
Fabricante de medicamentos: Bioverativ Therapeutics Inc. (Updated: 2023-05-05)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
ELOCTATE® [Factor antihemofílico (recombinante), proteína de fusión Fc], polvo liofilizado para solución para inyección intravenosa
Aprobación inicial en EE. UU.: 2014
INDICACIONES Y USO
ELOCTATE, Factor antihemofílico (recombinante), proteína de fusión Fc, es un factor antihemofílico derivado de ADN recombinante indicado en adultos y niños con hemofilia A (deficiencia congénita del factor VIII) para:
- Tratamiento a demanda y control de episodios hemorrágicos
- Manejo perioperatorio del sangrado
- Profilaxis de rutina para reducir la frecuencia de episodios hemorrágicos.
Limitación de uso
ELOCTATE no está indicado para el tratamiento de la enfermedad de von Willebrand. (1)
DOSIFICACIÓN Y ADMINISTRACIÓN
Solo para uso intravenoso después de la reconstitución.
- Cada vial de ELOCTATE está etiquetado con la cantidad de factor VIII recombinante en unidades internacionales (UI o unidad). Una unidad por kilogramo de peso corporal elevará el nivel de factor VIII en un 2% (UI/dL). (2.1)
- Para el tratamiento a demanda y el control de episodios hemorrágicos y el manejo perioperatorio, calcule la dosis utilizando las siguientes fórmulas:
Incremento estimado del factor VIII (UI/dL o % de lo normal) = [Dosis total (UI)/peso corporal (kg)] x 2 (UI/dL por UI/kg)
O
Dosis requerida (UI) = Peso corporal (kg) x Aumento deseado del factor VIII (UI/dL o % de lo normal) x 0,5 (UI/kg por UI/dL) - Para profilaxis de rutina: 50 UI/kg cada 4 días. Ajuste la dosis según la respuesta del paciente con dosis en el rango de 25-65 UI/kg a intervalos de 3-5 días.
- Para profilaxis de rutina en niños menores de 6 años: 50 UI/kg dos veces por semana. Ajuste la dosis según la respuesta del paciente con dosis en el rango de 25-65 UI/kg a intervalos de 3-5 días. Es posible que se requieran dosis más frecuentes o más altas de hasta 80 UI/kg. (2.1)
FORMAS DE DOSIFICACIÓN Y FUERZAS
Para inyección: nominalmente 250, 500, 750, 1000, 1500, 2000, 3000, 4000, 5000 o 6000 UI, polvo liofilizado en viales de dosis única para reconstitución. (3)
CONTRAINDICACIONES
No lo use en pacientes que hayan tenido reacciones de hipersensibilidad potencialmente mortales, incluida la anafilaxia, a ELOCTATE o excipientes de ELOCTATE (sacarosa, cloruro de sodio, L-histidina, cloruro de calcio y polisorbato 20). (4)
ADVERTENCIAS Y PRECAUCIONES
- Las reacciones de hipersensibilidad, incluida la anafilaxia, son posibles. Si ocurren síntomas, suspenda inmediatamente ELOCTATE e inicie el tratamiento apropiado. (5.1)
- Se han reportado anticuerpos neutralizantes (inhibidores) al factor VIII. Si no se alcanzan los niveles esperados de actividad del factor VIII en plasma, o si el sangrado no se controla con una dosis adecuada, realice un ensayo que mida la concentración del inhibidor del factor VIII. (5.2, 5.5)
REACCIONES ADVERSAS
Pacientes previamente tratados (PPT): Las reacciones adversas que ocurren con mayor frecuencia (>0,5% de los sujetos) en los ensayos clínicos fueron artralgia, malestar general, mialgia, dolor de cabeza y erupción cutánea. (6)
Pacientes previamente no tratados (PUP): Las reacciones adversas que ocurren con mayor frecuencia (incidencia ≥1%) en los ensayos clínicos fueron inhibición del factor VIII, trombosis relacionada con el dispositivo y erupción papular. (6)
Para reportar REACCIONES ADVERSAS SOSPECHOSAS, comuníquese con Bioverativ Therapeutics Inc. al 1-855-693-5628 o con la FDA al 1-800-FDA-1088 o www.fda.gov/medwatch.
USO EN POBLACIONES ESPECÍFICAS
Pediátrico: La depuración (basada en el peso corporal por kg) es mayor (75%) en pacientes pediátricos de 1 a 5 años de edad. Es posible que se necesiten dosis más altas o más frecuentes. (8.4)
Consulte 17 para obtener INFORMACIÓN PARA EL PACIENTE y el etiquetado del paciente aprobado por la FDA.
Revisado: 5/2023
Tabla de Contenido
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDO*
1 INDICACIONES Y USO
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Dosis
2.2 Preparación y Reconstitución
2.3 Administración
3 FORMAS Y FUERZAS DE DOSIFICACIÓN
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Reacciones de Hipersensibilidad
5.2 Anticuerpos Neutralizantes
5.3 Factores de Riesgo Cardiovascular
5.4 Complicación Relacionada con el Catéter
5.5 Monitoreo de Pruebas de Laboratorio
6 REACCIONES ADVERSAS
6.1 Experiencia en Ensayos Clínicos
6.2 Experiencia Postcomercialización
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.4 Uso Pediátrico
8.5 Uso Geriátrico
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de Acción
12.2 Farmacodinamia
12.3 Farmacocinética
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
14 ESTUDIOS CLÍNICOS
15 REFERENCIAS
16 CÓMO SE SUMINISTRA/ALMACENAMIENTO Y MANEJO
17 INFORMACIÓN PARA EL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información completa de prescripción no están enumeradas.
1 INDICACIONES Y USO
ELOCTATE, Factor Antihemofílico (Recombinante), Proteína de Fusión Fc, es un factor antihemofílico derivado de ADN recombinante indicado en adultos y niños con Hemofilia A (deficiencia congénita del Factor VIII) para:
- Tratamiento a demanda y control de episodios hemorrágicos,
- Manejo perioperatorio de la hemorragia,
- Profilaxis de rutina para reducir la frecuencia de episodios hemorrágicos.
Limitación de Uso
ELOCTATE no está indicado para el tratamiento de la enfermedad de von Willebrand.
2 DOSIS Y ADMINISTRACIÓN
Para uso intravenoso después de la reconstitución únicamente.
2.1 Dosis
- La dosis y la duración del tratamiento dependen de la gravedad de la deficiencia del Factor VIII, la ubicación y la extensión del sangrado, y la condición clínica del paciente. Es necesario un control cuidadoso de la terapia de reemplazo en casos de cirugía mayor o episodios de sangrado potencialmente mortales.
- Cada etiqueta del vial de ELOCTATE indica la potencia del Factor VIII en unidades internacionales (UI). Una UI corresponde a la actividad del Factor VIII contenida en un mililitro de plasma humano normal.
- La asignación de potencia se determina utilizando un ensayo de sustrato cromogénico. Un estudio de campo1 ha indicado que los niveles de Factor VIII en plasma se pueden controlar utilizando un ensayo de sustrato cromogénico o un ensayo de coagulación de un solo paso que se utiliza habitualmente en los laboratorios clínicos de EE. UU.
- El cálculo de la dosis requerida de Factor VIII se basa en el hallazgo empírico de que 1 UI de Factor VIII por kg de peso corporal eleva el nivel de Factor VIII en plasma en 2 UI/dL.
El aumento máximo esperado in vivo en el nivel de Factor VIII expresado como UI/dL (o % de lo normal) se estima utilizando la siguiente fórmula:
Incremento Estimado de Factor VIII (UI/dL o % de lo normal) = [Dosis Total (UI)/peso corporal (kg)] × 2 (UI/dL por UI/kg)
La dosis para lograr un aumento máximo deseado in vivo en el nivel de Factor VIII se puede calcular utilizando la siguiente fórmula:
Dosis (UI) = peso corporal (kg) × Aumento Deseado de Factor VIII (UI/dL o % de lo normal) × 0,5 (UI/kg por UI/dL)
- Los pacientes pueden variar en sus respuestas farmacocinéticas (por ejemplo, vida media, recuperación in vivo) y clínicas. Base la dosis y la frecuencia de ELOCTATE en la respuesta clínica individual.
- Puede ser necesario ajustar la dosis en pacientes pediátricos menores de seis años de edad [ver Uso en poblaciones específicas (8.4)]. Para pacientes de seis años de edad o mayores, generalmente no se requiere ajuste de dosis.
Tratamiento a demanda y control de episodios de sangrado
Se proporciona una guía para la dosificación de ELOCTATE para el tratamiento a demanda y el control de los episodios de sangrado en Tabla 1. Se debe considerar mantener una actividad del Factor VIII en o por encima del rango objetivo.
| Tipo de sangrado | Nivel de Factor VIII requerido (UI/dL o % de lo normal) |
Dosis (UI/kg) |
Frecuencia de dosificación (horas) | Duración de la terapia (días) |
|---|---|---|---|---|
| Menor y moderado Articulación, músculo superficial/sin compromiso neurovascular (excepto iliopsoas), laceración profunda y renal, tejido blando superficial, membranas mucosas |
40-60 | 20-30 | Repetir cada 24-48 horas (12 a 24 horas para pacientes menores de 6 años de edad) |
Hasta que el episodio de sangrado se resuelva |
| Mayor Hemorragia potencialmente mortal o que pone en peligro una extremidad, iliopsoas y músculo profundo con lesión neurovascular, retroperitoneo, intracraneal o gastrointestinal |
80-100 | 40-50 | Repetir cada 12-24 horas (8 a 24 horas para pacientes menores de 6 años de edad) | Hasta que el sangrado se resuelva (aproximadamente 7-10 días) |
Manejo Perioperatorio
Se proporciona una guía para la dosificación de ELOCTATE durante la cirugía (manejo perioperatorio) en Tabla 2. Se debe considerar el mantenimiento de una actividad del Factor VIII en o por encima del rango objetivo.
| Tipo de Cirugía | Nivel de Factor VIII Requerido (UI/dL o % de lo normal) |
Dosis (UI/kg) |
Frecuencia de Dosificación (horas) |
Duración de la Terapia (días) |
|---|---|---|---|---|
| Menor Extracción dental sin complicaciones |
50-80 | 25-40 | Repetir cada 24 horas (12-24 horas para pacientes menores de 6 años de edad) | Al menos 1 día hasta que se logre la cicatrización |
| Mayor Cirugía intracraneal, intraabdominal o de reemplazo articular |
80-120 (pre y postoperatorio) |
Preoperatorio: 40-60 Repetir: 40-50 |
Dosis preoperatoria de 40 a 60 UI/kg seguida de una dosis de repetición de 40-50 UI/kg después de 8-24 horas (6 a 24 para pacientes menores de 6 años de edad) y luego cada 24 horas para mantener la actividad del FVIII dentro del rango objetivo | Hasta que se logre una cicatrización adecuada de la herida, luego continuar la terapia durante al menos 7 días para mantener una actividad del Factor VIII dentro del rango objetivo |
Profilaxis de rutina
- El régimen de inicio recomendado es de 50 UI/kg de ELOCTATE administrado cada 4 días. Ajuste el régimen según la respuesta del paciente con dosis en el rango de 25-65 UI/kg a intervalos de 3-5 días.
- Para niños <6 años de edad, el régimen de inicio recomendado es de 50 UI/kg de ELOCTATE administrado dos veces por semana. Ajuste el régimen según la respuesta del paciente con dosis en el rango de 25-65 UI/kg a intervalos de 3-5 días. Es posible que se requieran dosis más frecuentes o más altas de hasta 80 UI/kg [ver Uso en poblaciones específicas (8.4), Farmacología clínica (12.3)].
2.2 Preparación y reconstitución
- Utilice una técnica aséptica (limpia y libre de gérmenes) y una superficie de trabajo plana durante el procedimiento de reconstitución.
- Deje que el vial de ELOCTATE, que contiene el polvo liofilizado blanco a blanquecino, y la jeringa precargada con diluyente alcancen la temperatura ambiente antes de usar.
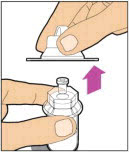
- Retire la tapa de plástico del vial y limpie el tapón de goma del vial con una toallita con alcohol. Deje que el tapón de goma se seque.
-
Retire completamente el respaldo del paquete del adaptador del vial pelando la tapa. No retire el adaptador del vial del paquete ni toque el interior del paquete del adaptador.
-
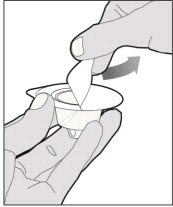
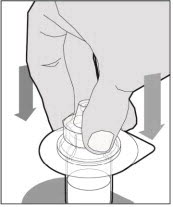
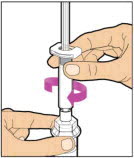
Coloque el vial sobre una superficie plana y sólida y use una mano para sostener el vial firmemente. Use la otra mano para colocar el adaptador del vial sobre el vial. Coloque la espiga del adaptador directamente sobre el centro del tapón de goma y empuje el adaptador hacia abajo hasta que la espiga perfore el centro del tapón del vial y esté completamente insertada.
-
Levante la cubierta del paquete lejos del adaptador del vial y deseche la cubierta.
-
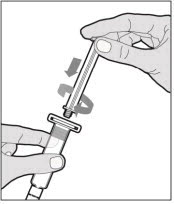
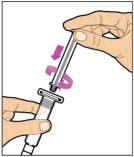
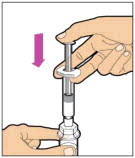
Sostenga la varilla del émbolo en el disco circular. Coloque la punta de la varilla del émbolo en el extremo de la jeringa. Gire en el sentido de las agujas del reloj hasta que esté firmemente unido. Solo use la jeringa con diluyente proporcionada en el paquete de ELOCTATE.
- Con una mano, sostenga la jeringa con diluyente por la parte estriada directamente debajo de la tapa, con la tapa apuntando hacia arriba. No use si la tapa se ha quitado o no está firmemente unida.
- Con la otra mano, agarre la tapa y dóblela en un ángulo de 90° hasta que se rompa. Después de que la tapa se rompa, verá la punta de vidrio de la jeringa. No toque la punta de vidrio de la jeringa ni el interior de la tapa.
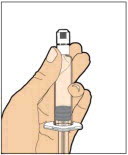
- Con el vial sentado sobre una superficie plana, inserte la punta de la jeringa en la abertura del adaptador. Gire la jeringa en el sentido de las agujas del reloj hasta que esté firmemente unida al adaptador.
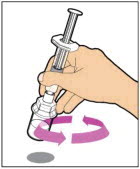
- Presione lentamente la varilla del émbolo para inyectar todo el diluyente en el vial. La varilla del émbolo puede subir ligeramente después de este proceso. Esto es normal.
- Con la jeringa aún conectada al adaptador, gire suavemente el vial hasta que el producto esté completamente disuelto. No agite. La solución reconstituida debe ser transparente a ligeramente opalescente e incolora. No use ELOCTATE reconstituido si contiene partículas visibles o está turbio.
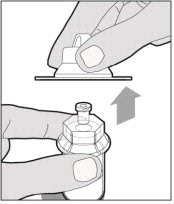
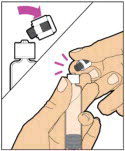
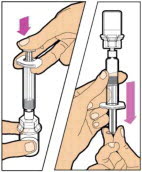
- Asegúrese de que la varilla del émbolo esté completamente presionada. Voltee el vial. Tire lentamente de la varilla del émbolo para extraer la solución en la jeringa. Tenga cuidado de no sacar completamente la varilla del émbolo de la jeringa.
- Desenrosque suavemente la jeringa del adaptador del vial y deseche el vial con el adaptador aún unido. No toque la punta de la jeringa ni el interior de la tapa.
- Use ELOCTATE reconstituido lo antes posible, pero no más tarde de 3 horas después de la reconstitución. No toque la punta de vidrio de la jeringa si no se usa inmediatamente después de la reconstitución. Proteger de la luz solar directa. No refrigerar después de la reconstitución.
Para combinar dos o más viales de ELOCTATE, después del paso 12 anterior, siga estos pasos de agrupación:
- Retire la jeringa con diluyente del adaptador del vial girándola en sentido contrario a las agujas del reloj hasta que esté completamente separada.
- Deje el adaptador del vial unido al vial, ya que es necesario para conectar una jeringa grande con cierre luer (no incluida en el kit). No separe la jeringa con diluyente hasta que esté listo para conectar la jeringa grande con cierre luer.
- Conecte una jeringa grande con cierre luer separada girando en el sentido de las agujas del reloj hasta que esté firmemente en su lugar.
- Tire lentamente de la varilla del émbolo para extraer la solución en la jeringa.
- Repita este procedimiento de agrupación con cada vial que sea necesario para obtener la dosis requerida. Al agrupar, no separe la jeringa grande con cierre luer hasta que esté listo para conectarla al siguiente vial (con el adaptador del vial conectado). Una vez que haya agrupado la dosis requerida, proceda a la administración utilizando la jeringa grande con cierre luer.
2.3 Administración
Solo para inyección intravenosa
- Inspeccione visualmente la solución de ELOCTATE reconstituida para detectar partículas y decoloración antes de la administración. No use si se observa materia particulada o decoloración.
- No administre ELOCTATE reconstituido en el mismo tubo o contenedor con otros medicamentos.
Pasos de administración:
- Conecte la jeringa al extremo del conector del tubo del equipo de infusión girando en el sentido de las agujas del reloj hasta que esté firmemente en su lugar.
- Presione el émbolo hasta que todo el aire se haya eliminado de la jeringa y ELOCTATE haya llegado al extremo del tubo del equipo de infusión. No empuje la solución de ELOCTATE a través de la aguja.
- Retire la cubierta protectora de la aguja del tubo del equipo de infusión.
- Realice una infusión intravenosa en bolo. La velocidad de administración debe determinarse por el nivel de comodidad del paciente y no más rápido que 10 ml por minuto. Después de infundir ELOCTATE, retire y deseche adecuadamente el equipo de infusión.
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
ELOCTATE se encuentra disponible en forma de polvo liofilizado de color blanco a blanquecino en frascos de dosis única que contienen nominalmente 250, 500, 750, 1000, 1500, 2000, 3000, 4000, 5000 o 6000 unidades internacionales (UI) por frasco. La potencia real del Factor VIII está etiquetada en cada frasco de ELOCTATE.
4 CONTRAINDICACIONES
ELOCTATE está contraindicado en pacientes que han tenido reacciones de hipersensibilidad potencialmente mortales a ELOCTATE o sus excipientes (sacarosa, cloruro de sodio, L-histidina, cloruro de calcio y polisorbato 20).
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Reacciones de hipersensibilidad
Se han notificado reacciones de hipersensibilidad con ELOCTATE. Se han notificado reacciones de hipersensibilidad de tipo alérgico, incluida la anafilaxia, con productos de reemplazo del Factor VIII. Los signos tempranos de reacciones de hipersensibilidad que pueden progresar a anafilaxia pueden incluir angioedema, opresión en el pecho, disnea, sibilancias, urticaria y prurito. Suspenda inmediatamente la administración e inicie el tratamiento adecuado si se producen reacciones de hipersensibilidad.
5.2 Anticuerpos neutralizantes
Se ha notificado la formación de anticuerpos neutralizantes (inhibidores) al Factor VIII después de la administración de ELOCTATE. Controle a todos los pacientes para detectar el desarrollo de inhibidores del Factor VIII mediante observaciones clínicas y pruebas de laboratorio apropiadas. Si el nivel de Factor VIII plasmático no aumenta como se esperaba o si el sangrado no se controla después de la administración de ELOCTATE, sospeche la presencia de un inhibidor (anticuerpo neutralizante) [ver Advertencias y precauciones (5.5)].
5.3 Factores de riesgo cardiovascular
Los pacientes hemofílicos con factores de riesgo cardiovascular o enfermedades pueden tener el mismo riesgo de desarrollar eventos cardiovasculares que los pacientes no hemofílicos cuando la coagulación se ha normalizado mediante el tratamiento con Factor VIII.
5.4 Complicación relacionada con el catéter
Si se requiere un dispositivo de acceso venoso central (CVAD), se debe considerar el riesgo de complicaciones relacionadas con el CVAD, incluidas las infecciones locales, la bacteriemia y la trombosis del sitio del catéter [ver Reacciones adversas (6)].
5.5 Control de las pruebas de laboratorio
Controle la actividad del Factor VIII plasmático realizando una prueba validada (por ejemplo, ensayo de coagulación de un solo paso), para confirmar que se han alcanzado y mantenido los niveles adecuados de Factor VIII [ver Dosificación y administración (2)].
Controle el desarrollo de inhibidores del Factor VIII. Realice un ensayo de inhibición de Bethesda si no se alcanzan los niveles plasmáticos esperados de Factor VIII, o si el sangrado no se controla con la dosis esperada de ELOCTATE. Utilice unidades de Bethesda (BU) para informar los niveles de inhibidores.
6 REACCIONES ADVERSAS
Las reacciones adversas más frecuentes (incidencia >0.5% de los sujetos) notificadas en los ensayos clínicos de pacientes previamente tratados (PPT) fueron artralgia, malestar, mialgia, cefalea y erupción cutánea. Las reacciones adversas más frecuentes (incidencia ≥1.0% de los sujetos) notificadas en los ensayos clínicos de pacientes previamente no tratados (PPN) fueron inhibición del factor VIII, trombosis relacionada con el dispositivo y erupción papular.
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se llevan a cabo en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no se pueden comparar directamente con las tasas en los ensayos clínicos de otro fármaco y pueden no reflejar las tasas observadas en la práctica.
Pacientes previamente tratados (PPT)
ELOCTATE se ha evaluado en 276 sujetos en cinco estudios completados en pacientes previamente tratados (PPT) con hemofilia A grave (<1% de actividad FVIII endógena o una mutación genética compatible con hemofilia A grave) que recibieron al menos una dosis de ELOCTATE como parte de la profilaxis de rutina, el tratamiento a demanda de episodios hemorrágicos o el manejo perioperatorio. Sesenta y nueve (25.0%) fueron sujetos pediátricos <12 años de edad, 25 (9.1%) fueron adolescentes (12 a <18 años de edad) y 182 (65.9%) fueron adultos (18 años de edad y mayores). Hubo 200 sujetos tratados durante al menos 104 semanas, 151 sujetos tratados durante al menos 156 semanas y 107 sujetos tratados durante al menos 208 semanas. El número total de días de exposición (DE) fue de 80.848 con una mediana de 294 (rango 1-735) días de exposición por sujeto. Los sujetos recibieron un total de 82.024 inyecciones con una mediana de 303.5 inyecciones de ELOCTATE (rango 1-755) por sujeto. Los eventos adversos (EA) se monitorearon durante un total de 893.72 años-sujeto. Dos sujetos (0.7% del total de 276) con factores de riesgo cardiovascular experimentaron cada uno una reacción adversa grave de infarto de miocardio durante el estudio.
Se notificaron reacciones adversas (RA) en 11 de 276 (4.0%) sujetos tratados con profilaxis de rutina o terapia episódica (a demanda). No se observaron diferencias específicas de edad en las RA entre los sujetos pediátricos y adultos. Tabla 3 resume las reacciones adversas más frecuentes en los PPT. Las reacciones adversas adicionales, cada una de las cuales ocurrió en un solo sujeto (incidencia 0.4%), incluyen mareos, disgeusia, bradicardia, hipertensión, sofocos, angiopatía (término del investigador: dolor vascular después de la inyección del fármaco del estudio), tos, dolor abdominal inferior, dolor de espalda, hinchazón de las articulaciones, dolor en el pecho, sensación de frío, sensación de calor e hipotensión procedimental. Dos sujetos fueron retirados del estudio debido a reacciones adversas de erupción cutánea y artralgia. En los estudios, no se detectaron inhibidores y no se notificaron eventos de anafilaxia.
| MedDRA* Sistema de órganos de clase | Reacciones adversas | Número de sujetos n (%) |
|---|---|---|
|
||
| Trastornos del sistema nervioso | Cefalea | 2 (0.7) |
| Trastornos de la piel y del tejido subcutáneo | Erupción cutánea | 2 (0.7) |
| Trastornos musculoesqueléticos y del tejido conjuntivo | Artralgia Mialgia |
2 (0.7) 2 (0.7) |
| Trastornos generales y condiciones del lugar de administración | Malestar | 2 (0.7) |
Pacientes previamente no tratados (PUP)
La seguridad de ELOCTATE también se evaluó en un estudio completado (estudio PUP) en 103 sujetos con hemofilia A grave (<1% de actividad FVIII endógena). En general, la mediana del número de semanas de tratamiento fue de 64,24 semanas (rango: 0,0–206,8 semanas). El número de sujetos con al menos 10 días de exposición (ED) fue de 87 (84,5%), al menos 20 ED fue de 85 (82,5%) y al menos 50 ED fue de 81 (78,6%).
Se informaron reacciones adversas a los medicamentos (RAM) en 29 de 103 (28,2%) sujetos tratados con ELOCTATE en profilaxis de rutina, episódica y/o terapias de inducción de tolerancia inmunitaria (ITI). Las RAM en PUP se resumen en Tabla 4.
| Clase de órgano del sistema MedDRA* | Reacciones adversas | Número de sujetos (%) N=103† |
|---|---|---|
|
||
| Trastornos de la sangre y del sistema linfático | Inhibición del factor VIII‡ | 28 (27.2) |
| Trastornos generales y condiciones del lugar de administración | Trombosis relacionada con el dispositivo§ | 2 (1.9) |
| Trastornos de la piel y del tejido subcutáneo | Erupción papular | 1 (1.0) |
Inmunogenicidad
Los sujetos de los ensayos clínicos fueron monitoreados para detectar anticuerpos neutralizantes contra el Factor VIII. Ningún PTP desarrolló anticuerpos neutralizantes confirmados contra el Factor VIII. Un sujeto de 25 años tuvo un anticuerpo neutralizante transitorio, positivo, de 0,73 BU en la semana 14, que no se confirmó en pruebas repetidas 18 días después y posteriormente.
En el Estudio PUP, se observó el desarrollo de anticuerpos neutralizantes (inhibidores) en 28 sujetos, 14 de ellos tenían un inhibidor de alto título. Con base en los sujetos con una prueba de inhibidor después de un hito de día de exposición (ED) o que desarrollaron un inhibidor en cualquier momento durante el estudio, la incidencia del desarrollo del inhibidor del Factor VIII fue:
- 28/90 sujetos (31,1%) con al menos 10 ED
- 28/86 sujetos (32,6%) con al menos 50 ED
El tiempo medio hasta el desarrollo del inhibidor para los 28 sujetos fue de 9 ED (rango intercuartílico: 6,5–12).
La detección de anticuerpos que reaccionan con el Factor VIII depende en gran medida de muchos factores, incluida la sensibilidad y especificidad del ensayo, el manejo de la muestra, el momento de la recolección de la muestra, los medicamentos concomitantes y la enfermedad subyacente. Por lo tanto, puede ser engañoso comparar la incidencia de anticuerpos a ELOCTATE con la incidencia de anticuerpos a otros productos.
6.2 Experiencia postcomercialización
Las siguientes reacciones adversas se han identificado durante el uso posterior a la aprobación de ELOCTATE. Debido a que estas reacciones se informan voluntariamente de una población de tamaño incierto, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal con la exposición al fármaco.
Trastornos de la sangre y del sistema linfático: desarrollo de inhibidores del factor VIII
Trastornos del sistema inmunitario: hipersensibilidad
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de Riesgos
No hay estudios sobre el uso de ELOCTATE en mujeres embarazadas para informar un riesgo asociado a los medicamentos. El riesgo de fondo de defectos de nacimiento mayores y aborto espontáneo en la población indicada es desconocido; sin embargo, el riesgo de fondo de defectos de nacimiento mayores en la población general de los EE. UU. es del 2-4% y de aborto espontáneo del 15-20% de los embarazos clínicamente reconocidos.
No se han realizado estudios de toxicidad reproductiva y del desarrollo en animales con ELOCTATE. En un estudio de transferencia placentaria, ELOCTATE se detectó en muestras de sangre fetal de ratón a aproximadamente el 1% de los niveles de sangre materna (rango, 0.2% a 1.9%), de 3 a 4 horas después de la administración de dosis a ratones preñados con 260 a 650 veces la dosis clínica de 20 a 50 UI/kg de ELOCTATE [ver Datos].
Se desconoce si ELOCTATE puede causar daño fetal cuando se administra a una mujer embarazada, o si puede afectar la capacidad reproductiva. Si ELOCTATE es claramente necesario para tratar a una mujer embarazada, avise al paciente que se desconocen los riesgos para la madre y para el feto.
Datos de animales
Ratones preñados, genéticamente modificados, deficientes en FVIII (ratones Hem A) fueron inyectados por vía intravenosa con una sola dosis de 400 UI (aproximadamente 13,000 UI/kg) de ELOCTATE al final del embarazo en el Día de Gestación 19. Se tomaron muestras de sangre de los ratones madres y los fetos de 3 a 4 horas después de la administración de la dosis, y se midió la actividad del FVIII en el plasma materno y fetal utilizando un ensayo cromogénico del FVIII. Después de administrar la dosis de ELOCTATE a ratones HemA preñados, la actividad del FVIII en la sangre fetal fue aproximadamente del 1% de los niveles de sangre materna, lo que sugiere que puede ocurrir la transferencia placentaria de ELOCTATE. La relevancia de estos datos para los humanos es desconocida.
8.2 Lactancia
Resumen de Riesgos
No hay información sobre la presencia de ELOCTATE en la leche materna, sus efectos en el lactante amamantado o sus efectos en la producción de leche. Los beneficios para el desarrollo y la salud de la lactancia materna deben considerarse junto con la necesidad clínica de la madre de ELOCTATE y cualquier posible efecto adverso en el lactante amamantado por ELOCTATE o por la condición materna subyacente.
8.4 Uso Pediátrico
Se han realizado estudios de seguridad y eficacia en 82 pacientes pediátricos (PTP) previamente tratados <18 años de edad que recibieron al menos una dosis de ELOCTATE como parte de la profilaxis de rutina, el tratamiento a demanda de episodios de sangrado o el manejo perioperatorio. Los sujetos adolescentes se inscribieron en el ensayo de seguridad y eficacia para adultos y adolescentes, y los sujetos <12 se inscribieron en un ensayo pediátrico. La seguridad y eficacia de ELOCTATE se han evaluado en 103 pacientes pediátricos previamente no tratados (PUP) <6 años de edad (mediana 0.58 años; rango: 0.02–4 años) en un estudio (Estudio PUP) [ver Reacciones adversas (6)].
Los datos farmacocinéticos de un estudio pediátrico de los 54 sujetos evaluables <12 años de edad mostraron que no se requirió ningún ajuste de dosis para pacientes ≥6 años de edad. Los niños de 1 a 5 años tuvieron una vida media más corta y un aclaramiento más alto (ajustado por peso corporal); por lo tanto, puede ser necesaria una dosis más alta o una dosificación más frecuente en este grupo de edad [ver Farmacología clínica (12.3)].
8.5 Uso Geriátrico
Los estudios clínicos de ELOCTATE no incluyeron un número suficiente de sujetos de 65 años o más para determinar si responden de manera diferente a los sujetos más jóvenes.
11 DESCRIPCIÓN
ELOCTATE, Factor Antihemofílico (Recombinante), Proteína de Fusión Fc, es un polvo liofilizado estéril, no pirogénico, de color blanco a blanquecino para reconstitución para inyección intravenosa. El producto se suministra en viales de dosis única que contienen potencias nominales de 250, 500, 750, 1000, 1500, 2000 3000, 4000, 5000 o 6000 unidades internacionales (UI). Cada vial de ELOCTATE está etiquetado con el contenido real en UI. El polvo para inyección se reconstituye con 3 ml de agua estéril para inyección (SWFI) suministrada en una jeringa precargada estéril. La solución reconstituida debe estar esencialmente libre de partículas. El producto final contiene los siguientes excipientes: sacarosa, cloruro de sodio, L-histidina, cloruro de calcio y polisorbato 20. ELOCTATE no contiene conservantes.
El ingrediente activo de ELOCTATE es el Factor VIII recombinante delecionado en el dominio B, proteína de fusión Fc (BDD-rFVIIIFc). BDD-rFVIIIFc es una proteína recombinante que consiste en un análogo delecionado en el dominio B del Factor de Coagulación VIII humano unido covalentemente a la secuencia del dominio Fc de la inmunoglobulina G1 (IgG1) humana. La porción del Factor VIII de la molécula tiene una cadena pesada de 90 kDa y una cadena ligera de 80 kDa (similar al Factor VIII endógeno), que están unidas por 14 (de 908) aminoácidos del dominio B central. La porción de FVIII tiene modificaciones postraduccionales comparables a las del Factor VIII endógeno. El dominio Fc de la molécula contiene las regiones de bisagra, CH2 y CH3 de la IgG1. BDD-rFVIIIFc contiene 1890 aminoácidos con un peso molecular aparente de 220 kDa. La mayor parte de la proteína expresada se procesa proteolíticamente a una molécula de dos cadenas; sin embargo, ELOCTATE también puede contener hasta un 39% de una forma no procesada de una sola cadena. Se ha demostrado que ambas moléculas tienen una actividad del Factor VIII comparable.
BDD-rFVIIIFc se produce mediante tecnología de ADN recombinante a partir de una línea celular de riñón embrionario humano (HEK), que ha sido ampliamente caracterizada. La línea celular HEK expresa BDD-rFVIIIFc en un medio de cultivo celular definido que no contiene ninguna proteína derivada de fuentes animales o humanas. BDD-rFVIIIFc se purifica mediante una serie de pasos de cromatografía, incluida la captura por afinidad con un fragmento de anticuerpo recombinante de cadena única producido en un sistema de expresión de levadura. No se utilizan proteínas de origen humano o animal en los procesos de purificación o formulación. El proceso de producción también incorpora dos pasos específicos de eliminación viral: un paso de tratamiento con detergente para la inactivación y un paso de filtración de 15 nm para la eliminación de virus.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
ELOCTATE es una proteína de fusión recombinante que reemplaza temporalmente el Factor VIII de coagulación faltante necesario para una hemostasia efectiva. ELOCTATE contiene la región Fc de la inmunoglobulina G1 humana (IgG1), que se une al receptor Fc neonatal (FcRn). FcRn es parte de una vía natural que retrasa la degradación lisosomal de las inmunoglobulinas al reciclarlas de vuelta a la circulación y prolongar su vida media plasmática.
12.2 Farmacodinamia
La hemofilia A es un trastorno hemorrágico caracterizado por una deficiencia de Factor VIII de coagulación funcional, lo que resulta en un tiempo de coagulación prolongado del plasma del paciente, medido mediante la prueba de tiempo de tromboplastina parcial activado (aPTT). El tratamiento con ELOCTATE normaliza el aPTT durante el período de dosificación efectivo.
12.3 Farmacocinética
La farmacocinética (PK) de ELOCTATE (rFVIIIFc) se evaluó en 28 sujetos después de una infusión intravenosa de 10 minutos de una dosis única de 50 UI/kg. Los parámetros de PK se basaron en la actividad del FVIII plasmático medida mediante el ensayo de coagulación de un solo paso. El perfil de PK obtenido en la semana 14, después de la dosificación repetida, fue comparable con el perfil de PK obtenido después de la primera dosis. Los datos de PK demuestran que ELOCTATE tiene una vida media de circulación prolongada. El tiempo al 1% fue de 5,10 días (IC del 95%: 4,54, 5,66). La vida media plasmática terminal de ELOCTATE en comparación con un Factor VIII recombinante comercializado actualmente (ADVATE®) fue 1,5 veces más larga.
Farmacocinética pediátrica y adolescente
Los parámetros farmacocinéticos (PK) de ELOCTATE se determinaron para adolescentes (de 12 a 17 años) en el estudio de adultos y adolescentes y para niños (de 1 a 5 años y de 6 a 11 años) en el estudio pediátrico. Tabla 5 presenta los parámetros de PK calculados a partir de los datos pediátricos de 65 sujetos, menores de 18 años, después de recibir una dosis única de 50 UI/kg.
En comparación con los adultos y adolescentes, el aclaramiento ajustado al peso corporal fue un 75% más alto en niños de 1 a 5 años. Estos resultados indican la necesidad de ajustes de dosis en niños de 1 a 5 años.
La evaluación de PK de los sujetos pediátricos, de 6 a 17 años, mostró que sus perfiles de PK y las medias aritméticas de los parámetros de PK son similares a los de los adultos. Por lo tanto, para los sujetos de 6 años o más, no se requiere un ajuste de dosis basado en la edad.
| Parámetros de PK* | Estudio pediátrico | Estudio de adultos y adolescentes | ||
|---|---|---|---|---|
| 1 a 5 años | 6 a 11 años | 12 a 17 años | Adultos† | |
| N=23 | N=31 | N=11 | N=28 | |
|
Abreviaturas: IC = intervalo de confianza; AUC = área bajo la curva tiempo de actividad del FVIII; t1/2 = vida media terminal; MRT = tiempo medio de residencia; CL = aclaramiento ajustado al peso corporal; Vss = volumen de distribución en estado estacionario ajustado al peso corporal |
||||
| Recuperación incremental (UI/dL por UI/kg) |
1.92 (1.80, 2.04) |
2.44 (2.07, 2.80) |
1.85 (1.58, 2.12) |
2.26 (2.13, 2.40) |
| AUC/Dosis (UI x h/dL por UI/kg) |
30.0 (26.5, 33.6) |
41.9 (34.0, 49.8) |
38.7 (34.3, 43.1) |
54.1 (47.0, 61.1) |
| t½ (h) | 12.7 (11.2, 14.1) |
14.9 (12.0, 17.8) |
16.4 (14.1, 18.6) |
19.7 (17.4, 22.0) |
| MRT (h) | 17.2 (15.4, 19.1) |
20.9 (17.1, 24.7) |
23.1 (19.9, 26.4) |
26.1 (23.2, 28.9) |
| CL (mL/h/kg) | 3.60 (3.13, 4.07) |
2.78 (2.44, 3.13) |
2.66 (2.34, 2.98) |
2.06 (1.78, 2.34) |
| Vss (mL/kg) | 58.6 (54.9, 62.3) |
52.1 (45.3, 59.0) |
60.3 (53.3, 67.3) |
49.5 (46.9, 52.2) |
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
No se han realizado estudios a largo plazo en animales para investigar los efectos carcinogénicos de ELOCTATE. In vitro e in vivo no se realizaron pruebas de ELOCTATE para mutagenicidad o efectos sobre la fertilidad.
14 ESTUDIOS CLÍNICOS
La seguridad y eficacia de ELOCTATE se evaluó en dos ensayos clínicos multicéntricos, prospectivos, de etiqueta abierta en pacientes previamente tratados (PPT) (estudio en adultos y adolescentes y estudio pediátrico), y en un estudio de extensión.
El estudio en adultos y adolescentes comparó la eficacia de cada uno de los dos regímenes de tratamiento profiláctico (individualizado y fijo semanal) con el tratamiento episódico (a demanda); determinó la eficacia hemostática en el tratamiento de episodios hemorrágicos; y determinó la eficacia hemostática durante el manejo perioperatorio en sujetos que se sometieron a procedimientos quirúrgicos mayores. El estudio incluyó a un total de 165 pacientes masculinos previamente tratados (PPT) con hemofilia A grave (<1% de actividad del factor VIII endógeno o una mutación genética consistente con hemofilia A grave). Los sujetos tenían entre 12 y 65 años, incluidos 13 sujetos pediátricos de 12 a 17 años. De los 165 sujetos incluidos, 164 recibieron al menos una dosis de ELOCTATE y 163 (98%) fueron evaluables para la eficacia. Un total de 153 sujetos (93%) completaron el estudio.
El estudio pediátrico evaluó la eficacia del tratamiento profiláctico individualizado; determinó la eficacia hemostática en el tratamiento de episodios hemorrágicos; y determinó la eficacia hemostática durante el manejo perioperatorio en sujetos que se sometieron a procedimientos quirúrgicos. El estudio incluyó a un total de 71 pacientes pediátricos masculinos previamente tratados con hemofilia A grave (<1% de actividad del FVIII endógeno o una mutación genética consistente con hemofilia A grave). De los 71 sujetos incluidos, 69 recibieron al menos 1 dosis de ELOCTATE y fueron evaluables para la eficacia. Todos los sujetos tenían menos de 12 años (35 tenían entre 1 y 5 años y 34 tenían entre 6 y 11 años).
El estudio de extensión evaluó la seguridad y eficacia de los regímenes de tratamiento profiláctico o el tratamiento a demanda; así como la eficacia hemostática durante el manejo perioperatorio en sujetos que se sometieron a procedimientos quirúrgicos. El estudio incluyó a un total de 240 pacientes masculinos previamente tratados (de 2 a 66 años de edad) con hemofilia A grave que completaron el estudio en adultos y adolescentes o el estudio pediátrico.
Tratamiento a demanda y control de episodios hemorrágicos
En el estudio en adultos y adolescentes, un total de 757 episodios hemorrágicos en 106 sujetos fueron tratados con ELOCTATE. La mayoría de los episodios hemorrágicos fueron espontáneos y localizados en las articulaciones. La dosis mediana por inyección utilizada para tratar un episodio hemorrágico fue de 27,35 (IQR 22,73, 32,71) UI/kg. La evaluación de la respuesta a cada inyección fue registrada por los sujetos a las 8-12 horas después del tratamiento. Se utilizó una escala de calificación de 4 puntos de excelente, buena, moderada y sin respuesta para evaluar la respuesta. La eficacia en el control de episodios hemorrágicos en sujetos ≥12 años de edad se resume en Tabla 6.
| Nuevos episodios hemorrágicos | (n=757) | |
|---|---|---|
|
||
| # de Inyecciones para tratar episodios hemorrágicos | ||
| 1 inyección | 661 (87.3%) | |
| 2 inyecciones | 79 (10.4%) | |
| >2 inyecciones | 17 (2.2%) | |
| Respuesta a la primera inyección* | (n=745) | |
| Excelente o buena | 78.1% | |
| Moderada | 21.2% | |
| Sin respuesta | 0.7% | |
En el estudio pediátrico, se trataron un total de 86 episodios de sangrado en 69 sujetos pediátricos con ELOCTATE. La evaluación de la respuesta a cada inyección fue registrada por los sujetos a las 8 a 12 horas posteriores al tratamiento. Se utilizó una escala de calificación de 4 puntos de excelente, buena, moderada y sin respuesta para evaluar la respuesta. La eficacia en el control de los episodios de sangrado en sujetos <12 años de edad se resume en Tabla 7.
La eficacia hemostática en el tratamiento de las hemorragias se calificó como excelente o buena en el 92,6% para todas las primeras inyecciones evaluables.
| 1-5 Años
(n=35) |
6 a 11 Años
(n=34) |
Total (<12 Años) (n=69) |
||
|---|---|---|---|---|
|
||||
| Nuevos episodios de sangrado | (n=38) | (n=48) | (n=86) | |
| # de Inyecciones para tratar episodios de sangrado | 1 inyección | 29 (76.3%) | 41 (85.4%) | 70 (81.4%) |
| 2 inyecciones | 7 (18.4%) | 3 (6.3%) | 10 (11.6%) | |
| >2 inyecciones | 2 (5.3%) | 4 (8.3%) | 6 (7.0%) | |
| Dosis mediana por inyección (UI/kg) para tratar un episodio de sangrado (IQR) | 51.35 (29.94, 59.52) |
48.15 (29.08, 55.97) |
49.69 (29.41, 56.82) |
|
| Dosis total mediana (UI/kg) para tratar un episodio de sangrado (IQR) | 56.40 (29.94, 72.46) |
53.49 (29.08, 66.80) |
54.90 (29.41, 71.09) |
|
| Respuesta a la primera inyección* | (n=35) | (n=46) | (n=81) | |
| Excelente o buena | 32 (91.4%) | 43 (93.5%) | 75 (92.6%) | |
| Moderada | 3 (8.6%) | 1 (2.2%) | 4 (4.9%) | |
| Sin respuesta | 0 (0.0%) | 2 (4.3%) | 2 (2.5%) | |
Cirugías mayores
La hemostasia se evaluó en cuarenta y cinco (45) cirugías en treinta y dos (32) sujetos del estudio de adultos y adolescentes y el estudio de extensión. No hubo cirugías mayores en el estudio pediátrico o los estudios de farmacocinética. De las 45 cirugías mayores, 36 cirugías (80.0%) requirieron una sola dosis perioperatoria para mantener la hemostasia. De las 42 cirugías mayores tratadas con al menos una dosis, la dosis promedio mediana por inyección para mantener la hemostasia durante la cirugía fue de 59.1 UI/kg (rango 35-111). El día de la cirugía, la mayoría de los sujetos recibieron una segunda inyección. La dosis total el día de la cirugía varió de 37.6 a 157.9 UI/kg.
La respuesta hemostática fue evaluada por el investigador utilizando escalas ordinales de la siguiente manera:
Excelente: Pérdida de sangre intraoperatoria y postoperatoria similar a (o menor que) la del paciente no hemofílico. No se necesitan dosis adicionales de rFVIIIFc y las transfusiones de componentes sanguíneos requeridas son similares a las del paciente no hemofílico
Bueno: La pérdida de sangre intraoperatoria y/o postoperatoria es ligeramente mayor que lo esperado para el paciente no hemofílico, pero la diferencia no fue clínicamente significativa. La pérdida de sangre intraoperatoria no es más de 250 mL mayor que lo esperado para un paciente no hemofílico y no se necesitan dosis adicionales de rFVIIIFc y las transfusiones de componentes sanguíneos requeridas son similares a las del paciente no hemofílico
Regular: La pérdida de sangre intraoperatoria y/o postoperatoria es mayor que lo esperado para el paciente no hemofílico y se necesitó tratamiento adicional. La pérdida de sangre intraoperatoria de 250 a 500 mL mayor que lo esperado para una persona sin hemofilia o dosis adicional de rFVIIIFc necesaria o aumento del requisito de transfusión de componentes sanguíneos
Pobre/ninguna: Sangrado intraoperatorio y/o postoperatorio significativo que fue sustancialmente mayor que lo esperado para el paciente no hemofílico, requirió intervención y no se explicó por un problema quirúrgico/médico que no fuera la hemofilia: Pérdida de sangre intraoperatoria >500 mL mayor que para el paciente no hemofílico o hipotensión inesperada o transferencia inesperada a la unidad de cuidados intensivos debido a sangrado o aumento sustancial del requisito de transfusión de componentes sanguíneos.
Para cuarenta y una (41) cirugías mayores en veintinueve (29) sujetos, la respuesta hemostática se evaluó y calificó como excelente en 38 (93%) cirugías y buena en 3 (7%) cirugías.
Los tipos de cirugías evaluadas incluyen procedimientos ortopédicos mayores como reemplazos articulares (rodilla bilateral, así como reemplazos unilaterales de codo, cadera y rodilla), fusión de tobillo y amputación. Otras cirugías mayores incluyen apendicectomía, artroscopia, cirugía espinal y reparación de hernia inguinal.
Cirugías menores
Se realizó una evaluación hemostática de 72 procedimientos quirúrgicos menores en 59 sujetos de los tres estudios, y todos (100%) tuvieron una respuesta excelente o buena (respuesta excelente [61 de 72; 84.7%] y respuesta buena [11 de 72; 15.3%]).
Estudio de adultos y adolescentes
La eficacia de la profilaxis de rutina se evaluó frente al tratamiento a demanda. Un total de 117 sujetos recibieron un régimen individualizado, dos veces por semana, que comenzó con 25 UI/kg el primer día seguido de 50 UI/kg el cuarto día. La dosis y el intervalo se ajustaron dentro del rango de 25-65 UI/kg cada 3-5 días para mantener los niveles valle entre 1% y 3% por encima del nivel basal, o más alto, según lo indicado clínicamente para prevenir el sangrado. El intervalo de dosificación mediano fue de 3.5 días. Entre los 112 sujetos tratados durante al menos 6 meses, 111 (99%) lograron un intervalo de dosificación de tres días o más, 39 (35%) lograron un intervalo de dosificación de 4 días o más, y 33 (29%) lograron un intervalo de dosificación de 5 días o más durante los últimos 3 meses del estudio. Veintitrés sujetos recibieron 65 UI/kg de ELOCTATE una vez por semana durante un período mediano de 28 semanas. Otros 23 sujetos recibieron dosis episódicas (a demanda) de ELOCTATE para el tratamiento de episodios de sangrado y estuvieron en el estudio durante un período mediano de 29 semanas. Utilizando un modelo binomial negativo para analizar la tasa de sangrado anualizada (ABR), hubo una reducción estadísticamente significativa en la ABR del 92% (p<0.001) para los sujetos en el brazo de profilaxis individualizada y una reducción estadísticamente significativa del 76% (p<0.001) para los sujetos en el brazo de profilaxis semanal en comparación con el brazo episódico (a demanda). Cincuenta y tres (53) de 117 (45%) sujetos no experimentaron episodios de sangrado mientras estaban en profilaxis individualizada y 4 de 23 (17%) sujetos no experimentaron episodios de sangrado mientras estaban en profilaxis semanal.
Las ABR medianas en sujetos evaluables para la eficacia se resumen en Tabla 8.
| Etiología del episodio de sangrado | Profilaxis individualizada (N=117) |
Episódica (a demanda) (N=23) |
|---|---|---|
|
||
| ABR general | 1.6 | 33.6 |
| (0.0, 4.7) | (21.1, 48.7) | |
| ABR espontánea | 0.0 | 20.2 |
| (0.0, 2.0) | (12.2, 36.8) | |
| Joint ABR | 0.0 | 22.8 |
| (0.0, 3.1) | (15.1, 39.0) | |
Estudio pediátrico
Sesenta y nueve (69) sujetos recibieron ELOCTATE en un régimen de dosis profiláctica individualizada que comienza con un régimen dos veces por semana que consiste en 25 UI/kg el primer día seguido de 50 UI/kg el cuarto día. La dosis podría ajustarse dentro del rango de 25-80 UI/kg con un intervalo de dosificación mínimo de cada 2 días para mantener un valle de 1% por encima del nivel basal o según lo indicado clínicamente para prevenir el sangrado. El intervalo de dosificación promedio fue de 3.49 días (rango intercuartílico, 3.46 a 3.51 días) sin diferencia en el intervalo de dosificación promedio entre los grupos de edad. El 89.9% de los sujetos permanecieron en un intervalo de dos veces por semana. La dosis semanal promedio de ELOCTATE para sujetos de 1 a 5 años de edad fue de 91.63 UI/kg (rango intercuartílico (IQR), 84.72 a 104.56 UI/kg). Para los sujetos en el grupo de edad de 6 a 11 años, la dosis semanal promedio fue de 86.88 UI/kg (IQR, 79.12 a 103.08 UI/kg).
De todos los sujetos, 32 (46.4%) no experimentaron episodios de sangrado (18 sujetos (51.4%) de 1 a 5 años de edad y 14 sujetos (41.2%) de 6 a 11 años de edad). Una presentación de las ABR promedio evaluables para la eficacia se resume en Tabla 9.
| Etiología del episodio de sangrado | 1-5 Años (N=35) |
6 a 11 Años (N=34) |
Total (<12 Años) (N=69) |
|---|---|---|---|
|
|||
| ABR general | 0.0 | 2.0 | 2.0 |
| (0.0, 4.0) | (0.0, 4.0) | (0.0, 4.0) | |
| ABR espontánea | 0.0 | 0.0 | 0.0 |
| (0.0, 0.0) | (0.0, 0.0) | (0.0, 0.0) | |
| Joint ABR | 0.0 | 0.0 | 0.0 |
| (0.0, 1.9) | (0.0, 2.0) | (0.0, 2.0) | |
15 REFERENCIAS
- Sommer JM, Moore N, McGuffie-Valentine B, et al. Estudio de campo comparativo que evalúa la actividad de la proteína de fusión de factor VIII recombinante Fc en muestras de plasma en laboratorios de hemostasia clínica. Haemophilia. 2014;20:294–300.
16 FORMA DE SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
Modo de Suministro
ELOCTATE se suministra en kits que comprenden un vial de dosis única que contiene nominalmente 250, 500, 750, 1000, 1500, 2000, 3000, 4000, 5000 o 6000 unidades internacionales (UI) de potencia de Factor VIII, una jeringa precargada con 3 ml de agua estéril para inyección y un adaptador de vial estéril (dispositivo de reconstitución). La cantidad real de ELOCTATE en UI se indica en la etiqueta y en la caja de cada vial.
No está fabricado con látex de caucho natural.
| Concentración | Código de Color de Potencia | Número NDC del Kit |
| 250 IU | Amarillo | 71104-801-01 |
| 500 IU | Rojo | 71104-802-01 |
| 750 IU | Granate | 71104-803-01 |
| 1000 IU | Verde | 71104-804-01 |
| 1500 IU | Verde Oscuro | 71104-805-01 |
| 2000 IU | Azul Real | 71104-806-01 |
| 3000 IU | Gris Niebla | 71104-807-01 |
| 4000 IU | Naranja | 71104-808-01 |
| 5000 IU | Marrón Niebla | 71104-809-01 |
| 6000 IU | Morado | 71104-810-01 |
Antes de la reconstitución:
- Guarde ELOCTATE en el envase original para proteger los viales de ELOCTATE de la luz.
- Guarde ELOCTATE en polvo a una temperatura de 2 °C a 8 °C (36 °F a 46 °F). No lo congele para evitar que se dañe la jeringa precargada con diluyente.
- ELOCTATE puede almacenarse a temperatura ambiente, sin exceder los 30 °C (86 °F), durante un único período de hasta 6 meses, dentro de la fecha de caducidad impresa en la etiqueta.
- Si se almacena a temperatura ambiente, registre la fecha en que se saca ELOCTATE del refrigerador en la caja, en el área provista. Después del almacenamiento a temperatura ambiente, no vuelva a colocar el producto en el refrigerador.
- No lo use después de la fecha de caducidad impresa en el vial o 6 meses después de la fecha que se escribió en la caja, lo que ocurra primero.
Después de la Reconstitución:
- El producto reconstituido puede almacenarse a temperatura ambiente, sin exceder los 30 °C (86 °F), hasta por 3 horas. Protéjalo de la luz solar directa. Después de la reconstitución, si el producto no se utiliza en un plazo de 3 horas, debe desecharse.
- No utilice ELOCTATE si la solución reconstituida está turbia o contiene partículas.
- Deseche cualquier cantidad de ELOCTATE que no se haya utilizado.
17 INFORMACIÓN PARA EL PACIENTE
Aconseje a los pacientes que:
- Lean el etiquetado del paciente aprobado por la FDA (Información para el paciente y Instrucciones de uso).
- Llame a su proveedor de atención médica o vaya al departamento de emergencias de inmediato si ocurre una reacción de hipersensibilidad. Los signos tempranos de reacciones de hipersensibilidad pueden incluir erupción cutánea, urticaria, picazón, hinchazón facial, opresión en el pecho y sibilancias.
- Comuníquese con su proveedor de atención médica o centro de tratamiento para recibir tratamiento y/o evaluación adicionales si experimenta una falta de respuesta clínica a la terapia con Factor VIII, ya que esto puede ser un signo de desarrollo de inhibidores.
Fabricado por:
Bioverativ Therapeutics Inc.
Waltham, MA 02451
A SANOFI COMPANY
Número de licencia de EE. UU. 2078
©2023 Bioverativ Therapeutics Inc. Todos los derechos reservados.
ELOCTATE® es una marca registrada de Bioverativ Therapeutics Inc.
Para información de patentes: https://www.sanofi.us/en/products-and-resources/patents
INSERTO PARA EL PACIENTE
Información para el paciente
ELOCTATE® /el’ ok’ tate /
[Antihemophilic Factor (Recombinant), Fc Fusion Protein]
Lea atentamente esta Información para el paciente antes de usar ELOCTATE y cada vez que le surtan la receta, ya que puede haber información nueva. Esta Información para el paciente no sustituye a las conversaciones con su proveedor de atención médica sobre su afección médica o su tratamiento.
¿Qué es ELOCTATE?
ELOCTATE es un medicamento inyectable que se usa para ayudar a controlar y prevenir el sangrado en personas con hemofilia A (deficiencia congénita del factor VIII).
Es posible que su proveedor de atención médica le administre ELOCTATE cuando se someta a una cirugía.
¿Quiénes no deben usar ELOCTATE?
No debe usar ELOCTATE si ha tenido una reacción alérgica al mismo en el pasado.
¿Qué debo decirle a mi proveedor de atención médica antes de usar ELOCTATE?
Hable con su proveedor de atención médica sobre:
- Cualquier problema médico que tenga o haya tenido.
- Todos los medicamentos recetados y de venta libre que tome, incluidos los medicamentos de venta libre, los suplementos o las hierbas medicinales.
- Embarazo o si está planeando quedar embarazada. Se desconoce si ELOCTATE puede dañar al feto.
- Lactancia materna. Se desconoce si ELOCTATE pasa a la leche materna y si puede dañar al bebé.
¿Cómo debo usar ELOCTATE?
Usted recibe ELOCTATE como una infusión en la vena. Su proveedor de atención médica le indicará cómo administrarse las infusiones usted mismo y puede observarlo mientras se administra la primera dosis de ELOCTATE.
Comuníquese con su proveedor de atención médica de inmediato si el sangrado no se controla después de usar ELOCTATE.
¿Cuáles son los posibles efectos secundarios de ELOCTATE?
Puede tener una reacción alérgica a ELOCTATE. Llame a su proveedor de atención médica o al servicio de urgencias de inmediato si tiene alguno de los siguientes síntomas: dificultad para respirar, opresión en el pecho, hinchazón de la cara, sarpullido o urticaria.
Su cuerpo también puede producir anticuerpos llamados “inhibidores” contra ELOCTATE. Esto puede impedir que ELOCTATE funcione correctamente. Es posible que su proveedor de atención médica le realice análisis de sangre para detectar inhibidores.
Otros efectos secundarios comunes de ELOCTATE son dolor de cabeza, sarpullido, dolor articular, dolor muscular y malestar general.
Si tiene factores de riesgo para desarrollar coágulos sanguíneos anormales en su cuerpo, como un catéter venoso permanente, el tratamiento con Factor VIII puede aumentar este riesgo.
Estos no son los únicos efectos secundarios posibles de ELOCTATE. Informe a su proveedor de atención médica sobre cualquier efecto secundario que le moleste o que no desaparezca.
¿Cómo debo almacenar ELOCTATE?
- Mantenga ELOCTATE en su envase original.
- Protéjalo de la luz.
- No lo congele.
- Guárdelo refrigerado (de 2 °C a 8 °C o de 36 °F a 46 °F) o a temperatura ambiente [sin exceder los 30 °C (86 °F)], hasta por seis meses.
- Cuando lo guarde a temperatura ambiente:
- –
- Anote en la caja la fecha en que el producto se sacó del refrigerador.
- –
- Utilice el producto antes de que finalice este período de 6 meses o deséchelo.
- –
- No vuelva a colocar el producto en el refrigerador.
No use ELOCTATE después de la fecha de caducidad impresa en el vial o, si lo sacó del refrigerador, después de la fecha que anotó en la caja, lo que ocurra primero.
Después de la reconstitución (mezcla con el diluyente):
- No use ELOCTATE si la solución reconstituida no es transparente a ligeramente opalescente e incolora.
- Utilice el producto reconstituido lo antes posible.
- Puede guardar la solución reconstituida a temperatura ambiente, sin exceder los 30 °C (86 °F), hasta por tres horas. Proteja el producto reconstituido de la luz solar directa. Deseche cualquier producto que no se utilice en un plazo de tres horas.
¿Qué más debo saber sobre ELOCTATE?
A veces se recetan medicamentos para fines distintos a los que se enumeran aquí. No use ELOCTATE para una afección para la que no fue recetado. No comparta ELOCTATE con otras personas, incluso si tienen los mismos síntomas que usted.
Esta Información para el paciente ha sido aprobada por la Administración de Alimentos y Medicamentos de los Estados Unidos.
Fabricado por:
Bioverativ Therapeutics Inc.
Waltham, MA 02451
A SANOFI COMPANY
US License Number 2078
©2023 Bioverativ Therapeutics Inc. Todos los derechos reservados.
ELOCTATE® es una marca registrada de Bioverativ Therapeutics Inc.
Revisado: mayo de 2023
INSTRUCCIONES DE USO
ELOCTATE®
Factor antihemofílico (recombinante), proteína de fusión Fc
INSTRUCCIONES DE USO
Lea las Instrucciones de Uso antes de comenzar a usar ELOCTATE y cada vez que reciba un reabastecimiento. Puede haber nueva información. Esta información no sustituye la conversación con su proveedor de atención médica acerca de su condición médica o su tratamiento.
Su proveedor de atención médica debe mostrarle a usted o a su cuidador cómo reconstituir y administrar ELOCTATE la primera vez que se use ELOCTATE.
Compruebe la fecha de caducidad del kit de ELOCTATE.
No use el producto si ha pasado la fecha de caducidad.
Permita que el vial de ELOCTATE y el diluyente lleguen a temperatura ambiente.
No use fuentes de calor externas como poner el vial y/o el diluyente en agua caliente.
Encuentre una superficie de trabajo limpia y plana y reúna todos los suministros que necesitará para reconstituir y administrar ELOCTATE.
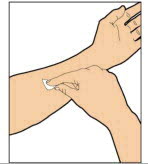
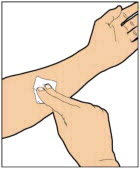
Lávese las manos con agua y jabón. Se debe utilizar la técnica aséptica (limpia y libre de gérmenes).
SU KIT CONTIENE:
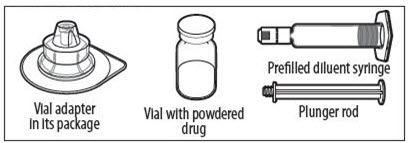
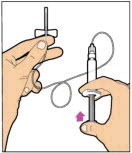
RECONSTITUCIÓN
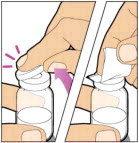
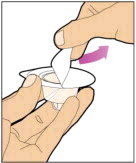
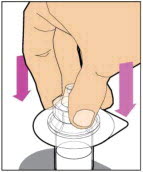

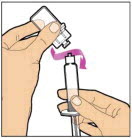


MEZCLA
MEZCLA es el proceso de combinar dos o más viales reconstituidos en una jeringa más grande (no en la jeringa de diluyente) antes de la administración intravenosa.
Si está utilizando dos o más viales, siga estos pasos de mezcla.
Asegúrese de dejar el adaptador del vial conectado al vial, ya que lo necesitará para conectar una jeringa grande con cierre Luer.
No desconecte la jeringa de diluyente o la jeringa grande con cierre Luer hasta que esté listo para conectar la jeringa grande con cierre Luer al siguiente vial (con el adaptador del vial conectado).
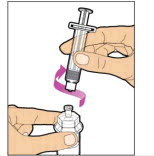
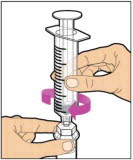
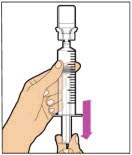
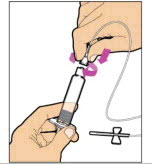
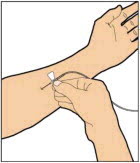
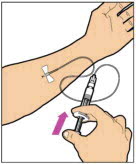
ADMINISTRACIÓN (Inyección intravenosa)
ELOCTATE se administra mediante infusión intravenosa después de reconstituir el polvo del medicamento con el diluyente.
Su proveedor de atención médica debe enseñarle cómo infundir ELOCTATE. Una vez que le hayan enseñado a autoinfundirse, puede seguir estas instrucciones.
No administre ELOCTATE reconstituido si contiene partículas, está descolorido o turbio.
CONDICIONES DE ALMACENAMIENTO – KIT DE PRODUCTO
Mantener refrigerado hasta su uso.
Mantener alejado de la luz solar directa.
CONDICIONES DE ALMACENAMIENTO – RECONSTITUIDO
ELOCTATE debe administrarse dentro de las 3 horas posteriores a la reconstitución.
No refrigerar después de la reconstitución.
Mantener alejado de la luz solar directa.
Estas Instrucciones de uso han sido aprobadas por la Administración de Alimentos y Medicamentos de los Estados Unidos.
Fabricado por:
Bioverativ Therapeutics Inc.
Waltham, MA 02451
A SANOFI COMPANY
Número de licencia de EE. UU. 2078
©2023 Bioverativ Therapeutics Inc. Todos los derechos reservados.
Para obtener más información, visite www.ELOCTATE.com o llame al 1-855-693-5628
Revisado en mayo de 2023
PANEL PRINCIPAL DE VISUALIZACIÓN – Caja de Kit – 250 UI
250 UI nominal
NDC 71104-801-01
Un frasco de dosis única
con 3 mL de jeringa precargada de diluyente
ELOCTATE®
Factor antihemofílico
(recombinante), proteína de fusión Fc
Para administración intravenosa
Únicamente con receta
Fabricado por:
Bioverativ Therapeutics Inc.
Waltham, MA 02451
Número de licencia de EE. UU. 2078
www.ELOCTATE.com
Bioverativ

PANEL PRINCIPAL DE EXHIBICIÓN – Caja del kit – 500 UI
500 UI nominales
NDC 71104-802-01
Un vial de dosis única
con una jeringa precargada de 3 mL de diluyente
ELOCTATE®
Factor antihemofílico
(Recombinante), Proteína de fusión Fc
Para administración intravenosa
Receta médica solamente
Fabricado por:
Bioverativ Therapeutics Inc.
Waltham, MA 02451
Número de licencia de EE.UU. 2078
www.ELOCTATE.com
Bioverativ

PANEL PRINCIPAL DE VISUALIZACIÓN – Caja de Kit – 750 UI
750 UI nominal
NDC 71104-803-01
Un vial de dosis única
con 3 mL de jeringa precargada con diluyente
ELOCTATE®
Factor antihemofílico
(recombinante), proteína de fusión Fc
Para administración intravenosa
Sólo receta médica
Fabricado por:
Bioverativ Therapeutics Inc.
Waltham, MA 02451
Número de licencia de EE. UU. 2078
www.ELOCTATE.com
Bioverativ

PANEL PRINCIPAL DE VISUALIZACIÓN – Caja de kit – 1000 UI
1000 UI nominal
NDC 71104-804-01
Un vial de dosis única
con una jeringa precargada de 3 mL
de diluyente
ELOCTATE®
Factor antihemofílico
( recombinante), proteína de fusión Fc
Para administración intravenosa
Sólo con receta
Fabricado por:
Bioverativ Therapeutics Inc.
Waltham, MA 02451
Número de licencia en EE. UU. 2078
www.ELOCTATE.com
Bioverativ

PANEL PRINCIPAL DE VISUALIZACIÓN – Caja de kit – 1500 UI
1500 UI Nominal
NDC 71104-805-01
Un vial de dosis única
con una jeringa precargada de diluyente de 3 mL
ELOCTATE®
Factor Antihemofílico
(Recombinante), Proteína de Fusión Fc
Para Administración Intravenosa
Solo con Receta Médica
Fabricado por:
Bioverativ Therapeutics Inc.
Waltham, MA 02451
Número de Licencia en EE. UU. 2078
www.ELOCTATE.com
Bioverativ

PANEL PRINCIPAL DE VISUALIZACIÓN – Caja de Kit – 2000 UI
2000 UI Nominal
NDC 71104-806-01
Un vial de dosis única
con 3 mL de jeringa precargada
de diluyente
ELOCTATE®
Factor Antihemofílico
(Recombinante), Proteína de Fusión Fc
Para administración intravenosa
Sólo con receta médica
Fabricado por:
Bioverativ Therapeutics Inc.
Waltham, MA 02451
Número de licencia de EE. UU. 2078
www.ELOCTATE.com
Bioverativ

PANEL PRINCIPAL DE VISUALIZACIÓN – Caja del kit – 3000 UI
3000 UI Nominal
NDC 71104-807-01
Un vial de dosis única
con jeringa precargada de 3 mL
de diluyente
ELOCTATE®
Factor Antihemofílico
(Recombinante), Proteína de Fusión Fc
Para Administración Intravenosa
Solo con receta médica
Fabricado por:
Bioverativ Therapeutics Inc.
Waltham, MA 02451
Número de Licencia en EE. UU. 2078
www.ELOCTATE.com
Bioverativ

Panel de visualización principal – Kit de cartón – 4000 UI
4000 UI Nominal
NDC 71104-808-01
Un vial de dosis única
con una jeringa precargada de diluyente de 3 mL
ELOCTATE®
Factor Antihemofílico
(Recombinante), Proteína de Fusión Fc
Para Administración Intravenosa
Solo con Receta Médica
Fabricado por:
Bioverativ Therapeutics Inc.
Waltham, MA 02451
Número de Licencia en EE. UU. 2078
www.ELOCTATE.com
Bioverativ

Panel principal de visualización – Kit de cartón – 5000 UI
5000 UI Nominal
NDC 71104-809-01
Un vial de dosis única
con una jeringa precargada de diluyente de 3 mL
ELOCTATE®
Factor Antihemofílico
(Recombinante), Proteína de Fusión Fc
Para Administración Intravenosa
Solo para uso con receta médica
Fabricado por:
Bioverativ Therapeutics Inc.
Waltham, MA 02451
Número de Licencia en EE. UU. 2078
www.ELOCTATE.com
Bioverativ

Panel de visualización principal – Kit de cartón – 6000 UI
6000 UI Nominal
NDC 71104-810-01
Un vial de dosis única
con 3 mL de jeringa precargada
de diluyente
ELOCTATE®
Factor Antihemofílico
(Recombinante), Proteína de Fusión Fc
Para administración intravenosa
Sólo con receta médica
Fabricado por:
Bioverativ Therapeutics Inc.
Waltham, MA 02451
Número de licencia de EE. UU. 2078
www.ELOCTATE.com
Bioverativ
