Fabricante de medicamentos: AstraZeneca Pharmaceuticals LP (Updated: 2024-11-04)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
BRILINTA® (ticagrelor) comprimidos, para administración oral
Aprobación inicial en EE. UU.: 2011
ADVERTENCIA: RIESGO DE HEMORRAGIA
Consulte la información completa de prescripción para ver la advertencia completa en recuadro.
- •
- BRILINTA, al igual que otros agentes antiplaquetarios, puede causar hemorragias significativas,
- •
- a veces mortales. (5.1, 6.1)
- •
- No use BRILINTA en pacientes con hemorragia patológica activa
- •
- o antecedentes de hemorragia intracraneal. (4.1, 4.2)
- •
- No inicie BRILINTA en pacientes que se sometan a una cirugía urgente de injerto de derivación de arteria coronaria
- •
- (CABG). (5.1, 6.1)
- •
- Si es posible, controle la hemorragia sin interrumpir el tratamiento con BRILINTA.
- •
- La interrupción de BRILINTA aumenta el riesgo de eventos cardiovasculares posteriores.
- •
- (5.2)
INDICACIONES Y USO
BRILINTA es un inhibidor plaquetario P2Y12 indicado
- •
- para reducir el riesgo de muerte cardiovascular (CV), infarto de miocardio (IM) y accidente cerebrovascular en pacientes con síndrome coronario agudo (SCA) o antecedentes de IM. Durante al menos los primeros 12 meses después del SCA, es superior al clopidogrel.
BRILINTA también reduce el riesgo de trombosis del stent en pacientes que han recibido un stent para el tratamiento del SCA. (1.1) - •
- para reducir el riesgo de un primer IM o accidente cerebrovascular en pacientes con enfermedad arterial coronaria (EAC) con alto riesgo de tales eventos. Si bien el uso no se limita a este contexto, la eficacia de BRILINTA se estableció en una población con diabetes mellitus tipo 2 (DM2). (1.2)
- •
- para reducir el riesgo de accidente cerebrovascular en pacientes con accidente cerebrovascular isquémico agudo (puntuación de la escala de accidente cerebrovascular del NIH ≤5) o ataque isquémico transitorio (AIT) de alto riesgo. (1.3)
POSOLOGÍA Y ADMINISTRACIÓN
- •
- SCA o antecedentes de IM
- •
- Inicie el tratamiento con una dosis de carga oral de 180 mg de BRILINTA. A continuación, administre 90 mg dos veces al día durante el primer año. Después de un año, administre 60 mg dos veces al día. (2.2)
- •
- Pacientes con EAC y sin antecedentes de accidente cerebrovascular o IM
- •
- Administre 60 mg de BRILINTA dos veces al día. (2.3)
- •
- Accidente cerebrovascular isquémico agudo
- •
- Inicie el tratamiento con una dosis de carga de 180 mg de BRILINTA y continúe con 90 mg dos veces al día durante un máximo de 30 días. (2.4)
Use BRILINTA con una dosis de mantenimiento diaria de aspirina de 75-100 mg. (2) Sin embargo, en pacientes que se han sometido a ICP, considere la terapia antiplaquetaria única con BRILINTA en función del riesgo cambiante de eventos trombóticos frente a hemorrágicos. (2.2)
FORMAS Y CONCENTRACIONES FARMACÉUTICAS
- •
- Comprimidos de 60 mg y 90 mg (3)
CONTRAINDICACIONES
ADVERTENCIAS Y PRECAUCIONES
- •
- Se informó con mayor frecuencia disnea con BRILINTA que con los agentes de control en los ensayos clínicos. La disnea por BRILINTA es autolimitada. (5.3)
- •
- Insuficiencia hepática grave: Probable aumento de la exposición al ticagrelor. (5.6)
- •
- Interferencia en las pruebas de laboratorio: Se han notificado resultados falsos negativos en las pruebas funcionales de plaquetas para la trombocitopenia inducida por heparina (TIH). No se espera que BRILINTA afecte a las pruebas de anticuerpos PF4 para la TIH. (5.8)
REACCIONES ADVERSAS
Las reacciones adversas más comunes (>5%) son hemorragia y disnea. (5.1, 5.3, 6.1)
Para notificar REACCIONES ADVERSAS SOSPECHOSAS, comuníquese con AstraZeneca al 1-800-236-9933 o con la FDA al 1-800-FDA-1088 o www.fda.gov/medwatch.
INTERACCIONES MEDICAMENTOSAS
- •
- Evitar el uso con inhibidores o inductores potentes del CYP3A. (7.1, 7.2)
- •
- Opioides: Disminución de la exposición a ticagrelor. Considere el uso de un agente antiplaquetario parenteral. (7.3)
- •
- Los pacientes que reciben más de 40 mg por día de simvastatina o lovastatina pueden tener un mayor riesgo de efectos adversos relacionados con las estatinas. (7.4)
- •
- Las concentraciones plasmáticas de rosuvastatina pueden aumentar. Controlar los efectos adversos relacionados con las estatinas. (7.4)
- •
- Controlar los niveles de digoxina con el inicio o cualquier cambio en BRILINTA. (7.5)
USO EN POBLACIONES ESPECÍFICAS
- •
- Lactancia: No se recomienda la lactancia materna. (8.2)
Ver 17 para INFORMACIÓN AL PACIENTE y Guía de medicamentos.
Revisado: 11/2024
Tabla de Contenido
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDO*
ADVERTENCIA: RIESGO DE HEMORRAGIA
1 INDICACIONES Y USO
1.1 Síndrome Coronario Agudo o Antecedentes de Infarto de Miocardio
1.2 Enfermedad de la Arteria Coronaria pero Sin Accidente Cerebrovascular o Infarto de Miocardio Previo
1.3 Accidente Cerebrovascular Isquémico Agudo o Ataque Isquémico Transitorio (AIT)
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Instrucciones Generales
2.2 Síndrome Coronario Agudo o Antecedentes de Infarto de Miocardio
2.3 Enfermedad de la Arteria Coronaria pero Sin Accidente Cerebrovascular o Infarto de Miocardio Previo
2.4 Accidente Cerebrovascular Isquémico Agudo o Ataque Isquémico Transitorio (AIT)
3 FORMAS Y CONCENTRACIONES FARMACÉUTICAS
4 CONTRAINDICACIONES
4.1 Antecedentes de Hemorragia Intracraneal
4.2 Hemorragia Activa
4.3 Hipersensibilidad
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Riesgo de Hemorragia
5.2 Suspensión de BRILINTA en Pacientes Tratados por Enfermedad de la Arteria Coronaria
5.3 Disnea
5.4 Bradiarritmias
5.5 Insuficiencia Hepática Grave
5.6 Apnea del Sueño Central
5.7 Interferencias en las Pruebas de Laboratorio
6 REACCIONES ADVERSAS
6.1 Experiencia en Ensayos Clínicos
6.2 Experiencia Postcomercialización
7 INTERACCIONES MEDICAMENTOSAS
7.1 Inhibidores Potentes del CYP3A
7.2 Inductores Potentes del CYP3A
7.3 Opioides
7.4 Simvastatina, Lovastatina, Rosuvastatina
7.5 Digoxina
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.4 Uso Pediátrico
8.5 Uso Geriátrico
8.6 Insuficiencia Hepática
8.7 Insuficiencia Renal
10 SOBREDOSIFICACIÓN
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de Acción
12.2 Farmacodinamia
12.3 Farmacocinética
12.5 Farmacogenómica
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
14 ESTUDIOS CLÍNICOS
14.1 Síndromes Coronarios Agudos y Prevención Secundaria después del Infarto de Miocardio
14.2 Enfermedad de la Arteria Coronaria pero Sin Accidente Cerebrovascular o Infarto de Miocardio Previo
14.3 Accidente Cerebrovascular Isquémico Agudo o Ataque Isquémico Transitorio (AIT)
16 PRESENTACIÓN/ALMACENAMIENTO Y MANIPULACIÓN
17 INFORMACIÓN PARA EL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información completa de prescripción no se enumeran.
ADVERTENCIA EN EL RECUADRO
ADVERTENCIA: RIESGO DE HEMORRAGIA
- •
- BRILINTA, al igual que otros agentes antiplaquetarios, puede causar hemorragias significativas, a veces mortales (5.1, 6.1).
- •
- No use BRILINTA en pacientes con hemorragia patológica activa o antecedentes de hemorragia intracraneal
- •
- (4.1, 4.2).
- •
- No inicie BRILINTA en pacientes que se sometan a una cirugía de injerto de derivación de arteria coronaria (CABG) urgente (5.1,
- •
- 6.1).
- •
- Si es posible, controle la hemorragia sin interrumpir el tratamiento con BRILINTA. La interrupción de BRILINTA aumenta el riesgo de
- •
- eventos cardiovasculares posteriores (5.2).
1 INDICACIONES Y USO
1.1 Síndrome Coronario Agudo o Antecedentes de Infarto de Miocardio
BRILINTA está indicado para reducir el riesgo de muerte cardiovascular (CV), infarto de miocardio (IM) y accidente cerebrovascular en pacientes con síndrome coronario agudo (SCA) o antecedentes de IM. Durante al menos los primeros 12 meses después del SCA, es superior al clopidogrel.
BRILINTA también reduce el riesgo de trombosis del stent en pacientes que han sido sometidos a implante de stent para el tratamiento del SCA [ver Estudios Clínicos (14.1)].
1.2 Enfermedad de la Arteria Coronaria pero Sin Accidente Cerebrovascular o Infarto de Miocardio Previo
BRILINTA está indicado para reducir el riesgo de un primer IM o accidente cerebrovascular en pacientes con enfermedad de la arteria coronaria (EAC) con alto riesgo de tales eventos [ver Estudios Clínicos (14.2)]. Si bien su uso no se limita a este contexto, la eficacia de BRILINTA se estableció en una población con diabetes mellitus tipo 2 (DM2).
1.3 Accidente Cerebrovascular Isquémico Agudo o Ataque Isquémico Transitorio (AIT)
BRILINTA está indicado para reducir el riesgo de accidente cerebrovascular en pacientes con accidente cerebrovascular isquémico agudo (puntuación en la escala NIH de accidente cerebrovascular ≤5) o ataque isquémico transitorio (AIT) de alto riesgo [ver Estudios Clínicos (14.3)].
2 DOSIS Y ADMINISTRACIÓN
2.1 Instrucciones generales
Aconseje a los pacientes que olviden una dosis de BRILINTA que tomen la siguiente dosis a la hora programada.
Para los pacientes que no pueden tragar los comprimidos enteros, los comprimidos de BRILINTA se pueden triturar, mezclar con agua y beber.
La mezcla también se puede administrar a través de una sonda nasogástrica (CH8 o superior) [ver Farmacología clínica (12.3)].
No administre BRILINTA con otro inhibidor plaquetario oral P2Y12.
Evite la aspirina en dosis superiores a las recomendadas [ver Estudios clínicos (14.1)].
2.2 Síndrome coronario agudo o antecedentes de infarto de miocardio
Inicie el tratamiento con una dosis de carga de 180 mg de BRILINTA. Administre la primera dosis de mantenimiento de 90 mg de
BRILINTA, de 6 a 12 horas después de la dosis de carga. Administre 90 mg de BRILINTA dos veces al día durante el primer año después de
un evento de SCA. Después de un año, administre 60 mg de BRILINTA dos veces al día.
Inicie BRILINTA con una dosis de mantenimiento diaria de aspirina de 75 mg a 100 mg. Sin embargo, en pacientes que han
sido sometidos a intervención coronaria percutánea (ICP), considere la terapia antiplaquetaria única con BRILINTA en función del
riesgo evolutivo de eventos trombóticos versus hemorrágicos [ver Advertencias y precauciones (5.1) y Estudios clínicos (14)].
2.3 Enfermedad de la arteria coronaria pero sin accidente cerebrovascular o infarto de miocardio previo
Administre 60 mg de BRILINTA dos veces al día.
Generalmente, use BRILINTA con una dosis de mantenimiento diaria de aspirina de 75 mg a 100 mg [ver Estudios clínicos (14)].
2.4 Accidente cerebrovascular isquémico agudo o accidente isquémico transitorio (AIT)
Inicie el tratamiento con una dosis de carga de 180 mg de BRILINTA y luego continúe con 90 mg dos veces al día durante un máximo de 30 días.
Administre la primera dosis de mantenimiento de 6 a 12 horas después de la dosis de carga.
Use BRILINTA con una dosis de carga de aspirina (300 mg a 325 mg) y una dosis de mantenimiento diaria de aspirina de 75 mg a
100 mg [ver Estudios clínicos (14)].
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
BRILINTA (ticagrelor) 90 mg se suministra como un comprimido recubierto con película, redondo, biconvexo, amarillo, marcado con un “90” sobre una “T” en un lado.
BRILINTA (ticagrelor) 60 mg se suministra como un comprimido recubierto con película, redondo, biconvexo, rosa, marcado con un “60” sobre una “T” en un lado.
4 CONTRAINDICACIONES
4.1 Antecedentes de Hemorragia Intracraneal
BRILINTA está contraindicado en pacientes con antecedentes de hemorragia intracraneal (HIC) debido al alto riesgo de HIC recurrente en esta población [ver Estudios Clínicos (14.1), (14.2)].
4.2 Hemorragia Activa
BRILINTA está contraindicado en pacientes con hemorragia patológica activa, como úlcera péptica o hemorragia intracraneal [ver Advertencias y Precauciones (5.1) y Reacciones Adversas (6.1)].
4.3 Hipersensibilidad
BRILINTA está contraindicado en pacientes con hipersensibilidad (p. ej., angioedema) al ticagrelor o a cualquier componente del producto.
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Riesgo de hemorragia
Los medicamentos que inhiben la función plaquetaria, incluido BRILINTA, aumentan el riesgo de hemorragia [ver Advertencias y precauciones
(5.2) y Reacciones adversas (6.1)].
Pacientes tratados por accidente cerebrovascular isquémico agudo o AIT
Los pacientes con NIHSS >5 y los pacientes que recibieron trombolisis fueron excluidos de THALES y no se recomienda el uso de BRILINTA en dichos pacientes.
5.2 Suspensión de BRILINTA en pacientes tratados por enfermedad coronaria
La suspensión de BRILINTA aumentará el riesgo de infarto de miocardio, accidente cerebrovascular y muerte en pacientes tratados
por enfermedad coronaria. Si BRILINTA debe suspenderse temporalmente (por ejemplo, para tratar una hemorragia o una cirugía importante),
reinícielo lo antes posible. Cuando sea posible, interrumpa el tratamiento con BRILINTA durante cinco días antes de la cirugía que
tenga un riesgo importante de hemorragia. Reanude BRILINTA tan pronto como se logre la hemostasia.
5.3 Disnea
En los ensayos clínicos, aproximadamente del 14% (PLATO y PEGASUS) al 21% (THEMIS) de los pacientes tratados con BRILINTA desarrollaron disnea. La disnea fue generalmente de intensidad leve a moderada y a menudo se resolvió durante el tratamiento continuo, pero provocó la interrupción del fármaco del estudio en el 0,9% (PLATO), el 1,0% (THALES), el 4,3% (PEGASUS) y el 6,9% (THEMIS) de los pacientes.
En un subestudio de PLATO, 199 sujetos se sometieron a pruebas de función pulmonar independientemente de si informaron disnea. No hubo indicios de un efecto adverso sobre la función pulmonar evaluada después de un mes o después de al menos 6 meses de tratamiento crónico.
Si un paciente desarrolla disnea nueva, prolongada o empeorada que se determine que está relacionada con BRILINTA, no se requiere ningún tratamiento específico; continúe con BRILINTA sin interrupción si es posible. En el caso de disnea intolerable que requiera la interrupción de BRILINTA, considere recetar otro agente antiplaquetario.
5.4 Bradiarritmias
BRILINTA puede causar pausas ventriculares [ver Reacciones adversas (6.1)]. Se han notificado bradiarritmias, incluido el bloqueo auriculoventricular, en la fase posterior a la comercialización. Los pacientes con antecedentes de síndrome del seno enfermo, bloqueo auriculoventricular de 2º o 3º grado o
síncope relacionado con bradicardia no protegido por un marcapasos fueron excluidos de los estudios clínicos y pueden tener un mayor
riesgo de desarrollar bradiarritmias con ticagrelor.
5.5 Insuficiencia hepática grave
Evite el uso de BRILINTA en pacientes con insuficiencia hepática grave. Es probable que la insuficiencia hepática grave aumente
la concentración sérica de ticagrelor. No hay estudios de pacientes con BRILINTA con insuficiencia hepática grave [ver
5.6 Apnea central del sueño
Se ha notificado apnea central del sueño (ACS), incluida la respiración de Cheyne-Stokes (RCS), en la fase posterior a la comercialización en
pacientes que toman ticagrelor, incluida la recurrencia o el empeoramiento de la ACS/RCS tras la reexposición. Si se sospecha apnea central del sueño,
considere una evaluación clínica adicional.
5.7 Interferencias en las pruebas de laboratorio
Falsos resultados negativos en las pruebas funcionales de trombocitopenia inducida por heparina (TIH)
Se ha notificado que BRILINTA causa resultados falsos negativos en las pruebas funcionales de plaquetas (incluido el ensayo de agregación plaquetaria inducida por heparina (HIPA)) para pacientes con trombocitopenia inducida por heparina (TIH). Esto está relacionado con
la inhibición del receptor P2Y12 en las plaquetas de donantes sanos en la prueba por ticagrelor en el suero/plasma del paciente afectado.
Se requiere información sobre el tratamiento concomitante con BRILINTA para la interpretación de las pruebas funcionales de TIH.
Según el mecanismo de interferencia de BRILINTA, no se espera que BRILINTA afecte las pruebas de anticuerpos PF4 para
TIH.
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas también se discuten en otras partes del etiquetado:
- •
- Hemorragia [ver Advertencias y precauciones (5.1)]
- •
- Disnea [ver Advertencias y precauciones (5.3)]
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se realizan en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no se pueden comparar directamente con las tasas en los ensayos clínicos de otro fármaco y pueden no reflejar las tasas observadas en la práctica.
BRILINTA se ha evaluado en cuanto a seguridad en más de 58.000 pacientes.
Hemorragia en PLATO (Reducción del riesgo de eventos trombóticos en el SCA)
La Figura 1 es una gráfica del tiempo hasta el primer evento de hemorragia mayor no relacionada con CABG.
Figura 1 – Estimación de Kaplan-Meier del tiempo hasta el primer evento de hemorragia mayor según la definición de PLATO no relacionado con CABG (PLATO)
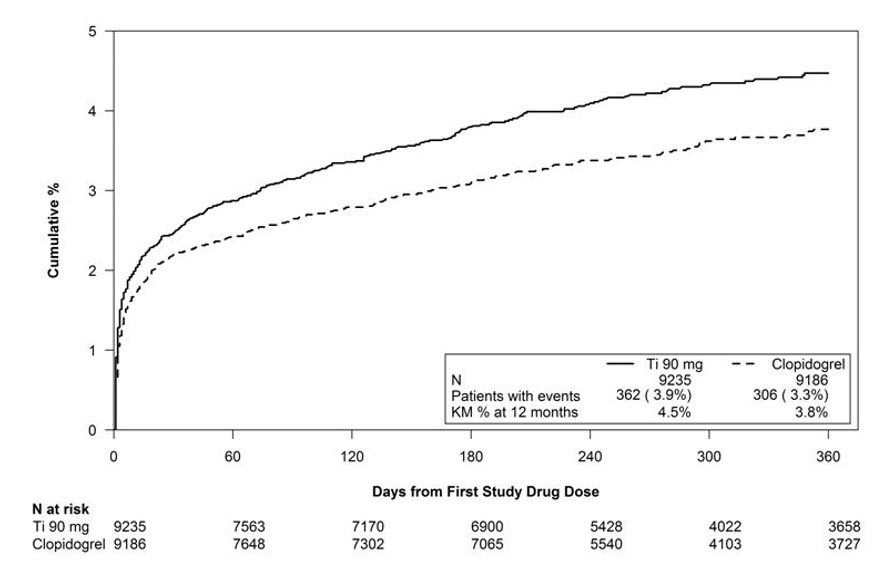
La frecuencia de hemorragia en PLATO se resume en las Tablas 1 y 2. Aproximadamente la mitad de los eventos de hemorragia mayor no relacionados con CABG ocurrieron en los primeros 30 días.
|
||
|
BRILINTA* |
Clopidogrel |
|
|
n (%) pacientes con evento |
n (%) pacientes con evento |
|
|
PLATO Mayor + Menor |
713 (7.7) |
567 (6.2) |
|
Mayor |
362 (3.9) |
306 (3.3) |
|
Fatal/Potencialmente mortal |
171 (1.9) |
151 (1.6) |
|
Fatal |
15 (0.2) |
16 (0.2) |
|
Hemorragia intracraneal (Fatal/Potencialmente mortal) |
26 (0.3) |
15 (0.2) |
|
Hemorragia menor según PLATO: requiere intervención médica para detener o tratar la hemorragia. Hemorragia mayor según PLATO: cualquiera de las siguientes: fatal; intracraneal; intrapericárdica con taponamiento cardíaco; shock hipovolémico o hipotensión grave que requiere intervención; significativamente incapacitante (p. ej., intraocular con pérdida permanente de la visión); asociada con una disminución de Hb de al menos 3 g/dL (o una caída en el hematocrito (Hct) de al menos 9 %); transfusión de 2 o más unidades. Hemorragia mayor según PLATO, fatal/potencialmente mortal: cualquier hemorragia mayor como se describe anteriormente y asociada con una disminución de Hb de más de 5 g/dL (o una caída en el hematocrito (Hct) de al menos 15 %); transfusión de 4 o más unidades. Fatal: Un evento de hemorragia que provocó directamente la muerte en un plazo de 7 días. |
||
Ningún factor demográfico basal alteró el riesgo relativo de hemorragia con BRILINTA en comparación con clopidogrel.
En PLATO, 1584 pacientes se sometieron a cirugía de CABG. Los porcentajes de esos pacientes que sangraron se muestran en la Figura 2 y la Tabla 2.
Figura 2 – Hemorragia relacionada con CABG ‘mayor, mortal/potencialmente mortal’ según los días transcurridos desde la última dosis del fármaco del estudio hasta el procedimiento de CABG (PLATO)
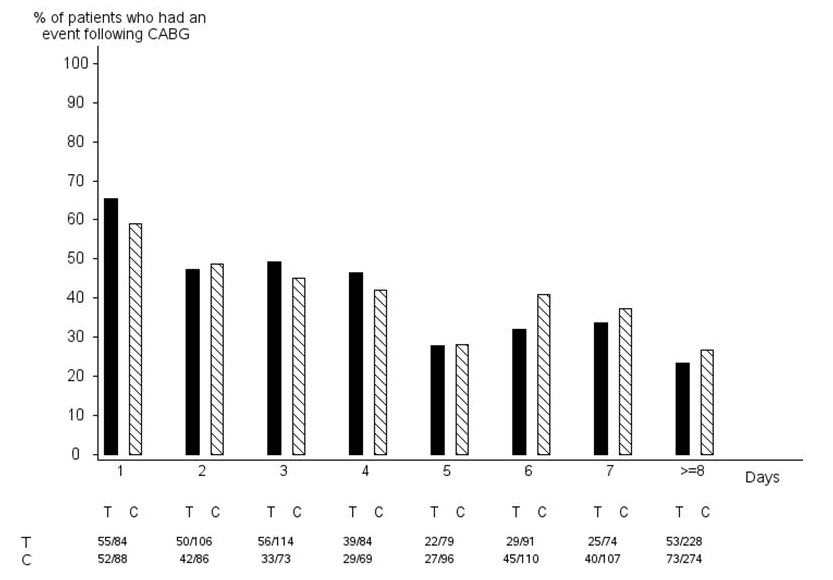
El eje X representa los días transcurridos desde la última dosis del fármaco del estudio antes de la CABG.
El protocolo PLATO recomendaba un procedimiento para suspender el fármaco del estudio antes de la CABG u otra cirugía mayor sin desvelar el tratamiento. Si la cirugía era electiva o no urgente, el fármaco del estudio se interrumpía temporalmente, de la siguiente manera: Si la práctica local permitía que los efectos antiplaquetarios se disiparan antes de la cirugía, las cápsulas (clopidogrel enmascarado) se suspendían 5 días antes de la cirugía y las tabletas (ticagrelor enmascarado) se suspendían durante un mínimo de 24 horas y un máximo de 72 horas antes de la cirugía. Si la práctica local era realizar la cirugía sin esperar a que se disiparan los efectos antiplaquetarios, las cápsulas y las tabletas se suspendían 24 horas antes de la cirugía y se permitía el uso de aprotinina u otros agentes hemostáticos. Si la práctica local era utilizar la monitorización de la agregación plaquetaria (IPA) para determinar cuándo se podía realizar la cirugía, tanto las cápsulas como las tabletas se suspendían al mismo tiempo y se seguían los procedimientos de monitorización habituales.
T Ticagrelor; C Clopidogrel.
|
||
|
BRILINTA* N=770 |
Clopidogrel N=814 |
|
|
n (%) pacientes con evento |
n (%) pacientes con evento |
|
|
Total de hemorragias mayores PLATO |
626 (81.3) |
666 (81.8) |
|
Mortal/potencialmente mortal |
337 (43.8) |
350 (43.0) |
|
Mortal |
6 (0.8) |
7 (0.9) |
|
Hemorragia mayor PLATO: cualquiera de las siguientes: mortal; intracraneal; intrapericárdica con taponamiento cardíaco; shock hipovolémico o hipotensión grave que requiera intervención; significativamente incapacitante (p. ej., intraocular con pérdida permanente de la visión); asociada con una disminución de Hb de al menos 3 g/dL (o una disminución del hematocrito (Hct) de al menos el 9 %); transfusión de 2 o más unidades. Hemorragia mayor, mortal/potencialmente mortal PLATO: cualquier hemorragia mayor como se describe anteriormente y asociada con una disminución de Hb de más de 5 g/dL (o una disminución del hematocrito (Hct) de al menos el 15 %); transfusión de 4 o más unidades. |
||
Cuando el tratamiento antiplaquetario se interrumpió 5 días antes de la CABG, se produjo una hemorragia mayor en el 75 % de los pacientes tratados con BRILINTA y en el 79 % de los tratados con clopidogrel.
Otras reacciones adversas en PLATO
Las reacciones adversas que ocurrieron a una tasa del 4 % o más en PLATO se muestran en la Tabla 3.
|
||
|
BRILINTA* |
Clopidogrel |
|
|
Disnea |
13.8 |
7.8 |
|
Mareo |
4.5 |
3.9 |
|
Náuseas |
4.3 |
3.8 |
Hemorragia en PEGASUS (Prevención secundaria en pacientes con antecedentes de infarto de miocardio)
El resultado general de los eventos hemorrágicos en el estudio PEGASUS se muestra en la Tabla 4.
|
||
|
BRILINTA* N=6958 |
Placebo N=6996 |
|
|
Eventos / 1000 años-paciente |
Eventos / 1000 años-paciente |
|
|
TIMI Mayor |
8 |
3 |
|
Fatal |
1 |
1 |
|
Hemorragia intracraneal |
2 |
1 |
|
TIMI Mayor o Menor |
11 |
5 |
|
TIMI Mayor: Hemorragia fatal, O cualquier hemorragia intracraneal, O signos clínicamente evidentes de hemorragia asociados con una disminución de hemoglobina (Hgb) de ≥5 g/dL, o una disminución del hematocrito (Hct) de ≥15%. Fatal: Un evento hemorrágico que condujo directamente a la muerte en un plazo de 7 días. TIMI Menor: Clínicamente aparente con una disminución de 3-5 g/dL en la hemoglobina. |
||
El perfil de hemorragia de BRILINTA 60 mg en comparación con la aspirina sola fue consistente en múltiples subgrupos predefinidos (por ejemplo, por edad, sexo, peso, raza, región geográfica, afecciones concomitantes, terapia concomitante, stent y antecedentes médicos) para eventos hemorrágicos TIMI Mayor y TIMI Mayor o Menor.
Otras reacciones adversas en PEGASUS
Las reacciones adversas que ocurrieron en PEGASUS a tasas del 3% o más se muestran en la Tabla 5.
|
||
|
BRILINTA* |
Placebo |
|
|
Disnea |
14.2% |
5.5% |
|
Mareo |
4.5% |
4.1% |
|
Diarrea |
3.3% |
2.5% |
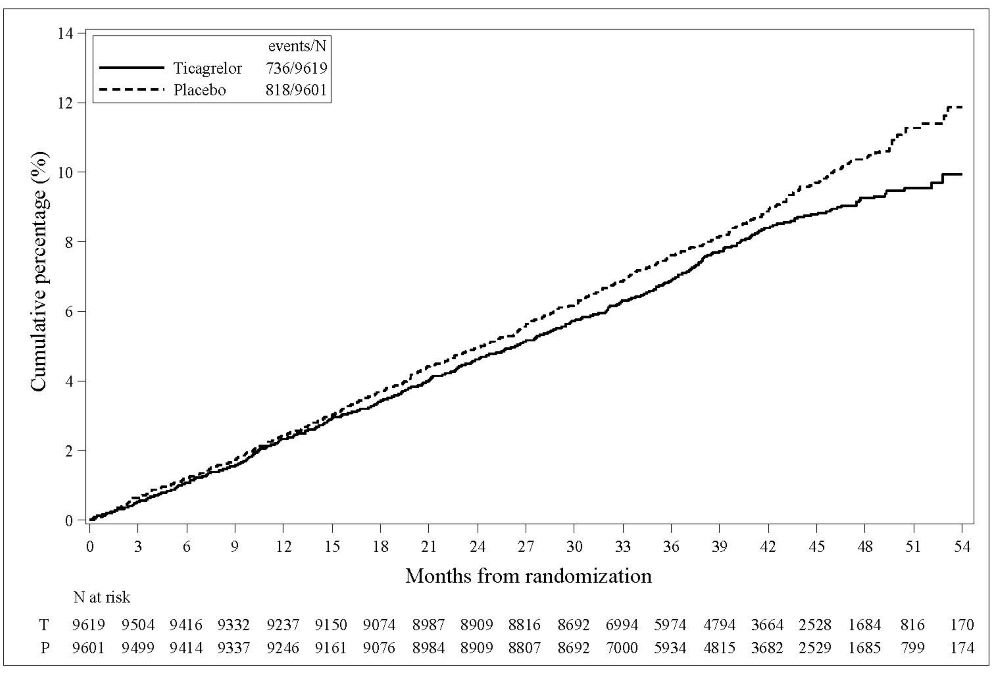
Hemorragia en THEMIS (Prevención de eventos cardiovasculares mayores en pacientes con CAD y diabetes mellitus tipo 2)
La curva de Kaplan-Meier del tiempo hasta el primer evento de hemorragia mayor TIMI se presenta en la Figura 3.
Figura 3 – Tiempo hasta el primer evento de hemorragia mayor TIMI (THEMIS)
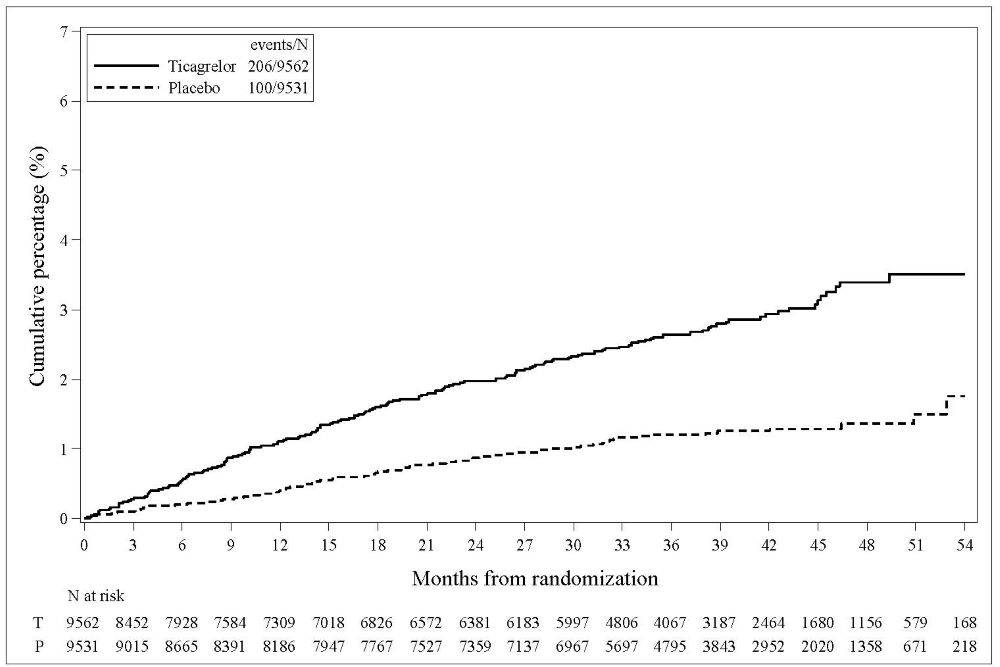
T = Ticagrelor; P = Placebo; N = Número de pacientes
Los eventos hemorrágicos en THEMIS se muestran a continuación en la Tabla 6.
|
BRILINTA N=9562 |
Placebo N=9531 |
|
|
Eventos / 1000 años-paciente |
Eventos / 1000 años-paciente |
|
|
TIMI Mayor |
9 |
4 |
|
TIMI Mayor o Menor |
12 |
5 |
|
TIMI Mayor o Menor o que requirió atención médica |
46 |
18 |
|
Hemorragia fatal |
1 |
0 |
|
Hemorragia intracraneal |
3 |
2 |
Hemorragia en THALES (Reducción del riesgo de accidente cerebrovascular en pacientes con accidente cerebrovascular isquémico agudo o AIT)
La curva de Kaplan-Meier del curso temporal de los eventos de hemorragia grave de GUSTO se presenta en la Figura 4.
Figura 4 – Curso temporal de los eventos de hemorragia grave de GUSTO
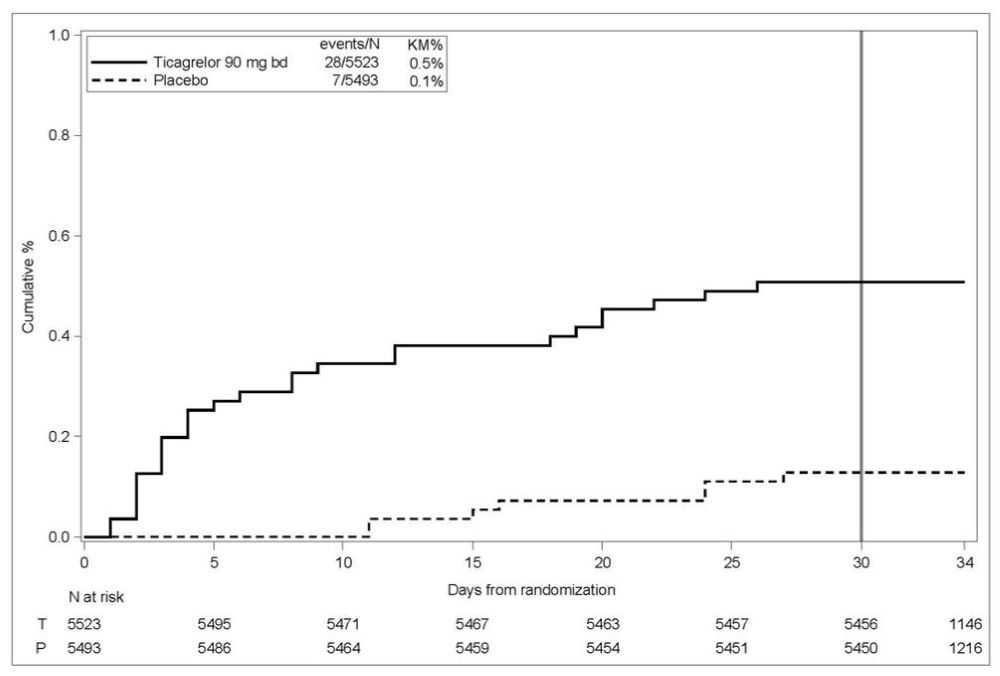
KM%: Porcentaje de Kaplan-Meier evaluado al día 30; T = Ticagrelor; P = placebo; N = Número de pacientes
GUSTO Grave: Cualquiera de los siguientes: hemorragia fatal, hemorragia intracraneal (excluyendo transformaciones hemorrágicas asintomáticas de infartos cerebrales isquémicos y excluyendo microhemorragias < 10 mm evidentes solo en imágenes de resonancia magnética de eco de gradiente), hemorragia que causó compromiso hemodinámico que requirió intervención (p. ej., presión arterial sistólica <90 mmHg que requirió reposición de sangre o líquidos, o soporte vasopresor/inotrópico, o intervención quirúrgica).
Hemorragia intracraneal y hemorragia fatal en THALES: En total, hubo 21 hemorragias intracraneales (HIC) para BRILINTA y 6 HIC para placebo. Las hemorragias fatales, casi todas HIC, ocurrieron en 11 pacientes con BRILINTA y en 2 con placebo.
Bradicardia
En un subestudio Holter de aproximadamente 3000 pacientes en PLATO, más pacientes tuvieron pausas ventriculares con BRILINTA (6,0%) que con clopidogrel (3,5%) en la fase aguda; las tasas fueron del 2,2% y el 1,6%, respectivamente, después de 1 mes. PLATO, PEGASUS, THEMIS y THALES excluyeron a pacientes con mayor riesgo de eventos bradicárdicos (p. ej., pacientes con síndrome del seno enfermo, bloqueo AV de 2nd o 3rd grado, o síncope relacionado con bradicardia y no protegidos con un marcapasos).
Anormalidades de laboratorio
Ácido úrico sérico:
En PLATO, los niveles de ácido úrico sérico aumentaron aproximadamente 0,6 mg/dL desde el inicio con BRILINTA 90 mg y aproximadamente 0,2 mg/dL con clopidogrel. La diferencia desapareció dentro de los 30 días de suspender el tratamiento. Los informes de gota no difirieron entre los grupos de tratamiento en PLATO (0,6% en cada grupo).
En PEGASUS, los niveles de ácido úrico sérico aumentaron aproximadamente 0,2 mg/dL desde el inicio con BRILINTA 60 mg y no se observó elevación con aspirina sola. La gota ocurrió con más frecuencia en pacientes con BRILINTA que en pacientes con solo aspirina (1,5%, 1,1%). Las concentraciones medias de ácido úrico sérico disminuyeron después de suspender el tratamiento.
Creatinina sérica:
En PLATO, se observó un aumento >50% en los niveles de creatinina sérica en el 7,4% de los pacientes que recibieron BRILINTA 90 mg en comparación con el 5,9% de los pacientes que recibieron clopidogrel. Los aumentos generalmente no progresaron con el tratamiento continuo y a menudo disminuyeron con la terapia continua. Se observó evidencia de reversibilidad al suspender el tratamiento incluso en aquellos con los mayores aumentos durante el tratamiento. Los grupos de tratamiento en PLATO no difirieron en cuanto a eventos adversos graves relacionados con el riñón, como insuficiencia renal aguda, insuficiencia renal crónica, nefropatía tóxica u oliguria.
En PEGASUS, la concentración de creatinina sérica aumentó en más del 50% en aproximadamente el 4% de los pacientes que recibieron BRILINTA 60 mg, similar a la aspirina sola. La frecuencia de eventos adversos relacionados con el riñón fue similar para ticagrelor y aspirina sola, independientemente de la edad y la función renal basal.
6.2 Experiencia postcomercialización
Las siguientes reacciones adversas se han identificado durante el uso posterior a la aprobación de BRILINTA. Debido a que estas reacciones se notifican voluntariamente a partir de una población de tamaño desconocido, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal con la exposición al fármaco.
Trastornos de la sangre y del sistema linfático: Se ha notificado raramente púrpura trombocitopénica trombótica (PTT) con el uso de BRILINTA. La PTT es una afección grave que puede ocurrir después de una breve exposición (<2 semanas) y requiere tratamiento inmediato.
Trastornos del sistema inmunitario: Reacciones de hipersensibilidad que incluyen angioedema [ver Contraindicaciones (4.3)].
Trastornos respiratorios: Apnea central del sueño, respiración de Cheyne-Stokes
Trastornos de la piel y del tejido subcutáneo: Erupción cutánea
7 INTERACCIONES MEDICAMENTOSAS
7.1 Inhibidores potentes del CYP3A
Los inhibidores potentes del CYP3A aumentan sustancialmente la exposición a ticagrelor y, por lo tanto, aumentan el riesgo de disnea, hemorragia y otros eventos adversos. Evite el uso de inhibidores potentes del CYP3A (p. ej., ketoconazol, itraconazol, voriconazol, claritromicina, nefazodona, ritonavir, saquinavir, nelfinavir, indinavir, atazanavir y telithromicina) [ver Farmacología clínica (12.3)].
7.2 Inductores potentes del CYP3A
Los inductores potentes del CYP3A reducen sustancialmente la exposición a ticagrelor y, por lo tanto, disminuyen la eficacia del ticagrelor. Evite el uso con inductores potentes del CYP3A (p. ej., rifampicina, fenitoína, carbamazepina y fenobarbital) [ver Farmacología clínica (12.3)].
7.3 Opioides
Al igual que con otros inhibidores orales de P2Y12, la administración conjunta de agonistas opioides retrasa y reduce la absorción de ticagrelor y su metabolito activo, presumiblemente debido a la ralentización del vaciado gástrico [ver Farmacología clínica (12.3)]. Considere el uso de un agente antiplaquetario parenteral en pacientes con síndrome coronario agudo que requieren la administración conjunta de morfina u otros agonistas opioides.
7.4 Simvastatina, Lovastatina, Rosuvastatina
BRILINTA aumenta las concentraciones séricas de simvastatina y lovastatina porque estos fármacos se metabolizan mediante el CYP3A4. Evite dosis de simvastatina y lovastatina superiores a 40 mg [ver Farmacología clínica (12.3)].
Brilinta aumenta la concentración sérica de rosuvastatina porque la rosuvastatina es un sustrato de BCRP [ver Farmacología clínica (12.3)].
7.5 Digoxina
BRILINTA inhibe el transportador de P-glucoproteína; controle los niveles de digoxina con el inicio o el cambio en el tratamiento con BRILINTA [ver Farmacología clínica (12.3)].
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de Riesgo
Los datos disponibles de informes de casos con el uso de BRILINTA en mujeres embarazadas no han identificado un riesgo asociado al fármaco de defectos congénitos importantes, aborto espontáneo o resultados adversos maternos o fetales. El ticagrelor administrado a ratas embarazadas y conejas embarazadas durante la organogénesis causó anomalías estructurales en la descendencia a dosis maternas aproximadamente 5 a 7 veces la dosis humana máxima recomendada (MRHD) basada en el área de superficie corporal. Cuando se administró ticagrelor a ratas durante la gestación tardía y la lactancia, se observó muerte de crías y efectos en el crecimiento de las crías a aproximadamente 10 veces la MRHD (ver Datos).
Se desconoce el riesgo de fondo estimado de defectos congénitos importantes y aborto espontáneo para la población indicada. Todos los embarazos tienen un riesgo de fondo de defectos congénitos, pérdida u otros resultados adversos. En la población general de EE. UU., el riesgo de fondo estimado de defectos congénitos importantes y aborto espontáneo en embarazos clínicamente reconocidos es del 2 al 4 % y del 15 al 20 %, respectivamente.
Datos
Datos en animales
En estudios de toxicología reproductiva, las ratas embarazadas recibieron ticagrelor durante la organogénesis en dosis de 20 a 300 mg/kg/día. 20 mg/kg/día es aproximadamente lo mismo que la MRHD de 90 mg dos veces al día para un humano de 60 kg en base a mg/m2. Los resultados adversos en la descendencia ocurrieron a dosis de 300 mg/kg/día (16,5 veces la MRHD en base a mg/m2) e incluyeron lóbulo hepático y costillas supernumerarias, osificación incompleta de las esternébras, articulación desplazada de la pelvis y esternébras deformadas/desalineadas. En la dosis intermedia de 100 mg/kg/día (5,5 veces la MRHD en base a mg/m2), se observó un retraso en el desarrollo del hígado y el esqueleto. Cuando las conejas embarazadas recibieron ticagrelor durante la organogénesis en dosis de 21 a 63 mg/kg/día, los fetos expuestos a la dosis materna más alta de 63 mg/kg/día (6,8 veces la MRHD en base a mg/m2) tuvieron un retraso en el desarrollo de la vesícula biliar y se produjo una osificación incompleta del hioides, el pubis y las esternébras.
En un estudio prenatal/posnatal, las ratas embarazadas recibieron ticagrelor en dosis de 10 a 180 mg/kg/día durante la gestación tardía y la lactancia. Se observó muerte de crías y efectos en el crecimiento de las crías a 180 mg/kg/día (aproximadamente 10 veces la MRHD en base a mg/m2). Se produjeron efectos relativamente menores, como retrasos en el despliegue del pabellón auricular y la apertura de los ojos, a dosis de 10 y 60 mg/kg (aproximadamente la mitad y 3,2 veces la MRHD en base a mg/m2).
8.2 Lactancia
Resumen de Riesgo
No hay datos sobre la presencia de ticagrelor o sus metabolitos en la leche materna, los efectos en el lactante o los efectos en la producción de leche. El ticagrelor y sus metabolitos estuvieron presentes en la leche de rata en concentraciones más altas que en el plasma materno. Cuando un fármaco está presente en la leche animal, es probable que el fármaco esté presente en la leche materna. No se recomienda la lactancia materna durante el tratamiento con BRILINTA.
8.4 Uso pediátrico
No se ha establecido la seguridad y eficacia de BRILINTA en pacientes pediátricos. La eficacia no se demostró en un estudio adecuado y bien controlado realizado en 101 pacientes pediátricos tratados con BRILINTA, de 2 a <18 años, para reducir la tasa de crisis vaso-oclusivas en la enfermedad de células falciformes.
8.5 Uso geriátrico
Aproximadamente la mitad de los pacientes en PLATO, PEGASUS, THEMIS y THALES tenían ≥65 años de edad y al menos el 15 % tenían ≥75 años de edad. No se observaron diferencias generales en la seguridad o la eficacia entre los pacientes ancianos y los pacientes más jóvenes.
8.6 Insuficiencia hepática
El ticagrelor se metaboliza en el hígado y la función hepática deteriorada puede aumentar los riesgos de hemorragia y otros eventos adversos. Evite el uso de BRILINTA en pacientes con insuficiencia hepática grave. Existe una experiencia limitada con BRILINTA en pacientes con insuficiencia hepática moderada; considere los riesgos y beneficios del tratamiento, teniendo en cuenta el probable aumento de la exposición al ticagrelor. No es necesario ningún ajuste de dosis en pacientes con insuficiencia hepática leve [ver Advertencias y precauciones (5.5) y Farmacología clínica (12.3)].
8.7 Insuficiencia renal
No es necesario ningún ajuste de dosis en pacientes con insuficiencia renal [ver Farmacología clínica (12.3)].
Pacientes con enfermedad renal en etapa terminal en diálisis
Los estudios de eficacia y seguridad clínica con BRILINTA no incluyeron pacientes con enfermedad renal en etapa terminal (ERET) en diálisis. En pacientes con ERET mantenidos con hemodiálisis intermitente, no se espera una diferencia clínicamente significativa en las concentraciones de ticagrelor y su metabolito y la inhibición plaquetaria en comparación con las observadas en pacientes con función renal normal [ver Farmacología clínica (12.3)]. No se sabe si estas concentraciones producirán una eficacia y seguridad similares en pacientes con ERET en diálisis a las observadas en PLATO, PEGASUS, THEMIS y THALES.
10 SOBREDOSIS
Actualmente no se conoce ningún tratamiento para revertir los efectos de BRILINTA, y el ticagrelor no es dializable. El tratamiento de la sobredosis debe seguir la práctica médica estándar local. El sangrado es el efecto farmacológico esperado de una sobredosis. Si se produce una hemorragia, se deben tomar las medidas de apoyo adecuadas.
La transfusión de plaquetas no revirtió el efecto antiplaquetario de BRILINTA en voluntarios sanos y es poco probable que sea de beneficio clínico en pacientes con hemorragia.
Otros efectos de la sobredosis pueden incluir efectos gastrointestinales (náuseas, vómitos, diarrea) o pausas ventriculares. Monitorizar el ECG.
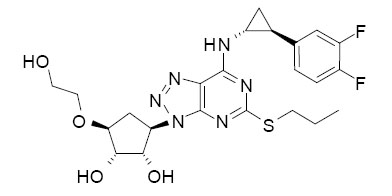
11 DESCRIPCIÓN
BRILINTA contiene ticagrelor, una ciclopentiltriazolopirimidina, inhibidor de la activación y agregación plaquetaria mediada por el receptor P2Y12 ADP. Químicamente es (1S, 2S, 3R, 5S)-3-[7-{[(1R, 2S)-2-(3,4-difluorofenil)ciclopropil]amino}-5-(propiltio)-3H-[1,2,3]-triazolo[4,5-d]pirimidina-3-il]-5-(2-hidroxietoxi)ciclopentano-1,2-diol. La fórmula empírica del ticagrelor es C23H28F2N6O4S y su peso molecular es 522.57. La estructura química del ticagrelor es:
El ticagrelor es un polvo cristalino con una solubilidad acuosa de aproximadamente 10 μg/mL a temperatura ambiente.
Las tabletas de BRILINTA 90 mg para administración oral contienen 90 mg de ticagrelor y los siguientes ingredientes: manitol, fosfato de calcio dibásico, glicolato de almidón sódico, hidroxipropilcelulosa, estearato de magnesio, hidroxipropilmetilcelulosa, dióxido de titanio, talco, polietilenglicol 400 y óxido de hierro amarillo.
Las tabletas de BRILINTA 60 mg para administración oral contienen 60 mg de ticagrelor y los siguientes ingredientes: manitol, fosfato de calcio dibásico, glicolato de almidón sódico, hidroxipropilcelulosa, estearato de magnesio, hidroxipropilmetilcelulosa, dióxido de titanio, polietilenglicol 400, óxido de hierro negro y óxido de hierro rojo.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
El ticagrelor y su metabolito principal interactúan de forma reversible con el receptor P2Y12 ADP plaquetario para prevenir la transducción de señales y la activación plaquetaria. El ticagrelor y su metabolito activo son aproximadamente equipotentes.
12.2 Farmacodinamia
La inhibición de la agregación plaquetaria (IPA) por ticagrelor y clopidogrel se comparó en un estudio de 6 semanas que examinó los efectos de la inhibición plaquetaria aguda y crónica en respuesta a 20 μM ADP como agonista de la agregación plaquetaria.
El inicio de la IPA se evaluó el día 1 del estudio después de dosis de carga de 180 mg de ticagrelor o 600 mg de clopidogrel. Como se muestra en la Figura 5, la IPA fue mayor en el grupo de ticagrelor en todos los puntos de tiempo. El efecto máximo de IPA del ticagrelor se alcanzó alrededor de las 2 horas y se mantuvo durante al menos 8 horas.
El desplazamiento de la IPA se examinó después de 6 semanas con ticagrelor 90 mg dos veces al día o clopidogrel 75 mg al día, nuevamente en respuesta a 20 μM ADP.
Como se muestra en la Figura 6, la IPA máxima media después de la última dosis de ticagrelor fue del 88% y del 62% para clopidogrel. El recuadro de la Figura 6 muestra que después de 24 horas, la IPA en el grupo de ticagrelor (58%) fue similar a la IPA en el grupo de clopidogrel (52%), lo que indica que los pacientes que omiten una dosis de ticagrelor aún mantendrían una IPA similar a la IPA mínima de los pacientes tratados con clopidogrel. Después de 5 días, la IPA en el grupo de ticagrelor fue similar a la IPA en el grupo placebo. Se desconoce cómo el riesgo de hemorragia o el riesgo trombótico siguen la IPA, tanto para ticagrelor como para clopidogrel.
Figura 5 – Inhibición media de la agregación plaquetaria (±SE) después de dosis orales únicas de placebo, 180 mg de ticagrelor o 600 mg de clopidogrel
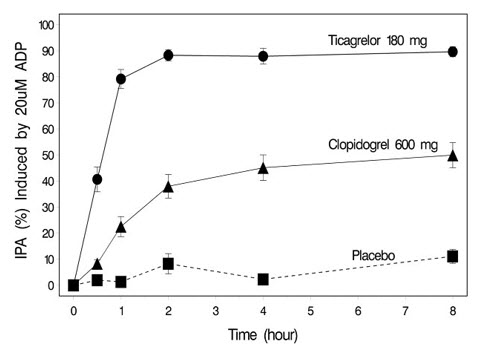
Figura 6 – Inhibición media de la agregación plaquetaria (IPA) después de 6 semanas con placebo, ticagrelor 90 mg dos veces al día o clopidogrel 75 mg al día
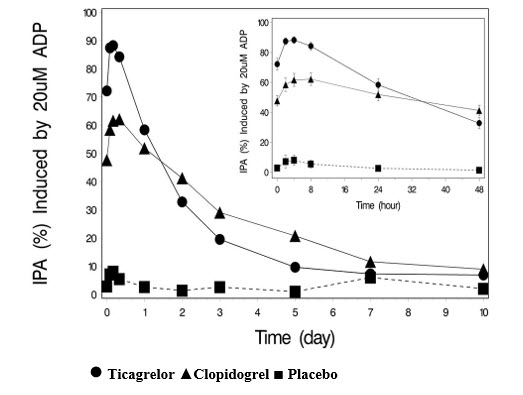
La transición de clopidogrel a BRILINTA resultó en un aumento absoluto de la IPA del 26.4% y de BRILINTA a clopidogrel resultó en una disminución absoluta de la IPA del 24.5%. Los pacientes pueden pasar de clopidogrel a BRILINTA sin interrupción del efecto antiplaquetario [ver Dosis y administración (2)].
12.3 Farmacocinética
El ticagrelor demuestra una farmacocinética proporcional a la dosis, que es similar en pacientes y voluntarios sanos.
Absorción
BRILINTA se puede tomar con o sin alimentos. La absorción de ticagrelor ocurre con una mediana tmáx de 1.5 h (rango 1.0–4.0). La formación del principal metabolito circulante AR-C124910XX (activo) a partir de ticagrelor ocurre con una mediana tmáx de 2.5 h (rango 1.5-5.0).
La biodisponibilidad absoluta media del ticagrelor es de aproximadamente el 36% (rango 30%-42%). La ingestión de una comida rica en grasas no tuvo ningún efecto sobre la Cmáx de ticagrelor, pero resultó en un aumento del 21% en el AUC. La Cmáx de su metabolito principal disminuyó en un 22% sin cambios en el AUC.
BRILINTA en comprimidos triturados mezclados en agua, administrados por vía oral o administrados a través de una sonda nasogástrica en el estómago, es bioequivalente a los comprimidos enteros (AUC y Cmáx dentro del 80-125% para ticagrelor y AR-C124910XX) con una mediana tmáx de 1.0 hora (rango 1.0 – 4.0) para ticagrelor y 2.0 horas (rango 1.0 – 8.0) para AR-C124910XX.
Distribución
El volumen de distribución en estado estacionario del ticagrelor es de 88 L. El ticagrelor y el metabolito activo se unen ampliamente a las proteínas plasmáticas humanas (>99%).
Metabolismo
CYP3A4 es la principal enzima responsable del metabolismo del ticagrelor y la formación de su principal metabolito activo. El ticagrelor y su principal metabolito activo son sustratos e inhibidores débiles de la P-glicoproteína. La exposición sistémica al metabolito activo es aproximadamente del 30 al 40% de la exposición al ticagrelor. El ticagrelor es un inhibidor de la BCRP.
Excreción
La principal vía de eliminación del ticagrelor es el metabolismo hepático. Cuando se administra ticagrelor radiomarcado, la recuperación media de radiactividad es de aproximadamente el 84% (58% en heces, 26% en orina). Las recuperaciones de ticagrelor y el metabolito activo en la orina fueron ambas inferiores al 1% de la dosis. La principal vía de eliminación del metabolito principal del ticagrelor es probablemente la secreción biliar. La t1/2 media es de aproximadamente 7 horas para el ticagrelor y 9 horas para el metabolito activo.
Poblaciones específicas
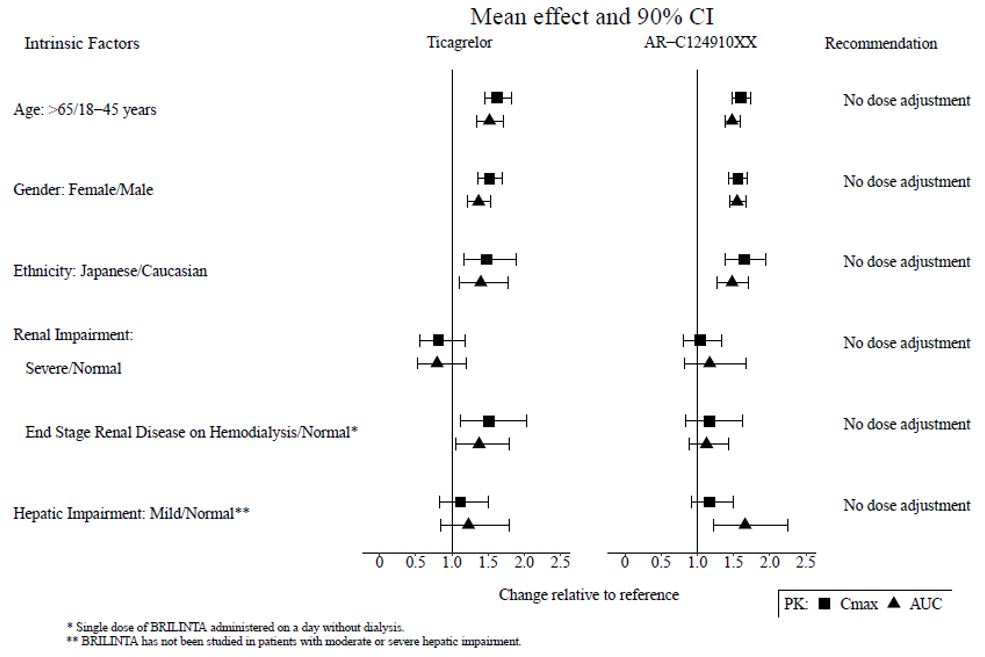
Los efectos de la edad, el sexo, el origen étnico, la insuficiencia renal y la insuficiencia hepática leve sobre la farmacocinética del ticagrelor se presentan en la Figura 7. Los efectos son modestos y no requieren ajuste de dosis.
Pacientes con enfermedad renal terminal en hemodiálisis
En pacientes con enfermedad renal terminal en hemodiálisis, el AUC y la Cmáx de BRILINTA 90 mg administrados en un día sin diálisis fueron un 38% y un 51% más altos, respectivamente, en comparación con los sujetos con función renal normal. Se observó un aumento similar en la exposición cuando BRILINTA se administró inmediatamente antes de la diálisis, lo que demuestra que BRILINTA no es dializable. La exposición del metabolito activo aumentó en menor medida. El efecto IPA de BRILINTA fue independiente de la diálisis en pacientes con enfermedad renal terminal y similar al de los adultos sanos con función renal normal.
Figura 7 – Impacto de los factores intrínsecos en la farmacocinética del ticagrelor
Efectos de otros medicamentos sobre BRILINTA
CYP3A4 es la principal enzima responsable del metabolismo de ticagrelor y la formación de su principal metabolito activo. Los efectos de otros fármacos sobre la farmacocinética de ticagrelor se presentan en la Figura 8 como cambio relativo a ticagrelor administrado solo (prueba/referencia). Los inhibidores potentes de CYP3A (p. ej., ketoconazol, itraconazol y claritromicina) aumentan sustancialmente la exposición a ticagrelor. Los inhibidores moderados de CYP3A tienen efectos menores (p. ej., diltiazem). Los inductores de CYP3A (p. ej., rifampicina) reducen sustancialmente los niveles sanguíneos de ticagrelor. Los inhibidores de la P-gp (p. ej., ciclosporina) aumentan la exposición a ticagrelor.
La coadministración de 5 mg de morfina intravenosa con una dosis de carga de 180 mg de ticagrelor disminuyó la exposición media observada a ticagrelor hasta en un 25 % en adultos sanos y hasta en un 36 % en pacientes con SCA sometidos a ICP. El Tmax se retrasó entre 1 y 2 horas. La exposición al metabolito activo disminuyó en una medida similar. La coadministración de morfina no retrasó ni disminuyó la inhibición plaquetaria en adultos sanos. La agregación plaquetaria media fue mayor hasta 3 horas después de la dosis de carga en pacientes con SCA coadministrados con morfina.
La coadministración de fentanilo intravenoso con una dosis de carga de 180 mg de ticagrelor en pacientes con SCA sometidos a ICP produjo efectos similares sobre la exposición a ticagrelor y la inhibición plaquetaria.
Figure 8 – Effect of co-administered drugs on the pharmacokinetics of ticagrelor
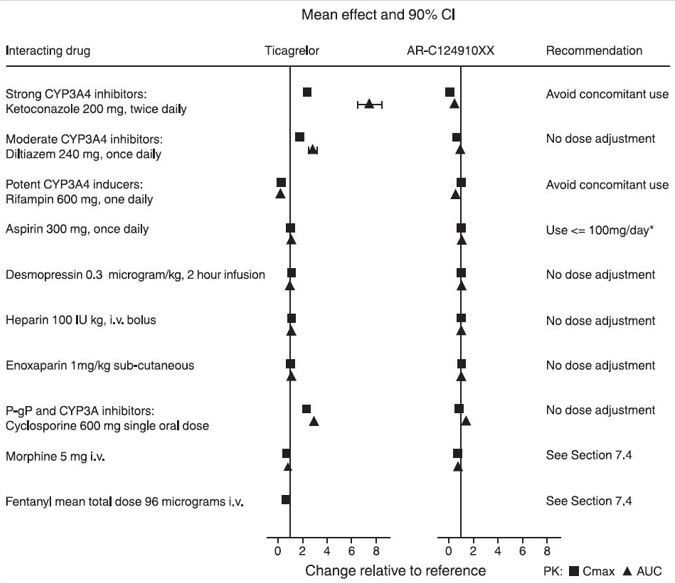
*See Dosage and Administration (2)
Efectos de BRILINTA sobre otros medicamentos
Los estudios de metabolismo in vitro demuestran que ticagrelor y su principal metabolito activo son inhibidores débiles de CYP3A4, activadores potenciales de CYP3A5 e inhibidores del transportador P-gp. Los estudios de metabolismo in vitro demuestran que ticagrelor es un inhibidor de BCRP. Se demostró que ticagrelor y AR-C124910XX no tienen ningún efecto inhibitorio sobre la actividad de CYP1A2, CYP2C19 y CYP2E1 humana. Para conocer los efectos específicos in vivo sobre la farmacocinética de simvastatina, atorvastatina, etinilestradiol, levonorgestrel, tolbutamida, digoxina y ciclosporina, consulte la Figura 9.
Figure 9 – Impact of BRILINTA on the pharmacokinetics of co-administered drugs
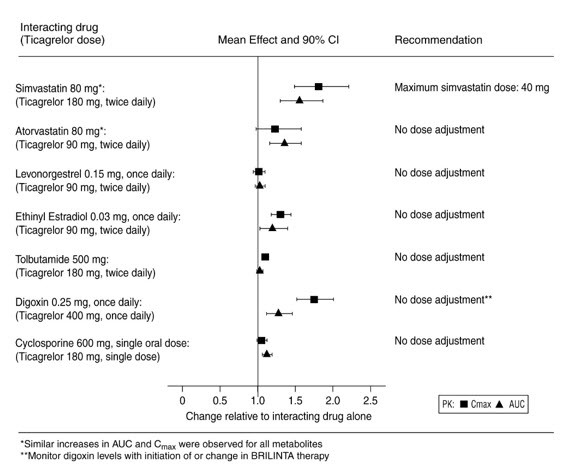
12.5 Farmacogenómica
En una cohorte de subestudio genético de PLATO, la tasa de eventos CV trombóticos en el grupo de BRILINTA no dependió del estado de pérdida de función de CYP2C19.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogenicidad, Mutagénesis, Deterioro de la Fertilidad
Carcinogenicidad
El ticagrelor no fue carcinogénico en ratones a dosis de hasta 250 mg/kg/día ni en ratas macho a dosis de hasta 120 mg/kg/día (19 y 15 veces la dosis máxima recomendada en humanos (MRHD) de 90 mg dos veces al día sobre la base del AUC, respectivamente). Se observaron carcinomas uterinos, adenocarcinomas uterinos y adenomas hepatocelulares en ratas hembras a dosis de 180 mg/kg/día (29 veces la dosis máxima recomendada de 90 mg dos veces al día sobre la base del AUC), mientras que 60 mg/kg/día (8 veces la MRHD basada en el AUC) no fue carcinogénico en ratas hembras.
Mutagénesis
El ticagrelor no demostró genotoxicidad cuando se probó en la prueba de mutagenicidad bacteriana de Ames, el ensayo de linfoma de ratón y la prueba de micronúcleos de rata. El metabolito activo O-desmetilado no demostró genotoxicidad en el ensayo de Ames y el ensayo de linfoma de ratón.
Deterioro de la Fertilidad
El ticagrelor no tuvo ningún efecto sobre la fertilidad masculina a dosis de hasta 180 mg/kg/día ni sobre la fertilidad femenina a dosis de hasta 200 mg/kg/día (>15 veces la MRHD sobre la base del AUC). Las dosis de ≥10 mg/kg/día administradas a ratas hembras causaron un aumento en la incidencia de ciclos estrales de duración irregular (1,5 veces la MRHD basada en el AUC).
14 ESTUDIOS CLÍNICOS
14.1 Síndromes coronarios agudos y prevención secundaria después de un infarto de miocardio
PLATO
PLATO (NCT00391872) fue un estudio aleatorizado doble ciego que comparó BRILINTA (N=9333) con clopidogrel (N=9291), ambos administrados en combinación con aspirina y otro tratamiento estándar, en pacientes con síndromes coronarios agudos (SCA), que se presentaron dentro de las 24 horas del inicio del episodio más reciente de dolor torácico o síntomas. El criterio principal de valoración del estudio fue la combinación de la primera aparición de muerte cardiovascular, IM no mortal (excluyendo el IM silencioso) o accidente cerebrovascular no mortal.
Los pacientes que ya habían sido tratados con clopidogrel podían ser incluidos y aleatorizados a cualquiera de los tratamientos del estudio. Se excluyeron los pacientes con hemorragia intracraneal previa, hemorragia gastrointestinal en los últimos 6 meses o con diátesis hemorrágica o trastorno de la coagulación conocidos. Se excluyó la participación de pacientes que tomaban anticoagulantes y los pacientes que desarrollaron una indicación de anticoagulación durante el ensayo fueron retirados del fármaco del estudio. Los pacientes podían incluirse independientemente de la intención de controlar el SCA médica o invasivamente, pero la aleatorización de los pacientes no se estratificó por esta intención.
Todos los pacientes aleatorizados a BRILINTA recibieron una dosis de carga de 180 mg seguida de una dosis de mantenimiento de 90 mg dos veces al día. Los pacientes del grupo de clopidogrel fueron tratados con una dosis de carga inicial de clopidogrel de 300 mg, si no se había administrado previamente clopidogrel. Los pacientes sometidos a ICP podían recibir 300 mg adicionales de clopidogrel a discreción del investigador. Se recomendó una dosis de mantenimiento diaria de aspirina de 75-100 mg, pero se permitieron dosis de mantenimiento más altas de aspirina según el criterio local. Los pacientes fueron tratados durante al menos 6 meses y hasta 12 meses.
Los pacientes de PLATO fueron predominantemente hombres (72%) y caucásicos (92%). Alrededor del 43% de los pacientes tenían >65 años y el 15% >75 años. La exposición mediana al fármaco del estudio fue de 276 días. Aproximadamente la mitad de los pacientes recibieron clopidogrel antes del estudio y aproximadamente el 99% de los pacientes recibieron aspirina en algún momento durante PLATO. Alrededor del 35% de los pacientes recibían una estatina al inicio del estudio y el 93% recibieron una estatina en algún momento durante PLATO.
La Tabla 7 muestra los resultados del estudio para el criterio principal de valoración compuesto y la contribución de cada componente al criterio principal de valoración. Se muestran análisis separados de criterios de valoración secundarios para la aparición general de muerte CV, IM y accidente cerebrovascular y mortalidad general.
|
||||||
|
BRILINTA* N=9333 |
Clopidogrel N=9291 |
Hazard Ratio (95% CI) |
p-value |
|||
|
Eventos / 1000 años-paciente |
Eventos / 1000 años-paciente |
|||||
|
Compuesto de muerte CV, IM o accidente cerebrovascular |
111 |
131 |
0.84 (0.77, 0.92) |
0.0003 |
||
|
Muerte CV |
32 |
43 |
0.74 |
|||
|
IM no mortal |
64 |
76 |
0.84 |
|||
|
Accidente cerebrovascular no mortal |
15 |
12 |
1.24 |
|||
|
Criterios de valoración secundarios† |
||||||
|
Muerte cardiovascular |
45 |
57 |
0.79 (0.69, 0.91) |
0.0013 |
|
Infarto de miocardio‡ |
65 |
76 |
0.84 (0.75, 0.95) |
0.0045 |
|
Accidente cerebrovascular‡ |
16 |
14 |
1.17 (0.91, 1.52) |
0.22 |
|
Mortalidad por todas las causas |
51 |
65 |
0.78 (0.69, 0.89) |
0.0003 |
La curva de Kaplan-Meier (Figura 10) muestra el tiempo hasta la primera aparición del criterio de valoración compuesto principal de muerte CV, IM no mortal o accidente cerebrovascular no mortal en el estudio general.
Figura 10 – Tiempo hasta la primera aparición de muerte CV, IM o accidente cerebrovascular (PLATO)
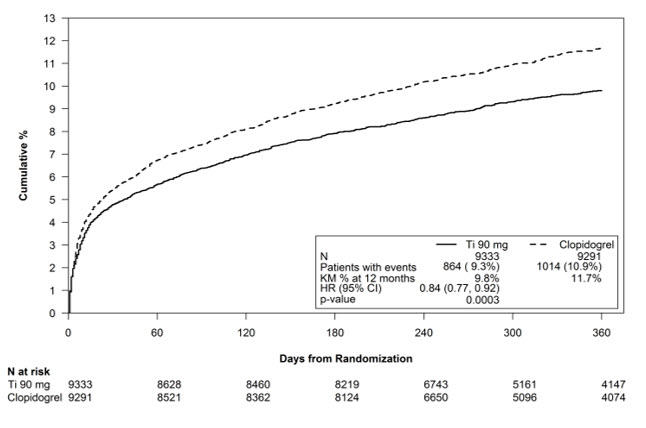
Las curvas se separan a los 30 días [reducción del riesgo relativo (RRR) 12%] y continúan divergiendo durante el período de tratamiento de 12 meses (RRR 16%).
Entre 11 289 pacientes con ICP que recibieron cualquier stent durante el PLATO, hubo un menor riesgo de trombosis del stent (1,3% para “definitiva” adjudicada) que con clopidogrel (1,9%) (HR 0,67, IC del 95% 0,50-0,91; p=0,009). Los resultados fueron similares para los stents farmacológicos y los stents metálicos desnudos.
Se examinó una amplia gama de diferencias demográficas, medicamentos concomitantes de referencia y otros tratamientos para determinar su influencia en el resultado. Algunos de ellos se muestran en la Figura 11. Estos análisis deben interpretarse con precaución, ya que las diferencias pueden reflejar el azar entre un gran número de análisis. La mayoría de los análisis muestran efectos compatibles con los resultados generales, pero hay dos excepciones: un hallazgo de heterogeneidad por región y una fuerte influencia de la dosis de mantenimiento de aspirina. Estos se consideran más adelante.
La mayoría de las características mostradas son características basales, pero algunas reflejan determinaciones posteriores a la aleatorización (por ejemplo, dosis de mantenimiento de aspirina, uso de ICP).
Figura 11 – Análisis de subgrupos de (PLATO)
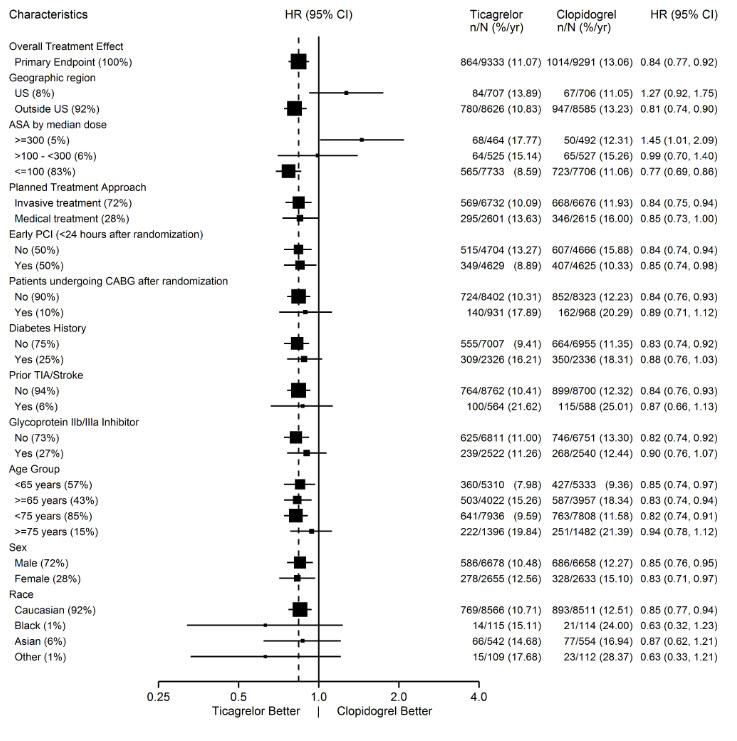
Nota: La figura anterior presenta los efectos en varios subgrupos, la mayoría de los cuales son características basales y la mayoría de los cuales fueron preespecificados. Los límites de confianza del 95% que se muestran no tienen en cuenta cuántas comparaciones se realizaron, ni reflejan el efecto de un factor particular después del ajuste para todos los demás factores. La homogeneidad o heterogeneidad aparente entre los grupos no debe sobreinterpretarse.
Diferencias regionales
Los resultados en el resto del mundo en comparación con los efectos en Norteamérica (EE. UU. y Canadá) muestran un efecto menor en Norteamérica, numéricamente inferior al control e impulsado por el subconjunto de EE. UU. La prueba estadística para la comparación EE. UU./no EE. UU. es estadísticamente significativa (p=0,009), y la misma tendencia está presente tanto para la muerte CV como para el IM no mortal. Los resultados individuales y los valores nominales de p, como todos los análisis de subgrupos, requieren una interpretación cautelosa, y podrían representar hallazgos casuales. Sin embargo, la consistencia de las diferencias tanto en la mortalidad CV como en los componentes de IM no mortal apoya la posibilidad de que el hallazgo sea fiable.
Se examinó una amplia variedad de diferencias basales y de procedimiento entre EE. UU. y países que no son EE. UU. (incluida la gestión invasiva prevista frente a la gestión médica planificada, el uso de inhibidores de GPIIb/IIIa, el uso de stents farmacológicos frente a stents metálicos desnudos) para ver si podían explicar las diferencias regionales, pero con una excepción, la dosis de mantenimiento de aspirina, estas diferencias no parecieron provocar diferencias en el resultado.
Dosis de aspirina
El protocolo PLATO dejó la elección de la dosis de mantenimiento de aspirina al investigador y los patrones de uso fueron diferentes en los sitios de EE. UU. que en los sitios fuera de EE. UU. Alrededor del 8% de los investigadores que no eran de EE. UU. administraron dosis de aspirina superiores a 100 mg, y alrededor del 2% administraron dosis superiores a 300 mg. En EE. UU., el 57% de los pacientes recibieron dosis superiores a 100 mg y el 54% recibieron dosis superiores a 300 mg. Los resultados generales favorecieron a BRILINTA cuando se usó con dosis de mantenimiento bajas (≤100 mg) de aspirina, y los resultados analizados por dosis de aspirina fueron similares en EE. UU. y en otros lugares. La Figura 10 muestra los resultados generales por dosis media de aspirina. La Figura 12 muestra los resultados por región y dosis.
Figura 12 – Muerte CV, IM, accidente cerebrovascular por dosis de mantenimiento de aspirina en EE. UU. y fuera de EE. UU. (PLATO)
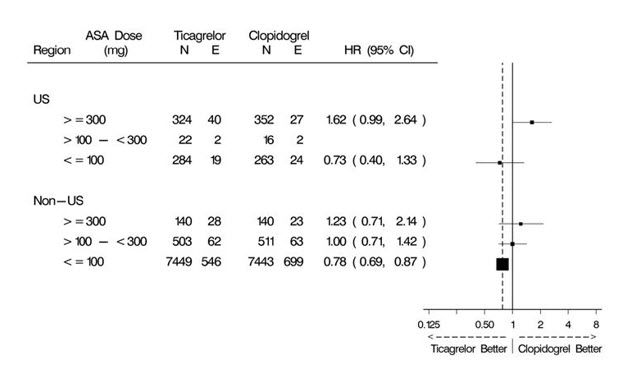
Como cualquier análisis de subgrupos no planificado, especialmente uno en el que la característica no es una característica basal verdadera (pero puede estar determinada por la práctica habitual del investigador), los análisis anteriores deben tratarse con precaución. Sin embargo, es notable que la dosis de aspirina predice el resultado en ambas regiones con un patrón similar, y que el patrón es similar para los dos componentes principales del criterio de valoración principal, la muerte CV y el IM no mortal.
A pesar de la necesidad de tratar estos resultados con precaución, parece haber una buena razón para restringir la dosis de mantenimiento de aspirina que acompaña al ticagrelor a 100 mg. Las dosis más altas no tienen un beneficio establecido en el contexto del SCA, y existe una fuerte indicación de que el uso de dichas dosis reduce la eficacia de BRILINTA.
PEGASUS
El estudio PEGASUS TIMI-54 (NCT01225562) fue un estudio de 21 162 pacientes, aleatorizado, doble ciego, controlado con placebo y de grupos paralelos. Se compararon dos dosis de ticagrelor, 90 mg dos veces al día o 60 mg dos veces al día, coadministradas con 75-150 mg de aspirina, con la terapia con aspirina sola en pacientes con antecedentes de IM. El criterio de valoración principal fue el compuesto de la primera aparición de muerte CV, IM no mortal y accidente cerebrovascular no mortal. La muerte CV y la mortalidad por todas las causas se evaluaron como criterios de valoración secundarios.
Los pacientes fueron elegibles para participar si tenían ≥50 años, con antecedentes de IM de 1 a 3 años antes de la aleatorización, y tenían al menos uno de los siguientes factores de riesgo para eventos cardiovasculares trombóticos: edad ≥65 años, diabetes mellitus que requería medicación, al menos un IM anterior, evidencia de enfermedad coronaria multivaso o aclaramiento de creatinina <60 mL/min. Los pacientes podían aleatorizarse independientemente de su terapia previa con bloqueadores del receptor ADP o una interrupción de la terapia. Se excluyeron los pacientes que requerían o que se esperaba que requirieran diálisis renal durante el estudio. Se excluyeron los pacientes con cualquier hemorragia intracraneal previa, hemorragia gastrointestinal en los últimos 6 meses o con diátesis hemorrágica o trastorno de la coagulación conocidos. Se excluyó la participación de pacientes que tomaban anticoagulantes y los pacientes que desarrollaron una indicación de anticoagulación durante el ensayo fueron retirados del estudio farmacológico. Se incluyó un pequeño número de pacientes con antecedentes de accidente cerebrovascular. Según información externa a PEGASUS, 102 pacientes con antecedentes de accidente cerebrovascular (90 de los cuales recibieron el fármaco del estudio) fueron dados de alta antes de tiempo y no se inscribieron más pacientes de este tipo.
Los pacientes fueron tratados durante al menos 12 meses y hasta 48 meses, con un tiempo medio de seguimiento de 33 meses.
Los pacientes fueron predominantemente hombres (76%), caucásicos (87%), con una edad media de 65 años, y el 99,8% de los pacientes recibieron tratamiento previo con aspirina.
La curva de Kaplan-Meier (Figura 13) muestra el tiempo hasta la primera aparición del criterio de valoración compuesto principal de muerte CV, IM no mortal o accidente cerebrovascular no mortal.
Figura 13 – Tiempo hasta la primera aparición de muerte CV, IM o accidente cerebrovascular (PEGASUS)
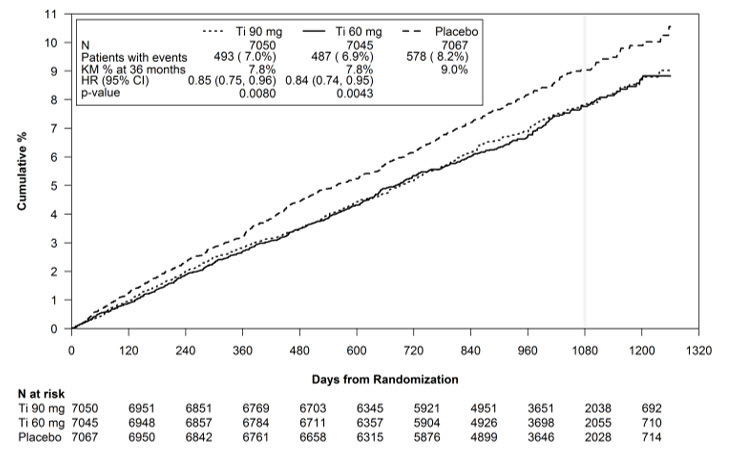
Ti = Ticagrelor BID, CI = Intervalo de confianza; HR = Razón de riesgo; KM = Kaplan-Meier; N = Número de pacientes.
Tanto los regímenes de 60 mg como de 90 mg de BRILINTA en combinación con aspirina fueron superiores a la aspirina sola en la reducción de la incidencia de muerte CV, IM o accidente cerebrovascular. Las reducciones absolutas del riesgo para BRILINTA más aspirina frente a aspirina sola fueron del 1,27% y del 1,19% para los regímenes de 60 y 90 mg, respectivamente. Aunque los perfiles de eficacia de los dos regímenes fueron similares, la dosis más baja tuvo menores riesgos de hemorragia y disnea.
La Tabla 8 muestra los resultados para el régimen de 60 mg más aspirina vs. aspirina sola.
|
BRILINTA* |
Placebo |
HR (IC 95%) |
p-valor |
|
|
Eventos / 1000 años-paciente |
Eventos / 1000 años-paciente |
|||
|
Tiempo hasta la primera muerte CV, IM o accidente cerebrovascular† |
26 |
31 |
0.84 (0.74, 0.95) |
0.0043 |
|
9 |
11 |
0.83 (0.68, 1.01) |
||
|
Infarto de miocardio§ |
15 |
18 |
0.84 (0.72, 0.98) |
|
|
Accidente cerebrovascular§ |
5 |
7 |
0.75 (0.57, 0.98) |
|
|
Mortalidad por todas las causas‡ |
16 |
18 |
0.89 (0.76, 1.04) |
|
|
IC = Intervalo de confianza; CV = Cardiovascular; HR = Razón de riesgo; IM = Infarto de miocardio; N = Número de pacientes. |
||||
En PEGASUS, la reducción relativa del riesgo (RRR) para el criterio de valoración compuesto de 1 a 360 días (17 % RRR) y de 361 días en adelante (16 % RRR) fueron similares.
El efecto del tratamiento con BRILINTA 60 mg sobre la aspirina pareció similar en la mayoría de los subgrupos predefinidos; véase la Figura 14.
Figura 14 – Análisis de subgrupos de ticagrelor 60 mg (PEGASUS)
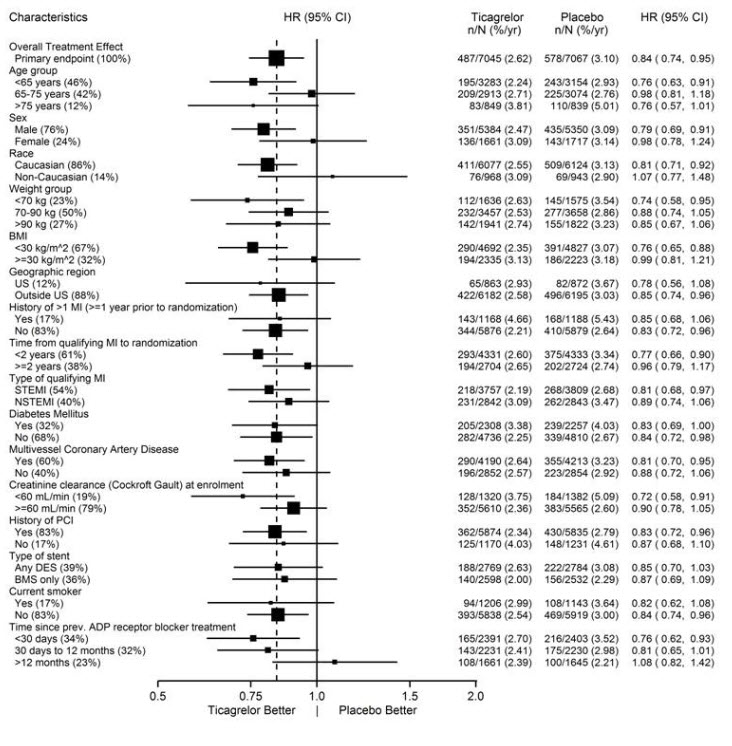
Nota: La figura anterior presenta los efectos en varios subgrupos, todos los cuales son características basales y la mayoría de los cuales fueron preespecificados. Los límites de confianza del 95 % que se muestran no tienen en cuenta cuántas comparaciones se realizaron, ni reflejan el efecto de un factor en particular después del ajuste para todos los demás factores. No se debe sobreinterpretar la aparente homogeneidad o heterogeneidad entre los grupos.
14.2 Cardiopatía coronaria pero sin accidente cerebrovascular o infarto de miocardio previo
THEMIS
El estudio THEMIS (NCT01991795) fue un estudio doble ciego, de grupos paralelos, en el que 19 220 pacientes con CCP y diabetes mellitus tipo 2 (DM2), pero sin antecedentes de IM o accidente cerebrovascular, se asignaron aleatoriamente a BRILINTA dos veces al día o placebo, con un tratamiento de fondo de 75-150 mg de aspirina. El criterio de valoración principal fue el compuesto de la primera aparición de muerte CV, IM y accidente cerebrovascular. La muerte CV, el IM, el accidente cerebrovascular isquémico y la muerte por todas las causas se evaluaron como criterios de valoración secundarios.
Los pacientes fueron elegibles para participar si tenían ≥ 50 años con CCP, definida como antecedentes de ICP o CABG, o evidencia angiográfica de ≥ 50 % de estenosis de la luz de al menos 1 arteria coronaria y DM2 tratada durante al menos 6 meses con medicamentos hipoglucemiantes. Se excluyeron los pacientes con hemorragia intracerebral previa, hemorragia gastrointestinal en los últimos 6 meses, diátesis hemorrágica conocida y trastorno de la coagulación. Se excluyó la participación de los pacientes que tomaban anticoagulantes o antagonistas del receptor ADP, y los pacientes que desarrollaron una indicación para esos medicamentos durante el ensayo fueron retirados del estudio farmacológico.
Los pacientes fueron tratados durante una mediana de 33 meses y hasta 58 meses.
Los pacientes fueron predominantemente hombres (69 %), con una edad media de 66 años. Al inicio del estudio, el 80 % tenía antecedentes de revascularización coronaria; el 58 % se había sometido a una ICP, el 29 % a una CABG y el 7 % a ambas. La proporción de pacientes estudiados en EE. UU. fue del 12 %. Los pacientes de THEMIS tenían CCP establecida y otros factores de riesgo que los colocaban en mayor riesgo cardiovascular.
BRILINTA fue superior al placebo en la reducción de la incidencia de muerte CV, IM o accidente cerebrovascular. El efecto sobre el criterio de valoración compuesto fue impulsado por los componentes individuales IM y accidente cerebrovascular; véase la Tabla 9.
|
BRILINTA |
Placebo |
HR (IC del 95 %) |
p-valor |
|||
|
Eventos / 1000 pacientes-año |
Eventos / 1000 pacientes-año |
|||||
|
Tiempo hasta la primera muerte CV, IM o accidente cerebrovascular* |
24 |
27 |
0,90 (0,81; 0,99) |
0,04 |
||
|
Muerte CV† |
12 |
11 |
1,02 (0,88; 1,18) |
|||
|
Infarto de miocardio† |
9 |
11 |
0,84 (0,71; 0,98) |
|||
|
Accidente cerebrovascular† |
6 |
7 |
0,82 (0,67; 0,99) |
|||
|
Criterios de valoración secundarios |
||||||
|
Muerte CV |
12 |
11 |
1.02 (0.88, 1.18) |
|
|
Infarto de miocardio |
9 |
11 |
0.84 (0.71, 0.98) |
|
|
Accidente isquémico cerebrovascular |
5 |
6 |
0.80 (0.64, 0.99) |
|
|
Muerte por todas las causas |
18 |
19 |
0.98 (0.87, 1.10) |
|
|
CI = Intervalo de confianza; CV = Cardiovascular; HR = Hazard ratio; MI = Infarto de miocardio. |
||||
La curva de Kaplan-Meier (Figura 15) muestra el tiempo hasta la primera aparición del criterio de valoración compuesto principal de muerte CV, IM o accidente cerebrovascular.
Figura 15 – Tiempo hasta la primera aparición de muerte CV, IM o accidente cerebrovascular (THEMIS)
T = Ticagrelor; P = Placebo; N = Número de pacientes.
El efecto del tratamiento con BRILINTA pareció similar en todos los subgrupos de pacientes, véase la Figura 16.
Figura 16 – Análisis de subgrupos de ticagrelor (THEMIS)

Nota: La figura anterior presenta los efectos en varios subgrupos, todos los cuales son características basales. Los límites de confianza del 95% que se muestran no tienen en cuenta cuántas comparaciones se realizaron, ni reflejan el efecto de un factor particular después del ajuste para todos los demás factores. No se debe sobreinterpretar la aparente homogeneidad o heterogeneidad entre los grupos.
14.3 Accidente cerebrovascular isquémico agudo o ataque isquémico transitorio (AIT)
THALES
El estudio THALES (NCT03354429) fue un estudio aleatorizado, doble ciego, de grupos paralelos, con 11 016 pacientes, de BRILINTA 90 mg dos veces al día frente a placebo en pacientes con accidente cerebrovascular isquémico agudo o ataque isquémico transitorio (AIT). El criterio de valoración principal fue la primera aparición del compuesto de accidente cerebrovascular y muerte hasta 30 días. El accidente cerebrovascular isquémico se evaluó como uno de los criterios de valoración secundarios.
Los pacientes fueron elegibles para participar si tenían ≥40 años de edad, con accidente cerebrovascular isquémico agudo no cardioembólico (puntuación NIHSS ≤5) o AIT de alto riesgo (definido como puntuación ABCD2 ≥6 o estenosis aterosclerótica ipsilateral ≥50% en la carótida interna o una arteria intracraneal). Los pacientes que recibieron trombólisis o trombectomía en las 24 horas previas a la aleatorización no fueron elegibles.
Los pacientes fueron aleatorizados dentro de las 24 horas posteriores al inicio de un accidente cerebrovascular isquémico agudo o AIT para recibir 30 días de BRILINTA (90 mg dos veces al día, con una dosis de carga inicial de 180 mg) o placebo, con un tratamiento de fondo de aspirina inicialmente de 300-325 mg y luego de 75-100 mg diarios. La duración media del tratamiento fue de 31 días.
BRILINTA fue superior al placebo en la reducción de la tasa del criterio de valoración principal (compuesto de accidente cerebrovascular y muerte), lo que corresponde a una reducción del riesgo relativo (RRR) del 17% y una reducción del riesgo absoluto (ARR) del 1,1% (Tabla 10). El efecto se debió principalmente a una reducción significativa en el componente de accidente cerebrovascular del criterio de valoración principal (19% RRR, 1,1% ARR).
|
||||||
|
BRILINTA N=5523 |
Placebo N=5493 |
HR (IC del 95%) |
p-valor |
|||
|
n (pacientes con evento) |
KM% |
n (pacientes con evento) |
KM% |
|||
|
Tiempo hasta el primer accidente cerebrovascular o muerte |
303 |
5,4% |
362 |
6,5% |
0,83 (0,71; 0,96) |
0,015 |
|
Tiempo hasta el primer accidente cerebrovascular* |
284 |
5,1% |
347 |
6,3% |
0,81 (0,69; 0,95) |
|
|
Tiempo hasta la muerte* |
36 |
0.6% |
27 |
0.5% |
1.33 (0.81, 2.19) |
|
|
Variable principal secundaria |
||||||
|
Tiempo hasta el primer accidente cerebrovascular isquémico |
276 |
5.0% |
345 |
6.2% |
0.79 (0.68, 0.93) |
0.004 |
|
IC = Intervalo de confianza; HR = Razón de riesgo; KM = Porcentaje de Kaplan-Meier calculado a los 30 días; N = Número de pacientes |
||||||
La curva de Kaplan-Meier (Figura 17) muestra el tiempo hasta la primera aparición del criterio principal de valoración compuesto de accidente cerebrovascular y muerte.
Figura 17 – Tiempo hasta la primera aparición de accidente cerebrovascular o muerte (THALES)
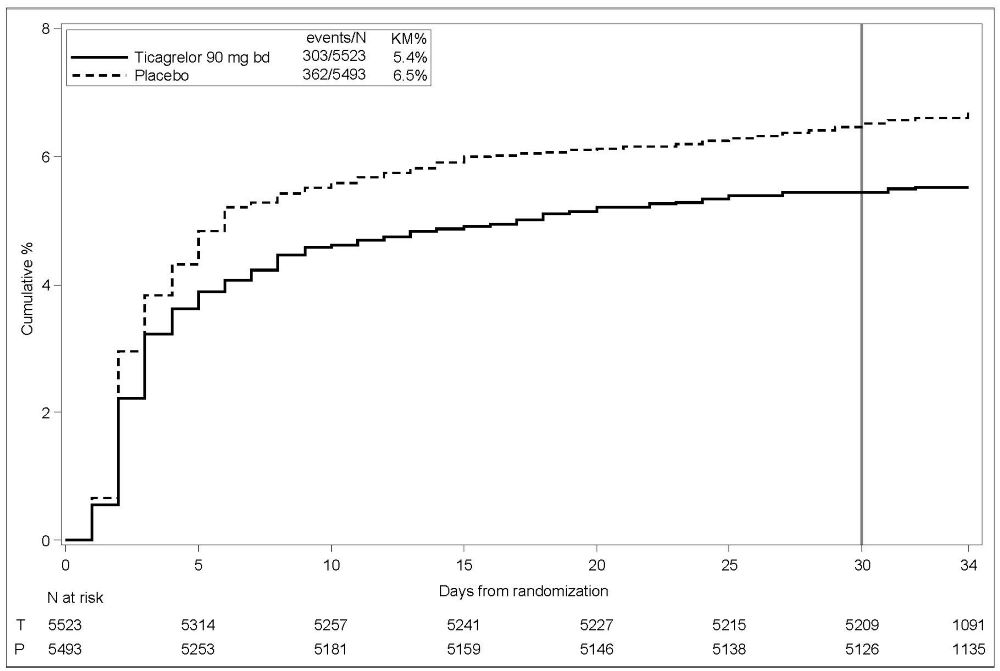
KM%: Porcentaje de Kaplan-Meier evaluado a los 30 días; T=Ticagrelor; P=placebo; N=Número de pacientes
El efecto del tratamiento de BRILINTA sobre el accidente cerebrovascular y la muerte se acumuló durante los primeros 10 días y se mantuvo a los 30 días. Aunque no se ha estudiado, esto sugiere que un tratamiento más corto podría producir un beneficio similar y reducir el riesgo de hemorragia.
El efecto del tratamiento de BRILINTA fue generalmente consistente en los subgrupos predefinidos (Figura 18).
Figura 18 – Análisis de subgrupos de ticagrelor 90 mg (THALES)
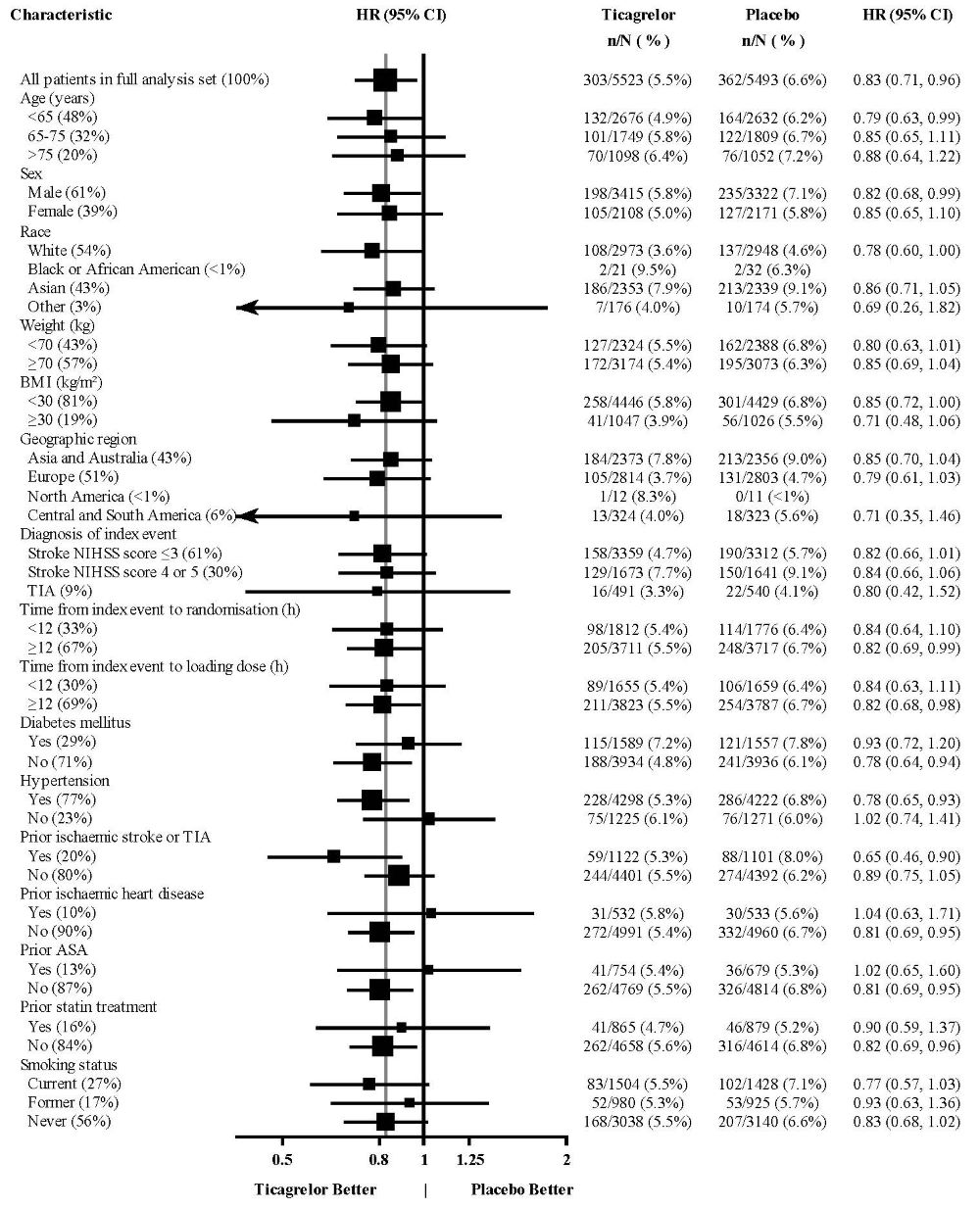
Nota: La figura anterior presenta los efectos en varios subgrupos, todos los cuales son características basales y fueron preespecificados. Los límites de confianza del 95% que se muestran no tienen en cuenta cuántas comparaciones se realizaron, ni reflejan el efecto de un factor particular después del ajuste para todos los demás factores. No se debe sobreinterpretar la aparente homogeneidad o heterogeneidad entre los grupos.
A los 30 días, hubo una reducción absoluta del 1,2% (IC del 95%: -2,1%, -0,3%) en la incidencia de accidente cerebrovascular no hemorrágico y muerte (excluyendo la hemorragia mortal) a favor del ticagrelor (294 eventos: 5,3%) sobre el placebo (359 eventos: 6,5%) en la población de intención de tratar. En la misma población, hubo un aumento absoluto del 0,4% (IC del 95%: 0,2%, 0,6%) en la incidencia de hemorragia grave según GUSTO desfavorable para el brazo de ticagrelor (28 eventos: 0,5%) en comparación con el brazo de placebo (7 eventos: 0,1%).
16 FORMA DE SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
BRILINTA (ticagrelor) 90 mg se suministra como un comprimido recubierto con película, redondo, biconvexo, amarillo, con un “90” sobre una “T” en un lado:
Frascos de 60 – NDC 0186-0777-60
Unidad de dosis hospitalaria de 100 unidades – NDC 0186-0777-39
BRILINTA (ticagrelor) 60 mg se suministra como un comprimido recubierto con película, redondo, biconvexo, rosa, con un “60” sobre una “T” en un lado:
Frascos de 60 – NDC 0186-0776-60
Almacenamiento y manipulación
Almacenar a 25°C (77°F); se permiten excursiones de 15 a 30°C (59 a 86°F) [véase temperatura ambiente controlada USP].
17 INFORMACIÓN PARA EL PACIENTE
Aconseje al paciente que lea el etiquetado del paciente aprobado por la FDA (Guía de medicamentos).
Aconseje a los pacientes que las dosis diarias de aspirina no deben exceder los 100 mg y que eviten tomar cualquier otro medicamento que contenga aspirina.
Aconseje a los pacientes que:
- •
- Sangrarán y se harán moretones más fácilmente
- •
- Tardarán más de lo habitual en detener el sangrado
- •
- Deben informar cualquier sangrado inesperado, prolongado o excesivo, o sangre en sus heces u orina.
Aconseje a los pacientes que contacten a su médico si experimentan dificultad respiratoria inesperada, especialmente si es grave.
Aconseje a los pacientes que informen a los médicos y dentistas que están tomando BRILINTA antes de cualquier cirugía o procedimiento dental.
Aconseje a las mujeres que no se recomienda la lactancia materna durante el tratamiento con BRILINTA [ver Uso en poblaciones específicas (8.2)].
BRILINTA® es una marca comercial del grupo de empresas AstraZeneca.
Distribuido por: AstraZeneca Pharmaceuticals LP, Wilmington, DE 19850
© AstraZeneca 2024
Guía de medicación
|
GUÍA DEL MEDICAMENTO BRILINTA® (brih-LIN-tah) (ticagrelor) Comprimidos |
|
¿Cuál es la información más importante que debo saber sobre BRILINTA? BRILINTA se usa para reducir su probabilidad de sufrir o morir de un ataque cardíaco o un accidente cerebrovascular. BRILINTA (y medicamentos similares) puede causar sangrado que puede ser grave y, a veces, provocar la muerte. En casos de sangrado grave, como sangrado interno, el sangrado puede requerir transfusiones de sangre o cirugía. Mientras toma BRILINTA:
Llame a su proveedor de atención médica de inmediato si tiene alguno de estos signos o síntomas de sangrado mientras toma BRILINTA:
No deje de tomar BRILINTA sin hablar con el proveedor de atención médica que se lo recetó. Las personas que reciben tratamiento con un stent y dejan de tomar BRILINTA demasiado pronto tienen un mayor riesgo de desarrollar un coágulo de sangre en el stent, sufrir un ataque cardíaco o morir. Si deja de tomar BRILINTA debido a una hemorragia o por otras razones, su riesgo de sufrir un ataque cardíaco o un accidente cerebrovascular puede aumentar. Su proveedor de atención médica puede indicarle que deje de tomar BRILINTA 5 días antes de la cirugía. Esto ayudará a disminuir su riesgo de sangrado durante la cirugía o el procedimiento. Su proveedor de atención médica debe indicarle cuándo debe volver a tomar BRILINTA, lo antes posible después de la cirugía. Tomar BRILINTA con aspirina BRILINTA se toma con aspirina, a menos que su proveedor de atención médica le indique lo contrario. Hable con su proveedor de atención médica sobre la dosis de aspirina que debe tomar con BRILINTA. En la mayoría de los casos, no debe tomar una dosis de aspirina superior a 100 mg diarios. No tome dosis de aspirina superiores a las que le indique su proveedor de atención médica. Informe a su proveedor de atención médica si toma otros medicamentos que contienen aspirina y no tome nuevos medicamentos de venta libre que contengan aspirina. |
|
¿Qué es BRILINTA? BRILINTA es un medicamento recetado que se usa para:
No se sabe si BRILINTA es seguro y eficaz en niños. |
|
No tome BRILINTA si:
|
|
Antes de tomar BRILINTA, informe a su proveedor de atención médica sobre todas sus afecciones médicas, si:
Informe a todos sus proveedores de atención médica y dentistas que está tomando BRILINTA. Deben hablar con el proveedor de atención médica que le recetó BRILINTA antes de que se someta a cualquier cirugía o procedimiento. Informe a su proveedor de atención médica sobre todos los medicamentos que toma, incluidos los medicamentos recetados y de venta libre, las vitaminas y los suplementos herbales. BRILINTA puede afectar la forma en que funcionan otros medicamentos, y otros medicamentos pueden afectar la forma en que funciona BRILINTA. Ciertos medicamentos pueden aumentar su riesgo de sangrado. Conozca los medicamentos que toma. Lleve una lista de ellos para mostrársela a su proveedor de atención médica y farmacéutico cuando obtenga un nuevo medicamento. |
¿Cómo debo tomar BRILINTA?
- •
- Tome BRILINTA exactamente como le indique su proveedor de atención médica.
- •
- Su proveedor de atención médica le indicará cuántas tabletas de BRILINTA debe tomar y cuándo tomarlas.
- •
- Tome BRILINTA con aspirina, a menos que su proveedor de atención médica le indique lo contrario. Consulte “¿Cuál es la información más importante que debo saber sobre BRILINTA?”
- •
- Puede tomar BRILINTA con o sin alimentos.
- •
- Tome BRILINTA dos veces al día, aproximadamente a la misma hora cada día.
- •
- Si se pierde la dosis programada de BRILINTA, tome la siguiente dosis a la hora programada. No tome 2 dosis al mismo tiempo a menos que su proveedor de atención médica le indique que lo haga.
- •
- Si toma demasiado BRILINTA, llame a su proveedor de atención médica o al centro de control de intoxicaciones local o vaya a la sala de emergencias más cercana de inmediato.
- Si no puede tragar la(s) tableta(s) entera(s), puede triturar la(s) tableta(s) de BRILINTA y mezclarla(s) con agua. Beba toda el agua de inmediato. Vuelva a llenar el vaso con agua, revuelva y beba toda el agua.
- BRILINTA también se puede administrar a través de ciertos tubos nasogástricos (NG). Pregunte a su proveedor de atención médica cómo tomar BRILINTA a través de un tubo NG.
¿Cuáles son los posibles efectos secundarios de BRILINTA?
BRILINTA puede causar efectos secundarios graves, incluyendo:
- •
- Consulte “¿Cuál es la información más importante que debo saber sobre BRILINTA?”
- •
-
Disnea. Informe a su proveedor de atención médica si tiene disnea nueva, empeoramiento o inesperada cuando está en reposo, por la noche o cuando realiza cualquier actividad.
Latido cardíaco lento o irregular.
- •
- Respiración irregular. Informe a su proveedor de atención médica si desarrolla patrones de respiración irregulares cuando está dormido o despierto, como aceleración, desaceleración o pausas breves en la respiración. Su proveedor de atención médica decidirá si necesita una evaluación adicional.
Estos no son todos los posibles efectos secundarios de BRILINTA.
Llame a su médico para obtener asesoramiento médico sobre los efectos secundarios. Puede reportar los efectos secundarios a la FDA al 1-800-FDA-1088.
¿Cómo debo almacenar BRILINTA?
- •
- Almacene BRILINTA a temperatura ambiente entre 68°F y 77°F (20°C y 25°C).
Mantenga BRILINTA y todos los medicamentos fuera del alcance de los niños.
Información general sobre el uso seguro y eficaz de BRILINTA.
A veces, los medicamentos se prescriben para fines distintos a los enumerados en una Guía de Medicación. No use BRILINTA para una condición para la que no fue prescrita. No le dé BRILINTA a otras personas, incluso si tienen los mismos síntomas que usted. Puede dañarlos. Puede pedir a su farmacéutico o proveedor de atención médica información sobre BRILINTA escrita para profesionales de la salud.
¿Cuáles son los ingredientes de BRILINTA?
Ingrediente activo: ticagrelor
Tabletas de 90 mg:
Ingredientes inactivos: manitol, fosfato de calcio dibásico, glicolato de almidón sódico, hidroxipropilcelulosa, estearato de magnesio, hidroxipropilmetilcelulosa, dióxido de titanio, talco, polietilenglicol 400 y óxido de hierro amarillo.
Tabletas de 60 mg:
Ingredientes inactivos: manitol, fosfato de calcio dibásico, glicolato de almidón sódico, hidroxipropilcelulosa, estearato de magnesio, hidroxipropilmetilcelulosa, dióxido de titanio, polietilenglicol 400, óxido de hierro negro y óxido de hierro rojo.
Distribuido por: AstraZeneca Pharmaceuticals LP, Wilmington, DE 19850
Para más información llame al 1-800-236-9933 o visite www.Brilinta.com.
Esta Guía de Medicación ha sido aprobada por la Administración de Alimentos y Medicamentos de los EE. UU. Revisada: 03/2024
PANEL PRINCIPAL DE VISUALIZACIÓN DEL ENVASE/ETIQUETA – 60 mg
NDC 0186-0776-60
60 comprimidos
BRILINTA®
ticagrelor comprimidos
60 mg
Rx solamente
Entregue la Guía del Medicamento adjunta a cada paciente.
AstraZeneca
PANEL PRINCIPAL DE VISUALIZACIÓN DEL ENVASE/ETIQUETA – 90 mg
NDC 0186-0777-60
60 tabletas
BRILINTA®
ticagrelor tabletas
90 mg
Rx only
Entregue la Guía de Medicamentos adjunta a cada paciente.
AstraZeneca