Fabricante de medicamentos: AstraZeneca Pharmaceuticals LP (Updated: 2025-01-16)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
CALQUENCE® (acalabrutinib) cápsulas, para administración oral
Aprobación inicial en EE. UU.: 2017
CAMBIOS IMPORTANTES RECIENTES
Indicaciones y uso, Linfoma de células del manto (1.1) 1/2025
Posología y administración, Posología recomendada (2.1) 1/2025
Posología y administración, Modificaciones de la posología para reacciones adversas (2.4) 1/2025
Advertencias y precauciones, Segundas neoplasias malignas (5.4) 1/2025
Advertencias y precauciones, Arritmias cardíacas (5.5) 6/2024
Advertencias y precauciones, Hepatotoxicidad, incluida la lesión hepática inducida por fármacos (5.6) 6/2024
INDICACIONES Y USO
CALQUENCE es un inhibidor de la cinasa indicado para:
• En combinación con bendamustina y rituximab para el tratamiento de pacientes adultos con linfoma de células del manto (LCM) previamente no tratado que no son candidatos a trasplante autólogo de células madre hematopoyéticas (TCMH). (1.1)
• Para el tratamiento de pacientes adultos con LCM que han recibido al menos un tratamiento previo. (1.2)
• Para el tratamiento de pacientes adultos con leucemia linfocítica crónica (LLC) o linfoma linfocítico pequeño (LLS). (1.3)
POSOLOGÍA Y ADMINISTRACIÓN
La dosis recomendada es de 100 mg por vía oral aproximadamente cada 12 horas; tragar entera con agua, con o sin alimentos. (2.1)
FORMAS Y CONCENTRACIONES FARMACÉUTICAS
Cápsulas: 100 mg. (3)
CONTRAINDICACIONES
Ninguna. (4)
ADVERTENCIAS Y PRECAUCIONES
- •
- Infecciones graves y oportunistas: Vigilar los signos y síntomas de infección y tratarlos con prontitud. (5.1)
- •
- Hemorragia: Vigilar las hemorragias y controlarlas adecuadamente. (5.2)
- •
- Citopenias: Vigilar regularmente los hemogramas completos. (5.3)
- •
- Segundas neoplasias malignas: Se han producido otras neoplasias malignas, incluidos cánceres de piel y otros tumores sólidos. Aconseje a los pacientes que utilicen protección solar. (5.4)
- •
- Arritmias cardíacas: Vigilar los síntomas de arritmias y controlarlos. (5.5)
- •
- Hepatotoxicidad, incluida la lesión hepática inducida por fármacos: Vigilar la función hepática durante todo el tratamiento. (5.6)
REACCIONES ADVERSAS
Las reacciones adversas más frecuentes (incidencia ≥ 30%) fueron: anemia, neutropenia, infección de las vías respiratorias superiores, trombocitopenia, cefalea, diarrea y dolor musculoesquelético. (6.1)
Para notificar SOSPECHAS DE REACCIONES ADVERSAS, póngase en contacto con AstraZeneca en el 1-800-236-9933 o con la FDA en el 1-800-FDA-1088 o www.fda.gov/medwatch.
INTERACCIONES FARMACOLÓGICAS
- •
- Inhibidores del CYP3A: Evitar la administración conjunta con inhibidores potentes del CYP3A. Pueden recomendarse ajustes de la dosis. (2.3, 7, 12.3)
- •
- Inductores del CYP3A: Evitar la administración conjunta con inductores potentes del CYP3A. Pueden recomendarse ajustes de la dosis. (2.3, 7, 12.3)
- •
- Agentes reductores del ácido gástrico: Evitar la administración conjunta con inhibidores de la bomba de protones (IBP). Espaciar la administración con antagonistas de los receptores H2 y antiácidos. (2.4, 7, 12.3)
USO EN POBLACIONES ESPECÍFICAS
Ver 17 para obtener INFORMACIÓN PARA EL PACIENTE.
Revisado: 1/2025
Tabla de contenido
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDO*
1 INDICACIONES Y USO
1.1 Linfoma de células del manto previamente no tratado
1.2 Linfoma de células del manto previamente tratado
1.3 Leucemia linfocítica crónica o linfoma linfocítico pequeño
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Dosis recomendada
2.2 Dosis recomendada para insuficiencia hepática
2.3 Dosis recomendada para interacciones medicamentosas
2.4 Modificaciones de la dosis para reacciones adversas
3 FORMAS Y CONCENTRACIONES FARMACÉUTICAS
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Infecciones graves y oportunistas
5.2 Hemorragia
5.3 Citopenias
5.4 Segundas neoplasias malignas
5.5 Arritmias cardíacas
5.6 Hepatotoxicidad, incluida la lesión hepática inducida por fármacos
6 REACCIONES ADVERSAS
6.1 Experiencia en ensayos clínicos
6.2 Experiencia postcomercialización
7 INTERACCIONES MEDICAMENTOSAS
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.3 Mujeres y hombres en edad fértil
8.4 Uso pediátrico
8.5 Uso geriátrico
8.6 Insuficiencia hepática
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
12.2 Farmacodinamia
12.3 Farmacocinética
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, mutagénesis, deterioro de la fertilidad
14 ESTUDIOS CLÍNICOS
14.1 Linfoma de células del manto previamente no tratado
14.2 Linfoma de células del manto previamente tratado
14.3 Leucemia linfocítica crónica
16 PRESENTACIÓN/ALMACENAMIENTO Y MANEJO
17 INFORMACIÓN PARA EL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información completa de prescripción no se enumeran.
1 INDICACIONES Y USO
1.1 Linfoma de células del manto previamente no tratado
CALQUENCE en combinación con bendamustina y rituximab está indicado para el tratamiento de pacientes adultos con linfoma de células del manto (LCM) previamente no tratado que no son candidatos a trasplante autólogo de células madre hematopoyéticas (TCMH).
1.2 Linfoma de células del manto previamente tratado
CALQUENCE está indicado para el tratamiento de pacientes adultos con LCM que han recibido al menos un tratamiento previo.
1.3 Leucemia linfocítica crónica o linfoma linfocítico pequeño
CALQUENCE está indicado para el tratamiento de pacientes adultos con leucemia linfocítica crónica (LLC) o linfoma linfocítico pequeño (LLP).
2 DOSIS Y ADMINISTRACIÓN
2.1 Dosis recomendada
Instrucciones de administración de CALQUENCE
Aconseje a los pacientes que traguen la cápsula entera con agua. Aconseje a los pacientes que no abran, rompan ni mastiquen las cápsulas. CALQUENCE se puede tomar con o sin alimentos. Si se olvida una dosis de CALQUENCE durante más de 3 horas, debe omitirse y tomar la siguiente dosis a la hora programada habitualmente. No se deben tomar cápsulas adicionales de CALQUENCE para compensar una dosis olvidada.
CALQUENCE como monoterapia
Para pacientes con LMC, LLC o LLG, la dosis recomendada de CALQUENCE es de 100 mg administrada por vía oral aproximadamente cada 12 horas hasta la progresión de la enfermedad o la toxicidad inaceptable.
CALQUENCE en combinación con bendamustina y rituximab
Para pacientes con LMC previamente no tratada, la dosis recomendada de CALQUENCE es de 100 mg administrada por vía oral aproximadamente cada 12 horas hasta la progresión de la enfermedad o la toxicidad inaceptable.
Comience CALQUENCE el día 1 del ciclo 1 (cada ciclo es de 28 días) y adminístrelo hasta la progresión de la enfermedad o la toxicidad inaceptable. Administre bendamustina 90 mg/m2 los días 1 y 2 y rituximab 375 mg/m2 el día 1 del ciclo 1 y continúe durante un total de 6 ciclos. Los pacientes que logren una respuesta (RP o RC) después de los primeros 6 ciclos pueden recibir rituximab de mantenimiento el día 1 de cada ciclo alternativo durante un máximo de 12 dosis adicionales, comenzando en el ciclo 8 hasta el ciclo 30 [ver Estudios clínicos (14.1)].
CALQUENCE en combinación con obinutuzumab
Para pacientes con LLC o LLG previamente no tratada, la dosis recomendada de CALQUENCE es de 100 mg administrada por vía oral aproximadamente cada 12 horas hasta la progresión de la enfermedad o la toxicidad inaceptable. Comience CALQUENCE en el ciclo 1 (cada ciclo es de 28 días). Comience obinutuzumab en el ciclo 2 durante un total de 6 ciclos y consulte la información de prescripción de obinutuzumab para la dosificación recomendada. Administre CALQUENCE antes que obinutuzumab cuando se administren el mismo día.
2.2 Dosis recomendada para insuficiencia hepática
Evite la administración de CALQUENCE en pacientes con insuficiencia hepática grave.
No se requieren modificaciones de la dosis para pacientes con insuficiencia hepática leve o moderada [ver Uso en poblaciones específicas (8.6) y Farmacología clínica (12.3)].
2.3 Dosis recomendada para interacciones medicamentosas
Modificaciones de la dosis para uso con inhibidores o inductores del CYP3A
Estas se describen en la Tabla 1 [ver Interacciones medicamentosas (7)].
|
CYP3A |
Medicamento coadministrado |
Uso recomendado de CALQUENCE |
|
Inhibición |
Inhibidor potente del CYP3A |
Evite el uso concomitante. Si estos inhibidores se van a utilizar a corto plazo (como antiinfecciosos durante un máximo de siete días), interrumpa CALQUENCE. |
|
Inhibidor moderado del CYP3A |
100 mg una vez al día. |
|
|
Inducción |
Inductores potentes del CYP3A |
Evite el uso concomitante. Si no se pueden evitar estos inductores, aumente la dosis de CALQUENCE a 200 mg aproximadamente cada 12 horas. |
Uso concomitante con agentes reductores del ácido gástrico
Inhibidores de la bomba de protones: Evite el uso concomitante [ver Interacciones medicamentosas (7)].
Antagonistas de los receptores H2: Tome CALQUENCE 2 horas antes de tomar un antagonista de los receptores H2 [ver Interacciones medicamentosas (7)].
Antiácidos: Separe la administración por al menos 2 horas [ver Interacciones medicamentosas (7)].
2.4 Modificaciones de la dosis para reacciones adversas
Las modificaciones de la dosis recomendadas se proporcionan en las Tablas 2 y 3.
|
Evento |
Aparición de la reacción adversa |
Modificación de la dosis (Dosis inicial = 100 mg aproximadamente cada 12 horas) |
|
Toxicidades no hematológicas de grado 3 o superior, Trombocitopenia de grado 3 con hemorragia, |
Primera y segunda |
Interrumpa CALQUENCE. Una vez que la toxicidad se haya resuelto a grado 1 o al nivel basal, CALQUENCE puede reanudarse a 100 mg aproximadamente cada 12 horas. |
|
Trombocitopenia de grado 4 o Neutropenia de grado 4 que dure más de 7 días |
Tercera |
Interrumpa CALQUENCE. Una vez que la toxicidad se haya resuelto a grado 1 o al nivel basal, CALQUENCE puede reanudarse a una frecuencia reducida de 100 mg una vez al día. |
|
Cuarta |
Suspenda CALQUENCE. |
|
|
Las reacciones adversas se clasifican según los Criterios de Terminología Común para Eventos Adversos del Instituto Nacional del Cáncer (NCI CTCAE). |
||
| Reacción adversa | Gravedada | Modificación de la dosis (Dosis inicial de CALQUENCE = 100 mg aproximadamente cada 12 horas) |
|---|---|---|
|
Neutropeniab[ver Advertencias y precauciones (5.4)] |
Recuento absoluto de neutrófilos inferior a 0,5 x 109 /L durante más de 7 días |
Interrupción de CALQUENCE. Una vez que la toxicidad se haya resuelto a Grado ≤ 2, reanudar CALQUENCE con la dosis inicial. Tras la 2ª o 3ª aparición, reducir la dosis de CALQUENCE a 100 mg una vez al día.c Interrumpir CALQUENCE en la 4ª aparición. Para bendamustina: Interrupción de bendamustina. Una vez que la toxicidad se haya resuelto a Grado ≤ 2, reanudar bendamustina y considerar la reducción de la dosis a 70 mg/m2.d,e |
|
Trombocitopeniaf[ver Advertencias y precauciones (5.4)] |
Recuento de plaquetas de 25 a 50 x 109/L con hemorragia clínicamente significativa o recuento de plaquetas inferior a 25 x 109/L |
Interrupción de CALQUENCE. Una vez que la toxicidad se haya resuelto a Grado ≤ 2 o basal, reanudar CALQUENCE con la dosis inicial. Si hay recurrencia, reducir la dosis de CALQUENCE a 100 mg una vez al día.c Considerar la interrupción de CALQUENCE en la 3ª aparición. Para bendamustinaf: Interrupción de bendamustina. Una vez que la toxicidad se haya resuelto a Grado ≤ 2 o basal, reanudar bendamustina y considerar la reducción de la dosis a 70 mg/m2.e |
|
Reacciones adversas no hematológicas [ver Advertencias y precauciones (5)] |
Grado 3 o superior |
Interrupción de CALQUENCE. Una vez que la toxicidad se haya resuelto a Grado ≤ 2 o basal, reanudar CALQUENCE con la dosis inicial. Si hay recurrencia, reducir la dosis de CALQUENCE a 100 mg una vez al día.c Interrumpir CALQUENCE en la 3ª aparición de toxicidad de Grado 4. Para la toxicidad de Grado 3, considerar los riesgos y beneficios de continuar con CALQUENCE. Para bendamustina: Interrupción de bendamustina. Una vez que la toxicidad se haya resuelto a Grado ≤ 2 o basal, reanudar bendamustina y considerar la reducción de la dosis a 70 mg/m2.e |
|
a Clasificado según los Criterios comunes de terminología para eventos adversos del Instituto Nacional del Cáncer (NCI CTCAE) versión 4.03. b Para la neutropenia con ANC inferior a 1 x 109/L, puede ser apropiada la consideración de la interrupción de la dosis de bendamustina y la reducción de la dosis a 70 mg/m2 en ciertas circunstancias. c La dosis puede volver a aumentarse a criterio del médico si el paciente tolera una dosis reducida durante ≥4 semanas. d Considerar el uso de factores de crecimiento mieloides antes de la reducción de la dosis de bendamustina. e Considerar la interrupción de bendamustina si se requiere una reducción adicional de la dosis. f Para la trombocitopenia, un recuento de plaquetas inferior a 50 x 109/L debe provocar la interrupción de la dosis de bendamustina incluso en ausencia de hemorragia clínicamente significativa. |
||
Consulte la información de prescripción de cada uno de los productos utilizados en combinación con CALQUENCE para obtener información adicional sobre el manejo de las toxicidades.
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
Cápsulas: cápsulas de gelatina dura de 100 mg de acalabrutinib tamaño 1, con cuerpo amarillo y tapa azul, marcadas con tinta negra con ‘ACA 100 mg’.
4 CONTRAINDICACIONES
Ninguna.
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Infecciones graves y oportunistas
Se han producido infecciones mortales y graves, incluidas infecciones oportunistas, en pacientes con neoplasias hematológicas tratados con CALQUENCE.
Se produjeron infecciones graves o de Grado 3 o superior (bacterianas, víricas o fúngicas) en el 32 % de los 1764 pacientes expuestos a CALQUENCE en ensayos clínicos, con mayor frecuencia debido a infecciones del tracto respiratorio (19 % de todos los pacientes, incluida la neumonía en el 9 %) [véase Reacciones adversas (6.1)]. Estas infecciones se produjeron predominantemente en ausencia de neutropenia de Grado 3 o 4, y se notificó infección neutropénica en el 2,7 % de todos los pacientes. Las infecciones oportunistas en los receptores de CALQUENCE han incluido, entre otras, la reactivación del virus de la hepatitis B, la neumonía fúngica, la neumonía por Pneumocystis jiroveci, la reactivación del virus de Epstein-Barr, el citomegalovirus y la leucoencefalopatía multifocal progresiva (LMP). Considere la profilaxis en pacientes con mayor riesgo de infecciones oportunistas. Controle a los pacientes para detectar signos y síntomas de infección y trátelos con prontitud.
5.2 Hemorragia
Se han producido acontecimientos hemorrágicos mortales y graves en pacientes tratados con CALQUENCE. Se produjo hemorragia mayor (hemorragia grave o de Grado 3 o superior o cualquier hemorragia del sistema nervioso central) en el 4,4 % de los pacientes, y hemorragia mortal en el 0,2 % de los 1764 pacientes expuestos a CALQUENCE en ensayos clínicos. Se produjeron acontecimientos hemorrágicos de cualquier grado, excluyendo hematomas y petequias, en el 40 % de los pacientes [véase Reacciones adversas (6.1)].
El uso de agentes antitrombóticos concomitantemente con CALQUENCE puede aumentar aún más el riesgo de hemorragia. En los ensayos clínicos, se produjo hemorragia mayor en el 7 % de los pacientes que tomaban CALQUENCE sin agentes antitrombóticos y en el 4 % de los pacientes que tomaban CALQUENCE con agentes antitrombóticos. Considere los riesgos y beneficios de los agentes antitrombóticos cuando se administren conjuntamente con CALQUENCE. Controle a los pacientes para detectar signos de hemorragia.
Considere la relación beneficio-riesgo de suspender CALQUENCE durante 3 a 7 días antes y después de la cirugía, dependiendo del tipo de cirugía y del riesgo de hemorragia.
5.3 Citopenias
CALQUENCE puede causar citopenias de Grado 3 o 4. Las citopenias de Grado 3 o 4 incluyeron disminución del recuento absoluto de neutrófilos (26 %), disminución de plaquetas (10 %), disminución de hemoglobina (10 %) y disminución del recuento absoluto de linfocitos (10 %) en pacientes tratados con CALQUENCE solo o en combinación con obinutuzumab; se desarrolló neutropenia de Grado 4 en el 14 % [véase Reacciones adversas (6.1)].
Controle los hemogramas completos regularmente durante el tratamiento. Interrumpa el tratamiento, reduzca la dosis o suspenda el tratamiento según sea necesario [véase Posología y administración (2.4)].
5.4 Segundas neoplasias malignas
Se produjeron segundas neoplasias malignas, incluidos cánceres de piel y otros tumores sólidos, en el 18 % de los 1764 pacientes expuestos a CALQUENCE en ensayos clínicos [véase Reacciones adversas (6.1)]. La neoplasia maligna secundaria más frecuente fue el cáncer de piel no melanoma, notificado en el 10 % de los pacientes, seguido de otros tumores sólidos en el 9 % (incluidos melanoma, cáncer de pulmón, cánceres gastrointestinales y cánceres genitourinarios) y neoplasias hematológicas (1 %). Controle a los pacientes para detectar el desarrollo de segundos cánceres y aconseje protección contra la exposición al sol.
5.5 Arritmias cardíacas
Se han producido arritmias cardíacas mortales y graves en pacientes tratados con CALQUENCE. Se notificó fibrilación o aleteo auricular de Grado 3 o 4 en el 2,6 % de los 1764 pacientes tratados con CALQUENCE, y todos los grados de fibrilación o aleteo auricular se notificaron en el 7 % de todos los pacientes [véase Reacciones adversas (6.1)]. Se notificaron acontecimientos de arritmia ventricular de Grado 3 o superior en el 0,6 % de los pacientes, incluidos casos mortales en el 0,3 % de todos los pacientes. El riesgo de arritmias puede aumentar en pacientes con factores de riesgo cardíaco, hipertensión, arritmias previas e infección aguda. Controle los síntomas de arritmia (p. ej., palpitaciones, mareos, síncope, disnea) y trátelos según corresponda.
5.6 Hepatotoxicidad, incluida la lesión hepática inducida por fármacos
Se ha producido hepatotoxicidad, incluidos casos graves, potencialmente mortales y potencialmente mortales de lesión hepática inducida por fármacos (LHIF), en pacientes tratados con inhibidores de la tirosina quinasa de Bruton, incluido CALQUENCE.
Evalúe la bilirrubina y las transaminasas al inicio y durante todo el tratamiento con CALQUENCE. En el caso de los pacientes que desarrollan pruebas hepáticas anormales después de CALQUENCE, controle con más frecuencia las anomalías de las pruebas hepáticas y los signos y síntomas clínicos de toxicidad hepática. Si se sospecha LHIF, suspenda CALQUENCE. Tras la confirmación de LHIF, suspenda CALQUENCE.
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas clínicamente significativas se describen con mayor detalle en otras secciones del etiquetado:
- •
- Infecciones graves y oportunistas [ver Advertencias y precauciones (5.1)]
- •
- Hemorragia [ver Advertencias y precauciones (5.2)]
- •
- Citopenias [ver Advertencias y precauciones (5.3)]
- •
- Neoplasias malignas secundarias [ver Advertencias y precauciones (5.4)]
- •
- Arritmias cardíacas [ver Advertencias y precauciones (5.5)]
- •
- Hepatotoxicidad, incluyendo DILI [ver Advertencias y precauciones (5.6)]
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se realizan en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no se pueden comparar directamente con las tasas en los ensayos clínicos de otro fármaco y pueden no reflejar las tasas observadas en la práctica.
Los datos en las Advertencias y precauciones reflejan la exposición a CALQUENCE 100 mg aproximadamente cada 12 horas en 1764 pacientes con neoplasias hematológicas malignas. El tratamiento incluye monoterapia con CALQUENCE en 1256 pacientes en 9 ensayos y combinaciones de CALQUENCE en 508 pacientes en 3 ensayos. Entre estos receptores de CALQUENCE, el 88 % estuvo expuesto durante al menos 6 meses y el 80 % estuvo expuesto durante al menos un año. En esta población de seguridad agrupada, las reacciones adversas en ≥ 30 % de 1764 pacientes, excluyendo las anormalidades de laboratorio, fueron diarrea (37 %), infección del tracto respiratorio superior (36 %), cefalea (35 %), dolor musculoesquelético (33 %), infección del tracto respiratorio inferior (32 %) y fatiga (32 %). Las anormalidades de laboratorio de grado 3 o 4 más comunes (≥ 10 %) fueron disminución del recuento absoluto de neutrófilos (31 %), disminución del recuento absoluto de linfocitos (23 %), disminución de plaquetas (11 %) y disminución de hemoglobina (10 %).
Linfoma de células del manto previamente no tratado
Los datos de seguridad que se describen a continuación reflejan la exposición a CALQUENCE (100 mg aproximadamente cada 12 horas, con o sin BR) en pacientes con LCM [ver Estudios clínicos (14.1)].
ECHO
La seguridad de CALQUENCE en combinación con bendamustina y rituximab (CALQUENCE más BR) se evaluó en 297 pacientes con LCM previamente no tratado en ECHO [ver Estudios clínicos (14.1)]. El ensayo incluyó pacientes con LCM previamente no tratado, ≥ 65 años de edad sin intención de trasplante, bilirrubina total ≤ 1,5 × LSN, AST o ALT ≤ 2,5 × LSN y aclaramiento de creatinina estimado de > 50 mL/min. Los pacientes recibieron 6 ciclos (como ciclos de 28 días) de CALQUENCE 100 mg por vía oral dos veces al día (n = 297) o placebo (n = 297) en combinación con bendamustina y rituximab. Luego, los pacientes recibieron CALQUENCE 100 mg por vía oral dos veces al día o placebo continuamente hasta la enfermedad progresiva o toxicidad inaceptable, con 12 dosis adicionales de rituximab cada dos ciclos hasta el ciclo 30.
La duración media del tratamiento con CALQUENCE fue de 28,6 meses. Un total de 171 (57,6 %) pacientes fueron tratados con CALQUENCE durante ˃ 24 meses y 122 (41,1 %) pacientes fueron tratados durante ˃ 36 meses.
Se produjeron reacciones adversas graves en el 69 % de los pacientes que recibieron CALQUENCE más BR. Las reacciones adversas graves notificadas en ≥ 2 % de los pacientes fueron neumonía (23 %; incluye neumonía por COVID-19), COVID-19 (20 %; incluye neumonía por COVID-19), pirexia (6 %), neoplasia maligna secundaria (7 %), erupción cutánea (3,4 %), neutropenia febril (3,4 %), fibrilación auricular (3 %), sepsis (2,7 %) y anemia (2,4 %). Las reacciones adversas mortales que ocurrieron en los 30 días posteriores al último tratamiento del estudio se notificaron en el 12 % de los que recibieron CALQUENCE más BR, incluida la COVID-19 (6 %; incluye neumonía por COVID-19), neumonía (1 %), sepsis (0,3 %), neoplasia maligna secundaria (0,7 %) y neumonitis (0,3 %).
Las reacciones adversas provocaron la interrupción permanente de CALQUENCE en el 43 %, las interrupciones de la dosis en el 74 % y las reducciones de la dosis en el 10 % de los pacientes. Las reacciones adversas que provocaron una modificación de la dosis en > 10 % incluyeron infecciones, citopenias, erupciones cutáneas y toxicidad gastrointestinal. Las reacciones adversas que provocaron la interrupción permanente de CALQUENCE en ≥ 4 % de los pacientes incluyeron COVID-19 (incluye neumonía por COVID-19) y neutropenia.
Las tablas 4 y 5 resumen las reacciones adversas y las anormalidades de laboratorio seleccionadas observadas en los pacientes tratados en ECHO.
|
Sistemas corporales Reacciones adversas* |
CALQUENCE más BR N = 297 |
Placebo más BR N = 297 |
||||
|
Todos los grados (%) |
Grado 3 o 4 (%) |
Todos los grados (%) |
Grado 3 o 4 (%) |
|||
|
Trastornos de la piel y del tejido subcutáneo |
||||||
|
Erupción cutáneaa |
47 |
12 |
31 |
3 |
||
|
Infecciones |
||||||
|
Covid-19b |
38 |
13 |
27 |
11 |
||
|
Infección del tracto respiratorio superiorc |
30 |
0.7 |
29 |
1 |
||
|
….Neumoníad |
31 |
17 |
25 |
14 |
||
|
Trastornos gastrointestinales |
||||||
|
Diarrea |
37 |
3 |
28 |
2.4 |
||
|
Vómitos |
26 |
0.7 |
14 |
1 |
||
|
Estreñimiento |
25 |
1 |
25 |
0.3 |
||
|
Trastornos generales |
||||||
|
Fatiga |
37 |
3.7 |
32 |
4.4 |
||
|
Pirexia |
29 |
2.4 |
24 |
1.3 |
||
|
Edema |
20 |
1.3 |
19 |
0 |
||
|
Trastornos del sistema nervioso |
||||||
|
Dolor de cabeza |
31 |
1.7 |
14 |
0.7 |
|
Mareo |
18 |
1 |
17 |
0.3 |
|
Trastornos respiratorios, torácicos y mediastínicos |
||||
|
Tos |
27 |
0 |
20 |
0.3 |
|
Disnea |
17 |
1 |
11 |
2.7 |
|
Neoplasias |
||||
|
Neoplasia maligna secundaria primaria |
19 |
7 |
15 |
7 |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
||||
|
Artralgia |
18 |
0.7 |
16 |
1 |
|
Trastornos vasculares |
||||
|
Hemorragiaf |
||||
|
*Excluye términos de laboratorio. a Incluye erupción cutánea, dermatitis y otros términos relacionados. b Incluye las siguientes reacciones adversas mortales: n=24 para COVID-19. c Incluye infección del tracto respiratorio superior, sinusitis, faringitis y términos relacionados. d Incluye neumonía, términos que contienen neumonía e infecciones relacionadas. La neumonía por COVID-19 se representa tanto en Neumonía como en COVID-19. e Incluye términos relacionados con neoplasias malignas, incluidas las neoplasias cutáneas. f Incluye todos los términos que contienen hematoma o hemorragia y términos relacionados que indican sangrado. |
||||
Las reacciones adversas clínicamente relevantes en < 15 % de los pacientes que recibieron CALQUENCE más BR incluyeron hematomas, dolor abdominal, fibrilación o aleteo auricular y síndrome de lisis tumoral.
|
Anormalidad de laboratorio |
CALQUENCE más BRa |
Placebo más BRa |
||||
|
Todos los grados (%) |
Grado 3 o 4 (%) |
Todos los grados (%) |
Grado 3 o 4 (%) |
|||
|
Anormalidades hematológicas |
||||||
|
Linfocitos disminuidos |
98 |
87 |
97 |
89 |
||
|
Hemoglobina disminuida |
80 |
11 |
65 |
11 |
||
|
Neutrófilos disminuidos |
76 |
56 |
77 |
51 |
||
|
Plaquetas disminuidas |
69 |
18 |
60 |
16 |
||
|
Anormalidades químicas |
||||||
|
AST aumentada |
53 |
5 |
50 |
3.4 |
||
|
Ácido úrico aumentado |
45 |
45 |
40 |
40 |
||
|
ALT aumentada |
44 |
7 |
41 |
2.4 |
||
|
Potasio aumentado |
40 |
2 |
38 |
2.7 |
||
|
Creatinina aumentada |
37 |
3 |
28 |
2.4 |
||
|
Fosfato disminuido |
36 |
4.4 |
30 |
4.7 |
|
Potasio disminuido |
29 |
7 |
23 |
6 |
|
Bilirrubina aumentada |
19 |
2 |
12 |
2 |
|
a El denominador utilizado para calcular la tasa varió entre 296 y 297 según el número de pacientes con un valor basal y al menos un valor posterior al tratamiento. |
||||
Las anormalidades de laboratorio de Grado 4 en > 15% de los pacientes tratados con CALQUENCE más BR incluyen disminución del recuento absoluto de linfocitos (26%), disminución del recuento absoluto de neutrófilos (36%) y aumento del ácido úrico (17%).
Linfoma de células del manto previamente tratado
ACE-LY-004
Los datos de seguridad descritos en esta sección reflejan la exposición a CALQUENCE (100 mg aproximadamente cada 12 horas) en 124 pacientes con MCL previamente tratados en el ensayo LY-004 [ver Estudios clínicos (14.2)]. La duración mediana del tratamiento con CALQUENCE fue de 16,6 (rango: 0,1 a 26,6) meses. Un total de 91 (73,4%) pacientes fueron tratados con CALQUENCE durante ≥ 6 meses y 74 (59,7%) pacientes fueron tratados durante ≥ 1 año.
Las reacciones adversas más comunes (≥ 20%) de cualquier grado fueron anemia, trombocitopenia, cefalea, neutropenia, diarrea, fatiga, mialgia y equimosis. La gravedad de Grado 1 para los eventos no hematológicos más comunes fue la siguiente: cefalea (25%), diarrea (16%), fatiga (20%), mialgia (15%) y equimosis (19%). La reacción adversa no hematológica más común de Grado ≥ 3 (informada en al menos el 2% de los pacientes) fue la diarrea.
Las reducciones de dosis y la interrupción debido a cualquier reacción adversa se informaron en el 1,6% y el 6,5% de los pacientes, respectivamente.
Los cuadros 6 y 7 presentan la categoría de frecuencia de las reacciones adversas observadas en pacientes con MCL tratados con CALQUENCE.
|
Sistema orgánico Reacciones adversas* |
Monoterapia con CALQUENCE N=124 |
|
|
Todos los grados (%) |
Grado ≥ 3 (%) |
|
|
Trastornos del sistema nervioso |
||
|
Cefalea |
39 |
1.6 |
|
Trastornos gastrointestinales |
||
|
Diarrea |
31 |
3.2 |
|
Náuseas |
19 |
0.8 |
|
Dolor abdominal |
15 |
1.6 |
|
Estreñimiento |
15 |
– |
|
Vómitos |
13 |
1.6 |
|
Trastornos generales |
||
|
Fatiga |
28 |
0.8 |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
||
|
Mialgia |
21 |
0.8 |
|
Trastornos de la piel y del tejido subcutáneo |
||
|
Equimosis† |
21 |
– |
|
Erupción‡ |
18 |
0.8 |
|
Trastornos vasculares |
||
|
Hemorragia§ |
8 |
0.8 |
|
Trastornos respiratorios, torácicos y mediastínicos |
||
|
Epistaxis |
6 |
– |
|
||
|
Hematología Reacciones adversas* |
Monoterapia con CALQUENCE N=124 |
|
|
Todos los grados (%) |
Grado ≥ 3 (%) |
|
|
Disminución de la hemoglobina |
46 |
10 |
|
Disminución de las plaquetas |
44 |
12 |
|
Disminución de neutrófilos |
36 |
15 |
Se produjeron aumentos de creatinina de 1,5 a 3 veces el límite superior de lo normal en el 4,8 % de los pacientes.
Leucemia Linfocítica Crónica
Los datos de seguridad que se describen a continuación reflejan la exposición a CALQUENCE (100 mg aproximadamente cada 12 horas, con o sin obinutuzumab) en 511 pacientes con LLC de dos ensayos clínicos controlados aleatorizados [véase Estudios clínicos (14.3)].
Las reacciones adversas más comunes (≥ 30%) de cualquier grado en pacientes con LLC fueron anemia, neutropenia, trombocitopenia, cefalea, infección del tracto respiratorio superior y diarrea.
ELEVATE-TN
La seguridad de CALQUENCE más obinutuzumab (CALQUENCE+G), CALQUENCE en monoterapia y obinutuzumab más clorambucilo (GClb) se evaluó en un ensayo aleatorizado, multicéntrico, abierto y controlado activamente en 526 pacientes con LLC previamente no tratada [véase Estudios clínicos (14.2)].
Las reacciones adversas más comunes (≥ 30%) de cualquier grado en pacientes con LLC fueron anemia, neutropenia, trombocitopenia, cefalea, infección del tracto respiratorio superior y diarrea.
ELEVATE-TN
La seguridad de CALQUENCE más obinutuzumab (CALQUENCE+G), CALQUENCE en monoterapia y obinutuzumab más clorambucilo (GClb) se evaluó en un ensayo aleatorizado, multicéntrico, abierto y controlado activamente en 526 pacientes con LLC previamente no tratada [véase Estudios clínicos (14.3)].
Los pacientes asignados al azar al brazo CALQUENCE+G fueron tratados con CALQUENCE y obinutuzumab en combinación durante seis ciclos, luego con CALQUENCE en monoterapia hasta la progresión de la enfermedad o toxicidad inaceptable. Los pacientes iniciaron obinutuzumab el día 1 del ciclo 2, continuando durante un total de 6 ciclos. El paciente aleatorizado a la monoterapia con CALQUENCE recibió CALQUENCE aproximadamente cada 12 horas hasta la progresión de la enfermedad o la toxicidad inaceptable. El ensayo requirió una edad ≥ 65 años o de 18 a < 65 años con una puntuación total de la Escala de Evaluación de Enfermedades Acumuladas (CIRS) > 6 o un aclaramiento de creatinina de 30 a 69 mL/min, transaminasas hepáticas ≤ 3 veces el LSN y bilirrubina total ≤ 1,5 veces el LSN, y permitió que los pacientes recibieran agentes antitrombóticos distintos de la warfarina o antagonistas de la vitamina K equivalentes.
Durante el tratamiento aleatorizado, la duración media de la exposición a CALQUENCE en los brazos de CALQUENCE+G y monoterapia con CALQUENCE fue de 27,7 meses (rango 0,3 a 40 meses), con un 95 % y un 92 % y un 89 % y un 86 % de los pacientes con al menos 6 meses y 12 meses de exposición, respectivamente. En el brazo de obinutuzumab y clorambucilo, el número medio de ciclos fue de 6, con un 84 % de los pacientes que recibieron al menos 6 ciclos de obinutuzumab, el 70 % de los pacientes recibieron al menos 6 ciclos de clorambucilo. El ochenta y cinco por ciento de los pacientes en el brazo CALQUENCE+G recibieron al menos 6 ciclos de obinutuzumab.
En los brazos de CALQUENCE+G y monoterapia con CALQUENCE, se informaron reacciones adversas mortales que ocurrieron en ausencia de progresión de la enfermedad y con inicio dentro de los 30 días posteriores al último tratamiento del estudio en un 2 % para cada brazo de tratamiento, con mayor frecuencia por infección. Se informaron reacciones adversas graves en el 39 % de los pacientes en el brazo CALQUENCE+G y en el 32 % en el brazo de monoterapia con CALQUENCE, con mayor frecuencia debido a eventos de neumonía (del 2,8 % al 7 %).
En el brazo CALQUENCE+G, las reacciones adversas provocaron la interrupción del tratamiento en el 11 % de los pacientes y una reducción de la dosis de CALQUENCE en el 7 % de los pacientes. En el brazo de monoterapia con CALQUENCE, las reacciones adversas provocaron la interrupción en el 10 % y la reducción de la dosis en el 4 % de los pacientes.
Los cuadros 8 y 9 presentan las reacciones adversas y las anormalidades de laboratorio identificadas en el ensayo ELEVATE-TN.
|
||||||
|
Sistema orgánico Reacción adversa*
|
CALQUENCE más Obinutuzumab |
Monoterapia con CALQUENCE N=179 |
Obinutuzumab más Clorambucil |
|||
|
Todos los grados (%) |
Grado ≥ 3 (%) |
Todos los grados (%) |
Grado ≥ 3 (%) |
Todos los grados (%) |
Grado ≥ 3 (%) |
|
|
||||||
|
69 |
22‡ |
65 |
14‡ |
46 |
13‡ |
|
39 |
2.8 |
35 |
0 |
17 |
1.2 |
|
24 |
8 |
18 |
4.5 |
7 |
1.8 |
|
15 |
1.7 |
15 |
2.8 |
5 |
0.6 |
|
||||||
|
53 |
37 |
23 |
13 |
78 |
50 |
|
52 |
12 |
53 |
10 |
54 |
14 |
|
51 |
12 |
32 |
3.4 |
61 |
16 |
|
12 |
11 |
16 |
15 |
0.6 |
0.6 |
|
||||||
|
40 |
1.1 |
39 |
1.1 |
12 |
0 |
|
20 |
0 |
12 |
0 |
7 |
0 |
|
||||||
|
39 |
4.5 |
35 |
0.6 |
21 |
1.8 |
|
20 |
0 |
22 |
0 |
31 |
0 |
|
||||||
|
37 |
2.2 |
32 |
1.1 |
16 |
2.4 |
|
22 |
1.1 |
16 |
0.6 |
4.7 |
1.2 |
|
||||||
|
34 |
2.2 |
23 |
1.1 |
24 |
1.2 |
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
20 |
1.7 |
20 |
1.7 |
6 |
0 |
Otras reacciones adversas clínicamente relevantes (incidencia de todos los grados < 15%) en los receptores de CALQUENCE (CALQUENCE en combinación con obinutuzumab y monoterapia) incluyeron:
- •
- Neoplasias: segunda neoplasia maligna (10%), cáncer de piel no melanoma (5%)
- •
- Trastornos cardíacos: fibrilación o aleteo auricular (3,6%), hipertensión (5%)
- •
- Infección: infección por herpesvirus (6%)
| Anormalidad de laboratorio*† | CALQUENCE más Obinutuzumab N=178 |
Monoterapia con CALQUENCE N=179 |
Obinutuzumab más Clorambucil N=169 |
|||
|---|---|---|---|---|---|---|
| Todos los grados (%) |
Grado ≥ 3 (%) | Todos los grados (%) |
Grado ≥ 3 (%) | Todos los grados (%) |
Grado ≥ 3 (%) | |
|
Aumento del ácido úrico |
29 |
29 |
22 |
22 |
37 |
37 |
|
Aumento de ALT |
30 |
7 |
20 |
1.1 |
36 |
6 |
|
Aumento de AST |
38 |
5 |
17 |
0.6 |
60 |
8 |
|
Aumento de bilirrubina |
13 |
0.6 |
15 |
0.6 |
11 |
0.6 |
Se produjeron aumentos de creatinina de 1,5 a 3 veces el LSN en el 3,9 % y el 2,8 % de los pacientes en el brazo de combinación de CALQUENCE y en el brazo de monoterapia, respectivamente.
ASCEND
La seguridad de CALQUENCE en pacientes con LMC recidivante o refractaria se evaluó en un estudio aleatorizado, abierto (ASCEND) [véase Estudios clínicos (14.3)]. El ensayo incluyó pacientes con LMC recidivante o refractaria después de al menos un tratamiento previo y requirió transaminasas hepáticas ≤ 2 veces el LSN, bilirrubina total ≤ 1,5 veces el LSN y un aclaramiento de creatinina estimado ≥ 30 mL/min. El ensayo excluyó a los pacientes que tenían un recuento absoluto de neutrófilos < 500/µL, un recuento de plaquetas < 30 000/µL, un tiempo de protrombina o un tiempo de tromboplastina parcial activado > 2 veces el LSN, una enfermedad cardiovascular significativa o la necesidad de inhibidores o inductores potentes del CYP3A. Se permitió que los pacientes recibieran agentes antitrombóticos que no fueran warfarina o un antagonista de la vitamina K equivalente.
En ASCEND, 154 pacientes recibieron CALQUENCE (100 mg aproximadamente cada 12 horas hasta la progresión de la enfermedad o la toxicidad inaceptable), 118 recibieron idelalisib (150 mg aproximadamente cada 12 horas hasta la progresión de la enfermedad o la toxicidad inaceptable) con hasta 8 infusiones de un producto de rituximab, y 35 recibieron hasta 6 ciclos de bendamustina y un producto de rituximab. La mediana de edad general fue de 68 años (rango: 32-90); el 67 % eran hombres; el 92 % eran blancos; y el 88 % tenían un estado de rendimiento ECOG de 0 o 1.
En el brazo de CALQUENCE, se produjeron reacciones adversas graves en el 29 % de los pacientes. Las reacciones adversas graves en > 5 % de los pacientes que recibieron CALQUENCE incluyeron infección del tracto respiratorio inferior (6 %). Las reacciones adversas mortales en los 30 días posteriores a la última dosis de CALQUENCE se produjeron en el 2,6 % de los pacientes, incluidas las de neoplasias malignas secundarias e infecciones.
En los receptores de CALQUENCE, la interrupción permanente debido a una reacción adversa se produjo en el 10 % de los pacientes, con mayor frecuencia debido a neoplasias malignas secundarias seguidas de infección. Las reacciones adversas provocaron interrupciones de la dosificación de CALQUENCE en el 34 % de los pacientes, con mayor frecuencia debido a infecciones del tracto respiratorio seguidas de neutropenia, y reducción de la dosis en el 3,9 % de los pacientes.
Las reacciones adversas seleccionadas se describen en la Tabla 10 y las anomalías de laboratorio no hematológicas se describen en la Tabla 11. Estas tablas reflejan la exposición a CALQUENCE con una duración mediana de 15,7 meses, con el 94 % de los pacientes en tratamiento durante más de 6 meses y el 86 % de los pacientes en tratamiento durante más de 12 meses. La duración mediana de la exposición a idelalisib fue de 11,5 meses, con el 72 % de los pacientes en tratamiento durante más de 6 meses y el 48 % de los pacientes en tratamiento durante más de 12 meses. El ochenta y tres por ciento de los pacientes completaron 6 ciclos de bendamustina y producto de rituximab.
|
||||||
|
Sistema orgánico Reacción adversa*
|
CALQUENCE N=154 |
Idelalisib más Rituximab Producto N=118 |
Bendamustina más producto de Rituximab N=35 |
|||
|
Todos |
Grado ≥ 3 (%) |
Todos |
Grado ≥ 3 (%) |
Todos |
Grado ≥ 3 (%) |
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
Otras reacciones adversas clínicamente relevantes (incidencia de todos los grados < 15%) en los receptores de CALQUENCE incluyeron:
- •
- Trastornos de la piel y del tejido subcutáneo: equimosis (10%), erupción cutánea (9%)
- •
- Neoplasias: segunda neoplasia maligna (12%), cáncer de piel no melanoma (6%)
- •
- Trastornos musculoesqueléticos y del tejido conjuntivo: artralgia (8%)
- •
- Trastornos cardíacos: fibrilación o aleteo auricular (5%), hipertensión (3.2%)
- •
- Infección: infección por herpesvirus (4.5%)
| Anomalía de laboratorio* | CALQUENCE N=154 |
Idelalisib más Rituximab N=118 |
Bendamustina más Rituximab N=35 |
|||
|---|---|---|---|---|---|---|
| Todos los grados (%) |
Grado ≥ 3 (%) | Todos los grados (%) |
Grado ≥ 3 (%) | Todos los grados (%) |
Grado ≥ 3 (%) | |
|
||||||
|
Aumento del ácido úrico |
15 |
15 |
11 |
11 |
23 |
23 |
|
Aumento de ALT |
15 |
1.9 |
59 |
23 |
26 |
2.9 |
|
Aumento de AST |
13 |
0.6 |
48 |
13 |
31 |
2.9 |
|
Aumento de bilirrubina |
13 |
1.3 |
16 |
1.7 |
26 |
11 |
|
Por NCI CTCAE versión 5 |
||||||
6.2 Experiencia postcomercialización
Las siguientes reacciones adversas se han identificado durante el uso de CALQUENCE después de la aprobación. Debido a que estas reacciones se notifican voluntariamente a partir de una población de tamaño incierto, no siempre es posible estimar su frecuencia de manera fiable o establecer una relación causal con la exposición al fármaco.
- •
- Trastornos cardíacos: arritmias ventriculares
- •
- Trastornos hepatobiliares: lesión hepática inducida por fármacos
7 INTERACCIONES MEDICAMENTOSAS
|
Inhibidores potentes del CYP3A |
||
|
Impacto clínico |
|
|
|
Prevención o manejo |
|
|
|
Inhibidores moderados del CYP3A |
||
|
Impacto clínico |
|
|
|
Prevención o manejo |
|
|
|
Inductores potentes del CYP3A |
||
|
Impacto clínico |
|
|
|
Prevención o manejo |
|
|
|
Agentes reductores del ácido gástrico |
||
|
Impacto clínico |
|
|
|
Prevención o manejo |
Antiácidos |
Separar la administración por al menos 2 horas [véase Dosis recomendada para interacciones medicamentosas (2.3)]. |
|
Antagonistas de los receptores H2 |
Tome CALQUENCE 2 horas antes de tomar el antagonista de los receptores H2 [véase Dosis recomendada para interacciones medicamentosas (2.3)]. |
|
|
Inhibidores de la bomba de protones |
Evite la administración conjunta. Debido al efecto duradero de los inhibidores de la bomba de protones, la separación de las dosis puede no eliminar la interacción con CALQUENCE. |
|
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de Riesgo
Basándose en los hallazgos en animales, CALQUENCE puede causar daño fetal y distocia cuando se administra a una mujer embarazada. No hay datos disponibles en mujeres embarazadas para informar sobre el riesgo asociado al fármaco. En estudios de reproducción animal, la administración de acalabrutinib a animales durante la organogénesis provocó distocia en ratas y reducción del crecimiento fetal en conejos con exposiciones maternas (AUC) 2 veces superiores a las exposiciones en pacientes a la dosis recomendada de 100 mg aproximadamente cada 12 horas (ver Datos). Se debe advertir a las mujeres embarazadas sobre el riesgo potencial para el feto.
Se desconoce el riesgo de fondo estimado de defectos congénitos importantes y aborto espontáneo para la población indicada. Todos los embarazos tienen un riesgo de fondo de defectos congénitos, pérdida u otros resultados adversos. En la población general de EE. UU., el riesgo de fondo estimado de defectos congénitos importantes y aborto espontáneo en embarazos clínicamente reconocidos es del 2-4 % y del 15-20 %, respectivamente.
Datos
Datos en animales
En un estudio combinado de fertilidad y desarrollo embriofetal en ratas hembra, se administró acalabrutinib por vía oral en dosis de hasta 200 mg/kg/día a partir de 14 días antes del apareamiento hasta el día de gestación [GD] 17. No se observaron efectos en el desarrollo y la supervivencia embriofetales. El AUC a 200 mg/kg/día en ratas embarazadas fue aproximadamente 9 veces el AUC en pacientes a la dosis recomendada de 100 mg aproximadamente cada 12 horas. Se confirmó la presencia de acalabrutinib y su metabolito activo en el plasma fetal de rata.
En un estudio de desarrollo embriofetal en conejos, se administró acalabrutinib por vía oral a animales preñados en dosis de hasta 200 mg/kg/día durante el período de organogénesis (desde el GD 6-18). La administración de acalabrutinib en dosis ≥ 100 mg/kg/día produjo toxicidad materna y 100 mg/kg/día provocó una disminución del peso corporal fetal y un retraso en la osificación esquelética. El AUC a 100 mg/kg/día en conejas preñadas fue aproximadamente 2 veces el AUC en pacientes a 100 mg aproximadamente cada 12 horas.
En un estudio de desarrollo pre y postnatal en ratas, se administró acalabrutinib por vía oral a animales preñados durante la organogénesis, el parto y la lactancia, en dosis de 50, 100 y 150 mg/kg/día. Se observó distocia (parto prolongado o difícil) y mortalidad de la descendencia en dosis ≥ 100 mg/kg/día. El AUC a 100 mg/kg/día en ratas preñadas fue aproximadamente 2 veces el AUC en pacientes a 100 mg aproximadamente cada 12 horas. También se observó una papila renal subdesarrollada en la descendencia de la generación F1 a 150 mg/kg/día con un AUC aproximadamente 5 veces superior al AUC en pacientes a 100 mg aproximadamente cada 12 horas.
8.2 Lactancia
Resumen de Riesgo
No hay datos disponibles sobre la presencia de acalabrutinib o su metabolito activo en la leche materna, sus efectos en el niño amamantado o en la producción de leche. Acalabrutinib y su metabolito activo estuvieron presentes en la leche de ratas lactantes. Debido al potencial de reacciones adversas en un niño amamantado por CALQUENCE, se debe aconsejar a las mujeres lactantes que no amamanten mientras toman CALQUENCE y durante al menos 2 semanas después de la dosis final.
8.3 Mujeres y hombres en edad fértil
CALQUENCE puede causar daño embriofetal y distocia cuando se administra a mujeres embarazadas [ver Uso en poblaciones específicas (8.1)].
Prueba de embarazo
Se recomienda realizar una prueba de embarazo a las mujeres en edad fértil antes de iniciar el tratamiento con CALQUENCE.
Anticoncepción
Mujeres
Se debe aconsejar a las pacientes en edad fértil que utilicen un método anticonceptivo eficaz durante el tratamiento con CALQUENCE y durante al menos 1 semana después de la última dosis de CALQUENCE. Si este medicamento se usa durante el embarazo, o si la paciente queda embarazada mientras toma este medicamento, se debe informar a la paciente sobre el posible peligro para el feto.
8.4 Uso pediátrico
No se ha establecido la seguridad y eficacia de CALQUENCE en pacientes pediátricos.
8.5 Uso en geriatría
LLC y MCL previamente tratados
De los 1467 pacientes tratados con CALQUENCE con LLC o MCL recidivante o refractario en ensayos clínicos, 977 (67 %) tenían 65 años o más, y 328 (22 %) tenían 75 años o más. Entre los pacientes de 65 años o más, el 74 % tuvo reacciones adversas de grado 3 o superior y el 58 % tuvo reacciones adversas graves. Entre los pacientes menores de 65 años, el 61 % tuvo reacciones adversas de grado 3 o superior y el 39 % tuvo reacciones adversas graves. No se observaron diferencias clínicamente relevantes en la eficacia entre los pacientes ≥ 65 años y los más jóvenes.
MCL previamente no tratado
De los 297 pacientes tratados con CALQUENCE con MCL previamente no tratado, 214 (72 %) tenían entre 65 y 74 años y 83 (28 %) tenían 75 años o más. No se observaron diferencias clínicamente relevantes en la seguridad o la eficacia entre los pacientes de 65 a 74 años y los que tenían 75 años o más.
8.6 Insuficiencia hepática
Evitar la administración de CALQUENCE en pacientes con insuficiencia hepática grave. No se ha evaluado la seguridad de CALQUENCE en pacientes con insuficiencia hepática moderada o grave [ver Dosis recomendada para insuficiencia hepática (2.2) y Farmacología clínica (12.3)].
11 DESCRIPCIÓN
CALQUENCE (acalabrutinib) es un inhibidor de la tirosina quinasa de Bruton (BTK). La fórmula molecular del acalabrutinib es C26H23N7O2, y el peso molecular es 465,51. El nombre químico es 4-{8-amino-3-[(2S)-1-(but-2-inoil)pirrolidin-2-il]imidazo[1,5-a]pirazin-1-il)}-N-(piridin-2-il)benzamida.
La estructura química del acalabrutinib se muestra a continuación:
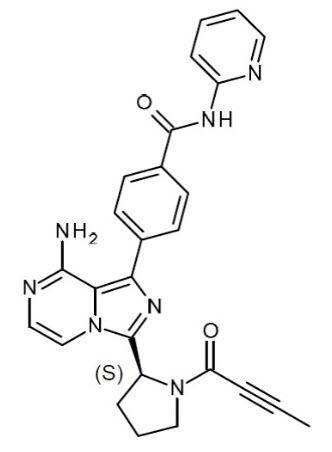
El acalabrutinib es un polvo blanco a amarillo con solubilidad dependiente del pH. Es libremente soluble en agua a valores de pH inferiores a 3 y prácticamente insoluble a valores de pH superiores a 6.
Las cápsulas CALQUENCE para administración oral contienen 100 mg de acalabrutinib y los siguientes ingredientes inactivos: celulosa microcristalina silicificada, almidón parcialmente pregelatinizado, estearato de magnesio y glicolato de almidón sódico. La cápsula contiene gelatina, dióxido de titanio, óxido de hierro amarillo, FD&C Blue 2 y está impresa con tinta negra comestible.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
Acalabrutinib es un inhibidor de BTK de molécula pequeña. Acalabrutinib y su metabolito activo, ACP-5862, forman un enlace covalente con un residuo de cisteína en el sitio activo de BTK, lo que lleva a la inhibición de la actividad enzimática de BTK. BTK es una molécula de señalización del receptor de antígeno de células B (BCR) y de las vías del receptor de citocinas. En las células B, la señalización de BTK da como resultado la activación de las vías necesarias para la proliferación, el tráfico, la quimiotaxis y la adhesión de las células B. En estudios no clínicos, acalabrutinib inhibió la activación mediada por BTK de las proteínas de señalización descendentes CD86 y CD69 e inhibió la proliferación de células B malignas y el crecimiento tumoral en modelos de xenotrasplante de ratón.
12.2 Farmacodinamia
En pacientes con neoplasias malignas de células B tratados con 100 mg aproximadamente cada 12 horas, se mantuvo una ocupación de BTK media en estado estacionario de ≥ 95% en sangre periférica durante 12 horas, lo que resultó en la inactivación de BTK durante todo el intervalo de dosificación recomendado.
Electrofisiología cardíaca
El efecto de acalabrutinib en el intervalo QTc se evaluó en un estudio de QTc completo de 4 vías con diseño cruzado, aleatorizado, doble ciego, doble placebo, controlado con placebo y positivo, en 48 sujetos adultos sanos. La administración de una dosis única de acalabrutinib que es la dosis única máxima recomendada multiplicada por 4 no prolongó el intervalo QTc en ninguna medida clínicamente relevante (es decir, ≥ 10 ms).
12.3 Farmacocinética
Acalabrutinib exhibe proporcionalidad a la dosis, y las exposiciones tanto de acalabrutinib como de su metabolito activo, ACP-5862, aumentan con la dosis en un rango de dosis de 75 a 250 mg (0,75 a 2,5 veces la dosis única recomendada aprobada) en pacientes con neoplasias malignas de células B. A la dosis recomendada de 100 mg dos veces al día, la media geométrica (% coeficiente de variación [CV]) del área bajo la curva de concentración plasmática del fármaco en función del tiempo (AUC24h) y la concentración plasmática máxima (Cmax) para acalabrutinib fueron 1843 (38%) ng•h/mL y 563 (29%) ng/mL, respectivamente, y para ACP-5862 fueron 3947 (43%) ng•h/mL y 451 (52%) ng/mL, respectivamente.
Absorción
La media geométrica de la biodisponibilidad absoluta de acalabrutinib fue del 25%. El tiempo medio [mín, máx] hasta alcanzar las concentraciones plasmáticas máximas de acalabrutinib (Tmax) fue de 0,9 [0,5, 1,9] horas, y de 1,6 [0,9, 2,7] horas para ACP-5862.
Efecto de los alimentos
En sujetos sanos, la administración de una dosis única de 75 mg de acalabrutinib (0,75 veces la dosis única recomendada aprobada) con una comida alta en grasas y calorías (aproximadamente 918 calorías, 59 gramos de carbohidratos, 59 gramos de grasa y 39 gramos de proteína) no afectó el AUC medio en comparación con la administración en ayunas. La Cmax resultante disminuyó en un 73% y la Tmax se retrasó entre 1 y 2 horas.
Distribución
La unión reversible a proteínas plasmáticas humanas fue del 97,5% para acalabrutinib y del 98,6% para ACP-5862. La relación sangre-plasma media in vitro fue de 0,8 para acalabrutinib y de 0,7 para ACP-5862. La media geométrica (% CV) del volumen de distribución en estado estacionario (Vss) fue de aproximadamente 101 (52%) L para acalabrutinib y de 67 (32%) L para ACP-5862.
Eliminación
La media geométrica (% CV) de la semivida de eliminación terminal (t1/2) fue de 1 (59%) hora para acalabrutinib y de 3,5 (24%) horas para ACP-5862. La media geométrica (%CV) del aclaramiento oral aparente (CL/F) fue de 71 (35%) L/h para acalabrutinib y de 13 (42%) L/h para ACP-5862.
Metabolismo
Acalabrutinib se metaboliza predominantemente mediante enzimas CYP3A y, en menor medida, mediante conjugación con glutatión e hidrólisis de amida, según estudios in vitro. ACP-5862 se identificó como el principal metabolito activo en plasma con una exposición media geométrica (AUC) que fue aproximadamente de 2 a 3 veces mayor que la exposición de acalabrutinib. ACP-5862 es aproximadamente un 50% menos potente que acalabrutinib con respecto a la inhibición de BTK.
Excreción
Tras la administración de una dosis única de 100 mg de acalabrutinib radiomarcado en sujetos sanos, se recuperó el 84% de la dosis en las heces y el 12% de la dosis en la orina, con menos del 2% de la dosis excretada como acalabrutinib inalterado en la orina y las heces.
Poblaciones específicas
Edad, raza y peso corporal
La edad (32 a 90 años), el sexo, la raza (caucásica, afroamericana) y el peso corporal (40 a 149 kg) no tuvieron efectos clínicamente significativos en la farmacocinética de acalabrutinib y su metabolito activo, ACP-5862.
Insuficiencia renal
No se observó ninguna diferencia farmacocinética clínicamente relevante en pacientes con insuficiencia renal leve o moderada (eGFR ≥ 30 mL/min/1,73 m2, según lo estimado por la ecuación MDRD (modificación de la dieta en la enfermedad renal)). La farmacocinética de acalabrutinib no se ha evaluado en pacientes con insuficiencia renal grave (eGFR < 29 mL/min/1,73 m2, MDRD) o insuficiencia renal que requiera diálisis.
Insuficiencia hepática
El AUC de acalabrutinib aumentó 1,9 veces en sujetos con insuficiencia hepática leve (clase A de Child-Pugh), 1,5 veces en sujetos con insuficiencia hepática moderada (clase B de Child-Pugh) y 5,3 veces en sujetos con insuficiencia hepática grave (clase C de Child-Pugh) en comparación con sujetos con función hepática normal. No se observó ninguna diferencia farmacocinética clínicamente relevante en ACP-5862 en sujetos con insuficiencia hepática grave (clase C de Child-Pugh) en comparación con sujetos con función hepática normal. No se observaron diferencias farmacocinéticas clínicamente relevantes en acalabrutinib y ACP-5862 en pacientes con insuficiencia hepática leve o moderada (bilirrubina total menor o igual al límite superior de la normalidad [LSN] y AST mayor que LSN, o bilirrubina total mayor que LSN y cualquier AST) en relación con pacientes con función hepática normal (bilirrubina total y AST dentro del LSN).
Estudios de interacción farmacológica
Efecto de los inhibidores de CYP3A en acalabrutinib
La administración conjunta con un inhibidor potente del CYP3A (200 mg de itraconazol una vez al día durante 5 días) aumentó la Cmax de acalabrutinib en 3,9 veces y el AUC en 5,1 veces en sujetos sanos.
Las simulaciones farmacocinéticas basadas en la fisiología (PBPK) con acalabrutinib e inhibidores moderados del CYP3A (eritromicina, fluconazol, diltiazem) mostraron que la administración conjunta aumentó la Cmax y el AUC de acalabrutinib aproximadamente de 2 a 3 veces.
Efecto de los inductores del CYP3A sobre el acalabrutinib
La administración conjunta con un inductor potente del CYP3A (600 mg de rifampicina una vez al día durante 9 días) disminuyó la Cmax de acalabrutinib en un 68 % y el AUC en un 77 % en sujetos sanos.
Agentes reductores del ácido gástrico
La solubilidad del acalabrutinib disminuye con el aumento del pH. La administración conjunta con un antiácido (1 g de carbonato de calcio) disminuyó el AUC de acalabrutinib en un 53 % en sujetos sanos. La administración conjunta con un inhibidor de la bomba de protones (40 mg de omeprazol durante 5 días) disminuyó el AUC de acalabrutinib en un 43 %.
Estudios In Vitro
Vías metabólicas
El acalabrutinib es un inhibidor débil del CYP3A4/5, CYP2C8 y CYP2C9, pero no inhibe el CYP1A2, CYP2B6, CYP2C19, CYP2D6, UGT1A1 y UGT2B7. El ACP-5862 es un inhibidor débil del CYP2C8, CYP2C9 y CYP2C19, pero no inhibe el CYP1A2, CYP2B6, CYP2D6, CYP3A4/5, UGT1A1 y UGT2B7.
El acalabrutinib es un inductor débil del CYP1A2, CYP2B6 y CYP3A4; el ACP-5862 induce débilmente el CYP3A4.
Según los datos in vitro y el modelado PBPK, no se espera interacción con los sustratos del CYP a concentraciones clínicamente relevantes.
Sistemas de transporte de fármacos
El acalabrutinib y su metabolito activo, ACP-5862, son sustratos de la glicoproteína P (P-gp) y la proteína de resistencia al cáncer de mama (BCRP). El acalabrutinib no es un sustrato de los transportadores de captación renal OAT1, OAT3 y OCT2, ni de los transportadores hepáticos OATP1B1 y OATP1B3. El ACP-5862 no es un sustrato de OATP1B1 u OATP1B3.
El acalabrutinib y el ACP-5862 no inhiben la P-gp, OAT1, OAT3, OCT2, OATP1B1, OATP1B3 y MATE2-K a concentraciones clínicamente relevantes.
El acalabrutinib puede aumentar la exposición a los sustratos de BCRP coadministrados (p. ej., metotrexato) mediante la inhibición de la BCRP intestinal. El ACP-5862 no inhibe la BCRP a concentraciones clínicamente relevantes. El acalabrutinib no inhibe la MATE1, mientras que el ACP-5862 puede aumentar la exposición a los sustratos de MATE1 coadministrados (p. ej., metformina) mediante la inhibición de la MATE1.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
No se han realizado estudios de carcinogenicidad con acalabrutinib.
Acalabrutinib no fue mutagénico en un ensayo in vitro de mutación inversa bacteriana (AMES) ni clastogénico en un ensayo in vitro de aberración cromosómica en linfocitos humanos o en un ensayo in vivo de micronúcleos de médula ósea de rata.
En un estudio de fertilidad en ratas, no hubo efectos de acalabrutinib sobre la fertilidad en ratas macho con exposiciones 11 veces mayores, o en ratas hembra con exposiciones 9 veces mayores que el AUC observado en pacientes a la dosis recomendada de 100 mg dos veces al día.
14 ESTUDIOS CLÍNICOS
14.1 Linfoma de células del manto previamente no tratado
ECHO
La eficacia de CALQUENCE en pacientes con LMC previamente no tratado se evaluó en un estudio aleatorizado, doble ciego, controlado con placebo y multicéntrico (ECHO; NCT02972840). El estudio incluyó a 598 pacientes de ≥ 65 años de edad y que no tenían intención de trasplante. El estudio excluyó a pacientes con bilirrubina total > 1,5 × límite superior de la normalidad (LSN), AST o ALT > 2,5 × LSN, o aclaramiento de creatinina estimado de ≤ 50 mL/min. Los pacientes se asignaron al azar en una proporción de 1:1 para recibir CALQUENCE más bendamustina y rituximab (CALQUENCE más BR) o placebo más BR. La dosificación para ambos brazos se administró en ciclos de 28 días de la siguiente manera:
- •
- CALQUENCE más BR se administró durante un máximo de 6 ciclos de tratamiento. CALQUENCE 100 mg por vía oral se administró dos veces al día a partir del Día 1 del Ciclo 1. La bendamustina se administró a 90 mg/m2 por vía intravenosa durante 30 minutos los Días 1 y 2 de cada uno de los 6 ciclos. El rituximab se administró a 375 mg/m2 por vía intravenosa el Día 1 de cada ciclo durante 6 ciclos.
- •
- Para los pacientes que lograron una respuesta (RP o RC), CALQUENCE 100 mg por vía oral dos veces al día se administró de forma continua, en combinación con rituximab administrado a 375 mg/m2 el Día 1 cada dos ciclos durante un máximo de 12 dosis adicionales hasta el Ciclo 30. Después de la interrupción del rituximab, los pacientes continuaron con la monoterapia con CALQUENCE a 100 mg por vía oral dos veces al día hasta la progresión de la enfermedad o la toxicidad inaceptable.
Los pacientes del brazo de control recibieron el mismo régimen, pero placebo en lugar de CALQUENCE. Se permitió el cambio a la monoterapia con CALQUENCE para los pacientes del brazo de placebo más BR en caso de progresión de la enfermedad.
De todos los pacientes aleatorizados, la mediana de edad fue de 71 años (rango: 65-86); el 71% eran hombres; el 78% eran blancos, el 16% asiáticos, el 0,5% eran negros o afroamericanos. En total, el 80% tenía histología clásica de LMC, el 7,7% tenía LMC blastica y el 5,5% tenía LMC pleomórfica. La puntuación MIPI (Índice pronóstico internacional del linfoma de células del manto) simplificada fue baja en el 33%, intermedia en el 43% y alta en el 24% de los pacientes. Un total del 38% de los pacientes tenían una masa tumoral ≥ 5 cm y el 86% tenían enfermedad en estadio IV de Ann Arbor.
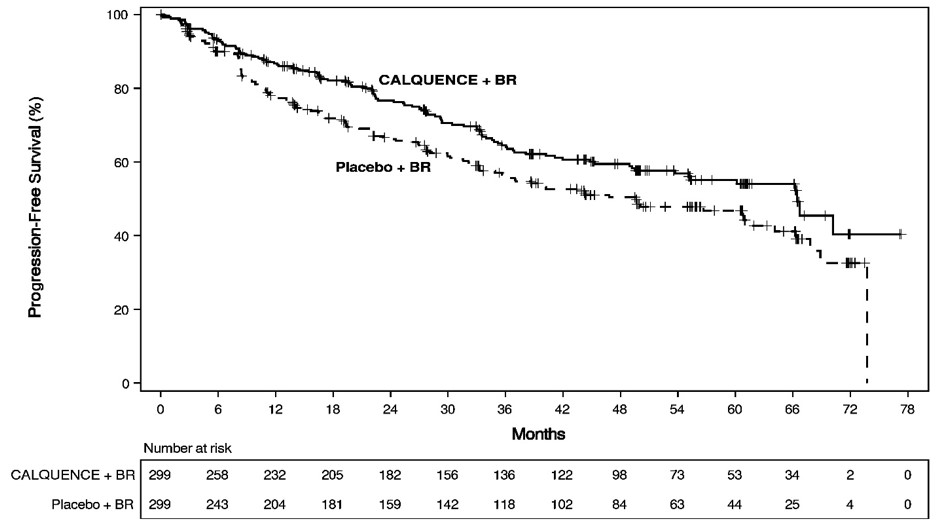
El principal resultado de eficacia fue la supervivencia libre de progresión (SLP) evaluada por un Comité de Revisión Independiente (CRI) utilizando la Clasificación de Lugano. Los resultados de eficacia se presentan en la Tabla 12. Las curvas de Kaplan-Meier para la SLP se muestran en la Figura 1. En este análisis intermedio preespecificado, la mediana de seguimiento para la SLP fue de 49,8 meses en ambos brazos.
|
||
|
Resultados por CRI |
CALQUENCE más BR N= 299 |
Placebo más BR N= 299 |
|
Supervivencia libre de progresión* |
||
|
66,4 (55,1, NE) |
49,6 (36,0, 64,1) |
|
0,73 (0,57, 0,94) |
|
|
0,016 |
|
|
Tasa de respuesta global (TRG) (RC + RP) |
||
|
272 (91) |
263 (88) |
|
87, 94 |
84, 91 |
|
199 (67) |
160 (54) |
|
73 (24) |
103 (34) |
|
0.220 |
|
|
HR = hazard ratio, CR = complete response, PR = partial response, NE – not evaluable |
||
Figura 1. Curva de Kaplan-Meier de la SPP evaluada por el IRC en pacientes con LMC previamente no tratados en ECHO
En el momento del análisis de la SPP, la mediana de la supervivencia general no se había alcanzado en ninguno de los brazos, con un total de 203 muertes: 97 (32%) pacientes en el brazo de CALQUENCE más BR y 106 (35%) pacientes en el brazo de placebo más BR habían fallecido.
14.2 Linfoma de células del manto previamente tratado
ACE-LY-004
La eficacia de CALQUENCE se basó en el ensayo LY-004 titulado “Un estudio abierto, de fase 2, de ACP-196 en sujetos con linfoma de células del manto” (NCT02213926). El ensayo LY-004 incluyó un total de 124 pacientes con LMC que habían recibido al menos un tratamiento previo.
La mediana de edad fue de 68 (rango de 42 a 90) años, el 80% eran hombres y el 74% eran caucásicos. Al inicio del estudio, el 93% de los pacientes tenían un estado de rendimiento ECOG de 0 o 1. La mediana del tiempo transcurrido desde el diagnóstico fue de 46,3 meses y la mediana del número de tratamientos previos fue de 2 (rango de 1 a 5), incluido el 18% con trasplante previo de células madre. Se excluyeron los pacientes que recibieron tratamiento previo con inhibidores de BTK. Los regímenes previos más comunes fueron los basados en CHOP (52%) y ARA-C (34%). Al inicio del estudio, el 37% de los pacientes tenían al menos un tumor con un diámetro mayor ≥ 5 cm, el 73% tenían afectación extranodal, incluido el 51% con afectación de la médula ósea. La puntuación del Índice Pronóstico Internacional Simplificado del Linfoma de Células del Manto (MIPI) (que incluye la edad, la puntuación ECOG y la lactato deshidrogenasa y el recuento de leucocitos basales) fue intermedia en el 44% y alta en el 17% de los pacientes.
CALQUENCE se administró por vía oral a 100 mg aproximadamente cada 12 horas hasta la progresión de la enfermedad o la toxicidad inaceptable. La intensidad de la dosis mediana fue del 98,5%. El principal resultado de eficacia del ensayo LY-004 fue la tasa de respuesta global y la mediana de seguimiento fue de 15,2 meses.
|
||
|
Evaluación del investigador N=124 |
Evaluación del Comité de Revisión Independiente (CRI) N=124 |
|
|
Tasa de respuesta global (TRG)* |
||
|
TRG (%) [IC del 95%] |
81 [73, 87] |
80 [72, 87] |
|
Respuesta completa (%) [IC del 95%] |
40 [31, 49] |
40 [31, 49] |
|
Respuesta parcial (%) [IC del 95%] |
41 [32, 50] |
40 [32, 50] |
|
Duración de la respuesta (DoR) |
||
|
DoR mediana en meses [rango] |
NE [1+ a 20+] |
NE [0+ a 20+] |
|
IC= Intervalo de confianza; NE=No estimable; + indica observaciones censuradas |
||
La mediana del tiempo hasta la mejor respuesta fue de 1,9 meses.
Linfocitosis
Tras la iniciación de CALQUENCE, se observó un aumento temporal en el recuento de linfocitos (definido como un recuento absoluto de linfocitos (RAL) aumentado ≥ 50 % respecto al valor basal y una evaluación posterior al valor basal ≥ 5 x 109) en el 31,5 % de los pacientes del ensayo LY-004. La mediana del tiempo hasta el inicio de la linfocitosis fue de 1,1 semanas y la mediana de la duración de la linfocitosis fue de 6,7 semanas.
14.3 Leucemia Linfocítica Crónica
La eficacia de CALQUENCE en pacientes con LLC se demostró en dos ensayos aleatorizados y controlados. La indicación de CALQUENCE incluye pacientes con LSL, ya que es la misma enfermedad.
ELEVATE-TN
La eficacia de CALQUENCE se evaluó en el ensayo ELEVATE-TN, un ensayo aleatorizado, multicéntrico, abierto, controlado activamente, de 3 brazos de CALQUENCE en combinación con obinutuzumab, CALQUENCE en monoterapia y obinutuzumab en combinación con clorambucil en 535 pacientes con leucemia linfocítica crónica previamente no tratada (NCT02475681). Se incluyeron pacientes de 65 años o más o de entre 18 y 65 años con una puntuación total de la Escala de Evaluación de Enfermedades Acumuladas (CIRS) > 6 o aclaramiento de creatinina de 30 a 69 mL/min. El ensayo también requirió transaminasas hepáticas ≤ 3 veces el límite superior de la normalidad (LSN) y bilirrubina total ≤ 1,5 veces el LSN, y excluyó a pacientes con transformación de Richter.
Los pacientes se aleatorizaron en una proporción de 1:1:1 en 3 brazos para recibir:
- •
- CALQUENCE más obinutuzumab (CALQUENCE+G): Se administraron 100 mg de CALQUENCE aproximadamente cada 12 horas a partir del día 1 del ciclo 1 hasta la progresión de la enfermedad o la toxicidad inaceptable. Se administró obinutuzumab a partir del día 1 del ciclo 2 durante un máximo de 6 ciclos de tratamiento. Se administraron 1000 mg de obinutuzumab los días 1 y 2 (100 mg el día 1 y 900 mg el día 2), 8 y 15 del ciclo 2, seguidos de 1000 mg el día 1 de los ciclos 3 a 7. Cada ciclo fue de 28 días.
- •
- Monoterapia con CALQUENCE: Se administraron 100 mg de CALQUENCE aproximadamente cada 12 horas hasta la progresión de la enfermedad o la toxicidad inaceptable.
- •
- Obinutuzumab más clorambucil (GClb): Se administraron obinutuzumab y clorambucil durante un máximo de 6 ciclos de tratamiento. Se administraron 1000 mg de obinutuzumab por vía intravenosa los días 1 y 2 (100 mg el día 1 y 900 mg el día 2), 8 y 15 del ciclo 1, seguidos de 1000 mg el día 1 de los ciclos 2 a 6. Se administraron 0,5 mg/kg de clorambucil por vía oral los días 1 y 15 de los ciclos 1 a 6. Cada ciclo fue de 28 días.
La aleatorización se estratificó por el estado de la mutación de deleción 17p, el estado de rendimiento ECOG (0 o 1 frente a 2) y la región geográfica. Se aleatorizaron un total de 535 pacientes, 179 a CALQUENCE+G, 179 a monoterapia con CALQUENCE y 177 a GClb. La mediana de edad general fue de 70 años (rango: 41 a 91 años), el 47 % tenía enfermedad en estadio III o IV de Rai, el 14 % tenía deleción 17p o mutación TP53, el 63 % de los pacientes tenía una IGVH no mutada y el 18 % tenía deleción 11q. Las características demográficas y de la enfermedad basales fueron similares entre los brazos de tratamiento.
La eficacia se basó en la supervivencia libre de progresión (SLP) evaluada por un Comité de Revisión Independiente (CRI). La mediana de la duración del seguimiento fue de 28,3 meses (rango: 0,0 a 40,8 meses). Los resultados de eficacia se presentan en la Tabla 10. Las curvas de Kaplan-Meier para la SLP se muestran en la Figura 1.
|
|||
|
CALQUENCE más Obinutuzumab N=179 |
Monoterapia con CALQUENCE N=179 |
Obinutuzumab más Clorambucil N=177 |
|
|
Supervivencia libre de progresión* |
|||
|
Número de eventos (%) |
14 (8) |
26 (15) |
93 (53) |
|
PD, n (%) |
9 (5) |
20 (11) |
82 (46) |
|
Eventos de muerte, n (%) |
5 (3) |
6 (3) |
11 (6) |
|
Mediana (IC 95%), meses† |
NE |
NE (34, NE) |
22.6 (20, 28) |
|
HR‡(IC 95%) |
0.10 (0.06, 0.17) |
0.20 (0.13, 0.30) |
– |
|
Valor p§ |
< 0.0001 |
< 0.0001 |
– |
|
Tasa general de respuesta* (CR + CRi + nPR + PR) |
|||
|
TRR, n (%) |
168 (94) |
153 (86) |
139 (79) |
|
(IC 95%) |
(89, 97) |
(80, 90) |
(72, 84) |
|
Valor p¶ |
< 0.0001 |
0.0763 |
– |
|
CR, n (%) |
23 (13) |
1 (1) |
8 (5) |
|
CRi, n (%) |
1 (1) |
0 |
0 |
|
nPR, n (%) |
1 (1) |
2 (1) |
3 (2) |
|
PR, n (%) |
143 (80) |
150 (84) |
128 (72) |
|
ITT=intención de tratar; IC=intervalo de confianza; HR=hazard ratio; NE=no estimable; CR=respuesta completa; CRi=respuesta completa con recuperación incompleta del recuento sanguíneo; nPR=respuesta parcial nodular; PR=respuesta parcial. |
|||
Figura 2: Curva de Kaplan-Meier de la SPP evaluada por el IRC en pacientes con LLC en ELEVATE-TN
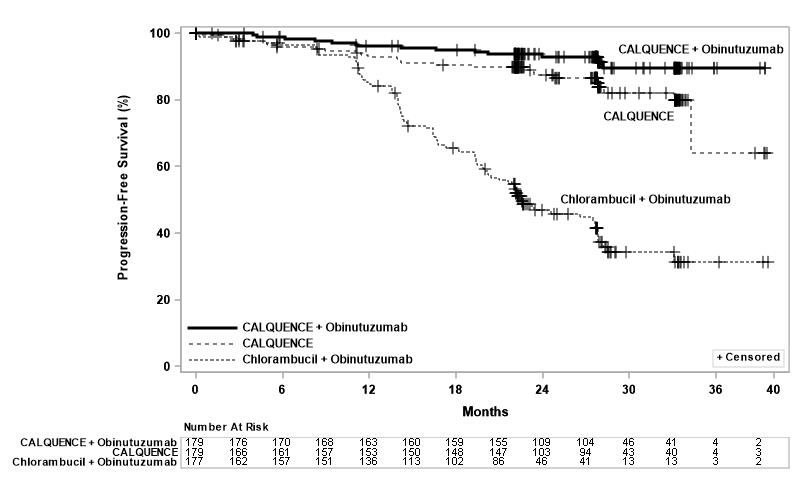
Con una mediana de seguimiento de 28,3 meses, no se alcanzó la mediana de supervivencia general en ningún brazo, con menos del 10 % de los pacientes que experimentaron un evento.
ASCEND
La eficacia de CALQUENCE en pacientes con LLC recidivante o refractaria se basó en un ensayo multicéntrico, aleatorizado y abierto (ASCEND; NCT02970318). El ensayo incluyó a 310 pacientes con LLC recidivante o refractaria después de al menos 1 terapia sistémica previa. El ensayo excluyó a pacientes con enfermedad transformada, leucemia prolinfocítica o tratamiento previo con venetoclax, un inhibidor de la tirosina quinasa de Bruton o un inhibidor de la quinasa de fosfoinosítido-3.
Los pacientes fueron aleatorizados en una proporción de 1:1 para recibir:
- •
- CALQUENCE 100 mg aproximadamente cada 12 horas hasta la progresión de la enfermedad o toxicidad inaceptable, o
- •
- A elección del investigador:
- o
- Idelalisib más un producto de rituximab (IR): Idelalisib 150 mg por vía oral aproximadamente cada 12 horas hasta la progresión de la enfermedad o toxicidad inaceptable, en combinación con 8 infusiones de un producto de rituximab (375 mg/m2 por vía intravenosa el día 1 del ciclo 1, seguido de 500 mg/m2 cada 2 semanas durante 4 dosis y luego cada 4 semanas durante 3 dosis), con una duración del ciclo de 28 días.
- o
- Bendamustina más un producto de rituximab (BR): Bendamustina 70 mg/m2 por vía intravenosa (días 1 y 2 de cada ciclo de 28 días), en combinación con un producto de rituximab (375 mg/m2 por vía intravenosa el día 1 del ciclo 1, luego 500 mg/m2 el día 1 de los ciclos posteriores), durante un máximo de 6 ciclos.
La aleatorización se estratificó por el estado de la mutación de deleción 17p, el estado de rendimiento ECOG (0 o 1 frente a 2) y el número de terapias previas (1 a 3 frente a ≥ 4). De los 310 pacientes en total, 155 fueron asignados a monoterapia con CALQUENCE, 119 a IR y 36 a BR. La mediana de edad general fue de 67 años (rango: 32 a 90 años), el 42 % tenía enfermedad en estadio III o IV de Rai, el 28 % tenía deleción 17p o mutación TP53, el 78 % de los pacientes tenía un IGVH no mutado y el 27 % tenía una deleción 11q. El brazo de CALQUENCE tuvo una mediana de 1 terapia previa (rango 1-8), con un 47 % que tuvo al menos 2 terapias previas. El brazo de elección del investigador tuvo una mediana de 2 terapias previas (rango 1-10), con un 57 % que tuvo al menos 2 terapias previas.
En el brazo de CALQUENCE, la mediana de duración del tratamiento fue de 15,7 meses, con el 94 % de los pacientes tratados durante al menos 6 meses y el 86 % de los pacientes tratados durante al menos 1 año. En el brazo de elección del investigador, la mediana de duración del tratamiento fue de 8,4 meses, con el 59 % de los pacientes tratados durante al menos 6 meses y el 37 % tratados durante al menos 1 año.
La eficacia se basó en la SPP evaluada por un IRC, con una mediana de seguimiento de 16,1 meses (rango 0,03 a 22,4 meses). Los resultados de eficacia se presentan en la Tabla 11. La curva de Kaplan-Meier para la SPP se muestra en la Figura 2. No hubo diferencias estadísticamente significativas en las tasas de respuesta general entre los dos brazos de tratamiento.
| Monoterapia con CALQUENCE N=155 |
Elección del investigador de Idelalisib + producto de Rituximab o Bendamustina + producto de Rituximab N=155 |
|
|---|---|---|
|
||
|
Supervivencia libre de progresión* |
||
|
Número de eventos, n (%) |
27 (17) |
68 (44) |
|
Progresión de la enfermedad, n |
19 |
59 |
|
Muerte, n |
8 |
9 |
|
Mediana (IC del 95 %), meses† |
NE (NE, NE) |
16,5 (14,0, 17,1) |
|
HR (IC del 95 %)‡ |
0,31 (0,20, 0,49) |
|
|
Valor de P§ |
< 0.0001 |
|
|
TRG, n (%)¶ |
126 (81) |
117 (75) |
|
(IC del 95%) |
(74, 87) |
(68, 82) |
|
CR, n (%) |
0 |
2 (1) |
|
CRi, n (%) |
0 |
0 |
|
nPR, n (%) |
0 |
0 |
|
PR, n (%) |
126 (81) |
115 (74) |
|
ITT=intención de tratar; IC=intervalo de confianza; HR=hazard ratio; NE=no estimable; CR=respuesta completa; CRi=respuesta completa con recuperación incompleta del recuento sanguíneo; nPR=respuesta parcial nodular; PR=respuesta parcial |
||
Figura 3: Curva de Kaplan-Meier de la SPP evaluada por IRC en pacientes con LLC en ASCEND
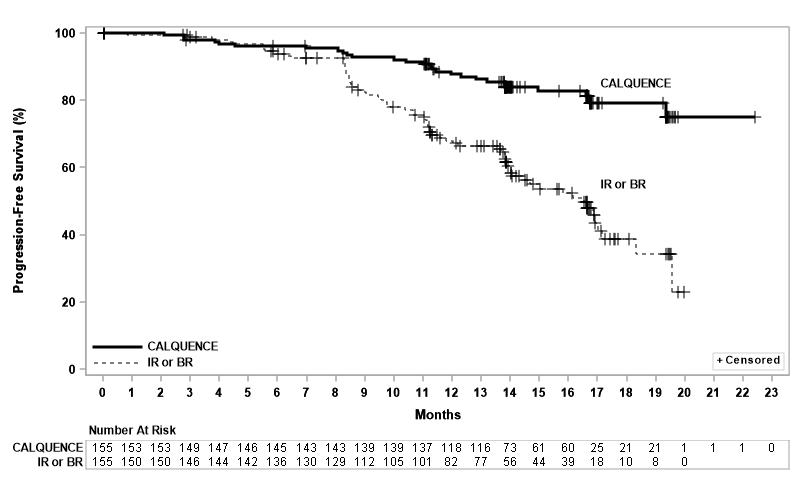
Con un seguimiento mediano de 16,1 meses, no se alcanzó la supervivencia general mediana en ninguno de los brazos, con menos del 11 % de los pacientes que experimentaron un evento.
16 SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
Presentación
|
Tamaño del envase |
Contenido |
Número NDC |
|
Frasco con 60 unidades |
Frasco que contiene 60 cápsulas 100 mg, cápsulas de gelatina dura con cuerpo amarillo y tapa azul, marcadas con tinta negra con ‘ACA 100 mg’ |
0310-0512-60 |
Almacenamiento
Conserve a 20°C-25°C (68°F-77°F); se permiten excursiones a 15°C-30°C (59°F-86°F) [véase USP Controlled Room Temperature].
17 INFORMACIÓN DE ASESORAMIENTO AL PACIENTE
Aconseje al paciente que lea el etiquetado del paciente aprobado por la FDA (Información para el paciente).
Infecciones graves y oportunistas
Informe a los pacientes de la posibilidad de infecciones graves y que informen sobre signos o síntomas que sugieran una infección [ver Advertencias y precauciones (5.1)].
Hemorragia
Informe a los pacientes que informen sobre signos o síntomas de sangrado. Informe a los pacientes que puede ser necesario interrumpir CALQUENCE para cirugías mayores [ver Advertencias y precauciones (5.2)].
Citopenias
Informe a los pacientes que necesitarán análisis de sangre periódicos para controlar los recuentos sanguíneos durante el tratamiento con CALQUENCE [ver Advertencias y precauciones (5.3)].
Segundas neoplasias malignas
Informe a los pacientes que se han notificado otras neoplasias malignas en pacientes tratados con CALQUENCE, incluido el cáncer de piel y otros tumores sólidos. Aconseje a los pacientes que usen protección solar [ver Advertencias y precauciones (5.4)].
Arritmias cardíacas
Aconseje a los pacientes que informen sobre cualquier signo de palpitaciones, mareos, desmayos, molestias en el pecho y dificultad para respirar [ver Advertencias y precauciones (5.5)].
Hepatotoxicidad, incluida la lesión hepática inducida por fármacos:
Informe a los pacientes que pueden aparecer problemas hepáticos, incluida la lesión hepática inducida por fármacos y anomalías en las pruebas hepáticas, durante el tratamiento con CALQUENCE. Aconseje a los pacientes que se pongan en contacto con su proveedor de atención médica inmediatamente si experimentan malestar abdominal, orina oscura o ictericia [ver Advertencias y precauciones (5.6)].
Complicaciones del embarazo
CALQUENCE puede causar daño fetal y distocia. Aconseje a las mujeres que eviten quedar embarazadas durante el tratamiento y durante al menos 1 semana después de la última dosis de CALQUENCE [ver Uso en poblaciones específicas (8.3)].
Lactancia
Aconseje a las mujeres que no amamanten durante el tratamiento con CALQUENCE y durante al menos 2 semanas después de la dosis final [ver Uso en poblaciones específicas (8.2)].
Instrucciones de dosificación
Indique a los pacientes que tomen CALQUENCE por vía oral dos veces al día, con aproximadamente 12 horas de diferencia. CALQUENCE se puede tomar con o sin alimentos. Aconseje a los pacientes que las cápsulas de CALQUENCE deben tragarse enteras con un vaso de agua, sin abrirse, romperse ni masticarse [ver Dosificación y administración (2.1)].
Dosis olvidada
Aconseje a los pacientes que si olvidan una dosis de CALQUENCE, aún pueden tomarla hasta 3 horas después de la hora en que normalmente la tomarían. Si han transcurrido más de 3 horas, se les debe indicar que se salten esa dosis y tomen la siguiente dosis de CALQUENCE a la hora habitual. Advierta a los pacientes que no deben tomar cápsulas adicionales para compensar la dosis que olvidaron [ver Dosificación y administración (2.1)].
Interacciones medicamentosas
Aconseje a los pacientes que informen a sus proveedores de atención médica sobre todos los medicamentos concomitantes, incluidos los medicamentos de venta libre, las vitaminas y los productos herbales [ver Interacciones medicamentosas (7)].
Distribuido por:
AstraZeneca Pharmaceuticals LP
Wilmington, DE 19850
CALQUENCE es una marca registrada del grupo de empresas AstraZeneca.
©AstraZeneca 2025
INFORMACIÓN PARA EL PACIENTE
|
INFORMACIÓN PARA EL PACIENTE CALQUENCE® (KAL-kwens) (acalabrutinib) Cápsulas |
|
¿Qué es CALQUENCE? CALQUENCE es un medicamento recetado que se usa para tratar a adultos con:
No se sabe si CALQUENCE es seguro y eficaz en niños. |
|
Antes de tomar CALQUENCE, informe a su proveedor de atención médica sobre todas sus afecciones médicas, incluso si:
Informe a su proveedor de atención médica sobre todos los medicamentos que toma, incluidos los medicamentos recetados y de venta libre, las vitaminas y los suplementos herbales. Tomar CALQUENCE con ciertos otros medicamentos puede afectar la forma en que funciona CALQUENCE y puede causar efectos secundarios. Informe especialmente a su proveedor de atención médica si toma un medicamento anticoagulante. |
|
¿Cómo debo tomar CALQUENCE?
|
|
¿Cuáles son los posibles efectos secundarios de CALQUENCE? CALQUENCE puede causar efectos secundarios graves, incluyendo:
Los efectos secundarios más comunes de CALQUENCE incluyen:
Estos no son todos los posibles efectos secundarios de CALQUENCE. Llame a su médico para obtener asesoramiento médico sobre los efectos secundarios. Puede informar sobre los efectos secundarios a la FDA al 1‑800‑FDA‑1088. |
|
¿Cómo debo guardar CALQUENCE?
Mantenga CALQUENCE y todos los medicamentos fuera del alcance de los niños. |
|
Información general sobre el uso seguro y eficaz de CALQUENCE. Los medicamentos a veces se recetan para fines distintos de los que figuran en un prospecto de información para el paciente. No use CALQUENCE para una afección para la que no se le haya recetado. No le dé CALQUENCE a otras personas, aunque tengan los mismos síntomas que usted. Puede perjudicarlas. Puede pedir más información sobre CALQUENCE a su proveedor de atención médica o farmacéutico, que está escrita para profesionales de la salud. |
|
¿Cuáles son los ingredientes de CALQUENCE? Principio activo: acalabrutinib Ingredientes inactivos: celulosa microcristalina silicificada, almidón pregelatinizado, estearato de magnesio y glicolato de almidón sódico. La cápsula contiene: gelatina, dióxido de titanio, óxido de hierro amarillo, FD&C Blue 2 y tinta negra. Distribuido por: AstraZeneca Pharmaceuticals LP, Wilmington, DE 19850 CALQUENCE es una marca registrada del grupo de empresas AstraZeneca. ©AstraZeneca 2025 Para obtener más información, visite www.CALQUENCE.com o llame al 1-800-236-9933. |
Esta información para el paciente ha sido aprobada por la Administración de Alimentos y Medicamentos de los EE. UU. Revisado: 1/2025
PANEL DE VISUALIZACIÓN PRINCIPAL DEL PAQUETE/ETIQUETA
NDC 0310-0512-60 60 cápsulas
CALQUENCE®
(acalabrutinib) cápsulas
100 mg
Rx only
Fabricado para:
AstraZeneca Pharmaceuticals LP
Wilmington, DE 19850
Por: AstraZeneca AB
SE-151 85 Södertàlje, Suecia
Producto de Suiza
AstraZeneca