
Fabricante de medicamentos: AbbVie Inc. (Updated: 2024-07-12)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
VENCLEXTA® (comprimidos de venetoclax), para administración oral
Aprobación inicial en EE. UU.: 2016
INDICACIONES Y USO
VENCLEXTA es un inhibidor de BCL-2 indicado:
- Para el tratamiento de pacientes adultos con leucemia linfocítica crónica (LLC) o linfoma linfocítico pequeño (LLS). (1.1)
- En combinación con azacitidina, o decitabina, o citarabina en dosis bajas para el tratamiento de la leucemia mieloide aguda (LMA) de novo en adultos de 75 años o más, o que tienen comorbilidades que impiden el uso de quimioterapia de inducción intensiva. (1.2)
DOSIFICACIÓN Y ADMINISTRACIÓN
- Consulte la información completa de prescripción para las dosis recomendadas de VENCLEXTA. (2.2, 2.3)
- Tome los comprimidos de VENCLEXTA por vía oral una vez al día con una comida y agua. No mastique, triture ni rompa los comprimidos. (2.8)
- Proporcione profilaxis para el síndrome de lisis tumoral. (2.1, 2.4)
FORMAS FARMACÉUTICAS Y CONCENTRACIONES
Comprimidos: 10 mg, 50 mg, 100 mg (3)
CONTRAINDICACIONES
ADVERTENCIAS Y PRECAUCIONES
- Síndrome de lisis tumoral (SLT): Anticipe el SLT; evalúe el riesgo en todos los pacientes. Premedique con antihiperuricémicos y asegúrese de una hidratación adecuada. Emplee medidas más intensivas (hidratación intravenosa, monitorización frecuente, hospitalización) a medida que aumenta el riesgo general. (2.4, 5.1)
- Neutropenia: Monitoree los recuentos sanguíneos. Interrumpa la dosificación y reanúdela a la misma dosis o una dosis reducida. Considere medidas de apoyo. (2.5, 5.2)
- Infecciones: Monitoree los signos y síntomas de infección y trátelos de inmediato. Suspenda el tratamiento para la infección de grado 3 y 4 hasta que se resuelva y reanúdelo a la misma dosis o una dosis reducida. (2.5, 5.3)
- Inmunización: No administre vacunas atenuadas vivas antes, durante o después del tratamiento con VENCLEXTA hasta que se recupere la población de células B. (5.4)
- Toxicidad embrio-fetal: Puede causar daño embrio-fetal. Avise a las mujeres en edad fértil del riesgo potencial para el feto y de la necesidad de utilizar métodos anticonceptivos eficaces. (5.5)
- No se recomienda el tratamiento de pacientes con mieloma múltiple con VENCLEXTA en combinación con bortezomib más dexametasona fuera de los ensayos clínicos controlados. (5.6)
REACCIONES ADVERSAS
En LLC/LLS, las reacciones adversas más frecuentes (≥20%) para VENCLEXTA cuando se administra en combinación con obinutuzumab o rituximab o como monoterapia son neutropenia, trombocitopenia, anemia, diarrea, náuseas, infección del tracto respiratorio superior, tos, dolor musculoesquelético, fatiga y edema. (6.1)
En LMA, las reacciones adversas más frecuentes (≥30%) en combinación con azacitidina o decitabina o citarabina en dosis bajas son náuseas, diarrea, trombocitopenia, estreñimiento, neutropenia, neutropenia febril, fatiga, vómitos, edema, pirexia, neumonía, disnea, hemorragia, anemia, erupción cutánea, dolor abdominal, sepsis, dolor musculoesquelético, mareos, tos, dolor orofaríngeo e hipotensión. (6.1)
Para informar sobre REACCIONES ADVERSAS SOSPECHOSAS, póngase en contacto con AbbVie Inc. al 1-800-633-9110 o con la FDA al 1-800-FDA-1088 o www.fda.gov/medwatch.
INTERACCIONES MEDICAMENTOSAS
USO EN POBLACIONES ESPECÍFICAS
Consulte 17 para obtener INFORMACIÓN PARA EL PACIENTE y la Guía de medicamentos.
Revisado: 7/2024
Tabla de Contenido
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDO*
1 INDICACIONES Y USO
1.1 Leucemia Linfocítica Crónica/Linfoma Linfocítico Pequeño
1.2 Leucemia Mieloide Aguda
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Información de Seguridad Importante
2.2 Dosis Recomendada para Leucemia Linfocítica Crónica/Linfoma Linfocítico Pequeño
2.3 Dosis Recomendada para Leucemia Mieloide Aguda
2.4 Evaluación de Riesgos y Profilaxis para el Síndrome de Lisis Tumoral
2.5 Modificaciones de la Dosis para Reacciones Adversas
2.6 Modificaciones de la Dosis para Interacciones Medicamentosas
2.7 Modificaciones de la Dosis para Pacientes con Insuficiencia Hepática Grave
2.8 Administración
3 FORMAS Y FUERZAS DE DOSIFICACIÓN
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Síndrome de Lisis Tumoral
5.2 Neutropenia
5.3 Infecciones
5.4 Inmunización
5.5 Toxicidad Embriofetal
5.6 Aumento de la Mortalidad en Pacientes con Mieloma Múltiple cuando VENCLEXTA se Agrega a Bortezomib y Dexametasona
6 REACCIONES ADVERSAS
6.1 Experiencia en Ensayos Clínicos
7 INTERACCIONES MEDICAMENTOSAS
7.1 Efectos de Otros Medicamentos en VENCLEXTA
7.2 Efecto de VENCLEXTA en Otros Medicamentos
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.3 Mujeres y Hombres en Potencial Reproductivo
8.4 Uso Pediátrico
8.5 Uso Geriátrico
8.6 Insuficiencia Renal
8.7 Insuficiencia Hepática
10 SOBREDOSIS
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de Acción
12.2 Farmacodinamia
12.3 Farmacocinética
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
13.2 Toxicología y/o Farmacología Animal
14 ESTUDIOS CLÍNICOS
14.1 Leucemia Linfocítica Crónica/Linfoma Linfocítico Pequeño
14.2 Leucemia Mieloide Aguda
16 CÓMO SE SUMINISTRA/ALMACENAMIENTO Y MANEJO
17 INFORMACIÓN PARA EL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información completa de prescripción no están enumeradas.
1 INDICACIONES Y USO
1.1 Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma
VENCLEXTA está indicado para el tratamiento de pacientes adultos con leucemia linfocítica crónica (LLC) o linfoma linfocítico pequeño (LLS).
1.2 Acute Myeloid Leukemia
VENCLEXTA está indicado en combinación con azacitidina, o decitabina, o citarabina en dosis bajas para el tratamiento de la leucemia mieloide aguda (LMA) de novo en adultos de 75 años o más, o que tienen comorbilidades que impiden el uso de quimioterapia de inducción intensiva.
2 DOSIS Y ADMINISTRACIÓN
2.1 Información de seguridad importante
Evalúe los factores específicos del paciente para el nivel de riesgo de síndrome de lisis tumoral (SLT) y proporcione hidratación profiláctica y antihiperuricémicos a los pacientes antes de la primera dosis de VENCLEXTA para reducir el riesgo de SLT [ver Dosificación y administración (2.4) y Advertencias y precauciones (5.1)].
2.2 Dosis recomendada para la leucemia linfocítica crónica/linfoma linfocítico pequeño
La dosificación de VENCLEXTA comienza con un aumento gradual de 5 semanas. El programa de dosificación de aumento gradual de 5 semanas está diseñado para reducir gradualmente la carga tumoral (reducción) y disminuir el riesgo de SLT.
Programa de aumento gradual de la dosis de VENCLEXTA de 5 semanas
Administre VENCLEXTA de acuerdo con el programa de dosificación de aumento gradual de 5 semanas hasta la dosis recomendada de 400 mg por vía oral una vez al día, como se muestra en Tabla 1.
| VENCLEXTA Dosis diaria oral |
|
| Semana 1 | 20 mg |
| Semana 2 | 50 mg |
| Semana 3 | 100 mg |
| Semana 4 | 200 mg |
| Semana 5 y posteriores | 400 mg |
El paquete inicial de LLC/SLL proporciona las primeras 4 semanas de VENCLEXTA de acuerdo con el programa de aumento gradual [ver Cómo se suministra/Almacenamiento y manipulación (16)].
En combinación con obinutuzumab
Comience la administración de obinutuzumab a 100 mg en el Día 1 del Ciclo 1, seguido de 900 mg en el Día 2 del Ciclo 1. Administre 1000 mg en los Días 8 y 15 del Ciclo 1 y en el Día 1 de cada ciclo posterior de 28 días durante un total de 6 ciclos. Consulte la información de prescripción de obinutuzumab para obtener información adicional sobre la dosificación.
En el Día 22 del Ciclo 1, comience VENCLEXTA de acuerdo con el programa de aumento gradual de 5 semanas (ver Tabla 1). Después de completar la fase de aumento gradual en el Día 28 del Ciclo 2, continúe con VENCLEXTA a una dosis de 400 mg por vía oral una vez al día desde el Día 1 del Ciclo 3 hasta el último día del Ciclo 12.
En combinación con rituximab
Comience la administración de rituximab después de que el paciente haya completado el programa de dosificación de aumento gradual de 5 semanas para VENCLEXTA (ver Tabla 1) y haya recibido VENCLEXTA a la dosis recomendada de 400 mg por vía oral una vez al día durante 7 días. Administre rituximab en el Día 1 de cada ciclo de 28 días durante 6 ciclos, a una dosis de 375 mg/m2 por vía intravenosa para el Ciclo 1 y 500 mg/m2 por vía intravenosa para los Ciclos 2-6. Continúe con VENCLEXTA 400 mg por vía oral una vez al día durante 24 meses desde el Día 1 del Ciclo 1 de rituximab.
Consulte la información de prescripción de rituximab para obtener información adicional sobre la dosificación.
Monoterapia
La dosis recomendada de VENCLEXTA es de 400 mg una vez al día después de completar el programa de dosificación de aumento gradual de 5 semanas (ver Tabla 1). Continúe con VENCLEXTA hasta la progresión de la enfermedad o la toxicidad inaceptable.
2.3 Dosis recomendada para la leucemia mieloide aguda
La dosis recomendada y el aumento gradual de VENCLEXTA dependen del agente de combinación. Siga el programa de dosificación, incluido el aumento gradual de la dosis de 3 o 4 días, como se muestra en la Tabla 2. Comience la administración de VENCLEXTA en el Día 1 del Ciclo 1 en combinación con:
- Azacitidina 75 mg/m2 por vía intravenosa o subcutánea una vez al día en los Días 1-7 de cada ciclo de 28 días; O
- Decitabina 20 mg/m2 por vía intravenosa una vez al día en los Días 1-5 de cada ciclo de 28 días; O
- Citarabina 20 mg/m2 por vía subcutánea una vez al día en los Días 1-10 de cada ciclo de 28 días.
| VENCLEXTA Dosis diaria oral |
||
| Día 1 | 100 mg | |
| Día 2 | 200 mg | |
| Día 3 | 400 mg | |
| Días 4 y posteriores | 400 mg por vía oral una vez al día de cada ciclo de 28 días en combinación con azacitidina o decitabina |
600 mg por vía oral una vez al día de cada ciclo de 28 días en combinación con citarabina de dosis baja |
Continúe con VENCLEXTA, en combinación con azacitidina o decitabina o citarabina en dosis bajas, hasta la progresión de la enfermedad o la toxicidad inaceptable.
Consulte Estudios clínicos (14.2) e Información de prescripción para azacitidina, decitabina o citarabina para obtener información adicional sobre la dosificación.
2.4 Evaluación del riesgo y profilaxis para el síndrome de lisis tumoral
Los pacientes tratados con VENCLEXTA pueden desarrollar síndrome de lisis tumoral (SLT). Consulte la sección correspondiente a continuación para obtener detalles específicos sobre el manejo. Evalúe los factores específicos del paciente para el nivel de riesgo de SLT y proporcione hidratación profiláctica y antihiperuricémicos a los pacientes antes de la primera dosis de VENCLEXTA para reducir el riesgo de SLT.
Leucemia linfocítica crónica/Linfoma linfocítico pequeño
VENCLEXTA puede causar una reducción rápida del tumor y, por lo tanto, representa un riesgo de SLT en la fase inicial de aumento gradual de 5 semanas. Los cambios en la química sanguínea compatibles con SLT que requieren un manejo inmediato pueden ocurrir tan pronto como 6 a 8 horas después de la primera dosis de VENCLEXTA y en cada aumento de dosis. El SLT también puede ocurrir al reanudar VENCLEXTA después de una interrupción de la dosis. Consulte Tabla 4 y Tabla 5 para las modificaciones de la dosis de VENCLEXTA después de la interrupción.
El riesgo de SLT es un continuo basado en múltiples factores, particularmente la función renal reducida (aclaramiento de creatinina [CLcr] <80 mL/min) y la carga tumoral; la esplenomegalia también puede aumentar el riesgo de SLT.
Realice evaluaciones de la carga tumoral, incluida la evaluación radiográfica (por ejemplo, tomografía computarizada), evalúe la química sanguínea (potasio, ácido úrico, fósforo, calcio y creatinina) en todos los pacientes y corrija las anomalías preexistentes antes de iniciar el tratamiento con VENCLEXTA. El riesgo puede disminuir a medida que disminuye la carga tumoral [consulte Advertencias y precauciones (5.1) y Uso en poblaciones específicas (8.6)].
La Tabla 3 a continuación describe la profilaxis y el monitoreo de SLT recomendados durante el tratamiento con VENCLEXTA según la determinación de la carga tumoral a partir de datos de ensayos clínicos. Considere todas las comorbilidades del paciente antes de la determinación final del régimen de profilaxis y monitoreo. Reevalúe el riesgo de SLT al reiniciar VENCLEXTA después de una interrupción de la dosis que dure más de 1 semana durante la fase de aumento gradual, o más de 2 semanas después de completar el aumento gradual. Instituya la profilaxis y el monitoreo según sea necesario.
| Carga tumoral | Profilaxis | Química sanguínea Monitoreoc,d |
||
| Hidratacióna | Anti- hiperuricémicosb |
Configuración y Frecuencia de Evaluaciones |
||
| Baja | Todos los LN <5 cm Y ALC <25 x109/L |
Oral (1.5 a 2 L) |
Alopurinol | Ambulatorio
|
| Media | Cualquier LN de 5 a <10 cm O ALC ≥25 x109/L |
Oral (1.5 a 2 L) y considerar intravenosa adicional |
Alopurinol | Ambulatorio
|
| Alta | Cualquier LN ≥10 cm O ALC ≥25 x109/L Y cualquier LN ≥5 cm |
Oral (1.5 a 2 L) y intravenosa (150 a 200 mL/hr según tolerancia) |
Alopurinol; considere rasburicasa si el ácido úrico basal está elevado | En el hospital
Ambulatorio
|
| ALC = recuento absoluto de linfocitos; CLcr = aclaramiento de creatinina; LN = ganglio linfático. aAdministrar hidratación intravenosa a cualquier paciente que no pueda tolerar la hidratación oral. bComenzar con alopurinol o un inhibidor de la xantina oxidasa de 2 a 3 días antes de iniciar VENCLEXTA. cEvaluar la química sanguínea (potasio, ácido úrico, fósforo, calcio y creatinina); revisar en tiempo real. dPara pacientes con riesgo de TLS, controlar la química sanguínea cada 6 a 8 horas y a las 24 horas en cada dosis de aumento posterior. |
||||
Leucemia mieloide aguda
- Todos los pacientes deben tener un recuento de glóbulos blancos inferior a 25 × 109/L antes de iniciar VENCLEXTA. Puede ser necesaria la citoreducción antes del tratamiento.
- Antes de la primera dosis de VENCLEXTA, proporcione a todos los pacientes medidas profilácticas que incluyen hidratación adecuada y agentes antihiperuricémicos y continúe durante la fase de aumento gradual.
- Evalúe la química sanguínea (potasio, ácido úrico, fósforo, calcio y creatinina) y corrija las anomalías preexistentes antes de iniciar el tratamiento con VENCLEXTA.
- Controle la química sanguínea para TLS antes de la dosis, de 6 a 8 horas después de cada nueva dosis durante el aumento gradual y 24 horas después de alcanzar la dosis final.
- Para los pacientes con factores de riesgo de TLS (por ejemplo, blastos circulantes, alta carga de afectación de la leucemia en la médula ósea, niveles elevados de lactato deshidrogenasa [LDH] previos al tratamiento o función renal reducida), considere medidas adicionales, incluida una mayor vigilancia de laboratorio y la reducción de la dosis inicial de VENCLEXTA.
2.5 Modificaciones de la dosis para reacciones adversas
Leucemia linfocítica crónica/Linfoma linfocítico pequeño
Las modificaciones de la dosis recomendadas para VENCLEXTA para reacciones adversas se proporcionan en la Tabla 4 y las reducciones de la dosis recomendadas para VENCLEXTA para reacciones adversas se proporcionan en la Tabla 5.
Para los pacientes que tienen una interrupción de la dosis que dura más de 1 semana durante la fase de aumento gradual, o más de 2 semanas después de completar el aumento gradual, vuelva a evaluar el riesgo de TLS para determinar si es necesario reiniciar con una dosis reducida (por ejemplo, todos o algunos niveles del programa de aumento gradual de la dosis) [ver Dosificación y administración (2.2, 2.4)].
| Reacción adversa | Ocurrencia | Modificación de la dosis |
| Síndrome de lisis tumoral | ||
| Cambios en la química sanguínea o síntomas sugestivos de TLS [ver Advertencias y precauciones (5.1)] | Cualquiera | Retenga la dosis del día siguiente. Si se resuelve dentro de las 24 a 48 horas de la última dosis, reanude a la misma dosis. |
| Para cualquier cambio en la química sanguínea que requiera más de 48 horas para resolverse, reanude a una dosis reducida (ver Tabla 5). | ||
| Para cualquier evento de TLS clínico,b reanude a una dosis reducida después de la resolución (ver Tabla 5). | ||
| Reacciones adversas no hematológicas | ||
| Toxicidades no hematológicas de grado 3 o 4 [ver Reacciones adversas (6.1)] | 1ra ocurrencia | Interrupción de VENCLEXTA. Una vez que se resuelva a un nivel de grado 1 o de referencia, reanude VENCLEXTA a la misma dosis. |
| 2da y ocurrencias subsiguientes | Interrupción de VENCLEXTA. Siga las pautas de reducción de la dosis en la Tabla 5 cuando reanude el tratamiento con VENCLEXTA después de la resolución. Puede ocurrir una reducción de la dosis mayor a discreción del médico. |
|
| Reacciones adversas hematológicas | ||
| Neutropenia de grado 3 con infección o fiebre; o toxicidades hematológicas de grado 4 (excepto linfopenia) [ver Advertencias y precauciones (5.2)] | 1ra ocurrencia | Interrupción de VENCLEXTA. Una vez que se resuelva a un nivel de grado 1 o de referencia, reanude VENCLEXTA a la misma dosis. |
| 2da y ocurrencias subsiguientes | Interrupción de VENCLEXTA. Siga las pautas de reducción de la dosis en la Tabla 5 cuando reanude el tratamiento con VENCLEXTA después de la resolución. Puede ocurrir una reducción de la dosis mayor a discreción del médico. |
|
| Considere la posibilidad de suspender VENCLEXTA para los pacientes que requieren reducciones de la dosis a menos de 100 mg durante más de 2 semanas. aLas reacciones adversas se clasificaron utilizando la versión 4.0 de NCI CTCAE. bEl TLS clínico se definió como TLS de laboratorio con consecuencias clínicas como insuficiencia renal aguda, arritmias cardíacas o muerte súbita y/o convulsiones [ver Reacciones adversas (6.1)]. |
||
| Dosis en la interrupción, mg | Dosis de reinicio, mga,b |
| 400 | 300 |
| 300 | 200 |
| 200 | 100 |
| 100 | 50 |
| 50 | 20 |
| 20 | 10 |
| aDurante la fase de aumento gradual, continúe con la dosis reducida durante 1 semana antes de aumentar la dosis. bSi una interrupción de la dosificación dura más de 1 semana durante la fase de aumento gradual o más de 2 semanas después de la finalización del aumento gradual, vuelva a evaluar el riesgo de TLS y determine si es necesario reiniciar a una dosis reducida [ver Dosificación y administración (2.2, 2.4)]. |
|
Leucemia mieloide aguda
Monitoree los recuentos sanguíneos con frecuencia hasta la resolución de las citopenias. La modificación de la dosis y las interrupciones para las citopenias dependen del estado de remisión. Las modificaciones de la dosis de VENCLEXTA para reacciones adversas se proporcionan en la Tabla 6.
| Reacción adversa | Ocurrencia | Modificación de la dosis |
| Reacciones adversas hematológicas | ||
| Neutropenia de grado 4 con o sin fiebre o infección; o trombocitopenia de grado 4 [ver Advertencias y precauciones (5.2)] | Ocurrencia antes de lograr la remisióna | En la mayoría de los casos, no interrumpa VENCLEXTA en combinación con azacitidina, decitabina o citarabina de dosis baja debido a citopenias antes de lograr la remisión. |
| Primera ocurrencia después de lograr la remisión y que dura al menos 7 días | Retrase el ciclo posterior de VENCLEXTA en combinación con azacitidina, decitabina o citarabina de dosis baja y controle los recuentos sanguíneos. Una vez que se resuelva a Grado 1 o 2, reanude VENCLEXTA a la misma dosis en combinación con azacitidina, decitabina o citarabina de dosis baja. |
|
| Ocurrencias posteriores en ciclos después de lograr la remisión y que duran 7 días o más | Retrase el ciclo posterior de VENCLEXTA en combinación con azacitidina o decitabina o citarabina de dosis baja y controle los recuentos sanguíneos. Una vez que se resuelva a Grado 1 o 2, reanude VENCLEXTA a la misma dosis en combinación con azacitidina, decitabina o citarabina de dosis baja, y reduzca la duración de VENCLEXTA en 7 días durante cada uno de los ciclos posteriores, como 21 días en lugar de 28 días. |
|
| Reacciones adversas no hematológicas | ||
| Toxicidades no hematológicas de grado 3 o 4 [ver Reacciones adversas (6.1)] | Cualquier ocurrencia | Interrumpa VENCLEXTA si no se resuelve con cuidados de apoyo. Una vez que se resuelva a Grado 1 o nivel basal, reanude VENCLEXTA a la misma dosis. |
| aRecomiende una evaluación de la médula ósea. | ||
2.6 Modificaciones de la Dosis para Interacciones Medicamentosas
Inhibidores Fuertes o Moderados de CYP3A o Inhibidores de P-gp
Tabla 7 describe la contraindicación o modificación de la dosis de VENCLEXTA basada en el uso concomitante con un inhibidor fuerte o moderado de CYP3A o un inhibidor de P-gp [ver Interacciones Medicamentosas (7.1)] al inicio, durante o después de la fase de aumento gradual.
Reanude la dosis de VENCLEXTA que se usó antes del uso concomitante de un inhibidor fuerte o moderado de CYP3A o un inhibidor de P-gp de 2 a 3 días después de la interrupción del inhibidor [ver Interacciones Medicamentosas (7.1)].
| Administrado Concomitantemente |
Inicio y Fase de Aumento Gradual |
Dosis Diaria Estable (Después de la Fase de Aumento Gradual)a |
|
| Posaconazol | CLL/SLL | Contraindicado | Reducir la dosis de VENCLEXTA a 70 mg. |
| AML | Día 1 – 10 mg Día 2 – 20 mg Día 3 – 50 mg Día 4 – 70 mg |
||
| Otro inhibidor fuerte de CYP3A | CLL/SLL | Contraindicado | Reducir la dosis de VENCLEXTA a 100 mg. |
| AML | Día 1 – 10 mg Día 2 – 20 mg Día 3 – 50 mg Día 4 – 100 mg |
||
| Inhibidor moderado de CYP3A | Reducir la dosis de VENCLEXTA al menos un 50%. | ||
| Inhibidor de P-gp | |||
| aEn pacientes con CLL/SLL, considere medicamentos alternativos o reduzca la dosis de VENCLEXTA como se describe en Tabla 7. | |||
2.7 Modificaciones de la Dosis para Pacientes con Insuficiencia Hepática Grave
Reducir la dosis de VENCLEXTA una vez al día en un 50% para pacientes con insuficiencia hepática grave (Child-Pugh C); monitorear a estos pacientes más de cerca para detectar reacciones adversas [ver Uso en Poblaciones Específicas (8.7)].
2.8 Administración
Instruya a los pacientes sobre lo siguiente:
- Tome VENCLEXTA con una comida y agua.
- Tome VENCLEXTA aproximadamente a la misma hora todos los días.
- Trague las tabletas de VENCLEXTA enteras. No mastique, triture o rompa las tabletas antes de tragarlas.
La dosis recomendada de VENCLEXTA se puede administrar utilizando cualquiera de las concentraciones de tabletas aprobadas (por ejemplo, los pacientes pueden tomar 2 x 50 mg tabletas o 10 x 10 mg tabletas en lugar de 1 x 100 mg tableta según sea necesario).
Si el paciente olvida una dosis de VENCLEXTA dentro de las 8 horas de la hora en que normalmente la toma, instruya al paciente a tomar la dosis olvidada lo antes posible y reanude el programa de dosificación diario normal. Si un paciente olvida una dosis por más de 8 horas, instruya al paciente a no tomar la dosis olvidada y reanude el programa de dosificación habitual al día siguiente.
Si el paciente vomita después de la dosificación, instruya al paciente a no tomar una dosis adicional ese día y a tomar la próxima dosis prescrita a la hora habitual.
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
| Fuerza de la Tableta | Descripción de la Tableta |
| 10 mg | Tableta redonda, biconvexa, de color amarillo pálido, recubierta con película, con “V” grabado en un lado y “10” en el otro lado |
| 50 mg | Tableta oblonga, biconvexa, de color beige, recubierta con película, con “V” grabado en un lado y “50” en el otro lado |
| 100 mg | Tableta oblonga, biconvexa, de color amarillo pálido, recubierta con película, con “V” grabado en un lado y “100” en el otro lado |
4 CONTRAINDICACIONES
El uso concomitante de VENCLEXTA con inhibidores fuertes de CYP3A al inicio y durante la fase de ajuste de dosis está contraindicado en pacientes con CLL/SLL debido al riesgo potencial de aumento del síndrome de lisis tumoral [ver Dosis y administración (2.6) e Interacciones medicamentosas (7.1)].
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Síndrome de lisis tumoral
Se ha producido síndrome de lisis tumoral (SLT), incluidos eventos fatales e insuficiencia renal que requieren diálisis, en pacientes tratados con VENCLEXTA [ver Reacciones adversas (6.1)].
VENCLEXTA puede causar una reducción rápida del tumor y, por lo tanto, representa un riesgo de SLT al inicio y durante la fase de aumento gradual en todos los pacientes, y durante la reiniciación después de la interrupción de la dosis en pacientes con CLL/SLL. Los cambios en la química sanguínea compatibles con SLT que requieren una gestión inmediata pueden ocurrir tan pronto como 6 a 8 horas después de la primera dosis de VENCLEXTA y en cada aumento de dosis. Se ha informado de SLT, incluidos casos fatales, después de una sola dosis de 20 mg de VENCLEXTA.
En pacientes con CLL/SLL que siguieron el aumento gradual de la dosis actual (5 semanas) y las medidas de profilaxis y monitorización del SLT, la tasa de SLT fue del 2% en los ensayos de monoterapia con VENCLEXTA CLL/SLL. La tasa de SLT se mantuvo constante con VENCLEXTA en combinación con obinutuzumab o rituximab. Con un aumento gradual de la dosis de 2 a 3 semanas y una dosis inicial más alta en pacientes con CLL/SLL, la tasa de SLT fue del 13% e incluyó muertes e insuficiencia renal [ver Reacciones adversas (6.1)].
En pacientes con AML que siguieron el programa de dosificación actual de aumento gradual de 3 días y las medidas de profilaxis y monitorización del SLT, la tasa de SLT fue del 1,1% en los pacientes que recibieron VENCLEXTA en combinación con azacitidina (VIALE-A). En pacientes con AML que siguieron un programa de dosificación de aumento gradual de 4 días y las medidas de profilaxis y monitorización del SLT, la tasa de SLT fue del 5,6% e incluyó muertes e insuficiencia renal en pacientes que recibieron VENCLEXTA en combinación con citarabina de dosis baja (VIALE-C) [ver Reacciones adversas (6.1)].
El riesgo de SLT es un continuo basado en múltiples factores, particularmente la función renal reducida, la carga tumoral y el tipo de malignidad. La esplenomegalia también puede aumentar el riesgo de SLT en pacientes con CLL/SLL.
Evalúe a todos los pacientes para determinar el riesgo y proporcione profilaxis adecuada para el SLT, incluida la hidratación y los antihiperuricémicos. Controle la química sanguínea y gestione las anomalías con prontitud. Emplee medidas más intensivas (hidratación intravenosa, monitorización frecuente, hospitalización) a medida que aumenta el riesgo general. Interrumpa la dosificación si es necesario; al reiniciar VENCLEXTA, siga las pautas de modificación de la dosis [ver Dosificación y administración (2.1, 2.2, 2.3, 2.4) y Uso en poblaciones específicas (8.6)].
El uso concomitante de VENCLEXTA con inhibidores de la P-gp o inhibidores fuertes o moderados del CYP3A aumenta la exposición a venetoclax, lo que puede aumentar el riesgo de SLT al inicio y durante la fase de aumento gradual de VENCLEXTA. Para los pacientes con CLL/SLL, la coadministración de VENCLEXTA con inhibidores fuertes del CYP3A al inicio y durante la fase de aumento gradual de 5 semanas está contraindicada [ver Contraindicaciones (4)]. Para los pacientes con AML, reduzca la dosis de VENCLEXTA cuando se administre conjuntamente con inhibidores fuertes del CYP3A al inicio y durante la fase de aumento gradual de 3 o 4 días. Para los pacientes con CLL/SLL o AML, reduzca la dosis de VENCLEXTA cuando se administre conjuntamente con inhibidores moderados del CYP3A4 o inhibidores de la P-gp [ver Dosificación y administración (2.6) e Interacciones medicamentosas (7.1)].
5.2 Neutropenia
En pacientes con CLL, la neutropenia de grado 3 o 4 se desarrolló en el 63% al 64% de los pacientes y la neutropenia de grado 4 se desarrolló en el 31% al 33% de los pacientes cuando se trataron con VENCLEXTA en estudios de combinación y monoterapia. La neutropenia febril se produjo en el 4% al 6% de los pacientes [ver Reacciones adversas (6.1)].
En pacientes con AML, los recuentos de neutrófilos basales empeoraron en el 95% al 100% de los pacientes tratados con VENCLEXTA en combinación con azacitidina, decitabina o citarabina de dosis baja. La neutropenia puede recurrir con ciclos posteriores.
Controle los hemogramas completos durante todo el período de tratamiento. Para la interrupción y la reanudación de la dosis de VENCLEXTA por neutropenia grave, consulte la Tabla 4 para CLL y la Tabla 6 para AML [ver Dosificación y administración (2.5)]. Considere medidas de apoyo, incluidos antimicrobianos y factores de crecimiento (por ejemplo, G-CSF).
5.3 Infecciones
Se han producido infecciones fatales y graves, como neumonía y sepsis, en pacientes tratados con VENCLEXTA [ver Reacciones adversas (6.1)].
Controle a los pacientes para detectar signos y síntomas de infección y trate con prontitud. Suspenda VENCLEXTA para la infección de grado 3 y 4 hasta que se resuelva. Para las reanudaciones de la dosis, consulte la Tabla 4 para CLL y la Tabla 6 para AML [ver Dosificación y administración (2.5)].
5.4 Inmunización
No administre vacunas atenuadas vivas antes, durante o después del tratamiento con VENCLEXTA hasta que se produzca la recuperación de las células B. No se ha estudiado la seguridad y eficacia de la inmunización con vacunas atenuadas vivas durante o después de la terapia con VENCLEXTA. Avise a los pacientes que las vacunas pueden ser menos eficaces.
5.5 Toxicidad embrionaria y fetal
Con base en los hallazgos en animales y su mecanismo de acción, VENCLEXTA puede causar daño embrionario y fetal cuando se administra a una mujer embarazada. En un estudio embrionario-fetal realizado en ratones, la administración de venetoclax a animales preñados a exposiciones equivalentes a las observadas en pacientes a una dosis de 400 mg diarios resultó en pérdida postimplantación y disminución del peso fetal.
Avise a las mujeres embarazadas del riesgo potencial para el feto. Avise a las mujeres en edad fértil que utilicen métodos anticonceptivos eficaces durante el tratamiento con VENCLEXTA y durante 30 días después de la última dosis [ver Uso en poblaciones específicas (8.1, 8.3)].
5.6 Aumento de la mortalidad en pacientes con mieloma múltiple cuando VENCLEXTA se añade a bortezomib y dexametasona
En un ensayo aleatorizado (BELLINI; NCT02755597) en pacientes con mieloma múltiple recidivante o refractario, la adición de VENCLEXTA a bortezomib más dexametasona, un uso para el que VENCLEXTA no está indicado, resultó en un aumento de la mortalidad. No se recomienda el tratamiento de pacientes con mieloma múltiple con VENCLEXTA en combinación con bortezomib más dexametasona fuera de los ensayos clínicos controlados.
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas clínicamente significativas se describen en otra parte de la etiqueta:
- Síndrome de lisis tumoral [ver Advertencias y precauciones (5.1)]
- Neutropenia [ver Advertencias y precauciones (5.2)]
- Infecciones [ver Advertencias y precauciones (5.3)]
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se llevan a cabo en condiciones ampliamente variables, las tasas de eventos adversos observadas en los ensayos clínicos de un fármaco no se pueden comparar directamente con las tasas de los ensayos clínicos de otro fármaco y pueden no reflejar las tasas observadas en la práctica.
En CLL/SLL, la población de seguridad refleja la exposición a VENCLEXTA como monoterapia en pacientes en M13-982, M14-032 y M12-175 y en combinación con obinutuzumab o rituximab en pacientes en CLL14 y MURANO. En esta población de seguridad de CLL/SLL, las reacciones adversas más comunes (≥20%) para VENCLEXTA fueron neutropenia, trombocitopenia, anemia, diarrea, náuseas, infección del tracto respiratorio superior, tos, dolor musculoesquelético, fatiga y edema.
En AML, la población de seguridad refleja la exposición a VENCLEXTA en combinación con decitabina, azacitidina o citarabina de dosis baja en pacientes en M14-358, VIALE-A y VIALE-C. En esta población de seguridad, las reacciones adversas más comunes (≥30% en cualquier ensayo) fueron náuseas, diarrea, trombocitopenia, estreñimiento, neutropenia, neutropenia febril, fatiga, vómitos, edema, pirexia, neumonía, disnea, hemorragia, anemia, erupción cutánea, dolor abdominal, sepsis, dolor musculoesquelético, mareos, tos, dolor orofaríngeo e hipotensión.
Leucemia linfocítica crónica/Linfoma linfocítico pequeño
VENCLEXTA en combinación con obinutuzumab
La seguridad de VENCLEXTA en combinación con obinutuzumab (VEN+G) (N=212) versus obinutuzumab en combinación con clorambucil (GClb) (N=214) se evaluó en CLL14, un ensayo aleatorizado, abierto, controlado activamente en pacientes con CLL no tratada previamente [ver Estudios clínicos (14.1)]. Los pacientes aleatorizados al brazo VEN+G fueron tratados con VENCLEXTA y obinutuzumab en combinación durante seis ciclos, luego con VENCLEXTA como monoterapia durante seis ciclos adicionales. Los pacientes iniciaron la primera dosis del aumento de 5 semanas para VENCLEXTA en el Día 22 del Ciclo 1 y una vez completado, continuaron con VENCLEXTA 400 mg por vía oral una vez al día durante un total de 12 ciclos. El ensayo requirió una Escala de calificación de enfermedad acumulativa total (CIRS) >6 o CLcr <70 mL/min, transaminasas hepáticas y bilirrubina total ≤2 veces el límite superior de lo normal y excluyó a los pacientes con cualquier puntuación de deterioro de órgano/sistema individual de 4 por CIRS excepto el órgano/sistema de ojos, oídos, nariz y garganta. La duración media de la exposición a VENCLEXTA fue de 10,5 meses (rango: 0 a 13,5 meses) y el número medio de ciclos de obinutuzumab fue de 6 en el brazo VEN+G.
Se informaron reacciones adversas graves en el 49% de los pacientes en el brazo VEN+G, con mayor frecuencia debido a neutropenia febril y neumonía (5% cada una). Se informaron reacciones adversas fatales que ocurrieron en ausencia de progresión de la enfermedad y con inicio dentro de los 28 días del último tratamiento del estudio en el 2% (4/212) de los pacientes, con mayor frecuencia por infección.
En el brazo VEN+G, las reacciones adversas llevaron a la interrupción del tratamiento en el 16% de los pacientes, a la reducción de la dosis en el 21% y a la interrupción de la dosis en el 74%. La neutropenia llevó a la interrupción de VENCLEXTA en el 2% de los pacientes, a la reducción de la dosis en el 13% y a la interrupción de la dosis en el 41%.
Tabla 9 presenta las reacciones adversas identificadas en CLL14.
| Reacción adversa | VENCLEXTA + Obinutuzumab (N = 212) |
Obinutuzumab + Clorambucil (N = 214) |
||
| Todos los grados (%) |
Grado ≥3 (%) |
Todos los grados (%) |
Grado ≥3 (%) |
|
| Trastornos de la sangre y del sistema linfático | ||||
| Neutropeniaa | 60 | 56 | 62 | 52 |
| Anemiaa | 17 | 8 | 20 | 7 |
| Trastornos gastrointestinales | ||||
| Diarrea | 28 | 4 | 15 | 1 |
| Náuseas | 19 | 0 | 22 | 1 |
| Constipación | 13 | 0 | 9 | 0 |
| Vómitos | 10 | 1 | 8 | 1 |
| Trastornos generales y condiciones del sitio de administración | ||||
| Fatigaa | 21 | 2 | 23 | 1 |
| Infecciones e infestaciones | ||||
| Infección del tracto respiratorio superiora |
17 | 1 | 17 | 1 |
| aIncluye múltiples términos de reacciones adversas. | ||||
Otras reacciones adversas clínicamente importantes (todos los grados) informadas en <10% de los pacientes tratados con VEN+G se presentan a continuación:
Trastornos de la sangre y del sistema linfático: neutropenia febril (6%)
Infección e infestaciones (todos incluyen múltiples términos de reacciones adversas): neumonía (9%), infección del tracto urinario (6%), sepsis (4%)
Trastorno del metabolismo y la nutrición: síndrome de lisis tumoral (1%)
Durante el tratamiento con VENCLEXTA en monoterapia después de completar VEN+G, la reacción adversa que ocurrió en ≥10% de los pacientes fue neutropenia (26%). Las reacciones adversas de grado ≥3 que ocurrieron en ≥2% de los pacientes fueron neutropenia (23%) y anemia (2%).
Tabla 10 presenta anormalidades de laboratorio CLL14.
| Anormalidad de laboratorioa | VENCLEXTA + Obinutuzumab (N = 212) |
Obinutuzumab + Clorambucil (N = 214) |
||
| Todos los grados (%) |
Grado 3 o 4 (%) |
Todos los grados (%) |
Grado 3 o 4 (%) |
|
| Hematología | ||||
| Leucopenia | 90 | 46 | 89 | 41 |
| Linfopenia | 87 | 57 | 87 | 51 |
| Neutropenia | 83 | 63 | 79 | 56 |
| Trombocitopenia | 68 | 28 | 71 | 26 |
| Anemia | 53 | 15 | 46 | 11 |
| Química | ||||
| Creatinina en sangre aumentada | 80 | 6 | 74 | 2 |
| Hipocalcemia | 67 | 9 | 58 | 4 |
| Hiperkalemia | 41 | 4 | 35 | 3 |
| Hiperuricemia | 38 | 38 | 38 | 38 |
| aIncluye anormalidades de laboratorio que fueron nuevas o que empeoraron, o con empeoramiento desde el inicio desconocido. | ||||
Las anormalidades de laboratorio de Grado 4 que se desarrollaron en ≥2% de los pacientes tratados con VEN+G incluyeron neutropenia (32%), leucopenia y linfopenia (10%), trombocitopenia (8%), hipocalcemia (8%), hiperuricemia (7%), aumento de la creatinina en sangre (3%), hipercalcemia (3%) e hipopotasemia (2%).
VENCLEXTA en combinación con Rituximab
La seguridad de VENCLEXTA en combinación con rituximab (VEN+R) (N=194) versus bendamustina en combinación con rituximab (B+R) (N=188) se evaluó en MURANO [ver Estudios Clínicos (14.1)]. Los pacientes aleatorizados a VEN+R completaron el aumento programado (5 semanas) y recibieron VENCLEXTA 400 mg una vez al día, en combinación con rituximab durante 6 ciclos seguidos de monoterapia con VENCLEXTA, durante un total de 24 meses después del aumento. En el momento del análisis, la duración media de la exposición a VENCLEXTA fue de 22 meses y el número medio de ciclos de rituximab fue de 6 en el brazo VEN+R.
Se informaron reacciones adversas graves en el 46% de los pacientes en el brazo VEN+R, siendo las más frecuentes (≥5%) la neumonía (9%). Se informaron reacciones adversas fatales que ocurrieron en ausencia de progresión de la enfermedad y dentro de los 30 días del último tratamiento con VENCLEXTA y/o 90 días del último rituximab en el 2% (4/194) de los pacientes.
En el brazo VEN+R, las reacciones adversas llevaron a la interrupción del tratamiento en el 16% de los pacientes, a la reducción de la dosis en el 15% y a la interrupción de la dosis en el 71%. La neutropenia y la trombocitopenia llevaron a la interrupción de VENCLEXTA en el 3% de los pacientes. La neutropenia llevó a la interrupción de la dosis de VENCLEXTA en el 46% de los pacientes.
Tabla 11 presenta las reacciones adversas identificadas en MURANO.
| Reacción adversa | VENCLEXTA + Rituximab (N = 194) |
Bendamustina + Rituximab (N = 188) |
||
| Todos los grados (%) |
Grado ≥3 (%) |
Todos los grados (%) |
Grado ≥3 (%) |
|
| Trastornos de la sangre y del sistema linfático | ||||
| Neutropeniaa | 65 | 62 | 50 | 44 |
| Anemiaa | 16 | 11 | 23 | 14 |
| Trastornos gastrointestinales | ||||
| Diarrea | 40 | 3 | 17 | 1 |
| Náuseas | 21 | 1 | 34 | 1 |
| Estreñimiento | 14 | <1 | 21 | 0 |
| Infecciones e infestaciones | ||||
| Infección de las vías respiratorias superioresa |
39 | 2 | 23 | 2 |
| Infección de las vías respiratorias inferioresa |
18 | 2 | 10 | 2 |
| Neumoníaa | 10 | 7 | 14 | 10 |
| Trastornos generales y condiciones del lugar de administración | ||||
| Fatigaa | 22 | 2 | 26 | <1 |
| aIncluye múltiples términos de reacción adversa. | ||||
Otras reacciones adversas clínicamente importantes (todos los grados) notificadas en <10% de los pacientes tratados con VEN+R se presentan a continuación:
Trastornos de la sangre y del sistema linfático: neutropenia febril (4%)
Trastornos gastrointestinales: vómitos (8%)
Infecciones e infestaciones: sepsis (<1%)
Trastornos del metabolismo y la nutrición: síndrome de lisis tumoral (3%)
Durante el tratamiento con VENCLEXTA en monoterapia después de completar el tratamiento de combinación VEN+R, las reacciones adversas que ocurrieron en ≥10% de los pacientes fueron infección del tracto respiratorio superior (21%), diarrea (19%), neutropenia (16%) e infecciones del tracto respiratorio inferior (11%). Las reacciones adversas de grado 3 o 4 que ocurrieron en ≥2% de los pacientes fueron neutropenia (12%) y anemia (3%).
Tabla 12 presenta las anormalidades de laboratorio identificadas en MURANO.
| Anormalidad de laboratorio | VENCLEXTA + Rituximab (N = 194) |
Bendamustina + Rituximab (N = 188) |
||
| Todos los gradosa (%) |
Grado 3 o 4 (%) |
Todos los gradosa (%) |
Grado 3 o 4 (%) |
|
| Hematología | ||||
| Leucopenia | 89 | 46 | 81 | 35 |
| Linfopenia | 87 | 56 | 79 | 55 |
| Neutropenia | 86 | 64 | 84 | 59 |
| Anemia | 50 | 12 | 63 | 15 |
| Trombocitopenia | 49 | 15 | 60 | 20 |
| Química | ||||
| Creatinina en sangre aumentada | 77 | <1 | 78 | 1 |
| Hipocalcemia | 62 | 5 | 51 | 2 |
| Hiperuricemia | 36 | 36 | 33 | 33 |
| Hiperkalemia | 24 | 3 | 19 | 2 |
| aIncluye anormalidades de laboratorio que fueron nuevas o que empeoraron, o con empeoramiento desde el inicio desconocido. | ||||
Las anormalidades de laboratorio de Grado 4 que se desarrollaron en ≥2% de los pacientes tratados con VEN+R incluyeron neutropenia (31%), linfopenia (16%), leucopenia (6%), trombocitopenia (6%), hiperuricemia (4%), hipocalcemia (2%), hipoglucemia (2%) e hipermagnesemia (2%).
VENCLEXTA como monoterapia
La seguridad de VENCLEXTA se evaluó en datos agrupados de tres ensayos de un solo brazo (M13-982, M14-032 y M12-175). Los pacientes recibieron VENCLEXTA 400 mg por vía oral una vez al día después de completar la fase de aumento gradual (N=352). La duración media del tratamiento con VENCLEXTA en el momento del análisis de datos fue de 14,5 meses (rango: 0 a 50 meses). El cincuenta y dos por ciento de los pacientes recibieron VENCLEXTA durante más de 60 semanas.
En el conjunto de datos agrupados, la edad media fue de 66 años (rango: 28 a 85 años), el 93% eran blancos y el 68% eran hombres. El número medio de terapias previas fue de 3 (rango: 0 a 15).
Se informaron reacciones adversas graves en el 52% de los pacientes, siendo las más frecuentes (≥5%) la neumonía (9%), la neutropenia febril (5%) y la sepsis (5%). Las reacciones adversas fatales que ocurrieron en ausencia de progresión de la enfermedad y dentro de los 30 días del tratamiento con venetoclax se informaron en el 2% de los pacientes en los estudios de monoterapia con VENCLEXTA, con mayor frecuencia (2 pacientes) por shock séptico.
Las reacciones adversas llevaron a la interrupción del tratamiento en el 9% de los pacientes, a la reducción de la dosis en el 13% y a la interrupción de la dosis en el 36%. Las reacciones adversas más frecuentes que llevaron a la interrupción del fármaco fueron la trombocitopenia y la anemia hemolítica autoinmune. La reacción adversa más frecuente (≥5%) que llevó a reducciones o interrupciones de la dosis fue la neutropenia (8%).
Tabla 13 presenta las reacciones adversas identificadas en estos ensayos.
| Reacción adversa | VENCLEXTA (N = 352) |
|
| Todos los grados (%) |
Grado ≥3 (%) |
|
| Trastornos de la sangre y del sistema linfático | ||
| Neutropeniaa | 50 | 45 |
| Anemiaa | 33 | 18 |
| Trombocitopeniaa | 29 | 20 |
| Linfopeniaa | 11 | 7 |
| Neutropenia febril | 6 | 6 |
| Trastornos gastrointestinales | ||
| Diarrea | 43 | 3 |
| Náuseas | 42 | 1 |
| Dolor abdominala | 18 | 3 |
| Vómitos | 16 | 1 |
| Estreñimiento | 16 | <1 |
| Mucositisa | 13 | <1 |
| Infecciones e infestaciones | ||
| Infección de las vías respiratorias altasa | 36 | 1 |
| Neumoníaa | 14 | 8 |
| Infección de las vías respiratorias bajasa | 11 | 2 |
| Trastornos generales y condiciones del lugar de administración | ||
| Fatigaa | 32 | 4 |
| Edemaa | 22 | 2 |
| Pirexia | 18 | <1 |
| Trastornos musculoesqueléticos y del tejido conjuntivo | ||
| Dolor musculoesqueléticoa | 29 | 2 |
| Artralgia | 12 | <1 |
| Trastornos respiratorios, torácicos y mediastínicos | ||
| Tosa | 22 | 0 |
| Disneaa | 13 | 1 |
| Trastornos del sistema nervioso | ||
| Cefalea | 18 | <1 |
| Vértigoa | 14 | 0 |
| Trastornos de la piel y del tejido subcutáneo | ||
| Erupcióna | 18 | <1 |
| Reacciones adversas clasificadas utilizando los Criterios de Terminología Común para Eventos Adversos del NCI versión 4.0. aIncluye múltiples términos de reacciones adversas. |
||
Tabla 14 presenta las anormalidades de laboratorio reportadas durante todo el tratamiento que fueron nuevas o que empeoraron desde el inicio. Las anormalidades de laboratorio de Grado 4 más comunes (>5%) observadas con la monoterapia con VENCLEXTA fueron anormalidades de laboratorio hematológicas, incluyendo neutropenia (33%), leucopenia (11%), trombocitopenia (15%) y linfopenia (9%).
| Anormalidad de laboratorio | VENCLEXTA (N = 352) |
|
| Todos los gradosa (%) |
Grado 3 o 4 (%) |
|
| Hematología | ||
| Leucopenia | 89 | 42 |
| Neutropenia | 87 | 63 |
| Linfopenia | 74 | 40 |
| Anemia | 71 | 26 |
| Trombocitopenia | 64 | 31 |
| Química | ||
| Hipocalcemia | 87 | 12 |
| Hiperglucemia | 67 | 7 |
| Hiperkalemia | 59 | 5 |
| AST aumentado | 53 | 3 |
| Hipoalbuminemia | 49 | 2 |
| Hipofosfatemia | 45 | 11 |
| Hiponatremia | 40 | 9 |
| aIncluye anormalidades de laboratorio que fueron nuevas o que empeoraron, o que empeoraron desde el inicio desconocido. | ||
Reacciones adversas importantes en CLL/SLL
Síndrome de lisis tumoral
El síndrome de lisis tumoral es un riesgo importante identificado al iniciar VENCLEXTA.
CLL14
La incidencia de TLS fue del 1% (3/212) en pacientes tratados con VEN+G [ver Advertencias y precauciones (5.1)]. Los tres eventos de TLS se resolvieron y no llevaron a la retirada del ensayo. La administración de obinutuzumab se retrasó en dos casos en respuesta a los eventos de TLS.
MURANO
La incidencia de TLS fue del 3% (6/194) en pacientes tratados con VEN+R. Después de que se inscribieran 77/389 pacientes en el ensayo, el protocolo se modificó para incorporar las medidas actuales de profilaxis y monitorización de TLS descritas en las secciones 2.2 y 2.4 [ver Dosis y administración (2.2, 2.4)]. Todos los eventos de TLS ocurrieron durante el período de aumento gradual de VENCLEXTA y se resolvieron en dos días. Los seis pacientes completaron el aumento gradual y alcanzaron la dosis diaria recomendada de 400 mg de VENCLEXTA. No se observó TLS clínico en pacientes que siguieron el programa actual de aumento gradual de 5 semanas y las medidas de profilaxis y monitorización de TLS [ver Dosis y administración (2.2, 2.4)]. Las tasas de anomalías de laboratorio relevantes para TLS para pacientes tratados con VEN+R se presentan en Tabla 12.
Estudios de monoterapia (M13-982 y M14-032)
En 168 pacientes con CLL tratados de acuerdo con las recomendaciones descritas en las secciones 2.1 y 2.2, la tasa de TLS fue del 2% [ver Dosis y administración (2.2, 2.4)]. Todos los eventos cumplieron con los criterios de laboratorio de TLS (anomalías de laboratorio que cumplieron con ≥2 de los siguientes dentro de las 24 horas entre sí: potasio >6 mmol/L, ácido úrico >476 µmol/L, calcio <1,75 mmol/L o fósforo >1,5 mmol/L), o se informaron como eventos de TLS. Los eventos ocurrieron en pacientes que tenían un ganglio linfático (s) ≥5 cm y/o un recuento absoluto de linfocitos (ALC) ≥25 x 109/L. Todos los eventos se resolvieron en 5 días. No se observó TLS con consecuencias clínicas como insuficiencia renal aguda, arritmias cardíacas o muerte súbita y/o convulsiones en estos pacientes. Todos los pacientes tenían CLcr ≥50 mL/min. Las anomalías de laboratorio relevantes para TLS fueron hiperkalemia (17% de todos los grados, 1% de grado ≥3), hiperfosfatemia (14% de todos los grados, 2% de grado ≥3), hipocalcemia (16% de todos los grados, 2% de grado ≥3) e hiperuricemia (10% de todos los grados, <1% de grado ≥3).
En los ensayos iniciales de fase 1 de búsqueda de dosis, que tenían una fase de aumento gradual más corta (2-3 semanas) y dosis iniciales más altas, la incidencia de TLS fue del 13% (10/77; 5 TLS de laboratorio, 5 TLS clínicos), incluidos 2 eventos fatales y 3 eventos de insuficiencia renal aguda, 1 que requirió diálisis. Después de esta experiencia, la evaluación del riesgo de TLS, el régimen de dosificación, la profilaxis de TLS y las medidas de monitorización se revisaron [ver Dosis y administración (2.2, 2.4)].
Leucemia mieloide aguda
VENCLEXTA en combinación con azacitidina
La seguridad de VENCLEXTA en combinación con azacitidina (VEN+AZA) (N=283) frente a placebo en combinación con azacitidina (PBO+AZA) (N=144) se evaluó en VIALE-A, un ensayo aleatorizado doble ciego, en pacientes con AML de novo [ver Estudios clínicos (14.2)]. En la línea de base, los pacientes tenían ≥75 años de edad o tenían comorbilidades que impedían el uso de quimioterapia de inducción intensiva basada en al menos uno de los siguientes criterios: estado de rendimiento ECOG de línea de base de 2-3, comorbilidad cardíaca o pulmonar grave, insuficiencia hepática moderada, CLcr <45 mL/min u otra comorbilidad. Los pacientes se asignaron aleatoriamente para recibir VENCLEXTA 400 mg por vía oral una vez al día después de completar la fase de aumento gradual en combinación con azacitidina (75 mg/m2 ya sea por vía intravenosa o subcutánea en los días 1-7 de cada ciclo de 28 días) o placebo en combinación con azacitidina. Entre los pacientes que recibieron VEN+AZA, la duración media de la exposición a VENCLEXTA fue de 7,6 meses (rango: <0,1 a 30,7 meses).
Se informaron reacciones adversas graves en el 83% de los pacientes que recibieron VEN+AZA, siendo las más frecuentes (≥5%) la neutropenia febril (30%), la neumonía (22%), la sepsis (excluyendo la micótica; 19%) y la hemorragia (6%). Las reacciones adversas fatales ocurrieron en el 23% de los pacientes que recibieron VEN+AZA, siendo las más frecuentes (≥2%) la neumonía (4%), la sepsis (excluyendo la micótica; 3%) y la hemorragia (2%).
Las reacciones adversas llevaron a la interrupción permanente de VENCLEXTA en el 24% de los pacientes, a reducciones de dosis en el 2% y a interrupciones de dosis en el 72%. Las reacciones adversas que llevaron a la interrupción de VENCLEXTA en ≥2% de los pacientes fueron sepsis (excluyendo la micótica; 3%) y neumonía (2%). La reacción adversa más frecuente que llevó a la reducción de la dosis fue la neumonía (0,7%). Las reacciones adversas que requirieron una interrupción de la dosis en ≥5% de los pacientes incluyeron neutropenia febril (20%), neutropenia (20%), neumonía (14%), sepsis (excluyendo la micótica; 11%) y trombocitopenia (10%). Entre los pacientes que lograron la eliminación de la leucemia de la médula ósea, el 53% sufrió interrupciones de la dosis por un recuento absoluto de neutrófilos (ANC) <500/microlitro.
Tabla 15 presenta las reacciones adversas identificadas en VIALE-A.
| Reacción adversa | VENCLEXTA + Azacitidina (N = 283) |
Placebo + Azacitidina (N = 144) |
||
| Todos los grados (%) |
Grado 3 o 4 (%) |
Todos los grados (%) |
Grado 3 o 4 (%) |
|
| Trastornos gastrointestinales | ||||
| Náuseas | 44 | 2 | 35 | <1 |
| Diarreaa | 43 | 5 | 33 | 3 |
| Vómitosb | 30 | 2 | 23 | <1 |
| Estomatitisc | 18 | 1 | 13 | 0 |
| Dolor abdominald | 18 | <1 | 13 | 0 |
| Trastornos de la sangre y del sistema linfático | ||||
| Neutropenia febril | 42 | 42 | 19 | 19 |
| Trastornos musculoesqueléticos y del tejido conjuntivo | ||||
| Dolor musculoesqueléticoe | 36 | 2 | 28 | 1 |
| Trastornos generales y condiciones del lugar de administración | ||||
| Fatigaf | 31 | 6 | 23 | 2 |
| Edemag | 27 | <1 | 19 | 0 |
| Trastornos vasculares | ||||
| Hemorragiah | 27 | 7 | 24 | 3 |
| Hipotensióni | 12 | 5 | 8 | 3 |
| Trastornos del metabolismo y la nutrición | ||||
| Disminución del apetitoj | 25 | 4 | 17 | <1 |
| Trastornos de la piel y del tejido subcutáneo | ||||
| Erupciónk | 25 | 1 | 15 | 0 |
| Infecciones e infestaciones | ||||
| Sepsisl (excluyendo hongos) | 22 | 22 | 16 | 14 |
| Infección del tracto urinariom | 16 | 6 | 9 | 6 |
| Trastornos respiratorios, torácicos y mediastínicos | ||||
| Disnean | 18 | 4 | 10 | 2 |
| Trastornos del sistema nervioso | ||||
| Mareoso | 17 | <1 | 8 | <1 |
| aIncluye diarrea y colitis. bIncluye vómitos y hematemesis. cIncluye estomatitis, ulceración bucal, inflamación de la mucosa, queilitis, úlcera aftosa, glositis y ulceración de la lengua. dIncluye dolor abdominal, dolor abdominal superior, molestia abdominal y dolor abdominal inferior. eIncluye artralgia, dolor de espalda, dolor en las extremidades, dolor musculoesquelético, dolor óseo, mialgia, dolor de cuello, dolor torácico no cardíaco, artritis, dolor torácico musculoesquelético, rigidez musculoesquelética, dolor espinal y malestar musculoesquelético. fIncluye fatiga y astenia. gIncluye edema periférico, edema, edema generalizado, edema de párpados, edema facial, edema peniano, edema periorbital e hinchazón. hIncluye epistaxis, hematuria, hemorragia conjuntival, hemoptisis, hemorragia hemorroidal, sangrado gingival, hemorragia bucal, hemorragia intracraneal, hemorragia vaginal, hemorragia cerebral, hemorragia gastrointestinal, hemorragia muscular, hemorragia cutánea, hemorragia gastrointestinal superior, hemorragia anal, hemorragia ocular, gastritis hemorrágica, hemorragia, hemorragia del tracto urinario, diátesis hemorrágica, accidente cerebrovascular hemorrágico, vasculitis hemorrágica, hemorragia gastrointestinal inferior, hemorragia de la mucosa, hemorragia peniana, hemorragia postprocedimiento, hemorragia rectal, hemorragia retiniana, shock hemorrágico, hemorragia de tejidos blandos, hemorragia subdural, hemorragia de la lengua, hemorragia uretral, hemorragia en el sitio de punción vascular, hemorragia vítrea y hemorragia de la herida. iIncluye hipotensión e hipotensión ortostática. jIncluye disminución del apetito e hiporexia. kIncluye erupción cutánea, erupción maculopapular, erupción macular, erupción medicamentosa, erupción papular, erupción pustular, eczema, erupción eritematosa, erupción pruriginosa, dermatitis acneiforme, erupción morbiliforme, dermatitis, eczema asteatósico, erupción exfoliativa y dermatitis perivascular. lIncluye sepsis, bacteriemia por escherichia, sepsis por escherichia, shock séptico, bacteriemia, bacteriemia estafilocócica, bacteriemia por klebsiella, sepsis estafilocócica, bacteriemia estreptocócica, bacteriemia enterococo, sepsis por klebsiella, bacteriemia por pseudomonas, sepsis por pseudomonas, urosepsis, sepsis bacteriana, sepsis por clostridios, sepsis por enterococo, sepsis neutropénica y sepsis estreptocócica. mIncluye infección del tracto urinario, infección del tracto urinario por escherichia, cistitis, infección del tracto urinario por enterococo, infección del tracto urinario bacteriana, pielonefritis aguda e infección del tracto urinario por pseudomonas. nIncluye disnea, disnea de esfuerzo y disnea en reposo. oIncluye mareos y vértigo. |
||||
Otras reacciones adversas clínicamente importantes (todos los grados) en ≥10% que no cumplieron con los criterios para Tabla 15 o <10% se presentan a continuación:
Trastornos hepatobiliares: colecistitis/colelitiasisa (4%)
Infecciones e infestaciones: neumoníab (33%)
Trastornos del metabolismo y la nutrición: síndrome de lisis tumoral (1%)
Trastornos del sistema nervioso: cefaleac (11%)
Investigaciones: disminución del peso (13%).
aIncluye colecistitis aguda, colelitiasis, colecistitis y colecistitis crónica.
bIncluye neumonía, infección pulmonar, neumonía fúngica, neumonía por klebsiella, neumonía atípica, infección del tracto respiratorio inferior, neumonía viral, infección del tracto respiratorio inferior fúngica, neumonía por hemophilus, neumonía neumocócica y neumonía por virus respiratorio sincitial.
cIncluye cefalea y cefalea tensional.
Tabla 16 presenta las anormalidades de laboratorio identificadas en VIALE-A.
| Anormalidad de laboratorio | VENCLEXTA + Azacitidina |
Placebo + Azacitidina |
||
| Todos los grados (%) |
Grado 3 o 4 (%) |
Todos los grados (%) |
Grado 3 o 4 (%) |
|
| Hematología | ||||
| Neutrófilos disminuidos | 98 | 98 | 88 | 81 |
| Plaquetas disminuidas | 94 | 88 | 94 | 80 |
| Linfocitos disminuidos | 91 | 71 | 72 | 39 |
| Hemoglobina disminuida | 61 | 57 | 56 | 52 |
| Química | ||||
| Bilirrubina aumentada | 53 | 7 | 40 | 4 |
| Calcio disminuido | 51 | 6 | 39 | 9 |
| Sodio disminuido | 46 | 14 | 47 | 8 |
| Fosfatasa alcalina aumentada | 42 | 1 | 29 | <1 |
| Bicarbonato en sangre disminuido | 31 | <1 | 25 | 0 |
| El denominador utilizado para calcular la tasa varió de 85 a 144 en el brazo PBO+AZA y de 125 a 283 en el brazo VEN+AZA en función del número de pacientes con al menos un valor posterior al tratamiento. | ||||
VENCLEXTA en combinación con azacitidina o decitabina
La seguridad de VENCLEXTA en combinación con azacitidina (N=67) o decitabina (N=13) se evaluó en M14-358, un ensayo no aleatorizado de pacientes con AML de novo. En la línea de base, los pacientes tenían ≥75 años de edad o tenían comorbilidades que impedían el uso de quimioterapia de inducción intensiva según al menos uno de los siguientes criterios: estado de rendimiento ECOG de línea de base de 2-3, comorbilidad cardíaca o pulmonar grave, insuficiencia hepática moderada, CLcr <45 mL/min u otra comorbilidad [ver Estudios clínicos (14.2)]. Los pacientes recibieron VENCLEXTA 400 mg por vía oral una vez al día después de completar la fase de aumento en combinación con azacitidina (75 mg/m2 ya sea por vía intravenosa o subcutánea en los días 1-7 de cada ciclo de 28 días) o decitabina (20 mg/m2 por vía intravenosa en los días 1-5 de cada ciclo de 28 días).
Azacitidina
La duración media de la exposición a VENCLEXTA cuando se administró en combinación con azacitidina fue de 6,5 meses (rango: 0,1 a 38,1 meses). La seguridad de VENCLEXTA en combinación con azacitidina en este ensayo es consistente con la de VIALE-A.
Decitabina
La duración media de la exposición a VENCLEXTA cuando se administró en combinación con decitabina fue de 8,4 meses (rango: 0,5 a 39 meses).
Se informaron reacciones adversas graves en el 85% de los pacientes que recibieron VENCLEXTA con decitabina, siendo las más frecuentes (≥10%) la sepsis (excluyendo la micótica; 46%), la neutropenia febril (38%) y la neumonía (31%). Una (8%) reacción adversa fatal de bacteriemia ocurrió dentro de los 30 días posteriores al inicio del tratamiento.
La interrupción permanente de VENCLEXTA debido a reacciones adversas ocurrió en el 38% de los pacientes. La reacción adversa más frecuente que condujo a la interrupción permanente (≥5%) fue la neumonía (8%).
Las reducciones de dosis de VENCLEXTA debido a reacciones adversas ocurrieron en el 15% de los pacientes. La reacción adversa más frecuente que condujo a la reducción de la dosis (≥5%) fue la neutropenia (15%).
Las interrupciones de la dosis de VENCLEXTA debido a reacciones adversas ocurrieron en el 69% de los pacientes. Las reacciones adversas más frecuentes que condujeron a la interrupción de la dosis (≥10%) fueron la neutropenia (38%), la neutropenia febril (23%), la leucopenia (15%) y la neumonía (15%).
Las reacciones adversas más comunes (≥30%) fueron la neutropenia febril (69%), la fatiga (62%), el estreñimiento (62%), el dolor musculoesquelético (54%), el mareo (54%), las náuseas (54%), el dolor abdominal (46%), la diarrea (46%), la neumonía (46%), la sepsis (excluyendo la micótica; 46%), la tos (38%), la pirexia (31%), la hipotensión (31%), el dolor orofaríngeo (31%), el edema (31%) y el vómito (31%). Las anomalías de laboratorio más comunes (≥30%) fueron los neutrófilos disminuidos (100%), los linfocitos disminuidos (100%), los glóbulos blancos disminuidos (100%), las plaquetas disminuidas (92%), el calcio disminuido (85%), la hemoglobina disminuida (69%), la glucosa aumentada (69%), el magnesio disminuido (54%), el potasio disminuido (46%), la bilirrubina aumentada (46%), la albúmina disminuida (38%), la fosfatasa alcalina aumentada (38%), el sodio disminuido (38%), la ALT aumentada (31%), la creatinina aumentada (31%) y el potasio aumentado (31%).
VENCLEXTA en combinación con citarabina de dosis baja
VIALE-C
La seguridad de VENCLEXTA en combinación con citarabina de dosis baja (VEN+LDAC) (N=142) versus placebo con citarabina de dosis baja (PBO+LDAC) (N=68) se evaluó en VIALE-C, un ensayo aleatorizado doble ciego en pacientes con AML de novo. En la línea de base, los pacientes tenían ≥75 años de edad o tenían comorbilidades que impedían el uso de quimioterapia de inducción intensiva según al menos uno de los siguientes criterios: estado de rendimiento ECOG de línea de base de 2-3, comorbilidad cardíaca o pulmonar grave, insuficiencia hepática moderada, CLcr <45 mL/min u otra comorbilidad [ver Estudios clínicos (14.2)]. Los pacientes fueron aleatorizados para recibir VENCLEXTA 600 mg por vía oral una vez al día después de completar una fase de aumento de 4 días en combinación con citarabina de dosis baja (20 mg/m2 por vía subcutánea una vez al día en los días 1-10 de cada ciclo de 28 días) o placebo en combinación con citarabina de dosis baja. Entre los pacientes que recibieron VEN+LDAC, la duración media de la exposición a VENCLEXTA fue de 3,9 meses (rango: <0,1 a 17,1 meses).
Se informaron reacciones adversas graves en el 65% de los pacientes que recibieron VEN+LDAC, siendo las más frecuentes (≥10%) la neumonía (17%), la neutropenia febril (16%) y la sepsis (excluyendo la micótica; 12%). Las reacciones adversas fatales ocurrieron en el 23% de los pacientes que recibieron VEN+LDAC, siendo las más frecuentes (≥5%) la neumonía (6%) y la sepsis (excluyendo la micótica; 7%).
Las reacciones adversas llevaron a la interrupción permanente de VENCLEXTA en el 25% de los pacientes, a reducciones de dosis en el 9% y a interrupciones de dosis en el 63%. La reacción adversa más frecuente (>2%) que resultó en la interrupción permanente de VENCLEXTA fue la neumonía (6%). Las reacciones adversas que requirieron una reducción de la dosis en ≥1% de los pacientes fueron la neumonía (1%) y la trombocitopenia (1%), y las reacciones adversas que requirieron una interrupción de la dosis en ≥5% de los pacientes incluyeron la neutropenia (20%), la trombocitopenia (15%), la neumonía (8%), la neutropenia febril (6%) y la sepsis (excluyendo la micótica; 6%). Entre los pacientes que lograron la eliminación de la leucemia de la médula ósea, el 32% sufrió interrupciones de la dosis por ANC <500/microlitro.
Tabla 17 presenta las reacciones adversas identificadas en VIALE-C.
| Reacción adversa | VENCLEXTA + Dosis baja Citarabina (N = 142) |
Placebo + Dosis baja Citarabina (N = 68) |
||
| Todos los grados (%) |
Grado 3 o 4 (%) |
Todos los grados (%) |
Grado 3 o 4 (%) |
|
| Trastornos gastrointestinales | ||||
| Náuseas | 42 | 1 | 31 | 0 |
| Diarrea | 28 | 3 | 16 | 0 |
| Vómitos | 25 | <1 | 13 | 0 |
| Dolor abdominala | 15 | <1 | 9 | 3 |
| Estomatitisb | 15 | 1 | 6 | 0 |
| Trastornos de la sangre y del sistema linfático | ||||
| Neutropenia febril | 32 | 32 | 29 | 29 |
| Infecciones e infestaciones | ||||
| Neumoníac | 29 | 19 | 21 | 21 |
| Trastornos vasculares | ||||
| Hemorragiad | 27 | 8 | 16 | 1 |
| Hipotensióne | 11 | 5 | 4 | 1 |
| Trastornos musculoesqueléticos y del tejido conjuntivo | ||||
| Dolor musculoesqueléticof | 23 | 3 | 18 | 0 |
| Trastornos generales y condiciones del lugar de administración | ||||
| Fatigag | 22 | 2 | 21 | 0 |
| Trastornos del sistema nervioso | ||||
| Dolor de cabeza | 11 | 0 | 6 | 0 |
| aIncluye dolor abdominal, dolor abdominal superior, malestar abdominal y dolor abdominal inferior. bIncluye estomatitis, ulceración bucal, úlcera aftosa, glositis, inflamación de la mucosa y ulceración de la lengua. cIncluye neumonía, infección pulmonar, infección del tracto respiratorio inferior, neumonía fúngica, infección del tracto respiratorio inferior fúngica, neumonía por Pneumocystis jirovecii, neumonía por aspiración, neumonía citomegálica y neumonía pseudomonal. dIncluye epistaxis, hemorragia conjuntival, hemoptisis, hemorragia gastrointestinal, sangrado gingival, hemorragia bucal, hemorragia gastrointestinal superior, hematuria, hemorragia retiniana, hemorragia en el sitio de catéter, hemorragia cerebral, hemorragia gástrica, gastritis hemorrágica, hemorragia intracraneal, hemorragia subcutánea, hemorragia labial, hemorragia de la mucosa, hemorragia faríngea, hemorragia postprocedimiento, hemorragia alveolar pulmonar, hemorragia pulmonar, hemorragia de la pulpa dental, hemorragia uterina y hemorragia en el sitio de acceso vascular. eIncluye hipotensión e hipotensión ortostática. fIncluye dolor de espalda, artralgia, dolor en las extremidades, dolor musculoesquelético, mialgia, dolor de cuello, dolor torácico no cardíaco, artritis, dolor óseo, dolor musculoesquelético torácico y dolor espinal. gIncluye fatiga y astenia. |
||||
Otras reacciones adversas clínicamente importantes (todos los grados) en ≥10% que no cumplieron con los criterios para Tabla 17 o <10% se presentan a continuación:
Trastornos hepatobiliares: colecistitis/colelitiasisa (1%)
Infecciones e infestaciones: sepsisb (excluyendo micótica; 15%), infección del tracto urinarioc (8%)
Trastornos del metabolismo y la nutrición: disminución del apetito (19%), síndrome de lisis tumoral (6%)
Trastornos del sistema nervioso: mareosd (9%)
Trastornos respiratorios, torácicos y mediastínicos: disneae (10%)
Investigaciones: disminución del peso (9%).
aIncluye colecistitis y colecistitis aguda.
bIncluye sepsis, bacteriemia, shock séptico, sepsis neutropénica, bacteriemia estafilocócica, bacteriemia estreptocócica, sepsis bacteriana, bacteriemia por Escherichia, bacteriemia por Pseudomonas y sepsis estafilocócica.
cIncluye infección del tracto urinario e infección del tracto urinario por Escherichia.
dIncluye mareos y vértigo.
eIncluye disnea y disnea de esfuerzo.
Tabla 18 describe las anormalidades de laboratorio identificadas en VIALE-C.
| Anormalidad de laboratorio | VENCLEXTA + Dosis baja Citarabina |
Placebo + Dosis baja Citarabina |
||
| Todos los grados (%) |
Grado 3 o 4 (%) |
Todos los grados (%) |
Grado 3 o 4 (%) |
|
| Hematología | ||||
| Plaquetas disminuidas | 97 | 95 | 92 | 90 |
| Neutrófilos disminuidos | 95 | 92 | 82 | 71 |
| Linfocitos disminuidos | 92 | 69 | 65 | 24 |
| Hemoglobina disminuida | 63 | 57 | 57 | 54 |
| Química | ||||
| Bilirrubina aumentada | 61 | 7 | 38 | 7 |
| Albúmina disminuida | 61 | 6 | 43 | 4 |
| Potasio disminuido | 56 | 16 | 42 | 14 |
| Calcio disminuido | 53 | 8 | 45 | 13 |
| Glucosa aumentada | 52 | 13 | 59 | 9 |
| AST aumentada | 36 | 6 | 37 | 1 |
| Fosfatasa alcalina aumentada | 34 | 1 | 26 | 1 |
| ALT aumentada | 30 | 4 | 26 | 1 |
| Sodio aumentado | 11 | 3 | 6 | 1 |
| El denominador utilizado para calcular la tasa varió de 38 a 68 en el brazo PBO+LDAC y de 65 a 142 en el brazo VEN+LDAC en función del número de pacientes con al menos un valor posterior al tratamiento. | ||||
M14-387
La seguridad de VENCLEXTA en combinación con citarabina de dosis baja (N=61) se evaluó en M14-387, un ensayo no aleatorizado, abierto de pacientes con AML de novo [ver Estudios clínicos (14.2)]. En la línea de base, los pacientes tenían ≥75 años de edad, o tenían comorbilidades que impedían el uso de quimioterapia de inducción intensiva basada en al menos uno de los siguientes criterios: estado de rendimiento ECOG de línea de base de 2-3, comorbilidad cardíaca o pulmonar grave, insuficiencia hepática moderada, CLcr <45 mL/min, u otra comorbilidad. Los pacientes recibieron VENCLEXTA 600 mg por vía oral una vez al día después de completar la fase de aumento en combinación con citarabina de dosis baja (20 mg/m2 por vía subcutánea los días 1-10 de cada ciclo de 28 días). La seguridad de VENCLEXTA en combinación con citarabina de dosis baja es consistente con la de VIALE-C.
7 INTERACCIONES MEDICAMENTOSAS
7.1 Efectos de otros medicamentos en VENCLEXTA
Inhibidores fuertes o moderados de CYP3A o inhibidores de P-gp
El uso concomitante con un inhibidor fuerte o moderado de CYP3A o un inhibidor de P-gp aumenta la Cmax y el AUC0-INF de venetoclax [ver Farmacología clínica (12.3)], lo que puede aumentar las toxicidades de VENCLEXTA, incluido el riesgo de TLS [ver Advertencias y precauciones (5.1)].
El uso concomitante con un inhibidor fuerte de CYP3A al inicio y durante la fase de aumento de la dosis en pacientes con CLL/SLL está contraindicado [ver Contraindicaciones (4)].
En pacientes con CLL/SLL que toman una dosis diaria constante (después de la fase de aumento de la dosis), considere medicamentos alternativos o ajuste la dosis de VENCLEXTA y controle con más frecuencia las reacciones adversas [ver Dosis y administración (2.5, 2.6)].
En pacientes con AML, ajuste la dosis de VENCLEXTA y controle con más frecuencia las reacciones adversas [ver Dosis y administración (2.5, 2.6)].
Reanude la dosis de VENCLEXTA que se usó antes del uso concomitante con un inhibidor fuerte o moderado de CYP3A o un inhibidor de P-gp de 2 a 3 días después de la interrupción del inhibidor [ver Dosis y administración (2.5, 2.6)].
Evite los productos de pomelo, las naranjas de Sevilla y la carambola durante el tratamiento con VENCLEXTA, ya que contienen inhibidores de CYP3A.
Inductores fuertes o moderados de CYP3A
El uso concomitante con un inductor fuerte de CYP3A disminuye la Cmax y el AUC0-INF de venetoclax [ver Farmacología clínica (12.3)], lo que puede disminuir la eficacia de VENCLEXTA. Evite el uso concomitante de VENCLEXTA con inductores fuertes de CYP3A o inductores moderados de CYP3A.
7.2 Efecto de VENCLEXTA en otros medicamentos
Warfarina
El uso concomitante de VENCLEXTA aumenta la Cmax y el AUC0-INF de warfarina [ver Farmacología clínica (12.3)], lo que puede aumentar el riesgo de sangrado. Controle la relación internacional normalizada (INR) con más frecuencia en pacientes que usan warfarina de forma concomitante con VENCLEXTA.
Sustratos de P-gp
El uso concomitante de VENCLEXTA aumenta la Cmax y el AUC0-INF de los sustratos de P-gp [ver Farmacología clínica (12.3)], lo que puede aumentar las toxicidades de estos sustratos. Evite el uso concomitante de VENCLEXTA con un sustrato de P-gp. Si el uso concomitante es inevitable, separe la dosificación del sustrato de P-gp al menos 6 horas antes de VENCLEXTA.
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de Riesgos
Basado en los hallazgos en animales y su mecanismo de acción [ver Farmacología Clínica (12.1)], VENCLEXTA puede causar daño embrionario-fetal cuando se administra a una mujer embarazada. No hay datos disponibles sobre el uso de VENCLEXTA en mujeres embarazadas para informar un riesgo asociado al medicamento. La administración de venetoclax a ratones embarazadas durante el período de organogénesis fue fetotóxica a exposiciones 1.2 veces la exposición humana a la dosis recomendada de 400 mg diarios basada en AUC. Advierta a las mujeres embarazadas sobre el riesgo potencial para un feto.
El riesgo de fondo estimado de defectos de nacimiento mayores y aborto espontáneo para la población indicada es desconocido. Todos los embarazos tienen un riesgo de fondo de defectos de nacimiento, pérdida u otros resultados adversos. En la población general de los EE. UU., el riesgo de fondo estimado de defectos de nacimiento mayores y aborto espontáneo en embarazos clínicamente reconocidos es del 2% al 4% y del 15% al 20%, respectivamente.
Datos
Datos de animales
En estudios de desarrollo embrionario-fetal, venetoclax se administró a ratones y conejos embarazadas durante el período de organogénesis. En ratones, venetoclax se asoció con un aumento de la pérdida postimplantación y una disminución del peso corporal fetal a 150 mg/kg/día (exposiciones maternas aproximadamente 1.2 veces la exposición humana a la dosis recomendada de 400 mg una vez al día). No se observó teratogenicidad ni en el ratón ni en el conejo.
8.2 Lactancia
Resumen de Riesgos
No hay datos sobre la presencia de VENCLEXTA en la leche materna o los efectos en el niño amamantado o la producción de leche. Venetoclax estuvo presente en la leche cuando se administró a ratas lactantes (ver Datos).
Debido al potencial de reacciones adversas graves en un niño amamantado, aconseje a las mujeres que no amamanten durante el tratamiento con VENCLEXTA y durante 1 semana después de la última dosis.
Datos
Datos de animales
Venetoclax se administró (dosis única; 150 mg/kg oral) a ratas lactantes de 8 a 10 días después del parto. Venetoclax en la leche fue 1.6 veces menor que en el plasma. El fármaco original (venetoclax) representó la mayoría del material total relacionado con el fármaco en la leche, con niveles traza de tres metabolitos.
8.3 Mujeres y Hombres en Potencial Reproductivo
VENCLEXTA puede causar daño fetal cuando se administra a mujeres embarazadas [ver Uso en Poblaciones Específicas (8.1)].
Prueba de Embarazo
Verifique el estado de embarazo en mujeres en potencial reproductivo antes de iniciar VENCLEXTA.
Anticoncepción
Aconseje a las mujeres en potencial reproductivo que usen métodos anticonceptivos efectivos durante el tratamiento con VENCLEXTA y durante 30 días después de la última dosis.
Infertilidad
Basado en los hallazgos en animales, VENCLEXTA puede afectar la fertilidad masculina [ver Toxicología No Clínica (13.1)].
8.4 Uso Pediátrico
La seguridad y eficacia de VENCLEXTA no se han establecido en pacientes pediátricos.
Datos de Toxicidad en Animales Juveniles
En un estudio de toxicología juvenil, se administró venetoclax a ratones a 10, 30 o 100 mg/kg/día por sonda gástrica oral desde los 7 hasta los 60 días de edad. Los signos clínicos de toxicidad incluyeron disminución de la actividad, deshidratación, palidez de la piel y postura encorvada a ≥30 mg/kg/día. Además, se produjeron efectos de mortalidad y peso corporal a 100 mg/kg/día. Otros efectos relacionados con venetoclax fueron disminuciones reversibles en los linfocitos a ≥10 mg/kg/día; una dosis de 10 mg/kg/día es aproximadamente 0.06 veces la dosis clínica de 400 mg en una base de mg/m2 para un niño de 20 kg.
8.5 Uso Geriátrico
Leucemia Linfocítica Crónica/Linfoma Linfocítico Pequeño
De los 352 pacientes con CLL/SLL previamente tratados evaluados para seguridad en 3 ensayos abiertos de monoterapia con VENCLEXTA, el 57% (201/352) tenían ≥65 años de edad y el 18% (62/352) tenían ≥75 años de edad.
No se observaron diferencias clínicamente significativas en seguridad y eficacia entre pacientes mayores y más jóvenes en los estudios de combinación y monoterapia.
Leucemia Mieloide Aguda
De los 283 pacientes que recibieron VENCLEXTA con azacitidina en VIALE-A, el 96% tenían ≥65 años de edad y el 60% tenían ≥75 años de edad.
De los 13 pacientes que recibieron VENCLEXTA en combinación con decitabina en M14-358, el 100% tenían ≥65 años de edad y el 62% tenían ≥75 años de edad.
De los 142 pacientes que recibieron VENCLEXTA en combinación con citarabina de dosis baja en VIALE-C, el 92% tenían ≥65 años de edad y el 57% tenían ≥75 años de edad.
Los estudios clínicos de VENCLEXTA en pacientes con AML no incluyeron un número suficiente de adultos más jóvenes para determinar si los pacientes de 65 años de edad o mayores responden de manera diferente a los adultos más jóvenes.
8.6 Deterioro Renal
Debido al mayor riesgo de TLS, los pacientes con función renal reducida (CLcr <80 mL/min, calculado por la fórmula de Cockcroft-Gault) requieren una profilaxis y un seguimiento más intensivos para reducir el riesgo de TLS al iniciar el tratamiento con VENCLEXTA [ver Dosificación y administración (2.1, 2.2, 2.3, 2.4) y Advertencias y precauciones (5.1)].
No se recomienda un ajuste de dosis para pacientes con deterioro renal leve, moderado o grave (CLcr ≥15 mL/min) [ver Farmacología clínica (12.3)].
8.7 Deterioro Hepático
No se recomienda un ajuste de dosis para pacientes con deterioro hepático leve (Child-Pugh A) o moderado (Child-Pugh B).
Reduzca la dosis de VENCLEXTA para pacientes con deterioro hepático grave (Child-Pugh C); controle a estos pacientes con más frecuencia para detectar reacciones adversas [ver Dosificación y administración (2.5, 2.7) y Farmacología clínica (12.3)].
10 SOBREDOSIS
No existe un antídoto específico para VENCLEXTA. Para los pacientes que experimenten sobredosis, monitorice de cerca y proporcione tratamiento de apoyo apropiado; durante la fase de aumento, interrumpa VENCLEXTA y controle cuidadosamente los signos y síntomas de TLS junto con otras toxicidades [ver Dosificación y administración (2.2, 2.3, 2.4, 2.5)]. Debido al gran volumen de distribución de venetoclax y su extensa unión a proteínas, es poco probable que la diálisis resulte en una eliminación significativa de venetoclax.
11 DESCRIPCIÓN
Venetoclax es un inhibidor de BCL-2. Es un sólido de color amarillo claro a amarillo oscuro con la fórmula empírica C45H50ClN7O7S y un peso molecular de 868.44. Venetoclax se describe químicamente como 4-(4-{[2-(4-clorofenil)-4,4-dimetilciclohex-1-en-1-il]metil}piperazina-1-il)-N-({3-nitro-4-[(tetrahidro-2H-pirano-4-ylmetil)amino]fenil}sulfonilo)-2-(1H-pirrolo[2,3-b]piridina-5-yloxi)benzamida) y tiene la siguiente estructura química:
![la siguiente estructura química para Venetoclax es un inhibidor de BCL-2. Es un sólido de color amarillo claro a amarillo oscuro con la fórmula empírica C45H50ClN7O7S y un peso molecular de 868.44. Venetoclax se describe químicamente como 4-(4-{[2-(4-clorofenil)-4,4-dimetilciclohex-1-en-1-il]metil}piperazina-1-il)-N-({3-nitro-4-[(tetrahidro-2H-pirano-4-ylmetil)amino]fenil}sulfonilo)-2-(1H-pirrolo[2,3-b]piridina-5-yloxi)benzamida).](/images/111/venetoclax-spl-01.jpg)
Venetoclax tiene una solubilidad acuosa muy baja.
Las tabletas VENCLEXTA para uso oral se suministran como tabletas de color amarillo pálido o beige que contienen 10, 50 o 100 mg de venetoclax como ingrediente activo. Cada tableta también contiene los siguientes ingredientes inactivos: copovidona, dióxido de silicio coloidal, polisorbato 80, fumarato de estearilo sódico y fosfato de calcio dibásico. Además, las tabletas recubiertas de 10 mg y 100 mg incluyen lo siguiente: óxido de hierro amarillo, alcohol polivinílico, polietilenglicol, talco y dióxido de titanio. Las tabletas recubiertas de 50 mg también incluyen lo siguiente: óxido de hierro amarillo, óxido de hierro rojo, óxido de hierro negro, alcohol polivinílico, talco, polietilenglicol y dióxido de titanio. Cada tableta está marcada con “V” en un lado y “10”, “50” o “100” correspondiente a la potencia de la tableta en el otro lado.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de Acción
Venetoclax es un inhibidor selectivo y bioavailable por vía oral de BCL-2, una proteína anti-apoptótica. Se ha demostrado la sobreexpresión de BCL-2 en células de CLL y AML, donde media la supervivencia de las células tumorales y se ha asociado con resistencia a los quimioterapéuticos. Venetoclax ayuda a restaurar el proceso de apoptosis al unirse directamente a la proteína BCL-2, desplazando proteínas pro-apoptóticas como BIM, desencadenando la permeabilización de la membrana externa mitocondrial y la activación de caspasas. En estudios no clínicos, venetoclax ha demostrado actividad citotóxica en células tumorales que sobreexpresan BCL-2.
12.2 Farmacodinámica
Basado en los análisis de respuesta a la exposición para la eficacia, se observó una relación entre la exposición al fármaco y una mayor probabilidad de respuesta en estudios clínicos en pacientes con CLL/SLL y en pacientes con AML. Basado en los análisis de respuesta a la exposición para la seguridad, se observó una relación entre la exposición al fármaco y una mayor probabilidad de algunos eventos de seguridad en estudios clínicos en pacientes con AML. No se observó relación de exposición-seguridad en pacientes con CLL/SLL a dosis de hasta 1200 mg administradas como monoterapia y hasta 600 mg administradas en combinación con rituximab.
Electrofisiología Cardíaca
El efecto de múltiples dosis de VENCLEXTA de hasta 1200 mg una vez al día (2 veces la dosis máxima recomendada aprobada) sobre el intervalo QTc fue evaluado en un ensayo abierto, de un solo brazo en 176 pacientes con malignidades hematológicas previamente tratadas. VENCLEXTA no tuvo un gran efecto en el intervalo QTc (es decir, >20 ms) y no hubo relación entre la exposición a venetoclax y el cambio en el intervalo QTc.
12.3 Farmacocinética
La concentración máxima media (± desviación estándar) en estado estacionario de Cmax de venetoclax fue de 2.1 ± 1.1 mcg/mL y AUC0-24h fue de 32.8 ± 16.9 mcg•h/mL tras la administración de 400 mg una vez al día con una comida baja en grasa. El AUC en estado estacionario de venetoclax aumentó proporcionalmente en el rango de dosis de 150 a 800 mg (0.25 a 1.33 veces la dosis máxima recomendada aprobada). La farmacocinética de venetoclax no cambia con el tiempo.
Absorción
La concentración máxima en plasma de venetoclax se alcanzó de 5 a 8 horas tras la administración oral múltiple en condiciones alimentarias.
Efecto de los Alimentos
La administración con una comida baja en grasa (aproximadamente 512 kilocalorías, 25% de calorías de grasa, 60% de calorías de carbohidratos y 15% de calorías de proteínas) aumentó la exposición a venetoclax aproximadamente 3.4 veces y la administración con una comida alta en grasa (aproximadamente 753 kilocalorías, 55% de calorías de grasa, 28% de calorías de carbohidratos y 17% de calorías de proteínas) aumentó la exposición a venetoclax de 5.1 a 5.3 veces en comparación con las condiciones de ayuno.
Distribución
Venetoclax se une altamente a las proteínas plasmáticas humanas con una fracción no unida en plasma <0.01 en un rango de concentración de 1-30 micromolar (0.87-26 mcg/mL). La relación media sangre-plasma fue de 0.57. El volumen aparente de distribución (Vdss/F) de venetoclax varió de 256-321 L en pacientes.
Eliminación
La vida media de eliminación terminal de venetoclax fue de aproximadamente 26 horas.
Metabolismo
Venetoclax es predominantemente metabolizado por CYP3A in vitro. El principal metabolito identificado en plasma, M27, tiene una actividad inhibitoria contra BCL-2 que es al menos 58 veces menor que la de venetoclax in vitro y su AUC representó el 80% de la AUC del fármaco original.
Excreción
Después de una dosis oral única de [14C]-venetoclax 200 mg a sujetos sanos, >99.9% de la dosis se recuperó en heces (21% como inalterado) y <0.1% en orina dentro de 9 días.
Poblaciones Específicas
No se observaron diferencias clínicamente significativas en la farmacocinética de venetoclax según la edad (19 a 93 años), sexo, peso, deterioro renal leve a severo (CLcr 15 a 89 mL/min, calculado por Cockcroft-Gault), o deterioro hepático leve a moderado (bilirrubina total normal y transaminasa aspartato (AST) > límite superior de normalidad (ULN) o bilirrubina total 1 a 3 veces ULN). Se desconoce el efecto de la enfermedad renal en etapa terminal (CLcr <15 mL/min) o la diálisis sobre la farmacocinética de venetoclax.
Grupos Raciales o Étnicos
No se observaron diferencias clínicamente significativas en la farmacocinética de venetoclax en pacientes blancos, negros y asiáticos inscritos en los Estados Unidos. De 771 pacientes con AML, los pacientes asiáticos de países asiáticos [China (5.6%), Japón (5.5%), Corea del Sur (2.1%) y Taiwán (0.9%)] tuvieron una exposición a venetoclax 63% mayor que las poblaciones no asiáticas.
Pacientes con Deterioro Hepático
Tras una dosis única de VENCLEXTA 50 mg, la exposición sistémica a venetoclax (AUC0-INF) fue 2.7 veces mayor en sujetos con deterioro hepático severo (Child-Pugh C) en comparación con sujetos con función hepática normal [ver Dosis y Administración (2.7) y Uso en Poblaciones Específicas (8.7)]. No se observaron diferencias clínicamente relevantes en la exposición sistémica a venetoclax entre sujetos con deterioro hepático leve o moderado y sujetos con función hepática normal.
Estudios de Interacciones Medicamentosas
Estudios Clínicos
No se observaron diferencias clínicamente significativas en la farmacocinética de venetoclax cuando se coadministró con azacitidina, azitromicina, citarabina, decitabina, agentes reductores de ácido gástrico, obinutuzumab o rituximab.
Ketoconazol
El uso concomitante de ketoconazol (un inhibidor potente de CYP3A, P-gp y BCRP) 400 mg una vez al día durante 7 días aumentó la Cmax de venetoclax en un 130% y el AUC0-INF en un 540% [ver Interacciones medicamentosas (7.1)].
Ritonavir
El uso concomitante de ritonavir (un inhibidor potente de CYP3A, P-gp y OATP1B1/B3) 50 mg una vez al día durante 14 días aumentó la Cmax de venetoclax en un 140% y el AUC en un 690% [ver Interacciones medicamentosas (7.1)].
Posaconazol
El uso concomitante de posaconazol (un inhibidor potente de CYP3A y P-gp) 300 mg con VENCLEXTA 50 mg y 100 mg durante 7 días dio como resultado un 61% y 86% más alto en la Cmax de venetoclax, respectivamente, en comparación con VENCLEXTA 400 mg administrado solo. El AUC0-24h de venetoclax fue un 90% y 144% más alto, respectivamente [ver Interacciones medicamentosas (7.1)].
Rifampicina
El uso concomitante de una sola dosis de rifampicina (un inhibidor de OATP1B1/1B3 y P-gp) 600 mg aumentó la Cmax de venetoclax en un 106% y el AUC0-INF en un 78%. El uso concomitante de múltiples dosis de rifampicina (como un inductor potente de CYP3A) 600 mg una vez al día durante 13 días disminuyó la Cmax de venetoclax en un 42% y el AUC0-INF en un 71% [ver Interacciones medicamentosas (7.1)].
Warfarina
El uso concomitante de una sola dosis de 400 mg de VENCLEXTA con 5 mg de warfarina dio como resultado un aumento del 18% al 28% en la Cmax y el AUC0-INF de R-warfarina y S-warfarina [ver Interacciones medicamentosas (7.2)].
Digoxina
El uso concomitante de una sola dosis de VENCLEXTA 100 mg con digoxina (un sustrato de P-gp) 0.5 mg aumentó la Cmax de digoxina en un 35% y el AUC0-INF en un 9% [ver Interacciones medicamentosas (7.2)].
Estudios in vitro
Venetoclax no es un inhibidor o inductor de CYP1A2, CYP2B6, CYP2C19, CYP2D6 o CYP3A4. Venetoclax es un inhibidor débil de CYP2C8, CYP2C9 y UGT1A1.
Venetoclax no es un inhibidor de UGT1A4, UGT1A6, UGT1A9 o UGT2B7.
Venetoclax es un inhibidor y sustrato de P-gp y BCRP y un inhibidor débil de OATP1B1.
Venetoclax no es un inhibidor de OATP1B3, OCT1, OCT2, OAT1, OAT3, MATE1 o MATE2K.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, mutagénesis, deterioro de la fertilidad
Ni venetoclax ni M27, un metabolito humano importante, fueron carcinógenos en un estudio de 6 meses en ratones transgénicos (Tg.rasH2) a dosis orales de hasta 400 mg/kg/día de venetoclax y a una dosis oral única de 250 mg/kg/día de M27.
Venetoclax no fue mutagénico en un ensayo de mutagenicidad bacteriana in vitro (Ames), no indujo aberraciones numéricas o estructurales en un ensayo de aberración cromosómica in vitro utilizando linfocitos de sangre periférica humana, y no fue clastogénico en un ensayo de micronúcleos de médula ósea de ratón in vivo a dosis de hasta 835 mg/kg. El metabolito M27 fue negativo para la actividad genotóxica en los ensayos in vitro de Ames y aberración cromosómica.
Se realizaron estudios de fertilidad y desarrollo embrionario temprano en ratones machos y hembras. Estos estudios evalúan el apareamiento, la fertilización y el desarrollo embrionario hasta la implantación. No hubo efectos de venetoclax en los ciclos de celo, el apareamiento, la fertilidad, el cuerpo lúteo, los implantes uterinos o los embriones vivos por camada a dosis de hasta 600 mg/kg/día. Sin embargo, existe un riesgo para la fertilidad masculina humana basado en la toxicidad testicular (pérdida de células germinales) observada en perros a exposiciones tan bajas como 0.5 veces la exposición al AUC humana a una dosis de 400 mg.
13.2 Toxicología y/o farmacología animal
En perros, venetoclax causó necrosis unicelular en varios tejidos, incluyendo la vesícula biliar, el páncreas exocrino y el estómago, sin evidencia de alteración de la integridad tisular o disfunción orgánica; estos hallazgos fueron de magnitud mínima a leve. Después de un período de dosificación de 4 semanas y un período de recuperación posterior de 4 semanas, la necrosis unicelular mínima todavía estaba presente en algunos tejidos y no se ha evaluado la reversibilidad después de períodos más largos de dosificación o recuperación.
Además, después de aproximadamente 3 meses de dosificación diaria en perros, venetoclax causó una decoloración blanca progresiva del pelaje debido a la pérdida de pigmento de melanina.
14 ESTUDIOS CLÍNICOS
14.1 Leucemia linfocítica crónica/linfoma linfocítico de células pequeñas
En combinación con obinutuzumab
CLL14 (BO25323) fue un ensayo aleatorizado (1:1), multicéntrico, abierto, con control activo (NCT02242942) que evaluó la eficacia y seguridad de VENCLEXTA en combinación con obinutuzumab (VEN+G) frente a obinutuzumab en combinación con clorambucilo (GClb) en pacientes con LLC no tratados previamente con afecciones médicas coexistentes (puntuación total de la escala de evaluación de la enfermedad acumulativa [CIRS] >6 o CLcr <70 ml/min). El ensayo requería transaminasas hepáticas y bilirrubina total ≤2 veces el límite superior de lo normal y excluía a los pacientes con transformación de Richter o cualquier puntuación de deterioro individual de órganos/sistemas de 4 según la CIRS, excepto en el sistema de órganos de ojos, oídos, nariz y garganta.
Todos los pacientes recibieron obinutuzumab a una dosis de 1000 mg los días 1 (la primera dosis pudo dividirse en 100 mg y 900 mg los días 1 y 2), 8 y 15 del ciclo 1, y el día 1 de cada ciclo posterior durante un total de 6 ciclos. Los pacientes del grupo de VEN+G comenzaron el esquema de ajuste gradual de la dosis de VENCLEXTA de 5 semanas [ver Posología y administración (2.2, 2.4)] el día 22 del ciclo 1 y recibieron 400 mg de VENCLEXTA por vía oral una vez al día desde el día 1 del ciclo 3 hasta el último día del ciclo 12. Los pacientes aleatorizados al grupo de GClb recibieron 0.5 mg/kg de clorambucilo por vía oral el día 1 y el día 15 de los ciclos 1 a 12. Cada ciclo tuvo una duración de 28 días.
Se aleatorizó a un total de 432 pacientes, 216 en cada grupo. Las características demográficas y de la enfermedad iniciales fueron similares entre los grupos. La mediana de edad fue de 72 años (rango: de 41 a 89 años), el 89% eran de raza blanca, el 67% eran hombres; el 36% y el 43% se encontraban en estadio B y C de Binet, respectivamente, y el 88% presentaba un estado funcional del Grupo Oncológico Cooperativo del Este (ECOG) <2. La mediana de la puntuación CIRS fue de 8.0 (rango: de 0 a 28) y el 58% de los pacientes tenían una CLcr <70 ml/min. Se detectó una deleción 17p en el 8% de los pacientes, mutaciones de TP53 en el 10%, deleción 11q en el 19% e IgVH no mutada en el 57%.
La eficacia se basó en la supervivencia sin progresión (SSP) según la evaluación de un Comité de Revisión Independiente (CRI). La mediana de la duración del seguimiento de la SSP fue de 28 meses (rango: de 0 a 36 meses). Los resultados de eficacia del CLL14 se muestran en la Tabla 19. La curva de Kaplan-Meier para la SSP se muestra en la Figura 1.
| Criterio de valoración | VENCLEXTA + obinutuzumab (N = 216) |
Obinutuzumab + clorambucilo (N = 216) |
| Supervivencia sin progresióna | ||
| Número de eventos, n (%) | 29 (13) | 79 (37) |
| Progresión de la enfermedad | 14 (6) | 71 (33) |
| Muerte | 15 (7) | 8 (4) |
| Mediana, meses | No alcanzada | No alcanzada |
| HR (IC del 95%)b | 0.33 (0.22, 0.51) | |
| Valor pb | <0.0001 | |
| Tasa de respuestac, n (%) | ||
| ORRd | 183 (85) | 154 (71) |
| IC del 95% | (79, 89) | (65, 77) |
| RC | 100 (46) | 47 (22) |
| RC+RCid | 107 (50) | 50 (23) |
| RP | 76 (35) | 104 (48) |
| IC = intervalo de confianza; RC = respuesta completa; RCi = respuesta completa con recuperación incompleta de la médula ósea; HR = razón de riesgo; ORR = tasa de respuesta global (RC + RCi + RP); RP = respuesta parcial. aDesde la aleatorización hasta el primer evento de progresión de la enfermedad o muerte por cualquier causa. Evaluación del CRI; estimación de Kaplan-Meier. bLa estimación de la HR se basa en el modelo de riesgos proporcionales de Cox estratificado por el estadio de Binet y la región geográfica; el valor p se basa en la prueba de rangos logarítmicos estratificada por los mismos factores. cSegún las directrices del Taller Internacional sobre Leucemia Linfocítica Crónica (IWCLL) de 2008. dValores p basados en la prueba de Cochran-Mantel-Haenszel; p=0.0007 para la ORR; p<0.0001 para la RC+RCi. |
||
Figura 1. Curva de Kaplan-Meier de la supervivencia libre de progresión evaluada por IRC en el CLL14
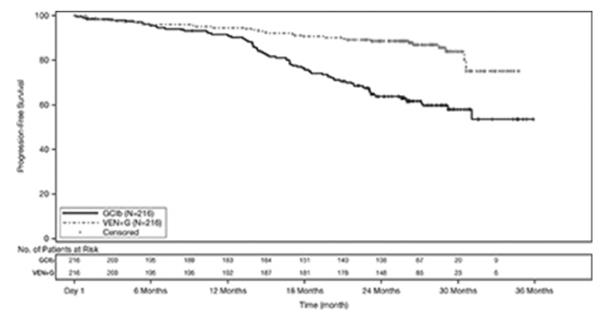
En el momento del análisis, no se había alcanzado la mediana de supervivencia global (SG), con menos del 10% de los pacientes que experimentaron un evento. La mediana de la duración del seguimiento para la SG fue de 28 meses.
La enfermedad mínima residual (EMR) se evaluó mediante la reacción en cadena de la polimerasa con oligonucleótidos específicos de alelos (ASO-PCR). La definición de estado negativo fue de menos de una célula CLL por cada 104 leucocitos. Las tasas de negatividad de la EMR 3 meses después de la finalización del tratamiento, independientemente de la respuesta y en los pacientes que lograron la RC, se muestran en la Tabla 20. En esta evaluación, se obtuvieron muestras de médula ósea coincidentes de 134 pacientes del grupo de VEN+G que eran negativos para la EMR en sangre periférica; de ellos, 122 pacientes (91%) fueron negativos para la EMR tanto en sangre periférica como en médula ósea.
| VENCLEXTA + Obinutuzumab |
Obinutuzumab + Clorambucilo |
|
| Tasa de negatividad de la EMR (población ITT) | ||
| N | 216 | 216 |
| Médula ósea, n (%) | 123 (57) | 37 (17) |
| 95% CI | (50, 64) | (12, 23) |
| valor pa | <0.0001 | |
| Sangre periférica, n (%) | 163 (76) | 76 (35) |
| 95% CI | (69, 81) | (29, 42) |
| valor pa | <0.0001 | |
| Tasa de negatividad de la EMR en pacientes con RC | ||
| N | 100 | 47 |
| Médula ósea, n (%) | 69 (69) | 21 (45) |
| 95% CI | (59, 78) | (30, 60) |
| valor pa | 0.0048 | |
| Sangre periférica, n (%) | 87 (87) | 29 (62) |
| 95% CI | (79, 93) | (46, 75) |
| valor pa | 0.0005 | |
| CI = intervalo de confianza; RC = remisión completa. aValor p basado en la prueba de chi-cuadrado. |
||
Doce meses después de la finalización del tratamiento, las tasas de MRD negativa en sangre periférica fueron del 58 % (126/216) en pacientes tratados con VEN+G y del 9 % (20/216) en pacientes tratados con GClb.
En combinación con rituximab
MURANO fue un ensayo aleatorizado (1:1), multicéntrico, abierto (NCT02005471) que evaluó la eficacia y seguridad de VENCLEXTA en combinación con rituximab (VEN+R) frente a bendamustina en combinación con rituximab (B+R) en pacientes con LLC que habían recibido al menos una línea de tratamiento previo. Los pacientes del grupo de VEN+R completaron el esquema de ajuste de dosis de VENCLEXTA de 5 semanas [ver Posología y administración (2.2, 2.4)] y recibieron 400 mg de VENCLEXTA por vía oral una vez al día durante 24 meses a partir del día 1 del ciclo 1 de rituximab en ausencia de progresión de la enfermedad o toxicidad inaceptable. El rituximab se inició después del ajuste de dosis de 5 semanas a una dosis de 375 mg/m2 por vía intravenosa el día 1 del ciclo 1 y 500 mg/m2 por vía intravenosa el día 1 de los ciclos 2-6. Los pacientes aleatorizados a B+R recibieron 70 mg/m2 de bendamustina por vía intravenosa los días 1 y 2 durante 6 ciclos en combinación con rituximab en la dosis y el esquema descritos anteriormente. Cada ciclo fue de 28 días.
Se aleatorizó a un total de 389 pacientes: 194 al grupo de VEN+R y 195 al grupo de B+R. Las características demográficas y de la enfermedad iniciales fueron similares entre los grupos de VEN+R y B+R. La mediana de edad fue de 65 años (rango: 22 a 85 años), el 97 % eran de raza blanca, el 74 % eran hombres y el 99 % tenían un estado funcional ECOG <2. La mediana de líneas de tratamiento previas fue de 1 (rango: 1 a 5); el 59 % había recibido 1 tratamiento previo, el 26 % había recibido 2 tratamientos previos y el 16 % había recibido 3 o más tratamientos previos. Los tratamientos previos incluyeron agentes alquilantes (94 %), anticuerpos anti-CD20 (77 %), inhibidores de la vía del receptor de células B (2 %) y análogos de purina previos (81 %, incluido fludarabina/ciclofosfamida/rituximab en el 55 %). Se detectó una deleción 17p en el 24 % de los pacientes, mutaciones de TP53 en el 25 %, deleción 11q en el 32 % y IgVH no mutada en el 63 %.
La eficacia se basó en la PFS según la evaluación de un IRC. La mediana de seguimiento para la PFS fue de 23,4 meses (rango: 0 a 37,4+ meses). Los resultados de eficacia para MURANO se muestran en la Tabla 21. La curva de Kaplan-Meier para la PFS se muestra en la Figura 2.
| Criterio de valoración | VENCLEXTA + Rituximab (N = 194) |
Bendamustina + Rituximab (N = 195) |
| Supervivencia sin progresióna | ||
| Número de eventos, n (%) | 35 (18) | 106 (54) |
| Progresión de la enfermedad, n | 26 | 91 |
| Eventos mortales, n | 9 | 15 |
| Mediana, meses (IC del 95 %) | No alcanzada | 18,1 (15,8, 22,3) |
| HR (IC del 95 %)b | 0,19 (0,13, 0,28) | |
| Valor pb | <0,0001 | |
| Tasa de respuestac, n (%) | ||
| ORR | 179 (92) | 141 (72) |
| IC del 95 % | (88, 96) | (65, 78) |
| RC+RCi | 16 (8) | 7 (4) |
| nPR | 3 (2) | 1 (1) |
| RP | 160 (82) | 133 (68) |
| IC = intervalo de confianza; RC = remisión completa; RCi = remisión completa con recuperación medular incompleta; HR = hazard ratio; nPR = remisión parcial nodular; ORR = tasa de respuesta global (RC + RCi + nPR + RP); RP = remisión parcial. aEstimación de Kaplan-Meier. bLa estimación de HR se basa en el modelo de riesgos proporcionales de Cox estratificado por deleción 17p, estado de riesgo y región geográfica; el valor p se basa en la prueba de log-rank estratificada por los mismos factores. cSegún las directrices del Taller Internacional sobre Leucemia Linfocítica Crónica (IWCLL) de 2008. |
||
Figura 2. Curva de Kaplan-Meier de la supervivencia sin progresión evaluada por el CRI en MURANO
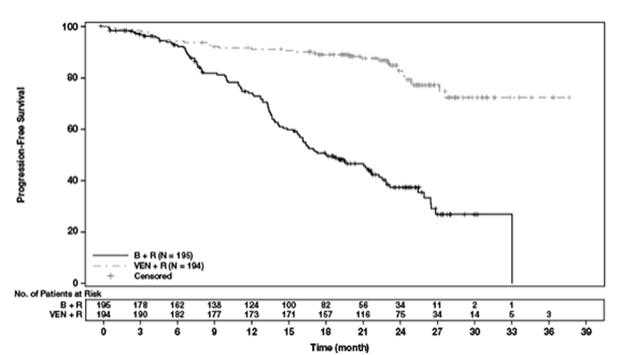
A los 3 meses después de la última dosis de rituximab, la tasa de EMR negativa en sangre periférica en pacientes que lograron RP o mejor fue del 54 % (104/194) en el grupo de VEN+R y del 12 % (23/195) en el grupo de B+R. La tasa de RC/RCi negativa para EMR en este punto temporal fue del 3 % (6/194) en el grupo de VEN+R y del 2 % (3/195) en el grupo de B+R.
Seguimiento de 71 meses
Con un seguimiento general de 71 meses, la mediana de la SSP evaluada por el investigador fue de 53.6 meses (IC del 95 %: 48.4, 57.0) en el grupo de VEN+R y de 17.0 meses (IC del 95 %: 15.5, 21.7) en el grupo de B+R. No se alcanzó la mediana de la SG en ninguno de los grupos. Se produjo la muerte en el 16 % (32/194) de los pacientes del grupo de VEN+R y en el 33 % (64/195) de los pacientes del grupo de B+R (HR estratificado 0.40; IC del 95 % [0.26, 0.62]). La curva de Kaplan-Meier para la SG se muestra en la Figura 3.
Figura 3. Curva de Kaplan-Meier de la supervivencia global en MURANO
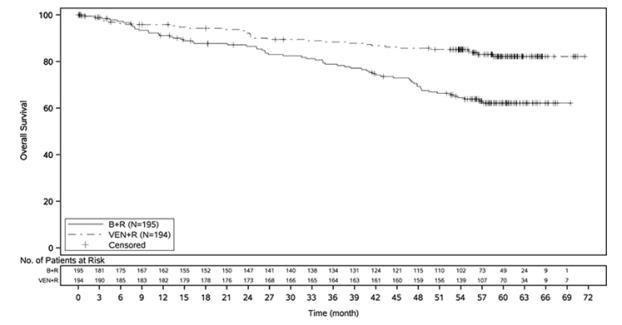
Monoterapia
La eficacia de la monoterapia con VENCLEXTA en la LLC o la LLS previamente tratadas se basa en tres ensayos de un solo grupo.
M13-982
M13-982 (NCT01889186) fue un ensayo abierto, multicéntrico, en el que se incluyeron 106 pacientes con LLC con deleción del 17p que habían recibido al menos un tratamiento previo. En el ensayo, se confirmó la deleción del 17p en muestras de sangre periférica de pacientes utilizando el kit de sondas FISH Vysis CLL, que está aprobado por la FDA para la selección de pacientes para el tratamiento con VENCLEXTA. Los pacientes recibieron 400 mg de VENCLEXTA por vía oral una vez al día después de completar el esquema de dosificación de escalada [véanse Dosificación y administración (2.2, 2.4)].
La eficacia se basó en la tasa de respuesta global (TRG) según la evaluación de un CRI.
La Tabla 22 resume las características demográficas y de la enfermedad al inicio del estudio en la población del ensayo.
| Característica | N = 106 |
| Edad, años; mediana (rango) | 67 (37-83) |
| Blanca; % | 97 |
| Hombre; % | 65 |
| Estado funcional ECOG; % 0 1 2 |
40 52 8 |
| Carga tumoral; % Recuento absoluto de linfocitos ≥25 x 109/L Uno o más ganglios ≥5 cm |
50 53 |
| Número de tratamientos previos; mediana (rango) | 2.5 (1-10) |
| Tiempo desde el diagnóstico, años; mediana (rango)a | 6.6 (0.1-32.1) |
| ECOG = Eastern Cooperative Oncology Group. aN=105. |
|
La mediana del tiempo de tratamiento en el momento de la evaluación fue de 12.1 meses (rango: de 0 a 21.5 meses). Los resultados de eficacia se muestran en la Tabla 23.
| Criterio de valoración | VENCLEXTA N = 106 |
| TRG, n (%)a (IC del 95 %) |
85 (80) (71, 87) |
| RC + RCi, n (%) RC, n (%) RCi, n (%) |
8 (8) 6 (6) 2 (2) |
| nPR, n (%) | 3 (3) |
| RP, n (%) | 74 (70) |
| IC = intervalo de confianza; RC = remisión completa; RCi = remisión completa con recuperación incompleta de la médula ósea; CRI = comité de revisión independiente; nPR = remisión parcial nodular; TRG = tasa de respuesta global (RC + RCi + nPR + RP); RP = remisión parcial. aSegún las directrices del IWCLL de 2008. |
|
El tiempo medio hasta la primera respuesta fue de 0.8 meses (rango: 0.1 a 8.1 meses).
Basado en una fecha de corte de datos posterior y la eficacia evaluada por el investigador, la duración de la respuesta (DOR) varió de 2.9 a 32.8+ meses. La DOR mediana no se ha alcanzado con un seguimiento mediano de 22 meses.
La enfermedad residual mínima se evaluó en sangre periférica y médula ósea para los pacientes que lograron CR o CRi, tras el tratamiento con VENCLEXTA. Tres por ciento (3/106) lograron negatividad de MRD en la sangre periférica y médula ósea (menos de una célula CLL por 104 leucocitos).
M12-175
M12-175 (NCT01328626) fue un ensayo multicéntrico, abierto, que inscribió a pacientes previamente tratados con CLL o SLL, incluidos aquellos con deleción 17p. La eficacia se evaluó en 67 pacientes (59 con CLL, 8 con SLL) que habían recibido VENCLEXTA 400 mg por vía oral una vez al día tras completar un esquema de dosificación escalonada. Los pacientes continuaron con esta dosis hasta la progresión de la enfermedad o toxicidad inaceptable. La duración media del tratamiento en el momento de la evaluación fue de 22.1 meses (rango: 0.5 a 71.7 meses).
La edad mediana fue de 65 años (rango: 42 a 84 años), el 78% eran hombres y el 87% eran blancos. El número mediano de tratamientos previos fue de 3 (rango: 1 a 11). En la línea base, el 67% de los pacientes tenía uno o más nódulos ≥5 cm, el 30% de los pacientes tenía ALC ≥25 x 109/L, el 33% tenía IgVH no mutado documentado, y el 21% tenía deleción 17p documentada.
La eficacia se basó en las pautas IWCLL de 2008 y fue evaluada por un IRC. La ORR fue del 76% (IC del 95%: 64%, 86%), con una tasa de CR + CRi del 10% y una tasa de PR del 66%. La DOR mediana fue de 36.2 meses (rango: 2.4 a 52.4 meses).
M14-032
M14-032 (NCT02141282) fue un ensayo multicéntrico, abierto, que inscribió a pacientes con CLL que habían sido tratados previamente y progresaron con o después de ibrutinib o idelalisib. Los pacientes recibieron VENCLEXTA 400 mg por vía oral una vez al día tras completar el esquema de dosificación escalonada [ver Dosificación y Administración (2.2, 2.4)]. Los pacientes continuaron con esta dosis hasta la progresión de la enfermedad o toxicidad inaceptable. En el momento del análisis, la duración mediana del tratamiento fue de 19.5 meses (rango: 0.1 a 39.5 meses).
De los 127 pacientes tratados (91 con ibrutinib previo, 36 con idelalisib previo), la edad mediana fue de 66 años (rango: 28 a 85 años), el 70% eran hombres y el 92% eran blancos. El número mediano de tratamientos previos fue de 4 (rango: 1 a 15). En la línea base, el 41% de los pacientes tenía uno o más nódulos ≥5 cm, el 31% tenía un ALC ≥25 x 109/L, el 57% tenía IgVH no mutado documentado, y el 39% tenía deleción 17p documentada.
La eficacia se basó en las pautas IWCLL de 2008 y fue evaluada por un IRC. La ORR fue del 70% (IC del 95%: 61%, 78%), con una tasa de CR + CRi del 5% y una tasa de PR del 65%. La DOR mediana no se alcanzó con un tiempo de seguimiento mediano de 19.9 meses (rango: 2.9 a 36 meses).
14.2 Leucemia Mieloide Aguda
VENCLEXTA fue estudiado en pacientes adultos con LMA recién diagnosticada que tenían 75 años o más, o tenían comorbilidades que impedían el uso de quimioterapia de inducción intensiva basada en al menos uno de los siguientes criterios: estado de rendimiento ECOG en línea base de 2-3, comorbilidad cardíaca o pulmonar severa, deterioro hepático moderado, CLcr <45 mL/min, u otra comorbilidad.
En combinación con Azacitidina o Decitabina
VIALE-A fue un ensayo aleatorizado (2:1), doble ciego, controlado con placebo, multicéntrico (NCT02993523) que evaluó la eficacia y seguridad de VENCLEXTA en combinación con azacitidina (VEN+AZA) frente a placebo con azacitidina (PBO+AZA).
Los pacientes recibieron VENCLEXTA 400 mg por vía oral una vez al día en los Días 1-28 tras completar el esquema de dosificación escalonada [ver Dosificación y Administración (2.3)] o placebo en combinación con azacitidina 75 mg/m2 ya sea por vía intravenosa o subcutánea en los Días 1-7 de cada ciclo de 28 días comenzando en el Día 1 del Ciclo 1. Durante el escalonamiento, los pacientes recibieron profilaxis TLS y fueron hospitalizados para monitoreo.
Una vez que la evaluación de la médula ósea confirmó una remisión, definida como menos del 5% de blastos leucémicos con citopenia tras el tratamiento del Ciclo 1, VENCLEXTA o placebo se interrumpió hasta 14 días o hasta que ANC ≥500/microlitro y el recuento de plaquetas ≥50 × 103/microlitro. Para los pacientes con enfermedad resistente al final del Ciclo 1, se realizó una evaluación de la médula ósea después del Ciclo 2 o 3 y según lo indicado clínicamente. La azacitidina se reanudó el mismo día que VENCLEXTA o placebo tras la interrupción. Se implementó una reducción de dosis de azacitidina en el ensayo clínico para el manejo de la toxicidad hematológica [ver Dosificación y Administración (2.5)]. Los pacientes continuaron el tratamiento hasta la progresión de la enfermedad o toxicidad inaceptable.
Un total de 431 pacientes fueron aleatorizados: 286 al brazo VEN+AZA y 145 al brazo PBO+AZA. Las características demográficas y de enfermedad en línea base se muestran en Tabla 24.
| Característica | VENCLEXTA + Azacitidina N = 286 |
Placebo + Azacitidina N = 145 |
| Edad, años; mediana (rango) | 76 (49, 91) | 76 (60, 90) |
| Raza | ||
| White; % | 76 | 75 |
| Black or African American; % | 1 | 1.4 |
| Asian; % | 23 | 23 |
| Males; % | 60 | 60 |
| ECOG performance status; % | ||
| 0-1 | 55 | 56 |
| 2 | 40 | 41 |
| 3 | 5.6 | 3.4 |
| Bone marrow blast; % | ||
| <30% | 30 | 28 |
| ≥30% to <50% | 21 | 23 |
| ≥50% | 49 | 49 |
| Disease history; % | ||
| De Novo AML | 75 | 76 |
| Secondary AML | 25 | 24 |
| Cytogenetic risk detecteda, % | ||
| Intermediate | 64 | 61 |
| Poor | 36 | 39 |
| Mutation analyses detected; n/Nb (%) | ||
| IDH1 or IDH2 | 61/245 (25) | 28/127 (22) |
| IDH1 | 23/245 (9.4) | 11/127 (8.7) |
| IDH2 | 40/245 (16) | 18/127 (14) |
| FLT3 | 29/206 (14) | 22/108 (20) |
| NPM1 | 27/163 (17) | 17/86 (20) |
| TP53 | 38/163 (23) | 14/86 (16) |
| aSegún las Pautas de la Red Nacional Integral del Cáncer (NCCN) de 2016. bNúmero de muestras de BMA evaluables recibidas al inicio. |
||
La eficacia se basó en la supervivencia general (SG), medida desde la fecha de aleatorización hasta la muerte por cualquier causa. La combinación de VEN+AZA fue superior en la SG a PBO+AZA.
La curva de Kaplan-Meier para la SG se muestra en la Figura 4. Los resultados de eficacia de VIALE-A se muestran en la Tabla 25.
Figura 4. Curva de Kaplan-Meier para la supervivencia general en VIALE-A
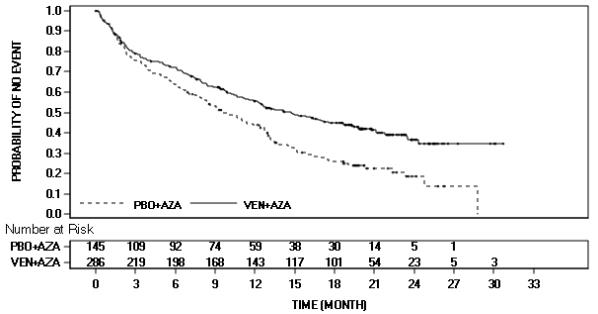
| Criterio de valoración | VENCLEXTA + Azacitidina (N = 286) |
Placebo + Azacitidina (N = 145) |
| Supervivencia general | ||
| Medianaa, meses (IC del 95%) |
14.7 (11.9, 18.7) |
9.6 (7.4, 12.7) |
| Hazard ratiob (IC del 95%) | 0.66 (0.52, 0.85) | |
| Valor de pb | <0.001 | |
| Tasa de respuesta | ||
| RC, n (%) | 105 (37) | 26 (18) |
| (IC del 95%) | (31, 43) | (12, 25) |
| Valor de pc | <0.001 | |
| Mediana de la DRRCa,d (meses) | 18.0 | 13.4 |
| IC del 95% | (15.3, -) | (8.7, 17.6) |
| RC+RCh, n (%) | 185 (65) | 33 (23) |
| (IC del 95%) | (59, 70) | (16, 30) |
| Valor de pc | <0.001 | |
| Mediana de la DRRC+RCha,e (meses) | 17.8 | 13.9 |
| IC del 95% | (15.3, -) | (10.4, 15.7) |
| IC = intervalo de confianza; RC = remisión completa; RCh = remisión completa con recuperación hematológica parcial; DRRC = duración de la RC; HR = hazard ratio; – = no alcanzado. La RC (remisión completa) se definió como un recuento absoluto de neutrófilos >1000/microlitro, plaquetas >100 000/microlitro, independencia de las transfusiones de glóbulos rojos y médula ósea con <5% de blastos. Ausencia de blastos circulantes y blastos con bastones de Auer; ausencia de enfermedad extramedular. La RCh (remisión completa con recuperación hematológica parcial) se definió como <5% de blastos en la médula ósea, sin evidencia de enfermedad y recuperación parcial de los recuentos sanguíneos periféricos (plaquetas >50 000/microlitro y RAN >500/microlitro). aEstimación de Kaplan-Meier. bLa estimación del hazard ratio (VEN+AZA frente a PBO+AZA) se basa en un modelo de riesgos proporcionales de Cox estratificado por citogenética (riesgo intermedio, riesgo bajo) y edad (de 18 a <75, ≥75 años) según la asignación aleatoria; el valor de p se basa en la prueba de log-rank estratificada por los mismos factores. cEl valor de p procede de la prueba de Cochran-Mantel-Haenszel estratificada por edad y riesgo citogenético. dLa duración de la RC se define como el número de días desde la fecha de la primera respuesta de la RC hasta la fecha de la primera evidencia de recaída morfológica confirmada, enfermedad progresiva confirmada o muerte debido a la progresión de la enfermedad. eLa duración de la RC+RCh se define como el número de días desde la fecha de la primera respuesta de la RC+RCh (la primera de RC o RCh) hasta la fecha de la primera evidencia de recaída morfológica confirmada, enfermedad progresiva confirmada o muerte debido a la progresión de la enfermedad. |
||
Entre los pacientes tratados con VEN+AZA, 155 eran dependientes de transfusiones de glóbulos rojos (RBC) o plaquetas al inicio del estudio; de estos pacientes, el 49 % (76/155) se volvieron independientes de las transfusiones de RBC y plaquetas durante cualquier período consecutivo ≥56 días después del inicio del estudio. De los pacientes tratados con VEN+AZA, 131 eran independientes de las transfusiones de RBC y plaquetas al inicio del estudio, el 69 % (90/131) permanecieron independientes de las transfusiones durante cualquier período consecutivo ≥56 días después del inicio del estudio. Entre los pacientes tratados con PBO+AZA, 81 eran dependientes de transfusiones de RBC o plaquetas al inicio del estudio; de estos pacientes, el 27 % (22/81) se volvieron independientes de las transfusiones de RBC y plaquetas durante cualquier período consecutivo ≥56 días después del inicio del estudio. De los pacientes tratados con PBO+AZA, 64 eran independientes de las transfusiones de RBC y plaquetas al inicio del estudio, el 42 % (27/64) permanecieron independientes de las transfusiones durante cualquier período consecutivo ≥56 días después del inicio del estudio.
La mediana del tiempo hasta la primera respuesta de CR o CRh fue de 1,0 meses (rango: 0,6 a 14,3 meses) con el tratamiento con VEN+AZA.
M14-358
M14-358 (NCT02203773) fue un ensayo no aleatorizado, abierto, que evaluó la eficacia de VENCLEXTA en combinación con azacitidina (N=84) o decitabina (N=31) en pacientes con AML recién diagnosticada. De esos pacientes, 67 que recibieron la combinación con azacitidina y 13 que recibieron la combinación con decitabina tenían 75 años o más, o tenían comorbilidades que impedían el uso de quimioterapia de inducción intensiva.
Los pacientes recibieron 400 mg de VENCLEXTA por vía oral una vez al día después de completar el esquema de dosificación de aumento gradual [ver Dosificación y administración (2.3)] en combinación con azacitidina (75 mg/m2 por vía intravenosa o subcutánea en los días 1 a 7 de cada ciclo de 28 días comenzando en el día 1 del ciclo 1) o decitabina (20 mg/m2 por vía intravenosa en los días 1 a 5 de cada ciclo de 28 días comenzando en el día 1 del ciclo 1). Durante la fase de aumento gradual, los pacientes recibieron profilaxis contra el síndrome de lisis tumoral y fueron hospitalizados para su control. Los pacientes continuaron el tratamiento hasta la progresión de la enfermedad o la aparición de toxicidad inaceptable. Una vez que la evaluación de la médula ósea confirmó una remisión, definida como menos del 5 % de blastocitos leucémicos, con citopenia después del tratamiento del ciclo 1, se interrumpió la administración de VENCLEXTA hasta 14 días o hasta que el RAN ≥500/microlitro y el recuento de plaquetas ≥50 × 103/microlitro. En el ensayo clínico se implementó la reducción de la dosis de azacitidina para el tratamiento de la toxicidad hematológica [ver Dosificación y administración (2.5)]. En el ensayo clínico no se implementaron reducciones de la dosis de decitabina. Las características demográficas y de la enfermedad al inicio del estudio se muestran en la Tabla 26.
| Característica | VENCLEXTA + Azacitidina N = 67 |
VENCLEXTA + Decitabina N = 13 |
| Edad, años; mediana (rango) | 76 (61-90) | 75 (68-86) |
| Raza; % | ||
| Blanca | 87 | 77 |
| Negra o afroamericana | 4.5 | 0 |
| Asiática | 1.5 | 0 |
| Nativo hawaiano o de las islas del Pacífico | 1.5 | 15 |
| Indígena estadounidense/nativo de Alaska | 0 | 7.7 |
| Otra no reportada | 6 | 0 |
| Masculino; % | 60 | 38 |
| Estado funcional ECOG; % 0-1 2 3 |
64 33 3 |
92 7.7 0 |
| Antecedentes de la enfermedad; % AML de novo AML secundaria |
73 27 |
85 15 |
| Análisis de mutaciones detectadasa; % | ||
| TP53 | 15 | 31 |
| IDH1 o IDH2 | 27 | 0 |
| FLT3 | 16 | 23 |
| NPM1 | 19 | 15 |
| Riesgo citogenético detectadob,c; % | ||
| Intermedio | 64 | 38 |
| Pobre | 34 | 62 |
| Comorbilidades de referenciad; % | ||
| Enfermedad cardíaca grave | 4.5 | 7.7 |
| Enfermedad pulmonar grave | 1.5 | 0 |
| Insuficiencia hepática moderada | 9 | 0 |
| Aclaramiento de creatinina <45 mL/min | 13 | 7.7 |
| ECOG = Eastern Cooperative Oncology Group. aIncluye 6 pacientes con muestra insuficiente para análisis en el grupo de azacitidina y 4 en el grupo de decitabina. bComo se define por la categorización de riesgo de la National Comprehensive Cancer Network (NCCN) v2014. cSin mitosis en 1 paciente en el grupo de azacitidina (riesgo favorable excluido por análisis de hibridación in situ fluorescente [FISH]). dLos pacientes pueden haber tenido más de una comorbilidad. |
||
Los resultados de eficacia se muestran en Tabla 27.
| Resultados de eficacia | VENCLEXTA + Azacitidina N = 67 |
VENCLEXTA + Decitabina N = 13 |
| CR, n (%) | 29 (43) | 7 (54) |
| (95% IC) | (31, 56) | (25, 81) |
| CRh, n (%) | 12 (18) | 1 (7.7) |
| (95% IC) | (9.6, 29) | (0.2, 36) |
| IC = intervalo de confianza; CR = remisión completa; CRh = remisión completa con recuperación hematológica parcial. | ||
La mediana de seguimiento fue de 15.9 meses (rango: 0.4 a 40.3 meses) para VENCLEXTA en combinación con azacitidina. La mediana de duración de la RC fue de 23.8 meses (IC del 95%: 15.4, -) y la mediana de duración de la RC+RCi fue de 26.5 meses (IC del 95%: 17.4, -).
La mediana de seguimiento fue de 11.0 meses (rango: 0.7 a 38.8 meses) para VENCLEXTA en combinación con decitabina. La mediana de duración de la RC fue de 12.7 meses (IC del 95%: 1.4, -) y la mediana de duración de la RC+RCi fue de 12.7 meses (IC del 95%: 1.4, 20.0).
La duración de la RC se define como el tiempo desde la primera documentación de la RC hasta la primera fecha de recaída, progresión clínica de la enfermedad o muerte debido a la progresión de la enfermedad, lo que ocurra primero. La duración de la RC+RCi se define como el tiempo desde la primera documentación de la RC o la RCi hasta la primera fecha de recaída, progresión clínica de la enfermedad o muerte debido a la progresión de la enfermedad, lo que ocurra primero.
La mediana del tiempo hasta la primera RC o RCi en pacientes tratados con VENCLEXTA en combinación con azacitidina fue de 1.0 mes (rango: 0.7 a 8.9 meses).
La mediana del tiempo hasta la primera RC o RCi en pacientes tratados con VENCLEXTA en combinación con decitabina fue de 1.9 meses (rango: 0.8 a 4.2 meses).
De los pacientes tratados con VENCLEXTA en combinación con azacitidina, el 12% (8/67) recibió posteriormente un trasplante de células madre.
En el ensayo se incluyeron 35 pacientes adicionales (rango de edad: 65 a 74 años) que no tenían comorbilidades conocidas que impidieran el uso de quimioterapia de inducción intensiva y fueron tratados con VENCLEXTA en combinación con azacitidina (N=17) o decitabina (N=18).
Para los 17 pacientes tratados con VENCLEXTA en combinación con azacitidina, la tasa de RC fue del 35% (IC del 95%: 14%, 62%). La tasa de RCi fue del 41% (IC del 95%: 18%, 67%). Nueve (53%) pacientes recibieron posteriormente un trasplante de células madre.
Para los 18 pacientes tratados con VENCLEXTA en combinación con decitabina, la tasa de RC fue del 56% (IC del 95%: 31%, 79%). La tasa de RCi fue del 22% (IC del 95%: 6.4%, 48%). Cuatro (22%) pacientes recibieron posteriormente un trasplante de células madre.
En combinación con dosis bajas de citarabina
VIALE-C fue un ensayo multicéntrico, aleatorizado (2:1), doble ciego, controlado con placebo (NCT03069352) que evaluó la eficacia y seguridad de VENCLEXTA en combinación con dosis bajas de citarabina (VEN+LDAC) frente a placebo con dosis bajas de citarabina (PBO+LDAC).
Los pacientes recibieron 600 mg de VENCLEXTA por vía oral una vez al día los días 1-28 después de completar el esquema de dosificación de escalada [ver Posología y administración (2.3)] o placebo en combinación con 20 mg/m2 de citarabina por vía subcutánea una vez al día los días 1-10 de cada ciclo de 28 días comenzando en el día 1 del ciclo 1. Durante la fase de escalada, los pacientes recibieron profilaxis para el SLT y fueron hospitalizados para su monitorización.
Una vez que la evaluación de la médula ósea confirmó una remisión, definida como menos del 5% de blastos leucémicos con citopenia después del tratamiento del ciclo 1, se interrumpió VENCLEXTA o el placebo hasta 14 días o hasta que el RAN ≥500/microlitro y el recuento de plaquetas ≥50 × 103/microlitro. En los pacientes con enfermedad resistente al final del ciclo 1, se realizó una evaluación de la médula ósea después del ciclo 2 o 3 y según estuviera clínicamente indicado. La LDAC se reanudó el mismo día que VENCLEXTA o el placebo después de la interrupción. Los pacientes continuaron recibiendo tratamiento hasta la progresión de la enfermedad o la aparición de toxicidad inaceptable. Las características demográficas y de la enfermedad al inicio del estudio se muestran en la Tabla 28.
| Característica | VENCLEXTA + Dosis bajas de citarabina N = 143 |
Placebo + Dosis bajas de citarabina N = 68 |
| Edad, años; mediana (rango) | 76 (36, 93) | 76 (41, 88) |
| Raza; % | ||
| Blanca | 71 | 69 |
| Negra o afroamericana | 1.4 | 1.5 |
| Asiática | 27 | 29 |
| Sexo masculino; % | 55 | 57 |
| Estado funcional ECOG; % 0-1 2 3 |
52 44 4.2 |
50 37 13 |
| Antecedentes de la enfermedad; % LMA de novo LMA secundaria |
59 41 |
66 34 |
| Análisis de mutaciones detectadas; n/Na (%) | ||
| TP53 | 22/112 (20) | 9/52 (17) |
| IDH1 o IDH2 | 21/112 (19) | 12/52 (23) |
| FLT3 | 20/112 (18) | 9/52 (17) |
| NPM1 | 18/112 (16) | 7/52 (13) |
| Riesgo citogenético detectadob; % | ||
| Favorable | <1 | 4 |
| Intermediate | 63 | 63 |
| Poor | 33 | 29 |
| aNúmero de muestras de BMA evaluables recibidas al inicio. bSegún las Pautas de la Red Nacional Integral del Cáncer (NCCN) de 2016. |
||
La eficacia se basó en la tasa de RC y la duración de la RC con evidencia de apoyo de la tasa de RC+RCi, la duración de la RC+RCi y la tasa de conversión de la dependencia de la transfusión a la independencia de la transfusión. La tasa de RC en el grupo de VEN+LDAC fue del 27 % (IC del 95 %: 20 %, 35 %) con una mediana de duración de la RC de 11.1 meses (IC del 95 %: 6.1, -), y la tasa de RC en el grupo de PBO+LDAC fue del 7.4 % (IC del 95 %: 2.4 %, 16 %) con una mediana de duración de la RC de 8.3 meses (IC del 95 %: 3.1, -). La tasa de RC+RCi en el grupo de VEN+LDAC fue del 47 % (IC del 95 %: 39 %, 55 %) y en el grupo de PBO+LDAC fue del 15 % (IC del 95 %: 7.3 %, 25 %) con una mediana de duración de la RC+RCi de 11.1 meses con el tratamiento con VEN+LDAC y de 6.2 meses con el tratamiento con PBO+LDAC. La mediana del tiempo hasta la primera respuesta de RC o RCi fue de 1.0 mes (rango: 0.7 a 5.8 meses) con el tratamiento con VEN+LDAC.
Entre los pacientes tratados con VEN+LDAC, 111 eran dependientes de transfusiones de GR y/o plaquetas al inicio; de estos pacientes, el 33 % (37/111) de los pacientes se volvieron independientes de las transfusiones de GR y plaquetas durante cualquier período consecutivo ≥56 días después del inicio. De los pacientes tratados con VEN+LDAC, 32 eran independientes de las transfusiones de GR y plaquetas al inicio, el 50 % (16/32) permanecieron independientes de las transfusiones durante cualquier período consecutivo ≥56 días después del inicio.
Entre los pacientes tratados con PBO+LDAC, 55 eran dependientes de transfusiones de GR y/o plaquetas al inicio; de estos pacientes, el 13 % (7/55) de los pacientes se volvieron independientes de las transfusiones de GR y plaquetas durante cualquier período consecutivo ≥56 días después del inicio. De los pacientes tratados con PBO+LDAC, 13 eran independientes de las transfusiones de GR y plaquetas al inicio, el 31 % (4/13) permanecieron independientes de las transfusiones durante cualquier período consecutivo ≥56 días después del inicio.
VEN+LDAC no mejoró significativamente la SG en comparación con PBO+LDAC. El índice de riesgo (HR) para la SG fue de 0.75 (IC del 95 %: 0.52, 1.07); valor p 0.114. La mediana de la SG para el grupo de VEN+LDAC fue de 7.2 meses (IC del 95 %: 5.6, 10.1) y para el grupo de PBO+LDAC fue de 4.1 meses (IC del 95 %: 3.1, 8.8).
M14-387
M14-387 (NCT02287233) fue un ensayo no aleatorizado, abierto, que evaluó la eficacia de VEN+LDAC (N=82) en pacientes con LMA recién diagnosticada, incluidos pacientes con exposición previa a un agente hipometilante para un trastorno hematológico antecedente. De esos pacientes, 61 tenían 75 años o más, o tenían comorbilidades que impedían el uso de quimioterapia de inducción intensiva.
Los pacientes recibieron 600 mg de VENCLEXTA por vía oral una vez al día en los Días 1-28 después de completar la fase de aumento gradual [ver Posología y administración (2.3)] en combinación con 20 mg/m2 de citarabina por vía subcutánea una vez al día en los Días 1-10 de cada ciclo de 28 días comenzando en el Día 1 del Ciclo 1. Durante el aumento gradual, los pacientes recibieron profilaxis de STL y fueron hospitalizados para su control. Una vez que la evaluación de la médula ósea confirmó una remisión, definida como menos del 5 % de blastocitos leucémicos con citopenia después del tratamiento del Ciclo 1, se interrumpió VENCLEXTA hasta 14 días o hasta que el RAN ≥500/microlitro y el recuento de plaquetas ≥50 × 103/microlitro. Los pacientes continuaron el tratamiento hasta la progresión de la enfermedad o la toxicidad inaceptable. Las características demográficas y de la enfermedad al inicio se muestran en la Tabla 29.
| Característica | VENCLEXTA en combinación con dosis bajas de citarabina N = 61 |
| Edad, años; mediana (rango) | 76 (63-90) |
| Raza; % | |
| Blanca | 92 |
| Negra o afroamericana | 1.6 |
| Asiática | 1.6 |
| No reportada | 4.9 |
| Hombre; % | 74 |
| Estado de desempeño ECOG; % 0-1 2 3 |
66 33 1.6 |
| Antecedentes de la enfermedad; % LMA de novo LMA secundaria |
54 46 |
| Análisis de mutaciones detectadasa; % | |
| TP53 | 8.2 |
| IDH1 o IDH2 | 23 |
| FLT3 | 21 |
| NPM1 | 9.8 |
| Riesgo citogenético detectadob; % | |
| Intermedio | 59 |
| Malo | 34 |
| Sin mitosis | 6.6 |
| Comorbilidades basalesc; % | |
| Enfermedad cardíaca grave | 9.8 |
| Insuficiencia hepática moderada | 4.9 |
| Creatinine clearance ≥30 or <45 mL/min | 3.3 |
| aIncluye 7 pacientes con muestra insuficiente para el análisis. bSegún la definición de la categorización de riesgo de la Red Nacional Integral del Cáncer (National Comprehensive Cancer Network, NCCN) v2014. cLos pacientes pueden haber tenido más de una comorbilidad. |
|
La mediana de seguimiento fue de 7.3 meses (rango: 0.3 a 54.0 meses). La tasa de RC fue del 21 % (IC del 95 %: 12, 34) y la tasa de RCp fue del 21 % (IC del 95 %: 12, 34).
La mediana de duración de la RC fue de 22.9 meses (IC del 95 %: 5.1, -) y la mediana de duración de la RC+RCp fue de 14.3 meses (IC del 95 %: 6.1, 31.2).
La mediana de tiempo hasta la primera RC o RCp para los pacientes tratados con VEN+LDAC fue de 1.0 mes (rango: 0.8 a 9.4 meses).
En el ensayo se inscribieron 21 pacientes adicionales (rango de edad: 67 a 74 años) que no presentaban comorbilidades conocidas que impidieran el uso de quimioterapia de inducción intensiva y fueron tratados con VEN+LDAC. La tasa de RC fue del 33 % (IC del 95 %: 15 %, 57 %). La tasa de RCp fue del 24 % (IC del 95 %: 8.2 %, 47 %). Un paciente (4.8 %) se sometió posteriormente a un trasplante de células madre.
16 FORMA DE SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
VENCLEXTA se dispensa de la siguiente manera:
| Presentación del empaque | Número de tabletas | Código Nacional de Medicamentos (NDC) |
| Paquete inicial para CLL/SLL | Cada paquete contiene cuatro paquetes de blíster semanales:
|
0074-0579-28 |
| Billetera que contiene tabletas de 10 mg | 14 tabletas de 10 mg | 0074-0561-14 |
| Billetera que contiene tabletas de 50 mg | 7 tabletas de 50 mg | 0074-0566-07 |
| Blíster de dosis unitaria que contiene tabletas de 10 mg | 2 tabletas de 10 mg | 0074-0561-11 |
| Blíster de dosis unitaria que contiene tabletas de 50 mg | 1 tableta de 50 mg | 0074-0566-11 |
| Blíster de dosis unitaria que contiene tabletas de 100 mg | 1 tableta de 100 mg | 0074-0576-11 |
| Frasco que contiene tabletas de 100 mg | 28 tabletas de 100 mg | 0074-0576-30 |
| Frasco que contiene tabletas de 100 mg | 120 tabletas de 100 mg | 0074-0576-22 |
Las tabletas recubiertas con película de VENCLEXTA de 10 mg son redondas, biconvexas, de color amarillo pálido, con “V” grabado en un lado y “10” en el otro.
Las tabletas recubiertas con película de VENCLEXTA de 50 mg son oblongas, biconvexas, de color beige, con “V” grabado en un lado y “50” en el otro.
Las tabletas recubiertas con película de VENCLEXTA de 100 mg son oblongas, biconvexas, de color amarillo pálido, con “V” grabado en un lado y “100” en el otro.
Almacenar en el envase original a una temperatura no superior a 86°F (30°C). Dispensar al paciente en el envase original para protegerlo de la humedad.
17 INFORMACIÓN PARA EL PACIENTE
Aconseje al paciente que lea el etiquetado del paciente aprobado por la FDA (Guía de medicamentos).
Síndrome de lisis tumoral
Avise a los pacientes del riesgo potencial de TLS, particularmente al inicio del tratamiento, durante la fase de aumento gradual y con la reanudación después de una interrupción, y que informen inmediatamente cualquier signo o síntoma asociado con este evento (fiebre, escalofríos, náuseas, vómitos, confusión, dificultad para respirar, convulsiones, latidos cardíacos irregulares, orina oscura o turbia, cansancio inusual, dolor muscular y/o molestias en las articulaciones) a su proveedor de atención médica (HCP) para su evaluación [ver Advertencias y precauciones (5.1)].
Aconseje a los pacientes que se mantengan adecuadamente hidratados todos los días cuando tomen VENCLEXTA para reducir el riesgo de TLS. El volumen recomendado es de 6 a 8 vasos (aproximadamente 56 onzas en total) de agua cada día. Los pacientes deben beber agua comenzando 2 días antes y el día de la primera dosis, y cada vez que se aumente la dosis [ver Dosificación y administración (2.4)].
Aconseje a los pacientes sobre la importancia de mantener las citas programadas para análisis de sangre u otras pruebas de laboratorio [ver Dosificación y administración (2.4)].
Aconseje a los pacientes que puede ser necesario tomar VENCLEXTA en el hospital o en el consultorio médico para permitir el monitoreo de TLS.
Neutropenia
Aconseje a los pacientes que se pongan en contacto con su HCP inmediatamente si desarrollan fiebre o cualquier signo de infección. Avise a los pacientes de la necesidad de un monitoreo periódico de los recuentos sanguíneos [ver Advertencias y precauciones (5.2)].
Infecciones
Aconseje a los pacientes que se pongan en contacto con su HCP inmediatamente si desarrollan fiebre o cualquier signo de infección [ver Advertencias y precauciones (5.3)].
Interacciones medicamentosas
Aconseje a los pacientes que eviten consumir productos de toronja, naranjas de Sevilla o carambola durante el tratamiento con VENCLEXTA. Avise a los pacientes que VENCLEXTA puede interactuar con algunos medicamentos; por lo tanto, aconseje a los pacientes que informen a su proveedor de atención médica sobre el uso de cualquier medicamento recetado, medicamentos de venta libre, vitaminas y productos herbales [ver Contraindicaciones (4) e Interacciones medicamentosas (7.1)].
Inmunizaciones
Aconseje a los pacientes que eviten la vacunación con vacunas vivas porque pueden no ser seguras o efectivas durante el tratamiento con VENCLEXTA [ver Advertencias y precauciones (5.4)].
Toxicidad embrionaria-fetal
Avise a las mujeres embarazadas del riesgo potencial para el feto. Aconseje a las mujeres o con potencial reproductivo que informen a su proveedor de atención médica sobre un embarazo conocido o sospechoso [ver Advertencias y precauciones (5.5) y Uso en poblaciones específicas (8.1)].
Aconseje a las pacientes con potencial reproductivo que usen métodos anticonceptivos efectivos durante la terapia y durante 30 días después de la última dosis [ver Uso en poblaciones específicas (8.3)].
Lactancia
Aconseje a las mujeres que no amamanten durante el tratamiento con VENCLEXTA y durante 1 semana después de la última dosis [ver Uso en poblaciones específicas (8.2)].
Infertilidad
Aconseje a los hombres con potencial reproductivo que VENCLEXTA puede afectar la fertilidad [ver Uso en poblaciones específicas (8.3)].
Administración
Aconseje a los pacientes que guarden VENCLEXTA en el envase original.
Aconseje a los pacientes que tomen VENCLEXTA exactamente como se lo recetaron y que no cambien su dosis ni dejen de tomar VENCLEXTA a menos que su HCP se lo indique. Aconseje a los pacientes que tomen VENCLEXTA por vía oral una vez al día, aproximadamente a la misma hora todos los días, de acuerdo con las instrucciones de su HCP y que las tabletas deben tragarse enteras con una comida y agua sin masticar, triturar o romper [ver Dosificación y administración (2.8)].
Aconseje a los pacientes con CLL/SLL que guarden VENCLEXTA en el envase original durante las primeras 4 semanas de tratamiento y que no transfieran las tabletas a otro envase.
Aconseje a los pacientes que si se olvida una dosis de VENCLEXTA por menos de 8 horas, tomen la dosis olvidada de inmediato y tomen la siguiente dosis como de costumbre. Si se olvida una dosis de VENCLEXTA por más de 8 horas, aconseje a los pacientes que esperen y tomen la siguiente dosis a la hora habitual [ver Dosificación y administración (2.8)].
Aconseje a los pacientes que no tomen ninguna dosis adicional ese día si vomitan después de tomar VENCLEXTA y que tomen la siguiente dosis a la hora habitual al día siguiente.
Fabricado y comercializado por:
AbbVie Inc.
North Chicago, IL 60064
y
Comercializado por:
Genentech USA, Inc.
Miembro del Grupo Roche
South San Francisco, CA 94080-4990
© 2024 AbbVie. Todos los derechos reservados.
Venclexta y su diseño son marcas comerciales de AbbVie Inc.
© 2024 Genentech, Inc. Todos los derechos reservados.
20087124 R1
Guía de medicación
| GUÍA DEL MEDICAMENTO VENCLEXTA® (ven-KLEKS-tuh) (comprimidos de venetoclax) |
|
| ¿Cuál es la información más importante que debo saber sobre VENCLEXTA? VENCLEXTA puede causar efectos secundarios graves, que incluyen:
|
|
| ○ fiebre ○ escalofríos ○ náuseas ○ vómitos ○ confusión ○ dificultad para respirar |
○ convulsiones ○ ritmo cardíaco irregular ○ orina oscura o turbia ○ cansancio inusual ○ dolor muscular o articular |
| Beba mucha agua durante el tratamiento con VENCLEXTA para ayudar a reducir su riesgo de desarrollar SLT. Beba de 6 a 8 vasos (aproximadamente 56 onzas en total) de agua cada día, comenzando 2 días antes de su primera dosis, el día de su primera dosis de VENCLEXTA y cada vez que se aumente su dosis. Su proveedor de atención médica puede retrasar, disminuir su dosis o suspender el tratamiento con VENCLEXTA si tiene efectos secundarios. Al reiniciar VENCLEXTA después de suspenderlo durante 1 semana o más, su proveedor de atención médica puede volver a verificar su riesgo de SLT y cambiar su dosis. Consulte “¿Cuáles son los posibles efectos secundarios de VENCLEXTA?” para obtener más información sobre los efectos secundarios. |
|
| ¿Qué es VENCLEXTA? VENCLEXTA es un medicamento recetado que se usa:
No se sabe si VENCLEXTA es seguro y eficaz en niños. |
|
| ¿Quién no debe tomar VENCLEXTA? Ciertos medicamentos no se deben tomar cuando comienza a tomar VENCLEXTA y mientras su dosis se aumenta lentamente debido al riesgo de aumento del síndrome de lisis tumoral (SLT).
|
|
Antes de tomar VENCLEXTA, informe a su proveedor de atención médica sobre todas sus afecciones médicas, incluso si:
Informe a su proveedor de atención médica sobre todos los medicamentos que toma, incluidos los medicamentos recetados y de venta libre, las vitaminas y los suplementos herbales. VENCLEXTA y otros medicamentos pueden afectar su efecto mutuo causando efectos secundarios graves. Consulte “¿Quién no debe tomar VENCLEXTA?” |
|
¿Cómo debo tomar VENCLEXTA?
|
|
| ¿Qué debo evitar mientras tomo VENCLEXTA? No debe beber jugo de toronja, comer toronja, naranjas de Sevilla (que se usan a menudo en mermeladas) ni carambola mientras esté tomando VENCLEXTA. Estos productos pueden aumentar la cantidad de VENCLEXTA en su sangre. |
|
| ¿Cuáles son los posibles efectos secundarios de VENCLEXTA? VENCLEXTA puede causar efectos secundarios graves, que incluyen:
Informe a su proveedor de atención médica de inmediato si tiene fiebre o cualquier signo de infección durante el tratamiento con VENCLEXTA. |
|
|
|
|
Los efectos secundarios más comunes de VENCLEXTA en combinación con azacitidina, o decitabina, o citarabina de dosis baja en personas con AML incluyen: |
|
|
|
| VENCLEXTA puede causar problemas de fertilidad en los hombres. Esto puede afectar su capacidad para engendrar un hijo. Hable con su proveedor de atención médica si tiene inquietudes sobre la fertilidad. Estos no son todos los posibles efectos secundarios de VENCLEXTA. Llame a su médico para obtener asesoramiento médico sobre los efectos secundarios. Puede informar los efectos secundarios a la FDA al 1-800-FDA-1088. |
|
¿Cómo debo almacenar VENCLEXTA?
Mantenga VENCLEXTA y todos los medicamentos fuera del alcance de los niños. |
|
| Información general sobre el uso seguro y eficaz de VENCLEXTA. Los medicamentos a veces se recetan para fines distintos de los que se enumeran en una Guía de medicamentos. No use VENCLEXTA para una afección para la que no fue recetado. No le dé VENCLEXTA a otras personas, incluso si tienen los mismos síntomas que usted. Puede hacerles daño. Puede pedirle a su proveedor de atención médica o farmacéutico información sobre VENCLEXTA que esté escrita para profesionales de la salud. |
|
| ¿Cuáles son los ingredientes en VENCLEXTA? Ingrediente activo: venetoclax Ingredientes inactivos: copovidona, dióxido de silicio coloidal, polisorbato 80, fumarato de estearilo sódico y fosfato de calcio dibásico. Las tabletas recubiertas de 10 mg y 100 mg también incluyen: óxido de hierro amarillo, alcohol polivinílico, polietilenglicol, talco y dióxido de titanio. Las tabletas recubiertas de 50 mg también incluyen: óxido de hierro amarillo, óxido de hierro rojo, óxido de hierro negro, alcohol polivinílico, talco, polietilenglicol y dióxido de titanio. |
|
| Fabricado y comercializado por: AbbVie Inc. North Chicago, IL 60064 © 2024 AbbVie. Todos los derechos reservados. Venclexta y su diseño son marcas registradas de AbbVie Inc. 20087124 R1 |
Comercializado por: Genentech USA, Inc. Un miembro del Grupo Roche South San Francisco, CA 94080-4990 © 2024 Genentech, Inc. Todos los derechos reservados. |
| Para más información, visite www.venclexta.com o llame al 1-800-633-9110 | |
| Esta Guía de Medicamentos ha sido aprobada por la Administración de Alimentos y Medicamentos de EE. UU. | Revisado: 7/2024 |
PANEL DE VISUALIZACIÓN PRINCIPAL
NDC 0074-0579-28
Paquete de Inicio para CLL/SLL
VENCLEXTA®
(comprimidos de venetoclax)
10 mg, 50 mg y 100 mg
Paquete de Inicio
! ADVERTENCIA
Comuníquese con su médico cuando reciba este medicamento.
Puede ser necesario tomar su primera dosis en presencia de su médico para prevenir un posible efecto secundario grave.
DISPENSADOR: Cada vez que se dispense VENCLEXTA, entregue al paciente la Guía del Medicamento adjunta.
abbvie
Sólo con receta médica
Genentech
PANEL DE VISUALIZACIÓN PRINCIPAL
NDC 0074-0561-14
Rx only
VENCLEXTA®
(venetoclax tablets)
10 mg per tablet
14 Tablets
Dispense the accompanying Medication Guide to each patient.
abbvie
Genentech

PANEL DE VISUALIZACIÓN PRINCIPAL
NDC 0074-0566-11
Rx only
VENCLEXTA®
(venetoclax tablets)
50 mg per tablet
1 Tablet
Dispense the accompanying Medication Guide to each patient.
abbvie
Genentech
PANEL DE VISUALIZACIÓN PRINCIPAL
NDC 0074-0576-22
Rx only
VENCLEXTA®
(venetoclax tablets)
100 mg
Atención: Dispensar y almacenar Venclexta en el envase original para protegerlo de la humedad.
120 Tabletas
Entregar la Guía del Medicamento que se acompaña a cada paciente.
abbvie
Genetech
