
Fabricante de medicamentos: Biogen (Updated: 2024-04-24)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
SPINRAZA® de forma segura y eficaz. Consulte la información completa de prescripción para SPINRAZA.
SPINRAZA (nusinersen) inyección, para uso intratecal
Aprobación inicial en EE. UU.: 2016
INDICACIONES Y USO
SPINRAZA es un oligonucleótido antisentido dirigido a la supervivencia de la neurona motora-2 (SMN2) indicado para el tratamiento de la atrofia muscular espinal (SMA) en pacientes pediátricos y adultos (1)
DOSIFICACIÓN Y ADMINISTRACIÓN
SPINRAZA se administra por vía intratecal (2.1)
Información sobre la dosificación (2.1)
- La dosis recomendada es de 12 mg (5 mL) por administración
- Inicie el tratamiento con SPINRAZA con 4 dosis de carga: las tres primeras dosis de carga deben administrarse a intervalos de 14 días; la 4ta dosis de carga debe administrarse 30 días después de la 3ra dosis. Una dosis de mantenimiento debe administrarse una vez cada 4 meses a partir de entonces.
Instrucciones importantes de preparación y administración (2.2)
- Deje que se caliente a temperatura ambiente antes de la administración
- Administre dentro de las 4 horas de la extracción del vial
- Antes de la administración, retire 5 mL de líquido cefalorraquídeo
- Administre como inyección intratecal en bolo durante 1 a 3 minutos
Pruebas de laboratorio y monitoreo para evaluar la seguridad (2.3)
- Al inicio y antes de cada dosis, obtenga un recuento de plaquetas, pruebas de laboratorio de coagulación y pruebas cuantitativas de proteína en orina puntual
FORMAS Y FUERZAS DE DOSIFICACIÓN
Inyección: 12 mg/5 mL (2,4 mg/mL) en un vial de dosis única (3)
CONTRAINDICACIONES
Ninguna.
(4)
ADVERTENCIAS Y PRECAUCIONES
- Trombocitopenia y anormalidades de la coagulación: Riesgo aumentado de complicaciones hemorrágicas; se requieren pruebas al inicio y antes de cada dosis y según sea clínicamente necesario (5.1, 2.3)
- Toxicidad renal: Se requieren pruebas cuantitativas de proteína en orina puntual al inicio y antes de cada dosis (5.2, 2.3)
REACCIONES ADVERSAS
Las reacciones adversas más comunes que ocurrieron en al menos el 20% de los pacientes tratados con SPINRAZA y ocurrieron al menos un 5% más frecuentemente que en los pacientes control fueron:
- infección respiratoria inferior y estreñimiento en pacientes con SMA de inicio infantil (6.1)
- pirexia, cefalea, vómitos y dolor de espalda en pacientes con SMA de inicio tardío (6.1)
Para informar sobre REACCIONES ADVERSAS SOSPECHOSAS, comuníquese con Biogen al 1-844-477-4672 o con la FDA al 1-800-FDA-1088 o www.fda.gov/medwatch.
Ver 17 para INFORMACIÓN PARA EL PACIENTE.
Revisado: 4/2024
Tabla de Contenido
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDO*
1 INDICACIONES Y USO
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Información sobre la dosificación
2.2 Instrucciones importantes de preparación y administración
2.3 Pruebas de laboratorio y monitoreo para evaluar la seguridad
3 FORMAS Y FUERZAS DE DOSIFICACIÓN
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Trombocitopenia y anormalidades de la coagulación
5.2 Toxicidad renal
6 REACCIONES ADVERSAS
6.1 Experiencia en ensayos clínicos
6.2 Inmunogenicidad
6.3 Experiencia postcomercialización
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.4 Uso pediátrico
8.5 Uso geriátrico
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
12.2 Farmacodinamia
12.3 Farmacocinética
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, mutagénesis, deterioro de la fertilidad
14 ESTUDIOS CLÍNICOS
14.1 SMA de inicio infantil
14.2 SMA de inicio tardío
14.3 SMA presintomática
16 CÓMO SE SUMINISTRA/ALMACENAMIENTO Y MANEJO
16.1 Cómo se suministra
16.2 Almacenamiento y manejo
17 INFORMACIÓN PARA EL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información completa de prescripción no están enumeradas.
1 INDICACIONES Y USO
SPINRAZA está indicado para el tratamiento de la atrofia muscular espinal (AME) en pacientes pediátricos y adultos.
2 DOSIS Y ADMINISTRACIÓN
2.1 Información de dosificación
SPINRAZA se administra por vía intratecal por profesionales de la salud con experiencia en la realización de punciones lumbares o bajo su dirección.
Dosis recomendada
La dosis recomendada es de 12 mg (5 ml) por administración.
Inicie el tratamiento con SPINRAZA con 4 dosis de carga. Las tres primeras dosis de carga deben administrarse a intervalos de 14 días. La cuarta dosis de carga debe administrarse 30 días después de la tercera dosis. Posteriormente, se debe administrar una dosis de mantenimiento una vez cada 4 meses.
Dosis olvidada
Dosis de carga olvidada
Si olvida una dosis de carga (cualquiera de las 4 dosis de carga), administre la dosis de carga olvidada lo antes posible; ajuste la fecha de las dosis posteriores para mantener el intervalo recomendado entre dosis.
Dosis de mantenimiento olvidada
Menos de 8 meses desde la última dosis
Administre la dosis de mantenimiento olvidada lo antes posible; luego administre la siguiente dosis de mantenimiento según la fecha programada originalmente, siempre que estas dos dosis se administren con al menos 14 días de diferencia.
Al menos 8 meses pero menos de 16 meses desde la última dosis
Administre la dosis de mantenimiento olvidada lo antes posible, seguida de una dosis adicional 14 días después, y luego administre la siguiente dosis de mantenimiento 4 meses después.
2.2 Instrucciones importantes de preparación y administración
SPINRAZA es solo para uso intratecal.
Prepare y use SPINRAZA de acuerdo con los siguientes pasos utilizando una técnica aséptica. Cada vial está destinado a una sola dosis.
Preparación
- Guarde SPINRAZA en la caja en un refrigerador hasta el momento de su uso.
- Deje que el vial de SPINRAZA alcance la temperatura ambiente (25o C/77o F) antes de la administración. No utilice fuentes de calor externas.
- Inspeccione el vial de SPINRAZA para detectar la presencia de partículas y decoloración antes de la administración. No administre SPINRAZA si se observan partículas visibles o si el líquido del vial está descolorido. No se requiere el uso de filtros externos.
- Extraiga 12 mg (5 ml) de SPINRAZA del vial de dosis única en una jeringa y deseche el contenido no utilizado del vial.
- Administre SPINRAZA dentro de las 4 horas posteriores a su extracción del vial.
Administración
- Considere la sedación según lo indique el estado clínico del paciente.
- Considere la posibilidad de utilizar ultrasonidos u otras técnicas de imagen para guiar la administración intratecal de SPINRAZA, especialmente en pacientes más jóvenes.
- Antes de la administración, extraiga 5 ml de líquido cefalorraquídeo.
- Administre SPINRAZA como una inyección intratecal en bolo durante 1 a 3 minutos utilizando una aguja de anestesia espinal [consulte Dosificación y administración (2.1)]. No administre SPINRAZA en áreas de la piel donde haya signos de infección o inflamación [consulte Reacciones adversas (6.3)].
2.3 Pruebas de laboratorio y monitorización para evaluar la seguridad
Realice las siguientes pruebas de laboratorio al inicio y antes de cada dosis de SPINRAZA y según sea clínicamente necesario [consulte Advertencias y precauciones (5.1, 5.2)]:
- Recuento de plaquetas
- Tiempo de protrombina; tiempo de tromboplastina parcial activada
- Prueba cuantitativa de proteínas en orina puntual
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
Inyección: 12 mg/5 mL (2.4 mg/mL) nusinersen como una solución transparente e incolora en un vial de dosis única.
4 CONTRAINDICACIONES
Ninguna.
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Trombocitopenia y Anomalías de la Coagulación
Se han observado anomalías de la coagulación y trombocitopenia, incluida la trombocitopenia aguda grave, después de la administración de algunos oligonucleótidos antisentido.
En los estudios controlados con placebo para pacientes con SMA de inicio infantil y de inicio tardío, 24 de 146 (16%) pacientes tratados con SPINRAZA con recuento de plaquetas alto, normal o desconocido en la línea de base desarrollaron un nivel de plaquetas por debajo del límite inferior de lo normal, en comparación con 10 de 72 (14%) pacientes controlados con placebo.
En el estudio controlado con placebo en pacientes con SMA de inicio tardío (Estudio 2), dos pacientes tratados con SPINRAZA desarrollaron recuentos de plaquetas inferiores a 50.000 células por microlitro, con un nivel más bajo de 10.000 células por microlitro registrado en el día 28 del estudio.
Debido al riesgo de trombocitopenia y anomalías de la coagulación por SPINRAZA, los pacientes pueden tener un mayor riesgo de complicaciones hemorrágicas.
Realice un recuento de plaquetas y pruebas de laboratorio de coagulación en la línea de base y antes de cada administración de SPINRAZA y según sea clínicamente necesario.
5.2 Toxicidad Renal
Se ha observado toxicidad renal, incluida la glomerulonefritis potencialmente mortal, después de la administración de algunos oligonucleótidos antisentido.
SPINRAZA está presente y se excreta por el riñón [ver Farmacología clínica (12.3)]. En los estudios controlados con placebo para pacientes con SMA de inicio infantil y de inicio tardío, 71 de 123 (58%) de los pacientes tratados con SPINRAZA tuvieron proteína en la orina elevada, en comparación con 22 de 65 (34%) pacientes controlados con placebo. Realice una prueba cuantitativa de proteína en la orina puntual (preferiblemente utilizando una muestra de orina de la primera mañana) en la línea de base y antes de cada dosis de SPINRAZA. Para una concentración de proteína en la orina superior a 0,2 g/L, considere repetir la prueba y realizar una evaluación adicional.
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas graves se describen en detalle en otras secciones del etiquetado:
- Trombocitopenia y anormalidades de la coagulación [ver Advertencias y precauciones (5.1)]
- Toxicidad renal [ver Advertencias y precauciones (5.2)]
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se llevan a cabo en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de SPINRAZA no se pueden comparar directamente con las tasas en ensayos clínicos de otros medicamentos y pueden no reflejar las tasas observadas en la práctica.
En estudios clínicos, 346 pacientes (47% hombres, 76% caucásicos) fueron tratados con SPINRAZA, incluidos 314 expuestos durante al menos 6 meses, 258 expuestos durante al menos 1 año y 138 expuestos durante al menos 2 años. La seguridad de SPINRAZA se estudió en lactantes presintomáticos con SMA; pacientes pediátricos (aproximadamente de 3 días a 16 años de edad en la primera dosis) con SMA sintomática; en un ensayo controlado con placebo en lactantes con SMA sintomática (Estudio 1; n=80 para SPINRAZA, n=41 para control); en un ensayo controlado con placebo en niños con SMA sintomática (Estudio 2; n=84 para SPINRAZA, n=42 para control); en un estudio abierto en lactantes presintomáticos (Estudio 3, n=25) y otros estudios en lactantes sintomáticos (n=54) y pacientes de aparición tardía (n=103). En el Estudio 1, 58 pacientes estuvieron expuestos durante al menos 6 meses y 28 pacientes estuvieron expuestos durante al menos 12 meses. En el Estudio 2, 84 pacientes estuvieron expuestos durante al menos 6 meses y 82 pacientes estuvieron expuestos durante al menos 12 meses.
Ensayo clínico en SMA de inicio infantil (Estudio 1)
En el Estudio 1, las características de la enfermedad de referencia fueron en gran medida similares en los pacientes tratados con SPINRAZA y los pacientes con control de placebo, excepto que los pacientes tratados con SPINRAZA en la línea de base tenían un porcentaje más alto en comparación con los pacientes con control de placebo de respiración paradójica (89% vs 66%), neumonía o síntomas respiratorios (35% vs 22%), dificultades para tragar o alimentarse (51% vs 29%) y necesidad de apoyo respiratorio (26% vs 15%).
Las reacciones adversas más comunes que ocurrieron en al menos el 20% de los pacientes tratados con SPINRAZA y ocurrieron al menos un 5% más frecuentemente que en los pacientes de control fueron infección respiratoria inferior y estreñimiento. Las reacciones adversas graves de atelectasia fueron más frecuentes en los pacientes tratados con SPINRAZA (18%) que en los pacientes de control (10%). Debido a que los pacientes del Estudio 1 eran lactantes, las reacciones adversas que se informan verbalmente no se pudieron evaluar en este estudio.
|
1 Dosis de carga seguidas de 12 mg (5 mL) una vez cada 4 meses |
||
|
2 Incluye infección por adenovirus, bronquiolitis, bronquitis, bronquitis viral, infección por coronavirus, influenza, infección del tracto respiratorio inferior, infección viral del tracto respiratorio inferior, infección pulmonar, infección por virus parainfluenzae, neumonía, neumonía bacteriana, neumonía influenzal, neumonía moraxella, neumonía viral parainfluenzae, neumonía neumocócica, neumonía pseudomonal, bronquiolitis por virus respiratorio sincitial, neumonía viral y bronquiolitis por virus respiratorio sincitial. |
||
| Reacciones adversas | SPINRAZA 12 mg1 N = 80 % |
Control de procedimiento simulado N = 41 % |
| Infección respiratoria inferior2 | 55 | 37 |
| Estreñimiento | 35 | 22 |
| Dentición | 18 | 7 |
| Infección del tracto urinario | 9 | 0 |
| Congestión del tracto respiratorio superior | 8 | 2 |
| Infección de oído | 6 | 2 |
| Flatulencia | 5 | 2 |
| Disminución de peso | 5 | 2 |
En un estudio clínico abierto en lactantes con SMA sintomática, se informó hiponatremia grave en un paciente tratado con SPINRAZA que requirió suplementación con sal durante 14 meses.
Se informaron casos de erupción cutánea en pacientes tratados con SPINRAZA. Un paciente, 8 meses después de comenzar el tratamiento con SPINRAZA, desarrolló lesiones maculares rojas indoloras en el antebrazo, la pierna y el pie durante un período de 8 semanas. Las lesiones se ulceraron y se cubrieron de costras en 4 semanas y se resolvieron en varios meses. Un segundo paciente desarrolló lesiones cutáneas maculares rojas en la mejilla y la mano diez meses después del inicio del tratamiento con SPINRAZA, que se resolvieron en 3 meses. Ambos casos continuaron recibiendo SPINRAZA y tuvieron una resolución espontánea de la erupción.
SPINRAZA puede causar una reducción en el crecimiento, medida por la altura, cuando se administra a lactantes, como sugieren las observaciones del estudio controlado. Se desconoce si algún efecto de SPINRAZA sobre el crecimiento sería reversible con la suspensión del tratamiento.
Ensayo clínico en SMA de aparición tardía (Estudio 2)
En el Estudio 2, las características de la enfermedad de referencia fueron en gran medida similares en los pacientes tratados con SPINRAZA y los pacientes con control simulado, excepto por la proporción de pacientes tratados con SPINRAZA que alguna vez habían logrado la capacidad de ponerse de pie sin apoyo (13% vs 29%) o caminar con apoyo (24% vs 33%).
Las reacciones adversas más comunes que ocurrieron en al menos el 20% de los pacientes tratados con SPINRAZA y ocurrieron al menos un 5% más frecuentemente que en los pacientes de control fueron pirexia, dolor de cabeza, vómitos y dolor de espalda.
|
1 Dosis de carga seguidas de 12 mg (5 mL) una vez cada 6 meses |
||
| Reacciones adversas | SPINRAZA 12 mg1 N=84 % |
Control de procedimiento simulado N=42 % |
| Pirexia | 43 | 36 |
| Dolor de cabeza | 29 | 7 |
| Vómitos | 29 | 12 |
| Dolor de espalda | 25 | 0 |
| Epistaxis | 7 | 0 |
| Caída | 5 | 0 |
| Congestión de las vías respiratorias | 5 | 2 |
| Alergia estacional | 5 | 2 |
También se ha observado el síndrome postpunción lumbar después de la administración de SPINRAZA.
6.2 Inmunogenicidad
Al igual que con todos los oligonucleótidos, existe la posibilidad de inmunogenicidad. La detección de la formación de anticuerpos depende en gran medida de la sensibilidad y la especificidad del ensayo. Además, la incidencia observada de positividad de anticuerpos (incluidos los anticuerpos neutralizantes) en un ensayo puede verse influenciada por varios factores, incluida la metodología del ensayo, el manejo de las muestras, el momento de la recolección de las muestras, los medicamentos concomitantes y la enfermedad subyacente. Por estas razones, la comparación de la incidencia de anticuerpos a nusinersen en los estudios descritos a continuación con la incidencia de anticuerpos en otros estudios o con otros productos puede ser engañosa.
La respuesta inmunogénica a nusinersen se evaluó en 294 pacientes con muestras de plasma posteriores a la referencia para anticuerpos contra el fármaco (ADA). Diecisiete pacientes (6%) desarrollaron ADA emergentes durante el tratamiento, de los cuales 5 fueron transitorios, 12 se consideraron persistentes. Persistente se definió como tener una prueba positiva seguida de otra más de 100 días después de la primera prueba positiva. Además, “persistente” también se define como tener una o más muestras positivas y ninguna muestra más de 100 días después de la primera muestra positiva. Transitorio se definió como tener uno o más resultados positivos y no se confirmó que fuera persistente. No hay datos suficientes para evaluar el efecto de los ADA en la respuesta clínica, los eventos adversos o el perfil farmacocinético de nusinersen.
6.3 Experiencia postcomercialización
Las siguientes reacciones adversas se han identificado durante el uso posterior a la aprobación de SPINRAZA. Debido a que estas reacciones se informan voluntariamente de una población de tamaño incierto, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal con la exposición al fármaco.
Se han informado infecciones graves asociadas con la punción lumbar, como la meningitis. También se han informado hidrocefalia, meningitis aséptica, reacciones de hipersensibilidad (por ejemplo, angioedema, urticaria, erupción cutánea) y aracnoiditis.
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de riesgos
No existen datos adecuados sobre el riesgo para el desarrollo asociado con el uso de SPINRAZA en mujeres embarazadas. Cuando se administró nusinersen por inyección subcutánea a ratones durante el embarazo y la lactancia, se observó toxicidad para el desarrollo (deterioro neuroconductual a largo plazo) en todas las dosis probadas (consulte Datos). En la población general de los EE. UU., el riesgo de base estimado de defectos de nacimiento importantes y aborto espontáneo en embarazos clínicamente reconocidos es del 2 al 4 % y del 15 al 20 %, respectivamente. Se desconoce el riesgo de base de defectos de nacimiento importantes y aborto espontáneo para la población indicada.
Datos
Datos en animales
Cuando se administró nusinersen (0, 3, 10 o 25 mg/kg) por vía subcutánea a ratones machos y hembras cada dos días antes y durante el apareamiento y continuando en las hembras durante la organogénesis, no se observaron efectos adversos sobre el desarrollo embriofetal. La administración subcutánea de nusinersen (0, 6, 12,6 o 25 mg/kg) a conejas preñadas cada dos días durante la organogénesis no produjo evidencia de toxicidad para el desarrollo embriofetal.
Cuando se administró nusinersen (1,4, 5,8 o 17,2 mg/kg) a ratones hembra preñadas mediante inyección subcutánea cada dos días durante la organogénesis y continuando una vez cada seis días durante el período de lactancia, se observaron efectos neuroconductuales adversos (alteraciones en la actividad locomotora, déficits de aprendizaje y memoria) cuando se evaluó a las crías después del destete o como adultos. No se estableció un nivel sin efecto para el deterioro neuroconductual.
8.2 Lactancia
Resumen de riesgos
No hay datos sobre la presencia de nusinersen en la leche humana, los efectos en el lactante o los efectos del fármaco en la producción de leche. Se detectó nusinersen en la leche de ratones lactantes cuando se administró mediante inyección subcutánea. Los beneficios para el desarrollo y la salud de la lactancia materna deben considerarse junto con la necesidad clínica de la madre de SPINRAZA y cualquier efecto adverso potencial sobre el lactante amamantado por SPINRAZA o por la condición materna subyacente.
8.4 Uso pediátrico
Se ha establecido la seguridad y eficacia de SPINRAZA en pacientes pediátricos desde recién nacidos hasta los 17 años [consulte Estudios clínicos (14.1)].
Datos de toxicidad animal juvenil
En estudios de toxicidad intratecal en monos juveniles, la administración de nusinersen (0, 0,3, 1 o 3 mg/dosis durante 14 semanas y 0, 0,3, 1 o 4 mg/dosis durante 53 semanas) provocó histopatología cerebral (vacuolización neuronal y necrosis/restos celulares en el hipocampo) a dosis medias y altas y déficits agudos y transitorios en los reflejos espinales inferiores a dosis altas en cada estudio. Además, se observaron posibles déficits neuroconductuales en una prueba de aprendizaje y memoria a la dosis alta en el estudio de 53 semanas en monos. La dosis sin efecto para la neurohistopatología en monos (0,3 mg/dosis) es aproximadamente equivalente a la dosis humana cuando se calcula anualmente y se corrige por la diferencia de especies en el volumen de LCR.
8.5 Uso geriátrico
Los estudios clínicos de SPINRAZA no incluyeron suficientes sujetos de 65 años o más para determinar si responden de manera diferente a los sujetos más jóvenes.
11 DESCRIPCIÓN
Nusinersen es un oligonucleótido antisentido modificado, donde los grupos 2′-hidroxi de los anillos ribofuranosil son reemplazados por grupos 2′-O-2-metoxietilo y los enlaces fosfato son reemplazados por enlaces fosforotioato. Nusinersen se une a una secuencia específica en el intrón aguas abajo del exón 7 del transcrito SMN2. La fórmula estructural es:
SPINRAZA se suministra como una solución estéril, sin conservantes, incolora para uso intratecal en un vial de vidrio de dosis única. Cada solución de 1 mL contiene 2.4 mg de nusinersen (equivalente a 2.53 mg de sal de sodio de nusinersen). Cada 1 mL también contiene cloruro de calcio dihidratado (0.21 mg) USP, cloruro de magnesio hexahidratado (0.16 mg) USP, cloruro de potasio (0.22 mg) USP, cloruro de sodio (8.77 mg) USP, fosfato dibásico de sodio anhidro (0.10 mg) USP, fosfato monobásico de sodio dihidratado (0.05 mg) USP y agua para inyección USP. El producto puede contener ácido clorhídrico o hidróxido de sodio para ajustar el pH. El pH es ~7.2.
La fórmula molecular de SPINRAZA es C234H323N61O128P17S17Na17 y el peso molecular es 7501.0 daltons.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
SPINRAZA es un oligonucleótido antisentido (ASO) diseñado para tratar la SMA causada por mutaciones en el cromosoma 5q que conducen a la deficiencia de proteína SMN. Usando ensayos in vitro y estudios en modelos animales transgénicos de SMA, se demostró que SPINRAZA aumenta la inclusión del exón 7 en los transcritos de ácido ribonucleico mensajero (ARNm) de SMN2 y la producción de proteína SMN de longitud completa.
12.2 Farmacodinamia
Las muestras de autopsia de pacientes (n=3) tuvieron niveles más altos de ácido ribonucleico mensajero (ARNm) de SMN2 que contiene el exón 7 en la médula espinal torácica en comparación con los bebés con SMA no tratados.
Electrofisiología cardíaca
En los estudios controlados con placebo en 247 pacientes con atrofia muscular espinal que recibieron SPINRAZA o placebo, se observaron valores de QTcF >500 ms y un cambio desde los valores basales >60 ms en 4 (2.4%) pacientes que recibieron SPINRAZA. En comparación con el placebo, no hubo un aumento en la incidencia de reacciones adversas cardíacas asociadas con la repolarización ventricular retardada en pacientes tratados con SPINRAZA.
12.3 Farmacocinética
Absorción
La inyección intratecal de SPINRAZA en el líquido cefalorraquídeo (LCR) permite que la nusinersen se distribuya desde el LCR a los tejidos diana del sistema nervioso central (SNC). Después de la administración intratecal, las concentraciones plasmáticas mínimas de nusinersen fueron relativamente bajas, en comparación con la concentración mínima del LCR. Los valores medios de Tmax plasmático oscilaron entre 1.7 y 6.0 horas. Los valores medios de Cmax plasmático y AUC aumentaron aproximadamente de forma proporcional a la dosis hasta una dosis de 12 mg.
Distribución
Los datos de autopsia de pacientes (n=3) mostraron que SPINRAZA administrado intratecalmente se distribuyó dentro del SNC y los tejidos periféricos, como el músculo esquelético, el hígado y el riñón.
Metabolismo
La nusinersen se metaboliza mediante hidrólisis mediada por exonucleasa (3′- y 5′) y no es un sustrato, ni un inhibidor o inductor de las enzimas CYP450.
Excreción
La vida media de eliminación terminal media se estima en 135 a 177 días en el LCR y de 63 a 87 días en plasma. La principal vía de eliminación es probablemente la excreción urinaria para la nusinersen y sus metabolitos de cadena corta. A las 24 horas, solo el 0.5% de la dosis administrada se recuperó en la orina.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
Carcinogénesis
La administración de nusinersen (0, 5, 15 o 50 mg/kg) a ratones machos y hembras mediante inyección subcutánea, una vez cada dos semanas durante 2 años, dio como resultado un aumento en la incidencia de tumores vasculares (hemangioma y hemangiosarcoma combinados) en la dosis más alta probada.
Mutagénesis
Nusinersen no demostró evidencia de genotoxicidad en ensayos in vitro (Ames y aberración cromosómica en células CHO) e in vivo (micronúcleo de ratón).
Deterioro de la Fertilidad
Cuando nusinersen (0, 3, 10 o 25 mg/kg) se administró mediante inyección subcutánea a ratones cada dos días antes y durante el apareamiento y continuando en las hembras durante toda la organogénesis, no se observaron efectos adversos sobre la fertilidad masculina o femenina.
14 ESTUDIOS CLÍNICOS
La eficacia de SPINRAZA se demostró en dos ensayos clínicos dobles ciegos, controlados con procedimiento ficticio en pacientes con SMA de inicio infantil sintomático y de inicio posterior (Estudio 1 y Estudio 2), y fue respaldada por ensayos clínicos abiertos realizados en pacientes presintomáticos y sintomáticos de SMA. Los hallazgos generales de estos ensayos respaldan la eficacia de SPINRAZA en todo el rango de pacientes con SMA y parecen apoyar el inicio temprano del tratamiento con SPINRAZA.
14.1 SMA de Inicio Infantil
El Estudio 1 (NCT02193074) fue un estudio multicéntrico, aleatorizado, doble ciego, controlado con procedimiento ficticio en 121 lactantes sintomáticos con una edad de hasta 7 meses en el momento de la primera dosis, diagnosticados con SMA (inicio de los síntomas antes de los 6 meses de edad). Los pacientes se asignaron aleatoriamente en una relación 2:1 para recibir bien 12 mg de SPINRAZA o una inyección ficticia como una serie de dosis de carga administradas intratecalmente seguidas de dosis de mantenimiento administradas cada 4 meses. Los pacientes en este estudio se consideraron con mayor probabilidad de desarrollar el Tipo 1 de SMA.
Se realizó un análisis de eficacia interino planificado basado en los pacientes que fallecieron, se retiraron o completaron al menos 183 días de tratamiento. De los 82 pacientes incluidos en el análisis interino (52 pacientes en el grupo tratado con SPINRAZA y 30 en el grupo de control ficticio), el 44% eran varones, el 87% eran caucásicos, el 2% eran negros y el 4% eran asiáticos. La edad en el primer tratamiento varió de 30 a 262 días (mediana 181). La duración del tratamiento varió de 6 a 442 días (mediana 261 días). Las características demográficas basales estaban equilibradas entre los grupos de SPINRAZA y de control, excepto por la edad en el primer tratamiento (mediana de 175 y 206 días, respectivamente). Los grupos de SPINRAZA y de control estaban equilibrados en relación con la edad gestacional, el peso al nacer, la duración de la enfermedad y el número de copias de SMN2. La mediana de la duración de la enfermedad fue de 14 semanas. Hubo cierto desequilibrio en la edad en el inicio de los síntomas, con un 88% de los sujetos en el grupo de SPINRAZA y un 77% en el grupo de control que presentaron síntomas en las primeras 12 semanas de vida.
El punto final primario evaluado en el momento del análisis interino fue la proporción de respondedores: pacientes con una mejora en los hitos motores de acuerdo con la Sección 2 del Examen Neurológico Infantil de Hammersmith (HINE). Este punto final evalúa siete áreas diferentes de desarrollo de hitos motores, con un puntaje máximo de entre 2-4 puntos para cada una, dependiendo del hito, y un puntaje máximo total de 26. Un respondedor del tratamiento se definió como cualquier paciente con al menos un aumento de 2 puntos (o puntaje máximo de 4) en la capacidad de patear (consistente con una mejora de al menos 2 hitos), o al menos un aumento de 1 punto en los hitos motores de control de la cabeza, rodar, sentarse, gatear, ponerse de pie o caminar (consistente con una mejora de al menos 1 hito). Para ser clasificados como respondedores, los pacientes necesitaban exhibir mejoría en más categorías de hitos motores que empeoramiento. De los 82 pacientes que eran elegibles para el análisis interino, un porcentaje estadísticamente significativamente mayor de pacientes logró la definición de respondedor de hito motor en el grupo de SPINRAZA (40%) en comparación con el grupo de control ficticio (0%). Los resultados del análisis final fueron consistentes con los del análisis interino (Tabla 3). El 51% de los pacientes en el grupo de SPINRAZA logró la definición de respondedor de hito motor en comparación con el 0% de los pacientes en el grupo de control ficticio. Figura 1 es una presentación descriptiva de la distribución del cambio neto desde la línea base en el puntaje total del hito motor de la Sección 2 del HINE para los pacientes en el conjunto de eficacia final que no murieron o se retiraron del estudio.
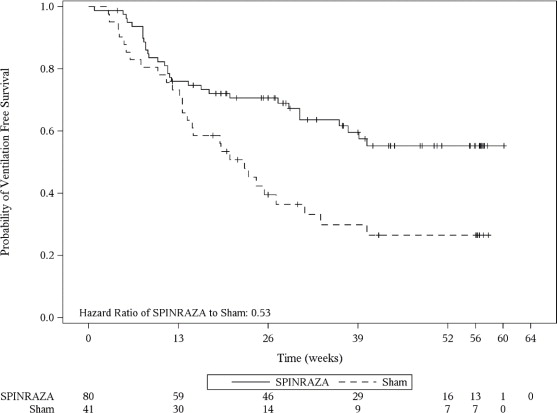
El punto final primario evaluado en el análisis final fue el tiempo hasta la muerte o la ventilación permanente (≥ 16 horas de ventilación/día continuamente durante > 21 días en ausencia de un evento reversible agudo o traqueotomía). Se observaron efectos estadísticamente significativos en la supervivencia libre de eventos y la supervivencia general en pacientes en el grupo de SPINRAZA en comparación con aquellos en el grupo de control ficticio (Tabla 4). Se observó una reducción del 47% en el riesgo de muerte o ventilación permanente en el grupo de SPINRAZA (p = 0.005) (Figura 2). El tiempo mediano hasta la muerte o la ventilación permanente no se alcanzó en el grupo de SPINRAZA y fue de 22.6 semanas en el grupo de control ficticio. También se observó una reducción estadísticamente significativa del 63% en el riesgo de muerte (p = 0.004).
En el análisis final, el estudio también evaluó los efectos del tratamiento en la Prueba Infantil de Hospital de Niños de Filadelfia de Trastornos Neuromusculares (CHOP-INTEND), que es una evaluación de las habilidades motoras en pacientes con SMA de inicio infantil. Los resultados de CHOP-INTEND se muestran en Tabla 3.
|
1En el análisis final, los análisis de CHOP-INTEND y de hito motor se realizaron utilizando el Conjunto de Eficacia (SPINRAZA n = 73; Control ficticio n = 37). |
||
|
2Evaluado en el más tarde de Día 183, Día 302 y Día 394 de la Visita del Estudio |
||
|
3Según la Sección 2 de HINE: ≥ 2 puntos de aumento [o puntaje máximo] en la capacidad de patear, o ≥ 1 punto de aumento en los hitos motores de control de la cabeza, rodar, sentarse, gatear, ponerse de pie o caminar, Y mejora en más categorías de hitos motores que empeoramiento), definido como un respondedor para este análisis primario. |
||
|
4No se controló estadísticamente para comparaciones múltiples |
||
| Punto final | Pacientes tratados con SPINRAZA (n = 73) | Pacientes de control ficticio (n = 37) |
| Función motora | ||
| Hitos motores1
Proporción que alcanzó los criterios de respondedor de hito motor predefinidos (Sección 2 de HINE)2,3 |
37 (51%) |
0 (0%)
|
| CHOP-INTEND1
Proportion achieving a 4-point improvement Proportion achieving a 4-point worsening4 |
52 (71%) 2 (3%) |
1 (3%) 17 (46%) |
|
1En el análisis final, la supervivencia libre de eventos y la supervivencia general se evaluaron utilizando la población de intención de tratar (ITT SPINRAZA n=80; Sham-control n=41). |
||
|
2Basado en la prueba de rango logarítmico estratificada por la duración de la enfermedad |
||
| Punto final | Pacientes tratados con SPINRAZA (n=80) | Pacientes de control Sham (n=41) |
| Supervivencia | ||
| Supervivencia libre de eventos1
Número de pacientes que murieron o recibieron ventilación permanente Hazard ratio (IC del 95%) p-valor2 |
31 (39%) |
28 (68%) |
|
0.53 (0.32 -0.89) p=0.005 |
||
| Supervivencia general1
Número de pacientes que murieron |
13 (16%) |
16 (39%) |
|
Hazard Ratio (IC del 95%) p-valor2 |
0.37 (0.18 – 0.77) p=0.004 |
|
Figura 1. Porcentaje de pacientes que fallecieron y cambio neto desde el punto de referencia en la puntuación total de hitos motores (HINE) entre pacientes vivos en el conjunto de eficacia final del Estudio 1 *
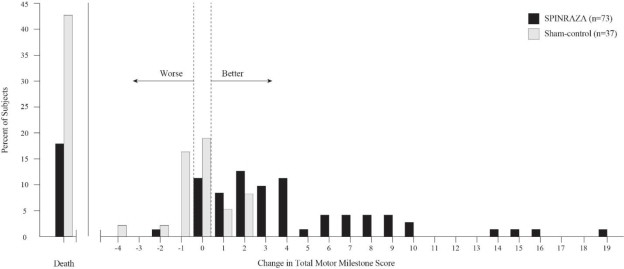
*Para los sujetos que estaban vivos y seguían en el estudio, el cambio en la puntuación total de hitos motores se calculó en el más tardío de los días 183, 302 o 394.
Figura 2. Supervivencia libre de eventos en el conjunto de intención de tratar
14.2 SMA de inicio tardío
El Estudio 2 (NCT02292537) fue un estudio multicéntrico, aleatorizado, doble ciego, controlado con procedimiento simulado en 126 niños sintomáticos con SMA de inicio tardío (inicio de síntomas después de los 6 meses de edad). Los pacientes se aleatorizaron en una proporción de 2:1 para recibir ya sea 12 mg de SPINRAZA o inyección simulada en forma de una serie de dosis de carga administradas intratecalmente, seguidas de dosis de mantenimiento administradas cada 6 meses.
La edad mediana en el cribado fue de 3 años (rango 2 – 9 años), y la edad mediana del inicio de signos y síntomas clínicos de SMA fue de 11 meses (rango 6 – 20 meses). Del total de 126 pacientes incluidos en el estudio, el 47% eran varones, el 75% eran caucásicos, el 2% eran negros y el 18% eran asiáticos. El período de tratamiento varió de 324 a 482 días (mediana 450 días). En el punto de referencia, los pacientes tenían una puntuación media en la Escala Funcional Motora Expandida de Hammersmith (HFMSE) de 21.6, todos habían alcanzado la sentada independiente y ninguno de los pacientes había alcanzado la marcha independiente. Los pacientes en este estudio se consideraron con mayor probabilidad de desarrollar el Tipo 2 o 3 de SMA.
El punto final primario evaluado fue el cambio en la puntuación respecto al punto de referencia en el mes 15 en la HFMSE. La HFMSE evalúa la función motora en pacientes con SMA que tienen ambulación limitada y comprende 33 actividades puntuadas que proporcionan información objetiva sobre la capacidad motora y la progresión clínica, como la capacidad de sentarse sin ayuda, ponerse en pie o caminar. Cada ítem se puntúa de 0 a 2, y la puntuación total máxima es 66. Las puntuaciones más altas indican una mejor función motora. El análisis primario se realizó en la población de intención de tratar (ITT), que incluía todos los sujetos que se aleatorizaron y recibieron al menos 1 dosis de SPINRAZA o al menos un procedimiento simulado. En el análisis final, se observó una mejora estadísticamente significativa en las puntuaciones en la HFMSE desde el punto de referencia hasta el mes 15 en el grupo tratado con SPINRAZA en comparación con el grupo control con simulación (Tabla 5).
|
1Evaluado utilizando la población de intención de tratar que recibió al menos una dosis de SPINRAZA o al menos un procedimiento simulado (SPINRAZA n=84; Control simulado n=42); los datos de pacientes sin una visita en el mes 15 se imputaron utilizando el método de imputación múltiple |
||
|
2Media aritmética mínima cuadrada |
||
|
3Un valor negativo indica empeoramiento, un valor positivo indica mejora. |
||
|
4Basado en la regresión logística con el efecto del tratamiento y el ajuste para la edad de cada sujeto en el cribado y la puntuación HFMSE en el punto de referencia |
||
| Punto final | Pacientes tratados con SPINRAZA (n=84) | Pacientes del control simulado (n=42) |
| Puntuación HFMSE
Cambio desde el punto de referencia en la puntuación total HFMSE en 15 meses1,2,3 Proporción de pacientes que lograron al menos una mejora de 3 puntos desde el punto de referencia hasta el mes 151 |
3,9 (95% CI: 3,0, 4,9) 56,8% (95% CI: 45,6, 68,1) |
-1,0 (95% CI: -2,5, 0,5) 26,3% (95% CI: 12,4, 40,2) |
14.3 SMA presintomática
Los resultados del ensayo controlado con placebo en pacientes con SMA de inicio infantil (Estudio 1) (NCT02193074) y de inicio tardío (Estudio 2) (NCT02292537) fueron respaldados por un ensayo abierto no controlado realizado en 25 pacientes con SMA presintomática que tenían un diagnóstico genético de SMA 5q y 2 o 3 copias de SMN2 (Estudio 3) (NCT02386553). En el Estudio 3, 15 pacientes (60%) que tenían 2 copias de SMN2 y 10 pacientes (40%) que tenían 3 copias de SMN2; 48% eran hombres, 56% eran caucásicos, 12% eran asiáticos, 4% eran indios americanos o nativos de Alaska y 28% eran de otra raza o no se informó la raza. Los pacientes tenían entre 3 días y 42 días de edad (mediana de 22 días) en el momento de la primera dosis. Los pacientes recibieron 12 mg de SPINRAZA como una serie de dosis de carga administradas intratecalmente, seguidas de dosis de mantenimiento administradas cada 4 meses. Los pacientes fueron evaluados con los hitos motores de la Organización Mundial de la Salud (OMS), un conjunto de 6 hitos en el desarrollo motor que se espera que se alcancen a los 24 meses de edad en niños sanos. Se realizó un análisis intermedio después de que todos los pacientes hubieran recibido SPINRAZA durante al menos 14 meses (mediana de 25 meses, rango de 14 a 34 meses). Los pacientes tenían entre 14 y 34 meses de edad (edad mediana de 26 meses) en el momento del análisis. En el momento del análisis intermedio (corte de datos mayo de 2018), todos los pacientes que recibieron SPINRAZA antes del inicio de los síntomas de SMA sobrevivieron sin necesidad de ventilación permanente y más allá de lo que se esperaría en función de su número de copias de SMN2. Los 25 pacientes (100%) habían alcanzado el hito motor de la OMS de sentarse sin apoyo y 22 pacientes (88%) habían alcanzado el hito de caminar con ayuda. De los 22 pacientes que tenían más edad que la esperada para haber alcanzado la capacidad de caminar de forma independiente (según la definición del percentil 95 de la edad esperada de logro de la OMS), 17 (77%) alcanzaron el hito de caminar solos (es decir, caminar de forma independiente).
16 FORMA DE SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
16.1 Cómo se suministra
SPINRAZA inyección es una solución estéril, clara e incolora que se suministra como una solución de 12 mg/5 mL (2,4 mg/mL) en un vial de vidrio de dosis única sin conservantes. El NDC es 64406-058-01.
16.2 Almacenamiento y manipulación
Almacenar en un refrigerador entre 2°C y 8°C (36°F y 46°F) en la caja original para proteger de la luz. No congelar.
SPINRAZA debe protegerse de la luz y mantenerse en la caja original hasta el momento de su uso.
Si no hay refrigeración disponible, SPINRAZA puede almacenarse en su caja original, protegido de la luz a una temperatura de 30oC (86oF) o menos durante un máximo de 14 días.
Antes de la administración, los viales sin abrir de SPINRAZA pueden retirarse y volver a colocarse en el refrigerador, si es necesario. Si se retira de la caja original, el tiempo total combinado fuera del refrigerador no debe exceder las 30 horas a una temperatura que no supere los 25oC (77oF).
17 INFORMACIÓN PARA EL PACIENTE
Trombocitopenia y Anomalías de la Coagulación
Informe a los pacientes y cuidadores que SPINRAZA podría aumentar el riesgo de sangrado. Informe a los pacientes y cuidadores de la importancia de obtener pruebas de laboratorio de sangre al inicio y antes de cada dosis para controlar los signos de un mayor potencial de sangrado. Indique a los pacientes y cuidadores que busquen atención médica si se produce un sangrado inesperado [ver Advertencias y precauciones (5.1)].
Toxicidad Renal
Informe a los pacientes y cuidadores que SPINRAZA podría causar toxicidad renal. Informe a los pacientes y cuidadores de la importancia de obtener pruebas de orina al inicio y antes de cada dosis para controlar los signos de posible toxicidad renal [ver Advertencias y precauciones (5.2)].
49655-10
Fabricado para:
Biogen
Cambridge, MA 02142
SPINRAZA es una marca registrada de Biogen.
© Biogen 2016-2024
PANEL DE VISUALIZACIÓN PRINCIPAL
Panel de visualización principal – Etiqueta de caja de Spinraza 12 mg/5 ml
NDC 64406-058-01
Spinraza™
(nusinersen)
Inyección
12 mg/5 mL
(2.4 mg/mL)
Solución estéril para
Inyección intratecal únicamente
Solo para uso con receta médica
Vial de un solo uso
Desechar la porción no utilizada

PANEL DE VISUALIZACIÓN PRINCIPAL
Panel de visualización principal – Etiqueta del frasco de Spinraza 12mg/5ml
NDC 64406-058-01
Spinraza™
(nusinersen) inyección
12 mg/5 mL (2,4 mg/mL)
Solo para inyección intratecal
