
Fabricante de medicamentos: GlaxoSmithKline LLC (Updated: 2023-03-08)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
NUCALA (mepolizumab) para inyección, para uso subcutáneo
NUCALA (mepolizumab) inyección, para uso subcutáneo
Aprobación inicial en EE. UU.: 2015
INDICACIONES Y USO
NUCALA es un anticuerpo monoclonal antagonista de la interleucina-5 (IL-5) (IgG1 kappa) indicado para:
- •
- Tratamiento de mantenimiento adicional de pacientes adultos y pediátricos de 6 años de edad o mayores con asma grave y con un fenotipo eosinofílico. (1.1)
- •
- Tratamiento de mantenimiento adicional de pacientes adultos de 18 años de edad o mayores con rinosinusitis crónica con pólipos nasales (CRSwNP). (1.2)
- •
- El tratamiento de pacientes adultos con granulomatosis eosinofílica con poliangiitis (EGPA). (1.3)
- •
- El tratamiento de pacientes adultos y pediátricos de 12 años de edad o mayores con síndrome hipereosinofílico (HES) durante ≥6 meses sin una causa secundaria no hematológica identificable. (1.4)
Limitaciones de uso: No para el alivio del broncoespasmo agudo o el estado asmático. (1.1)
DOSIFICACIÓN Y ADMINISTRACIÓN
- •
- Asma grave en pacientes de 12 años de edad o mayores: 100 mg administrados por vía subcutánea una vez cada 4 semanas. (2.1)
- •
- Asma grave en pacientes de 6 a 11 años: 40 mg administrados por vía subcutánea una vez cada 4 semanas. (2.1)
- •
- CRSwNP: 100 mg administrados por vía subcutánea una vez cada 4 semanas. (2.2)
- •
- EGPA: 300 mg como 3 inyecciones separadas de 100 mg administradas por vía subcutánea una vez cada 4 semanas. (2.3)
- •
- HES: 300 mg como 3 inyecciones separadas de 100 mg administradas por vía subcutánea una vez cada 4 semanas. (2.4)
FORMAS Y FUERZAS DE DOSIFICACIÓN
CONTRAINDICACIONES
Historia de hipersensibilidad a mepolizumab o excipientes en la formulación. (4)
ADVERTENCIAS Y PRECAUCIONES
- •
- Se han producido reacciones de hipersensibilidad (p. ej., anafilaxia, angioedema, broncoespasmo, hipotensión, urticaria, erupción cutánea) después de la administración de NUCALA. Suspenda NUCALA en caso de reacción de hipersensibilidad. (5.1)
- •
- No lo use para tratar el broncoespasmo agudo o el estado asmático. (5.2)
- •
- Se han producido infecciones por herpes zóster en pacientes que reciben NUCALA. Considere la vacunación si es médicamente apropiado. (5.3)
- •
- No suspenda los corticosteroides sistémicos o inhalados abruptamente al iniciar el tratamiento con NUCALA. Disminuya los corticosteroides gradualmente, si es apropiado. (5.4)
- •
- Trate a los pacientes con infecciones por helmintos preexistentes antes de la terapia con NUCALA. Si los pacientes se infectan mientras reciben tratamiento con NUCALA y no responden al tratamiento antihelmíntico, suspenda NUCALA hasta que la infección parasitaria se resuelva. (5.5)
REACCIONES ADVERSAS
Reacciones adversas más comunes (incidencia ≥5%):
- •
- Asma: dolor de cabeza, reacción en el sitio de inyección, dolor de espalda y fatiga. (6.1)
- •
- CRSwNP: dolor orofaríngeo y artralgia. (6.2)
- •
- EGPA y HES: las reacciones adversas más comunes son similares a las del asma. (6.3, 6.4)
Para informar sobre REACCIONES ADVERSAS SOSPECHOSAS, comuníquese con GlaxoSmithKline al 1-888-825-5249 o con la FDA al 1-800-FDA-1088 o www.fda.gov/medwatch.
Consulte 17 para obtener INFORMACIÓN PARA EL PACIENTE y el etiquetado del paciente aprobado por la FDA.
Revisado: 3/2023
Tabla de Contenido
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICACIONES Y USO
1.1 Mantenimiento del Tratamiento del Asma Grave
1.2 Mantenimiento del Tratamiento de la Rinosinusitis Crónica con Pólipos Nasales
1.3 Eosinophilic Granulomatosis with Polyangiitis
1.4 Hypereosinophilic Syndrome
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Asma Grave
2.2 Rinosinusitis Crónica con Pólipos Nasales
2.3 Eosinophilic Granulomatosis with Polyangiitis
2.4 Hypereosinophilic Syndrome
2.5 Preparación y Administración de NUCALA para Inyección en Vial
2.6 Preparación y Administración de la Inyección de NUCALA Autoinyector Precargado y Jeringas Precargadas
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Reacciones de Hipersensibilidad
5.2 Síntomas Agudos de Asma o Empeoramiento de la Enfermedad
5.3 Infecciones Oportunistas: Herpes Zóster
5.4 Reducción de la Dosis de Corticosteroides
5.5 Infección Parasitaria (Helmintos)
6 REACCIONES ADVERSAS
6.1 Experiencia en Ensayos Clínicos en Asma Grave
6.2 Experiencia en Ensayos Clínicos en Rinosinusitis Crónica con Pólipos Nasales
6.3 Clinical Trials Experience in Eosinophilic Granulomatosis with Polyangiitis
6.4 Clinical Trials Experience in Hypereosinophilic Syndrome
6.5 Inmunogenicidad
6.6 Experiencia Posterior a la Comercialización
7 INTERACCIONES MEDICAMENTOSAS
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.4 Uso Pediátrico
8.5 Uso Geriátrico
10 SOBREDOSIS
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de Acción
12.2 Farmacodinamia
12.3 Farmacocinética
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
14 ESTUDIOS CLÍNICOS
14.1 Asma Grave
14.2 Rinosinusitis Crónica con Pólipos Nasales
14.3 Eosinophilic Granulomatosis with Polyangiitis
14.4 Hypereosinophilic Syndrome
16 CÓMO SUMINISTRARLO/ALMACENAMIENTO Y MANIPULACIÓN
17 INFORMACIÓN DE ASESORAMIENTO AL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información de prescripción completa no están listadas.
1 INDICACIONES Y USO
1.1 Tratamiento de mantenimiento del asma grave
NUCALA está indicado para el tratamiento de mantenimiento adicional de pacientes adultos y pediátricos de 6 años de edad o mayores con asma grave y con un fenotipo eosinofílico [ver Uso en poblaciones específicas (8.4), Estudios clínicos (14.1)].
Limitaciones de uso
NUCALA no está indicado para el alivio del broncoespasmo agudo o el estado asmático.
1.2 Tratamiento de mantenimiento de la rinosinusitis crónica con pólipos nasales
NUCALA está indicado para el tratamiento de mantenimiento adicional de la rinosinusitis crónica con pólipos nasales (CRSwNP) en pacientes adultos de 18 años de edad o mayores con respuesta inadecuada a los corticosteroides nasales.
1.3 Granulomatosis eosinofílica con poliangiitis
NUCALA está indicado para el tratamiento de pacientes adultos con granulomatosis eosinofílica con poliangiitis (EGPA).
1.4 Síndrome hipereosinofílico
NUCALA está indicado para el tratamiento de pacientes adultos y pediátricos de 12 años de edad o mayores con síndrome hipereosinofílico (HES) durante ≥6 meses sin una causa secundaria no hematológica identificable.
2 DOSIS Y ADMINISTRACIÓN
NUCALA es solo para uso subcutáneo.
2.1 Asma grave
Adultos y adolescentes a partir de 12 años
La dosis recomendada de NUCALA en adultos y adolescentes a partir de 12 años es de 100 mg administrados una vez cada 4 semanas mediante inyección subcutánea en la parte superior del brazo, el muslo o el abdomen [consulte Dosificación y administración (2.5, 2.6)].
Pacientes pediátricos de 6 a 11 años
La dosis recomendada de NUCALA para inyección en pacientes pediátricos de 6 a 11 años es de 40 mg administrados una vez cada 4 semanas mediante inyección subcutánea en la parte superior del brazo, el muslo o el abdomen [consulte Dosificación y administración (2.5, 2.6)].
2.2 Rinosinusitis crónica con pólipos nasales
La dosis recomendada de NUCALA es de 100 mg administrados una vez cada 4 semanas mediante inyección subcutánea en la parte superior del brazo, el muslo o el abdomen [consulte Dosificación y administración (2.5, 2.6)].
2.3 Granulomatosis eosinofílica con poliangitis
La dosis recomendada de NUCALA es de 300 mg administrados una vez cada 4 semanas mediante inyección subcutánea en 3 inyecciones separadas de 100 mg en la parte superior del brazo, el muslo o el abdomen [consulte Dosificación y administración (2.5, 2.6)]. Administre inyecciones individuales de 100 mg con una separación de al menos 5 cm (aproximadamente 2 pulgadas).
2.4 Síndrome hipereosinofílico
La dosis recomendada de NUCALA es de 300 mg administrados una vez cada 4 semanas mediante inyección subcutánea en 3 inyecciones separadas de 100 mg en la parte superior del brazo, el muslo o el abdomen [consulte Dosificación y administración (2.5, 2.6)]. Administre inyecciones individuales de 100 mg con una separación de al menos 5 cm (aproximadamente 2 pulgadas).
2.5 Preparación y administración del vial de NUCALA para inyección
NUCALA para inyección debe ser reconstituido y administrado por un profesional de la salud. De acuerdo con la práctica clínica, se recomienda el seguimiento de los pacientes después de la administración de agentes biológicos [consulte Advertencias y precauciones (5.1)].
Instrucciones de reconstitución
- 1.
- Reconstituya NUCALA para inyección en el vial con 1.2 mL de agua estéril para inyección, USP, preferiblemente utilizando una jeringa de 2 o 3 mL y una aguja de calibre 21. La solución reconstituida tendrá una concentración de 100 mg/mL de mepolizumab. No mezcle con otros medicamentos.
- 2.
- Dirija el chorro de agua estéril para inyección verticalmente hacia el centro del polvo liofilizado, que puede tener una apariencia similar a la de un pastel. Haga girar suavemente el vial durante 10 segundos con un movimiento circular a intervalos de 15 segundos hasta que el polvo se disuelva.
Nota: No agite la solución reconstituida durante el procedimiento, ya que esto puede provocar la formación de espuma o la precipitación del producto. La reconstitución suele completarse en los 5 minutos posteriores a la adición del agua estéril para inyección, pero puede llevar más tiempo. - 3.
- Si se utiliza un dispositivo de reconstitución mecánica (agitador) para reconstituir NUCALA para inyección, agite a 450 rpm durante no más de 10 minutos. Alternativamente, es aceptable agitar a 1,000 rpm durante no más de 5 minutos.
- 4.
- Inspeccione visualmente la solución reconstituida para detectar la presencia de partículas y comprobar su claridad antes de su uso. La solución debe ser de transparente a opalescente e incolora a amarillo pálido o marrón pálido, esencialmente libre de partículas. Sin embargo, es de esperar que aparezcan pequeñas burbujas de aire, lo cual es aceptable. Si quedan partículas en la solución o si la solución parece turbia o lechosa, no debe administrarse.
- 5.
- Si la solución reconstituida no se utiliza inmediatamente:
- •
- guárdela a una temperatura inferior a 30 °C (86 °F),
- •
- no la congele, y
- •
- deséchela si no se utiliza en las 8 horas siguientes a su reconstitución.
Administración de la dosis de 100 mg
- 1.
- Para la administración subcutánea, preferiblemente utilizando una jeringa de polipropileno de 1 mL equipada con una aguja desechable de calibre 21 a 27 x 0.5 pulgadas (13 mm).
- 2.
- Justo antes de la administración, extraiga 1 mL de NUCALA reconstituido para inyección. No agite la solución reconstituida durante el procedimiento, ya que esto podría provocar la formación de espuma o la precipitación del producto.
- 3.
- Administre la inyección de 1 mL (equivalente a 100 mg de mepolizumab) por vía subcutánea en la parte superior del brazo, el muslo o el abdomen.
Administración de la dosis de 40 mg
- 1.
- Para la administración subcutánea, preferiblemente utilizando una jeringa de polipropileno de 1 mL equipada con una aguja desechable de calibre 21 a 27 x 0.5 pulgadas (13 mm).
- 2.
- Justo antes de la administración, extraiga 0.4 mL de NUCALA reconstituido para inyección. No agite la solución reconstituida durante el procedimiento, ya que esto podría provocar la formación de espuma o la precipitación del producto.
- 3.
- Administre la inyección de 0.4 mL (equivalente a 40 mg de mepolizumab) por vía subcutánea en la parte superior del brazo, el muslo o el abdomen.
Cada vial de NUCALA para inyección debe utilizarse para un solo paciente y debe desecharse cualquier resto del contenido.
2.6 Preparación y administración de la inyección de NUCALA con autoinyector precargado y jeringas precargadas
La inyección de NUCALA está diseñada para su uso bajo la supervisión de un profesional de la salud.
La autoinyectora precargada de 100 mg/ml y la jeringa precargada de 100 mg/ml solo se pueden usar en adultos y adolescentes a partir de los 12 años. El paciente puede autoinyectarse o la persona que lo cuida puede administrarle la inyección de NUCALA de 100 mg/ml por vía subcutánea después de que el profesional sanitario determine que es apropiado.
La jeringa precargada de 40 mg/0.4 ml solo se puede usar en niños de 6 a 11 años y debe ser administrada por un profesional sanitario o la persona que cuida al paciente. La persona que cuida al paciente puede administrar la inyección de NUCALA de 40 mg/0.4 ml por vía subcutánea después de que el profesional sanitario determine que es apropiado.
Antes de su uso, proporcione la formación adecuada sobre la técnica de inyección subcutánea y sobre la preparación y administración de la inyección de NUCALA [see Instructions for Use].
- 1.
- Saque la autoinyectora precargada o la jeringa precargada del refrigerador y déjela reposar a temperatura ambiente durante 30 minutos antes de la inyección. No caliente la inyección de NUCALA de ninguna otra manera.
- 2.
- Antes de la administración, inspeccione visualmente la ventana de la autoinyectora precargada o la jeringa precargada para detectar la presencia de partículas o cambio de color. La inyección de NUCALA debe tener un aspecto entre transparente y opalescente, y un color entre incoloro y amarillo pálido o marrón pálido. No utilice la inyección de NUCALA si el producto presenta cambio de color, turbidez o partículas. No utilice la autoinyectora precargada ni la jeringa precargada de NUCALA si se cae sobre una superficie dura.
- 3.
- Administre la inyección subcutánea en el muslo o el abdomen, evitando los 5 cm (aproximadamente 2 pulgadas) alrededor del ombligo. También se puede utilizar la parte superior del brazo si la inyección subcutánea la administra la persona que cuida al paciente.
- 4.
- Para su uso en EGPA y HES, asegúrese de que los puntos de inyección para cada inyección subcutánea estén separados por al menos 5 cm (aproximadamente 2 pulgadas).
- 5.
- Nunca administre inyecciones en zonas donde la piel esté sensible, amoratada, enrojecida o endurecida.
- 6.
- Si se olvida una dosis, adminístrela lo antes posible. A partir de entonces, el paciente puede reanudar la administración el día habitual. Si ya es el momento de la siguiente dosis, adminístrela según lo previsto.
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
Para inyección
- •
- 100 mg de polvo liofilizado blanco a blanquecino en un vial de una dosis única para reconstitución.
Inyección
- •
- 100 mg/mL como solución clara a opalescente, incolora a amarillo pálido a marrón pálido en un autoinyector precargado de una dosis única.
- •
- 100 mg/mL como solución clara a opalescente, incolora a amarillo pálido a marrón pálido en una jeringa de vidrio precargada de una dosis única.
- •
- 40 mg/0,4 mL como solución clara a opalescente, incolora a amarillo pálido a marrón pálido en una jeringa de vidrio precargada de una dosis única.
4 CONTRAINDICACIONES
NUCALA está contraindicado en pacientes con antecedentes de hipersensibilidad a mepolizumab o excipientes en la formulación [ver Advertencias y precauciones (5.1), Descripción (11)].
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Reacciones de hipersensibilidad
Se han producido reacciones de hipersensibilidad (p. ej., anafilaxia, angioedema, broncoespasmo, hipotensión, urticaria, erupción cutánea) tras la administración de NUCALA. Estas reacciones generalmente ocurren dentro de las horas posteriores a la administración, pero en algunos casos pueden tener una aparición tardía (es decir, días). En caso de una reacción de hipersensibilidad, se debe suspender el tratamiento con NUCALA [ver Contraindicaciones (4)].
5.2 Síntomas agudos de asma o deterioro de la enfermedad
NUCALA no debe utilizarse para tratar los síntomas agudos del asma ni las exacerbaciones agudas. No utilice NUCALA para tratar el broncoespasmo agudo o el estado asmático. Los pacientes deben buscar atención médica si su asma permanece sin control o empeora después del inicio del tratamiento con NUCALA.
5.3 Infecciones oportunistas: Herpes zóster
Se ha producido herpes zóster en sujetos que recibieron NUCALA 100 mg en ensayos clínicos controlados [ver Reacciones adversas (6.1)]. Considere la vacunación si es médicamente apropiado.
5.4 Reducción de la dosis de corticosteroides
No suspenda abruptamente los corticosteroides sistémicos o inhalados (ICS) al iniciar el tratamiento con NUCALA. Las reducciones en la dosis de corticosteroides, si corresponde, deben ser graduales y realizarse bajo la supervisión directa de un médico. La reducción de la dosis de corticosteroides puede asociarse con síntomas de abstinencia sistémicos y/o desenmascarar afecciones previamente suprimidas por la terapia con corticosteroides sistémicos.
5.5 Infección parasitaria (por helmintos)
Los eosinófilos pueden estar involucrados en la respuesta inmunológica a algunas infecciones por helmintos. Los pacientes con infecciones parasitarias conocidas fueron excluidos de la participación en ensayos clínicos. Se desconoce si NUCALA influirá en la respuesta de un paciente contra las infecciones parasitarias. Trate a los pacientes con infecciones por helmintos preexistentes antes de iniciar el tratamiento con NUCALA. Si los pacientes se infectan mientras reciben tratamiento con NUCALA y no responden al tratamiento antihelmíntico, suspenda el tratamiento con NUCALA hasta que la infección se resuelva.
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas se describen con mayor detalle en otras secciones:
- •
- Reacciones de hipersensibilidad [ver Advertencias y precauciones (5.1)]
- •
- Infecciones oportunistas: herpes zóster [ver Advertencias y precauciones (5.3)]
Debido a que los ensayos clínicos se llevan a cabo en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no se pueden comparar directamente con las tasas en los ensayos clínicos de otro fármaco y pueden no reflejar las tasas observadas en la práctica.
6.1 Experiencia en ensayos clínicos en asma grave
Pacientes adultos y adolescentes de 12 años o más
Se evaluó un total de 1.327 pacientes con asma grave en 3 ensayos aleatorizados, controlados con placebo y multicéntricos de 24 a 52 semanas de duración (Ensayo 1, NCT01000506; Ensayo 2, NCT01691521; y Ensayo 3, NCT01691508). De estos, 1.192 tenían antecedentes de 2 o más exacerbaciones en el año anterior a la inscripción a pesar del uso regular de ICS de dosis alta más controlador(es) adicional(es) (Ensayos 1 y 2), y 135 pacientes requirieron corticosteroides orales (OCS) diarios además del uso regular de ICS de dosis alta más controlador(es) adicional(es) para mantener el control del asma (Ensayo 3). Todos los pacientes tenían marcadores de inflamación eosinofílica de las vías respiratorias [ver Estudios clínicos (14.1)]. De los pacientes inscritos, el 59% eran mujeres, el 85% eran blancos y las edades oscilaban entre los 12 y los 82 años. Mepolizumab se administró por vía subcutánea o intravenosa una vez cada 4 semanas; 263 pacientes recibieron NUCALA (mepolizumab 100 mg subcutáneo) durante al menos 24 semanas. Los eventos adversos graves que ocurrieron en más de 1 paciente y en un porcentaje mayor de pacientes que recibieron NUCALA 100 mg (n = 263) que placebo (n = 257) incluyeron 1 evento, herpes zóster (2 pacientes frente a 0 pacientes, respectivamente). Aproximadamente el 2% de los pacientes que recibieron NUCALA 100 mg se retiraron de los ensayos clínicos debido a eventos adversos en comparación con el 3% de los pacientes que recibieron placebo.
La incidencia de reacciones adversas en las primeras 24 semanas de tratamiento en los 2 ensayos confirmatorios de eficacia y seguridad (Ensayos 2 y 3) con NUCALA 100 mg se muestra en Tabla 1.
|
Reacción adversa |
NUCALA (Mepolizumab 100 mg subcutáneo) (n = 263) % |
Placebo (n = 257) % |
|
Dolor de cabeza |
19 |
18 |
|
Reacción en el sitio de inyección |
8 |
3 |
|
Dolor de espalda |
5 |
4 |
|
Fatiga |
5 |
4 |
|
Influenza |
3 |
2 |
|
Infección del tracto urinario |
3 |
2 |
|
Dolor abdominal superior |
3 |
2 |
|
Prurito |
3 |
2 |
|
Eczema |
3 |
<1 |
|
Espasmos musculares |
3 |
<1 |
Ensayo de 52 semanas: Las reacciones adversas del ensayo 1 con 52 semanas de tratamiento con mepolizumab 75 mg intravenoso (IV) (n = 153) o placebo (n = 155) y con una incidencia ≥3% y más común que el placebo y no se muestran en la Tabla 1 fueron: dolor abdominal, rinitis alérgica, astenia, bronquitis, cistitis, mareos, disnea, infección del oído, gastroenteritis, infección del tracto respiratorio inferior, dolor musculoesquelético, congestión nasal, nasofaringitis, náuseas, faringitis, pirexia, erupción cutánea, dolor de muelas, infección viral, infección viral del tracto respiratorio e vómitos. Además, se produjeron 3 casos de herpes zóster en pacientes que recibieron mepolizumab 75 mg IV en comparación con 2 pacientes en el grupo placebo.
Reacciones sistémicas, incluidas las reacciones de hipersensibilidad: En los ensayos 1, 2 y 3 descritos anteriormente, el porcentaje de pacientes que experimentaron reacciones sistémicas (alérgicas y no alérgicas) fue del 3% en el grupo que recibió NUCALA 100 mg y del 5% en el grupo placebo. Las reacciones alérgicas/de hipersensibilidad sistémicas fueron informadas por el 1% de los pacientes en el grupo que recibió NUCALA 100 mg y el 2% de los pacientes en el grupo placebo. Las manifestaciones más comúnmente reportadas de reacciones alérgicas/de hipersensibilidad sistémicas reportadas en el grupo que recibió NUCALA 100 mg incluyeron erupción cutánea, prurito, dolor de cabeza y mialgia. Las reacciones sistémicas no alérgicas fueron informadas por el 2% de los pacientes en el grupo que recibió NUCALA 100 mg y el 3% de los pacientes en el grupo placebo. Las manifestaciones más comúnmente reportadas de reacciones sistémicas no alérgicas reportadas en el grupo que recibió NUCALA 100 mg incluyeron erupción cutánea, rubor y mialgia. La mayoría de las reacciones sistémicas en pacientes que recibieron NUCALA 100 mg (5/7) se experimentaron el día de la dosificación.
Reacciones en el sitio de inyección: Las reacciones en el sitio de inyección (por ejemplo, dolor, eritema, hinchazón, picazón, sensación de ardor) ocurrieron a una tasa del 8% en pacientes que recibieron NUCALA 100 mg en comparación con el 3% en pacientes que recibieron placebo.
Seguridad a largo plazo: Novecientos noventa y ocho pacientes recibieron NUCALA 100 mg en estudios de extensión de etiqueta abierta en curso, durante los cuales se informaron casos adicionales de herpes zóster. El perfil general de eventos adversos ha sido similar a los ensayos de asma grave descritos anteriormente.
Pacientes pediátricos de 6 a 11 años
Los datos de seguridad para NUCALA se basan en 1 ensayo clínico de etiqueta abierta que inscribió a 36 pacientes con asma grave de 6 a 11 años. Los pacientes recibieron 40 mg (para aquellos que pesan <40 kg) o 100 mg (para aquellos que pesan ≥40 kg) de NUCALA administrado por vía subcutánea una vez cada 4 semanas. Los pacientes recibieron NUCALA durante 12 semanas (fase inicial corta). Después de una interrupción del tratamiento de 8 semanas, 30 pacientes recibieron NUCALA durante 52 semanas adicionales (fase larga). El perfil de reacciones adversas para pacientes de 6 a 11 años fue similar al observado en pacientes de 12 años o más.
6.2 Experiencia en ensayos clínicos en rinosinusitis crónica con pólipos nasales
Se evaluó un total de 407 pacientes con CRSwNP en 1 ensayo de tratamiento aleatorizado, controlado con placebo, multicéntrico, de 52 semanas. Los pacientes recibieron NUCALA 100 mg o placebo por vía subcutánea una vez cada 4 semanas. Los pacientes tenían CRSwNP recurrente con antecedentes de cirugía previa y estaban en corticosteroides nasales durante al menos 8 semanas antes de la selección [ver Estudios clínicos (14.2)]. De los pacientes inscritos, el 35% eran mujeres, el 93% eran blancos y las edades oscilaban entre los 18 y los 82 años. Aproximadamente el 2% de los pacientes que recibieron NUCALA 100 mg se retiraron del tratamiento del estudio debido a eventos adversos en comparación con el 2% de los pacientes que recibieron placebo.
Tabla 2 resume las reacciones adversas que ocurrieron en ≥3% de los pacientes tratados con NUCALA y con mayor frecuencia que en los pacientes tratados con placebo en el ensayo CRSwNP.
| CRSwNP = Rinosinusitis crónica con pólipos nasales. | ||
|
Reacción adversa |
NUCALA (Mepolizumab 100 mg Subcutáneo) (n = 206) % |
Placebo (n = 201) % |
|
Dolor orofaríngeo |
8 |
5 |
|
Artralgia |
6 |
2 |
|
Dolor abdominal superior |
3 |
2 |
|
Diarrea |
3 |
2 |
|
Pirexia |
3 |
2 |
|
Sequedad nasal |
3 |
<1 |
|
Erupción cutánea |
3 |
<1 |
Reacciones sistémicas, incluidas reacciones de hipersensibilidad
En el ensayo de 52 semanas, el porcentaje de pacientes que experimentaron reacciones sistémicas (alérgicas [hipersensibilidad de tipo I] y otras) fue <1% en el grupo que recibió NUCALA 100 mg y <1% en el grupo placebo. Las reacciones alérgicas sistémicas (hipersensibilidad de tipo I) fueron reportadas por <1% de los pacientes en el grupo que recibió NUCALA 100 mg y ningún paciente en el grupo placebo. Las manifestaciones de las reacciones alérgicas sistémicas (hipersensibilidad de tipo I) incluyeron urticaria, eritema y erupción cutánea, y 1 de las 3 reacciones ocurrió el día de la dosificación. Otras reacciones sistémicas fueron reportadas por ningún paciente en el grupo que recibió NUCALA 100 mg y <1% de los pacientes en el grupo placebo.
Reacciones en el sitio de inyección
Las reacciones en el sitio de inyección (por ejemplo, eritema, prurito) ocurrieron a una tasa del 2% en pacientes que recibieron NUCALA 100 mg en comparación con <1% en pacientes que recibieron placebo.
6.3 Experiencia en ensayos clínicos en granulomatosis eosinofílica con poliangiitis
Se evaluó un total de 136 pacientes con EGPA en 1 ensayo de tratamiento aleatorizado, controlado con placebo, multicéntrico, de 52 semanas. Los pacientes recibieron 300 mg de NUCALA o placebo por vía subcutánea una vez cada 4 semanas. Los pacientes inscritos tenían un diagnóstico de EGPA durante al menos 6 meses antes de la inscripción con antecedentes de enfermedad recurrente o refractaria y estaban en una dosis estable de prednisolona oral o prednisona de mayor o igual a 7,5 mg/día (pero no mayor a 50 mg/día) durante al menos 4 semanas antes de la inscripción [ver Estudios clínicos (14.3)]. De los pacientes inscritos, el 59% eran mujeres, el 92% eran blancos y las edades oscilaron entre los 20 y los 71 años. No se identificaron reacciones adversas adicionales a las reportadas en los ensayos de asma grave.
Reacciones sistémicas, incluidas reacciones de hipersensibilidad
En el ensayo de 52 semanas, el porcentaje de pacientes que experimentaron reacciones sistémicas (alérgicas y no alérgicas) fue del 6% en el grupo que recibió 300 mg de NUCALA y del 1% en el grupo placebo. Las reacciones alérgicas/de hipersensibilidad sistémicas fueron reportadas por el 4% de los pacientes en el grupo que recibió 300 mg de NUCALA y el 1% de los pacientes en el grupo placebo. Las manifestaciones de las reacciones alérgicas/de hipersensibilidad sistémicas reportadas en el grupo que recibió 300 mg de NUCALA incluyeron erupción cutánea, prurito, rubor, fatiga, hipertensión, sensación de calor en el tronco y el cuello, extremidades frías, disnea y estridor. Las reacciones sistémicas no alérgicas fueron reportadas por 1 (1%) paciente en el grupo que recibió 300 mg de NUCALA y ningún paciente en el grupo placebo. La manifestación reportada de las reacciones sistémicas no alérgicas reportadas en el grupo que recibió 300 mg de NUCALA fue angioedema. La mitad de las reacciones sistémicas en pacientes que recibieron 300 mg de NUCALA (2/4) se experimentaron el día de la dosificación.
Reacciones en el sitio de inyección
Las reacciones en el sitio de inyección (por ejemplo, dolor, eritema, hinchazón) ocurrieron a una tasa del 15% en pacientes que recibieron 300 mg de NUCALA en comparación con el 13% en pacientes que recibieron placebo.
6.4 Experiencia en ensayos clínicos en síndrome hipereosinofílico
Se evaluó un total de 108 pacientes adultos y adolescentes de 12 años o más con HES en un ensayo de tratamiento aleatorizado, controlado con placebo, multicéntrico, de 32 semanas. Los pacientes con HES secundario no hematológico o HES positivo para la quinasa FIP1L1‑PDGFRα fueron excluidos del ensayo. Los pacientes recibieron 300 mg de NUCALA o placebo por vía subcutánea una vez cada 4 semanas. Los pacientes deben haber estado en una dosis estable de terapia de fondo para HES durante las 4 semanas previas a la aleatorización [ver Estudios clínicos (14.4)]. De los pacientes inscritos, el 53% eran mujeres, el 93% eran blancos y las edades oscilaron entre los 12 y los 82 años. No se identificaron reacciones adversas adicionales a las reportadas en los ensayos de asma grave.
Reacciones sistémicas, incluidas reacciones de hipersensibilidad
En el ensayo, no se reportaron reacciones alérgicas sistémicas (hipersensibilidad de tipo I). Otras reacciones sistémicas fueron reportadas por 1 (2%) paciente en el grupo que recibió 300 mg de NUCALA y ningún paciente en el grupo placebo. La manifestación reportada de otra reacción sistémica fue una reacción cutánea multifocal experimentada el día de la dosificación.
Reacciones en el sitio de inyección
Las reacciones en el sitio de inyección (por ejemplo, ardor, picazón) ocurrieron a una tasa del 7% en pacientes que recibieron 300 mg de NUCALA en comparación con el 4% en pacientes que recibieron placebo.
6.5 Inmunogenicidad
En pacientes adultos y adolescentes con asma grave que recibieron NUCALA 100 mg, 15/260 (6%) tuvieron anticuerpos anti-mepolizumab detectables. Se detectaron anticuerpos neutralizantes en 1 paciente con asma que recibió NUCALA 100 mg. Los anticuerpos anti-mepolizumab aumentaron ligeramente (aproximadamente un 20%) la eliminación de mepolizumab. No hubo evidencia de una correlación entre los títulos de anticuerpos anti-mepolizumab y el cambio en el nivel de eosinófilos. No se conoce la relevancia clínica de la presencia de anticuerpos anti-mepolizumab. En el ensayo clínico de niños de 6 a 11 años con asma grave que recibieron NUCALA 40 o 100 mg, 2/35 (6%) tuvieron anticuerpos anti-mepolizumab detectables durante la fase inicial corta del ensayo. Ningún niño tuvo anticuerpos anti-mepolizumab detectables durante la fase larga del ensayo.
En pacientes con CRSwNP que recibieron NUCALA 100 mg, 6/196 (3%) tuvieron anticuerpos anti-mepolizumab detectables. No se detectaron anticuerpos neutralizantes en ningún paciente con CRSwNP.
En pacientes con EGPA que recibieron 300 mg de NUCALA, 1/68 (<2%) tuvieron anticuerpos anti-mepolizumab detectables. No se detectaron anticuerpos neutralizantes en ningún paciente con EGPA.
En pacientes adultos y adolescentes con HES que recibieron 300 mg de NUCALA, 1/53 (2%) tuvieron anticuerpos anti-mepolizumab detectables. No se detectaron anticuerpos neutralizantes en ningún paciente con HES.
La frecuencia reportada de anticuerpos anti-mepolizumab puede subestimar la frecuencia real debido a una menor sensibilidad del ensayo en presencia de una alta concentración de fármaco. Los datos reflejan el porcentaje de pacientes cuyos resultados de las pruebas fueron positivos para anticuerpos contra mepolizumab en ensayos específicos. La incidencia observada de positividad de anticuerpos en un ensayo depende en gran medida de varios factores, incluida la sensibilidad y especificidad del ensayo, la metodología del ensayo, el manejo de las muestras, el momento de la recolección de las muestras, los medicamentos concomitantes y la enfermedad subyacente.
6.6 Experiencia Postcomercialización
Además de las reacciones adversas notificadas en los ensayos clínicos, se han identificado las siguientes reacciones adversas durante el uso posterior a la aprobación de NUCALA. Debido a que estas reacciones se notifican voluntariamente de una población de tamaño incierto, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal con la exposición al fármaco. Estos eventos se han elegido para su inclusión debido a su gravedad, frecuencia de notificación o conexión causal con NUCALA o una combinación de estos factores.
Trastornos del sistema inmunitario
Reacciones de hipersensibilidad, incluida la anafilaxia.
7 INTERACCIONES MEDICAMENTOSAS
No se han realizado ensayos formales de interacción medicamentosa con NUCALA.
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de Riesgos
Los datos sobre la exposición durante el embarazo son insuficientes para informar sobre el riesgo asociado al fármaco. Los anticuerpos monoclonales, como el mepolizumab, se transportan a través de la placenta de forma lineal a medida que avanza el embarazo; por lo tanto, es probable que los efectos potenciales sobre el feto sean mayores durante el segundo y tercer trimestre del embarazo. En un estudio de desarrollo prenatal y postnatal realizado en monos cynomolgus, no hubo evidencia de daño fetal con la administración IV de mepolizumab durante todo el embarazo a dosis que produjeron exposiciones hasta aproximadamente 9 veces la exposición a la dosis humana máxima recomendada (MRHD) de 300 mg subcutánea (ver Datos).
En la población general de los EE. UU., el riesgo de fondo estimado de defectos de nacimiento mayores y aborto espontáneo en embarazos clínicamente reconocidos es del 2% al 4% y del 15% al 20%, respectivamente.
Consideraciones Clínicas
Riesgo Materno y/o Embriofetal Asociado a la Enfermedad: En mujeres con asma mal controlada o moderadamente controlada, la evidencia demuestra que existe un mayor riesgo de preeclampsia en la madre y prematuridad, bajo peso al nacer y pequeño para la edad gestacional en el neonato. El nivel de control del asma debe controlarse estrechamente en las mujeres embarazadas y el tratamiento debe ajustarse según sea necesario para mantener un control óptimo.
Datos
Datos en Animales: En un estudio de desarrollo prenatal y postnatal, monos cynomolgus preñados recibieron mepolizumab desde el día 20 hasta el 140 de gestación a dosis que produjeron exposiciones hasta aproximadamente 9 veces las que se lograron con la MRHD (en base al AUC con dosis IV maternas de hasta 100 mg/kg una vez cada 4 semanas). El mepolizumab no provocó efectos adversos sobre el crecimiento fetal o neonatal (incluida la función inmunitaria) hasta 9 meses después del nacimiento. No se realizaron exámenes para detectar malformaciones internas o esqueléticas. El mepolizumab cruzó la placenta en monos cynomolgus. Las concentraciones de mepolizumab fueron aproximadamente 2,4 veces más altas en los bebés que en las madres hasta el día 178 posparto. Los niveles de mepolizumab en la leche fueron ≤0,5% de la concentración sérica materna.
En un estudio de fertilidad, desarrollo embrionario temprano y desarrollo embriofetal, ratones CD-1 preñados recibieron un anticuerpo análogo, que inhibe la actividad de la interleucina-5 (IL-5) murina, a una dosis IV de 50 mg/kg una vez por semana durante toda la gestación. El anticuerpo análogo no fue teratógeno en ratones. Se ha informado que el desarrollo embriofetal de ratones deficientes en IL-5 no se ve afectado en general en relación con los ratones de tipo salvaje.
8.2 Lactancia
Resumen de Riesgos
No hay información sobre la presencia de mepolizumab en la leche materna, los efectos en el lactante amamantado o los efectos sobre la producción de leche. Sin embargo, el mepolizumab es un anticuerpo monoclonal humanizado (IgG1 kappa), y la inmunoglobulina G (IgG) está presente en la leche materna en pequeñas cantidades. El mepolizumab estuvo presente en la leche de monos cynomolgus posparto después de la dosificación durante el embarazo [ver Uso en Poblaciones Específicas (8.1)]. Los beneficios para el desarrollo y la salud de la lactancia materna deben considerarse junto con la necesidad clínica de la madre de NUCALA y cualquier efecto adverso potencial en el lactante amamantado por el mepolizumab o por la condición materna subyacente.
8.4 Uso Pediátrico
Asma Grave
La seguridad y eficacia de NUCALA para el asma grave, y con un fenotipo eosinofílico, se han establecido en pacientes pediátricos de 6 años de edad o mayores.
El uso de NUCALA en adolescentes de 12 a 17 años está respaldado por evidencia de ensayos adecuados y bien controlados en adultos y adolescentes. Un total de 28 adolescentes de 12 a 17 años con asma grave fueron inscritos en los ensayos de asma de Fase 3. De estos, 25 fueron inscritos en el ensayo de exacerbación de 32 semanas (Ensayo 2, NCT01691521) y tenían una edad promedio de 14,8 años. Los pacientes tenían antecedentes de 2 o más exacerbaciones en el año anterior a pesar del uso regular de ICS de dosis media o alta más controladores adicionales con o sin OCS y tenían eosinófilos en sangre de ≥150 células/mcL en la selección o ≥300 células/mcL dentro de los 12 meses anteriores a la inscripción. [Ver Estudios Clínicos (14.1).] Los pacientes tuvieron una reducción en la tasa de exacerbaciones que tendió a favor de NUCALA. De los 19 adolescentes que recibieron NUCALA, 9 recibieron 100 mg y la depuración aparente media en estos pacientes fue un 35% menor que la de los adultos. El perfil de seguridad observado en adolescentes fue generalmente similar al de la población general en los estudios de Fase 3 [ver Reacciones Adversas (6.1)].
El uso de NUCALA en pacientes pediátricos de 6 a 11 años con asma grave, y con un fenotipo eosinofílico, está respaldado por evidencia de ensayos adecuados y bien controlados en adultos y adolescentes con datos farmacocinéticos, farmacodinámicos y de seguridad adicionales en niños de 6 a 11 años. Se llevó a cabo un solo ensayo clínico abierto (NCT02377427) en 36 niños de 6 a 11 años (edad promedio: 8,6 años, 31% mujeres) con asma grave. Los criterios de inclusión fueron los mismos que para los adolescentes en el ensayo de exacerbación de 32 semanas (Ensayo 2). Con base en los datos farmacocinéticos de este ensayo, se determinó que una dosis de 40 mg subcutánea cada 4 semanas tenía una exposición similar a la de los adultos y adolescentes que recibieron una dosis de 100 mg subcutánea [ver Farmacología Clínica (12.3)].
La eficacia de NUCALA en pacientes pediátricos de 6 a 11 años se extrapola de la eficacia en adultos y adolescentes con apoyo de análisis farmacocinéticos que muestran niveles de exposición a fármacos similares para 40 mg administrados por vía subcutánea cada 4 semanas en niños de 6 a 11 años en comparación con adultos y adolescentes [ver Farmacología Clínica (12.3)]. El perfil de seguridad y la respuesta farmacodinámica observados en este ensayo para niños de 6 a 11 años fueron similares a los observados en adultos y adolescentes [ver Reacciones Adversas (6.1), Farmacología Clínica (12.2)].
No se ha establecido la seguridad y eficacia en pacientes pediátricos menores de 6 años con asma grave.
Rinosinusitis crónica con pólipos nasales
No se ha establecido la seguridad y eficacia en pacientes menores de 18 años con CRSwNP.
Granulomatosis eosinofílica con poliangiitis
No se ha establecido la seguridad y eficacia en pacientes menores de 18 años con EGPA.
Síndrome hipereosinofílico
La seguridad y eficacia de NUCALA para HES se han establecido en pacientes adolescentes de 12 años o más. No se ha establecido la seguridad y eficacia en pacientes pediátricos menores de 12 años con HES.
El uso de NUCALA para esta indicación está respaldado por evidencia de un estudio adecuado y bien controlado (NCT02836496) en adultos y adolescentes y un estudio de extensión de etiqueta abierta (NCT03306043). Un adolescente recibió NUCALA durante el estudio controlado y este paciente y otros 3 adolescentes recibieron NUCALA durante el estudio de extensión de etiqueta abierta [ver Estudios clínicos (14.4)]. El 1 adolescente tratado con NUCALA en el ensayo de 32 semanas no tuvo un brote de HES ni se informó ningún evento adverso. Todos los adolescentes recibieron 300 mg de NUCALA durante 20 semanas en la extensión de etiqueta abierta.
8.5 Uso en geriatría
Los ensayos clínicos de NUCALA no incluyeron un número suficiente de pacientes de 65 años o más que recibieron NUCALA (n = 79) para determinar si responden de manera diferente a los pacientes más jóvenes. Otra experiencia clínica informada no ha identificado diferencias en las respuestas entre los pacientes ancianos y los más jóvenes. En general, la selección de la dosis para un paciente anciano debe ser cautelosa, generalmente comenzando en el extremo inferior del rango de dosificación, lo que refleja la mayor frecuencia de disminución de la función hepática, renal o cardíaca y de enfermedades concomitantes u otra terapia farmacológica. Con base en los datos disponibles, no es necesario ajustar la dosis de NUCALA en pacientes geriátricos, pero no se puede descartar una mayor sensibilidad en algunos individuos mayores.
10 SOBREDOSIS
No existe un tratamiento específico para una sobredosis con mepolizumab. Si ocurre una sobredosis, el paciente debe recibir tratamiento de apoyo con monitorización adecuada según sea necesario.
11 DESCRIPCIÓN
Mepolizumab es un anticuerpo monoclonal antagonista de IL-5 humanizado. Mepolizumab se produce mediante tecnología de ADN recombinante en células de ovario de hámster chino. Mepolizumab tiene un peso molecular aproximado de 149 kDa.
NUCALA para inyección es un polvo liofilizado estéril, libre de conservantes, blanco a blanquecino, en un vial de dosis única para inyección subcutánea después de la reconstitución. Tras la reconstitución con 1,2 mL de Agua estéril para inyección, USP, la concentración resultante es 100 mg/mL y proporciona 1 mL [véase Dosis y administración (2.5) ]. Cada vial proporciona 100 mg de mepolizumab, polisorbato 80 (0,67 mg), fosfato sódico dibásico heptahidrato (7,14 mg) y sacarosa (160 mg), con un pH de 7,0.
El tapón del vial no está fabricado con látex de caucho natural.
La inyección de NUCALA es una solución estéril, libre de conservantes, clara a opalescente, incolora a amarillo pálido a marrón pálido para uso subcutáneo.
La inyección de NUCALA se suministra en un autoinyector precargado de dosis única de 1 mL con una aguja fija de 29 gauge y media pulgada y en una jeringa precargada de dosis única de 1 mL con una aguja fija de 29 gauge y media pulgada con protector de aguja. Cada 1 mL proporciona 100 mg de mepolizumab, ácido cítrico monohidrato (0,95 mg), EDTA disódico dihidrato (0,019 mg), polisorbato 80 (0,20 mg), fosfato sódico dibásico heptahidrato (4,16 mg) y sacarosa (120 mg), con un pH de 6,3.
La inyección de NUCALA se suministra en una jeringa precargada de dosis única de 0,4 mL con una aguja fija de 29 gauge y media pulgada con protector de aguja. Cada 0,4 mL proporciona 40 mg de mepolizumab, ácido cítrico monohidrato (0,38 mg), EDTA disódico dihidrato (0,0074 mg), polisorbato 80 (0,08 mg), fosfato sódico dibásico heptahidrato (1,66 mg) y sacarosa (48 mg), con un pH de 6,3.
El autoinyector precargado y la jeringa precargada no están fabricados con látex de caucho natural.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
Mepolizumab es un antagonista de IL-5 (IgG1 kappa). IL-5 es la principal citocina responsable del crecimiento y la diferenciación, el reclutamiento, la activación y la supervivencia de los eosinófilos. Mepolizumab se une a IL-5 con una constante de disociación de 100 pM, inhibiendo la bioactividad de IL-5 al bloquear su unión a la cadena alfa del complejo receptor de IL-5 expresado en la superficie celular de los eosinófilos. La inflamación es un componente importante en la patogenia del asma, la CRSwNP, la EGPA y la HES. Múltiples tipos de células (por ejemplo, mastocitos, eosinófilos, neutrófilos, macrófagos, linfocitos) y mediadores (por ejemplo, histamina, eicosanoides, leucotrienos, citocinas) están involucrados en la inflamación. Mepolizumab, al inhibir la señalización de IL-5, reduce la producción y la supervivencia de los eosinófilos; sin embargo, el mecanismo de acción de mepolizumab en el asma, la CRSwNP, la EGPA y la HES no se ha establecido definitivamente.
12.2 Farmacodinamia
La respuesta farmacodinámica (reducción de eosinófilos en sangre) después de dosis repetidas de mepolizumab administradas por vía subcutánea o intravenosa se evaluó en sujetos adultos con asma y niveles de eosinófilos en sangre >200 células/mcL. Los sujetos recibieron 1 de 4 tratamientos con mepolizumab (administrados cada 28 días durante un total de 3 dosis): 12,5 mg subcutáneos, 125 mg subcutáneos, 250 mg subcutáneos o 75 mg IV. Sesenta y seis de los 70 sujetos aleatorizados completaron el ensayo. En comparación con los niveles basales, los eosinófilos en sangre disminuyeron de manera dependiente de la dosis. Se observó una reducción en los niveles de eosinófilos en sangre en todos los grupos de tratamiento para el día 3 (48 horas después de la dosis). En el día 84 (4 semanas después de la última dosis), la reducción media geométrica observada desde el inicio en los eosinófilos en sangre fue del 64%, 78%, 84% y 90% en los grupos de tratamiento subcutáneo de 12,5 mg, IV de 75 mg, subcutáneo de 125 mg y subcutáneo de 250 mg, respectivamente. Las dosis subcutáneas predichas por el modelo que proporcionan el 50% y el 90% de la reducción máxima de eosinófilos en sangre en el día 84 se estimaron en 11 y 99 mg, respectivamente. Estos resultados, junto con los datos de eficacia clínica del ensayo de rango de dosis de exacerbación en sujetos adultos y adolescentes con asma grave (Ensayo 1) apoyaron la evaluación de mepolizumab 75 mg IV y 100 mg subcutáneos en los ensayos confirmatorios de asma grave [ver Estudios clínicos (14.1)]. Después de la administración subcutánea de mepolizumab 100 mg cada 4 semanas durante 32 semanas en sujetos adultos y adolescentes con asma grave (Ensayo 2), los eosinófilos en sangre se redujeron a un recuento medio geométrico de 40 células/mcL, lo que corresponde a una reducción media geométrica del 84% en comparación con el placebo.
La respuesta farmacodinámica (reducción de eosinófilos en sangre) también se evaluó en niños de 6 a 11 años con asma grave. Después de la administración subcutánea de mepolizumab 40 mg cada 4 semanas durante 52 semanas, los eosinófilos en sangre se redujeron a un recuento medio geométrico de 48 células/mcL. Esto corresponde a una reducción media geométrica desde el inicio del 85%.
La magnitud de la reducción en adultos, adolescentes y niños se observó dentro de las 4 semanas de tratamiento y se mantuvo durante los períodos de tratamiento.
Para los adultos con CRSwNP, después de la administración subcutánea de mepolizumab 100 mg cada 4 semanas durante 52 semanas, los eosinófilos en sangre se redujeron a un recuento medio geométrico de 60 células/mcL. Hubo una reducción media geométrica del 83% en comparación con el placebo. Esta magnitud de reducción se observó dentro de las 4 semanas de tratamiento y se mantuvo durante el período de tratamiento [ver Estudios clínicos (14.2)].
Para los adultos con EGPA, después de la administración subcutánea de mepolizumab 300 mg cada 4 semanas durante 52 semanas, los eosinófilos en sangre se redujeron a un recuento medio geométrico de 38 células/mcL. Hubo una reducción media geométrica del 83% en comparación con el placebo, y esta magnitud de reducción se observó dentro de las 4 semanas de tratamiento [ver Estudios clínicos (14.3)].
Para los adultos y adolescentes con HES, después de la administración subcutánea de mepolizumab 300 mg cada 4 semanas durante 32 semanas, los eosinófilos en sangre se redujeron a un recuento medio geométrico de 70 células/mcL. Hubo una reducción media geométrica del 92% en comparación con el placebo [ver Estudios clínicos (14.4)].
12.3 Farmacocinética
Después de la dosificación subcutánea en sujetos adultos con asma, mepolizumab exhibió una farmacocinética aproximadamente proporcional a la dosis en un rango de dosis de 12,5 a 250 mg. Las propiedades farmacocinéticas de mepolizumab observadas en sujetos con CRSwNP (adultos), EGPA (adultos) o HES (adultos y adolescentes) fueron similares a las propiedades farmacocinéticas observadas en sujetos con asma grave (adultos y adolescentes).
La administración subcutánea de mepolizumab 300 mg tuvo aproximadamente 3 veces la exposición sistémica de mepolizumab 100 mg.
Absorción
Después de la administración subcutánea de 100 mg en la parte superior del brazo de sujetos adultos y adolescentes con asma, la biodisponibilidad de mepolizumab se estimó en aproximadamente el 80%.
Después de la administración subcutánea repetida una vez cada 4 semanas, hubo aproximadamente una acumulación de 2 veces en estado estacionario.
Distribución
El volumen central de distribución de la población de mepolizumab en sujetos adultos con asma se estima en 3,6 L para un individuo de 70 kg.
Eliminación
Después de la administración subcutánea de mepolizumab en sujetos adultos con asma, la vida media terminal media (t1/2) osciló entre 16 y 22 días. El aclaramiento sistémico aparente de la población de mepolizumab en sujetos adultos y adolescentes con asma se estima en 0,28 L/día para un individuo de 70 kg.
Metabolismo: Mepolizumab es un anticuerpo monoclonal IgG1 humanizado que se degrada por las enzimas proteolíticas ampliamente distribuidas en el cuerpo y no se limita al tejido hepático.
Poblaciones específicas
Grupos raciales y pacientes masculinos y femeninos: Los análisis de farmacocinética poblacional indicaron que no hubo un efecto significativo de la raza y el sexo en el aclaramiento de mepolizumab.
Edad: Los análisis de farmacocinética poblacional indicaron que no hubo un efecto significativo de la edad en el aclaramiento de mepolizumab.
Pacientes pediátricos: La farmacocinética de mepolizumab después de la administración subcutánea en sujetos de 6 a 11 años con asma grave se investigó en la fase inicial de tratamiento de 12 semanas de un ensayo clínico abierto. Las exposiciones (AUC) después de la administración subcutánea de 40 mg (para niños que pesan <40 kg) o 100 mg (para niños que pesan ≥40 kg) fueron 1,32 y 1,97 veces más altas, respectivamente, en comparación con las observadas en adultos y adolescentes que recibieron 100 mg. Con base en estos resultados, la simulación de una dosis subcutánea de 40 mg cada 4 semanas en niños de 6 a 11 años, independientemente del peso corporal, resultó en exposiciones predichas similares a las observadas en adultos y adolescentes.
Pacientes con insuficiencia renal: No se han realizado ensayos clínicos para investigar el efecto de la insuficiencia renal en la farmacocinética de mepolizumab. Con base en los análisis farmacocinéticos poblacionales, la depuración de mepolizumab fue comparable entre los sujetos con valores de depuración de creatinina entre 50 y 80 mL/min y los pacientes con función renal normal. Hay datos limitados disponibles en sujetos con valores de depuración de creatinina <50 mL/min; sin embargo, mepolizumab no se elimina por vía renal.
Pacientes con insuficiencia hepática: No se han realizado ensayos clínicos para investigar el efecto de la insuficiencia hepática en la farmacocinética de mepolizumab. Dado que mepolizumab se degrada por enzimas proteolíticas ampliamente distribuidas, no restringidas al tejido hepático, es poco probable que los cambios en la función hepática tengan algún efecto en la eliminación de mepolizumab.
Estudios de interacción medicamentosa
No se han realizado estudios formales de interacción medicamentosa con mepolizumab. En los análisis farmacocinéticos poblacionales de los estudios de fase 3, no hubo evidencia de un efecto de los fármacos de molécula pequeña comúnmente coadministrados en la exposición a mepolizumab.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
No se han realizado estudios a largo plazo en animales para evaluar el potencial carcinogénico de mepolizumab. La literatura publicada utilizando modelos animales sugiere que IL-5 y los eosinófilos son parte de una reacción inflamatoria temprana en el sitio de la tumorogénesis y pueden promover el rechazo del tumor. Sin embargo, otros informes indican que la infiltración de eosinófilos en los tumores puede promover el crecimiento tumoral. Por lo tanto, se desconoce el riesgo de malignidad en humanos por un anticuerpo contra IL-5 como mepolizumab.
La fertilidad masculina y femenina no se vio afectada en base a la ausencia de hallazgos histopatológicos adversos en los órganos reproductivos de monos cynomolgus que recibieron mepolizumab durante 6 meses a dosis IV de hasta 100 mg/kg una vez cada 4 semanas (aproximadamente 20 veces la MRHD de 300 mg en base al AUC). El apareamiento y el rendimiento reproductivo no se vieron afectados en ratones CD-1 machos y hembras que recibieron un anticuerpo análogo, que inhibe la actividad de la IL-5 murina, a una dosis IV de 50 mg/kg una vez por semana.
14 ESTUDIOS CLÍNICOS
14.1 Asma grave
El programa de desarrollo del asma para NUCALA en pacientes de 12 años de edad o mayores incluyó 3 ensayos doble ciego, aleatorizados y controlados con placebo: 1 ensayo de rango de dosis y exacerbación (Ensayo 1, NCT01000506) y 2 ensayos confirmatorios (Ensayo 2, NCT01691521 y Ensayo 3, NCT01691508). Mepolizumab se administró cada 4 semanas en los 3 ensayos como complemento al tratamiento de fondo. Todos los pacientes continuaron con su terapia de fondo para el asma durante toda la duración de los ensayos.
Ensayo de rango de dosis y exacerbación
El ensayo 1 fue un ensayo de rango de dosis y reducción de exacerbaciones de 52 semanas en pacientes con asma grave con antecedentes de 2 o más exacerbaciones en el año anterior a pesar del uso regular de ICS de alta dosis más controlador(es) adicional(es) con o sin OCS. Los pacientes inscritos en este ensayo debían tener al menos 1 de los siguientes 4 criterios preespecificados en los 12 meses anteriores: recuento de eosinófilos en sangre ≥300 células/mcL, recuento de eosinófilos en esputo ≥3%, concentración de óxido nítrico exhalado ≥50 ppb, o deterioro del control del asma después de una reducción ≤25% en el ICS/OCS de mantenimiento regular. Se evaluaron tres dosis IV de mepolizumab (75, 250 y 750 mg) administradas una vez cada 4 semanas en comparación con placebo. Los resultados de este ensayo y el estudio farmacodinámico apoyaron la evaluación de mepolizumab 75 mg IV y 100 mg subcutáneo en los ensayos posteriores [ver Farmacología clínica (12.2)]. NUCALA no está indicado para uso IV y solo debe administrarse por vía subcutánea.
Ensayos confirmatorios
Se estudió un total de 711 pacientes con asma grave en los 2 ensayos confirmatorios (Ensayos 2 y 3). En estos 2 ensayos, los pacientes debían tener eosinófilos en sangre de ≥150 células/mcL en la selección (dentro de las 6 semanas de la dosificación) o eosinófilos en sangre de ≥300 células/mcL dentro de los 12 meses de la inscripción. El criterio de eosinófilos en sangre de ≥150 células/mcL en la selección se derivó de análisis exploratorios de datos del ensayo 1. El ensayo 2 fue un ensayo controlado con placebo y activo de 32 semanas en pacientes con asma grave con antecedentes de 2 o más exacerbaciones en el año anterior a pesar del uso regular de ICS de alta dosis más controlador(es) adicional(es) con o sin OCS. Los pacientes recibieron mepolizumab 75 mg IV (n = 191), NUCALA 100 mg (n = 194) o placebo (n = 191) una vez cada 4 semanas durante 32 semanas.
El ensayo 3 fue un ensayo de reducción de OCS de 24 semanas en pacientes con asma grave que requirieron OCS diarios además del uso regular de ICS de alta dosis más controlador(es) adicional(es) para mantener el control del asma. Los pacientes del ensayo 3 no estaban obligados a tener antecedentes de exacerbaciones en el año anterior. Los pacientes recibieron NUCALA 100 mg (n = 69) o placebo (n = 66) una vez cada 4 semanas durante 24 semanas. El uso medio de OCS al inicio fue similar en los 2 grupos de tratamiento: 13,2 mg en el grupo placebo y 12,4 mg en el grupo que recibió NUCALA 100 mg.
La demografía y las características al inicio de estos 3 ensayos se proporcionan en Tabla 3.
| FEV1 = volumen espiratorio forzado en 1 segundo, SABA = agonista beta2 de acción corta, FVC = capacidad vital forzada. | |||
|
Ensayo 1 (N = 616) |
Ensayo 2 (N = 576) |
Ensayo 3 (N = 135) |
|
|
Edad media, años |
49 |
50 |
50 |
|
Mujer, n (%) |
387 (63) |
328 (57) |
74 (55) |
|
Blanca, n (%) |
554 (90) |
450 (78) |
128 (95) |
|
Duración del asma, años, media |
19 |
20 |
19 |
|
Nunca fumó, n (%) |
483 (78) |
417 (72) |
82 (61) |
|
FEV1 al inicio, L |
1.88 |
1.82 |
1.95 |
|
% predicho de FEV1 al inicio |
60 |
61 |
59 |
|
Reversibilidad basal % |
25 |
27 |
26 |
|
FEV1/FVC post-SABA basal |
0.67 |
0.66 |
0.66 |
|
Media geométrica del recuento de eosinófilos en la línea de base, células/mcL |
250 |
290 |
240 |
|
Número medio de exacerbaciones en el año anterior |
3.6 |
3.6 |
3.1 |
Exacerbaciones
La eficacia se evaluó en los ensayos 1 y 2 utilizando un punto final de la frecuencia de exacerbaciones definido como empeoramiento del asma que requirió el uso de corticosteroides orales/sistémicos y/o hospitalización y/o visitas al departamento de emergencias. Para los pacientes que recibían OCS de mantenimiento, una exacerbación que requirió OCS se definió como el uso de corticosteroides orales/sistémicos al menos el doble de la dosis existente durante al menos 3 días. En comparación con el placebo, los pacientes que recibieron NUCALA 100 mg o mepolizumab 75 mg IV experimentaron significativamente menos exacerbaciones. Además, en comparación con el placebo, hubo menos exacerbaciones que requirieron hospitalización y/o visitas al departamento de emergencias y exacerbaciones que requirieron solo hospitalización en el paciente con NUCALA 100 mg (Tabla 4).
| IV = intravenoso, SC = subcutáneo. | ||||||||
|
Ensayo |
Tratamiento |
Exacerbaciones por año |
||||||
|
Tasa |
Diferencia |
Razón de tasas (IC del 95%) |
||||||
|
Todas las exacerbaciones |
||||||||
|
Ensayo 1 |
Placebo (n = 155) |
2.40 |
||||||
|
Mepolizumab 75 mg IV (n = 153) |
1.24 |
1.16 |
0.52 (0.39, 0.69) |
|||||
|
Ensayo 2 |
Placebo (n = 191) |
1.74 |
||||||
|
Mepolizumab 75 mg IV (n = 191) |
0.93 |
0.81 |
0.53 (0.40, 0.72) |
|||||
|
NUCALA 100 mg SC (n = 194) |
0.83 |
0.91 |
0.47 (0.35, 0.64) |
|||||
|
Exacerbaciones que requieren hospitalización/visita a la sala de emergencias |
||||||||
|
Ensayo 1 |
Placebo (n = 155) |
0.43 |
||||||
|
Mepolizumab 75 mg IV (n = 153) |
0.17 |
0.26 |
0.40 (0.19, 0.81) |
|||||
|
Ensayo 2 |
Placebo (n = 191) |
0.20 |
||||||
|
Mepolizumab 75 mg IV (n = 191) |
0.14 |
0.06 |
0.68 (0.33, 1.41) |
|||||
|
NUCALA 100 mg SC (n = 194) |
0.08 |
0.12 |
0.39 (0.18, 0.83) |
|||||
|
Exacerbaciones que requieren hospitalización |
||||||||
|
Ensayo 1 |
Placebo (n = 155) |
0.18 |
||||||
|
Mepolizumab 75 mg IV (n = 153) |
0.11 |
0.07 |
0.61 (0.28, 1.33) |
|||||
|
Trial 2 |
Placebo (n = 191) |
0.10 |
||||||
|
Mepolizumab 75 mg IV (n = 191) |
0.06 |
0.04 |
0.61 (0.23, 1.66) |
|||||
|
NUCALA 100 mg SC (n = 194) |
0.03 |
0.07 |
0.31 (0.11, 0.91) |
|||||
El tiempo hasta la primera exacerbación fue más largo para los grupos que recibieron NUCALA 100 mg y mepolizumab 75 mg IV en comparación con placebo en el ensayo 2 (Figura 1).
Figura 1. Curva de incidencia acumulativa de Kaplan-Meier para el tiempo hasta la primera exacerbación (Ensayo 2 de asma grave)
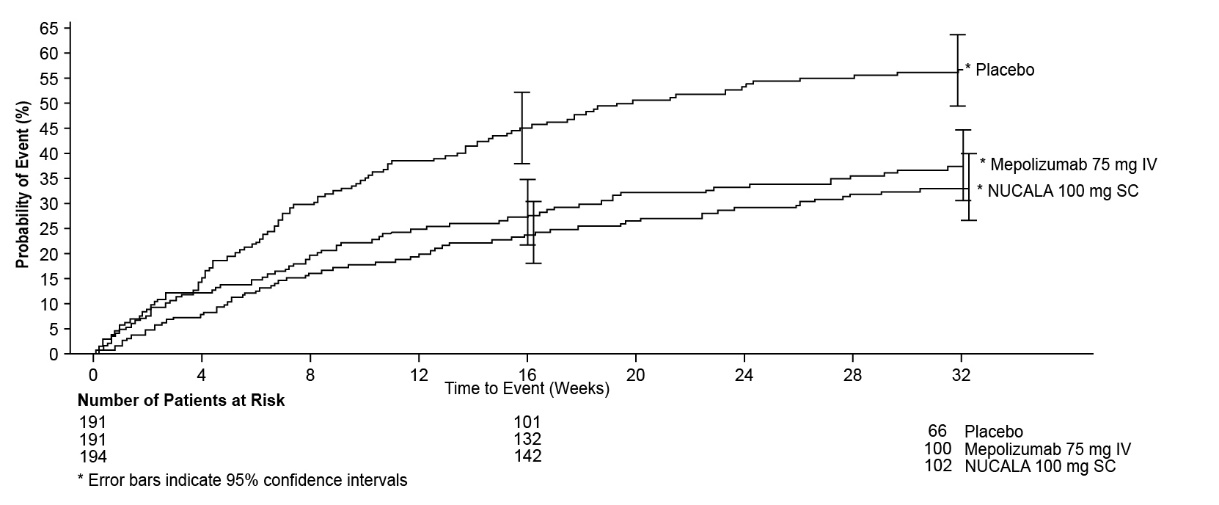
IV = intravenoso, SC = subcutáneo.
Los datos del ensayo 1 se exploraron para determinar los criterios que podrían identificar a los pacientes que probablemente se beneficiarían del tratamiento con NUCALA. El análisis exploratorio sugirió que el recuento de eosinófilos en sangre basal de ≥150 células/mcL era un predictor potencial del beneficio del tratamiento. El análisis exploratorio de los datos del ensayo 2 también sugirió que el recuento de eosinófilos en sangre basal (obtenido dentro de las 6 semanas posteriores al inicio de la dosificación) de ≥150 células/mcL era un predictor potencial de eficacia y mostró una tendencia de mayor beneficio en la exacerbación con el aumento del recuento de eosinófilos en sangre. En el ensayo 2, los pacientes inscritos únicamente sobre la base del recuento histórico de eosinófilos en sangre de ≥300 células/mcL en los 12 meses anteriores, pero que tenían un recuento de eosinófilos en sangre basal <150 células/mcL, prácticamente no tuvieron ningún beneficio en la exacerbación después del tratamiento con NUCALA 100 mg en comparación con placebo.
El Cuestionario de control del asma-5 (ACQ-5) se evaluó en los ensayos 1 y 2, y el Cuestionario respiratorio de St. George (SGRQ) se evaluó en el ensayo 2. En el ensayo 1, la tasa de respuesta del ACQ-5 (definida como una disminución en la puntuación de 0,5 o más como umbral) para el brazo de mepolizumab IV de 75 mg fue del 47% en comparación con el 50% para placebo con una razón de posibilidades (OR) de 1,1 (IC del 95%: 0,7, 1,7). En el ensayo 2, la tasa de respuesta del ACQ-5 para el brazo de tratamiento para NUCALA 100 mg fue del 57% en comparación con el 45% para placebo con una OR de 1,8 (IC del 95%: 1,2, 2,8). En el ensayo 2, la tasa de respuesta del SGRQ (definida como una disminución en la puntuación de 4 o más como umbral) para el brazo de tratamiento para NUCALA 100 mg fue del 71% en comparación con el 55% para placebo con una OR de 2,1 (IC del 95%: 1,3, 3,2).
Reducción de corticosteroides orales
El ensayo 3 evaluó el efecto de NUCALA 100 mg en la reducción del uso de OCS de mantenimiento. La eficacia se evaluó utilizando un punto final del porcentaje de reducción de la dosis de OCS durante las semanas 20 a 24 en comparación con la dosis basal, mientras se mantenía el control del asma. Los pacientes se clasificaron de acuerdo con su cambio en el uso de OCS durante el ensayo con las siguientes categorías: disminución del 90% al 100%, disminución del 75% a <90%, disminución del 50% a <75%, >0% a <50% de disminución y sin mejora (es decir, sin cambio o cualquier aumento o falta de control del asma o retiro del tratamiento). En comparación con placebo, los pacientes que recibieron NUCALA 100 mg lograron mayores reducciones en la dosis diaria de mantenimiento de OCS, mientras mantenían el control del asma. Dieciséis (23%) pacientes en el grupo que recibió NUCALA 100 mg frente a 7 (11%) en el grupo placebo tuvieron una reducción del 90% al 100% en su dosis de OCS. Veinticinco (36%) pacientes en el grupo que recibió NUCALA 100 mg frente a 37 (56%) en el grupo placebo se clasificaron como sin mejora para la dosis de OCS. Además, el 54% de los pacientes que recibieron NUCALA 100 mg lograron al menos una reducción del 50% en la dosis diaria de prednisona en comparación con el 33% de los pacientes que recibieron placebo (IC del 95% para la diferencia: 4%, 37%). También se realizó un análisis exploratorio en el subgrupo de 29 pacientes en el ensayo 3 que tenían un recuento promedio de eosinófilos en sangre basal y de detección <150 células/mcL. Cinco (29%) pacientes en el grupo que recibió NUCALA 100 mg frente a 0 (0%) en el grupo placebo tuvieron una reducción del 90% al 100% en su dosis. Cuatro (24%) pacientes en el grupo que recibió NUCALA 100 mg frente a 8 (67%) en el grupo placebo se clasificaron como sin mejora para la dosis de OCS. El ACQ y el SGRQ también se evaluaron en el ensayo 3 y mostraron resultados similares a los del ensayo 2.
Función pulmonar
El cambio desde el inicio en el volumen espiratorio forzado en 1 segundo (FEV1) medio se midió en los 3 ensayos y se presenta en Tabla 5. En comparación con placebo, NUCALA 100 mg no proporcionó mejoras consistentes en el cambio medio desde el inicio en FEV1.
| FEV1 = volumen espiratorio forzado en 1 segundo. a Dosis = 75 mg intravenoso. b Volumen espiratorio forzado en 1 segundo (FEV1) en la semana 52. c Dosis = 100 mg subcutáneo. d FEV1 en la semana 32. |
||||||||||||
|
Ensayo |
Diferencia de placebo en el cambio medio desde |
|||||||||||
|
Semana 12 |
Semana 24 |
Semanas 32/52 |
||||||||||
|
1a |
10 (-87, 108) |
5 (-98, 108) |
61 (-39, 161)b |
|||||||||
|
2c |
52 (-30, 134) |
76 (-6, 159) |
98 (11, 184)d |
|||||||||
|
3c |
56 (-91, 203) |
114 (-42, 271) |
NA |
|||||||||
El efecto de mepolizumab en la función pulmonar también se estudió en un ensayo controlado con placebo de 12 semanas que incluyó pacientes con asma que recibían una dosis moderada de ICS con evidencia de síntomas y deterioro de la función pulmonar. La inscripción no dependió de un historial de exacerbaciones o un recuento de eosinófilos preespecificado. El cambio desde el inicio en FEV1 en la semana 12 fue numéricamente menor en los grupos de tratamiento con mepolizumab que en el grupo placebo.
14.2 Sinusitis crónica con pólipos nasales
Se evaluó un total de 407 pacientes adultos con CRSwNP en un ensayo aleatorizado, doble ciego, controlado con placebo, multicéntrico de 52 semanas (NCT03085797). Los pacientes recibieron NUCALA 100 mg o placebo administrado por vía subcutánea una vez cada 4 semanas mientras continuaban con la terapia con corticosteroides nasales. Los pacientes deben haber recibido corticosteroides nasales de fondo durante ≥8 semanas antes de la selección. Los pacientes tenían CRSwNP recurrente y sintomático, y se habían sometido a al menos 1 cirugía para la extirpación de pólipos nasales en los 10 años anteriores. Se requirió que los pacientes tuvieran síntomas de obstrucción nasal con una puntuación de la escala analógica visual (VAS) de >5 de una puntuación máxima de 10. También se requirió que los pacientes tuvieran una puntuación de pólipo nasal endoscópico bilateral (NPS) de ≥5 de 8 con NPS ≥2 en cada cavidad nasal. Los pacientes informaron las puntuaciones de VAS de obstrucción nasal diariamente colocando una sola marca en una línea continua etiquetada de 0 (ninguna) a 100 (tan mal como puedas imaginar). La distancia a lo largo de la línea se convirtió a una escala de 0 a 10 puntos para la puntuación. Para NPS, los pólipos en cada lado de la nariz se calificaron en una escala categórica (0 = sin pólipos, 1 = pólipos pequeños en el meato medio que no llegan por debajo del borde inferior de la concha media, 2 = pólipos que llegan por debajo del borde inferior del cornete medio, 3 = pólipos grandes que llegan al borde inferior del cornete inferior o pólipos mediales a la concha media, 4 = pólipos grandes que causan casi una congestión/obstrucción completa del meato inferior) para una puntuación total de 0 a 8. No se realizaron tomografías computarizadas de los senos paranasales al inicio ni durante el tratamiento para evaluar la opacificación de los senos paranasales.
Los criterios de valoración coprimarios fueron el cambio desde el inicio hasta la semana 52 en el NPS endoscópico total (escala de 0 a 8) según lo evaluado por evaluadores ciegos independientes y el cambio desde el inicio en la puntuación de VAS de obstrucción nasal (escala de 0 a 10) durante las semanas 49 a 52. El criterio de valoración secundario clave fue el tiempo hasta la primera cirugía nasal (polipectonomía nasal) hasta la semana 52 en este ensayo. Otros criterios de valoración secundarios fueron el cambio desde el inicio en la puntuación de VAS de pérdida del olfato durante las semanas 49 a 52, y la proporción de pacientes que requirieron esteroides sistémicos para los pólipos nasales hasta la semana 52. Todos los puntajes de VAS fueron recopilados diariamente por los pacientes e informados en una escala de 0 a 10 (0 = ninguno, 10 = tan mal como puedas imaginar).
La demografía y las características basales de los pacientes en este ensayo se proporcionan en Tabla 6.
| CRSwNP = Sinusitis crónica con pólipos nasales, SD = desviación estándar, OCS = corticosteroide oral, NPS = puntuación de pólipo nasal, VAS = escala analógica visual, AERD = enfermedad respiratoria exacerbada por la aspirina. a Según lo evaluado por evaluadores ciegos independientes. |
|
|
N = 407 |
|
|
Edad media, años |
49 |
|
Mujer, n (%) |
143 (35) |
|
Blanco, n (%) |
379 (93) |
|
Duración media de CRSwNP en años (SD) |
11.4 (8.4) |
|
Pacientes con ≥1 cirugía en los últimos 10 años (%) |
407 (100) |
|
Pacientes con ≥3 cirugías en los últimos 10 años (%) |
124 (30) |
|
Uso de OCS (≥1 curso) en los últimos 12 meses, n (%) |
197 (48) |
|
NPS endoscópico bilateral medioa, (SD), rango 0-8 |
5.5 (1.29) |
|
Puntuación media de VAS de obstrucción nasal, (SD), rango 0-10 |
9.0 (0.83) |
|
Media geométrica de células eosinófilas sanguíneas/mcL (IC del 95%) |
390 (360, 420) |
|
Asma, n (%) |
289 (71) |
|
AERD, n (%) |
108 (27) |
Puntuación endoscópica del pólipo nasal y puntuaciones de la escala visual analógica de obstrucción nasal
Los pacientes que recibieron NUCALA 100 mg tuvieron una mejora estadísticamente significativa (disminución) en la NPS bilateral en la semana 52 y en la puntuación de la escala VAS de obstrucción nasal de las semanas 49 a 52 al final del período de tratamiento de 52 semanas (Tabla 7).
| CRSwNP = Rinosinusitis crónica con pólipos nasales, SD = desviación estándar, SE = error estándar, NPS = puntuación del pólipo nasal en la semana 52. a Los pacientes con cirugía nasal fueron asignados a la peor puntuación posible para el período posterior a la cirugía nasal. Los datos faltantes se imputaron en función de los datos disponibles fuera del tratamiento en todos los brazos de tratamiento. Las imputaciones se realizaron paso a paso por visita y se condicionaron a los datos de las visitas anteriores con las mismas covariables utilizadas en el modelo de análisis. b Medias de mínimos cuadrados del análisis utilizando medidas repetidas de modelos mixtos con covariables de grupo de tratamiento, región geográfica, puntuación basal y log(e) conteo de eosinófilos en sangre basal, visita, términos de interacción para visita por basal y visita por tratamiento. |
|||||||
|
Puntuacionesa (rango) |
Placebo n = 201 |
NUCALA 100 mg n = 206 |
Diferencia media vs. Placebo |
||||
|
Media basal (SD) |
Cambio mediob (SE) |
Media basal (SD) |
Cambio mediob (SE) |
||||
|
NPS (0-8) |
5.6 (1.41) |
0.06 (0.14) |
5.4 (1.17) |
-0.87 (0.14) |
-0.93 (-1.31, -0.55) |
||
|
Obstrucción nasal (0-10) |
9.02 (0.83) |
-2.54 (0.25) |
8.92 (0.83) |
-4.40 (0.25) |
-1.86 (-2.53, -1.19) |
||
Polipectómia nasal
El parámetro secundario clave fue el tiempo hasta la primera cirugía nasal (polipectómia nasal) hasta la semana 52. La proporción de pacientes que se sometieron a cirugía se redujo significativamente en un 57% (razón de riesgo: 0,43, IC del 95%: 0,25, 0,76) en el grupo tratado con NUCALA 100 mg en comparación con el grupo placebo (Figura 2). En la semana 52, 18 (9%) pacientes que recibieron NUCALA 100 mg se sometieron a cirugía en comparación con 46 (23%) pacientes en el grupo placebo.
Figura 2. Gráfico de Kaplan-Meier del tiempo hasta la primera cirugía nasal en CRSwNP
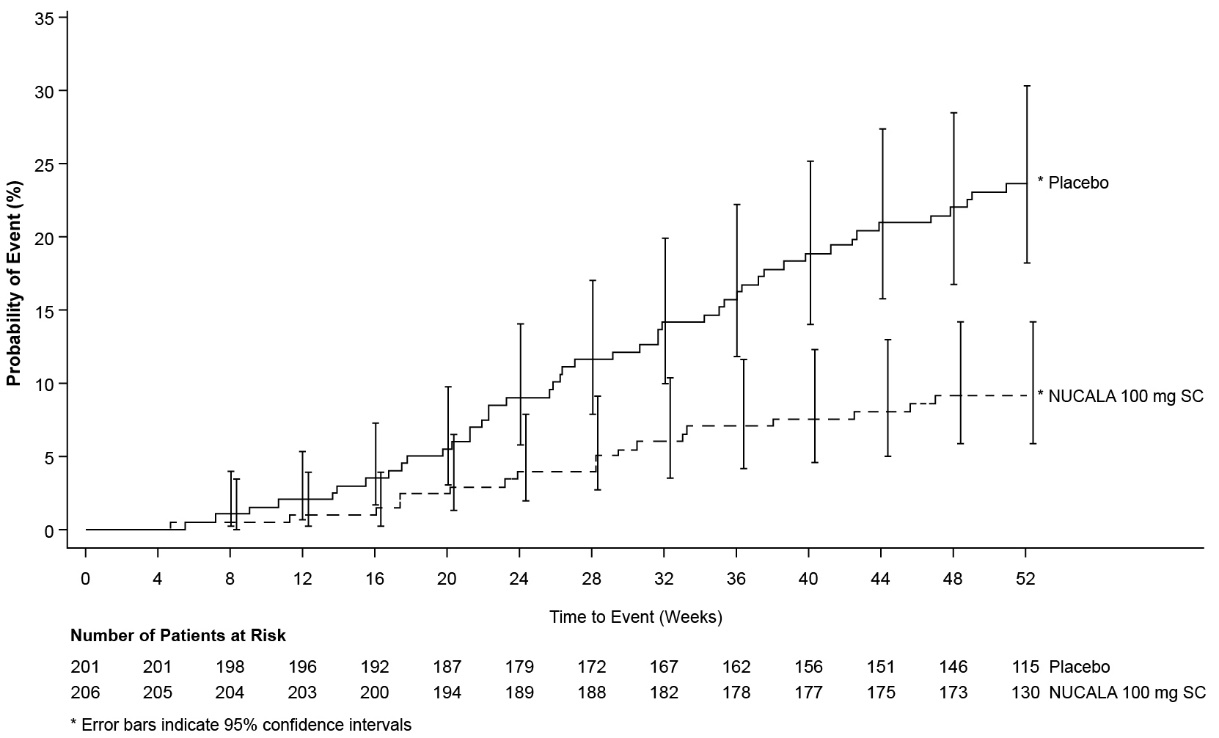
CRSwNP = Rinosinusitis crónica con pólipos nasales, SC = subcutánea.
Puntuaciones adicionales de los síntomas de CRSwNP
Para los pacientes que recibieron NUCALA 100 mg, se observó una mejora estadísticamente significativa en la pérdida del olfato en comparación con el placebo y también se observaron mejoras en las puntuaciones individuales de los síntomas de la EVA en comparación con los pacientes del grupo placebo en las 4 semanas previas al final del período de tratamiento de 52 semanas (Tabla 8).
| EVA = escala analógica visual; DE = desviación estándar; EE = error estándar. a Los pacientes con cirugía nasal recibieron la peor puntuación posible para el período posterior a la cirugía nasal. Los datos faltantes se imputaron en función de los datos disponibles fuera del tratamiento en todos los brazos de tratamiento. Las imputaciones se realizaron paso a paso por visita y se condicionaron a los datos de las visitas anteriores con las mismas covariables utilizadas en el modelo de análisis. b Medias de mínimos cuadrados de un análisis utilizando medidas repetidas de modelos mixtos con covariables de grupo de tratamiento, región geográfica, puntuación basal y recuento de eosinófilos en sangre basal log(e) visita, términos de interacción para visita por basal y visita por tratamiento. c Este parámetro no se preespecificó en el plan de análisis para ajustar la multiplicidad. |
|||||||||||||
|
Puntuaciones de la EVAa (rango) |
Placebo n = 201 |
NUCALA 100 mg n = 206 |
Diferencia media frente a placebo (IC del 95%) |
||||||||||
|
Media basal (DE) |
Cambio mediob (EE) |
Media basal (DE) |
Cambio mediob (EE) |
||||||||||
|
Pérdida del olfato (0-10) |
9,68 (0,60) |
-1,46 (0,24) |
9,63 (0,83) |
-2,92 (0,24) |
-1,46 (-2,11, -0,81) |
||||||||
|
Secreción nasalc (0-10) |
8,78 (1,25) |
-2,49 (0,26) |
8,78 (1,07) |
-4,38 (0,25) |
-1,89 (-2,58, -1,20) |
||||||||
|
Moco en la gargantac (0-10) |
8,58 (1,63) |
-2,37 (0,26) |
8,51 (1,61) |
-4,07 (0,26) |
-1,70 (-2,41, -0,99) |
||||||||
|
Dolor facialc (0-10) |
7,77 (2,72) |
-2,04 (0,28) |
7,76 (2,51) |
-3,73 (0,27) |
-1,69 (-2,43, -0,95) |
||||||||
Reducción de corticosteroides
El tratamiento con NUCALA 100 mg redujo significativamente la necesidad de esteroides sistémicos para pólipos nasales en comparación con placebo hasta la semana 52 (odds ratio: 0,58, IC del 95%: 0,36, 0,92). En los pacientes que recibieron NUCALA 100 mg, 52 (25%) requirieron ≥1 ciclo de esteroides sistémicos en comparación con 74 (37%) en el grupo placebo durante el período de tratamiento de 52 semanas.
Resultados en pacientes con asma concomitante
En 289 (71%) pacientes con asma concomitante, los análisis preespecificados mostraron mejoras en los criterios de valoración coprimarios consistentes con los observados en la población general en los pacientes que recibieron NUCALA 100 mg en comparación con placebo. Además, según un análisis post hoc en estos pacientes, hubo una mayor respuesta desde el inicio en la semana 52 en el control del asma medido por el ACQ‑5 para NUCALA 100 mg en comparación con placebo (el 57% de los pacientes con NUCALA cumplieron con el umbral de respuesta de reducción ≥0,5, en comparación con el 35% en el grupo placebo, con una odds ratio de 2,42 [IC del 95% 1,43, 4,11]).
14.3 Granulomatosis eosinofílica con poliangiitis
Se evaluó un total de 136 pacientes adultos con EGPA en un ensayo aleatorizado, controlado con placebo, multicéntrico de 52 semanas (NCT02020889). Los pacientes recibieron 300 mg de NUCALA o placebo administrados por vía subcutánea una vez cada 4 semanas mientras continuaban con su terapia estable con OCS. A partir de la semana 4, los OCS se redujeron gradualmente durante el período de tratamiento a discreción del investigador. La eficacia se evaluó en este ensayo utilizando criterios de valoración coprimarios de la duración total acumulada de la remisión durante el período de tratamiento de 52 semanas, definida como Puntuación de Actividad de Vasculitis de Birmingham (BVAS) = 0 (sin vasculitis activa) más dosis de prednisolona o prednisona menor o igual a 4 mg/día, y la proporción de pacientes en remisión tanto en la semana 36 como en la semana 48 de tratamiento. La BVAS es una herramienta completada por el clínico para evaluar la vasculitis clínicamente activa que probablemente requeriría tratamiento, después de excluir otras causas.
Las características demográficas y de referencia de los pacientes en este ensayo se proporcionan en Tabla 9.
| EGPA = Granulomatosis eosinofílica con poliangiitis, SD = desviación estándar. a Equivalente a prednisona o prednisolona. b p. ej., azatioprina, metotrexato, ácido micofenólico. |
|
|
N = 136 |
|
|
Edad media, años |
48,5 |
|
Mujer, n (%) |
80 (59) |
|
Blanca, n (%) |
125 (92) |
|
Duración de la EGPA, años, media (SD) |
5,5 (4,63) |
|
Historia de >1 recaída confirmada en los últimos 2 años, n (%) |
100 (74) |
|
Enfermedad refractaria, n (%) |
74 (54) |
|
Recurrencia de los síntomas de EGPA, n (%) |
68 (50) |
|
Tratamiento de inducción fallido, n (%) |
6 (4) |
|
Dosis diaria de corticosteroides oralesa de referencia, mg, mediana (rango) |
12 (7,5-50) |
|
Recibiendo terapia inmunosupresorab, n (%) |
72 (53) |
Remisión
Los pacientes que recibieron 300 mg de NUCALA lograron un tiempo acumulado significativamente mayor en remisión en comparación con el placebo. Una proporción significativamente mayor de pacientes que recibieron 300 mg de NUCALA lograron la remisión tanto en la semana 36 como en la semana 48 en comparación con el placebo (Tabla 10). Los resultados de los componentes de la remisión también se muestran en Tabla 10. Además, significativamente más pacientes que recibieron 300 mg de NUCALA lograron la remisión dentro de las primeras 24 semanas y permanecieron en remisión durante el resto del período de tratamiento del ensayo de 52 semanas en comparación con el placebo (19% para 300 mg de NUCALA versus 1% para placebo; OR 19.7; 95% CI: 2.3, 167.9).
| EGPA = Granulomatosis eosinofílica con poliangiitis, OCS = corticosteroide oral, BVAS = Puntuación de actividad de vasculitis de Birmingham. a Una razón de posibilidades >1 favorece a NUCALA. |
||||||||
|
Remisión (OCS ≤4 mg/día + BVAS = 0) |
OCS ≤4 mg/día |
BVAS = 0 |
||||||
|
Placebo n = 68 |
NUCALA 300 mg n = 68 |
Placebo n = 68 |
NUCALA 300 mg n = 68 |
Placebo n = 68 |
NUCALA 300 mg n = 68 |
|||
|
Duración acumulada durante 52 semanas, n (%) |
||||||||
|
0 |
55 (81) |
32 (47) |
46 (68) |
27 (40) |
6 (9) |
3 (4) |
||
|
>0 a <12 semanas |
8 (12) |
8 (12) |
12 (18) |
5 (7) |
15 (22) |
13 (19) |
||
|
12 a <24 semanas |
3 (4) |
9 (13) |
6 (9) |
12 (18) |
11 (16) |
5 (7) |
||
|
24 a <36 semanas |
0 |
10 (15) |
2 (3) |
10 (15) |
17 (25) |
2 (3) |
||
|
≥36 semanas |
2 (3) |
9 (13) |
2 (3) |
14 (21) |
19 (28) |
45 (66) |
||
|
Razón de posibilidades (NUCALA/placebo)a |
5.9 |
5.1 |
3.7 |
|||||
|
(95% CI) |
(2.7, 13.0) |
(2.5, 10.4) |
(1.8, 7.6) |
|||||
|
Proporción de pacientes en las semanas 36 y 48 |
||||||||
|
Pacientes, n (%) |
2 (3) |
22 (32) |
7 (10) |
28 (41) |
23 (34) |
34 (50) |
||
|
Odds ratio (NUCALA/placebo)a |
16.7 |
6.6 |
1.9 |
|||||
|
(95% CI) |
(3.6, 77.6) |
(2.6, 17.1) |
(0.9, 4.2) |
|||||
Además, se demostró un beneficio estadísticamente significativo para estos criterios de valoración utilizando la remisión definida como BVAS = 0 más prednisolona/prednisona ≤7.5 mg/día.
Recaída
El tiempo hasta la primera recaída (definida como empeoramiento relacionado con vasculitis, asma o síntomas sinonasales que requieren un aumento de la dosis de corticosteroides o terapia inmunosupresora u hospitalización) fue significativamente más largo para los pacientes que recibieron 300 mg de NUCALA en comparación con el placebo con una razón de riesgo de 0.32 (95% CI: 0.21, 0.5) (Figura 3). Además, los pacientes que recibieron 300 mg de NUCALA tuvieron una reducción en la tasa de recaída en comparación con los pacientes que recibieron placebo (razón de tasa 0.50; 95% CI: 0.36, 0.70 para 300 mg de NUCALA en comparación con el placebo). La incidencia y el número de tipos de recaída (vasculitis, asma, sinonasal) fueron numéricamente más bajos con NUCALA en comparación con el placebo.
Figura 3. Gráfico de Kaplan-Meier del tiempo hasta la primera recaída en EGPA
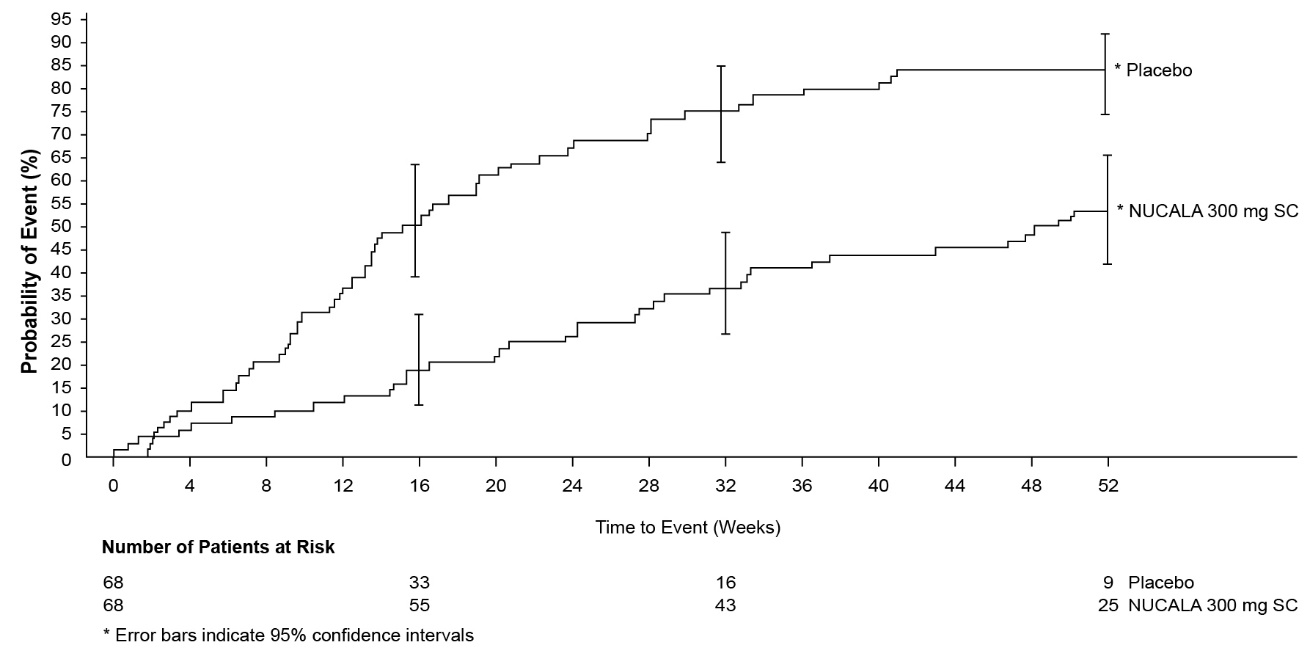
EGPA = Granulomatosis eosinofílica con poliangitis, SC = subcutánea.
Reducción de corticosteroides
Los pacientes que recibieron 300 mg de NUCALA tuvieron una reducción significativamente mayor en la dosis diaria promedio de OCS en comparación con los pacientes que recibieron placebo durante las semanas 48 a 52 (Tabla 11).
| EGPA = Granulomatosis eosinofílica con poliangitis. a Analizado utilizando un modelo de probabilidades proporcionales con covariables de grupo de tratamiento, dosis diaria de corticosteroides orales de referencia, puntuación de actividad de vasculitis de Birmingham de referencia y región. b Una razón de posibilidades <1 favorece a NUCALA. |
|||||||||||
|
Número (%) de pacientes |
|||||||||||
|
Placebo n = 68 |
NUCALA 300 mg n = 68 |
||||||||||
|
0 |
2 (3) |
12 (18) |
|||||||||
|
>0 to ≤4.0 mg |
3 (4) |
18 (26) |
|||||||||
|
>4.0 to ≤7.5 mg |
18 (26) |
10 (15) |
|||||||||
|
>7.5 mg |
45 (66) |
28 (41) |
|||||||||
|
Comparación: NUCALA/placeboa |
|||||||||||
|
Odds ratiob |
0.20 |
||||||||||
|
95% CI |
0.09, 0.41 |
||||||||||
Cuestionario de Control del Asma-6 (ACQ-6)
El ACQ-6, un cuestionario de 6 ítems completado por el paciente, fue desarrollado para medir la adecuación del control del asma y el cambio en el control del asma. La tasa de respuesta del ACQ-6 en tratamiento durante las semanas 48 a 52 (definida como una disminución en la puntuación de 0,5 o más en comparación con la línea de base) fue del 22% para 300 mg de NUCALA y del 16% para placebo (OR 1,56; IC del 95%: 0,63, 3,88 para 300 mg de NUCALA en comparación con placebo).
14.4 Síndrome Hipereosinofílico
Se evaluó un total de 108 pacientes adultos y adolescentes de 12 años o más con SHE durante al menos 6 meses en un ensayo aleatorizado, doble ciego, controlado con placebo, multicéntrico de 32 semanas (NCT02836496). Los pacientes con SHE secundaria no hematológica (p. ej., hipersensibilidad a medicamentos, infección por helmintos parásitos, infección por VIH, neoplasia maligna no hematológica) o SHE positiva para la quinasa FIP1L1-PDGFRα fueron excluidos del ensayo. Los pacientes recibieron 300 mg de NUCALA o placebo por vía subcutánea una vez cada 4 semanas mientras continuaban con su terapia estable para la SHE. Los pacientes que ingresaron al ensayo habían experimentado al menos 2 brotes de SHE en los últimos 12 meses y un recuento de eosinófilos en sangre de 1000 células/mcL o más durante la selección. Los brotes históricos de SHE para los criterios de inclusión en el ensayo se definieron como un empeoramiento de los síntomas clínicos relacionados con la SHE o un aumento del recuento de eosinófilos en sangre que requirió una escalada en la terapia. Los pacientes deben haber estado en terapia estable para la SHE durante las 4 semanas previas a la aleatorización. La terapia para la SHE podría incluir OCS crónicas o episódicas, terapia inmunosupresora o citotóxica.
La eficacia de NUCALA en la SHE se estableció en función de la proporción de pacientes que experimentaron un brote de SHE durante el período de tratamiento de 32 semanas. Un brote de SHE se definió como un empeoramiento de los signos y síntomas clínicos de la SHE o un aumento de los eosinófilos (en al menos 2 ocasiones), lo que resultó en la necesidad de aumentar los OCS o aumentar/agregar terapia citotóxica o inmunosupresora para la SHE.
Las características demográficas y de línea de base de los pacientes en este ensayo se proporcionan en Tabla 12.
| SHE = Síndrome Hipereosinofílico, DE = desviación estándar. | |
|
N = 108 |
|
|
Edad media, años (DE) |
46,0 (15,78) |
|
Mujer, n (%) |
57 (53) |
|
Blanca, n (%) |
100 (93) |
|
Duración media de la SHE, años |
5,55 |
Brotes
El ensayo comparó la proporción de pacientes que experimentaron un brote de SHE o se retiraron del ensayo en los grupos de tratamiento con NUCALA y placebo (Tabla 13). Durante el período de tratamiento de 32 semanas, la incidencia de brotes de SHE durante el período de tratamiento fue del 56% para el grupo placebo y del 28% para el grupo tratado con NUCALA (reducción del 50%).
| SHE = Síndrome Hipereosinofílico, CMH = Cochran-Mantel-Haenszel. a El análisis comparó el número de pacientes que experimentaron ≥1 brote de SHE y/o se retiraron del ensayo prematuramente. b Una razón de posibilidades <1 favorece a NUCALA. |
||||
|
Número (%) de pacientes |
||||
|
Placebo n = 54 |
NUCALA 300 mg n = 54 |
|||
|
Pacientes con ≥1 brote de SHE o que se retiraron del ensayo |
30 (56) |
15 (28) |
||
|
Pacientes con ≥1 brote de SHE |
28 (52) |
14 (26) |
||
|
Pacientes sin brote de SHE que se retiraron del ensayo |
2 (4) |
1 (2) |
||
|
Comparación: NUCALA/placeboa |
||||
|
Valor P de CMH |
0,002 |
|||
|
Razón de posibilidadesb |
0,28 |
|||
|
IC del 95% |
(0,12, 0,64) |
|||
Tiempo hasta la primera exacerbación
Se observó una diferencia entre los brazos de NUCALA y placebo en el tiempo hasta la primera exacerbación de HES (Figura 4). El riesgo de primera exacerbación de HES durante el período de tratamiento fue un 66% menor para los pacientes tratados con NUCALA en comparación con placebo (razón de riesgo: 0.34; IC del 95%: 0.18, 0.67, P = 0.002).
Figura 4. Curva de Kaplan-Meier para el tiempo hasta la primera exacerbación de HES
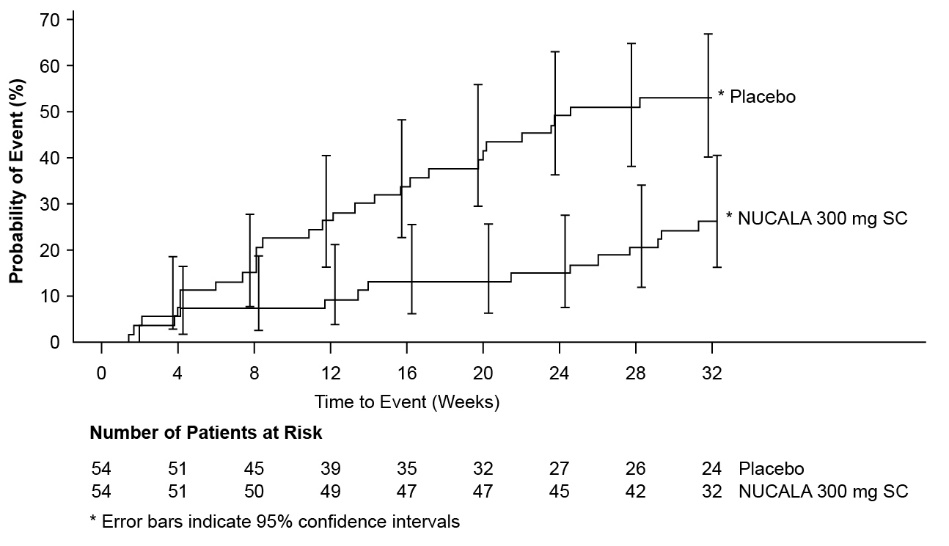
HES = Síndrome hipereosinofílico, SC = subcutáneo.
Proporción de pacientes que experimentaron exacerbaciones durante la semana 20 hasta la semana 32
Desde la semana 20 hasta la semana 32, significativamente menos pacientes experimentaron una exacerbación de HES o se retiraron del ensayo cuando fueron tratados con 300 mg de NUCALA en comparación con placebo (17% vs. 35%, respectivamente, P = 0.020; OR: 0.33; IC del 95%: 0.13, 0.85).
Tasa de exacerbaciones
Los pacientes que recibieron NUCALA experimentaron significativamente menos exacerbaciones de HES durante el período de tratamiento de 32 semanas en comparación con el grupo placebo (Tabla 14). El tratamiento con NUCALA resultó en una reducción estadísticamente significativa del 66% en la tasa anualizada de exacerbaciones de HES en comparación con placebo.
| a Valores de P ajustados basados en la jerarquía preestablecida de puntos finales. b Una razón de tasas <1 favorece a NUCALA. |
|||||||
|
Número (%) de pacientes |
|||||||
|
Placebo n = 54 |
NUCALA 300 mg n = 54 |
||||||
|
0 |
26 (48) |
40 (74) |
|||||
|
1 |
15 (28) |
11 (20) |
|||||
|
2 |
7 (13) |
3 (6) |
|||||
|
3 |
5 (9) |
0 |
|||||
|
4 |
1 (2) |
0 |
|||||
|
≥5 |
0 |
0 |
|||||
|
Comparación: NUCALA/placebo |
|||||||
|
Valor de P de Wilcoxon (no ajustado/ajustado)a |
0.002/0.02 |
||||||
|
Tasa/año |
1.46 |
0.50 |
|||||
|
Razón de tasasb |
0.34 |
||||||
|
IC del 95% |
(0.19, 0.63) |
||||||
Inventario breve de fatiga
El ítem 3 del Inventario breve de fatiga (BFI) pide a los pacientes que registren su peor nivel de gravedad de cansancio/fatiga durante las últimas 24 horas (escala: 0 = ninguna fatiga a 10 = tan malo como puedas imaginar). En la línea de base, las puntuaciones medianas del ítem 3 del BFI fueron similares entre los grupos de tratamiento (4.46 para NUCALA 300 mg y 4.69 para placebo). En la semana 32, las puntuaciones del ítem 3 del BFI mejoraron con NUCALA en comparación con placebo (P = 0.036). El cambio mediano desde la puntuación de línea de base para el ítem 3 del BFI en la semana 32 fue -0.66 en el grupo tratado con NUCALA y 0.32 en el grupo placebo.
16 FORMA DE SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
NUCALA para inyección
NUCALA (mepolizumab) para inyección es un polvo liofilizado estéril, libre de conservantes, de color blanco a blanquecino, para reconstitución e inyección subcutánea en un vial de vidrio de dosis única con un sello abatible. El tapón del vial no está hecho con látex de caucho natural.
NUCALA para inyección se suministra como:
Viales de dosis única de 100 mg en cajas de 1 (NDC 0173-0881-01).
Almacenar los viales a menos de 77 °F (25 °C). No congelar. Almacenar en la caja original para proteger de la luz.
Inyección de NUCALA
La inyección de NUCALA (mepolizumab) es una solución estéril, libre de conservantes, transparente a opalescente, incolora a amarillo pálido a marrón pálido para uso subcutáneo. Cada autoinyector prellenado de dosis única administra 100 mg de mepolizumab en 1 mL de solución. Cada jeringa prellenada de dosis única administra 100 mg de mepolizumab en 1 mL de solución o 40 mg de mepolizumab en 0,4 mL de solución. Los autoinyectores y las jeringas no están hechos con látex de caucho natural.
La inyección de NUCALA se suministra como:
- •
- 100 mg/mL, dosis única, autoinyector prellenado con aguja de 29-gauge, de media pulgada, en cajas de 1 (NDC 0173-0892-01).
- •
- 100 mg/mL, dosis única, jeringa de vidrio prellenada con aguja de 29-gauge, de media pulgada, con protector de aguja en cajas de 1 (NDC 0173-0892-42).
- •
- 40 mg/0,4 mL, dosis única, jeringa de vidrio prellenada con aguja de 29-gauge, de media pulgada, con protector de aguja en cajas de 1 (NDC 0173-0904-42).
Antes de la dispensación: Refrigerar los autoinyectores prellenados y las jeringas prellenadas a 36 °F a 46 °F (2 °C a 8 °C). Mantener el producto en la caja original para proteger de la luz. No congelar. No agitar. Evitar la exposición al calor.
Después de la dispensación: Refrigerar los autoinyectores prellenados y las jeringas prellenadas a 36 °F a 46 °F (2 °C a 8 °C). Mantener el producto en la caja original para proteger de la luz hasta el momento de su uso. No congelar. No agitar. Evitar la exposición al calor.
Si es necesario, una caja sin abrir se puede almacenar fuera del refrigerador a una temperatura de hasta 86 °F (30 °C) durante un máximo de 7 días. Desechar si se deja fuera del refrigerador durante más de 7 días.
La inyección de NUCALA debe administrarse dentro de las 8 horas posteriores a su extracción de la caja. Desechar si no se administra dentro de las 8 horas.
17 INFORMACIÓN PARA EL PACIENTE
Aconseje al paciente que lea el etiquetado del paciente aprobado por la FDA (Información para el paciente e Instrucciones de uso).
Reacciones de hipersensibilidad
Informe a los pacientes que se han producido reacciones de hipersensibilidad (por ejemplo, anafilaxia, angioedema, broncoespasmo, hipotensión, urticaria, erupción cutánea) después de la administración de NUCALA. Indique a los pacientes que se pongan en contacto con su médico si se producen tales reacciones.
No para síntomas agudos o enfermedad en deterioro
Informe a los pacientes que NUCALA no trata los síntomas agudos del asma ni las exacerbaciones agudas. Informe a los pacientes que busquen atención médica si su asma permanece sin control o empeora después de iniciar el tratamiento con NUCALA.
Infecciones oportunistas: Herpes Zoster
Informe a los pacientes que se han producido infecciones por herpes zóster en pacientes que reciben NUCALA y, cuando sea médicamente apropiado, informe a los pacientes que se debe considerar la vacunación.
Reducción de la dosis de corticosteroides
Informe a los pacientes que no deben interrumpir los corticosteroides sistémicos o inhalados excepto bajo la supervisión directa de un médico. Informe a los pacientes que la reducción de la dosis de corticosteroides puede estar asociada con síntomas de abstinencia sistémica y/o desenmascarar afecciones previamente suprimidas por la terapia con corticosteroides sistémicos.
Las marcas comerciales son propiedad o están licenciadas al grupo de empresas GSK.
Fabricado por
GlaxoSmithKline LLC
Filadelfia, PA 19104
Licencia de EE. UU. No. 1727
Distribuido por
GlaxoSmithKline
Durham, NC 27701
©2023 Grupo de empresas GSK o su licenciante.
NCL:10PI
INSERTO PARA EL PACIENTE
|
Información para el paciente |
|
|
NUCALA (new KAH la) (mepolizumab) para inyección, para uso subcutáneo |
NUCALA (new KAH la) (mepolizumab) inyección, para uso subcutáneo |
|
¿Qué es NUCALA?
No se sabe si NUCALA es seguro y eficaz en niños con asma grave menores de 6 años de edad. No se sabe si NUCALA es seguro y eficaz en niños y adolescentes con CRSwNP o EGPA menores de 18 años de edad. No se sabe si NUCALA es seguro y eficaz en niños con HES menores de 12 años de edad. |
|
|
No use NUCALA si es alérgico a mepolizumab o a cualquiera de los ingredientes de NUCALA. Consulte el final de este folleto para obtener una lista completa de los ingredientes de NUCALA. |
|
|
Antes de recibir NUCALA, informe a su proveedor de atención médica sobre todas sus afecciones médicas, incluso si:
|
|
|
¿Cómo recibiré NUCALA? Su proveedor de atención médica le recetará la dosis adecuada para usted, dependiendo de lo que esté siendo tratado. Cuando la inyección la administra un proveedor de atención médica:
Cuando la inyección la administra un paciente o un cuidador del paciente con una jeringa precargada o un autoinyector precargado:
|
|
|
¿Cuáles son los posibles efectos secundarios de NUCALA? NUCALA puede causar efectos secundarios graves, que incluyen:
ο hinchazón de la cara, la boca y la lengua ο dificultad para respirar ο desmayo, mareos, sensación de mareo (presión arterial baja) ο erupción cutánea ο urticaria
Los efectos secundarios más comunes de NUCALA incluyen: dolor de cabeza, reacciones en el sitio de inyección (dolor, enrojecimiento, hinchazón, picazón o sensación de ardor en el sitio de inyección), dolor de espalda y cansancio (fatiga). Se ha informado dolor en la boca/garganta y dolor en las articulaciones con CRSwNP. Estos no son todos los posibles efectos secundarios de NUCALA. Llame a su médico para obtener asesoramiento médico sobre los efectos secundarios. Puede informar los efectos secundarios a la FDA al 1-800-FDA-1088. |
|
|
¿Cómo debo almacenar NUCALA?
Mantenga NUCALA y todos los medicamentos fuera del alcance de los niños. |
|
|
Información general sobre el uso seguro y eficaz de NUCALA. Los medicamentos a veces se recetan para fines distintos de los que se enumeran en un Folleto de información para el paciente. No le dé NUCALA a otras personas, incluso si tienen los mismos síntomas que usted. Puede hacerles daño. Puede pedirle a su farmacéutico o proveedor de atención médica información sobre NUCALA que esté escrita para profesionales de la salud. |
|
|
¿Cuáles son los ingredientes de NUCALA? Ingrediente activo: mepolizumab. Ingredientes inactivos (viales): polisorbato 80, fosfato disódico heptahidratado y sacarosa. Ingredientes inactivos (autoinyectores precargados y jeringas precargadas): ácido cítrico monohidratado, EDTA disódico dihidratado, polisorbato 80, fosfato disódico heptahidratado y sacarosa. Para obtener más información sobre NUCALA, llame al 1-888-825-5249 o visite nuestro sitio web en www.NUCALA.com. Las marcas comerciales son propiedad o están licenciadas al grupo de empresas GSK. Fabricado por: GlaxoSmithKline LLC, Filadelfia, PA 19104, EE. UU. Licencia No. 1727 Distribuido por: GlaxoSmithKline, Durham, NC 27701 ©2023 Grupo de empresas GSK o su licenciante. NCL:9PIL |
|
- Esta información para el paciente ha sido aprobada por la Administración de Alimentos y Medicamentos de los EE. UU. Revisado: marzo de 2023
INSTRUCCIONES DE USO
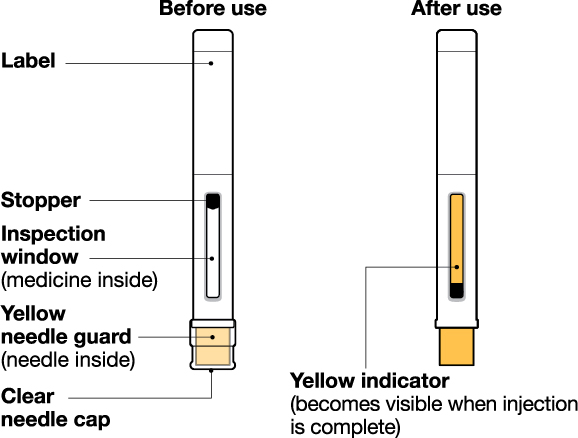
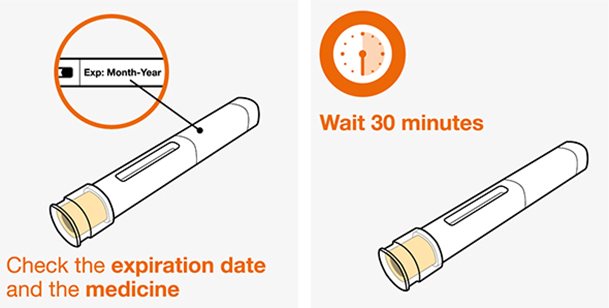
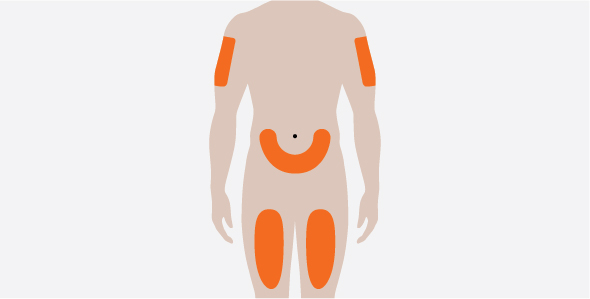
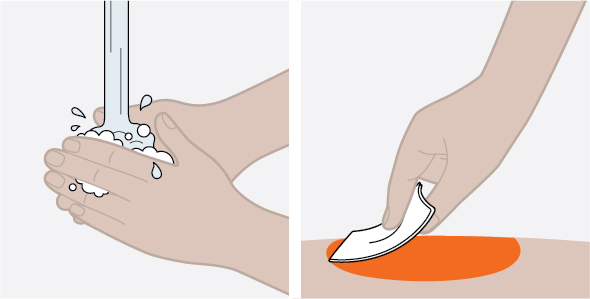
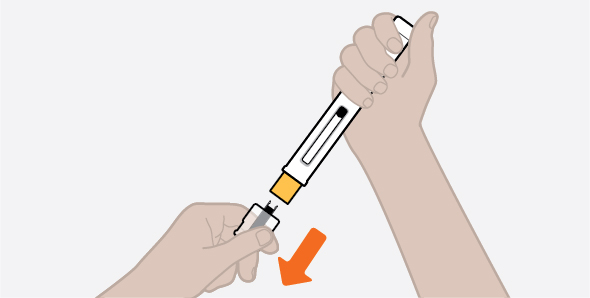
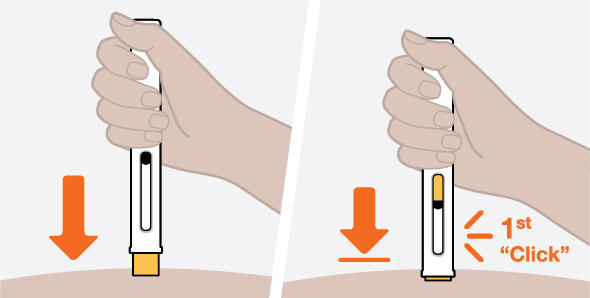
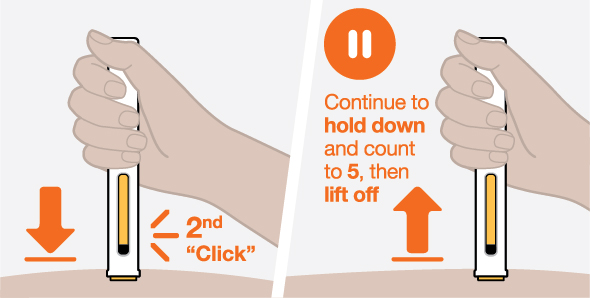
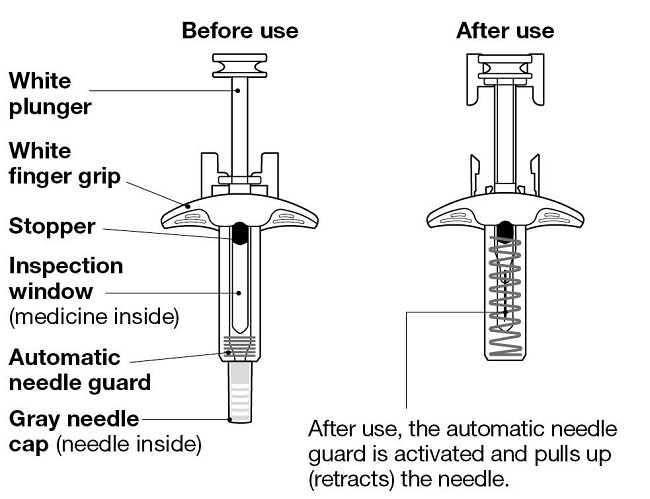
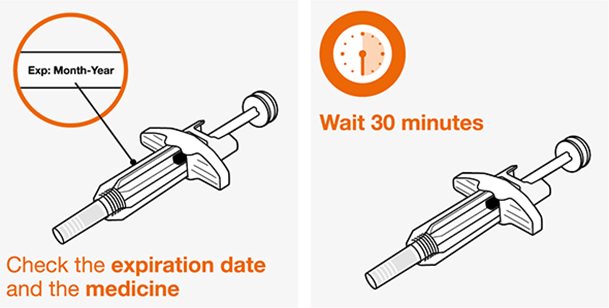
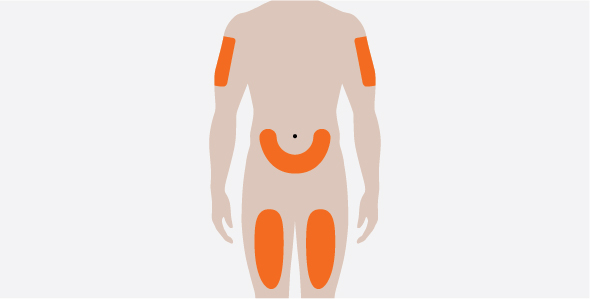
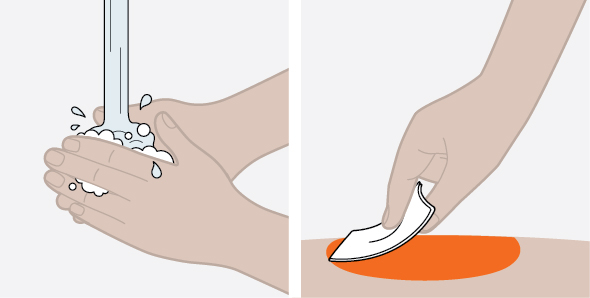
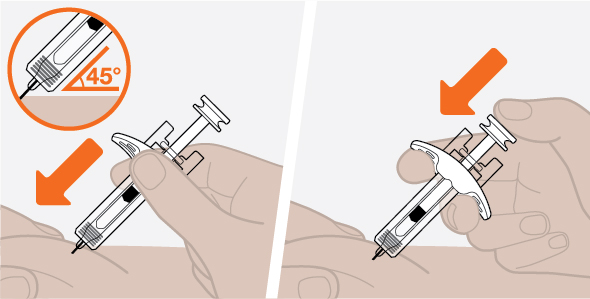
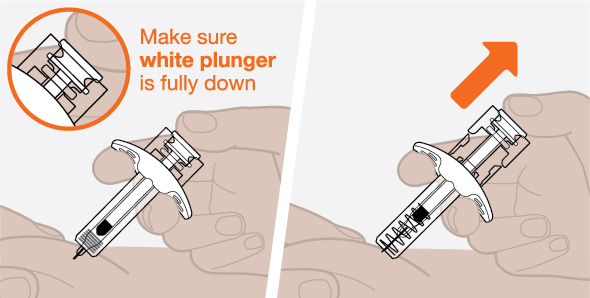
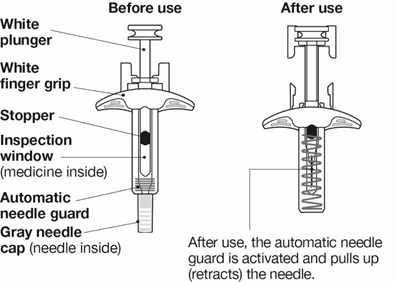
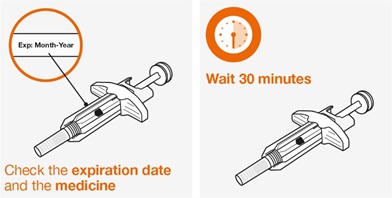
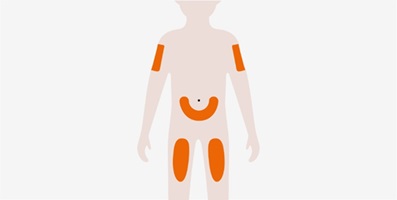
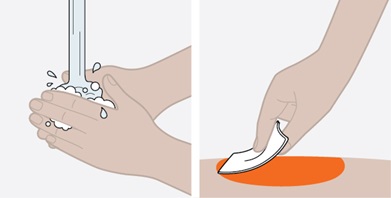
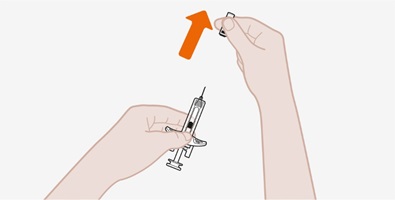
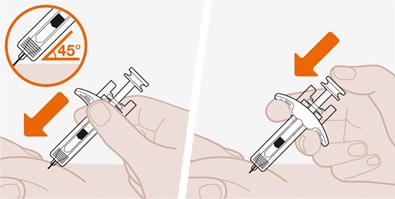
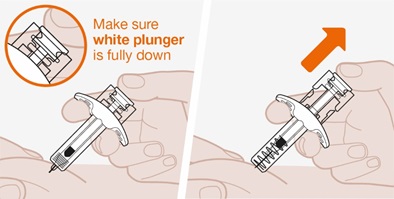

Panel de visualización del paquete/etiqueta
PANEL DE EXHIBICIÓN PRINCIPAL
NDC 0173-0881-01
Nucala
(mepolizumab)
para Inyección
100 mg/vial
Solo para uso con receta médica
GSK
Para inyección subcutánea después de la reconstitución.
La solución reconstituida contiene 100 mg/mL.
Vial de dosis única. Deseche la porción no utilizada.
Contenido: Cada vial contiene 100 mg de mepolizumab, polisorbato 80 (0.67 mg), fosfato de sodio dibásico heptahidratado (7.14 mg) y sacarosa (160 mg).
Sin conservantes.
No hay estándar de potencia en los Estados Unidos.
1 vial
No acepte si los sellos de la caja están rotos.
©2023 Grupo de empresas GSK o su licenciante.
- Rev. 4/23
- 62000000087874

PANEL DE VISUALIZACIÓN PRINCIPAL
PANEL DE EXHIBICIÓN PRINCIPAL
NDC 0173-0892-01
Nucala
(mepolizumab)
100 mg/mL
Sólo con receta médica
GSK
Autoinyector precargado
Únicamente para uso subcutáneo.
Contenidos:
-1 Dosis única de 1 mL. Autoinyector precargado
-Instrucciones de uso
-Información de prescripción
-Información para el paciente
©2023 Grupo de empresas GSK o su licenciante.
No acepte si el sello de seguridad está faltando o roto.
- 62000000087671 Rev. 5/23
PANEL DE VISUALIZACIÓN PRINCIPAL
PANEL DE EXHIBICIÓN PRINCIPAL
NDC 0173-0904-42
Nucala
(mepolizumab)
Inyección
40mg/0.4 mL
Sólo con receta médica
GSK
Jeringa precargada
Para uso subcutáneo únicamente.
Contenido:
– 1 Jeringa precargada de dosis única de 0.4 mL
– Instrucciones de uso
– Información de prescripción
– Información para pacientes
©2023 Grupo de compañías GSK o su licenciante.
No acepte si el sello de seguridad está faltando o roto.
Rev. 4/23
62000000087781
