en pacientes pediátricos con XLH que reciben CRYSVITA.
7 INTERACCIONES MEDICAMENTOSAS
7.1 Fosfato Oral y Análogos Activos de Vitamina D
El uso concomitante de CRYSVITA con fosfato oral y/o análogos activos de vitamina D aumentará las concentraciones de fosfato más de lo esperado con CRYSVITA solo. Este aumento puede resultar en hiperfosfatemia, que puede inducir nefrocalcinosis.
El uso concomitante de CRYSVITA con fosfato oral y/o análogos activos de vitamina D está contraindicado.
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de Riesgos
No hay datos disponibles sobre el uso de CRYSVITA en mujeres embarazadas para informar un riesgo asociado a la droga de resultados adversos en el desarrollo. En el útero, la exposición a burosumab-twza en monos cynomolgus no resultó en efectos teratógenos. Se observaron efectos adversos como pérdida fetal tardía y parto prematuro en monos cynomolgus embarazadas, sin embargo, es poco probable que estos efectos indiquen un riesgo clínico porque ocurrieron a una exposición a la droga que fue 15 veces mayor, por AUC, que la exposición humana a la dosis humana máxima recomendada (MRHD) de 2 mg/kg cada 2 semanas y se acompañaron de hiperfosfatemia materna y mineralización placentaria (ver Datos). Los niveles de fósforo en suero deben controlarse durante todo el embarazo [ver Dosificación y administración (2.2)]. Reporte los embarazos a la línea de informes de eventos adversos de Kyowa Kirin, Inc. al 1-844-768-3544.
El riesgo de fondo de defectos de nacimiento mayores y aborto espontáneo para la población indicada es desconocido; sin embargo, el riesgo de fondo estimado en la población general de EE. UU. de defectos de nacimiento mayores es del 2% al 4% y de aborto espontáneo del 15% al 20% de los embarazos clínicamente reconocidos.
Datos
Datos de animales
En un estudio de toxicidad reproductiva en monos cynomolgus embarazadas, se administró burosumab-twza por vía intravenosa una vez cada dos semanas desde el día 20 del embarazo hasta el parto o la cesárea en el día 133, que incluye el período de organogénesis, a dosis de 0.2-, 2- y 15 veces la exposición humana a la MRHD adulta de 2 mg/kg cada 2 semanas. El tratamiento no resultó en efectos teratógenos en los fetos o la descendencia. Se observó un aumento en la pérdida fetal tardía, un período de gestación más corto y una mayor incidencia de nacimientos prematuros a 15 veces la exposición humana a la MRHD adulta de 2 mg/kg cada 2 semanas, de forma concomitante con hiperfosfatemia materna y mineralización placentaria. Se detectó burosumab-twza en el suero de los fetos, lo que indica el transporte a través de la placenta. La hiperfosfatemia, pero no la mineralización ectópica, estuvo presente en los fetos y la descendencia de las madres expuestas a 15 veces la exposición humana a la MRHD de 2 mg/kg de dosis cada 2 semanas. Burosumab-twza no afectó el crecimiento pre y posnatal, incluida la supervivencia de la descendencia.
8.2 Lactancia
Resumen de Riesgos
No hay información sobre la presencia de burosumab-twza en la leche materna, o los efectos de burosumab-twza en la producción de leche o el lactante. La IgG materna está presente en la leche materna. Sin embargo, se desconocen los efectos de la exposición gastrointestinal local y la exposición sistémica limitada a burosumab-twza en el lactante. La falta de datos clínicos durante la lactancia impide una determinación clara del riesgo de CRYSVITA para un lactante durante la lactancia. Por lo tanto, se deben considerar los beneficios para el desarrollo y la salud de la lactancia materna junto con la necesidad clínica de la madre de CRYSVITA y cualquier posible efecto adverso en el lactante amamantado por CRYSVITA o por la condición materna subyacente.
8.4 Uso pediátrico
La seguridad y eficacia de CRYSVITA se han establecido en pacientes pediátricos de 6 meses de edad o mayores. La seguridad y eficacia en pacientes pediátricos de 1 año de edad o mayores con XLH se basan en un estudio de fase 3, abierto, de control activo [61 pacientes de 1 a 12 años de edad (Estudio 1)] y dos estudios abiertos [52 pacientes de 5 a 12 años de edad (Estudio 2), y 13 pacientes de 1 a 4 años de edad (Estudio 3)] que evalúan el fósforo en suero y los hallazgos radiográficos. La seguridad y eficacia en pacientes de 6 meses a 1 año y adolescentes están respaldadas por evidencia de los estudios en pacientes pediátricos de 1 año a menos de 13 años de edad con modelado y simulación adicionales de datos farmacocinéticos (PK) y farmacodinámicos (PD) de adultos y pediátricos para informar la dosificación [ver Reacciones adversas (6.1) y Estudios clínicos (14)].
No se ha establecido la seguridad y eficacia de CRYSVITA en pacientes pediátricos con XLH menores de 6 meses de edad.
La seguridad y eficacia de CRYSVITA en pacientes pediátricos de 2 años de edad o mayores con TIO están respaldadas por evidencia de los estudios en pacientes adultos con TIO con modelado y simulación adicionales de datos de PK de pacientes adultos y pediátricos con XLH y pacientes adultos con TIO para informar la dosificación.
No se ha establecido la seguridad y eficacia de CRYSVITA en pacientes pediátricos con TIO menores de 2 años de edad.
8.5 Uso geriátrico
Los estudios clínicos de CRYSVITA no incluyeron un número suficiente de pacientes de 65 años de edad o mayores para determinar si responden de manera diferente a los pacientes más jóvenes. Otra experiencia clínica reportada no ha identificado diferencias en las respuestas entre los pacientes de edad avanzada y los pacientes más jóvenes. En general, la selección de la dosis para un paciente de edad avanzada debe ser cautelosa, generalmente comenzando en el extremo inferior del rango de dosificación, reflejando la mayor frecuencia de disminución de la función hepática, renal o cardíaca, y de enfermedades concomitantes u otra terapia farmacológica.
8.6 Insuficiencia renal
Se desconoce el efecto de la insuficiencia renal en la farmacocinética de burosumab-twza. Sin embargo, la insuficiencia renal puede inducir un metabolismo mineral anormal que aumentará las concentraciones de fosfato más de lo esperado con CRYSVITA solo. Este aumento puede resultar en hiperfosfatemia que puede inducir nefrocalcinosis.
CRYSVITA está contraindicado en pacientes con insuficiencia renal grave, definida como:
- pacientes pediátricos con tasa de filtración glomerular estimada (eGFR) de 15 mL/min/1.73m2 a 29 mL/min/1.73m2 o enfermedad renal en etapa terminal (eGFR < 15 mL/min/1.73m2)
- pacientes adultos con aclaramiento de creatinina (CLcr) de 15 mL/min a 29 mL/min o enfermedad renal en etapa terminal (CLcr < 15 mL/min).
10 SOBREDOSIS
No se han reportado casos de sobredosis con CRYSVITA. CRYSVITA se ha administrado en ensayos clínicos pediátricos sin toxicidad limitante de la dosis utilizando dosis de hasta 2 mg/kg de peso corporal con una dosis máxima de 90 mg, administrada cada dos semanas. En ensayos clínicos en adultos con HLD, no se ha observado toxicidad limitante de la dosis utilizando dosis de hasta 1 mg/kg o una dosis total máxima de 128 mg cada 4 semanas. En conejos y monos cynomolgus no HLD, se observó mineralización ectópica en múltiples tejidos y órganos a dosis de burosumab-twza que dieron como resultado niveles de fosfato sérico supra-fisiológicos. También se observaron efectos adversos en el hueso, incluidas reducciones en la densidad mineral ósea, la mineralización ósea y la resistencia ósea a una exposición mayor que la exposición humana [ver Toxicología no clínica (13.2)].
En caso de sobredosis, se recomienda medir inmediatamente los niveles de fósforo sérico, los niveles de calcio sérico y la función renal y controlarlos periódicamente hasta que se resuelvan a los niveles normales/basales. En caso de hiperfosfatemia, suspenda CRYSVITA e inicie el tratamiento médico adecuado.
11 DESCRIPCIÓN
Burosumab-twza es una inmunoglobulina G humana subclase 1 (IgG1), anticuerpo anti-factor de crecimiento de fibroblastos humanos 23 (FGF23) producido por tecnología de ADN recombinante utilizando células de ovario de hámster chino. Burosumab-twza está compuesto por dos moléculas de cadena pesada (cadena γ1) y dos moléculas de cadena ligera (cadena κ). Cada cadena pesada tiene una fracción de carbohidrato ligada a N en asparagina 297 (Asn297). El peso molecular de burosumab-twza determinado por espectrometría de masas es de aproximadamente 147.000.
La inyección de CRYSVITA (burosumab-twza) para administración subcutánea se suministra como una solución estéril, sin conservantes, de transparente a ligeramente opalescente e incolora a amarillo-marrón pálido en un vial de dosis única.
Cada 1 mL de solución contiene 10 mg, 20 mg o 30 mg de burosumab-twza, L-histidina (1,55 mg), L-metionina (1,49 mg), polisorbato 80 (0,5 mg), D-sorbitol (45,91 mg) en Agua para Inyección, USP. Se puede usar ácido clorhídrico para ajustar a un pH de 6,25.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
La hipofosfatemia ligada al cromosoma X es causada por un exceso de factor de crecimiento fibroblástico 23 (FGF23) que suprime la reabsorción tubular renal de fosfato y la producción renal de 1,25 dihidroxivitamina D. El burosumab-twza se une e inhibe la actividad biológica del FGF23, restaurando la reabsorción renal de fosfato y aumentando la concentración sérica de 1,25 dihidroxivitamina D.
12.2 Farmacodinamia
Tras la administración SC en pacientes con XLH y TIO, las concentraciones más altas de burosumab-twza se asociaron con un mayor aumento de los niveles de fósforo en suero. El aumento del fósforo en suero fue reversible y volvió a la línea de base con la eliminación del burosumab-twza sistémico.
La relación entre la tasa máxima de reabsorción tubular renal de fosfato y la tasa de filtración glomerular (TmP/GFR) mostró aumentos dependientes de la dosis desde la línea de base [ver Estudios clínicos (14)].
Se observó una elevación en el FGF23 total en suero después del inicio del tratamiento con burosumab-twza, sin embargo, la implicación clínica es desconocida.
12.3 Farmacocinética
Los siguientes parámetros farmacocinéticos se observaron en pacientes con XLH a los que se administró la dosis inicial recomendada aprobada basada en un paciente de 70 kg, a menos que se especifique lo contrario. Según el análisis de PK de la población, las características de PK del burosumab-twza fueron similares entre los pacientes con XLH y TIO.
El burosumab-twza exhibió una farmacocinética lineal después de las inyecciones SC dentro del rango de dosis de 0,1 a 1 mg/kg (0,08 a 0,8 veces la dosis máxima recomendada aprobada basada en un paciente de 70 kg con XLH).
La concentración media en el valle en estado estacionario (± DE) del burosumab-twza fue de 5,8 (± 3,4) mcg/mL en pacientes adultos con XLH.
Absorción
Los valores medios de Tmax del burosumab-twza oscilaron entre 8 y 11 días.
Distribución
El volumen aparente de distribución del burosumab-twza es de 8 L.
Eliminación
El aclaramiento aparente es de 0,290 L/día. La vida media del burosumab-twza es de aproximadamente 19 días.
Metabolismo
La vía exacta para el metabolismo del burosumab-twza no se ha caracterizado. Se espera que el burosumab-twza se degrade en pequeños péptidos y aminoácidos a través de vías catabólicas.
Poblaciones específicas
No se observó ninguna diferencia clínicamente significativa en la farmacocinética del burosumab-twza en función de la edad.
Se desconoce el efecto del deterioro renal o hepático en la farmacocinética del burosumab-twza.
Pacientes pediátricos
La concentración en el valle en estado estacionario fue de 15,8 (± 9,4) mcg/mL en pacientes con XLH de 5 a 12 años, y de 11,2 (± 4,6) mcg/mL en pacientes con XLH de 1 a 4 años.
Peso corporal
El aclaramiento y el volumen de distribución del burosumab-twza aumentan con el peso corporal.
Estudios de interacción medicamentosa
No se han realizado estudios de interacción medicamentosa con CRYSVITA.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
El potencial carcinogénico de burosumab-twza no se ha evaluado en estudios a largo plazo en animales.
No se han realizado estudios para evaluar el potencial mutagénico de burosumab-twza.
No se han realizado estudios específicos de fertilidad en animales para evaluar los efectos de burosumab-twza.
Los estudios de toxicología con burosumab-twza de hasta 40 semanas de duración en monos cynomolgus no mostraron efectos adversos significativos en los órganos reproductores femeninos a dosis de hasta 16 veces la exposición humana a la dosis humana máxima recomendada (MRHD) de 2 mg/kg cada 2 semanas. En los monos machos, se observó una mineralización mínima de la rete testis o los túbulos seminíferos asociada con hiperfosfatemia a 3- a 9-veces la exposición humana a la MRHD de 2 mg/kg cada 2 semanas, pero el análisis de semen no mostró ningún efecto adverso.
13.2 Toxicología y/o Farmacología Animal
En conejos y monos cynomolgus, la inhibición de la señalización de FGF23 por burosumab-twza aumentó el fosfato sérico y la 1,25 dihidroxivitamina D. Se observó mineralización ectópica en múltiples tejidos y órganos a dosis de burosumab-twza que dieron como resultado niveles de fosfato sérico supra-fisiológicos. En un estudio en ratones Hyp hipofosfatémicos de tipo salvaje (WT), un modelo murino de XLH, la mineralización ectópica fue marcadamente menor en los ratones Hyp.
En monos cynomolgus adultos, burosumab-twza aumentó la renovación ósea, el contenido mineral y/o la densidad mineral y el grosor cortical a 9- a 16-veces la exposición humana a la MRHD de 2 mg/kg cada 2 semanas. Se observaron efectos adversos en el hueso, incluidas reducciones en la densidad mineral ósea, la mineralización ósea y la resistencia ósea en monos machos adultos a 9- a 11-veces la exposición humana a la MRHD de 2 mg/kg cada 2 semanas.
En monos cynomolgus juveniles, burosumab-twza aumentó la renovación ósea, el contenido mineral y/o la densidad mineral y/o el grosor cortical a 0,2- a 2-veces la exposición clínica pediátrica. La mineralización ósea disminuyó en un mono macho a 2 veces la exposición pediátrica, pero no hubo efecto en la resistencia ósea. Burosumab-twza no afectó el desarrollo óseo en monos juveniles a dosis de hasta 2 veces la exposición pediátrica.
14 ESTUDIOS CLÍNICOS
14.1 Hipo fosfatemia ligada al cromosoma X pediátrica
CRYSVITA se ha evaluado en tres estudios que reclutaron un total de 126 pacientes pediátricos con XLH.
El estudio 1 (NCT 02915705) es un estudio aleatorizado, abierto de 64 semanas en 61 pacientes pediátricos con XLH, de 1 a 12 años de edad, que comparó el tratamiento con CRYSVITA con un control activo (fosfato oral y vitamina D activa). En el momento de la primera dosis, la edad media de los pacientes fue de 6,3 años y el 44% eran hombres. Todos los pacientes tenían evidencia radiográfica de raquitismo en la línea de base, con una puntuación RSS de ≥ 2,0 y habían recibido fosfato oral y análogos de vitamina D activa durante una duración media (DE) de 4 (3,1) años. El fosfato oral y los análogos de vitamina D activa se suspendieron antes de la inscripción en el estudio durante un período de lavado de 7 días y luego se reiniciaron para los pacientes del grupo de control activo. Los pacientes fueron aleatorizados para recibir CRYSVITA a una dosis inicial de 0,8 mg/kg cada dos semanas o fosfato oral (dosis recomendada 20-60 mg/kg/día) y vitamina D activa (dosis recomendadas calcitriol 20-30 ng/kg/día o alfacalcidol 40-60 ng/kg/día). Los pacientes aleatorizados al control activo recibieron una dosis media de fosfato oral de aproximadamente 41 mg/kg/día (rango 18 a 110 mg/kg/día) en la semana 40 y aproximadamente 46 mg/kg/día (rango 18 mg/kg/día a 166 mg/kg/día) en la semana 64. También recibieron una dosis media de calcitriol oral de 26 ng/kg/día en la semana 40 y 27 ng/kg/día en la semana 64 o una cantidad terapéuticamente equivalente de alfacalcidol. Ocho pacientes en el brazo de CRYSVITA aumentaron la dosis a 1,2 mg/kg según las mediciones de fósforo en suero. Todos los pacientes completaron al menos 64 semanas en el estudio.
Fósforo en suero
En el estudio 1, CRYSVITA aumentó los niveles medios (DE) de fósforo en suero de 2,4 (0,24) mg/dL en la línea de base a 3,3 (0,43) mg/dL en la semana 40 y a 3,3 (0,42) mg/dL en la semana 64. En el grupo de control activo, las concentraciones medias (DE) de fósforo en suero aumentaron de 2,3 (0,26) mg/dL en la línea de base a 2,5 (0,34) mg/dL en la semana 40 y a 2,5 (0,39) mg/dL en la semana 64. La capacidad de reabsorción de fosfato renal, evaluada mediante TmP/GFR, aumentó en los pacientes tratados con CRYSVITA de una media (DE) de 2,2 (0,37) mg/dL en la línea de base a 3,4 (0,67) mg/dL y 3,3 (0,65) mg/dL en la semana 40 y la semana 64, respectivamente. En el grupo de control activo, la media (DE) de TmP/GFR disminuyó de 2,0 (0,33) mg/dL en la línea de base a 1,8 (0,35) mg/dL en la semana 40 y se mantuvo por debajo de la línea de base en la semana 64 a 1,9 (0,49) mg/dL.
Figura 1: Concentración de fósforo en suero y cambio desde la línea de base (mg/dL) (Media ± DE) por grupo de tratamiento en niños de 1 a 12 años en el estudio 1
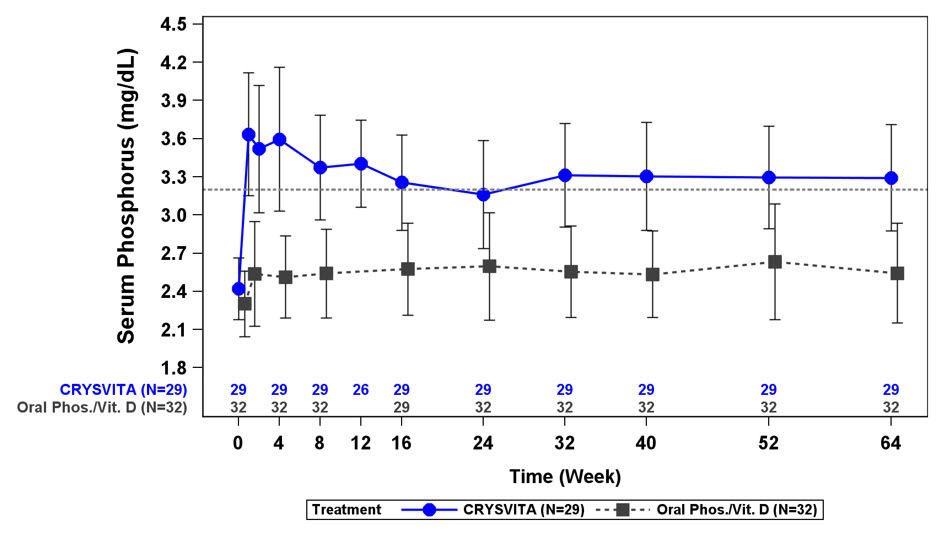
La línea punteada representa el límite inferior de lo normal (3,2 mg/dL) para los pacientes del estudio 1.
Evaluación radiográfica del raquitismo
Las radiografías se examinaron para evaluar el raquitismo relacionado con XLH utilizando la puntuación de gravedad del raquitismo de Thacher de 10 puntos (RSS) y la impresión global radiográfica de cambio de 7 puntos (RGI-C). La puntuación RSS se asigna en función de las imágenes de la muñeca y la rodilla de un solo punto de tiempo, con puntuaciones más altas que indican una mayor gravedad del raquitismo. La puntuación RGI-C se asigna en función de las comparaciones lado a lado de las radiografías de la muñeca y la rodilla de dos puntos de tiempo, con puntuaciones más altas que indican una mayor mejora en la evidencia radiográfica de raquitismo. Una puntuación RGI-C de +2,0 se definió como evidencia radiográfica de curación sustancial.
En el estudio 1, la media (DE) total de RSS en la línea de base fue de 3,2 (0,98) en el grupo de CRYSVITA y de 3,2 (1,14) en el grupo de control activo. Después de 40 semanas de tratamiento con CRYSVITA, la media total de RSS disminuyó de 3,2 a 1,1 (0,72) y de 3,2 a 2,5 (1,09) en el grupo de control activo. La media LS (EE) de la puntuación global RGI-C fue de +1,9 (0,11) en el grupo de CRYSVITA y de +0,8 (0,11) en el grupo de control activo en la semana 40 (ver Tabla 10). En la semana 40, 21 de los 29 pacientes del grupo de CRYSVITA y 2 de los 32 pacientes del brazo de control activo lograron una puntuación global RGI-C ≥ +2,0. Estos hallazgos se mantuvieron en la semana 64, como se muestra en la Tabla 10.
| Punto final Punto de tiempo |
CRYSVITA cada 2 semanas (N=29) |
Control activo (N=32) |
|---|---|---|
|
||
| Puntuación total de RSS | ||
| Media (DE) de la línea de base | 3,2 (0,98) | 3,2 (1,14) |
| Cambio medio LS desde la línea de base en la puntuación total* (la reducción indica mejora) con IC del 95% | ||
| Semana 40 | -2,0 (-2,33, -1,75) | -0,7 (-0,98, -0,43) |
| Semana 64 | -2,2 (-2,46, -2,00) | -1,0 (-1,31, -0,72) |
| Puntuación global RGI-C† | ||
| Puntuación media LS* (positivo indica curación) con IC del 95% | ||
| Semana 40 | +1.9 (+1.70, +2.14) | +0.8 (+0.56, +0.99) |
| Semana 64 | +2.06 (+1.91, +2.20) | +1.03 (+0.77, +1.30) |
Anomalía Esquelética de las Extremidades Inferiores
En el Estudio 1, las anomalías esqueléticas de las extremidades inferiores se evaluaron mediante RGI-C en radiografías de piernas largas de pie. En la Semana 64, el grupo CRYSVITA mantuvo una mayor mejora en comparación con el grupo de control activo (media LS [EE]: +1,25 [0,17] frente a +0,29 [0,12]; diferencia de +0,97 (IC del 95%: +0,57, +1,37, modelo GEE)).
Actividad de la Fosfatasa Alcalina Sérica
Para el Estudio 1, la actividad media (DE) de la fosfatasa alcalina sérica total disminuyó de 511 (125) en el inicio a 337 (86) U/L en el grupo CRYSVITA (cambio medio: -33%) y de 523 (154) en el inicio a 495 (182) U/L en el grupo de control activo (cambio medio: -5%) en la Semana 64.
Crecimiento
En el Estudio 1, el tratamiento con CRYSVITA durante 64 semanas aumentó la puntuación Z de altura media (DE) de pie de -2,32 (1,17) en el inicio a -2,11 (1,11) en la Semana 64 (cambio medio LS (EE) de +0,17 (0,07)). En el grupo de control activo, la puntuación Z de altura media (DE) aumentó de -2,05 (0,87) en el inicio a -2,03 (0,83) en la Semana 64 (cambio medio LS (EE) de +0,02 (0,04)). La diferencia entre los grupos de tratamiento en la Semana 64 fue de +0,14 (IC del 95%: 0,00, +0,29).
El Estudio 2 (NCT 02163577) es un estudio aleatorizado, abierto, en 52 pacientes prepubescentes con XLH, de 5 a 12 años de edad, que comparó el tratamiento con CRYSVITA administrado cada 2 semanas frente a cada 4 semanas. Tras una fase inicial de titulación de la dosis de 16 semanas, los pacientes completaron 48 semanas de tratamiento con CRYSVITA cada 2 semanas. Los 52 pacientes completaron al menos 64 semanas en el estudio; ningún paciente abandonó el estudio. La dosis de burosumab-twza se ajustó para alcanzar una concentración sérica de fósforo en ayunas de 3,5 a 5,0 mg/dL en función del nivel de fósforo en ayunas el día de la administración de la dosis. Veintiséis de los 52 pacientes recibieron CRYSVITA cada dos semanas hasta una dosis máxima de 2 mg/kg. La dosis media fue de 0,73 mg/kg (rango: 0,3, 1,5) en la Semana 16, 0,98 mg/kg (rango: 0,4, 2,0) en la Semana 40 y 1,04 mg/kg (rango: 0,4, 2,0) en la Semana 60. Los 26 pacientes restantes recibieron CRYSVITA cada cuatro semanas. Al inicio del estudio, la edad media de los pacientes era de 8,5 años y el 46% eran varones. El noventa y seis por ciento había recibido fosfato oral y análogos de la vitamina D activa durante una duración media (DE) de 7 (2,4) años. El fosfato oral y los análogos de la vitamina D activa se suspendieron antes de la inscripción en el estudio. El noventa y cuatro por ciento de los pacientes presentaban evidencia radiográfica de raquitismo en el inicio.
El Estudio 3 (NCT 02750618) es un estudio abierto de 64 semanas en 13 pacientes pediátricos con XLH, de 1 a 4 años de edad. Los pacientes recibieron CRYSVITA a una dosis de 0,8 mg/kg cada dos semanas, con 3 pacientes que aumentaron la dosis hasta 1,2 mg/kg en función de las mediciones de fósforo sérico. Todos los pacientes completaron al menos 40 semanas en el estudio; ningún paciente abandonó el estudio. Al inicio del estudio, la edad media de los pacientes era de 2,9 años y el 69% eran varones. Todos los pacientes presentaban evidencia radiográfica de raquitismo en el inicio y 12 pacientes habían recibido fosfato oral y análogos de la vitamina D activa durante una duración media (DE) de 16,7 (14,4) meses. El fosfato oral y los análogos de la vitamina D activa se suspendieron antes de la inscripción en el estudio.
Fósforo Sérico
En el Estudio 2, CRYSVITA aumentó los niveles medios (DE) de fósforo sérico de 2,4 (0,40) en el inicio a 3,3 (0,40) y 3,4 (0,45) mg/dL en la Semana 40 y la Semana 64 en los pacientes que recibieron CRYSVITA cada 2 semanas. La relación entre la tasa máxima de reabsorción tubular renal de fosfato y la tasa de filtración glomerular (TmP/TFG) aumentó en estos pacientes de una media (DE) de 2,2 (0,49) en el inicio a 3,3 (0,60) y 3,4 (0,53) mg/dL en la Semana 40 y la Semana 64.
En el Estudio 3, CRYSVITA aumentó los niveles medios (DE) de fósforo sérico de 2,5 (0,28) mg/dL en el inicio a 3,5 (0,49) mg/dL en la Semana 40.
Evaluación Radiográfica del Raquitismo
En el Estudio 2, la puntuación total media (DE) de RSS en el inicio fue de 1,9 (1,17) en los pacientes que recibieron CRYSVITA cada dos semanas. Tras 40 semanas de tratamiento con CRYSVITA, la puntuación total media de RSS disminuyó de 1,9 a 0,8 (véase Tabla 11). Tras 40 semanas de tratamiento con CRYSVITA, la puntuación global media de RGI-C fue de +1,7 en los pacientes que recibieron CRYSVITA cada dos semanas. Dieciocho de los 26 pacientes alcanzaron una puntuación de RGI-C ≥ +2,0. Estos hallazgos se mantuvieron en la Semana 64, como se muestra en la Tabla 11.
En el Estudio 3, la puntuación total media (DE) de RSS en el inicio fue de 2,9 (1,37) en 13 pacientes. Tras 40 semanas de tratamiento con CRYSVITA, la puntuación total media de RSS disminuyó de 2,9 a 1,2 y la puntuación global media (EE) de RGI-C fue de +2,3 (0,08) (véase Tabla 11). Los 13 pacientes alcanzaron una puntuación global de RGI-C ≥ +2,0.
| Variable Tiempo |
CRYSVITA Cada 2 Semanas | |
|---|---|---|
| Estudio 2* (N=26) |
Estudio 3† (N=13) |
|
|
||
| Puntuación Total de RSS | ||
| Media (DE) en el inicio | 1,9 (1,17) | 2,9 (1,37) |
| Cambio medio LS desde el inicio en la puntuación total (la reducción indica mejora) con IC del 95% | ||
| Semana 40 | -1,1 (-1,28, -0,85) | -1,7 (-2,03, -1,44) |
| Semana 64 | -1,0 (-1,2, -0,79) | |
| Puntuación global RGI-C | ||
| Puntuación media de LS (positiva indica curación) con IC del 95% | ||
| Semana 40 | +1.7 (+1.48, +1.84) | +2.3 (+2.16, +2.51) |
| Semana 64 | +1.6 (+1.34, +1.78) | |
Anomalía esquelética de las extremidades inferiores
En el Estudio 3, el cambio medio (EE) en la deformidad de las extremidades inferiores, según la evaluación de RGI-C, utilizando radiografías de pie de pierna larga, fue de +1.3 (0.14) en la semana 40.
Actividad de la fosfatasa alcalina sérica
Para el Estudio 2, la actividad media (DE) de la fosfatasa alcalina sérica total fue de 462 (110) U/L en el inicio y disminuyó a 354 (73) U/L en la semana 64 (-23%) en los pacientes que recibieron CRYSVITA cada 2 semanas.
Para el Estudio 3, la actividad media (DE) de la fosfatasa alcalina sérica total fue de 549 (194) U/L en el inicio y disminuyó a 335 (88) U/L en la semana 40 (cambio medio: -36%).
Crecimiento
En el Estudio 2, el tratamiento con CRYSVITA durante 64 semanas aumentó la puntuación Z de altura media (DE) de pie de -1.72 (1.03) en el inicio a -1.54 (1.13) en los pacientes que recibieron CRYSVITA cada dos semanas (cambio medio de LS de +0.19 (IC del 95%: 0.09 a 0.29).
14.2 Hipo fosfatemia ligada al cromosoma X en adultos
El Estudio 4 (NCT 02526160) es un estudio aleatorizado, doble ciego, controlado con placebo en 134 pacientes adultos con XLH. El estudio comprende una fase de tratamiento controlada con placebo de 24 semanas seguida de un período de tratamiento abierto de 24 semanas en el que todos los pacientes recibieron CRYSVITA. CRYSVITA se administró a una dosis de 1 mg/kg cada 4 semanas. Al inicio del estudio, la edad media de los pacientes fue de 40 años (rango de 19 a 66 años) y el 35% eran hombres. Todos los pacientes tenían dolor esquelético asociado con XLH/osteomalacia en el inicio. La concentración media (DE) de fósforo sérico en el inicio fue inferior al límite inferior de lo normal a 1.98 (0.31) mg/dL. No se permitieron los análogos de fosfato oral y vitamina D activa durante el estudio. De los 134 pacientes inscritos en el estudio, un paciente del grupo CRYSVITA interrumpió el tratamiento durante el período de tratamiento controlado con placebo de 24 semanas, y 7 pacientes interrumpieron CRYSVITA durante el período de tratamiento abierto.
El Estudio 5 (NCT 02537431) es un estudio de 48 semanas, abierto, de un solo brazo en 14 pacientes adultos con XLH para evaluar los efectos de CRYSVITA en la mejora de la osteomalacia, según la evaluación histológica e histomorfométrica de las biopsias de hueso de la cresta ilíaca. Los pacientes recibieron 1 mg/kg de CRYSVITA cada cuatro semanas. Al inicio del estudio, la edad media de los pacientes fue de 40 años (rango de 25 a 52 años) y el 43% eran hombres. No se permitieron los análogos de fosfato oral y vitamina D activa durante el estudio.
Fósforo sérico
En el Estudio 4 en el inicio, el fósforo sérico medio (DE) fue de 1.9 (0.32) y 2.0 (0.30) mg/dL en los grupos placebo y CRYSVITA, respectivamente. Durante el período inicial de 24 semanas, doble ciego, controlado con placebo, el fósforo sérico medio (DE) a través de los puntos medios de los intervalos de dosis (2 semanas después de la dosis) fue de 2.1 (0.30) y 3.2 (0.53) mg/dL en los grupos placebo y CRYSVITA, y el fósforo sérico medio (DE) a través de los finales de los intervalos de dosis fue de 2.0 (0.30) y 2.7 (0.45) mg/dL en los grupos placebo y CRYSVITA.
Un total del 94% de los pacientes tratados con CRYSVITA lograron un nivel de fósforo sérico por encima del límite inferior de lo normal (LLN) en comparación con el 8% en el grupo placebo hasta la semana 24 (ver Tabla 12).
| Placebo (N = 66) |
CRYSVITA (N = 68) |
|
|---|---|---|
| Los IC del 95% se calculan utilizando el método de puntuación de Wilson. | ||
|
||
| Logró fósforo sérico medio > LLN a través de los puntos medios de los intervalos de dosis hasta la semana 24 – n (%) | 5 (8%) | 64 (94%) |
| IC del 95% | (3.3, 16.5) | (85.8, 97.7) |
| valor de p* | < 0.0001 | |
Durante el período de tratamiento abierto, el fósforo sérico se mantuvo durante la terapia continua con CRYSVITA, sin evidencia de pérdida de efecto hasta la semana 48.
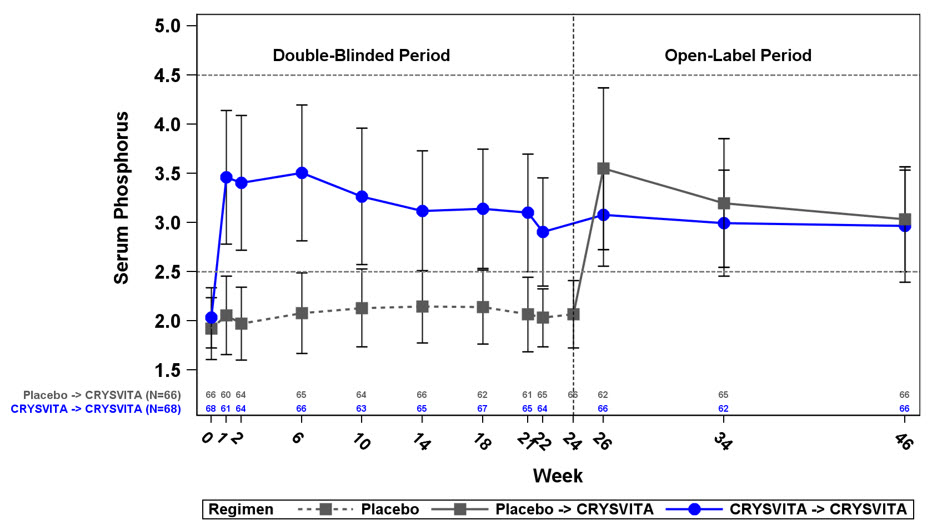
En el inicio del estudio, la proporción media (DE) de la tasa máxima de reabsorción tubular renal de fosfato a la tasa de filtración glomerular (TmP/GFR) fue de 1,60 (0,37) y 1,68 (0,40) mg/dL en los grupos de placebo y CRYSVITA, respectivamente. En la semana 22 (punto medio de un intervalo de dosis), la TmP/GFR media (DE) fue de 1,69 (0,37) y 2,73 (0,75) mg/dL en los grupos de placebo y CRYSVITA. En la semana 24 (final de un intervalo de dosis), la TmP/GFR media (DE) fue de 1,73 (0,42) y 2,21 (0,48) mg/dL en los grupos de placebo y CRYSVITA. Durante el período de tratamiento abierto, la TmP/GFR se mantuvo estable durante la terapia continua con CRYSVITA hasta la semana 48.
Evaluación radiográfica de la osteomalacia
En el estudio 4, se realizó una radiografía esquelética al inicio del estudio para identificar fracturas y pseudofracturas relacionadas con la osteomalacia. Las fracturas relacionadas con la osteomalacia se definen como lucencias atraumáticas que se extienden a través de ambas corticales óseas y las pseudofracturas se definen como lucencias atraumáticas que se extienden a través de una cortical. El 52% de los pacientes tenían fracturas activas (no curadas) (12%) o pseudofracturas activas (47%) al inicio del estudio. Las fracturas y pseudofracturas activas se localizaron principalmente en los fémures, la tibia/peroné y los metatarsianos de los pies. La evaluación de estos sitios de fractura/pseudofractura activa en la semana 24 demostró una mayor tasa de curación completa en el grupo de CRYSVITA en comparación con el placebo, como se muestra en la Tabla 13. Durante el período de tratamiento doble ciego controlado con placebo hasta la semana 24, aparecieron un total de 6 nuevas fracturas o pseudofracturas en 68 pacientes que recibieron CRYSVITA, en comparación con 8 nuevas anomalías en 66 pacientes que recibieron placebo (ver Tabla 13).
| Fracturas activas | Pseudofracturas activas | Fracturas totales | ||||
|---|---|---|---|---|---|---|
| Placebo n (%) |
CRYSVITA n (%) |
Placebo n (%) |
CRYSVITA n (%) |
Placebo n (%) |
CRYSVITA n (%) |
|
| No. de fracturas al inicio del estudio | 13 | 14 | 78 | 51 | 91 | 65 |
| Curadas en la semana 24 | 0 (0%) | 7 (50%) | 7 (9%) | 21 (41%) | 7 (8%) | 28 (43%) |
Durante el período de tratamiento abierto, los pacientes que continuaron recibiendo CRYSVITA mostraron una curación continua de las fracturas en la semana 48 [fracturas activas (n = 8, 57%), pseudofracturas activas (n = 33, 65%)]. En el grupo ‘placebo a CRYSVITA’, se observó la curación de las fracturas en la semana 48 para las fracturas activas (n = 6, 46%) y las pseudofracturas activas (n = 26, 33%).
Resultados informados por el paciente
El estudio 4 evaluó los síntomas relacionados con XLH informados por los pacientes (dolor, rigidez articular y función física).
A las 24 semanas, el brazo de CRYSVITA mostró una mejora media desde el inicio (-7,9) en comparación con el brazo de placebo (+0,3) en la puntuación de gravedad de la rigidez (rango de 0 a 100; las puntuaciones más bajas reflejan una mejora de los síntomas).
A las 24 semanas, no se demostró ninguna diferencia significativa entre CRYSVITA y el placebo en la intensidad del dolor informada por el paciente o la puntuación de la función física.
Histomorfometría ósea
En el estudio 5, después de 48 semanas de tratamiento, se observó la curación de la osteomalacia en diez pacientes, como lo demuestra la disminución del volumen de osteoide/volumen óseo (OV/BV) de una puntuación media (DE) del 26% (12,4) en el inicio al 11% (6,5), un cambio del -57%. El grosor del osteoide (O.Th) disminuyó en once pacientes de una media (DE) de 17 (4,1) micrómetros a 12 (3,1) micrómetros, un cambio del -33%. El tiempo de retraso de la mineralización (MLt) disminuyó en 6 pacientes de una media (DE) de 594 (675) días a 156 (77) días, un cambio medio del -74%.
14.3 Osteomalacia inducida por tumor
CRYSVITA se ha evaluado en dos estudios que reclutaron un total de 27 pacientes con TIO.
El estudio 6 (NCT 02304367) es un estudio abierto de un solo brazo que reclutó a 14 pacientes adultos con un diagnóstico confirmado de hipofosfatemia relacionada con FGF23 producida por un tumor subyacente que no era susceptible de extirpación quirúrgica o no se pudo localizar. De los 14 pacientes con TIO reclutados en el estudio 6, ocho eran hombres, y los pacientes tenían entre 33 y 68 años de edad (mediana de 59,5 años). Los análogos de fosfato oral y vitamina D activa se suspendieron dos semanas antes de la inscripción en el estudio. Los pacientes recibieron CRYSVITA cada 4 semanas a una dosis inicial basada en el peso de 0,3 mg/kg que se tituló para alcanzar un nivel de fósforo sérico en ayunas de 2,5 a 4,0 mg/dL. La dosis media fue de 0,83 mg/kg en la semana 20, 0,87 mg/kg en la semana 48, 0,77 mg/kg en la semana 96 y 0,71 mg/kg en la semana 144.
El estudio 7 (NCT 02722798) es un estudio abierto de un solo brazo. En el estudio 7, 13 pacientes adultos con un diagnóstico confirmado de TIO recibieron CRYSVITA. De los 13 pacientes con TIO que recibieron tratamiento en el estudio 7, seis eran hombres, y los pacientes tenían entre 41 y 73 años de edad (mediana de 58,0 años). Los análogos de fosfato oral y vitamina D activa se suspendieron dos semanas antes de la inscripción en el estudio. Los pacientes recibieron CRYSVITA cada 4 semanas a una dosis inicial basada en el peso de 0,3 mg/kg que se tituló para alcanzar un nivel de fósforo sérico en ayunas de 2,5 a 4,0 mg/dL. La dosis media (DE) fue de 0,91 (0,59) mg/kg en la semana 48 y de 0,96 (0,70) mg/kg en la semana 88.
Fósforo sérico
En el estudio 6, CRYSVITA aumentó los niveles medios (DE) de fósforo sérico de 1,60 (0,47) mg/dL en el inicio a 2,64 (0,76) mg/dL promediados en el punto medio de los intervalos de dosis hasta la semana 24, con el 50% de los pacientes (7/14) alcanzando un nivel medio de fósforo sérico por encima del LLN promediado en el punto medio de los intervalos de dosis hasta la semana 24. El aumento de las concentraciones medias de fósforo sérico se mantuvo cerca o por encima del LLN hasta la semana 144 (Figura 3). La relación entre la tasa máxima de reabsorción tubular renal de fosfato y la tasa de filtración glomerular (TmP/GFR) aumentó en estos pacientes de una media (DE) de 1,12 (0,54) mg/dL en el inicio a 2,12 (0,64) mg/dL en la semana 48, y se mantuvo estable hasta la semana 144.
Figura 3: Concentración de fósforo sérico y cambio desde el inicio en el estudio 6 (mg/dL)
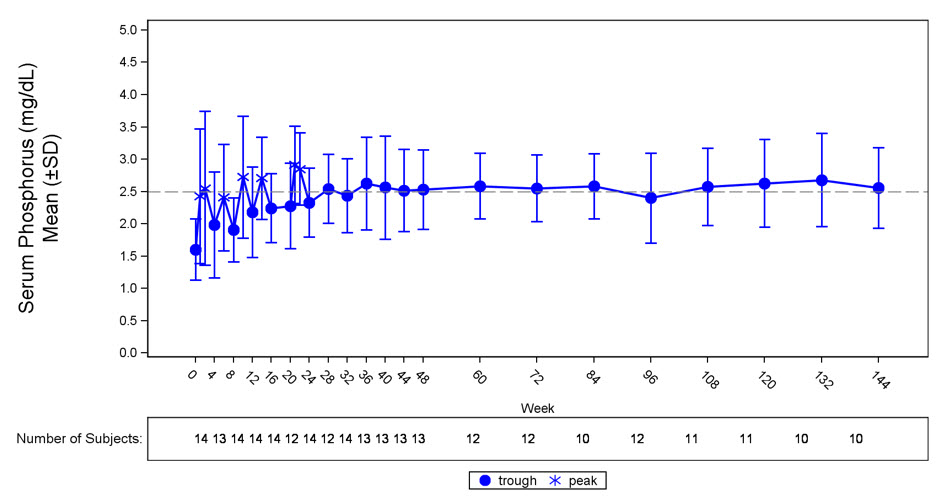
La línea punteada representa el límite inferior de lo normal (2,5 mg/dL) para los pacientes del estudio 6.
En el estudio 7, CRYSVITA aumentó los niveles medios (DE) de fósforo sérico de 1,62 (0,49) mg/dL en el inicio a 2,63 (0,87) mg/dL promediados en el punto medio de los intervalos de dosis hasta la semana 24, con el 69% de los pacientes (9/13) alcanzando un nivel medio de fósforo sérico por encima del LLN promediado en el punto medio del intervalo de dosis hasta la semana 24. Las concentraciones medias de fósforo sérico se mantuvieron por encima del LLN hasta la semana 88. La capacidad de reabsorción renal de fosfato, evaluada mediante TmP/GFR, aumentó de una media (DE) de 1,15 (0,43) mg/dL en el inicio a 2,30 mg/dL (0,48) mg/dL en la semana 48.
Histomorfometría ósea
En el estudio 6, la osteomalacia estaba presente en el inicio en nueve de los 11 pacientes con biopsias óseas pareadas, y la curación se evaluó después de 48 semanas de tratamiento. En estos 9 pacientes con osteomalacia en el inicio, OV/BV disminuyó de una puntuación media (DE) del 21,2% (19,9) en el inicio al 13,9% (16,7), un cambio del -34%. O.Th disminuyó de una media (DE) de 18,9 (11,9) micrómetros a 12,1 (10,1) micrómetros, un cambio del -36%. MLt disminuyó en 3 pacientes de una media (DE) de 667 (414) días a 331 (396) días, un cambio del -50%.
En el estudio 7, la osteomalacia estaba presente en el inicio en los 3 pacientes con biopsias óseas pareadas, y la curación se evaluó después de 48 semanas de tratamiento. En estos 3 pacientes, OV/BV disminuyó de una puntuación media (DE) del 14,0% (15,2) en el inicio al 9,2% (5,5), un cambio del -34%. O.Th disminuyó de una media (DE) de 16,0 (13,7) micrómetros a 13,5 (7,1) micrómetros, un cambio del -16%.
Evaluación radiográfica de la osteomalacia
En el estudio 6, se realizaron gammagrafías óseas de cuerpo entero con 99mtecnecio en el inicio y en puntos de tiempo posteriores durante el estudio en los 14 pacientes. Las gammagrafías óseas permiten evaluar los sitios de mayor captación de trazador en una amplia gama de afecciones óseas, incluida la osteomalacia. En pacientes con TIO, se presume que la mayor captación de trazador en la gammagrafía ósea son fracturas y pseudofracturas no traumáticas. En el inicio, todos los pacientes tenían áreas de captación de trazador con un total de 249 anomalías óseas en 14 pacientes. El número de áreas de captación de trazador disminuyó de la semana 48 a la semana 144, lo que sugiere la curación de las anomalías óseas.
16 FORMA DE SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
La inyección de CRYSVITA (burosumab-twza) para administración subcutánea se suministra como una solución estéril, sin conservantes, transparente a ligeramente opalescente e incolora a amarillo-marrón pálido. El producto está disponible como un vial de dosis única por caja en las siguientes concentraciones:
10 mg/mL (NDC# 42747-102-01)
20 mg/mL (NDC# 42747-203-01)
30 mg/mL (NDC# 42747-304-01)
Los viales de CRYSVITA deben almacenarse en la caja original hasta el momento de su uso en condiciones refrigeradas a 36°F a 46°F (2°C a 8°C). Mantenga el vial de CRYSVITA en la caja original para protegerlo de la luz hasta el momento de su uso.
No congele ni agite CRYSVITA.
No use CRYSVITA después de la fecha de vencimiento estampada en la caja.
Los viales de CRYSVITA son de dosis única. Deseche cualquier producto no utilizado.
17 INFORMACIÓN PARA EL PACIENTE
Interacciones medicamentosas
Avise a los pacientes que no usen ningún producto oral de fosfato y/o análogo de vitamina D activa [ver Contraindicaciones (4)].
Reacciones de hipersensibilidad
Avise a los pacientes que CRYSVITA puede causar eventos de hipersensibilidad como erupción cutánea, erupción cutánea en el sitio de inyección y urticaria. Indique a los pacientes que se pongan en contacto con su médico si se producen tales reacciones [ver Reacciones adversas (6.1)].
Reacciones en el sitio de inyección
Informe a los pacientes que se han producido reacciones en el sitio de inyección (por ejemplo, eritema, erupción cutánea, hinchazón, hematomas, dolor, prurito, urticaria y hematoma) en el sitio de inyección de CRYSVITA. Indique a los pacientes que se pongan en contacto con su médico si se producen tales reacciones [ver Reacciones adversas (6.1)].
Síndrome de piernas inquietas
Avise a los pacientes que CRYSVITA puede inducir el síndrome de piernas inquietas o empeorar los síntomas del síndrome de piernas inquietas existente. Indique a los pacientes que se pongan en contacto con su médico si se produce tal reacción [ver Reacciones adversas (6.1)].
Embarazo
Informe los embarazos a la línea de informes de eventos adversos de Kyowa Kirin, Inc. al 1-844-768-3544 [ver Uso en poblaciones específicas (8.1)].
Fabricado por:
Kyowa Kirin, Inc.
Princeton, NJ 08540
Licencia de EE. UU. No. 2077
PANEL PRINCIPAL DE VISUALIZACIÓN – Vial de 10 mg/mL
NDC 42747-102-01
CRYSViTA®
(burosumab-twza)
Inyección
10 mg/mL
Vial de dosis única de 1 mL
Kyowa Kirin, Inc.
Licencia de EE. UU. No. 2077

PANEL PRINCIPAL DE VISUALIZACIÓN – Vial Cartón de 10 mg/mL
NDC 42747-102-01
CRYSViTA®
(burosumab-twza)
Inyección
10 mg/mL
Sólo para uso subcutáneo
Vial de dosis única
Deseche la porción no utilizada
Sólo con receta médica
1 vial
PANEL PRINCIPAL DE VISUALIZACIÓN – Vial de 20 mg/mL
NDC 42747-203-01
CRYSViTA®
(burosumab-twza)
Inyección
20 mg/mL
Vial de dosis única de 1 mL
Kyowa Kirin, Inc.
Licencia de EE. UU. No. 2077

PANEL PRINCIPAL DE EXHIBICIÓN – Caja de vial de 20 mg/mL
NDC 42747-203-01
CRYSViTA®
(burosumab-twza)
Inyección
20 mg/mL
Sólo para uso subcutáneo
Vial de dosis única
Deseche la porción no utilizada
Sólo con receta médica
1 vial
PANEL PRINCIPAL DE VISUALIZACIÓN – Vial de 30 mg/mL
NDC 42747-304-01
CRYSViTA®
(burosumab-twza)
Inyección
30 mg/mL
Vial de dosis única de 1 mL
Kyowa Kirin, Inc.
Licencia de EE. UU. No. 2077

PANEL PRINCIPAL DE VISUALIZACIÓN – Vial Cartón de 30 mg/mL
NDC 42747-304-01
CRYSViTA®
(burosumab-twza)
Inyección
30 mg/mL
Sólo para uso subcutáneo
Vial de dosis única
Deseche la porción no utilizada
Rx solamente
1 vial