
Fabricante de medicamentos: UCB, Inc. (Updated: 2024-09-23)
ASPECTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
VIMPAT® (lacosamida) tableta recubierta con película, para uso oral, CV
VIMPAT® (lacosamida) inyección, para uso intravenoso, CV
VIMPAT® (lacosamida) solución oral, CV
Aprobación inicial en EE. UU.: 2008
CAMBIOS RECIENTES IMPORTANTES
INDICACIONES Y USO
DOSIFICACIÓN Y ADMINISTRACIÓN
-
Adultos (17 años y mayores):
- La dosis inicial para monoterapia para el tratamiento de las convulsiones de inicio parcial es de 100 mg dos veces al día (2.1)
- La dosis inicial para terapia adyuvante para el tratamiento de las convulsiones de inicio parcial o las convulsiones tónico-clónicas generalizadas primarias es de 50 mg dos veces al día (2.1)
- La dosis máxima recomendada para monoterapia y terapia adyuvante es de 200 mg dos veces al día (2.1)
- Pacientes pediátricos de 1 mes a menos de 17 años: La dosis recomendada se basa en el peso corporal y se administra por vía oral dos veces al día (2.1)
- Aumente la dosis en función de la respuesta clínica y la tolerabilidad, no más de una vez por semana (2.1)
- Inyección: solo para uso intravenoso cuando la administración oral no es temporalmente factible; la dosis recomendada se basa en el peso corporal y se administra dos o tres veces al día durante 15 a 60 minutos; se recomienda obtener un ECG antes de iniciar en ciertos pacientes (2.7, 5.3)
- Se recomienda el ajuste de la dosis para la insuficiencia renal grave (2.4, 12.3)
- Se recomienda el ajuste de la dosis para la insuficiencia hepática leve o moderada; no se recomienda el uso en pacientes con insuficiencia hepática grave (2.5, 12.3)
FORMAS DE DOSIFICACIÓN Y FUERZAS
CONTRAINDICACIONES
Ninguna (4)
ADVERTENCIAS Y PRECAUCIONES
- Monitorear a los pacientes para detectar comportamiento suicida e ideación (5.1)
- VIMPAT puede causar mareos y ataxia (5.2)
- Anomalías del ritmo y la conducción cardíacos: se recomienda obtener un ECG antes de comenzar y después de la titulación a un estado de mantenimiento estable en pacientes con afecciones proarrítmicas subyacentes o que reciben medicamentos concomitantes que afectan la conducción cardíaca; monitorear de cerca a estos pacientes (5.3, 7.2)
- VIMPAT puede causar síncope (5.4)
- VIMPAT debe retirarse gradualmente para minimizar el potencial de aumento de la frecuencia de las convulsiones (5.5)
- Reacción medicamentosa con eosinofilia y síntomas sistémicos (DRESS)/ Hipersensibilidad multiorgánica: suspender si no hay otra etiología (5.6)
REACCIONES ADVERSAS
- Terapia adyuvante: las reacciones adversas más comunes en adultos (≥10% y mayores que el placebo) son diplopía, dolor de cabeza, mareos, náuseas y somnolencia (6.1)
- Monoterapia: las reacciones adversas más comunes son similares a las observadas en los estudios de terapia adyuvante (6.1)
- Pacientes pediátricos: las reacciones adversas son similares a las observadas en pacientes adultos (6.1)
Para informar sobre REACCIONES ADVERSAS SOSPECHOSAS, comuníquese con UCB, Inc. al 1-844-599-2273 o con la FDA al 1-800-FDA-1088 o www.fda.gov/medwatch
Ver 17 para INFORMACIÓN DE ASESORAMIENTO AL PACIENTE y Guía de medicamentos.
Revisado: 10/2023
Tabla de Contenido
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: CONTENIDO*
1 INDICACIONES Y USO
1.1 Convulsiones de inicio parcial
1.2 Convulsiones tónico-clónicas generalizadas primarias
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Información de dosificación
2.2 Información de dosificación inicial alternativa para lograr la dosificación de mantenimiento en un plazo más corto
2.3 Conversión de un solo antiepiléptico (AED) a VIMPAT en monoterapia para el tratamiento de las convulsiones de inicio parcial
2.4 Información de dosificación para pacientes con insuficiencia renal
2.5 Información de dosificación para pacientes con insuficiencia hepática
2.6 Instrucciones de administración para las tabletas y la solución oral de VIMPAT
2.7 Información de preparación y administración para la inyección de VIMPAT
2.8 Suspensión de VIMPAT
3 FORMAS Y FUERZAS DE DOSIFICACIÓN
4 CONTRAINDICACIONES
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Comportamiento e ideación suicida
5.2 Mareos y ataxia
5.3 Anomalías del ritmo y la conducción cardíacos
5.4 Síncope
5.5 Retirada de fármacos antiepilépticos (AED)
5.6 Reacción medicamentosa con eosinofilia y síntomas sistémicos (DRESS)/Hipersensibilidad multiorgánica
5.7 Riesgos en pacientes con fenilcetonuria
6 REACCIONES ADVERSAS
6.1 Experiencia en ensayos clínicos
6.2 Experiencia postcomercialización
7 INTERACCIONES MEDICAMENTOSAS
7.1 Inhibidores potentes de CYP3A4 o CYP2C9
7.2 Medicamentos concomitantes que afectan la conducción cardíaca
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.2 Lactancia
8.4 Uso pediátrico
8.5 Uso geriátrico
8.6 Insuficiencia renal
8.7 Insuficiencia hepática
9 ABUSO Y DEPENDENCIA DE DROGAS
9.1 Sustancia controlada
9.2 Abuso
9.3 Dependencia
10 SOBREDOSIS
11 DESCRIPCIÓN
11.1 Tabletas de VIMPAT
11.2 Inyección de VIMPAT
11.3 Solución oral de VIMPAT
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
12.2 Farmacodinamia
12.3 Farmacocinética
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, mutagénesis, deterioro de la fertilidad
14 ESTUDIOS CLÍNICOS
14.1 Monoterapia en pacientes con convulsiones de inicio parcial
14.2 Terapia adyuvante en pacientes con convulsiones de inicio parcial
14.3 Terapia adyuvante en pacientes con convulsiones tónico-clónicas generalizadas primarias
16 CÓMO SE SUMINISTRA/ALMACENAMIENTO Y MANEJO
16.1 Cómo se suministra
16.2 Almacenamiento y manejo
17 INFORMACIÓN PARA EL PACIENTE
- *
- Las secciones o subsecciones omitidas de la información completa de prescripción no están enumeradas.
1 INDICACIONES Y USO
1.1 Crisis parciales
VIMPAT está indicado para el tratamiento de crisis parciales en pacientes de 1 mes de edad o mayores.
1.2 Crisis tónico-clónicas generalizadas primarias
VIMPAT está indicado como terapia adyuvante en el tratamiento de crisis tónico-clónicas generalizadas primarias en pacientes de 4 años de edad o mayores.
2 DOSIS Y ADMINISTRACIÓN
2.1 Información sobre la dosificación
La dosis recomendada para la monoterapia y la terapia adyuvante de las crisis parciales en pacientes de 1 mes de edad o mayores y para la terapia adyuvante de las crisis tónico-clónicas generalizadas primarias en pacientes de 4 años de edad o mayores se incluye en la Tabla 1. En pacientes pediátricos, el régimen de dosificación recomendado depende del peso corporal. La dosis debe aumentarse en función de la respuesta clínica y la tolerabilidad, no más de una vez por semana. Los incrementos de titulación no deben exceder los que se muestran en la Tabla 1.
| Edad y peso corporal | Dosis inicial | Régimen de titulación | Dosis de mantenimiento |
|---|---|---|---|
|
|||
| Adultos (17 años o mayores) | Monoterapia†: 100 mg dos veces al día (200 mg por día) Terapia adyuvante: 50 mg dos veces al día (100 mg por día) |
Aumentar en 50 mg dos veces al día (100 mg por día) cada semana | Monoterapia†: 150 mg a 200 mg dos veces al día (300 mg a 400 mg por día) Terapia adyuvante: 100 mg a 200 mg dos veces al día (200 mg a 400 mg por día) |
| Pacientes pediátricos que pesan al menos 50 kg | 50 mg dos veces al día (100 mg por día) |
Aumentar en 50 mg dos veces al día (100 mg por día) cada semana | Monoterapia†: 150 mg a 200 mg dos veces al día (300 mg a 400 mg por día) Terapia adyuvante: 100 mg a 200 mg dos veces al día (200 mg a 400 mg por día) |
| Pacientes pediátricos que pesan de 30 kg a menos de 50 kg | 1 mg/kg dos veces al día (2 mg/kg/día) |
Aumentar en 1 mg/kg dos veces al día (2 mg/kg/día) cada semana | 2 mg/kg a 4 mg/kg dos veces al día (4 mg/kg/día a 8 mg/kg/día) |
| Pacientes pediátricos que pesan de 11 kg a menos de 30 kg | 1 mg/kg dos veces al día (2 mg/kg/día) |
Aumentar en 1 mg/kg dos veces al día (2 mg/kg/día) cada semana | 3 mg/kg a 6 mg/kg dos veces al día (6 mg/kg/día a 12 mg/kg/día) |
| Pacientes pediátricos que pesan de 6 kg a menos de 11 kg ‡ | |||
| Pacientes pediátricos que pesan menos de 6 kg ‡ | Intravenoso: 0.66 mg/kg tres veces al día (2 mg/kg/día) |
Intravenoso: Aumentar en 0.66 mg/kg tres veces al día (2 mg/kg/día) cada semana |
Intravenoso: 2.5 mg/kg a 5 mg/kg tres veces al día (7.5 mg/kg/día a 15 mg/kg/día) |
| Oral: 1 mg/kg dos veces al día (2 mg/kg/día) |
Oral: Aumentar en 1 mg/kg dos veces al día (2 mg/kg/día) cada semana |
Oral: 3.75 mg/kg a 7.5 mg/kg dos veces al día (7.5 mg/kg/día a 15 mg/kg/día) |
|
En ensayos clínicos adyuvantes en pacientes adultos con convulsiones de inicio parcial, una dosis superior a 200 mg dos veces al día (400 mg por día) no fue más efectiva y se asoció con una tasa sustancialmente mayor de reacciones adversas [ver Reacciones adversas (6.1) y Estudios clínicos (14.2)].
Dosis de VIMPAT inyección
VIMPAT inyección se puede usar cuando la administración oral no es temporalmente factible [ver Dosis y administración (2.6) y Advertencias y precauciones (5.3)]. VIMPAT inyección se puede administrar por vía intravenosa a pacientes adultos y pediátricos que pesan 6 kg o más con los mismos regímenes de dosificación descritos para la dosificación oral. Para pacientes pediátricos que pesan menos de 6 kg, VIMPAT inyección se puede iniciar con una dosis de 0,66 mg/kg tres veces al día (ver Tabla 1).
La experiencia del estudio clínico de VIMPAT intravenoso se limita a 5 días de tratamiento consecutivo.
2.2 Información de dosis inicial alternativa para lograr la dosis de mantenimiento en un plazo más corto
Para monoterapia y terapia adyuvante para convulsiones de inicio parcial en pacientes de 1 mes de edad o más y para terapia adyuvante para convulsiones tónico-clónicas generalizadas primarias en pacientes de 4 años de edad o más, se puede administrar un régimen de dosificación inicial alternativo para la semana 1 (por ejemplo, que incluye una dosis de carga y/o una dosis inicial más alta) en pacientes para quienes lograr la dosis de mantenimiento recomendada en un plazo más corto está clínicamente indicado (ver Tabla 2). El régimen de dosificación inicial alternativo debe continuarse durante una semana. VIMPAT luego se puede titular en función de la respuesta clínica y la tolerabilidad, no más frecuentemente que una vez por semana, si es necesario. La dosis de carga debe administrarse con supervisión médica debido a la posibilidad de un aumento de la incidencia de reacciones adversas, incluidas las reacciones adversas del sistema nervioso central (SNC) y cardiovasculares [ver Advertencias y precauciones (5.2, 5.3), Reacciones adversas (6.1), y Farmacología clínica (12.3)]. Los incrementos de titulación no deben exceder los que se muestran en la Tabla 2.
| Edad y peso corporal | Dosis inicial alternativa | Régimen de titulación | Dosis de mantenimiento |
|---|---|---|---|
|
|||
| Adultos (17 años y mayores) | Dosis de carga única: 200 mg 12 horas después iniciar: 100 mg dos veces al día (200 mg por día) |
Aumentar en 50 mg dos veces al día (100 mg por día) a intervalos semanales, si es necesario | Monoterapia†: 150 mg a 200 mg dos veces al día (300 mg a 400 mg por día) Terapia adyuvante: 100 mg a 200 mg dos veces al día (200 mg a 400 mg por día) |
| Pacientes pediátricos que pesan al menos 50 kg | Dosis de carga única: 200 mg 12 horas después iniciar: 100 mg dos veces al día (200 mg por día) |
Aumentar en 50 mg dos veces al día (100 mg por día) a intervalos semanales, si es necesario | Monoterapia†: 150 mg a 200 mg dos veces al día (300 mg a 400 mg por día) Terapia adyuvante: 100 mg a 200 mg dos veces al día (200 mg a 400 mg por día) |
| Pacientes pediátricos que pesan de 30 kg a menos de 50 kg | Dosis de carga única: 4 mg/kg 12 horas después iniciar: 2 mg/kg dos veces al día (4 mg/kg/día) |
Aumentar en 1 mg/kg dos veces al día (2 mg/kg/día) a intervalos semanales, si es necesario | 2 mg/kg a 4 mg/kg dos veces al día (4 mg/kg/día a 8 mg/kg/día) |
| Pacientes pediátricos que pesan de 11 kg a menos de 30 kg | Dosis de carga única: 4.5 mg/kg 12 horas después iniciar: 3 mg/kg dos veces al día (6 mg/kg/día) |
Aumentar en 1 mg/kg dos veces al día (2 mg/kg/día) a intervalos semanales, si es necesario | 3 mg/kg a 6 mg/kg dos veces al día (6 mg/kg/día a 12 mg/kg/día) |
| Pacientes pediátricos que pesan de 6 kg a menos de 11 kg ‡ | |||
| Pacientes pediátricos que pesan menos de 6 kg ‡ | Intravenosa: No se requiere dosis de carga 2.5 mg/kg tres veces al día (7.5 mg/kg/día) |
Intravenosa: Aumentar en 0.66 mg/kg tres veces al día (2 mg/kg/día) a intervalos semanales, si es necesario |
Intravenosa: 2.5 mg/kg a 5 mg/kg tres veces al día (7.5 mg/kg/día a 15 mg/kg/día) |
| Oral: No se requiere dosis de carga 3.75 mg/kg dos veces al día (7.5 mg/kg/día) |
Oral: Aumentar en 1 mg/kg dos veces al día (2 mg/kg/día) a intervalos semanales, si es necesario |
Oral: 3.75 mg/kg a 7.5 mg/kg dos veces al día (7.5 mg/kg/día a 15 mg/kg/día) |
|
2.3 Conversión de un solo antiepiléptico (AED) a monoterapia con VIMPAT para el tratamiento de las convulsiones de inicio parcial
Para los pacientes que ya están tomando un solo AED y se convertirán a monoterapia con VIMPAT, la retirada del AED concomitante no debe producirse hasta que se alcance la dosis terapéutica de VIMPAT y se haya administrado durante al menos 3 días. Se recomienda una retirada gradual del AED concomitante durante al menos 6 semanas.
2.4 Información sobre la dosis para pacientes con insuficiencia renal
Para los pacientes con insuficiencia renal leve o moderada, no es necesario ajustar la dosis. Para los pacientes con insuficiencia renal grave [aclaramiento de creatinina (CLCR) inferior a 30 mL/min según la ecuación de Cockcroft-Gault para adultos; CLCR inferior a 30 mL/min/1,73 m2 según la ecuación de Schwartz para pacientes pediátricos] o enfermedad renal en estadio terminal, se recomienda una reducción del 25% de la dosis máxima.
En todos los pacientes con insuficiencia renal, el inicio y la titulación de la dosis deben basarse en la respuesta clínica y la tolerabilidad.
Hemodiálisis
VIMPAT se elimina eficazmente del plasma mediante hemodiálisis. Tras un tratamiento de hemodiálisis de 4 horas, debe considerarse la posibilidad de suplementar la dosis hasta un 50%.
Inhibidores potentes concomitantes de CYP3A4 o CYP2C9
Puede ser necesaria una reducción de la dosis en pacientes con insuficiencia renal que estén tomando inhibidores potentes de CYP3A4 y CYP2C9 [véase Interacciones medicamentosas (7.1), Uso en poblaciones específicas (8.6) y Farmacología clínica (12.3)].
2.5 Información sobre la dosis para pacientes con insuficiencia hepática
Para los pacientes con insuficiencia hepática leve o moderada, se recomienda una reducción del 25% de la dosis máxima. El inicio y la titulación de la dosis deben basarse en la respuesta clínica y la tolerabilidad en pacientes con insuficiencia hepática.
No se recomienda el uso de VIMPAT en pacientes con insuficiencia hepática grave.
Inhibidores potentes concomitantes de CYP3A4 y CYP2C9
Puede ser necesaria una reducción de la dosis en pacientes con insuficiencia hepática que estén tomando inhibidores potentes de CYP3A4 y CYP2C9 [véase Interacciones medicamentosas (7.1), Uso en poblaciones específicas (8.7) y Farmacología clínica (12.3)].
2.6 Instrucciones de administración para las tabletas y la solución oral de VIMPAT
Las tabletas y la solución oral de VIMPAT pueden tomarse con o sin alimentos.
Tabletas de VIMPAT
Las tabletas de VIMPAT deben tragarse enteras con líquido. No divida las tabletas de VIMPAT.
Solución oral de VIMPAT
Se recomienda un dispositivo de medición calibrado para medir y administrar la dosis prescrita con precisión. Una cucharadita o cucharada de uso doméstico no es un dispositivo de medición adecuado.
La solución oral de VIMPAT también puede administrarse mediante una sonda nasogástrica o una sonda de gastrostomía.
Deseche cualquier solución oral de VIMPAT no utilizada que quede después de 6 meses de la primera apertura del frasco.
2.7 Información sobre la preparación y la administración de la inyección de VIMPAT
Preparación
La inyección de VIMPAT puede administrarse por vía intravenosa sin dilución adicional o puede mezclarse con los diluyentes que se enumeran a continuación. La solución diluida no debe almacenarse durante más de 4 horas a temperatura ambiente.
Diluyentes:
Solución inyectable de cloruro de sodio al 0,9% (p/v)
Solución inyectable de dextrosa al 5% (p/v)
Solución inyectable de Ringer lactato
Los productos farmacéuticos parenterales deben inspeccionarse visualmente para detectar partículas y decoloración antes de su administración, siempre que la solución y el envase lo permitan. No debe utilizarse el producto que contenga partículas o decoloración.
La inyección de VIMPAT es para un solo uso. Cualquier porción no utilizada de la inyección de VIMPAT debe desecharse.
Administración
La duración de la infusión recomendada es de 30 a 60 minutos; sin embargo, las infusiones tan rápidas como 15 minutos pueden administrarse en adultos si es necesario [véase Reacciones adversas (6.1) y Farmacología clínica (12.3)]. Las duraciones de la infusión inferiores a 30 minutos generalmente no se recomiendan en pacientes pediátricos [véase Reacciones adversas (6.1)].
La infusión intravenosa de VIMPAT puede causar bradicardia, bloqueos AV y taquiarritmias ventriculares [véase Advertencias y precauciones (5.3)]. Se recomienda obtener un ECG antes de comenzar con VIMPAT y después de que VIMPAT se haya titulado a la dosis de mantenimiento en estado estacionario en pacientes con condiciones proarrítmicas subyacentes o que estén tomando medicamentos concomitantes que afecten la conducción cardíaca [véase Interacciones medicamentosas (7.2)].
2.8 Suspensión de VIMPAT
Se recomienda una retirada gradual de VIMPAT durante al menos 1 semana [ver Advertencias y precauciones (5.5)].
3 FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
VIMPAT Tablets
- 50 mg: rosa, oval, recubierta con película, con “SP” grabado en un lado y “50” en el otro
- 100 mg: amarillo oscuro, oval, recubierta con película, con “SP” grabado en un lado y “100” en el otro
- 150 mg: salmón, oval, recubierta con película, con “SP” grabado en un lado y “150” en el otro
- 200 mg: azul, oval, recubierta con película, con “SP” grabado en un lado y “200” en el otro
VIMPAT Oral Solution
- 10 mg/mL: líquido transparente, incoloro a amarillo o amarillo-marrón, con sabor a fresa
4 CONTRAINDICACIONES
Ninguna.
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Comportamiento y pensamientos suicidas
Los fármacos antiepilépticos (FAE), incluido VIMPAT, aumentan el riesgo de pensamientos o comportamiento suicidas en pacientes que toman estos fármacos para cualquier indicación. Los pacientes tratados con cualquier FAE para cualquier indicación deben ser monitoreados para la aparición o el empeoramiento de la depresión, los pensamientos o el comportamiento suicidas, y/o cualquier cambio inusual en el estado de ánimo o el comportamiento.
Los análisis agrupados de 199 ensayos clínicos controlados con placebo (monoterapia y terapia adyuvante) de 11 FAE diferentes mostraron que los pacientes asignados al azar a uno de los FAE tuvieron aproximadamente el doble de riesgo (riesgo relativo ajustado 1.8, IC del 95%: 1.2, 2.7) de pensamientos o comportamiento suicidas en comparación con los pacientes asignados al azar a placebo. En estos ensayos, que tuvieron una duración media de tratamiento de 12 semanas, la incidencia estimada de comportamiento o ideación suicida entre 27.863 pacientes tratados con FAE fue del 0.43%, en comparación con el 0.24% entre 16.029 pacientes tratados con placebo, lo que representa un aumento de aproximadamente un caso de pensamientos o comportamiento suicidas por cada 530 pacientes tratados. Hubo cuatro suicidios en pacientes tratados con fármacos en los ensayos y ninguno en pacientes tratados con placebo, pero el número de eventos es demasiado pequeño para permitir cualquier conclusión sobre el efecto del fármaco en el suicidio.
El aumento del riesgo de pensamientos o comportamiento suicidas con FAE se observó tan pronto como una semana después de comenzar el tratamiento con FAE y persistió durante la duración del tratamiento evaluado. Debido a que la mayoría de los ensayos incluidos en el análisis no se extendieron más allá de las 24 semanas, no se pudo evaluar el riesgo de pensamientos o comportamiento suicidas más allá de las 24 semanas.
El riesgo de pensamientos o comportamiento suicidas fue generalmente consistente entre los fármacos en los datos analizados. El hallazgo de un mayor riesgo con FAE de diferentes mecanismos de acción y en una gama de indicaciones sugiere que el riesgo se aplica a todos los FAE utilizados para cualquier indicación. El riesgo no varió sustancialmente según la edad (5-100 años) en los ensayos clínicos analizados.
La Tabla 3 muestra el riesgo absoluto y relativo por indicación para todos los FAE evaluados.
| Indicación | Pacientes con placebo con eventos por cada 1000 pacientes | Pacientes con fármacos con eventos por cada 1000 pacientes | Riesgo relativo: Incidencia de eventos en pacientes con fármacos/Incidencia en pacientes con placebo |
Diferencia de riesgo: Pacientes con fármacos adicionales con eventos por cada 1000 pacientes |
|---|---|---|---|---|
| Epilepsia | 1.0 | 3.4 | 3.5 | 2.4 |
| Psiquiátrica | 5.7 | 8.5 | 1.5 | 2.9 |
| Otra | 1.0 | 1.8 | 1.9 | 0.9 |
| Total | 2.4 | 4.3 | 1.8 | 1.9 |
El riesgo relativo de pensamientos o conductas suicidas fue mayor en los ensayos clínicos para la epilepsia que en los ensayos clínicos para condiciones psiquiátricas u otras, pero las diferencias en el riesgo absoluto fueron similares.
Cualquiera que considere prescribir VIMPAT o cualquier otro AED debe equilibrar este riesgo con el riesgo de la enfermedad no tratada. La epilepsia y muchas otras enfermedades para las que se prescriben antiepilépticos están asociadas por sí mismas con morbilidad y mortalidad y un mayor riesgo de pensamientos y conductas suicidas. Si surgen pensamientos y conductas suicidas durante el tratamiento, el prescriptor debe considerar si la aparición de estos síntomas en un paciente dado puede estar relacionada con la enfermedad que se trata.
5.2 Mareos y ataxia
VIMPAT puede causar mareos y ataxia en pacientes adultos y pediátricos. En pacientes adultos con crisis de inicio parcial que tomaban de 1 a 3 AED concomitantes, el mareo fue experimentado por el 25% de los pacientes aleatorizados a las dosis recomendadas (200 a 400 mg/día) de VIMPAT (en comparación con el 8% de los pacientes con placebo) y fue la reacción adversa que con más frecuencia condujo a la suspensión (3%). La ataxia fue experimentada por el 6% de los pacientes aleatorizados a las dosis recomendadas (200 a 400 mg/día) de VIMPAT (en comparación con el 2% de los pacientes con placebo). El inicio de los mareos y la ataxia se observó más comúnmente durante la titulación. Hubo un aumento sustancial en estas reacciones adversas en dosis superiores a 400 mg/día [ver Reacciones adversas (6.1)]. Si una dosis de carga está clínicamente indicada, administrar con supervisión médica debido a la posibilidad de un aumento en la incidencia de reacciones adversas, incluidas las reacciones adversas del SNC como mareos y ataxia.
5.3 Anomalías del ritmo y conducción cardíaca
Prolongación del intervalo PR, bloqueo auriculoventricular y taquiarritmias ventriculares
Se han observado prolongaciones dependientes de la dosis en el intervalo PR con VIMPAT en estudios clínicos en pacientes adultos y en voluntarios sanos [ver Farmacología clínica (12.2)]. En ensayos clínicos adyuvantes en pacientes adultos con crisis de inicio parcial, el bloqueo auriculoventricular asintomático de primer grado se observó como una reacción adversa en el 0,4% (4/944) de los pacientes aleatorizados para recibir VIMPAT y en el 0% (0/364) de los pacientes aleatorizados para recibir placebo. Un caso de bradicardia profunda se observó en un paciente durante una infusión de 15 minutos de 150 mg de VIMPAT. Cuando VIMPAT se administra con otros fármacos que prolongan el intervalo PR, es posible una mayor prolongación del PR.
En el entorno postcomercialización, se han reportado arritmias cardíacas en pacientes tratados con VIMPAT, incluyendo bradicardia, bloqueo auriculoventricular y taquiarritmias ventriculares, que rara vez han resultado en asístole, paro cardíaco y muerte. La mayoría, aunque no todos, los casos han ocurrido en pacientes con condiciones proarritmogénicas subyacentes o en aquellos que toman medicamentos concomitantes que afectan la conducción cardíaca o prolongan el intervalo PR. Estos eventos han ocurrido tanto con las vías de administración oral como intravenosa y en las dosis prescritas, así como en el entorno de sobredosis [ver Sobredosis (10)]. En todos los pacientes para los que una dosis de carga está clínicamente indicada, administrar la dosis de carga con supervisión médica debido a la posibilidad de un aumento en la incidencia de reacciones adversas, incluidas las reacciones adversas cardiovasculares.
VIMPAT debe usarse con precaución en pacientes con condiciones proarritmogénicas subyacentes como problemas conocidos de conducción cardíaca (por ejemplo, bloqueo auriculoventricular de primer grado marcado, bloqueo auriculoventricular de segundo grado o superior y síndrome del seno enfermo sin marcapasos), enfermedades cardíacas graves (como isquemia miocárdica o insuficiencia cardíaca o enfermedad estructural del corazón) y canalesopatías cardíacas de sodio (por ejemplo, síndrome de Brugada). VIMPAT también debe usarse con precaución en pacientes que toman medicamentos concomitantes que afectan la conducción cardíaca, incluidos bloqueadores de canales de sodio, betabloqueantes, bloqueadores de canales de calcio, bloqueadores de canales de potasio y medicamentos que prolongan el intervalo PR [ver Interacciones medicamentosas (7.2)]. En tales pacientes, se recomienda obtener un ECG antes de iniciar VIMPAT y después de que VIMPAT se titule a la dosis de mantenimiento en estado estacionario. Además, estos pacientes deben ser monitorizados estrechamente si se les administra VIMPAT por vía intravenosa [ver Reacciones adversas (6.1) y Interacciones medicamentosas (7.2)].
Fibrilación auricular y flutter auricular
En los ensayos investigacionales a corto plazo de VIMPAT en pacientes adultos con crisis de inicio parcial no hubo casos de fibrilación auricular o flutter auricular. Tanto la fibrilación auricular como el flutter auricular han sido reportados en ensayos de crisis de inicio parcial de etiqueta abierta y en la experiencia postcomercialización. En pacientes adultos con neuropatía diabética, para la que VIMPAT no está indicado, el 0,5% de los pacientes tratados con VIMPAT experimentó una reacción adversa de fibrilación auricular o flutter auricular, en comparación con el 0% de los pacientes tratados con placebo. La administración de VIMPAT puede predisponer a arritmias auriculares (fibrilación auricular o flutter auricular), especialmente en pacientes con neuropatía diabética y/o enfermedad cardiovascular.
5.4 Síncope
En los ensayos controlados a corto plazo de VIMPAT en pacientes adultos con convulsiones de inicio parcial sin enfermedades sistémicas significativas, no hubo un aumento en la síncope en comparación con el placebo. En los ensayos controlados a corto plazo en pacientes adultos con neuropatía diabética, para los cuales VIMPAT no está indicado, el 1.2% de los pacientes que fueron tratados con VIMPAT reportaron una reacción adversa de síncope o pérdida del conocimiento, en comparación con el 0% de los pacientes tratados con placebo con neuropatía diabética. La mayoría de los casos de síncope se observaron en pacientes que recibieron dosis superiores a 400 mg/día. La causa de la síncope no se determinó en la mayoría de los casos. Sin embargo, varios se asociaron con cambios en la presión arterial ortostática, aleteo/fibrilación auricular (y taquicardia asociada) o bradicardia. También se han observado casos de síncope en estudios clínicos abiertos de convulsiones de inicio parcial en pacientes adultos y pediátricos. Estos casos se asociaron con antecedentes de factores de riesgo para la enfermedad cardíaca y el uso de medicamentos que ralentizan la conducción AV.
5.5 Retirada de medicamentos antiepilépticos (AED)
Al igual que con todos los AED, VIMPAT debe retirarse gradualmente (durante un mínimo de 1 semana) para minimizar el potencial de aumento de la frecuencia de las convulsiones en pacientes con trastornos convulsivos.
5.6 Reacción medicamentosa con eosinofilia y síntomas sistémicos (DRESS)/Hipersensibilidad multiorgánica
Se ha informado de reacción medicamentosa con eosinofilia y síntomas sistémicos (DRESS), también conocida como hipersensibilidad multiorgánica, en pacientes que toman medicamentos antiepilépticos, incluido VIMPAT. Algunos de estos eventos han sido fatales o potencialmente mortales. DRESS típicamente, aunque no exclusivamente, se presenta con fiebre, erupción cutánea, linfadenopatía y/o hinchazón facial, en asociación con la participación de otros sistemas orgánicos, como hepatitis, nefritis, anomalías hematológicas, miocarditis o miositis, a veces parecidas a una infección viral aguda. La eosinofilia suele estar presente. Este trastorno es variable en su expresión, y otros sistemas orgánicos no mencionados aquí pueden estar involucrados. Es importante tener en cuenta que las manifestaciones tempranas de hipersensibilidad (por ejemplo, fiebre, linfadenopatía) pueden estar presentes aunque la erupción cutánea no sea evidente. Si estos signos o síntomas están presentes, el paciente debe ser evaluado inmediatamente. VIMPAT debe suspenderse si no se puede establecer una etiología alternativa para los signos o síntomas.
5.7 Riesgos en pacientes con fenilcetonuria
La fenilalanina puede ser dañina en pacientes con fenilcetonuria (PKU). La solución oral de VIMPAT contiene aspartamo, una fuente de fenilalanina. Una dosis de 200 mg de solución oral de VIMPAT (equivalente a 20 mL) contiene 0.32 mg de fenilalanina. Antes de recetar la solución oral de VIMPAT a un paciente con PKU, considere la cantidad diaria combinada de fenilalanina de todas las fuentes, incluida la solución oral de VIMPAT.
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas graves se describen a continuación y en otras partes del etiquetado:
- Comportamiento y pensamientos suicidas [ver Advertencias y precauciones (5.1)]
- Mareos y ataxia [ver Advertencias y precauciones (5.2)]
- Anomalías del ritmo y la conducción cardíacos [ver Advertencias y precauciones (5.3)]
- Síncope [ver Advertencias y precauciones (5.4)]
- Reacción medicamentosa con eosinofilia y síntomas sistémicos (DRESS)/Reacciones de hipersensibilidad multiorgánicas [ver Advertencias y precauciones (5.6)]
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se llevan a cabo en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un medicamento no se pueden comparar directamente con las tasas en los ensayos clínicos de otro medicamento y pueden no reflejar las tasas observadas en la práctica.
VIMPAT Comprimidos y Solución Oral en Adultos
En el desarrollo precomercialización de la terapia adyuvante para las convulsiones de inicio parcial, 1327 pacientes adultos recibieron VIMPAT comprimidos en ensayos controlados y no controlados, de los cuales 1000 fueron tratados durante más de 6 meses y 852 durante más de 12 meses. El programa de desarrollo de monoterapia para las convulsiones de inicio parcial incluyó 425 pacientes adultos, 310 de los cuales fueron tratados durante más de 6 meses y 254 durante más de 12 meses.
Convulsiones de inicio parcial
Ensayo de control histórico de monoterapia (Estudio 1)
En el ensayo de monoterapia para las convulsiones de inicio parcial, el 16% de los pacientes aleatorizados para recibir VIMPAT a las dosis recomendadas de 300 y 400 mg/día abandonaron el ensayo como resultado de una reacción adversa. La reacción adversa que con mayor frecuencia (≥1% en VIMPAT) condujo a la interrupción fue el mareo.
Las reacciones adversas que ocurrieron en este estudio fueron generalmente similares a las que ocurrieron en estudios adyuvantes controlados con placebo. Una reacción adversa, el insomnio, ocurrió a una tasa de ≥2% y no se informó a una tasa similar en estudios previos. Esta reacción adversa también se ha observado en la experiencia postcomercialización [ver Reacciones adversas (6.2)]. Debido a que este estudio no incluyó un grupo de control con placebo, no se pudo establecer la causalidad.
Los mareos, el dolor de cabeza, las náuseas, la somnolencia y la fatiga ocurrieron todos a incidencias más bajas durante la fase de retirada del AED y la fase de monoterapia, en comparación con la fase de titulación [ver Estudios clínicos (14.1)].
Ensayos controlados de terapia adyuvante (Estudios 2, 3 y 4)
En los ensayos clínicos controlados de terapia adyuvante para las convulsiones de inicio parcial, la tasa de interrupción como resultado de una reacción adversa fue del 8% y el 17% en los pacientes aleatorizados para recibir VIMPAT a las dosis recomendadas de 200 y 400 mg/día, respectivamente, el 29% a 600 mg/día (1,5 veces mayor que la dosis máxima recomendada) y el 5% en los pacientes aleatorizados para recibir placebo. Las reacciones adversas que con mayor frecuencia (>1% en VIMPAT y mayor que placebo) condujeron a la interrupción fueron los mareos, la ataxia, los vómitos, la diplopía, las náuseas, el vértigo y la visión borrosa.
La Tabla 4 muestra la incidencia de las reacciones adversas que ocurrieron en ≥2% de los pacientes adultos con convulsiones de inicio parcial en el grupo total de VIMPAT y para las cuales la incidencia fue mayor que placebo.
| Reacción adversa | Placebo N=364 % |
VIMPAT 200 mg/día N=270 % |
VIMPAT 400 mg/día N=471 % |
VIMPAT 600 mg/día* N=203 % |
VIMPAT Total N=944 % |
|---|---|---|---|---|---|
|
|||||
| Trastornos del oído y el laberinto | |||||
| Vértigo | 1 | 5 | 3 | 4 | 4 |
| Trastornos oculares | |||||
| Diplopía | 2 | 6 | 10 | 16 | 11 |
| Visión borrosa | 3 | 2 | 9 | 16 | 8 |
| Trastornos gastrointestinales | |||||
| Nausea | 4 | 7 | 11 | 17 | 11 |
| Vomiting | 3 | 6 | 9 | 16 | 9 |
| Diarrhea | 3 | 3 | 5 | 4 | 4 |
| General disorders and administration site conditions | |||||
| Fatigue | 6 | 7 | 7 | 15 | 9 |
| Gait disturbance | <1 | <1 | 2 | 4 | 2 |
| Asthenia | 1 | 2 | 2 | 4 | 2 |
| Injury, poisoning and procedural complications | |||||
| Contusion | 3 | 3 | 4 | 2 | 3 |
| Skin laceration | 2 | 2 | 3 | 3 | 3 |
| Nervous system disorders | |||||
| Dizziness | 8 | 16 | 30 | 53 | 31 |
| Headache | 9 | 11 | 14 | 12 | 13 |
| Ataxia | 2 | 4 | 7 | 15 | 8 |
| Somnolence | 5 | 5 | 8 | 8 | 7 |
| Tremor | 4 | 4 | 6 | 12 | 7 |
| Nystagmus | 4 | 2 | 5 | 10 | 5 |
| Balance disorder | 0 | 1 | 5 | 6 | 4 |
| Memory impairment | 2 | 1 | 2 | 6 | 2 |
| Psychiatric disorders | |||||
| Depression | 1 | 2 | 2 | 2 | 2 |
| Skin and subcutaneous disorders | |||||
| Pruritus | 1 | 3 | 2 | 3 | 2 |
La tasa general de reacciones adversas fue similar en pacientes hombres y mujeres. Aunque hubo pocos pacientes no caucásicos, no se observaron diferencias en las incidencias de reacciones adversas en comparación con los pacientes caucásicos.
VIMPAT Tableta y Solución Oral en Pacientes Pediátricos
La seguridad de VIMPAT se evaluó en estudios clínicos de pacientes pediátricos de 1 mes a menos de 17 años de edad para el tratamiento de las convulsiones de inicio parcial. En todos los estudios en pacientes pediátricos con convulsiones de inicio parcial, 847 pacientes de 1 mes a menos de 17 años de edad recibieron solución oral o tableta de VIMPAT, de los cuales 596 recibieron VIMPAT durante al menos 1 año. Las reacciones adversas notificadas en estudios clínicos de pacientes pediátricos de 1 mes a menos de 17 años de edad fueron similares a las observadas en pacientes adultos.
Convulsiones tónico-clónicas generalizadas primarias en pacientes (de 4 años de edad o mayores) Ensayo de terapia adyuvante (Estudio 5)
En el ensayo controlado con placebo de terapia adyuvante para convulsiones tónico-clónicas generalizadas primarias, las reacciones adversas que ocurrieron en el estudio fueron generalmente similares a las que ocurrieron en los estudios controlados con placebo de convulsiones de inicio parcial. Las reacciones adversas más comunes (≥ 10% con VIMPAT) notificadas en pacientes tratados con VIMPAT fueron mareos (23%), somnolencia (17%), dolor de cabeza (14%) y náuseas (10%), en comparación con 7%, 14%, 10% y 6%, respectivamente, de los pacientes que recibieron placebo. Además, se notificó una reacción adversa no notificada previamente de epilepsia mioclónica en el 3% de los pacientes tratados con VIMPAT en comparación con el 1% de los pacientes que recibieron placebo. También se observa que 2 pacientes que recibieron VIMPAT tuvieron un empeoramiento agudo de las convulsiones poco después del inicio del medicamento, incluido un episodio de estado epiléptico, en comparación con ningún paciente que recibió placebo.
Anormalidades de laboratorio
Se han producido anormalidades en las pruebas de función hepática en ensayos controlados con VIMPAT en pacientes adultos con convulsiones de inicio parcial que estaban tomando de 1 a 3 medicamentos antiepilépticos concomitantes. Las elevaciones de ALT a ≥3× ULN ocurrieron en el 0,7% (7/935) de los pacientes con VIMPAT y el 0% (0/356) de los pacientes con placebo. Un caso de hepatitis con transaminasas >20× ULN ocurrió en un sujeto sano 10 días después de la finalización del tratamiento con VIMPAT, junto con nefritis (proteinuria y cilindros urinarios). Los estudios serológicos fueron negativos para hepatitis viral. Las transaminasas volvieron a la normalidad en un mes sin tratamiento específico. En el momento de este evento, la bilirrubina era normal. La hepatitis/nefritis se interpretó como una reacción de hipersensibilidad retardada a VIMPAT.
Otras reacciones adversas
La siguiente es una lista de reacciones adversas notificadas por pacientes tratados con VIMPAT en todos los ensayos clínicos en pacientes adultos, incluidos los ensayos controlados y los ensayos de extensión de etiqueta abierta a largo plazo. Las reacciones adversas abordadas en otras tablas o secciones no se enumeran aquí.
Trastornos de la sangre y del sistema linfático: neutropenia, anemia
Trastornos cardíacos: palpitaciones
Trastornos del oído y del laberinto: tinnitus
Trastornos gastrointestinales: estreñimiento, dispepsia, boca seca, hipoestesia oral
Trastornos generales y condiciones del lugar de administración: irritabilidad, pirexia, sensación de embriaguez
Lesiones, envenenamiento y complicaciones de procedimientos: caída
Trastornos musculoesqueléticos y del tejido conjuntivo: espasmos musculares
Trastornos del sistema nervioso: parestesia, trastorno cognitivo, hipoestesia, disartria, trastorno de la atención, síndrome cerebeloso
Trastornos psiquiátricos: estado confusional, estado de ánimo alterado, estado de ánimo deprimido
VIMPAT Inyección
Pacientes adultos (17 años o mayores)
Las reacciones adversas con la administración intravenosa a pacientes adultos con convulsiones de inicio parcial generalmente fueron similares a las que ocurrieron con la formulación oral, aunque la administración intravenosa se asoció con reacciones adversas locales como dolor o molestia en el sitio de inyección (2,5%), irritación (1%) y eritema (0,5%). Un caso de bradicardia profunda (26 lpm: PA 100/60 mmHg) ocurrió en un paciente durante una infusión de 15 minutos de 150 mg de VIMPAT. Este paciente estaba tomando un betabloqueante. La infusión se interrumpió y el paciente experimentó una recuperación rápida.
La seguridad de una dosis de carga de 15 minutos de VIMPAT Inyección 200 mg a 400 mg seguida de la administración oral de VIMPAT administrada dos veces al día a la misma dosis diaria total que la infusión intravenosa inicial se evaluó en un estudio abierto en pacientes adultos con convulsiones de inicio parcial. Los pacientes debían haber mantenido un régimen de dosis estable de 1 a 2 antiepilépticos comercializados durante al menos 28 días antes de la asignación del tratamiento. Los grupos de tratamiento fueron los siguientes:
- Dosis única de VIMPAT Inyección intravenosa 200 mg seguida de VIMPAT oral 200 mg/día (100 mg cada 12 horas)
- Dosis única de VIMPAT Inyección intravenosa 300 mg seguida de VIMPAT oral 300 mg/día (150 mg cada 12 horas)
- Dosis única de VIMPAT Inyección intravenosa 400 mg seguida de VIMPAT oral 400 mg/día (200 mg cada 12 horas).
La Tabla 5 muestra la incidencia de reacciones adversas que ocurrieron en ≥5% de los pacientes adultos en cualquier grupo de dosificación de VIMPAT.
| Reacción adversa | VIMPAT 200 mg N=25 % |
VIMPAT 300 mg N=50 % |
VIMPAT 400 mg N=25 % |
VIMPAT Total N=100 % |
|---|---|---|---|---|
| Trastornos oculares | ||||
| Diplopía | 4 | 6 | 20 | 9 |
| Visión borrosa | 0 | 4 | 12 | 5 |
| Trastornos gastrointestinales | ||||
| Náuseas | 0 | 16 | 24 | 14 |
| Boca seca | 0 | 6 | 12 | 6 |
| Vómitos | 0 | 4 | 12 | 5 |
| Parestesia oral | 4 | 4 | 8 | 5 |
| Hipoestesia oral | 0 | 6 | 8 | 5 |
| Diarrea | 0 | 8 | 0 | 4 |
| Trastornos generales y condiciones del lugar de administración | ||||
| Fatiga | 0 | 18 | 12 | 12 |
| Trastorno de la marcha | 8 | 2 | 0 | 3 |
| Dolor de pecho | 0 | 0 | 12 | 3 |
| Trastornos del sistema nervioso | ||||
| Mareos | 20 | 46 | 60 | 43 |
| Somnolencia | 0 | 34 | 36 | 26 |
| Dolor de cabeza | 8 | 4 | 16 | 8 |
| Parestesia | 8 | 6 | 4 | 6 |
| Temblor | 0 | 6 | 4 | 4 |
| Coordinación anormal | 0 | 6 | 0 | 3 |
| Trastornos de la piel y del tejido subcutáneo | ||||
| Prurito | 0 | 6 | 4 | 4 |
| Hiperhidrosis | 0 | 0 | 8 | 2 |
Pacientes pediátricos (de 1 mes a menos de 17 años de edad)
La seguridad de la inyección de VIMPAT se evaluó en un estudio multicéntrico, abierto, de 103 pacientes pediátricos de 1 mes a menos de 17 años de edad con epilepsia. Las infusiones se administraron principalmente durante un período de 30 a 60 minutos; los tiempos de infusión inferiores a 30 minutos no se estudiaron adecuadamente en pacientes pediátricos [ver Dosificación y administración (2.7)]. Aunque no se observaron reacciones adversas graves o severas en el momento de la infusión en este pequeño estudio, se espera que las reacciones adversas asociadas con la inyección de VIMPAT en pacientes pediátricos sean similares a las observadas en adultos.
En un estudio de cohorte retrospectivo de registros electrónicos de atención médica de 686 pacientes pediátricos que comenzaron VIMPAT con dosificación intravenosa, la incidencia de erupción cutánea dentro de los 30 días de la interrupción de VIMPAT intravenoso fue dos veces mayor en la cohorte de pacientes que iniciaron con el régimen de dosificación inicial alternativo para lograr la dosis de mantenimiento en un período de tiempo más corto [ver Dosificación y administración (2.2)] en comparación con la cohorte de pacientes que iniciaron con un régimen de dosificación inicial más bajo [ver Dosificación y administración (2.1)].
6.2 Experiencia postcomercialización
Las siguientes reacciones adversas se han identificado durante el uso posterior a la aprobación de VIMPAT. Debido a que estas reacciones se informan voluntariamente de una población de tamaño incierto, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal con la exposición al fármaco.
Trastornos de la sangre y del sistema linfático: Agranulocitosis
Trastornos psiquiátricos: Agresión, agitación, alucinación, insomnio, trastorno psicótico
Trastornos de la piel y del tejido subcutáneo: Angioedema, erupción cutánea, urticaria, síndrome de Stevens-Johnson, necrólisis epidérmica tóxica.
Trastornos neurológicos: Disquinesia, convulsiones nuevas o que empeoran
7 INTERACCIONES MEDICAMENTOSAS
7.1 Inhibidores fuertes de CYP3A4 o CYP2C9
Los pacientes con insuficiencia renal o hepática que están tomando inhibidores fuertes de CYP3A4 y CYP2C9 pueden tener un aumento significativo en la exposición a VIMPAT. Puede ser necesaria la reducción de la dosis en estos pacientes.
7.2 Medicamentos concomitantes que afectan la conducción cardíaca
VIMPAT debe usarse con precaución en pacientes que toman medicamentos concomitantes que afectan la conducción cardíaca (bloqueadores de los canales de sodio, betabloqueantes, bloqueadores de los canales de calcio, bloqueadores de los canales de potasio), incluidos aquellos que prolongan el intervalo PR (incluidos los AED bloqueadores de los canales de sodio), debido a un riesgo de bloqueo AV, bradicardia o taquiarritmia ventricular. En tales pacientes, se recomienda obtener un ECG antes de comenzar VIMPAT y después de que VIMPAT se titule a un estado estable. Además, estos pacientes deben ser monitoreados de cerca si se les administra VIMPAT por vía intravenosa [ver Advertencias y precauciones (5.3)].
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Registro de Exposición al Embarazo
Existe un registro de exposición al embarazo que monitorea los resultados del embarazo en mujeres expuestas a fármacos antiepilépticos (FAE), como VIMPAT, durante el embarazo. Anime a las mujeres que toman VIMPAT durante el embarazo a que se inscriban en el registro de embarazo de la North American Antiepileptic Drug (NAAED) llamando al 1-888-233-2334 o visitando http://www.aedpregnancyregistry.org/.
Resumen de Riesgos
Los datos disponibles del registro de embarazo de la North American Antiepileptic Drug (NAAED), un estudio de cohorte prospectivo, informes de casos y una serie de casos con el uso de VIMPAT en mujeres embarazadas son insuficientes para identificar un riesgo asociado al fármaco de defectos de nacimiento importantes, aborto espontáneo u otros resultados adversos maternos o fetales. La lacosamida produjo toxicidad del desarrollo (aumento de la mortalidad embrionaria y perinatal, déficit de crecimiento) en ratas después de la administración durante el embarazo. Se observó neurotoxicidad del desarrollo en ratas después de la administración durante un período de desarrollo postnatal correspondiente al tercer trimestre del embarazo humano. Estos efectos se observaron a dosis asociadas con exposiciones plasmáticas clínicamente relevantes (ver Datos).
El riesgo de fondo de defectos de nacimiento importantes y aborto espontáneo para la población indicada es desconocido. Todos los embarazos tienen un riesgo de fondo de defectos de nacimiento, pérdida u otros resultados adversos. En la población general de los EE. UU., el riesgo de fondo estimado de defectos de nacimiento importantes y aborto espontáneo en embarazos clínicamente reconocidos es del 2-4% y del 15-20%, respectivamente.
Datos
Datos en Animales
La administración oral de lacosamida a ratas embarazadas (20, 75 o 200 mg/kg/día) y conejos (6,25, 12,5 o 25 mg/kg/día) durante el período de organogénesis no produjo ningún efecto en la incidencia de anomalías estructurales fetales. Sin embargo, las dosis máximas evaluadas se vieron limitadas por la toxicidad materna en ambas especies y la muerte embrionaria fetal en ratas. Estas dosis se asociaron con exposiciones plasmáticas maternas de lacosamida (AUC) aproximadamente 2 y 1 veces (rata y conejo, respectivamente) que en humanos a la dosis humana máxima recomendada (MRHD) de 400 mg/día.
En dos estudios en los que se administró lacosamida (25, 70 o 200 mg/kg/día y 50, 100 o 200 mg/kg/día) por vía oral a ratas durante todo el embarazo y la lactancia, se observó un aumento de la mortalidad perinatal y una disminución del peso corporal en la descendencia a la dosis más alta probada. La dosis sin efecto para la toxicidad del desarrollo pre y postnatal en ratas (70 mg/kg/día) se asoció con un AUC plasmático materno de lacosamida similar al de los humanos a la MRHD.
La administración oral de lacosamida (30, 90 o 180 mg/kg/día) a ratas durante los períodos neonatal y juvenil de desarrollo resultó en una disminución del peso cerebral y cambios neuroconductuales a largo plazo (rendimiento alterado en campo abierto, déficits en el aprendizaje y la memoria). Generalmente se considera que el período postnatal temprano en ratas corresponde al embarazo tardío en humanos en términos de desarrollo cerebral. La dosis sin efecto para la neurotoxicidad del desarrollo en ratas se asoció con un AUC plasmático de lacosamida menor que el de los humanos a la MRHD.
Datos In Vitro
Se ha demostrado que la lacosamida in vitro interfiere con la actividad de la proteína mediadora de la respuesta al colapso-2 (CRMP-2), una proteína involucrada en la diferenciación neuronal y el control del crecimiento axonal. No se pueden descartar posibles efectos adversos en el desarrollo del SNC relacionados con esta actividad.
8.2 Lactancia
Resumen de Riesgos
Los datos de la literatura publicada indican que la lacosamida está presente en la leche materna. Hay informes de aumento de la somnolencia en bebés amamantados expuestos a la lacosamida (ver Consideraciones Clínicas). No hay información sobre los efectos de la lacosamida en la producción de leche.
Los beneficios para el desarrollo y la salud de la lactancia materna deben considerarse junto con la necesidad clínica de la madre de VIMPAT y cualquier posible efecto adverso en el lactante amamantado por VIMPAT o por la condición materna subyacente.
8.4 Uso Pediátrico
Convulsiones de Inicio Parcial
La seguridad y eficacia de VIMPAT para el tratamiento de las convulsiones de inicio parcial se han establecido en pacientes pediátricos de 1 mes a menos de 17 años de edad. El uso de VIMPAT en este grupo de edad está respaldado por evidencia de estudios adecuados y bien controlados de VIMPAT en adultos con convulsiones de inicio parcial, datos farmacocinéticos de pacientes adultos y pediátricos, y datos de seguridad en 847 pacientes pediátricos de 1 mes a menos de 17 años de edad [ver Reacciones Adversas (6.1), Farmacología Clínica (12.3), y Estudios Clínicos (14.1, 14.2)].
La seguridad y eficacia en pacientes pediátricos menores de 1 mes de edad no se han establecido.
Convulsiones Tónico-Clónicas Generalizadas Primarias
La seguridad y eficacia de VIMPAT como terapia adyuvante en el tratamiento de las convulsiones tónico-clónicas generalizadas primarias en pacientes pediátricos con epilepsia generalizada idiopática de 4 años de edad o más se estableció en un estudio multicéntrico, aleatorizado, doble ciego, controlado con placebo, de grupos paralelos de 24 semanas (Estudio 5), que incluyó a 37 pacientes pediátricos de 4 años a menos de 17 años de edad [ver Reacciones adversas (6.1) y Estudios clínicos (14.3)].
No se ha establecido la seguridad y eficacia en pacientes pediátricos menores de 4 años.
Datos en animales
Se ha demostrado que la lacosamida in vitro interfiere con la actividad de la proteína mediadora de la respuesta al colapso-2 (CRMP-2), una proteína involucrada en la diferenciación neuronal y el control del crecimiento axonal. No se pueden descartar posibles efectos adversos relacionados con el desarrollo del SNC. La administración de lacosamida a ratas durante los períodos neonatal y juvenil de desarrollo postnatal (aproximadamente equivalente al desarrollo neonatal hasta la adolescencia en humanos) resultó en una disminución del peso cerebral y cambios neuroconductuales a largo plazo (rendimiento alterado en campo abierto, déficits en el aprendizaje y la memoria). La dosis sin efecto para la neurotoxicidad del desarrollo en ratas se asoció con una exposición plasmática a la lacosamida (AUC) menor que la de los humanos a la dosis máxima recomendada en humanos de 400 mg/día.
8.5 Uso en geriatría
No hubo un número suficiente de pacientes de edad avanzada inscritos en los ensayos de convulsiones de inicio parcial (n=18) para determinar adecuadamente si responden de manera diferente a los pacientes más jóvenes.
No es necesario ningún ajuste de la dosis de VIMPAT en función de la edad. En pacientes de edad avanzada, la titulación de la dosis debe realizarse con precaución, generalmente comenzando en el extremo inferior del rango de dosificación, lo que refleja la mayor frecuencia de disminución de la función hepática, disminución de la función renal, aumento de las anomalías de la conducción cardíaca y polifarmacia [ver Dosificación y administración (2.1, 2.4, 2.5) y Farmacología clínica (12.3)].
8.6 Insuficiencia renal
No es necesario ningún ajuste de la dosis en pacientes con insuficiencia renal leve a moderada (CLCR ≥30 mL/min). En pacientes con insuficiencia renal grave (CLCR <30 mL/min según lo estimado por la ecuación de Cockcroft-Gault para adultos; CLCR <30 mL/min/1.73m2 según lo estimado por la ecuación de Schwartz para pacientes pediátricos) y en aquellos con enfermedad renal en etapa terminal, se recomienda una reducción del 25% de la dosis máxima [ver Dosificación y administración (2.4) y Farmacología clínica (12.3)].
En todos los pacientes con insuficiencia renal, el inicio y la titulación de la dosis deben basarse en la respuesta clínica y la tolerabilidad.
VIMPAT se elimina eficazmente del plasma mediante hemodiálisis. Se debe considerar la suplementación de la dosis hasta un 50% después de la hemodiálisis.
8.7 Insuficiencia hepática
Para pacientes adultos y pediátricos con insuficiencia hepática leve a moderada, se recomienda una reducción del 25% de la dosis máxima. Los pacientes con insuficiencia hepática leve a moderada deben ser observados de cerca para detectar reacciones adversas, y el inicio y la titulación de la dosis deben basarse en la respuesta clínica y la tolerabilidad [ver Dosificación y administración (2.5), Farmacología clínica (12.3)].
La farmacocinética de la lacosamida no se ha evaluado en insuficiencia hepática grave. No se recomienda el uso de VIMPAT en pacientes con insuficiencia hepática grave.
9 ABUSO DE DROGAS Y DEPENDENCIA
9.2 Abuso
El abuso es el uso intencional y no terapéutico de un fármaco, incluso una sola vez, por sus efectos psicológicos o fisiológicos deseables. En un estudio de potencial de abuso en humanos, dosis únicas de 200 mg (equivalente a la dosis única máxima) y 800 mg de lacosamida (equivalente al doble de la dosis de mantenimiento diaria recomendada) produjeron respuestas subjetivas de tipo euforia que se diferenciaron estadísticamente del placebo; a 800 mg, estas respuestas de tipo euforia fueron estadísticamente indistinguibles de las producidas por alprazolam, un fármaco de la Lista IV. La duración de las respuestas de tipo euforia después de la lacosamida fue menor que la que siguió al alprazolam. También se informó una alta tasa de euforia como evento adverso en el estudio de potencial de abuso en humanos después de dosis únicas de 800 mg de lacosamida (15% [5/34]) en comparación con el placebo (0%) y en dos estudios farmacocinéticos después de dosis únicas y múltiples de 300-800 mg de lacosamida (que van del 6% [2/33] al 25% [3/12]) en comparación con el placebo (0%). Sin embargo, la tasa de euforia informada como evento adverso en el programa de desarrollo de VIMPAT a dosis terapéuticas fue inferior al 1%.
9.3 Dependencia
La dependencia física es un estado que se desarrolla como resultado de la adaptación fisiológica en respuesta al uso repetido de drogas, que se manifiesta por signos y síntomas de abstinencia después de la interrupción abrupta o una reducción significativa de la dosis de un fármaco. La interrupción abrupta de la lacosamida en los ensayos clínicos con pacientes con dolor neuropático diabético no produjo signos o síntomas asociados con un síndrome de abstinencia indicativo de dependencia física. Sin embargo, la dependencia psicológica no puede excluirse debido a la capacidad de la lacosamida para producir eventos adversos de tipo euforia en humanos.
10 SOBREDOSIS
Los eventos reportados después de la ingesta de más de 800 mg (el doble de la dosis diaria máxima recomendada) de VIMPAT incluyen mareos, náuseas y convulsiones (convulsiones tónico-clónicas generalizadas, estado epiléptico). También se han observado trastornos de la conducción cardíaca, confusión, disminución del nivel de conciencia, shock cardiogénico, paro cardíaco y coma. Se han producido muertes tras sobredosis de lacosamida de varios gramos.
No existe un antídoto específico para la sobredosis de VIMPAT. Se deben seguir los procedimientos estándar de descontaminación. Se indica el cuidado de apoyo general del paciente, incluida la monitorización de los signos vitales y la observación del estado clínico del paciente. Se debe contactar con un Centro de Control de Envenenamiento Certificado para obtener información actualizada sobre el manejo de la sobredosis de VIMPAT.
Los procedimientos estándar de hemodiálisis dan como resultado una eliminación significativa de VIMPAT (reducción de la exposición sistémica en un 50% en 4 horas). La hemodiálisis puede estar indicada en función del estado clínico del paciente o en pacientes con insuficiencia renal significativa.
11 DESCRIPCIÓN
El nombre químico de la lacosamida, el enantiómero (R) único, es (R)-2-acetamido-N-benzil-3-metoxipropionamida (IUPAC). La lacosamida es un aminoácido funcionalizado. Su fórmula molecular es C13H18N2O3 y su peso molecular es 250.30. La estructura química es:
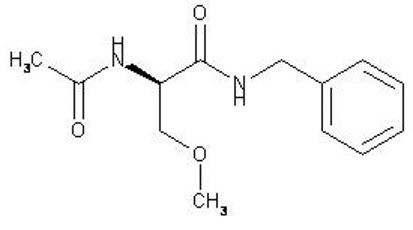
La lacosamida es un polvo blanco a amarillo claro. Es poco soluble en agua y ligeramente soluble en acetonitrilo y etanol.
11.1 Comprimidos VIMPAT
Los comprimidos VIMPAT para administración oral contienen lacosamida y los siguientes ingredientes inactivos: dióxido de silicio coloidal, crospovidona, hidroxipropilcelulosa, estearato de magnesio, celulosa microcristalina, polietilenglicol, alcohol polivinílico, talco, dióxido de titanio y pigmentos de color especificados a continuación:
Los comprimidos VIMPAT se suministran como comprimidos grabados y contienen los siguientes agentes colorantes:
Comprimidos de 50 mg: óxido de hierro rojo, óxido de hierro negro, FD&C Blue #2/azul índigo carmín lago de aluminio
Comprimidos de 100 mg: óxido de hierro amarillo
Comprimidos de 150 mg: óxido de hierro amarillo, óxido de hierro rojo, óxido de hierro negro
Comprimidos de 200 mg: FD&C Blue #2/azul índigo carmín lago de aluminio
11.2 Inyección VIMPAT
La inyección VIMPAT es una solución estéril, clara e incolora que contiene 10 mg de lacosamida por mL para perfusión intravenosa. Un vial de 20 mL contiene 200 mg de sustancia farmacológica de lacosamida. Los ingredientes inactivos son cloruro de sodio (7.62 mg/mL) y agua para inyección. El ácido clorhídrico se utiliza para el ajuste del pH. La inyección VIMPAT tiene un pH de 3.8 a 5.0.
11.3 Solución oral VIMPAT
La solución oral VIMPAT contiene 10 mg de lacosamida por mL. Los ingredientes inactivos son agua purificada, solución de sorbitol, glicerina, polietilenglicol, carboximetilcelulosa sódica, acesulfamo potásico, metilparabeno, saborizante (incluyendo sabores naturales y artificiales, propilenglicol, aspartamo y maltol), ácido cítrico anhidro y cloruro de sodio.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
El mecanismo preciso por el cual VIMPAT ejerce sus efectos antiepilépticos en humanos aún no se ha dilucidado por completo. Los estudios electrofisiológicos in vitro han demostrado que la lacosamida mejora selectivamente la inactivación lenta de los canales de sodio dependientes de voltaje, lo que da como resultado la estabilización de las membranas neuronales hiperexcitables y la inhibición del disparo neuronal repetitivo.
12.2 Farmacodinamia
Se realizó un análisis farmacocinético-farmacodinámico (eficacia) basado en los datos agrupados de los 3 ensayos de eficacia para las convulsiones de inicio parcial. La exposición a la lacosamida se correlaciona con la reducción de la frecuencia de las convulsiones. Sin embargo, las dosis superiores a 400 mg/día no parecen conferir un beneficio adicional en los análisis de grupo.
Electrofisiología cardíaca
Los efectos electrocardiográficos de VIMPAT se determinaron en un ensayo de farmacología clínica aleatorizado, doble ciego, de 247 sujetos sanos. Las dosis orales crónicas de 400 y 800 mg/día (equivalentes a la dosis diaria máxima recomendada y dos veces la dosis diaria máxima recomendada, respectivamente) se compararon con placebo y un control positivo (400 mg de moxifloxacina). VIMPAT no prolongó el intervalo QTc y no tuvo un efecto relacionado con la dosis o clínicamente importante en la duración del QRS. VIMPAT produjo un pequeño aumento relacionado con la dosis en el intervalo PR medio. En estado estacionario, el tiempo del intervalo PR medio máximo observado correspondió con tmax. El aumento máximo del intervalo PR (en tmax) restado del placebo fue de 7,3 ms para el grupo de 400 mg/día y de 11,9 ms para el grupo de 800 mg/día. Para los pacientes que participaron en los ensayos controlados, el aumento máximo medio del intervalo PR restado del placebo para una dosis de VIMPAT de 400 mg/día fue de 3,1 ms en pacientes con convulsiones de inicio parcial y de 9,4 ms para pacientes con neuropatía diabética.
12.3 Farmacocinética
La farmacocinética de VIMPAT se ha estudiado en sujetos adultos sanos (rango de edad de 18 a 87 años), adultos con convulsiones de inicio parcial, adultos con neuropatía diabética y sujetos con insuficiencia renal y hepática.
La farmacocinética de VIMPAT es similar en sujetos sanos, pacientes con convulsiones de inicio parcial y pacientes con convulsiones tónico-clónicas generalizadas primarias.
VIMPAT se absorbe completamente después de la administración oral con un efecto de primer paso insignificante con una alta biodisponibilidad absoluta de aproximadamente el 100%. Las concentraciones plasmáticas máximas de lacosamida se producen aproximadamente de 1 a 4 horas después de la dosis después de la administración oral, y la vida media de eliminación es de aproximadamente 13 horas. Las concentraciones plasmáticas en estado estacionario se alcanzan después de 3 días de administración repetida dos veces al día. La farmacocinética de VIMPAT es proporcional a la dosis (100-800 mg) e invariante en el tiempo, con baja variabilidad inter e intraindividual. En comparación con la lacosamida, el metabolito principal, el metabolito O-desmetilado, tiene un Tmax más largo (0,5 a 12 horas) y una vida media de eliminación (15-23 horas).
Absorción y biodisponibilidad
VIMPAT se absorbe completamente después de la administración oral. La biodisponibilidad oral de las tabletas de VIMPAT es de aproximadamente el 100%. Los alimentos no afectan la velocidad y el grado de absorción.
Después de la administración intravenosa, Cmax se alcanza al final de la infusión. Las infusiones intravenosas de 30 y 60 minutos son bioequivalentes a la tableta oral. Para la infusión intravenosa de 15 minutos, se cumplió la bioequivalencia para AUC(0-tz) pero no para Cmax. La estimación puntual de Cmax fue un 20% más alta que Cmax para la tableta oral y el 90% de IC para Cmax excedió el límite superior del rango de bioequivalencia.
En un ensayo que comparó la tableta oral con una solución oral que contenía 10 mg/mL de lacosamida, se demostró la bioequivalencia entre ambas formulaciones.
Una sola dosis de carga de 200 mg se aproxima a las concentraciones en estado estacionario comparables a la administración oral de 100 mg dos veces al día.
Distribución
El volumen de distribución es de aproximadamente 0,6 L/kg y, por lo tanto, cercano al volumen del agua corporal total. VIMPAT está menos del 15% unido a las proteínas plasmáticas.
Metabolismo y eliminación
VIMPAT se elimina principalmente de la circulación sistémica por excreción renal y biotransformación.
Después de la administración oral e intravenosa de 100 mg de [14C]-lacosamida, aproximadamente el 95% de la radiactividad administrada se recuperó en la orina y menos del 0,5% en las heces. Los principales compuestos excretados fueron la lacosamida sin cambios (aproximadamente el 40% de la dosis), su metabolito O-desmetilado (aproximadamente el 30%) y una fracción polar estructuralmente desconocida (~20%). La exposición plasmática del principal metabolito humano, la O-desmetil-lacosamida, es de aproximadamente el 10% de la de la lacosamida. Este metabolito no tiene actividad farmacológica conocida.
Las isoformas CYP principalmente responsables de la formación del metabolito principal (O-desmetilado) son CYP3A4, CYP2C9 y CYP2C19. La vida media de eliminación del fármaco sin cambios es de aproximadamente 13 horas y no se altera por diferentes dosis, administración múltiple o administración intravenosa.
No hay interconversión enantiomérica de la lacosamida.
Poblaciones específicas
Insuficiencia renal
La lacosamida y su principal metabolito se eliminan de la circulación sistémica principalmente por excreción renal.
El AUC de VIMPAT aumentó aproximadamente un 25 % en pacientes con insuficiencia renal leve (CLCR 50-80 mL/min) y moderada (CLCR 30-50 mL/min) y un 60 % en pacientes con insuficiencia renal grave (CLCR≤30 mL/min) en comparación con sujetos con función renal normal (CLCR>80 mL/min), mientras que Cmax no se vio afectada. VIMPAT se elimina eficazmente del plasma mediante hemodiálisis. Después de un tratamiento de hemodiálisis de 4 horas, el AUC de VIMPAT se reduce aproximadamente en un 50% [véase Dosis y administración (2.4)].
Insuficiencia hepática
Lacosamida se metaboliza. Los sujetos con insuficiencia hepática moderada (Child-Pugh B) mostraron concentraciones plasmáticas más altas de lacosamida (aproximadamente un 50-60 % más alto AUC en comparación con sujetos sanos). La farmacocinética de la lacosamida no se ha evaluado en insuficiencia hepática grave [véase Dosis y administración (2.5)].
Pacientes pediátricos (1 mes a menos de 17 años de edad)
Se realizó un estudio multicéntrico, doble ciego, aleatorizado, controlado con placebo, en grupo paralelo con un período de titulación de 20 días y un período de mantenimiento de 7 días utilizando solución oral de VIMPAT (8mg/kg/día a 12mg/kg/día) en 255 (128 fueron aleatorizados a VIMPAT y 127 fueron aleatorizados a placebo) pacientes pediátricos con epilepsia de 1 mes a menos de 4 años de edad con crisis parciales de inicio no controladas. El perfil farmacocinético pediátrico de VIMPAT se determinó en un análisis farmacocinético poblacional utilizando datos de concentración plasmática escasos obtenidos en seis estudios controlados con placebo y cinco estudios de etiqueta abierta en 1655 pacientes adultos y pediátricos con epilepsia de 1 mes a menos de 17 años que recibieron formulaciones intravenosas, solución oral o comprimidos orales.
Se necesita un régimen de dosificación basado en el peso para lograr exposiciones de lacosamida en pacientes pediátricos de 1 mes a menos de 17 años de edad similares a las observadas en adultos tratados con dosis eficaces de VIMPAT [véase Dosis y administración (2.1)]. Para pacientes que pesan 10 kg, 28,9 kg (el peso corporal promedio de la población) y 70 kg, la vida media plasmática típica (t1/2) es de 7,2 horas, 10,6 horas y 14,8 horas, respectivamente. Las concentraciones plasmáticas en estado estacionario se alcanzan después de 3 días de administración repetida dos veces al día.
La farmacocinética de VIMPAT en pacientes pediátricos es similar cuando se usa como monoterapia o como terapia adyuvante para el tratamiento de crisis parciales de inicio y como terapia adyuvante para el tratamiento de crisis tónico-clónicas generalizadas primarias.
Pacientes geriátricos
En los ancianos (>65 años), el AUC y Cmax normalizados por dosis y peso corporal aumentan aproximadamente un 20 % en comparación con sujetos jóvenes (18-64 años). Esto puede estar relacionado con el peso corporal y la disminución de la función renal en los sujetos ancianos.
Género
Los ensayos clínicos de VIMPAT indican que el género no tiene una influencia clínicamente relevante en la farmacocinética de VIMPAT.
Raza
No hay diferencias clínicamente relevantes en la farmacocinética de VIMPAT entre sujetos asiáticos, negros y caucásicos.
Polimorfismo CYP2C19
No hay diferencias clínicamente relevantes en la farmacocinética de VIMPAT entre metabolizadores pobres y extensos de CYP2C19. Los resultados de un ensayo en metabolizadores pobres (PM) (N=4) y metabolizadores extensos (EM) (N=8) del citocromo P450 (CYP) 2C19 mostraron que las concentraciones plasmáticas de lacosamida fueron similares en PM y EM, pero las concentraciones plasmáticas y la cantidad excretada en la orina del metabolito O-desmetilado se redujeron aproximadamente en un 70 % en PM en comparación con EM.
Interacciones medicamentosas
Evaluación in vitro de interacciones medicamentosas
Los estudios de metabolismo in vitro indican que lacosamida no induce la actividad enzimática de las isoformas del citocromo P450 que metabolizan medicamentos CYP1A2, 2B6, 2C9, 2C19 y 3A4. Lacosamida no inhibió CYP 1A1, 1A2, 2A6, 2B6, 2C8, 2C9, 2D6, 2E1, 3A4/5 en las concentraciones plasmáticas observadas en estudios clínicos.
Los datos in vitro sugieren que lacosamida tiene el potencial de inhibir CYP2C19 en concentraciones terapéuticas. Sin embargo, un estudio in vivo con omeprazol no mostró un efecto inhibitorio en la farmacocinética de omeprazol.
Lacosamida no fue un sustrato o inhibidor de la P-glicoproteína.
Lacosamida es un sustrato de CYP3A4, CYP2C9 y CYP2C19. Los pacientes con insuficiencia renal o hepática que están tomando potentes inhibidores de CYP3A4 y CYP2C9 pueden tener una exposición aumentada a lacosamida.
Como <15% de la lacosamida se une a las proteínas plasmáticas, es poco probable una interacción clínicamente relevante con otros fármacos a través de la competencia por los sitios de unión de proteínas.
Evaluación in vivo de interacciones medicamentosas
- Estudios de interacción medicamentosa con AED
-
Efecto de VIMPAT en AED concomitantes
VIMPAT 400 mg/día no tuvo influencia en la farmacocinética de 600 mg/día de ácido valproico y 400 mg/día de carbamazepina en sujetos sanos.
Los estudios clínicos controlados con placebo en pacientes con convulsiones de inicio parcial demostraron que las concentraciones plasmáticas en estado estacionario de levetiracetam, carbamazepina, epóxido de carbamazepina, lamotrigina, topiramato, derivado monohidroxilado (MHD) de oxcarbazepina, fenitoína, ácido valproico, fenobarbital, gabapentina, clonazepam y zonisamida no se vieron afectadas por la ingesta concomitante de VIMPAT a ninguna dosis. -
Efecto de los AED concomitantes en VIMPAT
Los estudios de interacción fármaco-fármaco en sujetos sanos demostraron que 600 mg/día de ácido valproico no tuvieron influencia en la farmacocinética de 400 mg/día de VIMPAT. Del mismo modo, 400 mg/día de carbamazepina no tuvieron influencia en la farmacocinética de VIMPAT en un estudio en sujetos sanos. Los resultados de la farmacocinética poblacional en pacientes con convulsiones de inicio parcial mostraron pequeñas reducciones (15% a 20% menos) en las concentraciones plasmáticas de lacosamida cuando VIMPAT se administró conjuntamente con carbamazepina, fenobarbital o fenitoína.
-
Efecto de VIMPAT en AED concomitantes
- Estudios de interacción medicamentosa con otros medicamentos
-
Digoxina
No hubo efecto de VIMPAT (400 mg/día) en la farmacocinética de digoxina (0,5 mg una vez al día) en un estudio en sujetos sanos. -
Metformina
No hubo cambios clínicamente relevantes en los niveles de metformina después de la coadministración de VIMPAT (400 mg/día).
La metformina (500 mg tres veces al día) no tuvo efecto en la farmacocinética de VIMPAT (400 mg/día). -
Omeprazol
El omeprazol es un sustrato e inhibidor del CYP2C19.
No hubo efecto de VIMPAT (600 mg/día) en la farmacocinética de omeprazol (40 mg dosis única) en sujetos sanos. Los datos indicaron que la lacosamida tuvo poco efecto inhibidor o inductor in vivo en el CYP2C19.
El omeprazol a una dosis de 40 mg una vez al día no tuvo efecto en la farmacocinética de VIMPAT (300 mg dosis única). Sin embargo, los niveles plasmáticos del metabolito O-desmetil se redujeron aproximadamente un 60% en presencia de omeprazol. -
Midazolam
El midazolam es un sustrato del 3A4.
No hubo efecto de VIMPAT (200 mg dosis única o dosis repetidas de 400 mg/día administradas como 200 mg BID) en la farmacocinética de midazolam (dosis única, 7,5 mg), lo que indica que no hay efectos inhibidores o inductores en el CYP3A4. -
Anticonceptivos orales
No hubo influencia de VIMPAT (400 mg/día) en la farmacodinámica y farmacocinética de un anticonceptivo oral que contenía 0,03 mg de etinilestradiol y 0,15 mg de levonorgestrel en sujetos sanos, excepto que se observó un aumento del 20% en la Cmax de etinilestradiol. -
Warfarina
La coadministración de VIMPAT (400 mg/día) con warfarina (25 mg dosis única) no provocó un cambio clínicamente relevante en los efectos farmacocinéticos y farmacodinámicos de la warfarina en un estudio en sujetos masculinos sanos.
-
Digoxina
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
Carcinogénesis
No hubo evidencia de carcinogenicidad relacionada con el fármaco en ratones o ratas. Los ratones y las ratas recibieron lacosamida una vez al día por administración oral durante 104 semanas a dosis que produjeron exposiciones plasmáticas (AUC) hasta aproximadamente 1 y 3 veces, respectivamente, el AUC plasmático en humanos a la dosis humana máxima recomendada (MRHD) de 400 mg/día.
14 ESTUDIOS CLÍNICOS
14.1 Monoterapia en pacientes con convulsiones de inicio parcial
La eficacia de VIMPAT en monoterapia se estableció en un ensayo multicéntrico, aleatorizado, con control histórico que incluyó a 425 pacientes, de 16 a 70 años, con convulsiones de inicio parcial (Estudio 1). Para ser incluidos en el Estudio 1, los pacientes debían estar tomando dosis estables de 1 o 2 medicamentos antiepilépticos comercializados. Este tratamiento continuó durante el período de referencia de 8 semanas. Para permanecer en el estudio, los pacientes debían tener al menos 2 convulsiones de inicio parcial por 28 días durante el período de referencia de 8 semanas. El período de referencia fue seguido por un período de titulación de 3 semanas, durante el cual se agregó VIMPAT al régimen antiepiléptico en curso. Esto fue seguido por un período de mantenimiento de 16 semanas (es decir, un período de retiro de 6 semanas para los medicamentos antiepilépticos de fondo, seguido de un período de monoterapia de 10 semanas). Los pacientes fueron aleatorizados 3 a 1 para recibir VIMPAT 400 mg/día o VIMPAT 300 mg/día. Las asignaciones de tratamiento fueron ciegas. La respuesta al tratamiento se basó en una comparación del número de pacientes que cumplieron con los criterios de salida durante la fase de mantenimiento, en comparación con los controles históricos. El control histórico consistió en un análisis agrupado de los grupos de control de 8 estudios de diseño similar, que utilizaron una dosis sub-terapéutica de un medicamento antiepiléptico. Se consideró que la superioridad estadística al control histórico se demostró si el límite superior de un intervalo de confianza del 95% de dos caras para el porcentaje de pacientes que cumplieron con los criterios de salida en pacientes que recibieron VIMPAT permaneció por debajo del límite de predicción del 95% inferior del 65% derivado de los datos de control histórico.
Los criterios de salida fueron uno o más de los siguientes: (1) duplicación de la frecuencia promedio de convulsiones mensuales durante cualquier período de 28 días consecutivos, (2) duplicación de la frecuencia más alta de convulsiones de 2 días consecutivos, (3) aparición de una sola convulsión tónico-clónica generalizada, (4) prolongación clínicamente significativa o empeoramiento de la duración general de las convulsiones, frecuencia, tipo o patrón considerado por el investigador que requiera la interrupción del ensayo, (5) estado epiléptico o aparición de nuevas convulsiones en serie/racimo. El perfil de la población del estudio pareció comparable al de la población de control histórico.
Para el grupo de VIMPAT 400 mg/día, la estimación del porcentaje de pacientes que cumplieron con al menos 1 criterio de salida fue del 30% (IC del 95%: 25%, 36%). El límite superior del IC del 95% de dos caras (36%) estuvo por debajo del umbral del 65% derivado de los datos de control histórico, cumpliendo con los criterios preestablecidos para la eficacia. VIMPAT 300 mg/día también cumplió con los criterios preestablecidos para la eficacia.
14.2 Terapia adyuvante en pacientes con convulsiones de inicio parcial
La eficacia de VIMPAT como terapia adyuvante en convulsiones de inicio parcial se estableció en tres ensayos multicéntricos, aleatorizados, doble ciego, controlados con placebo de 12 semanas en pacientes adultos (Estudio 2, Estudio 3 y Estudio 4). Los pacientes inscritos tenían convulsiones de inicio parcial con o sin generalización secundaria, y no estaban adecuadamente controlados con 1 a 3 AED concomitantes. Durante un período de referencia de 8 semanas, los pacientes debían tener un promedio de ≥4 convulsiones de inicio parcial por 28 días sin un período libre de convulsiones que excediera los 21 días. En estos 3 ensayos, los pacientes tenían una duración media de la epilepsia de 24 años y una frecuencia media de convulsiones de referencia que oscilaba entre 10 y 17 por 28 días. El 84% de los pacientes estaban tomando de 2 a 3 AED concomitantes con o sin estimulación concomitante del nervio vago.
El Estudio 2 comparó dosis de VIMPAT 200, 400 y 600 mg/día con placebo. El Estudio 3 comparó dosis de VIMPAT 400 y 600 mg/día con placebo. El Estudio 4 comparó dosis de VIMPAT 200 y 400 mg/día con placebo. En los tres ensayos, después de una fase de referencia de 8 semanas para establecer la frecuencia de convulsiones de referencia antes de la aleatorización, los pacientes fueron aleatorizados y titulados a la dosis aleatorizada (se permitió una retrotitulación de 1 paso de VIMPAT 100 mg/día o placebo en caso de reacciones adversas intolerables al final de la fase de titulación). Durante la fase de titulación, en los 3 ensayos de terapia adyuvante, el tratamiento se inició a 100 mg/día (50 mg dos veces al día) y se incrementó en incrementos semanales de 100 mg/día hasta la dosis objetivo. La fase de titulación duró 6 semanas en el Estudio 2 y el Estudio 3, y 4 semanas en el Estudio 4. En los tres ensayos, la fase de titulación fue seguida por una fase de mantenimiento que duró 12 semanas, durante la cual los pacientes debían permanecer en una dosis estable de VIMPAT.
Una reducción en la frecuencia de convulsiones de 28 días (de referencia a la fase de mantenimiento), en comparación con el grupo placebo, fue la variable principal en los tres ensayos de terapia adyuvante. Se observó un efecto estadísticamente significativo con el tratamiento con VIMPAT (Figura 1) a dosis de 200 mg/día (Estudio 4), 400 mg/día (Estudios 2, 3 y 4) y 600 mg/día (Estudios 2 y 3).
Las evaluaciones de subgrupos de VIMPAT no demuestran diferencias importantes en el control de las convulsiones en función del sexo o la raza, aunque los datos sobre la raza fueron limitados (alrededor del 10% de los pacientes no eran caucásicos).
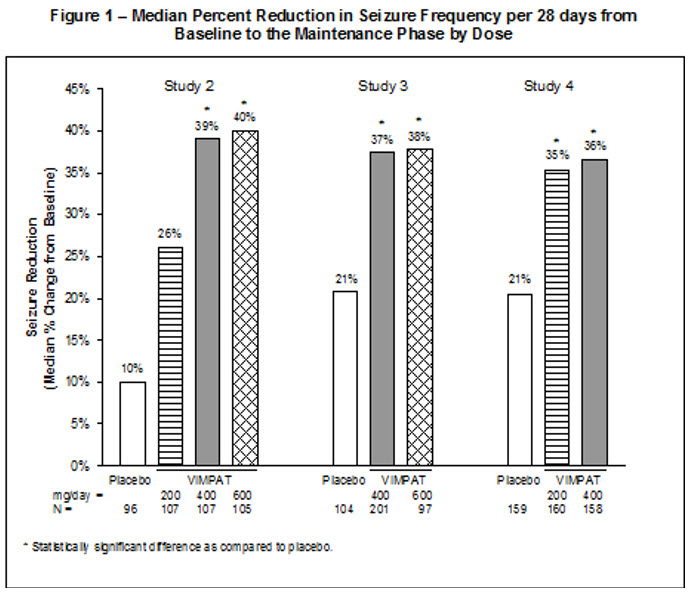
La Figura 2 presenta el porcentaje de pacientes (eje X) con una reducción porcentual en la frecuencia de convulsiones parciales (tasa de respuesta) desde la referencia hasta la fase de mantenimiento al menos tan grande como la representada en el eje Y. Un valor positivo en el eje Y indica una mejora desde la referencia (es decir, una disminución en la frecuencia de convulsiones), mientras que un valor negativo indica un empeoramiento desde la referencia (es decir, un aumento en la frecuencia de convulsiones). Por lo tanto, en una visualización de este tipo, una curva para un tratamiento efectivo se desplaza hacia la izquierda de la curva para el placebo. La proporción de pacientes que lograron cualquier nivel particular de reducción en la frecuencia de convulsiones fue consistentemente mayor para los grupos de VIMPAT, en comparación con el grupo placebo. Por ejemplo, el 40% de los pacientes aleatorizados a VIMPAT (400 mg/día) experimentaron una reducción del 50% o más en la frecuencia de convulsiones, en comparación con el 23% de los pacientes aleatorizados a placebo. Los pacientes con un aumento en la frecuencia de convulsiones >100% se representan en el eje Y como igual o mayor que -100%.
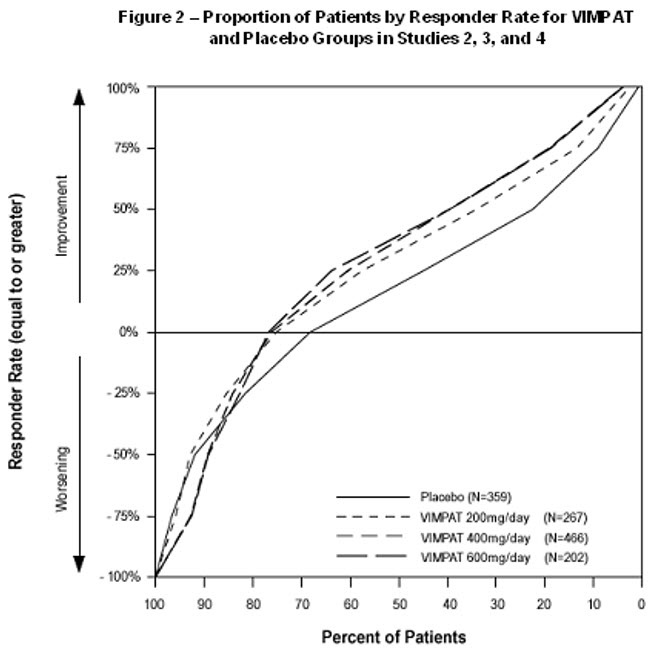
14.3 Terapia Adyuvante en Pacientes con Crisis Tonico-Clónicas Generalizadas Primarias
La eficacia de VIMPAT como terapia adyuvante en pacientes de 4 años de edad o mayores con epilepsia generalizada idiopática que experimentan crisis tonic-clónicas generalizadas primarias (PGTC) se estableció en un estudio multicéntrico, de grupos paralelos, aleatorizado, doble ciego, controlado con placebo de 24 semanas (Estudio 5). El estudio consistió en un período de referencia histórico de 12 semanas, un período de referencia prospectivo de 4 semanas y un período de tratamiento de 24 semanas (que incluyó un período de titulación de 6 semanas y un período de mantenimiento de 18 semanas). Los pacientes elegibles con una dosis estable de 1 a 3 fármacos antiepilépticos que experimentaron al menos 3 crisis PGTC documentadas durante el período de referencia combinado de 16 semanas se aleatorizaron 1:1 para recibir VIMPAT (n=121) o placebo (n=121).
Los pacientes recibieron dosis en un régimen de dosis fija. La dosificación se inició a una dosis de 2 mg/kg/día en pacientes que pesaban menos de 50 kg o 100 mg/día en pacientes que pesaban 50 kg o más en 2 dosis divididas. Durante el período de titulación, las dosis de VIMPAT se ajustaron en incrementos de 2 mg/kg/día en pacientes que pesaban menos de 50 kg o 100 mg/día en pacientes que pesaban 50 kg o más a intervalos semanales para alcanzar la dosis objetivo del período de mantenimiento de 12 mg/kg/día en pacientes que pesaban menos de 30 kg, 8 mg/kg/día en pacientes que pesaban de 30 a menos de 50 kg, o 400 mg/día en pacientes que pesaban 50 kg o más.
El criterio de valoración principal de eficacia (pacientes en el conjunto de análisis completo modificado: VIMPAT n=118, placebo n=121) fue el tiempo hasta la segunda crisis PGTC durante el período de tratamiento de 24 semanas (Figura 3). El riesgo de desarrollar una segunda crisis PGTC fue estadísticamente significativamente menor en el grupo VIMPAT que en el grupo placebo durante el período de tratamiento de 24 semanas (razón de riesgo=0.548, IC del 95% de la razón de riesgo: 0.381, 0.788, valor p = 0.001), con la correspondiente reducción del riesgo siendo del 45.2%.
El criterio de valoración secundario clave fue el porcentaje de pacientes que no experimentaron una crisis PGTC durante el período de tratamiento de 24 semanas. Las estimaciones ajustadas de Kaplan-Meier de la libertad de crisis PGTC a las 24 semanas fueron del 31.3% en el grupo VIMPAT y del 17.2% en el grupo placebo. La diferencia ajustada entre los grupos de tratamiento fue del 14.1% (IC del 95%: 3.2, 25.1, valor p=0.011).
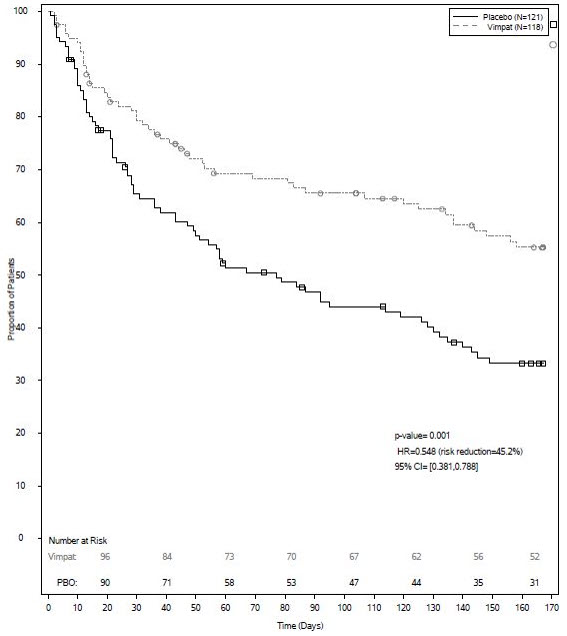 |
| Los números en la parte inferior de la figura son para los pacientes que aún están en riesgo en el estudio en un momento dado (es decir, los pacientes que continúan en el estudio sin un evento o censura antes del momento dado). |
16 FORMA DE SUMINISTRO/ALMACENAMIENTO Y MANIPULACIÓN
16.1 Cómo se suministra
VIMPAT (lacosamida) Tabletas
- 50 mg son tabletas rosadas, ovaladas, recubiertas con película, grabadas con “SP” en un lado y “50” en el otro. Se suministran de la siguiente manera:
Frascos de 60 NDC 0131-2477-35 Cartón de dosis unitaria de 60 tabletas [6 tarjetas, cada tarjeta contiene 10 tabletas] NDC 0131-2477-60 - 100 mg son tabletas ovaladas, recubiertas con película, de color amarillo oscuro, grabadas con “SP” en un lado y “100” en el otro. Se suministran de la siguiente manera:
Frascos de 60 NDC 0131-2478-35 Cartón de dosis unitaria de 60 tabletas [6 tarjetas, cada tarjeta contiene 10 tabletas] NDC 0131-2478-60 - 150 mg son tabletas ovaladas, recubiertas con película, de color salmón, grabadas con “SP” en un lado y “150” en el otro. Se suministran de la siguiente manera:
Frascos de 60 NDC 0131-2479-35 Cartón de dosis unitaria de 60 tabletas [6 tarjetas, cada tarjeta contiene 10 tabletas] NDC 0131-2479-60 - 200 mg son tabletas ovaladas, recubiertas con película, de color azul, grabadas con “SP” en un lado y “200” en el otro. Se suministran de la siguiente manera:
Frascos de 60 NDC 0131-2480-35 Cartón de dosis unitaria de 60 tabletas [6 tarjetas, cada tarjeta contiene 10 tabletas] NDC 0131-2480-60
16.2 Almacenamiento y manipulación
Almacenar a 20°C a 25°C (68°F a 77°F); se permiten excursiones entre 15°C a 30°C (59°F a 86°F). [Ver Temperatura ambiente controlada USP]
No congelar la inyección o la solución oral de VIMPAT. Deseche cualquier solución oral de VIMPAT no utilizada que quede después de seis (6) meses de la primera apertura del frasco.
17 INFORMACIÓN PARA EL PACIENTE
Aconseje al paciente o cuidador que lea el etiquetado del paciente aprobado por la FDA (Guía de medicamentos). La Guía de medicamentos acompaña al producto y también se puede acceder en www.vimpat.com o llamando al 1-844-599-2273.
Pensamientos y comportamiento suicidas
Se debe aconsejar a los pacientes, sus cuidadores y familias que los AED, incluido VIMPAT, pueden aumentar el riesgo de pensamientos y comportamiento suicidas y se les debe aconsejar sobre la necesidad de estar alerta ante la aparición o el empeoramiento de los síntomas de depresión, cualquier cambio inusual en el estado de ánimo o el comportamiento, o la aparición de pensamientos suicidas, comportamiento o pensamientos sobre autolesión. Los comportamientos preocupantes deben informarse inmediatamente a los profesionales de la salud [ver Advertencias y precauciones (5.1)].
Mareos y ataxia
Se debe aconsejar a los pacientes que el uso de VIMPAT puede causar mareos, visión doble, coordinación y equilibrio anormales y somnolencia. Se debe aconsejar a los pacientes que toman VIMPAT que no conduzcan, operen maquinaria compleja o participen en otras actividades peligrosas hasta que se hayan acostumbrado a cualquier efecto de este tipo asociado con VIMPAT [ver Advertencias y precauciones (5.2)].
Anomalías del ritmo y la conducción cardíacos
Se debe aconsejar a los pacientes que VIMPAT está asociado con cambios electrocardiográficos que pueden predisponer a latidos cardíacos irregulares y síncope. Se ha informado de paro cardíaco. Este riesgo aumenta en pacientes con enfermedad cardiovascular subyacente, con problemas de conducción cardíaca o que están tomando otros medicamentos que afectan el corazón. Se debe informar a los pacientes y deben informar a su proveedor de atención médica de inmediato cualquier signo o síntoma cardíaco. Los pacientes que desarrollen síncope deben acostarse con las piernas levantadas y comunicarse con su proveedor de atención médica [ver Advertencias y precauciones (5.3)].
Reacción medicamentosa con eosinofilia y síntomas sistémicos (DRESS)/Hipersensibilidad multiorgánica
Los pacientes deben ser conscientes de que VIMPAT puede causar reacciones de hipersensibilidad graves que afectan a múltiples órganos como el hígado y los riñones. VIMPAT debe suspenderse si se sospecha una reacción de hipersensibilidad grave. Los pacientes también deben recibir instrucciones para informar a sus médicos de inmediato cualquier síntoma de toxicidad hepática (por ejemplo, fatiga, ictericia, orina oscura) [ver Advertencias y precauciones (5.6)].
Registro de embarazo
Aconseje a los pacientes que notifiquen a su proveedor de atención médica si quedan embarazadas o tienen la intención de quedar embarazadas durante la terapia con VIMPAT. Anime a los pacientes a inscribirse en el registro de embarazo de medicamentos antiepilépticos de América del Norte (NAAED) si quedan embarazadas. Este registro está recopilando información sobre la seguridad de los AED durante el embarazo [ver Uso en poblaciones específicas (8.1)].
Lactancia
Aconseje a las mujeres lactantes que usan VIMPAT que monitoreen a los bebés en busca de exceso de somnolencia y que busquen atención médica si notan este signo [ver Uso en poblaciones específicas (8.2)].
SECCIÓN NO CLASIFICADA DE SPL
Fabricado para: UCB, Inc. Smyrna, GA 30080
VIMPAT® es una marca registrada bajo licencia de Harris FRC Corporation y está cubierta por una o más reclamaciones de la patente de EE. UU. 38,551.
©2023, UCB, Inc., Smyrna, GA 30080
Todos los derechos reservados.
Guía de medicación
PANEL PRINCIPAL DE VISUALIZACIÓN – Caja de cartón con blíster de tabletas de 50 mg
NDC 0131-2477-60
Rx only
VIMPAT®
(lacosamida) tabletas CV
50 mg
ATENCIÓN FARMACÉUTICO:
Cada paciente debe recibir
la Guía del Medicamento adjunta.
Dosis Unitaria
60 Tabletas
(6 tarjetas, cada tarjeta
contiene 10 tabletas)
Fabricado para:
UCB, Inc.
Smyrna, GA 30080
PANEL PRINCIPAL DE VISUALIZACIÓN – Caja de cartón con blíster de tabletas de 100 mg
NDC 0131-2478-60
Rx only
VIMPAT®
(lacosamide) tablets CV
100 mg
ATENCIÓN FARMACÉUTICO:
Cada paciente debe recibir
la Guía del Medicamento que se acompaña.
Dosis Unitaria
60 Tabletas
(6 tarjetas, cada tarjeta
contiene 10 tabletas)
Fabricado para:
UCB, Inc.
Smyrna, GA 30080
PANEL PRINCIPAL DE EXHIBICIÓN – Caja con blíster de tabletas de 150 mg
NDC 0131-2479-60
Rx only
VIMPAT®
(lacosamide) tablets CV
150 mg
ATENCIÓN FARMACÉUTICO:
Cada paciente debe recibir
la Guía de Medicamentos adjunta.
Dosis Unitaria
60 Tabletas
(6 tarjetas, cada tarjeta
contiene 10 tabletas)
Manufacturado para:
UCB, Inc.
Smyrna, GA 30080
PANEL PRINCIPAL DE VISUALIZACIÓN – Caja de blíster de tabletas de 200 mg
NDC 0131-2480-60
Rx only
VIMPAT®
(lacosamida) tabletas CV
200 mg
ATENCIÓN FARMACÉUTICO:
Cada paciente debe recibir
la Guía del Medicamento adjunta.
Dosis Unitaria
60 Tabletas
(6 tarjetas, cada tarjeta
contiene 10 tabletas)
Fabricado para:
UCB, Inc.
Smyrna, GA 30080
PANEL PRINCIPAL DE VISUALIZACIÓN – Vial de 20 mL en caja
NDC 0131-1810-67
Rx Only
Uso intravenoso únicamente
10 viales de dosis única
VIMPAT®
(lacosamida) inyección CV
200 mg /20 mL
(10 mg/mL)
Fabricado para:
UCB, Inc.
Smyrna, GA 30080

PANEL PRINCIPAL DE VISUALIZACIÓN – Etiqueta de botella de 200 mL – NDC 0131-5410-72
NDC 0131-5410-72
VIMPAT®
(lacosamida) solución oral CV
10 mg / mL
Rx only
ATENCIÓN FARMACÉUTICO:
Cada paciente debe recibir la
Guía del Medicamento que se acompaña.
Fabricado para:
UCB, Inc.
Smyrna, GA 30080
200 mL
