
Fabricante de medicamentos: Genentech, Inc. (Updated: 2024-04-30)
INFORMACIÓN DESTACADA DE LA PRESCRIPCIÓN
ALECENSA® (alectinib) cápsulas, para uso oral
Aprobación inicial en los EE. UU.: 2015
CAMBIOS IMPORTANTES RECIENTES
INDICACIONES Y USO
ALECENSA es un inhibidor de la cinasa indicado para:
- el tratamiento adyuvante en pacientes adultos después de la resección tumoral de cáncer de pulmón de células no pequeñas (NSCLC, por sus siglas en inglés) positivo para la cinasa del linfoma anaplásico (ALK) (tumores ≥ 4 cm o con nodos positivos) según lo detectado por una prueba aprobada por la FDA. (1.1)
- el tratamiento de pacientes adultos con NSCLC metastásico positivo para ALK según lo detectado por una prueba aprobada por la FDA. (1.2)
DOSIFICACIÓN Y ADMINISTRACIÓN
600 mg por vía oral dos veces al día. Administrar ALECENSA con alimentos. (2.2)
FORMAS FARMACÉUTICAS Y CONCENTRACIONES
Cápsulas: 150 mg (3)
CONTRAINDICACIONES
Ninguna. (4)
ADVERTENCIAS Y PRECAUCIONES
- Hepatotoxicidad: Monitorear las pruebas de laboratorio hepáticas cada 2 semanas durante los primeros 3 meses de tratamiento, luego una vez al mes y según esté clínicamente indicado, con pruebas más frecuentes en pacientes que desarrollen elevaciones de transaminasas y bilirrubina. En caso de elevaciones graves de ALT, AST o bilirrubina, suspender, luego reducir la dosis o discontinuar permanentemente ALECENSA. (2.4, 5.1)
- Enfermedad pulmonar intersticial (ILD)/Neumonitis: Suspender inmediatamente ALECENSA en pacientes diagnosticados con ILD/neumonitis y discontinuar permanentemente si no se han identificado otras causas potenciales de ILD/neumonitis. (2.4, 5.2)
- Insuficiencia renal: Suspender ALECENSA por insuficiencia renal grave, luego reanudar ALECENSA a dosis reducida tras la recuperación o discontinuar permanentemente (2.4, 5.3).
- Bradicardia: Monitorear regularmente la frecuencia cardíaca y la presión arterial. Si es sintomática, suspender ALECENSA, luego reducir la dosis o discontinuar permanentemente. (2.4, 5.4)
- Mialgia grave y elevación de la creatina fosfocinasa (CPK): Evaluar la CPK cada 2 semanas durante el primer mes de tratamiento y en pacientes que informen dolor muscular, sensibilidad o debilidad inexplicables. En caso de elevaciones graves de CPK, suspender, luego reanudar o reducir la dosis. (2.4, 5.5)
- Anemia hemolítica: Si se sospecha anemia hemolítica, suspender ALECENSA. Si se confirma la anemia hemolítica, considerar reanudar a una dosis reducida tras la resolución o discontinuar permanentemente. (5.6)
- Toxicidad embrionaria y fetal: ALECENSA puede causar daño fetal. Advertir a las mujeres con potencial reproductivo sobre el riesgo potencial para el feto y usar anticoncepción efectiva. (5.7, 8.1, 8.3)
REACCIONES ADVERSAS
Las reacciones adversas más comunes (incidencia ≥20%) fueron hepatotoxicidad, estreñimiento, fatiga, mialgia, edema, sarpullido y tos. (6.1)
Para reportar REACCIONES ADVERSAS SOSPECHADAS, comunicarse con Genentech al 1-888-835-2555 o con la FDA al 1-800-FDA-1088 o www.fda.gov/medwatch.
Ver 17 para INFORMACIÓN DE ASESORAMIENTO AL PACIENTE e información para pacientes aprobada por la FDA.
Revisado: 4/2024
Tabla de Contenidos
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDOS*
1 INDICACIONES Y USO
1.1 Tratamiento Adyuvante del CPNM ALK-Positivo Resecado
1.2 Tratamiento del CPNM ALK-Positivo Metastásico
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Selección del Paciente
2.2 Dosificación y Administración
2.3 Dosificación Recomendada para Insuficiencia Hepática
2.4 Modificaciones de la Dosis por Reacciones Adversas
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Hepatotoxicidad
5.2 Enfermedad Pulmonar Intersticial (EPI)/Neumonitis
5.3 Insuficiencia Renal
5.4 Bradicardia
5.5 Mialgia Grave y Elevación de Creatina Fosfoquinasa (CPK)
5.6 Anemia Hemolítica
5.7 Toxicidad Embrio-Fetal
6 REACCIONES ADVERSAS
6.1 Experiencia en Ensayos Clínicos
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.3 Mujeres y Hombres con Potencial Reproductivo
8.4 Uso Pediátrico
8.5 Uso Geriátrico
8.6 Insuficiencia Renal
8.7 Insuficiencia Hepática
10 SOBREDOSIS
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de Acción
12.2 Farmacodinámica
12.3 Farmacocinética
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
14 ESTUDIOS CLÍNICOS
14.1 Tratamiento Adyuvante del CPNM ALK-Positivo Resecado
14.2 Tratamiento del CPNM ALK-Positivo Metastásico
16 PRESENTACIÓN/ALMACENAMIENTO Y MANEJO
17 INFORMACIÓN PARA EL CONSEJERO DEL PACIENTE
- *
- No se enumeran las secciones o subsecciones omitidas de la información completa de prescripción.
1 INDICACIONES Y USO
1.1 Tratamiento adyuvante del NSCLC ALK-positivo resecado
ALECENSA está indicado como tratamiento adyuvante en pacientes adultos después de la resección tumoral de cáncer de pulmón de células no pequeñas (NSCLC) positivo para la quinasa del linfoma anaplásico (ALK) (tumores ≥ 4 cm o con ganglios positivos), según lo detectado por una prueba aprobada por la FDA [ver Dosificación y administración (2.1)].
1.2 Tratamiento del NSCLC ALK-positivo metastásico
ALECENSA está indicado para el tratamiento de pacientes adultos con NSCLC ALK-positivo metastásico según lo detectado por una prueba aprobada por la FDA [ver Dosificación y administración (2.1)].
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Selección de pacientes
Seleccione a los pacientes con tumores resecables para el tratamiento adyuvante del cáncer de pulmón de célula no pequeña (CPNCP) con ALECENSA basándose en la presencia de positividad de ALK en el tejido tumoral [véase Indicaciones y uso (1.1) y Estudios clínicos (14.1)].
Seleccione a los pacientes para el tratamiento del CPNCP metastásico con ALECENSA basándose en la presencia de positividad de ALK en el tejido tumoral o en muestras de plasma [véase Indicaciones y uso (1.2) y Estudios clínicos (14.2)]. Si no se detectan reordenamientos de ALK en una muestra de plasma, analice el tejido tumoral si es posible.
La información sobre las pruebas aprobadas por la FDA para la detección de reordenamientos de ALK en el CPNCP está disponible en http://www.fda.gov/CompanionDiagnostics.
2.2 Dosificación y administración
La información sobre la dosificación recomendada para ALECENSA se proporciona en la Tabla 1.
| Indicación | Dosificación recomendada de ALECENSA | Duración |
|---|---|---|
| Tratamiento adyuvante del CPNCP resecado | 600 mg por vía oral dos veces al día con alimentos [véase Farmacología clínica (12.3)] |
Durante un total de 2 años o hasta la reaparición de la enfermedad o toxicidad inaceptable |
| CPNCP metastásico | Hasta progresión de la enfermedad o toxicidad inaceptable | |
|
||
2.3 Dosificación recomendada para insuficiencia hepática
La dosis recomendada de ALECENSA en pacientes con insuficiencia hepática grave (Child-Pugh C) es de 450 mg por vía oral dos veces al día [véase Uso en poblaciones específicas (8.7) y Farmacología clínica (12.3)].
2.4 Modificaciones de dosis por reacciones adversas
El programa de reducción de dosis para ALECENSA se proporciona en la Tabla 2.
| Programa de reducción de dosis | Nivel de dosis |
|---|---|
| Dosis inicial | 600 mg por vía oral dos veces al día |
| Primera reducción de dosis | 450 mg por vía oral dos veces al día |
| Segunda reducción de dosis | 300 mg por vía oral dos veces al día |
Suspenda si los pacientes no pueden tolerar la dosis de 300 mg dos veces al día.
Las recomendaciones para las modificaciones de dosis de ALECENSA en caso de reacciones adversas se proporcionan en la Tabla 3.
| Criterios* | Modificación de dosis de ALECENSA |
|---|---|
| Elevación de ALT o AST mayor de 5 veces el límite superior de lo normal (ULN) con bilirrubina total menor o igual a 2 veces ULN | Suspender temporalmente hasta recuperación a los niveles basales o a menor o igual a 3 veces ULN, luego reanudar con dosis reducida según la Tabla 2. |
| Elevación de ALT o AST mayor de 3 veces ULN con elevación de bilirrubina total mayor de 2 veces ULN en ausencia de colestasis o hemólisis | Suspender permanentemente ALECENSA. |
| Elevación de bilirrubina total mayor de 3 veces ULN | Suspender temporalmente hasta recuperación a los niveles basales o a menor o igual a 1.5 veces ULN, luego reanudar con dosis reducida según la Tabla 2. |
| Cualquier grado de enfermedad pulmonar intersticial (ILD)/neumonitis relacionada con el tratamiento | Suspender permanentemente ALECENSA. |
| Insuficiencia renal grado 3 | Suspender temporalmente hasta que la creatinina sérica se recupere a menor o igual a 1.5 veces ULN, luego reanudar con dosis reducida. |
| Insuficiencia renal grado 4 | Suspender permanentemente ALECENSA. |
| Bradicardia sintomática | Suspender ALECENSA hasta recuperación a bradicardia asintomática o a una frecuencia cardíaca de 60 lpm o superior. Si se identifica un medicamento concomitante contribuyente y se suspende, o se ajusta su dosis, reanudar ALECENSA a la dosis previa tras recuperación a bradicardia asintomática o a una frecuencia cardíaca de 60 lpm o superior. Si no se identifica un medicamento concomitante contribuyente, o si los medicamentos concomitantes contribuyentes no se suspenden o no se modifica su dosis, reanudar ALECENSA con dosis reducida (ver Tabla 2) tras recuperación a bradicardia asintomática o a una frecuencia cardíaca de 60 lpm o superior. |
| Bradicardia† (consecuencias potencialmente mortales, intervención urgente indicada) | Suspender permanentemente ALECENSA si no se identifica un medicamento concomitante contribuyente. Si se identifica un medicamento concomitante contribuyente y se suspende, o se ajusta su dosis, reanudar ALECENSA con dosis reducida (ver Tabla 2) tras recuperación a bradicardia asintomática o a una frecuencia cardíaca de 60 lpm o superior, con monitorización frecuente según indicación clínica. Suspender permanentemente ALECENSA en caso de recurrencia. |
| Elevación de CPK mayor de 5 veces ULN | Suspender temporalmente hasta recuperación a los niveles basales o a menor o igual a 2.5 veces ULN, luego reanudar con la misma dosis. |
| Elevación de CPK mayor de 10 veces ULN o segunda ocurrencia de elevación de CPK mayor de 5 veces ULN | Suspender temporalmente hasta recuperación a los niveles basales o a menor o igual a 2.5 veces ULN, luego reanudar con dosis reducida según la Tabla 2. |
| Anemia hemolítica | Suspender ALECENSA si se sospecha anemia hemolítica. Tras resolución, reanudar con dosis reducida o suspender permanentemente. |
3 FORMAS POSOLÓGICAS Y CONCENTRACIONES
Cápsulas duras de 150 mg, blancas, con “ALE” impreso en tinta negra en la tapa y “150 mg” impreso en tinta negra en el cuerpo.
4 CONTRAINDICACIONES
Ninguna.
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Hepatotoxicidad
Se produjo hepatotoxicidad grave, incluida la lesión hepática inducida por medicamentos, en pacientes tratados con ALECENSA.
En la población agrupada de seguridad [ver Reacciones adversas (6.1)] de pacientes que recibieron ALECENSA, la hepatotoxicidad ocurrió en el 41% de los pacientes y la incidencia de hepatotoxicidad de Grado ≥ 3 fue del 8%. En el estudio ALINA, la hepatotoxicidad ocurrió en el 61% de los pacientes tratados con ALECENSA y la incidencia de hepatotoxicidad de Grado ≥ 3 fue del 4,7%. La mayoría (72% de 136 pacientes) de las elevaciones de transaminasas ocurrieron durante los primeros 3 meses de tratamiento. La interrupción del tratamiento debido a hepatotoxicidad ocurrió en el 3,6% de los pacientes que recibieron ALECENSA en la población agrupada de seguridad y en el 1,6% de los pacientes tratados en el estudio ALINA.
En la población agrupada de seguridad, elevaciones concurrentes de ALT o AST superiores o iguales a 3 veces el límite superior normal (LSN) y bilirrubina total superior o igual a 2 veces el LSN, con fosfatasa alcalina normal, ocurrieron en menos del 1% de los pacientes tratados con ALECENSA. Tres pacientes con elevaciones de AST/ALT de Grados 3-4 tuvieron lesión hepática inducida por medicamentos (documentada por biopsia hepática en dos casos).
Monitorear las pruebas de función hepática, incluyendo ALT, AST y bilirrubina total, cada 2 semanas durante los primeros 3 meses de tratamiento, luego una vez al mes y según lo indicado clínicamente, con pruebas más frecuentes en pacientes que desarrollen elevaciones de transaminasas y bilirrubina. Según la gravedad de la reacción adversa al medicamento, suspender ALECENSA y reanudar con una dosis reducida o descontinuar permanentemente ALECENSA como se describe en Tabla 3 [ver Dosificación y Administración (2.4)].
5.2 Enfermedad pulmonar intersticial (EPI)/Neumonitis
La EPI/neumonitis ocurrió en pacientes tratados con ALECENSA.
En la población agrupada de seguridad [ver Reacciones adversas (6.1)], la EPI/neumonitis ocurrió en el 1,3% de los pacientes tratados con ALECENSA, con un 0,4% de pacientes con EPI/neumonitis de Grado 3.
Cinco pacientes (0,9%) en la población agrupada de seguridad discontinuaron ALECENSA debido a EPI/neumonitis. El tiempo medio hasta la aparición de EPI/neumonitis de Grado 3 o superior fue de 2,1 meses (rango: 0,6 meses a 3,6 meses).
Investigar de inmediato la EPI/neumonitis en cualquier paciente que presente un empeoramiento de los síntomas respiratorios indicativos de EPI/neumonitis (p. ej., disnea, tos y fiebre). Suspender inmediatamente el tratamiento con ALECENSA en pacientes diagnosticados con EPI/neumonitis y discontinuar permanentemente ALECENSA si no se han identificado otras causas potenciales de EPI/neumonitis [ver Dosificación y Administración (2.4) y Reacciones adversas (6)].
5.3 Insuficiencia renal
Se produjo insuficiencia renal, incluidos casos fatales, en pacientes tratados con ALECENSA.
En la población agrupada de seguridad [ver Reacciones adversas (6.1)], la insuficiencia renal ocurrió en el 12% de los pacientes tratados con ALECENSA, incluido el Grado ≥ 3 en el 1,7% de los pacientes, de los cuales el 0,4% fueron eventos fatales. El tiempo medio hasta la insuficiencia renal de Grado ≥ 3 fue de 3,7 meses (rango de 0,5 a 31,8 meses). Se requirieron modificaciones de la dosis por insuficiencia renal en el 2,4% de los pacientes.
Discontinuar permanentemente ALECENSA para toxicidad renal de Grado 4. Suspender ALECENSA para toxicidad renal de Grado 3 hasta la recuperación a menos o igual a 1,5 veces el LSN, luego reanudar con dosis reducida [ver Dosificación y Administración (2.4)].
5.4 Bradicardia
Ocurrió bradicardia sintomática en pacientes tratados con ALECENSA.
En la población agrupada de seguridad [ver Reacciones adversas (6.1)], la bradicardia ocurrió en el 11% de los pacientes tratados con ALECENSA. El veinte por ciento de 521 pacientes tratados con ALECENSA, a quienes se les realizaron electrocardiogramas (ECG) en serie, tuvieron frecuencias cardíacas posdesarrolladas de menos de 50 latidos por minuto (lpm).
Monitorear regularmente la frecuencia cardíaca y la presión arterial. Para la bradicardia asintomática, no se requiere modificación de la dosis. Para la bradicardia sintomática que no ponga en riesgo la vida, suspender ALECENSA hasta que se recupere a bradicardia asintomática o a una frecuencia cardíaca ≥ 60 lpm y evaluar los medicamentos concomitantes conocidos por causar bradicardia, así como los medicamentos antihipertensivos. Si la bradicardia es atribuible a un medicamento concomitante, reanudar ALECENSA con una dosis reducida (ver Tabla 2) luego de recuperarse a bradicardia asintomática o a una frecuencia cardíaca de ≥ 60 lpm, con monitoreo frecuente según lo indicado clínicamente.
Discontinuar permanentemente ALECENSA en casos de bradicardia potencialmente mortal si no se identifica ningún medicamento concomitante contribuyente [ver Dosificación y Administración (2.4)]. Discontinuar permanentemente ALECENSA por reaparición de bradicardia potencialmente mortal.
5.5 Mialgia grave y elevación de creatina fosfoquinasa (CPK)
Ocurrió mialgia grave y elevación de creatina fosfoquinasa (CPK) en pacientes tratados con ALECENSA.
En la población agrupada de seguridad [ver Reacciones adversas (6.1)], la mialgia (incluyendo reacciones musculares y musculoesqueléticas) ocurrió en el 31% de los pacientes tratados con ALECENSA, incluido el Grado ≥ 3 en el 0,8% de los pacientes. Se requirieron modificaciones de dosis por eventos de mialgia en el 2,1% de los pacientes.
En la población agrupada de seguridad, de los 491 pacientes con datos analíticos de CPK disponibles, la elevación de CPK ocurrió en el 56% de los pacientes tratados con ALECENSA, incluido el 6% de Grado ≥ 3. El tiempo medio hasta la elevación de CPK de Grado ≥ 3 fue de 15 días (rango intercuartílico – 15–337 días). Se produjeron modificaciones de dosis por elevación de CPK en el 5% de los pacientes.
En el estudio ALINA, la elevación de CPK ocurrió en el 77% de 128 pacientes con datos analíticos de CPK, incluidas elevaciones de Grado ≥ 3 en el 6%.
Aconsejar a los pacientes que reporten cualquier dolor muscular, sensibilidad o debilidad inexplicables. Evaluar los niveles de CPK cada 2 semanas durante el primer mes de tratamiento y según lo indicado clínicamente en pacientes que reporten síntomas. Según la gravedad de la elevación de CPK, suspender ALECENSA, luego reanudar o reducir la dosis [ver Dosificación y Administración (2.4)].
5.6 Anemia hemolítica
Ocurrió anemia hemolítica en pacientes tratados con ALECENSA.
La anemia hemolítica fue inicialmente reportada con ALECENSA en el entorno posterior a la comercialización, incluyendo casos asociados con un resultado negativo de la prueba de antiglobulina directa (DAT). Las evaluaciones para la determinación de anemia hemolítica fueron posteriormente recolectadas en el estudio ALINA, donde se observó anemia hemolítica en el 3.1% de los pacientes tratados con ALECENSA. Si se sospecha anemia hemolítica, suspenda ALECENSA e inicie las pruebas de laboratorio apropiadas. Si se confirma anemia hemolítica, considere reanudar con una dosis reducida tras la resolución o suspender permanentemente ALECENSA [ver Dosificación y Administración (2.4)].
5.7 Toxicidad Embrio-Fetal
Basado en hallazgos de estudios en animales y su mecanismo de acción, ALECENSA puede causar daño fetal cuando se administra a mujeres embarazadas. La administración oral de alectinib a ratas y conejas embarazadas durante el período de organogénesis resultó en toxicidad embrio-fetal y aborto en dosis tóxicas maternas con exposiciones aproximadamente 2.7 veces las observadas en humanos con alectinib 600 mg dos veces al día. Advierta a las mujeres embarazadas y mujeres con potencial reproductivo sobre el riesgo potencial para el feto.
Aconseje a las mujeres con potencial reproductivo que utilicen anticonceptivos efectivos durante el tratamiento con ALECENSA y durante 5 semanas después de la última dosis [ver Uso en Poblaciones Específicas (8.1 y 8.3) y Farmacología Clínica (12.1)].
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas se discuten con más detalle en otras secciones de la etiqueta:
- Hepatotoxicidad [ver Advertencias y precauciones (5.1)]
- Enfermedad pulmonar intersticial (ILD)/Neumonitis [ver Advertencias y precauciones (5.2)]
- Insuficiencia renal [ver Advertencias y precauciones (5.3)]
- Bradicardia [ver Advertencias y precauciones (5.4)]
- Mialgia severa y elevación de creatina fosfoquinasa (CPK) [ver Advertencias y precauciones (5.5)]
- Anemia hemolítica [ver Advertencias y precauciones (5.6)]
- Toxicidad embriofetal [ver Advertencias y precauciones (5.7)]
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se realizan bajo condiciones muy variadas, las tasas de reacciones adversas observadas en los ensayos clínicos de un medicamento no pueden compararse directamente con las tasas en los ensayos clínicos de otro medicamento y es posible que no reflejen las tasas observadas en la práctica.
La población de seguridad agrupada descrita en las ADVERTENCIAS Y PRECAUCIONES refleja la exposición a ALECENSA como agente único a 600 mg por vía oral dos veces al día en 533 pacientes en los estudios NP28761, NP28673, ALEX y ALINA [ver Estudios clínicos (14)]. Entre los 533 pacientes que recibieron ALECENSA, el 75% estuvo expuesto durante 6 meses o más y el 64% estuvo expuesto durante más de un año. En esta población de seguridad agrupada, las reacciones adversas más comunes (≥ 20%) fueron hepatotoxicidad (41%), estreñimiento (39%), fatiga (36%), mialgia (31%), edema (29%), erupción cutánea (23%) y tos (21%). Las anormalidades analíticas de Grado 3 o 4 más comunes (≥ 2%) fueron aumento de CPK (6%), disminución de hemoglobina (4,4%), aumento de ALT (4,2%), aumento de bilirrubina (4,0%) y aumento de AST (3,4%).
Tratamiento adyuvante de CPCNP ALK-positivo resecado
La seguridad de ALECENSA se evaluó en ALINA, un ensayo multicéntrico, abierto, aleatorizado para el tratamiento adyuvante de pacientes con CPCNP ALK-positivo resecado [ver Estudios clínicos (14.1)]. En el momento del análisis de SLP, la duración media de exposición fue de 23,9 meses para ALECENSA y 2,1 meses para la quimioterapia a base de platino.
Las reacciones adversas graves ocurrieron en el 13% de los pacientes tratados con ALECENSA; las reacciones adversas graves más frecuentes (≥ 1%) fueron neumonía (3,9%), apendicitis (3,1%) e infarto agudo de miocardio (1,6%). La interrupción permanente de ALECENSA debido a un evento adverso ocurrió en el 5% de los pacientes; las reacciones adversas más frecuentes (≥ 1%) que llevaron a la interrupción del tratamiento fueron neumonitis y hepatotoxicidad.
Las interrupciones de dosis de ALECENSA debido a una reacción adversa ocurrieron en el 27% de los pacientes. Las reacciones adversas que requirieron interrupción de la dosis en ≥ 2% de los pacientes incluyeron hepatotoxicidad, aumento de CPK en sangre, COVID-19, mialgia, dolor abdominal y neumonía.
Las reducciones de dosis de ALECENSA debido a una reacción adversa ocurrieron en el 26% de los pacientes. Las reacciones adversas que requirieron reducciones de dosis en ≥ 2% de los pacientes incluyeron hepatotoxicidad, aumento de CPK en sangre, erupción cutánea, bradicardia y mialgia.
Tabla 4 y 5 resumen las reacciones adversas y anormalidades analíticas comunes observadas en ALINA.
| Reacción adversa | ALECENSA N=128 |
Quimioterapia N=120 |
||
|---|---|---|---|---|
| Todos los grados (%) | Grados 3-4 (%) | Todos los grados (%) | Grados 3-4 (%) | |
| Basado en NCI CTCAE v5.0 | ||||
|
||||
| Reacción adversa | ALECENSA | Crizotinib | ||
|---|---|---|---|---|
| Todos los Grados (%) | Grados ≥3 (%) | Todos los Grados (%) | Grados ≥3 (%) | |
| Trastornos del sistema hepatobiliar | ||||
| Hepatotoxicidad* | 61 | 4.7† | 13 | 0 |
| Trastornos gastrointestinales | ||||
| Estreñimiento | 42 | 0.8† | 25 | 0.8† |
| Dolor abdominal‡ | 13 | 0 | 10 | 1.7† |
| Diarrea§ | 13 | 0.8† | 9 | 1.7† |
| Musculoesquelético | ||||
| Mialgia¶ | 34 | 0.8† | 1.7 | 0 |
| Infecciones e infestaciones | ||||
| COVID-19 | 29 | 0 | 0.8 | 0 |
| Trastornos generales y alteraciones en el lugar de administración | ||||
| Fatiga# | 25 | 0.8† | 28 | 4.2† |
| EdemaÞ | 16 | 0 | 1.7 | 0 |
| Trastornos de la piel y del tejido subcutáneo | ||||
| Erupciónß | 23 | 1.6† | 10 | 0 |
| Trastornos del sistema respiratorio | ||||
| Tosà | 20 | 0.8† | 3.3 | 0 |
| Disneaè | 13 | 0.8† | 2.5 | 0 |
| Renal | ||||
| Insuficiencia renalð | 16 | 0.8† | 9 | 0 |
| Trastornos del sistema nervioso | ||||
| Disgeusiaø | 13 | 0 | 3.3 | 0 |
| Cefalea | 11 | 0 | 7 | 0 |
| Exploraciones complementarias | ||||
| Aumento de peso | 13 | 0.8† | 0.8 | 0 |
| Trastornos cardiacos | ||||
| Bradicardiaý | 12 | 0 | 0 | 0 |
Reacciones adversas clínicamente significativas en < 10% de los pacientes que recibieron ALECENSA en ALINA: náuseas (8%), vómitos (7%), trastornos de la visión (4,7%; incluye visión borrosa, agudeza visual reducida y fotopsia), estomatitis (4,7%; incluye estomatitis y úlcera bucal), reacción de fotosensibilidad (3,9%) y neumonitis (2,3%).
| Parámetro | ALECENSA N=128 |
Quimioterapia N=120 |
||
|---|---|---|---|---|
| Todos los grados (%) | Grados 3–4 (%) | Todos los grados (%) | Grados 3–4 (%) | |
| Basado en NCI CTCAE v5.0 | ||||
|
||||
| Química | ||||
| Aumento de CPK | 77 | 8 | 8 | 1.7* |
| Aumento de AST | 75 | 0.8* | 25 | 0 |
| Aumento de bilirrubina | 68 | 2.3* | 4.2 | 0 |
| Aumento de fosfatasa alcalina | 64 | 0 | 14 | 0 |
| Aumento de ALT | 57 | 2.3* | 28 | 0 |
| Aumento de creatinina | 41 | 0 | 23 | 0 |
| Aumento de ácido úrico | 30 | 0 | 19 | 0 |
| Hematología | ||||
| Disminución de hemoglobina | 69 | 0 | 67 | 0.8* |
NSCLC metastásico ALK-positivo previamente no tratado
La seguridad de ALECENSA se evaluó en 152 pacientes con NSCLC ALK-positivo en el estudio ALEX. La duración mediana de exposición a ALECENSA fue de 17,9 meses. Las características de los pacientes de la población del estudio ALEX (n=303) fueron: mediana de edad 56 años, edad menor de 65 (77%), mujeres (56%), raza caucásica (50%), asiáticos (46%), histología de adenocarcinoma (92%), nunca fumadores (63%) y estado funcional ECOG 0 o 1 (93%).
Se produjeron reacciones adversas graves en el 28% de los pacientes tratados con ALECENSA; las reacciones adversas graves notificadas en el 2% o más de los pacientes tratados con ALECENSA fueron neumonía (4,6%) y alteración renal (3,9%). Se notificaron eventos adversos de grado ≥3 para el 41% de los pacientes en el grupo de ALECENSA. Ocurrieron reacciones adversas fatales en el 3,3% de los pacientes tratados con ALECENSA; estos fueron insuficiencia renal (2 pacientes), muerte súbita, paro cardíaco y neumonía (1 paciente cada uno). La interrupción permanente de ALECENSA debido a reacciones adversas se produjo en el 11% de los pacientes. Las reacciones adversas a medicamentos que llevaron a la interrupción de ALECENSA en el 1% o más de los pacientes fueron insuficiencia renal (2,0%), hiperbilirrubinemia (1,3%), aumento de ALT (1,3%) y aumento de AST (1,3%). Las interrupciones de la dosis de ALECENSA debido a una reacción adversa ocurrieron en el 20% de los pacientes. Las reacciones adversas que requirieron interrupción de la dosis en >2% de los pacientes incluyeron aumento de ALT y neumonía. Las reducciones de dosis de ALECENSA debido a una reacción adversa ocurrieron en el 17% de los pacientes. Las reacciones adversas que requirieron reducciones de dosis en >2% de los pacientes incluyeron hiperbilirrubinemia, aumento de AST y aumento de ALT.
Las tablas 6 y 7 resumen las reacciones adversas comunes y las anomalías de laboratorio observadas en ALEX.
| Reacción adversa | ALECENSA N=152 |
Crizotinib N=151 |
||
|---|---|---|---|---|
| Todos los grados (%) | Grados 3-4 (%) | Todos los grados (%) | Grados 3-4 (%) | |
| NCI CTCAE = Criterios Terminológicos Comunes para Eventos Adversos del Instituto Nacional del Cáncer; MedDRA = Diccionario Médico para Actividades Regulatorias; SOC = Sistema de Clasificación de Órganos. | ||||
|
||||
| Gastrointestinal | ||||
| Estreñimiento | 34 | 0 | 33 | 0 |
| Náuseas | 14 | 0,7 | 48 | 3,3 |
| Diarrea | 12 | 0 | 45 | 2,0 |
| General | ||||
| Fatiga* | 26 | 1,3 | 23 | 0,7 |
| Edema† | 22 | 0,7 | 34 | 0,7 |
| Musculoesquelético | ||||
| Mialgia‡ | 23 | 0 | 4,0 | 0 |
| Piel | ||||
| Erupción§ | 15 | 0,7 | 13 | 0 |
| Cardíaco | ||||
| Bradicardia¶ | 11 | 0 | 15 | 0 |
| Renal | ||||
| Insuficiencia renal# | 12 | 3.9Þ | 0 | 0 |
Las siguientes reacciones adversas a medicamentos clínicamente significativas adicionales se observaron en pacientes tratados con ALECENSA: aumento de peso (9,9%), vómitos (7%), reacción de fotosensibilidad (5,3%), trastornos de la visión (4,6%; incluye visión borrosa, deterioro visual, cuerpos flotantes en el vítreo, disminución de la agudeza visual y diplopía), estomatitis (3,3%), disgeusia (3,3%; incluye hipogeusia), enfermedad pulmonar intersticial (1,3%) y lesión hepática inducida por fármacos (1,3%).
| Parámetro | ALECENSA N=152 |
Crizotinib N=151 |
||
|---|---|---|---|---|
| Todos los grados (%) | Grados 3–4 (%) | Todos los grados (%) | Grados 3–4 (%) | |
| Nota: Basado en los Criterios Terminológicos Comunes para Eventos Adversos del Instituto Nacional del Cáncer v4.03. Excluye pacientes sin evaluaciones de laboratorio posteriores a la línea de base. |
||||
|
||||
| Química | ||||
| Hiperbilirrubinemia* | 54 | 5 | 4.7 | 0 |
| Aumento de AST† | 50 | 6 | 56 | 11 |
| Aumento de fosfatasa alcalina‡ | 50 | 0 | 44 | 0 |
| Aumento de ALT‡ | 40 | 6 | 62 | 16 |
| Aumento de creatinina‡,§ | 38 | 4.1 | 23 | 0.7 |
| Aumento de CPK¶ | 37 | 2.8 | 52 | 1.4 |
| Hipocalcemia* | 29 | 0 | 61 | 1.4 |
| Hiperglucemia# | 22 | 2.2 | 19 | 2.3 |
| HiponatremiaÞ | 18 | 6 | 20 | 4.1 |
| Hipopotasemia‡ | 17 | 2 | 12 | 0.7 |
| Hipoalbuminemiaß | 14 | 0 | 57 | 3.4 |
| Hiperpotasemia‡ | 12 | 1.4 | 16 | 1.4 |
| Hipofosfatemiaà | 9 | 1.4 | 25 | 2.7 |
| Aumento de gamma glutamil transferasaè | 7 | 0.7 | 39 | 4.1 |
| Hematología | ||||
| Anemia‡ | 62 | 7 | 36 | 0.7 |
| Linfopenia* | 14 | 1.4 | 34 | 4.1 |
| Neutropenia‡ | 14 | 0 | 36 | 7 |
Metastásico ALK-Positivo CPCNP Previamente Tratado con Crizotinib
La seguridad de ALECENSA fue evaluada en 253 pacientes con cáncer de pulmón de célula no pequeña (CPCNP) ALK-positivo tratados con ALECENSA en dos ensayos clínicos, los Estudios NP28761 y NP28673. La duración media de la exposición a ALECENSA fue de 9.3 meses. Ciento sesenta y nueve pacientes (67%) fueron expuestos a ALECENSA por más de 6 meses, y 100 pacientes (40%) por más de un año. Las características de la población fueron: mediana de edad 53 años, edad menor de 65 (86%), sexo femenino (55%), raza blanca (74%), asiática (18%), histología de adenocarcinoma de CPCNP (96%), nunca o ex fumador (98%), Estado de Rendimiento ECOG (ER) 0 o 1 (91%), y tratamiento previo con quimioterapia (78%).
Las reacciones adversas graves ocurrieron en el 19% de los pacientes; las reacciones adversas graves más frecuentemente reportadas fueron embolia pulmonar (1.2%), disnea (1.2%), e hiperbilirrubinemia (1.2%). Las reacciones adversas fatales ocurrieron en el 2.8% de los pacientes e incluyeron hemorragia (0.8%), perforación intestinal (0.4%), disnea (0.4%), embolia pulmonar (0.4%), y endocarditis (0.4%). La interrupción permanente de ALECENSA debido a reacciones adversas ocurrió en el 6% de los pacientes. Las reacciones adversas más frecuentes que llevaron a la interrupción permanente fueron hiperbilirrubinemia (1.6%), aumento de los niveles de ALT (1.6%), y aumento de los niveles de AST (1.2%). En general, el 23% de los pacientes que iniciaron el tratamiento con la dosis recomendada requirieron al menos una reducción de dosis. El tiempo medio hasta la primera reducción de dosis fue de 48 días. Las reacciones adversas más frecuentes que llevaron a reducciones o interrupciones de la dosis fueron elevaciones en bilirrubina (6%), CPK (4.3%), ALT (4.0%), AST (2.8%), y vómitos (2.8%).
Las Tablas 8 y 9 resumen las reacciones adversas comunes y las anormalidades de laboratorio observadas en los Estudios NP28761 y NP28673.
| Reacciones Adversas | ALECENSA N=253 |
|
|---|---|---|
| Todos los Grados (%) | Grados 3–4 (%)* | |
|
||
| Fatiga† | 41 | 1.2 |
| Estreñimiento | 34 | 0 |
| Edema‡ | 30 | 0.8 |
| Mialgia§ | 29 | 1.2 |
| Tos | 19 | 0 |
| Erupción¶ | 18 | 0.4 |
| Náuseas | 18 | 0 |
| Cefalea | 17 | 0.8 |
| Diarrea | 16 | 1.2 |
| Disnea | 16 | 3.6# |
| Dolor de espalda | 12 | 0 |
| Vómitos | 12 | 0.4 |
| Aumento de peso | 11 | 0.4 |
| Trastorno visualÞ | 10 | 0 |
Una reacción adversa al medicamento clínicamente significativa adicional fue la fotosensibilidad, que ocurrió en el 9,9% de los pacientes expuestos a ALECENSA en los Estudios NP28761 y NP28673. Se aconsejó a los pacientes que evitaran la exposición al sol y que usaran protector solar de amplio espectro. La incidencia de fotosensibilidad de Grado 2 fue del 0,4%; el resto de los eventos fueron de Grado 1 en gravedad.
| Parámetro | ALECENSA N=250 | |
|---|---|---|
| Todos los Grados (%) | Grados 3–4 (%)* | |
|
||
| Química | ||
| Aumento de AST | 51 | 3.6 |
| Aumento de fosfatasa alcalina | 47 | 1.2 |
| Aumento de CPK† | 43 | 4.6 |
| Hiperbilirrubinemia | 39 | 2.4 |
| Hiperglucemia‡ | 36 | 2.0 |
| Aumento de ALT | 34 | 4.8 |
| Hipocalcemia | 32 | 0.4 |
| Hipopotasemia | 29 | 4.0 |
| Aumento de creatinina§ | 28 | 0 |
| Hipofosfatemia | 21 | 2.8 |
| Hiponatremia | 20 | 2.0 |
| Hematología | ||
| Anemia | 56 | 2.0 |
| Linfopenia¶ | 22 | 4.6 |
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de riesgos
Con base en los hallazgos de estudios en animales y su mecanismo de acción, ALECENSA puede causar daño fetal cuando se administra a una mujer embarazada [ver Farmacología clínica (12.1)]. No hay datos disponibles sobre el uso de ALECENSA en mujeres embarazadas.
La administración de alectinib a ratas y conejas preñadas por vía de sonda oral durante el período de organogénesis resultó en toxicidad embrionaria y fetal y aborto en dosis tóxicas para la madre con exposiciones aproximadamente 2.7 veces mayores que las observadas en humanos tratados con alectinib a 600 mg dos veces al día (ver Datos). Advertir a las mujeres embarazadas sobre el riesgo potencial para el feto.
En la población general de los EE.UU., el riesgo estimado de defectos de nacimiento mayores y aborto espontáneo en embarazos clínicamente reconocidos es del 2% al 4% y del 15% al 20%, respectivamente.
Datos
Datos de animales
En un estudio preliminar en conejas de desarrollo embrionario y fetal, la administración de alectinib por sonda oral durante el período de organogénesis resultó en aborto o muerte embrionaria y fetal completa a una dosis tóxica para la madre de 27 mg/kg/día (aproximadamente 2.9 veces el área estimada bajo la curva (AUC0-24h,ss) en humanos tratados con alectinib 600 mg dos veces al día) en tres de seis conejas preñadas. Las tres conejas restantes en este grupo tenían pocos fetos vivos, disminución del peso fetal y placentario, y arteria subclavia retroesofágica. En un estudio preliminar de desarrollo embrionario y fetal en ratas, la administración de alectinib durante la organogénesis resultó en pérdida total de la camada en todas las ratas preñadas a 27 mg/kg/día (aproximadamente 4.5 veces la AUC0-24h,ss estimada en humanos tratados con alectinib 600 mg dos veces al día). Las dosis mayores o iguales a 9 mg/kg/día (aproximadamente 2.7 veces la AUC0-24h,ss estimada en humanos tratados con alectinib 600 mg dos veces al día) resultaron en toxicidad materna, así como toxicidades en el desarrollo que incluyen disminución del peso fetal, uréter dilatado, cordón tímico, ventrículo pequeño y pared ventricular delgada, y número reducido de vértebras sacras y caudales.
8.2 Lactancia
Resumen de riesgos
No hay datos sobre la presencia de alectinib o sus metabolitos en la leche materna humana, los efectos de alectinib sobre el niño lactante o sus efectos sobre la producción de leche. Debido al potencial de reacciones adversas graves en niños lactantes por alectinib, se debe advertir a una mujer lactante que no amamante durante el tratamiento con ALECENSA y hasta 1 semana después de la última dosis.
8.3 Mujeres y hombres con potencial reproductivo
ALECENSA puede causar daño fetal cuando se administra a una mujer embarazada [ver Uso en poblaciones específicas (8.1)].
Prueba de embarazo
Verificar el estado de embarazo en mujeres con potencial reproductivo antes de iniciar ALECENSA [ver Uso en poblaciones específicas (8.1)].
Anticonceptivos
Mujeres
Aconsejar a las mujeres con potencial reproductivo que usen anticonceptivos efectivos durante el tratamiento con ALECENSA y hasta 5 semanas después de la última dosis [ver Uso en poblaciones específicas (8.1)].
Hombres
Con base en los hallazgos de genotoxicidad, aconsejar a los hombres con parejas femeninas con potencial reproductivo que usen anticonceptivos efectivos durante el tratamiento con ALECENSA y durante 3 meses después de la última dosis [ver Toxicología no clínica (13.1)].
8.4 Uso pediátrico
No se ha establecido la seguridad y eficacia de ALECENSA en pacientes pediátricos.
Datos de animales
No se han realizado estudios en animales jóvenes utilizando alectinib. En general, los estudios de toxicología con dosis de alectinib que resultaron en exposiciones mayores o iguales a aproximadamente 4.5 veces las de los humanos tratados con alectinib a 600 mg dos veces al día en ratas produjeron cambios en los dientes y huesos en crecimiento. Los hallazgos en los dientes incluyeron decoloración y cambios en el tamaño del diente junto con alteraciones histopatológicas de las capas de ameloblastos y odontoblastos. También hubo disminuciones en el hueso trabecular y aumento de la actividad osteoclástica en el fémur y el esternón.
8.5 Uso geriátrico
El diecinueve por ciento de los 533 pacientes estudiados en NP28761, NP28673, ALEX y ALINA tenían 65 años de edad o más (el 3.2% tenían 75 años de edad o más). No se observaron diferencias generales en la efectividad según la edad. El análisis exploratorio sugiere una mayor incidencia de eventos adversos graves (38% vs 25%), más eventos adversos que conducen a la interrupción del tratamiento (18% vs 6%) y modificaciones de dosis (48% vs 35%) en pacientes de 65 años o más en comparación con aquellos menores de 65 años.
8.6 Insuficiencia renal
No se recomienda ajuste de dosis para pacientes con insuficiencia renal leve o moderada. No se ha estudiado la seguridad de ALECENSA en pacientes con insuficiencia renal grave (aclaramiento de creatinina menor de 30 mL/min) o enfermedad renal terminal [ver Farmacología clínica (12.3)].
8.7 Insuficiencia hepática
No se recomienda ajuste de dosis para pacientes con insuficiencia hepática leve (Child-Pugh A) o moderada (Child-Pugh B). Se produjo un aumento de la exposición de alectinib en pacientes con insuficiencia hepática grave (Child-Pugh C). La dosis recomendada de ALECENSA en pacientes con insuficiencia hepática grave (Child-Pugh C) es de 450 mg por vía oral dos veces al día [ver Dosificación y administración (2.3) y Farmacología clínica (12.3)].
10 SOBREDOSIS
No se dispone de experiencia con sobredosis. No existe un antídoto específico para la sobredosis con ALECENSA. Alectinib y su principal metabolito activo M4 están unidos a las proteínas plasmáticas en > 99%; por lo tanto, la hemodiálisis probablemente sea ineficaz en el tratamiento de la sobredosis.
11 DESCRIPCIÓN
ALECENSA (alectinib) es un inhibidor de la quinasa para administración oral. La fórmula molecular para alectinib es C30H34N4O2 ∙ HCl. El peso molecular es 482,62 g/mol (forma de base libre) y 519,08 g/mol (sal de clorohidrato). Alectinib se describe químicamente como clorohidrato de 9-etil-6, 6-dimetil-8-[4-(morfolin-4-il)piperidin-1-il]-11-oxo-6, 11-dihidro-5H-benzo[b]carbazol-3-carbonitrilo. La estructura química de alectinib se muestra a continuación:
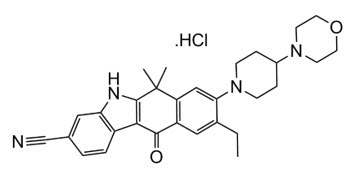
Alectinib HCl es un polvo blanco a blanco amarillento o un polvo con grumos con un pKa de 7,05 (base).
ALECENSA se suministra en cápsulas duras que contienen 150 mg de alectinib (equivalente a 161,33 mg de clorohidrato de alectinib) y los siguientes ingredientes inactivos: monohidrato de lactosa, hidroxipropilcelulosa, laurilsulfato de sodio, estearato de magnesio y carboximetilcelulosa cálcica. La cápsula contiene hipromelosa, carragenina, cloruro de potasio, dióxido de titanio, almidón de maíz y cera de carnauba. La tinta de impresión contiene óxido de hierro rojo (E172), óxido de hierro amarillo (E172), laca de aluminio azul FD&C No. 2 (E132), cera de carnauba, goma laca blanca y monooleato de glicerilo.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
Alectinib es un inhibidor de la tirosina quinasa que actúa sobre ALK y RET. En estudios no clínicos, alectinib inhibió la fosforilación de ALK y la activación mediada por ALK de las proteínas de señalización descendente STAT3 y AKT, y disminuyó la viabilidad de las células tumorales en múltiples líneas celulares que albergan fusiones, amplificaciones o mutaciones activadoras de ALK. El principal metabolito activo de alectinib, M4, mostró una potencia y actividad similar in vitro.
Alectinib y M4 demostraron actividad in vitro e in vivo contra múltiples formas mutantes de la enzima ALK, incluyendo algunas mutaciones identificadas en tumores de CPNCP en pacientes que han progresado con crizotinib.
En modelos de ratón implantados con tumores portadores de fusiones ALK, la administración de alectinib resultó en actividad antitumoral y prolongó la supervivencia, incluso en modelos de ratón implantados intracranialmente con líneas celulares tumorales impulsadas por ALK.
12.2 Farmacodinámica
Electrofisiología cardiaca
La capacidad de alectinib para prolongar el intervalo QT se evaluó en 221 pacientes a los que se administró ALECENSA 600 mg dos veces al día en estudios clínicos. ALECENSA no prolongó el intervalo QTc (QT corregido por frecuencia cardíaca) en ninguna extensión clínicamente relevante. Un paciente tuvo un valor máximo postbasal de QTcF superior a 500 mseg, y un paciente tuvo un cambio máximo de QTcF desde el basal de más de 60 mseg.
12.3 Farmacocinética
La farmacocinética de alectinib y su principal metabolito activo M4 se ha caracterizado en pacientes con CPNCP ALK-positivo y sujetos sanos.
En pacientes con CPNCP ALK-positivo, la concentración máxima media geométrica (coeficiente de variación %) en estado estacionario (Cmax,ss) para alectinib fue 665 ng/mL (44%) y para M4 fue 246 ng/mL (45%) con una relación de concentración pico a valle de 1.2. El área media geométrica bajo la curva en estado estacionario de 0 a 12 horas (AUC0-12h,ss) para alectinib fue 7,430 ng*h/mL (46%) y para M4 fue 2,810 ng*h/mL (46%). La exposición a alectinib es proporcional a la dosis en el rango de 460 mg a 900 mg (es decir, de 0.75 a 1.5 veces la dosis recomendada aprobada) en condiciones de alimentación. Alectinib y M4 alcanzaron concentraciones en estado estacionario en el día 7. La acumulación media geométrica fue aproximadamente 6 veces para alectinib y M4.
Absorción
Alectinib alcanzó concentraciones máximas a las 4 horas después de la administración de ALECENSA 600 mg dos veces al día en condiciones de alimentación en pacientes con CPNCP ALK-positivo.
La biodisponibilidad absoluta de alectinib fue del 37% (IC 90%: 34%, 40%) en condiciones de alimentación.
Una comida alta en grasas y calorías aumentó la exposición combinada (AUC0-inf) de alectinib más M4 por 3.1 veces (IC 90%: 2.7, 3.6) después de la administración oral de una dosis única de 600 mg de ALECENSA.
Distribución
El volumen aparente de distribución es 4,016 L para alectinib y 10,093 L para M4.
Alectinib y M4 se unen a proteínas plasmáticas humanas en más del 99%, independientemente de la concentración del fármaco.
Las concentraciones de alectinib en el líquido cefalorraquídeo en pacientes con CPNCP ALK-positivo se aproximan a las concentraciones libres estimadas de alectinib en el plasma.
Estudios in vitro sugieren que alectinib no es sustrato de la P-glicoproteína (P-gp), pero M4 sí es sustrato de P-gp. Alectinib y M4 no son sustratos de la proteína de resistencia del cáncer de mama (BCRP), la polipeptida transportadora de aniones orgánicos (OATP) 1B1 o OATP1B3.
Eliminación
El aclaramiento aparente (CL/F) es 81.9 L/hora para alectinib y 217 L/hora para M4. La semivida de eliminación media geométrica es 33 horas para alectinib y 31 horas para M4 en pacientes con CPNCP ALK-positivo.
Metabolismo
Alectinib es metabolizado por CYP3A4 a su principal metabolito activo M4. La relación de exposición media geométrica metabolito/parental en estado estacionario es 0.40. M4 es posteriormente metabolizado por CYP3A4. Alectinib y M4 fueron las principales moléculas circulantes en plasma, constituyendo el 76% de la radioactividad total.
Excreción
El 98% de la radioactividad se excretó en las heces después de la administración oral de una dosis única de alectinib radiomarcado en condiciones de alimentación. El 84% de la dosis se excretó en las heces como alectinib sin cambios, y el 6% de la dosis se excretó como M4. La excreción de radioactividad en orina fue inferior al 0.5% de la dosis administrada de alectinib radiomarcado.
Poblaciones específicas
La edad (21 a 83 años), el peso corporal (38 a 128 kg), el deterioro hepático leve (bilirrubina total ≤ LSN y AST > LSN o bilirrubina total 1 a ≤ 1.5 × LSN y AST cualquier valor), el deterioro renal leve a moderado (depuración de creatinina 30 a 89 mL/min), la raza (blanca, asiática y otras), y el sexo no tuvieron ningún efecto clínicamente significativo sobre la exposición sistémica de alectinib y M4. La farmacocinética de alectinib no se ha estudiado en pacientes con deterioro renal grave (depuración de creatinina < 30 mL/min) o enfermedad renal en etapa terminal.
Deterioro hepático: Después de la administración de una dosis oral única de 300 mg de ALECENSA, la relación media geométrica [intervalo de confianza del 90%] para el AUCinf combinada de alectinib y M4 en sujetos con deterioro hepático moderado (Child-Pugh B) fue 1.36 [0.947, 1.96] y en sujetos con deterioro hepático grave (Child-Pugh C) fue 1.76 [0.984, 3.15] en comparación con la de sujetos con función hepática normal. La Cmax combinada de alectinib y M4 fue comparable entre los tres grupos. No se recomienda un ajuste de dosis para pacientes con deterioro hepático leve o moderado. La dosis recomendada de ALECENSA en pacientes con deterioro hepático grave es 450 mg por vía oral dos veces al día [ver Dosificación y administración (2.3) y Uso en poblaciones específicas (8.7)].
Interacciones farmacológicas
Efecto de otros fármacos sobre alectinib
No se observó ningún efecto clínicamente significativo sobre la exposición combinada de alectinib más M4 en estudios clínicos después de la coadministración de ALECENSA con un inhibidor potente de CYP3A (posaconazol), un inductor potente de CYP3A (rifampicina) o un agente reductor de ácido (esomeprazol).
Efecto de alectinib sobre otros fármacos
No se espera un efecto clínicamente significativo sobre la exposición de midazolam (sustrato sensible de CYP3A) o repaglinida (sustrato sensible de CYP2C8) después de la coadministración con ALECENSA.
Estudios in vitro sugieren que alectinib y M4 no inhiben CYP1A2, 2B6, 2C9, 2C19 o 2D6.
Estudios in vitro sugieren que alectinib y M4 inhiben P-gp y BCRP. Alectinib no inhibió la actividad de transporte de OATP1B1, OATP1B3, OAT1, OAT3 u OCT2 in vitro.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
No se han realizado estudios de carcinogenicidad con alectinib.
Alectinib no fue mutagénico in vitro en el ensayo de mutación reversa en bacterias (Ames), pero fue positivo con un aumento en el número de micronúcleos en una prueba de micronúcleos de médula ósea en ratas. El mecanismo de inducción de micronúcleos fue la segregación cromosómica anormal (aneugénica) y no un efecto clastogénico en los cromosomas.
No se han realizado estudios en animales para evaluar el efecto de alectinib sobre la fertilidad. No se observaron efectos adversos en los órganos reproductivos masculinos y femeninos en estudios de toxicología general realizados en ratas y monos.
14 ESTUDIOS CLÍNICOS
14.1 Tratamiento adyuvante de CPNM ALK-positivo resecado
La eficacia de ALECENSA para el tratamiento adyuvante de pacientes con CPNM ALK-positivo después de la resección tumoral completa se evaluó en un ensayo clínico global, aleatorizado, abierto (ALINA: NCT03456076). Los pacientes elegibles debían tener CPNM ALK-positivo resecable, Estadio IB (tumores ≥ 4 cm) – IIIA según el Sistema de Estadificación de la Unión Internacional Contra el Cáncer/Comité Conjunto Estadounidense sobre el Cáncer (UICC/AJCC), 7ª Edición. Las reordenaciones de ALK se identificaron mediante una prueba de ALK aprobada por la FDA realizada localmente o mediante un ensayo VENTANA ALK (D5F3) CDx realizado de forma centralizada.
La aleatorización se estratificó por raza (asiática frente a otras razas) y estadio de la enfermedad (IB frente a II frente a IIIA). Los pacientes fueron aleatorizados (1:1) para recibir ALECENSA 600 mg por vía oral dos veces al día o quimioterapia basada en platino después de la resección tumoral. El tratamiento con ALECENSA continuó durante un total de 2 años, o hasta la recurrencia de la enfermedad o toxicidad inaceptable. La quimioterapia basada en platino se administró por vía intravenosa durante 4 ciclos, con cada ciclo durando 21 días, según uno de los siguientes regímenes:
- Cisplatino 75 mg/m2 el Día 1 más vinorelblina 25 mg/m2 los Días 1 y 8
- Cisplatino 75 mg/m2 el Día 1 más gemcitabina 1250 mg/m2 los Días 1 y 8
- Cisplatino 75 mg/m2 el Día 1 más pemetrexed 500 mg/m2 el Día 1
En caso de intolerancia a un régimen a base de cisplatino, se administró carboplatino en lugar de cisplatino en las combinaciones anteriores a una dosis de AUC 5 mg/mL/min o 6 mg/mL/min.
Las principales medidas de eficacia fueron la supervivencia libre de enfermedad (SLE) en pacientes con CPNM de estadio II-IIIA y la SLE en pacientes con CPNM de estadio IB-IIIA (población de intención de tratar [ITT]) según lo evaluado por el investigador. La SLE se definió como el tiempo desde la fecha de aleatorización hasta la fecha de ocurrencia de cualquiera de los siguientes eventos: primera recurrencia documentada de la enfermedad, nuevo CPNM primario o muerte por cualquier causa, lo que ocurriera primero. Una medida adicional de eficacia fue la supervivencia global (SG) en la población ITT.
Un total de 257 pacientes fueron aleatorizados a ALECENSA (N=130) o a quimioterapia (N=127). La mediana de edad fue de 56 años (rango: 26 a 87), el 24% tenía ≥ 65 años; el 52% eran mujeres; el 56% eran asiáticos, el 42% eran blancos, el 0.4% eran negros o afroamericanos, el 2.3% eran de raza desconocida; el 0.4% eran hispanos o latinos; el 60% nunca habían fumado; el 53% tenían un PS ECOG de 0; el 10% de los pacientes tenían enfermedad de Estadio IB, el 35% tenían Estadio II y el 55% tenían Estadio IIIA.
ALINA demostró una mejora estadísticamente significativa en la SLE para los pacientes tratados con ALECENSA en comparación con los pacientes tratados con quimioterapia. Los datos de SG no estaban maduros en el momento del análisis de SLE, con un 2.3% de muertes reportadas en la población ITT.
Los resultados de eficacia de ALINA se resumen en la Tabla 10 y la Figura 1.
| Parámetro de eficacia | Población Estadio II-IIIA | Población ITT | ||
|---|---|---|---|---|
| ALECENSA N=116 |
Quimioterapia N=115 |
ALECENSA N=130 |
Quimioterapia N=127 |
|
| SLE = Supervivencia Libre de Enfermedad; ITT = Intención de Tratar; CI = Intervalo de Confianza; NR = No Alcanzado; NE = No Estimable. | ||||
|
||||
| Eventos de SLE (%) | 14 (12) | 45 (39) | 15 (12) | 50 (39) |
| Recurrencia de la enfermedad (%) | 14 (12) | 44 (38) | 15 (12) | 49 (38) |
| Muerte | 0 | 1 (0,9) | 0 | 1 (0,8) |
| Mediana de SLE, meses (IC 95%)* |
NR (NE, NE) |
44,4 (27,8, NE) |
NR (NE, NE) |
41,3 (28,5, NE) |
| Cociente de riesgo (IC 95%)† | 0,24 (0,13, 0,45) | 0,24 (0,13, 0,43) | ||
| Valor p‡ | <0,0001 | <0,0001 | ||
Figura 1: Curvas de Kaplan-Meier de la SLP evaluada por el investigador (población ITT) en ALINA
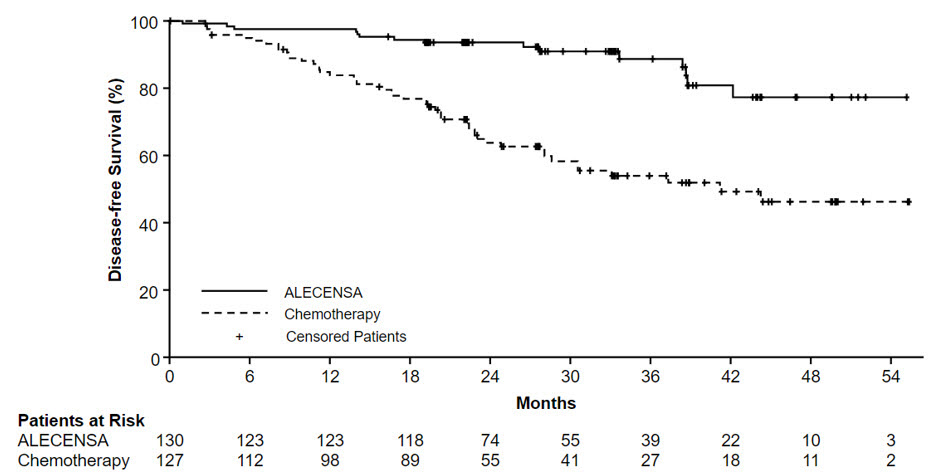
En un análisis exploratorio de los sitios de recaída, la proporción de pacientes con afectación cerebral en el momento de la recurrencia de la enfermedad fue de 4 pacientes (3,1%) en el brazo de ALECENSA y 14 pacientes (11%) en el brazo de quimioterapia.
14.2 Tratamiento del CPCNP ALK-positivo metastásico
CPCNP ALK-positivo metastásico no tratado previamente
La eficacia de ALECENSA para el tratamiento de pacientes con CPCNP ALK-positivo que no habían recibido terapia sistémica previa para la enfermedad metastásica se estableció en un estudio multicéntrico, abierto, aleatorizado, controlado con fármaco activo (ALEX: NCT02075840). Se requería que los pacientes tuvieran un estado funcional ECOG de 0-2 y CPCNP ALK-positivo según lo identificado por el ensayo VENTANA ALK (D5F3) CDx. Eran elegibles los pacientes neurológicamente estables con metástasis en el sistema nervioso central (SNC) tratadas o no tratadas, incluyendo metástasis leptomeníngeas; los pacientes con signos y síntomas neurológicos debido a metástasis en el SNC debían haber completado la radioterapia cerebral completa o la radiocirugía al menos 14 días antes de la inscripción y estar clínicamente estables. Los pacientes con un QTc basal >470 ms no eran elegibles.
Los pacientes se aleatorizaron 1:1 para recibir ALECENSA 600 mg por vía oral dos veces al día o crizotinib 250 mg por vía oral dos veces al día. La aleatorización se estratificó por estado funcional ECOG (0/1 vs. 2), raza (asiática vs. otras razas) y presencia o ausencia de metástasis en el SNC al inicio. El tratamiento en ambos brazos se continuó hasta la progresión de la enfermedad o toxicidad inaceptable. La principal medida de resultado de eficacia fue la supervivencia sin progresión (SLP) determinada por la evaluación del investigador según RECIST v1.1. Las medidas adicionales de resultado de eficacia fueron la SLP determinada por un comité independiente de revisión (CIR), el tiempo hasta la progresión del SNC por CIR según RECIST v1.1, la tasa de respuesta objetiva (TRO) y la duración de la respuesta (DR), y la supervivencia global (SG). Las medidas de resultado exploratorias adicionales fueron la tasa de respuesta objetiva del SNC (TRO-SNC) y la duración de la respuesta del SNC (DR-SNC) por CIR en pacientes con metástasis en el SNC al inicio.
Un total de 303 pacientes fueron aleatorizados a ALECENSA (n=152) o crizotinib (n=151). Las características demográficas de la población del estudio fueron 56% mujeres, edad mediana 56 años (rango: 18 a 91 años), 50% blancos, 46% asiáticos, 1% negros y 3% otras razas. La mayoría de los pacientes tenían adenocarcinoma (92%) y nunca habían fumado (63%). Las metástasis en el SNC estaban presentes en el 40% (n=122) de los pacientes: de estos, 43 pacientes tenían lesiones en el SNC medibles según lo determinado por un CIR. El estudio ALEX demostró una mejora significativa en la SLP. El tiempo hasta la progresión específica en el SNC evaluada por el CIR también mejoró significativamente; hubo una menor incidencia de progresión en el SNC como primer sitio de progresión de la enfermedad, sola o con progresión sistémica concurrente, en el brazo de ALECENSA (12%) en comparación con el brazo de crizotinib (45%). Los resultados de eficacia de ALEX se resumen en la Tabla 11 y la Figura 2.
| ALECENSA N=152 |
Crizotinib N=151 |
|
|---|---|---|
| SNC: sistema nervioso central, TRO: tasa de respuesta objetiva, CIR: comité independiente de revisión, IC: intervalo de confianza, NE: no estimable. | ||
| Supervivencia sin progresión | ||
| Número de eventos (%) | 63 (41%) | 92 (61%) |
| Enfermedad progresiva (%) | 51 (34%) | 82 (54%) |
| Muerte (%) | 12 (8%) | 10 (7%) |
| Mediana en meses (IC 95%) | 25,7 (19,9, NE) | 10,4 (7,7, 14,6) |
| Razón de riesgo (IC 95%) * | 0,53 (0,38, 0,73) | |
| Valor p * | < 0,0001 | |
| Tasa de respuesta objetiva | ||
| Tasa de respuesta objetiva, % (IC 95%) † | 79% (72, 85) | 72% (64, 79) |
| Valor p * | 0,1652 | |
| Respuesta completa, % | 13% | 6% |
| Respuesta parcial, % | 66% | 66% |
| Duración de la respuesta | ||
| Número de respondedores | n=120 | n=109 |
| Duración de la respuesta ≥6 meses | 82% | 57% |
| Duración de la respuesta ≥12 meses | 64% | 36% |
| Duración de la respuesta ≥18 meses | 37% | 14% |
Figura 2: Gráfico de Kaplan-Meier de supervivencia libre de progresión (IRC) en ALEX

Los resultados para SLP según lo determinado por la evaluación del investigador (HR=0,48 [IC del 95%: 0,35-0,66], log-rank estratificado p<0,0001) fueron similares a los observados por el IRC. En el punto de corte de datos, los datos de supervivencia global no estaban maduros.
Los resultados de los análisis exploratorios predefinidos de la tasa de respuesta del SNC en pacientes con lesiones del SNC medibles al inicio se resumen en la Tabla 12.
| ALECENSA | Crizotinib | |
|---|---|---|
| IRC: Comité de Revisión Independiente; IC: Intervalo de Confianza; NE: No Estimable | ||
|
||
| Evaluación de la respuesta tumoral en el SNC | N = 21 | N = 22 |
| Tasa de respuesta objetiva en el SNC, % (IC del 95%*) | 81% (58, 95) | 50% (28,72) |
| Respuesta completa | 38% | 5% |
| Duración de la respuesta en el SNC | ||
| Número de respondedores | 17 | 11 |
| Duración de la respuesta en el SNC ≥ 12 meses | 59% | 36% |
Cáncer de Pulmón No Microcítico ALK-Positivo Metastásico Previamente Tratado con Crizotinib
La seguridad y eficacia de ALECENSA se establecieron en dos estudios clínicos multicéntricos, de un solo brazo: NP28761 (NCT01588028) y NP28673 (NCT01801111). Se inscribieron pacientes con cáncer de pulmón no microcítico ALK-positivo localmente avanzado o metastásico, que habían progresado con crizotinib, con cáncer de pulmón no microcítico ALK-positivo documentado según una prueba aprobada por la FDA, y un puntaje de ECOG de 0-2 en ambos estudios. Los criterios de elegibilidad permitieron la inscripción de pacientes con quimioterapia previa y radioterapia previa en el SNC, siempre que las metástasis en el SNC estuvieran estables durante al menos dos semanas. Todos los pacientes recibieron ALECENSA 600 mg por vía oral dos veces al día. La principal medida de eficacia en ambos estudios fue la tasa de respuesta objetiva (ORR) según los Criterios de Evaluación de Respuesta en Tumores Sólidos (RECIST v1.1) según lo evaluado por un Comité de Revisión Independiente (IRC). Las medidas de desenlace adicionales evaluadas por el IRC incluyeron la duración de la respuesta (DOR), la ORR del SNC y la DOR del SNC.
El estudio NP28761 se realizó en América del Norte e inscribió a 87 pacientes. Las características demográficas y de la enfermedad basales en NP28761 fueron: mediana de edad 54 años (rango 29 a 79, 18% 65 años y más), 84% blancos y 8% asiáticos, 55% mujeres, 35% ECOG PS 0 y 55% ECOG PS 1, 100% nunca o ex fumadores, 99% Etapa IV, 94% adenocarcinoma y 74% quimioterapia previa. Los sitios más comunes de metástasis extratóraxica incluyeron 60% SNC (de los cuales el 65% había recibido radiación en el SNC), 43% ganglios linfáticos, 36% hueso y 34% hígado.
El estudio NP28673 se realizó internacionalmente e inscribió a 138 pacientes. Las características demográficas y de la enfermedad basales en NP28673 fueron: mediana de edad 52 años (rango 22 a 79, 10% 65 años y más), 67% blancos y 26% asiáticos, 56% mujeres, 32% ECOG PS 0 y 59% ECOG PS 1, 98% nunca o ex fumadores, 99% Etapa IV, 96% adenocarcinoma y 80% quimioterapia previa. Los sitios más comunes de metástasis extratóraxica incluyeron 61% SNC (de los cuales el 73% había recibido radiación en el SNC), 51% hueso, 38% ganglios linfáticos y 30% hígado.
Los resultados de eficacia de NP28761 y NP28673 en todos los pacientes tratados se resumen en la Tabla 13. La mediana de duración del seguimiento en el Estudio NP28761 fue de 4,8 meses tanto para la evaluación del IRC como del Investigador, y en el Estudio NP28673, 10,9 meses para la evaluación del IRC y 7,0 meses para la evaluación del Investigador. Todas las respuestas fueron respuestas parciales.
| Parámetro de Eficacia | NP28761 (N=87) | NP28673 (N=138) | ||
|---|---|---|---|---|
| Evaluación del IRC* | Evaluación del Investigador | Evaluación del IRC* | Evaluación del Investigador | |
|
||||
| Tasa de Respuesta Objetiva (95% IC) | 38% (28; 49) |
46% (35; 57) |
44% (36; 53) |
48% (39; 57) |
| Número de Respondedores | 33 | 40 | 61 | 66 |
| Duración de la Respuesta, mediana en meses (95% IC) | 7,5 (4,9, No Estimable) |
NE (4,9, No Estimable) |
11,2 (9,6, No Estimable) |
7,8 (7,4, 9,2) |
Se realizó una evaluación de la ORR y la duración de la respuesta para las metástasis en el SNC en el subgrupo de 51 pacientes en NP28761 y NP28673 con lesiones medibles en el SNC al inicio según RECIST v1.1, que se resumen en la Tabla 14. Treinta y cinco (69%) pacientes con lesiones medibles en el SNC habían recibido radiación cerebral previa, incluyendo 25 (49%) que completaron el tratamiento de radiación al menos 6 meses antes de comenzar el tratamiento con ALECENSA. Se observaron respuestas independientemente del estado de radiación cerebral previa.
| Parámetro de Eficacia | N=51 |
|---|---|
| Tasa de Respuesta Objetiva en el SNC (95% IC) | 61% (46, 74) |
| Respuesta Completa | 18% |
| Respuesta Parcial | 43% |
| Duración de la Respuesta en el SNC, mediana en meses (95% IC) | 9,1 (5,8, No Estimable) |
16 PRESENTACIÓN/ALMACENAMIENTO Y MANEJO
Cápsulas duras, cápsulas blancas de 150 mg con “ALE” impreso en tinta negra en la tapa y “150 mg” impreso en tinta negra en el cuerpo, disponibles en:
| 240 cápsulas por botella: | NDC 50242-130-01 |
Almacenamiento y estabilidad: No almacenar a más de 30°C (86°F). Almacenar en el envase original para proteger de la luz y la humedad.
17 INFORMACIÓN PARA EL PACIENTE
Aconseje al paciente que lea la información aprobada por la FDA para el paciente (Información para el paciente).
Informe a los pacientes lo siguiente:
Hepatotoxicidad
Informe a los pacientes sobre los signos y síntomas de elevaciones de bilirrubina y transaminasas hepáticas. Aconseje a los pacientes que se comuniquen con su proveedor de atención médica inmediatamente si presentan signos o síntomas de elevaciones de bilirrubina y transaminasas hepáticas [ver Advertencias y precauciones (5.1)].
Enfermedad pulmonar intersticial (ILD)/Neumonitis
Informe a los pacientes sobre los riesgos de ILD/neumonitis grave. Aconseje a los pacientes que se comuniquen con su proveedor de atención médica inmediatamente para informar sobre nuevos síntomas respiratorios o el empeoramiento de los mismos [ver Advertencias y precauciones (5.2)].
Insuficiencia renal
Informe a los pacientes sobre el riesgo de insuficiencia renal grave y potencialmente mortal. Aconseje a los pacientes que se comuniquen con su proveedor de atención médica si presentan cambios en el color de la orina, disminución del volumen urinario o hinchazón en las piernas y los pies [ver Advertencias y precauciones (5.3)].
Bradicardia
Informe a los pacientes que pueden presentar síntomas de bradicardia, como mareos, vértigo y síncope, mientras toman ALECENSA. Aconseje a los pacientes que se comuniquen con su proveedor de atención médica para informar estos síntomas y para informar sobre el uso de cualquier medicamento para el corazón o la presión arterial [ver Advertencias y precauciones (5.4)].
Mialgia grave/Elevación de CPK
Informe a los pacientes sobre los signos y síntomas de mialgia, incluyendo dolor muscular inexplicable y/o persistente, sensibilidad o debilidad. Aconseje a los pacientes que se comuniquen con su proveedor de atención médica inmediatamente para informar sobre nuevos síntomas o el empeoramiento de los síntomas de dolor o debilidad muscular [ver Advertencias y precauciones (5.5)].
Anemia hemolítica
Aconseje a los pacientes que se comuniquen con su proveedor de atención médica si desarrollan signos o síntomas de anemia hemolítica [ver Advertencias y precauciones (5.6)].
Fotosensibilidad
Informe a los pacientes sobre los signos y síntomas de fotosensibilidad. Aconseje a los pacientes que eviten la exposición prolongada al sol mientras toman ALECENSA y durante al menos 7 días después de la interrupción del medicamento del estudio y que utilicen la protección adecuada contra el sol. Aconseje a los pacientes que utilicen un protector solar de amplio espectro con protección contra los rayos ultravioleta A (UVA)/ultravioleta B (UVB) y un bálsamo labial (FPS ≥ 50) para ayudar a protegerse contra posibles quemaduras solares [ver Reacciones adversas (6.1)].
Toxicidad embrio-fetal
ALECENSA puede causar daño fetal si se toma durante el embarazo. Aconseje a las mujeres embarazadas y a las mujeres con potencial reproductivo sobre el posible riesgo para el feto [ver Advertencias y precauciones (5.6) y Uso en poblaciones específicas (8.1, 8.3)].
Aconseje a las mujeres con potencial reproductivo que utilicen un método anticonceptivo efectivo durante el tratamiento con ALECENSA y durante 5 semanas después de la última dosis de ALECENSA. Aconseje a los pacientes que informen a su proveedor de atención médica sobre un embarazo conocido o sospechado [ver Advertencias y precauciones (5.6) y Uso en poblaciones específicas (8.1, 8.3)].
Aconseje a los pacientes masculinos con parejas femeninas con potencial reproductivo que utilicen un método anticonceptivo efectivo durante el tratamiento con ALECENSA y durante 3 meses después de la última dosis [ver Uso en poblaciones específicas (8.3) y Toxicología no clínica (13.1)].
Lactancia
Aconseje a las mujeres que no amamanten durante el tratamiento con ALECENSA y durante 1 semana después de la última dosis [ver Uso en poblaciones específicas (8.2)].
Administración
Indique a los pacientes que tomen ALECENSA dos veces al día. Aconseje a los pacientes que tomen ALECENSA con alimentos y que traguen las cápsulas de ALECENSA enteras [ver Dosificación y administración (2.2)].
Dosis omitida
Aconseje a los pacientes que si omiten una dosis de ALECENSA o si vomitan después de tomar una dosis de ALECENSA, no deben tomar una dosis adicional, sino tomar la siguiente dosis a la hora habitual [ver Dosificación y administración (2.2)].
SECCIÓN SIN CLASIFICAR DEL SPL
Distribuido por:
Genentech USA, Inc.
Un miembro del Grupo Roche
1 DNA Way
South San Francisco, CA 94080-4990
ALECENSA® es una marca registrada de
Chugai Pharmaceutical Co., Ltd., Tokio, Japón
©2024 Genentech, Inc. Todos los derechos reservados.
PROSPECTO PARA EL PACIENTE
| Esta información para el paciente ha sido aprobada por la Administración de Alimentos y Medicamentos de los Estados Unidos. | Revisado: 04/2024 | ||
| INFORMACIÓN PARA EL PACIENTE ALECENSA® (a-le-sen-sah) (alectinib) cápsulas |
|||
| ¿Cuál es la información más importante que debo saber sobre ALECENSA? ALECENSA puede causar efectos secundarios graves, incluyendo:
|
|||
|
|
||
Consulte “¿Cuáles son los posibles efectos secundarios de ALECENSA?” para obtener más información sobre los efectos secundarios. |
|||
| ¿Qué es ALECENSA? ALECENSA es un medicamento recetado utilizado para tratar a adultos con cáncer de pulmón de células no pequeñas (CPNCP) causado por un gen anaplásico anormal de la linfoma quinasa (ALK):
Su proveedor de atención médica realizará una prueba para asegurarse de que ALECENSA es adecuado para usted. |
|||
Antes de tomar ALECENSA, informe a su proveedor de atención médica sobre todas sus afecciones médicas, incluyendo si usted:
Informe a su proveedor de atención médica sobre todos los medicamentos que toma, incluyendo medicamentos recetados, medicamentos de venta libre, vitaminas o suplementos herbales. |
|||
¿Cómo debo tomar ALECENSA?
|
|||
| ¿Qué debo evitar mientras tomo ALECENSA? Evite exponerse al sol durante el tratamiento con ALECENSA y durante 7 días después de la última dosis de ALECENSA. Su piel puede ser sensible al sol (fotosensibilidad) y es posible que se queme con más facilidad y sufra quemaduras solares graves. Use medidas de protección solar, como protector solar y bálsamo labial con un FPS 50 o superior para ayudar a protegerse de las quemaduras solares. |
|||
| ¿Cuáles son los posibles efectos secundarios de ALECENSA? ALECENSA puede causar efectos secundarios graves, incluyendo: Los efectos secundarios más comunes de ALECENSA incluyen: |
|||
|
|
||
| Estos no son todos los posibles efectos secundarios de ALECENSA. Para obtener más información, consulte a su proveedor de atención médica o farmacéutico. Llame a su médico para obtener asesoramiento médico sobre los efectos secundarios. Puede reportar efectos secundarios a la FDA al 1-800-FDA-1088. |
|||
¿Cómo debo almacenar ALECENSA?
Mantenga ALECENSA y todos los medicamentos fuera del alcance de los niños. |
|||
| Información general sobre el uso seguro y eficaz de ALECENSA. A veces se prescriben medicamentos para fines distintos a los enumerados en un prospecto de información para el paciente. No use ALECENSA para una afección para la cual no fue prescrita. No le dé ALECENSA a otras personas, incluso si tienen los mismos síntomas que usted. Puede causarles daño. Puede solicitar a su farmacéutico o proveedor de atención médica información sobre ALECENSA escrita para profesionales de la salud. |
|||
| ¿Cuáles son los ingredientes de ALECENSA? Ingrediente activo: alectinib Ingredientes inactivos: lactosa monohidrato, hidroxipropilcelulosa, lauril sulfato de sodio, estearato de magnesio y carboximetilcelulosa cálcica. La cubierta de la cápsula contiene: hipromelosa, carragenina, cloruro de potasio, dióxido de titanio, almidón de maíz y cera de carnauba. La tinta de impresión contiene: óxido de hierro rojo (E172), óxido de hierro amarillo (E172), laca de aluminio azul FD&C Nº 2 (E132), cera de carnauba, goma laca blanca y monooleato de glicerilo. Distribuido por: Genentech, Inc., A Member of the Roche Group, 1 DNA Way, South San Francisco, CA 94080-4990 ALECENSA® es una marca registrada de Chugai Pharmaceutical Co., Ltd., Tokio, Japón ©2024 Genentech, Inc. Para obtener más información, visite www.ALECENSA.com o llame al 1-800-253-2367. |
|||
PANEL DE VISUALIZACIÓN PRINCIPAL – Caja de 240 cápsulas
NDC 50242-130-01
Alecensa®
(alectinib)
cápsulas
150 mg
Cada cápsula contiene 150 mg de alectinib
(equivalente a 161,33 mg de clorhidrato de alectinib).
Sólo con receta
240 cápsulas
Genentech
10166066
