Fabricante de medicamentos: AstraZeneca Pharmaceuticals LP (Updated: 2024-09-17)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
FASENRA (benralizumab) inyección, para uso subcutáneo
Aprobación inicial en EE. UU.: 2017
CAMBIOS RECIENTES IMPORTANTES
INDICACIONES Y USO
FASENRA es un anticuerpo monoclonal citolítico dirigido al receptor alfa de la interleucina-5 (IgG1, kappa) indicado para:
- •
- tratamiento de mantenimiento complementario de pacientes adultos y pediátricos de 6 años de edad o mayores con asma grave y con un fenotipo eosinofílico. (1.1)
- •
- tratamiento de pacientes adultos con granulomatosis eosinofílica con poliangiitis (EGPA). (1.2)
Limitaciones de uso:
No para el alivio del broncoespasmo agudo o el estado asmático. (1.1)
DOSIFICACIÓN Y ADMINISTRACIÓN
Administrar mediante inyección subcutánea. (2.3)
Asma
Pacientes adultos y adolescentes de 12 años de edad o mayores:
- •
- La dosis recomendada es de 30 mg cada 4 semanas durante las primeras 3 dosis, seguidas de una vez cada 8 semanas a partir de entonces. (2.1)
Pacientes pediátricos de 6 a 11 años de edad:
- •
- Con un peso inferior a 35 kg: la dosis recomendada es de 10 mg cada 4 semanas durante las primeras 3 dosis, seguidas de una vez cada 8 semanas a partir de entonces. (2.1)
- •
- Con un peso de 35 kg o más: la dosis recomendada es de 30 mg cada 4 semanas durante las primeras 3 dosis, seguidas de una vez cada 8 semanas a partir de entonces. (2.1)
EGPA
La dosis recomendada es de 30 mg cada 4 semanas. (2.2)
Consulte la información completa de prescripción para obtener instrucciones de administración de la jeringa precargada de FASENRA y FASENRA PEN. (2.4, 2.5)
FORMAS Y FUERZAS DE DOSIFICACIÓN
CONTRAINDICACIONES
Hipersensibilidad conocida a benralizumab o excipientes. (4)
ADVERTENCIAS Y PRECAUCIONES
- •
- Reacciones de hipersensibilidad: Se han producido reacciones de hipersensibilidad (p. ej., anafilaxia, angioedema, urticaria, erupción cutánea) después de la administración de FASENRA. Suspenda el tratamiento en caso de reacción de hipersensibilidad. (5.1)
- •
- Reducción de la dosis de corticosteroides: No suspenda los corticosteroides sistémicos o inhalados de forma abrupta al iniciar el tratamiento con FASENRA. Reduzca los corticosteroides gradualmente, si procede. (5.3)
- •
- Infección parasitaria (helmintos): Trate a los pacientes con infecciones por helmintos preexistentes antes de la terapia con FASENRA. Si los pacientes se infectan mientras reciben FASENRA y no responden al tratamiento antihelmíntico, suspenda FASENRA hasta que la infección parasitaria se resuelva. (5.4)
REACCIONES ADVERSAS
Las reacciones adversas más frecuentes (incidencia mayor o igual al 5%) incluyen cefalea y faringitis. (6.1, 6.2)
Para informar sobre REACCIONES ADVERSAS SOSPECHOSAS, póngase en contacto con AstraZeneca al 1-800-236-9933 o la FDA al 1-800-FDA-1088 o www.fda.gov/medwatch.
Consulte el punto 17 para obtener INFORMACIÓN PARA EL PACIENTE y el etiquetado del paciente aprobado por la FDA.
Revisado: 9/2024
Tabla de Contenido
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDO*
1 INDICACIONES Y USO
1.1 Asma
1.2 Granulomatosis con Poliangiitis Eosinofílica
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Dosis Recomendada para Asma
2.2 Dosis Recomendada para EGPA
2.3 Instrucciones Generales de Administración
2.4 Instrucciones para la Administración de la Jeringa Prellenada FASENRA (Profesionales de la Salud)
2.5 Instrucciones para la Administración de FASENRA PEN
3 FORMAS Y FUERZAS DE DOSIFICACIÓN
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Reacciones de Hipersensibilidad
5.2 Síntomas Agudos de Asma o Deterioro de la Enfermedad
5.3 Reducción de la Dosis de Corticosteroides
5.4 Infección Parasitaria (Helminto)
6 REACCIONES ADVERSAS
6.1 Experiencia en Ensayos Clínicos
6.2 Experiencia Postcomercialización
7 INTERACCIONES MEDICAMENTOSAS
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.4 Uso Pediátrico
8.5 Uso Geriátrico
10 SOBREDOSIS
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de Acción
12.2 Farmacodinamia
12.3 Farmacocinética
12.6 Inmunogenicidad
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
14 ESTUDIOS CLÍNICOS
14.1 Estudios Clínicos en Pacientes con Asma
14.2 Estudios Clínicos en Pacientes con Granulomatosis con Poliangiitis Eosinofílica
16 CÓMO SE SUMINISTRA/ALMACENAMIENTO Y MANEJO
17 INFORMACIÓN PARA EL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información completa de prescripción no están enumeradas.
1 INDICACIONES Y USO
1.1 Asma
FASENRA está indicado para el tratamiento de mantenimiento adicional de pacientes adultos y pediátricos de 6 años de edad o mayores con asma grave y con un fenotipo eosinofílico [ver Uso en poblaciones específicas (8.4), Estudios clínicos (14.1)].
Limitaciones de uso:
- •
- FASENRA no está indicado para el alivio del broncoespasmo agudo o el estado asmático.
1.2 Granulomatosis eosinofílica con poliangitis
FASENRA está indicado para el tratamiento de pacientes adultos con granulomatosis eosinofílica con poliangitis (EGPA).
2 DOSIS Y ADMINISTRACIÓN
2.1 Dosis recomendada para el asma
Pacientes adultos y adolescentes de 12 años de edad o mayores
La dosis recomendada de FASENRA es de 30 mg (una inyección) administrada por vía subcutánea cada 4 semanas durante las primeras 3 dosis, y luego cada 8 semanas a partir de entonces.
Pacientes pediátricos de 6 a 11 años de edad
La dosis recomendada de FASENRA para pacientes pediátricos de 6 a 11 años de edad se basa en el peso corporal como se proporciona en la Tabla 1.
|
Peso corporal |
Dosis recomendada |
|
Menos de 35 kg |
10 mg (una inyección) administrada por vía subcutánea cada 4 semanas durante las primeras 3 dosis, y luego cada 8 semanas a partir de entonces. |
|
35 kg o más |
30 mg (una inyección) administrada por vía subcutánea cada 4 semanas durante las primeras 3 dosis, y luego cada 8 semanas a partir de entonces. |
2.2 Dosis recomendada para EGPA
La dosis recomendada de FASENRA es de 30 mg (una inyección) administrada una vez cada 4 semanas por inyección subcutánea.
2.3 Instrucciones generales de administración
FASENRA es solo para uso subcutáneo.
FASENRA está destinado a ser utilizado bajo la guía de un profesional de la salud. De acuerdo con la práctica clínica, se recomienda el seguimiento de los pacientes después de la administración de agentes biológicos [ver Advertencias y precauciones (5.1)].
Administre FASENRA en el muslo o el abdomen. El brazo superior también se puede utilizar si un profesional de la salud o un cuidador administra la inyección. Antes de la administración, caliente FASENRA dejando el cartón a temperatura ambiente durante unos 30 minutos. Inspeccione visualmente FASENRA para detectar partículas y decoloración antes de la administración. FASENRA es transparente a opalescente, incoloro a ligeramente amarillo, y puede contener algunas partículas traslúcidas o blancas a blanquecinas. No use FASENRA si el líquido está turbio, decolorado o si contiene partículas grandes o materia extraña.
Jeringa precargada
La jeringa precargada es para administración por un profesional de la salud.
Autoinyector (FASENRA PEN™)
FASENRA PEN está destinado a ser administrado por pacientes/cuidadores. Los pacientes/cuidadores pueden inyectarse después de recibir una capacitación adecuada en la técnica de inyección subcutánea y después de que el profesional de la salud determine que es apropiado.
En pacientes asmáticos de 6 a 11 años que pesan 35 kg o más, FASENRA PEN solo debe ser administrado por un cuidador o un profesional de la salud.
2.4 Instrucciones para la administración de la jeringa precargada de FASENRA (profesionales de la salud)
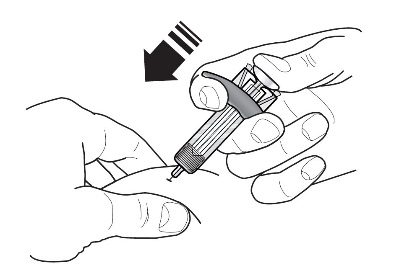
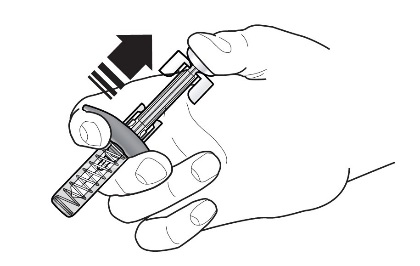
- Figura 1 Jeringas precargadas de FASENRA

Componentes de la jeringa precargada
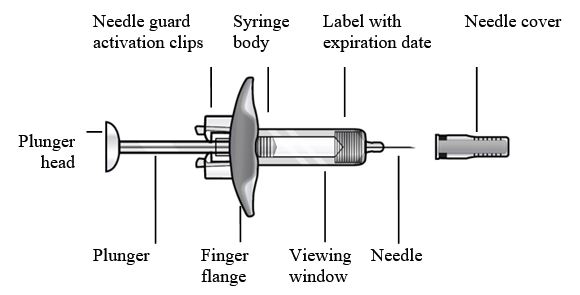
Para preparar la jeringa precargada de FASENRA para la administración subcutánea, lea atentamente y siga estas instrucciones de uso. FASENRA está disponible en una jeringa precargada de 10 mg y una de 30 mg. Compruebe las etiquetas del cartón de FASENRA y la jeringa precargada para asegurarse de que se está utilizando el producto correcto de 10 mg o 30 mg (Figura 1). Consulte la Figura 1 para identificar los componentes de la jeringa precargada que se utilizarán en los pasos de administración.
No toque los clips de activación del protector de la aguja para evitar la activación prematura del protector de la aguja.
2.5 Instrucciones para la Administración de FASENRA PEN
Consulte las “Instrucciones de uso” de FASENRA PEN para obtener instrucciones más detalladas sobre la preparación y administración de FASENRA PEN [ver Instrucciones de uso]. Un paciente puede autoinyectarse o el cuidador del paciente puede administrar FASENRA PEN por vía subcutánea después de que el profesional sanitario determine que es apropiado. En pacientes de 6 a 11 años que pesen 35 kg o más, FASENRA PEN solo debe ser administrado por un cuidador o profesional sanitario.
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
Inyección: solución de transparente a opalescente, de incolora a ligeramente amarilla y puede contener algunas partículas translúcidas o de blancas a blanquecinas disponible como:
- •
- Solución de 10 mg/0.5 mL en una jeringa precargada de dosis única.
- •
- Solución de 30 mg/mL en una jeringa precargada de dosis única.
- •
- Solución de 30 mg/mL en un autoinyector de dosis única FASENRA PEN.
4 CONTRAINDICACIONES
FASENRA está contraindicado en pacientes con hipersensibilidad conocida a benralizumab o a cualquiera de sus excipientes [ver Advertencias y precauciones (5.1)].
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Reacciones de Hipersensibilidad
Se han producido reacciones de hipersensibilidad (por ejemplo, anafilaxia, angioedema, urticaria, erupción cutánea) después de la administración de FASENRA. Estas reacciones generalmente ocurren dentro de las horas posteriores a la administración, pero en algunos casos tienen un inicio tardío (es decir, días). En caso de una reacción de hipersensibilidad, FASENRA debe suspenderse [ver Contraindicaciones (4)].
5.2 Síntomas Agudos de Asma o Deterioro de la Enfermedad
FASENRA no debe utilizarse para tratar los síntomas agudos del asma o las exacerbaciones agudas. No utilice FASENRA para tratar el broncoespasmo agudo o el estado asmático. Los pacientes deben buscar consejo médico si su asma permanece sin control o empeora después de iniciar el tratamiento con FASENRA.
5.3 Reducción de la Dosis de Corticosteroides
No suspenda abruptamente los corticosteroides sistémicos o inhalados (ICS) al iniciar la terapia con FASENRA. Las reducciones en la dosis de corticosteroides, si son apropiadas, deben ser graduales y realizarse bajo la supervisión directa de un médico. La reducción en la dosis de corticosteroides puede estar asociada con síntomas de abstinencia sistémica y/o desenmascarar afecciones previamente suprimidas por la terapia con corticosteroides sistémicos.
5.4 Infección Parasitaria (Helminto)
Los eosinófilos pueden estar involucrados en la respuesta inmunológica a algunas infecciones por helmintos. Los pacientes con infecciones conocidas por helmintos fueron excluidos de la participación en los ensayos clínicos. Se desconoce si FASENRA influirá en la respuesta de un paciente contra las infecciones por helmintos.
Trate a los pacientes con infecciones preexistentes por helmintos antes de iniciar la terapia con FASENRA. Si los pacientes se infectan mientras reciben tratamiento con FASENRA y no responden al tratamiento antihelmíntico, suspenda el tratamiento con FASENRA hasta que la infección se resuelva.
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas se describen con mayor detalle en otras secciones:
- •
- Reacciones de hipersensibilidad [ver Advertencias y precauciones (5.1)]
Debido a que los ensayos clínicos se llevan a cabo en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no se pueden comparar directamente con las tasas en los ensayos clínicos de otro fármaco y pueden no reflejar las tasas observadas en la práctica.
6.1 Experiencia en ensayos clínicos
Pacientes adultos y adolescentes de 12 años de edad o mayores con asma
En tres ensayos clínicos (SIROCCO, CALIMA y ZONDA) para el asma, 1.808 pacientes recibieron al menos 1 dosis de FASENRA [ver Estudios clínicos (14.1)]. Los datos descritos a continuación reflejan la exposición a FASENRA en 1.663 pacientes, incluidos 1.556 expuestos durante al menos 24 semanas y 1.387 expuestos durante al menos 48 semanas. La exposición a la seguridad de FASENRA se deriva de dos ensayos de fase 3 controlados con placebo (SIROCCO y CALIMA) de 48 semanas de duración [FASENRA cada 4 semanas (n=841), FASENRA cada 4 semanas durante 3 dosis, luego cada 8 semanas (n=822) y placebo (n=847)]. Si bien se incluyó un régimen de dosificación de FASENRA cada 4 semanas en los ensayos clínicos, FASENRA administrado cada 4 semanas durante 3 dosis, luego cada 8 semanas a partir de entonces es la dosis recomendada [ver Dosificación y administración (2.1)]. La población estudiada tenía entre 12 y 75 años de edad, de los cuales el 64% eran mujeres y el 79% eran blancos.
Las reacciones adversas que ocurrieron con una incidencia mayor o igual al 3% se muestran en la Tabla 2.
|
||
|
Reacciones adversas |
FASENRA (N=822) % |
Placebo (N=847) % |
|
Dolor de cabeza |
8 |
6 |
|
Fiebre |
3 |
2 |
|
Faringitis* |
5 |
3 |
|
Reacciones de hipersensibilidad† |
3 |
3 |
Ensayo de 28 semanas
Las reacciones adversas de ZONDA con 28 semanas de tratamiento con FASENRA (n=73) o placebo (n=75) en las que la incidencia fue más común en FASENRA que en placebo incluyen dolor de cabeza (8.2% en comparación con 5.3%, respectivamente) y pirexia (2.7% en comparación con 1.3%, respectivamente) [ver Estudios clínicos (14.1)]. Las frecuencias para las reacciones adversas restantes con FASENRA fueron similares al placebo.
Reacciones en el sitio de inyección en pacientes con asma
En SIROCCO y CALIMA, la jeringa precargada de FASENRA 30 mg se administró a la dosis recomendada y las reacciones en el sitio de inyección (por ejemplo, dolor, eritema, prurito, pápula) ocurrieron a una tasa del 2.2% en pacientes tratados con FASENRA en comparación con el 1.9% en pacientes tratados con placebo.
Pacientes pediátricos de 6 a 11 años de edad con asma
Los datos de seguridad para FASENRA se basan en un ensayo farmacocinético y farmacodinámico de 48 semanas, abierto, de grupos paralelos (TATE) de 28 pacientes pediátricos de 6 a 11 años con asma grave y con un fenotipo eosinofílico [ver Uso en poblaciones específicas (8.4) y Farmacología clínica (12.2,12.3)]. Los pacientes recibieron una dosis subcutánea de 10 mg (para aquellos que pesan menos de 35 kg) o 30 mg (para aquellos que pesan 35 kg o más) de FASENRA administrada cada 4 semanas durante las primeras 3 dosis, luego cada 8 semanas a partir de entonces [ver Dosificación y administración (2.1)]. No se observaron nuevas señales de seguridad en estos pacientes.
Pacientes adultos con EGPA
La seguridad de FASENRA se basa en 70 pacientes adultos que recibieron al menos 1 dosis de FASENRA 30 mg administrada por vía subcutánea cada 4 semanas en un estudio controlado activo de 52 semanas de duración (MANDARA) [ver Estudios clínicos (14.2)]. La incidencia de reacciones adversas fue consistente con las reportadas en asma, con la excepción del dolor de cabeza, que ocurrió en el 17% de los pacientes con EGPA tratados con FASENRA. No se identificaron nuevas reacciones adversas.
6.2 Experiencia postcomercialización
Además de las reacciones adversas informadas en los ensayos clínicos, se han identificado las siguientes reacciones adversas durante el uso posterior a la aprobación de FASENRA. Debido a que estas reacciones se informan voluntariamente de una población de tamaño incierto, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal con la exposición al fármaco. Estos eventos se han elegido para su inclusión debido a su gravedad, frecuencia de notificación o conexión causal con FASENRA o una combinación de estos factores.
Trastornos del sistema inmunitario: reacciones de hipersensibilidad, incluida la anafilaxis.
7 INTERACCIONES MEDICAMENTOSAS
No se han realizado estudios formales de interacción medicamentosa.
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de riesgo
Los datos sobre la exposición durante el embarazo de los ensayos clínicos son insuficientes para informar sobre el riesgo asociado al fármaco. Los anticuerpos monoclonales como el benralizumab se transportan a través de la placenta durante el tercer trimestre del embarazo; por lo tanto, los efectos potenciales en el feto son probablemente mayores durante el tercer trimestre del embarazo. En un estudio de desarrollo prenatal y postnatal realizado en monos cynomolgus, no hubo evidencia de daño fetal con la administración IV de benralizumab durante todo el embarazo en dosis que produjeron exposiciones de hasta aproximadamente 310 veces la exposición en la dosis humana máxima recomendada (MRHD) de 30 mg SC [ver Datos].
En la población general de EE. UU., el riesgo estimado de fondo de defectos de nacimiento mayores y abortos espontáneos en embarazos clínicamente reconocidos es del 2% al 4% y del 15% al 20%, respectivamente.
Consideraciones clínicas
Riesgo materno y/o del embrión/feto asociado a la enfermedad
En mujeres con asma poco o moderadamente controlada, las evidencias demuestran que existe un mayor riesgo de preeclampsia en la madre y prematuridad, bajo peso al nacer y peso bajo para la edad gestacional en el neonato. El nivel de control del asma debe ser monitoreado estrechamente en mujeres embarazadas y el tratamiento ajustado según sea necesario para mantener un control óptimo.
Datos
Datos animales
En un estudio de desarrollo prenatal y postnatal, monos cynomolgus embarazados recibieron benralizumab desde el inicio en GD20 hasta GD22 (dependiendo de la determinación del embarazo), en GD35, una vez cada 14 días a partir de entonces durante todo el período de gestación y 1 mes después del parto (máximo 14 dosis) en dosis que produjeron exposiciones de hasta aproximadamente 310 veces las alcanzadas con la MRHD (en base a AUC con dosis IV maternas de hasta 30 mg/kg una vez cada 2 semanas). Benralizumab no provocó efectos adversos en el crecimiento fetal o neonatal (incluyendo la función inmune) hasta 6,5 meses después del nacimiento. No hubo evidencia de malformaciones externas, viscerales o esqueléticas relacionadas con el tratamiento. Benralizumab no fue teratogénico en monos cynomolgus. Benralizumab cruzó la placenta en monos cynomolgus. Las concentraciones de benralizumab fueron aproximadamente iguales en madres e infantes en el día 7 postparto, pero fueron más bajas en los infantes en puntos de tiempo posteriores. Los recuentos de eosinófilos se suprimieron en los monos infantiles con recuperación gradual a los 6 meses postparto; sin embargo, la recuperación de los recuentos de eosinófilos no se observó en un mono infantil durante este período.
8.2 Lactancia
Resumen de riesgo
No hay información sobre la presencia de benralizumab en la leche humana o animal, y los efectos del benralizumab en el lactante amamantado y en la producción de leche no se conocen. Sin embargo, el benralizumab es un anticuerpo monoclonal humanizado (clase IgG1/κ), y la inmunoglobulina G (IgG) está presente en la leche humana en pequeñas cantidades. Si el benralizumab se transfiere a la leche humana, los efectos de la exposición local en el tracto gastrointestinal y la posible exposición sistémica limitada en el lactante al benralizumab son desconocidos. Se deben considerar los beneficios del desarrollo y la salud de la lactancia materna junto con la necesidad clínica de la madre de benralizumab y cualquier efecto adverso potencial en el niño amamantado por el benralizumab o por la condición materna subyacente.
8.4 Uso pediátrico
Asma
La seguridad y eficacia de FASENRA para el tratamiento de mantenimiento adicional de pacientes con asma grave y con un fenotipo eosinofílico se han establecido en pacientes pediátricos de 6 años o más. El uso de FASENRA para esta indicación está respaldado por la evidencia de lo siguiente:
Pacientes adolescentes de 12 a 17 años de edad
El uso de FASENRA en adolescentes con asma grave y con un fenotipo eosinofílico está respaldado por la evidencia de SIROCCO (n = 53) y CALIMA (n = 55) que incluyó 108 adolescentes de 12 a 17 años (edad media 14 años, 42% mujeres, 82% blancos, 2% asiáticos, 4% negros o afroamericanos) con asma. De estos pacientes, 46 recibieron placebo, 40 recibieron 30 mg de FASENRA cada 4 semanas durante 3 dosis, seguidas de cada 8 semanas después de eso, y 22 recibieron 30 mg de FASENRA cada 4 semanas. Los pacientes debían pesar 40 kg o más y tener un historial de 2 o más exacerbaciones de asma que requirieran tratamiento con corticosteroides orales o sistémicos en los últimos 12 meses y una función pulmonar reducida en la línea de base (FEV1 pre-broncodilatador <90%) a pesar del tratamiento regular con dosis media o alta de ICS y LABA con o sin OCS u otra terapia controladora [ver Estudios clínicos (14)]. La farmacocinética del benralizumab en adolescentes de 12 a 17 años de edad fue consistente con la de los adultos basada en el análisis farmacocinético poblacional y la reducción en los recuentos de eosinófilos en sangre fue similar a la observada en adultos después del mismo tratamiento de FASENRA. El perfil de reacciones adversas en adolescentes fue generalmente similar a la población general en los ensayos clínicos [ver Reacciones adversas (6.1)].
Pacientes pediátricos de 6 a 11 años de edad
El uso de FASENRA en pacientes pediátricos de 6 a 11 años con asma grave y con un fenotipo eosinofílico está respaldado por evidencias de ensayos adecuados y bien controlados en adultos y adolescentes, y por datos adicionales de farmacocinética, farmacodinámica y seguridad en pacientes pediátricos de 6 a 11 años. La eficacia de FASENRA en pacientes pediátricos de 6 a 11 años se extrapola de la eficacia en tres ensayos clínicos (SIROCCO, CALIMA y ZONDA) [ver Estudios Clínicos (14)] con el apoyo del análisis farmacocinético y la respuesta farmacodinámica en pacientes pediátricos de 6 a 11 años en comparación con adultos y adolescentes. TATE es un ensayo abierto de 48 semanas de farmacocinética y farmacodinámica que se realizó en 28 pacientes de 6 a 11 años (edad media 9 años; 6-8 años, n = 11; 9-11 años, n = 17; 32% mujeres, blancos 29%, asiáticos 32%, negros o afroamericanos 29%) con asma grave y con un fenotipo eosinofílico.
Según los datos farmacocinéticos de TATE, se determinó que una dosis subcutánea de 10 mg (pacientes < 35 kg) y una dosis subcutánea de 30 mg (pacientes ≥ 35 kg) de benralizumab administrada cada 4 semanas durante las primeras 3 dosis y luego cada 8 semanas a partir de entonces en pacientes de 6 a 11 años tuvo una exposición similar o mayor, respectivamente, en comparación con adultos y adolescentes que recibieron una dosis subcutánea de 30 mg con el mismo régimen de dosificación [ver Farmacología Clínica (12.3)]. La respuesta farmacodinámica observada en TATE en pacientes pediátricos de 6 a 11 años fue similar a la observada en adultos y adolescentes [ver Farmacología Clínica (12.2)]. No se observaron señales de seguridad nuevas en TATE y la seguridad para la mayor exposición al fármaco está respaldada por los datos de seguridad de SIROCCO y CALIMA en adultos y adolescentes, y ZONDA en adultos, que recibieron 30 mg de FASENRA cada 4 semanas durante 1 año.
La seguridad y la eficacia en pacientes menores de 6 años de edad no se han establecido.
EGPA
La seguridad y la eficacia de FASENRA en pacientes con EGPA menores de 18 años de edad no se han establecido.
8.5 Uso geriátrico
Asma
Del número total de pacientes en ensayos clínicos de asma de benralizumab, el 13% (n = 320) tenían 65 años o más, mientras que el 0,4% (n = 9) tenían 75 años o más. No se observaron diferencias generales en la seguridad o la eficacia entre estos pacientes y los más jóvenes, y la experiencia clínica reportada no ha identificado diferencias en las respuestas entre los pacientes ancianos y los más jóvenes, pero no se puede descartar una mayor sensibilidad de algunos individuos mayores.
EGPA
De los 70 pacientes con EGPA expuestos a FASENRA, un total de 13 (19%) tenían 65 años o más. Los estudios clínicos de FASENRA para EGPA no incluyeron un número suficiente de sujetos de 65 años o más para determinar si responden de manera diferente a los sujetos más jóvenes.
10 SOBREDOSIS
No existe un tratamiento específico para una sobredosis con benralizumab. Si ocurre una sobredosis, el paciente debe recibir tratamiento de apoyo con monitorización apropiada según sea necesario.
11 DESCRIPCIÓN
Benralizumab es un anticuerpo monoclonal humanizado (clase IgG1/κ) selectivo para la subunidad alfa del receptor de interleucina-5 (IL-5Rα). Benralizumab se produce en células ováricas de hámster chino mediante tecnología de ADN recombinante. Benralizumab tiene un peso molecular de aproximadamente 150 kDa.
FASENRA (benralizumab) inyección es una solución estéril, sin conservantes, transparente a opalescente, incolora a ligeramente amarilla para inyección subcutánea. Dado que benralizumab es una proteína, pueden estar presentes algunas partículas traslúcidas o blancas a blanquecinas en la solución.
Cada jeringa precargada de dosis única de 10 mg proporciona 0.5 mL que contienen 10 mg de benralizumab, L-histidina (0.7 mg); L-histidina clorhidrato monohidratado (1.2 mg); polisorbato 20 (0.03 mg); α,α‑trehalosa dihidratada (47 mg); y Agua para inyección, USP, a un pH de 5.5 – 6.5. La jeringa precargada de dosis única contiene una jeringa de vidrio de 1 mL con una aguja de acero inoxidable de calibre 29 ½ pulgada estacada.
Cada jeringa precargada de dosis única de 30 mg o autoinyector de dosis única proporciona 1 mL que contienen 30 mg de benralizumab, L-histidina (1.4 mg); L-histidina clorhidrato monohidratado (2.3 mg); polisorbato 20 (0.06 mg); α,α‑trehalosa dihidratada (95 mg); y Agua para inyección, USP, a un pH de 5.5 – 6.5. La jeringa precargada de dosis única o el autoinyector de dosis única contiene una jeringa de vidrio de 1 mL con una aguja de acero inoxidable de calibre 29 ½ pulgada estacada.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
Benralizumab es un anticuerpo monoclonal humanizado afucosilado (IgG1, kappa) que se une directamente a la subunidad alfa del receptor humano de la interleucina-5 (IL-5Rα) con una constante de disociación de 11 pM. El receptor de IL-5 se expresa en la superficie de los eosinófilos y los basófilos. En un entorno in vitro, la ausencia de fucosa en el dominio Fc de benralizumab facilita la unión (45,5 nM) a los receptores FcɣRIII en las células efectoras inmunitarias, como las células asesinas naturales (NK), lo que lleva a la apoptosis de los eosinófilos y los basófilos a través de la citotoxicidad celular dependiente de anticuerpos (ADCC).
La inflamación es un componente importante en la patogenia del asma y la EGPA. Múltiples tipos de células (por ejemplo, mastocitos, eosinófilos, neutrófilos, macrófagos, linfocitos) y mediadores (por ejemplo, histamina, eicosanoides, leucotrienos, citocinas) están involucrados en la inflamación. Benralizumab, al unirse a la cadena IL-5Rα, reduce los eosinófilos a través de ADCC; sin embargo, el mecanismo de acción de benralizumab en el asma y la EGPA no se ha establecido definitivamente.
12.2 Farmacodinamia
En el ensayo de rango de dosis de fase 2 de 52 semanas, los pacientes con asma recibieron 1 de 3 dosis de benralizumab [2 mg (n = 81), 20 mg (n = 81) o 100 mg (n = 222)] o placebo (n = 222). Todas las dosis se administraron cada 4 semanas durante las primeras 3 dosis, seguidas de cada 8 semanas a partir de entonces. Los niveles medianos de eosinófilos en sangre en el inicio del estudio fueron 310, 280, 190 y 190 células/μL en los grupos de 2, 20 y 100 mg de benralizumab y placebo, respectivamente. Se observaron reducciones dependientes de la dosis en los eosinófilos en sangre. En el momento de la última dosis (semana 40), los recuentos medianos de eosinófilos en sangre fueron 100, 50, 40, 170 células/μL en los grupos de 2, 20 y 100 mg de benralizumab y placebo, respectivamente.
Se observó una reducción en los recuentos de eosinófilos en sangre 24 horas después de la administración en un ensayo de fase 2 de asma.
En SIROCCO y CALIMA, después de la administración SC de benralizumab en la dosis recomendada, los eosinófilos en sangre se redujeron a un recuento medio absoluto de eosinófilos en sangre de 0 células/μL [ver Estudios clínicos (14)]. Esta magnitud de reducción se observó en el primer punto de tiempo, 4 semanas de tratamiento, y se mantuvo durante todo el período de tratamiento.
El tratamiento con benralizumab también se asoció con reducciones en los basófilos en sangre, lo que se observó constantemente en todos los estudios clínicos de asma. En el ensayo de rango de dosis de fase 2 de asma, los recuentos de basófilos en sangre se midieron mediante citometría de flujo. Los recuentos medianos de basófilos en sangre fueron 45, 52, 46 y 40 células/µL en los grupos de 2 mg, 20 mg y 100 mg de benralizumab y placebo, respectivamente. A las 52 semanas (12 semanas después de la última dosis), los recuentos medianos de basófilos en sangre fueron 42, 18, 17 y 46 células/µL en los grupos de 2 mg, 20 mg y 100 mg de benralizumab y placebo, respectivamente.
En TATE, un ensayo de 48 semanas con pacientes de 6 a 11 años que tenían asma grave y con un fenotipo eosinofílico [ver Uso en poblaciones específicas (8.4)], la magnitud de la reducción de eosinófilos en sangre fue similar a la observada en adultos y adolescentes. Los niveles medianos de eosinófilos en sangre en el inicio del estudio fueron 400 y 340 células/μL en pacientes que pesaban <35 kg y ≥35 kg, respectivamente. En todos los puntos de tiempo posteriores a la dosis, los recuentos medianos de eosinófilos se redujeron a 10 a 20 células/μL en pacientes que pesaban <35 kg y a 20 a 30 células/μL en pacientes que pesaban ≥35 kg. La reducción de eosinófilos en sangre se observó en el primer punto de tiempo, 4 semanas de tratamiento, y se mantuvo durante todo el período de tratamiento.
En pacientes con EGPA, la reducción de eosinófilos en sangre fue consistente con el efecto observado en los ensayos de asma. Los niveles medianos absolutos de eosinófilos en sangre en el grupo de benralizumab en el inicio del estudio fueron 240 células/μL. Después de la administración SC de benralizumab en la dosis recomendada en pacientes con EGPA, los eosinófilos en sangre se redujeron a un recuento medio absoluto de eosinófilos en sangre de 20 a 30 células/μL. La reducción de eosinófilos en sangre se observó en el primer punto de tiempo observado, 1 semana de tratamiento, y se mantuvo durante todo el período de tratamiento de 52 semanas.
12.3 Farmacocinética
Las propiedades farmacocinéticas de benralizumab que se indican a continuación se basan en los análisis farmacocinéticos poblacionales de los ensayos de asma. Los hallazgos en EGPA fueron generalmente consistentes con los del asma, aunque se predice una menor depuración para los pacientes con EGPA en relación con los pacientes con asma [ver Eliminación].
La farmacocinética de benralizumab fue aproximadamente proporcional a la dosis en pacientes adultos y adolescentes con asma después de la administración subcutánea en un rango de dosis de 20 a 200 mg.
Absorción
Después de la administración subcutánea a pacientes con asma, la semivida de absorción fue de aproximadamente 3,5 días. Según el análisis farmacocinético poblacional, la biodisponibilidad absoluta estimada fue de aproximadamente el 59% y no hubo diferencias clínicamente relevantes en la biodisponibilidad relativa en la administración al abdomen, muslo o brazo.
Distribución
Según el análisis farmacocinético poblacional, el volumen de distribución central y periférico de benralizumab fue de 3,1 L y 2,5 L, respectivamente, para un individuo de 70 kg.
Eliminación
A partir del análisis farmacocinético poblacional, benralizumab exhibió farmacocinética lineal y no hubo evidencia de una vía de depuración mediada por el receptor diana. La depuración sistémica típica estimada (CL) para benralizumab fue de 0,29 L/d para un paciente con asma que pesaba 70 kg. La CL típica estimada fue de 0,22 L/d para pacientes con EGPA. Después de la administración subcutánea en pacientes con asma, la semivida de eliminación fue de aproximadamente 15,5 días.
Metabolismo
Benralizumab es un anticuerpo monoclonal IgG1 humanizado que se degrada por enzimas proteolíticas ampliamente distribuidas en el cuerpo y no restringidas al tejido hepático.
Poblaciones específicas:
Edad
Según el análisis farmacocinético poblacional, la edad no afectó la depuración de benralizumab.
Género, Raza
Un análisis farmacocinético poblacional indicó que no hubo un efecto significativo del género y la raza en la depuración de benralizumab.
Pacientes con insuficiencia renal
No se han realizado estudios clínicos formales para investigar el efecto de la insuficiencia renal en benralizumab. Con base en el análisis farmacocinético poblacional, la depuración de benralizumab fue comparable en sujetos con valores de depuración de creatinina entre 30 y 80 mL/min y pacientes con función renal normal. Hay datos limitados disponibles en sujetos con valores de depuración de creatinina inferiores a 30 mL/min; sin embargo, benralizumab no se elimina por vía renal.
Pacientes con insuficiencia hepática
No se han realizado estudios clínicos formales para investigar el efecto de la insuficiencia hepática en benralizumab. Los anticuerpos monoclonales IgG no se eliminan principalmente a través de la vía hepática; no se espera que el cambio en la función hepática influya en la depuración de benralizumab. Con base en el análisis farmacocinético poblacional, los biomarcadores de función hepática basal (ALT, AST y bilirrubina) no tuvieron un efecto clínicamente relevante en la depuración de benralizumab.
Pacientes pediátricos
Se investigó la farmacocinética de benralizumab después de la administración subcutánea de 10 mg o 30 mg en pacientes de 6 a 11 años con asma grave y con un fenotipo de asma eosinofílico en la fase inicial de tratamiento de 16 semanas de TATE, un ensayo abierto de 48 semanas. Entre los pacientes de 6 a 11 años que pesaban <35 kg que recibieron 10 mg, la concentración media en el valle en la semana 16 fue similar a la de los adultos y adolescentes que recibieron 30 mg. Entre los pacientes de 6 a 11 años que pesaban ≥35 kg que recibieron 30 mg, la concentración media en el valle en la semana 16 fue un 62% más alta en relación con los adultos y adolescentes que recibieron la misma dosis, debido al menor peso corporal en los pacientes pediátricos.
Estudios de interacción medicamentosa
No se han realizado estudios formales de interacción fármaco-fármaco.
Las enzimas del citocromo P450, las bombas de eflujo y los mecanismos de unión a proteínas no están involucrados en la depuración de benralizumab. No hay evidencia de expresión de IL-5Rα en los hepatocitos y la depleción de eosinófilos no produce alteraciones sistémicas crónicas de las citocinas proinflamatorias.
No se espera un efecto de benralizumab en la farmacocinética de los medicamentos administrados conjuntamente. Con base en el análisis poblacional, los medicamentos que se administran comúnmente de forma conjunta no tuvieron ningún efecto en la depuración de benralizumab en pacientes con asma.
12.6 Inmunogenicidad
La incidencia observada de anticuerpos anti-fármaco (ADA) depende en gran medida de la sensibilidad y la especificidad del ensayo. Las diferencias en los métodos de ensayo excluyen comparaciones significativas de la incidencia de ADA en los estudios descritos a continuación con la incidencia de ADA en otros estudios, incluidos los de benralizumab o de otros productos de benralizumab.
En pacientes adultos y adolescentes con asma, la incidencia de ADA a benralizumab en pacientes tratados con FASENRA en el régimen de dosificación recomendado durante el período de tratamiento de 48 a 56 semanas fue del 13%. Un total del 12% de los pacientes tratados con FASENRA desarrollaron anticuerpos neutralizantes.
En pacientes pediátricos de 6 a 11 años con asma grave y con un fenotipo eosinofílico, la incidencia de ADA a benralizumab durante un período de tratamiento abierto de 48 semanas fue comparable a la de los pacientes adultos y adolescentes.
En pacientes con EGPA, la incidencia de ADA a benralizumab durante el período de tratamiento controlado activo de 52 semanas fue del 9% (6 de 67 pacientes). Se detectó actividad de anticuerpos neutralizantes en uno de los pacientes positivos a ADA.
Efectos de los anticuerpos anti-fármaco en la farmacocinética y la farmacodinamia
Los anticuerpos anti-benralizumab se asociaron con un aumento de la depuración de benralizumab y un aumento de los niveles de eosinófilos en sangre en pacientes con asma con títulos altos de ADA en comparación con los pacientes negativos a anticuerpos. Se observaron concentraciones en el valle de benralizumab reducidas en un paciente con EGPA con títulos altos de ADA.
En los ensayos de asma con pacientes adultos y adolescentes y en el ensayo de EGPA, no se observó evidencia de una asociación de ADA con la eficacia o la seguridad.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No se han realizado estudios a largo plazo en animales para evaluar el potencial carcinogénico de benralizumab. La literatura publicada que utiliza modelos animales sugiere que la IL-5 y los eosinófilos son parte de una reacción inflamatoria temprana en el sitio de la tumorigénesis y pueden promover el rechazo tumoral. Sin embargo, otros informes indican que la infiltración de eosinófilos en los tumores puede promover el crecimiento tumoral. Por lo tanto, se desconoce el riesgo de malignidad en humanos por un anticuerpo que se une a IL-5Rα como benralizumab.
La fertilidad masculina y femenina no se vio afectada en base a la ausencia de hallazgos histopatológicos adversos en los órganos reproductivos de los monos cynomolgus tratados con benralizumab durante 9 meses a dosis IV de hasta 25 mg/kg o a dosis SC de hasta 30 mg/kg una vez cada 2 semanas (aproximadamente 400 y 270 veces la MRHD en base al AUC).
14 ESTUDIOS CLÍNICOS
14.1 Estudios clínicos en pacientes con asma
La eficacia de FASENRA para el tratamiento de mantenimiento complementario del asma grave con fenotipo eosinofílico se evaluó en dos ensayos aleatorizados, doble ciego, de grupos paralelos, controlados con placebo y de exacerbación, SIROCCO (NCT01928771) y CALIMA (NCT01914757), durante 48 y 56 semanas de duración, respectivamente. Además, los efectos de FASENRA en la reducción del uso de corticosteroides orales y el efecto sobre la función pulmonar se evaluaron en ensayos clínicos, ZONDA (NCT02075255) y un ensayo de función pulmonar de 12 semanas (NCT02322775), respectivamente.
SIROCCO y CALIMA fueron ensayos aleatorizados, doble ciego, de grupos paralelos, controlados con placebo y de exacerbación en pacientes de 12 años o mayores y de 48 y 56 semanas de duración, respectivamente. En los ensayos se aleatorizaron un total de 2510 pacientes. Los pacientes debían tener antecedentes de 2 o más exacerbaciones del asma que requirieran tratamiento con corticosteroides orales o sistémicos en los últimos 12 meses, una puntuación del ACQ‑6 de 1,5 o más en el cribado y una función pulmonar reducida al inicio [FEV1 prebroncodilatador inferior al 80 % en adultos e inferior al 90 % en adolescentes] a pesar del tratamiento regular con dosis altas de ICS (SIROCCO) o con dosis medias o altas de ICS (CALIMA) más un agonista beta de acción prolongada (LABA) con o sin corticosteroides orales (OCS) y medicamentos adicionales para el control del asma. Los pacientes se estratificaron por geografía, edad y recuento de eosinófilos en sangre (≥300 células/μL o <300 células/μL). Se evaluó FASENRA administrado una vez cada 4 semanas durante las primeras 3 dosis, y luego cada 4 u 8 semanas a partir de entonces como complemento del tratamiento de fondo en comparación con placebo.
Todos los sujetos continuaron con su tratamiento de fondo para el asma durante todo el tiempo que duraron los ensayos.
ZONDA fue un ensayo de reducción de OCS aleatorizado, doble ciego, de grupos paralelos, en 220 pacientes adultos con asma. Los pacientes fueron requeridos tratamiento con OCS diarios (7,5 a 40 mg por día) además del uso regular de ICS y LABA en dosis altas con o sin controlador(es) adicional(es). El ensayo incluyó un período de preinclusión de 8 semanas durante el cual se ajustó la dosis de OCS a la dosis mínima eficaz sin perder el control del asma. A efectos del ajuste de la dosis de OCS, el investigador evaluó el control del asma en función del FEV1, el flujo espiratorio máximo, los despertares nocturnos, el uso de medicación de rescate con broncodilatadores de acción corta o cualquier otro síntoma que requiriera un aumento de la dosis de OCS. La mediana de la dosis inicial de OCS fue similar en todos los grupos de tratamiento. Los pacientes debían tener recuentos de eosinófilos en sangre superiores o iguales a 150 células/μL y antecedentes de al menos una exacerbación en los últimos 12 meses. La mediana de la dosis inicial de OCS fue de 10 mg (rango: de 8 a 40 mg) para los 3 grupos de tratamiento (placebo, FASENRA cada 4 semanas y FASENRA cada 4 semanas durante las primeras 3 dosis y, a continuación, una vez cada 8 semanas).
Si bien se estudiaron 2 regímenes de dosificación en SIROCCO, CALIMA y ZONDA, el régimen de dosificación recomendado es de 30 mg de FASENRA administrados cada 4 semanas durante las primeras 3 dosis, y luego cada 8 semanas a partir de entonces [ver Dosificación y administración (2.1)].
| Población total | |||
|---|---|---|---|
|
SIROCCO (N=1204) |
CALIMA (N=1306) |
ZONDA (N=220) |
|
|
Media de edad (años) |
49 |
49 |
51 |
|
Mujeres (%) |
66 |
62 |
61 |
|
Blanca (%) |
73 |
84 |
93 |
|
Duración del asma, mediana (años) |
15 |
16 |
12 |
|
Nunca fumó (%) |
80 |
78 |
79 |
|
FEV1 inicial medio prebroncodilatador (L) |
1.67 |
1.76 |
1.85 |
|
% predicho promedio inicial de FEV1 |
57 |
58 |
60 |
|
FEV1/FVC (%) medio posterior a SABA |
66 |
65 |
62 |
|
Recuento de eosinófilos inicial promedio (células/μL) |
472 |
472 |
575 |
|
Número promedio de exacerbaciones en el año anterior |
3 |
3 |
3 |
Exacerbaciones
El criterio principal de valoración para SIROCCO y CALIMA fue la tasa de exacerbaciones del asma en pacientes con recuentos basales de eosinófilos en sangre superiores o iguales a 300 células/μL que estaban tomando dosis altas de ICS y LABA. La exacerbación del asma se definió como un empeoramiento del asma que requirió el uso de corticosteroides orales/sistémicos durante al menos 3 días, y/o visitas al departamento de emergencias que requirieron el uso de corticosteroides orales/sistémicos y/u hospitalización. Para los pacientes que recibían corticosteroides orales de mantenimiento, una exacerbación del asma que requirió corticosteroides orales se definió como un aumento temporal de los corticosteroides orales/sistémicos estables durante al menos 3 días o una dosis única de corticosteroides inyectables de depósito. En SIROCCO, el 35 % de los pacientes que recibieron FASENRA experimentaron una exacerbación del asma en comparación con el 51 % que recibieron placebo. En CALIMA, el 40 % de los pacientes que recibieron FASENRA experimentaron una exacerbación del asma en comparación con el 51 % que recibieron placebo (Tabla 4).
|
Ensayo |
Tratamiento |
Exacerbaciones por año |
||
|
Tasa |
Diferencia |
Razón de tasas (IC del 95 %) |
||
|
Todas las exacerbaciones |
||||
|
SIROCCO |
FASENRA† (n=267) |
0.74 |
-0.78 |
0.49 (0.37, 0.64) |
|
Placebo (n=267) |
1.52 |
— |
— |
|
|
CALIMA |
FASENRA† (n=239) |
0.73 |
-0.29 |
0.72 (0.54, 0.95) |
|
Placebo (n=248) |
1.01 |
— |
— |
|
|
||||
|
SIROCCO |
FASENRA† (n=267) |
0.09 |
-0.16 |
0.37 (0.20, 0.67) |
|
Placebo (n=267) |
0.25 |
— |
— |
|
|
CALIMA |
FASENRA† (n=239) |
0.12 |
0.02 |
1.23 (0.64, 2.35) |
|
Placebo (n=248) |
0.10 |
— |
— |
|
|
Exacerbaciones que requieren hospitalización |
||||
|
SIROCCO |
FASENRA† (n=267) |
0.07 |
-0.07 |
0.48 (0.22, 1.03) |
|
Placebo (n=267) |
0.14 |
— |
— |
|
|
CALIMA |
FASENRA† (n=239) |
0.07 |
0.02 |
1.48 (0.65, 3.37) |
|
Placebo (n=248) |
0.05 |
— |
— |
|
El tiempo hasta la primera exacerbación fue más prolongado en los pacientes que recibieron FASENRA en comparación con el placebo en el estudio SIROCCO (Figura 2). Se observaron hallazgos similares en el estudio CALIMA.
Figura 2. Curvas de Incidencia Acumulada de Kaplan-Meier para el Tiempo hasta la Primera Exacerbación, SIROCCO
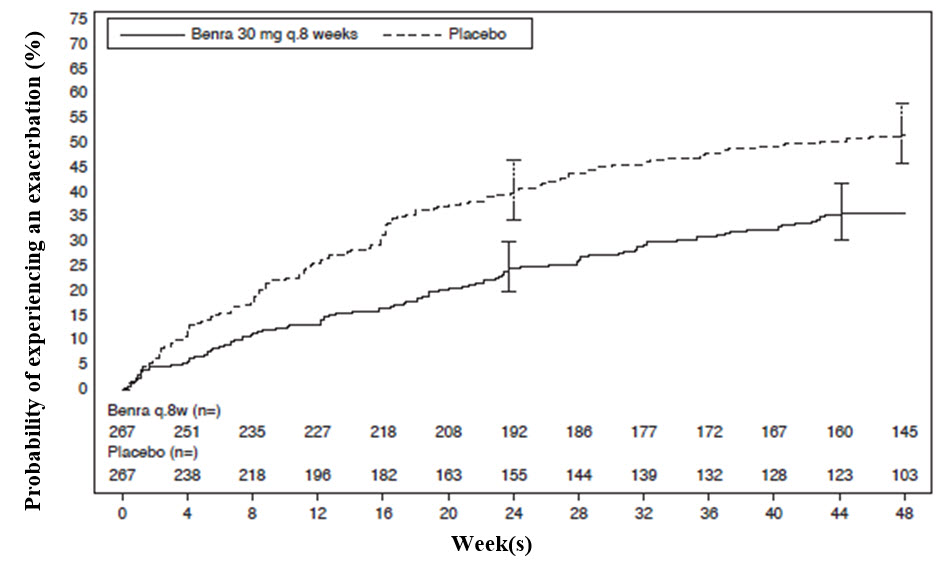
Los análisis de subgrupos de los estudios SIROCCO y CALIMA identificaron a los pacientes con un mayor historial de exacerbaciones previas y un recuento inicial de eosinófilos en sangre como posibles factores predictivos de una mejor respuesta al tratamiento. Se observaron reducciones en las tasas de exacerbación independientemente de los recuentos iniciales de eosinófilos periféricos; sin embargo, los pacientes con un recuento inicial de eosinófilos en sangre ≥300 células/μL mostraron una respuesta numéricamente mayor que aquellos con recuentos <300 células/μL. En ambos ensayos, los pacientes con un historial de 3 o más exacerbaciones en los 12 meses previos a la aleatorización a FASENRA mostraron una respuesta de exacerbación numéricamente mayor que aquellos con menos exacerbaciones previas.
Reducción de los Corticosteroides Orales
El estudio ZONDA evaluó el efecto de FASENRA en la reducción del uso de corticosteroides orales de mantenimiento en pacientes adultos con asma. El criterio principal de valoración fue el porcentaje de reducción con respecto al valor inicial de la dosis final de OCS durante las semanas 24 a 28, mientras se mantenía el control del asma (véase la definición de control del asma en la descripción del ensayo). En comparación con el placebo, los pacientes que recibieron FASENRA lograron mayores reducciones en la dosis diaria de corticosteroides orales de mantenimiento, al tiempo que mantuvieron el control del asma. La mediana del porcentaje de reducción en la dosis diaria de OCS con respecto al valor inicial fue del 75 % en los pacientes que recibieron FASENRA (IC del 95 %: 60, 88) en comparación con el 25 % en los pacientes que recibieron placebo (IC del 95 %: 0, 33). Se observaron reducciones del 50 % o más en la dosis de OCS en 48 (66 %) pacientes que recibieron FASENRA en comparación con los que recibieron placebo 28 (37 %). La proporción de pacientes con una dosis final media inferior o igual a 5 mg en las semanas 24 a 28 fue del 59 % para FASENRA y del 33 % para el placebo (odds ratio 2,74, IC del 95 %: 1,41, 5,31). Solo los pacientes con una dosis inicial optimizada de OCS de 12,5 mg o menos fueron elegibles para lograr una reducción del 100 % en la dosis de OCS durante el estudio. De esos pacientes, el 52 % (22 de 42) que recibieron FASENRA y el 19 % (8 de 42) que recibieron placebo lograron una reducción del 100 % en la dosis de OCS. Las exacerbaciones que dieron lugar a hospitalización o visita a urgencias también se evaluaron como criterio de valoración secundario. En este ensayo de 28 semanas, los pacientes que recibieron FASENRA tuvieron 1 evento, mientras que los que recibieron placebo tuvieron 14 eventos (tasa anualizada de 0,02 y 0,32, respectivamente; razón de tasas de 0,07, IC del 95 %: 0,01, 0,63).
Función Pulmonar
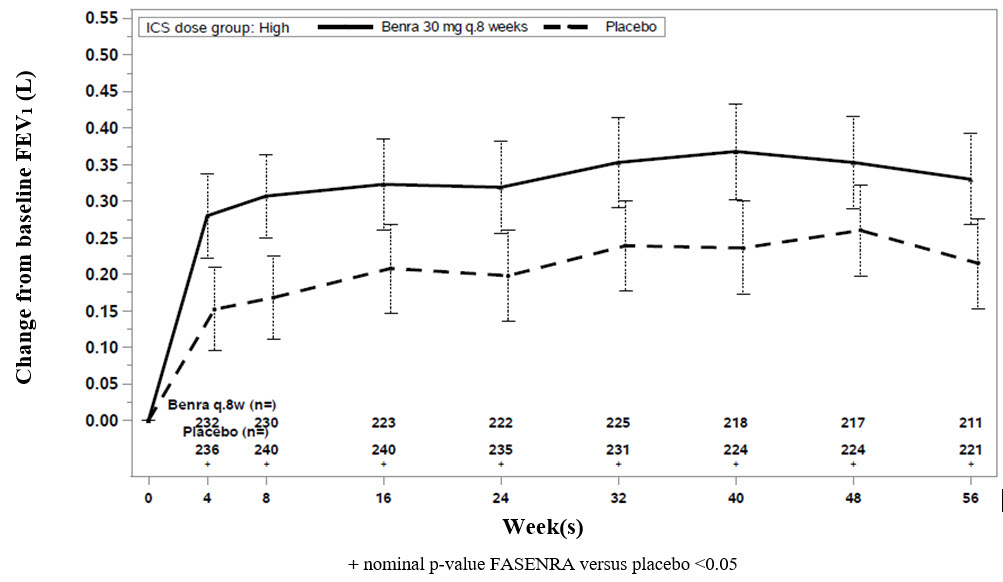
El cambio con respecto al valor inicial en el FEV1 medio se evaluó en los estudios SIROCCO, CALIMA y ZONDA como criterio de valoración secundario. En comparación con el placebo, FASENRA proporcionó mejoras constantes a lo largo del tiempo en el cambio medio con respecto al valor inicial en el FEV1 (Figura 3 y Tabla 5).
Figura 3. Cambio Medio con respecto al Valor Inicial en el FEV1 Previo al Broncodilatador (L), CALIMA
|
|
|
Ensayo |
Diferencia con respecto al Placebo en el Cambio Medio con respecto al Valor Inicial del FEV1 Previo al Broncodilatador (L) (IC del 95 %) |
|
SIROCCO |
0.159 (0.068, 0.249) |
|
CALIMA |
0.116 (0.028, 0.204) |
|
ZONDA |
0.112 (-0.033, 0.258) |
Los análisis de subgrupos también mostraron mayores mejoras en el VEF1 en pacientes con recuentos de eosinófilos en sangre basales más altos y antecedentes de exacerbaciones más frecuentes.
El programa clínico para FASENRA también incluyó un ensayo de función pulmonar de 12 semanas, aleatorizado, doble ciego, controlado con placebo, realizado en 211 pacientes adultos con asma de leve a moderada. Los pacientes fueron tratados con placebo o benralizumab 30 mg SC cada 4 semanas durante 3 dosis. La función pulmonar, medida por el cambio desde el inicio en el VEF1 en la semana 12, mejoró en el grupo de tratamiento con benralizumab en comparación con el placebo.
Resultados informados por el paciente
El Cuestionario de control del asma-6 (ACQ-6) y el Cuestionario estandarizado de calidad de vida del asma para mayores de 12 años (AQLQ(S)+12) se evaluaron en SIROCCO, CALIMA y ZONDA. La tasa de respondedores para ambas medidas se definió como una mejora en la puntuación de 0,5 o más como umbral al final de SIROCCO, CALIMA y ZONDA (48, 56 y 28 semanas, respectivamente). En SIROCCO, la tasa de respondedores al ACQ-6 para FASENRA fue del 60 % frente al 50 % del placebo (odds ratio 1,55; IC del 95 %: 1,09 a 2,19). En CALIMA, la tasa de respondedores al ACQ-6 para FASENRA fue del 63 % frente al 59 % del placebo (odds ratio 1,16; IC del 95 %: 0,80 a 1,68). En SIROCCO, la tasa de respondedores para AQLQ(S)+12 para FASENRA fue del 57 % frente al 49 % del placebo (odds ratio 1,42; IC del 95 %: 0,99 a 2,02), y en CALIMA, 60 % FASENRA frente al 59 % del placebo (odds ratio de 1,03; IC del 95 %: 0,70 a 1,51). Se observaron resultados similares en ZONDA.
14.2 Estudios clínicos en pacientes con granulomatosis eosinofílica con poliangeítis
La eficacia de FASENRA para la granulomatosis eosinofílica con poliangeítis (GEPA) se evaluó en un ensayo clínico aleatorizado, doble ciego, controlado con activo, de no inferioridad (MANDARA [NCT04157348]) de 52 semanas de duración. En el ensayo participaron un total de 140 adultos mayores de 18 años con GEPA. Los pacientes debían tener asma, eosinofilia (1000 células/uL o >10 % de leucocitos) y antecedentes de enfermedad recidivante o refractaria tratada con prednisolona/prednisona de fondo con o sin terapia inmunosupresora. Los pacientes fueron aleatorizados para recibir FASENRA 30 mg administrados por vía subcutánea cada 4 semanas o mepolizumab 300 mg administrados por vía subcutánea cada 4 semanas, además de la terapia de fondo continua. A partir de la semana 4, la dosis de corticosteroides orales (OCS) se redujo a criterio del investigador. El ensayo MANDARA fue un ensayo de no inferioridad y no se diseñó para evaluar si FASENRA era superior a mepolizumab. El margen de no inferioridad (NI) preespecificado fue una diferencia de tratamiento del -25 %. Los criterios de valoración secundarios (duración acumulada de la remisión, recaída, reducción de OCS y el cuestionario de control del asma-6) no se incluyeron en el procedimiento de pruebas múltiples preespecificado para determinar la significancia estadística.
Las características demográficas y basales de los pacientes en MANDARA se proporcionan en la Tabla 6.
| MANDARA (N=140) | |
|---|---|
|
Edad media (años) |
52 |
|
Mujeres (%) |
60 |
|
Blanca (%) |
79 |
|
Asiática (%) |
12 |
|
Otra (%) |
4 |
|
Hispana o latina (%) |
3 |
|
Tiempo desde el diagnóstico de GEPA, años, media (DE) |
5,2 (5,6) |
|
Antecedentes de ≥1 recaída confirmada en los últimos 2 años (%) |
79 |
|
Enfermedad refractaria (%) |
60 |
|
Dosis diaria basal de corticosteroides orales*, mg, mediana (rango) |
10 (5 – 40) |
|
BVAS basal, mediana (rango) |
0 (0 – 18) |
|
BVAS=0 (%) |
52 |
|
Recibir terapia inmunosupresora† (%) |
36 |
|
ANCA positivo‡ (%) |
29 |
|
Evidencia de biopsia de vasculitis/inflamación eosinofílica (%) |
38 |
|
SD=desviación estándar; BVAS=puntuación de actividad de la vasculitis de Birmingham. |
|
Remisión
El criterio principal de valoración en MANDARA fue la proporción de pacientes en remisión, definida como una puntuación de actividad de la vasculitis de Birmingham (BVAS, por sus siglas en inglés) = 0 (sin vasculitis activa) más una dosis de prednisolona/prednisona ≤ 4 mg/día, tanto en la semana 36 como en la semana 48. La BVAS es una herramienta completada por el médico, que se divide en 9 sistemas basados en órganos, para evaluar la vasculitis clínicamente activa que probablemente requeriría tratamiento, después de la exclusión de otras causas. Como se muestra en la Tabla 7, FASENRA demostró no inferioridad a mepolizumab para el criterio principal de valoración de la remisión y los componentes de la remisión.
|
||||||
|
Remisión
(OCS≤4 mg/día + |
OCS≤4 mg/día |
BVAS=0 |
||||
|
FASENRA* N=70 |
Mepo† N=70 |
FASENRA* N=70 |
Mepo† N=70 |
FASENRA* N=70 |
Mepo† N=70 |
|
|
Pacientes en remisión en las semanas 36 y 48 |
||||||
|
Pacientes, n (%)‡ |
41 (59) |
40 (57) |
43 (62) |
41 (58) |
58 (83) |
59 (84) |
|
Diferencias en la tasa de remisión (%)‡ (95% CI) |
2.7 |
— |
4.1 |
— |
-1.2 |
— |
|
(-13, 18)§ |
— |
(-11, 19) |
— |
(-13, 11) |
— |
|
|
N=número de pacientes en el análisis. |
||||||
Utilizando una definición alternativa de remisión de BVAS=0 más prednisolona/prednisona ≤7,5 mg/día, se observó una eficacia consistente entre los grupos para estos criterios de valoración.
Duración acumulada de la remisión
La duración total acumulada de la remisión fue similar en FASENRA en comparación con mepolizumab (odds ratio 1,4, IC del 95%: 0,75, 2,5). Los resultados de la duración acumulada de la remisión se muestran en la Tabla 8. La proporción de pacientes que lograron la remisión dentro de las primeras 24 semanas de tratamiento y que permanecieron en remisión hasta la semana 52 fue del 42% para FASENRA y del 37% para mepolizumab (diferencia en la tasa de respuesta del 5,5%, IC del 95%: -9,3, 20). Este resultado no fue estadísticamente significativo, ya que no se especificó previamente ningún procedimiento de prueba múltiple.
|
||||||
|
Remisión (OCS≤4 mg/día + BVAS=0) |
OCS≤4 mg/día |
BVAS=0 |
||||
|
FASENRA* N=70 |
Mepo† N=70 |
FASENRA* N=70 |
Mepo† N=70 |
FASENRA* N=70 |
Mepo† N=70 |
|
|
Duración acumulada durante 52 semanas‡, n (%) |
||||||
|
0 semanas§ >0 a <12 semanas 12 a <24 semanas 24 a <36 semanas ≥36 semanas |
9 (13) 12 (17) 8 (11) 21 (30) 20 (29) |
15 (21) 10 (14) 8 (11) 19 (27) 18 (26) |
9 (13) 10 (14) 9 (13) 19 (27) 23 (33) |
12 (17) 12 (17) 8 (11) 18 (26) 20 (29) |
0 0 2 (3) 6 (9) 62 (89) |
0 2 (3) 2 (3) 7 (10) 59 (84) |
|
Odds ratio¶ |
1.4 (0.75, 2.5) |
— — |
1.4 (0.74, 2.5) |
— — |
1.5 (0.54, 4.2) |
— — |
Recaída
El índice de riesgo para el tiempo hasta la primera recaída (definido como empeoramiento relacionado con vasculitis, asma o síntomas nasosinusales que requieren un aumento en la dosis de corticosteroides o terapia inmunosupresora u hospitalización) fue de 0.98 (IC del 95%: 0.53, 1.8). Se observó recaída en el 30% de los pacientes con FASENRA y en el 30% de los pacientes con mepolizumab. La tasa de recaída anualizada fue de 0.50 para los pacientes que recibieron FASENRA frente a 0.49 para los pacientes que recibieron mepolizumab (razón de tasas 1.0, IC del 95%: 0.56, 1.9). Los tipos de recaída fueron consistentes para los pacientes que recibieron FASENRA o mepolizumab.
Reducción de corticosteroides orales
Durante las semanas 48 a 52, se observó una reducción del 100% en la dosis de OCS en el 41% de los pacientes que recibieron FASENRA en comparación con el 26% de los que recibieron mepolizumab (diferencia del 16%, IC del 95%: 0.67, 31). Durante las semanas 48 a 52, se observaron reducciones del 50% o más en el 86% de los pacientes que recibieron FASENRA en comparación con el 74% de los que recibieron mepolizumab (diferencia del 12%, IC del 95%: -0.57, 25). Estos resultados no fueron estadísticamente significativos ya que no hubo un procedimiento de prueba múltiple preespecificado.
Cuestionario de control del asma-6 (ACQ-6)
El ACQ-6 es un cuestionario de 6 ítems que completa el paciente para medir la idoneidad del control del asma y el cambio en el control del asma. La tasa de respondedores al ACQ-6 durante las semanas 48 a 52 (definida como una disminución en la puntuación de 0.5 o más en comparación con el valor inicial) fue del 42% para FASENRA y del 48% para mepolizumab (diferencia -6.2%, IC del 95%: -19, 6.2).
16 FORMA DE SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
Presentación
FASENRA (benralizumab) inyección es una solución estéril, sin conservantes, transparente a opalescente, incolora a ligeramente amarilla y puede contener algunas partículas traslúcidas o blancas a blanquecinas, para inyección subcutánea suministrada como una jeringa precargada de dosis única o un autoinyector de dosis única. La jeringa precargada (incluido el tapón y la tapa) y el autoinyector no están hechos con látex de caucho natural.
FASENRA está disponible como:
Jeringa precargada de dosis única
- •
- La caja contiene una jeringa precargada de dosis única de 10 mg/0,5 mL con una varilla de émbolo gris: NDC 0310-1745-01
- •
- La caja contiene una jeringa precargada de dosis única de 30 mg/mL con una varilla de émbolo azul: NDC 0310-1730-30
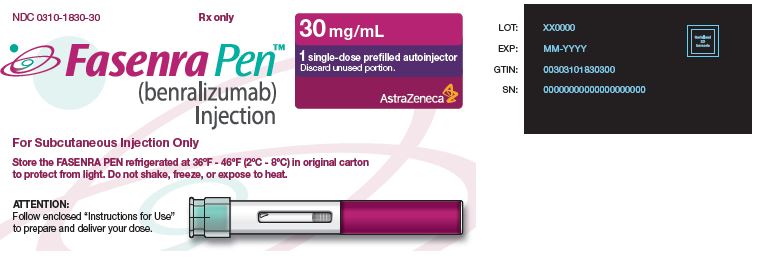
Autoinyector de dosis única FASENRA PEN
- •
- La caja contiene un autoinyector de dosis única de 30 mg/mL: NDC 0310-1830-30
Almacenamiento y manipulación
Almacenar refrigerado a 36°F a 46°F (2°C a 8°C) en la caja original para proteger de la luz.
Si es necesario, la jeringa precargada y el autoinyector se pueden almacenar a temperatura ambiente hasta 77°F (25°C) durante un máximo de 14 días en la caja original para proteger de la luz. Una vez que se retira del refrigerador y se lleva a temperatura ambiente (hasta 77°F [25°C]), la jeringa precargada y el autoinyector deben usarse dentro de los 14 días o desecharse.
No congelar. No agitar. No exponer al calor.
17 INFORMACIÓN PARA EL PACIENTE
Avise a los pacientes y/o cuidadores que lean el etiquetado del paciente aprobado por la FDA (Información para el paciente e Instrucciones de uso para FASENRA PEN) antes de que el paciente comience a usar FASENRA y cada vez que se renueve la receta, ya que puede haber nueva información que necesiten saber.
Instrucciones de administración
Instruya a los pacientes y/o cuidadores sobre la técnica adecuada de inyección subcutánea utilizando FASENRA PEN, incluida la técnica aséptica, y la preparación y administración de FASENRA PEN antes de su uso. Avise a los pacientes que sigan las recomendaciones de eliminación de objetos punzantes [ver Instrucciones de uso].
Reacciones de hipersensibilidad
Informe a los pacientes que se han producido reacciones de hipersensibilidad (por ejemplo, anafilaxia, angioedema, urticaria, erupción cutánea) después de la administración de FASENRA. Estas reacciones generalmente ocurrieron dentro de las horas posteriores a la administración de FASENRA, pero en algunos casos tuvieron un inicio tardío (es decir, días). Instruya a los pacientes que se pongan en contacto con su proveedor de atención médica si experimentan síntomas de una reacción alérgica [ver Advertencias y precauciones (5.1)].
No para síntomas agudos o enfermedad en deterioro
Informe a los pacientes que FASENRA no trata los síntomas agudos del asma o las exacerbaciones agudas. Informe a los pacientes que busquen atención médica si su asma permanece sin control o empeora después del inicio del tratamiento con FASENRA [ver Advertencias y precauciones (5.2)].
Reducción de la dosis de corticosteroides
Informe a los pacientes que no deben suspender los corticosteroides sistémicos o inhalados excepto bajo la supervisión directa de un médico. Informe a los pacientes que la reducción de la dosis de corticosteroides puede estar asociada con síntomas de abstinencia sistémica y/o desenmascarar afecciones previamente suprimidas por la terapia con corticosteroides sistémicos [ver Advertencias y precauciones (5.3)].
Fabricado por
AstraZeneca AB
Södertälje, Suecia SE-15185
Licencia de EE. UU. No. 2059
Distribuido por
AstraZeneca Pharmaceuticals LP,
Wilmington, DE 19850
FASENRA es una marca comercial del grupo de empresas AstraZeneca.
©AstraZeneca 2024
INSERTO PARA EL PACIENTE
|
Información para el paciente FASENRA® (fas-en-rah) (benralizumab) inyección, para uso subcutáneo |
|
¿Qué es FASENRA? FASENRA es un medicamento recetado que se usa:
No se sabe si FASENRA es seguro y eficaz en niños con asma menores de 6 años. No se sabe si FASENRA es seguro y eficaz en niños con EGPA menores de 18 años. |
|
No use FASENRA si es alérgico a benralizumab o a cualquiera de los ingredientes de FASENRA. Consulte el final de este folleto para obtener una lista completa de los ingredientes de FASENRA. |
|
Antes de usar FASENRA, informe a su médico sobre todas sus afecciones médicas, incluso si:
Informe a su médico sobre todos los medicamentos que toma, incluidos los medicamentos recetados y de venta libre, las vitaminas y los suplementos herbales. No deje de tomar sus otros medicamentos para su condición a menos que su médico se lo indique. |
|
¿Cómo usaré FASENRA?
|
|
¿Cuáles son los posibles efectos secundarios de FASENRA? FASENRA puede causar efectos secundarios graves, que incluyen:
Los efectos secundarios más comunes de FASENRA incluyen dolor de cabeza y dolor de garganta. Estos no son todos los posibles efectos secundarios de FASENRA. Llame a su médico para obtener asesoramiento médico sobre los efectos secundarios. Puede informar los efectos secundarios a la FDA al 1-800-FDA-1088. |
|
¿Cómo debo guardar FASENRA?
|
|
Información general sobre el uso seguro y eficaz de FASENRA. Los medicamentos a veces se recetan para fines distintos de los que se enumeran en un folleto de información para el paciente. No use FASENRA para una condición para la que no fue recetado. No le dé FASENRA a otras personas, incluso si tienen los mismos síntomas que usted. Puede hacerles daño. Puede pedirle a su médico o farmacéutico información sobre FASENRA que esté escrita para profesionales de la salud. |
|
¿Cuáles son los ingredientes de FASENRA? Ingrediente activo: benralizumab Ingredientes inactivos: L-histidina, L-histidina clorhidrato monohidratado, polisorbato 20, α,α-trehalosa dihidratada y Agua para inyección Fabricado por: AstraZeneca AB, Södertälje, Suecia SE-15185 Número de licencia de EE. UU. 2059 Distribuido por: AstraZeneca Pharmaceuticals LP, Wilmington, DE 19850 FASENRA es una marca comercial del grupo de empresas AstraZeneca. ©AstraZeneca 2024 Para obtener más información, visite https://www.FASENRA.com o llame al 1-800-236-9933. |
Esta información para el paciente ha sido aprobada por la Administración de Alimentos y Medicamentos de los EE. UU. Revisado: septiembre de 2024
INSTRUCCIONES DE USO
|
Instrucciones de uso FASENRA PEN™ (fas-en-rah) (benralizumab) para inyección subcutánea Autoinyector de dosis única |
|||||||||
|
Antes de usar su FASENRA PEN, su proveedor de atención médica debe mostrarle a usted o a su cuidador cómo usarlo correctamente. En personas de 6 a 11 años, FASENRA solo debe ser administrado por un cuidador o proveedor de atención médica. Lea estas Instrucciones de uso antes de comenzar a usar su FASENRA PEN y cada vez que reciba una recarga. Puede haber nueva información. Esta información no reemplaza hablar con su proveedor de atención médica sobre su condición médica o su tratamiento. Si usted o su cuidador tienen alguna pregunta, hable con su proveedor de atención médica. Información importante:
|
|||||||||
|
No use su FASENRA PEN si:
|
No:
|
||||||||
|
Si ocurre alguna de estas situaciones, deseche el FASENRA PEN en un contenedor de eliminación de objetos punzantes resistente a las perforaciones y use un FASENRA PEN nuevo. Cada FASENRA PEN contiene 1 dosis de FASENRA que es para un solo uso. Mantenga FASENRA y todos los medicamentos fuera de la vista y el alcance de los niños. |
|||||||||
|
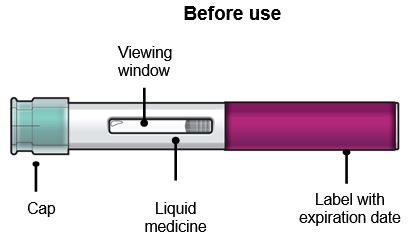
Su FASENRA PEN No quite la tapa hasta que haya llegado al Paso 6 de estas instrucciones y esté listo para inyectar FASENRA. |
|||||||||
 |
 |
||||||||
|
Paso 1 – Reúna los suministros |
|||||||||
|
|||||||||
 |
|||||||||
|
Paso 2 – Prepárese para usar su FASENRA PEN Compruebe la fecha de vencimiento (EXP). No lo use si la fecha de vencimiento ha pasado. Deje que FASENRA se caliente a temperatura ambiente entre 68°F a 77°F (20°C a 25°C) durante unos 30 minutos antes de administrar la inyección. No caliente el FASENRA PEN de ninguna otra manera. Por ejemplo, no lo caliente en un microondas o agua caliente, ni lo coloque cerca de otras fuentes de calor. Use FASENRA dentro de los 14 días de retirarlo del refrigerador. Después de 14 días, deseche el FASENRA PEN. No quite la tapa hasta que haya llegado al Paso 6. |
 |
||||||||
|
Paso 3 – Compruebe el líquido |
|||||||||
 |
Observe el líquido en el FASENRA PEN a través de la ventana de visualización. El líquido debe ser transparente e incoloro a ligeramente amarillo. Puede contener pequeñas partículas blancas. No inyecte FASENRA si el líquido está turbio, descolorido o contiene partículas grandes. Puede ver pequeñas burbujas de aire en el líquido. Esto es normal. No necesita hacer nada al respecto. |
||||||||
|
Paso 4 – Elija el sitio de inyección |
|||||||||
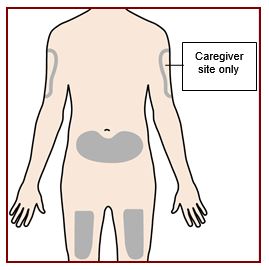 |
Si usted se está administrando la inyección, el sitio de inyección recomendado es la parte delantera del muslo o la parte inferior del estómago (abdomen). Un cuidador puede inyectarlo en la parte superior del brazo, el muslo o el abdomen. No intente inyectarse en el brazo. Para cada inyección, elija un sitio diferente que esté a por lo menos 1 pulgada (3 cm) de distancia del lugar donde se inyectó por última vez. No inyecte:
|
||||||||
|
Paso 5 – Limpie el sitio de inyección |
|||||||||
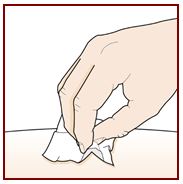 |
Lávese bien las manos con agua y jabón. Limpie el sitio de inyección con una toallita con alcohol en movimientos circulares. Deje que se seque al aire. No toque el área limpia antes de inyectar. No abanique ni sople sobre el área limpia. |
||||||||
|
Paso 6 – Retire la tapa |
|||||||||
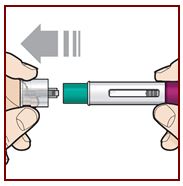 |
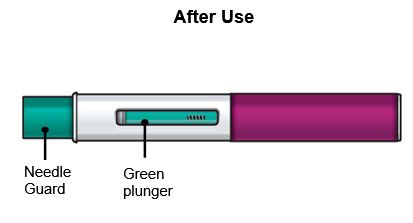
Sostenga el FASENRA PEN con una mano. Retire cuidadosamente la tapa con la otra mano. Deje la tapa a un lado para tirarla más tarde. Ahora está expuesto el protector de la aguja verde. Está ahí para evitar que toque la aguja. No intente tocar la aguja ni presionar el protector de la aguja con el dedo. No intente volver a colocar la tapa en el FASENRA PEN. Podría hacer que la inyección se produzca demasiado pronto o dañar la aguja. Complete los siguientes pasos inmediatamente después de quitar la tapa. |
||||||||
|
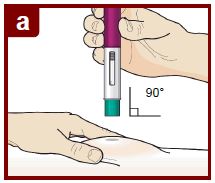
Paso 7 – Inyecte FASENRA |
|||||||||
|
Siga las instrucciones de su médico sobre cómo inyectar. Puede pellizcar suavemente el sitio de inyección o administrar la inyección sin pellizcar la piel. Inyecte FASENRA siguiendo los pasos de las figuras a, b, c, y d. Mantenga el FASENRA PEN en su lugar durante toda la inyección. No cambie la posición del FASENRA PEN después de que la inyección haya comenzado. |
|||||||||
 |
 |
 |
 |
||||||
|
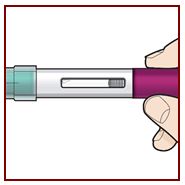
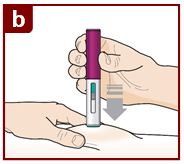
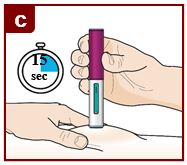
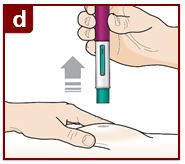
Coloque el FASENRA PEN en el sitio de inyección. Coloque el protector de la aguja del FASENRA PEN plano contra la piel (ángulo de 90 grados). Asegúrese de que pueda ver la ventana de visualización. |
Presione firmemente. Oirá un clic. Un ‘clic’ le indica que la inyección ha comenzado. El émbolo verde se moverá hacia abajo en la ventana de visualización durante la inyección. |
Mantenga presionado firmemente durante 15 segundos. Oirá un segundo ‘clic’. El segundo clic le indica que la inyección ha terminado. El émbolo verde llenará la ventana de visualización. |
Levante el FASENRA PEN en línea recta. El protector de la aguja se deslizará hacia abajo y se bloqueará en su lugar sobre la aguja. |
||||||
|
Paso 8 – Compruebe la ventana de visualización |
|||||||||
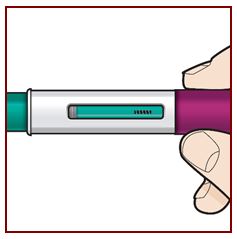 |
Compruebe la ventana de visualización para asegurarse de que se ha inyectado todo el líquido. Si el émbolo verde no llena la ventana de visualización, es posible que no haya recibido la dosis completa. Si esto sucede o si tiene alguna otra duda, llame a su médico. |
||||||||
|
 |
 |
Después de la inyección |
||||||
|
Paso 9 – Verifique el sitio de inyección |
|||||||||
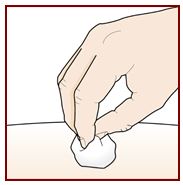 |
Puede haber una pequeña cantidad de sangre o líquido donde se inyectó. Esto es normal. Presione suavemente sobre su piel con una bola de algodón o gasa hasta que deje de sangrar. No frote el sitio de inyección. Si es necesario, cubra el sitio de inyección con una venda pequeña. |
||||||||
|
Paso 10 – Deseche el FASENRA PEN usado de forma segura |
|||||||||
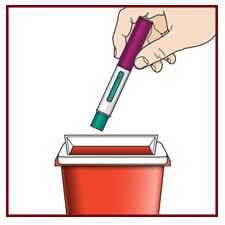 |
No tire el FASENRA PEN a la basura de su casa. Tire la tapa y otros suministros usados a la basura de su casa. |
||||||||
|
Pautas de eliminación Si no tiene un contenedor de eliminación de objetos punzantes aprobado por la FDA, puede usar un contenedor doméstico que sea:
Cuando su contenedor de eliminación de objetos punzantes esté casi lleno, deberá seguir las pautas de su comunidad para la forma correcta de desechar su contenedor de eliminación de objetos punzantes. Puede haber leyes estatales o locales sobre cómo debe desechar las agujas y jeringas usadas. Para obtener más información sobre la eliminación segura de objetos punzantes y para obtener información específica sobre la eliminación de objetos punzantes en el área donde vive, visite el sitio web de la FDA en: http://www.fda.gov/safesharpsdisposal. No deseche su contenedor de eliminación de objetos punzantes usado en la basura de su casa a menos que las pautas de su comunidad lo permitan. No recicle su contenedor de eliminación de objetos punzantes usado. Para obtener más información, visite www.FasenraPen.com o llame al 1-800-236-9933. Si aún tiene preguntas, llame a su proveedor de atención médica. Fabricado por: AstraZeneca AB, Södertälje, Suecia SE-15185 Número de licencia de EE. UU. 2059 Distribuido por: AstraZeneca Pharmaceuticals LP, Wilmington, DE 19850 FASENRA es una marca comercial del grupo de empresas AstraZeneca. ©AstraZeneca 2024 Estas Instrucciones de uso han sido aprobadas por la Administración de Alimentos y Medicamentos de los EE. UU. |
|||||||||
|
Emitido: abril de 2024 |
|||||||||
PANEL DE VISUALIZACIÓN PRINCIPAL DEL PAQUETE/ETIQUETA
NDC 0310-1730-30 Rx only
|
FASENRA® (benralizumab) Injection Para inyección subcutánea únicamente Almacenar la jeringa precargada refrigerada a 36°F – 46°F (2°C – 8°C) en el envase original para proteger de la luz. No agitar, congelar ni exponer al calor. |
30 mg/mL 1 jeringa precargada de dosis única Deseche la porción no utilizada. AstraZeneca |

PANEL DE VISUALIZACIÓN PRINCIPAL DEL PAQUETE/ETIQUETA
NDC 0310-1745-01 Rx only
|
FASENRA® (benralizumab) Inyección Solo para inyección subcutánea Conserve la jeringa precargada refrigerada a 36 °F – 46 °F (2 °C – 8 °C) en el envase original para protegerla de la luz. No agitar, congelar ni exponer al calor. |
10 mg/0.5 mL 1 jeringa precargada de dosis única Deseche la porción no utilizada. AstraZeneca |

Panel de visualización del paquete/etiqueta
NDC 0310-1830-30 Rx only
|
FASENRA PEN™ (benralizumab) Injection Para inyección subcutánea únicamente Almacene el FASENRA PEN refrigerado a 36°F – 46°F (2°C – 8°C) en el envase original para protegerlo de la luz. No agite, congele ni exponga al calor. |
30 mg/mL 1 autoinyector prellenado de dosis única Deseche la porción no utilizada. AstraZeneca |