Fabricante de medicamentos: E.R. Squibb & Sons, L.L.C. (Updated: 2024-07-31)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
SPRYCEL (dasatinib) tabletas, para uso oral
Aprobación inicial en EE. UU.: 2006
INDICACIONES Y USO
SPRYCEL es un inhibidor de la cinasa indicado para el tratamiento de
- •
- adultos recién diagnosticados con leucemia mieloide crónica (LMC) con cromosoma Filadelfia positivo (Ph+). (1, 14)
- •
- adultos con LMC Ph+ en fase crónica, acelerada o blástica mieloide o linfoide con resistencia o intolerancia a la terapia previa, incluido imatinib. (1, 14)
- •
- adultos con leucemia linfoblástica aguda con cromosoma Filadelfia positivo (Ph+ ALL) con resistencia o intolerancia a la terapia previa. (1, 14)
- •
- pacientes pediátricos de 1 año de edad o mayores con LMC Ph+ en fase crónica. (1, 14)
- •
- pacientes pediátricos de 1 año de edad o mayores con ALL Ph+ recién diagnosticada en combinación con quimioterapia. (1, 14)
DOSIFICACIÓN Y ADMINISTRACIÓN
- •
- LMC en fase crónica en adultos: 100 mg una vez al día. (2)
- •
- LMC en fase acelerada, LMC en fase blástica mieloide o linfoide o ALL Ph+ en adultos: 140 mg una vez al día. (2)
- •
- LMC y ALL en fase crónica en pediatría: dosis inicial basada en el peso corporal. (2)
- •
- Administrar por vía oral, con o sin alimentos. No triturar, cortar ni masticar las tabletas. (2)
FORMAS Y FUERZAS DE DOSIFICACIÓN
Tabletas: 20 mg, 50 mg, 70 mg, 80 mg, 100 mg y 140 mg. (3)
CONTRAINDICACIONES
Ninguna. (4)
ADVERTENCIAS Y PRECAUCIONES
- •
- Mielosupresión y eventos hemorrágicos: Puede ocurrir trombocitopenia, neutropenia y anemia graves. Use con precaución si se usa concomitantemente con medicamentos que inhiben la función plaquetaria o anticoagulantes. Monitorear los recuentos sanguíneos completos regularmente. Transfundir e interrumpir SPRYCEL cuando esté indicado. (2.5, 5.1, 5.2)
- •
- Retención de líquidos: Retención de líquidos, a veces grave, incluidos derrames pleurales. Manejar con medidas de apoyo y/o modificación de la dosis. (2.5, 5.3)
- •
- Toxicidad cardiovascular: Monitorear a los pacientes en busca de signos o síntomas y tratarlos de manera adecuada. (5.4)
- •
- Hipertensión arterial pulmonar (HAP): SPRYCEL puede aumentar el riesgo de desarrollar HAP, que puede ser reversible al suspender el tratamiento. Considere el riesgo basal y evalúe a los pacientes en busca de signos y síntomas de HAP durante el tratamiento. Suspenda SPRYCEL si se confirma la HAP. (5.5)
- •
- Prolongación del intervalo QT: Use SPRYCEL con precaución en pacientes que tienen o pueden desarrollar prolongación del intervalo QT. (5.6)
- •
- Reacciones dermatológicas graves: Se han notificado casos individuales de reacciones dermatológicas mucocutáneas graves. (5.7)
- •
- Síndrome de lisis tumoral: Se ha notificado el síndrome de lisis tumoral. Mantenga una hidratación adecuada y corrija los niveles de ácido úrico antes de iniciar el tratamiento con SPRYCEL. (5.8)
- •
- Toxicidad embrio-fetal: Puede causar daño fetal. Avise a los pacientes de potencial reproductivo del riesgo potencial para el feto y de usar métodos anticonceptivos eficaces. (5.9, 8.1, 8.3)
- •
- Efectos sobre el crecimiento y el desarrollo en pacientes pediátricos: Se han notificado fusión retardada de las epífisis, osteopenia, retraso del crecimiento y ginecomastia. Monitorear el crecimiento óseo y el desarrollo en pacientes pediátricos. (5.10)
- •
- Hepatotoxicidad: Evaluar la función hepática antes de iniciar el tratamiento y mensualmente a partir de entonces o según esté clínicamente indicado. Monitorear la función hepática cuando se combina con quimioterapia conocida por estar asociada con disfunción hepática. (5.11)
REACCIONES ADVERSAS
Las reacciones adversas más comunes (≥15%) en pacientes que recibieron SPRYCEL como terapia de un solo agente incluyeron mielosupresión, eventos de retención de líquidos, diarrea, dolor de cabeza, erupción cutánea, hemorragia, disnea, fatiga, náuseas y dolor musculoesquelético. (6)
Las reacciones adversas más comunes (≥30%) en pacientes pediátricos que recibieron SPRYCEL en combinación con quimioterapia incluyeron mucositis, neutropenia febril, pirexia, diarrea, náuseas, vómitos, dolor musculoesquelético, dolor abdominal, tos, dolor de cabeza, erupción cutánea, fatiga, estreñimiento, arritmia, hipertensión, edema, infecciones (bacterianas, virales y fúngicas), hipotensión, disminución del apetito, hipersensibilidad, disnea, epistaxis, neuropatía periférica y alteración del estado de conciencia. (6)
Para informar sobre REACCIONES ADVERSAS SOSPECHOSAS, comuníquese con Bristol-Myers Squibb al 1-800-721-5072 o con la FDA al 1-800-FDA-1088 o www.fda.gov/medwatch.
INTERACCIONES MEDICAMENTOSAS
- •
- Inhibidores fuertes de CYP3A4: Puede ser necesaria una reducción de la dosis. (2.3, 7.1)
- •
- Inductores fuertes de CYP3A4: Puede ser necesario un aumento de la dosis. (2.3, 7.1)
- •
- Antiácidos: Evite la administración simultánea. (7.1)
- •
- Antagonistas H2 e inhibidores de la bomba de protones: Evite la coadministración. (7.1)
Ver 17 para INFORMACIÓN PARA EL PACIENTE y etiquetado del paciente aprobado por la FDA.
Revisado: 7/2024
Tabla de Contenido
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICACIONES Y USO
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Dosis de SPRYCEL en pacientes adultos
2.2 Dosis de SPRYCEL en pacientes pediátricos con LMC o LLA Ph+
2.3 Modificación de la dosis
2.4 Aumento de la dosis en adultos con LMC y LLA Ph+, y pacientes pediátricos con LMC
2.5 Ajuste de la dosis por reacciones adversas
2.6 Duración del tratamiento
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Myelosuppression
5.2 Eventos relacionados con hemorragias
5.3 Retención de líquidos
5.4 Toxicidad cardiovascular
5.5 Hipertensión arterial pulmonar
5.6 QT Prolongation
5.7 Reacciones dermatológicas graves
5.8 Tumor Lysis Syndrome
5.9 Toxicidad embriofetal
5.10 Efectos sobre el crecimiento y el desarrollo en pacientes pediátricos
5.11 Hepatotoxicity
6 REACCIONES ADVERSAS
6.1 Experiencia en ensayos clínicos
6.2 Experiencia posterior a la comercialización
7 INTERACCIONES MEDICAMENTOSAS
7.1 Efecto de otros medicamentos sobre dasatinib
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.3 Mujeres y hombres con potencial reproductivo
8.4 Uso pediátrico
8.5 Uso geriátrico
10 SOBREDOSIS
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
12.2 Farmacodinámica
12.3 Farmacocinética
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 ESTUDIOS CLÍNICOS
14.1 LMC en fase crónica recién diagnosticada en adultos
14.2 LMC o LLA Ph+ resistente o intolerante a imatinib en adultos
14.3 LMC en pacientes pediátricos
14.4 LLA Ph+ en pacientes pediátricos
15 REFERENCIAS
16 CÓMO SUMINISTRAR/ALMACENAMIENTO Y MANIPULACIÓN
17 INFORMACIÓN DE ASESORAMIENTO AL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información de prescripción completa no se enumeran.
1 INDICACIONES Y USO
SPRYCEL (dasatinib) está indicado para el tratamiento de pacientes adultos con
- •
- leucemia mieloide crónica (LMC) de novo con cromosoma Filadelfia positivo (Ph+).
- •
- LMC Ph+ crónica, acelerada o en fase blástica mieloide o linfoide con resistencia o intolerancia a la terapia previa, incluyendo imatinib.
- •
- leucemia linfoblástica aguda con cromosoma Filadelfia positivo (Ph+ ALL) con resistencia o intolerancia a la terapia previa.
SPRYCEL (dasatinib) está indicado para el tratamiento de pacientes pediátricos de 1 año de edad o mayores con
- •
- LMC Ph+ en fase crónica.
- •
- Ph+ ALL de novo en combinación con quimioterapia.
2 DOSIS Y ADMINISTRACIÓN
2.1 Dosificación de SPRYCEL en pacientes adultos
La dosis inicial recomendada de SPRYCEL para la LMC en fase crónica en adultos es de 100 mg administrada por vía oral una vez al día. La dosis inicial recomendada de SPRYCEL para la LMC en fase acelerada, fase blástica mieloide o linfoide de la LMC o ALL Ph+ en adultos es de 140 mg administrada por vía oral una vez al día. Las tabletas no deben triturarse, cortarse o masticarse; deben tragarse enteras. SPRYCEL puede tomarse con o sin alimentos, ya sea por la mañana o por la noche.
2.2 Dosificación de SPRYCEL en pacientes pediátricos con LMC o ALL Ph+
La dosis inicial recomendada para pediatría se basa en el peso corporal como se muestra en la Tabla 1. La dosis recomendada debe administrarse por vía oral una vez al día con o sin alimentos. Recalcule la dosis cada 3 meses en función de los cambios en el peso corporal, o con más frecuencia si es necesario.
No triture, corte o mastique las tabletas. Trague las tabletas enteras. Hay consideraciones de administración adicionales para pacientes pediátricos que tienen dificultad para tragar las tabletas enteras [véase Uso en poblaciones específicas (8.4) y Farmacología clínica (12.3)].
| Peso corporal (kg)b | Dosis diaria (mg) |
|---|---|
| a Para pacientes pediátricos con ALL Ph+, comience la terapia con SPRYCEL el día 15 o antes de la quimioterapia de inducción, cuando se confirma el diagnóstico y continúe durante 2 años. b No se recomienda la dosificación en tabletas para pacientes que pesen menos de 10 kg. |
|
|
10 a menos de 20 |
40 mg |
|
20 a menos de 30 |
60 mg |
|
30 a menos de 45 |
70 mg |
|
al menos 45 |
100 mg |
Consulte Sección 2.4 para obtener recomendaciones sobre la escalada de dosis en adultos con LMC y ALL Ph+ y pacientes pediátricos con LMC.
2.3 Modificación de la dosis
Inductores fuertes de CYP3A4
Evite el uso concomitante de inductores fuertes de CYP3A4 y hierba de San Juan. Si los pacientes deben recibir un inductor fuerte de CYP3A4 de forma concomitante, considere aumentar la dosis de SPRYCEL. Si se aumenta la dosis de SPRYCEL, controle cuidadosamente al paciente para detectar toxicidad [ver Interacciones medicamentosas (7.1)].
Inhibidores fuertes de CYP3A4
Evite el uso concomitante de inhibidores fuertes de CYP3A4 y zumo de pomelo. Se recomienda seleccionar un medicamento concomitante alternativo con un potencial de inhibición enzimática nulo o mínimo, si es posible. Si SPRYCEL debe administrarse con un inhibidor fuerte de CYP3A4, considere una reducción de la dosis a:
• 40 mg diarios para pacientes que toman SPRYCEL 140 mg diarios.
• 20 mg diarios para pacientes que toman SPRYCEL 100 mg diarios.
• 20 mg diarios para pacientes que toman SPRYCEL 70 mg diarios.
Para los pacientes que toman SPRYCEL 60 mg o 40 mg diarios, considere interrumpir SPRYCEL hasta que se suspenda el inhibidor. Permita un período de lavado de aproximadamente 1 semana después de que se suspenda el inhibidor antes de reiniciar SPRYCEL.
Se predice que estas dosis reducidas de SPRYCEL ajustarán el área bajo la curva (AUC) al rango observado sin inhibidores de CYP3A4; sin embargo, no hay datos clínicos disponibles con estos ajustes de dosis en pacientes que reciben inhibidores fuertes de CYP3A4. Si SPRYCEL no se tolera después de la reducción de la dosis, suspenda el inhibidor fuerte de CYP3A4 o interrumpa SPRYCEL hasta que se suspenda el inhibidor. Permita un período de lavado de aproximadamente 1 semana después de que se suspenda el inhibidor antes de aumentar la dosis de SPRYCEL [ver Interacciones medicamentosas (7.1)].
2.4 Escalada de dosis en adultos con LMC y ALL Ph+, y pacientes pediátricos con LMC
Para los pacientes adultos con LMC y ALL Ph+, considere la escalada de dosis a 140 mg una vez al día (fase crónica de la LMC) o 180 mg una vez al día (fase avanzada de la LMC y ALL Ph+) en los pacientes que no alcancen una respuesta hematológica o citogenética con la dosis inicial recomendada. Para los pacientes pediátricos con LMC, considere la escalada de dosis a 120 mg una vez al día (ver Tabla 2 a continuación). No se recomienda la escalada de dosis para los pacientes pediátricos con ALL Ph+, donde SPRYCEL se administra en combinación con quimioterapia.
Escala la dosis de SPRYCEL como se muestra en la Tabla 2 en pacientes pediátricos con fase crónica de la LMC que no alcancen una respuesta hematológica o citogenética con la dosis inicial recomendada.
|
Formulación |
Dosis (dosis máxima por día) |
|
|
Dosis inicial |
Escalada |
|
|
Tabletas |
40 mg |
50 mg |
|
60 mg |
70 mg |
|
|
70 mg |
90 mg |
|
|
100 mg |
120 mg |
|
2.5 Ajuste de la dosis para reacciones adversas
Mielosupresión
En estudios clínicos, la mielosupresión se manejó mediante la interrupción de la dosis, la reducción de la dosis o la interrupción del tratamiento del estudio. El factor de crecimiento hematopoyético se ha utilizado en pacientes con mielosupresión resistente. Las pautas para las modificaciones de la dosis para pacientes adultos y pediátricos se resumen en las Tablas 3 y 4, respectivamente.
| * ANC: recuento absoluto de neutrófilos | ||
|
Fase crónica de la LMC (dosis inicial 100 mg una vez al día) |
ANC* <0.5 × 109/L |
|
|
Fase acelerada de la LMC, fase blástica de la LMC y ALL Ph+ (dosis inicial 140 mg una vez al día) |
ANC* <0.5 × 109/L |
|
| *ANC: recuento absoluto de neutrófilos ** dosis de tableta más baja no disponible |
||||
|
||||
|
1. Si la citopenia persiste durante más de 3 semanas, compruebe si la citopenia está relacionada con la leucemia (aspiración o biopsia de médula ósea). 2. Si la citopenia no está relacionada con la leucemia, suspenda SPRYCEL hasta que el ANC* ≥1.0 × 109/L y las plaquetas ≥75 × 109/L y reanude a la dosis inicial original o a una dosis reducida. 3. Si la citopenia vuelve a aparecer, repita la aspiración/biopsia de médula ósea y reanude SPRYCEL a una dosis reducida. |
|
|
|
|
|
Tabletas |
|
|
|
|
|
|
|
||
|
|
|
||
|
|
|
||
Para pacientes pediátricos con LMC en fase crónica, si la neutropenia o trombocitopenia de grado ≥ 3 se repite durante la respuesta hematológica completa (CHR), interrumpa SPRYCEL y reanúdelo a una dosis reducida. Implemente reducciones de dosis temporales para grados intermedios de citopenia y respuesta a la enfermedad según sea necesario.
Para pacientes pediátricos con ALL Ph+, si la neutropenia y/o trombocitopenia provocan un retraso en el siguiente bloque de tratamiento de más de 14 días, interrumpa SPRYCEL y reanúdelo al mismo nivel de dosis una vez que se inicie el siguiente bloque de tratamiento. Si la neutropenia y/o trombocitopenia persisten y el siguiente bloque de tratamiento se retrasa otros 7 días, realice una evaluación de la médula ósea para evaluar la celularidad y el porcentaje de blastos. Si la celularidad de la médula ósea es <10%, interrumpa el tratamiento con SPRYCEL hasta que el ANC >500/μL (0.5 x 109/L), momento en el que se puede reanudar el tratamiento a la dosis completa. Si la celularidad de la médula ósea es >10%, se puede considerar la reanudación del tratamiento con SPRYCEL.
Reacciones adversas no hematológicas
Para adultos con LMC y ALL Ph+ y pacientes pediátricos con LMC Ph+, si se desarrolla una reacción adversa no hematológica grave con el uso de SPRYCEL, el tratamiento debe suspenderse hasta que la reacción adversa se resuelva o mejore. Posteriormente, el tratamiento puede reanudarse según sea apropiado a una dosis reducida dependiendo de la gravedad y la recurrencia [ver Advertencias y precauciones (5)].
Para pacientes pediátricos con ALL Ph+, interrumpa el tratamiento en casos de reacciones adversas no hematológicas de grado ≥ 3, con la excepción de las anomalías en las pruebas de función hepática, y reanúdelo a una dosis reducida cuando se resuelva a grado ≤1. Para la bilirrubina directa elevada por encima de 5 veces el límite superior institucional de lo normal (ULN), interrumpa el tratamiento hasta que mejore a la línea de base o grado ≤1. Para AST/ALT elevado por encima de 15 veces el ULN institucional, interrumpa el tratamiento hasta que mejore a la línea de base o grado <1. Para las anomalías recurrentes en las pruebas de función hepática como las mencionadas anteriormente, reduzca la dosis si esta reacción adversa se repite después de la reiniciación de SPRYCEL. Las recomendaciones de reducción de dosis se describen en la Tabla 5.
| ** dosis de tableta más baja no disponible | ||||
|
Dosis (dosis máxima por día) |
||||
|
1. Si se produce una toxicidad no hematológica de grado 2, considere interrumpir SPRYCEL si no hay recuperación a pesar de la terapia sintomática; una vez recuperado a grado ≤1, reanude a la dosis inicial original. Reanude SPRYCEL a una dosis reducida para eventos recurrentes. 2. Si se produce una toxicidad no hematológica de grado 3, suspenda SPRYCEL hasta que se recupere a grado ≤1 y luego reanúdelo a una dosis reducida. 3. Si la bilirrubina directa es >5 ULN o AST/ALT >15 ULN, interrumpa SPRYCEL hasta que se recupere a grado ≤1 y luego reanude SPRYCEL a la dosis inicial original. Reanude SPRYCEL a una dosis reducida para la hepatotoxicidad recurrente. |
Dosis inicial original |
Reducción de dosis de un nivel |
Reducción de dosis de dos niveles |
|
|
Tabletas |
40 mg |
20 mg |
** |
|
|
60 mg |
40 mg |
20 mg |
||
|
70 mg |
60 mg |
50 mg |
||
|
100 mg |
80 mg |
70 mg |
||
2.6 Duración del tratamiento
En estudios clínicos, el tratamiento con SPRYCEL en adultos y en pacientes pediátricos con LMC en fase crónica se continuó hasta la progresión de la enfermedad o hasta que el paciente ya no lo toleró. No se ha establecido el efecto de la interrupción del tratamiento en el resultado a largo plazo de la enfermedad después del logro de una respuesta citogenética (incluida la respuesta citogenética completa [CCyR]) o una respuesta molecular mayor (MMR y MR4.5).
En estudios clínicos, el tratamiento con SPRYCEL en pacientes pediátricos con ALL Ph+ se administró durante un período máximo de 2 años [ver Dosificación y administración (2.2) y Estudios clínicos (14.4)].
SPRYCEL es un producto peligroso. Siga los procedimientos especiales de manipulación y eliminación aplicables.1
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
Las tabletas de SPRYCEL (dasatinib) están disponibles en tabletas recubiertas con película, biconvexas, de color blanco a blanquecino de 20 mg, 50 mg, 70 mg, 80 mg, 100 mg y 140 mg.
4 CONTRAINDICACIONES
Ninguna.
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Mielosupresión
El tratamiento con SPRYCEL se asocia con trombocitopenia, neutropenia y anemia graves (NCI CTCAE Grado 3 o 4), que ocurren más temprano y con más frecuencia en pacientes con LMC en fase avanzada o ALL Ph+ que en pacientes con LMC en fase crónica [ver Reacciones adversas (6.1)].
En pacientes con LMC en fase crónica, realice hemogramas completos (CBC) cada 2 semanas durante 12 semanas y luego cada 3 meses a partir de entonces, o según lo indique clínicamente. En pacientes con LMC en fase avanzada o ALL Ph+, realice CBC semanalmente durante los primeros 2 meses y luego mensualmente a partir de entonces, o según lo indique clínicamente.
En pacientes pediátricos con ALL Ph+ tratados con SPRYCEL en combinación con quimioterapia, realice CBC antes del inicio de cada bloque de quimioterapia y según lo indique clínicamente. Durante los bloques de consolidación de la quimioterapia, realice CBC cada 2 días hasta la recuperación.
La mielosupresión es generalmente reversible y normalmente se maneja reteniendo temporalmente SPRYCEL y/o reduciendo la dosis [ver Dosis y administración (2.5)].
5.2 Eventos relacionados con hemorragias
SPRYCEL puede causar hemorragias graves y fatales. En todos los estudios clínicos de LMC o ALL Ph+, hemorragias del sistema nervioso central (SNC) de Grado ≥3, incluidas las mortales, ocurrieron en <1% de los pacientes que recibieron SPRYCEL. La incidencia de hemorragia de Grado 3/4 ocurrió en el 5,8% de los pacientes adultos y generalmente requirió interrupciones del tratamiento y transfusiones. La incidencia de hemorragia de Grado 5 ocurrió en el 0,4% de los pacientes adultos. El sitio más frecuente de hemorragia fue el gastrointestinal [ver Reacciones adversas (6.1)]. La mayoría de los eventos de hemorragia en los estudios clínicos estaban asociados con trombocitopenia grave. Además de causar trombocitopenia en sujetos humanos, la dasatinib causó disfunción plaquetaria in vitro.
Los medicamentos concomitantes que inhiben la función plaquetaria o los anticoagulantes pueden aumentar el riesgo de hemorragia.
5.3 Retención de líquidos
SPRYCEL puede causar retención de líquidos [ver Reacciones adversas (6.1)]. Después de 5 años de seguimiento en el estudio aleatorizado de adultos con LMC en fase crónica recién diagnosticada (n = 258), se informó retención de líquidos de Grado 3 o 4 en el 5% de los pacientes, incluyendo el 3% de los pacientes con derrame pleural de Grado 3 o 4. En pacientes adultos con LMC en fase crónica recién diagnosticada o resistente o intolerante a imatinib, la retención de líquidos de Grado 3 o 4 ocurrió en el 6% de los pacientes tratados con SPRYCEL en la dosis recomendada (n = 548). En pacientes adultos con LMC en fase avanzada o ALL Ph+ tratados con SPRYCEL en la dosis recomendada (n = 304), se informó retención de líquidos de Grado 3 o 4 en el 8% de los pacientes, incluyendo derrame pleural de Grado 3 o 4 informado en el 7% de los pacientes. En pacientes pediátricos con LMC en fase crónica, se informaron casos de retención de líquidos de Grado 1 o 2 en el 10,3% de los pacientes.
Evalue a los pacientes que desarrollen síntomas de derrame pleural u otra retención de líquidos, como disnea nueva o empeorada con el esfuerzo o en reposo, dolor torácico pleurítico o tos seca, rápidamente con una radiografía de tórax o imágenes diagnósticas adicionales según sea apropiado. Los eventos de retención de líquidos generalmente se manejaron con medidas de cuidado de apoyo que pueden incluir diuréticos o cursos cortos de esteroides. El derrame pleural severo puede requerir toracocentesis y terapia de oxígeno. Considere la reducción de la dosis o la interrupción del tratamiento [ver Dosis y administración (2.5)].
5.4 Toxicidad cardiovascular
SPRYCEL puede causar disfunción cardiaca [ver Reacciones adversas (6.1)]. Después de 5 años de seguimiento en el ensayo aleatorizado de adultos con LMC en fase crónica recién diagnosticada (n = 258), ocurrieron las siguientes reacciones adversas cardíacas: eventos isquémicos cardíacos (3,9% dasatinib vs 1,6% imatinib), retención de líquidos cardíaca relacionada (8,5% dasatinib vs 3,9% imatinib) y anormalidades del sistema de conducción, más comúnmente arritmias y palpitaciones (7,0% dasatinib vs 5,0% imatinib). Se presentaron 2 casos (0,8%) de enfermedad oclusiva arterial periférica con imatinib y 2 (0,8%) ataques isquémicos transitorios con dasatinib. Monitoree a los pacientes en busca de signos o síntomas consistentes con disfunción cardiaca y trátelos adecuadamente.
5.5 Hipertensión arterial pulmonar
SPRYCEL puede aumentar el riesgo de desarrollar hipertensión arterial pulmonar (HAP) en pacientes adultos y pediátricos, que puede ocurrir en cualquier momento después del inicio, incluso después de más de 1 año de tratamiento. Las manifestaciones incluyen disnea, fatiga, hipoxia y retención de líquidos [ver Reacciones adversas (6.1)]. La HAP puede ser reversible al interrumpir SPRYCEL. Evalúe a los pacientes en busca de signos y síntomas de enfermedades cardiopulmonares subyacentes antes de iniciar SPRYCEL y durante el tratamiento. Si se confirma HAP, SPRYCEL debe suspenderse permanentemente.
5.6 Prolongación del QTc
SPRYCEL puede aumentar el riesgo de prolongación del QTc en pacientes, incluidos aquellos con hipokalemia o hipomagnesemia, pacientes con síndrome de QT largo congénito, pacientes que toman medicamentos antiarrítmicos u otros productos medicinales que provocan prolongación del QTc y terapia con dosis altas acumulativas de antraciclinas [ver Reacciones adversas (6.1)]. Corrija la hipokalemia o la hipomagnesemia antes y durante la administración de SPRYCEL.
5.7 Reacciones Dermatológicas Graves
Se han notificado casos de reacciones dermatológicas mucocutáneas graves, incluido el síndrome de Stevens-Johnson [ver Reacciones adversas (6.2)] y el eritema multiforme, en pacientes tratados con SPRYCEL. Suspenda permanentemente en pacientes que experimenten una reacción mucocutánea grave durante el tratamiento si no se puede identificar otra etiología.
5.8 Síndrome de Lisis Tumoral
Se ha notificado el síndrome de lisis tumoral en pacientes con resistencia a la terapia previa con imatinib, principalmente en la enfermedad en fase avanzada. Debido al potencial de síndrome de lisis tumoral, mantenga una hidratación adecuada, corrija los niveles de ácido úrico antes de iniciar la terapia con SPRYCEL y controle los niveles de electrolitos. Los pacientes con enfermedad en estadio avanzado y/o alta carga tumoral pueden tener un riesgo mayor y deben controlarse con más frecuencia [ver Reacciones adversas (6.1)].
5.9 Toxicidad Embriofetal
Con base en datos humanos limitados, SPRYCEL puede causar daño fetal cuando se administra a una mujer embarazada. Se han notificado efectos farmacológicos adversos de SPRYCEL, incluidos el hidropesía fetal, la leucopenia fetal y la trombocitopenia fetal, con la exposición materna a SPRYCEL. Avise a las mujeres en edad fértil y a los hombres con parejas femeninas en edad fértil que usen métodos anticonceptivos efectivos durante el tratamiento con SPRYCEL y durante 30 días después de la última dosis [ver Uso en poblaciones específicas (8.1, 8.3)].
5.10 Efectos sobre el Crecimiento y el Desarrollo en Pacientes Pediátricos
En ensayos pediátricos de SPRYCEL en CML en fase crónica después de al menos 2 años de tratamiento, se notificaron reacciones adversas asociadas con el crecimiento óseo y el desarrollo en 5 (5.2%) pacientes, uno de los cuales fue de intensidad grave (Retraso del crecimiento Grado 3). Estos 5 casos incluyeron casos de fusión retardada de epífisis, osteopenia, retraso del crecimiento y ginecomastia [ver Reacciones adversas (6.1) y Uso en poblaciones específicas (8.4)]. De estos 5 casos, 1 caso de osteopenia y 1 caso de ginecomastia se resolvieron durante el tratamiento.
Controle el crecimiento óseo y el desarrollo en pacientes pediátricos.
5.11 Hepatotoxicidad
SPRYCEL puede causar hepatotoxicidad, medida por elevaciones en bilirrubina, aspartato aminotransferasa (AST), alanina aminotransferasa (ALT) y fosfatasa alcalina [ver Reacciones adversas (6.1)]. Controle las transaminasas al inicio y mensualmente o según esté clínicamente indicado durante el tratamiento. Reduzca la dosis, suspenda o suspenda permanentemente SPRYCEL según la gravedad [ver Dosificación y administración (2.5)]. Cuando SPRYCEL se administra en combinación con quimioterapia, se ha observado toxicidad hepática en forma de elevación de las transaminasas e hiperbilirrubinemia. Controle la función hepática cuando SPRYCEL se use en combinación con quimioterapia.
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas clínicamente significativas se analizan con mayor detalle en otras secciones del etiquetado:
- •
- Mielosupresión [ver Dosificación y administración (2.5) y Advertencias y precauciones (5.1)].
- •
- Eventos relacionados con el sangrado [ver Advertencias y precauciones (5.2)].
- •
- Retención de líquidos [ver Advertencias y precauciones (5.3)].
- •
- Toxicidad cardiovascular [ver Advertencias y precauciones (5.4)].
- •
- Hipertensión arterial pulmonar [ver Advertencias y precauciones (5.5)].
- •
- Prolongación del QT [ver Advertencias y precauciones (5.6)].
- •
- Reacciones dermatológicas graves [ver Advertencias y precauciones (5.7)].
- •
- Síndrome de lisis tumoral [ver Advertencias y precauciones (5.8)].
- •
- Efectos sobre el crecimiento y el desarrollo en pacientes pediátricos [ver Advertencias y precauciones (5.10)].
- •
- Hepatotoxicidad [ver Advertencias y precauciones (5.11)].
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se llevan a cabo en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no se pueden comparar directamente con las tasas en los ensayos clínicos de otro fármaco y pueden no reflejar las tasas observadas en la práctica.
Los datos descritos a continuación reflejan la exposición a SPRYCEL administrado como terapia de un solo agente en todas las dosis probadas en estudios clínicos (n=2809), incluidos 324 pacientes adultos con LMC en fase crónica de nuevo diagnóstico, 2388 pacientes adultos con LMC en fase crónica o avanzada resistente o intolerante a imatinib o ALL Ph+ y 97 pacientes pediátricos con LMC en fase crónica. La duración media de la terapia en un total de 2712 pacientes adultos fue de 19,2 meses (rango de 0 a 93,2 meses). En un ensayo aleatorizado en pacientes con LMC en fase crónica de nuevo diagnóstico, la duración media de la terapia fue de aproximadamente 60 meses. La duración media de la terapia en 1618 pacientes adultos con LMC en fase crónica fue de 29 meses (rango de 0 a 92,9 meses).
La duración media de la terapia en 1094 pacientes adultos con LMC en fase avanzada o ALL Ph+ fue de 6,2 meses (rango de 0 a 93,2 meses).
En dos ensayos no aleatorizados en 97 pacientes pediátricos con LMC en fase crónica (51 pacientes de nuevo diagnóstico y 46 pacientes resistentes o intolerantes al tratamiento previo con imatinib), la duración media de la terapia fue de 51,1 meses (rango de 1,9 a 99,6 meses).
En la población general de 2712 pacientes adultos, el 88% de los pacientes experimentaron reacciones adversas en algún momento y el 19% experimentaron reacciones adversas que llevaron a la interrupción del tratamiento.
En el ensayo aleatorizado en pacientes adultos con LMC en fase crónica de nuevo diagnóstico, el fármaco se suspendió por reacciones adversas en el 16% de los pacientes con un seguimiento mínimo de 60 meses. Después de un seguimiento mínimo de 60 meses, la tasa de interrupción acumulativa fue del 39%. Entre los 1618 pacientes con LMC en fase crónica, se informaron reacciones adversas relacionadas con el fármaco que llevaron a la interrupción en 329 (20,3%) pacientes; entre los 1094 pacientes con LMC en fase avanzada o ALL Ph+, se informaron reacciones adversas relacionadas con el fármaco que llevaron a la interrupción en 191 (17,5%) pacientes.
Entre los 97 sujetos pediátricos, se informaron reacciones adversas relacionadas con el fármaco que llevaron a la interrupción en 1 paciente (1%).
Las reacciones adversas notificadas en ≥10% de los pacientes adultos, y otras reacciones adversas de interés, en un ensayo aleatorizado en pacientes con LMC en fase crónica de nuevo diagnóstico con un seguimiento medio de aproximadamente 60 meses se presentan en la Tabla 6.
Las reacciones adversas notificadas en ≥10% de los pacientes adultos tratados con la dosis recomendada de 100 mg una vez al día (n=165), y otras reacciones adversas de interés, en un ensayo aleatorizado de optimización de la dosis de pacientes con LMC en fase crónica resistente o intolerante a la terapia previa con imatinib con un seguimiento medio de aproximadamente 84 meses se presentan en la Tabla 8.
Las reacciones adversas notificadas en ≥10% de los pacientes pediátricos con un seguimiento medio de aproximadamente 51,1 meses se presentan en la Tabla 11.
Se informaron reacciones adversas graves (SAR) relacionadas con el fármaco para el 16,7% de los pacientes adultos en el ensayo aleatorizado de pacientes con LMC en fase crónica de nuevo diagnóstico. Las reacciones adversas graves notificadas en ≥5% de los pacientes incluyeron derrame pleural (5%).
Se informaron SAR relacionadas con el fármaco para el 26,1% de los pacientes tratados con la dosis recomendada de 100 mg una vez al día en el ensayo aleatorizado de optimización de la dosis de pacientes adultos con LMC en fase crónica resistente o intolerante a la terapia previa con imatinib. Las reacciones adversas graves notificadas en ≥5% de los pacientes incluyeron derrame pleural (10%).
Se informaron SAR relacionadas con el fármaco para el 14,4% de los pacientes pediátricos.
Leucemia mieloide crónica (LMC)
Las reacciones adversas (excluyendo las anomalías de laboratorio) que se notificaron en al menos el 10% de los pacientes adultos se muestran en la Tabla 6 para los pacientes de nuevo diagnóstico con LMC en fase crónica y las Tablas 8 y 10 para los pacientes con LMC con resistencia o intolerancia a la terapia previa con imatinib.
| Todos los grados | Grado 3/4 | |||
|---|---|---|---|---|
| SPRYCEL (n=258) |
Imatinib (n=258) |
SPRYCEL (n=258) |
Imatinib (n=258) |
|
| Reacción adversa | Porcentaje (%) de pacientes | |||
| a Incluye insuficiencia cardíaca aguda, insuficiencia cardíaca congestiva, miocardiopatía, disfunción diastólica, disminución de la fracción de eyección y disfunción ventricular izquierda. b Incluye eritema, eritema multiforme, erupción, erupción generalizada, erupción macular, erupción papular, erupción pustular, exfoliación de la piel y erupción vesicular. c Reacción adversa de interés especial con <10% de frecuencia. d Incluye hemorragia conjuntival, hemorragia del oído, equimosis, epistaxis, hemorragia ocular, hemorragia gingival, hematoma, hematuria, hemoptisis, hematoma intraabdominal, petequias, hemorragia escleral, hemorragia uterina y hemorragia vaginal. |
||||
|
Retención de líquidos |
38 |
45 |
5 |
1 |
|
Derrame pleural |
28 |
1 |
3 |
0 |
|
Edema localizado superficial |
14 |
38 |
0 |
<1 |
|
Hipertensión pulmonar |
5 |
<1 |
1 |
0 |
|
Edema generalizado |
4 |
7 |
0 |
0 |
|
Derrame pericárdico |
4 |
1 |
1 |
0 |
|
Insuficiencia cardíaca congestiva/disfunción cardíacaa |
2 |
1 |
<1 |
<1 |
|
Edema pulmonar |
1 |
0 |
0 |
0 |
|
Diarrea |
22 |
23 |
1 |
1 |
|
Dolor musculoesquelético |
14 |
17 |
0 |
<1 |
|
Erupciónb |
14 |
18 |
0 |
2 |
|
Dolor de cabeza |
14 |
11 |
0 |
0 |
|
Dolor abdominal |
11 |
8 |
0 |
1 |
|
Fatiga |
11 |
12 |
<1 |
0 |
|
Náuseas |
10 |
25 |
0 |
0 |
|
Mialgia |
7 |
12 |
0 |
0 |
|
Artralgia |
7 |
10 |
0 |
<1 |
|
Hemorragiac |
8 |
8 |
1 |
1 |
|
Hemorragia gastrointestinal |
2 |
2 |
1 |
0 |
|
Otra hemorragiad |
6 |
6 |
0 |
<1 |
|
Hemorragia del SNC |
<1 |
<1 |
0 |
<1 |
|
Vómitos |
5 |
12 |
0 |
0 |
|
Espasmos musculares |
5 |
21 |
0 |
<1 |
Una comparación de las tasas acumuladas de reacciones adversas notificadas en ≥10% de los pacientes con un seguimiento mínimo de 1 y 5 años en un ensayo aleatorizado de pacientes recién diagnosticados con CML en fase crónica tratados con SPRYCEL se muestra en la Tabla 7.
| Seguimiento mínimo de 1 año | Seguimiento mínimo de 5 años | |||
|---|---|---|---|---|
| Todos los grados | Grado 3/4 | Todos los grados | Grado 3/4 | |
| Reacción adversa | Porcentaje (%) de pacientes | |||
| a Incluye insuficiencia cardíaca aguda, insuficiencia cardíaca congestiva, miocardiopatía, disfunción diastólica, disminución de la fracción de eyección y disfunción ventricular izquierda. b Incluye eritema, eritema multiforme, erupción, erupción generalizada, erupción macular, erupción papular, erupción pustular, exfoliación de la piel y erupción vesicular. |
||||
|
Retención de líquidos |
19 |
1 |
38 |
5 |
|
Derrame pleural |
10 |
0 |
28 |
3 |
|
Edema localizado superficial |
9 |
0 |
14 |
0 |
|
Hipertensión pulmonar |
1 |
0 |
5 |
1 |
|
Edema generalizado |
2 |
0 |
4 |
0 |
|
Derrame pericárdico |
1 |
<1 |
4 |
1 |
|
Insuficiencia cardíaca congestiva/cardíaca |
2 |
<1 |
2 |
<1 |
|
Edema pulmonar |
<1 |
0 |
1 |
0 |
|
Diarrea |
17 |
<1 |
22 |
1 |
|
Dolor musculoesquelético |
11 |
0 |
14 |
0 |
|
Erupciónb |
11 |
0 |
14 |
0 |
|
Dolor de cabeza |
12 |
0 |
14 |
0 |
|
Dolor abdominal |
7 |
0 |
11 |
0 |
|
Fatiga |
8 |
<1 |
11 |
<1 |
|
Náuseas |
8 |
0 |
10 |
0 |
A los 60 meses, hubo 26 muertes en pacientes tratados con dasatinib (10.1%) y 26 muertes en pacientes tratados con imatinib (10.1%); 1 muerte en cada grupo fue evaluada por el investigador como relacionada con la terapia del estudio.
| a Incluye erupción medicamentosa, eritema, eritema multiforme, eritrosis, erupción exfoliativa, eritema generalizado, erupción genital, erupción por calor, milia, erupción, erupción eritematosa, erupción folicular, erupción generalizada, erupción macular, erupción maculopapular, erupción papular, erupción pruriginosa, erupción pustular, exfoliación de la piel, irritación de la piel, urticaria vesiculosa y erupción vesicular. | ||
|
|
100 mg una vez al día |
|
|
|
Crónica |
|
|
Reacción adversa |
Todos los grados |
Grado 3/4 |
|
Porcentaje (%) de pacientes |
||
|
Retención de líquidos |
48 |
7 |
|
Edema localizado superficial |
22 |
0 |
|
Derrame pleural |
28 |
5 |
|
Edema generalizado |
4 |
0 |
|
Derrame pericárdico |
3 |
1 |
|
Hipertensión pulmonar |
2 |
1 |
|
Dolor de cabeza |
33 |
1 |
|
Diarrea |
28 |
2 |
|
Fatiga |
26 |
4 |
|
Disnea |
24 |
2 |
|
Dolor musculoesquelético |
22 |
2 |
|
Náuseas |
18 |
1 |
|
Erupción cutáneaa |
18 |
2 |
|
Mialgia |
13 |
0 |
|
Artralgia |
13 |
1 |
|
Infección (incluidas bacterianas, virales, fúngicas, |
13 |
1 |
|
Dolor abdominal |
12 |
1 |
|
Hemorragia |
12 |
1 |
|
Hemorragia gastrointestinal |
2 |
1 |
|
Prurito |
12 |
1 |
|
Dolor |
11 |
1 |
|
Estreñimiento |
10 |
1 |
Las tasas acumulativas de reacciones adversas seleccionadas que se informaron con el tiempo en pacientes tratados con la dosis inicial recomendada de 100 mg una vez al día en un ensayo aleatorizado de optimización de dosis de pacientes con CML en fase crónica resistente o intolerante a imatinib se muestran en la Tabla 9.
| Seguimiento mínimo de 2 años | Seguimiento mínimo de 5 años | Seguimiento mínimo de 7 años | ||||
|---|---|---|---|---|---|---|
| Reacción adversa | Todos los grados | Grado 3/4 | Todos los grados | Grado 3/4 | Todos los grados | Grado 3/4 |
| Porcentaje (%) de pacientes | ||||||
| a Resultados del ensayo aleatorizado de optimización de dosis notificados en la población de dosis inicial recomendada de 100 mg una vez al día (n=165). | ||||||
|
Diarrea |
27 |
2 |
28 |
2 |
28 |
2 |
|
Retención de líquidos |
34 |
4 |
42 |
6 |
48 |
7 |
|
Edema superficial |
18 |
0 |
21 |
0 |
22 |
0 |
|
Derrame pleural |
18 |
2 |
24 |
4 |
28 |
5 |
|
Edema generalizado |
3 |
0 |
4 |
0 |
4 |
0 |
|
Derrame pericárdico |
2 |
1 |
2 |
1 |
3 |
1 |
|
Hipertensión pulmonar |
0 |
0 |
0 |
0 |
2 |
1 |
|
Hemorragia |
11 |
1 |
11 |
1 |
12 |
1 |
|
Hemorragia gastrointestinal |
2 |
1 |
2 |
1 |
2 |
1 |
| a Incluye disfunción ventricular, insuficiencia cardíaca, insuficiencia cardíaca congestiva, miocardiopatía, miocardiopatía congestiva, disfunción diastólica, disminución de la fracción de eyección e insuficiencia ventricular. b Incluye erupción medicamentosa, eritema, eritema multiforme, eritrosis, erupción exfoliativa, eritema generalizado, erupción genital, erupción por calor, milia, erupción, erupción eritematosa, erupción folicular, erupción generalizada, erupción macular, erupción maculopapular, erupción papular, erupción pruriginosa, erupción pustular, exfoliación de la piel, irritación de la piel, urticaria vesiculosa y erupción vesicular. |
||||||
|
140 mg una vez al día |
||||||
|
Blasto mieloide acelerado |
Blasto mieloide |
Blasto linfoide |
||||
|
Reacción adversa |
Todos los |
Grado |
Todos los |
Grado |
Todos los |
Grado |
|
Porcentaje (%) de pacientes |
||||||
|
Retención de líquidos |
35 |
8 |
34 |
7 |
21 |
6 |
|
Edema localizado superficial |
18 |
1 |
14 |
0 |
3 |
0 |
|
Derrame pleural |
21 |
7 |
20 |
7 |
21 |
6 |
|
Edema generalizado |
1 |
0 |
3 |
0 |
0 |
0 |
|
Derrame pericárdico |
3 |
1 |
0 |
0 |
0 |
0 |
|
Insuficiencia cardíaca congestiva/disfunción |
0 |
0 |
4 |
0 |
0 |
0 |
|
Edema pulmonar |
1 |
0 |
4 |
3 |
0 |
0 |
|
Dolor de cabeza |
27 |
1 |
18 |
1 |
15 |
3 |
|
Diarrea |
31 |
3 |
20 |
5 |
18 |
0 |
|
Fatiga |
19 |
2 |
20 |
1 |
9 |
3 |
|
Disnea |
20 |
3 |
15 |
3 |
3 |
3 |
|
Dolor musculoesquelético |
11 |
0 |
8 |
1 |
0 |
0 |
|
Náuseas |
19 |
1 |
23 |
1 |
21 |
3 |
|
Erupción cutáneab |
15 |
0 |
16 |
1 |
21 |
0 |
|
Artralgia |
10 |
0 |
5 |
1 |
0 |
0 |
|
Infección (incluyendo bacteriana, viral, micótica, |
10 |
6 |
14 |
7 |
9 |
0 |
|
Hemorragia |
26 |
8 |
19 |
9 |
24 |
9 |
|
Hemorragia gastrointestinal |
8 |
6 |
9 |
7 |
9 |
3 |
|
Hemorragia del SNC |
1 |
1 |
0 |
0 |
3 |
3 |
|
Vómitos |
11 |
1 |
12 |
0 |
15 |
0 |
|
Pirexia |
11 |
2 |
18 |
3 |
6 |
0 |
|
Neutropenia febril |
4 |
4 |
12 |
12 |
12 |
12 |
| Todos los grados | Grado 3/4 | |
|---|---|---|
| Reacción adversa | Porcentaje (%) de pacientes | |
|
Dolor de cabeza |
28 |
3 |
|
Náuseas |
20 |
0 |
|
Diarrea |
21 |
0 |
|
Erupción cutánea |
19 |
0 |
|
Vómitos |
13 |
0 |
|
Dolor en la extremidad |
19 |
1 |
|
Dolor abdominal |
16 |
0 |
|
Fatiga |
10 |
0 |
|
Artralgia |
10 |
1 |
Se notificaron reacciones adversas asociadas con el crecimiento y desarrollo óseo en 5 (5.2%) de los pacientes pediátricos con CML en fase crónica [ver Advertencias y precauciones (5.10)].
Anormalidades de laboratorio
La mielosupresión se notificó comúnmente en todas las poblaciones de pacientes. La frecuencia de neutropenia, trombocitopenia y anemia de grado 3 o 4 fue mayor en pacientes con CML en fase avanzada que en CML en fase crónica (Tablas 12 y 13). La mielosupresión se notificó en pacientes con valores de laboratorio basales normales, así como en pacientes con anormalidades de laboratorio preexistentes.
En los pacientes que experimentaron mielosupresión grave, la recuperación generalmente ocurrió después de la interrupción o reducción de la dosis; la interrupción permanente del tratamiento ocurrió en el 2% de los pacientes adultos con CML en fase crónica de nuevo diagnóstico y el 5% de los pacientes adultos con resistencia o intolerancia a la terapia previa con imatinib [ver Advertencias y precauciones (5.1)].
Se notificaron elevaciones de grado 3 o 4 de transaminasas o bilirrubina y hipocalcemia, hipopotasemia e hipofosfatemia de grado 3 o 4 en pacientes con todas las fases de CML, pero se notificaron con una frecuencia mayor en pacientes con CML en fase blástica mieloide o linfoide. Las elevaciones de transaminasas o bilirrubina generalmente se manejaron con reducción o interrupción de la dosis. Los pacientes que desarrollaron hipocalcemia de grado 3 o 4 durante la terapia con SPRYCEL a menudo se recuperaron con la suplementación oral de calcio.
Las anormalidades de laboratorio notificadas en pacientes adultos con CML en fase crónica de nuevo diagnóstico se muestran en la Tabla 12. No hubo interrupciones de la terapia con SPRYCEL en esta población de pacientes debido a parámetros bioquímicos de laboratorio.
| SPRYCEL (n=258) |
Imatinib (n=258) |
|
|---|---|---|
| Porcentaje (%) de pacientes | ||
| Grados de CTC: neutropenia (grado 3 ≥0.5–<1.0 × 109/L, grado 4 <0.5 × 109/L); trombocitopenia (grado 3 ≥25–<50 × 109/L, grado 4 <25 × 109/L); anemia (hemoglobina grado 3 ≥65–<80 g/L, grado 4 <65 g/L); creatinina elevada (grado 3 >3–6 × límite superior del rango normal (ULN), grado 4 >6 × ULN); bilirrubina elevada (grado 3 >3–10 × ULN, grado 4 >10 × ULN); SGOT o SGPT elevado (grado 3 >5–20 × ULN, grado 4 >20 × ULN); hipocalcemia (grado 3 <7.0–6.0 mg/dL, grado 4 <6.0 mg/dL); hipofosfatemia (grado 3 <2.0–1.0 mg/dL, grado 4 <1.0 mg/dL); hipopotasemia (grado 3 <3.0–2.5 mmol/L, grado 4 <2.5 mmol/L). | ||
|
Parámetros hematológicos |
|
|
|
Neutropenia |
29 |
24 |
|
Trombocitopenia |
22 |
14 |
|
Anemia |
13 |
9 |
|
Parámetros bioquímicos |
|
|
|
Hipofosfatemia |
7 |
31 |
|
Hipopotasemia |
0 |
3 |
|
Hipocalcemia |
4 |
3 |
|
SGPT (ALT) elevado |
<1 |
2 |
|
SGOT (AST) elevado |
<1 |
1 |
|
Bilirrubina elevada |
1 |
0 |
|
Creatinina elevada |
1 |
1 |
Las anormalidades de laboratorio reportadas en pacientes con CML resistente o intolerante al imatinib que recibieron las dosis iniciales recomendadas de SPRYCEL se muestran por fase de la enfermedad en la Tabla 13.
| Grados CTC: neutropenia (Grado 3 ≥0.5–<1.0 × 109/L, Grado 4 <0.5 × 109/L); trombocitopenia (Grado 3 ≥25–<50 × 109/L, Grado 4 <25 × 109/L); anemia (hemoglobina Grado 3 ≥65–<80 g/L, Grado 4 <65 g/L); creatinina elevada (Grado 3 >3–6 × límite superior del rango normal (ULN), Grado 4 >6 × ULN); bilirrubina elevada (Grado 3 >3–10 × ULN, Grado 4 >10 × ULN); SGOT o SGPT elevados (Grado 3 >5–20 × ULN, Grado 4 >20 × ULN); hipocalcemia (Grado 3 <7.0–6.0 mg/dL, Grado 4 <6.0 mg/dL); hipofosfatemia (Grado 3 <2.0–1.0 mg/dL, Grado 4 <1.0 mg/dL); hipokalemia (Grado 3 <3.0–2.5 mmol/L, Grado 4 <2.5 mmol/L). * Los parámetros hematológicos para la dosificación de 100 mg una vez al día en la fase crónica de la CML reflejan un seguimiento mínimo de 60 meses. |
||||
|
Fase Crónica de la CML |
Fase Avanzada de la CML |
|||
|
Fase Acelerada |
Fase de Blastos Mieloides |
Fase de Blastos Linfoides |
||
|
(n=165) |
(n=157) |
(n=74) |
(n=33) |
|
|
|
Porcentaje (%) de Pacientes |
|||
|
Parámetros Hematológicos* |
|
|
|
|
|
Neutropenia |
36 |
58 |
77 |
79 |
|
Trombocitopenia |
24 |
63 |
78 |
85 |
|
Anemia |
13 |
47 |
74 |
52 |
|
Parámetros Bioquímicos |
|
|
|
|
|
Hipofosfatemia |
10 |
13 |
12 |
18 |
|
Hipokalemia |
2 |
7 |
11 |
15 |
|
Hipocalcemia |
<1 |
4 |
9 |
12 |
|
SGPT (ALT) Elevado |
0 |
2 |
5 |
3 |
|
SGOT (AST) Elevado |
<1 |
0 |
4 |
3 |
|
Bilirrubina Elevada |
<1 |
1 |
3 |
6 |
|
Creatinina Elevada |
0 |
2 |
8 |
0 |
Entre los pacientes adultos con CML en fase crónica con resistencia o intolerancia a la terapia previa con imatinib, las citopenias acumulativas de Grado 3 o 4 fueron similares a los 2 y 5 años, incluyendo: neutropenia (36% vs 36%), trombocitopenia (23% vs 24%) y anemia (13% vs 13%).
En los estudios pediátricos en CML, las tasas de anormalidades de laboratorio fueron consistentes con el perfil conocido para los parámetros de laboratorio en adultos.
Leucemia linfoblástica aguda con cromosoma Filadelfia positivo (Ph+ ALL) en adultos
Un total de 135 pacientes adultos con Ph+ ALL fueron tratados con SPRYCEL en estudios clínicos. La duración media del tratamiento fue de 3 meses (rango 0,03–31 meses). El perfil de seguridad de los pacientes con Ph+ ALL fue similar al de los pacientes con CML en fase blástica linfoide. Las reacciones adversas más frecuentemente reportadas incluyeron eventos de retención de líquidos, como derrame pleural (24%) y edema superficial (19%), y trastornos gastrointestinales, como diarrea (31%), náuseas (24%) y vómitos (16%). También se reportaron con frecuencia hemorragia (19%), pirexia (17%), erupción cutánea (16%) y disnea (16%). Las reacciones adversas graves reportadas en ≥5% de los pacientes incluyeron derrame pleural (11%), hemorragia gastrointestinal (7%), neutropenia febril (6%) e infección (5%).
Leucemia linfoblástica aguda con cromosoma Filadelfia positivo (Ph+ ALL) en pacientes pediátricos
La seguridad de SPRYCEL administrado de forma continua en combinación con quimioterapia multiagente se determinó en un estudio multicóhorte de 81 pacientes pediátricos con Ph+ ALL de nuevo diagnóstico. [ver Estudios clínicos (14.4)]. La duración media de la terapia fue de 24 meses (rango 2 a 27 meses).
Las reacciones adversas fatales ocurrieron en 3 pacientes (4%), todas las cuales fueron debidas a infecciones. Ocho (10%) pacientes experimentaron reacciones adversas que llevaron a la interrupción del tratamiento, incluyendo sepsis por hongos, hepatotoxicidad en el contexto de enfermedad de injerto contra huésped, trombocitopenia, infección por CMV, neumonía, náuseas, enteritis e hipersensibilidad a los medicamentos.
Las reacciones adversas graves más comunes (incidencia ≥10%) fueron pirexia, neutropenia febril, mucositis, diarrea, sepsis, hipotensión, infecciones (bacterianas, virales y fúngicas), hipersensibilidad, vómitos, insuficiencia renal, dolor abdominal y dolor musculoesquelético.
La incidencia de reacciones adversas comunes (incidencia ≥20%) en el estudio se muestra en la Tabla 14:
|
Porcentaje (%) de pacientes |
||
|
Reacción adversa |
Todos los grados |
Grado 3/4 |
|
Mucositis |
93 |
60 |
|
Neutropenia febril |
86 |
86 |
|
Pirexia |
85 |
17 |
|
Diarrea |
84 |
31 |
|
Náuseas |
84 |
11 |
|
Vómitos |
83 |
17 |
|
Dolor musculoesquelético |
83 |
25 |
|
Dolor abdominal |
78 |
17 |
|
Tos |
78 |
1 |
|
Dolor de cabeza |
77 |
15 |
|
Rash |
68 |
7 |
|
Fatigue |
59 |
3 |
|
Constipation |
57 |
1 |
|
Arrhythmia |
47 |
12 |
|
Hypertension |
47 |
10 |
|
Edema |
47 |
6 |
|
Viral infection |
40 |
12 |
|
Hypotension |
40 |
26 |
|
Decreased appetite |
38 |
22 |
|
Hypersensitivity |
36 |
20 |
|
Upper respiratory tract infection |
36 |
10 |
|
Dyspnea |
35 |
10 |
|
Epistaxis |
31 |
6 |
|
Peripheral neuropathy |
31 |
7 |
|
Sepsis (excluding fungal) |
n/a |
31 |
|
Altered state of consciousness |
30 |
4 |
|
Fungal infection |
30 |
11 |
|
Pneumonia (excluding fungal) |
28 |
25 |
|
Pruritus |
28 |
– |
|
Clostridial infection (excluding sepsis) |
25 |
14 |
|
Urinary Tract Infection |
24 |
14 |
|
Bacteremia (excluding fungal) |
22 |
20 |
|
Eritema |
22 |
6 |
|
Escalofríos |
21 |
– |
|
Derrame pleural |
21 |
9 |
|
Sinusitis |
21 |
10 |
|
Deshidratación |
20 |
9 |
|
Insuficiencia renal |
20 |
9 |
|
Deterioro visual |
20 |
– |
La incidencia de reacciones adversas comunes atribuidas por el investigador a SPRYCEL (informadas con una frecuencia de ≥10%, todos los grados y grado 3/4, respectivamente) en el estudio (N=81), incluyó neutropenia febril (23%, 23%), náuseas (21%, 4%), vómitos (19%, 4%), mucositis (17%, 6%), dolor musculoesquelético (17%, 2%), dolor abdominal (16%, 5%), diarrea (16%, 7%), erupción cutánea (15%, 0%), fatiga (12%, 0%), pirexia (12%, 6%) y cefalea (12%, 5%).
Las anormalidades de laboratorio de grado 3/4 de CTCAE en pacientes pediátricos con ALL Ph+ tratados con SPRYCEL en combinación con quimioterapia se muestran en la Tabla 15.
| Porcentaje (%) de pacientes | |
|---|---|
|
Parámetros hematológicos |
|
|
Neutropenia |
96 |
|
Trombocitopenia |
88 |
|
Anemia |
82 |
|
Parámetros bioquímicos |
|
|
SGPT (ALT) elevado |
47 |
|
Hipokalemia |
40 |
|
SGOT (AST) elevado |
26 |
|
Hipocalcemia |
19 |
|
Hiponatremia |
19 |
|
Bilirrubina elevada |
11 |
|
Hipofosfatemia |
11 |
|
La clasificación de la toxicidad es según la versión 4 de CTCAE. |
|
Datos agrupados adicionales de los ensayos clínicos
Las siguientes reacciones adversas adicionales se informaron en pacientes adultos y pediátricos (n=2809) en estudios clínicos de SPRYCEL CML y pacientes adultos en estudios clínicos de ALL Ph+ con una frecuencia de ≥10%, 1%– <10%, 0.1%– <1%, o < 0.1%. Estas reacciones adversas se incluyen en función de la relevancia clínica.
Trastornos gastrointestinales: 1%–<10% – inflamación de la mucosa (incluida la mucositis/estomatitis), dispepsia, distensión abdominal, estreñimiento, gastritis, colitis (incluida la colitis neutropénica), trastorno de los tejidos blandos de la boca; 0.1%–<1% – ascitis, disfagia, fisura anal, úlcera gastrointestinal superior, esofagitis, pancreatitis, enfermedad por reflujo gastroesofágico; <0.1% – gastroenteropatía con pérdida de proteínas, íleo, pancreatitis aguda, fístula anal.
Trastornos generales y condiciones del lugar de administración: ≥10% – edema periférico, edema facial; 1%–<10% – astenia, dolor en el pecho, escalofríos; 0.1%–<1% – malestar general, otro edema superficial, hinchazón periférica; <0.1% – trastorno de la marcha.
Trastornos de la piel y del tejido subcutáneo: 1%–<10% – alopecia, acné, piel seca, hiperhidrosis, urticaria, dermatitis (incluido el eccema); 0.1%–<1% – trastorno de la pigmentación, úlcera cutánea, condiciones bulbosas, fotosensibilidad, trastorno de las uñas, dermatosis neutrófila, paniculitis, síndrome de eritrodisestesia palmar-plantar, trastorno del cabello; <0.1% – vasculitis leucocitoclástica, fibrosis cutánea.
Trastornos respiratorios, torácicos y mediastínicos: 1%–<10% – infiltración pulmonar, neumonitis, tos; 0.1%– <1% – asma, broncoespasmo, disfonía, hipertensión arterial pulmonar; <0.1% – síndrome de dificultad respiratoria aguda, embolia pulmonar.
Trastornos del sistema nervioso: 1%–<10% – neuropatía (incluida la neuropatía periférica), mareos, disgeusia, somnolencia; 0.1%–<1% – amnesia, temblor, síncope, trastorno del equilibrio; <0.1% – convulsión, accidente cerebrovascular, ataque isquémico transitorio, neuritis óptica, parálisis del VII par craneal, demencia, ataxia.
Trastornos de la sangre y del sistema linfático: 0.1%–<1% – linfadenopatía, linfopenia; <0.1% – aplasia pura de células rojas.
Trastornos musculoesqueléticos y del tejido conjuntivo: 1%–<10% – debilidad muscular, rigidez musculoesquelética; 0.1%–<1% – rabdomiólisis, tendinitis, inflamación muscular, osteonecrosis, artritis; <0.1% – fusión retardada de epífisis (informada en 1%–<10% en los estudios pediátricos), retraso del crecimiento (informado en 1%–<10% en los estudios pediátricos).
Pruebas de laboratorio: 1%–<10% – aumento de peso, disminución de peso; 0.1%–<1% – aumento de la creatina fosfoquinasa en sangre, aumento de la gamma-glutamiltransferasa.
Infecciones e infestaciones: 1%–<10% – neumonía (incluidas las bacterianas, virales y fúngicas), infección/inflamación de las vías respiratorias superiores, infección por virus del herpes, infección por enterocolitis, sepsis (incluidos los resultados fatales [0.2%]).
Trastornos del metabolismo y la nutrición: 1%–<10% – alteraciones del apetito, hiperuricemia; 0.1%–<1% – hipoalbuminemia, síndrome de lisis tumoral, deshidratación, hipercolesterolemia; <0.1% – diabetes mellitus.
Trastornos cardíacos: 1%–<10% – arritmia (incluida la taquicardia), palpitaciones; 0.1%–<1% – angina de pecho, cardiomegalia, pericarditis, arritmia ventricular (incluida la taquicardia ventricular), electrocardiograma con onda T anormal, aumento de la troponina; <0.1% – cor pulmonale, miocarditis, síndrome coronario agudo, paro cardíaco, prolongación del intervalo PR en el electrocardiograma, enfermedad de la arteria coronaria, pleuropericarditis.
Trastornos oculares: 1%–<10% – trastorno visual (incluida la alteración visual, visión borrosa y reducción de la agudeza visual), ojo seco; 0.1%–<1% – conjuntivitis, deterioro de la visión, aumento de la lagrimación, <0.1% – fotofobia.
Trastornos vasculares: 1%–<10% – rubor, hipertensión; 0.1%–<1% – hipotensión, tromboflebitis, trombosis; <0.1% – livedo reticularis, trombosis venosa profunda, embolia.
Trastornos psiquiátricos: 1%–<10% – insomnio, depresión; 0.1%–<1% – ansiedad, labilidad afectiva, estado confusional, disminución de la libido.
Embarazo, puerperio y condiciones perinatales: <0.1% – aborto.
Trastornos del sistema reproductor y de las mamas: 0.1%–<1% – ginecomastia, trastorno menstrual.
Lesiones, envenenamiento y complicaciones de procedimientos: 1%–<10% – contusión.
Trastornos del oído y del laberinto: 1%–<10% – tinnitus; 0.1%–<1% – vértigo, pérdida de audición.
Trastornos hepatobiliares: 0.1%–<1% – colestasis, colecistitis, hepatitis.
Trastornos renales y urinarios: 0.1%–<1% – frecuencia urinaria, insuficiencia renal, proteinuria; <0.1% – deterioro de la función renal.
Trastornos del sistema inmunitario: 0.1%–<1% – hipersensibilidad (incluido el eritema nudoso).
Trastornos endocrinos: 0.1%–<1% – hipotiroidismo; <0.1% – hipertiroidismo, tiroiditis.
6.2 Experiencia postcomercialización
Las siguientes reacciones adversas adicionales se han identificado durante el uso posterior a la aprobación de SPRYCEL. Debido a que estas reacciones se reportan voluntariamente de una población de tamaño incierto, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal con la exposición al fármaco.
Infecciones: reactivación del virus de la hepatitis B
Trastornos cardíacos: fibrilación auricular/aleteo auricular
Trastornos respiratorios, torácicos y mediastínicos: enfermedad pulmonar intersticial, quilotórax
Trastornos de la piel y del tejido subcutáneo: síndrome de Stevens-Johnson
Trastornos renales y urinarios: síndrome nefrótico
Trastornos de la sangre y del sistema linfático: microangiopatía trombótica
Trastornos hepatobiliares: hepatotoxicidad
7 INTERACCIONES MEDICAMENTOSAS
7.1 Efecto de otros medicamentos en Dasatinib
Inhibidores fuertes de CYP3A4
La coadministración con inhibidores fuertes de CYP3A4 puede aumentar las concentraciones de dasatinib [ver Farmacología clínica (12.3)]. El aumento de las concentraciones de dasatinib puede aumentar el riesgo de toxicidad. Evite el uso concomitante de inhibidores fuertes de CYP3A4. Si no se puede evitar la administración concomitante de un inhibidor fuerte de CYP3A4, considere una reducción de la dosis de SPRYCEL [ver Dosificación y administración (2.5)].
Inductores fuertes de CYP3A4
La coadministración de SPRYCEL con inductores fuertes de CYP3A4 puede disminuir las concentraciones de dasatinib [ver Farmacología clínica (12.3)]. La disminución de las concentraciones de dasatinib puede reducir la eficacia. Considere medicamentos alternativos con un menor potencial de inducción enzimática. Si no se puede evitar la administración concomitante de un inductor fuerte de CYP3A4, considere un aumento de la dosis de SPRYCEL.
Agentes reductores del ácido gástrico
La coadministración de SPRYCEL con un agente reductor del ácido gástrico puede disminuir las concentraciones de dasatinib. La disminución de las concentraciones de dasatinib puede reducir la eficacia.
No administre antagonistas de H2 o inhibidores de la bomba de protones con SPRYCEL. Considere el uso de antiácidos en lugar de antagonistas de H2 o inhibidores de la bomba de protones. Administre el antiácido al menos 2 horas antes o 2 horas después de la dosis de SPRYCEL. Evite la administración simultánea de SPRYCEL con antiácidos.
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de Riesgos
Basado en datos humanos limitados, SPRYCEL puede causar daño fetal cuando se administra a una mujer embarazada. Se han reportado efectos farmacológicos adversos incluyendo hidropesía fetal, leucopenia fetal y trombocitopenia fetal con exposición materna a SPRYCEL. Los estudios de reproducción en animales en ratas han demostrado una mortalidad extensa durante la organogénesis, el período fetal y en los neonatos. Se observaron malformaciones esqueléticas en un número limitado de conceptus de rata y conejo sobrevivientes. Estos hallazgos ocurrieron a concentraciones plasmáticas de dasatinib por debajo de las que se encuentran en humanos que reciben dosis terapéuticas de dasatinib [ver Datos]. Advierta a una mujer embarazada del riesgo potencial para un feto.
El riesgo de fondo estimado en la población general de EE. UU. de defectos de nacimiento mayores es del 2% al 4% y de aborto espontáneo del 15% al 20% de los embarazos clínicamente reconocidos.
Consideraciones Clínicas
Reacciones Adversas Fetales/Neonatales
Se ha reportado la transferencia transplacentaria de dasatinib. Dasatinib se ha medido en plasma fetal y líquido amniótico a concentraciones comparables a las del plasma materno. Se han reportado hidropesía fetal, leucopenia fetal y trombocitopenia fetal con exposición materna a dasatinib. Estos efectos farmacológicos adversos en el feto son similares a las reacciones adversas observadas en pacientes adultos y pueden resultar en daño fetal o muerte neonatal [ver Advertencias y Precauciones (5.1, 5.3)].
Datos
Datos Humanos
Basado en la experiencia humana, se sospecha que dasatinib causa malformaciones congénitas, incluyendo defectos del tubo neural, y efectos farmacológicos dañinos en el feto cuando se administra durante el embarazo.
Datos en Animales
En estudios no clínicos a concentraciones plasmáticas por debajo de las observadas en humanos que reciben dosis terapéuticas de dasatinib, se observaron toxicidades embrio-fetales en ratas y conejos. Se observó muerte fetal en ratas. Tanto en ratas como en conejos, las dosis más bajas de dasatinib probadas (rata: 2.5 mg/kg/día [15 mg/m2/día] y conejo: 0.5 mg/kg/día [6 mg/m2/día]) dieron como resultado toxicidades embrio-fetales. Estas dosis produjeron AUC maternas de 105 ng•h/mL y 44 ng•h/mL (0.1 veces el AUC humano) en ratas y conejos, respectivamente. Las toxicidades embrio-fetales incluyeron malformaciones esqueléticas en múltiples sitios (escápula, húmero, fémur, radio, costillas y clavícula), reducción de la osificación (esternón; vértebras torácicas, lumbares y sacras; falanges de la pata delantera; pelvis; y cuerpo hioides), edema y microhepatia. En un estudio de desarrollo pre y postnatal en ratas, la administración de dasatinib desde el día 16 de gestación (GD) hasta el día 20 de lactancia (LD), GD 21 hasta LD 20, o LD 4 hasta LD 20 resultó en una mortalidad extensa de cachorros a exposiciones maternas que estaban por debajo de las exposiciones en pacientes tratados con dasatinib a la dosis de etiquetado recomendada.
8.2 Lactancia
Resumen de Riesgos
No hay datos disponibles sobre la presencia de dasatinib en la leche materna, los efectos del fármaco en el niño amamantado o los efectos del fármaco en la producción de leche. Sin embargo, dasatinib está presente en la leche de ratas lactantes. Debido al potencial de reacciones adversas graves en los niños lactantes por SPRYCEL, no se recomienda la lactancia durante el tratamiento con SPRYCEL y durante 2 semanas después de la última dosis.
8.3 Mujeres y Hombres en Potencial Reproductivo
SPRYCEL puede causar daño fetal cuando se administra a una mujer embarazada [ver Uso en Poblaciones Específicas (8.1)].
Anticoncepción
Aconseje a las mujeres en potencial reproductivo y a los hombres con parejas femeninas en potencial reproductivo que utilicen métodos anticonceptivos efectivos durante el tratamiento con SPRYCEL y durante 30 días después de la última dosis.
Infertilidad
Basado en datos en animales, dasatinib puede resultar en daño a los tejidos reproductivos femeninos y masculinos [ver Toxicología No Clínica (13.1)].
8.4 Uso Pediátrico
CML Ph+ en Fase Crónica
La seguridad y eficacia de SPRYCEL en monoterapia se han demostrado en pacientes pediátricos con CML en fase crónica de nuevo diagnóstico [ver Estudios Clínicos (14.3)]. No hay datos en niños menores de 1 año de edad. Se reportaron reacciones adversas asociadas con el crecimiento y desarrollo óseo en 5 (5.2%) de los pacientes [ver Advertencias y Precauciones (5.10)].
ALL Ph+
La seguridad y eficacia de SPRYCEL en combinación con quimioterapia se han demostrado en pacientes pediátricos de un año de edad o más con ALL Ph+ de nuevo diagnóstico. El uso de SPRYCEL en pacientes pediátricos está respaldado por evidencia de un estudio pediátrico. No hay datos en niños menores de 1 año. Se reportó un caso de osteopenia de grado 1.
El perfil de seguridad de SPRYCEL en sujetos pediátricos fue comparable al reportado en estudios en sujetos adultos [ver Reacciones adversas (6.1) y Estudios clínicos (14.3, 14.4)].
Monitorear el crecimiento y desarrollo óseo en pacientes pediátricos [ver Advertencias y precauciones (5.10)].
Pacientes pediátricos con dificultad para tragar tabletas
Cinco pacientes con ALL Ph+ de 2 a 10 años de edad recibieron al menos una dosis de SPRYCEL tableta dispersada en jugo en el Estudio CA180372. La exposición para las tabletas dispersas fue 36% menor en comparación con las tabletas intactas en pacientes pediátricos [ver Farmacología clínica (12.3)]. Debido a los datos clínicos limitados disponibles, no está claro si la dispersión de las tabletas de SPRYCEL altera significativamente la seguridad y/o eficacia de SPRYCEL.
8.5 Uso en geriatría
De los 2712 pacientes en estudios clínicos de SPRYCEL, 617 (23%) tenían 65 años de edad o más, y 123 (5%) tenían 75 años de edad o más. No se observaron diferencias en la Respuesta Citogenética Completa Confirmada (cCCyR) y MMR entre los pacientes mayores y más jóvenes. Si bien el perfil de seguridad de SPRYCEL en la población geriátrica fue similar al de la población más joven, los pacientes de 65 años de edad o más tienen más probabilidades de experimentar las reacciones adversas comúnmente reportadas de fatiga, derrame pleural, diarrea, disnea, tos, hemorragia gastrointestinal inferior y alteración del apetito, y más probabilidades de experimentar las reacciones adversas menos frecuentes de distensión abdominal, mareos, derrame pericárdico, insuficiencia cardíaca congestiva, hipertensión, edema pulmonar y disminución de peso, y deben ser monitoreados de cerca.
10 SOBREDOSIS
La experiencia con sobredosis de SPRYCEL en estudios clínicos se limita a casos aislados. La sobredosis más alta de 280 mg por día durante 1 semana se informó en dos pacientes y ambos desarrollaron mielosupresión grave y sangrado. Dado que SPRYCEL está asociado con mielosupresión grave [ver Advertencias y precauciones (5.1) y Reacciones adversas (6.1)], controle de cerca a los pacientes que ingieran más de la dosis recomendada para detectar mielosupresión y brinde el tratamiento de apoyo adecuado.
La sobredosis aguda en animales se asoció con cardiotoxicidad. La evidencia de cardiotoxicidad incluyó necrosis ventricular y hemorragia valvular/ventricular/auricular a dosis únicas ≥100 mg/kg (600 mg/m2) en roedores. Hubo una tendencia a aumentar la presión arterial sistólica y diastólica en monos a dosis únicas ≥10 mg/kg (120 mg/m2).
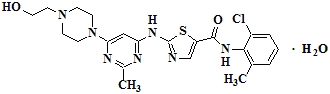
11 DESCRIPCIÓN
SPRYCEL (dasatinib) es un inhibidor de la cinasa. El nombre químico del dasatinib es N-(2-cloro-6-metilfenil)-2-[[6-[4-(2-hidroxietil)-1-piperazinil]-2-metil-4-pirimidinil]amino]-5-tiazolcarboxamida, monohidrato. La fórmula molecular es C22H26ClN7O2S • H2O, que corresponde a un peso de fórmula de 506.02 (monohidrato). La base libre anhidra tiene un peso molecular de 488.01. El dasatinib tiene la siguiente estructura química:
El dasatinib es un polvo de color blanco a blanquecino. La sustancia farmacológica es insoluble en agua y ligeramente soluble en etanol y metanol.
Los comprimidos de SPRYCEL son comprimidos recubiertos con película, biconvexos, de color blanco a blanquecino, que contienen dasatinib, con los siguientes ingredientes inactivos: lactosa monohidrato, celulosa microcristalina, croscarmelosa sódica, hidroxipropilcelulosa y estearato de magnesio. El recubrimiento del comprimido contiene hipromelosa, dióxido de titanio y polietilenglicol.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
Dasatinib, en concentraciones nanomolares, inhibe las siguientes quinasas: BCR-ABL, familia SRC (SRC, LCK, YES, FYN), c-KIT, EPHA2 y PDGFRβ. Según estudios de modelado, se predice que dasatinib se une a múltiples conformaciones de la quinasa ABL.
In vitro, dasatinib fue activo en líneas celulares leucémicas que representan variantes de enfermedades sensibles y resistentes al mesilato de imatinib. Dasatinib inhibió el crecimiento de líneas celulares de leucemia mieloide crónica (CML) y leucemia linfoblástica aguda (ALL) que sobreexpresaban BCR-ABL. En las condiciones de los ensayos, dasatinib pudo superar la resistencia a imatinib resultante de mutaciones en el dominio quinasa de BCR-ABL, la activación de vías de señalización alternativas que involucran las quinasas de la familia SRC (LYN, HCK) y la sobreexpresión del gen de resistencia a múltiples fármacos.
12.2 Farmacodinámica
Electrofisiología cardíaca
De 2440 pacientes tratados con SPRYCEL en todas las dosis probadas en ensayos clínicos, 16 pacientes (<1%) tuvieron prolongación del QTc reportada como reacción adversa. Veintidós pacientes (1%) experimentaron un QTcF > 500 ms. En 865 pacientes con leucemia tratados con SPRYCEL 70 mg BID en cinco estudios de fase 2, los cambios medios máximos en QTcF (límite superior del IC al 90%) desde el punto de partida oscilaron entre 7 ms y 13,4 ms.
Un análisis de los datos de cinco estudios de fase 2 en pacientes (70 mg BID) y un estudio de fase 1 en sujetos sanos (dosis única de 100 mg) sugiere que hay un aumento máximo de 3 a 6 milisegundos en el intervalo QTc corregido por Fridericia desde el punto de partida para los sujetos que reciben dosis terapéuticas de dasatinib, con intervalos de confianza superiores del 95% asociados <10 mseg.
12.3 Farmacocinética
La farmacocinética de dasatinib presenta aumentos proporcionales a la dosis en AUC y características de eliminación lineal en el rango de dosis de 15 mg/día (0,15 veces la dosis recomendada aprobada más baja) a 240 mg/día (1,7 veces la dosis recomendada aprobada más alta).
Con 100 mg QD, la concentración máxima en estado estacionario (Cmax) es de 82,2 ng/mL (CV% 69%), el área bajo la curva de concentración plasmática del fármaco en función del tiempo (AUC) es de 397 ng/mL*hr (CV% 55%). Se ha encontrado que la depuración de dasatinib es invariable en el tiempo. Cuando se administra a sujetos adultos sanos como comprimidos dispersos en jugo, la relación geométrica media ajustada fue de 0,97 (IC 90%: 0,85, 1,10) para Cmax y 0,84 (IC 90%: 0,78, 0,91) para AUC en comparación con los comprimidos intactos.
Absorción
Las concentraciones plasmáticas máximas (Cmax) de dasatinib se observan entre 0,5 horas y 6 horas (Tmax) después de la administración oral.
Efecto de la alimentación
Una comida rica en grasas aumentó el AUC medio de dasatinib después de una dosis única de 100 mg en un 14%. El contenido calórico total de la comida rica en grasas fue de 985 kcal. Las calorías derivadas de grasas, carbohidratos y proteínas fueron 52%, 34% y 14% para la comida rica en grasas.
Distribución
El volumen aparente de distribución es de 2505 L (CV% 93%).
La unión de dasatinib a las proteínas plasmáticas humanas in vitro fue de aproximadamente 96% y de su metabolito activo fue de 93%, sin dependencia de la concentración en el rango de 100 ng/mL a 500 ng/mL.
Dasatinib es un sustrato de P-gp in vitro.
Eliminación
La vida media terminal media de dasatinib es de 3 horas a 5 horas. La depuración oral aparente media es de 363,8 L/hora (CV% 81,3%).
Metabolismo
Dasatinib se metaboliza en humanos, principalmente por CYP3A4. CYP3A4 es la enzima principal responsable de la formación del metabolito activo. La flavina que contiene la monooxigenasa 3 (FMO-3) y las enzimas uridina difosfato-glucuronosiltransferasa (UGT) también están involucradas en la formación de los metabolitos de dasatinib.
La exposición del metabolito activo, que es equipotente a dasatinib, representa aproximadamente el 5% del AUC de dasatinib. El metabolito activo de dasatinib es poco probable que juegue un papel importante en la farmacología observada del fármaco. Dasatinib también tiene varios otros metabolitos oxidativos inactivos.
Excreción
La eliminación es principalmente a través de las heces. Después de una dosis radiomarcada única oral de dasatinib, el 4% de la radiactividad administrada se recuperó en la orina y el 85% en las heces en 10 días. Dasatinib sin cambios representó el 0,1% de la dosis administrada en la orina y el 19% de la dosis administrada en las heces, y el resto de la dosis fueron metabolitos.
Poblaciones específicas
La edad (de 15 a 86 años), el sexo y el deterioro renal (depuración de creatinina de 21,6 mL/min a 342,3 mL/min estimada por Cockcroft Gault) no tienen un efecto clínicamente relevante en la farmacocinética de dasatinib.
Pacientes pediátricos
La farmacocinética de dasatinib se evaluó en 43 pacientes pediátricos con leucemia o tumores sólidos a dosis orales que van desde 60 mg/m2 a 120 mg/m2 una vez al día, tomadas con o sin alimentos. La farmacocinética mostró proporcionalidad a la dosis con un aumento de la exposición relacionado con la dosis. La Tmax media se observó entre 0,5 horas y 6 horas y la vida media media fue de 2 horas a 5 horas. La media geométrica (CV%) del aclaramiento normalizado por peso corporal en estos 43 pacientes pediátricos es de 5,98 (41,5%) L/h/kg. En pacientes pediátricos con un régimen de dosificación de 60 mg/m2, las concentraciones plasmáticas promedio de estado estacionario de dasatinib simuladas por el modelo de media geométrica (CV%) fueron de 14,7 (64,6%) ng/mL (para 2 a <6 años de edad), 16,3 (97,5%) ng/mL (para 6 a <12 años de edad), y 18,2 (67,7%) ng/mL (para 12 años y mayores) [ver Dosificación y administración (2.2)]. El aclaramiento y el volumen de distribución de dasatinib cambian con el peso corporal en pacientes pediátricos. Dasatinib no se ha estudiado en pacientes < 1 año de edad.
La biodisponibilidad de las tabletas dispersas en pacientes pediátricos se estimó que era un 36% menor que la de las tabletas intactas.
Pacientes con insuficiencia hepática
En comparación con los sujetos con función hepática normal, los pacientes con insuficiencia hepática moderada (Child Pugh B) tuvieron disminuciones en la Cmax media en un 47% y en el AUC media en un 8%. Los pacientes con insuficiencia hepática grave (Child Pugh C) tuvieron disminuciones en la Cmax media en un 43% y en el AUC media en un 28% en comparación con los sujetos con función hepática normal.
Estudios de interacción medicamentosa
Enzimas del citocromo P450
La coadministración de ketoconazol (inhibidor potente de CYP3A4) dos veces al día aumentó la Cmax media de dasatinib en 4 veces y el AUC media de dasatinib en 5 veces después de una dosis oral única de 20 mg.
La coadministración de rifampicina (inductor potente de CYP3A4) una vez al día disminuyó la Cmax media de dasatinib en un 81% y el AUC media de dasatinib en un 82%.
Dasatinib es un inhibidor dependiente del tiempo de CYP3A4. Dasatinib no inhibe CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6 o 2E1. Dasatinib no induce enzimas CYP.
Agentes reductores del ácido gástrico
La administración de 30 mL de hidróxido de aluminio/hidróxido de magnesio 2 horas antes de una dosis única de SPRYCEL se asoció con ningún cambio relevante en el AUC media de dasatinib; sin embargo, la Cmax media de dasatinib aumentó en un 26%.
La administración simultánea de 30 mL de hidróxido de aluminio/hidróxido de magnesio con una dosis única de SPRYCEL se asoció con una reducción del 55% en el AUC media de dasatinib y una reducción del 58% en la Cmax media de dasatinib.
La administración de una dosis única de SPRYCEL 10 horas después de famotidina (antagonista de H2) redujo el AUC media de dasatinib en un 61% y la Cmax media de dasatinib en un 63%.
La administración de una dosis única de 100 mg de SPRYCEL 22 horas después de una dosis de 40 mg de omeprazol (inhibidor de la bomba de protones) en estado estacionario redujo el AUC media de dasatinib en un 43% y la Cmax media de dasatinib en un 42%.
Transportadores
Dasatinib no es un inhibidor de P-gp in vitro.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
En un estudio de carcinogenicidad de 2 años, se administraron dosis orales de dasatinib a ratas a 0.3, 1 y 3 mg/kg/día. La dosis más alta resultó en un nivel de exposición al fármaco en plasma (AUC) aproximadamente 60% de la exposición humana a 100 mg una vez al día. Dasatinib indujo un aumento estadísticamente significativo en la incidencia combinada de carcinomas de células escamosas y papilomas en el útero y el cuello uterino de las hembras de dosis alta y adenoma de próstata en los machos de dosis baja.
Dasatinib fue clastogénico cuando se probó in vitro en células de ovario de hámster chino, con y sin activación metabólica. Dasatinib no fue mutagénico cuando se probó en un ensayo de células bacterianas in vitro (prueba de Ames) y no fue genotóxico en un estudio de micronúcleos de rata in vivo.
Dasatinib no afectó el apareamiento o la fertilidad en ratas macho y hembra a una exposición al fármaco en plasma (AUC) similar a la exposición humana a 100 mg diarios. En estudios de dosis repetidas, la administración de dasatinib resultó en una reducción del tamaño y la secreción de las vesículas seminales, y próstata, vesículas seminales y testículos inmaduros. La administración de dasatinib resultó en inflamación uterina y mineralización en monos, y ovarios quísticos e hipertrofia ovárica en roedores.
14 ESTUDIOS CLÍNICOS
14.1 CML en Fase Crónica Recién Diagnosticada en Adultos
DASISION (Estudio de Dasatinib vs Imatinib en Pacientes con Leucemia Mieloide Crónica de Novo) (NCT00481247) fue un ensayo abierto, multicéntrico, internacional y aleatorizado realizado en pacientes adultos con CML en fase crónica recién diagnosticada. Un total de 519 pacientes fueron aleatorizados para recibir SPRYCEL 100 mg una vez al día o imatinib 400 mg una vez al día. Los pacientes con antecedentes de enfermedad cardíaca fueron incluidos en este ensayo, excepto aquellos que habían tenido un infarto de miocardio en los 6 meses anteriores, insuficiencia cardíaca congestiva en los 3 meses anteriores, arritmias significativas o prolongación del QTc. El criterio de valoración principal fue la tasa de respuesta citogenética completa confirmada (CCyR) dentro de los 12 meses. La CCyR confirmada se definió como una CCyR observada en dos ocasiones consecutivas (con al menos 28 días de diferencia).
La edad media fue de 46 años en el grupo de SPRYCEL y de 49 años en los grupos de imatinib, con un 10% y un 11% de pacientes ≥65 años de edad, respectivamente. Hubo ligeramente más pacientes masculinos que femeninos en ambos grupos (59% vs 41%). El 53% de todos los pacientes eran caucásicos y el 39% eran asiáticos. En la línea de base, la distribución de las puntuaciones de Hasford fue similar en los grupos de tratamiento con SPRYCEL e imatinib (riesgo bajo: 33% y 34%; riesgo intermedio: 48% y 47%; riesgo alto: 19% y 19%, respectivamente). Con un seguimiento mínimo de 12 meses, el 85% de los pacientes aleatorizados a SPRYCEL y el 81% de los pacientes aleatorizados a imatinib seguían en el estudio.
Con un seguimiento mínimo de 24 meses, el 77% de los pacientes aleatorizados a SPRYCEL y el 75% de los pacientes aleatorizados a imatinib seguían en el estudio y con un seguimiento mínimo de 60 meses, el 61% y el 62% de los pacientes, respectivamente, seguían en tratamiento en el momento del cierre del estudio.
Los resultados de eficacia se resumen en la Tabla 16.
| SPRYCEL (n=259) |
Imatinib (n=260) |
|
|---|---|---|
| a La CCyR confirmada se define como una CCyR observada en dos ocasiones consecutivas con al menos 28 días de diferencia. b La respuesta molecular mayor (en cualquier momento) se definió como una proporción BCR-ABL ≤0,1% mediante RQ-PCR en muestras de sangre periférica estandarizadas en la escala internacional. Estas son tasas acumulativas que representan un seguimiento mínimo para el período de tiempo especificado. * Ajustado para la puntuación de Hasford e indicó significación estadística a un nivel nominal predefinido de significación. IC = intervalo de confianza. |
||
|
CCyR Confirmadaa |
||
|
Dentro de los 12 meses (IC del 95%) |
76,8% (71,2–81,8) |
66,2% (60,1–71,9) |
|
Valor P |
0,007* |
|
|
Respuesta Molecular Mayorb |
||
|
12 meses (IC del 95%) |
52,1% (45,9–58,3) |
33,8% (28,1–39,9) |
|
Valor P |
<0,0001 |
|
|
60 meses (IC del 95%) |
76,4% (70,8–81,5) |
64,2% (58,1–70,1) |
Las tasas confirmadas de CCyR en 24, 36 y 60 meses para los brazos de SPRYCEL frente a imatinib fueron 80% frente a 74%, 83% frente a 77% y 83% frente a 79%, respectivamente. Las tasas de MMR a los 24 y 36 meses para los brazos de SPRYCEL frente a imatinib fueron 65% frente a 50% y 69% frente a 56%, respectivamente.
Después de 60 meses de seguimiento, el tiempo medio hasta la CCyR confirmada fue de 3.1 meses en 215 respondientes de SPRYCEL y de 5.8 meses en 204 respondientes de imatinib. El tiempo medio hasta la MMR después de 60 meses de seguimiento fue de 9.3 meses en 198 respondientes de SPRYCEL y de 15.0 meses en 167 respondientes de imatinib.
A los 60 meses, 8 pacientes (3%) en el brazo de dasatinib progresaron a fase acelerada o crisis blástica, mientras que 15 pacientes (6%) en el brazo de imatinib progresaron a fase acelerada o crisis blástica.
Las tasas estimadas de supervivencia a 60 meses para los pacientes tratados con SPRYCEL e imatinib fueron 90.9% (IC: 86.6%–93.8%) y 89.6% (IC: 85.2%–92.8%), respectivamente. Según los datos 5 años después de que el último paciente se inscribió en el ensayo, el 83% y el 77% de los pacientes estaban vivos en los grupos de tratamiento con dasatinib e imatinib, respectivamente; el 10% se sabía que habían fallecido en ambos grupos de tratamiento y el 7% y el 13% tenían un estado de supervivencia desconocido en los grupos de tratamiento con dasatinib e imatinib, respectivamente.
En el seguimiento de 60 meses en el brazo de SPRYCEL, la tasa de MMR en cualquier momento en cada grupo de riesgo determinado por la puntuación de Hasford fue 90% (riesgo bajo), 71% (riesgo intermedio) y 67% (riesgo alto). En el brazo de imatinib, la tasa de MMR en cualquier momento en cada grupo de riesgo determinado por la puntuación de Hasford fue 69% (riesgo bajo), 65% (riesgo intermedio) y 54% (riesgo alto).
La secuenciación de BCR-ABL se realizó en muestras de sangre de pacientes en el ensayo de diagnóstico reciente que interrumpieron el tratamiento con dasatinib o imatinib. Entre los pacientes tratados con dasatinib, las mutaciones detectadas fueron T315I, F317I/L y V299L.
Según datos in vitro, parece que dasatinib no es activo contra la mutación T315I.
14.2 CML resistente o intolerante a imatinib o ALL Ph+ en adultos
Se investigaron la eficacia y la seguridad de SPRYCEL en pacientes adultos con CML o ALL Ph+ cuya enfermedad era resistente o intolerante a imatinib: 1158 pacientes tenían CML en fase crónica, 858 pacientes tenían CML en fase acelerada, fase blástica mieloide o fase blástica linfocítica, y 130 pacientes tenían ALL Ph+. En un ensayo clínico en CML en fase crónica, la resistencia a imatinib se definió como el fracaso para lograr una respuesta hematológica completa (CHR; después de 3 meses), una respuesta citogenética mayor (MCyR; después de 6 meses) o una respuesta citogenética completa (CCyR; después de 12 meses); o la pérdida de una respuesta molecular previa (con un aumento concurrente ≥10% en metafases Ph+), una respuesta citogenética o una respuesta hematológica. La intolerancia a imatinib se definió como la incapacidad de tolerar 400 mg o más de imatinib al día o la interrupción de imatinib debido a toxicidad.
Los resultados descritos a continuación se basan en un mínimo de 2 años de seguimiento después del inicio del tratamiento con SPRYCEL en pacientes con un tiempo medio desde el diagnóstico inicial de aproximadamente 5 años. En todos los estudios, el 48% de los pacientes eran mujeres, el 81% eran blancos, el 15% eran negros o asiáticos, el 25% tenían 65 años o más, y el 5% tenían 75 años o más. La mayoría de los pacientes tenían historias de enfermedad prolongadas con tratamiento previo extenso, incluyendo imatinib, quimioterapia citotóxica, interferón y trasplante de células madre. En general, el 80% de los pacientes tenían enfermedad resistente a imatinib y el 20% de los pacientes eran intolerantes a imatinib. La dosis máxima de imatinib había sido de 400–600 mg/día en aproximadamente el 60% de los pacientes y >600 mg/día en el 40% de los pacientes.
El punto final de eficacia primario en CML en fase crónica era la MCyR, definida como la eliminación (CCyR) o la disminución sustancial (en al menos un 65%, respuesta citogenética parcial) de las células hematopoyéticas Ph+. El punto final de eficacia primario en CML en fase acelerada, fase blástica mieloide, fase blástica linfocítica y ALL Ph+ era la respuesta hematológica mayor (MaHR), definida como una CHR o ninguna evidencia de leucemia (NEL).
CML en fase crónica
Ensayo de optimización de dosis: Se realizó un ensayo aleatorizado, de etiqueta abierta (NCT00123474) en pacientes adultos con CML en fase crónica para evaluar la eficacia y la seguridad de SPRYCEL administrado una vez al día en comparación con SPRYCEL administrado dos veces al día. Los pacientes con enfermedades cardíacas significativas, incluyendo infarto de miocardio en los últimos 6 meses, insuficiencia cardíaca congestiva en los últimos 3 meses, arritmias significativas o prolongación del QTc, fueron excluidos del ensayo. El punto final de eficacia primario era la MCyR en pacientes con CML resistente a imatinib. Un total de 670 pacientes, de los cuales 497 tenían enfermedad resistente a imatinib, fueron aleatorizados al grupo de SPRYCEL 100 mg una vez al día, 140 mg una vez al día, 50 mg dos veces al día o 70 mg dos veces al día. La duración media del tratamiento fue de 22 meses.
Se logró la eficacia en todos los grupos de tratamiento con SPRYCEL, y el régimen de una vez al día demostró una eficacia comparable (no inferioridad) al régimen de dos veces al día en el punto final de eficacia primario (diferencia en MCyR 1.9%; IC 95% [−6.8%–10.6%]); sin embargo, el régimen de 100 mg una vez al día demostró una mejor seguridad y tolerabilidad.
Los resultados de eficacia se presentan en las Tablas 17 y 18 para pacientes adultos con CML en fase crónica que recibieron la dosis inicial recomendada de 100 mg una vez al día.
| Todos los pacientes | 100 mg Una vez al día (n=167) |
|---|---|
| a CHR (respuesta confirmada después de 4 semanas): WBC ≤ ULN institucional, plaquetas <450.000/mm3, sin blastos o promielocitos en sangre periférica, <5% mielocitos más metamilocitos en sangre periférica, basófilos en sangre periférica <20% y sin afectación extramedular. b MCyR combina tanto las respuestas completas (0% metafases Ph+) como parciales (>0%–35%). |
|
|
Tasa de respuesta hematológica % (IC 95%) |
|
|
CHRa |
92% (86–95) |
|
Cytogenetic Response Rate % (95% CI) |
|
|
MCyRb |
63% (56–71) |
|
CCyR |
50% (42–58) |
| Período mínimo de seguimiento | |||
|---|---|---|---|
| 2 años | 5 años | 7 años | |
| a Resultados informados en la dosis inicial recomendada de 100 mg una vez al día. b Criterios de respuesta molecular mayor: Definidos como transcritos BCR-ABL/control ≤0,1% por RQ-PCR en muestras de sangre periférica. |
|||
|
Respuesta molecular mayorb % (n/N) |
|||
|
Todos los pacientes aleatorizados |
34% (57/167) |
43% (71/167) |
44% (73/167) |
|
Pacientes resistentes a imatinib |
33% (41/124) |
40% (50/124) |
41% (51/124) |
|
Pacientes intolerantes a imatinib |
37% (16/43) |
49% (21/43) |
51% (22/43) |
Basándose en los datos 7 años después de que el último paciente se inscribió en el ensayo, se sabía que el 44% estaba vivo, el 31% se sabía que había muerto y el 25% tenía un estado de supervivencia desconocido.
A los 7 años, la transformación a fase acelerada o blástica se produjo en nueve pacientes en tratamiento en el grupo de tratamiento de 100 mg una vez al día.
CML en fase avanzada y ALL Ph+
Ensayo de optimización de dosis: Se realizó un ensayo aleatorizado abierto (NCT00123487) en pacientes con CML en fase avanzada (CML en fase acelerada, CML en fase blástica mieloide o CML en fase blástica linfoide) para evaluar la eficacia y seguridad de SPRYCEL administrado una vez al día en comparación con SPRYCEL administrado dos veces al día. El criterio de valoración principal de eficacia fue MaHR. Se aleatorizaron un total de 611 pacientes a los grupos de SPRYCEL 140 mg una vez al día o 70 mg dos veces al día. La duración media del tratamiento fue de aproximadamente 6 meses para ambos grupos de tratamiento. El régimen una vez al día demostró una eficacia comparable (no inferioridad) al régimen dos veces al día en el criterio de valoración principal de eficacia; sin embargo, el régimen de 140 mg una vez al día demostró una mejor seguridad y tolerabilidad.
Las tasas de respuesta para los pacientes en el grupo de 140 mg una vez al día se presentan en la Tabla 19.
| 140 mg una vez al día | ||||
|---|---|---|---|---|
| Acelerada (n=158) |
Blástica mieloide (n=75) |
Blástica linfoide (n=33) |
ALL Ph+ (n=40) |
|
| a Criterios de respuesta hematológica (todas las respuestas confirmadas después de 4 semanas): Respuesta hematológica mayor: (MaHR) = respuesta hematológica completa (CHR) + sin evidencia de leucemia (NEL). CHR: WBC ≤ límite superior del rango normal institucional, ANC ≥1000/mm3, plaquetas ≥100.000/mm3, sin blastos o promielocitos en sangre periférica, blastos de médula ósea ≤5%, <5% mielocitos más metamielocitos en sangre periférica, basófilos en sangre periférica <20% y sin afectación extramedular. NEL: mismos criterios que para CHR pero ANC ≥500/mm3 y ≤1000/mm3, o plaquetas ≥20.000/mm3 y ≤100.000/mm3. b MCyR combina respuestas completas (0% de metafases Ph+) y parciales (>0%–35%). IC = intervalo de confianza LSN = límite superior del rango normal. |
||||
|
MaHRa |
66% |
28% |
42% |
38% |
|
(95% IC) |
(59–74) |
(18–40) |
(26–61) |
(23–54) |
|
CHRa |
47% |
17% |
21% |
33% |
|
(95% IC) |
(40–56) |
(10–28) |
(9–39) |
(19–49) |
|
NELa |
19% |
11% |
21% |
5% |
|
(95% IC) |
(13–26) |
(5–20) |
(9–39) |
(1–17) |
|
MCyRb |
39% |
28% |
52% |
70% |
|
(95% IC) |
(31–47) |
(18–40) |
(34–69) |
(54–83) |
|
CCyR |
32% |
17% |
39% |
50% |
|
(95% CI) |
(25–40) |
(10–28) |
(23–58) |
(34–66) |
En el grupo de SPRYCEL 140 mg una vez al día, el tiempo medio hasta la MaHR fue de 1,9 meses (mínimo-máximo: 0,7-14,5) para los pacientes con LMC en fase acelerada, 1,9 meses (mínimo-máximo: 0,9-6,2) para los pacientes con LMC en fase mieloblástica y 1,8 meses (mínimo-máximo: 0,9-2,8) para los pacientes con LMC en fase linfoblástica.
En pacientes con LMC en fase mieloblástica, la duración media de la MaHR fue de 8,1 meses (mínimo-máximo: 2,7-21,1) y 9,0 (mínimo-máximo: 1,8-23,1) meses para el grupo de 140 mg una vez al día y el grupo de 70 mg dos veces al día, respectivamente. En pacientes con LMC en fase linfoblástica, la duración media de la MaHR fue de 4,7 meses (mínimo-máximo: 3,0-9,0) y 7,9 meses (mínimo-máximo: 1,6-22,1) para el grupo de 140 mg una vez al día y el grupo de 70 mg dos veces al día, respectivamente. En pacientes con ALL Ph + que fueron tratados con SPRYCEL 140 mg una vez al día, la duración media de la MaHR fue de 4,6 meses (mínimo-máximo: 1,4-10,2). Las medianas de supervivencia libre de progresión para pacientes con ALL Ph + tratados con SPRYCEL 140 mg una vez al día y 70 mg dos veces al día fueron de 4,0 meses (mínimo-máximo: 0,4-11,1) y 3,1 meses (mínimo-máximo: 0,3-20,8), respectivamente.
14.3 LMC en pacientes pediátricos
La eficacia de SPRYCEL en pacientes pediátricos se evaluó en dos estudios pediátricos de 97 pacientes con LMC en fase crónica. Entre los 97 pacientes con LMC en fase crónica tratados en dos estudios pediátricos, un ensayo abierto, no aleatorizado de rango de dosis (NCT00306202) y un ensayo abierto, no aleatorizado, de un solo brazo (NCT00777036), 51 pacientes (exclusivamente del ensayo de un solo brazo) tenían LMC en fase crónica recién diagnosticada y 46 pacientes (17 del ensayo de rango de dosis y 29 del ensayo de un solo brazo) eran resistentes o intolerantes al tratamiento previo con imatinib. Noventa y uno de los 97 pacientes pediátricos fueron tratados con comprimidos de SPRYCEL 60 mg/m2 una vez al día (dosis máxima de 100 mg una vez al día para pacientes con alto BSA). Los pacientes fueron tratados hasta la progresión de la enfermedad o toxicidad inaceptable.
Las características demográficas basales de los 46 pacientes resistentes o intolerantes a imatinib fueron: edad media 13,5 años (rango 2 a 20 años), 78,3% blancos, 15,2% asiáticos, 4,4% negros, 2,2% otros y 52% mujeres. Las características basales de los 51 pacientes recién diagnosticados fueron: edad media 12,8 años (rango 1,9 a 17,8 años), 60,8% blancos, 31,4% asiáticos, 5,9% negros, 2% otros y 49% mujeres.
La duración media de seguimiento fue de 5,2 años (rango 0,5 a 9,3 años) para los pacientes resistentes o intolerantes a imatinib y 4,5 años (rango 1,3 a 6,4 años) para los pacientes recién diagnosticados, respectivamente. Los resultados de eficacia de los dos estudios pediátricos se resumen en la Tabla 20.
La Tabla 20 muestra una tendencia creciente en la respuesta para CCyR, MCyR y MMR a lo largo del tiempo (de 3 meses a 24 meses). La tendencia creciente en la respuesta para los tres puntos finales se observa tanto en los pacientes recién diagnosticados como en los pacientes resistentes o intolerantes a imatinib.
Tabla 20: Eficacia de SPRYCEL en pacientes pediátricos con LMC CP-CML Respuesta acumulativa a lo largo del tiempo por período mínimo de seguimiento
| 3 meses | 6 meses | 12 meses | 24 meses | |
|---|---|---|---|---|
| a Pacientes del estudio pediátrico de LMC CP-CML recién diagnosticada que recibieron la formulación en comprimidos orales b Pacientes de los estudios pediátricos de LMC CP-CML resistentes o intolerantes a imatinib que recibieron la formulación en comprimidos orales |
||||
|
CCyR |
||||
|
Recién diagnosticados |
43,1% |
66,7% |
96,1% |
96,1% |
|
Anterior imatinib |
45,7% |
71,7% |
78,3% |
82,6% |
|
MCyR |
||||
|
Recién diagnosticados |
60,8% |
90,2% |
98,0% |
98,0% |
|
Anterior imatinib |
60,9% |
82,6% |
89,1% |
89,1% |
|
MMR |
||||
|
Recién diagnosticado |
7.8% |
31.4% |
56.9% |
74.5% |
|
Imatinib previo |
15.2% |
26.1% |
39.1% |
52.2% |
Con un seguimiento mediano de 4.5 años en pacientes recién diagnosticados, las duraciones medianas de CCyR, MCyR, MMR no pudieron estimarse ya que más de la mitad de los pacientes que respondieron no habían progresado al momento del corte de datos. El rango de duración de la respuesta fue (2.5+ a 66.5+ meses para CCyR), (1.4 a 66.5+ meses para MCyR) y (5.4+ a 72.5+ meses para los sujetos que lograron MMR para el mes 24 y 0.03+ a 72.5+ meses para los sujetos que lograron MMR en cualquier momento), donde ‘+’ indica una observación censurada.
Con un seguimiento mediano de 5.2 años en pacientes resistentes o intolerantes a imatinib, las duraciones medianas de CCyR, MCyR y MMR no pudieron estimarse ya que más de la mitad de los pacientes que respondieron no habían progresado al momento del corte de datos. El rango de duración de la respuesta fue (2.4 a 86.9+ meses para CCyR), (2.4 a 86.9+ meses para MCyR) y (2.6+ a 73.6+ meses para MMR), donde ‘+’ indica una observación censurada.
El tiempo mediano hasta la respuesta para MCyR fue de 2.9 meses (IC del 95%: 2.8 meses, 3.5 meses) en los pacientes con CP-CML resistentes/intolerantes a imatinib agrupados. El tiempo mediano hasta la respuesta para CCyR fue de 3.3 meses (IC del 95%: 2.8 meses, 4.7 meses) en los pacientes con CP-CML resistentes/intolerantes a imatinib agrupados. El tiempo mediano hasta la respuesta para MMR fue de 8.3 meses (IC del 95%: 5.0 meses, 11.8 meses) en los pacientes con CP-CML resistentes/intolerantes a imatinib agrupados.
El tiempo mediano hasta la respuesta para MCyR fue de 3.0 meses (IC del 95%: 2.8 meses, 4.3 meses) en los pacientes con CP-CML recién diagnosticados y sin tratamiento previo. El tiempo mediano hasta la respuesta para CCyR fue de 5.5 meses (IC del 95%: 3.0 meses, 5.7 meses) en los pacientes con CP-CML recién diagnosticados y sin tratamiento previo. El tiempo mediano hasta la respuesta para MMR fue de 8.9 meses (IC del 95%: 6.2 meses, 11.7 meses) en los pacientes con CP-CML recién diagnosticados y sin tratamiento previo.
En el estudio pediátrico de fase II, 1 paciente recién diagnosticado y 2 pacientes resistentes o intolerantes a imatinib progresaron a la fase blástica de la LMC.
14.4 Ph+ ALL en pacientes pediátricos
La eficacia de SPRYCEL en combinación con quimioterapia se evaluó en una sola cohorte (cohorte 1) del Estudio CA180372 (NCT01460160), un estudio multicéntrico de múltiples cohortes de pacientes pediátricos con ALL Ph+ de precursor de células B recién diagnosticado. Los 78 pacientes de la cohorte 1 recibieron SPRYCEL a una dosis diaria de 60 mg/m2 durante un máximo de 24 meses, en combinación con quimioterapia. El régimen de quimioterapia de base fue el protocolo de quimioterapia multiagente AIEOP-BFM ALL 2000.
Los pacientes tenían una edad mediana de 10.4 años (rango de 2.6 a 17.9 años) e incluían 20 pacientes (25%) de 2 a 6 años de edad, 37 pacientes (46%) de 7 a 12 años de edad y 24 pacientes (30%) de 13 a 17 años de edad. El 82% de los pacientes eran blancos y el 55% eran hombres. Treinta y dos pacientes (41%) tenían un recuento de glóbulos blancos (WBC) de ≥50,000 mcl al momento del diagnóstico y 17 pacientes (22%) tenían enfermedad extramedular.
La eficacia se estableció en base a la supervivencia libre de eventos (EFS) a 3 años, definida como el tiempo desde el inicio de SPRYCEL hasta la falta de respuesta completa al final del tercer bloque de alto riesgo, recaída, neoplasia maligna secundaria o muerte por cualquier causa. La tasa binaria de EFS a 3 años para los pacientes del Estudio CA180372 fue del 64.1% (IC del 95%: 52.4, 74.7). Al final de la inducción, 75 pacientes (96%) tenían una médula ósea con <5% de linfoblastos y 76 pacientes (97%) lograron esto al final de la consolidación.
15 REFERENCIAS
1. http://www.osha.gov/SLTC/hazardousdrugs/index.html
16 FORMA DE SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
Cómo se suministra
Las tabletas de SPRYCEL® (dasatinib) están disponibles como se describe en la Tabla 21.
|
Número NDC |
Strength |
Descripción |
Tabletas por frasco |
|
0003-0527-11 |
20 mg |
tableta recubierta con película, redonda, biconvexa, de color blanco a blanquecino, con «BMS» grabado en una cara y «527» en la otra |
60 |
|
0003-0528-11 |
50 mg |
tableta recubierta con película, ovalada, biconvexa, de color blanco a blanquecino, con «BMS» grabado en una cara y «528» en la otra |
60 |
|
0003-0524-11 |
70 mg |
tableta recubierta con película, redonda, biconvexa, de color blanco a blanquecino, con «BMS» grabado en una cara y «524» en la otra |
60 |
|
0003-0855-22 |
80 mg |
tableta recubierta con película, triangular, biconvexa, de color blanco a blanquecino, con «BMS» y «80» (BMS sobre 80) grabado en una cara y «855» en la otra |
30 |
|
0003-0852-22 |
100 mg |
tableta recubierta con película, ovalada, biconvexa, de color blanco a blanquecino, con «BMS 100» grabado en una cara y «852» en la otra |
30 |
|
0003-0857-22 |
140 mg |
tableta recubierta con película, redonda, biconvexa, de color blanco a blanquecino, con «BMS» y «140» (BMS sobre 140) grabado en una cara y «857» en la otra |
30 |
Almacenamiento
Las tabletas de SPRYCEL deben almacenarse a una temperatura de 20 °C a 25 °C (68 °F a 77 °F); se permiten excursiones entre 15 °C y 30 °C (59 °F y 86 °F) [ver USP Temperatura ambiente controlada].
Manipulación y eliminación
SPRYCEL es un producto antineoplásico. Siga los procedimientos especiales de manipulación y eliminación.1
El personal que esté embarazado debe evitar la exposición a las tabletas trituradas o rotas.
Las tabletas de SPRYCEL consisten en un núcleo de tableta, rodeado de una capa de película para evitar la exposición de los profesionales de la salud al principio activo. Se recomienda el uso de guantes de látex o nitrilo para una eliminación adecuada cuando se manipulen tabletas que se hayan triturado o roto accidentalmente, a fin de minimizar el riesgo de exposición dérmica.
17 INFORMACIÓN PARA EL PACIENTE
Aconseje al paciente que lea el etiquetado del paciente aprobado por la FDA (Información para el paciente).
Mielosupresión
Informe a los pacientes de la posibilidad de desarrollar recuentos bajos de células sanguíneas. Aconseje a los pacientes que informen inmediatamente la fiebre, particularmente en asociación con cualquier sugerencia de infección [ver Advertencias y precauciones (5.1)].
Hemorragia
Informe a los pacientes de la posibilidad de hemorragia grave y que informen inmediatamente cualquier signo o síntoma sugestivo de hemorragia (sangrado inusual o hematomas fáciles) [ver Advertencias y precauciones (5.2)].
Retención de líquidos
Se debe informar a los pacientes de la posibilidad de desarrollar retención de líquidos (hinchazón, aumento de peso, tos seca, dolor en el pecho al respirar o dificultad para respirar) y se les debe aconsejar que busquen atención médica de inmediato si aparecen esos síntomas [ver Advertencias y precauciones (5.3)].
Toxicidad cardiovascular
Informe a los pacientes de la posibilidad de desarrollar toxicidad cardiovascular, incluidos eventos isquémicos cardíacos, retención de líquidos relacionada con el corazón, anomalías de la conducción y AIT. Aconseje a los pacientes que busquen atención médica inmediata si ocurren síntomas sugestivos de toxicidad cardiovascular, como dolor en el pecho, dificultad para respirar, palpitaciones, problemas de visión transitorios o dificultad para hablar [ver Advertencias y precauciones (5.4)].
Hipertensión arterial pulmonar
Informe a los pacientes de la posibilidad de desarrollar hipertensión arterial pulmonar (disnea, fatiga, hipoxia y retención de líquidos) y aconseje a los pacientes que busquen atención médica de inmediato si aparecen esos síntomas [ver Advertencias y precauciones (5.5)].
Síndrome de lisis tumoral
Informe a los pacientes que informen inmediatamente y busquen atención médica por cualquier síntoma como náuseas, vómitos, debilidad, edema, dificultad para respirar, calambres musculares y convulsiones, que pueden indicar síndrome de lisis tumoral [ver Advertencias y precauciones (5.8)].
Crecimiento y desarrollo en pacientes pediátricos
Informe a los pacientes pediátricos y sus cuidadores de la posibilidad de desarrollar anomalías del crecimiento óseo, dolor óseo o ginecomastia y aconseje a los pacientes que busquen atención médica de inmediato si aparecen esos síntomas [ver Advertencias y precauciones (5.10)].
Toxicidad embrio-fetal
- •
- Aconseje a las mujeres embarazadas sobre el riesgo potencial para un feto [ver Advertencias y precauciones (5.9) y Uso en poblaciones específicas (8.1)].
- •
- Aconseje a las mujeres en edad fértil y a los hombres con parejas femeninas en edad fértil que utilicen métodos anticonceptivos eficaces durante el tratamiento con SPRYCEL y durante 30 días después de la última dosis. Aconseje a las mujeres que se pongan en contacto con su proveedor de atención médica si quedan embarazadas o si sospechan de un embarazo mientras toman SPRYCEL [ver Advertencias y precauciones (5.9) y Uso en poblaciones específicas (8.1, 8.3)].
Lactancia
- •
- Aconseje a las mujeres que no se recomienda la lactancia durante el tratamiento con SPRYCEL y durante 2 semanas después de la dosis final [ver Uso en poblaciones específicas (8.2)].
Quejas gastrointestinales
Informe a los pacientes que pueden experimentar náuseas, vómitos o diarrea con SPRYCEL. Aconseje a los pacientes que busquen atención médica si estos síntomas son molestos o persistentes.
Aconseje a los pacientes que usan antiácidos que eviten tomar SPRYCEL y antiácidos con menos de 2 horas de diferencia [ver Interacciones medicamentosas (7.1)].
Dolor
Informe a los pacientes que pueden experimentar dolor de cabeza o dolor musculoesquelético con SPRYCEL. Aconseje a los pacientes que busquen atención médica si estos síntomas son molestos o persistentes.
Fatiga
Informe a los pacientes que pueden experimentar fatiga con SPRYCEL. Aconseje a los pacientes que busquen atención médica si este síntoma es molesto o persistente.
Erupción cutánea
Informe a los pacientes que pueden experimentar erupción cutánea con SPRYCEL. Aconseje a los pacientes que busquen atención médica si este síntoma es molesto o persistente.
Lactosa
Informe a los pacientes que SPRYCEL contiene 135 mg de lactosa monohidrato en una dosis diaria de 100 mg y 189 mg de lactosa monohidrato en una dosis diaria de 140 mg.
Hepatotoxicidad
Avise a los pacientes que SPRYCEL puede causar hepatotoxicidad y que los pacientes con antecedentes de enfermedades hepáticas pueden estar en riesgo. Avise a los pacientes que busquen atención médica inmediata si se presenta algún síntoma sugestivo de hepatotoxicidad, como dolor abdominal, ictericia y ictericia escleral, anorexia, sangrado, hematomas y orina de color oscuro [ver Advertencias y precauciones (5.11)].
Instrucciones para tomar SPRYCEL
- •
- Dosis olvidada
- Avise a los pacientes que si olvidan una dosis de SPRYCEL, deben tomar la siguiente dosis programada a su hora habitual. El paciente no debe tomar dos dosis al mismo tiempo.
- •
- Jugo de toronja
- Avise a los pacientes que no deben beber jugo de toronja ya que puede aumentar la cantidad de SPRYCEL en su sangre y, por lo tanto, aumentar su riesgo de reacciones adversas.
Distribuido por:
Bristol-Myers Squibb Company
Princeton, NJ 08543 USA
INFORMACIÓN PARA EL PACIENTE SPRYCEL® (Dasatinib) comprimidos
|
¿Qué es SPRYCEL? SPRYCEL es un medicamento recetado que se usa para tratar:
Se desconoce si SPRYCEL es seguro y eficaz en niños menores de 1 año. |
||
|
Antes de tomar SPRYCEL, informe a su proveedor de atención médica sobre todas sus afecciones médicas, incluso si usted:
Informe a su proveedor de atención médica sobre todos los medicamentos que toma, incluidos los medicamentos recetados y de venta libre, las vitaminas, los antiácidos y los suplementos a base de hierbas. Si toma un antiácido, tómelo 2 horas antes o 2 horas después de su dosis de SPRYCEL. |
||
|
¿Cómo debo tomar SPRYCEL?
|
||
|
¿Cuáles son los posibles efectos secundarios de SPRYCEL? SPRYCEL puede causar efectos secundarios graves, incluyendo:
|
||
|
|
|
|
||
|
|
|
|
Los efectos secundarios más comunes de SPRYCEL en adultos y niños que reciben SPRYCEL solo incluyen: |
||
|
|
|
|
Los efectos secundarios más comunes de SPRYCEL en niños que reciben SPRYCEL con quimioterapia incluyen: |
||
|
|
|
|
SPRYCEL puede causar problemas de fertilidad en hombres y mujeres. Hable con su proveedor de atención médica si esto le preocupa. Informe a su proveedor de atención médica si tiene algún efecto secundario que le moleste o que no desaparezca. Estos no son todos los posibles efectos secundarios de SPRYCEL. Llame a su médico para obtener asesoramiento médico sobre los efectos secundarios. Puede informar los efectos secundarios a la FDA al 1-800-FDA-1088. |
||
|
¿Cómo debo almacenar SPRYCEL?
Mantenga SPRYCEL y todos los medicamentos fuera del alcance de los niños. |
||
|
Información general sobre el uso seguro y eficaz de SPRYCEL. Los medicamentos a veces se recetan para fines distintos a los que se enumeran en un folleto de información para el paciente. No use SPRYCEL para una condición para la que no está recetado. No le dé SPRYCEL a otras personas, incluso si tienen los mismos síntomas que usted. Puede dañarlos. Puede pedirle a su proveedor de atención médica o farmacéutico información sobre SPRYCEL que esté escrita para profesionales de la salud. |
||
|
¿Cuáles son los ingredientes de SPRYCEL? Ingrediente activo: dasatinib Ingredientes inactivos: lactosa monohidrato, celulosa microcristalina, croscarmelosa sódica, hidroxipropilcelulosa y estearato de magnesio. El recubrimiento de la tableta consiste en hipromelosa, dióxido de titanio y polietilenglicol. Distribuido por: Bristol-Myers Squibb Company, Princeton, NJ 08543 USA |
||
Esta información para el paciente ha sido aprobada por la Administración de Alimentos y Medicamentos de los Estados Unidos.
Revisado: febrero de 2023
SPRYCEL 20 mg comprimidos Embalaje Representativo
Ver la sección Presentación para una lista completa de los paquetes disponibles de SPRYCEL.
60 Tabletas NDC 0003-0527-11
SPRYCEL®
(dasatinib)
Tabletas
20 mg
Rx solamente
No triture, corte ni mastique las tabletas.
Trague las tabletas enteras.
Bristol Myers Squibb
SPRYCEL 50 mg comprimidos Embalaje Representativo
60 Tabletas NDC 0003-0528-11
SPRYCEL®
(dasatinib)
Tabletas
50 mg
Rx only
No triture, corte o mastique las tabletas.
Trague las tabletas enteras.
Bristol Myers Squibb
Envase representativo de SPRYCEL 70 mg comprimidos
60 Tabletas NDC 0003-0524-11
SPRYCEL®
(dasatinib)
Tabletas
70 mg
Rx solamente
No triture, corte ni mastique las tabletas.
Trague las tabletas enteras.
Bristol Myers Squibb
SPRYCEL 80 mg comprimidos Envase Representativo
30 Tabletas NDC 0003-0855-22
SPRYCEL®
(dasatinib)
Tabletas
80 mg
Rx only
No triture, corte ni mastique las tabletas.
Trague las tabletas enteras.
Bristol Myers Squibb
SPRYCEL 100 mg comprimidos Embalaje Representativo
30 Tabletas NDC 0003-0852-22
SPRYCEL®
(dasatinib)
Tabletas
100 mg
Rx only
No triture, corte o mastique las tabletas.
Trague las tabletas enteras.
Bristol Myers Squibb

Envase Representativo de SPRYCEL 140 mg comprimidos
30 Tabletas NDC 0003-0857-22
SPRYCEL®
(dasatinib)
Tabletas
140 mg
Rx only
No triture, corte ni mastique las tabletas.
Trague las tabletas enteras.
Bristol Myers Squibb