ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
Estos puntos destacados no incluyen toda la información necesaria para usar BIKTARVY de forma segura y eficaz. Consulte la información de prescripción completa de BIKTARVY.
BIKTARVY® (bictegravir, emtricitabina y tenofovir alafenamida) comprimidos, para uso oral Primera aprobación en EE. UU.: 2018
ADVERTENCIA: EXACERBACIÓN AGUDA POSTTRATAMIENTO DE LA HEPATITIS B
Consulte la información de prescripción completa para la advertencia en caja completa.
Se han notificado exacerbaciones agudas graves de la hepatitis B en pacientes coinfectados con VIH-1 y VHB que han suspendido productos que contienen emtricitabina (FTC) y/o tenofovir disoproxil fumarato (TDF), y pueden ocurrir con la suspensión de BIKTARVY. Vigile estrechamente la función hepática en estos pacientes. Si es apropiado, puede ser necesario un tratamiento antihepatitis B. (5.1)
BIKTARVY es una combinación de tres fármacos: bictegravir (BIC), un inhibidor de la transferencia de la cadena de la integrasa del virus de la inmunodeficiencia humana tipo 1 (VIH-1) (INSTI), y emtricitabina (FTC) y tenofovir alafenamida (TAF), ambos inhibidores de la transcriptasa inversa análogos de nucleósidos del VIH-1 (ITIN), e está indicado como un régimen completo para el tratamiento de la infección por VIH-1 en adultos y pacientes pediátricos que pesan al menos 14 kg:
que no tienen antecedentes de tratamiento antirretroviral o
para reemplazar el régimen antirretroviral actual en aquellos que están virologicamente suprimidos (ARN del VIH-1 inferior a 50 copias por mL) en un régimen antirretroviral estable sin sustituciones conocidas o sospechosas asociadas con resistencia a bictegravir o tenofovir. (1)
DOSIFICACIÓN Y ADMINISTRACIÓN
Pruebas: Antes o al iniciar BIKTARVY, realizar pruebas para la infección por el virus de la hepatitis B. Antes o al iniciar BIKTARVY, y durante el tratamiento, evaluar la creatinina sérica, el aclaramiento de creatinina estimado, la glucosa en orina y la proteína en orina en todos los pacientes según sea clínicamente apropiado. En pacientes con enfermedad renal crónica, también evaluar el fósforo sérico. (2.1)
Dosis recomendada en adultos y pacientes pediátricos que pesan al menos 25 kg, o adultos virologicamente suprimidos con aclaramiento de creatinina estimado inferior a 15 mL/min que reciben hemodiálisis crónica: Un comprimido que contiene 50 mg de BIC, 200 mg de FTC y 25 mg de TAF tomado una vez al día con o sin alimentos. (2.2)
Dosis recomendada en pacientes pediátricos que pesan al menos 14 kg y menos de 25 kg: Un comprimido que contiene 30 mg de BIC, 120 mg de FTC y 15 mg de TAF tomado una vez al día con o sin alimentos. (2.3)
Dosis recomendada en individuos embarazadas que están virologicamente suprimidos (ARN del VIH-1 inferior a 50 copias por mL) en un régimen antirretroviral estable sin sustituciones conocidas asociadas con resistencia a los componentes individuales de BIKTARVY: Un comprimido que contiene 50 mg de BIC, 200 mg de FTC y 25 mg de TAF tomado por vía oral una vez al día con o sin alimentos. (2.4)
Insuficiencia renal: No se recomienda BIKTARVY en pacientes con aclaramiento de creatinina estimado de 15 a menos de 30 mL/min, o menos de 15 mL/min que no reciben hemodiálisis crónica, o menos de 15 mL/min que no tienen antecedentes de tratamiento antirretroviral. (2.5)
Insuficiencia hepática: No se recomienda BIKTARVY en pacientes con insuficiencia hepática grave. (2.6)
FORMAS Y FUERZAS DE DOSIFICACIÓN
Comprimidos: 50 mg de BIC, 200 mg de FTC y 25 mg de TAF. (3)
Comprimidos: 30 mg de BIC, 120 mg de FTC y 15 mg de TAF. (3)
CONTRAINDICACIONES
Está contraindicada la administración conjunta de BIKTARVY con:
Síndrome de reconstitución inmunitaria: Puede requerir una evaluación y un tratamiento adicionales. (5.3)
Aparición o empeoramiento de la insuficiencia renal: Evaluar la creatinina sérica, el aclaramiento de creatinina estimado, la glucosa en orina y la proteína en orina al iniciar BIKTARVY y durante la terapia según sea clínicamente apropiado en todos los pacientes. También evaluar el fósforo sérico en pacientes con enfermedad renal crónica. (5.4)
Acidosis láctica/hepatomegalia grave con esteatosis: Suspender el tratamiento en pacientes que desarrollen síntomas o hallazgos de laboratorio sugestivos de acidosis láctica o hepatotoxicidad pronunciada. (5.5)
REACCIONES ADVERSAS
Las reacciones adversas más comunes (incidencia mayor o igual al 5%, todos los grados) son diarrea, náuseas y dolor de cabeza. (6.1)
Para informar sobre REACCIONES ADVERSAS SOSPECHOSAS, comuníquese con Gilead Sciences, Inc. al 1-800-GILEAD-5 o con la FDA al 1-800-FDA-1088 o www.fda.gov/medwatch.
INTERACCIONES MEDICAMENTOSAS
Debido a que BIKTARVY es un régimen completo, no se recomienda la administración conjunta con otros medicamentos antirretrovirales para el tratamiento de la infección por VIH-1. (7.1)
Consulte la Información de Prescripción Completa antes y durante el tratamiento para conocer las interacciones medicamentosas importantes. (4, 5.2, 7, 12.3)
USO EN POBLACIONES ESPECÍFICAS
Pediatría: No se recomienda para pacientes que pesan menos de 14 kg. (8.4)
Ver 17 para INFORMACIÓN PARA EL PACIENTE y el etiquetado del paciente aprobado por la FDA.
Las secciones o subsecciones que se omiten de la información completa de prescripción no se enumeran.
ADVERTENCIA EN EL RECUADRO
ADVERTENCIA: EXACERBACIÓN AGUDA POSTTRATAMIENTO DE LA HEPATITIS B
Se han notificado exacerbaciones agudas graves de la hepatitis B en pacientes coinfectados con VIH-1 y VHB que han suspendido productos que contienen emtricitabina (FTC) y/o tenofovir disoproxil fumarato (TDF), y puede ocurrir con la suspensión de BIKTARVY.
Monitoree estrechamente la función hepática con seguimiento clínico y de laboratorio durante al menos varios meses en pacientes coinfectados con VIH-1 y VHB que suspendan BIKTARVY. Si es apropiado, puede ser necesaria la terapia antihepatitis B [ver Advertencias y Precauciones (5.1)].
1 INDICACIONES Y USO
BIKTARVY está indicado como un régimen completo para el tratamiento de la infección por el virus de la inmunodeficiencia humana tipo 1 (VIH-1) en:
adultos y pacientes pediátricos que pesen al menos 14 kg:
que no tienen antecedentes de tratamiento antirretroviral o
para reemplazar el régimen antirretroviral actual en aquellos que están virológicamente suprimidos (ARN del VIH-1 inferior a 50 copias por mL) en un régimen antirretroviral estable sin sustituciones conocidas o sospechosas asociadas con resistencia a bictegravir o tenofovir [ver Dosificación y administración (2.4), y Uso en poblaciones específicas (8.1)].
2 DOSIS Y ADMINISTRACIÓN
2.1 Pruebas al inicio y durante el tratamiento con BIKTARVY
Antes de iniciar o al iniciar BIKTARVY, realice a los pacientes la prueba de infección por el virus de la hepatitis B [consulte Advertencias y precauciones (5.1)].
Antes de iniciar o al iniciar BIKTARVY, y durante el tratamiento con BIKTARVY, evalúe la creatinina sérica, el aclaramiento de creatinina estimado, la glucosa en orina y la proteína en orina en todos los pacientes según sea clínicamente apropiado. En pacientes con enfermedad renal crónica, también evalúe el fósforo sérico [consulte Advertencias y precauciones (5.4)].
2.2 Dosis recomendada en adultos y pacientes pediátricos que pesan al menos 25 kg
BIKTARVY es un producto combinado de dosis fija de tres fármacos que contiene bictegravir (BIC), emtricitabina (FTC) y tenofovir alafenamida (TAF). La dosis recomendada de BIKTARVY es una tableta que contiene 50 mg de BIC, 200 mg de FTC y 25 mg de TAF, que se toma por vía oral una vez al día con o sin alimentos en [consulte Dosis y administración (2.4)]:
adultos y pacientes pediátricos que pesan al menos 25 kg con un aclaramiento de creatinina estimado mayor o igual a 30 ml/min; o
adultos virológicamente suprimidos con un aclaramiento de creatinina estimado inferior a 15 ml/min que reciben hemodiálisis crónica. Los días de hemodiálisis, administre la dosis diaria de BIKTARVY después de finalizar el tratamiento de hemodiálisis [consulte Uso en poblaciones específicas (8.4, 8.6) y Farmacología clínica (12.3)].
2.3 Dosis recomendada en pacientes pediátricos que pesan al menos 14 kg y menos de 25 kg
La dosis recomendada de BIKTARVY es una tableta que contiene 30 mg de BIC, 120 mg de FTC y 15 mg de TAF, que se toma por vía oral una vez al día con o sin alimentos en:
En el caso de los niños que no puedan tragar una tableta entera, esta se puede partir y cada parte se puede tomar por separado, siempre que todas las partes se ingieran en un plazo aproximado de 10 minutos.
2.4 Dosis recomendada en embarazadas
La dosis recomendada de BIKTARVY en embarazadas es de un comprimido que contiene 50 mg de BIC, 200 mg de FTC y 25 mg de TAF, administrado por vía oral una vez al día con o sin alimentos en embarazadas con supresión virológica (ARN del VIH-1 inferior a 50 copias por ml) en un régimen antirretroviral estable sin sustituciones conocidas asociadas a la resistencia a ninguno de los componentes individuales de BIKTARVY. Se observaron exposiciones más bajas a BIKTARVY durante el embarazo; por lo tanto, la carga viral debe ser monitoreada de cerca [véase Interacciones medicamentosas (7.5), Uso en poblaciones específicas (8.1) y Farmacología clínica (12.3)].
2.5 No se recomienda en pacientes con insuficiencia renal grave
Las comprimidos de BIKTARVY están disponibles en dos concentraciones de dosis:
Comprimidos de 50 mg/200 mg/25 mg: 50 mg de bictegravir (BIC) (equivalente a 52.5 mg de bictegravir sodium), 200 mg de emtricitabina (FTC) y 25 mg de tenofovir alafenamida (TAF) (equivalente a 28 mg de tenofovir alafenamide fumarate). Estos comprimidos son de color marrón violáceo, con forma de cápsula, recubiertos con película y grabados con “GSI” en un lado y “9883” en el otro.
Comprimidos de 30 mg/120 mg/15 mg: 30 mg de BIC (equivalente a 31.5 mg de bictegravir sodium), 120 mg de FTC y 15 mg de TAF (equivalente a 16.8 mg de tenofovir alafenamide fumarate). Estos comprimidos son de color rosa, con forma de cápsula, recubiertos con película y grabados con “GSI” en un lado y “B” en el otro; o con una línea divisoria no funcional en un lado y “BVY” en el otro.
4 CONTRAINDICACIONES
BIKTARVY está contraindicado para ser coadministrado con:
dofetilida debido al potencial de aumento de las concentraciones plasmáticas de dofetilida y eventos graves y/o potencialmente mortales [ver Interacciones medicamentosas (7.5)].
rifampicina debido a la disminución de las concentraciones plasmáticas de BIC, lo que puede resultar en la pérdida del efecto terapéutico y el desarrollo de resistencia a BIKTARVY [ver Interacciones medicamentosas (7.5)].
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Exacerbación Aguda Grave de la Hepatitis B en Pacientes Coinfectados con VIH-1 y VHB
Los pacientes con VIH-1 deben ser examinados para detectar la presencia de infección crónica por el virus de la hepatitis B (VHB) antes o al iniciar el tratamiento antirretroviral [ver Dosificación y Administración (2.1)].
Se han notificado exacerbaciones agudas graves de la hepatitis B (por ejemplo, descompensación hepática e insuficiencia hepática) en pacientes que están coinfectados con VIH-1 y VHB y han interrumpido los productos que contienen FTC y/o tenofovir disoproxil fumarato (TDF), y pueden ocurrir con la interrupción de BIKTARVY. Los pacientes coinfectados con VIH-1 y VHB que interrumpen BIKTARVY deben ser controlados estrechamente con seguimiento clínico y de laboratorio durante al menos varios meses después de interrumpir el tratamiento. Si es apropiado, puede estar justificado el tratamiento contra la hepatitis B, especialmente en pacientes con enfermedad hepática avanzada o cirrosis, ya que la exacerbación de la hepatitis después del tratamiento puede provocar descompensación hepática e insuficiencia hepática.
5.2 Riesgo de Reacciones Adversas o Pérdida de la Respuesta Virológica Debido a Interacciones Medicamentosas
El uso concomitante de BIKTARVY con ciertos otros medicamentos puede resultar en interacciones medicamentosas conocidas o potencialmente significativas, algunas de las cuales pueden conducir a [ver Contraindicaciones (4), y Interacciones Medicamentosas (7.5)]:
Pérdida del efecto terapéutico de BIKTARVY y posible desarrollo de resistencia.
Posibles reacciones adversas clínicamente significativas por mayores exposiciones a medicamentos concomitantes.
Ver Tabla 3 para los pasos para prevenir o controlar estas posibles e importantes interacciones medicamentosas conocidas, incluidas las recomendaciones de dosificación. Considere el potencial de interacciones medicamentosas antes y durante el tratamiento con BIKTARVY; revise los medicamentos concomitantes durante el tratamiento con BIKTARVY; y controle las reacciones adversas asociadas con los medicamentos concomitantes.
5.3 Síndrome de Reconstitución Inmunitaria
Se ha notificado el síndrome de reconstitución inmunitaria en pacientes tratados con terapia antirretroviral combinada. Durante la fase inicial del tratamiento antirretroviral combinado, los pacientes cuyo sistema inmunitario responde pueden desarrollar una respuesta inflamatoria a infecciones oportunistas indolentes o residuales [como Mycobacterium avium infección, citomegalovirus, Pneumocystis jirovecii neumonía (PCP), o tuberculosis], que pueden requerir una evaluación y tratamiento adicionales.
También se ha notificado que los trastornos autoinmunitarios (como la enfermedad de Graves, la polimiositis, el síndrome de Guillain-Barré y la hepatitis autoinmune) ocurren en el contexto de la reconstitución inmunitaria; sin embargo, el tiempo de inicio es más variable y puede ocurrir muchos meses después del inicio del tratamiento.
5.4 Nuevo Inicio o Empeoramiento del Deterioro Renal
Se han notificado casos postcomercialización de deterioro renal, incluida la insuficiencia renal aguda, la tubulopatía renal proximal (PRT) y el síndrome de Fanconi, con productos que contienen TAF; si bien la mayoría de estos casos se caracterizaron por posibles factores de confusión que pueden haber contribuido a los eventos renales notificados, también es posible que estos factores puedan haber predispuesto a los pacientes a eventos adversos relacionados con tenofovir [ver Reacciones Adversas (6.1, 6.2)]. BIKTARVY no se recomienda en pacientes con deterioro renal grave (aclaramiento de creatinina estimado de 15 a menos de 30 mL/min), o pacientes con ESRD (aclaramiento de creatinina estimado inferior a 15 mL/min) que no reciben hemodiálisis crónica, o pacientes sin antecedentes de tratamiento antirretroviral y ESRD que reciben hemodiálisis crónica [ver Dosificación y Administración (2.4), y Uso en Poblaciones Específicas (8.6)].
Los pacientes que toman profármacos de tenofovir que tienen función renal deteriorada y los que toman agentes nefrotóxicos, incluidos los antiinflamatorios no esteroideos, tienen un mayor riesgo de desarrollar reacciones adversas relacionadas con el riñón.
Antes o al iniciar BIKTARVY, y durante el tratamiento con BIKTARVY, evalúe la creatinina sérica, el aclaramiento de creatinina estimado, la glucosa en orina y la proteína en orina en todos los pacientes según sea clínicamente apropiado. En pacientes con enfermedad renal crónica, también evalúe el fósforo sérico. Suspenda BIKTARVY en pacientes que desarrollen disminuciones clínicamente significativas en la función renal o evidencia de síndrome de Fanconi.
5.5 Acidosis Láctica/Hepatomegalia Grave con Esteatosis
Se ha notificado acidosis láctica y hepatomegalia grave con esteatosis, incluidos casos mortales, con el uso de análogos de nucleósidos, incluida la emtricitabina, un componente de BIKTARVY, y tenofovir DF, otro profármaco de tenofovir, solo o en combinación con otros antirretrovirales. El tratamiento con BIKTARVY debe suspenderse en cualquier paciente que desarrolle hallazgos clínicos o de laboratorio sugestivos de acidosis láctica o hepatotoxicidad pronunciada (que puede incluir hepatomegalia y esteatosis incluso en ausencia de elevaciones marcadas de las transaminasas).
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas se discuten en otras secciones del etiquetado:
Debido a que los ensayos clínicos se realizan en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un medicamento no pueden compararse directamente con las tasas en los ensayos clínicos de otro medicamento y pueden no reflejar las tasas observadas en la práctica.
Ensayos clínicos en adultos sin antecedentes de tratamiento antirretroviral
La evaluación de seguridad primaria de BIKTARVY se basó en datos de dos ensayos aleatorizados, doble ciego y controlados con activo, el ensayo 1489 y el ensayo 1490, en los que se reclutó a 1274 sujetos adultos infectados por el VIH-1 sin antecedentes de tratamiento antirretroviral hasta la semana 144. Después de la semana 144, los sujetos recibieron BIKTARVY de etiqueta abierta en una fase de extensión opcional durante 96 semanas adicionales (fin del estudio). Un total de 634 y 1025 sujetos recibieron un comprimido de BIKTARVY una vez al día durante las fases doble ciego (semana 144) y de extensión, respectivamente [ver Estudios clínicos (14.2)].
Las reacciones adversas más comunes (todos los grados) notificadas en al menos el 5 % de los sujetos del grupo de BIKTARVY en el ensayo 1489 o en el ensayo 1490 fueron diarrea, náuseas y cefalea. La proporción de sujetos que interrumpieron el tratamiento hasta la semana 144 con BIKTARVY, abacavir [ABC]/dolutegravir [DTG]/ lamivudina [3TC]) o DTG + FTC/TAF, debido a acontecimientos adversos, independientemente de la gravedad, fue del 1 %, 2 % y 2 %, respectivamente. En la Tabla 1 se muestra la frecuencia de las reacciones adversas (todos los grados) mayores o iguales al 2 % en el grupo de BIKTARVY.
Tabla 1 Reacciones adversas* (todos los grados) notificadas en ≥ 2 % de los adultos infectados por el VIH-1 sin antecedentes de tratamiento antirretroviral que recibieron BIKTARVY en los ensayos 1489 o 1490 (análisis de la semana 144)
Las frecuencias de las reacciones adversas se basan en todos los acontecimientos adversos atribuidos a los fármacos del ensayo por el investigador. No se produjeron reacciones adversas de grado 2 o superior en > 1 % de los sujetos tratados con BIKTARVY.
Diarrea
6%
4%
3%
3%
Náuseas
6%
18%
3%
5%
Cefalea
5%
5%
4%
3%
Fatiga
3%
4%
2%
2%
Sueños anormales
3%
3%
<1%
1%
Mareos
2%
3%
2%
1%
Insomnio
2%
3%
2%
<1%
Distensión abdominal
2%
2%
1%
2%
Las reacciones adversas adicionales (todos los Grados) que ocurrieron en menos del 2% de los sujetos a los que se les administró BIKTARVY en los Ensayos 1489 y 1490 incluyeron vómitos, flatulencia, dispepsia, dolor abdominal, erupción cutánea y depresión.
La ideación suicida, el intento de suicidio y la depresión suicida ocurrieron en el 2% de los sujetos a los que se les administró BIKTARVY; estos eventos ocurrieron principalmente en sujetos con antecedentes de depresión, intento de suicidio previo o enfermedad psiquiátrica.
La mayoría (84%) de los eventos adversos asociados con BIKTARVY fueron de Grado 1.
Las reacciones adversas en las fases de extensión abierta de los Ensayos 1489 y 1490 fueron similares a las observadas en los sujetos a los que se les administró BIKTARVY en el análisis de la Semana 144.
Ensayos clínicos en adultos con supresión virológica
La seguridad de BIKTARVY en adultos con supresión virológica se basó en los datos de la semana 48 de 282 sujetos en un ensayo aleatorizado, doble ciego, controlado con activo (Ensayo 1844) en el que los sujetos con supresión virológica cambiaron de DTG + ABC/3TC o ABC/DTG/3TC a BIKTARVY; datos de la semana 48 de 290 sujetos en un ensayo abierto, controlado con activo en el que los sujetos con supresión virológica cambiaron de un régimen que contenía atazanavir (ATV) (administrado con cobicistat o ritonavir) o darunavir (DRV) (administrado con cobicistat o ritonavir) más FTC/TDF o ABC/3TC, a BIKTARVY (Ensayo 1878); y datos de la semana 48 de un ensayo aleatorizado, doble ciego, controlado con activo en el que 284 sujetos con supresión virológica cambiaron de DTG más FTC/TAF o FTC/TDF, a BIKTARVY (Ensayo 4030). En general, el perfil de seguridad en sujetos adultos con supresión virológica en los Ensayos 1844, 1878 y 4030 fue similar al de los sujetos sin antecedentes de tratamiento antirretroviral [ver Estudios clínicos (14.3)].
Ensayo clínico en adultos con enfermedad renal terminal (ERT) que reciben hemodiálisis crónica
La seguridad de FTC y TAF (componentes de BIKTARVY) se evaluó en un ensayo abierto de un solo brazo (Ensayo 1825) en adultos con supresión virológica con ERT (aclaramiento estimado de creatinina inferior a 15 ml/min) en hemodiálisis crónica tratados con FTC+TAF en combinación con elvitegravir y cobicistat como un comprimido de combinación de dosis fija durante 96 semanas (N=55). La reacción adversa notificada con mayor frecuencia (evento adverso evaluado como causalmente relacionado por el investigador y todos los grados) fueron las náuseas (7%). Se notificaron eventos adversos graves en el 65% de los sujetos y los eventos adversos graves más frecuentes fueron neumonía (15%), sobrecarga de líquidos (7%), hiperpotasemia (11%) y osteomielitis (7%). En general, el 7% de los sujetos interrumpieron permanentemente el tratamiento debido a un evento adverso. En una fase de extensión del Ensayo 1825 en la que 10 sujetos cambiaron a BIKTARVY durante 48 semanas, los hallazgos de seguridad fueron similares a los de la fase inicial del ensayo abierto [ver Uso en poblaciones específicas (8.6) y Estudios clínicos (14.3)].
Anomalías de laboratorio
La frecuencia de anomalías de laboratorio (Grados 3-4) que ocurren en al menos el 2% de los sujetos que reciben BIKTARVY en los Ensayos 1489 y 1490 se presentan en la Tabla 2.
Tabla 2 Anomalías de laboratorio (Grados 3-4) informadas en ≥ 2% de los sujetos que recibieron BIKTARVY en los ensayos 1489 o 1490 (análisis de la semana 144)
Cambios en la creatinina sérica: Se ha demostrado que BIC aumenta la creatinina sérica debido a la inhibición de la secreción tubular de creatinina sin afectar la función glomerular renal [ver Farmacología clínica (12.2)]. Los aumentos en la creatinina sérica ocurrieron en la semana 4 de tratamiento y se mantuvieron estables hasta la semana 144. En los ensayos 1489 y 1490, la mediana (Q1, Q3) de creatinina sérica aumentó 0.11 (0.03, 0.19) mg por dL desde el inicio hasta la semana 144 en el grupo de BIKTARVY y fue similar a los grupos de comparación que recibieron ABC/DTG/3TC o DTG + FTC/TAF. No hubo interrupciones debido a eventos adversos renales y se encontraron eventos adversos renales graves en menos del 1% de los participantes tratados con BIKTARVY hasta la semana 144 en ensayos clínicos.
Cambios en la bilirrubina: En los ensayos 1489 y 1490, se observaron aumentos de la bilirrubina total en el 17 % de los sujetos a los que se les administró BIKTARVY hasta la semana 144. Los aumentos fueron principalmente de grado 1 (1.0 a 1.5 × ULN) (12 %) y de grado 2 (1.5 a 2.5 × ULN) (4 %). Los aumentos de bilirrubina graduados en los grupos de ABC/DTG/3TC y DTG + FTC/TAF fueron del 7 % y el 8 %, respectivamente. Los aumentos fueron principalmente de grado 1 (5 % ABC/DTG/3TC y 7 % DTG + FTC/TAF) o de grado 2 (2 % ABC/DTG/3TC y 2 % DTG + FTC/TAF). No hubo interrupciones debido a eventos adversos hepáticos hasta la semana 144 en los estudios clínicos de BIKTARVY.
Ensayos clínicos en sujetos pediátricos
Se evaluó la seguridad de BIKTARVY en sujetos infectados por el VIH-1 con supresión virológica de entre 12 y menos de 18 años de edad y un peso mínimo de 35 kg (N=50) hasta la semana 48 (cohorte 1), en sujetos con supresión virológica de entre 6 y menos de 12 años de edad y un peso mínimo de 25 kg (N=50) hasta la semana 24 (cohorte 2), y en sujetos con supresión virológica de al menos 2 años de edad y un peso mínimo de 14 a menos de 25 kg (N=22) hasta la semana 24 (cohorte 3) en un ensayo clínico abierto (ensayo 1474) [ver Estudios clínicos (14.4)]. No se identificaron nuevas reacciones adversas ni anomalías de laboratorio en comparación con las observadas en adultos. Se notificaron reacciones adversas en el 11 % de los sujetos pediátricos. La mayoría (76 %) de las reacciones adversas fueron de grado 1. No se notificaron reacciones adversas de grado 3 o 4. La reacción adversa notificada por más de un sujeto (independientemente de la gravedad) fue molestias abdominales (n=2). Un sujeto (1 %) presentó reacciones adversas de grado 2 de insomnio y ansiedad que provocaron la interrupción de BIKTARVY. Las demás reacciones adversas que se produjeron en sujetos individuales fueron similares a las observadas en adultos.
6.2 Experiencia posterior a la comercialización
Los siguientes eventos se han identificado durante el uso posterior a la aprobación de BIKTARVY o productos que contienen TAF. Debido a que estos eventos se notifican de forma voluntaria a partir de una población de tamaño incierto, no siempre es posible estimar de forma fiable su frecuencia o establecer una relación causal con la exposición al fármaco.
Trastornos renales y urinarios
Insuficiencia renal aguda, necrosis tubular aguda, tubulopatía renal proximal y síndrome de Fanconi
Trastornos de la piel y del tejido subcutáneo
Angioedema, síndrome de Stevens-Johnson/necrólisis epidérmica tóxica y urticaria
Investigaciones
Aumento de peso
7 INTERACCIONES MEDICAMENTOSAS
7.1 Otros medicamentos antirretrovirales
Debido a que BIKTARVY es un régimen completo, no se recomienda la coadministración con otros medicamentos antirretrovirales para el tratamiento de la infección por VIH-1 [ver Indicaciones y uso (1)]. No se proporciona información completa sobre las posibles interacciones medicamentosas con otros medicamentos antirretrovirales porque se desconoce la seguridad y eficacia de la terapia antirretroviral concomitante para el VIH-1.
7.2 Posibilidad de que BIKTARVY afecte otros medicamentos
BIC inhibe el transportador 2 de cationes orgánicos (OCT2) y el transportador de extrusora de toxinas y multidrogas 1 (MATE1) in vitro. La coadministración de BIKTARVY con medicamentos que son sustratos de OCT2 y MATE1 (por ejemplo, dofetilida) puede aumentar sus concentraciones plasmáticas (ver Tabla 3).
7.3 Posible efecto de otros medicamentos en uno o más componentes de BIKTARVY
BIC es un sustrato de CYP3A y UGT1A1. Un medicamento que es un inductor fuerte de CYP3A y también un inductor de UGT1A1 puede disminuir sustancialmente las concentraciones plasmáticas de BIC, lo que puede provocar la pérdida del efecto terapéutico de BIKTARVY y el desarrollo de resistencia [ver Farmacología clínica (12.3)].
El uso de BIKTARVY con un medicamento que es un inhibidor fuerte de CYP3A y también un inhibidor de UGT1A1 puede aumentar significativamente las concentraciones plasmáticas de BIC.
TAF es un sustrato de la glicoproteína P (P-gp) y la proteína de resistencia al cáncer de mama (BCRP). La coadministración de medicamentos que inhiben P-gp y BCRP puede aumentar la absorción y las concentraciones plasmáticas de TAF [ver Farmacología clínica (12.3)]. Se espera que la coadministración de medicamentos que inducen la actividad de P-gp disminuya la absorción de TAF, lo que provocará una disminución de la concentración plasmática de TAF, lo que puede provocar la pérdida del efecto terapéutico de BIKTARVY y el desarrollo de resistencia (ver Tabla 3).
7.4 Medicamentos que afectan la función renal
Debido a que FTC y tenofovir se excretan principalmente por los riñones mediante una combinación de filtración glomerular y secreción tubular activa, la coadministración de BIKTARVY con medicamentos que reducen la función renal o compiten por la secreción tubular activa puede aumentar las concentraciones de FTC, tenofovir y otros medicamentos eliminados por vía renal, y esto puede aumentar el riesgo de reacciones adversas. Algunos ejemplos de medicamentos que se eliminan por secreción tubular activa incluyen, entre otros, aciclovir, cidofovir, ganciclovir, valaciclovir, valganciclovir, aminoglucósidos (por ejemplo, gentamicina) y AINE de dosis alta o múltiples [ver Advertencias y precauciones (5.4)].
7.5 Interacciones medicamentosas establecidas y potencialmente significativas
La Tabla 3 proporciona una lista de interacciones medicamentosas establecidas o potencialmente clínicamente significativas con estrategias de prevención o manejo recomendadas. Las interacciones medicamentosas descritas se basan en estudios realizados con BIKTARVY, los componentes de BIKTARVY (BIC, FTC y TAF) como agentes individuales, o son interacciones medicamentosas que pueden ocurrir con BIKTARVY [ver Contraindicaciones (4), Advertencias y precauciones (5.2), y Farmacología clínica (12.3)].
Tabla 3 Interacciones medicamentosas establecidas y potencialmente significativas*: Puede recomendarse una alteración en el régimen
Clase de medicamento concomitante: Nombre del medicamento
La potencia de inducción de la hierba de San Juan puede variar ampliamente según la preparación.
Antiarrítmicos:
dofetilida
↑ Dofetilida
La coadministración está contraindicada debido al potencial de eventos graves y/o potencialmente mortales asociados con la terapia con dofetilida [ver Contraindicaciones (4)].
La coadministración con rifampicina está contraindicada debido al efecto de la rifampicina sobre el componente BIC de BIKTARVY [ver Contraindicaciones (4)]. No se recomienda la coadministración con rifabutina o rifapentina.
BIKTARVY se puede tomar al menos 2 horas antes o 6 horas después de tomar antiácidos que contienen Al/Mg. No se recomienda la administración rutinaria de BIKTARVY junto con, o 2 horas después, antiácidos que contienen Al/Mg. Suplementos o antiácidos que contienen calcio o hierro:
BIKTARVY y los suplementos o antiácidos que contienen calcio o hierro se pueden tomar junto con los alimentos. No se recomienda la administración rutinaria de BIKTARVY en ayunas junto con, o 2 horas después, suplementos o antiácidos que contienen calcio o hierro. En individuos embarazadas:
Antiácidos que contienen Al/Mg:
BIKTARVY se puede tomar al menos 2 horas antes o 6 horas después de los antiácidos que contienen Al/Mg independientemente de la ingesta de alimentos. Suplementos o antiácidos que contienen calcio o hierro:
BIKTARVY y los suplementos o antiácidos que contienen calcio o hierro se pueden tomar junto con los alimentos; pero cuando se toman con el estómago vacío, BIKTARVY debe tomarse al menos 2 horas antes o 6 horas después de los suplementos o antiácidos que contienen calcio o hierro.
Metformina
↑ Metformina
Consulte la información de prescripción de metformina para evaluar el beneficio y el riesgo del uso concomitante de BIKTARVY y metformina.
7.6 Medicamentos sin interacciones clínicamente significativas con BIKTARVY
Con base en los estudios de interacción medicamentosa realizados con BIKTARVY o los componentes de BIKTARVY, no se han observado interacciones medicamentosas clínicamente significativas cuando BIKTARVY se combina con los siguientes medicamentos: etinilestradiol, ledipasvir/sofosbuvir, midazolam, norgestimato, sertralina, sofosbuvir, sofosbuvir/velpatasvir y sofosbuvir/velpatasvir/voxilaprevir.
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Registro de exposición durante el embarazo
Existe un registro de exposición durante el embarazo que monitoriza los resultados del embarazo en individuos expuestos a BIKTARVY durante el embarazo. Se anima a los profesionales de la salud a registrar a los pacientes llamando al Registro de Embarazo con Antiretrovirales (APR) al 1-800-258-4263.
Resumen de riesgos
Los datos disponibles de estudios observacionales y el APR con el uso de BIC, FTC y TAF durante el embarazo no han establecido un riesgo asociado a los medicamentos de defectos de nacimiento importantes, aborto espontáneo u otros resultados adversos maternos o fetales. Los informes de individuos embarazadas tratados con productos que contienen BIC, FTC o TAF contribuyen a la evaluación general de riesgos del APR para estos componentes. Los datos disponibles del APR no muestran ninguna diferencia estadísticamente significativa en el riesgo general de defectos de nacimiento importantes para BIC, FTC o TAF en comparación con la tasa de fondo de defectos de nacimiento importantes del 2.7% en una población de referencia de EE. UU. del Programa de Defectos Congénitos del Área Metropolitana de Atlanta (MACDP) (ver Datos). La tasa de aborto espontáneo no se informa en el APR. La tasa de fondo estimada de aborto espontáneo en los embarazos clínicamente reconocidos en la población general de EE. UU. es del 15–20%.
La seguridad de BIKTARVY también se ha evaluado en un ensayo abierto que demostró hallazgos de seguridad que fueron consistentes con otros ensayos en adultos (ver Datos).
En estudios de reproducción animal, no se observó evidencia de resultados de desarrollo adversos con los componentes de BIKTARVY a exposiciones que no fueron tóxicas para la madre (conejos) o mayores que (ratas y ratones) las de los humanos a la dosis humana recomendada (RHD) (ver Datos). Durante la organogénesis, las exposiciones sistémicas (AUC) a BIC fueron aproximadamente 36 (ratas) y 0.6 veces (conejos), a FTC fueron aproximadamente 60 (ratones) y 108 veces (conejos), y a TAF fueron aproximadamente 2 (ratas) y 78 veces (conejos) la exposición a la RHD de BIKTARVY. En estudios de desarrollo pre/postnatal en ratas, las exposiciones sistémicas maternas (AUC) fueron 30 veces (BIC), 60 veces (FTC) y 19 veces (TDF) las exposiciones de cada componente en humanos a la RHD.
Datos
Datos humanos
BIKTARVY se evaluó en un ensayo clínico abierto de 33 adultos embarazadas con VIH-1 virológicamente suprimidas (ARN del VIH-1 < 50 copias/mL) y sin sustituciones conocidas asociadas con resistencia a BIC, FTC o TAF. Los adultos embarazadas recibieron BIKTARVY (que contiene 50 mg de BIC, 200 mg de FTC y 25 mg de TAF) una vez al día desde el segundo o tercer trimestre hasta el posparto. Las exposiciones de BIC, FTC y TAF fueron más bajas durante el embarazo en comparación con el posparto [ver Farmacología clínica (12.3)]. Los 32 participantes adultos que completaron el estudio mantuvieron la supresión viral durante el embarazo, en el parto y hasta la semana 18 posparto. La mediana del recuento de células CD4+ en el inicio del estudio fue de 558 células/µL, y el cambio mediano en el recuento de células CD4+ desde el inicio del estudio hasta la semana 12 posparto fue de 159 células/μL. Los 29 participantes neonatos tuvieron resultados negativos/indetectables de PCR del VIH-1 al nacer y/o a las 4 a 8 semanas después del nacimiento. Los hallazgos de seguridad en este ensayo fueron consistentes con otros ensayos en adultos.
BIKTARVY solo se estudió en individuos embarazadas que estaban virológicamente suprimidas, y se observaron exposiciones plasmáticas más bajas de BIKTARVY durante el embarazo en comparación con el posparto. Por lo tanto, BIKTARVY se recomienda en individuos embarazadas que están virológicamente suprimidas con un régimen antirretroviral estable sin sustituciones conocidas asociadas con resistencia a ninguno de los componentes individuales de BIKTARVY.
Bictegravir (BIC):
Con base en informes prospectivos al APR de más de 500 exposiciones a un régimen que contiene BIC durante el embarazo que resultaron en nacimientos vivos (incluidos 423 expuestos en el primer trimestre y 113 expuestos en el segundo/tercer trimestre), la prevalencia de defectos de nacimiento en nacimientos vivos fue del 4.3% (IC del 95%: 2.5% a 6.6%) y 1.8% (0.2%, 6.2%) después de la exposición al primer y segundo/tercer trimestre, respectivamente, a un régimen que contiene BIC.
Emtricitabina (FTC):
Con base en informes prospectivos al APR de más de 6,500 exposiciones a regímenes que contienen FTC durante el embarazo que resultaron en nacimientos vivos (incluidos más de 4,800 expuestos en el primer trimestre y más de 1,700 expuestos en el segundo/tercer trimestre), la prevalencia de defectos de nacimiento en nacimientos vivos fue del 2.9% (IC del 95%: 2.5% a 3.4%) y 2.8% (IC del 95%: 2.1% a 3.7%) después de la exposición al primer y segundo/tercer trimestre, respectivamente, a regímenes que contienen FTC.
Tenofovir Alafenamida (TAF):
Con base en informes prospectivos al APR de más de 1,200 exposiciones a regímenes que contienen TAF durante el embarazo que resultaron en nacimientos vivos (incluidos más de 1000 expuestos en el primer trimestre y más de 200 expuestos en el segundo/tercer trimestre), la prevalencia de defectos de nacimiento en nacimientos vivos fue del 3.9% (IC del 95%: 2.8% a 5.2%) y 4.8% (IC del 95%: 2.5% a 8.3%) después de la exposición al primer y segundo/tercer trimestre, respectivamente, a regímenes que contienen TAF.
Las limitaciones metodológicas del APR incluyen el uso de MACDP como grupo de comparación externo. La población de MACDP no es específica de la enfermedad, evalúa a individuos y bebés de un área geográfica limitada y no incluye resultados para nacimientos que ocurrieron en menos de 20 semanas de gestación.
Datos animales
Bictegravir: BIC se administró por vía oral a ratas preñadas (5, 30 o 300 mg/kg/día) y conejos (100, 300 o 1000 mg/kg/día) en los días de gestación 7 a 17 y 7 a 19, respectivamente. No se observaron efectos adversos embrio-fetales en ratas y conejos a exposiciones de BIC (AUC) de hasta aproximadamente 36 (ratas) y 0,6 (conejos) veces la exposición en humanos a la dosis diaria recomendada de BIKTARVY. Se observaron aborto espontáneo, aumento de los signos clínicos [cambios fecales, cuerpo delgado y frío al tacto] y disminución del peso corporal a una dosis tóxica para la madre en conejos (1000 mg/kg/día; aproximadamente 1,4 veces mayor que la exposición humana a la dosis diaria recomendada).
En un estudio de desarrollo pre/postnatal, BIC se administró por vía oral a ratas preñadas (hasta 300 mg/kg/día) desde los días de gestación 6 hasta el día 24 de lactancia/posparto. No se observaron efectos adversos significativos en la descendencia expuesta diariamente desde antes del nacimiento (in útero) hasta la lactancia a exposiciones maternas y de crías (AUC) de aproximadamente 30 y 11 veces más altas, respectivamente, que las exposiciones humanas a la dosis diaria recomendada.
Emtricitabina: FTC se administró por vía oral a ratones preñados (250, 500 o 1000 mg/kg/día) y conejos (100, 300 o 1000 mg/kg/día) durante la organogénesis (en los días de gestación 6 a 15 y 7 a 19, respectivamente). No se observaron efectos toxicológicos significativos en los estudios de toxicidad embrio-fetal realizados con emtricitabina en ratones a exposiciones aproximadamente 60 veces más altas y en conejos a exposiciones aproximadamente 108 veces más altas que las exposiciones humanas a la dosis diaria recomendada.
En un estudio de desarrollo pre/postnatal con FTC, se administraron dosis a ratones de hasta 1000 mg/kg/día; no se observaron efectos adversos significativos directamente relacionados con el fármaco en la descendencia expuesta diariamente desde antes del nacimiento (in útero) hasta la madurez sexual a exposiciones diarias (AUC) de aproximadamente 60 veces más altas que las exposiciones humanas a la dosis diaria recomendada.
Tenofovir alafenamida: TAF se administró por vía oral a ratas preñadas (25, 100 o 250 mg/kg/día) y conejos (10, 30 o 100 mg/kg/día) durante la organogénesis (en los días de gestación 6 a 17 y 7 a 20, respectivamente). No se observaron efectos adversos embrio-fetales en ratas y conejos a exposiciones de TAF de aproximadamente 2 (ratas) y 78 (conejos) veces más altas que la exposición en humanos a la dosis diaria recomendada de BIKTARVY. TAF se convierte rápidamente en tenofovir; la exposición a tenofovir observada en ratas y conejos fue 55 (ratas) y 86 (conejos) veces más alta que las exposiciones a tenofovir en humanos a la dosis diaria recomendada. Dado que TAF se convierte rápidamente en tenofovir y se observaron exposiciones más bajas a tenofovir en ratas y ratones después de la administración de TAF en comparación con la administración de TDF, se realizó un estudio de desarrollo pre/postnatal en ratas solo con TDF. Se administraron dosis de hasta 600 mg/kg/día durante la lactancia; no se observaron efectos adversos en la descendencia en el día 7 de gestación [y el día 20 de lactancia] a exposiciones a tenofovir de aproximadamente 12 [19] veces más altas que las exposiciones en humanos a la dosis diaria recomendada de BIKTARVY.
8.2 Lactancia
Resumen de riesgos
Los datos de la literatura publicada informan sobre la presencia de BIC, FTC, TAF y tenofovir en la leche materna. No hay datos sobre los efectos de BIC en el niño amamantado. Los datos de la literatura publicada no han informado de efectos adversos de FTC o TAF en un niño amamantado. No hay datos sobre los efectos de BIC, FTC o TAF en la producción de leche.
Los riesgos potenciales de la lactancia incluyen: (1) transmisión del VIH-1 a bebés VIH-1 negativos; (2) desarrollo de resistencia viral en bebés VIH-1 positivos; y (3) reacciones adversas en un bebé amamantado similares a las observadas en adultos.
8.4 Uso pediátrico
La seguridad y eficacia de BIKTARVY se han establecido como un régimen completo para el tratamiento de la infección por el virus de la inmunodeficiencia humana tipo 1 (VIH-1) en pacientes pediátricos que pesan al menos 14 kg:
que no tienen antecedentes de tratamiento antirretroviral o
para reemplazar el régimen antirretroviral actual en aquellos que están virologicamente suprimidos (ARN del VIH-1 inferior a 50 copias por mL) en un régimen antirretroviral estable sin resistencia conocida o sospechada a bictegravir o tenofovir [ver Indicaciones y uso (1), y Dosificación y administración (2.2, 2.3)].
El uso de BIKTARVY en pacientes pediátricos que pesan al menos 14 kg está respaldado por lo siguiente:
un ensayo abierto en tres cohortes de edad de sujetos pediátricos virologicamente suprimidos [ver Estudios clínicos (14.4)]
Cohorte 1: de 12 a menos de 18 años de edad y que pesan al menos 35 kg que reciben BIKTARVY hasta la semana 48 (N=50),
Cohorte 2: de 6 a menos de 12 años de edad y que pesan al menos 25 kg que reciben BIKTARVY hasta la semana 24 (N=50), y
Cohorte 3: al menos 2 años de edad y que pesan al menos 14 a menos de 25 kg hasta la semana 24 (N=22). No se inscribieron sujetos pediátricos de 2 años de edad; de los 6 sujetos pediátricos que tenían 3 años de edad al momento de la inscripción, 3 sujetos pesaron entre 14 y menos de 15 kg.
No se ha establecido la seguridad y eficacia de BIKTARVY en pacientes pediátricos que pesan menos de 14 kg.
8.5 Uso en poblaciones específicas
Los ensayos clínicos en sujetos con supresión virológica (Ensayos 4449, 1844 y 1878) incluyeron 111 sujetos de 65 años o más que recibieron BIKTARVY, incluidos 86 pacientes de un ensayo de un solo brazo abierto de sujetos de 65 años o más que cambiaron de su régimen antirretroviral anterior a BIKTARVY [ver Estudios clínicos (14.3)]. Del número total de pacientes tratados con BIKTARVY en estos ensayos, 100 (90%) tenían entre 65 y 74 años, y 11 (10%) tenían entre 75 y 84 años. No se observaron diferencias generales en seguridad o eficacia entre los sujetos de edad avanzada y los adultos entre 18 y menos de 65 años, y otras experiencias clínicas reportadas no han identificado diferencias en las respuestas entre los pacientes de edad avanzada y los más jóvenes, pero no se puede descartar una mayor sensibilidad de algunos individuos mayores.
8.6 Insuficiencia renal
La farmacocinética, la seguridad, las respuestas virológicas e inmunológicas de FTC y TAF (componentes de BIKTARVY) se evaluaron en un ensayo de un solo brazo, abierto (Ensayo 1825) en adultos con supresión virológica con ESRD (aclaramiento de creatinina estimado de menos de 15 mL/min) en hemodiálisis crónica tratados con FTC+TAF en combinación con elvitegravir y cobicistat como una tableta de dosis fija durante 96 semanas (N=55). En una fase de extensión del Ensayo 1825, 10 sujetos con supresión virológica cambiaron a BIKTARVY y todos permanecieron con supresión virológica durante 48 semanas [ver Reacciones adversas (6.1), Farmacología clínica (12.3), y Estudios clínicos (14.3)].
No se recomienda ningún ajuste de dosis de BIKTARVY en pacientes con aclaramiento de creatinina estimado mayor o igual a 30 mL/min, o en adultos con supresión virológica (aclaramiento de creatinina estimado por debajo de 15 mL/min) que reciben hemodiálisis crónica. Los días de hemodiálisis, administre la dosis diaria de BIKTARVY después de completar el tratamiento de hemodiálisis [ver Dosificación y administración (2.2)]
BIKTARVY no se recomienda en pacientes con aclaramiento de creatinina estimado por debajo de 30 mL/min, según Cockcroft-Gault, o pacientes con ESRD (aclaramiento de creatinina estimado por debajo de 15 mL/min) que no reciben diálisis crónica, o pacientes sin antecedentes de tratamiento antirretroviral y ESRD que reciben diálisis crónica, ya que la seguridad y/o eficacia de BIKTARVY no se ha establecido en estas poblaciones [ver Dosificación y administración (2.4), Advertencias y precauciones (5.4), y Farmacología clínica (12.3)].
8.7 Insuficiencia hepática
No se recomienda ningún ajuste de dosis de BIKTARVY en pacientes con insuficiencia hepática leve (Clase A de Child-Pugh) o moderada (Clase B de Child-Pugh). BIKTARVY no se ha estudiado en pacientes con insuficiencia hepática grave (Clase C de Child-Pugh). Por lo tanto, BIKTARVY no se recomienda para su uso en pacientes con insuficiencia hepática grave [ver Dosificación y administración (2.4), y Farmacología clínica (12.3)].
10 SOBREDOSIS
No se dispone de datos sobre sobredosis de BIKTARVY en pacientes. Si se produce una sobredosis, controle al paciente para detectar evidencia de toxicidad. El tratamiento de la sobredosis con BIKTARVY consiste en medidas generales de apoyo, incluida la monitorización de los signos vitales, así como la observación del estado clínico del paciente.
El tratamiento con hemodiálisis elimina aproximadamente el 30% de la dosis de FTC durante un período de diálisis de 3 horas que comienza dentro de las 1,5 horas posteriores a la administración de FTC (tasa de flujo sanguíneo de 400 mL/min y una tasa de flujo de dializado de 600 mL/min). No se sabe si la FTC se puede eliminar mediante diálisis peritoneal.
Tenofovir se elimina eficazmente mediante hemodiálisis con un coeficiente de extracción de aproximadamente el 54%.
11 DESCRIPCIÓN
BIKTARVY (bictegravir, emtricitabine y tenofovir alafenamida) es una tableta de combinación de dosis fija que contiene bictegravir (BIC), emtricitabine (FTC) y tenofovir alafenamida (TAF) para administración oral.
BIC es un inhibidor de la transferencia de cadena de la integrasa (INSTI).
FTC, un análogo nucleósido sintético de la citidina, es un inhibidor de la transcriptasa inversa análogo nucleósido del VIH (HIV NRTI).
TAF, un HIV NRTI, se convierte in vivo en tenofovir, un análogo fosfonato (nucleótido) acíclico de la 5′-monofosfato de adenosina.
Las tabletas de BIKTARVY están disponibles en dos concentraciones de dosis:
Tableta de 50 mg/200 mg/25 mg que contiene 50 mg de BIC (equivalente a 52,5 mg de bictegravir sódico), 200 mg de FTC y 25 mg de TAF (equivalente a 28 mg de tenofovir alafenamida fumarato).
Tableta de 30 mg/120 mg/15 mg que contiene 30 mg de BIC (equivalente a 31,5 mg de bictegravir sódico), 120 mg de FTC y 15 mg de TAF (equivalente a 16,8 mg de tenofovir alafenamida fumarato).
Ambas concentraciones de dosis de las tabletas de BIKTARVY incluyen los siguientes ingredientes inactivos: sodio croscarmelosa, estearato de magnesio y celulosa microcristalina. Las tabletas de ambas concentraciones de dosis están recubiertas con un material de recubrimiento que contiene óxido de hierro negro, óxido de hierro rojo, polietilenglicol, alcohol polivinílico, talco y dióxido de titanio.
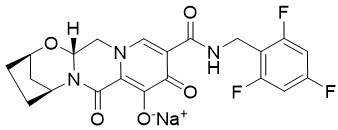
Bictegravir: El nombre químico del bictegravir sódico es 2,5-Metanopirido[1′,2′:4,5]pirazino[2,1-b][1,3]oxazepina-10-carboxamida, 2,3,4,5,7,9,13,13a-octahidro-8-hidroxi-7,9-dioxo-N-[(2,4,6-trifluorofenil)metil]-, sal sódica (1:1), (2R,5S,13aR)-.
El bictegravir sódico tiene una fórmula molecular de C21H17F3N3NaO5 y un peso molecular de 471,4 y tiene la siguiente fórmula estructural:
El bictegravir sódico es un sólido de color entre blanco amarillento y amarillo, con una solubilidad de 0,1 mg por mL en agua a 20 °C.
Emtricitabine: El nombre químico de la FTC es 4-amino-5-fluoro-1-(2R-hidroximetil-1,3-oxatiazolán-5S-il)-(1H)-pirimidin-2-ona. La FTC es el enantiómero (-) de un análogo tio de la citidina, que se diferencia de otros análogos de la citidina en que tiene un flúor en la posición 5.
La FTC tiene una fórmula molecular de C8H10FN3O3S y un peso molecular de 247,2 y tiene la siguiente fórmula estructural:
La FTC es un polvo de color blanco a blanco amarillento, con una solubilidad de aproximadamente 112 mg por mL en agua a 25 °C.
Tenofovir alafenamida: El nombre químico de la sustancia farmacéutica de tenofovir alafenamida fumarato es L-alanina, N-[(S)-[[(1R)-2-(6-amino-9H-purin-9-il)-1-metiletoxi]metil]fenoxifosfinil]-, éster isopropílico, (2E)-2-butenedioato (2:1).
El tenofovir alafenamida fumarato tiene una fórmula empírica de C21H29O5N6P∙½(C4H4O4) y un peso molecular de 534,5 y tiene la siguiente fórmula estructural:
El tenofovir alafenamida fumarato es un polvo de color blanco a blanco amarillento o marrón claro, con una solubilidad de 4,7 mg por mL en agua a 20 °C.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
BIKTARVY es una combinación de dosis fija de los fármacos antirretrovirales bictegravir (BIC), emtricitabina (FTC) y tenofovir alafenamida (TAF) [ver Microbiología (12.4)].
12.2 Farmacodinamia
Electrofisiología cardíaca
En un ensayo exhaustivo de QT/QTc en 48 sujetos sanos, BIC a dosis 1.5 y 6 veces la dosis recomendada no afectó el intervalo QT/QTc y no prolongó el intervalo PR. En un ensayo exhaustivo de QT/QTc en 48 sujetos sanos, TAF a la dosis recomendada o a una dosis 5 veces la dosis recomendada, no afectó el intervalo QT/QTc y no prolongó el intervalo PR. Se desconoce el efecto de FTC sobre el intervalo QT.
Efectos sobre la creatinina sérica
El cambio medio desde el inicio en la creatinina sérica en sujetos sanos que recibieron BIC 75 mg (1.5 veces la dosis recomendada aprobada) una vez al día con alimentos durante 14 días fue de 0.1 mg por dL en los días 7 y 14 en comparación con el placebo. BIC no tuvo un efecto significativo en el aclaramiento de creatinina estimado o en la tasa de filtración glomerular real (determinada por el aclaramiento del fármaco de prueba, iohexol).
12.3 Farmacocinética
Las propiedades farmacocinéticas (PK) de los componentes de BIKTARVY se proporcionan en la Tabla 4. Los parámetros PK de dosis múltiples de los componentes de BIKTARVY (basados en el análisis farmacocinético poblacional) se proporcionan en la Tabla 5.
Tabla 4 Propiedades farmacocinéticas de los componentes de BIKTARVY
Bictegravir (BIC)
Emtricitabina (FTC)
Tenofovir Alafenamida (TAF)
PBMCs=células mononucleares de sangre periférica; CES1=carboxilesterasa 1
Los valores se refieren a la razón media geométrica [comida rica en grasas/ayuno] en los parámetros PK y (intervalo de confianza del 90%). La comida rica en grasas es aproximadamente 800 kcal, 50% de grasa.
Los valores de t1/2 se refieren a la vida media plasmática terminal mediana (Q1, Q3). Tenga en cuenta que el metabolito activo de TAF, tenofovir difosfato, tiene una vida media de 150–180 horas dentro de los PBMC.
In vivo, TAF se hidroliza dentro de las células para formar tenofovir (metabolito principal), que se fosforila al metabolito activo, tenofovir difosfato. In vitro los estudios han demostrado que TAF se metaboliza a tenofovir por la catepsina A en los PBMC y los macrófagos; y por CES1 en los hepatocitos.
Dosificación en estudios de balance de masa: administración de una sola dosis de [14C] BIC; administración de una sola dosis de [14C] FTC después de la administración múltiple de FTC durante diez días; administración de una sola dosis de [14C] TAF.
Tabla 5 Parámetros de FC de dosis múltiples de BIC, FTC y TAF después de la administración oral de BIKTARVY en adultos infectados por VIH
Parámetro Media (CV%)
Bictegravir
Emtricitabina
Tenofovir Alafenamida
CV=Coeficiente de variación; NA=No aplicable
Cmax
(microgramo por mL)
6.15 (22.9)
2.13 (34.7)
0.121 (15.4)
AUCtau
(microgramo∙h por mL)
102 (26.9)
12.3 (29.2)
0.142 (17.3)
Ctrough
(microgramo por mL)
2.61 (35.2)
0.096 (37.4)
NA
Poblaciones específicas
Pacientes con insuficiencia renal
No se observaron diferencias clínicamente relevantes en la farmacocinética de BIC, TAF o su metabolito tenofovir entre sujetos con insuficiencia renal grave (aclaramiento de creatinina estimado de 15 a menos de 30 mL/min, por el método de Cockcroft-Gault) y sujetos sanos en estudios de Fase 1. En un estudio de Fase 1 separado de FTC solo, las exposiciones a FTC aumentaron en sujetos con insuficiencia renal grave.
La farmacocinética de BIC, FTC y TAF se evaluó en un subconjunto de sujetos infectados por VIH-1 con supresión virológica y ESRD (aclaramiento de creatinina estimado menor de 15 mL/min, por el método de Cockcroft-Gault) que recibieron hemodiálisis crónica en el ensayo 1825. La farmacocinética de TAF fue similar entre los sujetos sanos y los sujetos con ESRD que recibieron hemodiálisis crónica; los aumentos en las exposiciones a FTC y tenofovir en sujetos con ESRD no se consideraron clínicamente relevantes. Los valores medianos (mínimo, máximo) de Ctrough de BIC en sujetos (n=7) con ESRD que recibieron BIKTARVY fueron 846 ng/mL (288, 1810) en comparación con 2540 ng/mL (757, 6499) en sujetos (N=584) con función renal normal. A pesar de los valores de Ctrough de BIC significativamente más bajos en la población con ESRD con supresión virológica, se mantuvo la supresión virológica [ver Uso en poblaciones específicas (8.6), y Estudios clínicos (14.3)].
Pacientes con insuficiencia hepática
Bictegravir: No se observaron cambios clínicamente relevantes en la farmacocinética de BIC en sujetos con insuficiencia hepática moderada (Clase B de Child-Pugh).
Emtricitabina: La farmacocinética de FTC no se ha estudiado en sujetos con insuficiencia hepática; sin embargo, FTC no se metaboliza significativamente por las enzimas hepáticas, por lo que el impacto de la insuficiencia hepática debería ser limitado.
Tenofovir alafenamida: No se observaron cambios clínicamente relevantes en la farmacocinética de TAF o su metabolito tenofovir en sujetos con insuficiencia hepática leve o moderada (Clase A y B de Child-Pugh) [ver Uso en poblaciones específicas (8.7)].
Coinfección por virus de la hepatitis B y/o C
La farmacocinética de BIC, FTC y TAF no se ha evaluado en sujetos coinfectados con virus de la hepatitis B y/o C.
Pacientes geriátricos
La farmacocinética de BIC, FTC y TAF no se ha evaluado completamente en personas mayores (65 años de edad o más). El análisis de farmacocinética poblacional de sujetos infectados por VIH en ensayos de Fase 3 de BIKTARVY mostró que la edad no tuvo un efecto clínicamente relevante en las exposiciones a BIC y TAF hasta los 74 años de edad [ver Uso en poblaciones específicas (8.5)].
Pacientes pediátricos
La Ctrough media de BIC fue menor en 50 pacientes pediátricos de 12 a menos de 18 años y con un peso de al menos 35 kg que recibieron BIKTARVY en el ensayo 1474 en relación con los adultos después de la administración de BIKTARVY, pero no se consideró clínicamente significativa en función de las relaciones exposición-respuesta; las exposiciones a FTC y TAF en estos pacientes pediátricos fueron similares a las de los adultos (Tabla 6).
Tabla 6 Parámetros de PK de dosis múltiples de BIC, FTC y TAF después de la administración oral de BIKTARVY en sujetos pediátricos infectados por VIH de 12 a menos de 18 años
Del análisis de PK intensivo de la cohorte 1 del ensayo 1474 (n=24).
Cmax
(microgramo por mL)
6.24 (27.1)
2.69 (34.0)
0.133 (70.2)
AUCtau
(microgramo∙h por mL)
89.1 (31.0)
13.6 (21.7)
0.196 (50.3)
Ctrough
(microgramo por mL)
1.78 (44.4)
0.064 (25.0)
NA
La Cmax media de BIC y las exposiciones de FTC y TAF (AUCtau y Cmax) alcanzadas en 50 pacientes pediátricos de entre 6 y menos de 12 años de edad y con un peso de al menos 25 kg, y en 22 pacientes pediátricos de al menos 2 años de edad y con un peso de al menos 14 y menos de 25 kg que recibieron BIKTARVY en el ensayo 1474 fueron más altas que las exposiciones en adultos; sin embargo, los aumentos no se consideraron clínicamente significativos ya que los perfiles de seguridad fueron similares en pacientes adultos y pediátricos (Tablas 7 y 8) [ver Uso en poblaciones específicas (8.4)].
Tabla 7 Parámetros de PK de dosis múltiples de BIC, FTC y TAF después de la administración oral de BIKTARVY en sujetos pediátricos infectados por el VIH de 6 a menos de 12 años de edad
Del análisis de PK intensivo de la cohorte 2 del ensayo 1474 (n=25 excepto n=24 para Ctrough).
Cmax
(microgramo por mL)
9.46 (24.3)
3.89 (31.0)
0.205 (44.6)
AUCtau
(microgramo∙h por mL)
128 (27.8)
17.6 (36.9)
0.278 (40.3)
Ctrough
(microgramo por mL)
2.36 (39.0)
0.227 (323)
NA
Tabla 8 Parámetros de PK de dosis múltiples de BIC, FTC y TAF después de la administración oral de BIKTARVY en sujetos pediátricos infectados por el VIH de al menos 2 años de edad* y con un peso de al menos 14 y menos de 25 kg
Del análisis de PK intensivo de la cohorte 3 del ensayo 1474 (n=12 excepto n=11 para Ctrough para FTC).
Cmax
(microgramo por mL)
9.15 (44.8)
3.85 (34.7)
0.414 (31.0)
AUCtau
(microgramo∙h por mL)
126 (42.4)
15.0 (21.9)
0.305 (42.6)
Ctrough
(microgramo por mL)
2.43 (40.1)
0.210 (243)
NA
Raza y género
No se observaron cambios clínicamente relevantes en la farmacocinética de BIC, FTC y TAF en función del género o la raza.
Embarazo
Las exposiciones plasmáticas (Ctrough y AUCtau) de BIC, FTC y TAF después de la ingesta de BIKTARVY 50 mg/200 mg/25 mg fueron más bajas durante el embarazo en comparación con el posparto (Tabla 9). Los cambios en la exposición durante el embarazo no se consideran clínicamente significativos en las personas embarazadas con supresión virológica [ver Interacciones medicamentosas (7.5)].
Tabla 9 Parámetros farmacocinéticos en estado estacionario de BIC, FTC y TAF tras la administración oral de BIKTARVY en adultos embarazadas con supresión virológica del VIH-1 en el segundo y tercer trimestre y la semana 12 posparto
Como BIKTARVY es un régimen completo para el tratamiento de la infección por VIH-1, no se proporciona información completa sobre las posibles interacciones medicamentosas con otros agentes antirretrovirales.
BIC es un sustrato de CYP3A y UGT1A1.
BIC es un inhibidor de OCT2 y MATE1. A concentraciones clínicamente relevantes, BIC no es un inhibidor de los transportadores hepáticos OATP1B1, OATP1B3, OCT1, BSEP, los transportadores renales OAT1 y OAT3, o las enzimas CYP (incluida CYP3A) o UGT1A1.
TAF es un sustrato de P-gp y BCRP.
A concentraciones clínicamente relevantes, TAF no es un inhibidor de los transportadores de fármacos P-gp, BCRP, los transportadores hepáticos OATP1B1, OATP1B3, OCT1, BSEP, los transportadores renales OAT1, OAT3, OCT2, MATE1, o las enzimas CYP (incluida CYP3A) o UGT1A1.
Se realizaron estudios de interacción medicamentosa con BIKTARVY o sus componentes. Las Tablas 10 y 11 resumen los efectos farmacocinéticos de otros medicamentos sobre BIC y TAF, respectivamente. La Tabla 12 resume los efectos farmacocinéticos de BIKTARVY o sus componentes sobre otros medicamentos.
Efecto de Otros Medicamentos sobre los Componentes de BIKTARVY
Bictegravir: BIC inhibe la actividad de transferencia de cadena de la integrasa del VIH-1 (inhibidor de la transferencia de cadena de la integrasa; INSTI), una enzima codificada por el VIH-1 que es necesaria para la replicación viral. La inhibición de la integrasa evita la integración del ADN lineal del VIH-1 en el ADN genómico del huésped, bloqueando la formación del provirus del VIH-1 y la propagación del virus.
Emtricitabina: FTC, un análogo nucleósido sintético de la citidina, es fosforilado por enzimas celulares para formar emtricitabina 5′-trifosfato. La emtricitabina 5′-trifosfato inhibe la actividad de la RT del VIH-1 al competir con el sustrato natural desoxicitidina 5′-trifosfato y al incorporarse al ADN viral naciente, lo que da como resultado la terminación de la cadena. La emtricitabina 5′-trifosfato es un inhibidor débil de las ADN polimerasas mamíferas α, β, Ɛ y la ADN polimerasa mitocondrial γ.
Tenofovir Alafenamida: TAF es un profármaco fosfonamidato de tenofovir (análogo de la monofosfato de 2′-desoxiadenosina). La exposición plasmática a TAF permite la permeación a las células y luego TAF se convierte intracelularmente en tenofovir mediante la hidrólisis por catepsina A. El tenofovir se fosforila posteriormente por las quinasas celulares al metabolito activo tenofovir difosfato. El tenofovir difosfato inhibe la replicación del VIH-1 mediante la incorporación al ADN viral por la RT del VIH, lo que da como resultado la terminación de la cadena de ADN. El tenofovir difosfato es un inhibidor débil de las ADN polimerasas mamíferas que incluyen la ADN polimerasa mitocondrial γ y no hay evidencia de toxicidad para las mitocondrias en cultivo celular.
Actividad antiviral en cultivo celular
La combinación triple de BIC, FTC y TAF no fue antagónica con respecto a la actividad antiviral en cultivo celular.
Bictegravir: La actividad antiviral de BIC contra aislados de laboratorio y clínicos de VIH-1 se evaluó en líneas celulares linfoblastoides, PBMC, células monocíticas/macrofágicas primarias y linfocitos T CD4+. En células MT-4 (línea celular T linfoblástica humana) infectadas agudamente con VIH-1 IIIB, la concentración efectiva media del 50% (EC50) fue 2.4±0.4 nM, y el valor EC95 ajustado a la proteína fue 361 nM (0.162 microgramos por mL). BIC mostró actividad antiviral en PBMC activadas contra aislados clínicos de VIH-1 que representan los grupos M, N y O, incluidos los subtipos A, B, C, D, E, F y G, con un valor EC50 mediano de 0.55 nM (rango <0.05 a 1.71 nM). El valor EC50 contra un solo aislado de VIH-2 fue de 1.1 nM.
Emtricitabina: La actividad antiviral de FTC contra aislados de laboratorio y clínicos de VIH-1 se evaluó en líneas celulares linfoblastoides T, la línea celular MAGI-CCR5 y PBMC. En PBMC infectadas agudamente con subtipos A, B, C, D, E, F y G del VIH-1, el valor EC50 mediano para FTC fue de 9.5 nM (rango de 1 a 30 nM) y contra el VIH-2 fue de 7 nM.
Tenofovir Alafenamida: La actividad antiviral de TAF contra aislados de laboratorio y clínicos del VIH-1 subtipo B se evaluó en líneas celulares linfoblastoides, PBMC, células monocíticas/macrofágicas primarias y linfocitos T CD4. Los valores EC50 para TAF oscilaron entre 2.0 y 14.7 nM. TAF mostró actividad antiviral en cultivo celular contra todos los grupos de VIH-1 (M, N, O), incluidos los subtipos A, B, C, D, E, F y G (los valores EC50 oscilaron entre 0.1 y 12 nM) y actividad específica de la cepa contra el VIH-2 (los valores EC50 oscilaron entre 0.9 y 2.6 nM).
Resistencia
En cultivo celular
Bictegravir: Se han seleccionado aislados de VIH-1 con susceptibilidad reducida a BIC en cultivo celular. En una selección con BIC, surgió un pool de virus que expresaba sustituciones de aminoácidos M50I y R263K en la integrasa del VIH-1. Las sustituciones M50I, R263K y M50I+R263K, cuando se introdujeron en un virus de tipo salvaje mediante mutagénesis dirigida al sitio, conferían una reducción de 1.3-, 2.2- y 2.9 veces en la susceptibilidad a BIC, respectivamente. En una segunda selección, se detectó la aparición de sustituciones de aminoácidos T66I y S153F, y se observaron reducciones de 0.4-, 1.9- y 0.5 veces en la susceptibilidad a BIC con T66I, S153F y T66I+S153F, respectivamente. Además, las sustituciones S24G y E157K emergieron durante el proceso de selección.
Emtricitabina: Se seleccionaron aislados de VIH-1 con susceptibilidad reducida a FTC en cultivo celular y en sujetos tratados con FTC. La susceptibilidad reducida a FTC se asoció con sustituciones M184V o I en la RT del VIH-1.
Tenofovir Alafenamida: Se seleccionaron aislados de VIH-1 con susceptibilidad reducida a TAF en cultivo celular. Los aislados de VIH-1 seleccionados por TAF expresaron una sustitución K65R en la RT del VIH-1, a veces en presencia de sustituciones S68N o L429I; además, se observó una sustitución K70E en la RT del VIH-1.
En ensayos clínicos
En Sujetos Sin Historia de Tratamiento Antirretroviral: Los resultados agrupados de los análisis de resistencia genotípica se realizaron en aislados de VIH-1 emparejados de referencia y en tratamiento de sujetos que recibieron BIKTARVY en los ensayos 1489 y 1490 hasta la semana 144 de la fase de doble ciego (N=634) o la semana 96 de la fase de extensión (n=1025) [ver Estudios Clínicos (14.2)] que tenían ARN del VIH-1 mayor o igual a 200 copias/mL en el momento de la falla virológica confirmada o la interrupción temprana del fármaco del estudio. En la población final de análisis de resistencia, no surgieron sustituciones específicas de aminoácidos de manera consistente en los 11 sujetos con falla en el tratamiento con datos de resistencia genotípica evaluables y no se logró establecer una asociación con la resistencia genotípica a BIC. No se detectaron sustituciones asociadas a resistencia a NRTI emergentes del tratamiento en los 11 aislados de falla en el tratamiento evaluados. Los análisis de resistencia fenotípica de los aislados de falla encontraron cambios en la susceptibilidad a los fármacos por debajo de los límites biológicos o clínicos para BIC, FTC y TFV, en comparación con el VIH-1 de referencia de tipo salvaje.
En Sujetos Adultos con Supresión Virológica: En 2 de los ensayos de cambio, los ensayos 1844 y 1878, de sujetos infectados por VIH-1 con supresión virológica (n=572), solo un sujeto con rebote virológico en la población de análisis de resistencia tenía datos genotípicos y fenotípicos de IN, y 2 con rebote tenían datos genotípicos y fenotípicos de RT. Ningún sujeto tenía VIH-1 con resistencia genotípica o fenotípica emergente del tratamiento a BIC, FTC o TAF. En el ensayo 4030, ningún sujeto que recibió BIKTARVY tuvo resistencia fenotípica emergente del tratamiento a BIC, FTC o TAF.
En Sujetos Pediátricos con Supresión Virológica: En el ensayo 1474 [ver Estudios Clínicos (14.4)], dos de los 50 sujetos de la cohorte 1 fueron evaluados para el desarrollo de resistencia hasta la semana 48; no se detectaron sustituciones de aminoácidos conocidas por estar asociadas con resistencia a BIC, FTC o TFV. Ningún sujeto de la cohorte 2 o 3 cumplió con los criterios para los análisis de resistencia hasta la semana 24.
Resistencia Cruzada
Bictegravir: Se ha observado resistencia cruzada entre los INSTI. La susceptibilidad de BIC se probó frente a 64 aislados clínicos que expresan sustituciones asociadas a resistencia a INSTI conocidas enumeradas por IAS-USA (20 con sustituciones únicas y 44 con 2 o más sustituciones). Los aislados con una sola sustitución de resistencia a INSTI, incluidas E92Q, T97A, Y143C/R, Q148R y N155H, mostraron una susceptibilidad reducida a BIC de menos de 2 veces. Todos los aislados (n=14) con una susceptibilidad reducida a BIC de más de 2,5 veces (por encima del límite biológico para BIC) contenían sustituciones G140A/C/S y Q148H/R/K; la mayoría (64,3%, 9/14) tenía un patrón complejo de resistencia a INSTI con una sustitución adicional de resistencia a INSTI L74M, T97A o E138A/K. De esos aislados evaluados que contenían sustituciones G140A/C/S y Q148H/R/K en ausencia de sustituciones adicionales de resistencia a INSTI, el 38,5% (5/13) mostró una reducción de más de 2,5 veces. Además, los virus mutantes dirigidos al sitio con G118R (sustitución emergente del tratamiento con dolutegravir y raltegravir) y G118R+T97A tuvieron una susceptibilidad reducida a BIC de 3,4 y 2,8 veces, respectivamente.
BIC demostró una actividad antiviral equivalente con reducciones de susceptibilidad de menos de 2 veces frente a variantes del VIH-1 que expresan sustituciones asociadas con resistencia a NNRTI, NRTI y PI, en comparación con el virus de tipo salvaje.
Emtricitabina: Se ha observado resistencia cruzada entre los NRTI. Los virus resistentes a FTC con una sustitución M184V/I en la RT del VIH-1 fueron resistentes cruzados a la lamivudina. Los aislados del VIH-1 que contienen la sustitución K65R de la RT, seleccionados in vivo por abacavir, didanosina y tenofovir, demostraron una susceptibilidad reducida a la inhibición por FTC.
Tenofovir Alafenamida: Se ha observado resistencia cruzada entre los NRTI. Las sustituciones de resistencia a tenofovir K65R y K70E dan como resultado una susceptibilidad reducida a abacavir, didanosina, emtricitabina, lamivudina y tenofovir. El VIH-1 con múltiples sustituciones de análogos de timidina (M41L, D67N, K70R, L210W, T215F/Y, K219Q/E/N/R), o VIH-1 resistente a multinucleósidos con una mutación de inserción doble T69S o con un complejo de sustitución Q151M que incluye K65R, mostró una susceptibilidad reducida a TAF en cultivo celular.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
Bictegravir
BIC no fue carcinogénico en un estudio de 6 meses con ratones transgénicos rasH2 a dosis de hasta 100 mg/kg/día en machos y 300 mg/kg/día en hembras. BIC no fue carcinogénico en un estudio de 2 años con ratas a dosis de hasta 300 mg/kg/día, lo que resultó en exposiciones aproximadamente 31 veces la exposición en humanos a la dosis recomendada de BIKTARVY.
BIC no fue genotóxico en la prueba bacteriana de mutación inversa (prueba de Ames), ensayos de linfoma de ratón o micronúcleos de rata.
BIC no afectó la fertilidad, el rendimiento reproductivo o la viabilidad embrionaria en ratas macho y hembra a exposiciones 29 veces más altas (AUC) que en humanos a la dosis recomendada de BIKTARVY.
Emtricitabina
En estudios de carcinogenicidad a largo plazo de FTC, no se encontraron aumentos relacionados con el fármaco en la incidencia tumoral en ratones a dosis de hasta 750 mg por kg por día (25 veces la exposición sistémica humana a la dosis recomendada de BIKTARVY) o en ratas a dosis de hasta 600 mg por kg por día (30 veces la exposición sistémica humana a la dosis recomendada de BIKTARVY).
FTC no fue genotóxico en la prueba bacteriana de mutación inversa (prueba de Ames), ensayos de linfoma de ratón o micronúcleos de ratón.
FTC no afectó la fertilidad en ratas macho a aproximadamente 140 veces o en ratones macho y hembra a aproximadamente 60 veces exposiciones más altas (AUC) que en humanos que recibieron la dosis recomendada de BIKTARVY. La fertilidad fue normal en la descendencia de ratones expuestos diariamente desde antes del nacimiento (in útero) hasta la madurez sexual a exposiciones diarias (AUC) de aproximadamente 60 veces más altas que las exposiciones humanas a la dosis recomendada de BIKTARVY.
Tenofovir Alafenamida
Dado que TAF se convierte rápidamente en tenofovir y se observó una exposición más baja a tenofovir en ratas y ratones después de la administración de TAF en comparación con la administración de TDF, los estudios de carcinogenicidad se realizaron solo con TDF. Se llevaron a cabo estudios de carcinogenicidad oral a largo plazo de TDF en ratones y ratas a exposiciones de hasta aproximadamente 10 veces (ratones) y 4 veces (ratas) las observadas en humanos después de una dosis de 300 mg de TDF. La exposición a tenofovir en estos estudios fue aproximadamente 151 veces (ratones) y 51 veces (rata) las observadas en humanos después de la administración de la dosis diaria recomendada de BIKTARVY. A la dosis alta en ratonas, los adenomas hepáticos aumentaron a exposiciones a tenofovir aproximadamente 151 veces la exposición observada en humanos a la dosis recomendada de BIKTARVY. En ratas, el estudio fue negativo para hallazgos carcinogénicos.
TAF no fue genotóxico en la prueba bacteriana de mutación inversa (prueba de Ames), ensayos de linfoma de ratón o micronúcleos de rata.
No hubo efectos sobre la fertilidad, el rendimiento del apareamiento o el desarrollo embrionario temprano cuando se administró TAF a ratas macho a una dosis equivalente a 155 veces la dosis humana de BIKTARVY según las comparaciones de superficie corporal durante 28 días antes del apareamiento y a ratas hembra durante 14 días antes del apareamiento hasta el día 7 de gestación.
13.2 Toxicología y/o Farmacología Animal
Se observó una infiltración mínima a leve de células mononucleares en la úvea posterior en perros con gravedad similar después de la administración de TAF durante tres y nueve meses; se observó reversibilidad después de un período de recuperación de tres meses. No se observó toxicidad ocular en el perro a exposiciones sistémicas de 7 (TAF) y 14 (tenofovir) veces la exposición observada en humanos con la dosis diaria recomendada de BIKTARVY.
14 ESTUDIOS CLÍNICOS
14.1 Descripción de los Ensayos Clínicos
La eficacia y seguridad de BIKTARVY fueron evaluadas en los ensayos clínicos que se resumen en la Tabla 13.
Tabla 13 Ensayos Clínicos Realizados con BIKTARVY en Sujetos con Infección por VIH-1
Fase controlada activa, doble ciego, de 144 semanas, seguida de una fase de extensión en la que 1025 sujetos de los Ensayos Clínicos 1489 y 1490 recibieron BIKTARVY de forma abierta durante 96 semanas.
Los sujetos recibieron FTC+TAF en combinación con elvitegravir y cobicistat durante 96 semanas, seguido de una fase de extensión en la que 10 sujetos recibieron BIKTARVY durante 48 semanas.
Adultos virológicamente suprimidos‡ con ERT# que reciben hemodiálisis crónica
FTC+TAF en combinación con elvitegravir y cobicistat como combinación de dosis fija (55). En una fase de extensión del Ensayo Clínico 1825, 10 sujetos virológicamente suprimidos cambiaron a BIKTARVY.
Niños virológicamente suprimidos‡ de al menos 2 años de edad (al menos de 14 a menos de 25 kg)
BIKTARVY (22)
24
14.2 Resultados de los ensayos clínicos en adultos con VIH-1 y sin antecedentes de tratamiento antirretroviral
En el Ensayo 1489, los adultos fueron aleatorizados en una proporción 1:1 para recibir ya sea BIKTARVY (que contiene 50 mg de BIC, 200 mg de FTC y 25 mg de TAF) (N = 314) o ABC/DTG/3TC (600 mg/50 mg/300 mg) (N = 315) una vez al día. En el Ensayo 1490, los sujetos fueron aleatorizados en una proporción 1:1 para recibir ya sea BIKTARVY (N = 320) o DTG + FTC/TAF (50 mg + 200 mg/25 mg) (N = 325) una vez al día.
En el Ensayo 1489, la edad media fue de 34 años (rango 18 – 71), el 90% eran hombres, el 57% eran blancos, el 36% eran negros y el 3% eran asiáticos. El 22% de los pacientes se identificaron como hispanos / latinos. La carga viral media basal de VIH-1 en plasma fue de 4.4 log10 copias/mL (rango 1.3 – 6.5). El recuento medio basal de células CD4 + fue de 464 células por mm3 (rango 0 – 1424) y el 11% tenía recuentos de células CD4 + inferiores a 200 células por mm3. El 16% de los sujetos tenía cargas virales basales superiores a 100.000 copias por mL.
En el Ensayo 1490, la edad media fue de 37 años (rango 18 – 77), el 88% eran hombres, el 59% eran blancos, el 31% eran negros y el 3% eran asiáticos. El 25% de los pacientes se identificaron como hispanos / latinos. La carga viral media basal de VIH-1 en plasma fue de 4.4 log10 copias/mL (rango 2.3 – 6.6). El recuento medio basal de células CD4 + fue de 456 células por mm3 (rango 2 – 1636) y el 12% tenía recuentos de células CD4 + inferiores a 200 células por mm3. El 19% de los sujetos tenía cargas virales basales superiores a 100.000 copias por mL.
En ambos ensayos, los sujetos se estratificaron por la carga viral basal de VIH-1 (menor o igual a 100.000 copias por mL, mayor de 100.000 copias por mL a menor o igual a 400.000 copias por mL o mayor de 400.000 copias por mL), por el recuento de CD4 (menor de 50 células por mm3, 50 – 199 células por mm3 o mayor o igual a 200 células por mm3) y por la región (EE. UU. o fuera de EE. UU.).
Los resultados del tratamiento de los Ensayos 1489 y 1490 hasta la Semana 144 se presentan en la Tabla 14.
Tabla 14 Resultados virológicos del tratamiento aleatorizado en los Ensayos 1489 y 1490 en la Semana 144* en adultos sin antecedentes de tratamiento antirretroviral
Incluye sujetos que tuvieron ≥ 50 copias/mL en la ventana de la Semana 144; sujetos que suspendieron el tratamiento temprano debido a falta o pérdida de eficacia; sujetos que suspendieron por razones distintas a un evento adverso (EA), muerte o falta o pérdida de eficacia y en el momento de la suspensión tenían un valor viral de ≥ 50 copias/mL.
Incluye sujetos que suspendieron debido a un EA o muerte en cualquier punto en el tiempo desde el Día 1 hasta la ventana de tiempo si esto resultó en no tener datos virológicos sobre el tratamiento durante la ventana especificada.
Incluye sujetos que suspendieron por razones distintas a un EA, muerte o falta o pérdida de eficacia, por ejemplo, retiraron el consentimiento, se perdieron en el seguimiento, etc.
VIH-1 RNA < 50 copias/mL
82%
84%
82%
84%
Diferencia de tratamiento (95% IC) BIKTARVY vs. Comparador
No hay datos virológicos en la ventana de la Semana 144
18%
13%
13%
13%
Suspendió el medicamento del estudio debido a un EA o muerte‡
1%
2%
3%
3%
Suspendió el medicamento del estudio debido a otras razones y el último VIH-1 RNA disponible < 50 copias/mL§
16%
11%
11%
9%
Datos faltantes durante la ventana pero con el medicamento del estudio
1%
< 1%
0%
1%
Los resultados del tratamiento fueron similares en los subgrupos por edad, sexo, raza, carga viral basal y recuento de células CD4+ basal.
En los Ensayos 1489 y 1490, el aumento medio desde el valor basal en el recuento de células CD4+ en la semana 144 fue de 299 y 317 células por mm3 en los grupos BIKTARVY y ABC/DTG/3TC, respectivamente, y de 278 y 289 células por mm3 en los grupos BIKTARVY y DTG + FTC/TAF, respectivamente.
14.3 Resultados de los Ensayos Clínicos en Adultos con VIH-1 Virológicamente Suprimido que Cambiaron a BIKTARVY
En el Ensayo 1844, se evaluó la eficacia y seguridad de cambiar de un régimen de DTG + ABC/3TC o ABC/DTG/3TC a BIKTARVY en un ensayo aleatorizado, doble ciego de adultos infectados con VIH-1 virológicamente suprimidos (ARN de VIH-1 menor de 50 copias por mL) (N = 563, aleatorizados y dosificados). Los sujetos debían haber estado estabilmente suprimidos (ARN de VIH-1 menor de 50 copias por mL) con su régimen basal durante al menos 3 meses antes de la entrada en el ensayo y no tener antecedentes de fracaso del tratamiento. Los sujetos fueron aleatorizados en una proporción de 1:1 para cambiar a BIKTARVY (que contiene 50 mg de BIC, 200 mg de FTC y 25 mg de TAF) en el momento basal (N = 282), o permanecer en su régimen antirretroviral basal (N = 281). Los sujetos tenían una edad media de 45 años (rango 20 – 71), el 89% eran hombres, el 73% eran blancos y el 22% eran negros. El 17% de los sujetos se identificó como hispano / latino. El recuento medio basal de células CD4+ era de 723 células por mm3 (rango 124 – 2444).
En el Ensayo 1878, se evaluó la eficacia y seguridad de cambiar de ABC/3TC o FTC/TDF (200/300 mg) más ATV o DRV (administrados con cobicistat o ritonavir) a BIKTARVY (que contiene 50 mg de BIC, 200 mg de FTC y 25 mg de TAF) en un estudio aleatorizado, abierto de adultos infectados con VIH-1 virológicamente suprimidos (N = 577, aleatorizados y dosificados). Los sujetos debían haber estado estabilmente suprimidos con su régimen basal durante al menos 6 meses, no haber sido previamente tratados con ningún INSTI y no tener antecedentes de fracaso del tratamiento. Los sujetos fueron aleatorizados en una proporción de 1:1 para cambiar a BIKTARVY (N = 290) o permanecer en su régimen antirretroviral basal (N = 287). Los sujetos tenían una edad media de 46 años (rango 20 – 79), el 83% eran hombres, el 66% eran blancos y el 26% eran negros. El 19% de los sujetos se identificó como hispano / latino. El recuento medio basal de células CD4+ era de 663 células por mm3 (rango 62 – 2582). Los sujetos fueron estratificados por el régimen de tratamiento previo. En el cribado, el 15% de los sujetos estaban recibiendo ABC/3TC más ATV o DRV (administrados con cobicistat o ritonavir) y el 85% de los sujetos estaban recibiendo FTC/TDF más ATV o DRV (administrados con cobicistat o ritonavir).
Los resultados del tratamiento de los Ensayos 1844 y 1878 hasta la semana 48 se presentan en la Tabla 15.
Tabla 15 Resultados Virológicos de los Ensayos 1844 y 1878 en la Semana 48* en Adultos Virológicamente Suprimidos que Cambiaron a BIKTARVY
Incluye sujetos que tuvieron ≥ 50 copias/mL en la ventana de la semana 48; sujetos que descontinuaron temprano debido a falta o pérdida de eficacia; sujetos que descontinuaron por razones distintas a la falta o pérdida de eficacia y en el momento de la discontinuación tenían un valor viral de ≥ 50 copias/mL.
Incluye sujetos que descontinuaron por razones distintas a un AE, muerte o falta o pérdida de eficacia, por ejemplo, retiraron el consentimiento, se perdieron en el seguimiento, etc.
Sin datos virológicos en la ventana de la semana 48
5%
5%
6%
9%
Descontinuaron el fármaco del estudio debido a AE o muerte y el último ARN de VIH-1 disponible < 50 copias/mL
2%
1%
1%
1%
Descontinuaron el fármaco del estudio por otras razones y el último ARN de VIH-1 disponible < 50 copias/mL§
2%
3%
3%
7%
Datos faltantes durante la ventana pero con el fármaco del estudio
2%
1%
2%
2%
En el Ensayo 1844, los resultados del tratamiento entre los grupos de tratamiento fueron similares en los subgrupos por edad, sexo, raza y región. El cambio medio desde el punto de referencia en el recuento de CD4+ a la semana 48 fue de -31 células por mm3 en los sujetos que cambiaron a BIKTARVY y de 4 células por mm3 en los sujetos que permanecieron en ABC/DTG/3TC.
En el Ensayo 1878, los resultados del tratamiento entre los grupos de tratamiento fueron similares en los subgrupos por edad, sexo, raza y región. El cambio medio desde el punto de referencia en el recuento de CD4+ a la semana 48 fue de 25 células por mm3 en los pacientes que cambiaron a BIKTARVY y de 0 células por mm3 en los pacientes que permanecieron en su régimen de base.
En el Ensayo 4030, se evaluó la eficacia y seguridad de cambiar de DTG más ya sea FTC/TAF o FTC/TDF a BIKTARVY (que contiene 50 mg de BIC, 200 mg de FTC y 25 mg de TAF) en un estudio aleatorizado, doble ciego de adultos infectados por el VIH-1 con supresión viral. Los sujetos debían haber estado estabilmente suprimidos (ARN del VIH-1 menor de 50 copias/mL) en su régimen de base durante al menos 6 meses (si se documentó o se sospechó resistencia a NRTI) o al menos 3 meses (si no se documentó o se sospechó resistencia a NRTI) antes de la entrada al ensayo. Los sujetos fueron aleatorizados para cambiar a BIKTARVY (N = 284) o para continuar con su régimen de tratamiento previo, DTG + F/TAF (N = 281). El punto final primario fue la proporción de sujetos con ARN del VIH-1 ≥ 50 copias/mL a la semana 48. A la semana 48, la proporción de sujetos con ARN del VIH-1 ≥ 50 copias/mL fue del 0,4% (1/284) en el grupo BIKTARVY y del 1,1% (3/281) en el grupo DTG + F/TAF (diferencia -0,7% [95% CI: -2,8%, 1,0%]).
De los sujetos que recibieron BIKTARVY, 47 tenían VIH-1 con sustituciones de resistencia M184V o I preexistentes (M184M/V, M184M/I, M184V/I, M184V) en la RT del VIH-1. El ochenta y nueve por ciento (42/47) de los sujetos con M184V o I permanecieron suprimidos (ARN del VIH-1 < 50 copias/mL) y el 11% (5/47 sujetos) no tuvieron datos virológicos en el punto de tiempo de la semana 48 debido a la interrupción del fármaco del estudio.
En el Ensayo 1825, un ensayo abierto de un solo brazo, se evaluaron la eficacia, seguridad y farmacocinética de FTC y TAF (componentes de BIKTARVY) en adultos con ESRD (aclaramiento de creatinina estimado de menos de 15 mL/min) con supresión viral en hemodiálisis crónica tratados con FTC + TAF en combinación con elvitegravir y cobicistat como una tableta de combinación de dosis fija durante 96 semanas (N = 55). En una fase de extensión del Ensayo 1825, 10 sujetos con supresión viral cambiaron a BIKTARVY y todos los sujetos permanecieron con supresión viral (ARN del VIH-1 < 50 copias/mL) durante 48 semanas.
En el Ensayo 4449, se evaluó la eficacia y seguridad de cambiar de un régimen antirretroviral estable a BIKTARVY (que contiene 50 mg de BIC, 200 mg de FTC y 25 mg de TAF) en un ensayo abierto, de un solo brazo de adultos infectados por el VIH-1 con supresión viral (ARN del VIH-1 menor de 50 copias por mL) de 65 años o más (N = 86). Los sujetos tratados con BIKTARVY tenían una edad media de 70 años (rango: 65 a 80). El punto final primario fue la proporción de sujetos con ARN del VIH > 50 copias/mL a la semana 48. Ningún sujeto tuvo ARN del VIH > 50 copias/mL. El noventa y un por ciento (78/86) de los sujetos permanecieron suprimidos (ARN del VIH-1 < 50 copias/mL) a la semana 48. Ocho sujetos no tuvieron datos virológicos en el punto de tiempo de la semana 48 debido a la interrupción o datos faltantes.
14.4 Resultados de los Ensayos Clínicos en Sujetos Pediátricos con VIH-1
En el Ensayo 1474, un ensayo abierto de un solo brazo, se evaluó la eficacia, seguridad y farmacocinética de BIKTARVY en sujetos pediátricos infectados por el VIH-1 en adolescentes con supresión viral de entre 12 y menos de 18 años con un peso de al menos 35 kg (N = 50), en niños con supresión viral de entre 6 y menos de 12 años con un peso de al menos 25 kg (N = 50) y en niños con al menos 2 años de edad y un peso de al menos 14 y menos de 25 kg (N = 22).
Cohorte 1: Adolescentes con supresión viral (12 a menos de 18 años; al menos 35 kg)
Los sujetos de la cohorte 1 tratados con BIKTARVY (que contiene 50 mg de BIC, 200 mg de FTC y 25 mg de TAF) una vez al día tenían una edad media de 14 años (rango: 12 a 17) y un peso basal medio de 51,7 kg (rango: 35 a 123), el 64% eran mujeres, el 27% eran asiáticos y el 65% eran negros. Al inicio, la mediana del recuento de células CD4+ era de 750 células por mm3 (rango: 337 a 1207) y la mediana del porcentaje de CD4+ era del 33% (rango: 19% a 45%).
Después de cambiar a BIKTARVY, el 98% (49/50) de los sujetos de la cohorte 1 permanecieron suprimidos (ARN del VIH-1 < 50 copias/mL) a la semana 48. El cambio medio desde el punto de referencia en el recuento de células CD4+ a la semana 48 fue de -22 células por mm3.
Cohorte 2: Niños con supresión viral (6 a menos de 12 años; al menos 25 kg)
Los sujetos de la cohorte 2 tratados con BIKTARVY una vez al día tenían una edad media de 10 años (rango: 6 a 11) y un peso basal medio de 31,9 kg (rango: 25 a 69), el 54% eran mujeres, el 22% eran asiáticos y el 72% eran negros. Al inicio, la mediana del recuento de células CD4+ era de 898 células por mm3 (rango 390 a 1991) y la mediana del porcentaje de CD4+ era del 37% (rango: 19% a 53%).
Después de cambiar a BIKTARVY (que contiene 50 mg de BIC, 200 mg de FTC y 25 mg de TAF), el 100% (50/50) de los sujetos de la cohorte 2 permanecieron suprimidos (ARN del VIH-1 < 50 copias/mL) a la semana 24. El cambio medio desde el punto de referencia en el recuento de células CD4+ a la semana 24 fue de -24 células por mm3.
Cohorte 3: Niños con supresión viral (al menos 2 años; al menos 14 a menos de 25 kg)
Los sujetos de la cohorte 3 tratados con BIKTARVY (que contiene 30 mg de BIC, 120 mg de FTC y 15 mg de TAF) una vez al día tenían una edad media de 5 años (rango: 3 a 9) y un peso basal medio de 18,8 kg (rango: 14 a 24), el 50% eran mujeres, el 23% eran asiáticos y el 73% eran negros. Al inicio, el recuento medio de células CD4+ (DE) era de 1104 (440) y el porcentaje medio de CD4+ (DE) era del 33,4% (6,0%).
Después de cambiar a BIKTARVY, el 91% (20/22) de los sujetos de la cohorte 3 permanecieron suprimidos (ARN del VIH-1 < 50 copias/mL) a la semana 24. El ARN del VIH-1 no se recogió en la semana 24 para 2 sujetos debido a la interrupción del estudio relacionada con la pandemia de COVID-19. El cambio medio desde el punto de referencia hasta la semana 24 en el recuento de células CD4+ (DE) fue de -126 (264,2) células por mm3; y el cambio medio en el porcentaje de CD4+ (DE) desde el punto de referencia hasta la semana 24 fue del 0,2% (4,4%).
16 FORMA DE SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
Las tabletas de BIKTARVY están disponibles en frascos y blísteres:
Frasco
Las tabletas de 50 mg/200 mg/25 mg contienen 50 mg de bictegravir (BIC), 200 mg de emtricitabina (FTC) y 25 mg de tenofovir alafenamida (TAF). Estas tabletas son de color marrón púrpura, con forma de cápsula y recubiertas con película, con “GSI” grabado en relieve en un lado y “9883” en el otro lado (NDC 61958-2501-1).
Las tabletas de 30 mg/120 mg/15 mg contienen 30 mg de BIC, 120 mg de FTC y 15 mg de TAF. Estas tabletas son de color rosa, con forma de cápsula y recubiertas con película, con “GSI” grabado en relieve en un lado y “B” en el otro lado (NDC 61958-2505-1).
Las tabletas de 30 mg/120 mg/15 mg contienen 30 mg de BIC, 120 mg de FTC y 15 mg de TAF. Estas tabletas son de color rosa, con forma de cápsula y recubiertas con película, con una línea de división no funcional en un lado y “BVY” en el otro lado (NDC 61958-2506-1).
Cada frasco contiene 30 tabletas, un desecante de gel de sílice, una bobina de poliéster y se cierra con un cierre a prueba de niños. No retire el paquete desecante.
Almacene el frasco a menos de 30 °C (86 °F).
Mantenga el frasco bien cerrado.
Blíster
Las tabletas de 50 mg/200 mg/25 mg contienen 50 mg de BIC, 200 mg de FTC y 25 mg de TAF. Estas tabletas son de color marrón púrpura, con forma de cápsula y recubiertas con película, con “GSI” grabado en relieve en un lado y “9883” en el otro lado (NDC 61958-2501-3).
Cada blíster contiene 30 tabletas (4 tiras de 7 tabletas cada una y 1 tira de 2 tabletas). Los blísteres están sellados con un material de cubierta de lámina laminada a prueba de niños (pelar-empujar), y cada cavidad del blíster contiene una película desecante troquelada que está termosellada al material de cubierta de lámina.
Almacene el blíster a 25 °C (77 °F), se permiten excursiones a 15–30 °C (59–86 °F) (ver Temperatura ambiente controlada USP).
Dispense solo en los envases originales.
17 INFORMACIÓN PARA EL PACIENTE
Aconseje al paciente que lea el etiquetado del paciente aprobado por la FDA (Información para el paciente).
Exacerbación aguda post-tratamiento de la hepatitis B en pacientes con coinfección por VHB
Se han notificado exacerbaciones agudas graves de la hepatitis B en pacientes que están coinfectados con VHB y VIH-1 y que han interrumpido los productos que contienen FTC y/o TDF, y pueden ocurrir igualmente con la interrupción de BIKTARVY [ver Advertencias y precauciones (5.1)]. Aconseje al paciente que no interrumpa BIKTARVY sin informar primero a su médico.
Interacciones medicamentosas
BIKTARVY puede interactuar con ciertos medicamentos; por lo tanto, aconseje a los pacientes que informen a su médico sobre el uso de cualquier otro medicamento recetado o de venta libre o productos herbales, incluida la hierba de San Juan [ver Contraindicaciones (4) y Interacciones medicamentosas (7)].
Síndrome de reconstitución inmunitaria
Aconseje a los pacientes que informen a su médico inmediatamente de cualquier síntoma de infección, ya que en algunos pacientes con infección avanzada por VIH (SIDA), los signos y síntomas de inflamación de infecciones previas pueden ocurrir poco después de comenzar el tratamiento anti-VIH [ver Advertencias y precauciones (5.3)].
Insuficiencia renal
Aconseje a los pacientes que eviten tomar BIKTARVY con el uso concurrente o reciente de agentes nefrotóxicos. Se han notificado casos postcomercialización de insuficiencia renal, incluida la insuficiencia renal aguda [ver Advertencias y precauciones (5.4)].
Acidosis láctica y hepatomegalia grave
Se han notificado acidosis láctica y hepatomegalia grave con esteatosis, incluidos casos mortales, con el uso de medicamentos similares a BIKTARVY. Aconseje a los pacientes que deben dejar de tomar BIKTARVY si desarrollan síntomas clínicos sugestivos de acidosis láctica o hepatotoxicidad pronunciada [ver Advertencias y precauciones (5.5)].
Dosis olvidada
Informe a los pacientes que es importante tomar BIKTARVY en un horario de dosificación regular con o sin alimentos y evitar perder dosis, ya que puede provocar el desarrollo de resistencia [ver Dosificación y administración (2.2)].
División de tabletas
Aconseje a los cuidadores que, para los niños que no pueden tragar una tableta entera, la tableta se puede dividir y cada parte se toma por separado siempre que todas las partes se ingieran en aproximadamente 10 minutos [ver Dosificación y administración (2.3)].
Registro de embarazo
Informe a los pacientes que existe un registro de embarazo antirretroviral para monitorear los resultados fetales de las personas embarazadas expuestas a BIKTARVY [ver Uso en poblaciones específicas (8.1)].
Lactancia
Informe a las personas con infección por VIH-1 que los riesgos potenciales de la lactancia materna incluyen: (1) transmisión del VIH-1 a bebés VIH-1 negativos, (2) desarrollo de resistencia viral en bebés VIH-1 positivos y (3) reacciones adversas en un bebé amamantado similares a las observadas en adultos [ver Uso en poblaciones específicas (8.2)].
SECCIÓN NO CLASIFICADA DE SPL
BIKTARVY es una marca comercial de Gilead Sciences, Inc., o sus empresas relacionadas. Todas las demás marcas comerciales mencionadas en este documento son propiedad de sus respectivos dueños.
¿Cuál es la información más importante que debo saber sobre BIKTARVY?
BIKTARVY puede causar efectos secundarios graves, que incluyen:
Empeoramiento de la infección por el virus de la hepatitis B (VHB). Su proveedor de atención médica le hará una prueba para detectar la infección por VHB antes o cuando comience el tratamiento con BIKTARVY. Si tiene infección por VHB y toma BIKTARVY, su VHB puede empeorar (brote) si deja de tomar BIKTARVY. Un “brote” es cuando su infección por VHB regresa repentinamente de una manera peor que antes.
No se quede sin BIKTARVY. Vuelva a surtir su receta o hable con su proveedor de atención médica antes de que se le acabe BIKTARVY.
No deje de tomar BIKTARVY sin antes hablar con su proveedor de atención médica.
Si deja de tomar BIKTARVY, su proveedor de atención médica deberá controlar su salud con frecuencia y hacerle análisis de sangre regularmente durante varios meses para controlar su hígado, y puede darle un medicamento para tratar la hepatitis B. Informe a su proveedor de atención médica sobre cualquier síntoma nuevo o inusual que pueda tener después de dejar de tomar BIKTARVY.
BIKTARVY es un medicamento recetado que se usa sin otros medicamentos para el virus de la inmunodeficiencia humana-1 (VIH-1) para tratar la infección por VIH-1 en adultos y niños que pesan al menos 31 libras (14 kg):
que no han recibido medicamentos para el VIH-1 en el pasado, o
para reemplazar sus medicamentos actuales para el VIH-1 para las personas cuyo proveedor de atención médica determina que cumplen con ciertos requisitos.
El VIH-1 es el virus que causa el síndrome de inmunodeficiencia adquirida (SIDA). BIKTARVY contiene los medicamentos bictegravir, emtricitabina y tenofovir alafenamida. No se sabe si BIKTARVY es seguro y eficaz en niños que pesan menos de 31 libras (14 kg).
No tome BIKTARVY si también toma un medicamento que contiene:
dofetilide
rifampicina
¿Qué debo decirle a mi proveedor de atención médica antes de tomar BIKTARVY? Antes de tomar BIKTARVY, informe a su proveedor de atención médica sobre todas sus afecciones médicas, incluso si:
tiene problemas hepáticos, incluida la infección por VHB
tiene problemas renales
está embarazada o planea quedar embarazada. Informe a su proveedor de atención médica si queda embarazada durante el tratamiento con BIKTARVY. Registro de embarazo: Existe un registro de embarazo para quienes toman BIKTARVY durante el embarazo. El propósito de este registro es recopilar información sobre la salud de usted y su bebé. Hable con su proveedor de atención médica sobre cómo puede participar en este registro.
está amamantando o planea amamantar. BIKTARVY pasa a su bebé en la leche materna. Hable con su proveedor de atención médica sobre los siguientes riesgos para su bebé de amamantar durante el tratamiento con BIKTARVY:
el virus VIH-1 puede pasar a su bebé si su bebé no tiene infección por VIH-1.
el virus VIH-1 puede volverse más difícil de tratar si su bebé tiene infección por VIH-1.
su bebé puede tener efectos secundarios de BIKTARVY.
Informe a su proveedor de atención médica sobre todos los medicamentos que toma, incluidos los medicamentos recetados y de venta libre, antiácidos, laxantes, vitaminas y suplementos herbales. Algunos medicamentos pueden interactuar con BIKTARVY. Mantenga una lista de sus medicamentos y muéstresela a su proveedor de atención médica y farmacéutico cuando obtenga un medicamento nuevo.
Puede pedirle a su proveedor de atención médica o farmacéutico una lista de medicamentos que interactúan con BIKTARVY.
No empiece a tomar un medicamento nuevo sin decirle a su proveedor de atención médica. Su proveedor de atención médica puede decirle si es seguro tomar BIKTARVY con otros medicamentos.
¿Cómo debo tomar BIKTARVY?
Tome BIKTARVY exactamente como le indique su proveedor de atención médica. BIKTARVY se toma solo (no con otros medicamentos contra el VIH-1) para tratar la infección por VIH-1.
Tome BIKTARVY 1 vez al día con o sin alimentos.
Para los niños que no pueden tragar una tableta entera, la tableta se puede dividir y cada parte se puede tomar por separado siempre y cuando todas las partes se traguen en aproximadamente 10 minutos.
Si está en diálisis, tome su dosis diaria de BIKTARVY después de la diálisis.
No cambie su dosis ni deje de tomar BIKTARVY sin hablar primero con su proveedor de atención médica. Permanezca bajo el cuidado de un proveedor de atención médica durante el tratamiento con BIKTARVY.
Si toma antiácidos que contienen aluminio o magnesio, tome BIKTARVY al menos 2 horas antes o 6 horas después de tomar estos antiácidos.
Si toma suplementos o antiácidos que contienen hierro o calcio, tome BIKTARVY con alimentos al mismo tiempo que tome estos suplementos o antiácidos.
Si está embarazada y toma suplementos o antiácidos que contienen aluminio, magnesio, hierro o calcio, hable con su proveedor de atención médica sobre cómo tomar BIKTARVY junto con estos suplementos o antiácidos.
No se salte una dosis de BIKTARVY.
Si toma demasiado BIKTARVY, llame a su proveedor de atención médica o vaya inmediatamente a la sala de emergencias del hospital más cercano.
Cuando su suministro de BIKTARVY esté empezando a disminuir, obtenga más de su proveedor de atención médica o farmacia. Esto es muy importante porque la cantidad de virus en su sangre puede aumentar si se detiene el medicamento incluso por un corto tiempo. El virus puede desarrollar resistencia a BIKTARVY y ser más difícil de tratar.
¿Cuáles son los posibles efectos secundarios de BIKTARVY?
BIKTARVY puede causar efectos secundarios graves, incluyendo:
Cambios en su sistema inmunológico (Síndrome de Reconstitución Inmunológica) pueden ocurrir cuando comienza a tomar medicamentos contra el VIH-1. Su sistema inmunológico puede fortalecerse y comenzar a combatir infecciones que han estado ocultas en su cuerpo durante mucho tiempo. Informe de inmediato a su proveedor de atención médica si comienza a tener nuevos síntomas después de comenzar su medicamento contra el VIH-1.
Nuevos o peores problemas renales, incluyendo insuficiencia renal. Su proveedor de atención médica debe hacer análisis de sangre y orina para revisar sus riñones al comenzar y durante el tratamiento con BIKTARVY. Su proveedor de atención médica puede indicarle que deje de tomar BIKTARVY si desarrolla nuevos o peores problemas renales.
Demasiado ácido láctico en la sangre (acidosis láctica). Demasiado ácido láctico es una emergencia médica grave pero rara que puede llevar a la muerte. Informe de inmediato a su proveedor de atención médica si tiene estos síntomas: debilidad o estar más cansado de lo habitual, dolor muscular inusual, falta de aire o respiración rápida, dolor de estómago con náuseas y vómitos, manos y pies fríos o azulados, sentirse mareado o aturdido, o un latido cardíaco rápido o anormal.
Problemas graves en el hígado. En raras ocasiones, pueden ocurrir problemas graves en el hígado que pueden llevar a la muerte. Informe de inmediato a su proveedor de atención médica si tiene estos síntomas: la piel o la parte blanca de los ojos se vuelve amarilla, orina de color “té oscuro”, heces de color claro, pérdida del apetito durante varios días o más, náuseas o dolor en el área del estómago.
Los efectos secundarios más comunes de BIKTARVY son diarrea, náuseas y dolor de cabeza.
Estos no son todos los posibles efectos secundarios de BIKTARVY.
Llame a su médico para obtener consejos médicos sobre los efectos secundarios. Puede reportar los efectos secundarios a la FDA al 1-800-FDA-1088.
¿Cómo debo almacenar BIKTARVY?
Almacene la botella de BIKTARVY por debajo de 86°F (30°C).
Mantenga la botella bien cerrada.
BIKTARVY contiene un paquete desecante para ayudar a mantener su medicina seca (protegerla de la humedad). Mantenga el paquete desecante en la botella. No coma el paquete desecante.
Almacene el blíster de BIKTARVY a temperatura ambiente entre 68°F y 77°F (20°C y 25°C).
Mantenga BIKTARVY en su botella original o blíster.
BIKTARVY viene en un envase resistente a los niños.
Mantenga BIKTARVY y todos los medicamentos fuera del alcance de los niños.
Información general sobre el uso seguro y eficaz de BIKTARVY.
A veces, los medicamentos se recetan para fines distintos a los enumerados en un prospecto de información para el paciente. No use BIKTARVY para una condición para la que no fue prescrito. No le dé BIKTARVY a otras personas, incluso si tienen los mismos síntomas que usted. Puede hacerles daño. Si desea más información, hable con su proveedor de atención médica. Puede pedir a su proveedor de atención médica o farmacéutico información sobre BIKTARVY que esté escrita para profesionales de la salud.
¿Cuáles son los ingredientes de BIKTARVY?
Ingredientes activos: bictegravir, emtricitabina y tenofovir alafenamida.
Ingredientes inactivos: croscarmelosa sódica, estearato de magnesio y celulosa microcristalina.
Las tabletas están recubiertas con un material de recubrimiento que contiene óxido de hierro negro, óxido de hierro rojo, polietilenglicol, polivinil alcohol, talco y dióxido de titanio.
Fabricado y distribuido por: Gilead Sciences, Inc. Foster City, CA 94404
BIKTARVY es una marca comercial de Gilead Sciences, Inc., o sus empresas relacionadas. Todas las demás marcas comerciales mencionadas en este documento son propiedad de sus respectivos dueños.
Fabricante de medicamentos: Janssen Products, LP (Updated: 2024-08-09) ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓNEstos aspectos destacados no incluyen toda la información necesaria para usar ERLEADA de forma segura y eficaz. Consulte la información completa de prescripción de ERLEADA. ERLEADA ®(apalutamida) tabletas, para uso oral Aprobación inicial en…
Fabricante de medicamentos: Aurobindo Pharma Limited (Updated: 2024-10-11) Tags: DHODHEsclerosis Múltiple (EM) ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓNEstos aspectos destacados no incluyen toda la información necesaria para usar las TABLETAS DE TERIFLUNOMIDA de forma segura y eficaz. Consulte la información completa de prescripción para las TABLETAS DE…
Fabricante de medicamentos: GlaxoSmithKline Biologicals SA (Updated: 2023-05-22) Tags: 5-HT7 ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓNEstos puntos destacados no incluyen toda la información necesaria para usar SHINGRIX de manera segura y eficaz. Consulte la información de prescripción completa para SHINGRIX.SHINGRIX (vacuna recombinante contra el zóster, adyuvada), suspensión…